

Abstract
Human macrophages are protected by intrinsic antiviral defenses that provide moderate protection against HIV-1 infection. Macrophages that do become infected can serve as long-lived reservoirs, to disseminate HIV-1 to CD4+ T cells. Infection of macrophages with HIV-1 and HIV-2 is inhibited by constitutive mobilization of antioxidant response master transcription regulator Nrf2. The downstream mediator of this restriction was not identified. Among the tens of genes controlled directly by Nrf2 in macrophages, we found that xCT/SLC7A11, a 12-transmembrane, cystine-glutamate antiporter promotes antiretroviral activity. We show here that depletion of xCT mRNA increases HIV-1 infection. Reconstitution of xCT knock out cells with wild-type xCT but not a transport-deficient mutant restores anti-HIV-1 activity. Pharmacological inhibitors of xCT amino acid transport also increase infection. The block is independent of known restriction factors and acts against HIV-1 and HIV-2. Like the block triggered through Nrf2, xCT function impedes infection immediately before 2-LTR circle formation.
Introduction
Nrf2, a master transcription regulator of the cellular antioxidant response, controls a restriction to HIV infection in primary human monocyte-derived macrophages (hMDM) and in PMA-differentiated human monocytic cell lines [1]. The inhibition can be reduced or amplified by controlling Nrf2 function. Inhibition of HIV-1 occurs after reverse transcription but before 2-LTR circle formation, which is a hallmark of viral cDNA entry into the cell nucleus [2].
Cellular oxidative stress can be triggered by accumulation of reactive oxygen species (ROS) from exogenous sources like UV radiation or toxins like heavy metals, through endogenous sources such as biproducts of the cellular respiration, or in the case of macrophages and other phagocytes, through diffusion from within their phagosomes [3, 4]. Overaccumulation of ROS including superoxide or hydroxyl radical and downstream products like hydrogen peroxide are hazardous in cells where they can cause oxidative damage to membranes, proteins, and nucleic acids [5–7]. As a result, the clearance of ROS by enzymatic and nonenzymatic antioxidants is required for maintaining redox equilibrium and cell health [8].
Intracellular redox conditions are known to impact HIV infection. HIV transcription is modulated by reducing agents that alter the function of the transcription factor NF-κB [9–12]. Glutathione and N-acetyl cysteine treatments decrease HIV reverse transcriptase activity [13]. Finally, SAMHD1, a dNTPase that strongly inhibits HIV-1 infection in non-cycling cells, is redox sensitive [14–16].
Nrf2 is constitutively produced in macrophages. When cells are at redox equilibrium, most Nrf2 is marked for proteasomal degradation by KEAP1/CUL3 ubiquitin ligase complexes [17]. Oxidation of the KEAP1 sensor protein blocks Nrf2 binding and ubiquitination [18]. This allows Nrf2 accumulation in the cell nucleus and a subsequent increase in transcription of antioxidant response element-controlled (ARE) genes [19, 20]. ARE-bearing promoters control the production of proteins that counter oxidation to restore redox equilibrium [21].
xCT (SLC7A11), upregulated by Nrf2, is a twelve-pass, cytoplasmic membrane antiporter that exports one molecule of glutamate for each imported molecule of cystine [22–25]. In the cytoplasm, each cystine is reduced to two cysteine molecules. Cysteine is a limiting component of the tripeptide antioxidant glutathione, of proteins, post-translational modifications, and reducing agents. xCT has become the focus of cancer research. It is often overexpressed in cancer cells where it supports high rates of cell replication and helps to neutralize chemotherapeutic agents [26–29].
xCT impacts several virus types. It forms a multimolecular complex with integrins to allow binding of the Kaposi Sarcoma-Associated Herpes Virus (KSHV) [30–32]. KSHV encoded microRNA upregulates xCT expression to promote virus dissemination and protection from reactive nitrogen species [33]. xCT also acts as a cofactor for Vesicular Stomatitis Virus infection (VSV) through an as yet unidentified mechanism [34]. In mouse macrophages, VSV infection increases transcription of xCT and deletion of xCT in HAP-1 cells decreases infection. The impact of xCT on HIV-1 infection has not been determined.
Here we show that xCT inhibits HIV infection in primary human monocyte-derived macrophages, in both differentiated and undifferentiated human monocytic THP-1 cells, and in other cell types as well. This viral inhibition relies on amino acid transport. xCT mediates a block that is independent of the HIV-1 envelope glycoprotein and does not impact expression from the provirus after infection. Inducing xCT expression restores the block in xCT KO macrophages and the restriction, like that controlled by Nrf2, acts after reverse transcription but before 2-LTR circle formation.
Results
Constitutive levels of xCT expression inhibit HIV-1 in primary hMDMs and differentiated human monocytic cells.
HIV-1 infection increases when the level of Nrf2 is reduced and decreases when Nrf2 is mobilized [1]. We therefore expected that reduction or elimination of cellular proteins that are upregulated by Nrf2 and required for the restriction would boost macrophage infection.
Expression arrays were used to determine whether mRNAs coding for known antiviral proteins are upregulated after Nrf2 mobilization by the isothiocyanate sulforaphane (SFN) in primary hMDM or whether known viral cofactors are downregulated (File S1). No changes were noted in messages coding for proteins that are known to impact HIV-1 infection. As expected, messages coding for known antioxidant response proteins were upregulated. This suggested that an SFN-induced redox response, rather than upregulation of a known antiviral factor, promotes the observed infection block.
SFN inhibited infection of hMDMs, but not of CD4+ T cells [1]. This led us to speculate that SFN alters the transcriptional profile of these cell types differently. mRNA coding for xCT was the most highly upregulated message in SFN-treated primary hMDMs (File S1 and Fig S1). There was no apparent change in xCT expression in CD4+ T cells treated with SFN.
To determine the impact of xCT on infection, we reduced xCT protein levels by transfecting primary hMDMs with either non-targeting or xCT-specific siRNA. Following transfection, the macrophages were infected with VSV-G pseudotyped env(−), nef(−) HIV-1 luciferase reporter virus. Cells were lysed 24 hours after infection and luciferase activity was measured as a readout for expression from proviral DNA. The cultures transfected with xCT-specific siRNA showed 3-fold more luciferase activity than those transfected with non-targeting siRNA (Fig 1A). The decrease in xCT in response to siRNA transfection was confirmed by western blotting. xCT protein levels were reduced by about 60% in the cells transfected with xCT-specific siRNA (Fig 1B and C). To further confirm these results, we transfected PMA-differentiated THP-1 cells with siRNA before infecting them with the HIV-1 luciferase reporter virus pseudotyped with VSV-G. As in the hMDM cultures, we found a significant increase in luciferase expression following xCT-targeting siRNA transfection compared with the non-targeting control (Fig 1D). We also tested whether the difference in infection efficiency between non-targeting and xCT-specific siRNA depends on the mode of viral entry. We infected PMA-differentiated THP-1 cells with env(−) reporter virus pseudotyped with HIV-1 Env from the dual-tropic 89.6 virus. The xCT-specific siRNA transfected group showed three-fold greater infection than the non-targeting siRNA transfected group (Fig 1E). We confirmed depletion of xCT in the cells transfected with xCT-specific siRNA (Fig 1F).
Fig 1: Constitutive levels of xCT expression inhibit HIV-1 in primary hMDMs and differentiated human monocytic cells.
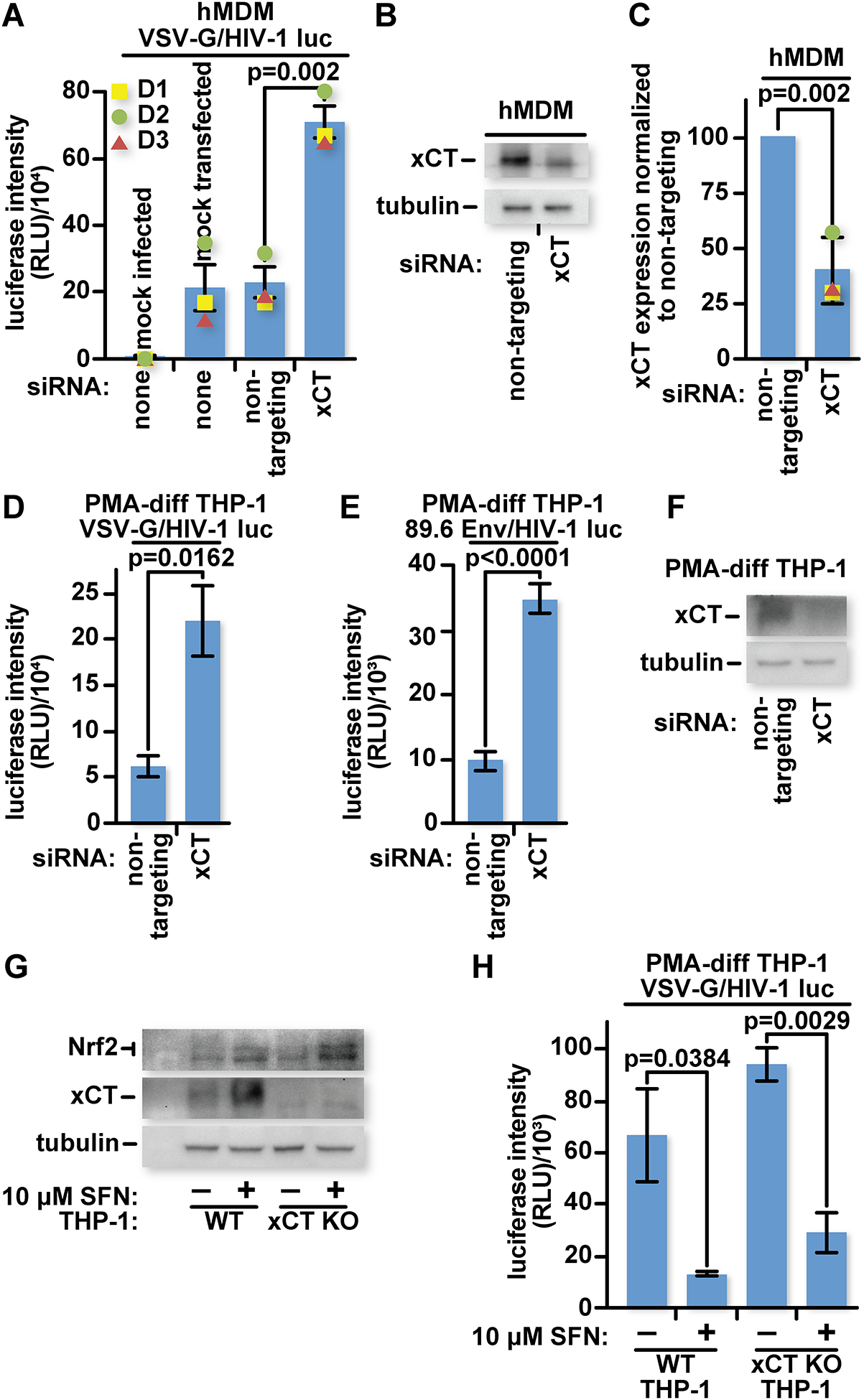
(A) 2.5×105 hMDMs from three different donors (D1–3) were mock-transfected or transfected with a non-targeting siRNA or xCT-targeting siRNA twice in four days. Forty-eight hours after the second transfection, the cultures were mock infected or infected with VSV-G pseudotyped HIV-1 luciferase reporter virus. Twenty-four hours after infection, cells were lysed, and luciferase activity was measured. (B) Representative samples from (A) were lysed in Laemmli buffer and run on an SDS-PAGE gel for characterization by western blot with antibodies specific for xCT or tubulin. (C) Densitometry was performed on the bands from (B) and signals were normalized to the non-targeting siRNA group. (D) 5×104 PMA-Differentiated THP-1s were transfected with a non-targeting siRNA or xCT-targeting siRNA once and infected three days later with VSV-G pseudotyped HIV-1 luciferase reporter virus. Twenty-four hours after infection, cells were lysed, and luciferase activity was measured. (E) Similar to (D) except cultures were infected with HIV-1 dual tropic 89.6 Env pseudotyped HIV-1 luciferase reporter virus. (F) Representative samples from (D) were lysed in Laemmli buffer and run on an SDS-PAGE gel for characterization by western blot with antibodies specific for xCT or tubulin. (G) 5×104 WT or xCT KO PMA-differentiated THP-1 cells were treated with 10 μM SFN or an equivalent volume of DMSO in complete RPMI-1640 for twenty-four hours before being lysed in Laemmli buffer and run on an SDS-PAGE gel for characterization by western blot with antibodies specific for xCT, Nrf2, or tubulin. (H) 5×104 WT or xCT KO THP-1 cells were treated as in (G) for twenty-four hours before infection with VSV-G pseudotyped HIV-1 luciferase reporter virus. Twenty-four hours later, cells were lysed and luciferase activity was measured. The data represent mean values ± SEM from three independent experiments.
