

Hydrogen peroxide mediates the toxicity of phagocytes, lactic acid bacteria, redox-cycling antibiotics, and photochemistry. The underlying mechanisms all involve its reaction with iron atoms, whether in enzymes or on the surface of DNA. Accordingly, when bacteria perceive toxic H2O2, they activate defensive tactics that are focused on iron metabolism. In this study, we identify a conundrum: DNA is best protected by the removal of iron from the cytoplasm, but this action impairs the ability of the cell to reactivate its iron-dependent enzymes. The actions of the Clp proteins appear to hedge against the oversequestration of iron by the miniferritin Dps. This buffering effect is important, because E. coli seeks not just to survive H2O2 but to grow in its presence.
KEYWORDS: Fenton reaction, OxyR, ferritins, iron-sulfur clusters
ABSTRACT
Hydrogen peroxide (H2O2) is formed in natural environments by both biotic and abiotic processes. It easily enters the cytoplasms of microorganisms, where it can disrupt growth by inactivating iron-dependent enzymes. It also reacts with the intracellular iron pool, generating hydroxyl radicals that can lethally damage DNA. Therefore, virtually all bacteria possess H2O2-responsive transcription factors that control defensive regulons. These typically include catalases and peroxidases that scavenge H2O2. Another common component is the miniferritin Dps, which sequesters loose iron and thereby suppresses hydroxyl-radical formation. In this study, we determined that Escherichia coli also induces the ClpS and ClpA proteins of the ClpSAP protease complex. Mutants that lack this protease, plus its partner, ClpXP protease, cannot grow when H2O2 levels rise. The growth defect was traced to the inactivity of dehydratases in the pathway of branched-chain amino acid synthesis. These enzymes rely on a solvent-exposed [4Fe-4S] cluster that H2O2 degrades. In a typical cell the cluster is continuously repaired, but in the clpSA clpX mutant the repair process is defective. We determined that this disability is due to an excessively small iron pool, apparently due to the oversequestration of iron by Dps. Dps was previously identified as a substrate of both the ClpSAP and ClpXP proteases, and in their absence its levels are unusually high. The implication is that the stress response to H2O2 has evolved to strike a careful balance, diminishing iron pools enough to protect the DNA but keeping them substantial enough that critical iron-dependent enzymes can be repaired.
IMPORTANCE Hydrogen peroxide mediates the toxicity of phagocytes, lactic acid bacteria, redox-cycling antibiotics, and photochemistry. The underlying mechanisms all involve its reaction with iron atoms, whether in enzymes or on the surface of DNA. Accordingly, when bacteria perceive toxic H2O2, they activate defensive tactics that are focused on iron metabolism. In this study, we identify a conundrum: DNA is best protected by the removal of iron from the cytoplasm, but this action impairs the ability of the cell to reactivate its iron-dependent enzymes. The actions of the Clp proteins appear to hedge against the oversequestration of iron by the miniferritin Dps. This buffering effect is important, because E. coli seeks not just to survive H2O2 but to grow in its presence.
INTRODUCTION
Contemporary organisms inherited their biochemical pathways from ancestors that evolved in an anoxic, iron-rich world (1). Those primordial organisms selected iron to be an enzyme cofactor because it is facile at both redox and ligand exchange processes. However, in contemporary oxic environments, this reliance upon iron-based enzymes makes cells vulnerable to reactive oxygen species (ROS). When intracellular oxygen collides with redox cofactors, electrons can be adventitiously transferred. If a single electron is transferred, superoxide is formed. If two electrons are transferred, hydrogen peroxide is produced. Both species can disrupt iron-dependent enzymes (2).
The toxicity of these oxidants was first inferred from the chance discoveries that organisms contain enzymes dedicated to scavenging them (3, 4). The model bacterium Escherichia coli contains two superoxide dismutases (MnSOD and FeSOD) in its cytoplasm and one (CuZnSOD) in its periplasm. The cytoplasm also includes an NADH peroxidase (AhpCF) and two catalases, KatG and KatE, which degrade H2O2. The importance of these enzymes was established by genetic studies. Mutants that lack either superoxide or H2O2 scavengers grow at wild-type rates under anoxic conditions, but in oxic media they struggle to synthesize branched-chain and aromatic amino acids. They also cannot catabolize any carbon source, such as acetate, that is processed by the tricarboxylic acid (TCA) cycle (5–8).
Biochemical studies uncovered the molecular bases of these phenotypes. Both O2− and H2O2 bind and univalently oxidize enzymatic iron cofactors that are accessible to solvent. The branched-chain biosynthetic and TCA cycle defects result when these oxidants oxidize the [4Fe-4S] clusters of dehydratases to an unstable [4Fe-4S]3+ valence; that species quickly disintegrates to a [3Fe-4S]+ form that lacks the critical substrate-binding iron atom (9). The aromatic biosynthetic problem ensues when O2− or H2O2 oxidizes the Fe(II) cofactor of the first enzyme in that pathway, DAHP synthase. The resultant Fe(III) atom dissociates, inactivating the enzyme. A variety of other mononuclear enzymes suffer the same fate (10, 11).
The basal level of scavenging enzymes keeps endogenous O2− and H2O2 levels low enough that these oxidation events are rare and the enzymes remain functional (2). However, a problem arises when environmental ROS are present. They are formed in habitats by a number of mechanisms: in reactions between sulfur and oxygen at oxic-anoxic interfaces, through H2O2 excretion by lactic acid bacteria, and as products of the oxidative burst of macrophages. Superoxide is an anion and cannot penetrate membranes (12, 13). However, H2O2 is small and uncharged, and it diffuses into cells (14). When it does so, it can push intracellular H2O2 concentrations into the micromolar range, which is enough to create the metabolic problems described above.
Even more ominously, H2O2 can also react with the cytoplasmic pool of loose iron whose physiological purpose is to metallate nascent metalloenzymes. This iron pool, functionally defined as the iron that can be quickly captured by the cell-permeable chelator desferrioxamine, ranges from ∼15 to 50 μm, depending upon the iron content of the medium (15–17). The Fenton reaction between Fe(II) and H2O2 generates hydroxyl radicals that damage DNA and other biomolecules (18). The DNA damage imperils the survival of the cell. Both excision and recombinational repair pathways are required for H2O2-stressed cells to remain viable (19, 20).
Because of these threats, most microbes respond to environmental H2O2 by activating a suite of defensive strategies. E. coli senses cytoplasmic H2O2 when it creates an activating disulfide bond in the OxyR transcription factor (21). OxyR is activated by as little as 0.2 μM cytoplasmic H2O2 (14); it then stimulates the expression of an extensive defensive regulon. Its strong induction of NADH peroxidase (AhpCF) and catalase (KatG) serves to push down the cytoplasmic H2O2 concentration (22). Dps, a miniferritin, sequesters loose iron (23–26); simultaneously, the induction of the Fur repressor inhibits the synthesis of iron importers (27, 28). Together, these actions suppress the rate of DNA damage. As the level of iron falls, the induction of MntH, a manganese importer, enables Fe(II)-based mononuclear enzymes to become metallated by manganese instead of iron; because manganese does not react with H2O2, this shift allows these enzymes to maintain their activity (11, 29), and induction of the suf operon (sufABCDSE) allows the synthesis of an iron-sulfur assembly system that supplants the housekeeping Isc system (30). The Suf system appears to be more resistant to H2O2, and it repairs the H2O2-damaged clusters that have fully disintegrated (31).
In this study, we pursued the observation that the clpSA operon is also induced during H2O2 stress. OxyR was confirmed to be its activator. We found that, in the face of iron restriction by Dps and Fur, the ClpSAP and ClpXP complexes provide a countervailing effect that helps maintain a fine balance between too much and too little intracellular iron. By doing so, they ensure that iron pools remain large enough to reactivate the damaged iron-sulfur clusters of key enzymes.
RESULTS
During H2O2 stress, OxyR induces the clpSA operon.
Our laboratory has performed transcriptomic analysis of Hpx− (catalase/peroxidase-deficient) strains (32) to identify the genes that are important when the cell experiences H2O2 stress. When these cells are aerated, the intracellular level of H2O2 rises to about 1 μm. This concentration can induce members of the OxyR, Fur, and IscR regulons. For context, the set point for OxyR activation appears to be about 0.2 μm cytoplasmic H2O2, and 0.5 μm is sufficient to impair some cell processes (2). Thus, this dose comprises a real stress to the cell.
The data set shows 4-fold increases in the levels of clpS and clpA mRNA in the aerated Hpx− mutants compared to those of wild-type cells. These two genes form an operon (Fig. 1A) and encode the adaptor proteins that facilitate protein degradation by the ClpSAP protease (33). We inserted a clpS′-lacZ fusion into the λ attachment site of the Hpx− strain and observed an approximately 10-fold induction when the bacterium was aerated (Fig. 1B). No induction occurred in the scavenger-proficient strain, confirming that the trigger was endogenous H2O2 (Fig. 1C).
FIG 1.

OxyR transcription factor drives expression of clpSA during H2O2 stress. (A) Genomic context of clpSA in E. coli (19.9 centisomes). RNA-Seq data, analyzed by Rockhopper, indicated that clpSA is an operon. (B) Induction of the clpS′-lacZ transcriptional fusion in Hpx− cells (ASE155) after aeration began at time zero. (C) Expression of the clpS′-lacZ fusion in WT (ASE149) and Hpx− (ASE155) cultures grown for at least 4 generations in anoxic (−O2) or oxic (+O2) medium. In this and other figures, error bars represent the standard errors of the means. (D, left) Aeration (2 h) does not induce fusion in an Hpx− strain that lacks OxyR. (Right) The fusion is induced in wild-type cells if they express a constitutively activated form of OxyR (oxyR-A233V). Strains: ASE157, ASE173, and ASE175.
