

Abstract
The N6‐methyladenosine (m6A) RNA modification serves crucial functions in RNA metabolism; however, the molecular mechanisms underlying the regulation of m6A are not well understood. Here, we establish arginine methylation of METTL14, a component of the m6A methyltransferase complex, as a novel pathway that controls m6A deposition in mammalian cells. Specifically, protein arginine methyltransferase 1 (PRMT1) interacts with, and methylates the intrinsically disordered C terminus of METTL14, which promotes its interaction with RNA substrates, enhances its RNA methylation activity, and is crucial for its interaction with RNA polymerase II (RNAPII). Mouse embryonic stem cells (mESCs) expressing arginine methylation‐deficient METTL14 exhibit significantly reduced global m6A levels. Transcriptome‐wide m6A analysis identified 1,701 METTL14 arginine methylation‐dependent m6A sites located in 1,290 genes involved in various cellular processes, including stem cell maintenance and DNA repair. These arginine methylation‐dependent m6A sites are associated with enhanced translation of genes essential for the repair of DNA interstrand crosslinks; thus, METTL14 arginine methylation‐deficient mESCs are hypersensitive to DNA crosslinking agents. Collectively, these findings reveal important aspects of m6A regulation and new functions of arginine methylation in RNA metabolism.
Keywords: arginine methylation, DNA repair, PRMT1, RGG motif, RNA m6A methylation
Subject Categories: Post-translational Modifications, Proteolysis & Proteomics; RNA Biology; Protein Biosynthesis & Quality Control
Post‐translational modification of METTL14 enhances its RNA methylation activity and promotes its interaction with RNA polymerase II and RNA substrates.
Introduction
N6‐methyladenosine (m6A) is the most abundant internal modification in cellular mRNA (Desrosiers et al, 1974; Adams & Cory, 1975; Dubin & Taylor, 1975). This chemical modification has emerged as a key regulator of mRNA metabolic processes, including transport, translation, splicing, and decay (Yue et al, 2015; Roignant & Soller, 2017; Roundtree et al, 2017). In mammals, m6A is deposited by a methyltransferase complex consisting of METTL3, METTL14, and WTAP (Liu et al, 2014; Ping et al, 2014; Wang et al, 2014b) and is actively removed from transcripts by the demethylases FTO and ALKBH5 (Jia et al, 2011; Zheng et al, 2013). The biological functions of m6A are generally carried out by its “reader” proteins, which include the YT521‐B homology (YTH) domain containing proteins YTHDF1–3 and YTHDC1–2 (Shi et al, 2019; Zaccara et al, 2019). For example, YTHDF1 and YTHDF3 recognize m6A‐modified mRNAs and promote their translation by recruiting translation initiation factors (Wang et al, 2015; Li et al, 2017), whereas YTHDF2 promotes the degradation of its target transcripts by recruiting the CCR4‐NOT deadenylase complex (Wang et al, 2014a). m6A homeostasis is crucial for normal development, and its dysregulation has been linked to the pathogenesis of many human diseases, including neurological disorders and cancer (Jaffrey & Kharas, 2017; Noack & Calegari, 2018). During early embryonic development, deposition of m6A provides an “identity” to transcripts encoding pluripotency transcription factors, such as Nanog, and promotes their expedited decay (Batista et al, 2014; Wang et al, 2014b; Geula et al, 2015). In neuronal progenitor cells, METTL14 knockout causes premature differentiation and delayed specification of neuronal subtypes (Yoon et al, 2017; Wang et al, 2018). In hematologic malignancies, METTL3 and METTL14 are both highly expressed, leading to increases in m6A and tumor cell proliferation (Barbieri et al, 2017; Vu et al, 2017; Weng et al, 2018). Additionally, reduced m6A has been shown to stabilize the mRNA levels of NANOG and KLF4, the key pluripotency factors required for the maintenance of breast cancer tumor‐initiating cells (Zhang et al, 2016). Together, these genetic knockout studies have provided valuable information in understanding the role of m6A in mRNA metabolism; however, the molecular mechanisms underlying the regulation of m6A are largely unknown.
Arginine methylation is a critical post‐translational modification (PTM) that regulates protein functions in mRNA metabolism (Bedford & Clarke, 2009; Yang & Bedford, 2013; Blanc & Richard, 2017). The human genome encodes nine protein arginine methyltransferases (PRMTs), which catalyze three types of arginine methylation: monomethylation (MMA), asymmetric dimethylation (ADMA), and symmetric dimethylation (SDMA) (Bedford & Clarke, 2009; Yang & Bedford, 2013). Proteomic studies revealed that RNA‐binding proteins form the largest group of PRMT substrates (Boisvert et al, 2003; Guo et al, 2014; Geoghegan et al, 2015), and motif analysis uncovered a conserved arginine/glycine‐rich (RGG/RG) polypeptide sequence as the preferred site for methylation (Thandapani et al, 2013; Guo et al, 2014; Geoghegan et al, 2015). These RGG/RG motifs are often located within the unstructured, intrinsically disordered regions (IDRs) of proteins and can mediate protein–protein and protein–nucleic acid interactions (Rajyaguru & Parker, 2012; Thandapani et al, 2013; Ozdilek et al, 2017; Chong et al, 2018). Arginine methylation of the RGG/RG motifs does not neutralize the cationic charge of the arginine residue but removes its potential hydrogen bond donors and imparts hydrophobicity of the protein (Tripsianes et al, 2011). Thus, arginine methylation has emerged as an important PTM that regulates the biochemical activity and the biological function of RGG/RG motif‐containing proteins.
Here, we identify PRMT1‐catalyzed arginine methylation of the RGG/RG motif‐containing IDR in the C‐terminal region of METTL14 as a novel molecular mechanism that controls m6A deposition in mammalian cells. Specifically, arginine methylation of the IDR enhances the interactions of METTL14 with RNA substrates and with RNA polymerase II (RNAPII), which are fundamental for catalyzing m6A deposition in vitro and in cells. We performed transcriptome‐wide m6A analysis in mouse embryonic stem cells and identified 1,701 arginine methylation‐dependent m6A sites located in 1,290 genes that function in various cellular processes. We focused on the DNA interstrand crosslink (ICL) repair pathway, in which the arginine methylation‐dependent m6A sites are significantly enriched, and demonstrated that these m6A sites are associated with enhanced translation of DNA repair genes. Consequently, mESCs expressing arginine methylation‐deficient METTL14 are hypersensitive to DNA crosslinking agents. Thus, our study reveals arginine methylation of METTL14 as a novel molecular mechanism underlying the regulation of m6A deposition.
Results
C‐terminal IDR of METTL14 is arginine methylated
RGG/RG motifs, located in the context of IDRs, are often in multiple copies (Thandapani et al, 2013). We found that the C terminus of METTL14 harbors an array of RGG/RG motifs, ranging from five in flies to ten in humans (Fig 1A, Appendix Fig S1). Consistent with the low complexity of these motifs, this region of METTL14 is predicted to be highly disordered (Fig EV1A and B). To determine the extent to which the C‐terminal IDR contributes to the RNA methylation activity of the METTL3/METTL14 complex, we performed in vitro RNA methylation assays and found that this region is essential for catalyzing m6A deposition in vitro (Fig EV1C), consistent with a recent report (Scholler et al, 2018). As RGG/RG motifs are the preferred methylation substrates for PRMTs (Thandapani et al, 2013), we hypothesized that arginine methylation of the C‐terminal IDR regulates the function of METTL14 in m6A RNA modification. To test whether METTL14 is arginine methylated, we performed in vitro methylation assays by incubating recombinant GST‐tagged METTL14 with PRMTs (PRMT1–8) in the presence of 3H‐labeled S‐adenosyl methionine (3H‐SAM). Among the PRMTs tested, we found that METTL14 can be methylated by PRMT1 and, to a much lesser extent, PRMT6 and PRMT3 (Fig 1B, Appendix Fig S2A). To determine whether arginine methylation occurs at the C‐terminal IDR of METTL14, we performed in vitro methylation assays with full‐length (FL) and C‐terminal IDR‐truncated mutant (1–400) METTL14. The arginine methylation of METTL14 was completely abolished after deleting the C‐terminal IDR (Fig 1C), suggesting that the RGG/RG motifs are indeed the sites of methylation.
Figure 1. METTL14 C‐terminal IDR is arginine methylated in vitro and in cells.
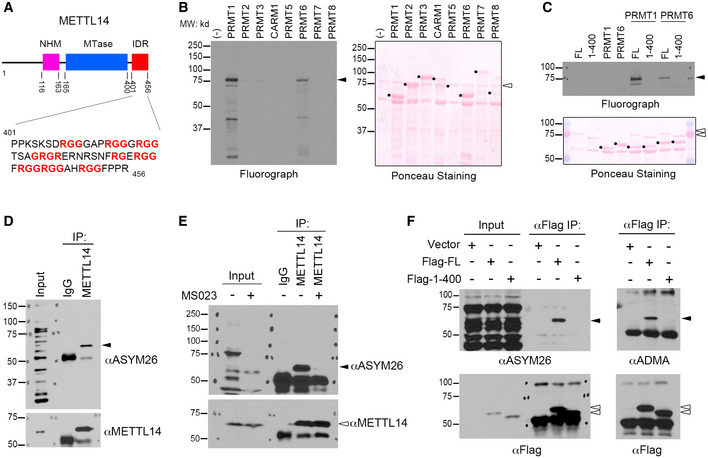
- Schematic representation of the domain structure of METTL14. The C‐terminal IDR, containing an array of RGG motifs, is highlighted. NHM: N‐terminal α‐helical motif; MTase: Methyltransferase domain; IDR: Intrinsically disordered region.
- METTL14 is arginine methylated in vitro. In vitro methylation assays were performed by incubating recombinant PRMTs (1–8) with purified GST‐tagged METTL14.
- METTL14 is arginine methylated at its C‐terminal IDR in vitro. GST‐tagged full‐length (FL) and C‐terminal IDR‐truncated (1–400) METTL14 were incubated with recombinant PRMT1 and PRMT6.
- METTL14 is arginine methylated in cells. Endogenous METTL14 was immunoprecipitated from HEK293 cells under denaturing conditions and detected using the ADMA antibody ASYM26.
- Inhibiting type I PRMT activity reduces METTL14 arginine methylation. HEK293 cells were treated with the type I PRMT inhibitor MS023 (1 μM, 48 h). METTL14 was immunoprecipitated from the cells and detected by Western blot analysis using anti‐METTL14 and anti‐ASYM26 antibodies.
- METTL14 is arginine methylated at its C‐terminal IDR in cells. HEK293 cells expressing Flag‐tagged FL or C‐terminal IDR‐truncated (1–400) METTL14 were lysed and immunoprecipitated with an anti‐Flag antibody. Arginine methylation of immunoprecipitated METTL14 was analyzed by Western blot using two different antibodies that recognize ADMA.
Data information: In both (B) and (C), Ponceau staining shows the loading of the recombinant proteins. The black dots indicate PRMT enzymes; black triangles indicate arginine methylated‐METTL14; open triangles indicate recombinant METTL14 proteins. In (D–F), the black triangles indicate arginine methylated‐METTL14; open triangles indicate the immunoprecipitated METTL14 proteins.
Source data are available online for this figure.
Figure EV1. The RG‐rich C terminus of METTL14 is intrinsically disordered and is essential for the RNA methyltransferase activity of METTL3/METTL14 complex.
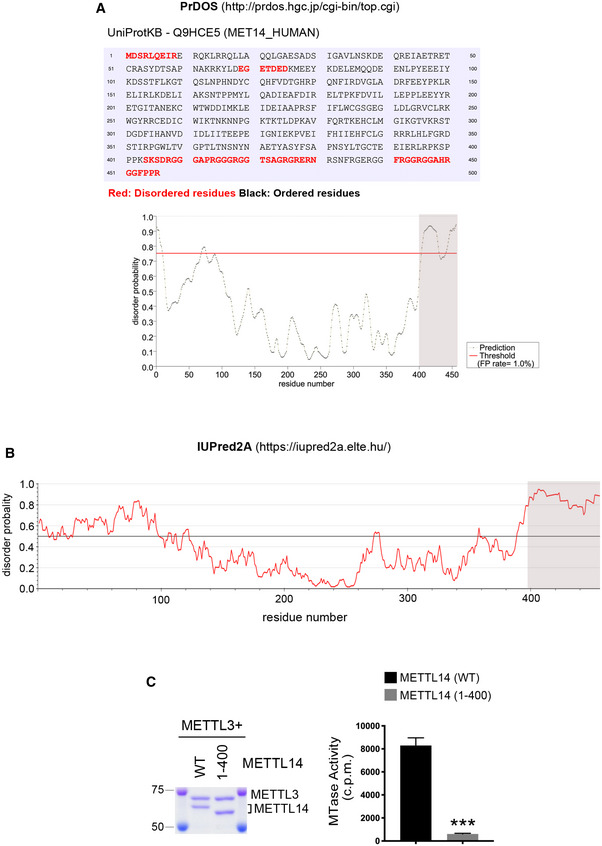
- Prediction of METTL14 IDRs and disorder probability using PrDOS. Disordered amino acids are highlighted in red. FP: false positive.
- Prediction of METTL14 disorder probability using IUPred2A.
- The C‐terminal IDR of METTL14 is essential for the RNA methylation activity of the METTL3/METTL14 complex. In vitro RNA methylation assays were performed by incubating biotin‐labeled RNA substrates with METTL3/METTL14 methyltransferase complexes containing WT and C‐terminal IDR‐truncated mutant (1–400) METTL14. The Coomassie staining shows the purification of the enzyme complex. The enzymatic activity was measured in counts per minute (c.p.m.) using a scintillation counter. Data from three independent replicates were analyzed by Student’s t‐test and shown as mean ± SD. ***P < 0.001.
Data information: In both (A) and (B), the shaded area indicates the most significantly disordered region.
Source data are available online for this figure.
To assess METTL14 arginine methylation in vivo, we immunoprecipitated endogenous METTL14 from HEK293 cells and confirmed its methylation using an antibody (ASYM26) that specifically recognizes ADMA, a modification catalyzed by type I PRMTs, including PRMT1, PRMT3, and PRMT6 (Fig 1D). Next, we treated HEK293 cells with a potent type I PRMT inhibitor MS023 (Eram et al, 2016) to inhibit cellular ADMA. The level of arginine methylated METTL14 was dramatically reduced upon treatment with the inhibitor (Fig 1E), further confirming that METTL14 is arginine methylated in cells. Additionally, METTL14 arginine methylation can also be detected in various human cancer cell lines, including those derived from cervical cancer (HeLa), lung cancer (A549 and H1299), and breast cancer (MDA‐MB‐231 and MCF7), and MS023 treatment reduces METTL14 arginine methylation in all cell lines tested (Appendix Fig S2B–D). Importantly, consistent with our in vitro methylation results, deleting the C‐terminal IDR completely abolished METTL14 methylation in vivo, as detected using two different ADMA antibodies (Fig 1F). Altogether, these results demonstrate that the C‐terminal IDR of METTL14 is arginine methylated.