To investigate whether SFN-mediated infection inhibition acts through xCT expression, we generated xCT knock-out (KO) THP-1 monocytes. We transduced a lentiviral CRISPR/Cas9 expression construct with a guide RNA specific for xCT into THP-1 cells and selected for transduced clones using an antibiotic. Frame shifts in both alleles of a selected clone were confirmed by DNA sequencing of PCR-amplified genomic target DNA (Fig S2). In contrast to the parental wildtype (WT) THP-1 cells which upregulate xCT in response to SFN, the xCT KO THP-1 cells produced no detectable xCT, even after SFN treatment (Fig 1G). SFN treatment, as expected, still increased Nrf2 levels in the xCT KO cultures.
WT and xCT KO THP-1 macrophages were infected with VSV-G pseudotyped HIV-1 luciferase reporter virus. Interestingly, SFN still inhibited HIV-1 in xCT KO THP-1 macrophages, suggesting that multiple anti-HIV-1 mechanisms are upregulated by SFN (Fig 1H).
Thus, in contrast to KSHV and VSV which benefit from xCT, HIV-1, in single-round infections, was inhibited in the presence of xCT. Depleting xCT with siRNA in primary MDMs or PMA-differentiated THP-1 macrophages increased infectivity. The block is also independent of the entry pathway as it was observed with virus bearing either VSV-G or HIV-1 Env.
Reintroduction of wild-type, but not functionally defective xCT, inhibits HIV-1 in xCT KO THP-1 macrophages.
Reintroduction of xCT into xCT KO cells provided an opportunity to confirm that the increased infectability of these cells is attributable specifically to xCT. Expression of exogenous xCT in xCT KO cells also allowed us to test whether xCT transport function is required for the block. We transduced xCT KO THP-1 cells with either empty lentiviral vector or vectors coding for xCT, or xCTC327L under the control of a doxycycline inducible promoter [35]. Cysteine 327 is located within the xCT substrate-binding region and is critical for efficient amino acid transport [36] (Fig 2A). Importantly, we made synonymous mutations in the xCT coding regions to assure that these no longer match the guide RNA used to target CRISPR/Cas9 to xCT.
Fig 2: Reintroduction of wild-type xCT, but not a transport defective xCT mutant, inhibits HIV-1 in xCT KO THP-1 macrophages.
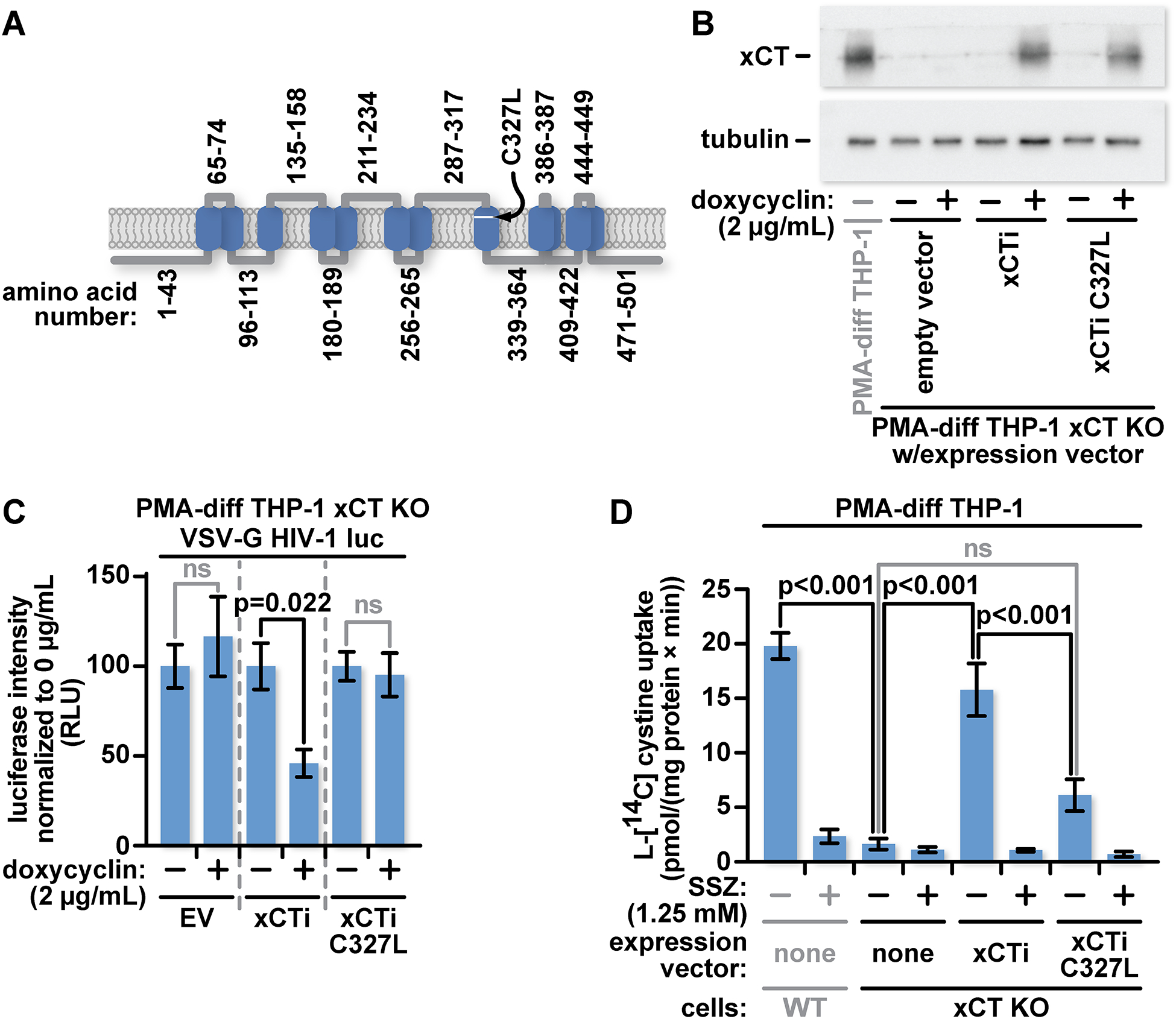
(A) Graphical representation of xCT showing the amino acid residue numbers of key segments and the relative location of the C327L mutation. (B) xCT KO THP-1 cells were transduced with either an empty lentiviral vector or lentiviral vectors coding for WT xCT (xCTi) or xCTC327L (C327L xCTi) with expression controlled by a doxycycline-inducible promoter. PMA-differentiated THP-1 cells were treated for twenty-four hours with media containing vehicle or 2 μg/mL doxycycline. Lysates from these cells were characterized by western blotting with antibodies specific for xCT or tubulin. (C) 5×104 PMA-differentiated THP-1 cells, as described in (A) were cultured for six days in complete RPMI-1640 supplemented with PMA, in the presence or absence of 2 μg/mL doxycycline before infection with VSV-G pseudotyped HIV-1 luciferase reporter virus. Twenty-four hours after infection cells were lysed and luciferase activity was measured. (D) Radiotracer assay of xCT-mediated cystine transport in PMA-differentiated WT, xCT KO, xCT KO xCTi, and xCT KO C327L xCTi THP-1 cells treated for forty-eight hours with 2 μg/mL doxycycline. xCT activity assays were performed in chemically defined medium containing 12.5 μM L-[14C] cystine and inhibitors of alternate cystine transporters as specified in Material and Methods. The xCT activity was quantitated by measuring intracellular L-[14C] cystine accumulation during a twenty-minute incubation at 37° C and further normalized to the specific activity of L-[14C] cystine and total protein content. The data represent mean values ± SEM from three independent experiments.
