

Abstract
In this study, we aim to investigate the regulation of specific long non-coding RNAs (lncRNAs) on the progression of ischemia/reperfusion (I/R) injury. We identified and characterized the exosomes derived from mouse primary aortic endothelial cells. Subsequently, we found that these exosomes expressed typical exosomal markers and high levels of LINC00174, which significantly ameliorated I/R-induced myocardial damage and suppressed the apoptosis, vacuolation, and autophagy of myocardial cells. Mechanistic approaches revealed that LINC00174 directly interacted with SRSF1 to suppress the expression of p53, thus restraining the transcription of myocardin and repressing the activation of the Akt/AMPK pathway that was crucial for autophagy initiation in I/R-induced myocardial damage. Moreover, this molecular mechanism was verified by in vivo study. In summary, exosomal LINC00174 generated from vascular endothelial cells repressed p53-mediated autophagy and apoptosis to mitigate I/R-induced myocardial damage, suggesting that targeting LINC00174 may be a novel strategy to treat I/R-induced myocardial infarction.
Keywords: LINC00174, vascular endothelial cells, ischemia/reperfusion injury, p53, autophagy, apoptosis
Graphical Abstract
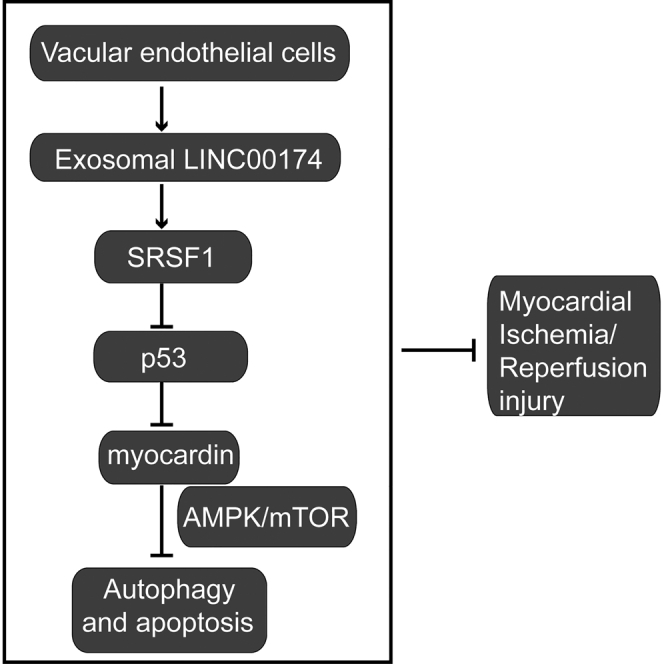
In the present study, Su and colleagues show that exosomal LINC00174 derived from vascular endothelial cells attenuates myocardial ischemia-reperfusion (I/R) injury by suppressing p53-mediated autophagy and apoptosis. This discovery reveals a new regulation mechanism for I/R-induced injury and provides a potential therapeutic strategy to treat I/R-induced myocardial infarction.
Introduction
Exosomes are nano-sized extracellular vesicles derived from endosomes in most eukaryotic cells, which were first described in 1987 by Rose Johnstone.1 After decades of investigation, the composition of exosomes is clear, which includes proteins, lipids and nucleic acids.2 Exosomes have been implicated in various physiological processes, such as cell-cell communication,3 the mediator of immune responses,4 and the secretion of active molecules.5 Moreover, exosomes play crucial roles in the pathogenesis of multiple diseases, such as cancer,6 inflammatory diseases,7 neurodegenerative diseases,8 and cardiovascular diseases.9 Emerging evidence demonstrates that exosomes could harness the canonical Wnt/β-catenin signaling pathway,5 the epidermal growth factor receptor (EGFR) signaling pathway,10 the Notch signaling pathway,11 and the transforming growth factor β (TGF-β) signaling pathway12 to influence the progression of distinct diseases. However, the explicit mechanism for the regulation of exosomes in myocardial ischemia-reperfusion (I/R) injury is little known.
Myocardial I/R-elicited tissue damage is a major cause of myocardial infarction (MI), which further contributes to the mortality and disability worldwide.13 The death of cardiomyocytes and the necrosis of affected tissues due to oxygen deprivation are the most remarkable features in the myocardium during MI progression. Thus, protecting cardiomyocytes from I/R-induced cell death is the most critical strategy to cure MI in the clinic.14 A related study reveals that exosomes secreted by vascular endothelial cells can regulate the contractile phenotype of adjacent smooth muscle cells;15 moreover, cardiac progenitor-derived exosomes have been recognized as the one potent protector of cardiomyocytes from acute I/R-induced injury.16 However, few reports have investigated the function and underlying mechanism of vascular endothelial cell-derived exosomes in the modulation of I/R-induced myocardial damage.
Long non-coding RNA (lncRNA) is a family of non-coding RNAs with a length of >200 nt. Emerging studies have elucidated that lncRNAs participate in the regulation of numerous biological activities, such as stem cell pluripotency, neurogenesis, immune response, and oncogenesis.17 In particular, recent studies have manifested that lncRNA wrapped in exosomes was secreted and transported to distal target cells to exert their function. For example, Gao and colleagues18 reported that exosomal lncRNA 91H was associated with the poor prognosis of colorectal cancer, and Chen et al.19 found that exosomal lncRNA-GAS5 enhanced the apoptosis of both vascular endothelial cells and macrophages in atherosclerosis. Although previous studies mainly found that exosomal microRNAs (miRNAs) were involved in the regulation of atherosclerosis and myocardial I/R injury,20 we speculate that exosomal lncRNA may play crucial roles in the modulation of myocardial injury. Long intergenic non-protein coding RNA 174 (LINC00174) is a newly discovered lncRNA, which is upregulated in colorectal cancer (CRC),21 glioma,22 and hepatocellular carcinoma (HCC).23 Previous studies reveal that LINC00174 functions as an oncogene to facilitate the progression of CRC, glioma, and HCC via distinct mechanisms,21, 22, 23 whereas our lab has found that vascular endothelial cells could secret exosomes that contain large quantities of LINC00174. However, the function of exosomal LINC00174 in myocardial I/R injury is still elusive.
Autophagy is a intracellular degradation process that destroys misfolded or dysfunctional components, which is crucial for cells to supply energy in response to nutrient deprivation,24 whereas apoptosis is a process of programmed cell death predominantly mediated by caspases.25 Accumulative evidence has demonstrated that autophagy usually blocks the initiation of apoptosis, and apoptosis-related caspase activation inhibits the autophagic process; moreover, the crosstalk between autophagy and apoptosis plays a fundamental role in numerous pathophysiological processes.26 For instance, autophagy has been recognized as the pro-survival mechanism for cardiomyocytes in I/R-induced damage;27, 28, 29, 30 furthermore, targeting autophagy using various agents has become an appealing strategy for the treatment of cardiorenal metabolic disorders.31,32 However, autophagy activation also inhibits apoptosis and ameliorates tissue damage in chronic ischemia.33 Beclin-1 is a potent suppressor of autophagy; partial knockdown of this gene reinstates autophagosome formation and protects cardiomyocytes from I/R-induced cell death.34 By contrast, apoptosis is a major cause of the death of cardiomyocytes during I/R injury.35 Some studies have found that ubiquitin-specific peptidase 49 (USP49) and 2,3,5,4-tetrahydroxystilbene-2-O-β-d-glucoside (TSG) could eliminate the I/R-induced apoptosis of H9c2 cardiomyocytes,36 while miR-181c-5p exacerbated the hypoxia/reoxygenation (H/R)-induced apoptosis of H9c2 cells by targeting PTPN4.37 Nevertheless, no significant progress has been achieved to eliminate I/R injury and MI after decades of investigation. Thus, the regulatory mechanism for autophagy and apoptosis in I/R-induced myocardial damage should be investigated to achieve a breakthrough in the treatment of I/R injury and MI.
In the present study, we investigated the function of exosomal LINC00174, which was secreted by vascular endothelial cells, on myocardial I/R injury. The results showed that LINC00174 expression in exosomes was reduced in I/R-induced mice, whereas the inoculation of exosomal LINC00174 could significantly ameliorate I/R-induced myocardial damage and suppress the apoptosis, vacuolation, and autophagy of myocardial cells. Mechanistically, LINC00174 could directly interact with serine and arginine rich splicing factor 1 (SRSF1), which then suppressed p53 expression, restrained the transcription of myocardin, and attenuated the activation of the Akt/AMP kinase (AMPK) pathway. In vivo experiments demonstrated that LINC00174 could downregulate the expression of myocardin, while LINC00174 knockdown evidently aggravated apoptosis and enhanced the autophagy activation of myocardial cells through augmented Akt/AMPK signaling. Our study revealed that exosomal LINC00174 secreted by vascular endothelial cells could inhibit p53-mediated autophagy and apoptosis in I/R-induced myocardial damage, which indicated that LINC00174 may be a novel target to mitigate I/R-induced myocardial infarction in the future.
Results
LINC00174 is expressed in exosomes derived from vascular endothelial cells
To obtain the exosomes derived from aortic endothelial cells, we isolated aortic endothelial cells from the aorta of C57BL/6 mice. After culturing for 12 days, these aortic endothelial cells formed tight connections and displayed the typical morphology of endothelial cells (Figure 1A). To further verify their identity, we then performed immunofluorescence staining for von Willebrand factor (vWF), which is a widely used marker protein of vascular endothelial cells, and the results demonstrated that all of the cells showed evident vWF expression in cytosol (Figure 1B). Next, we collected the culture medium and purified the secreted exosomes for subsequent analysis. The transmission electron microscopy (TEM) scanning image clearly showed that there were exosome-like vesicles in the supernatant, the diameter of which lay in the range of 60–90 nm (Figure 1C). Moreover, we measured the expression of exosomal markers such as CD9, CD63, and CD81 on the surface of isolated exosomes. The results demonstrated that all of these marker proteins were expressed on purified exosomes (Figure 1D). Western blot data further confirmed the high expression level of CD9 and CD63 in exosomes, whereas the marker proteins of mitochondria, the Golgi apparatus, and lysosomes were hardly detected in these exosomes (Figure 1E). Interestingly, subsequent qRT-PCR results elucidated that LINC00174 expression was significantly upregulated in isolated exosomes when compared with that in cell lysis (Figure 1F). These data demonstrated that exosomes secreted by vascular endothelial cells contained LINC00174.
Figure 1.

The isolation and characterization of vascular endothelial cell-derived exosomes
Mouse primary aortic endothelial cells were isolated from aorta of pathogen-free C57BL/6 mouse.
(A) The morphology of vascular endothelial cells was imaged after culturing for 3 days (left) and 12 days (right), respectively. Scale bar, 100 μm.
(B) The immunofluorescence staining of vWF in vascular endothelial cells. Scale bar, 100 μμm.