The same induction occurred in Hpx− Δfur and Hpx− ΔiscR strains (see Fig. S1 in the supplemental material), showing that these transcription factors did not mediate the effect. In contrast, no induction occurred in Hpx− ΔoxyR mutants (Fig. 1D). Further, the fusion was induced even in unstressed (scavenger-proficient) cells if they were transformed with a plasmid that encodes a constitutively active OxyR protein. Together, these results show that OxyR is both necessary and sufficient for H2O2-driven induction of clpSA.
The clpPX operon is not induced during H2O2 stress.
The Clp complexes consist of a barrel-shaped heptamer of ClpP that is capped by hexamers of the AAA+ proteins ClpX or ClpSA (34). The latter hexamers bind distinct recognition motifs in their substrate proteins and, in an ATP-driven process, denature those proteins and thread them into the protease core, where they are degraded. ClpSA and ClpX are also regarded as chaperones, because when they are dissociated from ClpP they can unfold and refold the same substrates (33, 35–39). Because the best-characterized role of ClpSA is to feed substrates to ClpP, we anticipated that during H2O2 stress OxyR might also induce clpP. ClpP is encoded by the clpPX operon, which is unlinked to clpSA (Fig. 2A). However, our transcriptome sequencing (RNA-Seq) data indicated that in the Hpx− strain the clpP and clpX mRNAs both remained at constitutive levels. A clpPX′-lacZ transcriptional fusion confirmed the absence of induction (Fig. 2B).
FIG 2.

clxPX operon does not respond to H2O2 stress. (A) Genomic context of clpPX in E. coli (9.84 centisomes). RNA-Seq data indicated that clpPX is an operon. (B) Expression of the clpPX′-lacZ fusion in WT (ASE252) and Hpx− (ASE254) strains grown in anoxic (−O2) or oxic (+O2) medium.
In an older microarray study, Zheng et al. reported an 11-fold induction of clpS (then denoted yljA) when 1 mM H2O2 was added to cultures of wild-type cells (40). Computational analysis predicted an OxyR binding site immediately upstream of the clpS promoter (41). Those data are congruent with ours. However, they also observed a 5-fold induction of clpS in a ΔoxyR strain. Further, moderate induction of clpA, clpX, and clpP transcripts also occurred in both the wild-type cell and the ΔoxyR mutant. A key difference between our experiments and theirs may be the dose that they used. One millimolar H2O2 easily saturates NADH peroxidase (AhpCF), the primary scavenger in the cell (14), and, thus, releases 1 mM H2O2 to the cytoplasm, about 1,000 times the dose that pertained to our Hpx− experiment. Millimolar H2O2 is enough to cause direct protein damage through sulfhydryl oxidation (42). In that situation, it is plausible that protein quality control mechanisms trigger unknown, OxyR-independent mechanisms of clpSA and clpPX induction. In pilot experiments, we did not observe induction of clpS′-lacZ (<1.5-fold) when 1 mM H2O2 was added to the oxyR mutant. We did not pursue this issue further, as in natural environments, even in phagocytes, it is unlikely that H2O2 concentrations exceed 10 μm (43, 44).
The clpSA and clpPX operons are both important in enabling E. coli to grow during H2O2 stress.
The fact that OxyR induces clpSA implies that these proteins serve a purpose in resisting H2O2 stress. Since ClpSA and ClpX might have overlapping functions, we deleted both members of the protease family and tested the growth of ΔclpSA ΔclpPX cells in wild-type and Hpx− backgrounds. Because complex media can obscure blocks in catabolic and biosynthetic pathways, most of the growth defects associated with H2O2 stress have been discovered in minimal media. In minimal glucose medium, the scavenger-proficient ΔclpSA ΔclpPX (unstressed) cells did not exhibit a significant growth defect (Fig. 3A). However, when scavenger-deficient Hpx− ΔclpSA ΔclpPX cells were diluted from anoxic to oxic media, their growth rate diminished, and when low-micromolar H2O2 was added, they struggled to grow at all (Fig. 3B). Thus, this phenotype emerges only when the cytoplasm experiences H2O2 stress.
FIG 3.

ΔclpSA ΔclpPX mutant is very sensitive to H2O2. (A) Growth of Clp+ and ΔclpSA ΔclpPX strains in an unstressed Hpx+ background. Strains: MG1655 and ASE217. (B) Growth of Clp+ and ΔclpSA ΔclpPX strains in an Hpx− background with 4 μM H2O2. Anaerobically grown exponential-phase cultures were diluted into aerobic medium containing H2O2 at time zero. Curves are representative of at least 3 experiments.
These experiments were repeated with Hpx− mutants lacking the specific protease components. Upon aeration into medium containing micromolar H2O2, the Hpx− ΔclpSA, Hpx− ΔclpX, and Hpx− ΔclpP strains each grew more poorly than Hpx− cells (Fig. 4). The transient phenotype of the Hpx− clpSA mutant is statistically robust (Fig. S2). We realized that the experimental design did not enable us to observe the impact of clpSA induction prior to acute H2O2 exposure. To address this shortcoming, cells were aerated and grown for four generations before the addition of exogenous H2O2, an arrangement that allowed the induction of the OxyR regulon before the greater stress was imposed. Under these conditions, the Hpx− ΔclpSA cells showed a severe growth defect but the Hpx− ΔclpX strain did not (Fig. 5). Together, the data suggest that when cells first encounter H2O2, the ClpX protein plays an important defensive role, but it is usurped by ClpSA during protracted stress.
FIG 4.

ClpSA, ClpX, and ClpP each are important during H2O2 stress. Anaerobically grown cultures of Hpx− strains were diluted into aerobic medium containing H2O2 at time zero. Strains: LC106, ASE11, ASE7, and ASE67. Curves are representative of at least 3 experiments. The transient phenotype of the Hpx− clpSA mutant is statistically robust (Fig. S2).
FIG 5.

When Hpx− strains are aerated prior to H2O2 addition, ClpSA but not ClpX is important for continued growth. Cultures were grown in aerated medium from an OD600 of 0.005 to 0.1. They were then (time zero) diluted into fresh medium, and 8 μM H2O2 was added. The superior growth of the ΔclpX strain is consistently observed. Strains: LC106, ASE11, and ASE7.
Li et al. determined the whole-cell ribosomal profile of log-phase E. coli in analogous minimal glucose medium (45). Their data reported the copies of various Clp proteins per cell: ClpS (514 copies/cell), ClpA (989), ClpP (5,854), and ClpX (6,334). Therefore, the 5- to 10-fold induction of ClpS and ClpA during H2O2 stress would cause these two proteins to become competitive with ClpX for binding ClpP. In principle, that effect could help the ClpSA adapters to lessen the activity of ClpXP protease, or it could enable ClpSA to provide a ClpP-independent (un)foldase function.
We found that Hpx− ΔclpSA ΔclpX cells exhibited a more severe growth phenotype than the Hpx− ΔclpX mutants, indicating that the value of clpSA induction is not simply to deactivate the ClpXP protease (Fig. 6). Strikingly, Hpx− ΔclpP cells grow substantially better than do Hpx− ΔclpSA ΔclpPX mutants, implying that ClpSA and/or ClpX do indeed supply protective functions in the absence of ClpP. However, the defect of the clpP mutant cannot be restored by a clpX-expressing plasmid, which suggests that ClpP also provides some authentic protection (Fig. S3). These comparative phenotypes were reproducible, recurring in rebuilt strains no matter the order in which mutations were introduced. In sum, ClpSA and ClpX seem to provide both ClpP protease-dependent and -independent benefits to H2O2-stressed cells.
FIG 6.
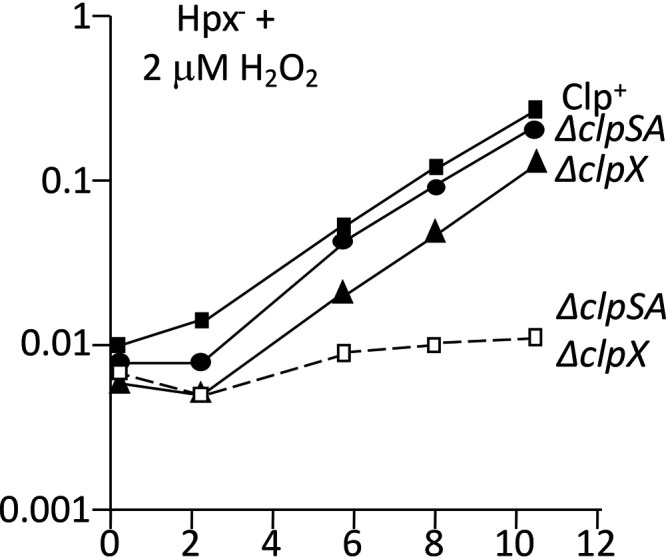
ClpSA and ClpX provide additive effects. Anaerobic cultures were diluted into aerobic medium containing 2 μM H2O2 at time zero. Strains: LC106, ASE11, ASE7, and ASE21.
Clp proteins facilitate the repair of damaged iron-sulfur clusters.
