

In this review, Lee and Olefsky discuss the characteristics of chronic inflammation in the major metabolic tissues and how obesity triggers these events, including a focus on the role of adipose tissue hypoxia and macrophage-derived exosomes.
Keywords: β-cell dysfunction, glucose intolerance, immunometabolism, inflammation, insulin resistance, macrophage, metaflammation
Abstract
Obesity is the most common cause of insulin resistance, and the current obesity epidemic is driving a parallel rise in the incidence of T2DM. It is now widely recognized that chronic, subacute tissue inflammation is a major etiologic component of the pathogenesis of insulin resistance and metabolic dysfunction in obesity. Here, we summarize recent advances in our understanding of immunometabolism. We discuss the characteristics of chronic inflammation in the major metabolic tissues and how obesity triggers these events, including a focus on the role of adipose tissue hypoxia and macrophage-derived exosomes. Last, we also review current and potential new therapeutic strategies based on immunomodulation.
Insulin resistance is a key feature of obesity and type 2 diabetes mellitus (T2DM). Since obesity is the most common cause of insulin resistance in humans, the obesity epidemic is the major cause of the rising global incidence of T2DM. In nondiabetic subjects with obesity, hyperinsulinemia usually compensates for the underlying insulin resistance, maintaining glucose homeostasis within normal or near normal levels. When compensatory hyperinsulinemia fails and insulin levels decline due to development of a β-cell defect, hyperglycemia and the typical T2DM state ensues, indicating that both insulin resistance and β-cell dysfunction are needed for the full development of this disease. In obesity, chronic tissue inflammation is a well-reported key contributor to decreased insulin sensitivity, particularly in adipose tissue and the liver. However, several recent reports have shown obesity-associated inflammatory responses in pancreatic islets (Zhao et al. 2003; Warren et al. 2006; Ehses et al. 2007; Böni-Schnetzler et al. 2008; Richardson et al. 2009; Mahdi et al. 2012; Jourdan et al. 2013; Kamada et al. 2013; Butcher et al. 2014; Hasnain et al. 2014; Ying et al. 2019), suggesting that chronic islet inflammation can also lead to β-cell dysfunction in the context of obese T2DM subjects.
Although there are a number of potential, and sometimes overlapping, mechanisms that can contribute to insulin resistance and β-cell dysfunction, in this current review we focus on the role of chronic tissue inflammation in these metabolic defects. The reader is referred to other excellent reviews on possible causes of insulin resistance and β-cell dysfunction, independent of chronic tissue inflammation (Halban et al. 2014; DeFronzo et al. 2015; Czech 2017; Newgard 2017; Guilherme et al. 2019; Kahn et al. 2019; Roden and Shulman 2019; Scherer 2019; Alonge et al. 2021; Sangwung et al. 2020).
The interconnections between inflammation and metabolic dysfunction are often described by the term immunometabolism (Hotamisligil 2017). Immunometabolism includes the effects of immune cells on the regulation of systemic metabolism, as well as the effects of metabolism within immune cells on inflammation. Both of these mechanisms are of importance, but in this review we focus our attention on the role of chronic inflammation in the etiology of disordered glucose homeostasis.
Chronic inflammation in adipose tissue, the liver, the central nervous system, the gastrointestinal (GI) tract, pancreatic islets, and muscle have all been described in obese/T2DM states, indicating the complex, multiorgan crosstalk network involved in these disorders. The general metabolic consequences of obesity-induced tissue inflammation are summarized in Table 1. These concepts are mostly derived from mouse studies, and among all these contributing tissues, adipose tissue inflammation in obesity has been the most extensively studied.
Table 1.
The metabolic effects of obesity-associated tissue inflammation

Although the contribution of chronic inflammation in T2DM in obesity has been suggested for many years, in the past two decades a number of studies have been published, bringing out underlying physiologic and molecular mechanisms as to how obesity-associated inflammation leads to insulin resistance and glucose intolerance. One of the earliest studies was reported by Grunfeld and Feingold (Feingold et al. 1989) who showed that treatment of rodents with TNFα leads to hyperglycemia. An additional key early study showed increased TNFα levels in the adipose tissue of obese mice and showed that neutralization of TNFα ameliorates insulin resistance (Hotamisligil et al. 1993). Numerous studies have identified various molecules within the inflammatory signaling cascade that can impair insulin signaling, such as JNK, IKKβ, and various cytokines (Xia et al. 2015; Goldfine and Shoelson 2017; Hotamisligil 2017; Zhao et al. 2018; Donath et al. 2019; Hall et al. 2020). Many reports have identified a mechanistic link between chronic inflammation and insulin resistance in rodent studies by use of various genetic gain- and loss-of-function studies (Lee et al. 2018). For example, mice engineered to globally deplete IKKβ, JNK, CCR2, or CCL2/MCP-1 show improved insulin sensitivity in obesity (Hirosumi et al. 2002; Arkan et al. 2005; Weisberg et al. 2006). Adipocyte or hepatocyte KO of JNK, HIF-1α, NEMO, or TLR4 also produce enhanced insulin sensitivity (Sabio et al. 2008; Wunderlich et al. 2008; Jia et al. 2014; Lee et al. 2014). In contrast, adipocyte-specific overexpression of Ccl2 or Hif1a induces insulin resistance and glucose intolerance on a normal chow diet (Kanda et al. 2006; Halberg et al. 2009). Most importantly, a large number of macrophage (or hematopoietic cell)-specific KOs of inflammatory molecules have been reported that all lead to protection from insulin resistance and glucose intolerance (Saberi et al. 2009; Olefsky and Glass 2010; Han et al. 2013; Hill et al. 2014; Takikawa et al. 2016; Desai et al. 2017). However, while there are a number of studies in human adipose tissue (Lofgren et al. 2000; Fabbrini et al. 2013; Hill et al. 2018), the liver (Senn et al. 2002; Ghazarian et al. 2017), primary adipocytes (Liu et al. 1998), skeletal muscle (Austin et al. 2008), and pancreatic islets (Maedler et al. 2002) that are supportive of this immunometabolism process (Pickup et al. 1997), the concept that chronic inflammation causes insulin resistance and β-cell dysfunction still remains to be validated in humans.
Association between T2DM and genetic variations associated with inflammation
While the contribution of chronic inflammation to the development of insulin resistance and T2DM remains to be validated in humans, recent advances in genome-wide association studies (GWAS) suggest that inflammation might be causally related to the incidence of T2DM in humans. While early GWASs showed that T2DM is associated with genetic variations in genes associated with insulin secretion and β-cell function (McCarthy et al. 2010; Dimas et al. 2014), later studies found that genes involved in peripheral insulin sensitivity and adipose tissue function such as PPARG and KLF14 are also associated with the incidence of T2DM (Voight et al. 2010). Moreover, variations in genes regulating T-cell (e.g., PTPRJ and CMIP) or macrophage (e.g., MAEA) function or inflammatory signaling pathways (e.g., WWOX, MAP8IP1, IFNGR1, ST6GAL1, JAZF1, MAP3K1, MACROD1, NFE2L3, and TLR4) are also associated with the incidence of T2DM (Waeber et al. 2000; Kooner et al. 2011; Cho et al. 2012; Manning et al. 2012; Locke et al. 2015; Shungin et al. 2015; Flannick et al. 2019; Liao et al. 2019; Diedisheim et al. 2020). While more inclusive GWAS and whole-exome sequencing studies are being updated (Mahajan et al. 2018; Flannick et al. 2019), many genetic variations associated with the incidence of T2DM are found in noncoding DNA regions. Moreover, heterogeneity in the etiology of T2DM (Philipson 2020) may dilute the contribution of specific genetic variations to disease pathology. One should take these factors into consideration when interpreting GWAS results.
Inflammation in obesity
Adipose tissue
Chronic adipose tissue inflammation is a characteristic feature of obesity, and two early studies published simultaneously in 2003 (Weisberg et al. 2003; Xu et al. 2003) showed that this was largely due to the increased accumulation of proinflammatory macrophages in both human and mouse adipose tissue. Many subsequent reports have documented this accumulation of adipose tissue macrophages (ATMs) in obese adipose tissue and have further characterized these macrophages with respect to surface markers, biological effects, and transcriptomic phenotypes. In addition to macrophages, other innate and adaptive immune cell types participate in obese adipose tissue inflammation (summarized in Fig. 1). However, macrophages are the dominant cell type mediating insulin resistance and metabolic dysfunction in obese adipose tissue. Thus, macrophages are the most abundant immune cell type accumulating in obese adipose tissue and liver. Moreover, these cells are the main immune cells secreting most of the inflammatory cytokines, galectin-3, and exosomes (Weisberg et al. 2003; Zeyda et al. 2007; Li et al. 2016; Ying et al. 2017a), and depletion of macrophages corrects insulin resistance and glucose tolerance in obese mice (Huang et al. 2010; Chatenoud et al. 2011; Lee et al. 2011b; Bu et al. 2013).
Figure 1.
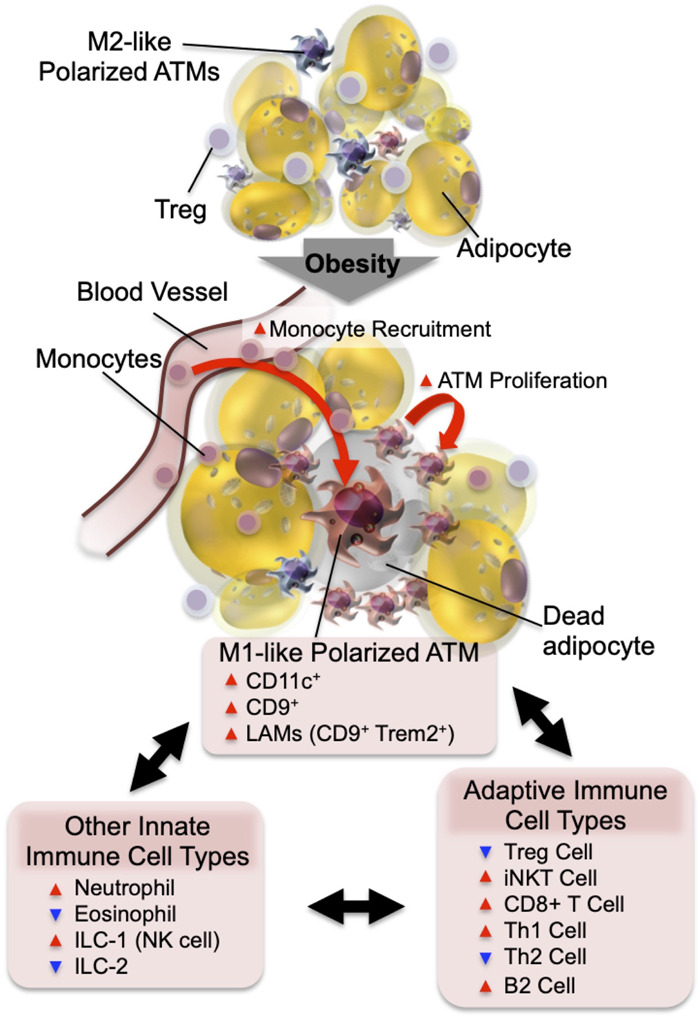
Adipose tissue inflammation in obesity. In the normal state, resident ATMs mostly show an M2-like polarized phenotype. Factors released from Tregs and eosinophils support ATMs to maintain this anti-inflammatory state. In obesity, increased adipocyte chemokine production induces increased blood monocyte recruitment, as well as ATM proliferation. The majority of monocyte-derived ATMs express CD11c and/or CD9 and the M1-like polarized phenotype. The decreased number of eosinophils and Tregs and the increased number of neutrophils, ILC1, CD8+ T cells, Th1 cells, and B2 cells enhance M1-like ATM polarization and adipose tissue inflammation.
Adipose tissue macrophages
In lean mice, macrophages account for 5%–10% of stromal vascular cells in visceral adipose tissue. As early as 3 d after feeding a high fat diet (HFD), increased proinflammatory cytokine expression and macrophage infiltration can be observed in visceral adipose tissue (Lee et al. 2011b). These changes gradually increase during the development of obesity, along with progressive deterioration of insulin sensitivity and glucose tolerance until obesity is fully stabilized and these changes start to plateau. In obesity, ATMs accumulate, becoming the most abundant immune cell type in adipose tissue, and can comprise up to 40% of all stromal vascular cells. Along with the obesity-induced quantitative changes in ATM content, ATMs also undergo phenotypic switching from M2-like polarized anti-inflammatory cells to the M1-like polarized proinflammatory state (Lumeng et al. 2007a). M1 macrophages are considered to represent the “classically activated” proinflammatory phenotype of macrophages, and these macrophages produce proinflammatory cytokines, reactive oxygen species, and nitric oxide that eliminate pathogens. M2 cells represent the “alternatively activated” anti-inflammatory phenotype of macrophages, which generally promote tissue repair and remodeling (Jakubzick et al. 2017; Koelwyn et al. 2018). In obesity, while the number of both M1- and M2-like polarized ATMs is increased, the increase in M1-like polarized ATMs is much greater, skewing the balance toward a tissue proinflammatory state. The majority of M1-like polarized ATMs are derived from circulating monocytes and express CD11c on the surface, whereas the majority of M2-like polarized ATMs are resident and do not display CD11c expression (Lumeng et al. 2007b). The inflammatory state of ATMs can also be modified by various lipid species. Saturated fatty acids (SFAs) derived from either dietary sources or adipocyte triglyceride hydrolysis can drive M1-like ATM polarization through a toll-like receptor 4 (TLR4)-dependent mechanism (Saberi et al. 2009; Holland et al. 2011; Orr et al. 2012; Tao et al. 2017). On the other hand, omega-3 FAs inhibit proinflammatory activation of ATMs through a GPR120-dependent mechanism (Oh et al. 2010).
The terms M1- and M2-like are often used to describe ATMs in lean and obese states as a matter of convenience. However, the concept of M1 and M2 macrophages was first introduced based on the immunologic differences between mouse strains that preferentially promote Th1 or Th2 responses. They are specifically defined as bone marrow progenitor cells polarized to an M1 or M2 state in culture by incubating with LPS/IFNγ or IL-4/IL-13, respectively (Mills et al. 2000). In the in vivo setting, ATMs do not exist within these narrow categories but actually span across a polarization spectrum similar to, but distinct from, M1 or M2 macrophages (Li et al. 2019). Thus, studies using single-cell RNA sequencing (scRNA-seq) analyses revealed that the transcriptomic profiles of ATMs are highly heterogeneous in obese adipose tissue. ATMs in obesity display a proinflammatory profile that is overlapping but different compared with in vitro generated M1 cells (Kratz et al. 2014; Hill et al. 2018; Jaitin et al. 2019). Moreover, they show unique metabolic gene expression profiles, including activation of lysosome-dependent lipid metabolism, collectively termed the “metabolic activation state” (Xu et al. 2013; Kratz et al. 2014; Coats et al. 2017). Hill et al. (2018) reported two subpopulations of ATMs depending on CD9 expression, and the number of CD9+ ATMs increases during the development of obesity. These CD9+ ATMs show unique functional, morphological, and locational features: They express high levels of proinflammatory cytokines, contain intracellular lipid droplets, secrete exosomes, and are located within crownlike structures surrounding dying adipocytes. In addition, a subset of ATMs expresses the monocyte marker Ly6C and also accumulates in obese adipose tissue. Since Ly6C is a marker for monocytes, the Ly6C+ ATMs most likely represent newly recruited monocyte-derived macrophages undergoing differentiation. As found in aortic wall macrophages, these Ly6C+ cells lose expression of Ly6C as they fully differentiate into mature macrophages (Jakubzick et al. 2017). The Ly6C+ ATMs are found outside of crownlike structures and can stimulate adipocyte differentiation (Hill et al. 2018). Many of the transcriptomic signature genes of CD9+ or Ly6C+ ATMs are distinct from in vitro differentiated M1 or M2 macrophages or from each other. Jaitin et al. (2019) conducted unbiased scRNA-seq analyses and found that ATMs can be divided into three subpopulations: one showing transcriptomic signatures of perivascular macrophages and another two subsets with CD9 expression. In line with the findings of Hill et al. (2018), they reported that the number of CD9+ ATMs expands in obesity and can be found within crownlike structures. They further characterized CD9+ ATMs using lineage tracing and found that ∼80% of the CD9+ ATMs are bone marrow derived. Interestingly, CD9+ ATMs can be divided into two subpopulations, depending on the expression of the lipid receptor Trem2. Trem2+ CD9+ ATMs abundantly express genes associated with phagocytosis and lipid metabolism. They termed these cells lipid-associated macrophages (LAMs), and showed that obesity-induced LAM expansion is TREM2 dependent and that Trem2 KO mice show increased weight gain, glucose intolerance, and dyslipidemia on a HFD, suggesting that TREM2-dependent LAM ATMs can mitigate obesity and metabolic dysregulation.
These CD9+ Trem2+ ATMs display functional similarities to CD11c+, M1-like polarized ATMs, and CD9 expression correlates with CD11c expression, although CD11c+ and CD9+ populations are not identical (Hill et al. 2018). This suggests that both the CD9+ and CD11c+ populations might be functionally heterogeneous. Additional studies are required to further define CD11c and CD9 double and single positive and negative populations.