(C) The exosomes derived from vascular endothelial cells were examined by transmission electron microscopy (TEM). Scale bar, 100 nm.
(D) The expression of surface markers (CD9, CD63, and CD81) on exosomes was assessed by flow cytometry.
(E) The expression of exosomal markers (CD9, CD63, and TSG101) and cellular organelle markers (cytochrome c, calnexin, and syntaxin 6) in vascular endothelial cell lysates and exosomes, respectively, was measured by western blotting.
(F) The relative expression of lncRNA LINC00174 in vascular endothelial cell lysates and exosomes was measured by PCR. The mRNAs were normalized to GAPDH mRNA; experiments were performed in triplicate.
(A–F) The data represented 1 of 3 independent experiments. (F) Data were represented as means ± SDs. p values were determined by unpaired 2-tailed Student’s t test. ∗p < 0.05 and ∗∗p < 0.01.
I/R-induced myocardial injury is relieved by exosomal LINC00174 by suppressing autophagy and apoptosis of myocardial cells
To explore the function of exosomal LINC00174, we then established a I/R-induced myocardial injury mouse model. As shown in Figure 2A, TEM scanning showed that there was much more myocardium vacuolation in the cardiomyocytes of I/R-treated mice; however, qRT-PCR analysis revealed that LINC00174 expression was downregulated in cardiomyocytes after I/R treatment. To verify the essential role of LINC00174, I/R-treated mice were intravenously administrated with LINC00174-containing exosomes that were derived from vascular endothelial cells. As expected, 2,3,5-triphenyltetrazoliumchloride (TTC) staining experiments demonstrated that LINC00174 injection significantly mitigated the myocardial infarction in I/R-treated mice (Figure 2B). To assess the injury status, we further measured the enzyme activity, including LDH (lactic dehydrogenase), CK (creatine kinase) and CK-MB (CK MB isoenzyme) in mice serum with commercial kits. The data demonstrated that I/R treatment significantly increased the activity of LDH and CK, as well as the CK-MB activity, which indicated that the death of myocardial cells was due to I/R-induced injury. However, the administration of LINC00174-containing exosomes derived from vascular endothelial cells potently repressed the death of myocardial cells, which was seen by the reduced activity of LDH, CK, and CK-MB (Figure 2C). Then, we performed color Doppler echocardiography in mice to monitor their cardiac function. The data showed abnormal echocardiographic parameters, including left ventricular end-diastolic dimension (LVEDD), left ventricular end systolic diameter (LVESD), left ventricular end-diastolic pressure (LVEDP), left ventricular fraction shortening (LVFS), left ventricular systolic pressure (LVSP), and left ventricular ejection fraction (LVEF) in the I/R group. As expected, the treatment of LINC00174-containing exosomes derived from vascular endothelial cells significantly ameliorated the I/R injury and improved the cardiac function, which was supported by the decreased LVEDD, LVESD, and LVEDP and the increased LVFS, LVSP, and LVEF (Figure 2D). Next, the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay showed that LINC00174 administration potently repressed the apoptosis of cardiomyocytes when compared with that in the control group mice (Figure 2E). We observed evident myocardium vacuolation in I/R-induced myocardial injury mice; however, exogenous LINC00174 treatment significantly ameliorated the myocardium vacuolation (Figure 2F). Using the TEM data, we also quantified the cell area occupied by autophagic vesicles in control mice or I/R-induced myocardial injury mice in the absence or presence of LINC00174. The result showed that I/R treatment induced potent upregulation of autophagic vesicle formation, which was then repressed by LINC00174 administration (Figure 2G). To confirm this finding, we further assessed the autophagy in cardiomyocytes using immunofluorescence staining of LC3 and the results demonstrated that I/R-induced myocardial injury obviously promoted autophagy activation and autophagosome formation, which was inhibited by exogenous LINC00174 injection (Figure 2H). In addition, we implemented western blotting to examine the expression of autophagy-related proteins such as Beclin1, p62, LC3-I, and LC3-II. The results illustrated that Beclin1 and the LC3-II:LC3-I ratio were both upregulated while p62 was downregulated upon I/R treatment; nevertheless, LINC00174 inoculation greatly reversed this phenotype (Figure 2I). Our data revealed that exosomal lncRNA LINC00174 derived from vascular endothelial cells may ameliorate I/R-induced myocardial injury by inhibiting autophagy and apoptosis.
Figure 2.

Exosomal LINC00174 can mitigate the I/R-induced myocardial injury
I/R-induced myocardial injury model was established with C57BL/6 mouse. They were then intravenously injected with PBS or isolated LINC00174-containing exosomes, respectively; n = 5.
(A) Left, the autophagosomes in myocardial cells were examined by TEM imaging. Scale bar, 1 μm. Right, the relative expression of LINC00174 in vascular endothelial cells was measured by qRT-PCR. The mRNAs were normalized to GAPDH mRNA; experiments were performed in triplicate.
(B) The size of myocardial infarction was measured by TTC staining; the statistic of the infarct area to left ventricle (LV) was shown in the right-hand chart. Scale bar, 1 cm.
(C) The activity of LDH, CK, and CK-MB in mice serum was measured by commercial kits.
(D) Key parameters were tested by color Doppler echocardiography to reflect the injury status of myocardium in mice.
(E) The apoptosis of myocardial cells was determined by TUNEL assay, the representative images were displayed at left, and the statistic of cell death was shown at right. Scale bar, 50 μm.
(F) The myocardium vacuolation was imaged by TEM. Scale bar, 1 μm.
(G) The cell area occupied by autophagic vesicles was quantified based on EM images with ImageJ software.
(H) The formation of autophagosomes was assessed by immunofluorescence staining for LC3; the cell nucleus was stained with DAPI. Scale bar, 10 μm.
(I) The expression of autophagy-related proteins in mice-derived myocardial cells was measured by western blotting; β-actin was used as the internal normalization control.
(A–I) The data represented 1 of 3 independent experiments. (A–E and G) Data were represented as means ± SDs. p values were determined by unpaired 2-tailed Student’s t test (A) or 1-way analysis of variance (ANOVA) followed by Tukey post hoc test (B–E and G). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. IFN, infarction area.
p53 downregulation is associated with the mitigation of I/R-induced myocardial injury by LINC00174
p53 has been reported as a cause of myocardial apoptosis during I/R injury.38,39 To ascertain whether p53 signaling was involved in the regulation of I/R injury by LINC00174, we established an H/R cell model with primary myocardial cells in vitro. The qRT-PCR results demonstrated that LINC00174 expression was downregulated after H/R treatment, although co-culturing with vascular endothelial cell-derived exosomes significantly augmented the expression of LINC00174 (Figure 3A). Next, we checked the expression of p53 in primary myocardial cells after H/R treatment. The western blotting result confirmed that p53 accumulation was significantly elevated, whereas the inoculation of LINC00174-containing exosomes potently repressed p53 expression (Figure 3B). To further verify the association between p53 and LINC00174 during I/R-induced injury, we knocked down the expression of LINC00174 by small hairpin RNA (shRNA) in primary myocardial cells. The qRT-PCR data showed that LINC00174 expression in myocardial cell-derived exosomes was reduced >50% (Figure 3C). After H/R treatment, p53 expression in myocardial cells was repressed by control exosomes, while LINC00174-silenced exosomes failed to downregulate the expression of p53 (Figure 3D). Moreover, the TUNEL assay showed that H/R treatment dramatically elevated the apoptosis of vascular endothelial cells, whereas this phenomenon was suppressed when excessive LINC00174-containing exosomes were supplemented in the co-culturing system. However, LINC00174 knockdown in the co-cultured exosomes aggravated the apoptosis. Interestingly, additional p53 knockdown counteracted the effects of LINC00174 silencing, which potently repressed the cell apoptosis; by contrast, simultaneous p53 overexpression led to much more cell death when LINC00174 expression was silenced (Figure 3E).
Figure 3.

LINC00174 modulates the I/R-induced myocardial injury by downregulating p53
(A and B) Mouse primary myocardial cells were pretreated as indicated before H/R treatment.
(A) The relative expression of LINC00174 in mouse primary myocardial cells after hypoxia treatment for 4 h was measured by qRT-PCR; n = 3.
(B) The expression of p53 in mouse primary myocardial cells after H/R treatment was measured by western blotting. β-Actin was used as the loading control.
(C) Negative control shRNA (shNC) or LINC00174 shRNA (shLINC00174) was transfected into mouse primary aortic endothelial cells; 48 h later, the exosomes in supernatant were harvested to determine the relative expression level of LINC00174 by qRT-PCR; n = 3.
(D) Mouse primary myocardial cells were pretreated as indicated before H/R treatment. The expression of p53 in mouse primary myocardial cells was measured by western blotting after H/R treatment for 4 h. β-Actin was used as the loading control.
(E–G) Mouse primary myocardial cells were pretreated as indicated before H/R treatment.
(E) The cell apoptosis after H/R treatment was determined by TUNEL assay; cell nuclei were stained by DAPI. Scale bar, 200 μm.
(F) The formation of autophagosomes was assessed by immunofluorescence staining; cell nucleus was stained with DAPI. Scale bar, 10 μm.
(G) The expression of autophagy-related proteins was measured by western blotting. β-Actin was used as the loading control.
(A–G) The data represented 1 of 3 independent experiments. (A and C) Data were represented as means ± SDs. p values were determined by 1-way ANOVA, followed by Tukey post hoc test. ∗p < 0.05 and ∗∗p < 0.01.
Next, we assessed the autophagy upon H/R treatment with the immunofluorescence staining of LC3 and the results illustrated that H/R treatment induced autophagy activation and autophagosome formation, which was inhibited by exogenous exosomes derived from vascular endothelial cells. However, LINC00174-silenced exosomes failed to suppress autophagosome formation. p53 knockdown in the myocardial cells potently inhibited the autophagosome formation in the presence of LINC00174-silenced exosomes, while p53 overexpression in myocardial cells further enhanced the autophagosome formation (Figure 3F). Consistently, the expression of Beclin1, LC3-I, and LC3-II in myocardial cells was enhanced after H/R treatment, repressed after co-culturing with LINC00174-containing exosomes, and elevated by co-culturing with LINC00174-knocking down exosomes. Although exogenous LINC00174-knocking down exosomes lost the protective function on H/R-induced myocardial injury, p53 knockdown in myocardial cells significantly suppressed the autophagy activation, while p53 overexpression in myocardial cells led to augmented autophagy activation. As a marker protein of autophagy activation, p62 showed a reversed expression pattern from that of Beclin1 and the LC3-II:LC3-I ratio (Figure 3G). Overall, these experiments demonstrated that p53 downregulation was involved in the mitigation of H/R-induced myocardial injury by LINC00174.