We tested xCT protein levels in the three transduced xCT KO groups in the presence or absence of doxycycline. We find similar levels of xCT protein in the xCT and xCTC327L inducible clones following induction. As expected, the cells transduced with empty vector do not express xCT after doxycycline treatment (Fig 2B).
To determine whether we could reconstitute the infection block in the xCT KO THP-1 cells, the three groups of cells were cultured in the presence or absence of doxycycline for 6 days before infection with VSV-G pseudotyped HIV-1 luciferase reporter virus. In contrast to the cultures transduced with empty vector or with xCTC327L, those with WT xCT showed significantly reduced luciferase activity (Fig 2C).
We measured the xCT-mediated cystine transport in xCT KO cells with inducible WT xCT or xCTC327L, comparing them against both the WT THP-1 and xCT KO THP-1 cells. We first measured the kinetics of L-[14C] cystine uptake in WT THP-1 cells to determine optimal incubation times for our assay. We minimized background cystine uptake by transporters other than xCT by supplementing the assay media with D-aspartate and acivicin to block the Na+-dependent L-cystine transport and γ-glutamyl transpeptidase activity, respectively [37]. Under these conditions L-[14C] cystine uptake was linear up to 40 min after initiation of the assay (Fig S3).
Based on this analysis, we selected a twenty-minute incubation time for all subsequent quantitative comparisons. Knocking out xCT expression eliminated all but background L-[14C] cystine uptake. Induction of WT xCT expression restored the transport activity to approximately 80% of that seen in WT cells, while induction of xCTC327L expression provided some, albeit significantly reduced, transport activity (Fig 2D). As an additional specificity control, we applied the xCT inhibitor, sulfasalazine (SSZ, 1.25 mM), to cultures and found that it blocked most L-[14C] cystine uptake in WT THP-1 cells, or doxycycline-inducible WT xCT or xCTC327L in xCT KO cells (Fig 2D).
In summary, we were able to restore cystine import and the HIV-1 infection block with inducible production of WT xCT. In contrast, transport defective xCTC327L failed to block HIV-1 infection and supported only a modest amount of cystine import.
Pharmacological inhibitors of xCT transport activity increase HIV infection efficiency.
Depletion of xCT protein increased the efficiency of HIV-1 infection while reintroduction of the transport defective xCT mutant failed to prevent HIV infection in xCT KO cells. These observations suggest that not just xCT, but also its function, is required to inhibit HIV-1. To confirm that amino acid transport is required for viral inhibition, we tested whether xCT transport inhibitors increase HIV-1 infection. Two structurally distinct inhibitors of cystine-glutamate transport were applied to cultures prior to HIV infection. We tested SSZ, an xCT cystine transport inhibitor and anti-inflammatory drug prescribed clinically to treat rheumatoid arthritis and inflammatory bowel disease [38, 39]. We also tested erastin, an xCT inhibitor that is in clinical trials against cancer [40, 41].
Wildtype and xCT KO THP-1 macrophages were treated with 0.625 mM SSZ, or with media containing equivalent amounts of vehicle (DMSO), 24 hours before infection with VSV-G pseudotyped HIV-1 luciferase reporter virus. Pretreatment of WT THP-1 cultures with SSZ resulted in a significant increase in luciferase activity over the DMSO control. In contrast, no increase of luciferase expression was observed in the xCT KO THP-1 cultures under the same conditions, where no xCT was present to be inhibited (Fig 3A). To confirm that pharmacological inhibition of xCT amino acid transport increases HIV infectability, we treated THP-1 macrophages with erastin which also resulted in a significant increase in luciferase activity (Fig 3B).
Fig 3: Pharmacological inhibitors of xCT transport activity increase HIV infection efficiency in differentiated human macrophages.
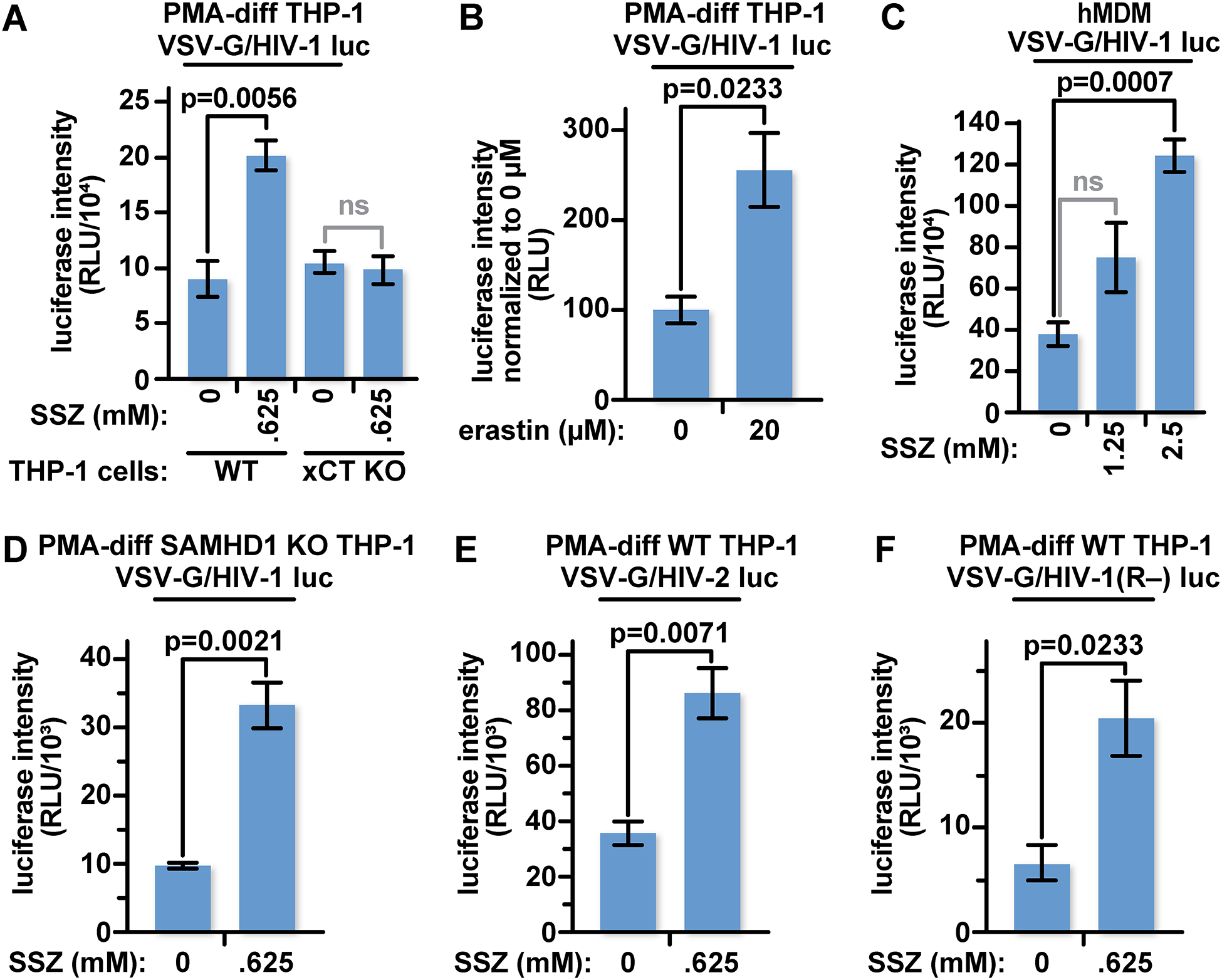
(A) 5×104 PMA-differentiated WT or xCT KO THP-1 cells were treated with 0.625 mM SSZ or an equivalent volume of DMSO in complete RPMI-1640 for twenty-four hours before infection with VSV-G pseudotyped HIV-1 luciferase reporter virus. Twenty-four hours later, cells were lysed and luciferase activity was measured. (B) 5×104 PMA-differentiated THP-1 cells were infected with VSV-G pseudotyped HIV-1 luciferase reporter virus as in panel (A) except the cultures were treated with 20 μM erastin. (C) 5×104 hMDMs were infected with VSV-G pseudotyped HIV-1 luciferase reporter virus as in (A) except cultures were treated with 1.25 or 2.5 mM SSZ. (D) 5×104 PMA-differentiated SAMHD1-KO THP-1 cells were treated and infected as in (A). (E) 5×104 PMA-differentiated WT were treated as in (A) twenty-four hours before infection with VSV-G pseudotyped HIV-2 luciferase reporter virus. (F) 5×104 PMA-differentiated WT were treated as in (A) twenty-four hours before infection with VSV-G pseudotyped HIV-1 vpr(−) luciferase reporter virus. The data represent mean values ± SEM from three independent experiments.
We also treated primary hMDM cultures with SSZ prior to infection with VSV-G pseudotyped HIV-1 luciferase reporter virus. We found a significant increase of luciferase activity in these cells, albeit at a higher SSZ concentration (Fig 3C).
To determine whether SSZ impacts the luciferase reporter itself, we tested the effect of SSZ on infections with a GFP reporter virus. We treated WT THP-1 cells with SSZ prior to infection with VSV-G pseudotyped HIV-1 GFP-reporter virus. We found, using flow cytometry, a dose-dependent increase in GFP positive cells in cultures treated with SSZ compared to the vehicle control (DMSO)(Fig S4). This suggests that the xCT-mediated block is independent of reporter identity. Because we were interrogating individual cells for GFP expression rather than cell lysates, we could also determine that more cells were becoming infected in the presence of SSZ rather than an equivalent number of cells expressing more of the reporter protein.
We tested whether the xCT-mediated restriction requires the potent anti-HIV-1 restriction factor, SAMHD1. The dNTPase activity of SAMHD1 has been shown to be enhanced by exogenous reducing agents and inhibited by oxidants [14, 16]. To test whether xCT can mediate a block in the absence of SAMHD1, we pretreated SAMHD1 KO THP-1 cells with 0.625 mM SSZ or the equivalent amount of DMSO [42]. As in the SSZ-treated WT THP-1 cultures (Fig 3A), we observed a significant increase in luciferase activity in the SSZ-treated group, despite the absence of SAMHD1 (Fig 3D and Fig S5). This suggests that xCT mediates a restriction that is independent of SAMHD1.