PRMT1 catalyzes METTL14 C‐terminal IDR arginine methylation
To determine which PRMT methylates METTL14 in cells, we examined the interactions of METTL14 with PRMT1, PRMT3, and PRMT6, the PRMTs that execute METTL14 arginine methylation in vitro (Fig 1B). HEK293 cells were transfected with GFP‐tagged PRMT1, PRMT3, and PRMT6, and co‐immunoprecipitation (co‐IP) assays were performed by immunoprecipitating endogenous METTL14 and detecting associated GFP‐PRMTs. Our results indicate that only PRMT1 interacts with METTL14 (Fig 2A). To further confirm this interaction, we performed reciprocal co‐IP assays of endogenously expressed METTL14 and PRMT1. We were able to detect PRMT1 in the METTL14‐immunoprecipitated protein complex (Fig 2B, left panel), and METTL14 in the PRMT1‐immunoprecipitated protein complex (Fig 2B, right panel). To determine whether METTL14 interacts with PRMT1 through its C‐terminal IDR, we compared the interaction of PRMT1 with full‐length (FL) and C‐terminal IDR truncation mutant (1–400) METTL14 using co‐IP assays. Deleting the C‐terminal IDR completely abolished METTL14 interaction with PRMT1 (Fig 2C). Furthermore, we also performed GST pull‐down assays and demonstrated that although full‐length recombinant GST‐METTL14 can pull down PRMT1 from HEK293 total cell lysates, C‐terminal IDR‐truncated METTL14 cannot (Fig 2D), further demonstrating that the C‐terminal IDR is essential for METTL14 interaction with PRMT1.
Figure 2. PRMT1 catalyzes METTL14 C‐terminal IDR arginine methylation.
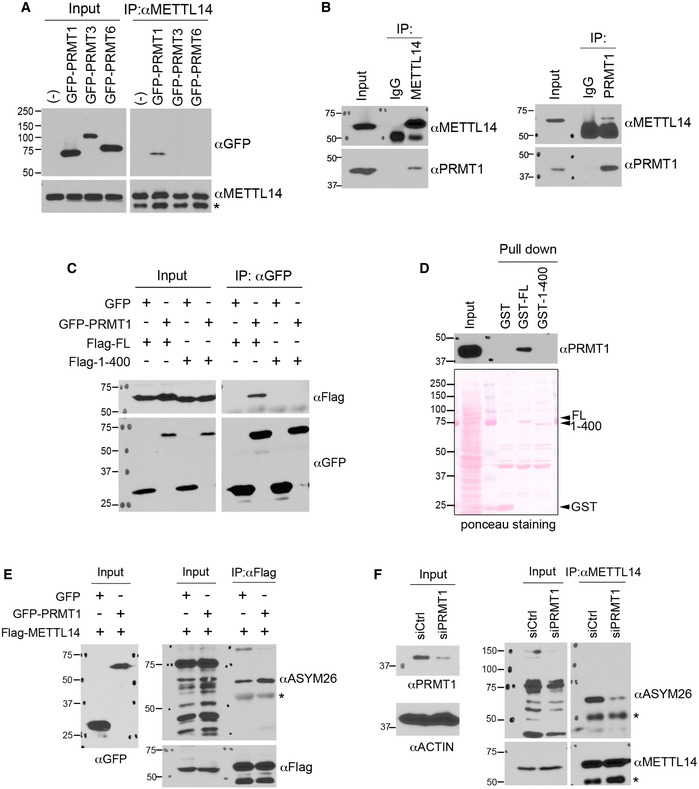
- PRMT1 interacts with METTL14 in cells. HEK293 cells expressing GFP‐tagged PRMT1, PRMT3, or PRMT6 were lysed and immunoprecipitated with an anti‐METTL14 antibody, followed by Western blot analysis using an anti‐GFP antibody. * indicates the location of the IgG heavy chain.
- Endogenous METTL14 interacts with PRMT1. The reciprocal endogenous co‐immunoprecipitation (IP) assays were performed using the METTL14 antibody for IP and the PRMT1 antibody for Western blot detection (left panel) and using the PRMT1 antibody for IP and the METTL14 antibody for Western blot detection (right panel).
- The C‐terminal IDR of METTL14 is essential for its interaction with PRMT1. HEK293 cells were transfected with GFP‐tagged PRMT1 and Flag‐tagged FL or C‐terminal IDR‐truncated (1–400) METTL14. IP was performed using an anti‐GFP antibody, and Western blot analysis was performed using anti‐GFP and anti‐Flag antibodies.
- GST pull‐down detection of the interactions of PRMT1 with GST‐tagged FL and truncated (1–400) recombinant METTL14. The black triangles indicate recombinant METTL14 proteins.
- Overexpression of PRMT1 enhances METTL14 arginine methylation. HEK293 cells were transfected with either GFP vector or GFP‐PRMT1, together with Flag‐METTL14. The level of METTL14 methylation was detected by IP using an anti‐Flag antibody, followed by Western blot analysis using an anti‐ASYM26 antibody. * indicates the location of the IgG heavy chain.
- Knockdown of PRMT1 expression reduces METTL14 arginine methylation. HEK293 cells were transfected with control siRNA (siCtrl) and the siRNA targeting PRMT1 (siPRMT1). METTL14 was immunoprecipitated from these cells, and its methylation level was detected by Western blot analysis using an anti‐ASYM26 antibody. * indicates the location of the IgG heavy chain.
Source data are available online for this figure.
To assess whether PRMT1 catalyzes METTL14 C‐terminal IDR arginine methylation in vivo, we assessed the level of METTL14 arginine methylation in HEK293 cells with altered PRMT1 expression. Overexpressing GFP‐PRMT1 increased METTL14 arginine methylation (Fig 2E) and knocking down the expression of PRMT1, but not PRMT3 or PRMT6, using siRNA reduced METTL14 arginine methylation (Fig 2F, Appendix Fig S3A), supporting the role of PRMT1 as a major PRMT catalyzing the arginine methylation of the METTL14 C‐terminal IDR in cells.
C‐terminal IDR arginine methylation enhances METTL14–RNA interaction and METTL3/METTL14 methyltransferase activity
To identify the arginine methylation site(s), we performed in vitro methylation assays after introducing a series of arginine (R)‐to‐lysine (K) mutations in the IDR, either individually or in various combinations. However, none of the mutations tested significantly reduced METTL14 methylation (Appendix Fig S3B), suggesting that multiple arginine residues are methylated but the combinations we selected were insufficient to cover them all. Thus, we immunoprecipitated METTL14 from HEK293 cells and performed mass spectrometry analysis to identify the methylation sites. Although five sites (Arg438, Arg442, Arg445, Arg450, and Arg456) were found to be both mono‐ and dimethylated (Figs 3A and EV2A), the mutation of all five arginine residues (5RK) only modestly reduced the methylation signal (by ~40%) in vitro and in cells (Fig 3B and C), suggesting that either additional arginine residues are methylated or compensatory methylation occurs when the preferred methylation sites are not available. We thus mutated all thirteen arginine residues in the IDR to lysine (13RK), which completely abolished METTL14 arginine methylation (Fig 3B and C, Appendix Fig S3C), and used this RK mutant as the arginine methylation‐deficient mutant METTL14 in the following studies.
Figure 3. C‐terminal IDR arginine methylation enhances METTL14–RNA interactions and METTL3/METTL14 RNA methylation activity.
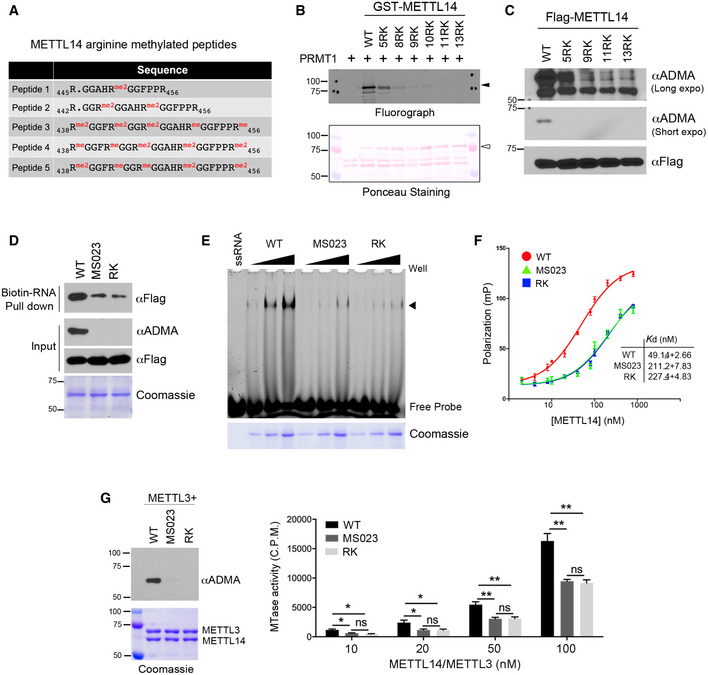
- Summary of METTL14 arginine‐methylated peptides identified by LC‐MS/MS.
- METTL14 IDR arginine methylation occurs at multiple arginine residues within RGG/RG motifs. Mutation of five arginine sites identified from mass spectrometry reduces METTL14 arginine methylation, but only mutation of all arginine residues to lysine completed blocked METTL14 methylation. Ponceau S staining shows the loading of the recombinant proteins. The black triangles indicate arginine methylated‐METTL14; open triangles indicate recombinant METTL14 proteins.
- METTL14 is methylated at multiple arginine residues in cells. HEK293 cells expressing Flag‐tagged WT or various R‐to‐K METTL14 mutants were lysed and immunoprecipitated with an anti‐Flag antibody. Arginine methylation of immunoprecipitated METTL14 was detected by Western blot analysis using an anti‐ADMA antibody. Both short and long exposures of the chemiluminescence signals are shown.
- Arginine methylation of the METTL14 IDR enhances its interaction with RNA substrates. RNA pull‐down assay was performed by incubating biotin‐labeled RNA with WT, hypomethylated (MS023), and arginine methylation‐deficient (RK) mutant METTL14. The pull‐down samples were detected by Western blot analysis using an anti‐Flag antibody. The methylation status of the recombinant proteins was confirmed by Western blot analysis using an anti‐ADMA antibody.
- EMSA was performed to compare the interactions of WT, hypomethylated (MS023), and arginine methylation‐deficient (RK) mutant METTL14 with 6‐FAM‐labeled RNA. Arrow indicates the shift of the RNA probe caused by the protein–RNA interaction. Coomassie staining shows the increasing amounts of recombinant proteins used in the assay.
- Fluorescence polarization assays were performed by incubating 6‐FAM‐labeled RNA with WT, hypomethylated (MS023), and arginine methylation‐deficient (RK) mutant METTL14. Each point represents the average of three independent replicates and error bars represent standard deviation (SD). The dissociation constant values (K d) were listed as mean ± SD.
- Arginine methylation of the C‐terminal IDR enhances the RNA methylation activity of the METTL14/METTL3 complex in vitro. In vitro RNA methylation assays were performed by incubating biotin‐labeled RNA substrates with METTL3/METTL14 methyltransferase complexes containing WT, hypomethylated (MS023), and arginine methylation‐deficient (RK) mutant METTL14 in various concentrations (10–100 nM). The methylation status of the METTL3/METTL14 complex was confirmed by Western blot analysis using an anti‐ADMA antibody. Coomassie staining shows the purification of the enzyme complex. Enzymatic activity was measured in counts per minute (c.p.m.) using a scintillation counter. Data from three replicates were analyzed by Student’s t‐test and shown as mean ± SD. *P < 0.05; **P < 0.01; ns, not significant.
Source data are available online for this figure.
Figure EV2. Characterization of the impacts of arginine methylation on METTL14–RNA interactions and RNA methylation activity.
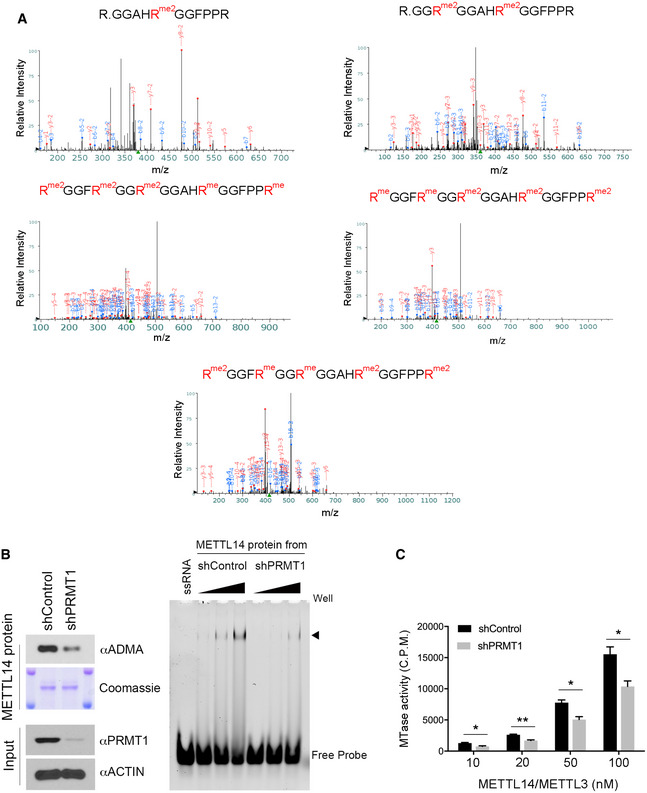
- Identification of METTL14 arginine methylation sites by mass spectrometry. LC‐MS/MS was performed on METTL14 proteins purified from HEK293 cells. Five peptides that are mono‐ or dimethylated were identified (R438, R442, R445, R450, and R456).
- Recombinant METTL14 proteins purified from PRMT1 knockdown HEK293 cells exhibits reduced RNA interactions. Flag‐METTL14 was expressed and purified from control (shControl) and PRMT1 knockdown (shPRMT1) HEK293 cells. The methylation level of METTL14 was detected by Western blot using an anti‐ADMA antibody. The amount of protein was visualized by Coomassie staining (left panel). EMSA was performed to compare the interaction of recombinant METTL14 purified from shControl and shPRMT1 HEK293 cells with 6‐FAM‐labeled RNA. Arrow indicates the shift of the RNA probe caused by the protein–RNA interaction (right panel).
- Recombinant METTL14 purified from PRMT1 knockdown HEK293 cells exhibits reduced RNA methylation activity. In vitro RNA methylation assays were performed by incubating biotin‐labeled RNA substrates with METTL3/METTL14 methyltransferase complexes purified from control (shControl) and PRMT1 knockdown (shPRMT1) HEK293 cells. The enzymatic activity was measured in counts per minute (c.p.m.) using a scintillation counter. Data from three independent replicates were analyzed by Student’s t‐test and shown as mean ± SD. *P < 0.05; **P < 0.01.
