

In this study, Shabtai et al. investigated the mechanism of thyroid hormone (TH)-dependent gene repression, generated a mouse line in which endogenous thyroid hormone receptor TRβ1 was epitope-tagged to allow precise chromatin immunoprecipitation at the low physiological levels of thyroid hormone receptors (TR), and defined high-confidence binding sites where TR functioned at enhancers regulated in the same direction as the nearest gene in a TRβ-dependent manner. Their results demonstrate that, in contrast to the canonical “all or none” coregulator switch model, TH regulates gene expression by orchestrating a shift in the relative binding of corepressors and coactivators.
Keywords: ligand regulation, negative regulation, nuclear receptor coregulators, nuclear receptors, thyroid receptor
Abstract
Thyroid hormones (THs) are powerful regulators of metabolism with major effects on body weight, cholesterol, and liver fat that have been exploited pharmacologically for many years. Activation of gene expression by TH action is canonically ascribed to a hormone-dependent “switch” from corepressor to activator binding to thyroid hormone receptors (TRs), while the mechanism of TH-dependent repression is controversial. To address this, we generated a mouse line in which endogenous TRβ1 was epitope-tagged to allow precise chromatin immunoprecipitation at the low physiological levels of TR and defined high-confidence binding sites where TRs functioned at enhancers regulated in the same direction as the nearest gene in a TRβ-dependent manner. Remarkably, although positive and negative regulation by THs have been ascribed to different mechanisms, TR binding was highly enriched at canonical DR4 motifs irrespective of the transcriptional direction of the enhancer. The canonical NCoR1/HDAC3 corepressor complex was reduced but not completely dismissed by TH and, surprisingly, similar effects were seen at enhancers associated with negatively as well as positively regulated genes. Conversely, coactivator CBP was found at all TH-regulated enhancers, with transcriptional activity correlating with the ratio of CBP to NCoR rather than their presence or absence. These results demonstrate that, in contrast to the canonical “all or none” coregulator switch model, THs regulate gene expression by orchestrating a shift in the relative binding of corepressors and coactivators.
Thyroid hormones (THs) have been known to be powerful regulators of metabolism for >100 yr (Tata 2013; Mullur et al. 2014). In addition to its levels being dysregulated in millions of people with thyroid disease, the hormone or related compounds have long been used pharmacologically to treat metabolic diseases (Kowalik et al. 2018). THs were once a preferred treatment for obesity, until side effects were deemed too harmful. More recently, TH analogs have demonstrated potential for treating fatty liver and nonalcoholic steatohepatitis (NASH) (Sinha et al. 2019). However, >30 yr after the cloning of TH receptors (TRs) as members of the nuclear receptor (NR) superfamily, the mechanism of TH action remains enigmatic.
TH executes its function on gene-network regulation by activating TH receptors (TRs) on chromatin (Sap et al. 1986; Weinberger et al. 1986; Glass et al. 1987; Murray et al. 1988; Hodin et al. 1989). Three functional TRs are encoded from two genes: TRα1 is encoded from the Thra gene, and TRβ1 and TRβ2 are splice variants of Thrb (Lazar 2003). The three proteins are homologous and their separate in vivo function is mostly explained by specificity in tissue expression rather than sequence differences (Forrest et al. 1990, 1991; Strait et al. 1990; Bradley et al. 1992). Liver is one of the most important targets of TH where TRβ1 is the predominant isoform (Weiss et al. 1998). While TH action in the liver includes carbohydrate, fatty acids, and cholesterol homeostasis, thyroid dysfunction can cause impairment of liver physiological function and, consequently, systemic dysregulation of the whole body metabolism (Sinha et al. 2019). Correlation was found between hypothyroidism and nonalcoholic fatty liver disease (NAFLD) (Chung et al. 2012; Mantovani et al. 2018). Moreover, TH treatment can decrease hepatosteatosis and inflammation and restore mitochondrial function in NAFLD (Sinha et al. 2012; Bruinstroop et al. 2018).
In contrast to steroid hormone receptors, TRs are not anchored to heat shock proteins in the cytoplasm (Carson-Jurica et al. 1990; Dalman et al. 1990). Rather, TRs are located in the cell nucleus and bind to DNA even in the absence of TH (Lazar et al. 1991; Brent et al. 1992; Forman et al. 1992; Ribeiro et al. 1992; Wahlström et al. 1992; Kim et al. 1996). TRs bind DNA with the highest affinity as a heterodimer with retinoid X receptor, favoring sites with a direct repeat of the sequence AGGTCA separated by 4 base pairs (bp) (DR4) (Yu et al. 1991; Kliewer et al. 1992; Wahlström et al. 1992).
While bound to chromatin TRs show the ability to activate or repress the expression of target genes (Lazar 2003; Shibusawa et al. 2003; Yen et al. 2003). For genes activated by TH, current dogma posits that TH binding acts as a switch between repressed and activated states. Unliganded TRs recruit the corepressor complex, mainly NCoR1 and SMRT (Chen and Evans 1995; Hörlein et al. 1995; Ishizuka and Lazar 2003; Choi et al. 2008), while TH binding releases the corepressors and leads to recruitment of coactivators that increase gene transcription (Chakravarti et al. 1996; Jeyakumar et al. 1997; Glass and Rosenfeld 2000). Recent studies suggest that this model is oversimplified and may only pertain to a subset of TH-regulated genes in mouse liver (Præstholm et al. 2020).
TH-dependent negative regulation is even less well understood, yet it is critical to physiology as repression of pituitary Tshb gene expression by TH is the critical physiological regulator of TH levels (Hoskins 1949; Gurr and Kourides 1983; Carr et al. 1985; Chin et al. 1985). Many other genes are repressed by TH in a variety of tissues, including the liver (Lompré et al. 1984; Thompson et al. 1987; Caubín et al. 1994; Feng et al. 1994; Iglesias et al. 1996; Dupré et al. 2004). Several potential mechanisms of TH-mediated repression have been postulated (Lazar 2003). One model, involving direct TR binding to a so-called negative TRE (nTRE) has been proposed to explain regulation of the Tshb (Wondisford et al. 1989; Carr et al. 1992; Shibusawa et al. 2003) and other genes (Chin et al. 1998; Kim et al. 2005; Nygård et al. 2006; Gillis et al. 2018). A competing model, known as transrepression, suggests that TR binding to nTREs is not via direct DNA binding but, rather, via tethering to another DNA-binding transcription factor such as the AP1 complex (Desbois et al. 1991; Zhang et al. 1991). The tethering mechanism has also been proposed for other NRs (Jonat et al. 1990; Yang-Yen et al. 1990; Schüle et al. 1991; Joseph et al. 2003).
In the past few years, as genome-wide approaches have been used to interrogate gene expression and enhancer activities, another model for negative regulation by NRs has gained wider acceptance. In this model, known as “squelching” or “coactivator redistribution,” NRs compete for limiting amounts of nuclear transcriptional cofactors without binding near the gene that is negatively regulated by a ligand (Schmidt et al. 2016). This concept was first introduced based on studies in yeast and then extended to NRs in mammalian cell lines (Meyer et al. 1989, 1992). The best evidence for this model in a chromatin context is based on studies of estrogen receptor (ER) (Carroll et al. 2006; Guertin et al. 2014) and PPARɣ. (Step et al. 2014; Nelson et al. 2018).
TRs are not highly expressed in tissues, and existing antibodies are suboptimal, which has hampered previous genome-wide studies relating endogenous TR binding to function. Here we describe a new mouse model with an epitope tag in frame with the first methionine of the TRβ1 protein, and use it to detect endogenous TRβ1 bound to chromatin at physiological expression levels. Up-regulation as well as down-regulation of liver gene expression was highly correlated with TRβ1 binding sites (TRBSs) on chromatin. TRBSs near both positively and negatively regulated genes demonstrated epigenomic characteristics of activating and repressive enhancers, respectively. While TH binding reduced NCoR/HDAC3 recruitment at activated enhancers, the corepressor complex was also found and destabilized at sites near down-regulated genes. In contrast, binding of the coactivator CBP was reduced specifically at repressed enhancers. Thus, unlike other NRs, ligand-dependent repression by TH involves direct TR binding through the canonical TR-binding motif. Furthermore, rather than the all or none canonical switch model, it is the direction of shift of coregulator association with TRβ enhancers that determines enhancer activity and transcriptional outcomes.
Results
An in vivo mouse model to interrogate TRβ function at physiological levels
TRβ is expressed at low levels in liver, estimated to be ∼4000–8000 receptors per rat liver nucleus (Oppenheimer et al. 1974; Samuels et al. 1974). To gain insight into the function of TRβ at physiological levels, we used CRISPR to insert a triple HA epitope tag in frame with the first methionine of the Thrb gene in C57BL/6J mice (Fig. 1A). Liver expression of Thrb and the TH target gene Dio1 was indistinguishable from that of WT mice (Supplemental Fig. S1A). Endogenous TRβ was readily detected immunohistologically, demonstrating nearly exclusive nuclear localization in liver (Supplemental Fig. S1B). HA-TRβ was readily detected by chromatin immunoprecipitation (ChIP) of liver using anti-HA antibody, with little or no background in livers from wild type (WT) mice (Supplemental Fig. S1C). Mice were rendered hypothyroid by treatment with anti-thyroid drug propylthiouracil (PTU) for 3 wk, leading to low serum levels of thyroid hormones T3 and T4 and high levels of thyroid stimulating hormone (TSH), which were reversed by treatment with a combination of T3 and T4 (Supplemental Fig. S1D). The systemic hypothroidism and reversal with T3/T4 were confirmed by analysis of canonical TH-responsive Dio1 and Thrsp genes (Supplemental Fig. S1E).