To simplify our analysis, we decided to focus on the behavior of cells when they were first stressed with H2O2 upon aeration. While H2O2 can damage DNA, the poisoned Hpx− ΔclpSA ΔclpX cells did not suffer any loss in cell viability (Fig. S4). Their failure to grow, then, most closely resembled the growth defects that occur when H2O2 inactivates enzymes in biosynthetic pathways. Indeed, although the Hpx− ΔclpSA ΔclpX strain grew poorly in H2O2-supplemented minimal medium, it could still grow in a rich medium, which provides biosynthetic end products (Fig. S5). Specific additions revealed that the growth defect was largely suppressed when the medium was supplemented with isoleucine, leucine, and valine (Fig. 7). This pathway is hypersensitive to H2O2, which can damage the catalytic [4Fe-4S] clusters in dehydratases (8). Branched-chain synthesis requires the function of two of these enzymes, isopropylmalate isomerase (IPMI) and dihydroxyacid dehydratase (DHAD). When Hpx− cells are exposed to excess H2O2, dehydratase activities dwindle. The OxyR response helps to alleviate this problem by inducing the Suf machinery (30, 31). The Suf system replaces damaged iron-sulfur clusters.
FIG 7.

Branched-chain amino acids suppress the H2O2 sensitivity of ΔclpSA ΔclpX cells. Anaerobic cultures were aerated at time zero in medium containing 8 μM H2O2. Where indicated, 0.5 mM Ile, Leu, and Val were supplied. Strains: LC106 and ASE21.
We initially considered the possibility that Clp protease activities enable amino acids to be harvested from dispensable proteins. Such a stop-gap measure would enable the induction of OxyR-driven proteins, such as Suf, during an emergency in which branched-chain amino acid synthesis was blocked. However, we did not find any evidence that ClpSA and ClpX help wild-type (Hpx+) cells to tolerate abrupt nutritional downshifts from rich to minimal media, which can create a similar biosynthetic crisis (Fig. S6). Further, in Hpx− cells the ΔclpSA ΔclpPX defect occurred independently of the Suf system, showing that the benefit of the Clp proteins was not simply to enable Suf induction or to assist its activity (Fig. S7).
The key observation was that in Hpx− Clp+ cells the activity of IPMI rebounded over the course of protracted H2O2 stress, whereas the activity remained low in the Hpx− ΔclpSA ΔclpPX mutant (Fig. 8A). Thus, the Clp proteins somehow help to restore the enzyme activity. We also confirmed that the IPMI activity of the Hpx− ΔclpSA ΔclpPX double mutant is lower than that of the Hpx− ΔclpSA and Hpx− ΔclpX single mutants, indicating that both sets of adapter proteins are important in maintaining the activity of IPMI (Fig. 8B). This pattern matches that of the overall growth defect.
FIG 8.

Clp proteins preserve the activity of isopropylmalate isomerase during H2O2 stress by enabling the repair of its oxidized iron-sulfur cluster. (A) The activity of IPMI was tracked after aeration of Hpx− and Hpx− ΔclpSA ΔclpX strains. (B) The IPMI activities of the various Hpx− mutants were determined under anaerobic conditions and after 4 h of aeration. Both ClpSA and ClpX can support enzyme activity. (C) Extracts were prepared from Hpx− Clp+ and Hpx− ΔclpSA ΔclpX strains under anaerobic conditions and after 4 h of aeration. Where indicated, extracts were treated with iron-dithiothreitol or iron-dithiothreitol-IscS-cysteine prior to assay. Successful reactivation by Fe/DTT indicates the clusters of the inactive enzymes were in the [3Fe-4S]+ state. Strains: LC106, ASE7, ASE11, and ASE125.
We characterized the status of the unrepaired clusters to discover which step of the repair process is deficient in ΔclpSA ΔclpPX mutants. When H2O2 reacts with the catalytic iron atom of the [4Fe-4S]2+ cluster in IPMI, the cluster is quickly converted to a [3Fe-4S]+ form (8). This form can be repaired by the addition of an electron and an Fe(II) atom; this process occurs in vivo on a scale of minutes. Occasionally, the [3Fe-4S]+ IPMI cluster disintegrates further, leaving behind an apoenzyme. Apoenzymes are repaired by the Suf system, which transfers to them a [4Fe-4S]2+ cluster that it first constructs on a scaffold protein. For any repair to occur, the damaged protein itself must be stable rather than aggregating or being degraded by general proteases.
The status of IPMI was tested by reactivating the enzyme in vitro. In cell extracts, the [3Fe-4S]+ form can be restored to the active [4Fe-4S]2+ state by simple incubation with ferrous iron and dithiothreitol (DTT). In contrast, reactivation of the apoenzyme form requires the addition of ferrous iron, cysteine, and IscS protein, which extracts sulfur atoms from cysteine and transfers them to the nascent cluster. We prepared cell extracts of Hpx− and Hpx− ΔclpSA ΔclpPX cells after they had been aerated for 4 h. Hpx− cell extracts exhibited approximately as much activity as did extracts from anoxic cells, and the activity did not increase upon incubation with either iron-dithiothreitol or iron-dithiothreitol-IscS-cysteine. These data indicate that in Clp-proficient cells, virtually all IPMI was present as a functional holoenzyme (Fig. 8C). Strikingly, the low IPMI activity of Hpx− ΔclpSA ΔclpPX extracts could be fully restored to the level of unstressed cells by the addition of iron/dithiothreitol. IscS-cysteine provided no further benefit. We conclude that the very low IPMI activity in this mutant is due to the accumulation of its cluster in the [3Fe-4S]+ state. The implication is that the Clp complexes somehow enable the reduction and/or remetallation of this partial cluster. The Clp proteins were evidently uninvolved in the stabilization or Suf-mediated reactivation of apoprotein.
Clp proteins increase intracellular free iron pools.
Calculations suggest that in wild-type cells, endogenous superoxide and H2O2 oxidize dehydratase cluster with a half time of about 30 min (2). Therefore, the reactivation of [3Fe-4S]+ clusters is expected to be a routine event for dehydratases, and their steady-state activities represent the dynamic equilibrium between damage and repair. Despite extensive effort, the research community has not identified any proteins that are involved in the repair process. By analogy with cluster reconstruction in extracts, it is possible that repair in vivo is solely a chemical process that relies upon intracellular Fe(II) pools and reductants.
Fortuitously, another project in our laboratory presented a clue as to the role of the Clp proteins. It is known that cysteine-rich cells are extremely vulnerable to H2O2. The lethal effects of H2O2 result from its reaction with the intracellular pool of loose iron; this Fenton reaction generates hydroxyl radicals that damage DNA according to the following reactions:
When cysteine is available, it quickly recycles the Fe(III) back to Fe(II) according to the following reaction:
(46). In this way each iron atom turns over many times, and the amount of DNA damage is amplified.
We performed transposon mutagenesis, and from 10,000 mutants we isolated ones that were unusually resistant to H2O2 exposure. Our intent was to identify mutations that affected the cysteine pools, but instead we recovered strains with insertions in clpX and clpP. The phenotype was reproduced by precise deletions in these genes (Fig. 9A). Relief from H2O2 lethality was also observed under conditions in which cysteine was not involved in the killing process (Fig. S8). We inferred that mutations in clpX and clpP are likely to diminish the intracellular pool of loose iron, thereby minimizing the yield of hydroxyl radicals.
FIG 9.

Clp proteins sustain the intracellular pools of free iron during H2O2 stress. (A) The ClpX protein increases the vulnerability of cells to oxidative DNA damage. Cells were aerated for 2 h and then exposed to cysteine and 2.5 mM H2O2. After 2 min of H2O2, cell survival was determined by colony formation. The combination of cysteine and millimolar H2O2 triggers the production of hydroxyl radicals by cellular pools of loose iron. The dashed line represents 100% survival. (B) The Clp proteins increase the intracellular pool of free iron that can be detected by EPR. Hpx− cells were aerated for 2 h before measurements. (C) The expression of the Fur:Fe(II)-repressed iucC-lacZ fusion was determined after 2 h of aeration. Elevated expression indicates that the intracellular loose-iron pools are inadequate for full protein metallation. Strains: LC106, ASE7, ASE11, ASE21, ASE67, ASE119, ASE125, MS100, ASE412, MS103, and ASE425.
The diminution of the iron pool could also be detected by monitoring the expression of the Fur regulon, which is repressed when intracellular iron pools are adequate. This regulon is largely turned off when wild-type cells grow in our glucose medium. Slight derepression occurs in Hpx− cells, but this effect becomes quite large if clpX is additionally deleted (Fig. 9C). Thus, the iron pool falls off enough that the cell perceives itself as being iron deficient.
To precisely quantify the iron pool, we compared the electron paramagnetic resonance (EPR) signals of Hpx− and Hpx− ΔclpSA ΔclpPX cells. This method visualizes the intracellular iron atoms that can be quickly captured by desferrioxamine, a cell-permeable iron chelator (17). Desferrioxamine does not extract iron from enzymes, so the signal is a measure of the loose iron pool. The EPR signal was 3-fold higher in Hpx− cells than in the isogenic ΔclpSA ΔclpPX strain. This result confirms that the Clp proteins ensure that intracellular iron pools remain large during periods of H2O2 stress (Fig. 9B). Thus, the diminished pool in the mutant strain has the effect of lowering the rate of DNA damage, but it also decreases the ability of the cell to remetallate its [3Fe-4S]+ dehydratase clusters.
Investigating the connection between Clp proteins and the loose-iron pool.
When H2O2 activates the OxyR transcription factor, the dps gene is induced (23). In our RNA-seq data, the dps mRNA is elevated 9-fold above that of unstressed cells (32). Dps is a miniferritin that sequesters loose iron in a ferric hydroxide crystal; it uses H2O2 as an oxidant so that Fe(II) can be stored (47). This action greatly diminishes the amount of hydroxyl radicals that are made in H2O2-stressed cells, thereby minimizing covalent damage to DNA and to proteins (26, 48, 49).