Source data are available online for this figure.
RGG/RG motifs can mediate protein–protein and protein–RNA interactions and are recognized as the second most common RNA‐binding domains in the human genome (Rajyaguru & Parker, 2012; Thandapani et al, 2013; Ozdilek et al, 2017; Chong et al, 2018). Therefore, to investigate the role of METTL14 C‐terminal IDR arginine methylation, we tested the hypothesis that arginine methylation of the RGG/RG motifs of METTL14 regulates its interactions with RNA substrates. Three independent assays were performed using recombinant METTL14 proteins purified from HEK293 cells. First, we carried out the RNA pull‐down assays by incubating recombinant arginine methylated (WT), hypomethylated (MS023‐treated), and RK mutant METTL14 proteins with biotin‐labeled RNA oligonucleotides harboring the consensus sequence motif for m6A modification, GGACU (Liu et al, 2014). The loss of arginine methylation, caused by either inhibitor treatment or by R‐to‐K mutation, dramatically reduced the interactions of METTL14 with the RNA substrates (Fig 3D). Next, we performed an Electrophoretic Mobility Shift Assay (EMSA) by incubating the 6‐carboxyfluorescein (6‐FAM)‐labeled RNA probe with increasing amounts of recombinant METTL14 proteins (as described in Fig 3D). Our results show that arginine methylated METTL14 exhibits much stronger binding to RNA substrates, compared with the hypomethylated and RK mutant METTL14 (Fig 3E). In addition, METTL14 protein purified from PRMT1 knockdown HEK293 cells also exhibited reduced interactions with RNA substrates, further supporting the role of PRMT1 in catalyzing METTL14 arginine methylation (Fig EV2B). Finally, to quantitatively compare the RNA‐binding affinity of these recombinant METTL14 proteins, we performed fluorescence polarization assays, which measure protein binding‐induced changes in the polarization of light emitted upon excitation of a fluorescence‐labeled RNA probe. Both hypomethylated and RK mutant METTL14 exhibited significantly lower RNA‐binding affinities than arginine methylated METTL14 (dissociation constant [Kd] values of 211.2 and 227.4 nM for hypomethylated and RK mutant METTL14, respectively, compared with 49.14 nM for arginine methylated METTL14; Fig 3F). Results from these three independent experiments demonstrate that C‐terminal IDR arginine methylation enhances the interactions of METTL14 with its RNA substrates.
To assess whether altered METTL14–RNA interaction affects the catalytic activity of the RNA methyltransferase complex, we compared the RNA methylation activity of the recombinant METTL14 proteins in vitro. The arginine methylated METTL14 (in complex with METTL3) exhibited significantly (~2 fold) higher m6A methyltransferase activity than the hypomethylated and arginine methylation‐deficient enzymes (Fig 3G). Similarly, METTL14 protein purified from PRMT1‐knockdown HEK293 cells also exhibited reduced RNA methylation activity (Fig EV2C). Note that the reduced RNA‐binding affinity (Fig 3D–F) and RNA methylation activity (Fig 3G) of RK mutant METTL14 was similar to that of the hypomethylated METTL14, suggesting that the effects observed were not artifacts caused by the R‐to‐K mutations. Altogether, these data demonstrate that arginine methylation of the C‐terminal IDR enhances the activity of the METTL3/METTL14 complex, likely by promoting the interactions of METTL14 with RNA substrates.
C‐terminal IDR arginine methylation enhances the METTL14–RNAPII interaction
We next investigate the role of C‐terminal IDR arginine methylation on the function of METTL14 in cells. To do so, we examined the impact of the loss of C‐terminal IDR arginine methylation on the subcellular localization of METTL14 and protein–protein interactions with its known partners. Immunofluorescence assays showed that neither the removal of the C‐terminal IDR nor the mutation of the arginine methylation sites affected the nuclear localization of METTL14 (Fig EV3A). Furthermore, consistent with previous crystal structure studies showing that the C terminus of METTL14 is not involved in its interaction with METTL3 (Sledz & Jinek, 2016; Wang et al, 2016a; Wang et al, 2016b), we observed that the interactions of METTL14 with other components of the m6A methyltransferase complex, including METTL3 and WTAP, were also unaffected by C‐terminal IDR truncation or RK mutation, as revealed by the GST pull‐down and co‐IP assays (Fig EV3, EV4, EV5).
Figure EV3. Localization and interaction analysis of arginine methylation‐deficient METTL14 in cells.
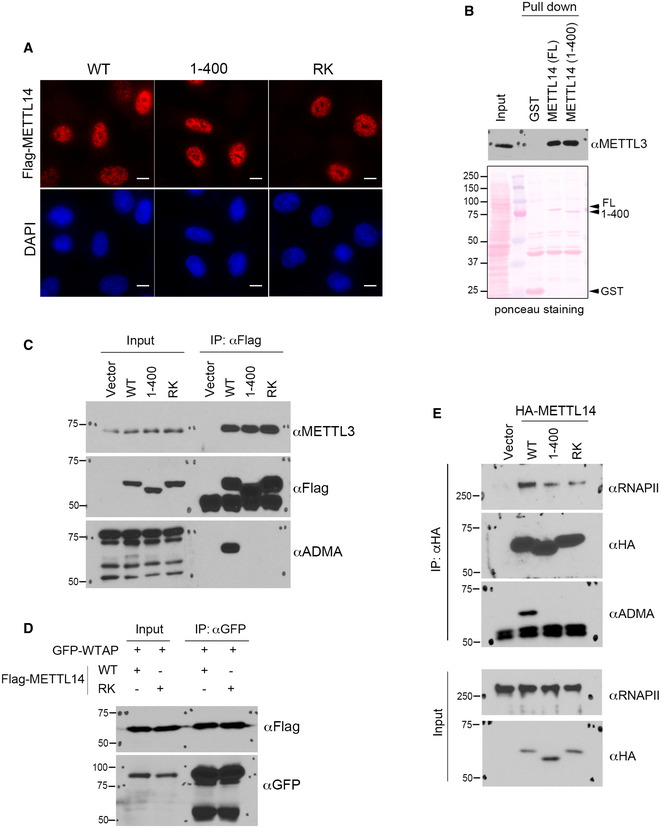
- Immunofluorescence assays were performed to examine the subcellular localization of WT, C‐terminal IDR‐truncated (1–400), and arginine methylation‐deficient (RK) mutant METTL14 in HeLa cells. The localizations of all three proteins were detected by using an anti‐Flag antibody. DAPI staining was performed to mark the cell nucleus. Scale bar: 10 µm.
- GST pull‐down assays were performed by incubating GST‐tagged full‐length (FL) and C‐terminal IDR‐truncated (1–400) METTL14 with HEK293 cell lysate. Western blot analysis was performed using an anti‐METTL3 antibody. Ponceau S staining shows the loading of the recombinant proteins in the pull‐down samples.
- Co‐IP assays were performed to detect the interactions of WT, C‐terminal IDR‐truncated (1–400), and arginine methylation‐deficient (RK) mutant METTL14 with METTL3. The methylation of METTL14 protein was confirmed by Western blot analysis using an anti‐ADMA antibody.
- Co‐IP assays were performed to detect the interactions of WT and arginine methylation‐deficient (RK) mutant METTL14 with GFP‐WTAP. The HEK293 cells were transiently transfected with the indicated plasmids 48 h before the assays were performed.
- Co‐IP assays were performed to detect the interactions of WT, C‐terminal IDR‐truncated (1–400), and arginine methylation‐deficient (RK) mutant METTL14 with RNAPII. The methylation of METTL14 protein was confirmed by Western blot analysis using an anti‐ADMA antibody.
Source data are available online for this figure.
Figure EV4. MeRIP‐seq (m6A‐seq) analysis of mESCs expressing WT and arginine methylation‐deficient METTL14.
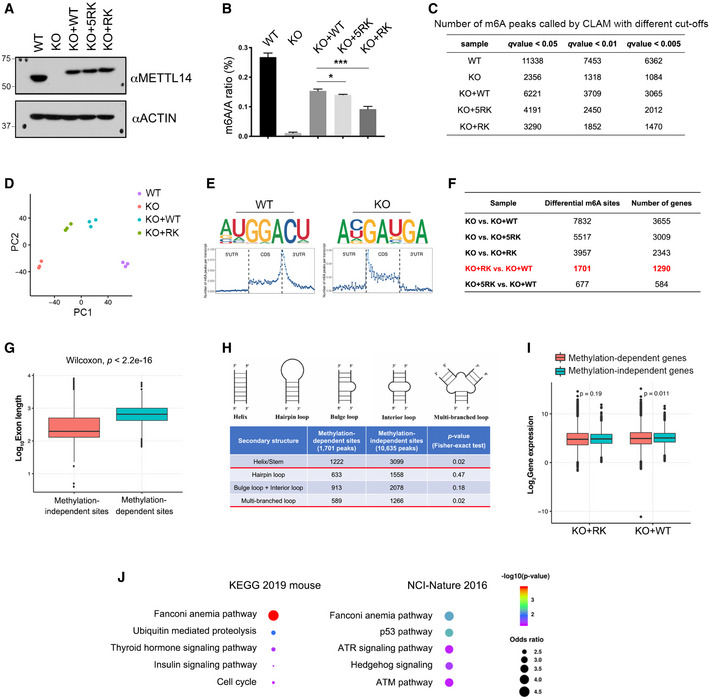
- Three isogenic mESC lines were established by re‐expressing WT (KO + WT), 5RK (KO + 5RK), and 13RK (KO + RK) mutant METTL14 in Mettl14 KO mESCs through lentivirus transduction. The expression of METTL14 in these cell lines was detected by Western blot analysis using an anti‐METTL14 antibody. ACTIN was used as a loading control.
- LC‐MS/MS was performed to quantify m6A levels (presented as the m6A/A ratio) in WT, Mettl14 KO, KO + WT, KO + 5RK, and KO + RK mESCs. The total RNA was extracted using the TRIzol reagent, and the poly(A) mRNA was purified for LC‐MS/MS analysis. Data from three biological replicates were analyzed by Student’s t‐test and shown as mean ± SD. *P < 0.05; ***P < 0.001.
- Summary of the numbers of m6A peaks in WT, Mettl14 KO, KO + WT, KO + 5RK, and KO + RK mESCs using different q‐value cutoffs.
- Principal component analysis (PCA) plot of m6A peaks in WT, Mettl14 KO, KO + WT, and KO + RK mESCs, each with three biological replicates. PC1 and PC2 are the top two principle components that explained the highest percentage of the variance.
- Sequence motifs of m6A‐enriched regions in WT and Mettl14 KO mESCs (upper panels). Topological distribution of normalized m6A peaks across the 5′UTR, CDS, and 3′UTR of mRNAs (lower panels).
- Summary of the numbers of differential m6A peaks and corresponding numbers of genes for each pair of comparison among all established mESC cell lines. The differential m6A sites and the number of genes harboring these sites compared between KO + RK and KO + WT mESCs were highlighted in red.
- Length comparison between internal exons harboring METTL14 arginine methylation‐independent and ‐dependent m6A sites. Statistical analysis was performed using the Wilcoxon test to measure the median difference between the two groups.
- Secondary structure prediction of RNA sequences harboring METTL14 arginine methylation‐dependent and ‐independent m6A sites. Statistical analysis was performed using Fisher’s exact test.
- Gene expression level comparison of genes harboring METTL14 arginine methylation‐independent and ‐dependent m6A sites in KO + RK and KO + WT mESCs. Statistical analysis was performed using the Wilcoxon test.
- GO pathway analysis using EnrichR (Kuleshov et al, 2016) reveals that genes harboring METTL14 arginine methylation‐dependent m6A peaks are enriched for the Fanconi anemia pathway. Examples of analysis using two‐pathway interaction annotation databases (KEGG 2019 mouse and NCI‐Nature 2016) are shown. Statistical analysis was performed using Fisher exact test, as defined in EnrichR.
Data information: In both (G) and (I), data are obtained from three biological replicates. The lower and upper hinges of the boxes are the 25th and 75th percentiles, and the central band corresponds to median. The upper whisker defines the value no larger than 1.5*distance between the first and third quartiles from the upper hinge, while the lower whisker defines the value no smaller than 1.5*distance between the first and third quartiles from the lower hinge.
Source data are available online for this figure.
Figure EV5. METTL14 C‐terminal IDR arginine methylation regulates m6A deposition on DNA repair genes.
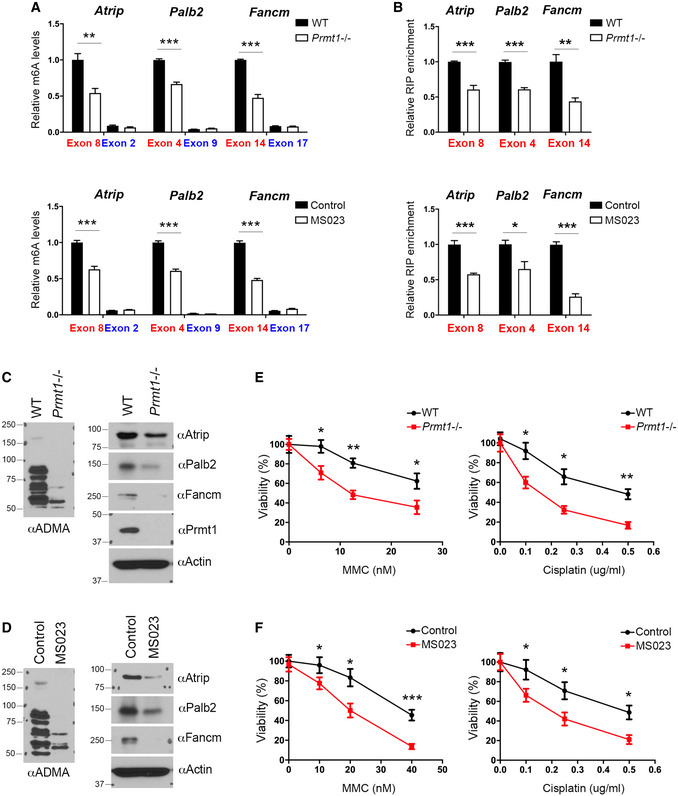
- MeRIP (m6A‐IP)‐qPCR assays were performed for WT and Prmt1 KO mESCs (upper panel), as well as for control and MS023‐treated mESCs (lower panel), to detect the impact of PRMT1 loss or inhibition on m6A deposition at targeted transcripts. m6A‐negative regions of the transcripts (blue) were included as negative controls.
- METTL14 RIP‐qPCR assays were performed for WT and Prmt1 KO mESCs (upper panel), as well as for control and MS023‐treated mESCs (lower panel), to detect the impact of PRMT1 loss or inhibition on the interactions of METTL14 with targeted transcripts.
- The expression of ICL repair genes is reduced in Prmt1 KO mESCs. Total cell lysates from WT and Prmt1 KO mESCs were subjected to Western blot analysis using the indicated antibodies.