Figure 1.
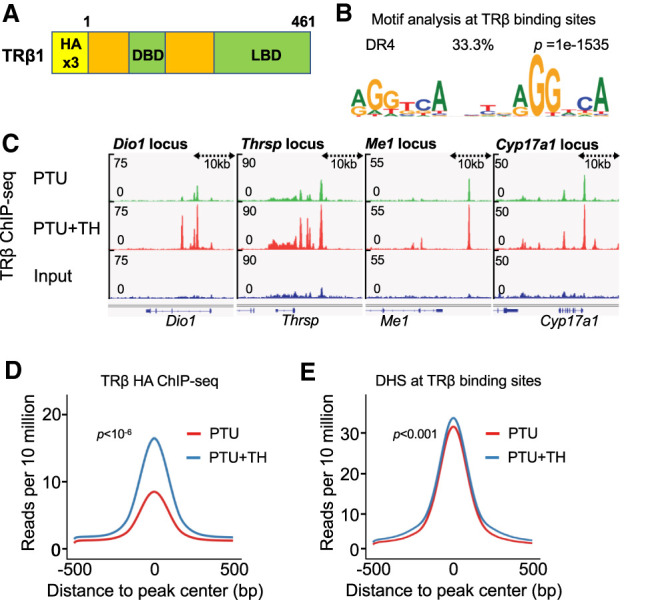
TRβ binding sites in hypothyroid and hyperthyroid mice with endogenous epitope-tagged TRβ1. (A) Schematic representation of the HA-TRβ protein. (B) Top hit from Homer de novo motif search at all TRβ binding sites identified the DR4 motif. (C) Examples of TRβ ChIP tracks for Dio1, Thrsp, Me1, and Cyp17a1 showing nearby TRBSs. (D) Average density profiles in reads per 10 million of TRβ ChIP-seq data (n = 3 biological replicates) at all TRBS in PTU- or PTU + TH-treated mice. (E) Average density profiles in reads per 10 million of DNase-seq data (n = 3 biological replicates) at all TRBS in PTU- or PTU + TH-treated mice.
HA-ChIP followed by deep sequencing (ChiP-seq) was performed on livers from PTU-treated (hypothyroid) livers as well as livers from PTU-treated mice given T3/T4. This analysis revealed 6500 TRβ binding sites (TRBS) using stringent cutoffs of P-value > 0.001, RPKM > 1 compared with input and were fourfold more than WT mice ChIP. De novo motif analysis of TRBS showed enrichment for the known functional TRBS, which is a direct motif of the AGGTCA sequence spaced by four bases, called the DR4 motif (Fig. 1B). In addition to the DR4, the DR1 motif was also enriched in the TRβ cistrome, albeit with a lower P value (Supplemental Fig. S1F). TRβ bound in the region of many canonical TH-activated genes including Dio1, Thrsp, Me1, and Cyp17a1 in both hypothyroid and hyperthyroid conditions (Fig. 1C).
Two prior studies evaluated TRBS in mouse liver: Ramadoss et al. (2014) performed ChIP-seq on biotinylated TRβ1 that has been overexpressed in mouse liver, while Grøntved et al. (2015) used an antibody to endogenous TRs that detects both TRα and β with relatively low specificity and sensitivity. There were numerous differences between the conclusions of the studies, likely related to the detection methods and overexpression model. For example, many more TRBSs were identified in the overexpression model. Moreover, Grøntved et al. (2015) detected little or no TRβ binding at TH-inducible genes in hypothyroid livers (e.g., black arrow in Supplemental Fig. S2A), while Ramadoss et al. (2014) reported TRβ binding at the sites that was nearly the same in hypothyroid and hyperthyroid livers (Supplemental Fig. S2A). Our results with endogenous HA-TRβ revealed binding that was equally or more robust with fewer nonspecific peaks (e.g., red arrow in Supplemental Fig. S2A) in both hypothyroid and TH-treated livers (Supplemental Fig. S2A).
We next performed global comparison of the TRβ cistromes using a single bioinformatics pipeline. As expected, the overexpression model of Ramadoss et al. (2014) had by far the most binding sites. The DR4 motif was enriched in all three studies, yet the HA-TRβ cistrome showed stronger enrichment, and sites common to all three studies and common to the HA and Grøntved et al. (2015) were the highest (10-fold and 15-fold, respectively) (Supplemental Fig. S2B). Focusing on these common sites, we noted that the effect of TH on TRβ binding was also notably different across studies. Ramadoss et al. (2014) observed a very small overall increase with TH treatment, while Grøntved et al. (2015) noted an increase with TH with binding strength that was generally quite low (Supplemental Fig. S2C). In contrast, the present studies using an antibody that was highly specific for endogenous tagged TRβ demonstrate robust binding that was markedly stimulated by TH (Supplemental Fig. S2C). A similar result was obtained when all of the HA-TRβ binding sites were included in the analysis (Fig. 1D). Interestingly, integrating the TRβ binding sites with published DNase-seq data generated from hypothyroid and hyperthyroid liver mice (Grøntved et al. 2015) revealed no significant difference between the openness of chromatin at these sites in the presence or absence of TH (Fig 1E).
TH alters chromatin structure at TR binding sites near TH-regulated genes
We next sought to understand the hepatic regulatory network and function of TRβ by integrating the TRβ cistromes with TH-dependent gene expression. Similar numbers of genes were up-regulated (777) and down-regulated (867) by TH (Fig. 2A). Each TRBS was mapped to the nearest gene then partitioned based on the effect of TH on the nearest gene relative to PTU-only treatment: down-regulated, no change in expression, and up-regulated (differential expression [DE]: |FC| > 1.5, adjusted FDR < 0.05). Remarkably, although we showed earlier that TH did not change overall chromatin accessibility on a genome-wide scale, TH increased chromatin accessibility at TRBSs near up-regulated genes (Fig 2B, red dots). Conversely, chromatin accessibility decreased upon TH treatment at TRBSs near down-regulated genes (Fig. 2B, blue dots). These findings suggest that TH binding to TRβ can alter chromatin structure in a manner that is associated with the outcome of gene regulation, with chromatin opening correlating with activation and closing being characteristic of ligand-induced repression. Since TRβ binding was globally increased by TH, we considered whether there might be a difference at sites associated with gene activation versus those associated with TH-dependent repression. However, despite the differences in chromatin accessibility, TH increased TRβ binding approximately twofold regardless of the relationship of the binding site to TH-dependent changes in chromatin structure and gene regulation (Fig. 2C).
Figure 2.
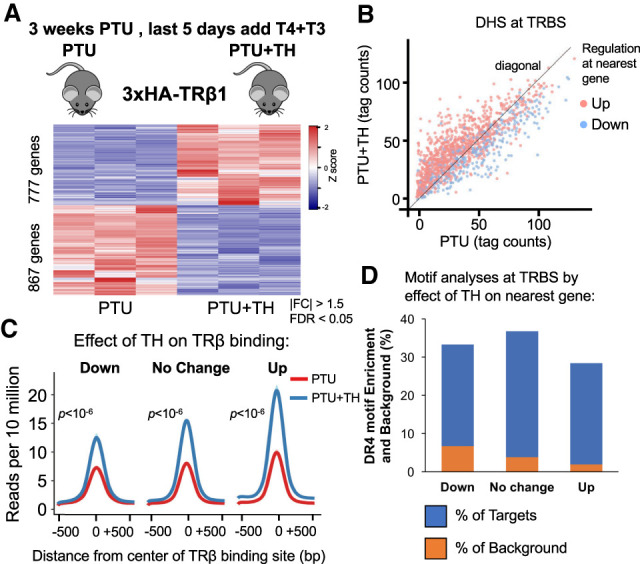
TRβ binding sites near genes that are induced or repressed by TH. (A) Heat map showing TH-dependent differentially regulated genes. n = 3 biological replicates, DE cutoff: |FC| > 1.5, two-sided Benjamini–Hochberg-adjusted FDR < 0.05 as determined by edgeR likelihood ratio test. (B) Scatter plot of DNase-seq data showing read counts centered to TRBSs from PTU-treated mice against PTU + TH-treated mice (n = 2 biological replicates). (Red) Up-regulated, (blue) down-regulated. DE cutoff taken from A. (C) Average density profiles in reads per 10 million of TRβ ChIP-seq data (n = 3 biological replicates) at TRBSs in PTU- or PTU + TH treated mice. TRBSs were categorized by the effect of TH on the nearest gene annotated to them. (D) DR4 motif enrichment (percentage) depicted as blue bars for each TRBSs group, categorized by the effect of TH on the nearest gene annotated to them. The orange bars overlay showing the (percentage) background level (Homer used for de novo motif search).
TR is known to bind preferentially in vitro as a heterodimer with RXR to DNA containing two directly repeated hexameric sequences related to AGGTCA spaced by 4 bp, which is referred to as a DR4 motif (Yu et al. 1991; Kliewer et al. 1992; Marks et al. 1992). While the DR4 motif has been shown to mediate TR binding and TH activation of gene expression (Umesono et al. 1991; Mader et al. 1993; Shulemovich et al. 1995), its role in TH-dependent repression is debated (Lazar 2003). Interestingly, de novo motif analysis revealed that the DR4 motif was enriched at TRBSs regardless of the relationship of the binding site to TH regulation of the nearest gene (Fig. 2D). These results suggest that TRβ binds directly to DR4 sites near both positively and negatively regulated genes. TH induces TRβ binding nearly equally at sites, which are distinguishable by the direction of chromatin accessibility induced by TH.