LINC00174 interacts with SRSF1
Up to now, no reports have studied the association between LINC00174 and p53 in the regulation of myocardial I/R injury. To address this question, we implemented an RNA pull-down assay with a biotinylated LINC00174 probe and mouse myocardial cell lysates. The proteins interacted with LINC00174 were then pulled down by streptavidin beads, which were then subjected to SDS-PAGE electrophoresis and mass spectrometry (MS) analysis (Figure 4A). MS data suggested that SRSF1 was a potential binding candidate for LINC00174. SRSF1 is a member of the SRSF family proteins that either activate or repress gene splicing, and it can also regulate the translation process in cell cytosol.40 To verify this interaction, we performed western blotting to check the existence of SRSF1 in the pull-down samples by LINC00174. The results showed that LINC00174 could interact with SRSF1, but not p53 (Figure 4B). To further confirm this finding, we performed an RNA immunoprecipitation (RIP) assay with SRSF1 antibody and p53 antibody, respectively. The data demonstrated that LINC00174 was precipitated and enriched by SRSF1 antibody when compared with that by control immunoglobulin G (IgG); however, LINC00174 was not recruited by the p53 antibody (Figure 4C). To map the binding site of LINC00174 on SRSF1, we designed and purified full length and several truncations of SRSF1 proteins (Figure 4D). The subsequent RIP experiments demonstrated that all 3 domains of SRSF1 (RRM1, RRM2, C-terminal domain) participated in the interaction between LINC00174 and SRSF1 (Figure 4E). Overall, these data elucidated that LINC00174 could bind to SRSF1, which may mediate the association between LINC00174 and p53 signaling.
Figure 4.

LINC00174 interacts with SRSF1
(A) Schematic illustration of RNA pulldown assay.
(B) Mouse primary myocardial cells were lysed and incubated with biotinylated LINC00174 sense or antisense strand; pulldown samples were then analyzed by western blotting.
(C) Mouse primary myocardial cells were lysed and incubated with indicated antibodies in RIP assay. Immunoprecipitated (IP)-enriched RNA was then analyzed by qRT-PCR; n = 5.
(D) Schematic illustration of SRSF1 full-length protein and truncations; their expression in E. coli was shown in the SDS-PAGE gel below.
(E) The binding of LINC00174 to SRSF1 full-length protein and truncated proteins was assessed by RIP.
(B–E) The data represented 1 of 3 independent experiments. (C and E) Data were represented as means ± SDs. p values were determined by 1-way ANOVA), followed by Tukey post hoc test. ∗p < 0.05 and ∗∗p < 0.01.
LINC00174 is implicated in the mitigation of myocardial I/R injury by regulating p53 signaling through SRSF1
Previous study has proved that SRSF1 can stabilize p53 via RPL5 and that SRSF1 is necessary for ribosomal stress-induced p53 activation in cancer cells.41 To assess the function of SRSF1 in the regulation of myocardial I/R injury, we conducted various in vitro assays with the H/R-induced cell injury model. The qRT-PCR results demonstrated that SRSF1 expression was downregulated after H/R treatment; however, co-culturing with vascular endothelial cell-derived exosomes significantly augmented the expression of SRSF1 (Figure 5A). Next, we assessed the expression of SRSF1 in primary myocardial cells after H/R treatment by western blotting. Consistently, SRSF1 expression was significantly reduced upon H/R treatment; however, co-culturing with LINC00174-containing exosomes potently elevated SRSF1 expression (Figure 5B). Strikingly, we found that SRSF1 overexpression in myocardial cells led to the downregulation of p53, while SRSF1 knockdown resulted in the elevation of p53 expression (Figure 5C). The subsequent TUNEL assay showed that SRSF1 overexpression repressed the apoptosis of mouse myocardial cells after H/R treatment in the presence of LINC00174-lacking exosomes, whereas SRSF1 knockdown conduced to enhanced apoptosis of myocardial cells under similar circumstances (Figure 5D). Moreover, the immunofluorescence experiments demonstrated that SRSF1 overexpression in myocardial cells evidently attenuated the autophagy activation and autophagosome formation induced by H/R treatment. By contrast, SRSF1 knockdown potently increased the autophagosome formation upon H/R treatment, even in the presence of LINC00174-lacking exosomes (Figure 5E). In accordance with the above functional data, the biochemical evidence also manifested that the expression of Beclin1 and the LC3-II:LC3-I ratio in myocardial cells was repressed after H/R treatment in the presence of excessive SRSF1. On the contrary, both of them were upregulated upon H/R treatment, when SRSF1 was silenced in myocardial cells. Consistently, p62 showed the reversed expression pattern. We note that the expression of p53 was negatively correlated with the expression of SRSF1 in the western blotting experiments (Figure 5F). These data illustrate that LINC00174 was implicated in the mitigation of myocardial I/R-induced injury by the negative regulation of p53 signaling by SRSF1.
Figure 5.

LINC00174 regulates p53 signaling through SRSF1
(A and B) Mouse primary myocardial cells were pretreated as indicated before H/R treatment. The relative expression of SRSF1 after H/R treatment for 4 h was measured by qRT-PCR; n = 3.
(B) The expression of SRSF1 after H/R treatment was measured by western blotting. β-Actin was used as the loading control.
(C) Mouse primary myocardial cells were transfected with indicated plasmids or siRNAs; 48 h later, the expression of p53 was measured by western blotting. β-Actin was used as the loading control.
(D–F) Mouse primary myocardial cells were pretreated as indicated. Then the cells were subjected to H/R treatment for 4 h.
(D) The apoptosis of myocardial cells was determined by TUNEL assay; cell nuclei were stained by DAPI; merged images are shown at the bottom. Scale bar, 200 μm.
(E) The level of LC3 in myocardial cells was examined by immunohistochemical staining. Scale bar, 10 μm.
(F) The expression of autophagy-related proteins, SRSF1, and p53 in myocardial cells was measured by western blotting; β-actin was used as the loading control.
(A–F) The data represented 1 of 3 independent experiments. (A) Data were represented as means ± SDs. p values were determined by 1-way ANOVA, followed by Tukey post hoc test; ∗∗p < 0.01.
P53 knockdown attenuates myocardial I/R injury via suppressing cell apoptosis and autophagy
To investigate the function and mechanism of p53 in I/R-induced myocardial injury, we knocked down p53 in vivo and established the I/R-induced myocardial injury mice model. The TUNEL assay showed that I/R treatment induced much higher apoptosis of myocardial cells than that in control mice; however, p53 knockdown significantly reduced the apoptosis of myocardial cells (Figure 6A). The immunohistochemical staining of LC3 in myocardium demonstrated that I/R treatment led to the activation of autophagy; however, p53 knockdown obviously restrained autophagy activation (Figure 6B). Consistently, the LC3-II:LC3-I ratio was significantly elevated upon I/R treatment; nevertheless, it declined when p53 expression was knocked down. Since previous studies have reported that both Akt42 and AMPK43 signaling are implicated in autophagy induction, we thus assessed the phosphorylation of Akt and AMPK in mouse myocardial cells after I/R treatment. The data showed that the phosphorylation of both Akt and AMPK was enhanced upon I/R-induced myocardial injury, which indicated that the autophagy process was activated. However, p53 knockdown attenuated their phosphorylation and repressed autophagy induction (Figure 6C). In summary, our data here suggest that the reduced cell apoptosis and attenuated Akt/AMPK signaling-mediated autophagy were the underlying mechanism for the mitigation of myocardial I/R injury by p53 knockdown.
Figure 6.

p53-knocking down mitigates myocardial I/R injury via suppressing cell apoptosis and autophagy
Mice were intravenously injected with adenovirus encoding control siRNA (siNC) or siRNA targeting p53 (sip53), and then subjected to I/R treatment; n = 5.
(A) The apoptosis of myocardial cells was determined by TUNEL assay; the representative images were displayed at left and the statistic of cell death was shown in the right-hand chart. Scale bar, 50 μm.
(B) The expression level of LC3 in myocardial cells was examined by immunohistochemical staining. Scale bar, 50 μm.
(C) The expression of autophagy-related proteins and the activation of Akt and AMPK pathways in myocardial cells was measured by western blotting; β-actin was used as the loading control.
(A–C) The data represented 1 of 3 independent experiments. (B) Data were represented as means ± SDs. p values were determined by 1-way ANOVA, followed by Tukey post hoc test (A and B)., ∗∗p < 0.01 and ∗∗∗p < 0.001.
The regulation of myocardin transcription by p53
Previous studies have found that myocardin can mediate autophagy and cell death in cardiomyocytes,44 which was essential for the survival and homeostasis of cardiomyocytes,45 and p53 can target myocardin to promote the differentiation of smooth muscle cells.46 To verify the implication of myocardin in the regulation of I/R-induced injury, we assessed the expression of myocardin in mouse cardiomyocytes that had been transfected with small interfering p53 (sip53) or control siRNA. The western blotting result demonstrated that myocardin was also upregulated after I/R treatment; however, p53 silencing led to the attenuation of myocardin (Figure 7A). Moreover, H/R treatment with mouse myocardial cells in vitro also led to the similar expression pattern of myocardin upon p53 knockdown (Figure 7B). Mechanistically, p53 was found to bind to the promoter region of myocardin in response to H/R treatment in the chromatin immunoprecipitation (ChIP) assay, and the accumulation of p53 on the promoter region of myocardin was gradually augmented with time after H/R treatment (Figure 7C). In addition, the dual luciferase assay further confirmed that p53 interacted with the promoter region of myocardin and facilitated its transcription upon H/R treatment, for p53 knockdown severely impaired the luciferase activity (Figure 7D). In summary, these experiments elucidated that p53 downregulation was crucial for mitigating I/R-induced myocardial injury, which was mediated by inhibiting the expression of myocardin.
Figure 7.

P53 regulates the transcription of myocardin
(A) Mice were intravenously injected with adenovirus encoding siNC or sip53 and then subjected to I/R treatment. Sham group mice were not treated by I/R; n = 5. The expression of myocardin in myocardial cells was measured by western blotting; β-actin was used as the loading control.
(B) Mouse primary myocardial cells were treated as indicated. Then, they were subjected to H/R treatment for 4 h. The expression of myocardin was measured by western blotting; β-actin was used as the loading control.
(C) Mouse primary myocardial cells were un-transfected or transfected with the indicated plasmids, which were then treated with H/R for the indicated time. Cells were subjected to ChIP assay with p53 antibody, and the IP DNA was analyzed by PCR.
(D) 293T cells were transfected with the indicated plasmids or siRNAs. Then, the cells were subjected to H/R treatment. The luciferase activity was measured with the dual luciferase assay kit.