- The expression of ICL repair genes is reduced in MS023‐treated mESCs. Total cell lysates from control and MS023‐treated mESCs were subjected to Western blot analysis using the indicated antibodies.
- Knockout of Prmt1 sensitizes mESCs to ICL damage. WT and Prmt1 KO mESCs were treated with various concentrations of MMC (left panel) or cisplatin (right panel) for 4 days before cell viability was measured.
- Similar to (E), except that mESCs were treated with MS023, to inhibit type I PRMT activity, while they were treated with MMC (left panel) or cisplatin (right panel).
Data information: In (A), (B), (E), and (F), data from three independent replicates were analyzed by Student’s t‐test and shown as mean ± SD. In both (E) and (F), each point represents the average of three independent replicates and error bars represent standard deviation. *P < 0.05; **P < 0.01, ***P < 0.001.
Source data are available online for this figure.
Because m6A deposition is co‐transcriptional and the RNA methyltransferase complex has been shown to associate with RNAPII (Ke et al, 2017; Slobodin et al, 2017; Huang et al, 2019), we tested whether arginine methylation regulates the interaction of METTL14 with RNAPII. First, we performed reciprocal co‐IP assays of endogenous METTL14 and RNAPII and detected RNAPII in the METTL14‐immunoprecipitated protein complex (Fig 4A, left panel), and METTL14 in the RNAPII‐immunoprecipitated protein complex (Fig 4A, right panel), consistent with a recent report (Huang et al, 2019). Next, to determine whether C‐terminal IDR arginine methylation contributes to this interaction, we transfected HA‐tagged WT and arginine methylation‐deficient (RK) mutant METTL14 and compared their interactions with RNAPII. Although the loss of C‐terminal IDR methylation does not affect the interaction of METTL14 with METTL3, its interaction with RNAPII was dramatically impaired (Fig 4B). Note that the deletion of the C‐terminal IDR also reduced the METTL14–RNAPII interaction (Fig EV3E), indicating that this region contributes to their interaction. Furthermore, we treated HEK293 cells with MS023 to inhibit METTL14 arginine methylation and observed a similarly reduced interaction between METTL14 and RNAPII (Fig 4C), suggesting that the METTL14–RNAPII interaction is regulated by C‐terminal IDR arginine methylation. Because the C‐terminal IDR arginine methylation enhances METTL14–RNA interaction (Figs 3D–F and EV2B), we next tested if RNA is involved in the METTL14–RNAPII interaction. We performed the co‐IP assays in the presence of RNase A, which led to a significantly reduced amount of METTL14‐associated RNAPII (Fig 4D). These results suggest that METTL14–RNA interaction contributes, at least in part, to the METTL14–RNAPII interaction. Collectively, these data show that arginine methylation of the C‐terminal IDR is critical for regulating the association of METTL14 with RNAPII.
Figure 4. Arginine methylation of the C‐terminal IDR enhances the interaction of METTL14 with RNAPII in cells.
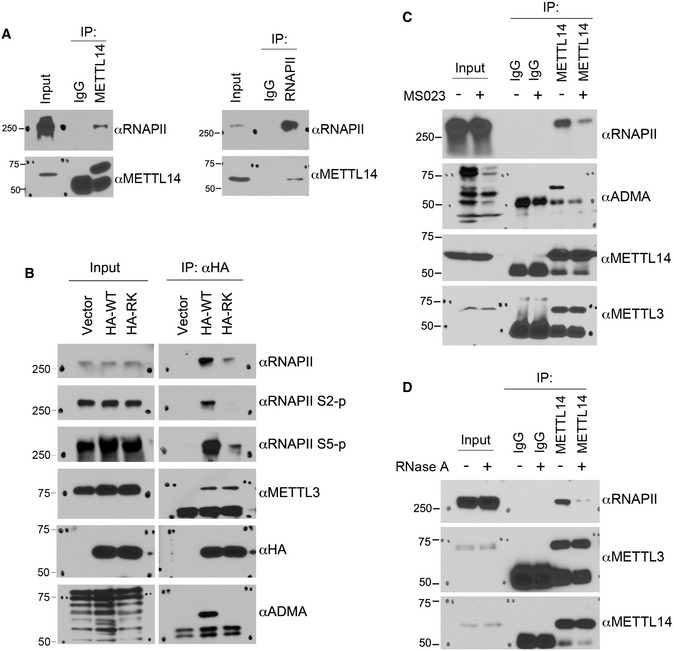
- Endogenous METTL14 interacts with RNAPII. Endogenous co‐immunoprecipitation (IP) was performed using the METTL14 antibody for IP and the RNAPII antibody for Western blot detection (left panel) and using the RNAPII antibody for IP and the METTL14 antibody for Western blot detection (right panel).
- Arginine methylation of the METTL14 C‐terminal IDR enhances its interaction with RNAPII. HEK293 cells were transfected with HA‐tagged WT or arginine methylation‐deficient (RK) mutant METTL14. IP was performed using an anti‐HA antibody, and Western blot analysis was performed using the indicated antibodies.
- Co‐IP assays were performed to compare the interactions between METTL14 and RNAPII in control and MS023‐treated HEK293 cells. Cells were treated with either DMSO or MS023 (1 μM) for 48 h before they were lysed. IP was performed using control IgG and METTL14 antibodies, respectively. Western blot analysis was performed using anti‐RNAPII, anti‐ADMA, anti‐METTL14, and anti‐METTL3 antibodies.
- Co‐IP assays were performed to examine the involvement of RNA in the METTL14–RNAPII interaction. Total cell lysates were either left untreated or treated with RNase A to remove the RNA component before IP. Western blot analysis was performed using anti‐RNAPII, anti‐METTL3, and anti‐METTL14 antibodies.
Source data are available online for this figure.
METTL14 arginine methylation regulates m6A deposition in vivo
To investigate the cellular function of METTL14 arginine methylation, we used mouse embryonic stem cells (mESCs), a model system in which we confirmed the arginine methylation of METTL14 (Appendix Fig S4A). We established three isogenic mESC lines by stably transfecting Mettl14 knockout mESCs (Geula et al, 2015) with human WT METTL14 (KO + WT), 5RK mutant METTL14 (KO + 5RK), and 13RK mutant METTL14 (KO + RK) (Figs 5A and EV4A, Appendix Fig S4B). Polyadenylated mRNA was purified from these cells and analyzed using liquid chromatography–tandem mass spectrometry (LC‐MS/MS). The m6A to A ratio (m6A/A) was reduced by ~40% in KO + RK mESCs compared with KO + WT mESCs (Fig 5B). Importantly, the reduction in m6A correlated with the degree of METTL14 methylation loss, as KO + 5RK mESCs exhibited a significant but relatively modest (~10%) reduction (Fig EV4B). Similar to what has previously been reported for the Mettl14 KO mESCs (Wang et al, 2014b; Geula et al, 2015), the KO + RK mESCs exhibited reduced pluripotency and proliferation compared with WT and KO + WT mESCs (Fig 5C–E).
Figure 5. Analysis of METTL14 arginine methylation‐dependent m6A sites.
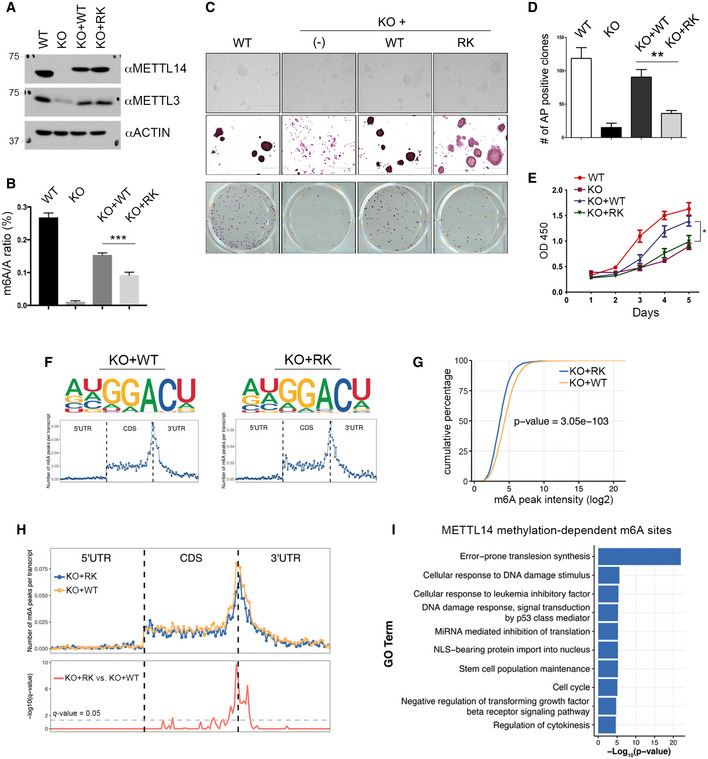
- Generation of isogenic mESC lines expressing WT and arginine methylation‐deficient mutant (RK) METTL14. Mettl14 KO mESCs were transfected with Flag‐tagged WT or RK mutant METTL14 using a lentivirus expression system. The expression of METTL14 and METTL3 in these cells was detected by Western blot analysis using anti‐METTL14 and anti‐METTL3 antibodies. ACTIN was used as a loading control.
- m6A levels are reduced in mESCs expressing arginine methylation‐deficient (RK) mutant METTL14. The mRNA purified from WT, Mettl14 KO, KO + WT, and KO + RK mESCs was subjected to LC‐MS/MS analysis to quantify m6A levels (presented as the m6A/A ratio).
- Morphology and alkaline phosphatase (AP) staining of mESCs expressing WT, Mettl14 KO, KO + WT, and KO + RK METTL14. Scale bar: 400 µm.
- Quantification of AP‐positive clones in (C).
- Proliferation of mESCs expressing WT, Mettl14 KO, KO + WT, and KO + RK METTL14 over a 5‐day period. Each point represents the average of three independent replicates, and error bars represent standard deviation (SD).
- Sequence motifs of m6A‐enriched regions in KO + WT and KO + RK mESCs (upper panels). Topological distribution of normalized m6A peaks across the 5′UTR, CDS, and 3′UTR of mRNAs (lower panels).
- Cumulative distribution of log2 m6A peak intensity in KO + WT and KO + RK mESCs. Statistical analysis was performed using the Wilcoxon test to measure the median difference of peak intensities between the two groups.
- Overlay of m6A distributions across the 5′UTR, CDS, and 3′UTR of mRNAs in KO + WT and KO + RK mESCs (upper panel). Statistical analysis of differential m6A peaks in KO + RK versus KO + WT mESCs (lower panel). The y‐axis represents the q‐value (−log10). The dashed gray line indicates q‐value = 0.05.
- Gene Ontology (GO) analysis of genes harboring METTL14 arginine methylation‐dependent m6A sites. Statistical analysis was performed using Hypergeometric test. The P‐value for the enrichment of each biological process (GO term) is shown.
Data information: In (B), (D), and (E), data from three independent replicates were analyzed by Student’s t‐test and shown as mean ± SD. *P < 0.05; **P < 0.01, ***P < 0.001.
Source data are available online for this figure.
To identify transcriptome‐wide m6A sites that are regulated by METTL14 arginine methylation, we performed methylated RNA‐IP (RIP) followed by high‐throughput sequencing (MeRIP‐seq or m6A‐seq) in WT, Mettl14 KO, KO + WT, KO + 5RK, and KO + RK mESCs (Fig EV4A). Crosslinking IP (CLIP)‐seq analysis of multi‐mapped reads (CLAM) (Zhang & Xing, 2017) was used to identify m6A peaks using different q‐value cutoffs (0.05, 0.01, and 0.005). Consistent with our LC‐MS/MS‐based m6A/A quantification (Figs 5B and EV4B), the number of m6A peaks identified by CLAM using all three cutoffs positively correlated with the degree of METTL14 arginine methylation (Fig EV4C). Principal component analysis (PCA) of the m6A peaks in each cell line demonstrated strong reproducibility (low variation) among three biological replicates (Fig EV4D). We choose 0.05 as the q‐value cutoff for further analysis because the number of m6A peaks identified in WT mESCs using that cutoff (11,338; Dataset EV1) was similar to the number reported for mESCs in other studies (Wang et al, 2014b; Aguilo et al, 2015; Geula et al, 2015). De novo motif analysis identified the RRACU m6A sequence motif as enriched at m6A sites, and distribution analysis revealed that the m6A sites in protein‐coding genes are enriched near the stop codon and at the beginning of the 3′UTR (Figs 5F and EV4E), both as previously described (Wang et al, 2014b; Aguilo et al, 2015; Geula et al, 2015). The comparison of m6A peaks in KO + WT and KO + RK mESCs revealed a significant decrease in m6A peak density upon the loss of METTL14 arginine methylation (Fig 5G). We next performed differential m6A analysis and identified 1,701 METTL14 arginine methylation‐dependent m6A sites in 1,290 genes and 10,635 methylation‐independent m6A sites in 5,764 genes between KO + WT and KO + RK mESCs (Fig EV4F, Dataset EV2). Although the majority of METTL14 arginine methylation‐dependent m6A sites are found near the stop codon and 3′ UTR, some of them are located in the internal exons (Fig 5H). We found that internal exons harboring these methylation‐dependent m6A sites are significantly longer than the exons harboring methylation‐independent m6A sites (Fig EV4G), indicating that m6A deposition in long exons is more dependent on METTL14 methylation than m6A deposition in short exons, consistent with the preference of m6A enrichment in long internal exons (Dominissini et al, 2012; Batista et al, 2014; Geula et al, 2015). Additionally, RNA sequences in the vicinity of these m6A sites are predicted to be more likely to form secondary structures, such as Helix/Stems or multi‐branched loops (Fig EV4H). Although the deposition of m6A has been linked to gene expression and mRNA stability, a comparison of the expression of genes with versus without methylation‐dependent m6A sites did not reveal any significant differences (Fig EV4I).
To determine the biological function of arginine methylation‐dependent m6A sites, we performed Gene Ontology (GO) analysis. Several known m6A‐regulated cellular processes are significantly enriched, such as stem cell population maintenance and regulation of the TGF‐β signaling pathway (Fig 5I), which might explain the changes in the morphology and pluripotency of KO + RK mESCs (Fig 5C–E). Unexpectedly, GO analysis also revealed a strong enrichment of METTL14 arginine methylation‐dependent genes in DNA repair pathways, particularly in error‐prone translesion synthesis and the Fanconi anemia pathway (Figs 5I and EV4J). Because the role of m6A‐mediated RNA metabolism in DNA repair is largely unknown, we aimed to investigate how METTL14 arginine methylation‐dependent m6A sites function in regulating DNA repair.