ATMs and the control of local catecholamine levels
Chawla and his colleagues suggested that M2-like polarized ATMs synthesize catecholamines (Nguyen et al. 2011; Qiu et al. 2014). However, this concept was challenged by other groups who reported that ATMs do not produce physiologically relevant levels of catecholamines and do not express the enzyme tyrosine hydroxylase, which is necessary for catecholamine synthesis. Moreover, these groups found IL-4 treatment does not alter energy expenditure and body weight either in cold, ambient, or thermoneutral temperatures (Fischer et al. 2017). As an alternative mechanism for how macrophages might modulate local catecholamine levels, a subset of macrophages, termed sympathetic neuron-associated macrophages (SAMs), are capable of extracellular catecholamine uptake and catabolism (Pirzgalska et al. 2017). SAMs represent another subset of ATMs found in proximity to sympathetic nerves in adipose tissue. In obesity, the number of SAMs increases, contributing to reduced adaptive thermogenic activity. The concept of macrophage-mediated catecholamine catabolism in adipose tissue was also reported by Dixit and his colleagues (Camell et al. 2017), who showed that aging-induced catecholamine resistance is associated with increased ATM catecholamine catabolism. In support of the concept that ATMs regulate local catecholamine levels, systemic depletion of tissue macrophages using clodronate liposomes, but not sympathetic denervation, blocks inguinal WAT beiging in adipocyte Fasn KO mice (Henriques et al. 2020).
Mechanisms of macrophage accumulation
The mechanism by which macrophages accumulate in obese adipose tissue involves (1) increased monocyte/macrophage chemotaxis (increased blood monocyte immigration) and decreased ATM emigration (or increased retention), and (2) higher rates of resident ATM proliferation. In vivo labeling of blood monocytes or resident ATMs have shown that blood monocyte migration into adipose tissue increases in obesity (Lumeng et al. 2007a; Oh et al. 2012). This increase is associated with monocytosis (Nagareddy et al. 2014) and increased production of chemokines such as CCL2/MCP-1 and leukotriene B4 (LTB4) in visceral adipose tissue (Li et al. 2015). Adipocyte-specific depletion of CCL2 reduces obesity-induced ATM accumulation (Kanda et al. 2006). Moreover, genetic deletion or pharmacological inhibition of the receptors for the chemokines CCL2 (CCR2) or LTB4 (LTB4R) blocks obesity-induced ATM accumulation (Lumeng et al. 2007a; Li et al. 2015). This shows that monocyte chemotaxis into adipose tissue is largely mediated by the CCL2/CCR2 and LTB4/LTB4R systems. In addition to increased chemotaxis, the levels of factors associated with macrophage retention, such as Netrin1 and Sema3E, are increased in obese adipose tissue, inhibiting emigration of ATMs (Shimizu et al. 2013; Wanschel et al. 2013; Ramkhelawon et al. 2014).
In addition to chemotaxis into adipose tissue, ATM accumulation is also due to increased resident ATM proliferation, which can be stimulated by CCL2 (Amano et al. 2014). In subsequent studies, increased ATM proliferation in obese adipose tissue was confirmed by separate groups (Zheng et al. 2016).
Other innate immune cells
Although relatively small in number, changes in other innate immune cell types also contribute to the development of adipose tissue inflammation, mainly by regulating ATM polarization and function. For example, innate lymphoid cell type 1 (ILC1) cells, including natural killer cells, reside in normal/lean adipose tissue and contribute to adipose tissue homeostasis (Lee et al. 2016; Boulenouar et al. 2017; Theurich et al. 2017). In obesity, the number of ILC1 cells increases and ILC1-derived cytokines, including IFNγ, stimulate proinflammatory activation of ATMs. On the other hand, the number of ILC2 cells producing Th2 cytokines decreases in obesity (Molofsky et al. 2013; Nussbaum et al. 2013). ILC2-derived cytokines support immigration and maturation of eosinophils in adipose tissue, which produce IL-4 and IL-13 that stimulate M2-like macrophage polarization (Wu et al. 2011). In obese adipose tissue, decreased ILC2 cells are associated with decreased local eosinophil numbers. Finally, the recruitment of neutrophils into adipose tissue increases in obesity, and factors released from neutrophil granules, such as neutrophil elastase and myeloperoxidase, can contribute to the development of insulin resistance (Talukdar et al. 2012; Tam et al. 2020).
Adaptive immune cells
Several types of adaptive immune cells are also involved in adipose tissue inflammation. In obesity, the number of adipose tissue CD8+ T cells increases, and factors released from these cells can facilitate ATM differentiation and chemotaxis (Nishimura et al. 2009). Among CD4+ T cells, the number of proinflammatory T helper (Th) 1 cells increases (Nishimura et al. 2009), whereas the adipose tissue content of anti-inflammatory Th2 cells decreases in obesity, contributing to increased adipose tissue interferon γ (IFNγ) levels and inflammation (Winer et al. 2009). Tregs (CD3+CD4+FOXP3+) are another CD4+ T-cell subset well studied in adipose tissue (Feuerer et al. 2009; Li et al. 2020). There are only a small number of these cells in young mice, but they start to accumulate in visceral adipose tissue at the age of 10–15 wk, due to increased proliferation and decreased turnover. Tregs represent the major CD4+ T-cell subset (40%–80% of total CD4+ T cells) by 20–30 wk of age in normal mice (Bapat et al. 2015). Tregs control the activity of other T cells and restrain the innate immune system by inhibiting monocyte immigration and proinflammatory polarization. In obesity, the number, or proportion, of adipose tissue Treg cells decreases, contributing to increased adipose tissue inflammation (Feuerer et al. 2009). Adipose tissue Tregs show a unique transcriptomic profile distinct from Tregs in other sites, which is largely regulated by peroxisome proliferator-activated receptor γ (PPARγ). Indeed, >80% of adipose tissue Tregs express PPARγ, which is unique to adipose Tregs, and Treg PPARγ mediates a component of the anti-inflammatory effects of thiazolidinediones (TZDs) in obese mice (Cipolletta et al. 2012). γδ T cells abundant in adipose tissue of normal mice support sympathetic innervation and Treg expansion by releasing IL-17 (Kohlgruber et al. 2018; Hu et al. 2020).
Invariant natural killer T (iNKT) cells account for 10%–30% of all adipose tissue-resident T cells (Huh et al. 2013). These are innate-like αβ T cells that are specifically stimulated by lipid antigens through the major histocompatibility complex (MHC)-like molecule CD1d (Huh et al. 2013; Lynch 2014). iNKT cells release IL-10, IL-4, IL13, and IL-2, potentiating M2-like polarization of ATMs. The number of adipose tissue iNKT cells decreases in obesity, and this could contribute to the development of inflammation. However, other reports cast doubt on the beneficial effects of iNKT cells. For example, Satoh et al. (2016) and Wu et al. (2012b) have shown proinflammatory and negative metabolic effects of iNKT cells in obese mice. As a possible explanation for these contradicting results, scRNA-seq analysis found that adipose tissue iNKT cells can be functionally divided into two subsets, depending on NK1.1 expression: NK1.1+ iNKTs are proinflammatory and produce IFNγ, whereas NK1.1− iNKTs are anti-inflammatory and produce IL-10 (LaMarche et al. 2020).
B lymphocytes account for 3%–10% of SVCs in normal adipose tissue and increase up to twofold in obesity or 10-fold in aged mice (Camell et al. 2019). Compared with the spleen and bone marrow, adipose tissue B cells show a higher B1:B2 ratio in normal mice. However, in obesity, the B1:B2 ratio decreases with increased recruitment of proinflammatory B2 cells (Ying et al. 2017b). B2 recruitment into obese visceral adipose tissue is mainly mediated by the LTB4/LTB4R1 system (Ying et al. 2017b; Srikakulapu and McNamara 2020), and these cells can contribute to adipose tissue inflammation and metabolic dysfunction in obesity.
Acute versus chronic inflammation
While numerous studies have shown the detrimental effects of chronic adipose tissue inflammation on the development of systemic insulin resistance in obesity, inflammation is a physiological process necessary for defense against pathogenic invasion or tissue damage and repair. Acute inflammation is necessary to maintain normal adipose tissue function by mediating healthy adipose tissue remodeling to safely accommodate excess lipids into adipose tissue (Wernstedt Asterholm et al. 2014). For example, Zhu et al. (2020) reported that inhibition of inflammatory pathways by inducible overexpression of an adenoviral protein, called RIDα/β (which blocks several inflammatory signaling pathways) reduces weight gain and adiposity and exacerbates glucose and insulin intolerance in HFD/obese mice. Therefore, it is likely that acute adipose tissue inflammation is necessary for adipose tissue homeostasis, whereas chronic inflammation contributes to the development of insulin resistance and adipose tissue dysfunction.
Liver
Hepatic macrophages
Obesity, or a HFD, leads to hepatic steatosis commonly referred to as nonalcoholic fatty liver disease (NAFLD), which can proceed to a condition termed nonalcoholic steatohepatitis (NASH). Obesity is also associated with increased hepatic inflammation, and macrophages are the major source of the proinflammatory cytokines (Stienstra et al. 2010; Xu et al. 2014). Injection of gadolinium or intravenous clodronate liposomes selectively depletes hepatic macrophages owing to the fenestrated hepatic sinusoidal structure. Selective depletion of hepatic macrophages improves insulin resistance and liver steatosis in obese mice or rats (Huang et al. 2010; Lanthier et al. 2010; Lee et al. 2011b; Reid et al. 2016). Liver macrophages can be divided into two major subsets based on their developmental origin: These include yolk-sac-derived resident Kupffer cells (KCs) and bone marrow-derived recruited hepatic macrophages (RHMs). In normal liver, hepatic macrophages account for ∼10% of all hepatic cells, and the majority of them are KCs (Bouwens et al. 1986). In obesity, the number of KCs is unchanged, whereas circulating monocyte chemotaxis and differentiation into macrophages (RHMs) increase, largely through the CCL2/CCR2 system (Obstfeld et al. 2010; Morinaga et al. 2015). While both KCs and RHMs are highly heterogeneous, RHMs express higher levels of M1-like polarized macrophage markers and proinflammatory gene expression, which is exacerbated in obesity (Arkan et al. 2005; Morinaga et al. 2015; Seidman et al. 2020). These results suggest that a major component of obesity-induced liver inflammation can be attributed to increased monocyte infiltration and macrophage differentiation (summarized in Fig. 2).
Figure 2.
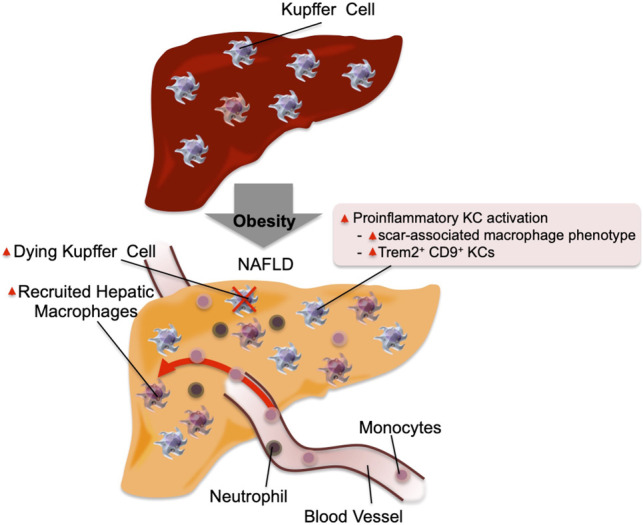
Inflammation in the liver. In the normal physiological state, KCs account for ∼10% of all liver cells. In addition, they scavenge pathogens and show M2-like polarized anti-inflammatory phenotype. In obese steatotic livers, KCs can show increased expression of genes associated with tissue repair and inflammation. These genes include Cd9 and Trem2. This is accompanied by increased KC apoptosis, and a component of KC death is compensated for by increased recruitment of blood monocytes, which differentiate into KC-like macrophages. Chemokines released by steatotic hepatocytes cause increased recruitment of blood monocytes into the liver, which differentiate into proinflammatory macrophages (RHMs) and produce factors that can cause insulin resistance. The recruitment of neutrophils also increases, and the molecules released from neutrophil granules such as neutrophil elastase and myeloperoxidase can induce insulin resistance in hepatocytes.
Recently, single-cell analyses of transcriptomic and epigenetic changes in hepatic macrophages (e.g., scRNA-seq and scATAC-seq) combined with monocyte tracking and KC depletion studies have further elucidated hepatic macrophage phenotypes (Seidman et al. 2020). Thus, in mice fed a NASH-prone HFD, KCs undergo partial loss of their identifying gene expression patterns while gaining a scar-associated macrophage phenotype with Trem2 and Cd9 expression. These changes are accompanied by increased KC apoptosis. As these KC numbers decline, they are replaced by monocyte-derived macrophages that undergo convergent epigenomic and transcriptomic changes and become new KC-like cells.
Other hepatic immune cells
The number of neutrophils is also increased in the liver of obese mice compared with lean/normal mice (Talukdar et al. 2012), and neutrophil-derived cytokines and chemokines contribute to the development of obesity-induced liver inflammation. Moreover, NE released from neutrophils can be taken up by hepatocytes and cause degradation of IRS-2, promoting decreased insulin sensitivity.
Among adaptive immune components, the number of CD8+ cytotoxic T cells increases in the liver of obese mice compared with lean mice through an IFNγ-dependent signaling pathway (Ghazarian et al. 2017). On other hand, the numbers of CD4+ T cells, Tregs, and γδ T cells decrease or remain unchanged in obesity.
Communication between liver cell types
Obesity-induced changes in the liver leading to the development of insulin resistance and NASH involve interactions between intrahepatic cell types such as hepatocytes, various immunocytes, and hepatic stellate cells (Koyama and Brenner 2017). For example, activation of hepatocyte JNK, IKKβ, or IKKε or the unfolded protein response (ER stress) increases production of chemokines and other hepatokines, stimulating KC proinflammatory activation and circulating monocyte and neutrophil recruitment (Arkan et al. 2005; Seki et al. 2012; Reilly et al. 2013; Lee et al. 2018). The incoming immune cells are highly proinflammatory (Morinaga et al. 2015; Seidman et al. 2020), enhancing liver inflammation. Hepatocyte and immunocyte-derived factors such as TGFβ can activate quiescent stellate cells and induce secretion of collagen, leading to liver fibrosis (Hellerbrand et al. 1999; Koyama and Brenner 2017). Moreover, activated stellate cell-derived cytokines and chemokines can enhance liver inflammation, exacerbating liver steatosis and fibrosis (Weiskirchen and Tacke 2014). Please see Koyama and Brenner (2017) and Arab et al. (2018) for excellent reviews on the pathophysiology of NAFLD and NASH.
Muscle
Skeletal muscle accounts for the majority of in vivo insulin-stimulated glucose disposal, and this is decreased in obesity/T2DM, contributing to the development of compensatory hyperinsulinemia and glucose intolerance. Several mechanisms such as mitochondrial dysfunction, microvascular dysfunction, and ectopic fat accumulation (or lipotoxicity) have been proposed as causes of this defect and are well reviewed elsewhere (Serné et al. 2007; Roden and Shulman 2019; Sergi et al. 2019). While relatively less studied compared with adipose tissue and liver, proinflammatory pathways are enhanced with increased macrophage infiltration in skeletal muscle of obese mice and T2DM patients compared with normal mice or human subjects (Fink et al. 2014). Systemic depletion of macrophages or genetic modifications suppressing proinflammatory activation of macrophages improves insulin sensitivity in skeletal muscle in mice (Patsouris et al. 2008; Han et al. 2013), suggesting that inflammation plays a causative role in the development of muscle insulin resistance. Consistent with this, treatment with proinflammatory cytokines induces insulin resistance in cultured myocytes or skeletal muscle in vivo (de Alvaro et al. 2004; Plomgaard et al. 2005). On the other hand, the anti-inflammatory cytokine, IL-10, can play a protective role. For example, muscle-specific overexpression of IL-10 mitigates and muscle-specific deletion of Il10 exacerbates obesity-induced muscle inflammation and insulin resistance (Hong et al. 2009; Dagdeviren et al. 2016).
With respect to mechanisms of muscle inflammation, intermyocellular adipose tissue (IMAT) or perimuscular adipose tissue (PMAT) have been implicated (Khan et al. 2015). Thus, in obesity, IMAT and PMAT expand and show macrophage accumulation, similar to visceral adipose tissue. As in visceral adipose tissue, the majority of macrophages infiltrated into obese skeletal muscle are CD11c+ M1-like polarized macrophages. Moreover, the changes in the proportion of different T-cell subsets (e.g., increased Th1 cells and decreased Tregs) show similar patterns as seen in visceral adipose tissue (Wu and Ballantyne 2017).