(A–D) The data represented 1 of 3 independent experiments. (D) Data were represented as means ± SDs. p values were determined by 1-way ANOVA, followed by Tukey post hoc test (A–C). ∗∗p < 0.01 and ∗∗∗p < 0.001.
p53 and myocardin-mediated myocardial I/R injury was modulated by LINC00174 in vivo
To further investigate the association of LINC00174, p53, and myocardin in the modulation of I/R injury in vivo, we injected LINC00174-containing exosomes into I/R-treated mice. The subsequent western blotting experiment using freshly isolated cardiomyocytes demonstrated that I/R treatment significantly increased the expression of myocardin, whereas LINC00174 inoculation robustly suppressed its expression (Figure 8A). Next, the recipient mice were administrated with LINC00174-knocking down exosomes, or with p53-knocking down adenovirus plus LINC00174-knocking down exosomes ahead of I/R treatment. Then, the serum was obtained for LHD, CK, and CK-MB measurement. In Figure 8B, we can see that LINC00174 knockdown in exosomes further enhanced the activity of LDH, CK, and CK-MB; however, simultaneous p53 knockdown partially reduced the increased activity of LDH, CK, and CK-MB. Moreover, the cardiac function determined by color Doppler echocardiography showed that LVEDD, LVESD, and LVEDP were even larger in mice administrated with LINC00174-lacking exosomes than those in control mice, accompanied by even lower LVFS, LVSP, and LVEF. However, administration with LINC00174-lacking exosomes plus p53 siRNAs potently reduced LVEDD, LVESD, and LVEDP and increased LVFS, LVSP, and LVEF (Figure 8C), which indicated the improved cardiac function when inhibiting p53 signaling. Next, the primary cardiomyocytes were isolated from mice and subjected to TUNEL assay analysis. Consistently, cardiomyocytes from mice receiving LINC00174-knocking down exosomes showed enhanced cell apoptosis when compared with those receiving LINC00174-sufficient exosomes. It was noted that additional p53 knockdown evidently reduced the apoptosis (Figure 8D). We performed immunohistochemical staining to assess the autophagy induction. As expected, it exhibited similar responses as cell apoptosis upon I/R treatment, augmented by LINC00174-downregulation while impaired by p53 knockdown (Figure 8E). Finally, we checked the activation status of AMPK and Akt in cardiomyocytes by western blotting. The data clearly demonstrated that LINC00174 silencing in vascular cell-derived cells significantly enhanced the activation of AMPK and Akt signaling in cardiomyocytes. However, simultaneous p53 knockdown impaired the signaling strength of AMPK and Akt pathways, as well as the expression of myocardin (Figure 8F). Our ex vivo data suggested that the LINC00174-p53-myocardin axis participated in the amelioration of myocardial I/R injury.
Figure 8.

p53 and myocardin-mediated myocardial I/R injury was modulated by LINC00174 in vivo
(A) I/R-induced myocardial injury mice were treated as indicated; n = 5. The expression level of myocardin in mouse primary myocardial cells was measured 24 h later by western blotting (upper panel) and qRT-PCR (lower panel). β-Actin was used as the loading control in western blotting. GAPDH was used as the normalization control in qRT-PCR.
(B–D) Mice were treated as indicated. The mice were subjected to I/R treatment 4 h later. Sham group mice were not treated by I/R; n = 5.
(B) The activity of LDH, CK, and CK-MB in mice serum was measured by commercial kits.
(C) Key parameters were tested by color Doppler echocardiography to reflect the injury status of myocardium in mice.
(D) The apoptosis of myocardial cells was determined by TUNEL assay; the representative images were displayed in the upper panel, and the statistic of cell death was shown in the lower chart. Scale bar, 50 μm.
(E) The expression level of LC3 in myocardial cells was examined by immunohistochemical staining. Scale bar, 50 μm.
(F) The expression of autophagy-related proteins and the activation of Akt and AMPK pathways in myocardial cells was measured by western blotting; β-actin was used as the loading control.
(A–F) The data represented 1 of 3 independent experiments. (A–D) Data were represented as means ± SDs. p values were determined by 1-way ANOVA, followed by Tukey post hoc test. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Discussion
Recently, non-coding RNAs packed in exosomes have been intensively studied for their sophisticated function in various biological activities,47 which suggested that these RNA molecules may be a crucial mediator for exosomes to exert their function. In the present study, we find that exosomal LINC00174, which is generated from vascular endothelial cells, mitigates the IR-induced myocardial injury by repressing p53-mediated autophagy and apoptosis. Mechanistically, LINC00174 directly targets SRSF1 and then reduces the expression of p53 in cardiomyocytes, which in turn resulted in restrained cell apoptosis, improved cardiac function, impaired myocardin expression, and repressed myocardin-induced autophagy.
Previous studies reveal that LINC00174 functions as an oncogene to facilitate the progression of CRC, glioma, and HCC through various mechanisms;21, 22, 23 however, no report has been published to investigate the function of LINC00174 in myocardial I/R injury. In the present study, we revealed that LINC00174 was enriched in exosomes derived from vascular endothelial cells. Although its expression in cardiomyocytes was impaired after I/R induction, the injection of exogenous LINC00174-containing exosomes successfully mitigated the myocardial I/R injury in the mouse model, including a reduced size of myocardial infarction, lessened cell apoptosis, improved cardiac function, decreased myocardium vacuolation, and attenuated autophagy activation. These data support the versatile roles of LINC00174 in tumor and cardiovascular diseases. When myocardial I/R occurs, the cellular LINC00174 in cardiomyocytes may be downregulated and cannot suppress p53-mediated autophagy activation and apoptosis. We speculate that exosomal LINC00174 derived from nearby vascular endothelial cells can reach the I/R site quickly and fuses with the affected cardiomyocytes, providing enough exogenous LINC00174 to combat p53-mediated autophagy activation and apoptosis. This mechanism supplies a quick response to rescue the affected cardiomyocytes during myocardial I/R injury. A similar mechanism is also found in I/R-induced kidney injury. Chen et al.48 found that epithelium-derived exosomal ATF3 RNA attenuated the transcription of the pro-inflammatory gene MCP-1 in renal I/R and attenuated I/R-induced kidney injury. Accumulative evidence shows that besides LINC00174, more and more lncRNAs are implicated in the modulation of myocardial I/R injury, although most of them are not wrapped and transported in exosomes. For example, Liu et al.44 found that lncRNA cardiac autophagy inhibitory factor (CAIF) was downregulated upon myocardial I/R injury, whereas enforced expression of CAIF led to repressed autophagy and reduced cell death. Chen and colleagues19 have reported that exosomal lncRNA-growth arrest-specific 5 (GAS5) derived from GAS5-overexpressing macrophages enhanced the apoptosis of vascular endothelial cells. Li et al.49 revealed that the inhibition of lncRNA X-inactive specific transcript (XIST) can improve myocardial I/R injury by repressing autophagy and the regulation of SOCS2. Moreover, exosomal miRNAs such as miR-21 and miR-451, derived from cardiac progenitor cells, also mitigate oxidative stress-induced apoptosis in myocardial I/R injury.16,50 Although the underlying mechanism is different, exosomal non-coding RNAs play vital roles in myocardial protection during I/R injury. Moreover, mechanistic approaches revealed that LINC00174 could directly interact with the SRSF1 protein and then obstruct the expression of p53 and its association with the myocardin promotor, which thus repressed the activation of AMPK and Akt and the downstream autophagy. This is a novel regulatory mechanism in the regulation of I/R injury for lncRNA. However, other mechanisms may be involved in the regulation of I/R injury by LINC00174, which still requires more investigation.
The implication of autophagy in the pathogenesis of cardiovascular diseases has been extensively studied, and shows both cardiac benefit and detrimental effects in various cardiopathologies, depending on the upstream signaling pathways and cell conditions.51 Autophagy is enhanced in both ischemia and reperfusion phases during cardiac I/R injury,52 with the aim of controlling the extent of cell damage and maintaining cell survival. In the cardiac ischemia phase, AMPK is activated in response to the low level of ATP and the high ratio of AMP:ATP as a nutrient sensor, which subsequently phosphorylates Unc-51-like autophagy activating kinase 1 (ULK1) at Ser 317 and Ser 777 in a direct or an indirect manner. It then induces autophagy, while the mammalian target of rapamycin (mTOR) can phosphorylate ULK1 at Ser 757 and abolishes its association with AMPK and then inhibits autophagy.53,54 However, Beclin1 steps onto center stage, so to speak, to mediate the autophagy process in the cardiac reperfusion phase, whereas AMPK is no longer activated in the stage.28 The upregulation of Beclin1 results in autophagy activation, probably induced by Bcl-2 interaction55 or increased reactive oxygen species (ROS) generation56 in the reperfusion process. Canonical phosphatidylinositol 3-kinase (PI3K)/Akt signaling can activate mTOR and thus suppress autophagy;57 meanwhile, Akt can directly phosphorylate Beclin1 to repress autophagy.58 However, in the present study, we observed the augmented Akt phosphorylation upon autophagy activation in H/R-treated myocardial cells, which contradicted the previous studies, but more studies are required to confirm this unusual phenotype. In the present study, we also found that autophagy was upregulated in cardiomyocytes after I/R treatment, as shown by the significantly increased autophagosomes, augmented AMPK phosphorylation, enhanced Beclin1 expression, and elevated LC3-II:LC3-I ratio. However, exosomal LINC00174 can suppress the activation of autophagy, according to our experiments, which were carried out by the LINC0014/SRSF1/p53/myocardin axis. However, the regulation of autophagy and cell death by p53 has been extensively studied, as a Bcl-2 family member, Bcl-2 19-kDa interacting protein (Bnip3) has been recognized as a crucial mediator in p53-induced autophagy and cell death. One report found that cardiac stress-induced p53 activation led to the upregulation of Bnip3, which triggered mitochondrial perturbations, autophagy induction, and cell death.59 By contrast, other groups reported that p53 repressed Bnip3 transcription and expression, resulting in mitophagy arrest, mitochondrial dysfunction,60 or reduced hypoxia-induced cell death.61 The contradictory results indicate that the role of p53 in I/R-induced autophagy and cell death may be complicated. Nevertheless, our study revealed that myocardin worked as another effector of p53 in autophagy activation and cell death during I/R injury, which further clarified the versatile roles of p53 in this process. lncRNA FOXD3-AS1 has been reported to aggravate the I/R injury of cardiomyocytes by promoting autophagy by activating nuclear factor κB/inducible nitric oxide synthase/cyclooxygenase 2 (NF-κB/iNOS/COX2) signaling,62 while lncRNA XIST has been proven to mitigate myocardial I/R injury by targeting miR-133a, suppressing autophagy, and regulating SOCS2.49 Although multiple pathways have been used by different lncRNAs, autophagy is still the crucial process for regulating I./R injury by lncRNAs. Therefore, inhibiting autophagy is a feasible approach to relieve or cure myocardial I/R injury.