Finally, we investigated whether targeting xCT-mediated amino acid transport increases HIV-2 infections or HIV-1 infection in the absence of the viral accessory protein Vpr. Both Vpr and the HIV-2 accessory protein Vpx interact with the CRL4 ubiquitin ligase complex to target host proteins for proteasomal degradation [43–46]. We found however, that SSZ pretreatment increases infection with these viruses at similar relative levels as HIV-1 (Fig 3E and F). This suggests that xCT is not targeting these accessory proteins, nor are these accessory proteins counteracting the xCT block within the afferent stage of infection.
In summary, xCT transporter function is important for the xCT-mediated restriction. Functionally impaired xCTC327L failed to restore the infection block in xCT KO cells and xCT inhibitors boosted HIV-1 and HIV-2 infection. The infection increase, tested with HIV-1, was only seen in cells expressing xCT. This effect was independent of the potent anti-retroviral factor, SAMHD1 and the viral accessory protein, Vpr.
SSZ increases infection efficiency only when applied before HIV-1.
NF-κB controls HIV-1 transcription and its activation can be regulated by intracellular glutathione and cysteine [9–11, 47–50]. This raised the possibility that at least a portion of the infection increase that we observed after modulating xCT function could be at the level of transcription. Thus, we wished to determine when SSZ must be applied, relative to the virus, to relieve the xCT-mediated restriction. In contrast to previous experiments in which cells were harvested 24 hours after infection, here we harvested cells 72 hours after HIV-1 challenge to allow time for the post-infection treatments to impact infectivity or expression from the provirus. We differentiated THP-1 cells with PMA and treated the cells at various timepoints from 72 hours before infection (2 hours after plating) to 24 hours after infection. We observed the greatest positive impact on infection when cultures were treated 72 hours before addition of virus. This benefit diminished almost linearly to 6 hours before infection, when the difference between treated and mock-treated cells became insignificant (Fig 4). Similarly, SSZ added at or after virus application did not significantly impact luciferase activity (Fig 4). Thus, it appears that SSZ requires time to alleviate the restriction and that the restriction cannot be reversed by application of SSZ after infection. Further, SSZ does not increase expression from proviruses as application after infection did not impact reporter levels detectably. This is consistent with other work that showed no impact of SSZ on LTR activation compared to negative controls [51].
Fig 4: SSZ increases infection only when it is applied before the virus.
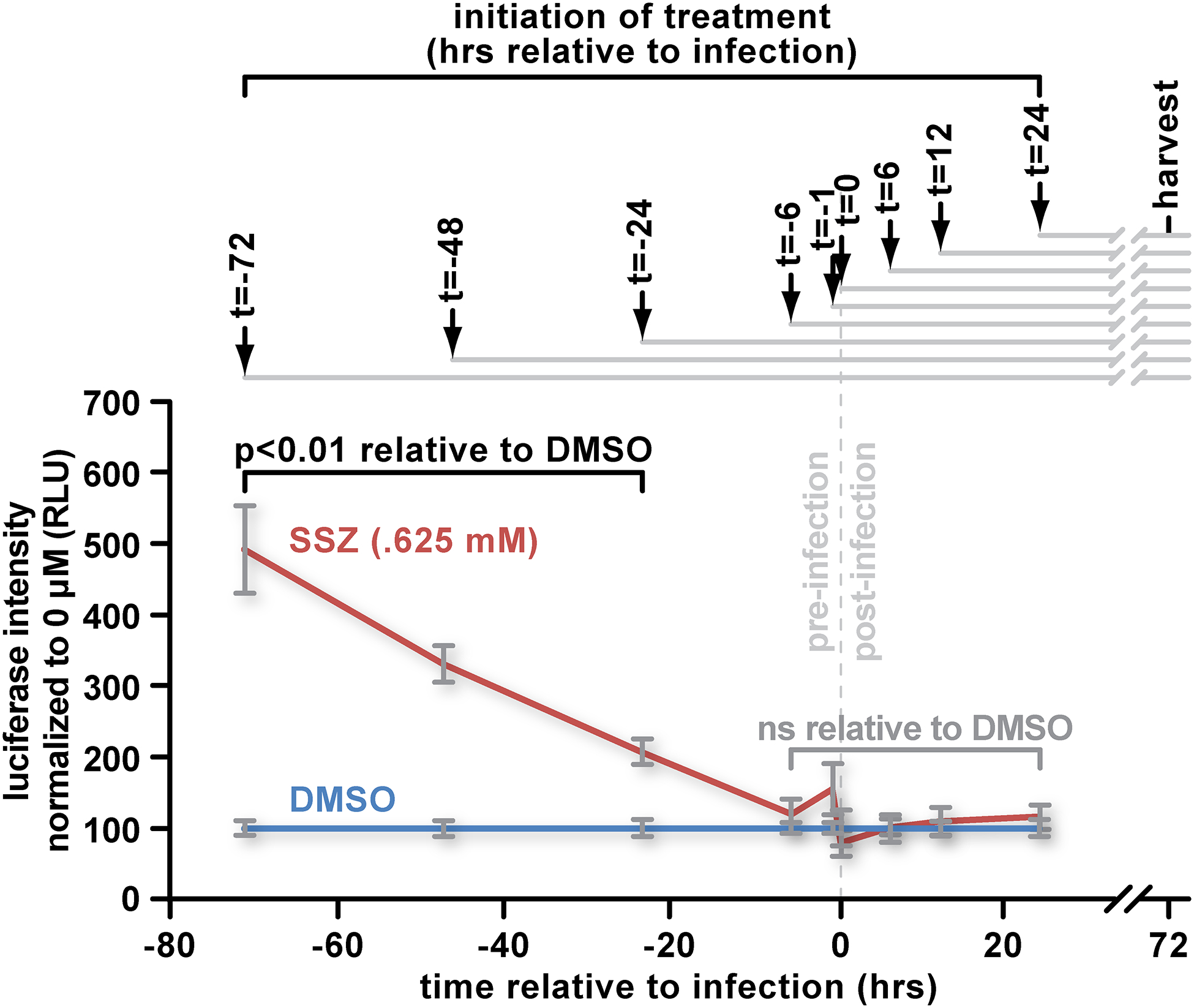
(A) Cultures of 5×104 PMA-differentiated WT THP-1 cells were treated with 0.625 mM SSZ or an equivalent amount of DMSO in complete RPMI-1640 containing PMA at the indicated timepoints. Cell cultures were infected with VSV-G pseudotyped HIV-1 luciferase reporter virus. Seventy-two hours later, cells were lysed, and luciferase activity was measured. The data represent mean values ± SEM from three independent experiments.
xCT mediates a block to HIV-1 that is active in dividing cells.
xCT-mediated amino acid transport activity is required for HIV-1 inhibition in hMDMs and in PMA-differentiated THP-1 cultures. We tested whether cystine-glutamate transport can also block HIV-1 in actively dividing cells. We determined the relative xCT protein levels in undifferentiated THP-1, HeLa, HEK293T, and SupT1 cells (Fig 5A). All but the xCT KO cells expressed xCT protein at levels that were detectable by western blotting, albeit at varying levels. We pretreated the THP-1 monocytes, HeLa, HEK293T, and SupT1 cells with 0.625 mM SSZ or the equivalent volume of DMSO for 24 hours before challenging the cultures with VSV-G pseudotyped HIV-1 luciferase reporter virus. Interestingly, despite expressing varying levels of xCT, THP-1 monocytes, HeLa, and HEK293T treated with SSZ expressed significantly greater luciferase activity relative to the DMSO-treated control groups (Fig 5B – D). SupT1 cells expressed less xCT than WT THP-1, HeLa and HEK293T cells and did not show a significant increase in luciferase expression in SSZ-treated groups compared to the DMSO controls (Fig 5E). To confirm that SSZ inhibits cystine-glutamate transport in these cells, we tested glutathione levels within each cell type. As expected, pretreatment with SSZ decreased cellular glutathione concentrations in each of these cell types compared to the equivalent amount of DMSO (Fig S6). Thus, the xCT-mediated restriction does not appear to be specific to differentiated macrophages.
Fig 5: xCT mediates a block to HIV that is active in dividing cells.