METTL14 arginine methylation‐dependent m6A sites are associated with enhanced translation of DNA repair genes
As demonstrated in the UCSC Genome Browser custom tracks (Fig 6A, Appendix Fig S4C), there was a significant reduction in m6A signals in the transcripts of error‐prone translesion synthesis and Fanconi anemia genes, including Atrip, Palb2, Fancm, Blm, Brca1, and Brca2 due to Mettl14 KO or expression of arginine methylation‐deficient (KO + RK) METTL14. Interestingly, most of these METTL14 arginine methylation‐dependent m6A sites are located in long internal exons, consistent with the transcriptome‐wide analysis showing that internal exons harboring arginine methylation‐dependent m6A sites are significantly longer than exons harboring arginine methylation‐independent m6A sites (Fig EV4G). These results were confirmed by m6A‐IP, followed by quantitative reverse transcription PCR (RT–qPCR) (Fig 6B, Appendix Fig S4D). Furthermore, consistent with our in vitro data showing an important role of arginine methylation in promoting METTL14–RNA interaction (Fig 3D–F), METTL14 RIP‐qPCR revealed that arginine methylation deficiency dramatically reduced the interactions of METTL14 with m6A‐positive regions of the target transcripts in vivo (Fig 6C, Appendix Fig S4E and F). Knocking out Prmt1 (Pawlak et al, 2000) or treating mESCs with the type I PRMT inhibitor MS023, which inhibits METTL14 arginine methylation (Figs 1E and 2F, Appendix Fig S2B–D), also reduced m6A deposition (Fig EV5A) and the interactions of METTL14 with target transcripts (Fig EV5B, Appendix Fig S6A and B), further supporting the role of PRMT1‐catalyzed METTL14 arginine methylation in regulating m6A deposition.
Figure 6. METTL14 arginine methylation‐dependent m6A sites are associated with enhanced translation of DNA repair genes.
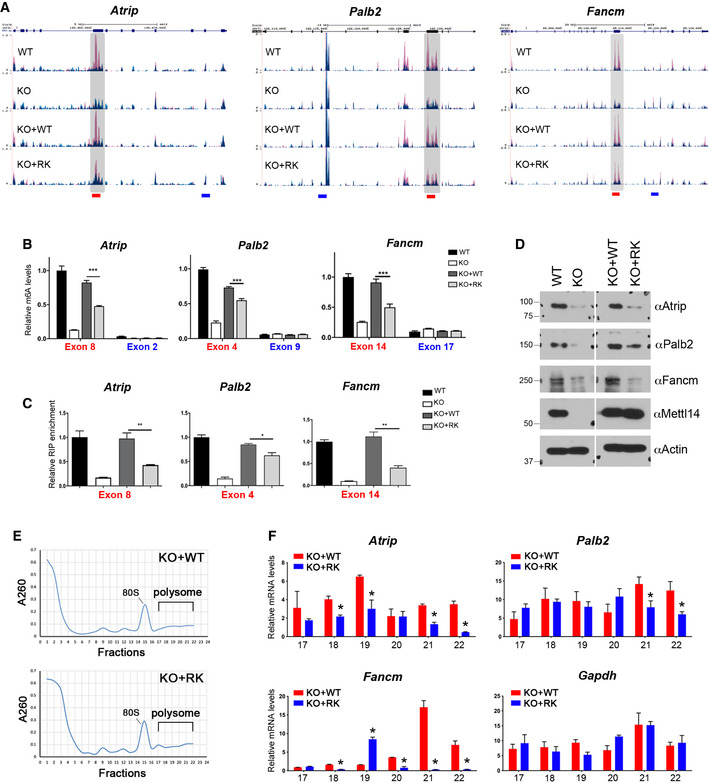
- UCSC Genome Browser custom tracks of m6A‐seq reads along the indicated mRNAs in WT, Mettl14 KO, KO + WT, and KO + RK mESCs. The y‐axis represents the normalized number of reads. Blue reads are from non‐immunoprecipitated input libraries, and red reads are from m6A‐IP libraries. Above the custom tracks, the thick blue boxes represent the protein‐coding regions (CDSs), the thin blue boxes represent the untranslated regions (UTRs), and the blue lines represent introns. The bars at the bottom of the custom tracks indicate the amplicon locations for MeRIP (m6A‐IP)‐qPCR assays (B) and METTL14 RIP‐qPCR assays (C) to detect m6A‐positive (red) and negative (blue) regions.
- MeRIP (m6A‐IP)‐qPCR assays were performed for WT, Mettl14 KO, KO + WT, and KO + RK mESCs to validate the MeRIP‐seq results. m6A‐negative regions of the transcripts (blue) were included as negative controls.
- METTL14 RIP‐qPCR assays were performed for WT, Mettl14 KO, KO + WT, and KO + RK mESCs to compare the binding of WT and RK mutant METTL14 to mRNA targets. Primers (red color) that amplify m6A positive regions of the transcripts were used.
- The expression of ICL repair genes is reduced in mESCs expressing arginine methylation‐deficient mutant (RK) METTL14. Total cell lysates from WT, Mettl14 KO, KO + WT, and KO + RK mESCs were subjected to Western blot analysis using the indicated antibodies.
- Polysome profiling was performed for KO + WT and KO + RK mESCs. Whole‐cell extracts were fractionated through centrifugation in a sucrose density gradient. Optical scans (OD260) of the collected fractions are shown.
- Quantification of ribosome‐bound mRNA for the indicated genes from individual fractions (as in (E)), relative to the amount of the total mRNA in all fractions. Gapdh was included as a negative control.
Data information: In (B), (C), and (F), data from three independent replicates were analyzed by Student’s t‐test and shown as mean ± SD. *P < 0.05; **P < 0.01, ***P < 0.001.
Source data are available online for this figure.
The deposition of m6A is tightly linked to gene expression (Shi et al, 2019; Zaccara et al, 2019). Therefore, we next investigated how changes in m6A deposition due to loss of METTL14 arginine methylation affect the expression of DNA repair genes. Western blot analysis revealed that the expression of Atrip, Palb2, and Fancm was reduced upon Mettl14 KO or expression of arginine methylation‐deficient (KO + RK) METTL14 (Fig 6D). A similar reduction in protein expression was also detected in Prmt1 KO and MS023‐treated mESCs (Fig EV5C and D). This reduction in protein expression was not due to reduced mRNA production (Appendix Fig S5A and B) or increased mRNA degradation (Appendix Fig S5C). Instead, polysome profiling analysis revealed a significant reduction in the association of these DNA repair transcripts with polyribosomes in KO + RK mESCs compared with KO + WT mESCs, suggesting a reduction in protein translation upon the loss of METTL14 arginine methylation (Fig 6E and F). Together, these results reveal that METTL14 arginine methylation is important for the efficient translation of DNA repair genes, likely through an m6A‐dependent mechanism.
Loss of METTL14 arginine methylation sensitizes mESCs to DNA damage
The expression of genes involved in error‐prone translesion synthesis and the Fanconi anemia pathway is essential for the repair of DNA interstrand crosslinks (ICLs), which form when both strands of DNA are covalently linked (Deans & West, 2011). ICLs prevent DNA strand separation, blocking DNA replication and transcription, and thus exerting potent biological effects. We next examined if METTL14 arginine methylation loss impairs cellular responses to ICLs. Consistent with our observation that Mettl14 KO and KO + RK mESCs exhibited reduced expression of ICL repair genes (Fig 6D), we found that these mESCs were significantly more sensitive than WT and KO + WT mESCs to treatment with mitomycin C (MMC) and cisplatin, two chemotherapeutic agents that kill cancer cells by inducing ICLs (Fig 7A and B). Similar sensitivity was also observed in Prmt1 KO and MS023‐treated mESCs (Fig EV5E and F). However, these cells were not differentially sensitive to ionizing radiation, which causes DNA double‐strand breaks (Appendix Fig S5D). Importantly, increasing the expression of Fanconi anemia pathway genes, such as Palb2, by transient transfection can partially reduce the sensitivity of KO + RK mESCs to MMC (Appendix Fig S5E).
Figure 7. Loss of METTL14 arginine methylation sensitizes mESCs to DNA damage.
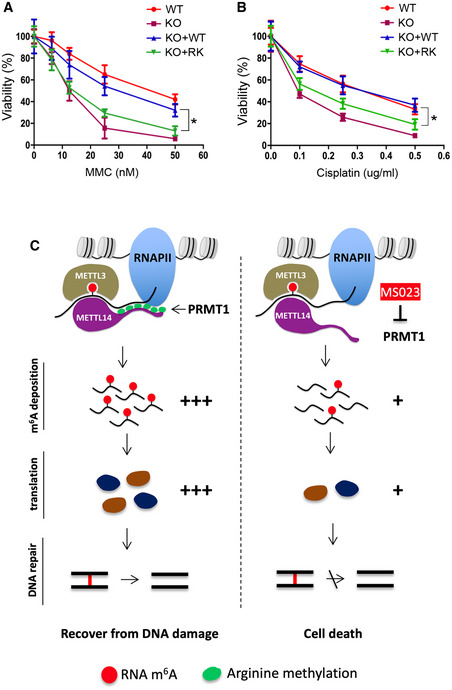
- mESCs expressing arginine methylation‐deficient mutant (RK) METTL14 are sensitive to ICL damage induced by MMC. WT, Mettl14 KO, KO + WT, and KO + RK mESCs were treated with various concentrations of MMC for 4 days before cell viability was measured.
- Similar to (A), except that mESCs were treated with cisplatin, another ICL‐inducing chemical, at various concentrations.
- Proposed model for METTL14 C‐terminal IDR arginine methylation‐mediated regulation of m6A RNA modification and its effects on ICL DNA repair. PRMT1‐mediated arginine methylation of the C‐terminal IDR of METTL14 promotes its interactions with RNA substrates and RNAPII, which enables efficient m6A deposition on transcripts involved in ICL repair. The deposition of m6A enhances the translation efficiency of these DNA repair genes, promoting the recovery of mESCs from DNA damage. Inhibiting METTL14 arginine methylation using the type I PRMT inhibitor MS023 reduces m6A deposition and the protein synthesis of ICL repair genes, thus sensitizing mESCs to DNA damage‐induced cell death.
Data information: In both (A) and (B), each point represents the average of three independent replicates and error bars represent standard deviation (SD). Statistical analysis was performed using Student’s t‐test. *P < 0.05.
Source data are available online for this figure.
Discussion
This study identified a unique functional role of arginine methylation in RNA m6A modification and gene expression through the regulation of IDR‐mediated protein–RNA and protein–protein interactions (Fig 7C), expanding our current knowledge about the role of arginine methylation in RNA metabolism. Considering the widespread impact of m6A‐mediated regulation across the human genome, the discovery of m6A regulation by METTL14 arginine methylation has broad implications in normal development and human diseases.
Arginine methylation as a regulator of RGG/RG motif‐containing IDRs
Although most protein domains must adopt a well‐defined structure to function, a large fraction of the proteome consists of IDRs that do not form defined three‐dimensional structures yet exhibit biological activity (Oldfield & Dunker, 2014; Protter et al, 2018). Specifically, IDR‐mediated liquid–liquid phase separation (LLPS) has emerged as a fundamental biophysical process governing the organization of high‐order chromatin architecture (Strom et al, 2017; Gibson et al, 2019; Zhang & Kutateladze, 2019), transcription (Boija et al, 2018; Cho et al, 2018; Sabari et al, 2018), and DNA repair (Kilic et al, 2019; Pessina et al, 2019), as well as many other membraneless organelles, such as stress granules and P‐bodies (Molliex et al, 2015; Boeynaems et al, 2017). IDRs exhibit a marked bias in their amino acid composition, including a high proportion of charged residues, such as arginine and lysine, and are predicted to be enriched for methylation (Oldfield & Dunker, 2014). Arginine can mediate multivalent interactions with nucleotides and proteins through hydrogen bonding and π‐stacking. The RGG/RG motif‐containing IDR of the METTL14 C terminus is conserved from flies to humans (Appendix Fig S1) and is crucial for METTL3/METTL14 RNA methyltransferase activity by contributing to RNA substrate binding (Scholler et al, 2018). Although it is yet to be determined if the C‐terminal IDR can promote METTL14 LLPS, our study reveals, for the first time, that PRMT1 can catalyze the arginine methylation of this IDR and regulate METTL14 protein function and m6A deposition.
Arginine methylation imparts bulkiness and hydrophobicity of a protein and can either positively or negatively affect protein–RNA and protein–protein interactions. We show that arginine methylation of the C‐terminal IDR of METTL14 enhances its interactions with RNA substrates and RNAPII (Figs 3 and 4). Although our data suggest that RNA is involved in mediating the METTL14–RNAPII interaction, arginine methylation may regulate this interaction through other mechanisms. For example, arginine methylation may promote the interaction with methylarginine “reader” proteins, such as the Tudor‐domain containing protein 3 (TDRD3) and the survival motor neuron (SMN) protein, both of which have been reported to interact with RNAPII (Sims et al, 2011; Yang et al, 2014; Zhao et al, 2016). Alternatively, this modification could enhance the interaction of the RGG/RG motif with RNAPII, as a recent report demonstrated that hnRNPG can directly bind to the phosphorylated carboxy‐terminal domain (CTD) of RNAPII through its RGG/RG motifs (Zhou et al, 2019). While beyond the scope of this study, testing these two hypotheses will provide additional insights into the molecular mechanisms by which arginine methylation regulates METTL14 function.
METTL14 arginine methylation and co‐transcriptional m6A deposition
m6A has been identified in chromatin‐associated pre‐mRNA (Ke et al, 2017; Louloupi et al, 2018), suggesting that its deposition is co‐transcriptional. However, it is still unclear how transcription machinery modulates the activity and specificity of the METTL3/METTL14 methyltransferase complex to control m6A deposition. The interaction of the METTL3/METTL14 complex with RNAPII, as shown in this study (Fig 4) and reported by others (Slobodin et al, 2017; Huang et al, 2019), provides a molecular basis for this co‐transcriptional RNA modification. Surprisingly, this interaction is dramatically reduced upon loss of METTL14 methylation (Fig 4B and C), indicating that arginine methylation of METTL14 could be an important molecular mechanism regulating co‐transcriptional m6A deposition. Indeed, mESCs expressing arginine methylation‐deficient METTL14 exhibited a significant reduction in global m6A levels (~40%, Fig 5B), particularly near the stop codon and at the beginning of the 3′ UTR of protein‐coding genes (Fig 5H). Although the loss of METTL14 arginine methylation reduces its interaction with all forms of RNAPII (Fig 4B), it is possible that elongating RNAPII (S2‐p) prefers to interact with hypermethylated METTL14 for m6A deposition in the coding sequence (CDS) and the 3′ UTR, because PRMT1, the enzyme that catalyzes METTL14 arginine methylation, has been found in the RNAPII elongation complex through interacting with the transcription elongating factor SPT5 (Kwak et al, 2003). Furthermore, histone H3 trimethylation at lysine 36 (H3K36me3), a histone mark that is tightly associated with transcription elongation, was recently shown to guide co‐transcriptional m6A deposition (Huang et al, 2019). H3K36me3 recruits METTL14 through a direct interaction, thus enriches the METTL3/METTL14 methyltransferase complex at this histone mark. Although the H3K36me3‐interacting region of METTL14 was mapped to its N‐terminal α‐helical motif (Huang et al, 2019), it remains possible that arginine methylation of the C‐terminal RGG/RG motif‐containing IDR could enhance METTL14–H3K36me3 engagement through processes such as LLPS in vivo. Alternatively, H3K36me3 could directly or indirectly promote PRMT1‐catalyzed METTL14 arginine methylation, thus enabling the enrichment of hypermethylated METTL14 in the vicinity of this elongation‐associated histone mark for enhanced m6A deposition.
m6A RNA methylation in the regulation of DNA repair
Loss of METTL14 arginine methylation leads to ~40% m6A reduction on cellular mRNAs (Fig 5B), an effect likely caused by overall reduced METTL14/METTL3 methyltransferase activity (Fig 3G) and/or uncoupling of co‐transcriptional m6A deposition (Fig 4B). Our GO analysis of METTL14 arginine methylation‐dependent m6A sites not only identified known m6A‐regulated cellular processes, such as stem cell population maintenance, but also revealed a previously underappreciated role of m6A in regulating DNA repair gene expression (Fig 5I). Interestingly, these m6A sites are mainly located in the long internal exons of DNA repair genes (Fig 6A and Appendix Fig S4C). Using polysome profiling analysis, we demonstrate that the METTL14 arginine methylation‐dependent m6A modification of these transcripts is essential for promoting their efficient protein translation (Fig 6D–F). It was recently reported that m6A in mRNA coding regions can promote translation by recruiting m6A reader YTHDC2 (Mao et al, 2019). Therefore, it is possible that YTHDC2 is involved in the translation of these DNA repair genes.