Another interesting finding in our study was the downregulation of SRSF1 upon H/R treatment, and the upregulation of SRSF1 after the inoculation of LINC00174-sufficient exosomes. The consistent expression pattern between SRSF1 and LINC00174 suggested that they may have a close relationship during the modulation of autophagy in I/R-induced injury. Our study for the first time reveals the interaction between SRSF1 and LINC00174 in cardiomyocytes, and the SRSF1 truncation experiments further showed that all three domains of SRSF1 contributed to the interaction with LINC00174. However, we still do not know the detailed effects of this interaction on the stability or function of SRSF1, but the association between SRSF1 and p53 has been investigated by the Krainer group,41 which showed that SRSF1 overexpression stabilized p53 via the RPL5-MDM2 complex and thus induced cellular senescence. Their work demonstrated that p53 expression was positively regulated by SRSF1 in fibroblasts by blocking the ubiquitylation of p53. Our study revealed the reversed expression pattern in I/R-induced cardiomyocytes, which indicated that other regulators may become involved in the modulation of p53 by SRSF1. As for exosomal LINC00174, which was endocytosed by encountered cardiomyocytes and functioned as an inhibitor for p53 protein in cell cytosol, it exhibited a unique feature of lncRNA in the modulation of gene expression. For most of the identified lncRNAs, they exert their function by targeting the other miRNAs and influencing the downstream proteins. For example, lncRNA MALAT1 targets miR-204 and miR-558 to augment LC3-II expression or ULK1 phosphorylation, which in turn promotes autophagy activation.63,64 Another lncRNA, AK088388, targets miR-30a to enhance the expression of Beclin1, which thus facilitates the autophagy induction and increases cell apoptosis.65 The novel mechanism revealed here indicates that exosomal LINC00174 probably regulates p53 and its substrate protein myocardin through indirect methods by harnessing miR-125b as the bridge.
In summary, our study revealed that elevated exosomal LINC00174, which was secreted by vascular endothelial cells, can protect cardiomyocytes from I/R-induced cell damage. Mechanistically, LINC00174 repressed p53-myocardin-mediated autophagy and apoptosis in an SRSF1-dependent manner. These data indicated that the LINC00174-SRSF1-P53-myocardin pathway functioned as a novel regulator of autophagy and apoptosis in cardiomyocytes, which could be used as a therapeutic target to attenuate the excessive autophagy and improve cardiac function after I/R.
Materials and Methods
Mice
All of the male C57/BL6 mice used in this study were obtained from the Institute of Laboratory Animal Science of the Chinese Academy of Medical Sciences (Beijing, P.R. China). They were maintained in a pathogen-free animal facility at Guilin Medical University. All of the protocols for animal experiments were approved by the Ethics Review Board for Animal Studies of Guilin Medical University.
Myocardial I/R injury mouse model
Male mice 10 weeks of age were used in myocardial I/R surgery. Briefly, mice were anesthetized and intubated with polyethylene-90 tubing and then ventilated with 95% oxygen/5% CO2 with a rodent ventilator (model 683, Harvard Apparatus, Holliston, MA, USA). Mouse body temperature was maintained between 34°C and 37°C on a warm pad. Afterward, an oblique incision was performed to expose the left anterior descending (LAD) coronary artery, which was then ligated with a 6-0 silk suture at the 1-mm position just below the tip of the left atrial appendage. After 30 min of coronary occlusion, the ligation was released to allow tissue reperfusion, which could be visualized. Next, the chest wall was closed, and the mice were sacrificed 24 h later for TTC staining or tissue harvesting.
To assess the influence of exosomal LINC00174 on I/R-induced myocardial injury, 50 μ purified exosomes or an equal volume of PBS was intravenously injected into recipient mice at 4 h before the myocardial I/R injury.
Enzyme activity measurement
A total of 10 μL serum samples was obtained from the indicated mice for the measurement of enzyme activity, including LDH, CK, and CK-MB. LDH activity was measured by a commercial kit (ab-102526, Abcam, Cambridge, UK). Briefly, 50 μL standards, a mice serum sample, and positive control samples were loaded into a flat-bottom 96-well plate and then 50 μL reaction mix (LDH assay buffer, LDH substrate mix) was added to each well. After mixing, the plate was subjected to OD450 measurement with a plate reader (NEO, Bio-Tek, Henrico, VA, USA) using a kinetic mode. The OD450 was repeatedly measured every 3 min for 60 min at 37°C. The LDH activity was then calculated following the instructions in the manual. Total CK was measured using a commercial kit (ab-155901, Abcam). Briefly, 50 μL standards, mice serum sample, and positive control samples were loaded into a flat-bottom 96-well plate. Then, 50 μL reaction mix (CK assay buffer, CK enzyme mix, CK developer, ATP, CK substrate) was added to each well. After mixing, the plate was subjected to OD450 measurement with a plate reader (NEO, Bio-Tek) using a kinetic mode. The OD450 was repeatedly measured every 2 min for 40 min at 37°C. The CK activity was then calculated following the instructions in the manual. CK-MB was measured by a commercial ELISA kit (ab-193696, Abcam). Briefly, 50 μL mice serum samples and standards were loaded into pre-coated ELISA plates and incubated at 4°C overnight. After washing 3 times, the plate was loaded with 100 μL biotinylated human CK-MB detection antibody and then incubated at room temperature for 1 h. After washing 3 times, the plate was loaded with 100 μL 1× horseradish peroxidase (HRP)-streptavidin solution and then incubated at room temperature for 45 min. After washing 3 times, the plate was loaded with 100 μL TMB One-Step Substrate (Thermo Fisher Scientific, Carlsbad, CA, USA) and incubated for 30 min at room temperature in the dark with gentle shaking. Finally, 50 μL stop solution was added to each well. Then, the OD450 nm was measured immediately on a plate reader (NEO, Bio-Tek). The CK-MB activity was calculated following the instructions in the manual.
Color Doppler echocardiography for mice
Color Doppler echocardiography was used to evaluate the performance of myocardium in mice after I/R surgery according to the standard operation protocol.66 The evaluvation parameters included cardiac blood flow parameters (e.g., LVEDD, LVESD, LVEF, LVFS, LVSP, LVEDP) and ± dP/dtmax.
The isolation and culture of mouse primary aortic endothelial cells
After the sacrifice of the C57/BL6 mouse, the thoracic aorta was quickly dissected and immersed in PBS. Then, the connective tissue attached to adventitia was removed under a stereomicroscope. Subsequently, the aorta was cut into 1- to 2-mm cross-sectional rings, which were then opened and placed on Matrigel with the endothelium layer facing down. The aorta tissue was covered with several drops of Matrigel and gently washed once using pre-warmed PBS. After loading pre-warmed endothelial cell growth medium (Ham’s F12/DMEM supplemented with 5% fetal bovine serum [FBS], 50 μg/mL ECGS [endothelial cell growth supplement, Sigma-Aldrich, St. Louis, MO, USA], 10 U/mL heparin [Sigma-Aldrich], 50 μg/mL penicillin, and 50 μg/mL streptomycin), the aorta tissue was cultured at 37°C in a humid incubator chamber with 5% CO2 for 5–7 days, allowing the endothelial cells to sprout and proliferate. When a large number of endothelial cells were observed, the aorta tissues were removed, and the cells were detached with dispase treatment for 20 min at 37°C. After centrifugation, the supernatant was discarded and the pelleted cells were re-suspended in pre-warmed endothelial cell growth medium, which were then cultured in a T75 flask pre-coated with fibronectin. Endothelial cells were split when they reached 90% confluence.
The isolation and culture of mouse primary myocardial cells
Mouse primary myocardial cells were isolated and cultured as previously described.67 Briefly, the C57/BL6 mouse was pre-injected with heparin (1,000 μL/kg body weight) 30 min before anesthetization by pentobarbital injection. After that, the chest cavity was opened and the heart was quickly dissected and then transferred into cold Ca2+-free 1× PBS with 20 mM 2,3-butanedione monoxime (BDM, Sigma-Aldrich) in a 10-cm dish on ice. After removing lung tissues and large blood vessels, the heart was washed with 1× PBS with 20 mM BDM to eliminate the blood. Next, the heart was immersed and washed in a second dish with Ca2+-free 1× PBS with 20 mM BDM. Then, the cleaned heart was immersed in 500 μL isolation medium in a 24-well plate and minced into small pieces (~1 mm3) using curved scissors. The heart tissues were collected in a 15-mL tube filled with 10 mL cold isolation medium and were gently agitated at 4°C overnight. On the second day, most of the supernatant was removed and 5 mL digestion medium (10 mL L15-medium supplemented with 20 mM BDM and 15 mg collagenase/dispase mixture [Roche, Basel, Switzerland]) was added. The cardiac tissue fragments were then digested at 37°C with gentle agitation for 30 min. Afterward, the tissue fragments were pipetted with a pre-wetted 10 mL pipette 20 times and the supernatant was then filtered through a 40-μm cell strainer to a new 50-mL tube, which was then centrifuged at 300 rpm for 5 min. After aspirating the supernatant, the cell pellet was re-suspended in plating medium (L-15 medium supplemented with 20 mM BDM) and then plated in a 10-cm dish for 3 h in a cell culture incubator, which could remove the fibroblasts and endothelial cells that attached to the uncovered dish. Then, the non-adherent cardiomyocytes in the supernatant were collected, counted, and re-plated into collagen-coated dishes at 2.0 × 105 cells/cm2. The cardiomyocytes were cultured in an incubator for 18 h without being disturbed.
One day after plating, the cardiomyocytes adhered to the culture dish and began to spontaneously contract. (When necessary, the culture medium should be replaced with fresh maintenance medium supplemented with proliferation inhibitors [10 μM cytosine-β-d-arabinofuranoside hydrochloride {Arac}; Sigma-Aldrich] and chronotropic agents [1 μM isoproterenol, Sigma-Aldrich].)
The establishment of the H/R cell model
The H/R model in mouse primary aortic endothelial cells was established by placing the cells in a Billups-Rothenberg modular incubator chamber (Billups-Rothenberg, San Diego, CA, USA), which was then saturated with 95% N2 and 5% CO2 and cultured at 37°C for 4 h. Afterward, the cells were subjected to reoxygenation by culturing cells at 37°C in a humidified incubator with 95% O2 and 5% CO2 for another 4 h. To assess the effect of exosomes on the expression of LINC00174 and p53 during the H/R process, 1 × 106 mouse primary aortic endothelial cells were seeded in 1 well of a 6-well plate, and then 50 μg purified exosomes or an equal volume of PBS was loaded into the one well of the 6-well plate before the H/R treatment.