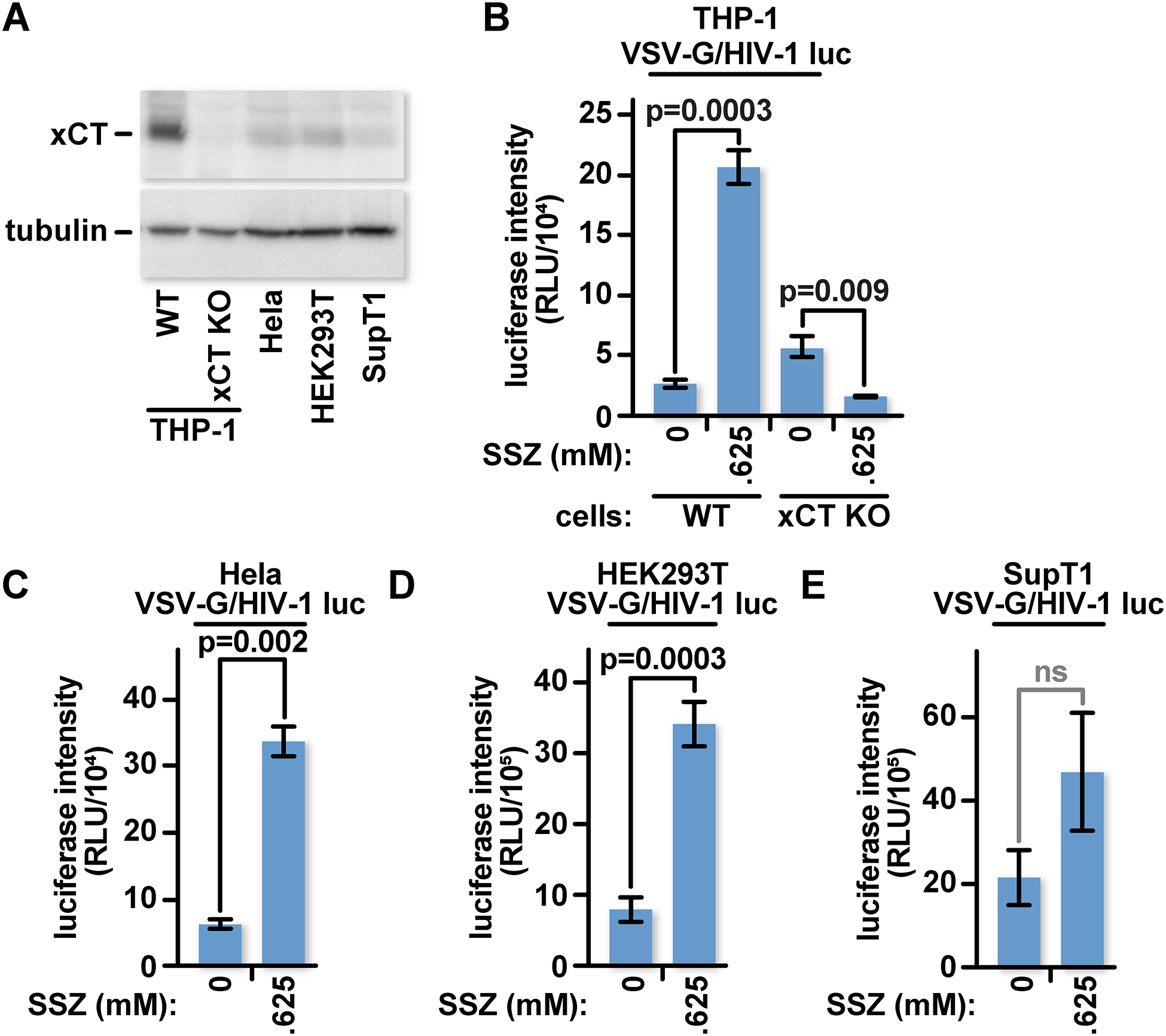
(A) WT THP-1 monocytes, xCT KO THP-1 monocytes, HeLa, HEK293T, and SupT1 cells were lysed and characterized by western blot with antibodies for xCT or tubulin. (B) 5×104 undifferentiated WT and xCT KO THP-1 cells were treated with 0.625 mM SSZ or an equivalent amount of DMSO in complete RPMI-1640 for twenty-four hours before infection with VSV-G pseudotyped HIV-1 luciferase reporter virus. Twenty-four hours later, cells were lysed and luciferase activity was measured. (C) 1×104 HeLa cells were treated and infected as in (B) except treated in complete DMEM. (D) As in (C) except with HEK293T. (E) As in (B) except with SupT1 cells. The data represent mean values ± SEM from three independent experiments.
xCT inhibits HIV-1 infection after reverse transcription but before 2-LTR circle formation.
In the cell, the RNA genome of HIV-1 is reverse transcribed into double-stranded cDNA which is imported into the nucleus as a nucleoprotein complex before integration into the host genome. We previously showed that SFN inhibits HIV-1 after reverse transcription, but before 2-LTR circle formation [1]. 2-LTR circles are a dead-end replication by-product that serves as a marker for the entry of the viral nucleoprotein complex into the cell nucleus. SFN mobilizes Nrf2 and upregulates xCT expression. However, SFN can inhibit HIV-1 infection in cells devoid of xCT (Fig 1H).
To identify which stage of infection is blocked by xCT, we used real time PCR with primers selected to detect late reverse transcription products, 2-LTR circles, and integrated proviral DNA. THP-1 monocytes were used here because they showed a larger infection increase in response to SSZ than the PMA-treated form (Fig 3A vs 5B). The cells were treated with treated with DMSO, 0.625 mM SSZ, or 10 μM AZT for 48 hours to optimize the efficacy of SSZ inhibition of the xCT-mediated block (Fig 4). The THP-1 monocytes were challenged with VSV-G pseudotyped HIV-1 GFP reporter virus. Heat-inactivated virus was added to separate cultures to test for plasmid contamination. Cells were lysed 8 hours after infection to detect late reverse transcription products or 24 hours after infection to detect 2-LTR circles or proviral DNA. Total genomic DNA was extracted from the cells, quantified, and diluted to 20 ng/mL prior to qPCR [52].
THP-1 cultures treated with SSZ had similar levels of late reverse transcription products as the DMSO-treated control cultures (Fig 6A). In contrast, we found a significant increase in 2-LTR circle products and integrated proviral DNA in the SSZ-treated cultures compared to the DMSO-treated cultures (Fig 6B and C). As expected, AZT treatment reduced levels at all three points of the viral replication cycle that we tested. We found a similar trend in HEK293T cells treated with SSZ compared to the DMSO control following 24-hour treatment with SSZ (Fig S7).
Fig 6: xCT inhibits HIV-1 following reverse transcription but before 2-LTR circle formation.
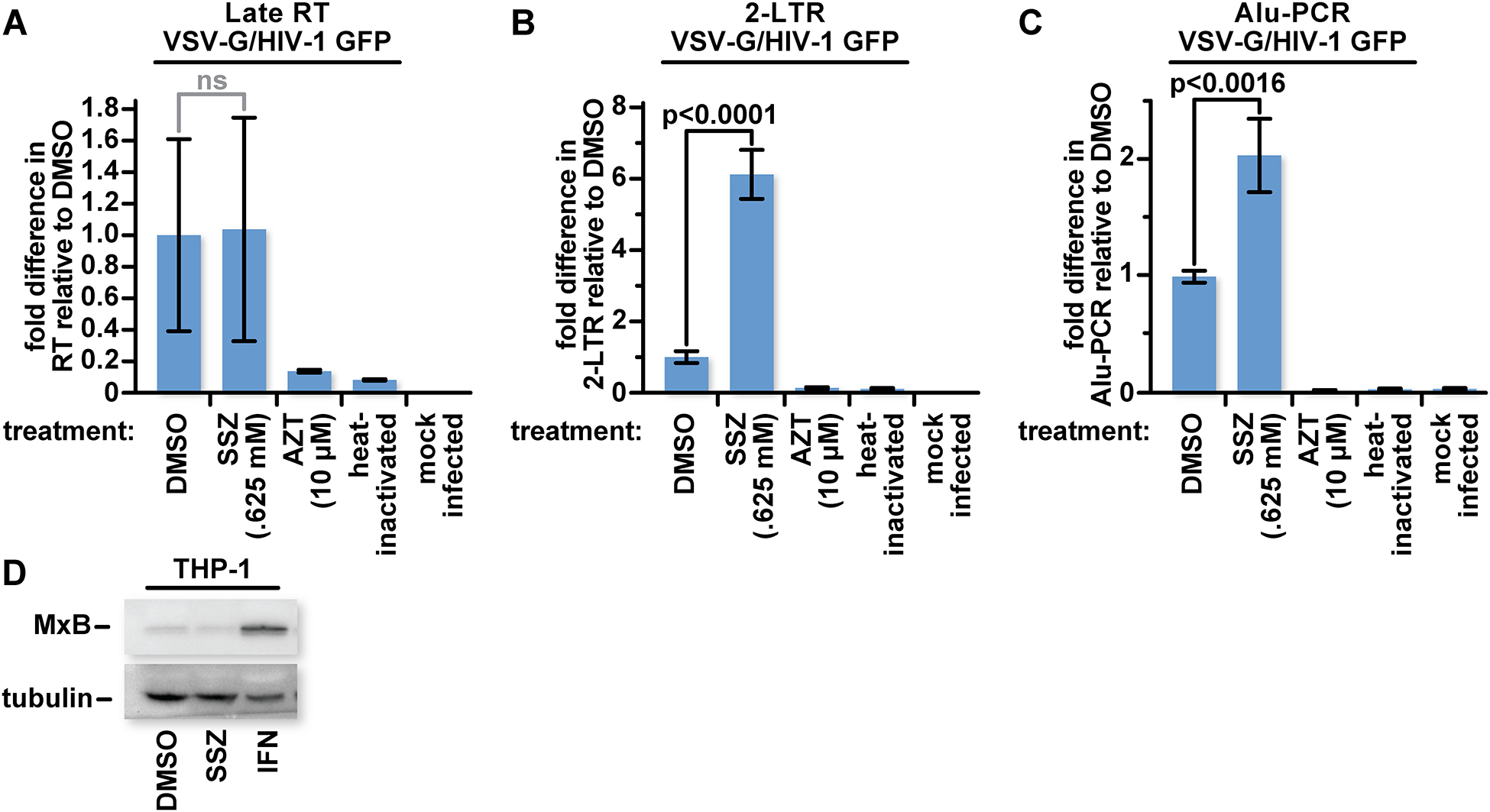
(A) 105 WT THP-1 cells were mock treated or treated with 0.625 mM SSZ an equivalent amount of DMSO in complete DMEM or with 10 μM AZT. Forty-eight hours after treatment with SSZ or DMSO, and twenty-four hours after AZT treatment, cells were mock infected or infected with VSV-G pseudotyped HIV-1 GFP reporter virus. Heat inactivated virus served as a control for plasmid carryover. Eight hours later, cells were lysed and total DNA was extracted. Samples were probed with primers targeting sequence markers for late reverse transcription by qPCR. (B) As with (A) except cells were lysed twenty-four hours post infection and probed for 2-LTR circles. (C) As with (B) except integrated HIV-1 gag and host genomic Alu sequences are amplified in the first round of PCR before probing for markers of integrated viral DNA by qPCR. (D) WT THP-1 monocytes were lysed and characterized by western blot with antibodies for MxB or tubulin. The data represent mean values ± SEM from three independent experiments.
Like the xCT-controlled restriction, the type 1 interferon (IFN)-inducible protein, MxB, inhibits HIV infection after reverse transcription but before 2-LTR circle formation [53]. To detect whether xCT is impacting MxB expression, we probed for MxB by western blot in THP-1 monocytes treated with .625 mM SSZ or the equivalent amount of DMSO. We found no changes to MxB expression in the SSZ-treated group. As expected, IFN treatment increased expression of MxB. SFN, like SSZ, did not impact MxB expression [1].