TH-dependent gene expression requires TRβ
We next studied Thrb knockout mice (Forrest et al. 1996; Amma et al. 2001) to determine the TH-regulated genes that require TRβ. Thrb−/− and Thrb+/+ mice were made hypothyroid by treatment with methimazole (MMI) for 4 wk in parallel with MMI-treated groups that were made hyperthyroid by additional treatment with T3 for the fourth week (MMI + T3), followed by RNA-seq analysis of the liver. Since this hypothyryoid/hyperthyroid protocol was different from that used for the HA-TRβ mice (3 wk PTU, with T4 + T3 given i.p. during the last 5 d), we compared the RNA-seq results from the control group with that of the HA-TRβ mice (performed at different institutions) (Supplemental Fig. S3A) and noted a strong correlation in TH-dependent gene expression (Supplemental Fig. S3B), which was even higher when differentially expressed genes were tested (Supplemental Fig. S3C). Additionally, >75% of both up-regulated and down-regulated genes in the HA-TRβ/PTU model (DE: |FC| > 1.5, adjusted FDR < 0.05) overlapped with the control of the methimazole model (Fig. 3A). Focusing on the genes that were TH-regulated in both models, genetic deletion of TRβ markedly reduced TH-responsiveness of both up-regulated (Fig. 3B) and down-regulated genes (Fig. 3C), consistent with TRβ being the dominant TR subtype in the liver (Weiss et al. 1998). This was observed in both models, validating with high confidence this TH/TR-dependent transcriptome in liver.
Figure 3.
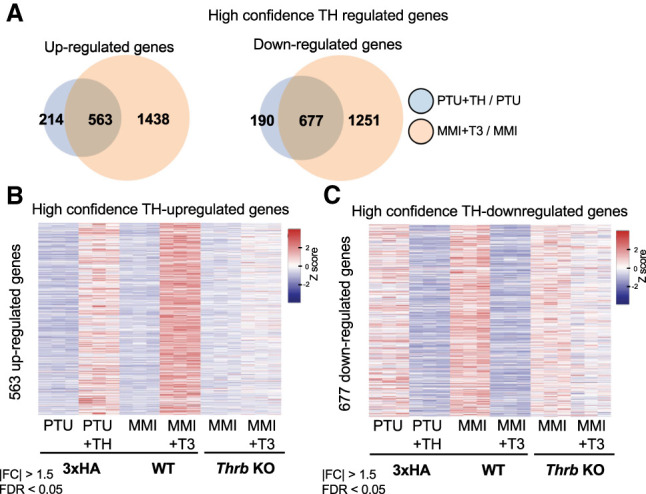
Defining high-confidence TH-regulated genes that require TRβ in liver. (A) Venn diagrams displaying overlap of differentially expressed genes identified in PTU + TH/PTU and MMI + T3/MMI experiments. Overlapped up-regulated genes on the left and down-regulated genes on the right. (B) Heat map showing relative expression of overlapped TH-dependent up-regulated genes between the two treatment systems and the treated Thrb−/− mice. (C) Heat map showing relative expression of overlapped TH-dependent down-regulated genes between the two treatment systems and the treated Thrb−/− mice. For all experiments, n = 3 biological replicates, DE cutoff: |FC| > 1.5, two-sided Benjamini–Hochberg-adjusted FDR < 0.05 as determined by edgeR likelihood ratio test.
TRβ binding sites near TH/TR-dependent genes are functional enhancers that depend on ligand receptor interaction
A large number of these TH/TR-dependent genes were highly associated with nearby TRβ binding sites and generated a greater overlap of differentially regulated genes between the two models (Supplemental Fig. S4). The effect of TRβ deletion on the magnitude of the TH-dependent change in gene expression (either up-regulation or down-regulation of expression) was >1.5-fold in >84% of the 505 TH-regulated, TRβ-dependent genes with proximal TRBSs (Fig. 4A). Moreover, one-third of these functional binding sites are near genes that are repressed by TH (Fig. 4B). For TRBS near both up-regulated and down-regulated genes, the most enriched DNA sequence motif was the DR4 (Fig. 4C), which thus did not discriminate between functional transcriptional outcome. Chromatin accessibility increased with TH at TRBS near high-confidence up-regulated genes and decreased near repressed genes (Fig. 4D), similar to what was observed for the cistrome near all differentially expressed genes.
Figure 4.
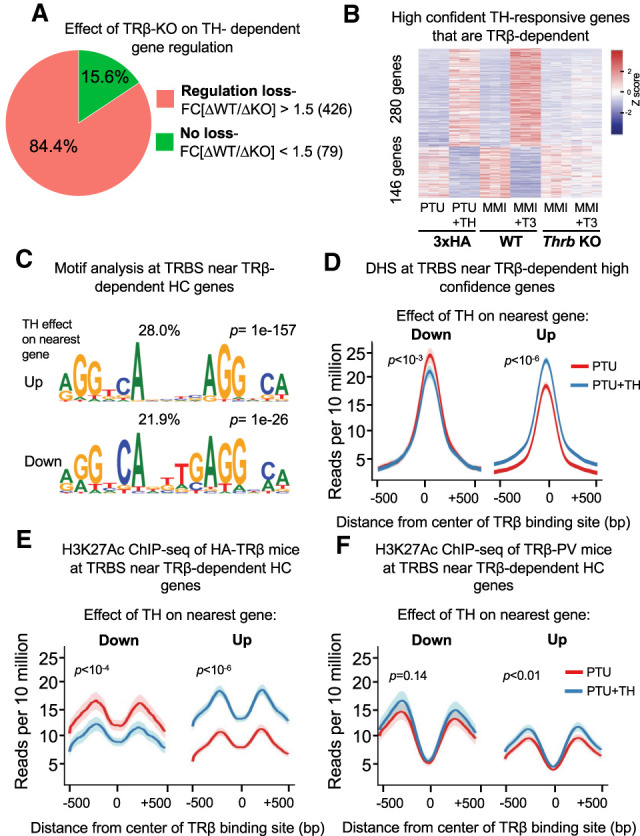
Functional TRBSs demonstrate enhancer characteristics that correlate with nearby gene expression. (A) Fraction of TH-regulated, TRβ-dependent genes with proximal TRBSs in which the effect of TRβ deletion on gene expression is >1.5-fold. (B) Heat map showing the effect of TRβ deletion on relative expression of TH-regulated, TRβ-dependent genes with proximal TRBS genes. n = 3 biological replicates, DE cutoff: |FC| > 1.5, two-sided Benjamini–Hochberg-adjusted FDR < 0.05 as determined by edgeR likelihood ratio test. (C) Homer de novo motif search at TRBS near high-confidence TH-regulated genes that are TRβ-dependent showing DR4 motif enrichment near both up-regulated and down-regulated genes. (D) Average density profiles in reads per 10 million of DNase-seq data (n = 3 biological replicates) at TRBSs near high-confidence TH up-regulated or down-regulated genes that are TRβ-dependent. The difference in reads between PTU and PTU + TH treatments is shown. (E) Average density profiles in reads per 10 million of H3K27Ac ChIP-seq data (n = 2 biological replicates) at TRBSs near high-confidence TH up- or down-regulated genes that are TRβ-dependent; showing the difference in reads between PTU and PTU + TH treatments. (F) Average density profiles in reads per 10 million of H3K27Ac ChIP-seq data in TRβ-PV mice lacking the ability to bind TH (n = 2 biological replicates) at TRBSs near high-confidence TH up-regulated or down-regulated genes that are TRβ-dependent. The difference in reads between PTU and PTU + TH treatments is shown.
Functional binding sites, commonly known as enhancers, are characterized by modification of histone H3 by acetylation at lysine 27 (H3K27Ac) that is related to the regulation of the gene whose transcription is being controlled. H3K27Ac was markedly increased by TH at TRBSs near TR-dependent up-regulated genes (Fig. 4E). In contrast, H3K27Ac went down in a TH-dependent manner at TRBSs near down-regulated genes (Fig. 4E). These results show that TRBSs near up-regulated as well as down-regulated genes have the characteristics of regulated enhancers. Moreover, the finding that TH reduces enhancer activity at TRBSs where TR is required for negative regulation strongly suggests that repression is mediated by liganded receptor binding directly at these sites. This is of great interest because the mechanism of TH-dependent down-regulation is controversial (Lazar 2003), and ligand-dependent negative regulation by other NRs including PPARɣ and the estrogen receptor has been shown to occur without direct binding to the target genes (Carroll et al. 2006; Step et al. 2014).
To better understand the role of TH binding to TRβ at TRBSs near negatively regulated genes, we integrated data from a mouse model in which a frameshift mutation in the ligand binding domain of TRβ (TRβ-PV) results in its inability to bind TH and to activate or to repress genes in a ligand-dependent manner (Kaneshige et al. 2000). A recent study showed blunted H3K27Ac at TRβ-PV binding sites after TH treatment (Præstholm et al. 2020), and we interrogated those ChIP-seq data with a focus on the TRBSs at high-confidence TR/TH-requiring genes as defined herein. Analysis of TH-induced genes at TRBS where H3K27Ac is normally stimulated by TH showed that these sites lost their ability to gain acetylation in the TRβ-PV livers, consistent with the recent study (Fig. 4F; Præstholm et al. 2020). Importantly, performing the same analysis on TRBSs near negatively regulated genes revealed that TH-dependent effects on H3K27Ac were lost as for positive regulated TRBSs (Fig. 4F). Furthermore, in TRβ-PV livers, the acetylation at these TRBSs was higher than at TRBSs near up-regulated genes and were comparable with acetylation levels noted in 3xHA-TRβ hypothyroid livers. Together, these results suggest that inability to bind ligand diminished TRβ/TH-dependent enhancer function for both direct activation and repression.