The isolation and identification of exosomes
Exosomes were isolated using the ExoQuick-TC ULTRA EV Isolation Kit for Tissue Culture Medium (System Biosciences, Palo Alto, CA, USA) according to the manufacturer’s instructions. Briefly, cell culture medium was collected and centrifuged at 3,000 × g for 15 min to remove cellular debris. The cleared supernatant was then transferred to a new tube and exosomes were precipitated by ExoQuick-TC solution (sample volume/ExoQuick-TC volume = 5/1) at 4°C overnight on ice. Afterward, the ExoQuick-TC/cell culture medium mixture was centrifuged at 3,000 × g for 10 min at 4°C. The supernatant was aspirated and pelleted exosomes were re-suspended in buffer B for protein concentration measurement using bicinchoninic acid (BCA) kits (Tiangen Biotech, Beijing, P.R. China). To purify the isolated exosomes, an equal amount of buffer A (with buffer B) was added to the re-suspended exosomal solution and then loaded onto the pre-equilibrated purification column by buffer B. After rotating for 5 min, the column was centrifuged at 1,000 × g for 30 s in a 2-mL Eppendorf tube. The eluted solution was purified exosomes, which were suitable for nanoparticle tracking analysis, TEM imaging, RNA extraction, fluorescence-activated cell sorting (FACS), and western blotting analysis.
To examine the specific exosomal surface markers (e.g., CD9, CD63, CD81) and marker proteins for other cellular organs (e.g., cytochrome c, TSG101, calnexin, syntaxin 6), FACS and western blotting were performed.
TEM imaging
Purified exosomes were fixed in 1% glutaraldehyde at room temperature for 2 h and then loaded onto a formvar-carbon-coated EM grid and left to dry at room temperature for 2–5 min. The samples were then negatively stained with phosphotungstic acid for 5 min. After drying for 10 min, the morphology of exosomes was visualized on FEI Tecnai F20 S TEM (FEI, Hillsboro, OR, USA) at 200 kV.
p53 siRNA and control
SRSF1 siRNA and control were ordered from Shanghai GenePharma (Shanghai, P.R. China) and their sequences are listed in Table 1.
Table 1.
p53 siRNA, SRSF1 siRNA, and control
| Name | Sequence (5′–3′) |
|---|---|
| p53 siRNA sense | CACCUCACUGCAUGGACGAUCUGUU |
| p53 siRNA antisense | AACAGAUCGUCCAUGCAGUGAGGUG |
| SRSF1 siRNA sense | CGGGUUAAAGUUGAUGGGCCCAGAA |
| SRSF1 siRNA antisense | UUCUGGGCCCAUCAACUUUAACCCG |
| siRNA control sense | ACUACUGAGUGACAGUAGA |
| siRNA control antisense | UCUACUGUCACUCAGUAGU |
Plasmid construction and transfection
Endogenous LINC00174 gene knockdown was implemented by shRNA. shRNA targeting LINC00174 (shLINC00174) and negative control shRNA (shNC) were purchased from GenePharma, and the sequence of shLINC00174 and shNC is shown in Table 2.
Table 2.
shRNA sequences used in plasmid construction
| shRNA name | Sequence (5′–3′) |
|---|---|
| shLINC00174 | CCGGTGCGTCCCGAAGG CTTCTCTACTCGAGTAGA GAAGCCTTCGGGACGCATTTTTG |
| shNC | CCGGCCTAAGGTTAAGTC GCCCTCGCTCGAGCGAGG GCGACTTAACCTTAGGTTTTT |
shLINC00174 and shNC were then subcloned into pLKO.1-puro vector (Sigma-Aldrich). Sequence-verified pLKO.1-shLINC00174 or pLKO.1-shNC plasmids were then used for the lentivirus package in HEK293T cells. Titer-determined lentivirus was then used to infect mouse primary vascular endothelial cells to obtain stable LINC00174-knocking down or NC cells.
RNA extraction, reverse-transcription, and quantitative real-time PCR
Small RNA and total RNA were extracted from tissues or cell lines using the mirVana miRNA isolation kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. They were then used for the first-strand cDNA generation with the TaqMan miRNA reverse transcription kit (Thermo Fisher Scientific) or with the high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific). qPCR was performed on the Mx3000P qPCR system (Agilent, Santa Clara, CA, USA). The expression of LINC00174 mRNAs was normalized to that of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA and calculated as 2−[(Ct of genes)−(Ct of GAPDH)]. All of the samples were prepared in triplicate. The primers used in qPCR are listed in Table 3.
Table 3.
The primers used in quantitative real-time PCR
| Primer name | Sequence (5′–3′) |
|---|---|
| LINC00174 forward | GGCCCAACACTTCCCTCAAA |
| LINC00174 reverse | CAGGGAGAAACGACCTGGAG |
| SRSF1 forward | CCGCAGGGAACAACGATTG |
| SRSF1 reverse | GCCGTATTTGTAGAACACGTCCT |
| GAPDH forward | GGACACAATGGATTGCAAGG |
| GAPDH reverse | TAACCACTGCTCCACTCTGG |
Co-immunoprecipitation assay
293T cells were transfected with SRSF1-overexpresing plasmids or knocking down siRNAs by lipofectamine 2000 (Thermo Fisher Scientific) for 48 h. After that, the cells were collected, rinsed in cold PBS, and lysed in 1 × cell lysis buffer (20 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 × Proteinase inhibitor cocktail) by rotation at 4°C for 1 h. After centrifugation at 12,000 × g, 4°C for 10 min, the supernatants were collected and incubated with 1 μg mouse anti-p53 antibody (CST-#2524S) or 1 μg mouse (G3A1) monoclonal antibody (mAb) IgG1 isotype control (CST-#5415), by gentle rotation at 4°C for 4 h, respectively. Next, 40 μL prewashed Protein A/G conjugated agarose beads were added to capture the precipitated antibody-antigen complex via 2 h incubation. After centrifugation and 3-round washing of the beads, protein samples were eluted by 1× reducing loading buffer and subjected to western blotting.
Western blotting
Tissues and cells were homogenized or lysed in RIPA buffer supplemented with a protease inhibitor cocktail (Selleck, Houston, TX, USA) and then subjected to protein amount quantification with a BCA kit (Tiangen Biotech). An equal amount of proteins was loaded and separated by SDS-PAGE and then transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, Bedford, MA), which was then blocked by 5% BSA in Tris-buffered saline with 0.1% Tween 20 (TBST) for 1 h at room temperature. Next, the PVDF membrane was rinsed and probed with the primary antibodies against the indicated target proteins at 4°C overnight: anti-CD9 antibody (1:2,000, Santa Cruz-sc-13118), anti-CD63 antibody (1:1,000, Santa Cruz-sc-365604), anti-TSG101 antibody (1:1,000, Santa Cruz-sc-7964), anti-cytochrome c antibody (1:2,000, Santa Cruz-sc-13561), anti-calnexin antibody (1:1,000, Santa Cruz-sc-23954), anti-syntaxin 6 antibody (1:1,000, Abcam-ab12370), anti-LC antibody (1:1,000, Cell Signaling Technology [Danvers, MA, USA], CST-#4108), anti-Beclin1 antibody (1:1,000, CST-#4122), anti-p62 antibody (1:1,000, CST-#8025), anti-p53 antibody (1:1,000, CST-#2524S), anti-β-actin antibody (1:5,000, CST-#4970), anti-SRSF1 antibody (1:1,000, Abcam-ab38017), anti-myocardin antibody (1:1,000, Abcam-ab107301), anti-Akt antibody (1:2,000, CST-#9272), anti-phospho Akt (Ser473) antibody (1:1,000, CST-#9271), anti-AMPK antibody (1:2,000, CST-#2532), and anti-phospho AMPK antibody (1:1,000, CST-#2535). After vigorous washing, PVDF membranes were re-probed with HRP-conjugated goat anti-mouse IgG (1:5,000, CST-#7076) or goat anti-rabbit IgG antibody (1:5,000, CST-#7074) for 1 h at room temperature. After washing, the PVDF membranes were incubated with ECL substrate to develop chemiluminescence on blots, which were captured by the ChemiDoc MP imaging system (Bio-Rad, Hercules, CA, USA) and normalized by ImageJ software (NIH, Bethesda, MD, USA).
Immunohistochemistry and immunofluorescent staining
For immunohistochemistry, the slides were incubated with antibody against LC3 (1:100, Cell Signaling Technology) at 4°C overnight and stained with diaminobenzidine (DAB) and counterstained with hematoxylin. The slides were then subjected to gradient ethanol dehydration and dimethyl benzene transparent and mounted with neutral resin cover slides. Images were captured using a Nikon (Tokyo, Japan) ECLIPSE 80i.
For immunofluorescence staining, cells cultured on poly-l-lysine-coated cover glasses were washed by PBS once and then fixed with 4% paraformaldehyde for 15 min at room temperature. After washing, the cells were permeabilized by 0.1% Triton X-100 for 15 min at room temperature. Next, the cells were blocked with 2% BSA for 30 min and then stained with rabbit anti-human LC3 antibody (1:200, Cell Signaling Technology) at 4°C for 6 h. After washing by PBS, the cells were probed with Alexa Fluor 594-conjugated goat anti-rabbit IgG (1:200, Thermo Fisher Scientific) at 4°C for 1 h. To display the cell nuclei, 100 μg/mL DAPI was applied to incubate with cells for 15 min at room temperature. After washing, the cells were subjected to an inverted fluorescence microscope (Olympus IX73, Olympus, Tokyo, Japan) for imaging. The LC3 puncta were measured by ImageJ software (NIH). Five individual pictures were analyzed for each kind of cell.
Cell apoptosis measurement by TUNEL assay
In-site cell apoptosis was determined by TUNEL assay (Abcam) according to the kit manual. Briefly, cells were dehydrated in xylene and ethanol gradient, which were then permeabilized in 1% Proteinase K for 20 min. After a TBS rinse, the samples were quenched by 3% H2O2 for 5 min. Next, the quenched samples were equilibrated with TdT equilibration buffer for 30 min, followed by incubation with TdT labeling reaction mix at 37°C for 1.5 h. Then, the stop solution was added and incubated with cells for 5 min. After rinsing and blocking, the samples were immersed in 1× conjugate for 30 min at room temperature. Afterward, the chemiluminescence was developed by covering the samples with DAB solution for 15 min. After washing, the samples were stained with Methyl Green counterstain solution for 3 min. Finally, the stained cell samples were dehydrated again in ethanol and xylene and then subjected to imaging on a fluorescence microscope (Olympus IX73). Five individual pictures were captured for each kind of cell.
RNA pull-down assay
Biotinylated LINC00174 or control lncRNA was synthesized (GenePharma) to pull down SRSF1 as a probe, which was then measured using western blotting. Briefly, 1 × 106 cells were transfected with biotinylated LINC00174 or control lncRNA by lipofectamine 2000 (Thermo Fisher Scientific) in a 10-cm dish. The cells were collected 48 h later and lysed in RIPA buffer, which was supplemented with RNase inhibitor (Selleck) and complete protease inhibitor (Selleck). Streptavidin agarose beads (Thermo Fisher Scientific) pre-blocked with yeast tRNA (Thermo Fisher Scientific) were used to incubate with cell lysates overnight at 4°C on a rotator. After centrifugation, the pelleted agarose beads were washed with cold lysis buffer 3 times, and the associated proteins were purified for subsequent western blotting analysis. The expression levels of p53 and SRSF1 were normalized to that of β-actin.