Pancreatic islets
β-Cell dysfunction is a critical etiologic component of T2DM, and a variety of mechanisms for this defect have been identified (for reviews, see Halban et al. 2014; Hudish et al. 2019). Several reports have shown islet inflammation can also be etiologically important in causing β-cell dysfunction in T2DM. The expression of proinflammatory cytokines (such as IL-1β, IL-33, IL-23, and IL-24) and macrophage infiltration is increased in the islets of obese/T2DM patients compared with normal subjects (Ehses et al. 2007 ; Böni-Schnetzler et al. 2008; Richardson et al. 2009; Mahdi et al. 2012; Butcher et al. 2014; Hasnain et al. 2014; Kamata et al. 2014; Schludi et al. 2017). Treatment of isolated human islets with proinflammatory cytokines reduces glucose-induced intracellular Ca2+ levels and GSIS (Corbett et al. 1993; O'Neill et al. 2013), and induces β-cell death (Brozzi et al. 2015). Unlike T1DM islets, obesity- or T2DM-associated islet inflammation is dominated by macrophages and does not involve T-cell infiltration (Ehses et al. 2007). The concept that islet inflammation induces β-cell dysfunction was more extensively tested in rodent studies. Systemic depletion of macrophages by intraperitoneal injection of clodronate liposomes potentiates glucose-stimulated insulin secretion (GSIS), as well as insulin sensitivity in obese mice (Eguchi et al. 2012). Ex vivo depletion of macrophages within primary islets isolated from HFD/obese mice enhances GSIS (Eguchi et al. 2012; Ying et al. 2019). Treatment of primary mouse islets with proinflammatory cytokines (e.g., IL-1β, IL-23, IL-24) reduces GSIS (Mahdi et al. 2012; Jourdan et al. 2013; Hasnain et al. 2014). Moreover, Myd88 or TLR4 KO mice are protected from β-cell dysfunction induced by subchronic lipid infusion (lipotoxicity) (Eguchi et al. 2012), while β-cell-specific IL-1 receptor antagonist (IL-1Ra) KO mice develop β-cell dysfunction (Böni-Schnetzler et al. 2018).
While similar to adipose tissue and liver inflammation in causing functional defects in parenchymal cells (in this case, β cells), there is a sharp difference in the mechanism for macrophage accumulation in obese islets compared with liver and adipose tissue. Thus, in the liver and adipose tissue, macrophage expansion largely involves recruitment of circulating monocytes (Weisberg et al. 2003; Morinaga et al. 2015; Jaitin et al. 2019; Seidman et al. 2020a), with a smaller component due to resident macrophage proliferation (Amano et al. 2014). In islets, macrophage accumulation is largely mediated by resident islet macrophage proliferation (Ying et al. 2019). Blood monocytes are recruited to the peri-islet region but do not enter the islet capsule (Eguchi et al. 2012; Ying et al. 2019).
In islets, there are two resident macrophage populations (intraislet and peri-islet macrophages) showing distinct anatomical distribution, phenotypes, and functional properties (Ying et al. 2019). These islet macrophages show a unique transcriptomic profile distinct from M1- or M2-like polarized macrophages or other tissue resident macrophages. Intraislet macrophages substantially proliferate in obesity, whereas peri-islet macrophages lining the islet capsule do not. Moreover, intraislet macrophages, but not peri-islet macrophages, inhibit β-cell insulin secretion. This effect of intraislet macrophages is mediated by a cell–cell contact-dependent mechanism, involving the engulfment of β-cell insulin secretory vesicles. In addition, proinflammatory cytokines produced from islet macrophages also suppress β-cell GSIS (Maedler et al. 2002; Eguchi et al. 2012). For example, IL-1β produced from islet macrophages binds to the IL-1 receptor (IL-1R), which is abundantly expressed on β cells, leading to decreased GSIS through stimulation of the JNK and NF-κB signaling pathways (Kwon et al. 1998; Larsen et al. 2005; Ortis et al. 2006; Wang et al. 2009). Furthermore, islet macrophages (both intraislet and peri-islet) release growth factors such as PDGF and IGF-1, simulating β-cell proliferation (Ying et al. 2019; Nackiewicz et al. 2020). Therefore, it is likely that obesity-associated islet inflammation induces decreased β-cell GSIS and promotes the β-cell proliferation that exists in obese islets. This results in changes to islet function favoring increased basal insulin secretion and decreased GSIS.
One can question whether increased β-cell proliferation by islet macrophages is necessarily detrimental to metabolic homeostasis. Indeed, although adverse effects of islet inflammation on GSIS and β function were reported in chronic obese inflammatory states, inflammation is a physiological process, and acute islet inflammation may be beneficial for the maintenance of islet function in the normal state (Banaei-Bouchareb et al. 2004; Geutskens et al. 2005; Yano et al. 2007; Kayali et al. 2012; Lee et al. 2013; Dror et al. 2017; Burke et al. 2018; Riopel et al. 2018; Ying et al. 2019). Therefore, it is possible that acute inflammation is necessary to maintain islet function, whereas chronic inflammation contributes to the development of β-cell dysfunction.
GI tract
Recent advances in our understanding of the gut microbiota have provided potential new insights as to how obesity affects tissue inflammation and metabolic defects. In both human and rodent obesity, the composition of the gut microbiota changes (dysbiosis), which can modify the host immune system and metabolism. HFD/obese WT mice display increased proinflammatory cytokine expression and IL17-producing γδ T cells, Th1 cells, CD8+ T cells, macrophages, and dendritic cells, along with decreased Tregs, ILC3 cells, and eosinophils in the intestinal lamina propria (Johnson et al. 2015; Monteiro-Sepulveda et al. 2015; Winer et al. 2016). Fecal transplantation experiments and clinical trials further showed that dysbiosis contributes to the development of inflammation and insulin resistance in obesity. For example, transplantation of germ-free mice with fecal preparations from human twins discordant for obesity showed that the obese gut microbiota can induce increased weight gain, adipose tissue mass, and glucose intolerance (Ridaura et al. 2013). Moreover, a clinical study found that administration of intestinal microbiota from lean donors to recipients with metabolic syndrome improved insulin sensitivity (Vrieze et al. 2012). These studies suggest that a component of the obesity-associated metabolic phenotype is due to dysbiosis. Nonetheless, it should be noted that the effects of fecal transplantation were small, and healthy gut microbiome transfer does not normalize the metabolic defects induced by obesity.
In this context, several gut microbiota-derived molecules/mechanisms that can modify host immune tone and metabolism have been reported. For example, in obesity or shortly after a HFD, gut permeability increases, allowing bacterial translocation across the intestinal barrier (Neal et al. 2006; Amar et al. 2011; Ha et al. 2020). Moreover, increased gut permeability also allows gut microbiota-derived molecules, such as lipopolysaccharide (LPS), to leak into the blood circulation (Erridge et al. 2007; Kumar et al. 2011). Increased LPS-stimulated TLR4 induces proinflammatory responses in various immunocytes, gut epithelial cells, and insulin target cells, resulting in insulin resistance and glucose intolerance (Shi et al. 2006; Saberi et al. 2009; Guo et al. 2013; Tao et al. 2017). Plasma levels of another gut microbiota-derived molecule, formyl-methionyl-leucyl-phenylalanine (N-formyl peptides), are also elevated in HFD/obese mice. Genetic deletion or pharmacological inhibition of the N-formyl peptide receptor Fpr1 improves glucose tolerance by increasing glucagonlike peptide 1 section from enteroendocrine L cells with a subsequent increase in β-cell GSIS. Of the bioactive metabolites produced by the gut microbiota, short chain fatty acids (SCFAs) are among the most abundant (Stevens and Hume 1998). SCFAs activate G-protein-coupled receptors (GPCRs), including GPR41 and GPR43, which are expressed in adipocytes, enterocytes, immunocytes, and pancreatic β cells. Several reports showed beneficial effects of GPR43 activation to reduce adiposity and improve insulin sensitivity, as well as β-cell GSIS in obese mice (Tolhurst et al. 2012; Kimura et al. 2013; McNelis et al. 2015). However, conflicting results were also reported (for review, see Ang and Ding 2016): For example, β-cell-specific depletion of both GPR41 and GPR43 increases GSIS and improves glucose tolerance in mice (Tang et al. 2015). In addition to SFAs, secondary and tertiary bile acids produced by gut microbes alter the composition of the bile acid pool (Brufau et al. 2010; Suhre et al. 2010; Haeusler et al. 2013; Ridaura et al. 2013; Yoshimoto et al. 2013) and modulate inflammation and metabolism by stimulating the bile acid receptor 1 (TGR5) and farnesoid X receptor (FXR) (Pols et al. 2011; Wollam and Antebi 2011).
Taken together, the GI tract plays a role in obesity-associated chronic tissue inflammation and metabolic dysregulation. One obvious mechanism is that the development of obesity leads to increased gastrointestinal permeability, such that proinflammatory bacterial products (e.g., LPS), and even bacteria themselves, gain access to surrounding lymph nodes and the systemic circulation. In addition, the dysbiosis of obesity provides another potential mechanism in which specific GI bacterial species, or combinations of species, can contribute to metainflammation. The latter is clear in mice but will require further translational validation in humans. This is a promising field, since further studies could identify specific factors that might be useful in drug discovery.
Hypoxia as initiating event
As noted above, it is well known that obesity leads to an accumulation of proinflammatory macrophages in adipose tissue in both mice and humans. These ATMs are key drivers of the development of insulin resistance and glucose intolerance. Areas of current active interest focus on how macrophage accumulation is initiated at the onset of obesity and how do proinflammatory ATMs cause systemic metabolic dysfunction. Several studies have been published, providing important insights into these questions. In this section, we attempt to integrate these new insights into an overall sequential hypothesis.
For years, it has been known that adipose tissue oxygen tension is decreased in obesity in both mice and humans (Hosogai et al. 2007; Halberg et al. 2009; Pasarica et al. 2009, 2010; Lawler et al. 2016; Seo et al. 2019; Smith et al. 2019). The initial view was that this adipose tissue hypoxic state was due to an imbalance between expanding adipose tissue mass and the available oxygen delivery. However, tissue oxygen tension is maintained by the balance between oxygen supply and demand, and more recent data show that obesity leads to increased adipocyte oxygen consumption (Lee et al. 2014). This increased consumption accounts for ∼40% of the decrease in interstitial oxygen tension in obesity (Seo et al. 2019). The remaining component of the decreased interstitial oxygen tension is not due to decreased arterial oxygen delivery (blood flow into adipose depot and blood O2 saturation) but is explained by decreased functional capillary density in obese adipose tissue (Lee et al. 2014). Therefore, there are two components, decreased capillary density and increased adipocyte oxygen consumption, that contribute to the relative hypoxia of adipose tissue in obesity. As a general matter, hypoxic conditions lead to greater expression of the adipocyte HIF-1α transcription factor, triggering the downstream hypoxia transcriptome response. However, the induction of HIF-1α does not incur until oxygen tensions fall to ≤1% (Seo et al. 2019). Therefore, the decrease in extracellular adipose tissue oxygen tension described in obesity is probably not enough to trigger the HIF-1α hypoxic response. On the other hand, the increase in adipocyte oxygen consumption is quite marked in obesity, bringing the intra-adipocyte oxygen concentration down to levels below ∼1.4%. This is adequate to induce adipocyte HIF-1α expression and the downstream effects mediated by this transcription factor (Hosogai et al. 2007; Halberg et al. 2009; Lee et al. 2014). This view is supported by the finding that hypoxia-induced pimonidazole adducts in obese adipose tissue are confined to adipocytes (Lee et al. 2014). Taken together, obesity-induced increased adipocyte oxygen consumption is likely the major contributor to the intracellular adipocyte hypoxic response, whereas the decreased interstitial oxygen tension probably plays a contributory or permissive role to exaggerate the impact of increased adipocyte oxygen consumption.
The mechanisms by which intracellular oxygen levels regulate HIF-1α have been well described and occur mainly at the protein level with smaller effects on mRNA expression (Ivan and Kaelin 2017; Schödel and Ratcliffe 2019; Semenza 2019). In normoxic conditions, prolyl hydroxylase domain proteins (PHDs) bind to HIF-1α, and PHD-dependent hydroxylation of HIF-1α leads to ubiquitin-dependent proteasomal degradation of the protein. Under hypoxia, PHDs are inactivated, which results in HIF-1α protein stabilization, causing increased HIF-1α protein expression. Although this is the major driver for regulation of HIF-1α expression, in obesity, modest increases in HIF-1α mRNA have also been reported as a contributing factor (Lee et al. 2014).
This induction of adipocyte HIF-1α plays an important role in mediating overall adipose tissue inflammation (Lee et al. 2014). This can occur through multiple mechanisms. For example, several chemokines are downstream transcription targets of HIF-1α, and expression of these chemokines is increased in obesity and other hypoxic conditions. These chemokines serve to recruit monocytes into the hypoxic adipose tissue, which then predominantly differentiate into proinflammatory, M1-like ATMs. As discussed in more detail above, the increase in M1-like ATMs is a key mechanism for the metabolic dysfunction characterized by insulin resistance and glucose intolerance in obesity (Fig. 3). There are many studies in which various proinflammatory genes have been deleted in macrophages, and all of these studies consistently show amelioration of obesity-induced metabolic dysfunction (Arkan et al. 2005; Weisberg et al. 2006; Saberi et al. 2009; Han et al. 2013; Li et al. 2015). Consistent with this formulation, the central role of HIF-1α in the proinflammatory responses leading to insulin resistance has been shown through cell type-specific knockout studies (Jiang et al. 2011; Lee et al. 2011a, 2014; Krishnan et al. 2012; Sun et al. 2013). Thus, adipocyte-specific HIF-1α knockout largely prevents obesity-induced ATM accumulation, adipose tissue inflammation, glucose intolerance, and insulin resistance but does not cause any change in overall body weight. This highlights the importance of HIF-1α in the overall adipose tissue inflammatory response, consistent with the idea that obesity-induced intracellular adipocyte hypoxia leads to HIF-1α induction, which in turn promotes ATM accumulation, inflammation, and subsequent glucose intolerance and insulin resistance.
Figure 3.
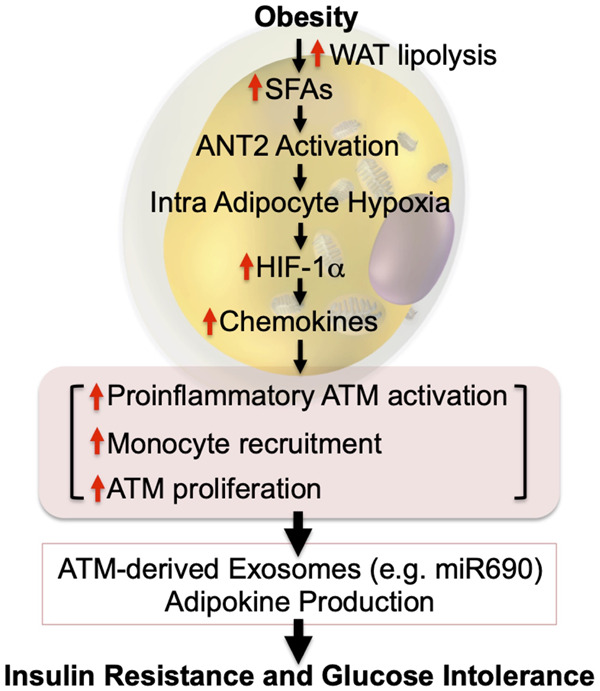
Intracellular adipocyte hypoxia triggers inflammation and insulin resistance. In obesity, increased intracellular FFAs stimulate ANT2-dependent increased mitochondrial uncoupled respiration in adipocytes. Combined with decreased functional capillary density, this leads to intracellular hypoxia and HIF-1α stabilization. HIF-1α induces increased chemokine production, leading to increased immune cell infiltration, including monocytes, and increased ATM proliferation. These changes critically affect ATM exosome secretion, as well as adipocyte exosome and adipokine production, causing systemic insulin resistance. Most of these sequential events have been shown in mouse models. This hypothesis remains to be validated in humans.
The mechanisms underlying obesity-induced increased adipocyte oxygen consumption are also of interest. Adenine nucleotide translocase (ANT) 2 is a mitochondrial inner-membrane protein that pumps protons from the intermembrane space into the mitochondrial matrix (Bertholet et al. 2019). Therefore, an increase in ANT2 activity results in proton leakage from the intermembrane space, leading to uncoupled mitochondrial respiration with enhanced O2 consumption. It should be noted that another important uncoupling protein, UCP1, is not expressed in white adipocytes (Cousin et al. 1992; Wu et al. 2012a). Adipose tissue SFA levels are elevated in obesity, and SFAs can activate ANT2-dependent uncoupling of oxidative metabolism, leading to increased adipocyte oxygen consumption (Lee et al. 2014). In theory, increased fatty acid oxidation could also lead to greater oxygen consumption, but more detailed studies have shown that this component is rather negligible compared with the direct effect of SFAs to activate ANT2 protein. To further solidify this overall concept, deletion of ANT2 in adipocytes can reverse all of these hypoxic responses. Thus, adipocyte-specific ANT2 KO is sufficient to block the obesity-induced increased adipocyte O2 consumption, ameliorating the adipocyte hypoxia response and preventing HIF-1α induction (Seo et al. 2019). This mitigates the development of adipose tissue inflammation with reduced proinflammatory ATM content and decreased expression of inflammatory pathway genes. This is accompanied by a robust improvement in glucose tolerance and insulin sensitivity in obese mice without any changes in body weight.