Thus, we find that inhibition of xCT function relieves an infection block after reverse transcription and before 2-LTR circle formation. This likely reflects alleviation of a block that interferes with entry of the viral DNA into the nucleus. The location of the restriction matches that of the SFN block through Nrf2. This again suggests that xCT is an antiviral factor directly downstream of the Nrf2-mediated block.
Discussion
In summary, we show for the first time that xCT function helps to determine the permissiveness of various cell types to HIV-1 and HIV-2 infection. Depletion of xCT with siRNA and blocking function with pharmaceutical inhibitors both boosted infection. Conversely, induction of only WT xCT expression in xCT KO cells was sufficient to block infection. Blocking xCT function with SSZ prevented a restriction, after late reverse transcription and before 2-LTR circle formation, like one that SFN-treatment enhanced.
SSZ and SFN thus have opposite effects on the same stage of the replication cycle. This raises the possibility that SFN, mobilizing Nrf2, increases xCT levels, and xCT function then acts to inhibit HIV-1 infection. Interestingly, we still observed an infection decrease in xCT KO THP-1 cells after SFN treatment. This suggests that while xCT function can itself hinder HIV infection, SFN, through Nrf2 has additional mechanisms for arming the cell against infection. xCT is of course one among tens of proteins upregulated by Nrf2 mobilization. Multiple antiviral mechanisms would not be surprising as SFN and Nrf2 inhibit infection from multiple types of viruses [54–57].
xCT spans the cytoplasmic membrane and serves as a fusion/entry receptor for KSHV in adherent cells. We considered the possibility that xCT could be engaging and blocking HIV-1 directly, either through the viral envelope glycoprotein or through other viral proteins. We found however, that single-round infections with VSV-G or dual-tropic HIV Env-pseudotyped HIV-1 were both increased after depletion of xCT expression with siRNA transfection (Fig 1D–F). VSV-G binds LDL-R and promotes fusion from endosomes at low pH while HIV engages CD4 and CCR5 as receptors, likely at the cell surface, and does not require low pH for fusion [58, 59]. This suggests that the block is independent of the entry pathway. A block after entry is further supported by our data showing that the infection is inhibited after reverse transcription, a process that cannot be completed outside of the cell.
SSZ treatment increases HIV infection in both nondividing and dividing cell types. hMDMs remain in the G0 phase of the cell cycle. PMA-differentiated THP-1 macrophages have a block to S phase and M phase [60]. In contrast, HeLa, HEK293T, and THP-1 monocyte cultures divide continuously. We can therefore discount a connection to regulation or requirements of the S and M phases.
Within the cell types tested, the relative increase in infection following SSZ treatment is similar, between 2- and 6-fold. There is however, a difference in xCT expression levels. Interestingly, while HeLa and HEK293T cells express lower levels of xCT than THP-1 monocytes, the relative increase in infection from SSZ treatment is similar. This could be due to differences in expression or regulation of a downstream factor impacted by xCT activity which acts directly on the virus.
xCT could, alternatively, bind to HIV structural, enzymatic or accessory proteins. The observed reliance on amino acid transport makes an interaction between xCT and viral proteins tenable only if transport and an xCT conformation required for that are linked in each scenario that we explored. However, if this were the case, we would expect xCT expression to be correlated with the relative increase in infection from SSZ treatments. As already discussed, this is not the case as HeLa and HEK293T have lower levels of xCT expression, yet the relative increase in infection following SSZ treatment is similar to that in THP-1 cells (Fig 5).
The xCTC327L mutant, an inefficient cystine transporter, did not inhibit HIV-1 like WT xCT (Fig 2C and D). This is also consistent with the observation that xCT inhibitors , SSZ and erastin, each boosted HIV-1 infection. Furthermore, we observed a time-dependent increase in infectivity in macrophages treated with SSZ where longer treatments increased infectivity more than shorter treatments (Fig 4). This supports a model in which xCT is not inhibiting HIV-1 directly but that its function is priming a downstream restriction. Cystine import boosts limiting intracellular levels of cysteine to supply both protein synthesis and production of glutathione. Specific levels of cysteine or glutathione, completely lost upon long-term loss of cystine import, could be required to modify the virus to inhibit the release of the viral genome into the nucleoplasm. Alternatively, the loss of critical levels of cysteine or glutathione may alter the cellular redox state to inactivate a yet unidentified restriction factor that blocks infection directly. Like xCT function, the interferon-inducible restriction MxB blocks HIV immediately before 2-LTR circle formation [61]. However, MxB is not known to be regulated by the redox state and neither SFN or SSZ treatment transcriptionally regulate MxB [1](Fig 6C). Identifying the target of xCT function that controls HIV-1 infectivity could provide new options for controlling HIV infection.
NF-κB is a redox-sensitive protein that regulates transcription of HIV [9, 11, 47, 62]. Exogenous antioxidant treatments decrease HIV expression through the NF-κB pathway [63, 64]. Our results however show that initiation of SSZ treatment of THP-1 cells in the interval between 6 hours before infection to 24 hours after infection does not increase luciferase expression as might be expected if SSZ increases expression from proviruses. It does increase infection when cultures are treated 72 to 24 hours before virus addition. This suggests that SSZ, and by extension xCT and cystine-glutamate transport, do not significantly impact expression from the provirus. This point is further underscored by our data showing that SSZ relieves a block before 2-LTR circle formation and integration of viral DNA (Fig 6B and C).
2-LTR circle formation is a marker for nuclear entry of the preintegration complex [2]. SSZ treatment resulted in an increase in 2-LTR circles, suggesting that xCT is causing a block before nuclear entry of the viral DNA. As the requirements for nuclear import are specific to the cargo being transported, the oxidating conditions caused by SSZ treatment are unlikely to impact gross effects of nuclear import. For example, NF-κB activation and nuclear import are inhibited by SSZ treatment through an xCT-independent mechanism [65]. Furthermore, nuclear import of the glucocorticoid receptor is decreased following exogenous oxidants [66]. In contrast, exogenous oxidants result in nuclear accumulation of the transcription factor, Nrf2 [25].
Accessory proteins, HIV-1 and HIV-2 Vpr and HIV-2 Vpx, are known to target host proteins for proteosomal degradation, to improve infectivity in the early stages of infection [43–45, 67]. We however, infected macrophages with HIV-2 or HIV-1 vpr(−) in the presence or absence of SSZ and found in the SSZ treated groups show a similar increase in infection as macrophages infected with HIV-1 (Fig 3A vs 3E and F). This suggests that the xCT block is not impacting Vpr or Vpx, nor are these proteins neutralizing the xCT-mediated block within the early single round infection. However, it is possible that upon multiple rounds of infection the virus may become resistant to the xCT-mediated block or a viral protein may target the block. We aimed here to focus on the block within the afferent stage of infection but understanding the response of the virus will be a future focus of our research.
SSZ is a sulfa drug that is used clinically to treat rheumatoid arthritis (RA). SSZ impacts several mechanisms found to suppress the immune response activated during RA, including the NF-κB pathway and the adenosine metabolism pathway [68, 69]. Off target effects of SSZ could impact HIV infection. xCT KO THP-1 monocytes treated with SSZ showed lower levels of luciferase expression than DMSO controls (Fig 5B). However, we speculate that relief of the block by SSZ treatment may enhance infection even more when we compensate for this xCT-independent inhibitory effect. Further, treatment with erastin, a molecularly divergent xCT inhibitor, also increased infection.
The findings described here could provide new therapeutic options against HIV-1 infection. Blocking xCT function in dendritic cells (DC) could promote their infection and thus their ability to present viral antigen to CD8+ T+ T cells for more effective vaccine strategies [70]. While we have not determined whether xCT inhibits HIV in DCs, we hypothesize that it does as in other monocyte-lineage cells. Alternatively, increasing xCT expression could enhance the constitutive inhibitory effect.
In its capacity as an arthritis treatment, SSZ has been used in individuals living with HIV-1 [39, 71]. Erastin is currently being investigated and optimized as an xCT-targeting chemotherapy against several types of cancers [40, 41]. The incidence of cancer is higher in individuals living with HIV-1 [72] so this population could also be considered for erastin treatment. Our work indicates that the use of xCT inhibitors may be detrimental in persons living with HIV-1 and should thus be carefully considered.
Materials and Methods
Cell Culture
Human leukocytes were isolated from whole blood of healthy donors as buffy coats following Ficoll-Paque™ (GE Healthcare, 17-1440-02) density gradient centrifugation. Cells were washed three times in phosphate buffered saline (PBS) (Corning, 21-040-CV) to remove residual Ficoll-Paque™. Adherent monocytes were isolated from lymphocytes and differentiated into macrophages over ten days in Dulbecco’s Modified Eagle Medium (DMEM) (Corning, #13619003) supplemented with 10% human AB serum (Corning, # 35060CI).
HEK293T (ATCC) and HeLa (ATCC) cell lines were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) (Sigma, #F4135), 100 I.U. pencillin (per mL), 100 μg/mL streptomycin and 292 μg/mL L-glutamine (100X stock, Corning, #30-009-CI). SupT1 (ATCC), THP-1 (ATCC) and SAMHD1 KO THP-1 (a gift from Baek Kim) cell lines were cultured in Roswell Park Memorial Institute 1640 media (RPMI-1640) (Corning, #10-040-CV) supplemented with 10% fetal bovine serum (FBS), 100 I.U. penicillin (per mL), 100 μg/mL streptomycin and 292 μg/mL L-glutamine. Where noted, THP-1 cells were differentiated into a macrophage-like state by treatment with 120 ng/mL phorbol myristate acetate (PMA) (Fisher, #16561-29-8) for 2 days before described treatments. Treatments were applied in the presence of PMA. All cells were cultured at 37°C in a humidified chamber with 5% CO2.