RIP
The RNA-binding protein immunoprecipitation kit (Millipore) was used in the RIP assay to assess the interaction between SRSF1 and LINC00174 following the manufacturer’s instructions. Briefly, cells were harvested and lysed by RIP lysis buffer and then incubated with magnetic beads conjugated with anti-p53 antibody, anti-SRSF1 antibody, or mouse IgG as NC in RIP immunoprecipitation buffer at 4°C overnight. Next, the beads were pelleted with a magnetic separator and washed by RIP wash buffer 3 times to remove non-specific materials, which were then digested by Proteinase K at 55°C for 30 min. Afterward, the RNAs binding to SRSF1, p53, or mouse IgG were extracted and quantified and then subjected to qRT-PCR analysis.
ChIP
Cells were harvested, washed, and fixed with 4% paraformaldehyde (PFA) for 15 min. Then, the cells were washed by PBS and lysed in ChIP lysis buffer (50 mM HEPES-KOH [pH 7.5], 140 mM NaCl, 1 mM EDTA [pH 8], 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, and 1× protease inhibitors) on ice. Afterward, the lysate was sonicated to shear chromatin DNA into 200- to 800-bp fragments. After centrifugation, the supernatant was collected, and the chromatin fragments were incubated with p53 antibody or normal mouse IgG at 4°C overnight. Pre-blocked protein A/G beads (by salmon sperm DNA) were added to capture immunoprecipitants at 4°C for 2 h. The precipitated DNA was eluted and analyzed by PCR to detect a 349-bp fragment in the promoter region of myocardin with the following primers: myocardin-F, 5′-ACAAATAACTCTGGGTCGGT-3′ and myocardin-R, 5′-AGACGATTCTGTATCTCGCA-3′. p53 antibody captured DNA was normalized to that precipitated by normal mouse IgG.
Dual-luciferase reporting assay
The promotor region for the transcription of myocardin was cloned and inserted into the upstream of the luciferase 2 gene in pGL4.17 vector, which was transfected into 293T cells together with pGL4.73 Renilla luciferase control plasmid by lipofectamine 2000 (Thermo Fisher Scientific). At the same time, the cells were un-transfected or transfected with control siRNA or siRNA targeting p53. The Renilla luciferase provided a normalization reference. At 48 h after transfection, the 293T cells were lysed and subjected to luciferase activity measurement with the Dual Luciferase Assay Kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Firefly and Renilla luciferase activities were determined by a plate reader (NEO, Bio-Tek) and normalized to Renilla luciferase data.
Data analysis
Each experiment was repeated at least 3 times, and 1 representative experiment shown. Data were shown as means ± standard deviations (SDs), which were analyzed by GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA). An unpaired 2-tailed Student’s t test was used to compare the difference between the 2 groups. One-way analysis of variance (ANOVA) followed by Tukey post hoc test was used for multiple comparison. Statistical significance was determined as indicated in the figure legends. Significance was noted at ∗p = 0.05, ∗∗p = 0.01, and ∗∗∗p = 0.001, respectively.
Acknowledgments
This work was supported by the Guangxi Natural Science Foundation (grant nos. 2020GXNSFDA238007 and 2018GXNSFAA050110) and the Guangxi BaGui Scholars Special Project.
Author contributions
Q.S. designed the study. Q.S., R.-P.C., and R.-X.D. contributed to the drafting the manuscript. X.-H.Y., R.-X.D., and W.-K.Z. performed the clinical and experimental studies. Y.-L.X. conducted the data acquisition. X.-W.L. and B.-H.K. carried out the data analysis. B.-H.K. performed the literature search. All of the authors read and approved the final submitted manuscript.
Declaration of interests
The authors declare no competing interests.
References
- 1.Johnstone R.M., Adam M., Hammond J.R., Orr L., Turbide C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes) J. Biol. Chem. 1987;262:9412–9420. [PubMed] [Google Scholar]
- 2.Colombo M., Raposo G., Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014;30:255–289. doi: 10.1146/annurev-cellbio-101512-122326. [DOI] [PubMed] [Google Scholar]
- 3.Raposo G., Nijman H.W., Stoorvogel W., Liejendekker R., Harding C.V., Melief C.J., Geuze H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996;183:1161–1172. doi: 10.1084/jem.183.3.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zitvogel L., Regnault A., Lozier A., Wolfers J., Flament C., Tenza D., Ricciardi-Castagnoli P., Raposo G., Amigorena S. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat. Med. 1998;4:594–600. doi: 10.1038/nm0598-594. [DOI] [PubMed] [Google Scholar]
- 5.Gross J.C., Chaudhary V., Bartscherer K., Boutros M. Active Wnt proteins are secreted on exosomes. Nat. Cell Biol. 2012;14:1036–1045. doi: 10.1038/ncb2574. [DOI] [PubMed] [Google Scholar]
- 6.Zhang L., Yu D. Exosomes in cancer development, metastasis, and immunity. Biochim. Biophys. Acta Rev. Cancer. 2019;1871:455–468. doi: 10.1016/j.bbcan.2019.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buzas E.I., György B., Nagy G., Falus A., Gay S. Emerging role of extracellular vesicles in inflammatory diseases. Nat. Rev. Rheumatol. 2014;10:356–364. doi: 10.1038/nrrheum.2014.19. [DOI] [PubMed] [Google Scholar]
- 8.Jan A.T., Malik M.A., Rahman S., Yeo H.R., Lee E.J., Abdullah T.S., Choi I. Perspective Insights of Exosomes in Neurodegenerative Diseases: A Critical Appraisal. Front. Aging Neurosci. 2017;9:317. doi: 10.3389/fnagi.2017.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jia G., Sowers J.R. Targeting endothelial exosomes for the prevention of cardiovascular disease. Biochim. Biophys. Acta Mol. Basis Dis. 2020;1866:165833. doi: 10.1016/j.bbadis.2020.165833. [DOI] [PubMed] [Google Scholar]
- 10.Higginbotham J.N., Demory Beckler M., Gephart J.D., Franklin J.L., Bogatcheva G., Kremers G.-J., Piston D.W., Ayers G.D., McConnell R.E., Tyska M.J., Coffey R.J. Amphiregulin exosomes increase cancer cell invasion. Curr. Biol. 2011;21:779–786. doi: 10.1016/j.cub.2011.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGough I.J., Vincent J.-P. Exosomes in developmental signalling. Development. 2016;143:2482–2493. doi: 10.1242/dev.126516. [DOI] [PubMed] [Google Scholar]
- 12.Languino L.R., Singh A., Prisco M., Inman G.J., Luginbuhl A., Curry J.M., South A.P. Exosome-mediated transfer from the tumor microenvironment increases TGFβ signaling in squamous cell carcinoma. Am. J. Transl. Res. 2016;8:2432–2437. [PMC free article] [PubMed] [Google Scholar]
- 13.Eltzschig H.K., Eckle T. Ischemia and reperfusion--from mechanism to translation. Nat. Med. 2011;17:1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hausenloy D.J., Yellon D.M. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J. Clin. Invest. 2013;123:92–100. doi: 10.1172/JCI62874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin X., He Y., Hou X., Zhang Z., Wang R., Wu Q. Endothelial Cells Can Regulate Smooth Muscle Cells in Contractile Phenotype through the miR-206/ARF6&NCX1/Exosome Axis. PLoS ONE. 2016;11:e0152959. doi: 10.1371/journal.pone.0152959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen L., Wang Y., Pan Y., Zhang L., Shen C., Qin G., Ashraf M., Weintraub N., Ma G., Tang Y. Cardiac progenitor-derived exosomes protect ischemic myocardium from acute ischemia/reperfusion injury. Biochem. Biophys. Res. Commun. 2013;431:566–571. doi: 10.1016/j.bbrc.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quinn J.J., Chang H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016;17:47–62. doi: 10.1038/nrg.2015.10. [DOI] [PubMed] [Google Scholar]
- 18.Gao T., Liu X., He B., Nie Z., Zhu C., Zhang P., Wang S. Exosomal lncRNA 91H is associated with poor development in colorectal cancer by modifying HNRNPK expression. Cancer Cell Int. 2018;18:11. doi: 10.1186/s12935-018-0506-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen L., Yang W., Guo Y., Chen W., Zheng P., Zeng J., Tong W. Exosomal lncRNA GAS5 regulates the apoptosis of macrophages and vascular endothelial cells in atherosclerosis. PLoS ONE. 2017;12:e0185406. doi: 10.1371/journal.pone.0185406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian J., Popal M.S., Zhao Y., Liu Y., Chen K., Liu Y. Interplay between Exosomes and Autophagy in Cardiovascular Diseases: Novel Promising Target for Diagnostic and Therapeutic Application. Aging Dis. 2019;10:1302–1310. doi: 10.14336/AD.2018.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen Y., Gao X., Tan W., Xu T. STAT1-mediated upregulation of lncRNA LINC00174 functions a ceRNA for miR-1910-3p to facilitate colorectal carcinoma progression through regulation of TAZ. Gene. 2018;666:64–71. doi: 10.1016/j.gene.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 22.Shi J., Zhang Y., Qin B., Wang Y., Zhu X. Long non-coding RNA LINC00174 promotes glycolysis and tumor progression by regulating miR-152-3p/SLC2A1 axis in glioma. J. Exp. Clin. Cancer Res. 2019;38:395. doi: 10.1186/s13046-019-1390-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao J.-T., Chi B.-J., Sun Y., Chi N.-N., Zhang X.-M., Sun J.-B., Chen Y., Xia Y. LINC00174 is an oncogenic lncRNA of hepatocellular carcinoma and regulates miR-320/S100A10 axis. Cell Biochem. Funct. 2020;38:859–869. doi: 10.1002/cbf.3498. [DOI] [PubMed] [Google Scholar]
- 24.Glick D., Barth S., Macleod K.F. Autophagy: cellular and molecular mechanisms. J. Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagata S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018;36:489–517. doi: 10.1146/annurev-immunol-042617-053010. [DOI] [PubMed] [Google Scholar]
- 26.Mariño G., Niso-Santano M., Baehrecke E.H., Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valentim L., Laurence K.M., Townsend P.A., Carroll C.J., Soond S., Scarabelli T.M., Knight R.A., Latchman D.S., Stephanou A. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2006;40:846–852. doi: 10.1016/j.yjmcc.2006.03.428. [DOI] [PubMed] [Google Scholar]