Stephani et al. demonstrated that ClpXP proteolyzes Dps in exponentially growing cells (50). The level of Dps is high in clpPX mutants. This relationship suggested that the elevated levels of Dps in clpPX mutants explains the unusually small loose-iron pool and the resultant deficiency in cluster repair. To probe further, we tested the sensitivity of dps mutants to H2O2 killing. These mutants were far more sensitive than wild-type cells, suggesting an enlarged iron pool during the period of H2O2 exposure (Fig. 10). In this background, the mutation of clpX had only a small impact on H2O2 sensitivity, in contrast to the large effect that is observed in Dps-proficient cells. These data suggest that an important action of ClpX during protracted stress is to ensure that Dps does not too completely sequester iron. One would then predict that the cluster repair defects of Hpx− ΔclpSA ΔclpPX cells would be suppressed by the addition of a mutation in dps. Unfortunately, this experiment is not possible, as dps null mutations block the growth of Hpx− strains due to profuse DNA damage (26).
FIG 10.
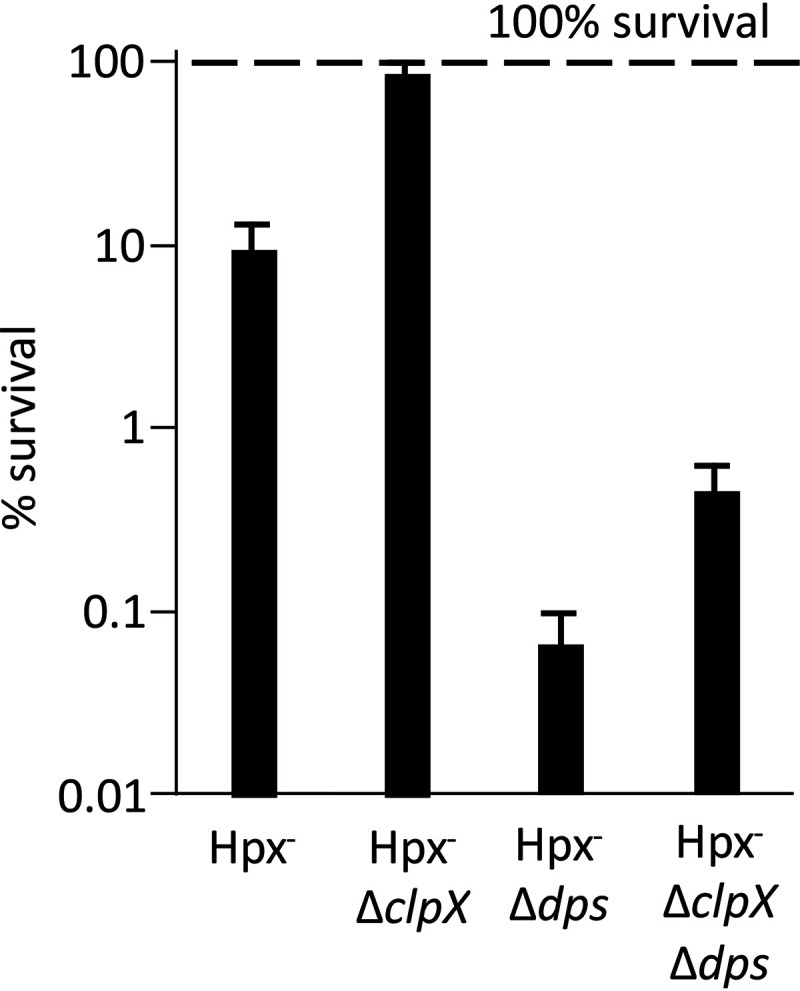
ClpX apparently affects iron pools through interactions with Dps. Cells were aerated for 2 h and then exposed to cysteine and 2.5 mM H2O2, as shown in Fig. 9A. A defect in iron sequestration by Dps elevates sensitivity to DNA damage. A clpX mutation suppresses the killing rate by 95% in a Dps+ strain but only marginally (25%) in a dps mutant. Strains: LC106, ASE7, SP66, and ASE117.
We also tested whether the pace of Fenton chemistry is suppressed by ferritin or bacterioferritin, the other two iron storage proteins of E. coli. We did not see any difference in cell killing in these mutants compared to that of their Hpx− parent (Fig. S9).
The Hpx− ΔclpSA ΔclpPX mutant is hypersensitive to manganese toxicity.
During H2O2 stress the OxyR transcription factor strongly induces the synthesis of MntH, the sole manganese importer of E. coli (29). In Hpx− strains the transcription of mntH rises sharply, and the intracellular level of manganese increases 10-fold (49). The combination of manganese import and iron sequestration allows manganese to replace iron in mononuclear enzymes, thereby ensuring their continued activity. However, manganese can compete with iron for other processes that require iron, including heme synthesis and potentially iron-sulfur cluster assembly; therefore, an excessively high manganese-to-iron ratio can inhibit growth (51). MntH-dependent manganese import was limited in our standard medium, which contains only ∼0.3 μm manganese. The addition of 4 μm manganese to the medium improved the growth of Hpx− Clp+ cells, as expected, but it created a growth defect in Hpx− ΔclpSA ΔclpPX cells (Fig. 11). Growth was restored when the medium was supplemented with branched-chain amino acids. These data suggest that when the iron pool is abnormally low, as it is in Hpx− Clp− cells, the loss of metal balance causes imported manganese to interfere with the repair of the damaged clusters in this pathway.
FIG 11.

Clp proteins are needed to avoid manganese toxicity during H2O2 stress. Anaerobic cultures were diluted into aerobic medium at time zero, and 4 μM manganese was added. The addition of branched-chain amino acids (ILV) suppressed the manganese-dependent growth defect. Growth inhibition by manganese was not observed in Hpx+ or Clp+ strains (Fig. S10). Strain: ASE125.
DISCUSSION
Intracellular iron pools pose a dilemma during H2O2 stress.
Oxidative stress is a pathology of iron metabolism, driven by the ability of ROS to bind and oxidize ferrous iron. When H2O2 inactivates iron-sulfur and mononuclear iron enzymes, it causes multiple pathway failures; when it reacts with DNA-bound iron, it generates mutagenic and even lethal DNA lesions. Air-tolerant bacteria contain enough scavenging activity that endogenous ROS are kept at low steady-state levels, but they are still vulnerable if H2O2 accumulates in the environment and diffuses into the cell. As little as 0.2 μM extracellular H2O2 creates an influx rate that exceeds the rate of endogenous H2O2 production, and 3 μM extracellular H2O2 generates enough stress that E. coli activates its OxyR response (52).
The greatest threat is that incoming H2O2 will create enough DNA damage that the cell dies. The amount of DNA damage depends directly upon the amount of loose iron inside the cell, and, unfortunately, stress threatens to further enlarge this pool in two ways. When oxidants damage iron enzymes, enough iron is released that the pool swells severalfold (15). Further, H2O2 oxidizes the Fe(II) cofactor of Fur, thereby deactivating this repressor and causing the inappropriate synthesis of iron-import systems (28). Thus, the OxyR system has evolved to shrink the iron pool in several ways. It induces the synthesis of the Fur protein itself, which, by sheer quantity, helps to restore repression of its regulon. It turns on the synthesis of YaaA, a protein whose molecular mechanism is unknown but which somehow diminishes the iron pool (Fig. 12B) (16). Most importantly, OxyR induces the synthesis of the specialized miniferritin Dps. Dps and its homologs, such as MrgA, in Gram-positive bacteria, seem to be universal members of H2O2 response systems (53). Dps sequesters enough iron that bacteria remain viable even if tens of micromolar H2O2 perfuse the E. coli cytoplasm. Without it, even 1 μM is lethal (26). This amount is very small; less, in fact, than what is contained in standard LB medium, which is why oxyR and dps mutants cannot form isolated colonies on conventional plates (52, 54).
FIG 12.

Model that integrates ClpX and ClpSA into the response to H2O2 stress. (A) Iron pools are a focus of how OxyR equips E. coli to cope with H2O2 stress. Oxidative enzyme damage releases iron, which catalyzes DNA oxidation. Proteins in blue are induced by OxyR. Dps, YaaA, and Fur collaborate to lower iron levels. MntH, Suf, and HemH ensure the continued activation of mononuclear, iron-sulfur, and heme enzymes when iron is scant. Clp proteins moderate iron sequestration by Dps so that damaged iron-sulfur clusters can be repaired. (B) Possible mechanism by which ClpX(P) and ClpSA(P) ensure Fe sufficiency during H2O2 stress. Proteins in blue are induced by OxyR. Nascent Dps is processed by protein deformylase (PDF) and methionine amino peptidase (MAP) and more slowly by an unknown protease. Using H2O2 as a coreactant, Dps stores iron. ClpX disassembles and/or degrades the longer form of Dps while ClpSA acts on the shorter form, thereby limiting iron sequestration. Loose iron is essential for the synthesis and repair of enzymatic cofactors. PDF and MAP are mononuclear Fe(II) enzymes that are inactivated by H2O2 but later reactivated by thioredoxin (Trx) and manganese imported by MntH. We speculate that fMet-Dps accumulates when PDF and MAP are damaged; this form does not present a recognition tag for ClpX or ClpSA. PDF and MAP activities are restored by thiol reduction and manganese insertion.