How does adipose tissue inflammation cause insulin resistance?
The fact that increased obesity-induced proinflammatory ATM content can directly cause insulin resistance is well established, at least in mice, but precisely how M1-like macrophages lead to decreased insulin sensitivity is not entirely clear and has been the subject of intense investigation. A logical presumption is that proinflammatory macrophages release factors that can cause paracrine or systemic effects on insulin target cells to impair insulin signaling. Classically, M1-like polarized macrophages release a variety of cytokines and chemokines. Chemokines act by providing a concentration gradient in which monocytes and other types of immune cells can migrate down that gradient toward the chemokine source; this is the operational definition of chemotaxis. In obese states, some of these chemokines produced in adipose tissue can leak into the circulation. However, circulating chemokines are not able to generate a differential concentration gradient to promote chemotaxis of monocytes out of the circulation into inflamed tissues. In addition, the circulating levels of chemokines due to this “spillover” are generally much lower than the biologically effective concentrations, so they will not have meaningful systemic physiologic effects. Even if a particular chemokine can cause decreased insulin signaling in vitro (Sartipy and Loskutoff 2003), it would still be a challenge to conclude that such a chemokine could act systemically, since the circulating systemic concentrations are so low.
More attention has been placed on secreted cytokines, since it is well known that certain cytokines (e.g., TNFα) have potent effects to cause decreased insulin signaling. TNFα can cause local tissue effects to decrease insulin action (Hotamisligil et al. 1994) by several mechanisms. TNFα can reduce Irs2 and Glut4 expression, promote inhibitory phosphorylation of insulin receptor substrate (IRS) proteins, enhance adipocyte lipolysis (FFA release) and ceramide synthesis, and inhibit PPARγ expression (Stephens et al. 1997; Guilherme et al. 2008). This phenomenon could occur in the various adipose tissue depots, which become enlarged and inflamed during the course of obesity (TNFα is used as an example, since it is the most well studied immune cell-derived cytokine). However, systemic insulin resistance requires decreased insulin sensitivity in muscle and the liver, as well as adipose tissue, and unless muscle and the liver are generating high levels of tissue cytokines, it is unlikely that TNFα or other cytokines that leak out of obese adipose tissue could cause systemic effects. Thus, the circulating concentrations of TNFα and other cytokines are substantially below the levels needed to exert biologic effects in tissues (Stephens et al. 1997; Amar et al. 2007; McGillicuddy et al. 2011). In contrast, adipose tissue levels of cytokines can be quite elevated in obesity, and this could cause local effects. If one administers large amounts of TNFα in vivo, raising circulating levels far higher than they normally exist in obesity, then an insulin-resistant state ensues (Lang et al. 1992). However, this does not mirror the actual pathophysiologic state in obesity in which cytokine levels in the blood, while higher than in normals, are not elevated to levels at which they exert substantial biologic effects. IL-6 may be an exception to this general principle, since the levels of IL-6 that enter the circulation in obesity have been reported to be biologically active. However, studies of IL-6's metabolic activities are mixed with some studies showing insulin-like actions to improve insulin sensitivity, while others report that it inhibits insulin action (Carey et al. 2006; Franckhauser et al. 2008).
Given the above discussion, many laboratories have been trying to identify other factors that are elaborated from M1-like macrophages that could cause metabolic dysfunction. One such factor is Galectin-3. Galectin-3 is produced almost exclusively from macrophages, and in both human and mouse obesity, blood levels are quite high compared with normals and are in the range where they exert biologic effects (Li et al. 2016). Indeed, in humans, circulating levels of Galectin-3 are elevated compared with normal subjects, and the degree of the increase in Galectin-3 levels is correlated with the magnitude of obesity and the degree of insulin resistance. In vitro studies show that Galectin-3 treatment of insulin target cells can directly cause decreased insulin signaling by interfering with the ability of insulin to properly activate the insulin receptor at the cell surface. Loss-of-function studies have been performed by analyzing macrophage-specific Galectin-3 KO mice. Depletion of macrophage Galectin-3 protects HFD mice from the development of glucose intolerance and insulin resistance, while weight gain is entirely normal. Additional gain- and loss-of-function studies with Galectin-3 are consistent with this formulation, suggesting that macrophage-derived circulating Galectin-3 can contribute to the development of insulin resistance in obesity.
The above considerations have led to a search for additional factors that can enter the circulation from obese adipose tissue and cause systemic insulin resistance. Indeed, recent studies suggest that ATM-derived exosomes might fulfill this role. Exosomes are small nanoparticles (∼150 nm) produced by most cell types (Pegtel and Gould 2019). They can be secreted into the interstitial space where they then enter the circulation leading to both paracrine and endocrine-like effects. Exosomes contain a variety of cargo constituents including proteins, lipids, microRNAs (miRNAs), and mRNAs, as well as a number of other RNA species. Recent studies have shown that exosomes derived from ATMs in obese mice can directly cause insulin resistance in vitro when applied to adipocytes, primary hepatocytes, or myocytes (Ying et al. 2017a). These “obese” ATM-derived exosomes can be harvested and administered intravenously to lean mice. When this was done, the lean recipient mice developed glucose intolerance, hyperinsulinemia, and insulin resistance comparable with that seen in the obese state. Importantly, eating behavior and body weight do not change as a result of in vivo exosome treatment. On the other side of this coin, ATM exosomes harvested from chow-fed lean mice produce the opposite phenotype. Treatment of adipocytes, myocytes, or primary hepatocytes with “lean” ATM exosomes directly leads to increased insulin signaling. More importantly, when obese mice were treated with “lean” ATM-derived exosomes for a period of 3 wk, these mice showed markedly improved glucose tolerance with an increase in insulin sensitivity. In a very real sense, these “lean” ATM exosomes produce a therapeutic response, mitigating metabolic dysfunction in obesity. With respect to mechanisms, these studies also showed that the biologic effects of obese and lean ATM exosomes are entirely mediated by their miRNA cargo, since depletion of exosomal miRNAs prevents all of these exosome-mediated actions (Fig. 4).
Figure 4.
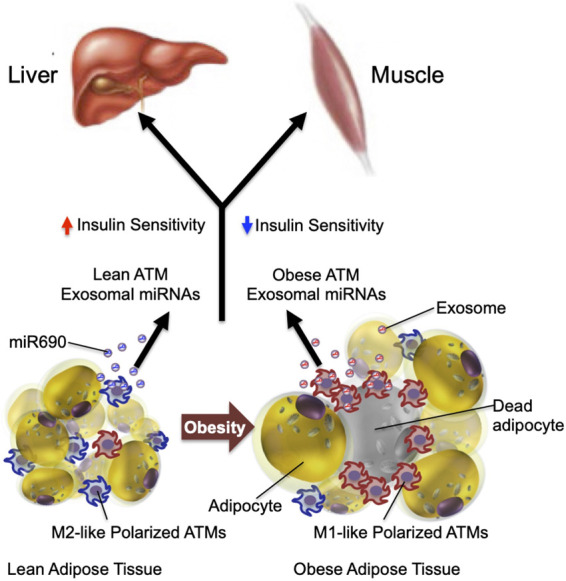
Systemic effect of ATM exosomes. ATMs can regulate systemic insulin sensitivity by releasing miRNA-containing exosomes. Injection of exosomes released from lean mouse ATMs increases insulin sensitivity in obese mice, whereas injection of obese mouse ATM-derived exosomes induces insulin resistance in lean mice. In addition, direct in vitro treatment of insulin target cells with “lean” or “obese” exosomes directly causes insulin sensitivity or resistance, respectively. Most of these sequential events have been shown in mouse models. This hypothesis remains to be validated in humans.
Recent studies have honed in on the specific miRNAs that can produce these beneficial “therapeutic” effects in mice. By conducting complete sequencing of M2-like ATM-derived exosomes, coupled with a bioinformatics approach and screening of specific mRNAs upregulated in lean ATM-derived exosomes, miR-690 was identified as an insulin-sensitizing miRNA. Thus, deletion of miR-690 prevents the beneficial effects of M2-like macrophage exosomes, and an artificial miR-690 mimic reproduces the beneficial effects of lean ATM exosomes on insulin sensitivity and glucose intolerance. In addition, an inhibitor (antagomir) of miR-690 completely blocks the effects of the miR-690 mimic. Thus, miR-690 emerges as a bona fide insulin-sensitizing molecule in both mouse and human cells, and if translational studies are successful, this molecule could potentially lead to an insulin-sensitizing therapeutic.
A comprehensive study from Thomou et al. (2017) identified the presence of several hundred miRNAs in blood exosomes in WT mice. When they studied adipocyte-specific Dicer KO mice, to deplete all miRNAs from any adipocyte-derived exosomes that enter the circulation, they found that ∼60% of the circulating miRNAs were reduced, indicating that adipocytes make important quantitative contributions to the circulating exosome pool. This study also found that miR-99b is highly expressed in exosomes released from brown adipocytes and that miR-99b can inhibit hepatic expression of FGF21, and suggested that the decrease in circulating FGF21 levels is linked to the improved glucose tolerance they observed in the adipocyte Dicer KO mice. Based on miRNA sequencing studies, it is important to note that there is a marked difference in miRNA composition between ATM versus adipocyte-derived exosomes, with only an ∼20% overlapping concordance (Thomou et al. 2017; Ying et al. 2017a). This further emphasizes the necessity to understand the function of cell type-specific exosomes in metabolic disease.
Exosomes are a component of a more diverse group of secreted particles termed extracellular vesicles (EVs). There are several additional published studies showing results consistent with the view that EVs/exosomes-derived from obese adipose tissue can affect insulin sensitivity (Kita et al. 2019). For example, obese human adipose tissue EV/exosome preparations displayed differential expression of several miRNAs that can impact insulin signaling. Exosomes harvested directly from obese adipose tissue impair insulin sensitivity, and one study implicated miR141-3P as playing a causative role (Kranendonk et al. 2014; Dang et al. 2019). It should be noted that in studies using adipose tissue-derived exosomes, one does not know which cell type within adipose tissue is producing the biologically active particles.
Taken together, the above studies lead to the model depicted in Figure 3. This model shows a general sequential hypothesis in which obesity leads to increased SFA activation of ANT2, causing uncoupled respiration with enhanced adipocyte oxygen consumption. This leads to a state of relative hypoxia within the adipocyte, causing HIF-1α induction and downstream inflammatory responses, including recruitment and accumulation of ATMs and increased expression of a variety of proinflammatory genes. This inflammatory state then can drive the generation of insulin and glucose intolerance, and ATM-derived exosomal miRNAs (particularly miR-690) are at least one mechanism leading to the metabolic dysfunction that characterizes the obese state.
Clinical implications
While various genetic gain- and loss-of-function experiments in mice have clearly shown the importance of chronic tissue inflammation in the etiology of insulin resistance and glucose intolerance, the concept remains to be definitively validated in humans. Since these kinds of gene manipulation studies are not possible in humans, we will need to rely on various anti-inflammatory pharmacologic treatment modalities to definitively test this overall concept in humans.
As we refine our mechanistic understanding of the effects of chronic inflammation, more precise tools to test these overall concepts in humans will hopefully emerge. While this remains to be done, there are currently a number of clinical studies providing data that relate to this issue. For example, there are several reports using inhibitory antibodies directed against TNFα in patients with metabolic disease. These agents obviously have great clinical success in the treatment of rheumatoid arthritis and inflammatory bowel disease (Lin et al. 2020; Vulliemoz et al. 2020), and many patients treated for these other conditions are obese or have T2DM. Some of these patients have been analyzed, and small-scale clinical studies have been reported. Unfortunately, the results of these studies have been variable with no clear-cut conclusions emerging. Some reports note modest improvement in glycemia or insulin sensitivity (Kiortsis et al. 2005; Gonzalez-Gay et al. 2006), whereas others find no effects (Ofei et al. 1996; Paquot et al. 2000; Dominguez et al. 2005; Bernstein et al. 2006; Wascher et al. 2011). Large-scale, randomized, double-blind clinical trials will be necessary to sort out any possible effects of TNFα inhibition in metabolic disease. Perhaps the most promising findings are from a meta-analysis study showing that TNF-α antibody treatment might reduce the risk of developing T2DM (Burska et al. 2015), although, again, this would have to be demonstrated in a prospective study for translational validation. Based on the available data, it is reasonable to suggest that the effects of anti-TNFα treatment may not be clinically robust in metabolic disease, with a number of studies showing marginal or no effects. In the absence of large prospective studies, it is useful to consider why TNFα inhibition might not be highly effective in humans, as it has been in obese mice. One possibility is that in humans, TNFα is not as important for insulin resistance as it appears to be in mice, and this remains to be tested. Another scenario is that cytokines like TNFα work predominantly at the tissue level, rather than through circulating TNFα effects, and it is possible that anti-TNFα biologics do not penetrate adipose tissue at high enough concentrations to sufficiently block the high tissue levels of TNFα in obesity.
Inhibitory antibodies directed against another inflammatory cytokine, IL-1β, have also been reported. Some of these studies have been larger scale and prospective and have shown small, but consistent, decreases in glucose and HbA1c levels as a result of IL-1β inhibition (Larsen et al. 2007; Everett et al. 2018). However, these beneficial effects appear to be due to enhanced insulin secretion and β-cell function, rather than an improvement in insulin sensitivity. This may point to the fact that IL-1β plays a significant role in the development of β-cell dysfunction in T2DM but is not mechanistically relevant to the generation of inflammation-induced insulin resistance. A large-scale prospective clinical study, termed the Cantos trial, has been reported for the treatment of CVD, this study showed clinically meaningful reductions in CV events in the IL-1β antibody-treated patients (Ridker et al. 2017). Although there were some significant unwanted side effects related to infections, this study does highlight the potential usefulness of anti-inflammatory therapy in atherosclerosis.
Another potential anti-inflammatory mechanism using salicylic acid derivatives has also been studied in the context of insulin resistance and T2DM. Early smaller-scale studies showed promising effects of salsalate treatment to improve insulin sensitivity and reduce glycemia in T2DM (Goldfine et al. 2008). A subsequent larger-scale prospective study has also shown modest reductions on the order of 0.3%–0.5% in HbA1c in diabetes (Goldfine et al. 2010). While these were encouraging, there has been little progress in clinical studies over the past several years with salicylic acid derivative treatment. Furthermore, it should be noted that while salicylates were initially thought to be anti-inflammatory by inhibiting IKKβ (Yuan et al. 2001), other studies have questioned whether this is the mechanism of action of this class of drugs. Thus, some reports have shown that salicylates may activate AMPK, accounting for metabolic improvement (Hawley et al. 2012).
Other possible approaches have been suggested. These include development of a small molecule agonist for GPR120 to harness the anti-inflammatory, insulin-sensitizing effects of this signaling cascade. Perhaps inhibitors of circulating Galectin-3 or selective inhibition of monocyte chemotaxis into adipose tissue and/or the liver might also prove useful. Although possible in the future, no clinical development programs related to these mechanisms have yet appeared.
Since hepatic inflammation is an important component of NASH pathophysiology, it is useful to briefly summarize the status of specific anti-inflammatory treatments in this disease. There have been two anti-inflammatory mechanisms tested in NASH: ASK1 inhibition (Harrison et al. 2020) and CCR2/5 inhibition (Ratziu et al. 2020). Although the long-term phase 3 clinical studies failed to show any improvement in fibrosis or NASH scores, it is important to point out that while these might be “potential anti-inflammatory” treatments, in fact, they did not cause any anti-inflammatory effects in the clinical trials. In other words, the NASH scores, which include lobular inflammation, were unchanged in the ASK1 and CCR2/5 inhibitor-treated patients. In grading NASH, inflammation is scored separately on a scale of 1–4 and the average value in these studies was ∼2. An effective therapy should lower the inflammation score by at least one point in treated subjects, and in these reports, it did not. Therefore, just as in the failure of “anti-inflammatory” treatments for insulin resistance, one must understand that simply referring to a treatment as anti-inflammatory and finding no effect on NASH or insulin resistance does not mean that the treatment was actually anti-inflammatory. The published clinical data simply have not provided an effective anti-inflammatory mechanism to test the hypothesis. In other words, the absence of positive data is not the same as the presence of negative data.
From a translational point of view, it is important to keep in mind that inhibiting inflammatory responses might lead to suppression of specific aspects of the immune system, which might lead to unwanted safety consequences. This needs to be carefully monitored in any clinical development program involving an anti-inflammatory approach to treat metabolic disease. This also informs the kinds of strategies used to identify drug targets, or drugs, that produce insulin-sensitizing, anti-inflammatory effects. Proximal proinflammatory signals typically activate a number of overlapping and interconnected inflammatory pathways, raising the possibility that targeting a specific distal inflammatory event might have inadequate efficacy if redundant proinflammatory signaling systems are still intact. On the other side of the coin, inhibiting the wider network of inflammatory pathways would probably carry the risk of increased unwanted side effects. In this context, it would be more advantageous to direct a therapy at a specific aspect of the inflammatory response critical to the development of insulin resistance. Obviously, if a drug delivery method were developed that provides tissue selectivity, this would be of great advantage in preventing insulin resistance with less risk of unwanted side effects.