TRβ canonically activates genes by dismissing the NCoR/HDAC3 corepressor complex while recruiting coactivators such as CBP. Analysis of available ChIP-seq data for NCoR1, HDAC3, and CBP (Præstholm et al. 2020) showed enrichment for expected transcription factors (Supplemental Fig. S5A–C) and binding motifs (Supplemental Fig. S5D–F), and thus we integrated these data with the high-confidence TRBSs. NCoR and HDAC3 binding was reduced by hyperthyroid conditions at TRBSs near TH-induced genes, as expected (Fig. 5A,B). However, TH also reduced NCoR and HDAC3 binding at TRBSs near down-regulated genes (Fig. 5A,B). Thus, TH-dependent transcriptional outcomes were not necessarily correlated with reduced binding of NCoR/HDAC3. A recent study proposed two distinct mechanisms of hyperacetylation at TR-occupied enhancers, distinguished by the presence of HDAC3 in the hypothyroid condition (Præstholm et al. 2020). However, rather than two distinct kinds of TRBS, analysis of the high-confidence TRBS revealed a continuous range of occupancy states (Supplemental Fig. S6A,B), with high correlation between NCoR1 and HDAC3 binding (Supplemental Fig. S6C).
Figure 5.
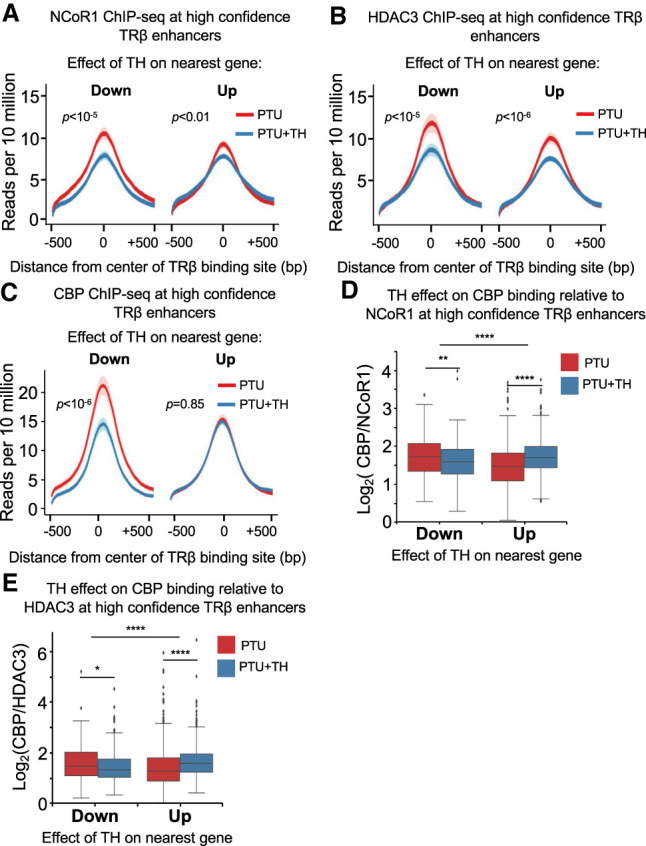
TH action involves a shift between corepressor and coactivator occupancy at enhancers of both positively and negatively regulated genes. (A) Average density profiles in reads per 10 million of NCoR1 ChIP-seq data (n = 2 biological replicates) at TRBSs near high-confidence TH-regulated and TRβ-dependent genes; showing the difference in reads between PTU and PTU + TH treatments. (B) Average density profiles in reads per 10 million of HDAC3 ChIP-seq data (n = 2 biological replicates) at TRBSs near high-confidence TH-regulated and TRβ-dependent genes; showing the difference in reads between PTU and PTU + TH treatments. (C) Average density profiles in reads per 10 million of CBP ChIP-seq data (n = 2 biological replicates) at TRBSs near high-confidence TH-regulated and TRβ-dependent genes; showing the difference in reads between PTU and PTU + TH treatments. (D) Box plot showing the effect of TH on CBP binding normalized to NCoR1. Value at each peak is log ratio of CPB to NCoR1 ChIP-seq signal. The effect is shown separately for active and repressive enhancers. (E) Box plot showing the effect of TH on CBP binding normalized to HDAC3. Value at each peak is log ratio of CPB to HDAC3 ChIP-seq signal. The effect is shown separately for active and repressive enhancers. For D and E, within the same region: paired t-test; different regions: unpaired t test. P-values are shown. (*) P < 0.05, (**) P < 0.01, (****) P < 0.0001.
We next considered the binding of coactivator CBP, which has intrinsic histone acetyltransferase (HAT) activity and is recruited as part of a coactivator containing SRC (Li et al. 2000; Sheppard et al. 2001). CBP also showed a continuous range of occupancy rather than bimodal binding (Supplemental Fig. S6D). Interestingly, CBP binding was robust but not changed between hypothyroid and hyperthyroid conditions at positively regulated genes. However, at enhancers near negatively regulated genes, CBP binding was high in the hypothyroid state and reduced by TH (Fig. 5C).
To further interrogate the role of coregulators in positive and negative regulation by TH, we normalized CBP binding to that of NCoR1 or HDAC3 at TRBSs of high-confidence TR/TH-requiring genes between hypothyroid and hyperthyroid conditions. Genome-wide, for up-regulated genes, TH increased the ratio of CBP to NCoR (or HDAC3) binding at nearby enhancers whereas, in contrast, the CBP to NCoR (or HDAC3) ratio was greater at down-regulated TR-bound enhancers under hypothyroid conditions (Fig. 5D,E). This is illustrated at specific TRBSs of genes that are positively regulated (Dio1, Idh3, and Gdp2) and negatively regulated (Gys2, Gsta2, and Etnppl) by TH (Supplemental Fig. S7).
We next investigated potential mechanisms that might dictate the coregulator shifts distinguishing positive and negative enhancers. Epigenetic landscape in silico deletion analysis (LISA) (Qin et al. 2020) did not uncover differences in regulatory candidate transcription factors between up-regulated and down-regulated genes (Supplemental Fig. S8A), and thus we turned our attention to whether three-dimensional chromatin architecture could influence TRβ-dependent gene activation or repression. Integration of our data with HiC data from liver tissue (Kim et al. 2018) revealed a small fraction of genes topological-associated domains (TADs) could have a regulatory function, but no difference between up-regulated or down-regulated genes (Supplemental Fig. S8B,C). Of note, however, positively regulated genes were more frequently positioned in the same TAD than negatively regulated (Supplemental Fig. S8D,E). Also, TRBSs where the CBP/NCOR1 ratio goes up or down with TH had a tendency to cluster in the same TAD (Supplemental Fig. S8F,G), while TAD-TAD interaction did not effect this propensity (Supplemental Fig. S8H,I). These results show that, in some cases, positive regulation by TH was favored by TADs, whereas negative regulation was less influenced by TAD structures. Together, the results show that transcriptional outcomes of TH binding to TR correlated with the relative recruitment of corepressor or coactivator rather than an absolute switch in their presence at enhancers, with similar principles pertaining to positively and negatively regulated genes.
Discussion
For many years, TH has served as a model for hormone action and TRβ as a model representative of the nuclear receptors family (Tata 2013). However, the sparse number of TRs in the cell and the unavailability of high-quality antibodies generated discrepancies of in vivo experiments (Ramadoss et al. 2014; Grøntved et al. 2015). Here, we have introduced an HA-TRβ-tagged mouse model, where TRβ is expressed under its endogenous promoter and can be detected using the highly sensitive antibody, and used the model to better understand the mechanism of TH action and the role of TRβ in chromatin.
Our data showed consistency with previous studies demonstrating TRβ binding to TRBSs at enhancers near up-regulated genes. These enhancers exhibited higher H3K27ac and chromatin accessibility upon TH treatment. Moreover, de novo motif analysis revealed enrichment for the DR4 motif at these sites. TRBSs related to the well-accepted hepatic TH responsive genes Dio1, Thrsp, Me1, and Cyp17a1 were occupied with TRβ in the unliganded state as well as in the liganded state. This is in accordance with early in vitro studies (Lazar et al. 1991; Brent et al. 1992; Forman et al. 1992; Ribeiro et al. 1992; Wahlström et al. 1992) and with the ChIP-seq results of Ramadoss et al. (2014), using TRβ1 overexpression in liver, but not with the results of Grøntved et al. (2015), which suggested that TR binding was ligand-dependent and nearly undetectable in the absence of TH. This is probably related to the ChIP-seq detection method, which used a less specific antibody requiring very stringent bioinformatic cutoffs (Grøntved et al. 2015).
Importantly, in this study we show that TH gene repression mediated by TRβ in the liver correlates with direct binding to a DR4-like sequence on a genome-wide scale. This is in contrast to ligand-dependent repression by other NRs using tethering or squelching mechanisms (Meyer et al. 1992; Joseph et al. 2003; Carroll et al. 2006; Step et al. 2014). While a previous ChIP-seq study of endogenous TRβ did not find TRBSs near TH-dependent repressed genes (Grøntved et al. 2015), a ChIP-seq study on overexpressed TRβ in the liver did observe such binding (Ramadoss et al. 2014). However, the overexpression study suggested that TH-facilitated gene repression is associated with reduction in TRβ occupancy and thus alluded to an indirect mechanism. Rather, our data show that, in general, TH increases endogenous TRβ binding regardless of its effect on the nearest gene. Moreover, the DR4 motif was enriched in these TRBSs, and the causal role of TRβ was demonstrated by the abrogation of TH-mediated repression in Thrb knockout mice.