The goal of the OxyR system is to enable E. coli not just to survive H2O2 but to grow in its presence. Indeed, OxyR even induces a periplasmic cytochrome c peroxidase that allows the cell to employ extracellular H2O2 as a terminal oxidant for respiration (55). However, if the cell is to grow, it must be able to metallate its ca. 200 iron-requiring enzymes (56), a task that would become difficult as Dps, Fur, and YaaA decrease the size of the loose-iron pool. Accordingly, several members of the OxyR regulon help to solve this problem. The induction of the MntH manganese importer allows manganese to supplant iron in mononuclear enzymes (11). The induced Suf system is better than the housekeeping Isc system at building iron-sulfur clusters when iron pools are very low (57), and the induction of ferrochelatase boosts the ability of the cell to insert scarce iron into porphyrins and thereby ensure continued heme synthesis (32). Thus, the OxyR response includes secondary adjustments, which allow the cell to continue charging nascent enzymes with the three basic types of iron cofactors.
The present work shows that these adaptations still fall short of addressing one special problem, the repair of damaged iron-sulfur clusters. The dehydratases that rely on these clusters are involved in key pathways, the TCA cycle, the catabolism of preferred carbon sources like serine and gluconate, and the synthesis of branched-chain amino acids; thus, it is crucial that repair occurs if the cell is to continue to grow in H2O2-containing habitats. When oxidized, these clusters accumulate in the [3Fe-4S] form and, therefore, are not substrates for the Suf and Isc systems, which assemble clusters de novo and transfer them to apoproteins. The rate of enzyme repair is limited by the size of the iron pool (58). We now see that the ClpXP and ClpSAP systems are important in ensuring that during H2O2 stress, the iron-sequestering systems do not drive iron levels so low that repair is impossible.
How do the Clp proteins sustain the iron pools?
Bacteria employ special AAA+ proteases to continuously turn over proteins whose persistence may have adverse consequences. SulA, which postpones cell septation as cells recover from DNA damage, and UmuD′, which is an error-prone polymerase that bypasses unfixed DNA lesions, are examples of protease substrates whose quick degradation allows the cell to return to normal behavior once the stress has ended (59, 60). Another example is SoxS, an inducible transcription factor that elaborates emergency responses to redox-cycling antibiotics (61).
ClpSAP and ClpXP belong to this protease class. ClpX and ClpS confer specificity, as they bind distinctive short N-terminal recognition motifs in their substrate proteins. Fishing methods have enabled workers to identify a few dozen proteins in E. coli that are substrates for them (62, 63). Three of them belong to the OxyR regulon: catalase G, which is a substrate of ClpSA; the cluster-assembly proteins SufS and SufU, which are also recognized by ClpSA; and Dps, which is uniquely bound by both ClpX and ClpSA. Our data show that the impact of Clp proteins on free-iron levels is independent of catalase G and the Suf system, but it does appear to depend on the presence of Dps. The simplest explanation, then, is that it is through their control of Dps activity that the Clp proteins ensure an adequate level of intracellular iron during H2O2 stress.
Figure 12B depicts the events involved in the posttranslational processing of Dps and how Clp proteases may integrate into our larger view of oxidative stress. In the absence of stress, Dps is expressed at a significant level, as indicated by its ability to defend the cell from a sudden bolus of H2O2 even if chloramphenicol is first added to block further induction. Upon synthesis, the formyl-methionine of the translated product is deformylated, and the terminal methionine is then removed to reveal an N-terminal sequence that is recognized by ClpX (62). Stephani et al. determined that in unstressed, log-phase cells the half-life of Dps was 10 min, but it was 4-fold longer in clpX and clpP mutants (50). Notably, some Dps is truncated by an unknown protease that removes the next five amino acid residues, including the recognition motif for ClpX binding. However, the new N-terminal sequence of this short form is recognized by ClpS (63). Thus, it is apparent how both ClpX and ClpSA can influence the cytoplasmic iron level. In growing cells, Dps(2–167) is 4-fold more abundant than Dps(6–167) (64). However, we suspect that this ratio will shift when H2O2 slows protein synthesis, and the average Dps protein is older, which may explain why ClpX initially has the larger phenotype but ClpSA is more influential after protracted stress.
When H2O2 levels rise, activated OxyR induces dps transcription by an order of magnitude. The quality of Dps that sets it apart from ferritin and bacterioferritin is that it uses H2O2 rather than oxygen as a coreactant when it transfers free ferrous iron to its internal ferric hydroxide crystal (47). This arrangement may ensure that Dps sequesters iron only during H2O2 stress; when the H2O2 level drops, Dps may stop storing it. ClpSA and ClpX can manifest chaperone activities without the involvement of ClpP, and this has prompted workers to speculate that they disassemble Dps dodecamers even without attendant proteolysis (65). We observed, in fact, that during H2O2 stress the phenotypes of clpX and clpSA exceeded that of clpP. In principle, Dps disassembly could elevate iron levels either by releasing stored iron directly or else simply by diminishing the number of Dps complexes that are actively sequestering iron. More work will be required to test the details of these ideas.
An intriguing wrinkle is that the maturation of Dps requires the actions of deformylase- and methionine-removing enzymes that both belong to the mononuclear Fe(II) family (11, 66). The deformylase is inactivated by as little as 1 μM intracellular H2O2; H2O2 directly oxidizes the iron atom to the Fe(III) form, which dissociates (11). This reaction includes the concurrent oxidation of a coordinating cysteine residue to a disulfide form, which cannot rebind metal. If these two enzymes stall, the newly induced Dps would accumulate in its full-length Dps1–167 form, which is not a substrate for either Clp complex (64). A potential upshot is that Dps will be stabilized. The retention of the Met residue is unlikely to impede the assembly of Dps into its active dodecamer structure, as the N-terminal thread is positioned outside the complex. The situation would persist until an influx of manganese reactivates the deformylase and peptidase.
If true, the scheme shows a notable parallel to the Dps (MrgA) control in Bacillus subtilis. In that bacterium, the stress response to H2O2 is controlled by the PerR repressor (53, 67). In normal iron-rich/manganese-poor cells, PerR is activated by Fe(II), and in this form it represses the synthesis of Dps. When H2O2 levels rise, the iron cofactor is oxidized, PerR loses activity, and the regulon is induced. However, in manganese-rich cells, Mn(II) occupies the metal-binding site of PerR, and the regulon, including Dps, remains repressed even if H2O2 stress is imposed. This arrangement between the metalation state of PerR and the synthesis of Dps has been interpreted as a design to ensure that H2O2-stressed cells are manganese rich. The scheme shown in Fig. 12A would achieve the same end in E. coli.
In sum, the data indicate that by sequestering iron during H2O2 stress, Dps protects DNA but potentially inhibits the repair of key iron-sulfur enzymes. The presence of the Clp complexes is needed to avoid complete iron depletion; therefore, the OxyR-driven induction of clpSA is a useful response (Fig. 12A). In the larger view, this tension between DNA protection and enzyme repair helps explain why the defensive tactics of the OxyR system are not constitutively expressed. Nevertheless, after adapting to H2O2 stress, E. coli is capable of almost full-speed growth. Evidently, it can do so only due to the iron-balancing act that is achieved by a cohort of OxyR-controlled proteins: Dps, YaaA, and Fur on the one hand and MntH, Suf, HemH, and ClpSA on the other.
MATERIALS AND METHODS
Bacterial strains.
The strains and plasmids used in this study are listed in Table S1 in the supplemental material. Gene deletions were created by the λ Red recombinase method on LB plus chloramphenicol (20 μg/ml) (68) and were confirmed by PCR analysis. Mutations were introduced into new strains by P1 transduction (69), selected on LB plus chloramphenicol (20 μg/ml), and verified by PCR analysis. When necessary, the chloramphenicol marker was removed by transformation with pCP20, followed by the removal of the temperature-sensitive plasmid. The loss of the marker was confirmed by PCR. All Hpx− strains were constructed in an anaerobic chamber to avoid selective pressure for the outgrowth of suppressors.
Growth conditions.
The standard growth medium contained minimal A salts (69) (10.5 g/liter dipotassium hydrogen phosphate, 4.5 g/liter potassium dihydrogen phosphate, 1 g/liter ammonium sulfate, and 0.5 g/liter sodium citrate, adjusted to pH 7), 0.2% glucose, 1 mM MgCl2, 5 mg/liter thiamine, and 0.5 mM histidine, phenylalanine, tryptophan, and tyrosine. Histidine was routinely added to the medium because the parent strain, MG1655, is a histidine bradytroph under anaerobic conditions; phenylalanine, tryptophan, and tyrosine were included because H2O2 poisons the pathway of aromatic amino acid synthesis (10). The media were made anoxic by autoclaving the medium components and moving them while hot into the anaerobic chamber (Coy Laboratory Products, Inc.). Thiamine, which was filter sterilized, was moved into the chamber and kept there for 2 days before use to ensure that it was anoxic. Specified l-amino acids were added at 0.5 mM.
Most experiments investigated the behavior of log-phase cells upon their initial exposure to H2O2. Exponential growth was first established under anoxic conditions to avoid the exposure and adaptation to endogenous H2O2 that would occur if the cells were aerated. The Hpx− cells were grown overnight in anoxic medium. They were then diluted to an optical density at 600 nm (OD600) of 0.005 in fresh anoxic media and were grown to an OD600 of ∼0.1. These log-phase cells then were diluted into aerobic media containing specific amounts of H2O2, and their growth was tracked. Other experiments examined the behavior of Hpx− cells that had first been cultured under oxic conditions, which establishes low-grade H2O2 stress (∼1 μM) that ensures the induction of the OxyR regulon. The cells were grown as described above to an OD600 of 0.1 anaerobically, diluted to an OD600 of 0.005 in oxic media, grown with robust shaking to an OD600 of 0.1, and then diluted again to an OD600 of 0.005 in oxic media to which a specified concentration of H2O2 had been added. Growth was then monitored.