Although our study uncovered a novel function of m6A in promoting DNA repair gene expression, a recent study by Xiang and colleagues reported the rapid, reversible accumulation of m6A RNA at the sites of UV irradiation, which recruits DNA polymerase κ (POLK) as an early response for DNA repair (Xiang et al, 2017). Interestingly, we found that the POLK transcript is also decorated with m6A, and loss of METTL14 arginine methylation caused a ~50% reduction in its m6A levels (Dataset EV2), indicating that POLK expression may also be subjected to arginine methylation‐dependent m6A regulation. Recently, METTL3 was reported to be recruited to DNA damage sites through ATM‐mediated phosphorylation at S43, which enhances m6A deposition on DNA damage‐associated RNAs to facilitate DNA repair (Zhang et al, 2020). Together, these studies highlight a crucial function of the m6A RNA modification in the regulation of DNA repair through the direct recruitment of DNA repair machinery as an early response to DNA damage and the enhancement of DNA repair gene expression as a sustained, long‐term response.
Consistent with these findings, genetic knockout and inhibition of PRMT1, which dampens METTL14 arginine methylation, also sensitized mESCs to MMC‐ and cisplatin‐induced cell death (Fig EV5E–F). Of particular relevance to this observation, Musiani and colleagues reported that, in response to cisplatin treatment, PRMT1 is recruited to chromatin to activate the transcription of genes involved in the senescence‐associated secretory phenotype by methylating histone H4 (Musiani et al, 2020). This finding suggests that PRMT1 functions through multiple pathways to promote cell survival in response to DNA damage. Recently, PRMTs have emerged as promising therapeutic targets for treating human malignancies, including solid tumors and blood cancers (Yang & Bedford, 2013; Guccione & Richard, 2019). Our work reveals that deficiencies in the repair of ICLs could be a specific vulnerability of PRMT inhibitor‐treated cells, suggesting that PRMT inhibition may be a promising strategy to sensitize cancer cells to existing chemotherapy drugs.
Materials and Methods
Plasmids and antibodies
Flag‐METTL3 (#53739), Flag‐METTL14 (#53740), Flag‐RNA Pol II (#35175), pMD2.G (#12259), and pSPAX2 (#12260) were purchased from Addgene. GST‐tagged PRMT1, PRMT2, PRMT3, CARM1, PRMT6, PRMT7, and PRMT8, as well as Myc‐PRMT5, plasmids were used to purify recombinant enzymes and have been described before (Huang et al, 2018). GFP‐tagged PRMT1, PRMT3, and PRMT6 were used for mammalian expression and have been described before (Yang et al, 2015). Human METTL14 cDNA was cloned into pGEX‐6P‐1 (GE Healthcare Life Sciences), pCMV‐HA (Clontech), p3xFlag‐CMV‐7.1 (Sigma), and pLV‐EF1a‐IRES‐Blast (Addgene, #85133) vectors. All R‐to‐K mutants of METTL14 were generated using a site‐directed mutagenesis kit (Agilent Technologies). The sequences of all primers used in this study are listed in Appendix Table S1.
The following antibodies were used for either IP or Western blot analysis: anti‐METTL14 (HPA038002, Sigma), anti‐METTL3 (A301‐567A, Bethyl), anti‐PRMT1 (A300‐722A, Bethyl), anti‐PRMT6 (IMG‐506, IMGENEX), anti‐Atrip (A7139, ABClonal), anti‐Fancm (12954‐1‐AP, Proteintech), anti‐Palb2 (14340‐1‐AP, Proteintech), anti‐Flag (F3165, Sigma), rabbit anti‐GFP (A6455), mouse anti‐GFP (sc9996, Santa Cruz Biotechnology), anti‐β‐ACTIN (A5441, Sigma), anti‐RNAPII (39097, Active motif), anti‐RNAPII S2p (91115, Active motif), anti‐RNAPII S5p (sc‐47701, Santa Cruz Biotechnology), mouse anti‐HA (901501, BioLegend), rabbit anti‐HA (3724S, Cell Signaling Technology), and anti‐ADMA (13522S, Cell Signaling Technology). The ASYM26 antibody was kindly provided by Dr. Stéphane Richard (McGill University). The PRMT3 antibody was kindly provided by Dr. Mark T. Bedford (MD Anderson Cancer Center). Detailed reagent information is listed in Appendix Table S2.
In vitro methylation assays
For the in vitro protein methylation assay, the reactions were carried out in 30 μl of phosphate‐buffered saline (PBS; pH 7.4) containing 0.5–1.0 μg substrate, 3 μg recombinant enzymes, and 0.42 μM 3H‐SAM (79 Ci/mmol from 7.5 μM stock solution; PerkinElmer Life Sciences). Each reaction was incubated at 30°C for 1 h, separated by SDS–PAGE, transferred to a PVDF membrane, and exposed to film for 1 day at −80°C. After exposure, the membrane was washed with methanol and stained with Ponceau S to visualize total protein loaded.
For the in vitro RNA methylation assay, reactions were carried out in a 96‐well Streptavidin FlashPlate (#SM9103001PK, PerkinElmer). In each well, the 20‐μl reaction mixture contained 200 nM biotin‐labeled RNA oligonucleotides (5′UACACUCGAUCUGGACUAAAGCUGCUC3′), 20 mM Tris (pH 7.5), 0.01% Triton X‐100, 1 mM DTT, 0.2 U/ml RNasin, 1% glycerol, 420 nM 3H‐SAM, and the indicated amounts of recombinant Flag‐METTL3 and Flag‐METTL14. Each in vitro methylation reaction was incubated at room temperature for 2 h. Enzymatic activity was measured in counts per min using a scintillation counter (PerkinElmer).
Immunoprecipitation of arginine methylated proteins
To detect arginine methylated proteins, cells were either left untreated or treated with the Type I PRMT inhibitor MS023 (1 μM) for 2 days. Cell pellets were lysed in 1x RIPA buffer (20 mM Tris–HCl [pH 7.5], 150 mM NaCl, 1% NP‐40, 0.5% sodium deoxycholate, 0.1% SDS, and protease inhibitor) for 1 h at 4°C. The lysates were sonicated on ice and clarified by centrifugation, followed by pre‐clearing with protein G agarose. The lysates were subsequently immunoprecipitated with specific antibodies, as indicated. Immunoprecipitated proteins were analyzed by Western blot using arginine methylation‐specific antibodies.
Recombinant protein purification
GST‐tagged proteins were purified from Escherichia coli strain BL21(DE3). A single colony of indicated plasmids was picked and cultured in 10ml LB Broth with 100 μg/ml ampicillin overnight. 40 ml fresh LB Broth with 100 μg/ml ampicillin was added the next day. The protein expression was induced with 1 mM IPTG at 30°C for 4 h. The cells were sonicated in PBS on ice and clarified by centrifugation. The lysates were subsequently incubated with Glutathione Sepharose 4B resin (GE Healthcare Life Sciences) overnight at 4°C. The GST‐tagged proteins were eluted with 10 mg/ml reduced L‐Glutathione in elution buffer (100 mM Tris–HCl, pH 7.4, with 150 mM NaCl) after washing three times with PBS buffer.
For the purification of Flag‐tagged recombinant proteins, HEK293 cells were transfected with indicated plasmids for 48 h and lysed in Co‐IP buffer (20 mM Tris–HCl [pH 7.5], 150 mM NaCl, 1% NP‐40, and protease inhibitor) at 4°C for 1 h. The lysates were briefly sonicated on ice and clarified by centrifugation. The lysates were subsequently incubated with anti‐Flag M2 magnetic beads overnight at 4°C. The Flag‐tagged proteins were eluted with 200 μg/ml 3xFlag peptide in TBS buffer (50 mM Tris–HCl, pH 7.4, with 150 mM NaCl) after washing three times with Co‐IP buffer.
GST pull‐down
All GST‐tagged proteins used in this study were purified from Escherichia coli strain BL21(DE3). Cells were lysed in lysis buffer containing 20 mM Tris–HCl (pH 7.4), 150 mM NaCl, 0.1% NP‐40, and protease inhibitors, and the cell lysates were incubated with purified GST‐tagged recombinant proteins with gentle rocking overnight at 4°C. Glutathione Sepharose beads (GE Healthcare Life Sciences) were added to the protein and lysate mixture and incubated with gentle rocking for 2 h at 4°C. The mixture was centrifuged, the supernatant was discarded, and the beads were washed three times with the cell lysis buffer. After centrifuging again, the pellet was resuspended in 30 μl 2X SDS sample buffer and heated at 95°C for 5 min. The samples were loaded on SDS–PAGE gels and analyzed by Western blot using the indicated antibodies.
Co‐IP assay
Cells were lysed in Co‐IP buffer (20 mM Tris–HCl [pH 7.4], 150 mM NaCl, 0.1% NP‐40, and protease inhibitors). After brief sonication, the lysate was centrifuged at 13,800 g for 10 min at 4°C. For each IP, the supernatant was incubated with 2 μg of the indicated antibody with gentle rocking overnight at 4°C. The next day, protein A/G beads (Thermo Scientific) were added to the antibody–cell lysate mixture and incubated with gentle rocking for 2 h at 4°C. The immunocomplex was precipitated by centrifugation and washed three times with the cell lysis buffer. The samples were loaded on SDS–PAGE gels and analyzed by Western blot using the indicated antibodies.
Immunofluorescence
The HeLa cells transfected with the indicated plasmids were grown on glass coverslips to the desired confluence (85%) before fixation. First, the cells were rinsed with PBS and were fixed with ice‐cold methanol for 20 min at room temperature. After blocking with 20% newborn calf serum for 1 h, the cells were incubated with the indicated antibodies at 4°C overnight. The cells were then stained with a fluorescence‐labeled secondary antibody and stained with 4′,6‐diamidino‐2‐phenylindole (DAPI). The coverslips were then sealed and examined using an Olympus BX50 fluorescence microscope.
Electrophoretic mobility shift assay (EMSA)
The 5′ 6‐FAM‐labeled ssRNA oligonucleotide (5′UACACUCGAUCUGGACUAAAGCUGCUC3′) was incubated with increasing amounts of indicated proteins at 4°C in 10 ul reaction buffer containing 50 mM Tris (pH 7.9), 250 mM KCl, 50 mM MgCl2, 0.5 mM EDTA, and 0.2 U/ml RNasin for 1 h. The reactions were then resolved on 6% native acrylamide gels (37.5:1 acrylamide:bis‐acrylamide) in 0.5x TBE buffer. The mobility shift of oligonucleotides was detected using Bio‐Rad ChemiDoc Imaging System.
Identification of METTL14 arginine methylation sites by LC‐MS/MS
Flag‐tagged recombinant METTL14 protein purified from HEK293 cells was resolved on an 8% SDS–PAGE gel and stained with SimplyBlue™ SafeStain (Invitrogen™, cat. no. LC6065). The protein band was excised and de‐stained, followed by in‐gel digestion using Trypsin/Lys‐C Mix (Promega, cat. no. V5073), according to the manufacturer’s instructions. After overnight digestion, the peptides were extracted three times by adding 50% ACN/0.1% TFA solution, 60% ACN/0.1% TFA solution, and 80% ACN/0.1% TFA solution to the gel pieces. The combined peptide extracts were evaporated using a Savant SpeedVac SVC 100H Centrifugal Evaporator. The peptides were dissolved in 1% formic acid (Fisher Chemical, cat. no. A11750) and analyzed by reversed‐phase LC/MS. The mass spectrometric analysis was carried out using a Thermo Scientific Orbitrap Fusion Mass Spectrometer equipped with an Easy Spray source and an Easy‐nLC1000 system. The raw spectra files were searched using both Proteome Discoverer Software with Sequest (Version 2.0) and the Mascot algorithm (Mascot 2.5.1).
Fluorescence polarization assay
Fluorescence polarization assays were performed in black, low‐flange, flat‐bottom 384‐well microplates with a nonbinding surface (Corning, MA). Various amounts of recombinant WT and mutant METTL14 proteins were incubated with 1 nM of a 5′ 6‐FAM‐labeled RNA probe (5′UACACUCGAUCUGGACUAAAGCUGCUC3′) in the binding buffer containing 20 mM Tris (pH 7.5), 0.01% Triton X‐100, 1 mM DTT, 0.2 U/ml RNasin, and 1% glycerol. Binding was performed for 15 min at 37°C. Fluorescence polarization was measured using a Biotech Synergy H4 plate reader (excitation 485 nm, emission 528 nm), and dissociation constants were calculated using GraphPad Prism 8.0.
Lentivirus packaging and stable mESC line generation
Lentiviruses were made by co‐transfecting each pLV‐EF1a‐IRES‐Blast METTL14 overexpression vector (WT and RK) with pSPAX2 and pMD2.G at a 4:3:1 ratio into 293T cells. The supernatant was harvested 48 h after transfection and filtered through a 0.45 μm filter. The virus was concentrated using PEG‐it Virus Precipitation Solution (#LV810A‐1, System Biosciences). Mettl14 KO mESCs, kindly provided by Dr. Jacob H. Hanna (Weizmann Institute of Science, Israel), were seeded in 6‐well plates and infected with the packaged lentiviruses in the presence of 5 μg/ml polybrene (Sigma). 24 h after infection, the mESCs were treated with 5 μg/ml blasticidin for 1 week to select those expressing WT or RK mutant METTL14 (KO + WT and KO + RK, respectively). mESCs were cultured under feeder‐free conditions supplemented with mouse leukemia inhibitory factor (GeminiBio).