The canonical coregulator switch model describes corepressor binding to TR at functional enhancers, with ligand binding triggering the dismissal of corepressor and recruitment of coactivator (Glass and Rosenfeld 2000). Mutational studies have demonstrated that NCoR and SRC1 both contribute to the magnitude of TH effects but do not provide evidence for an “either/or” mechanism of coregulator binding (Vella et al. 2014). A recent study investigated the switch model by focusing on the extremities of TH-dependent histone acetylation at TRBSs and suggested that the switch does occur but only at a subset of TH-activated genes (Præstholm et al. 2020). Here, however, focusing on functional enhancers in the context of a highly specific TRβ1 cistrome, we found that TH globally reduced without completely dismissing NCoR/HDAC3 binding at regulated enhancers correlating with down-regulated as well as up-regulated genes. Indeed, despite the presence of HDAC3, at down-regulated enhancers we observed elevated H3K27 acetylation in hypothyroid conditions. This is likely because of the abundance of CBP, a powerful HAT, at enhancers associated with TH-repression or other coactivators with redundant activity. It should be recognized that NCoR1 and CBP are not unique but rather representative of TRs corepressors and coactivators, respectively.
Together, these results lead us to suggest a novel view of gene regulation by TH, particularly in its negative transcriptional effects. Rather than a switch, TH functions to shift the ratio of corepressor to coactivator at both positive and negative enhancers that are bound and regulated by TR. On a genome-wide basis, repression of gene expression by TH involves direct binding of TRβ to repressive enhancers containing canonical DR4 motifs. The recruitment of activating versus repressive coregulator complexes could be mediated by several layers of regulation. We did not detect major differences in motifs found at TRBSs related to up-regulated versus down-regulation, suggesting that factors in addition to the TRBS must specify whether a given site functions as an activating or repressive enhancer. We did note some influence of three-dimensional architecture, particularly for positively regulated genes, but this could not fully explain the differences between genes that were up-regulated versus down-regulated by TH. Thus, other factors such as response type-specific enhancer-to-enhancer interactions and protein recruitment are likely to explain the direction of regulation by TH at a given TRBS.
Materials and methods
Animals and treatments
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Pennsylvania. Mice were housed in a temperature-controlled specific pathogen-free facility under 12-h light/dark cycles. Adult male mice of 12–14 wk were used in all experiments. For HA-TRβ mice, controls were age-matched WT C57BL/6 mice that were purchased from Jackson Laboratories. Thrb−/− mice (Forrest et al. 1996), originally on a mixed C57BL/6 × 129/Sv background, were back-crossed for multiple generations (more than nine) onto C57BL/6J; wild-type controls were C57BL/6J mice. For PTU treatment, mice were provided with drinking water containing 1% perchlorate and 0.05% propylthiouracil for 3 wk. In the last 5 d, all animals were intraperitoneally injected with vehicle (0.9% saline in 100-µL volume) for the hypothyroid group or 40 µg/100 g T4 with 4 µg/100 g T3 for the hyperthyroid group. For MMI treatment, mice were provided with 0.05% methimazole and 1% potassium perchlorate in the drinking water for 4 wk, inducing hypothyroidism, adapted from established methods (Amma et al. 2001). To induce hyperthyroidism, half of the groups were given T3 at 5 µg/mL in the same drinking water (MMI + T3) during the fourth week of treatment.
Generation of HA epitope-tagged TRβ1 mice
To generate Cas9 mRNA, a plasmid containing Cas9-HA-2NLS was linearized with XbaI (gift from Jorge Henao-Mejia, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA). Approximately 1 μg of linearized plasmid was incubated with a HiScribeTM T7 quick high-yield RNA synthesis kit (NEB E2050S). RNA was purified using RNeasy mini columns (Qiagen 74106), and the capping reaction used the Vaccinia capping system (NEB M2080S). RNA was purified using RNeasy micro clean-up columns (Qiagen 74004). Capped Cas9 mRNA was then subjected to polyadenylation (NEB M0276S) and purified over an RNeasy micro clean-up column and eluted in RNase-free water. Cas9 mRNAntegrity was validated using RNA BioAnalyzer. T7 promoter was added onto the gRNA template targeting the ATG start site by PCR amplification using specific primers (targeting guide sequence: TCATAGGTTAGTAGTCATGC). The T7-sgRNA product was purified using a PCR purification kit (Qiagen) and used as the template for in vitro transcription using the MegaShortScript kit (Life Technologies) following the manufacturer's instructions. Subsequent sgRNA was purified using the MegaClear kit (Life Technologies) and verified by RNA BioAnalyzer before dilution for microinjection. The following ssDNA homology donor (IDT) containing the 3×HA tag was resuspended in water and prepared using DNA Clean and Concentrator (Zymo): C*A*C*AGTGTAAATCAGGAAAGGACACAAAGATACCTGTCATACTGTTAGGAGTGCCAGCGTAATCTGGAACGTCATAAGGATACGATCCTGCATAGTCCGGGACGTCATAGGGATAGCCCGCATAGTCAGGAACATCGTATGGATACATAGGTTAGTAGTCATGCTGCGTCATCTTCTGTACTGGCATTCCCTAAAA*G*A*G. Microinjection was performed by the Transgenic and Chimeric Mouse Facility at the University of Pennsylvania using C57BL/6J mice from Jackson Laboratories. The microinjection buffer consisted of 1 mM Tris (pH 8.0), 0.1 mM EDTA, 100 ng/μL Cas9 mRNA, 50 ng/μL sgRNA, and 100 ng/μL of ssDNA homology donor. Insertion of epitope tag coding sequence was detected by PCR and confirmed by Sanger sequencing. HA-TRβ mice were backcrossed to the C57BL/6J genetic background for at least four to five generations and genotyped using the PCR primers: 5′-TGTCCACATCGCAAGTTCCAG-3′ and 3′-ATGAGTTCAGGGCATAGGGGTG-5′.
RNA extraction and gene expression measurement
Total RNA was extracted from snap-frozen liver tissues using TRIzol (Invitrogen) followed by an RNeasy mini kit (Qiagen). For RNA-seq, all samples were sequenced separately for three biological replicates. RNA integrity was examined using an Agilent RNA 6000 Nano kit. RNA samples with RNA integrity number >7 (1 μg) were used for RNA cleanup, library preparation, and sequencing by Novogene. For MMI-treated mice, total RNA was prepared from frozen liver, using TRIzol reagent (Invitrogen 15596-026), then mRNA-enriched from 5-μg aliquots of total RNA using a Dynabeads mRNA purification kit (Ambion # 61006) and quantified using a Qubit RNA broad-range assay kit (Thermo Fischer Scientific). Double-stranded cDNA was prepared using SuperScript double-stranded cDNA synthesis kit (Invitrogen), and cDNA libraries for sequencing were generated using a ThruPLEX DNA-seq kit (Takara R400428). Multiplex sequencing was performed, yielding single-end 50-base-length reads, at the NIDDK Genomic Core using an Illumina HiSeq 2500 instrument. All RNA-seq experiments were performed with three biological replicates per condition.
RNA-seq analysis
Reads were mapped to the mm9 reference genome using STAR (Dobin et al. 2013), selecting only uniquely mapped reads. Expression was quantified at the gene level with featureCounts (Liao et al. 2014), using Ensembl v.67 gene annotations. Only genes that have at least one read per million in at least two samples are considered in the following analysis. We then used edgeR (McCarthy et al. 2012) to find genes differentially expressed between two conditions. A gene is considered differentially expressed if the corresponding P-value (Benjamini-Hochberg-adjusted) is <0.05 and the fold change between conditions is at least 1.5.
For visualization purposes, we use FPKM values computed by edgeR. Heat maps show gene-wise z-scores of FPKM across all shown samples, unless indicated otherwise.
Gene ontology term enrichment in gene sets is computed using the g:Profiler web service (Raudvere et al. 2019). Benjamini-Hochberg-adjusted P-value must be <0.05 and enrichment >2 for a term to be considered enriched.
Chromatin immunoprecipitation
HA-TRβ ChIP-seq was performed as previously described (Armour et al. 2017). HA magnetic beads (Pierce) were used to perform the immunoprecipitation. For ChIP-seq of HA-TRβ, three biological replicates were used for sequencing, and WT mice were used as a negative control. The ChIP-seq library has been performed as previously described (Armour et al. 2017).
ChIP-seq analysis
ChIP-seq experiments on PTU mice and PTU + TH mice were performed independently on three biological replicates (n = 3), except for H3K27ac ChIPseq experiments in PTU- and PTU + TH mice for which there were two independent biological replicates (n = 2) per condition. All sequencing reads of biological replicates were aligned to the mm9 genome using Bowtie2 (v.2.2.6) (Langmead and Salzberg 2012) allowing for one mismatch (-N1). Duplicate reads were removed, and unique reads were extracted using SAMtools (v.1.7) (Li et al. 2009). Tag directories were generated from alignment files using HOMER (v.4.9.1) (Heinz et al. 2010).