Thiazolidinediones (TZDs) are a highly efficacious class of anti-diabetic drugs that stimulate PPARγ and ameliorate insulin resistance. TZDs have anti-inflammatory effects by directly inhibiting proinflammatory activation of macrophages, promoting differentiation toward the anti-inflammatory M2-like state (Jiang et al. 1998; Ricote et al. 1998; Bouhlel et al. 2007; Nelson et al. 2018). In addition, TZDs can act on PPARγ within Tregs, stimulating these cells to restrict inflammation induced by the innate immune system (Cipolletta et al. 2012). On the other hand, TZDs have been studied for many years, and a number of additional mechanisms for TZD/PPARγ-induced insulin sensitivity have been identified (Tontonoz and Spiegelman 2008). These include stimulation of adiponectin release (Yu et al. 2002; Xia et al. 2018), redistribution of adipose tissue fat storage (de Souza et al. 2001), and induction of direct PPARγ genes (such as Glut-4) (Young et al. 1995), which mediate insulin signaling. In the in vivo clinical situation, it is currently not possible to identify the degree to which TZD-mediated anti-inflammatory effects contribute to the overall insulin-sensitizing, anti-diabetic actions of these agents.
Since obesity is a key pathophysiologic factor in the development of chronic tissue inflammation, insulin resistance, and ultimately T2DM and/or NASH, it is evident that sustained weight loss is a straightforward preventative or treatment modality for these conditions. Unfortunately, while this method of treatment may be straightforward, it is far from easy. There are bariatric surgical approaches, several anti-obesity drug therapies, and numerous forms of lifestyle intervention combining various diets with exercise. Unfortunately, weight loss effects are often short lived, and weight regain (recidivism) is a major unresolved problem with current pharmacologic and lifestyle approaches. Even weight loss after bariatric surgery can wane over time, although, in general, the weight reduction achieved through surgical approaches is far more durable than with lifestyle and drug treatment. Despite the above-mentioned difficulties of sustaining long-term weight loss, it is abundantly clear that weight reduction leads to large therapeutic effects, with respect to decreased tissue inflammation, amelioration of T2DM, and either prevention or reversal of NAFLD/NASH. Since weight loss leads to these multiple beneficial effects concomitantly, it is not possible to dissect out the contributions of tissue inflammation to the overall metabolic improvement induced by weight loss, since multiple factors are changed with any reduction in adiposity.
As discussed earlier, the presence of increased proinflammatory ATMs, as well as other immune cells, has been well documented in human obesity, as is the presence of increased inflammatory responses in the liver, pancreatic islets, and gastrointestinal tract. At this point, the translational findings are associations and correlations and do not prove causality. Since genetic manipulations are not possible in humans, an effective anti-inflammatory modality will be necessary to test this overall concept. As summarized above, a small number of potential anti-inflammatory approaches have been tried, with limited success. Therefore, the field awaits further specific anti-inflammatory treatments to better understand the role of chronic tissue inflammation in the etiology of metabolic dysregulation in obese humans.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This study was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health (R01DK033651, P01DK074868, P30DK063491, and DK09062 to J.M.O. and R01DK124298 to Y.S.L.) and a grant from the Janssen Pharmaceuticals, Inc.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.346312.120.
Freely available online through the Genes & Development Open Access option.
References
- Alonge KM, D'Alessio DA, Schwartz MW. 2021. Brain control of blood glucose levels: implications for the pathogenesis of type 2 diabetes. Diabetologia 64: 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano SU, Cohen JL, Vangala P, Tencerova M, Nicoloro SM, Yawe JC, Shen Y, Czech MP, Aouadi M. 2014. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab 19: 162–171. 10.1016/j.cmet.2013.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amar S, Zhou Q, Shaik-Dasthagirisaheb Y, Leeman S. 2007. Diet-induced obesity in mice causes changes in immune responses and bone loss manifested by bacterial challenge. Proc Natl Acad Sci 104: 20466–20471. 10.1073/pnas.0710335105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amar J, Chabo C, Waget A, Klopp P, Vachoux C, Bermúdez-Humarán LG, Smirnova N, Bergá M, Sulpice T, Lahtinen S, et al. 2011. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol Med 3: 559–572. 10.1002/emmm.201100159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang Z, Ding JL. 2016. GPR41 and GPR43 in obesity and inflammation: protective or causative? Front Immunol 7: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arab JP, Arrese M, Trauner M. 2018. Recent insights into the pathogenesis of nonalcoholic fatty liver disease. Annu Rev Pathol 13: 321–350. 10.1146/annurev-pathol-020117-043617 [DOI] [PubMed] [Google Scholar]
- Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. 2005. IKK-β links inflammation to obesity-induced insulin resistance. Nat Med 11: 191–198. 10.1038/nm1185 [DOI] [PubMed] [Google Scholar]
- Austin RL, Rune A, Bouzakri K, Zierath JR, Krook A. 2008. siRNA-mediated reduction of inhibitor of nuclear factor-κB kinase prevents tumor necrosis factor-α-induced insulin resistance in human skeletal muscle. Diabetes 57: 2066–2073. 10.2337/db07-0763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaei-Bouchareb L, Gouon-Evans V, Samara-Boustani D, Castellotti MC, Czernichow P, Pollard JW, Polak M. 2004. Insulin cell mass is altered in Csf1op/Csf1op macrophage-deficient mice. J Leukoc Biol 76: 359–367. 10.1189/jlb.1103591 [DOI] [PubMed] [Google Scholar]
- Bapat SP, Myoung Suh J, Fang S, Liu S, Zhang Y, Cheng A, Zhou C, Liang Y, LeBlanc M, Liddle C, et al. 2015. Depletion of fat-resident Treg cells prevents age-associated insulin resistance. Nature 528: 137–141. 10.1038/nature16151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein LE, Berry J, Kim S, Canavan B, Grinspoon SK. 2006. Effects of etanercept in patients with the metabolic syndrome. Arch Intern Med 166: 902–908. 10.1001/archinte.166.8.902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertholet AM, Chouchani ET, Kazak L, Angelin A, Fedorenko A, Long JZ, Vidoni S, Garrity R, Cho J, Terada N, et al. 2019. H+ transport is an integral function of the mitochondrial ADP/ATP carrier. Nature 571: 515–520. 10.1038/s41586-019-1400-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böni-Schnetzler M, Thorne J, Parnaud G, Marselli L, Ehses JA, Kerr-Conte J, Pattou F, Halban PA, Weir GC, Donath MY. 2008. Increased interleukin (IL)-1β messenger ribonucleic acid expression in β-cells of individuals with type 2 diabetes and regulation of IL-1β in human islets by glucose and autostimulation. J Clin Endocrinol Metab 93: 4065–4074. 10.1210/jc.2008-0396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böni-Schnetzler M, Häuselmann SP, Dalmas E, Meier DT, Thienel C, Traub S, Schulze F, Steiger L, Dror E, Martin P, et al. 2018. β cell-specific deletion of the IL-1 receptor antagonist impairs β cell proliferation and insulin secretion. Cell Rep 22: 1774–1786. 10.1016/j.celrep.2018.01.063 [DOI] [PubMed] [Google Scholar]
- Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, et al. 2007. PPARγ activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab 6: 137–143. 10.1016/j.cmet.2007.06.010 [DOI] [PubMed] [Google Scholar]
- Boulenouar S, Michelet X, Duquette D, Alvarez D, Hogan AE, Dold C, O'Connor D, Stutte S, Tavakkoli A, Winters D, et al. 2017. Adipose type one innate lymphoid cells regulate macrophage homeostasis through targeted cytotoxicity. Immunity 46: 273–286. 10.1016/j.immuni.2017.01.008 [DOI] [PubMed] [Google Scholar]
- Bouwens L, Baekeland M, De Zanger R, Wisse E. 1986. Quantitation, tissue distribution and proliferation kinetics of Kupffer cells in normal rat liver. Hepatology 6: 718–722. 10.1002/hep.1840060430 [DOI] [PubMed] [Google Scholar]
- Brozzi F, Nardelli TR, Lopes M, Millard I, Barthson J, Igoillo-Esteve M, Grieco FA, Villate O, Oliveira JM, Casimir M, et al. 2015. Cytokines induce endoplasmic reticulum stress in human, rat and mouse β cells via different mechanisms. Diabetologia 58: 2307–2316. 10.1007/s00125-015-3669-6 [DOI] [PubMed] [Google Scholar]
- Brufau G, Stellaard F, Prado K, Bloks VW, Jonkers E, Boverhof R, Kuipers F, Murphy EJ. 2010. Improved glycemic control with colesevelam treatment in patients with type 2 diabetes is not directly associated with changes in bile acid metabolism. Hepatology 52: 1455–1464. 10.1002/hep.23831 [DOI] [PubMed] [Google Scholar]
- Bu L, Gao M, Qu S, Liu D. 2013. Intraperitoneal injection of clodronate liposomes eliminates visceral adipose macrophages and blocks high-fat diet-induced weight gain and development of insulin resistance. AAPS J 15: 1001–1011. 10.1208/s12248-013-9501-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke SJ, Batdorf HM, Burk DH, Martin TM, Mendoza T, Stadler K, Alami W, Karlstad MD, Robson MJ, Blakely RD, et al. 2018. Pancreatic deletion of the interleukin-1 receptor disrupts whole body glucose homeostasis and promotes islet β-cell de-differentiation. Mol Metab 14: 95–107. 10.1016/j.molmet.2018.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burska AN, Sakthiswary R, Sattar N. 2015. Effects of tumour necrosis factor antagonists on insulin sensitivity/resistance in rheumatoid arthritis: a systematic review and meta-analysis. PLoS One 10: e0128889. 10.1371/journal.pone.0128889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher MJ, Hallinger D, Garcia E, Machida Y, Chakrabarti S, Nadler J, Galkina EV, Imai Y. 2014. Association of proinflammatory cytokines and islet resident leucocytes with islet dysfunction in type 2 diabetes. Diabetologia 57: 491–501. 10.1007/s00125-013-3116-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camell CD, Sander J, Spadaro O, Lee A, Nguyen KY, Wing A, Goldberg EL, Youm Y-H, Brown CW, Elsworth J, et al. 2017. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature 550: 119–123. 10.1038/nature24022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camell CD, Günther P, Lee A, Goldberg EL, Spadaro O, Youm YH, Bartke A, Hubbard GB, Ikeno Y, Ruddle NH, et al. 2019. Aging induces an Nlrp3 inflammasome-dependent expansion of adipose B cells that impairs metabolic homeostasis. Cell Metab 30: 1024–1039.e6. 10.1016/j.cmet.2019.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey AL, Steinberg GR, Macaulay SL, Thomas WG, Holmes AG, Ramm G, Prelovsek O, Hohnen-Behrens C, Watt MJ, James DE, et al. 2006. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes 55: 2688–2697. 10.2337/db05-1404 [DOI] [PubMed] [Google Scholar]
- Chatenoud L, Feng B, Jiao P, Nie Y, Kim T, Jun D, van Rooijen N, Yang Z, Xu H. 2011. Clodronate liposomes improve metabolic profile and reduce visceral adipose macrophage content in diet-induced obese mice. PLoS One 6: e24358. 10.1371/journal.pone.0024358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Chen C-H, Hu C, Long J, Hee Ong RT, Sim X, Takeuchi F, Wu Y, Go MJ, Yamauchi T, et al. 2012. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet 44: 67–72. 10.1038/ng.1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, Mathis D. 2012. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 486: 549–553. 10.1038/nature11132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coats BR, Schoenfelt KQ, Barbosa-Lorenzi VC, Peris E, Cui C, Hoffman A, Zhou G, Fernandez S, Zhai L, Hall BA, et al. 2017. Metabolically activated adipose tissue macrophages perform detrimental and beneficial functions during diet-induced obesity. Cell Rep 20: 3149–3161. 10.1016/j.celrep.2017.08.096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett JA, Sweetland MA, Wang JL, Lancaster JR, McDaniel ML. 1993. Nitric oxide mediates cytokine-induced inhibition of insulin secretion by human islets of Langerhans. Proc Natl Acad Sci 90: 1731–1735. 10.1073/pnas.90.5.1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin B, Cinti S, Morroni M, Raimbault S, Ricquier D, Penicaud L, Casteilla L. 1992. Occurrence of brown adipocytes in rat white adipose tissue: molecular and morphological characterization. J Cell Sci 103(Pt 4): 931–942. [DOI] [PubMed] [Google Scholar]
- Czech MP. 2017. Insulin action and resistance in obesity and type 2 diabetes. Nat Med 23: 804–814. 10.1038/nm.4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagdeviren S, Jung DY, Lee E, Friedline RH, Noh HL, Kim JH, Patel PR, Tsitsilianos N, Tsitsilianos AV, Tran DA, et al. 2016. Altered interleukin-10 signaling in skeletal muscle regulates obesity-mediated inflammation and insulin resistance. Mol Cell Biol 36: 2956–2966. 10.1128/MCB.00181-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang SY, Leng Y, Wang ZX, Xiao X, Zhang X, Wen T, Gong HZ, Hong A, Ma Y. 2019. Exosomal transfer of obesity adipose tissue for decreased miR-141-3p mediate insulin resistance of hepatocytes. Int J Biol Sci 15: 351–368. 10.7150/ijbs.28522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Alvaro C, Teruel T, Hernandez R, Lorenzo M. 2004. Tumor necrosis factor α produces insulin resistance in skeletal muscle by activation of inhibitor κB kinase in a p38 MAPK-dependent manner. Journal of Biological Chemistry 279: 17070–17078. 10.1074/jbc.M312021200 [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Ferrannini E, Groop L, Henry RR, Herman WH, Holst JJ, Hu FB, Kahn CR, Raz I, Shulman GI, et al. 2015. Type 2 diabetes mellitus. Nat Rev Dis Primers 1: 15019. 10.1038/nrdp.2015.19 [DOI] [PubMed] [Google Scholar]
- Desai HR, Sivasubramaniyam T, Revelo XS, Schroer SA, Luk CT, Rikkala PR, Metherel AH, Dodington DW, Park YJ, Kim MJ, et al. 2017. Macrophage JAK2 deficiency protects against high-fat diet-induced inflammation. Sci Rep 7: 7653. 10.1038/s41598-017-07923-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza CJ, Eckhardt M, Gagen K, Dong M, Chen W, Laurent D, Burkey BF. 2001. Effects of pioglitazone on adipose tissue remodeling within the setting of obesity and insulin resistance. Diabetes 50: 1863–1871. 10.2337/diabetes.50.8.1863 [DOI] [PubMed] [Google Scholar]
- Diedisheim M, Carcarino E, Vandiedonck C, Roussel R, Gautier J-F, Venteclef N. 2020. Regulation of inflammation in diabetes: from genetics to epigenomics evidence. Mol Metab 41: 101041. 10.1016/j.molmet.2020.101041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimas AS, Lagou V, Barker A, Knowles JW, Mägi R, Hivert M-F, Benazzo A, Rybin D, Jackson AU, Stringham HM, et al. 2014. Impact of type 2 diabetes susceptibility variants on quantitative glycemic traits reveals mechanistic heterogeneity. Diabetes 63: 2158–2171. 10.2337/db13-0949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez H, Storgaard H, Rask-Madsen C, Steffen Hermann T, Ihlemann N, Baunbjerg Nielsen D, Spohr C, Kober L, Vaag A, Torp-Pedersen C. 2005. Metabolic and vascular effects of tumor necrosis factor-α blockade with etanercept in obese patients with type 2 diabetes. J Vasc Res 42: 517–525. 10.1159/000088261 [DOI] [PubMed] [Google Scholar]
- Donath MY, Dinarello CA, Mandrup-Poulsen T. 2019. Targeting innate immune mediators in type 1 and type 2 diabetes. Nat Rev Immunol 19: 734–746. 10.1038/s41577-019-0213-9 [DOI] [PubMed] [Google Scholar]
- Dror E, Dalmas E, Meier DT, Wueest S, Thüvenet J, Thienel C, Timper K, Nordmann TM, Traub S, Schulze F, et al. 2017. Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat Immunol 18: 283–292. 10.1038/ni.3659 [DOI] [PubMed] [Google Scholar]
- Eguchi K, Manabe I, Oishi-Tanaka Y, Ohsugi M, Kono N, Ogata F, Yagi N, Ohto U, Kimoto M, Miyake K, et al. 2012. Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metab 15: 518–533. 10.1016/j.cmet.2012.01.023 [DOI] [PubMed] [Google Scholar]
- Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R, Gueripel X, Ellingsgaard H, Schneider MK, Biollaz G, et al. 2007. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 56: 2356–2370. 10.2337/db06-1650 [DOI] [PubMed] [Google Scholar]
- Erridge C, Attina T, Spickett CM, Webb DJ. 2007. A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am J Clin Nutr 86: 1286–1292. 10.1093/ajcn/86.5.1286 [DOI] [PubMed] [Google Scholar]
- Everett BM, Donath MY, Pradhan AD, Thuren T, Pais P, Nicolau JC, Glynn RJ, Libby P, Ridker PM. 2018. Anti-Inflammatory therapy with canakinumab for the prevention and management of diabetes. J Am Coll Cardiol 71: 2392–2401. 10.1016/j.jacc.2018.03.002 [DOI] [PubMed] [Google Scholar]