RNA m6A quantification by LC‐MS/MS
Total RNA was isolated from the indicated cell lines using TRIzol reagent (Invitrogen). The polyadenylated RNA from these cells was isolated using two rounds of purification on oligo d(T)25 magnetic beads (Thermo Fisher). 25 ng of poly(A)+ RNA was digested using nuclease P1 (1 U, Sigma) in 20 μl of buffer containing 20 mM NH4OAc, (pH 5.5) at 42°C for 2 h, followed by the addition of FastAP buffer (2.3µl) and alkaline phosphatase (1 U, Thermo Fisher) and incubation at 37°C for 4 h. The sample was then filtered (0.22 μm pore size, 4 mm diameter, Millipore), and 5 μl of the solution was injected into a SCIEX Triple Quad 6500 + LC‐MS/MS system. The nucleosides were separated by reverse‐phase ultra‐performance liquid chromatography on a C18 column (Agilent) with online mass spectrometry detection performed in positive electrospray ionization mode. The nucleosides were quantified using the nucleoside‐to‐base ion mass transitions of 282 to 150 (m6A) and 268 to 136 (A). Nucleoside concentrations were determined by comparison to a standard curve obtained from pure nucleoside standards run with the same batch of samples. The m6A/A ratio was calculated based on the calibrated concentrations.
Colony formation and alkaline phosphatase staining assay
The mESCs were seeded at a 500 cells/well concentration in a 6‐well plate for 7 days. The cell culture medium was aspirated, and the cells were washed once with 1 ml of 1x PBST (1x PBS containing 0.05% Tween‐20). Subsequently, the cells were stained with an Alkaline Phosphatase Staining Kit (Biopioneer), according to the manufacturer’s instructions.
Proliferation and viability assay
To assess cell proliferation and viability, cells were cultured in 96‐well plates and counted at the indicated times using a CCK‐8 Cell Counting Kit‐8 (Dojindo), measuring the absorbance on a microplate reader using a 450‐nm filter.
Protein sequence alignment using ClustalW
The parameters for the alignment using ClustalW were the following: Gap Penalty: 10, Gap Length Penalty: 0.2, Delay Divergent Sequence: 30%, Protein Weight Matrix: Gonnet Series for multiple alignment parameters. For pairwise alignment: Gap Penalty: 10, Gap Length: 0.1, Protein Weight Matrix: Gonnet 250.
MeRIP‐seq (m6A‐seq)
Using a modified m6A‐seq protocol (Dominissini et al, 2013), we profiled the genome‐wide m6A methylomes of WT, Mettl14 KO, KO + WT, and KO + RK mESCs. For each sample, we analyzed three biological replicates using both RIP‐seq with an m6A‐specific antibody and standard RNA‐seq of the input control. Total RNA was extracted from the mESCs using TRIzol reagent. Enrichment of mRNA from total RNA was performed using a Dynabeads mRNA Purification Kit (Invitrogen), according to the manufacturer’s instructions. mRNA samples were chemically fragmented into ~100‐nucleotide‐long fragments by incubation with 10X RNA Fragmentation Reagent (Invitrogen) at 90°C for 90 s. The fragmentation reaction was stopped by adding 0.5 M EDTA, followed by standard ethanol precipitation. The fragmented RNA samples were resuspended in 10 mM Tris–HCl (pH 7.4). 20 ng of fragmented RNA to be used as input control was stored in −80°C in a final volume of 8.5 µl in FPF (Fragment, Prime, Finish) mix from the TruSeq Stranded Total RNA Kit (Illumina). The remaining RNA was subjected to m6A‐seq. Specifically, 2 µg of fragmented mRNA was incubated for 2 h at 4°C with 4 µg of affinity‐purified anti‐m6A polyclonal antibody (Synaptic Systems) in m6A‐IP buffer (150 mM NaCl, 0.1% NP‐40, 10 mM Tris–HCl, [pH 7.4], and 0.2 U/µl of RNasin). The RNA–antibody mixture was immunoprecipitated by incubation with protein A beads (Millipore) at 4°C for another 2 h. The beads were extensively washed with m6A‐IP buffer. The bound RNA was eluted with m6A elution buffer (150 mM NaCl, 6.7 mM m6A nucleotides, 0.1% NP‐40, 10 mM Tris–HCl, [pH 7.4], and 0.2 U/µl of RNasin) and elution wash buffer (150 mM NaCl, 0.1% NP‐40, 10 mM Tris–HCl, [pH 7.4], 0.2 U/µl of RNasin). The m6A eluate was purified using Agencourt AMPure XP beads (Beckman Coulter). The immunopurified RNA and non‐IP input control samples were used for library generation using the TruSeq Stranded Total RNA Kit (Illumina). Single‐end, 50‐nucleotide sequencing was performed on an Illumina HiSeq 4000 platform, according to the manufacturer’s instructions.
Gene expression quantification
The RNA‐seq reads of the input control RNA from WT, Mettl14 KO, KO + WT, and KO + RK mESCs were aligned to the mouse genome (GRCm38) with GENCODE annotation (release M13) using Kallisto (version 0.43.0) (Bray et al, 2016). Gene expression was reported in TPM (transcripts per million).
m6A peaks and differential m6A peaks calling procedure
We mapped the m6A‐seq reads to the mouse genome (GRCm38) with GENCODE annotation (release M13) using STAR (version 2.5.3a) (Dobin et al, 2013). Only uniquely mapped reads were used for m6A peak calling. Then, we ran CLAM (version 1.2.0) with local window size w = 100, P‐value correction using the Bonferroni correction, and m6A peaks were called as significant 100‐bp windows. To call METTL14 arginine methylation‐dependent (differential) m6A peaks between any two samples, we selected 100‐bp windows called as m6A peaks (peak window) in at least one replicate of one sample and compared the average peak window intensity signals for each sample (the ratio of reads per kilobase per million mapped reads [RPKM] in IP over input control). First, we calculated the fold change between samples for each peak window. Then, if the fold change was > 1.5 and the average RPKM of the peak window in the input controls was greater than 1 for both samples, t‐tests were performed to compare the peak intensities. Peak windows with P < 0.05 were considered as differential m6A sites. To call METTL14 arginine methylation‐independent (common) m6A peaks between any two samples, we followed a similar procedure as we did for differential m6A sites, expect that we required that 1/1.1 < fold change < 1.1, and that the P‐value ≥ 0.1.
PCA analysis of biological replicates based on m6A peaks
To investigate the reproducibility (variation) of the m6A peaks among biological replicates in WT, KO, KO + WT, and KO + RK mESCs, we performed unsupervised principal component analysis (PCA) of m6A peaks. First, we selected the peaks that were called by CLAM in at least one sample. Then, for each peak, we assigned 1 to the sample if the peaks were present and 0 if not. PCA was conducted via sklearn function in Python. The top two principle components that explained the highest percentage of the variance were chosen to visualize the m6A profiles of the twelve samples.
m6A motif finding, topological distribution, and composition analysis
Motifs were identified in the m6A peak windows for each sample using HOMER (Heinz et al, 2010) with the following parameters: motif_len = 5,6,7; size = 100; motif_num = 10. We performed a genome‐wide analysis to determine the topological distribution of m6A in the 5′UTR, coding sequence (CDS), and 3′UTR by splitting each transcript region into 50 bins with equal size. The frequency of m6A peaks in each bin was calculated as the number of m6A peaks per transcript. We then analyzed the composition of m6A peaks by looking at the proportions of m6A peaks in the 3′UTR, 5′UTR, CDS, other exons, and intron regions. Other exons were defined as the exons that could not be mapped to 3′UTR, 5′UTR, or CDS.
Differential topological distribution analysis of m6A peaks
Transcripts were first binned into 50 parts for the 5′UTR, CDS, and 3′UTR, respectively. To determine which sample of each comparison had more transcripts with m6A, the number of transcripts with or without m6A peaks were counted for each bin. Fisher’s exact tests were performed to evaluate statistical significance. A stringent FDR threshold of 0.01 was used to correct for multiple hypothesis testing.
RNA secondary structure analysis
To analyze the likelihood of RNA sequences to form secondary structures, we first obtained 200‐nt RNA sequences covering 100 nt of each putative m6A peak (including the RRACU motif), 50 nt upstream, and 50 nt downstream. The RNA secondary structures of differential and common m6A sites between the KO + WT and KO + RK mESCs were analyzed by using RNAfold (https://github.com/ViennaRNA/ViennaRNA) (Lorenz et al, 2011), which predicts RNA secondary structures and forgi (https://viennarna.github.io/forgi/graph_tutorial.html), a package developed by the RNAfold group to define the structure results predicted by RNAfold. Four RNA secondary structures (Helix/Stem, Hairpin loop, Bulge loop + Interior loop, and Multi‐branched loop) can be identified using the forgi software.
GO analysis of genes with differential m6A peaks
The GO annotation file was downloaded from ftp://ftp.ebi.ac.uk/pub/databases/GO/goa/MOUSE/. We analyzed the enrichment of GO terms among genes with differential m6A peaks and background genes (genes with expression levels TPM ≥ 1 in both samples) for KO + RK versus KO + WT. The hypergeometric test was performed to identify the significantly enriched GO terms for each comparison.
Reverse transcription‐quantitative PCR (RT–qPCR)
Total cellular RNA was extracted using TRIzol reagent and analyzed for integrity using the Agilent 2100 Bioanalyzer (Agilent Technologies). Total RNA (1 μg) was then used as a template to synthesize cDNA using the High‐Capacity cDNA Archive Kit (Applied Biosystems), according to the manufacturer’s instructions, and qPCR was subsequently performed on a CFX96 Real‐time System C1000 Touch Thermal Cycler (Bio‐Rad). RNA levels were normalized to the endogenous control gene Actb (ACTIN). Data analysis was performed using the Bio‐Rad CFX Manager 3.1. The experimental cycle threshold (Ct) was calibrated against the ACTIN control product. All amplifications were performed in triplicate.
RNA immunoprecipitation (RIP)‐qPCR
Cells were crosslinked with 1% formaldehyde for 10 min, and crosslinking was stopped by the addition of glycine to a final concentration of 0.25 M for 5 min. Cells were washed twice with cold PBS and lysed in RIP buffer (50 mM Tris–HCl [pH 7.5], 150 mM NaCl, 1% NP‐40, 0.5% sodium deoxycholate, 1 mM PMSF, 2 mM VRC, and protease inhibitors) with sonication. The cell lysates were centrifuged, and the supernatant was transferred to a clean tube. 2 µg METTL14 antibody was added to the supernatant and incubated overnight at 4°C with shaking. Dynabeads were added to the supernatant the next day and incubated at 4°C for 4 h with shaking. The beads were washed three times for 5 min with washing buffer I (50 mM Tris–HCl [pH 7.5], 1 M NaCl; 1% NP‐40, 1% sodium deoxycholate, and 2 mM VRC) and three times for 5 min with washing buffer II (50 mM Tris–HCl [pH 7.5], 1 M NaCl, 1% NP‐40, 1% sodium deoxycholate, 2 mM VRC, and 1 M urea). After washing, the beads were incubated in 100 μl of elution buffer (100 mM Tris–HCl [pH 8.0], 200 mM NaCl, 10 mM EDTA, 1% SDS, and 0.2 mg/ml Proteinase K) for 1 h at 42°C, followed by 1 h at 65°C. RNA was then extracted using TRIzol reagent and reverse transcribed into cDNA using the High‐Capacity cDNA Reverse Transcription Kit. The primers for RIP‐qPCR are listed in Appendix Table S1.
mRNA half‐life
Cells were treated with 5 µg/ml actinomycin D at different time points (0, 3, 6, and 9 h) before harvest. RNA was purified using TRIzol reagent and reverse transcribed into cDNA using the High‐Capacity cDNA Reverse Transcription Kit. The primers for the mRNA half‐life assay are listed in Appendix Table S1.
Polysome profiling
Cells were pre‐treated with 100 μg/ml cycloheximide for 5 min at 37°C, followed by washing using ice‐cold PBS containing 100 μg/ml cycloheximide. Cells were pelleted, lysed on ice in lysis buffer, then centrifuged. The supernatant was collected and loaded onto a 10/50% (w/v) sucrose gradient, followed by centrifugation at 270,860 g in an SW40 rotor (Beckman) for 3 h at 4°C. Sucrose solutions were freshly prepared in cell lysis buffer (20 mM HEPES [pH 7.6], 100 mM KCl, 5 mM MgCl2, 1% Triton X‐100, and 100 µg/ml cycloheximide, supplemented with protease inhibitor and RNase inhibitor). Separated samples were fractionated, and OD260 values were measured. An aliquot of the ribosome fraction was used to extract total RNA using TRIzol reagent for real‐time PCR analysis.
Statistical analysis
All experiments were performed at least three times. Statistical comparisons were performed using Student’s t‐tests. P < 0.05 was considered statistically significant.
Author contributions
Biochemical, molecular, and cellular experiments: ZW, LS, WY, TA, QA‐H, and JMS; Bioinformatics analysis: ZP; m6A‐seq: SA; Mass spectrometry analysis for m6A quantification: BTH; Supervision: CH; Experimental design, execution, data analysis, and manuscript preparation: ZW and ZP; Research supervision, data interpretation, and manuscript preparation: LL and YY.
Conflict of interest
C.H. is a scientific founder and a member of the scientific advisory board of Accent Therapeutics, Inc. All other authors declare no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Expanded View/Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Dr. Yi Xing (Children’s Hospital of Philadelphia) for discussion on the bioinformatics analysis. We thank Ross Tomaino (Taplin Mass Spectrometry Facility) for mass spectrometry analysis of METTL14 arginine methylation sites. We thank Dr. Jacob Hanna (Weizmann Institute of Science) for sharing WT and Mettl14 KO mESCs. We thank Dr. Stéphane Richard (McGill University) for sharing Prmt1 KO mESCs. B.T. Harada is supported by an NIH fellowship (CA221007); C. He is supported by an NIH grant (HG008935); Y.Z. Yang is supported by an NIH grant (GM133850); and L. Lin is supported by an NIH grant (GM121827). C. He. is an investigator of the Howard Hughes Medical Institute. We thank Dr. Kerin Higa for providing helpful comments on scientific writing.
The EMBO Journal (2021) 40: e106309.
Contributor Information
Lan Lin, Email: linlan@chop.edu.
Yanzhong Yang, Email: yyang@coh.org.