- 28.Matsui Y., Takagi H., Qu X., Abdellatif M., Sakoda H., Asano T., Levine B., Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 29.Troncoso R., Vicencio J.M., Parra V., Nemchenko A., Kawashima Y., Del Campo A., Toro B., Battiprolu P.K., Aranguiz P., Chiong M. Energy-preserving effects of IGF-1 antagonize starvation-induced cardiac autophagy. Cardiovasc. Res. 2012;93:320–329. doi: 10.1093/cvr/cvr321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang M., Linn B.S., Zhang Y., Ren J. Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochim. Biophys. Acta Mol. Basis Dis. 2019;1865:2293–2302. doi: 10.1016/j.bbadis.2019.05.007. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y., Whaley-Connell A.T., Sowers J.R., Ren J. Autophagy as an emerging target in cardiorenal metabolic disease: from pathophysiology to management. Pharmacol. Ther. 2018;191:1–22. doi: 10.1016/j.pharmthera.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu W., Sun S., Xu H., Li C., Ren J., Zhang Y. TBC1D15/RAB7-regulated mitochondria-lysosome interaction confers cardioprotection against acute myocardial infarction-induced cardiac injury. Theranostics. 2020;10:11244–11263. doi: 10.7150/thno.46883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan L., Vatner D.E., Kim S.J., Ge H., Masurekar M., Massover W.H., Yang G., Matsui Y., Sadoshima J., Vatner S.F. Autophagy in chronically ischemic myocardium. Proc. Natl. Acad. Sci. USA. 2005;102:13807–13812. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma X., Liu H., Foyil S.R., Godar R.J., Weinheimer C.J., Hill J.A., Diwan A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012;125:3170–3181. doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamacher-Brady A., Brady N.R., Gottlieb R.A. The interplay between pro-death and pro-survival signaling pathways in myocardial ischemia/reperfusion injury: apoptosis meets autophagy. Cardiovasc. Drugs Ther. 2006;20:445–462. doi: 10.1007/s10557-006-0583-7. [DOI] [PubMed] [Google Scholar]
- 36.Sun T., Liu H., Cheng Y., Yan L., Krittanawong C., Li S., Qian W., Su W., Chen X., Hou X. 2,3,5,4′-Tetrahydroxystilbene-2-O-β-d-glucoside eliminates ischemia/reperfusion injury–induced H9c2 cardiomyocytes apoptosis involving in Bcl-2, Bax, caspase-3, and Akt activation. J. Cell. Biochem. 2019;120:10972–10977. doi: 10.1002/jcb.27949. [DOI] [PubMed] [Google Scholar]
- 37.Ge L., Cai Y., Ying F., Liu H., Zhang D., He Y., Pang L., Yan D., Xu A., Ma H., Xia Z. miR-181c-5p Exacerbates Hypoxia/Reoxygenation-Induced Cardiomyocyte Apoptosis via Targeting PTPN4. Oxid. Med. Cell. Longev. 2019;2019:1957920. doi: 10.1155/2019/1957920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yano T., Abe K., Tanno M., Miki T., Kuno A., Miura T., Steenbergen C. Does p53 Inhibition Suppress Myocardial Ischemia-Reperfusion Injury? J. Cardiovasc. Pharmacol. Ther. 2018;23:350–357. doi: 10.1177/1074248418763612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naito A.T., Okada S., Minamino T., Iwanaga K., Liu M.L., Sumida T., Nomura S., Sahara N., Mizoroki T., Takashima A. Promotion of CHIP-mediated p53 degradation protects the heart from ischemic injury. Circ. Res. 2010;106:1692–1702. doi: 10.1161/CIRCRESAHA.109.214346. [DOI] [PubMed] [Google Scholar]
- 40.Maslon M.M., Heras S.R., Bellora N., Eyras E., Cáceres J.F. The translational landscape of the splicing factor SRSF1 and its role in mitosis. eLife. 2014;3:e02028. doi: 10.7554/eLife.02028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fregoso O.I., Das S., Akerman M., Krainer A.R. Splicing-factor oncoprotein SRSF1 stabilizes p53 via RPL5 and induces cellular senescence. Mol. Cell. 2013;50:56–66. doi: 10.1016/j.molcel.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan Q.-W., Cheng C., Hackett C., Feldman M., Houseman B.T., Nicolaides T., Haas-Kogan D., James C.D., Oakes S.A., Debnath J. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci. Signal. 2010;3:ra81. doi: 10.1126/scisignal.2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hardie D.G. AMPK and autophagy get connected. EMBO J. 2011;30:634–635. doi: 10.1038/emboj.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu C.-Y., Zhang Y.-H., Li R.-B., Zhou L.-Y., An T., Zhang R.-C., Zhai M., Huang Y., Yan K.-W., Dong Y.-H. LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53-mediated myocardin transcription. Nat. Commun. 2018;9:29. doi: 10.1038/s41467-017-02280-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang J., Lu M.M., Cheng L., Yuan L.-J., Zhu X., Stout A.L., Chen M., Li J., Parmacek M.S. Myocardin is required for cardiomyocyte survival and maintenance of heart function. Proc. Natl. Acad. Sci. USA. 2009;106:18734–18739. doi: 10.1073/pnas.0910749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tan Z., Li J., Zhang X., Yang X., Zhang Z., Yin K.J., Huang H. P53 Promotes Retinoid Acid-induced Smooth Muscle Cell Differentiation by Targeting Myocardin. Stem Cells Dev. 2018;27:534–544. doi: 10.1089/scd.2017.0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Silva M., Melo S.A. Non-coding RNAs in Exosomes: New Players in Cancer Biology. Curr. Genomics. 2015;16:295–303. doi: 10.2174/1389202916666150707154719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen H.-H., Lai P.-F., Lan Y.-F., Cheng C.-F., Zhong W.-B., Lin Y.-F., Chen T.-W., Lin H. Exosomal ATF3 RNA attenuates pro-inflammatory gene MCP-1 transcription in renal ischemia-reperfusion. J. Cell. Physiol. 2014;229:1202–1211. doi: 10.1002/jcp.24554. [DOI] [PubMed] [Google Scholar]
- 49.Li Z., Zhang Y., Ding N., Zhao Y., Ye Z., Shen L., Yi H., Zhu Y. Inhibition of lncRNA XIST Improves Myocardial I/R Injury by Targeting miR-133a through Inhibition of Autophagy and Regulation of SOCS2. Mol. Ther. Nucleic Acids. 2019;18:764–773. doi: 10.1016/j.omtn.2019.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiao J., Pan Y., Li X.H., Yang X.Y., Feng Y.L., Tan H.H., Jiang L., Feng J., Yu X.Y. Cardiac progenitor cell-derived exosomes prevent cardiomyocytes apoptosis through exosomal miR-21 by targeting PDCD4. Cell Death Dis. 2016;7:e2277. doi: 10.1038/cddis.2016.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delbridge L.M.D., Mellor K.M., Taylor D.J., Gottlieb R.A. Myocardial stress and autophagy: mechanisms and potential therapies. Nat. Rev. Cardiol. 2017;14:412–425. doi: 10.1038/nrcardio.2017.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ma H., Guo R., Yu L., Zhang Y., Ren J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. Eur. Heart J. 2011;32:1025–1038. doi: 10.1093/eurheartj/ehq253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim J., Kundu M., Viollet B., Guan K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Egan D., Kim J., Shaw R.J., Guan K.L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7:643–644. doi: 10.4161/auto.7.6.15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brady N.R., Hamacher-Brady A., Yuan H., Gottlieb R.A. The autophagic response to nutrient deprivation in the hl-1 cardiac myocyte is modulated by Bcl-2 and sarco/endoplasmic reticulum calcium stores. FEBS J. 2007;274:3184–3197. doi: 10.1111/j.1742-4658.2007.05849.x. [DOI] [PubMed] [Google Scholar]
- 56.Hariharan N., Zhai P., Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid. Redox Signal. 2011;14:2179–2190. doi: 10.1089/ars.2010.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shao X., Lai D., Zhang L., Xu H. Induction of Autophagy and Apoptosis via PI3K/AKT/TOR Pathways by Azadirachtin A in Spodoptera litura Cells. Sci. Rep. 2016;6:35482. doi: 10.1038/srep35482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang R.C., Wei Y., An Z., Zou Z., Xiao G., Bhagat G., White M., Reichelt J., Levine B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012;338:956–959. doi: 10.1126/science.1225967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang E.Y., Gang H., Aviv Y., Dhingra R., Margulets V., Kirshenbaum L.A. p53 mediates autophagy and cell death by a mechanism contingent on Bnip3. Hypertension. 2013;62:70–77. doi: 10.1161/HYPERTENSIONAHA.113.01028. [DOI] [PubMed] [Google Scholar]
- 60.Zhou H., Du W., Li Y., Shi C., Hu N., Ma S., Wang W., Ren J. Effects of melatonin on fatty liver disease: The role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. J. Pineal Res. 2018;64 doi: 10.1111/jpi.12450. [DOI] [PubMed] [Google Scholar]
- 61.Feng X., Liu X., Zhang W., Xiao W. p53 directly suppresses BNIP3 expression to protect against hypoxia-induced cell death. EMBO J. 2011;30:3397–3415. doi: 10.1038/emboj.2011.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tong G., Wang Y., Xu C., Xu Y., Ye X., Zhou L., Zhu G., Zhou Z., Huang J. Long non-coding RNA FOXD3-AS1 aggravates ischemia/reperfusion injury of cardiomyocytes through promoting autophagy. Am. J. Transl. Res. 2019;11:5634–5644. [PMC free article] [PubMed] [Google Scholar]
- 63.Guo X., Wu X., Han Y., Tian E., Cheng J. LncRNA MALAT1 protects cardiomyocytes from isoproterenol-induced apoptosis through sponging miR-558 to enhance ULK1-mediated protective autophagy. J. Cell. Physiol. 2019;234:10842–10854. doi: 10.1002/jcp.27925. [DOI] [PubMed] [Google Scholar]
- 64.Xiao J., Zhu X., He B., Zhang Y., Kang B., Wang Z., Ni X. MiR-204 regulates cardiomyocyte autophagy induced by ischemia-reperfusion through LC3-II. J. Biomed. Sci. 2011;18:35. doi: 10.1186/1423-0127-18-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang J.J., Bie Z.D., Sun C.F. Long noncoding RNA AK088388 regulates autophagy through miR-30a to affect cardiomyocyte injury. J. Cell. Biochem. 2019;120:10155–10163. doi: 10.1002/jcb.28300. [DOI] [PubMed] [Google Scholar]
- 66.Respress J.L., Wehrens X.H.T. Transthoracic Echocardiography in Mice. J. Vis. Exp. 2010;66:1738. doi: 10.3791/1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ehler E., Moore-Morris T., Lange S. Isolation and culture of neonatal mouse cardiomyocytes. J. Vis. Exp. 2013;67:50154. doi: 10.3791/50154. [DOI] [PMC free article] [PubMed] [Google Scholar]