Peaks were called using HOMER's findPeaks function with parameter “-style factor” (for transcription factors) or “-style histone” (for H3K27ac) for each individual replicate with a corresponding input directory as background, with default parameters. Each peak file was transposed to a bed format using pos2bed Perl script. The resulting replicate files were then sorted and merged to a single PTU and PTU + TH peak files using BEDOPS (v.2.4.30) (Neph et al. 2012). To find genes nearest to the peaks, we used HOMER's annotatePeaks command using mm9 genome. Tags were counted for each replicate and reported as reads per million (RPM). Annotated peaks were further filtered using R and RStudio (R v.3.6.3), leaving only peaks with reads >1 RPM in at least two replicates. To further increase the confidence in our peaks, we used annotatePeaks to calculate tag counts for each pooled condition and compared it with WT mice. We filtered out peaks for each pooled condition that had less than fourfold reads compared with WT mice. Peaks from each condition were finally merged to form the set of confident TRβ peaks.
Genome browser tracks were generated with HOMER function “makeUCSCfile -bigWig -fsize 1e20.” Track visualization was performed using the Integrated Genomics Viewer (IGV) (Thorvaldsdóttir et al. 2013). ChIP-seq signal densities at specific peak regions were obtained with annotatePeaks with a 1-kb window from the peak center, using the option “-size 1000 -hist” (for histograms) with 10-bp bin size. Average density profiles were generated with mean signal in normalized RPM and error bands for biological replicates. De novo motif analyses were performed with the HOMER function findMotifsGenome.pl using parameters “-len 8 to 18 and -p 4.”
Density profiles of H3K27ac PTU and PTU + TH aligned, pooled, tag directories around TRβ peak files were performed using annotatePeaks function with parameters “-size 1000, -rlog” and plotted using custom Rscript.
Statistical analysis
For analysis of data other than sequencing experiments’ output, the following procedures were used: Data are presented as means ± standard error of the mean. A two-tailed Student's t-test was used for comparison between two groups. All of the statistical analyses were performed with GraphPad Prism (not significant [ns], P < 0.5 [*], P < 0.01 [**], P < 0.001 [***], P < 0.0001 [****]).
Deep sequencing data
All data have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE159648.
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
We acknowledge the Functional Genomics Core (J. Schug and K. Kaestner) and the Biomarkers and RIA Core (H. Collins and M. Rickels) of the Penn Diabetes Research Center (DK19525) for next-generation sequencing, and hormone assays, respectively, and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Genomics Core (H. Smith) for next-generation sequencing at the National Institutes of Health. This work was supported by NIDDK grant DK43806, the Cox Institute for Medical Research (M.A.L.), and the intramural research program at NIDDK (Y.-W.C. and D.F.).
Author contributions: Y.S and M.A.L. conceived the project. Y.S. performed experiments and interpreted the data. Y.-W.C. performed Thrb−/− experiments. Y.G. performed the IF assay. J.R. generated the HA-TRβ mice. N.K.N. and K.B. analyzed the data. M.A.L. and D.F. provided supervision. Y.S. wrote the manuscript with input from Y.-W.C., D.F., and M.A.L.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.345686.120.
References
- Amma LL, Campos-Barros A, Wang Z, Vennström B, Forrest D. 2001. Distinct tissue-specific roles for thyroid hormone receptors β and α1 in regulation of type 1 deiodinase expression. Molecular Endocrinology 15: 467–475. [DOI] [PubMed] [Google Scholar]
- Armour SM, Remsberg JR, Damle M, Sidoli S, Ho WY, Li Z, Garcia BA, Lazar MA. 2017. An HDAC3-PROX1 corepressor module acts on HNF4α to control hepatic triglycerides. Nat Commun 8: 549. 10.1038/s41467-017-00772-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley DJ, Towle HC, Young WS. 1992. Spatial and temporal expression of α- and β-thyroid hormone receptor mRNAs, including the β 2-subtype, in the developing mammalian nervous system. J Neurosci 12: 2288–2302. 10.1523/JNEUROSCI.12-06-02288.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brent GA, Williams GR, Harney JW, Forman BM, Samuels HH, Moore DD, Larsen PR. 1992. Capacity for cooperative binding of thyroid hormone (T3) receptor dimers defines wild type T3 response elements. Mol Endocrinol 6: 502–514. [DOI] [PubMed] [Google Scholar]
- Bruinstroop E, Dalan R, Cao Y, Bee YM, Chandran K, Cho LW, Soh SB, Teo EK, Toh S-A, Leow MKS, et al. 2018. Low-dose levothyroxine reduces intrahepatic lipid content in patients with type 2 diabetes mellitus and NAFLD. J Clin Endocrinol Metab 103: 2698–2706. 10.1210/jc.2018-00475 [DOI] [PubMed] [Google Scholar]
- Carr FE, Ridgway EC, Chin WW. 1985. Rapid simultaneous measurement of rat α- and thyrotropin (TSH) β-subunit messenger ribonucleic acids (mRNAs) by solution hybridization: regulation of TSH subunit mRNAs by thyroid hormones. Endocrinology 117: 1272–1278. 10.1210/endo-117-3-1272 [DOI] [PubMed] [Google Scholar]
- Carr FE, Kaseem LL, Wong CWN. 1992. Thyroid hormone inhibits thyrotropin gene expression via a position-independent negative L-triiodothyronine-responsive element. J Biol Chem 267: 18689–18694. 10.1016/S0021-9258(19)37016-4 [DOI] [PubMed] [Google Scholar]
- Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, et al. 2006. Genome-wide analysis of estrogen receptor binding sites. Nat Genet 38: 1289–1297. 10.1038/ng1901 [DOI] [PubMed] [Google Scholar]
- Carson-Jurica MA, Schrader WT, O'Malley BW. 1990. Steroid receptor family: structure and functions. Endocr Rev 11: 201–220. 10.1210/edrv-11-2-201 [DOI] [PubMed] [Google Scholar]
- Caubín J, lglesias T, Bernal J, Muñoz A, Márquez G, Barbero JL, Zaballos A. 1994. Isolation of genomic DNA fragments corresponding to genes modulated in vivo by a transcription factor. Nucl Acids Res 22: 4132–4138. 10.1093/nar/22.20.4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarti D, LaMorte VJ, Nelson MC, Nakajima T, Schulman IG, Juguilon H, Montminy M, Evans RM. 1996. Role of CBP/P300 in nuclear receptor signalling. Nature 383: 99–103. 10.1038/383099a0 [DOI] [PubMed] [Google Scholar]
- Chen JDC, Evans RM. 1995. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 377: 454–457. 10.1038/377454a0 [DOI] [PubMed] [Google Scholar]
- Chin WW, Shupnik MA, Ross DS, Habener JF, Ridgway EC. 1985. Regulation of the α and thyrotropin β-subunit messenger ribonucleic acids by thyroid hormones. Endocrinology 116: 873–878. 10.1210/endo-116-3-873 [DOI] [PubMed] [Google Scholar]
- Chin S, Apriletti J, Gick G. 1998. Characterization of a negative thyroid hormone response element in the rat sodium, potassium-adenosine triphosphatase α3 gene promoter. Endocrinology 139: 3423–3431. 10.1210/endo.139.8.6161 [DOI] [PubMed] [Google Scholar]
- Choi K-C, Oh S-Y, Kang H-B, Lee Y-H, Haam S, Kim H-I, Kim K, Ahn Y-H, Kim K-S, Yoon H-G. 2008. The functional relationship between co-repressor N-CoR and SMRT in mediating transcriptional repression by thyroid hormone receptor α. Biochem J 411: 19–26. 10.1042/BJ20071393 [DOI] [PubMed] [Google Scholar]
- Chung GE, Kim D, Kim W, Yim JY, Park MJ, Kim YJ, Yoon J-H, Lee H-S. 2012. Non-alcoholic fatty liver disease across the spectrum of hypothyroidism. J Hepatol 57: 150–156. 10.1016/j.jhep.2012.02.027 [DOI] [PubMed] [Google Scholar]
- Dalman FC, Koenig RJ, Perdew GH, Massa E, Pratt WB. 1990. In contrast to the glucocorticoid receptor, the thyroid hormone receptor is translated in the DNA binding state and is not associated with hsp90. J Biol Chem 265: 3615–3618. 10.1016/S0021-9258(19)39636-X [DOI] [PubMed] [Google Scholar]
- Desbois C, Aubert D, Legrand C, Pain B, Samarut J. 1991. A novel mechanism of action for v-ErbA: abrogation of the inactivation of transcription factor AP-1 by retinoic acid and thyroid hormone receptors. Cell 67: 731–740. 10.1016/0092-8674(91)90068-A [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupré SM, Guissouma H, Flamant F, Seugnet I, Scanlan TS, Baxter JD, Samarut J, Demeneix BA, Becker N. 2004. Both thyroid hormone receptor (TR)β1 and TRβ2 isoforms contribute to the regulation of hypothalamic thyrotropin-releasing hormone. Endocrinology 145: 2337–2345. 10.1210/en.2003-1209 [DOI] [PubMed] [Google Scholar]
- Feng P, Li Q-L, Satoh T, Wilber JF. 1994. Ligand T3 dependent and independent effects of thyroid hormone receptors upon human TRH gene transcription in neuroblastoma cells. Biochem Biophys Res Commun 200: 171–177. 10.1006/bbrc.1994.1430 [DOI] [PubMed] [Google Scholar]