Data availability
The MeRIP‐seq (m6A‐seq) datasets produced in this study are available in the following database:
MeRIP‐seq (m6A‐seq) data: Gene Expression Omnibus GSE160108 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE160108
References
- Adams JM, Cory S (1975) Modified nucleosides and bizarre 5'‐termini in mouse myeloma mRNA. Nature 255: 28–33 [DOI] [PubMed] [Google Scholar]
- Aguilo F, Zhang F, Sancho A, Fidalgo M, Di Cecilia S, Vashisht A, Lee DF, Chen CH, Rengasamy M, Andino B et al (2015) Coordination of m(6)A mRNA methylation and gene transcription by ZFP217 regulates pluripotency and reprogramming. Cell Stem Cell 17: 689–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan‐Zambrano G, Robson SC, Aspris D, Migliori V, Bannister AJ, Han N et al (2017) Promoter‐bound METTL3 maintains myeloid leukaemia by m(6)A‐dependent translation control. Nature 552: 126–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K et al (2014) m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15: 707–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford MT, Clarke SG (2009) Protein arginine methylation in mammals: who, what, and why. Mol Cell 33: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc RS, Richard S (2017) Arginine methylation: the coming of age. Mol Cell 65: 8–24 [DOI] [PubMed] [Google Scholar]
- Boeynaems S, Bogaert E, Kovacs D, Konijnenberg A, Timmerman E, Volkov A, Guharoy M, De Decker M, Jaspers T, Ryan VH et al (2017) Phase separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Mol Cell 65: 1044–1055.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boija A, Klein IA, Sabari BR, Dall'Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM et al (2018) Transcription factors activate genes through the phase‐separation capacity of their activation domains. Cell 175: 1842–1855.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert FM, Cote J, Boulanger MC, Richard S (2003) A proteomic analysis of arginine‐methylated protein complexes. Mol Cell Proteomics 2: 1319–1330 [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L (2016) Near‐optimal probabilistic RNA‐seq quantification. Nat Biotechnol 34: 525–527 [DOI] [PubMed] [Google Scholar]
- Cho WK, Spille JH, Hecht M, Lee C, Li C, Grube V, Cisse II (2018) Mediator and RNA polymerase II clusters associate in transcription‐dependent condensates. Science 361: 412–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong PA, Vernon RM, Forman‐Kay JD (2018) RGG/RG motif regions in RNA binding and phase separation. J Mol Biol 430: 4650–4665 [DOI] [PubMed] [Google Scholar]
- Deans AJ, West SC (2011) DNA interstrand crosslink repair and cancer. Nat Rev Cancer 11: 467–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrosiers R, Friderici K, Rottman F (1974) Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci USA 71: 3971–3975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D, Moshitch‐Moshkovitz S, Schwartz S, Salmon‐Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob‐Hirsch J, Amariglio N, Kupiec M et al (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A‐seq. Nature 485: 201–206 [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch‐Moshkovitz S, Salmon‐Divon M, Amariglio N, Rechavi G (2013) Transcriptome‐wide mapping of N(6)‐methyladenosine by m(6)A‐seq based on immunocapturing and massively parallel sequencing. Nat Protoc 8: 176–189 [DOI] [PubMed] [Google Scholar]
- Dubin DT, Taylor RH (1975) The methylation state of poly A‐containing messenger RNA from cultured hamster cells. Nucleic Acids Res 2: 1653–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eram MS, Shen Y, Szewczyk M, Wu H, Senisterra G, Li F, Butler KV, Kaniskan HU, Speed BA, Dela Sena C et al (2016) A potent, selective, and cell‐active inhibitor of human type I protein arginine methyltransferases. ACS Chem Biol 11: 772–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan V, Guo A, Trudgian D, Thomas B, Acuto O (2015) Comprehensive identification of arginine methylation in primary T cells reveals regulatory roles in cell signalling. Nat Commun 6: 6758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geula S, Moshitch‐Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon‐Divon M, Hershkovitz V, Peer E, Mor N, Manor YS et al (2015) Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347: 1002–1006 [DOI] [PubMed] [Google Scholar]
- Gibson BA, Doolittle LK, Schneider MWG, Jensen LE, Gamarra N, Henry L, Gerlich DW, Redding S, Rosen MK (2019) Organization of chromatin by intrinsic and regulated phase separation. Cell 179: 470–484.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guccione E, Richard S (2019) The regulation, functions and clinical relevance of arginine methylation. Nat Rev Mol Cell Biol 20: 642–657 [DOI] [PubMed] [Google Scholar]
- Guo A, Gu H, Zhou J, Mulhern D, Wang Y, Lee KA, Yang V, Aguiar M, Kornhauser J, Jia X et al (2014) Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol Cell Proteomics 13: 372–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M, Chen Z, Deng X, Xiao G, Auer F et al (2019) Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co‐transcriptionally. Nature 567: 414–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Wang Z, Narayanan N, Yang Y (2018) Arginine methylation of the C‐terminus RGG motif promotes TOP3B topoisomerase activity and stress granule localization. Nucleic Acids Res 46: 3061–3074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffrey SR, Kharas MG (2017) Emerging links between m(6)A and misregulated mRNA methylation in cancer. Genome Med 9: 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG et al (2011) N6‐methyladenosine in nuclear RNA is a major substrate of the obesity‐associated FTO. Nat Chem Biol 7: 885–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S, Pandya‐Jones A, Saito Y, Fak JJ, Vagbo CB, Geula S, Hanna JH, Black DL, Darnell JE Jr, Darnell RB (2017) m(6)A mRNA modifications are deposited in nascent pre‐mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev 31: 990–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilic S, Lezaja A, Gatti M, Bianco E, Michelena J, Imhof R, Altmeyer M (2019) Phase separation of 53BP1 determines liquid‐like behavior of DNA repair compartments. EMBO J 38: e101379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A et al (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak YT, Guo J, Prajapati S, Park KJ, Surabhi RM, Miller B, Gehrig P, Gaynor RB (2003) Methylation of SPT5 regulates its interaction with RNA polymerase II and transcriptional elongation properties. Mol Cell 11: 1055–1066 [DOI] [PubMed] [Google Scholar]
- Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y, Sun HY, Zhu Q, Baidya P, Wang X et al (2017) Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation. Cell Res 27: 444–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X et al (2014) A METTL3‐METTL14 complex mediates mammalian nuclear RNA N6‐adenosine methylation. Nat Chem Biol 10: 93–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Höner zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL (2011) ViennaRNA package 2.0. Algorithms Mol Biol 6: 26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louloupi A, Ntini E, Conrad T, Orom UAV (2018) Transient N‐6‐Methyladenosine transcriptome sequencing reveals a regulatory role of m6A in splicing efficiency. Cell Rep 23: 3429–3437 [DOI] [PubMed] [Google Scholar]
- Mao Y, Dong L, Liu XM, Guo J, Ma H, Shen B, Qian SB (2019) m(6)A in mRNA coding regions promotes translation via the RNA helicase‐containing YTHDC2. Nat Commun 10: 5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP (2015) Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163: 123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiani D, Giambruno R, Massignani E, Ippolito MR, Maniaci M, Jammula S, Manganaro D, Cuomo A, Nicosia L, Pasini D et al (2020) PRMT1 is recruited via DNA‐PK to chromatin where it sustains the senescence‐associated secretory phenotype in response to cisplatin. Cell Rep 30: 1208–1222.e9 [DOI] [PubMed] [Google Scholar]
- Noack F, Calegari F (2018) Epitranscriptomics: a new regulatory mechanism of brain development and function. Front Neurosci 12: 85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield CJ, Dunker AK (2014) Intrinsically disordered proteins and intrinsically disordered protein regions. Annu Rev Biochem 83: 553–584 [DOI] [PubMed] [Google Scholar]
- Ozdilek BA, Thompson VF, Ahmed NS, White CI, Batey RT, Schwartz JC (2017) Intrinsically disordered RGG/RG domains mediate degenerate specificity in RNA binding. Nucleic Acids Res 45: 7984–7996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak MR, Scherer CA, Chen J, Roshon MJ, Ruley HE (2000) Arginine N‐methyltransferase 1 is required for early postimplantation mouse development, but cells deficient in the enzyme are viable. Mol Cell Biol 20: 4859–4869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessina F, Giavazzi F, Yin Y, Gioia U, Vitelli V, Galbiati A, Barozzi S, Garre M, Oldani A, Flaus A et al (2019) Functional transcription promoters at DNA double‐strand breaks mediate RNA‐driven phase separation of damage‐response factors. Nat Cell Biol 21: 1286–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS et al (2014) Mammalian WTAP is a regulatory subunit of the RNA N6‐methyladenosine methyltransferase. Cell Res 24: 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protter DSW, Rao BS, Van Treeck B, Lin Y, Mizoue L, Rosen MK, Parker R (2018) Intrinsically disordered regions can contribute promiscuous interactions to RNP granule assembly. Cell Rep 22: 1401–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajyaguru P, Parker R (2012) RGG motif proteins: modulators of mRNA functional states. Cell Cycle 11: 2594–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roignant JY, Soller M (2017) m(6)A in mRNA: an ancient mechanism for fine‐tuning gene expression. Trends Genet 33: 380–390 [DOI] [PubMed] [Google Scholar]
- Roundtree IA, Evans ME, Pan T, He C (2017) Dynamic RNA modifications in gene expression regulation. Cell 169: 1187–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall'Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC et al (2018) Coactivator condensation at super‐enhancers links phase separation and gene control. Science 361: eaar3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholler E, Weichmann F, Treiber T, Ringle S, Treiber N, Flatley A, Feederle R, Bruckmann A, Meister G (2018) Interactions, localization, and phosphorylation of the m(6)A generating METTL3‐METTL14‐WTAP complex. RNA 24: 499–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Wei J, He C (2019) Where, when, and how: context‐dependent functions of RNA methylation writers, readers, and erasers. Mol Cell 74: 640–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ 3rd, Rojas LA, Beck DB, Bonasio R, Schuller R, Drury WJ 3rd, Eick D, Reinberg D (2011) The C‐terminal domain of RNA polymerase II is modified by site‐specific methylation. Science 332: 99–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledz P, Jinek M (2016) Structural insights into the molecular mechanism of the m(6)A writer complex. Elife 5: e18434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slobodin B, Han R, Calderone V, Vrielink J, Loayza‐Puch F, Elkon R, Agami R (2017) Transcription impacts the efficiency of mRNA translation via co‐transcriptional N6‐adenosine methylation. Cell 169: 326–337.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, Karpen GH (2017) Phase separation drives heterochromatin domain formation. Nature 547: 241–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thandapani P, O'Connor TR, Bailey TL, Richard S (2013) Defining the RGG/RG motif. Mol Cell 50: 613–623 [DOI] [PubMed] [Google Scholar]
- Tripsianes K, Madl T, Machyna M, Fessas D, Englbrecht C, Fischer U, Neugebauer KM, Sattler M (2011) Structural basis for dimethylarginine recognition by the Tudor domains of human SMN and SPF30 proteins. Nat Struct Mol Biol 18: 1414–1420 [DOI] [PubMed] [Google Scholar]
- Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, Chou T, Chow A, Saletore Y, MacKay M et al (2017) The N(6)‐methyladenosine (m(6)A)‐forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med 23: 1369–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Doxtader KA, Nam Y (2016a) structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell 63: 306–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, Gong Z, Wang Q, Huang J, Tang C et al (2016b) Structural basis of N(6)‐adenosine methylation by the METTL3‐METTL14 complex. Nature 534: 575–578 [DOI] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G et al (2014a) N6‐methyladenosine‐dependent regulation of messenger RNA stability. Nature 505: 117–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C (2015) N(6)‐methyladenosine modulates messenger RNA translation efficiency. Cell 161: 1388–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC (2014b) N6‐methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 16: 191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Yue M, Wang J, Kumar S, Wechsler‐Reya RJ, Zhang Z, Ogawa Y, Kellis M, Duester G et al (2018) N(6)‐methyladenosine RNA modification regulates embryonic neural stem cell self‐renewal through histone modifications. Nat Neurosci 21: 195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, Shi H, Skibbe J, Shen C, Hu C et al (2018) METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell 22: 191–205.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Laurent B, Hsu CH, Nachtergaele S, Lu Z, Sheng W, Xu C, Chen H, Ouyang J, Wang S et al (2017) RNA m(6)A methylation regulates the ultraviolet‐induced DNA damage response. Nature 543: 573–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Bedford MT (2013) Protein arginine methyltransferases and cancer. Nat Rev Cancer 13: 37–50 [DOI] [PubMed] [Google Scholar]
- Yang Y, Hadjikyriacou A, Xia Z, Gayatri S, Kim D, Zurita‐Lopez C, Kelly R, Guo A, Li W, Clarke SG et al (2015) PRMT9 is a type II methyltransferase that methylates the splicing factor SAP145. Nat Commun 6: 6428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, McBride KM, Hensley S, Lu Y, Chedin F, Bedford MT (2014) Arginine methylation facilitates the recruitment of TOP3B to chromatin to prevent R loop accumulation. Mol Cell 53: 484–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KJ, Ringeling FR, Vissers C, Jacob F, Pokrass M, Jimenez‐Cyrus D, Su Y, Kim NS, Zhu Y, Zheng L et al (2017) Temporal control of mammalian cortical neurogenesis by m(6)A methylation. Cell 171: 877–889.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Liu J, He C (2015) RNA N6‐methyladenosine methylation in post‐transcriptional gene expression regulation. Genes Dev 29: 1343–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccara S, Ries RJ, Jaffrey SR (2019) Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol 20: 608–624 [DOI] [PubMed] [Google Scholar]
- Zhang C, Chen L, Peng D, Jiang A, He Y, Zeng Y, Xie C, Zhou H, Luo X, Liu H et al (2020) METTL3 and N6‐methyladenosine promote homologous recombination‐mediated repair of DSBs by modulating DNA‐RNA hybrid accumulation. Mol Cell 79: 425–442.e7 [DOI] [PubMed] [Google Scholar]
- Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, He X, Semenza GL (2016) Hypoxia induces the breast cancer stem cell phenotype by HIF‐dependent and ALKBH5‐mediated m(6)A‐demethylation of NANOG mRNA. Proc Natl Acad Sci USA 113: E2047–2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kutateladze TG (2019) Liquid‐liquid phase separation is an intrinsic physicochemical property of chromatin. Nat Struct Mol Biol 26: 1085–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Xing Y (2017) CLIP‐seq analysis of multi‐mapped reads discovers novel functional RNA regulatory sites in the human transcriptome. Nucleic Acids Res 45: 9260–9271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao DY, Gish G, Braunschweig U, Li Y, Ni Z, Schmitges FW, Zhong G, Liu K, Li W, Moffat J et al (2016) SMN and symmetric arginine dimethylation of RNA polymerase II C‐terminal domain control termination. Nature 529: 48–53 [DOI] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH et al (2013) ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49: 18–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou KI, Shi H, Lyu R, Wylder AC, Matuszek Z, Pan JN, He C, Parisien M, Pan T (2019) Regulation of co‐transcriptional pre‐mRNA splicing by m(6)A through the low‐complexity protein hnRNPG. Mol Cell 76: 70–81.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Expanded View/Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
The MeRIP‐seq (m6A‐seq) datasets produced in this study are available in the following database:
MeRIP‐seq (m6A‐seq) data: Gene Expression Omnibus GSE160108 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE160108