- Fabbrini E, Cella M, McCartney SA, Fuchs A, Abumrad NA, Pietka TA, Chen Z, Finck BN, Han DH, Magkos F, et al. 2013. Association between specific adipose tissue CD4+ T-cell populations and insulin resistance in obese individuals. Gastroenterology 145: 366–374.e3. 10.1053/j.gastro.2013.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feingold KR, Serio MK, Adi S, Moser AH, Grunfeld C. 1989. Tumor necrosis factor stimulates hepatic lipid synthesis and secretion. Endocrinology 124: 2336–2342. 10.1210/endo-124-5-2336 [DOI] [PubMed] [Google Scholar]
- Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, et al. 2009. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 15: 930–939. 10.1038/nm.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink LN, Costford SR, Lee YS, Jensen TE, Bilan PJ, Oberbach A, Blüher M, Olefsky JM, Sams A, Klip A. 2014. Pro-inflammatory macrophages increase in skeletal muscle of high fat-fed mice and correlate with metabolic risk markers in humans. Obesity (Silver Spring) 22: 747–757. 10.1002/oby.20615 [DOI] [PubMed] [Google Scholar]
- Fischer K, Ruiz HH, Jhun K, Finan B, Oberlin DJ, van der Heide V, Kalinovich AV, Petrovic N, Wolf Y, Clemmensen C, et al. 2017. Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nat Med 23: 623–630. 10.1038/nm.4316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannick J, Mercader JM, Fuchsberger C, Udler MS, Mahajan A, Wessel J, Teslovich TM, Caulkins L, Koesterer R, Barajas-Olmos F, et al. 2019. Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature 570: 71–76. 10.1038/s41586-019-1231-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franckhauser S, Elias I, Rotter Sopasakis V, Ferré T, Nagaev I, Andersson CX, Agudo J, Ruberte J, Bosch F, Smith U. 2008. Overexpression of Il6 leads to hyperinsulinaemia, liver inflammation and reduced body weight in mice. Diabetologia 51: 1306–1316. 10.1007/s00125-008-0998-8 [DOI] [PubMed] [Google Scholar]
- Geutskens SB, Otonkoski T, Pulkkinen MA, Drexhage HA, Leenen PJ. 2005. Macrophages in the murine pancreas and their involvement in fetal endocrine development in vitro. J Leukoc Biol 78: 845–852. 10.1189/jlb.1004624 [DOI] [PubMed] [Google Scholar]
- Ghazarian M, Revelo XS, Nøhr MK, Luck H, Zeng K, Lei H, Tsai S, Schroer SA, Park YJ, Chng MHY, et al. 2017. Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci Immunol 2: eaai7616. 10.1126/sciimmunol.aai7616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfine AB, Shoelson SE. 2017. Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk. J Clin Invest 127: 83–93. 10.1172/JCI88884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfine AB, Silver R, Aldhahi W, Cai D, Tatro E, Lee J, Shoelson SE. 2008. Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clin Transl Sci 1: 36–43. 10.1111/j.1752-8062.2008.00026.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE, Team T-TDS. 2010. The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann Intern Med 152: 346–357. 10.7326/0003-4819-152-6-201003160-00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Gay MA, De Matias JM, Gonzalez-Juanatey C, Garcia-Porrua C, Sanchez-Andrade A, Martin J, Llorca J. 2006. Anti-tumor necrosis factor-α blockade improves insulin resistance in patients with rheumatoid arthritis. Clin Exp Rheumatol 24: 83–86. [PubMed] [Google Scholar]
- Guilherme A, Virbasius JV, Puri V, Czech MP. 2008. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol 9: 367–377. 10.1038/nrm2391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilherme A, Henriques F, Bedard AH, Czech MP. 2019. Molecular pathways linking adipose innervation to insulin action in obesity and diabetes mellitus. Nat Rev Endocrinol 15: 207–225. 10.1038/s41574-019-0165-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Al-Sadi R, Said HM, Ma TY. 2013. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am J Pathol 182: 375–387. 10.1016/j.ajpath.2012.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha CWY, Martin A, Sepich-Poore GD, Shi B, Wang Y, Gouin K, Humphrey G, Sanders K, Ratnayake Y, Chan KSL, et al. 2020. Translocation of viable gut microbiota to mesenteric adipose drives formation of creeping fat in humans. Cell 183: 666–683.e17. 10.1016/j.cell.2020.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler RA, Astiarraga B, Camastra S, Accili D, Ferrannini E. 2013. Human insulin resistance is associated with increased plasma levels of 12α-hydroxylated bile acids. Diabetes 62: 4184–4191. 10.2337/db13-0639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halban PA, Polonsky KS, Bowden DW, Hawkins MA, Ling C, Mather KJ, Powers AC, Rhodes CJ, Sussel L, Weir GC. 2014. β-Cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. Diabetes Care 37: 1751–1758. 10.2337/dc14-0396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner-Eiger S, Dineen S, Magalang UJ, et al. 2009. Hypoxia-inducible factor 1α induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol 29: 4467–4483. 10.1128/MCB.00192-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JA, Ramachandran D, Roh HC, DiSpirito JR, Belchior T, Zushin PH, Palmer C, Hong S, Mina AI, Liu B, et al. 2020. Obesity-linked PPARγ S273 phosphorylation promotes insulin resistance through growth differentiation factor 3. Cell Metab 32: 665–675.e6. 10.1016/j.cmet.2020.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MS, Jung DY, Morel C, Lakhani SA, Kim JK, Flavell RA, Davis RJ. 2013. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 339: 218–222. 10.1126/science.1227568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SA, Wong VW, Okanoue T, Bzowej N, Vuppalanchi R, Younes Z, Kohli A, Sarin S, Caldwell SH, Alkhouri N, et al. 2020. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: results from randomized phase III STELLAR trials. J Hepatol 73: 26–39. 10.1016/j.jhep.2020.02.027 [DOI] [PubMed] [Google Scholar]
- Hasnain SZ, Borg DJ, Harcourt BE, Tong H, Sheng YH, Ng CP, Das I, Wang R, Chen AC, Loudovaris T, et al. 2014. Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate β cell stress. Nat Med 20: 1417–1426. 10.1038/nm.3705 [DOI] [PubMed] [Google Scholar]
- Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green KA, Mustard KJ, et al. 2012. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 336: 918–922. 10.1126/science.1215327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, Brenner DA. 1999. The role of TGFβ1 in initiating hepatic stellate cell activation in vivo. J Hepatol 30: 77–87. 10.1016/S0168-8278(99)80010-5 [DOI] [PubMed] [Google Scholar]
- Henriques F, Bedard AH, Guilherme A, Kelly M, Chi J, Zhang P, Lifshitz LM, Bellvé K, Rowland LA, Yenilmez B, et al. 2020. Single-cell RNA profiling reveals adipocyte to macrophage signaling sufficient to enhance thermogenesis. Cell Rep 32: 107998. 10.1016/j.celrep.2020.107998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AA, Reid Bolus W, Hasty AH. 2014. A decade of progress in adipose tissue macrophage biology. Immunol Rev 262: 134–152. 10.1111/imr.12216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DA, Lim HW, Kim YH, Ho WY, Foong YH, Nelson VL, Nguyen HCB, Chegireddy K, Kim J, Habertheuer A, et al. 2018. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc Natl Acad Sci 115: E5096–E5105. 10.1073/pnas.1802611115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang L, Görgün CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. 2002. A central role for JNK in obesity and insulin resistance. Nature 420: 333–336. 10.1038/nature01137 [DOI] [PubMed] [Google Scholar]
- Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, Knotts TA, Shui G, Clegg DJ, Wenk MR, et al. 2011. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest 121: 1858–1870. 10.1172/JCI43378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong EG, Ko HJ, Cho YR, Kim HJ, Ma Z, Yu TY, Friedline RH, Kurt-Jones E, Finberg R, Fischer MA, et al. 2009. Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes 58: 2525–2535. 10.2337/db08-1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, Furukawa S, Tochino Y, Komuro R, Matsuda M, et al. 2007. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 56: 901–911. 10.2337/db06-0911 [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. 2017. Inflammation, metaflammation and immunometabolic disorders. Nature 542: 177–185. 10.1038/nature21363 [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, Spiegelman BM. 1993. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science 259: 87–91. 10.1126/science.7678183 [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. 1994. Tumor necrosis factor α inhibits signaling from the insulin receptor. Proc Natl Acad Sci 91: 4854–4858. 10.1073/pnas.91.11.4854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Jin C, Zeng X, Resch JM, Jedrychowski MP, Yang Z, Desai BN, Banks AS, Lowell BB, Mathis D, et al. 2020. γδ T cells and adipocyte IL-17RC control fat innervation and thermogenesis. Nature 578: 610–614. 10.1038/s41586-020-2028-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube JJ, Scott DK, O'Doherty RM. 2010. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 59: 347–357. 10.2337/db09-0016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudish LI, Reusch JE, Sussel L. 2019. Β cell dysfunction during progression of metabolic syndrome to type 2 diabetes. J Clin Invest 129: 4001–4008. 10.1172/JCI129188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh JY, Kim JI, Park YJ, Hwang IJ, Lee YS, Sohn JH, Lee SK, Alfadda AA, Kim SS, Choi SH, et al. 2013. A novel function of adipocytes in lipid antigen presentation to iNKT cells. Mol Cell Biol 33: 328–339. 10.1128/MCB.00552-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan M, Kaelin WG Jr 2017. The EGLN-HIF O2-sensing system: multiple inputs and feedbacks. Mol Cell 66: 772–779. 10.1016/j.molcel.2017.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, Lundgren P, Bleriot C, Liu Z, Deczkowska A, et al. 2019. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell 178: 686–698.e14. 10.1016/j.cell.2019.05.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubzick CV, Randolph GJ, Henson PM. 2017. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol 17: 349–362. 10.1038/nri.2017.28 [DOI] [PubMed] [Google Scholar]
- Jia L, Vianna CR, Fukuda M, Berglund ED, Liu C, Tao C, Sun K, Liu T, Harper MJ, Lee CE, et al. 2014. Hepatocyte toll-like receptor 4 regulates obesity-induced inflammation and insulin resistance. Nat Commun 5: 3878. 10.1038/ncomms4878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B. 1998. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature 391: 82–86. 10.1038/34184 [DOI] [PubMed] [Google Scholar]
- Jiang C, Qu A, Matsubara T, Chanturiya T, Jou W, Gavrilova O, Shah YM, Gonzalez FJ. 2011. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes 60: 2484–2495. 10.2337/db11-0174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AM, Costanzo A, Gareau MG, Armando AM, Quehenberger O, Jameson JM, Olefsky JM. 2015. High fat diet causes depletion of intestinal eosinophils associated with intestinal permeability. PLoS One 10: e0122195. 10.1371/journal.pone.0122195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdan T, Godlewski G, Cinar R, Bertola A, Szanda G, Liu J, Tam J, Han T, Mukhopadhyay B, Skarulis MC, et al. 2013. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates β cell loss in type 2 diabetes. Nat Med 19: 1132–1140. 10.1038/nm.3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn CR, Wang G, Lee KY. 2019. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J Clin Invest 129: 3990–4000. 10.1172/JCI129187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada N, Seo SU, Chen GY, Núñez G. 2013. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol 13: 321–335. 10.1038/nri3430 [DOI] [PubMed] [Google Scholar]
- Kamata K, Mizukami H, Inaba W, Tsuboi K, Tateishi Y, Yoshida T, Yagihashi S. 2014. Islet amyloid with macrophage migration correlates with augmented β-cell deficits in type 2 diabetic patients. Amyloid 21: 191–201. 10.3109/13506129.2014.937857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, et al. 2006. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 116: 1494–1505. 10.1172/JCI26498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayali AG, Lopez AD, Hao E, Hinton A, Hayek A, King CC. 2012. The SDF-1α/CXCR4 axis is required for proliferation and maturation of human fetal pancreatic endocrine progenitor cells. PLoS One 7: e38721. 10.1371/journal.pone.0038721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan IM, Perrard XY, Brunner G, Lui H, Sparks LM, Smith SR, Wang X, Shi ZZ, Lewis DE, Wu H, et al. 2015. Intermuscular and perimuscular fat expansion in obesity correlates with skeletal muscle T cell and macrophage infiltration and insulin resistance. Int J Obes (Lond) 39: 1607–1618. 10.1038/ijo.2015.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T, Terasawa K, Kashihara D, Hirano K, Tani T, et al. 2013. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun 4: 1829. 10.1038/ncomms2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiortsis DN, Mavridis AK, Vasakos S, Nikas SN, Drosos AA. 2005. Effects of infliximab treatment on insulin resistance in patients with rheumatoid arthritis and ankylosing spondylitis. Ann Rheum Dis 64: 765–766. 10.1136/ard.2004.026534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita S, Maeda N, Shimomura I. 2019. Interorgan communication by exosomes, adipose tissue, and adiponectin in metabolic syndrome. J Clin Invest 129: 4041–4049. 10.1172/JCI129193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koelwyn GJ, Corr EM, Erbay E, Moore KJ. 2018. Regulation of macrophage immunometabolism in atherosclerosis. Nat Immunol 19: 526–537. 10.1038/s41590-018-0113-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlgruber AC, Gal-Oz ST, LaMarche NM, Shimazaki M, Duquette D, Koay HF, Nguyen HN, Mina AI, Paras T, Tavakkoli A, et al. 2018. γδ T cells producing interleukin-17A regulate adipose regulatory T cell homeostasis and thermogenesis. Nat Immunol 19: 464–474. 10.1038/s41590-018-0094-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooner JS, Saleheen D, Sim X, Sehmi J, Zhang W, Frossard P, Been LF, Chia K-S, Dimas AS, Hassanali N, et al. 2011. Genome-wide association study in individuals of south Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat Genet 43: 984–989. 10.1038/ng.921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama Y, Brenner DA. 2017. Liver inflammation and fibrosis. J Clin Invest 127: 55–64. 10.1172/JCI88881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranendonk ME, Visseren FL, van Herwaarden JA, Nolte-’t Hoen EN, de Jager W, Wauben MH, Kalkhoven E. 2014. Effect of extracellular vesicles of human adipose tissue on insulin signaling in liver and muscle cells. Obesity (Silver Spring) 22: 2216–2223. 10.1002/oby.20847 [DOI] [PubMed] [Google Scholar]
- Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, et al. 2014. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab 20: 614–625. 10.1016/j.cmet.2014.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan J, Danzer C, Simka T, Ukropec J, Walter KM, Kumpf S, Mirtschink P, Ukropcova B, Gasperikova D, Pedrazzini T, et al. 2012. Dietary obesity-associated Hif1α activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev 26: 259–270. 10.1101/gad.180406.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Akira S. 2011. Pathogen recognition by the innate immune system. Int Rev Immunol 30: 16–34. 10.3109/08830185.2010.529976 [DOI] [PubMed] [Google Scholar]
- Kwon G, Corbett JA, Hauser S, Hill JR, Turk J, McDaniel ML. 1998. Evidence for involvement of the proteasome complex (26S) and NFκB in IL-1β-induced nitric oxide and prostaglandin production by rat islets and RINm5F cells. Diabetes 47: 583–591. 10.2337/diabetes.47.4.583 [DOI] [PubMed] [Google Scholar]
- LaMarche NM, Kane H, Kohlgruber AC, Dong H, Lynch L, Brenner MB. 2020. Distinct iNKT cell populations use IFNγ or ER stress-induced IL-10 to control adipose tissue homeostasis. Cell Metab 32: 243–258.e6. 10.1016/j.cmet.2020.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang CH, Dobrescu C, Bagby GJ. 1992. Tumor necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose output. Endocrinology 130: 43–52. 10.1210/endo.130.1.1727716 [DOI] [PubMed] [Google Scholar]
- Lanthier N, Molendi-Coste O, Horsmans Y, van Rooijen N, Cani PD, Leclercq IA. 2010. Kupffer cell activation is a causal factor for hepatic insulin resistance. Am J Physiol Gastrointest Liver Physiol 298: G107–G116. 10.1152/ajpgi.00391.2009 [DOI] [PubMed] [Google Scholar]
- Larsen L, Størling J, Darville M, Eizirik DL, Bonny C, Billestrup N, Mandrup-Poulsen T. 2005. Extracellular signal-regulated kinase is essential for interleukin-1-induced and nuclear factor κB-mediated gene expression in insulin-producing INS-1E cells. Diabetologia 48: 2582–2590. 10.1007/s00125-005-0039-9 [DOI] [PubMed] [Google Scholar]
- Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY. 2007. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356: 1517–1526. 10.1056/NEJMoa065213 [DOI] [PubMed] [Google Scholar]