Inducible xCT Expression Vectors
The inducible xCT expression vectors were constructed as follows. We PCR-amplified xCT cDNA sequences from the clone EX-U1104-M06 (GeneCopoeia) and mutagenized the gRNA target sequence with synonymous substitutions so that these clones would not be targeted by residual CRISPR/Cas9 expression in the KO cells. We used primers 5’-CGATAGCGAATTCGTCGAACCATGGTCAGAAAGCCTG-3’ and 5’- GATATATCTGCCGAAGGCGAGGGATATCACAGCAGTAGCTG-3’ to amplify the 5’ portion of xCT. We used 5’-CTCGCCTTCGGCAGATATATCCTGGAACCATTTTTTATTCAATG-3’ and 5’- AGGCCACCGGTCAGCTAGCCATTCTAGATCGAACCACTTTGTACAAGAAAGCTGGGT-3’ to amplify the 3’ portion of xCT. We then mixed the 5’ and 3’ xCT amplification products, which overlap at the gRNA target area, and amplified the entire xCT reading frame with the first and last primers. The gRNA target, TGGCATTTGGACGCTACATT, was thus changed to TCGCCTTCGGCAGATATATC. The PCR products were cloned with a Zero Blunt™ PCR cloning kit (Thermo Fisher), and the sequences of the products were confirmed using Sanger sequencing. The codon for cysteine at amino acid 327 (GCCCTCTCCTGCTTTGGCTC) was mutagenized to a codon for leucine (GCCCTCTCCCTGTTTGGCTC) (GENEWIZ.). The xCT sequences were excised from the pCR-Blunt vector with EcoRI and AgeI and ligated into the same sites in pCW57-MCS1-P2A-MCS2 (Blast) (a gift from Adam Karpf (Addgene plasmid # 80921; http://n2t.net/addgene:80921; RRID:Addgene_80921)) and the sequences were reconfirmed.
Virus Preparation
3×106 HEK293T cells were plated in 10 cm plates (Corning, #430167) and allowed to adhere for 18 hours before transfection with expression vectors. Single-round infections: 10 μg HIV-1 pNL4.3 env(−) nef(−) luc(+), HIV-1 pNL4.3 env(−) nef(−) vpr(−)luc(+) (gifts from Nathaniel Landau), HIV-1 pNL4.3 env(−) nef(−) gfp(+) (a gift from Dana Gabuzda), , or HIV-2 ROD env(−) nef(−) luc(+) (a gift from Lee Ratner) were co-transfected with 10 μg pCL-VSV-G, a vesicular stomatitis virus G protein expression vector (a gift from Garry Nolan (Addgene plasmid # 1733 ; http://n2t.net/addgene:1733; RRID:Addgene_1733) or 10 μg 89.6 dual tropic HIV env (a gift from Kathleen Collins). Lentiviral transductions: xCT KO: 4 μg pCL-VSV-G, 8 μg pLP1 (Invitrogen™), 2 μg pLP2 (Invitrogen™), 8 μg lentiviral vector containing xCT (SLC7A11) gRNA and Cas9 (Transomic, #1254139). Empty vector/doxycycline-inducible xCT/xCTC327L: 4 μg pCL-VSV-G, 8 μg pLP1, 2 μg pLP2, 8 μg pCW57-blast-MCS1-P2A-MCS2/xCT/xCTC327L. These were transfected using a standard calcium phosphate transfection protocol. Five hours after the precipitate was added, the culture media was aspirated and replaced with fresh DMEM. Forty-eight hours later, virus-containing supernatant was harvested and centrifuged to remove producer cells and cellular debris. Viral stocks were stored and concentrated with Lenti-X Concentrator (Takara, # 631232) according to manufacturer’s protocol. HIV stocks were quantitated by HIV-1 p24 antigen capture ELISA (ABL, #5421) according to the manufacturer’s protocol and applied to cells at a concentration of 36 ng/mL. Converted substrate in the ELISA was measured with a SpectraMax iD3 Microplate Reader (Molecular Devices).
Stably Transduced Cell Lines
Lentivirus stocks were generated as described above. THP-1 monocytes were spinoculated at 1200 × g for one hour in V-bottom 96 well plates (Corning, #3894) and incubated at 37° C. xCT KO: Twenty-four hours after spincoculation, the supernatant was replaced with complete RPMI-1640 supplemented with 10 μg/ml puromycin (Sigma-Aldrich, #P8833) and 50 μM β-mercaptoethanol (Sigma-Aldrich, #M6250). Cells were distributed at one per well to generate clonal populations. Puromycin and β-mercaptoethanol-containing media was replaced every 5 days during the expansion of surviving cells. Empty vector/doxycycline-inducible xCT/xCTC327L: Twenty-four hours after spinoculation, transduction supernatant was replaced with complete RPMI-1640 supplemented with 7.5 μg/mL blasticidin (Sigma-Aldrich, #203350) and further replaced every five days. To induce xCT expression, transduced cells were incubated in complete RPMI-1640 supplemented with 2 μg/mL doxycycline.
Reagents
Sulfasalazine (SSZ) (Sigma-Aldrich, #S0883) and erastin (Selleckchem, #S7242) were dissolved in dimethyl sulfoxide (DMSO) to prepare 1.25 M or 100 mM stock solutions, respectively. Doxycycline (Dox) (Sigma, #D9891-5G) was dissolved in phosphate buffered saline (PBS) to prepare a 2 mg/mL stock solution. Azidothymidine (AZT) (NIH AIDS Reagent Program) was dissolved in PBS to prepare a 1 mM solution. Universal type 1 IFN (PBL Assay Science, #11200-2) was added to the complete cell culture media.
Luciferase Activity
Following a single-round HIV-1 luciferase-reporter virus infection, culture media was aspirated and cells were washed with PBS. Cells were then lysed and processed using Firefly Luciferase HS Assay Kit (Genecopoeia, #LF009) according to the manufacturer’s protocol. Relative light units were measured by CentralPRO Microplate Luminometer (Berthold Technologies, #LB962).
Flow Cytometry
Following a single-round HIV-1 GFP-reporter virus infection, culture media was aspirated, and cells were washed three times with PBS. Samples were fixed in 3.8% paraformaldehyde solution for ten minutes followed by three washes in PBS. Events were recorded on an LSR II Flow Cytometer (BD Biosciences).
siRNA Transfection
Non-targeting (GFP-Duplex, Dharmacon, #P-002048-01-20) and xCT-specific (Dharmacon, #J-007612-09-0020) short interfering RNAs (siRNA) were used to deplete target mRNA. Primary monocyte-derived macrophages were transfected with 36 nM siRNA using Viromer Green (Lipocalyx GmbH, #VG-01LB-01) and PMA-differentiated THP-1 macrophages were transfected with 54 nM siRNA using Viromer Blue (Lipocalyx GmbH, #VB-01LB-00) according to the manufacturer’s protocol. siRNA transfection was performed twice, forty-eight hours apart to ensure cell viability and optimal depletion of target mRNA in hMDMs and once in THP-1 macrophages.
Immunoblotting
Cells were lysed with Laemmli buffer (50 mM Tris-HCl (pH 6.8), 2% SDS, 10% glycerol, and 0.1% bromophenol blue) and resolved with 10% SDS-PAGE gels. Proteins were transferred onto PVDF membrane (MilliporeSigma, #IPVH00010), which were blocked with 5% non-fat dry milk dissolved in wash buffer (.1% Tween 20 in 1× PBS) before three washes in wash buffer. Membranes were then incubated, separately, overnight at 4° C on a rocking platform with the following primary antibodies: anti-tubulin (Cell Signaling Technologies, #3873S), anti-xCT (Cell Signaling Technologies, #12691S), anti-SAMHD1 (GeneTex, # GT851), anti-GCLM (GeneTex, # GTX114075), or anti-MxB (Santa Cruz, # 47197). Following incubation, the primary antibody was removed, the membranes were washed three times and then incubated with HRP-conjugated anti-mouse (ThermoFisher Scientific, #G21040), anti-rabbit (ThermoFisher Scientific, #G21234), or anti-goat (ThermoFisher Scientific, #81-1620) secondary antibody for two hours at room temperature. Chemiluminescent blot imaging was performed with a ChemiDoc Touch Imaging System (Bio-Rad). Densitometry was performed using the ImageJ analysis software from the National Institutes of Health.
xCT Activity Assay
PMA-differentiated WT, xCT KO, xCT KO xCTi, and xCT KO xCTC327Li THP-1 cells were cultured in 24-well culture plates with 2 μg/mL doxycycline. Differentiated WT THP-1 cells were washed three times to replace cell culture media with the chemically-defined basal medium containing 135 mM NaCl, 3.8 mM KCl, 1.2 mM MgSO4, 1.3 mM CaCl2, 1.2 mM KH2PO4, 10 mM D-Glucose, and 10 mM HEPES (pH 7.4, adjusted with NaOH) and preincubated in the same medium for 20 min at 37° C. Basal medium was then replaced with basal transport medium additionally containing, 2.5 μM L-[14C] cystine (0.5 μCi/mL), 10 μM of unlabeled L-cystine, 1 mM acivicin, and 1 mM D-aspartate. Isotope uptake was measured following a 37°C incubation for 5, 10, 20, or 40 minutes to establish cystine uptake kinetics (Fig S3). The experimental assays, based on the kinetics data, were incubated at 37° C for 20 min, and included parallel samples with 1.25 mM SSZ as a positive control for inhibition. The reactions were stopped by adding ice-cold basal medium followed by three additional washes with ice-cold basal media. Cells were then lysed in solution of 2% SDS containing 8 mM EDTA. [14C] content in cell lysates was determined with a TRICARB 2900 Liquid Scintillation Analyzer (PerkinElmer) and compared to total counts in the L-[14C] cystine-containing transport medium. L-[14C] cystine transport rates were normalized to the specific activity of the radiotracer in the transport media and protein content in each well. Protein concentration was quantified using a Bicinchoninic acid (BCA) assay kit (ThermoFisher Scientific, #23225) and measured on a SpectraMax iD3 Microplate Reader (Molecular Devices).
Glutathione Quantification
HeLa, HEK293T, SupT1 and THP-1 monocytes were treated with SSZ for 24 hours as indicated. Culture media was removed, and cells were washed three times with PBS. Cells were then lysed and processed according to the manufacturer’s protocol (Cayman Chemical, #600360). Fluorescence units were measured by SpectraMax iD3 Microplate Reader (Molecular Devices).