- Forman BM, Casanova J, Raaka BM, Ghysdael J, Samuels HH. 1992. Half-site spacing and orientation determines whether thyroid hormone and retinoic acid receptors and related factors bind to DNA response elements as monomers, homodimers, or heterodimers. Mol Endocrinol 6: 429–442. [DOI] [PubMed] [Google Scholar]
- Forrest D, Sjöberg M, Vennström B. 1990. Contrasting developmental and tissue-specific expression of α and β thyroid hormone receptor genes. EMBO J 9: 1519–1528. 10.1002/j.1460-2075.1990.tb08270.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest D, Hallböök F, Persson H, Vennström B. 1991. Distinct functions for thyroid hormone receptors α and β in brain development indicated by differential expression of receptor genes. EMBO J 10: 269–275. 10.1002/j.1460-2075.1991.tb07947.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest D, Hanebuth E, Smeyne RJ, Everds N, Stewart CL, Wehner JM, Curran T. 1996. Recessive resistance to thyroid hormone in mice lacking thyroid hormone receptor β: evidence for tissue-specific modulation of receptor function. EMBO J 15: 3006–3015. 10.1002/j.1460-2075.1996.tb00664.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis NE, Taber TH, Bolf EL, Beaudet CM, Tomczak JA, White JH, Stein JL, Stein GS, Lian JB, Frietze S, et al. 2018. Thyroid hormone receptor β suppression of RUNX2 is mediated by brahma-related gene 1-dependent chromatin remodeling. Endocrinology 159: 2484-2494. 10.1210/en.2018-00128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Rosenfeld MG. 2000. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev 14: 121–141. [PubMed] [Google Scholar]
- Glass CK, Franco R, Weinberger C, Albert VR, Evans RM, Rosenfeld MG. 1987. A c-erb-A binding site in rat growth hormone gene mediates trans-activation by thyroid hormone. Nature 329: 738–741. 10.1038/329738a0 [DOI] [PubMed] [Google Scholar]
- Grøntved L, Waterfall JJ, Kim DW, Baek S, Sung M-H, Zhao L, Park JW, Nielsen R, Walker RL, Zhu YJ, et al. 2015. Transcriptional activation by the thyroid hormone receptor through ligand-dependent receptor recruitment and chromatin remodelling. Nat Commun 6: 139. 10.1038/ncomms8048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin MJ, Zhang X, Coonrod SA, Hager GL. 2014. Transient estrogen receptor binding and p300 redistribution support a squelching mechanism for estradiol-repressed genes. Mol Endocrinol 28: 1522–1533. 10.1210/me.2014-1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurr JA, Kourides IA. 1983. Regulation of thyrotropin biosynthesis. Discordant effect of thyroid hormone on α and β subunit mRNA levels. J Biol Chem 258: 10208–10211. 10.1016/S0021-9258(17)44441-3 [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodin RA, Lazar MA, Wintman BI, Darling DS, Koenig RJ, Larsen PR, Moore DD, Chin WW. 1989. Identification of a thyroid hormone receptor that is pituitary-specific. Science 244: 76–79. 10.1126/science.2539642 [DOI] [PubMed] [Google Scholar]
- Hörlein AJ, Näär AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Söderström M, Glass CK, et al. 1995. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 377: 397–404. 10.1038/377397a0 [DOI] [PubMed] [Google Scholar]
- Hoskins RG. 1949. The thyroid-pituitary apparatus as a servo (feedback) mechanism. J Clin Endocrinol Metab 9: 1429–1431. 10.1210/jcem-9-12-1429 [DOI] [Google Scholar]
- Iglesias T, Caubín J, Stunnenberg HG, Zaballos A, Bernal J, Muñoz A. 1996. Thyroid hormone-dependent transcriptional repression of neural cell adhesion molecule during brain maturation. EMBO J 15: 4307–4316. 10.1002/j.1460-2075.1996.tb00805.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka T, Lazar MA. 2003. The N-CoR/histone deacetylase 3 complex is required for repression by thyroid hormone receptor. MCB 23: 5122–5131. 10.1128/MCB.23.15.5122-5131.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyakumar M, Tanen MR, Bagchi MK. 1997. Analysis of the functional role of steroid receptor coactivator-1 in ligand-induced transactivation by thyroid hormone receptor. Mol Endocrinol 11: 755–767. 10.1210/mend.11.6.0003 [DOI] [PubMed] [Google Scholar]
- Jonat C, Rahmsdorf HJ, Park K-K, Cato AC, Gebel S, Ponta H, Herrlich P. 1990. Antitumor promotion and antiinflammation: down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell 62: 1189–1204. 10.1016/0092-8674(90)90395-U [DOI] [PubMed] [Google Scholar]
- Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. 2003. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med 9: 213–219. 10.1038/nm820 [DOI] [PubMed] [Google Scholar]
- Kaneshige M, Kaneshige K, Zhu X-g, Dace A, Garrett L, Carter TA, Kazlauskaite R, Pankratz DG, Wynshaw-Boris A, Refetoff S, et al. 2000. Mice with a targeted mutation in the thyroid hormone β receptor gene exhibit impaired growth and resistance to thyroid hormone. Proc Natl Acad Sci 97: 13209–13214. 10.1073/pnas.230285997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S-W, Ahn I-M, Larsen PR. 1996. In vivo genomic footprinting of thyroid hormone-responsive genes in pituitary tumor cell lines. Mol Cell Biol 16: 4465–4477. 10.1128/MCB.16.8.4465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S-W, Ho S-C, Hong S-J, Kim KM, So EC, Christoffolete M, Harney JW. 2005. A novel mechanism of thyroid hormone-dependent negative regulation by thyroid hormone receptor, nuclear receptor corepressor (NCoR), and GAGA-binding factor on the rat CD44 promoter. J Biol Chem 280: 14545–14555. 10.1074/jbc.M411517200 [DOI] [PubMed] [Google Scholar]
- Kim YH, Marhon SA, Zhang Y, Steger DJ, Won K-J, Lazar MA. 2018. Rev-erbα dynamically modulates chromatin looping to control circadian gene transcription. Science 359: 1274–1277. 10.1126/science.aao6891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, Umesono K, Mangelsdorf DJ, Evans RM. 1992. Retinoid X receptor interacts with nuclear receptors in retinoic acid, thyroid hormone and vitamin D3 signalling. Nature 355: 446–449. 10.1038/355446a0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalik MA, Columbano A, Perra A. 2018. Thyroid hormones, thyromimetics and their metabolites in the treatment of liver disease. Front Endocrinol (Lausanne) 9: 382. 10.3389/fendo.2018.00382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar MA. 2003. Thyroid hormone action: a binding contract. J. Clin. Invest 112: 497–499. 10.1172/JCI19479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar MA, Berrodin TJ, Harding HP. 1991. Differential DNA binding by monomeric, homodimeric, and potentially heteromeric forms of the thyroid hormone receptor. Mol Cell Biol 11: 5005–5015. 10.1128/MCB.11.10.5005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, O'Malley BW, Wong J. 2000. P300 requires its histone acetyltransferase activity and SRC-1 interaction domain to facilitate thyroid hormone receptor activation in chromatin. Mol Cell Biol 20: 2031–2042. 10.1128/MCB.20.6.2031-2042.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- Lompré A-M, Nadal-Ginard B, Mahdavi V. 1984. Expression of the cardiac ventricular α- and β-myosin heavy chain genes is developmentally and hormonally regulated. J Biol Chem 259: 6437–6446. 10.1016/S0021-9258(20)82162-0 [DOI] [PubMed] [Google Scholar]
- Mader S, Chen J-Y, Chen Z, White J, Chambon P, Gronemeyer H. 1993. The patterns of binding of RAR, RXR and TR homo- and heterodimers to direct repeats are dictated by the binding specificites of the DNA binding domains. EMBO J 12: 5029–5041. 10.1002/j.1460-2075.1993.tb06196.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Nascimbeni F, Lonardo A, Zoppini G, Bonora E, Mantzoros CS, Targher G. 2018. Association between primary hypothyroidism and nonalcoholic fatty liver disease: a systematic review and meta-analysis. Thyroid 28: 1270–1284. 10.1089/thy.2018.0257 [DOI] [PubMed] [Google Scholar]
- Marks MS, Hallenbeck PL, Nagata T, Segars JH, Appella E, Nikodem VM, Ozato K. 1992. H-2RIIBP (RXR β) heterodimerization provides a mechanism for combinatorial diversity in the regulation of retinoic acid and thyroid hormone responsive genes. EMBO J 11: 1419–1435. 10.1002/j.1460-2075.1992.tb05187.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK. 2012. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40: 4288–4297. 10.1093/nar/gks042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M-E, Gronemeyer H, Turcotte B, Bocquel M-T, Tasset D, Chambon P. 1989. Steroid hormone receptors compete for factors that mediate their enhancer function. Cell 57: 433–442. 10.1016/0092-8674(89)90918-5 [DOI] [PubMed] [Google Scholar]
- Meyer M-E, Quirin-Stricker C, Lerouge T, Bocquel M-T, Gronemeyer H. 1992. A limiting factor mediates the differential activation of promoters by the human progesterone receptor isoforms. J Biol Chem 267: 10882–10887. 10.1016/S0021-9258(19)50100-4 [DOI] [PubMed] [Google Scholar]