Construction of transcriptional fusions and lacZ assays.
Promoter regions of genes were PCR amplified with primers (Table S2) and inserted into pSJ501 between KpnI and EcoRI restriction sites. The plasmid containing the transcriptional fusions were integrated at the attB site (attλ) on chromosomal DNA by pINTTS, which is a helper plasmid that carries lambda integration genes (70). Single-copy integrants were verified by PCR. The transcriptional fusions were introduced into desired strains by P1 transduction. Each fusion strain retained a working wild-type copy of the gene at its normal locus.
Gene expression under anaerobic conditions was measured by culturing the cells from an OD600 of 0.005 to an OD600 of ∼0.1 in 20 ml of anoxic growth medium in the anaerobic chamber. Expression under aerobic conditions was measured by diluting anaerobic log-phase cells to an OD600 of 0.005 in 20 ml of oxic medium and growing them to an OD600 of ∼0.1. To measure their β-galactosidase activities, cultures were then centrifuged at 7,000 × g for 5 min at 4°C, washed with ice-cold 50 mM Tris-HCl (pH 8), resuspended in 1 ml of the same buffer, and lysed by French press. The β-galactosidase was assayed at room temperature using Z buffer, 0.67 mg/ml O-nitrophenyl-β-d-galactopyranoside (ONPG) (Sigma) as a substrate, and 0.125 mg/ml extract (69). The final product, ortho-nitrophenol (ONP+), was detected at 420 nm. The molar extinction coefficient of ONP+ is 4,500 M−1 cm−1. One unit of activity was defined as the amount of enzyme that catalyzes the conversion of 1 μmol substrate/min. The total protein was measured using the Coomassie blue reagent from Thermo-Fisher.
IPMI enzyme assay.
Anaerobic IPMI activity was measured after culturing cells from an OD600 of 0.005 to an OD600 of ∼0.1 in 100 ml anoxic medium. The activity in aerated cells was measured after diluting log-phase anaerobic cells to an OD600 of 0.005 in 100 ml oxic medium and growing them for the specified time, typically to an OD600 of ∼0.1. Note that no H2O2 was added, as the phenotype was driven by endogenous H2O2 formation. In both cases, cells were harvested and activities were assayed under anoxic conditions to ensure that IPMI was not inactivated by oxidants in the lysate. The cultures were maintained in or returned to the anaerobic chamber, centrifuged at 7,000 × g for 5 min at 4°C, washed with ice-cold, anoxic 100 mM Tris-Cl (pH 7.6), and resuspended in 0.475 ml of the same cold buffer. Cell extracts were then prepared by sonication. IPMI activity was measured in the same buffer at room temperature by incubating the cell extract (protein concentration of 1.5 mg/ml) with 0.4 mM citraconic acid (Sigma) (71) in capped anoxic cuvettes, which were transferred out of the chamber to a spectrophotometer. The decrease in absorbance at 235 nm was monitored. Specific activity was calculated in terms of absorbance change per minute per milligram of protein.
The damaged [3Fe-4S] clusters of IPMI were reactivated in vitro (31) by incubating the cell extract (final concentration of 0.1 mg/ml) with 0.5 mM Fe(NH4)2(SO4)2 (Sigma) and 5 mM dithiothreitol (DTT) (Sigma) in anoxic 100 mM Tris-Cl (pH 7.6) at room temperature for 20 min. In vitro reconstitution of apoenzymes was carried out by incubating the cell extract with 0.5 mM Fe(NH4)2(SO4)2, 5 mM DTT, 2.5 mM cysteine, and 0.035 μg/ml purified IscS (0.008 U/mg) at room temperature for 2 h.
The activity of IscS was confirmed by using fumarase A-overexpressing cells (AW100/pFumA), which accumulate apo-enzyme because the native Isc system does not keep up with the rate of fumarase A synthesis. The cells were diluted to an OD600 of 0.03 in 30 ml of aerobic minimal A medium containing glucose, 0.2% Casamino Acids, 0.5 mM tryptophan, and 100 μg/ml ampicillin. The cells were grown to an OD600 of 0.5, and 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was added, followed by a 3-h incubation at 37°C. The cultures were transferred to the anaerobic chamber, centrifuged at 7,000 × g for 5 min at 4°C, washed with ice-cold anoxic 50 mM Tris-Cl (pH 7.65), and resuspended in 1 ml of the same buffer. The cell extracts were prepared by sonication. The activity of fumarase was measured by incubating the extracts at room temperature in anoxic 50 mM sodium phosphate buffer (pH 7.3) containing 0.05 M l-malate (Sigma). Specific activity was calculated in terms of absorbance change per minute per milligram of protein.
Purification of IscS.
E. coli BL21(DE3) cells containing pIsc-(His)6 were grown aerobically to an OD600 of 0.6 in 100 ml LB containing 100 μg/ml ampicillin. IPTG (1 mM) was added, and the culture was incubated at 37°C. Extracts were prepared and the overexpressed IscS-His6 was purified using His Gravitrap (GE Health Care) as described previously (31).
The desulfurase activity of IscS was assayed by measuring sulfide production under anoxic conditions (31). Purified IscS was incubated with 2.5 mM cysteine, 5 mM dithiothreitol, and 50 mM Tris-Cl–10 mM MgCl2, pH 7.65, at 37°C for 15 min. The reaction was stopped by the addition of 20% trichloroacetic acid. The precipitated proteins were removed by centrifugation, and the supernatant was incubated with 2 mM N,N-dimethyl-p-phenylenediamine (DPD) and 3 mM FeCl3 at room temperature for 30 min in the dark. FeCl3 was dissolved using 6 N HCl. The concentration of methylene blue that results from the reaction of DPD and sulfide was measured at 670 nm. The extinction coefficient is 11.8 mM−1 cm−1. One unit of activity creates 1 μmol sulfide per min. A standard curve was determined between 0.005 and 0.1 mM sodium sulfide.
Intracellular free-iron measurements.
Cells were grown in 0.5 liter oxic minimal glucose medium containing His, Phe, Trp, and Tyr to an OD600 of 0.1 and harvested by centrifugation at 7,000 × g for 5 min at 4°C. The aromatic amino acids were added, because endogenous H2O2 levels as low as 0.5 μM had been shown to poison the aromatic amino acid synthesis pathway (10). The cell pellet was resuspended in 10 ml minimal glucose medium containing His, Phe, Trp, and Tyr that was prewarmed to 37°C. The medium also contained 10 mM DETAPAC (diethylentriaminepentaacetic acid, pH 7.0) (Sigma) and 20 mM desferrioxamine (pH 8.0) (Sigma). DETAPAC blocks the further import of iron, while desferrioxamine diffuses into cells and binds unincorporated iron in an EPR-visible ferric form (17). The cells were incubated at 37°C for 15 min in a shaking water bath. The cells were then washed twice with 2 ml of ice-cold 20 mM Tris-Cl (pH 7.4). Cells were resuspended in 200 μl of ice cold 30% glycerol–20 mM Tris-Cl (pH 7.4), transferred into an EPR tube, and frozen in dry ice with ethanol. The OD600 of the final cell suspension was measured after a 1:1,000 dilution. Ferric chloride (Sigma) standards were first dissolved in water, mixed with desferrioxamine, and prepared in the same Tris buffer containing glycerol. The spectrometer settings were the following: microwave power, 10 mW; microwave frequency, 9.05 GHz; modulation amplitude, 12.5 Gauss at 100 KHz; time constant, 0.032; temperature, 15 K.
H2O2 killing assay.
The Hpx+ cells were grown in aerobic minimal A glucose medium containing 0.5 mM tryptophan, phenylalanine, tyrosine, and histidine from an OD600 of 0.005 to an OD600 of 0.1 before being treated with cysteine and H2O2 as described below. The Hpx− cells were precultured anaerobically in the same medium under anaerobic conditions to an OD600 of 0.1. The cells were then aerated for 2 h. The cells were then diluted to an OD600 of 0.025 in the same medium and incubated for 5 min with 0.5 mM cysteine. H2O2 (2.5 mM) was then added. At the indicated time points, aliquots were removed and 660 U/ml catalase (final concentration) was added to end the H2O2 stress. After serial dilution, samples were mixed with LB containing 0.8% agar and spread on LB plates. Numbers of CFU were determined after aerobic incubation at 37°C for 24 h.
To test the sensitivity to H2O2 of recA strains (17), the cells were precultured anaerobically to an OD600 of 0.1 in the medium described above. They were then diluted to an OD600 of 0.025 in the same oxic medium, and after 2 min H2O2 (2.5 mM) was added. After 3 min of H2O2 exposure, catalase was added to end the H2O2 stress. The cells were then plated on anaerobic LB plates.
H2O2 assays.
Aerobic Hpx− cells retain trace H2O2-scavenging activity due apparently to the weak ability of cytochrome oxidases to use H2O2 as a growth substrate (55). Experiments showed that clpSA clpX mutations had no effect on this residual scavenging activity. Cells were precultured from an OD600 of 0.005 to an OD600 of 0.1 in anoxic minimal A medium supplemented with 0.5 mM tryptophan, tyrosine, phenylalanine, and histidine. The cells were then diluted to an OD600 of 0.005 into aerobic medium of the same composition containing 8 μM H2O2. At intervals, 1-ml aliquots of the culture were centrifuged at 12,000 × g for 1 min, and the supernatants were transferred to fresh Eppendorf tubes and frozen on dry ice. The H2O2 concentrations of the supernatants were measured by the Amplex Red-horseradish peroxidase method (72). In the presence of horseradish peroxidase, Amplex Red reacts with hydrogen peroxide to produce resorufin, a highly fluorescent compound. The accumulation of this complex was monitored in a Shimadzu RF-Mini150 fluorometer with a 520-nm excitation filter and a 620-nm emission filter. A standard curve was generated with known amounts of H2O2.