- Lawler HM, Underkofler CM, Kern PA, Erickson C, Bredbeck B, Rasouli N. 2016. Adipose tissue hypoxia, inflammation, and fibrosis in obese insulin-sensitive and obese insulin-resistant subjects. J Clin Endocrinol Metab 101: 1422–1428. 10.1210/jc.2015-4125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Gesta S, Boucher J, Wang XL, Kahn CR. 2011a. The differential role of Hif1β/Arnt and the hypoxic response in adipose function, fibrosis, and inflammation. Cell Metab 14: 491–503. 10.1016/j.cmet.2011.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Li P, Huh JY, Hwang IJ, Lu M, Kim JI, Ham M, Talukdar S, Chen A, Lu WJ, et al. 2011b. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 60: 2474–2483. 10.2337/db11-0194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Morinaga H, Kim JJ, Lagakos W, Taylor S, Keshwani M, Perkins G, Dong H, Kayali AG, Sweet IR, et al. 2013. The fractalkine/CX3CR1 system regulates β cell function and insulin secretion. Cell 153: 413–425. 10.1016/j.cell.2013.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Kim JW, Osborne O, Oh DY, Sasik R, Schenk S, Chen A, Chung H, Murphy A, Watkins SM, et al. 2014. Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell 157: 1339–1352. 10.1016/j.cell.2014.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BC, Kim MS, Pae M, Yamamoto Y, Eberlé D, Shimada T, Kamei N, Park HS, Sasorith S, Woo JR, et al. 2016. Adipose natural killer cells regulate adipose tissue macrophages to promote insulin resistance in obesity. Cell Metab 23: 685–698. 10.1016/j.cmet.2016.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Wollam J, Olefsky JM. 2018. An integrated view of immunometabolism. Cell 172: 22–40. 10.1016/j.cell.2017.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Oh DY, Bandyopadhyay G, Lagakos WS, Talukdar S, Osborn O, Johnson A, Chung H, Maris M, Ofrecio JM, et al. 2015. LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat Med 21: 239–247. 10.1038/nm.3800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Liu S, Lu M, Bandyopadhyay G, Oh D, Imamura T, Johnson AM, Sears D, Shen Z, Cui B, et al. 2016. Hematopoietic-derived galectin-3 causes cellular and systemic insulin resistance. Cell 167: 973–984.e12. 10.1016/j.cell.2016.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Menoret A, Farragher C, Ouyang Z, Bonin C, Holvoet P, Vella AT, Zhou B. 2019. Single-cell transcriptomics–based MacSpectrum reveals macrophage activation signatures in diseases. JCI Insight 4: e126453. 10.1172/jci.insight.126453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Spallanzani RG, Mathis D. 2020. Visceral adipose tissue Tregs and the cells that nurture them. Immunol Rev 295: 114–125. 10.1111/imr.12850 [DOI] [PubMed] [Google Scholar]
- Liao ZZ, Wang YD, Qi XY, Xiao XH. 2019. JAZF1, a relevant metabolic regulator in type 2 diabetes. Diabetes Metab Res Rev 35: e3148. 10.1002/dmrr.3148 [DOI] [PubMed] [Google Scholar]
- Lin YJ, Anzaghe M, Schulke S. 2020. Update on the pathomechanism, diagnosis, and treatment options for rheumatoid arthritis. Cells 9: 880. 10.3390/cells9040880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LS, Spelleken M, Rohrig K, Hauner H, Eckel J. 1998. Tumor necrosis factor-α acutely inhibits insulin signaling in human adipocytes: implication of the p80 tumor necrosis factor receptor. Diabetes 47: 515–522. 10.2337/diabetes.47.4.515 [DOI] [PubMed] [Google Scholar]
- Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, Powell C, Vedantam S, Buchkovich ML, Yang J, et al. 2015. Genetic studies of body mass index yield new insights for obesity biology. Nature 518: 197–206. 10.1038/nature14177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lofgren P, van Harmelen V, Reynisdottir S, Naslund E, Ryden M, Rossner S, Arner P. 2000. Secretion of tumor necrosis factor-α shows a strong relationship to insulin-stimulated glucose transport in human adipose tissue. Diabetes 49: 688–692. 10.2337/diabetes.49.5.688 [DOI] [PubMed] [Google Scholar]
- Lumeng CN, Bodzin JL, Saltiel AR. 2007a. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117: 175–184. 10.1172/JCI29881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR. 2007b. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes 56: 16–23. 10.2337/db06-1076 [DOI] [PubMed] [Google Scholar]
- Lynch L. 2014. Adipose invariant natural killer T cells. Immunology 142: 337–346. 10.1111/imm.12269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA, Donath MY. 2002. Glucose-induced β cell production of IL-1β contributes to glucotoxicity in human pancreatic islets. J Clin Invest 110: 851–860. 10.1172/JCI200215318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, Payne AJ, Steinthorsdottir V, Scott RA, Grarup N, et al. 2018. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 50: 1505–1513. 10.1038/s41588-018-0241-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdi T, Hänzelmann S, Salehi A, Muhammed SJ, Reinbothe TM, Tang Y, Axelsson AS, Zhou Y, Jing X, Almgren P, et al. 2012. Secreted frizzled-related protein 4 reduces insulin secretion and is overexpressed in type 2 diabetes. Cell Metab 16: 625–633. 10.1016/j.cmet.2012.10.009 [DOI] [PubMed] [Google Scholar]
- Manning AK, Hivert M-F, Scott RA, Grimsby JL, Bouatia-Naji N, Chen H, Rybin D, Liu C-T, Bielak LF, Prokopenko I, et al. 2012. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet 44: 659–669. 10.1038/ng.2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MI, Feero WG, Guttmacher AE. 2010. Genomics, type 2 diabetes, and obesity. New England Journal of Medicine 363: 2339–2350. 10.1056/NEJMra0906948 [DOI] [PubMed] [Google Scholar]
- McGillicuddy FC, Harford KA, Reynolds CM, Oliver E, Claessens M, Mills KH, Roche HM. 2011. Lack of interleukin-1 receptor I (IL-1RI) protects mice from high-fat diet-induced adipose tissue inflammation coincident with improved glucose homeostasis. Diabetes 60: 1688–1698. 10.2337/db10-1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNelis JC, Lee YS, Mayoral R, van der Kant R, Johnson AM, Wollam J, Olefsky JM. 2015. GPR43 potentiates β-cell function in obesity. Diabetes 64: 3203–3217. 10.2337/db14-1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. 2000. M-1/M-2 macrophages and the Th1/Th2 paradigm. The J Immunol 164: 6166–6173. 10.4049/jimmunol.164.12.6166 [DOI] [PubMed] [Google Scholar]
- Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, Chawla A, Locksley RM. 2013. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med 210: 535–549. 10.1084/jem.20121964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro-Sepulveda M, Touch S, Mendes-Sá C, André S, Poitou C, Allatif O, Cotillard A, Fohrer-Ting H, Hubert EL, Remark R, et al. 2015. Jejunal T cell inflammation in human obesity correlates with decreased enterocyte insulin signaling. Cell Metab 22: 113–124. 10.1016/j.cmet.2015.05.020 [DOI] [PubMed] [Google Scholar]
- Morinaga H, Mayoral R, Heinrichsdorff J, Osborn O, Franck N, Hah N, Walenta E, Bandyopadhyay G, Pessentheiner AR, Chi TJ, et al. 2015. Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes 64: 1120–1130. 10.2337/db14-1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nackiewicz D, Dan M, Speck M, Chow SZ, Chen YC, Pospisilik JA, Verchere CB, Ehses JA. 2020. Islet macrophages shift to a reparative state following pancreatic β-cell death and are a major source of islet insulin-like growth factor-1. iScience 23: 100775. 10.1016/j.isci.2019.100775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagareddy PR, Kraakman M, Masters SL, Stirzaker RA, Gorman DJ, Grant RW, Dragoljevic D, Hong ES, Abdel-Latif A, Smyth SS, et al. 2014. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab 19: 821–835. 10.1016/j.cmet.2014.03.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal MD, Leaphart C, Levy R, Prince J, Billiar TR, Watkins S, Li J, Cetin S, Ford H, Schreiber A, et al. 2006. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J Immunol 176: 3070–3079. 10.4049/jimmunol.176.5.3070 [DOI] [PubMed] [Google Scholar]
- Nelson VL, Nguyen HCB, Garcìa-Cañaveras JC, Briggs ER, Ho WY, DiSpirito JR, Marinis JM, Hill DA, Lazar MA. 2018. PPARγ is a nexus controlling alternative activation of macrophages via glutamine metabolism. Genes Dev 32: 1035–1044. 10.1101/gad.312355.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard CB. 2017. Metabolomics and metabolic diseases: where do we stand? Cell Metab 25: 43–56. 10.1016/j.cmet.2016.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen KD, Qiu Y, Cui X, Goh YPS, Mwangi J, David T, Mukundan L, Brombacher F, Locksley RM, Chawla A. 2011. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 480: 104–108. 10.1038/nature10653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, et al. 2009. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 15: 914–920. 10.1038/nm.1964 [DOI] [PubMed] [Google Scholar]
- Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, Thornton EE, Krummel MF, Chawla A, Liang HE, et al. 2013. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 502: 245–248. 10.1038/nature12526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obstfeld AE, Sugaru E, Thearle M, Francisco AM, Gayet C, Ginsberg HN, Ables EV, Ferrante AW Jr. 2010. C-C chemokine receptor 2 (CCR2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes 59: 916–925. 10.2337/db09-1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofei F, Hurel S, Newkirk J, Sopwith M, Taylor R. 1996. Effects of an engineered human anti-TNF-α antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM. Diabetes 45: 881–885. 10.2337/diab.45.7.881 [DOI] [PubMed] [Google Scholar]
- Oh DY, Morinaga H, Talukdar S, Bae EJ, Olefsky JM. 2012. Increased macrophage migration into adipose tissue in obese mice. Diabetes 61: 346–354. 10.2337/db11-0860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM. 2010. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142: 687–698. 10.1016/j.cell.2010.07.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olefsky JM, Glass CK. 2010. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 72: 219–246. 10.1146/annurev-physiol-021909-135846 [DOI] [PubMed] [Google Scholar]
- O'Neill CM, Lu C, Corbin KL, Sharma PR, Dula SB, Carter JD, Ramadan JW, Xin W, Lee JK, Nunemaker CS. 2013. Circulating levels of IL-1B + IL-6 cause ER stress and dysfunction in islets from prediabetic male mice. Endocrinology 154: 3077–3088. 10.1210/en.2012-2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr JS, Puglisi MJ, Ellacott KL, Lumeng CN, Wasserman DH, Hasty AH. 2012. Toll-like receptor 4 deficiency promotes the alternative activation of adipose tissue macrophages. Diabetes 61: 2718–2727. 10.2337/db11-1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortis F, Cardozo AK, Crispim D, Störling J, Mandrup-Poulsen T, Eizirik DL. 2006. Cytokine-induced proapoptotic gene expression in insulin-producing cells is related to rapid, sustained, and nonoscillatory nuclear factor-κB activation. Mol Endocrinol 20: 1867–1879. 10.1210/me.2005-0268 [DOI] [PubMed] [Google Scholar]
- Paquot N, Castillo MJ, Lefebvre PJ, Scheen AJ. 2000. No increased insulin sensitivity after a single intravenous administration of a recombinant human tumor necrosis factor receptor: Fc fusion protein in obese insulin-resistant patients. J Clin Endocrinol Metab 85: 1316–1319. [DOI] [PubMed] [Google Scholar]
- Pasarica M, Sereda OR, Redman LM, Albarado DC, Hymel DT, Roan LE, Rood JC, Burk DH, Smith SR. 2009. Reduced adipose tissue oxygenation in human obesity: evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes 58: 718–725. 10.2337/db08-1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasarica M, Rood J, Ravussin E, Schwarz JM, Smith SR, Redman LM. 2010. Reduced oxygenation in human obese adipose tissue is associated with impaired insulin suppression of lipolysis. J Clin Endocrinol Metab 95: 4052–4055. 10.1210/jc.2009-2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. 2008. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab 8: 301–309. 10.1016/j.cmet.2008.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegtel DM, Gould SJ. 2019. Exosomes. Annu Rev Biochem 88: 487–514. 10.1146/annurev-biochem-013118-111902 [DOI] [PubMed] [Google Scholar]
- Philipson LH. 2020. Harnessing heterogeneity in type 2 diabetes mellitus. Nature Reviews Endocrinology 16: 79–80. 10.1038/s41574-019-0308-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickup JC, Mattock MB, Chusney GD, Burt D. 1997. NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia 40: 1286–1292. 10.1007/s001250050822 [DOI] [PubMed] [Google Scholar]
- Pirzgalska RM, Seixas E, Seidman JS, Link VM, Sánchez NM, Mahú I, Mendes R, Gres V, Kubasova N, Morris I, et al. 2017. Sympathetic neuron-associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat Med 23: 1309–1318. 10.1038/nm.4422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomgaard P, Bouzakri K, Krogh-Madsen R, Mittendorfer B, Zierath JR, Pedersen BK. 2005. Tumor necrosis factor-α induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes 54: 2939–2945. 10.2337/diabetes.54.10.2939 [DOI] [PubMed] [Google Scholar]
- Pols TW, Noriega LG, Nomura M, Auwerx J, Schoonjans K. 2011. The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. J Hepatol 54: 1263–1272. 10.1016/j.jhep.2010.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y, Nguyen Khoa D, Odegaard Justin I, Cui X, Tian X, Locksley Richard M, Palmiter Richard D, Chawla A. 2014. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell 157: 1292–1308. 10.1016/j.cell.2014.03.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramkhelawon B, Hennessy EJ, Ménager M, Ray TD, Sheedy FJ, Hutchison S, Wanschel A, Oldebeken S, Geoffrion M, Spiro W, et al. 2014. Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nat Med 20: 377–384. 10.1038/nm.3467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratziu V, Sanyal A, Harrison SA, Wong VW, Francque S, Goodman Z, Aithal GP, Kowdley KV, Seyedkazemi S, Fischer L, et al. 2020. Cenicriviroc treatment for adults with nonalcoholic steatohepatitis and fibrosis: final analysis of the phase 2b CENTAUR study. Hepatology 72: 892–905. 10.1002/hep.31108 [DOI] [PubMed] [Google Scholar]
- Reid DT, Reyes JL, McDonald BA, Vo T, Reimer RA, Eksteen B. 2016. Kupffer cells undergo fundamental changes during the development of experimental NASH and are critical in initiating liver damage and inflammation. PLoS One 11: e0159524. 10.1371/journal.pone.0159524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly SM, Chiang SH, Decker SJ, Chang L, Uhm M, Larsen MJ, Rubin JR, Mowers J, White NM, Hochberg I, et al. 2013. An inhibitor of the protein kinases TBK1 and IKK-ɛ improves obesity-related metabolic dysfunctions in mice. Nat Med 19: 313–321. 10.1038/nm.3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. 2009. Islet-associated macrophages in type 2 diabetes. Diabetologia 52: 1686–1688. 10.1007/s00125-009-1410-z [DOI] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. 1998. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature 391: 79–82. 10.1038/34178 [DOI] [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. 2013. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341: 1241214. 10.1126/science.1241214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. 2017. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 377: 1119–1131. 10.1056/NEJMoa1707914 [DOI] [PubMed] [Google Scholar]
- Riopel M, Seo JB, Bandyopadhyay GK, Li P, Wollam J, Chung H, Jung SR, Murphy A, Wilson M, de Jong R, et al. 2018. Chronic fractalkine administration improves glucose tolerance and pancreatic endocrine function. J Clin Invest 128: 1458–1470. 10.1172/JCI94330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden M, Shulman GI. 2019. The integrative biology of type 2 diabetes. Nature 576: 51–60. 10.1038/s41586-019-1797-8 [DOI] [PubMed] [Google Scholar]
- Saberi M, Woods NB, de Luca C, Schenk S, Lu JC, Bandyopadhyay G, Verma IM, Olefsky JM. 2009. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab 10: 419–429. 10.1016/j.cmet.2009.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, Barrett T, Kim JK, Davis RJ. 2008. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 322: 1539–1543. 10.1126/science.1160794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangwung P, Petersen KF, Shulman GI, Knowles JW. 2020. Mitochondrial dysfunction, insulin resistance, and potential genetic implications. Endocrinology 161. 10.1210/endocr/bqaa017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartipy P, Loskutoff DJ. 2003. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci 100: 7265–7270. 10.1073/pnas.1133870100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M, Hoshino M, Fujita K, Iizuka M, Fujii S, Clingan CS, Van Kaer L, Iwabuchi K. 2016. Adipocyte-specific CD1d-deficiency mitigates diet-induced obesity and insulin resistance in mice. Sci Rep 6: 28473. 10.1038/srep28473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer PE. 2019. The many secret lives of adipocytes: implications for diabetes. Diabetologia 62: 223–232. 10.1007/s00125-018-4777-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schludi B, Moin ASM, Montemurro C, Gurlo T, Matveyenko AV, Kirakossian D, Dawson DW, Dry SM, Butler PC, Butler AE. 2017. Islet inflammation and ductal proliferation may be linked to increased pancreatitis risk in type 2 diabetes. JCI Insight 2: e92282. 10.1172/jci.insight.92282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schödel J, Ratcliffe PJ. 2019. Mechanisms of hypoxia signalling: new implications for nephrology. Nat Rev Nephrol 15: 641–659. 10.1038/s41581-019-0182-z [DOI] [PubMed] [Google Scholar]
- Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H, Bruni CM, Ouyang Z, Li RZ, Sun X, et al. 2020. Niche-specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity 52: 1057–1074.e7. 10.1016/j.immuni.2020.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki E, Brenner DA, Karin M. 2012. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology 143: 307–320. 10.1053/j.gastro.2012.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. 2019. Pharmacologic targeting of hypoxia-inducible factors. Annu Rev Pharmacol Toxicol 59: 379–403. 10.1146/annurev-pharmtox-010818-021637 [DOI] [PubMed] [Google Scholar]
- Senn JJ, Klover PJ, Nowak IA, Mooney RA. 2002. Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes 51: 3391–3399. 10.2337/diabetes.51.12.3391 [DOI] [PubMed] [Google Scholar]
- Seo JB, Riopel M, Cabrales P, Huh JY, Bandyopadhyay GK, Andreyev AY, Murphy AN, Beeman SC, Smith GI, Klein S, et al. 2019. Knockdown of Ant2 reduces adipocyte hypoxia and improves insulin resistance in obesity. Nat Metab 1: 86–97. 10.1038/s42255-018-0003-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergi D, Naumovski N, Heilbronn LK, Abeywardena M, O'Callaghan N, Lionetti L, Luscombe-Marsh N. 2019. Mitochondrial (dys)function and insulin resistance: from pathophysiological molecular mechanisms to the impact of diet. Front Physiol 10: 532. 10.3389/fphys.2019.00532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serné EH, de Jongh RT, Eringa EC, IJzerman RG, Stehouwer CD. 2007. Microvascular dysfunction: a potential pathophysiological role in the metabolic syndrome. Hypertension 50: 204–211. 10.1161/HYPERTENSIONAHA.107.089680 [DOI] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. 2006. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116: 3015–3025. 10.1172/JCI28898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu I, Yoshida Y, Moriya J, Nojima A, Uemura A, Kobayashi Y, Minamino T. 2013. Semaphorin3E-induced inflammation contributes to insulin resistance in dietary obesity. Cell Metab 18: 491–504. 10.1016/j.cmet.2013.09.001 [DOI] [PubMed] [Google Scholar]
- Shungin D, Winkler TW, Croteau-Chonka DC, Ferreira T, Locke AE, Mägi R, Strawbridge RJ, Pers TH, Fischer K, Justice AE, et al. 2015. New genetic loci link adipose and insulin biology to body fat distribution. Nature 518: 187–196. 10.1038/nature14132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GI, Mittendorfer B, Klein S. 2019. Metabolically healthy obesity: facts and fantasies. J Clin Invest 129: 3978–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikakulapu P, McNamara CA. 2020. B lymphocytes and adipose tissue inflammation. Arterioscler Thromb Vasc Biol 40: 1110–1122. 10.1161/ATVBAHA.119.312467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens JM, Lee J, Pilch PF. 1997. Tumor necrosis factor-α-induced insulin resistance in 3T3-L1 adipocytes is accompanied by a loss of insulin receptor substrate-1 and GLUT4 expression without a loss of insulin receptor-mediated signal transduction. J Biol Chem 272: 971–976. 10.1074/jbc.272.2.971 [DOI] [PubMed] [Google Scholar]
- Stevens CE, Hume ID. 1998. Contributions of microbes in vertebrate gastrointestinal tract to production and conservation of nutrients. Physiol Rev 78: 393–427. 10.1152/physrev.1998.78.2.393 [DOI] [PubMed] [Google Scholar]
- Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JEM, van Rooijen N, Staels B, Kersten S, Müller M. 2010. Kupffer cells promote hepatic steatosis via interleukin-1β-dependent suppression of peroxisome proliferator-activated receptor α activity. Hepatology 51: 511–522. 10.1002/hep.23337 [DOI] [PubMed] [Google Scholar]
- Suhre K, Meisinger C, Döring A, Altmaier E, Belcredi P, Gieger C, Chang D, Milburn MV, Gall WE, Weinberger KM, et al. 2010. Metabolic footprint of diabetes: a multiplatform metabolomics study in an epidemiological setting. PLoS One 5: e13953. 10.1371/journal.pone.0013953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Halberg N, Khan M, Magalang UJ, Scherer PE. 2013. Selective inhibition of hypoxia-inducible factor 1α ameliorates adipose tissue dysfunction. Mol Cell Biol 33: 904–917. 10.1128/MCB.00951-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takikawa A, Mahmood A, Nawaz A, Kado T, Okabe K, Yamamoto S, Aminuddin A, Senda S, Tsuneyama K, Ikutani M, et al. 2016. HIF-1α in myeloid cells promotes adipose tissue remodeling toward insulin resistance. Diabetes 65: 3649–3659. 10.2337/db16-0012 [DOI] [PubMed] [Google Scholar]
- Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, et al. 2012. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med 18: 1407–1412. 10.1038/nm.2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam TH, Chan KL, Boroumand P, Liu Z, Brozinick JT, Bui HH, Roth K, Wakefield CB, Penuela S, Bilan PJ, et al. 2020. Nucleotides released from palmitate-activated murine macrophages attract neutrophils. J Biol Chem 295: 4902–4911. 10.1074/jbc.RA119.010868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C, Ahmed K, Gille A, Lu S, Gröne HJ, Tunaru S, Offermanns S. 2015. Loss of FFA2 and FFA3 increases insulin secretion and improves glucose tolerance in type 2 diabetes. Nat Med 21: 173–177. 10.1038/nm.3779 [DOI] [PubMed] [Google Scholar]
- Tao C, Holland WL, Wang QA, Shao M, Jia L, Sun K, Lin X, Kuo YC, Johnson JA, Gordillo R, et al. 2017. Short-term versus long-term effects of adipocyte toll-like receptor 4 activation on insulin resistance in male mice. Endocrinology 158: 1260–1270. 10.1210/en.2017-00024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theurich S, Tsaousidou E, Hanssen R, Lempradl AM, Mauer J, Timper K, Schilbach K, Folz-Donahue K, Heilinger C, Sexl V, et al. 2017. IL-6/Stat3-dependent induction of a distinct, obesity-associated NK cell subpopulation deteriorates energy and glucose homeostasis. Cell Metab 26: 171–184.e6. 10.1016/j.cmet.2017.05.018 [DOI] [PubMed] [Google Scholar]
- Thomou T, Mori MA, Dreyfuss JM, Konishi M, Sakaguchi M, Wolfrum C, Rao TN, Winnay JN, Garcia-Martin R, Grinspoon SK, et al. 2017. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 542: 450–455. 10.1038/nature21365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E, Cameron J, Grosse J, Reimann F, Gribble FM. 2012. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 61: 364–371. 10.2337/db11-1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P, Spiegelman BM. 2008. Fat and beyond: the diverse biology of PPARγ. Annu Rev Biochem 77: 289–312. 10.1146/annurev.biochem.77.061307.091829 [DOI] [PubMed] [Google Scholar]
- Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, et al. 2010. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet 42: 579–589. 10.1038/ng.609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrieze A, Van Nood E, Holleman F, Salojärvi J, Kootte RS, Bartelsman JF, Dallinga-Thie GM, Ackermans MT, Serlie MJ, Oozeer R, et al. 2012. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 143: 913–916.e7. 10.1053/j.gastro.2012.06.031 [DOI] [PubMed] [Google Scholar]
- Vulliemoz M, Brand S, Juillerat P, Mottet C, Ben-Horin S, Michetti P, on behalf of Swiss Ibdnet aowgotSSoG. 2020. TNF-α blockers in inflammatory bowel diseases: practical recommendations and a user's guide: an update. Digestion 101: 16–26. 10.1159/000506898 [DOI] [PubMed] [Google Scholar]
- Waeber G, Delplanque J, Bonny C, Mooser V, Steinmann M, Widmann C, Maillard A, Miklossy J, Dina C, H Hani El, et al. 2000. The gene MAPK8IP1, encoding islet-brain-1, is a candidate for type 2 diabetes. Nat Genet 24: 291–295. 10.1038/73523 [DOI] [PubMed] [Google Scholar]
- Wang Q, Zhang H, Zhao B, Fei H. 2009. IL-1β caused pancreatic β-cells apoptosis is mediated in part by endoplasmic reticulum stress via the induction of endoplasmic reticulum Ca2+ release through the c-Jun N-terminal kinase pathway. Mol Cell Biochem 324: 183–190. 10.1007/s11010-008-9997-9 [DOI] [PubMed] [Google Scholar]
- Wanschel A, Seibert T, Hewing B, Ramkhelawon B, Ray TD, van Gils JM, Rayner KJ, Feig JE, O'Brien ER, Fisher EA, et al. 2013. Neuroimmune guidance cue semaphorin 3E is expressed in atherosclerotic plaques and regulates macrophage retention. Arterioscler Thromb Vasc Biol 33: 886–893. 10.1161/ATVBAHA.112.300941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren RE, Zammitt NN, Deary IJ, Frier BM. 2006. The effects of acute hypoglycaemia on memory acquisition and recall and prospective memory in type 1 diabetes. Diabetologia 50: 178–185. 10.1007/s00125-006-0535-6 [DOI] [PubMed] [Google Scholar]
- Wascher TC, Lindeman JH, Sourij H, Kooistra T, Pacini G, Roden M. 2011. Chronic TNF-α neutralization does not improve insulin resistance or endothelial function in ‘healthy’ men with metabolic syndrome. Mol Med 17: 189–193. 10.2119/molmed.2010.00221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. 2003. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112: 1796–1808. 10.1172/JCI200319246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, Ferrante AW Jr. 2006. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest 116: 115–124. 10.1172/JCI24335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiskirchen R, Tacke F. 2014. Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg Nutr 3: 344–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernstedt Asterholm I, Tao C, Morley Thomas S, Wang Qiong A, Delgado-Lopez F, Wang Zhao V, Scherer Philipp E. 2014. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab 20: 103–118. 10.1016/j.cmet.2014.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, Dorfman R, Wang Y, Zielenski J, Mastronardi F, et al. 2009. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med 15: 921–929. 10.1038/nm.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer DA, Luck H, Tsai S, Winer S. 2016. The intestinal immune system in obesity and insulin resistance. Cell Metab 23: 413–426. 10.1016/j.cmet.2016.01.003 [DOI] [PubMed] [Google Scholar]
- Wollam J, Antebi A. 2011. Sterol regulation of metabolism, homeostasis, and development. Annu Rev Biochem 80: 885–916. 10.1146/annurev-biochem-081308-165917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Ballantyne CM. 2017. Skeletal muscle inflammation and insulin resistance in obesity. J Clin Invest 127: 43–54. 10.1172/JCI88880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, Chawla A, Locksley RM. 2011. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 332: 243–247. 10.1126/science.1201475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Boström P, Sparks Lauren M, Ye L, Choi Jang H, Giang A-H, Khandekar M, Virtanen Kirsi A, Nuutila P, Schaart G, et al. 2012a. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 150: 366–376. 10.1016/j.cell.2012.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Parekh VV, Gabriel CL, Bracy DP, Marks-Shulman PA, Tamboli RA, Kim S, Mendez-Fernandez YV, Besra GS, Lomenick JP, et al. 2012b. Activation of invariant natural killer T cells by lipid excess promotes tissue inflammation, insulin resistance, and hepatic steatosis in obese mice. Proc Natl Acad Sci 109: E1143–E1152. 10.1073/pnas.1200498109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich FT, Luedde T, Singer S, Schmidt-Supprian M, Baumgartl J, Schirmacher P, Pasparakis M, Bruning JC. 2008. Hepatic NF-κB essential modulator deficiency prevents obesity-induced insulin resistance but synergizes with high-fat feeding in tumorigenesis. Proc Nat Acad Sci 105: 1297–1302. 10.1073/pnas.0707849104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia JY, Holland WL, Kusminski CM, Sun K, Sharma AX, Pearson MJ, Sifuentes AJ, McDonald JG, Gordillo R, Scherer PE. 2015. Targeted induction of ceramide degradation leads to improved systemic metabolism and reduced hepatic steatosis. Cell Metab 22: 266–278. 10.1016/j.cmet.2015.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia JY, Sun K, Hepler C, Ghaben AL, Gupta RK, An YA, Holland WL, Morley TS, Adams AC, Gordillo R, et al. 2018. Acute loss of adipose tissue-derived adiponectin triggers immediate metabolic deterioration in mice. Diabetologia 61: 932–941. 10.1007/s00125-017-4516-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, et al. 2003. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112: 1821–1830. 10.1172/JCI200319451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, Ferrante AW Jr. 2013. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab 18: 816–830. 10.1016/j.cmet.2013.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu A, Negrin KA, Roth Flach RJ, DiStefano MT, Matevossian A, Friedline RH, Jung D, Kim JK, Czech MP. 2014. IL-1 signaling in obesity-induced hepatic lipogenesis and steatosis. PLoS One 9: e107265. 10.1371/journal.pone.0107265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano T, Liu Z, Donovan J, Thomas MK, Habener JF. 2007. Stromal cell derived factor-1 (SDF-1)/CXCL12 attenuates diabetes in mice and promotes pancreatic β-cell survival by activation of the prosurvival kinase Akt. Diabetes 56: 2946–2957. 10.2337/db07-0291 [DOI] [PubMed] [Google Scholar]
- Ying W, Riopel M, Bandyopadhyay G, Dong Y, Birmingham A, Seo JB, Ofrecio JM, Wollam J, Hernandez-Carretero A, Fu W, et al. 2017a. Adipose tissue macrophage-derived exosomal miRNAs can modulate in vivo and in vitro insulin sensitivity. Cell 171: 372–384.e12. 10.1016/j.cell.2017.08.035 [DOI] [PubMed] [Google Scholar]
- Ying W, Wollam J, Ofrecio JM, Bandyopadhyay G, El Ouarrat D, Lee YS, Oh DY, Li P, Osborn O, Olefsky JM. 2017b. Adipose tissue B2 cells promote insulin resistance through leukotriene LTB4/LTB4R1 signaling. J Clin Invest 127: 1019–1030. 10.1172/JCI90350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, Lee YS, Dong Y, Seidman JS, Yang M, Isaac R, Seo JB, Yang BH, Wollam J, Riopel M, et al. 2019. Expansion of islet-resident macrophages leads to inflammation affecting β cell proliferation and function in obesity. Cell Metab 29: 457–474.e5. 10.1016/j.cmet.2018.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. 2013. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499: 97–101. 10.1038/nature12347 [DOI] [PubMed] [Google Scholar]
- Young PW, Cawthorne MA, Coyle PJ, Holder JC, Holman GD, Kozka IJ, Kirkham DM, Lister CA, Smith SA. 1995. Repeat treatment of obese mice with BRL 49653, a new potent insulin sensitizer, enhances insulin action in white adipocytes. association with increased insulin binding and cell-surface GLUT4 as measured by photoaffinity labeling. Diabetes 44: 1087–1092. 10.2337/diab.44.9.1087 [DOI] [PubMed] [Google Scholar]
- Yu JG, Javorschi S, Hevener AL, Kruszynska YT, Norman RA, Sinha M, Olefsky JM. 2002. The effect of thiazolidinediones on plasma adiponectin levels in normal, obese, and type 2 diabetic subjects. Diabetes 51: 2968–2974. 10.2337/diabetes.51.10.2968 [DOI] [PubMed] [Google Scholar]
- Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, Shoelson SE. 2001. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkβ. Science 293: 1673–1677. 10.1126/science.1061620 [DOI] [PubMed] [Google Scholar]
- Zeyda M, Farmer D, Todoric J, Aszmann O, Speiser M, Györi G, Zlabinger GJ, Stulnig TM. 2007. Human adipose tissue macrophages are of an anti-inflammatory phenotype but capable of excessive pro-inflammatory mediator production. Int J Obes 31: 1420–1428. 10.1038/sj.ijo.0803632 [DOI] [PubMed] [Google Scholar]
- Zhao HL, Lai FM, Tong PC, Zhong DR, Yang D, Tomlinson B, Chan JC. 2003. Prevalence and clinicopathological characteristics of islet amyloid in Chinese patients with type 2 diabetes. Diabetes 52: 2759–2766. 10.2337/diabetes.52.11.2759 [DOI] [PubMed] [Google Scholar]
- Zhao P, Wong KI, Sun X, Reilly SM, Uhm M, Liao Z, Skorobogatko Y, Saltiel AR. 2018. TBK1 at the crossroads of inflammation and energy homeostasis in adipose tissue. Cell 172: 731–743 e712. 10.1016/j.cell.2018.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Yang Q, Cao J, Xie N, Liu K, Shou P, Qian F, Wang Y, Shi Y. 2016. Local proliferation initiates macrophage accumulation in adipose tissue during obesity. Cell Death Dis 7: e2167. 10.1038/cddis.2016.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, An YA, Kim M, Zhang Z, Zhao S, Zhu Y, Asterholm IW, Kusminski CM, Scherer PE. 2020. Suppressing adipocyte inflammation promotes insulin resistance in mice. Mol Metab 39: 101010. 10.1016/j.molmet.2020.101010 [DOI] [PMC free article] [PubMed] [Google Scholar]