- Mullur R, Liu Y-Y, Brent GA. 2014. Thyroid hormone regulation of metabolism. Physiol Rev 94: 355–382. 10.1152/physrev.00030.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray MB, Zilz ND, McCreary NL, MacDonald MJ, Towle HC. 1988. Isolation and characterization of rat cDNA clones for two distinct thyroid hormone receptors. J Biol Chem 263: 12770–12777. 10.1016/S0021-9258(18)37820-7 [DOI] [PubMed] [Google Scholar]
- Nelson VL, Nguyen HCB, Garcìa-Cañaveras JC, Briggs ER, Ho WY, DiSpirito JR, Marinis JM, Hill DA, Lazar MA. 2018. PPARγ is a nexus controlling alternative activation of macrophages via glutamine metabolism. Genes Dev 32: 1035–1044. 10.1101/gad.312355.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Kuehn MS, Reynolds AP, Haugen E, Thurman RE, Johnson AK, Rynes E, Maurano MT, Vierstra J, Thomas S, et al. 2012. BEDOPS: high-performance genomic feature operations. Bioinformatics 28: 1919–1920. 10.1093/bioinformatics/bts277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygård M, Becker N, Demeneix B, Pettersson K, Bondesson M. 2006. Thyroid hormone-mediated negative transcriptional regulation of Necdin expression. J Mol Endocrinol 36: 517–530. 10.1677/jme.1.01874 [DOI] [PubMed] [Google Scholar]
- Oppenheimer JH, Schwartz HL, Koerner D, Surks MI. 1974. Limited binding capacity sites for L-triiodothyronine in rat liver nuclei. Nuclear-cytoplasmic interrelation, binding constants, and cross-reactivity with L-thyroxine. J Clin Invest 53: 768–777. 10.1172/JCI107615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Præstholm SM, Siersbæk MS, Nielsen R, Zhu X, Hollenberg A, Cheng S-y, Grøntved L. 2020. Multiple mechanisms regulate H3 acetylation of enhancers in response to thyroid hormone. PLoS Genet 16: e1008770. 10.1371/journal.pgen.1008770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Fan J, Zheng R, Wan C, Mei S, Wu Q, Sun H, Brown M, Zhang J, Meyer CA, et al. 2020. Lisa: inferring transcriptional regulators through integrative modeling of public chromatin accessibility and ChIP-seq data. Genome Biol 21: 32. 10.1186/s13059-020-1934-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadoss P, Abraham BJ, Tsai L, Zhou Y, Costa-e-Sousa RH, Ye F, Bilban M, Zhao K, Hollenberg AN. 2014. Novel mechanism of positive versus negative regulation by thyroid hormone receptor β1 (TRβ1) identified by genome-wide profiling of binding sites in mouse liver. J Biol Chem 289: 1313–1328. 10.1074/jbc.M113.521450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, Vilo J. 2019. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 47: W191–W198. 10.1093/nar/gkz369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro RC, Kushner PJ, Apriletti JW, West BL, Baxter JD. 1992. Thyroid hormone alters in vitro DNA binding of monomers and dimers of thyroid hormone receptors. Mol Endocrinol 6: 1142–1152. [DOI] [PubMed] [Google Scholar]
- Samuels HH, Tsai JS, Casanova J, Stanley F. 1974. Thyroid hormone action: in vitro characterization of solubilized nuclear receptors from rat liver and cultured GH1 cells. J Clin Invest 54: 853–865. 10.1172/JCI107825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sap J, Muñoz A, Damm K, Goldberg Y, Ghysdael J, Leutz A, Beug H, Vennström B. 1986. The c-erb-A protein is a high-affinity receptor for thyroid hormone. Nature 324: 635–640. 10.1038/324635a0 [DOI] [PubMed] [Google Scholar]
- Schmidt SF, Larsen BD, Loft A, Mandrup S. 2016. Cofactor squelching: artifact or fact? Bioessays 38: 618–626. 10.1002/bies.201600034 [DOI] [PubMed] [Google Scholar]
- Schüle R, Rangarajan P, Yang N, Kliewer S, Ransone LJ, Bolado J, Verma IM, Evans RM. 1991. Retinoic acid is a negative regulator of AP-1-responsive genes. Proc Natl Acad Sci 88: 6092–6096. 10.1073/pnas.88.14.6092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard HM, Harries JC, Hussain S, Bevan C, Heery DM. 2001. Analysis of the steroid receptor coactivator 1 (SRC1)-CREB binding protein interaction interface and its importance for the function of SRC1. Mol Cell Biol 21: 39–50. 10.1128/MCB.21.1.39-50.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibusawa N, Hollenberg AN, Wondisford FE. 2003. Thyroid hormone receptor DNA binding is required for both positive and negative gene regulation. J Biol Chem 278: 732–738. 10.1074/jbc.M207264200 [DOI] [PubMed] [Google Scholar]
- Shulemovich K, Dimaculangan DD, Katz D, Lazer MA. 1995. DNA bending by thyroid hormone receptor: influence of half-site spacing and RXR. Nucleic Acids Res 23: 811–818. 10.1093/nar/23.5.811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha RA, You S-H, Zhou J, Siddique MM, Bay B-H, Zhu X, Privalsky ML, Cheng S-Y, Stevens RD, Summers SA, et al. 2012. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J. Clin. Invest 122: 2428–2438. 10.1172/JCI60580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha RA, Bruinstroop E, Singh BK, Yen PM. 2019. Nonalcoholic fatty liver disease and hypercholesterolemia: roles of thyroid hormones, metabolites, and agonists. Thyroid 29: 1173–1191. 10.1089/thy.2018.0664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Step SE, Lim H-W, Marinis JM, Prokesch A, Steger DJ, You S-H, Won K-J, Lazar MA. 2014. Anti-diabetic rosiglitazone remodels the adipocyte transcriptome by redistributing transcription to PPAR-driven enhancers. Genes Dev 28: 1018–1028. 10.1101/gad.237628.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strait KA, Schwartz HL, Perez-Castillo A, Oppenheimer JH. 1990. Relationship of c-erbA mRNA content to tissue triiodothyronine nuclear binding capacity and function in developing and adult rats. J Biol Chem 265: 10514–10521. 10.1016/S0021-9258(18)86977-0 [DOI] [PubMed] [Google Scholar]
- Tata JR. 2013. The road to nuclear receptors of thyroid hormone. Biochim Biophys Acta 1830: 3860–3866. 10.1016/j.bbagen.2012.02.017 [DOI] [PubMed] [Google Scholar]
- Thompson J, Moore SE, Walsh FS. 1987. Thyroid hormones regulate expression of the neural cell adhesion molecule in adult skeletal muscle. FEBS Lett 219: 135–138. 10.1016/0014-5793(87)81205-X [DOI] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umesono K, Murakami KK, Thompson CC, Evans RM. 1991. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell 65: 1255–1266. 10.1016/0092-8674(91)90020-Y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella KR, Ramadoss P, Costa-e-Sousa RH, Astapova I, Ye FD, Holtz KA, Harris JC, Hollenberg NA. 2014. Thyroid hormone signaling in vivo requires a balance between coactivators and corepressors. Mol Cell Biol 34: 1564–1575. 10.1128/MCB.00129-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlström GM, Sjöberg M, Andersson M, Nordström K, Vennström B. 1992. Binding characteristics of the thyroid hormone receptor homo- and heterodimers to consensus AGGTCA repeat motifs. Mol Endocrinol 6: 1013–1022. [DOI] [PubMed] [Google Scholar]
- Weinberger C, Thompson CC, Ong ES, Lebo R, Gruol DJ, Evans RM. 1986. The c-erb-A gene encodes a thyroid hormone receptor. Nature 324: 641–646. 10.1038/324641a0 [DOI] [PubMed] [Google Scholar]
- Weiss RE, Murata Y, Cua K, Hayashi Y, Seo H, Refetoff S. 1998. Thyroid hormone action on liver, heart, and energy expenditure in thyroid hormone receptor β-deficient mice. Endocrinology 139: 4945–4952. 10.1210/endo.139.12.6412 [DOI] [PubMed] [Google Scholar]
- Wondisford FE, Farr EA, Radovick S, Steinfelder HJ, Moates JM, McClaskey JH, Weintraub BD. 1989. Thyroid hormone inhibition of human thyrotropin β-subunit gene expression is mediated by a cis-acting element located in the first exon. J Biol Chem 264: 14601–14604. 10.1016/S0021-9258(18)63736-6 [DOI] [PubMed] [Google Scholar]
- Yang-Yen H-F, Chambard J-C, Sun Y-L, Smeal T, Schmidt TJ, Drouin J, Karin M. 1990. Transcriptional interference between c-Jun and the glucocorticoid receptor: mutual inhibition of DNA binding due to direct protein–protein interaction. Cell 62: 1205–1215. 10.1016/0092-8674(90)90396-V [DOI] [PubMed] [Google Scholar]
- Yen PM, Feng X, Flamant F, Chen Y, Walker RL, Weiss RE, Chassande O, Samarut J, Refetoff S, Meltzer PS. 2003. Effects of ligand and thyroid hormone receptor isoforms on hepatic gene expression profiles of thyroid hormone receptor knockout mice. EMBO Rep 4: 581–587. 10.1038/sj.embor.embor862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu VC, Delsert C, Andersen B, Holloway JM, Devary OV, Näär AM, Kim SY, Boutin J-M, Glass CK, Rosenfeld MG. 1991. RXR β: a coregulator that enhances binding of retinoic acid, thyroid hormone, and vitamin D receptors to their cognate response elements. Cell 67: 1251–1266. 10.1016/0092-8674(91)90301-E [DOI] [PubMed] [Google Scholar]
- Zhang X-K, Wills KN, Husmann M, Hermann T, Pfahl M. 1991. Novel pathway for thyroid hormone receptor action through interaction with jun and fos oncogene activities. Mol Cell Biol 11: 6016–6025. 10.1128/MCB.11.12.6016 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.