Cell viability assay.
Anaerobic overnight cultures of the Hpx− cells were diluted to an OD600 of 0.005 in anoxic minimal A medium containing 0.5 mM tryptophan, tyrosine, phenylalanine, and histidine. The cells were grown to an OD600 of ∼0.1 and then diluted to an OD600 of 0.005 into aerobic medium of the same composition containing 8 μM H2O2. Cultures were aerated by vigorous shaking. At the indicated time points, aliquots were removed to the anaerobic chamber, and 660 U/ml catalase (final concentration) was added to end the H2O2 stress. After serial dilution, samples were mixed with anoxic LB containing 0.8% agar and spread on anoxic LB plates. Colonies were determined after incubation at 37°C for 24 h in the anaerobic chamber.
Supplementary Material
ACKNOWLEDGMENTS
We thank Mark Nilges and Toby Woods of the UIUC EPR Center for experimental support. We are grateful to Stephen Finkel, Tricia Kiley, Eric Masse, Jeff Gardner, Jim Slauch, Soojin Jang, and Joe Nielands, who provided mutants and plasmids that were critical for the generation of strains used in this study.
This work was supported by grant GM049640 from the National Institutes of Health.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Anbar AD. 2008. Elements and evolution. Science 322:1481–1483. doi: 10.1126/science.1163100. [DOI] [PubMed] [Google Scholar]
- 2.Imlay JA. 2013. The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol 11:443–454. doi: 10.1038/nrmicro3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loew O. 1900. A new enzyme of general occurrence in organismis. Science 11:701–702. doi: 10.1126/science.11.279.701. [DOI] [PubMed] [Google Scholar]
- 4.McCord J, Fridovich I. 1969. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem 244:6049–6055. [PubMed] [Google Scholar]
- 5.Carlioz A, Touati D. 1986. Isolation of superoxide dismutase mutants in Escherichia coli: is superoxide dismutase necessary for aerobic life? EMBO J 5:623–630. doi: 10.1002/j.1460-2075.1986.tb04256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuo CF, Mashino T, Fridovich I. 1987. α,β-Dihydroxyisovalerate dehydratase: a superoxide-sensitive enzyme. J Biol Chem 262:4724–4727. [PubMed] [Google Scholar]
- 7.Gardner PR, Fridovich I. 1991. Superoxide sensitivity of the Escherichia coli aconitase. J Biol Chem 266:19328–19333. [PubMed] [Google Scholar]
- 8.Jang S, Imlay JA. 2007. Micromolar intracellular hydrogen peroxide disrupts metabolism by damaging iron-sulfur enzymes. J Biol Chem 282:929–937. doi: 10.1074/jbc.M607646200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flint DH, Tuminello JF, Emptage MH. 1993. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J Biol Chem 268:22369–22376. [PubMed] [Google Scholar]
- 10.Sobota JM, Gu M, Imlay JA. 2014. Intracellular hydrogen peroxide and superoxide poison 3-deoxy-D-arabinoheptulosonate 7-phosphate synthase, the first committed enzyme in the aromatic biosynthetic pathway of Escherichia coli. J Bacteriol 196:1980–1991. doi: 10.1128/JB.01573-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anjem A, Imlay JA. 2012. Mononuclear iron enzymes are primary targets of hydrogen peroxide stress. J Biol Chem 287:15544–15556. doi: 10.1074/jbc.M111.330365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lynch R, Fridovich I. 1978. Permeation of the erythrocyte stroma by superoxide radical. J Biol Chem 253:4697–4699. [PubMed] [Google Scholar]
- 13.Korshunov SS, Imlay JA. 2002. A potential role for periplasmic superoxide dismutase in blocking the penetration of external superoxide into the cytosol of phagocytosed bacteria. Mol Microbiol 43:95–106. doi: 10.1046/j.1365-2958.2002.02719.x. [DOI] [PubMed] [Google Scholar]
- 14.Seaver LC, Imlay JA. 2001. Hydrogen peroxide fluxes and compartmentalization inside growing Escherichia coli. J Bacteriol 183:7182–7189. doi: 10.1128/JB.183.24.7182-7189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keyer K, Imlay JA. 1996. Superoxide accelerates DNA damage by elevating free-iron levels. Proc Natl Acad Sci U S A 93:13635–13640. doi: 10.1073/pnas.93.24.13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Bauer SC, Imlay JA. 2011. The YaaA protein of the Escherichia coli OxyR regulon lessens hydrogen peroxide toxicity by diminishing the amount of intracellular unincorporated iron. J Bacteriol 193:2186–2196. doi: 10.1128/JB.00001-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woodmansee AL, Imlay JA. 2002. Quantitation of intracellular free iron by electron paramagnetic resonance spectroscopy. Methods Enzymol 349:3–9. doi: 10.1016/s0076-6879(02)49316-0. [DOI] [PubMed] [Google Scholar]
- 18.Imlay JA, Chin SM, Linn S. 1988. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science 240:640–642. doi: 10.1126/science.2834821. [DOI] [PubMed] [Google Scholar]
- 19.Demple B, Halbrook J, Linn S. 1983. Escherichia coli xth mutants are hypersensitive to hydrogen peroxide. J Bacteriol 153:1079–1082. doi: 10.1128/JB.153.2.1079-1082.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carlsson J, Carpenter VS. 1980. The recA+ gene product is more important that catalase and superoxide dismutase in protecting Escherichia coli against hydrogen peroxide toxicity. J Bacteriol 142:319–321. doi: 10.1128/JB.142.1.319-321.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng M, Aslund F, Storz G. 1998. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science 279:1718–1721. doi: 10.1126/science.279.5357.1718. [DOI] [PubMed] [Google Scholar]
- 22.Jacobson FS, Morgan RW, Christman MF, Ames BN. 1989. An alkyl hydroperoxide reductase from Salmonella typhimurium involved in the defense of DNA against oxidative damage. Purification and properties. J Biol Chem 264:1488–1496. [PubMed] [Google Scholar]
- 23.Altuvia S, Almiron M, Huisman G, Kolter R, Storz G. 1994. The dps promoter is activated by OxyR during growth and by IHF and sigma S in stationary phase. Mol Microbiol 13:265–272. doi: 10.1111/j.1365-2958.1994.tb00421.x. [DOI] [PubMed] [Google Scholar]
- 24.Grant RA, Filman DJ, Finkel SE, Kolter R, Hogle JM. 1998. The crystal structure of Dps, a ferritin homolog that binds and protects DNA. Nat Struct Biol 5:294–303. doi: 10.1038/nsb0498-294. [DOI] [PubMed] [Google Scholar]
- 25.Ilari A, Ceci P, Ferrari D, Rossi G, Chiancone E. 2002. Iron incorporation into E. coli Dps gives rise to a ferritin-like microcrystalline core. J Biol Chem 277:37619–37623. doi: 10.1074/jbc.M206186200. [DOI] [PubMed] [Google Scholar]
- 26.Park S, You X, Imlay JA. 2005. Substantial DNA damage from submicromolar intracellular hydrogen peroxide detected in Hpx− mutants of Escherichia coli. Proc Natl Acad Sci U S A 102:9317–9322. doi: 10.1073/pnas.0502051102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng M, Doan B, Schneider TD, Storz G. 1999. OxyR and SoxRS regulation of fur. J Bacteriol 181:4639–4643. doi: 10.1128/JB.181.15.4639-4643.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varghese S, Wu A, Park S, Imlay KRC, Imlay JA. 2007. Submicromolar hydrogen peroxide disrupts the ability of Fur protein to control free-iron levels in Escherichia coli. Mol Microbiol 64:822–830. doi: 10.1111/j.1365-2958.2007.05701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kehres DG, Janakiraman A, Slauch JM, Maguire ME. 2002. Regulation of Salmonella enterica serovar Typhimurium mntH transcription by H2O2, Fe2+, and Mn2+. J Bacteriol 184:3151–3158. doi: 10.1128/jb.184.12.3151-3158.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JH, Yeo WS, Roe JH. 2004. Induction of the sufA operon encoding Fe-S assembly proteins by superoxide generators and hydrogen peroxide: involvement of OxyR, IHF and an unidentified oxidant-responsive factor. Mol Microbiol 51:1745–1755. doi: 10.1111/j.1365-2958.2003.03946.x. [DOI] [PubMed] [Google Scholar]
- 31.Jang S, Imlay JA. 2010. Hydrogen peroxide inactivates the Escherichia coli Isc iron-sulphur assembly system, and OxyR induces the Suf system to compensate. Mol Microbiol 78:1448–1467. doi: 10.1111/j.1365-2958.2010.07418.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mancini S, Imlay JA. 2015. The induction of two biosynthetic enzymes helps Escherichia coli sustain heme synthesis and activate catalase during hydrogen peroxide stress. Mol Microbiol 96:744–763. doi: 10.1111/mmi.12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dougan DA, Reid BG, Horwich AL, Bukau B. 2002. ClpS, a substrate modulator of the ClpAP machine. Mol Cell 9:673–683. doi: 10.1016/s1097-2765(02)00485-9. [DOI] [PubMed] [Google Scholar]
- 34.Sauer RT, Bolon DN, Burton BM, Burton RE, Flynn JM, Grant RA, Hersch GL, Joshi SA, Kenniston JA, Levchenko I, Neher SB, Oakes ES, Siddiqui SM, Wah DA, Baker TA. 2004. Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell 119:9–18. doi: 10.1016/j.cell.2004.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoskins JR, Kim SY, Wickner S. 2000. Substrate recognition by the ClpA chaperone component of ClpAP protease. J Biol Chem 275:35361–35367. doi: 10.1074/jbc.M006288200. [DOI] [PubMed] [Google Scholar]
- 36.Hoskins JR, Wickner S. 2006. Two peptide sequences can function cooperatively to facilitate binding and unfolding by ClpA and degradation by ClpAP. Proc Natl Acad Sci U S A 103:909–914. doi: 10.1073/pnas.0509154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mhammedi-Alaoui A, Pato M, Gama MJ, Toussaint A. 1994. A new component of bacteriophage Mu replicative transposition machinery: the Escherichia coli ClpX protein. Mol Microbiol 11:1109–1116. doi: 10.1111/j.1365-2958.1994.tb00387.x. [DOI] [PubMed] [Google Scholar]
- 38.Wawrzynow A, Wojtkowiak D, Marszalek J, Banecki B, Jonsen M, Graves B, Georgopoulos C, Zylicz M. 1995. The ClpX heat-shock protein of Escherichia coli, the ATP-dependent substrate specificity component of the ClpP-ClpX protease, is a novel molecular chaperone. EMBO J 14:1867–1877. doi: 10.1002/j.1460-2075.1995.tb07179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wickner S, Gottesman S, Skowyra D, Hoskins J, McKenney K, Maurizi MR. 1994. A molecular chaperone, ClpA, functions like DnaK and DnaJ. Proc Natl Acad Sci U S A 91:12218–12222. doi: 10.1073/pnas.91.25.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng M, Wang X, Templeton LJ, Smulski DR, LaRossa RA, Storz G. 2001. DNA microarray-mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J Bacteriol 183:4562–4570. doi: 10.1128/JB.183.15.4562-4570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng M, Wang X, Doan B, Lewis KA, Schneider TD, Storz G. 2001. Computation-directed identification of OxyR DNA binding sites in Escherichia coli. J Bacteriol 183:4571–4579. doi: 10.1128/JB.183.15.4571-4579.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Winterbourn CC, Metodiewa D. 1999. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med 27:322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 43.Winterbourn CC, Hampton MB, Livesey JH, Kettle AJ. 2006. Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome. Implications for microbial killing. J Biol Chem 281:39860–39869. doi: 10.1074/jbc.M605898200. [DOI] [PubMed] [Google Scholar]
- 44.Slauch JM. 2011. How does the oxidative burst of macrophages kill bacteria? Mol Microbiol 80:580–583. doi: 10.1111/j.1365-2958.2011.07612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li G-W, Burkhardt D, Gross C, Weissman JS. 2014. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 157:624–635. doi: 10.1016/j.cell.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park S, Imlay JA. 2003. High levels of intracellular cysteine promote oxidative DNA damage by driving the Fenton reaction. J Bacteriol 185:1942–1950. doi: 10.1128/JB.185.6.1942-1950.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao G, Ceci P, Ilari A, Giangiacomo L, Laue TM, Chiancone E, Chasteen ND. 2002. Iron and hydrogen peroxide detoxification properties of DNA-binding protein from starved cells. A ferritin-like DNA-binding protein of Escherichia coli. J Biol Chem 277:27689–27696. doi: 10.1074/jbc.M202094200. [DOI] [PubMed] [Google Scholar]
- 48.Martinez A, Kolter R. 1997. Protection of DNA during oxidative stress by the nonspecific DNA-binding protein Dps. J Bacteriol 179:5188–5194. doi: 10.1128/jb.179.16.5188-5194.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anjem A, Varghese S, Imlay JA. 2009. Manganese import is a key element of the OxyR response to hydrogen peroxide in Escherichia coli. Mol Microbiol 72:844–858. doi: 10.1111/j.1365-2958.2009.06699.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stephani K, Weichart D, Hengge R. 2003. Dynamic control of Dps protein levels by ClpXP and ClpAP proteases in Escherichia coli. Mol Microbiol 49:1605–1614. doi: 10.1046/j.1365-2958.2003.03644.x. [DOI] [PubMed] [Google Scholar]
- 51.Martin JE, Waters LS, Storz G, Imlay JA. 2015. The Escherichia coli small protein MntS and exporter MntP optimize the intracellular concentration of manganese. PLoS Genet 11:e1004977. doi: 10.1371/journal.pgen.1004977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li X, Imlay JA. 2018. Improved measurements of scant hydrogen peroxide enable experiments that define its threshold of toxicity for Escherichia coli. Free Radic Biol Med 120:217–227. doi: 10.1016/j.freeradbiomed.2018.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen L, Helmann JD. 1995. Bacillus subtilis MrgA is a Dps (PexB) homologue: evidence for metalloregulation of an oxidative-stress gene. Mol Microbiol 18:295–300. doi: 10.1111/j.1365-2958.1995.mmi_18020295.x. [DOI] [PubMed] [Google Scholar]
- 54.Ezraty B, Henry C, Herisse M, Denamur E, Barras F. 2014. Commercial lysogeny broth culture media and oxidative stress: a cautious tale. Free Radic Biol Med 74:245–251. doi: 10.1016/j.freeradbiomed.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 55.Khademian M, Imlay JA. 2017. Escherichia coli cytochrome c peroxidase is a respiratory oxidase that enables the use of hydrogen peroxide as a terminal electron acceptor. Proc Natl Acad Sci U S A 114:E6922–E6931. doi: 10.1073/pnas.1701587114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rocha AG, Dancis A. 2016. Life without Fe-S clusters. Mol Microbiol 99:821–826. doi: 10.1111/mmi.13273. [DOI] [PubMed] [Google Scholar]
- 57.Outten FW, Djaman O, Storz G. 2004. A suf operon requirement for Fe-S cluster assembly during iron starvation in Escherichia coli. Mol Microbiol 52:861–872. doi: 10.1111/j.1365-2958.2004.04025.x. [DOI] [PubMed] [Google Scholar]
- 58.Maringanti S, Imlay JA. 1999. An intracellular iron chelator pleiotropically suppresses enzymatic and growth defects of superoxide dismutase-deficient Escherichia coli. J Bacteriol 181:3792–3802. doi: 10.1128/JB.181.12.3792-3802.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Higashitani A, Ishii Y, Kato Y, Koriuchi K. 1997. Functional dissection of a cell-division inhibitor, SulA, of Escherichia coli and its negative regulation by Lon. Mol Gen Genet 254:351–357. doi: 10.1007/s004380050426. [DOI] [PubMed] [Google Scholar]
- 60.Frank EG, Ennis DG, Gonzalez M, Levine AS, Woodgate R. 1996. Regulation of SOS mutagenesis by proteolysis. Proc Natl Acad Sci U S A 93:10291–10296. doi: 10.1073/pnas.93.19.10291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Griffith KL, Shah IM, Wolf RE, Jr. 2004. Proteolytic degradation of Escherichia coli transcription activators Sox and MarA as the mechanism for reversing the induction of the superoxide (SoxRS) and multiple antibiotic resistance (Mar) regulons. Mol Microbiol 51:1801–1816. doi: 10.1046/j.1365-2958.2003.03952.x. [DOI] [PubMed] [Google Scholar]
- 62.Flynn JM, Neher SB, Kim Y-I, Sauer RT, Baker TA. 2003. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol Cell 11:671–683. doi: 10.1016/s1097-2765(03)00060-1. [DOI] [PubMed] [Google Scholar]
- 63.Ninnis RL, Spall SK, Talbo GH, Truscott KN, Dougan DA. 2009. Modification of PATase by L/F-transferase generates a ClpS-dependent N-end rule substrate in Escherichia coli. EMBO J 28:1732–1744. doi: 10.1038/emboj.2009.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schmidt R, Zahn R, Bukau B, Mogk A. 2009. ClpS is the recognition component for Escherichia coli substrates of the N-end rule degradation pathway. Mol Microbiol 72:506–517. doi: 10.1111/j.1365-2958.2009.06666.x. [DOI] [PubMed] [Google Scholar]
- 65.LaBreck CJ, May S, Viola MG, Conti J, Camberg JL. 2017. The protein chaperon ClpX targets native and non-native aggregated substrates for remodeling, disassembly, and degradation with ClpP. Front Mol Biosci 4:1–13. doi: 10.3389/fmolb.2017.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chai SC, Wang WL, Ye ZQ. 2008. Fe(II) is the native cofactor for Escherichia coli methionine aminopeptidase. J Biol Chem 283:26879–26885. doi: 10.1074/jbc.M804345200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee JW, Helmann JD. 2006. The PerR transcription factor senses H2O2 by metal-catalyzed histidine oxidation. Nature 440:363–367. doi: 10.1038/nature04537. [DOI] [PubMed] [Google Scholar]
- 68.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 70.Haldimann A, Wanner BL. 2001. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol 183:6384–6393. doi: 10.1128/JB.183.21.6384-6393.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kohlhaw GB. 1988. Isopropylmalate dehydratase from yeast. Methods Enzymol 166:423–429. doi: 10.1016/s0076-6879(88)66055-1. [DOI] [PubMed] [Google Scholar]
- 72.Messner KR, Imlay JA. 2002. In vitro quantitation of biological superoxide and hydrogen peroxide generation. Methods Enzymol 349:354–361. doi: 10.1016/s0076-6879(02)49351-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.