Real-time PCR
Real-time PCR was performed as described [52]. Briefly, viral supernatant was treated with 20 U/mL DNase (Thermo Fisher Scientific, #AM2238) for thirty minutes prior to infection to minimize contamination from plasmid DNA that was transfected to produce the virus. To control for plasmid carryover, one group of cultures was incubated with viral stock heated at 65° for 10 minutes. Cells were lysed as indicated and total DNA was isolated using a DNeasy Blood and Tissue Kit (Qiagen, #69506). Isolated DNA was quantitated using a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific) and diluted to 20 ng/μL. DNA was amplified with TaqMan Fast Advanced Master Mix (Thermo Fisher, #4444557) in a StepOnePlus Real-Time PCR System (Applied Biosystems). The following primers were used:
- Late reverse transcription:
- Forward: 5’-TGTGTGCCCGTCTGTTGTGT-3’
- Reverse: 5’-GAGTCCTGCGTCGAGAGATC-3’
- Probe: 5’-FAM-CAGTGGCGCCCGAACAGGGA-TAMRA-3’
- 2-LTR:
- Forward: 5’-TGGTTAGACCAGATCTGAGCCT-3’
- Reverse: 5’-AGGTAGCCTTGTGTGTGGTAGATCC-3’
- Probe: 5’-FAM-TAGTGTGTGCCCGTCTGTTGTGTGAC-TAMRA-3’
- GAPDH (reference gene for Late RT and 2-LTR):
- Forward: 5’-GCATGGCCTTCCGTGTCCCC-3’
- Reverse: 5’-CCCTCCGACGCCTGCTTCAC-3’
- Probe: 5’-GGTGGACCTGACCTGCCGTCTAGA-3’
The ΔΔCt method was performed to calculate relative target gene copy numbers for late reverse transcription and 2-LTR products.
- Integrated Proviral DNA:
- 1st round PCR:
- Extracted genomic DNA was amplified by PCR with Platinum Taq DNA polymerase (Thermo Fisher, # 10966026) and dNTP mix (ThermoFisher, #18427013).
- First-Alu-F: 5’-AGCCTCCCGAGTAGCTGGGA-3’
- First-Alu-R: 5’- TTACAGGCATGAGCCACCG-3’
- First-gag-R: 5’- CAATATCATACGCCGAGAGTGCGCGCTTCAGCAAG-3’
- 2nd round PCR.
- Forward MH535: 5’- AACTAGGGAACCCACTGCTTAAG-3’
- Reverse tag-R: 5’-CAATATCATACGCCGAGAGTGC-3’
- Probe MH603: 5’-(FAM)-ACACTACTTGAAGCACTCAAGGCAAGCTTT-(TAMRA)-3’
- MH535: 5’-AACTAGGGAACCCACTGCTTAAG-3’
- MH536: 5’TCCACAGATCAAGGATATCTTGTC-3′
- Probe MH603: 5’-(FAM)-ACACTACTTGAAGCACTCAAGGCAAGCTTT-(TAMRA)-3’
Statistical analysis:
Data were analyzed using GraphPad Prism 8. The two-tailed student T-test was performed for single comparisons and the two-way ANOVA was used for multiple comparisons. P values are shown for each experiment.
Supplementary Material
Fig S3: L-[14C] cystine uptake is linear up to 40 minutes. PMA-differentiated WT THP-1 cells were washed and incubated in prewarmed Basal media for twenty minutes at 37°C followed by a five, ten, twenty or forty-minute incubation in [14C] cystine-containing transport media. The reaction was stopped by adding ice-cold basal media and DPMs were measured. Representative samples were lysed in 2% SDS solution and protein concentrations were measured by BCA assay. The graph includes data from one experiment. Error bars represent ± one SD from the mean.
Fig S1: SFN treatment causes upregulation of xCT in hMDM but not T cells. Primary CD4+ T cells and hMDMs were treated with 10 μM SFN or the equivalent amount of DMSO for twenty-four hours before lysis and characterization by western blot with antibodies for Nrf2, xCT or tubulin
Fig S2: CRISPR/Cas9-mediated frame-shifts in both alleles of xCT gene. WT THP-1 monocytes were transduced with a lentiviral vector that expresses CRISPR/Cas9 and xCT-specific gRNA. (A) PMA-differentiated WT and xCT KO THP-1 cells were lysed and characterized by western blotting with antibodies for xCT or tubulin. (B) The genomic DNA that includes the sequence targeted by the guide RNA was amplified using PCR and sequenced with the Sanger method. The upper chromatogram shows the mutant alleles and the lower shows the chromatogram from wild-type THP-1 cells for comparison. KO allele 1 (taller peaks) provided a stronger signal than KO allele 2 (minor peaks). KO allele 1 shows a single-residue deletion of a C (labeled Δ1) while KO allele 2 shows a 7-residue deletion, GATTTGG, (labeled Δ2). (C) The protein sequences, predicted from the sequencing data, for the xCT KO THP-1 line is shown.
Fig S4: xCT-mediated restriction is independent of reporter activity. PMA-differentiated WT THP-1 were treated with 5 μM AZT, 0.3125 mM, 0.625 mM or 1.25 mM SSZ or DMSO alone for twenty-four hours before being infected with VSV-G pseudotyped HIV-1 GFP reporter virus. Twenty-four hours post infection, viral supernatant was replaced with complete RPMI-1640 containing PMA. Twenty-four hours later, cells were fixed in 4% paraformaldehyde and GFP positive cells were measured by flow cytometry. The graphs represent data from one experiment. Error bars represent ± one SD from the mean.
Fig S5: SAMHD1 KO confirmation. WT and SAMHD1 KO THP-1 cells were lysed in Laemmli buffer and run on an SDS-PAGE gel for characterization by western blot with antibodies specific for SAMHD1, xCT or tubulin.
Fig S6: SSZ treatment decreases glutathione levels in all cell types tested. (A) THP-1 monocytes were treated with 0.625 mM or 1.25 mM SSZ or DMSO (solvent) alone in complete RPMI for twenty-four hours before being lysed and measured for glutathione concentrations according to manufacturer’s protocol. (B) Similar to (A) but HeLa treated in complete DMEM. (C) Similar to (B) but HEK293T. (D) Similar to (A) but SupT1. The graphs include data from one experiment. Error bars represent ± one SD from the mean.
Fig S7: SSZ treatment results in an increase in 2-LTR circle formation in HEK293T cells. HEK293T cells were treated with 0.625 mM SSZ, the equivalent amount of DMSO, or 10 μM AZT 24 hours before being infected with VSV-G pseudotyped HIV-1 GFP reporter virus. 8 and 24 hours post infection, cells were lysed and total DNA was extracted. Samples lysed at 8 hours post infection were probed with primers targeting sequence markers for late reverse transcription or 2-LTR circle formation for samples lysed 24 hours post infection by qPCR.
File S1: xCT is the highest upregulated transcript in hMDMs treated with SFN. Primary hMDMs were treated with 10, 20 or 30 μM SFN or the equivalent amount of DMSO prior to performing a microarray expression analysis. We compared the level of expression in SFN-treated samples versus the DMSO-treated controls and filtered for mRNAs that changed expression by at least 1.5-fold. xCT was expressed over 7-fold more than in DMSO-treated cells.
Acknowledgements
We thank Dawn Belville, Terra Cohn and Joanne George for administrative assistance; Jackson Maloney for assistance with flow cytometry; Emilee Patterson for laboratory assistance. This work was funded by NIH AI140993 to CN.
References
- 1.Furuya AK, et al. , Sulforaphane Inhibits HIV Infection of Macrophages through Nrf2. PLoS Pathog, 2016. 12(4): p. e1005581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mandal D and Prasad VR, Analysis of 2-LTR circle junctions of viral DNA in infected cells. Methods Mol Biol, 2009. 485: p. 73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moldovan L and Moldovan NI, Oxygen free radicals and redox biology of organelles. Histochemistry and Cell Biology, 2004. 122(4): p. 395–412. [DOI] [PubMed] [Google Scholar]
- 4.Dupré-Crochet S, Erard M, and Nüβe O, ROS production in phagocytes: why, when, and where? J Leukoc Biol, 2013. 94(4): p. 657–70. [DOI] [PubMed] [Google Scholar]
- 5.Ayala A, Muñoz MF, and Argüelles S, Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev, 2014. 2014: p. 360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jung T, Höhn A, and Grune T, The proteasome and the degradation of oxidized proteins: Part II - protein oxidation and proteasomal degradation. Redox Biol, 2014. 2: p. 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo C, et al. , Potential application of the oxidative nucleic acid damage biomarkers in detection of diseases. Oncotarget, 2017. 8(43): p. 75767–75777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birben E, et al. , Oxidative stress and antioxidant defense. World Allergy Organ J, 2012. 5(1): p. 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Staal FJ, et al. , Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proc Natl Acad Sci U S A, 1990. 87(24): p. 9943–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Asin S, et al. , NF-kappaB cis-acting motifs of the human immunodeficiency virus (HIV) long terminal repeat regulate HIV transcription in human macrophages. Journal of virology, 2001. 75(23): p. 11408–11416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mihm S, Galter D, and Droge W, Modulation of transcription factor NF kappa B activity by intracellular glutathione levels and by variations of the extracellular cysteine supply. Faseb j, 1995. 9(2): p. 246–52. [DOI] [PubMed] [Google Scholar]
- 12.Bai SK, et al. , beta-Carotene inhibits inflammatory gene expression in lipopolysaccharide-stimulated macrophages by suppressing redox-based NF-kappaB activation. Exp Mol Med, 2005. 37(4): p. 323–34. [DOI] [PubMed] [Google Scholar]
- 13.Kameoka M, et al. , Intracellular glutathione as a possible direct blocker of HIV type 1 reverse transcription. AIDS Res Hum Retroviruses, 1996. 12(17): p. 1635–8. [DOI] [PubMed] [Google Scholar]
- 14.Mauney CH, et al. , The SAMHD1 dNTP Triphosphohydrolase Is Controlled by a Redox Switch. Antioxid Redox Signal, 2017. 27(16): p. 1317–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patra KK, Bhattacharya A, and Bhattacharya S, Molecular dynamics investigation of a redox switch in the anti-HIV protein SAMHD1. Proteins, 2019. 87(9): p. 748–759. [DOI] [PubMed] [Google Scholar]
- 16.Wang Z, et al. , Functionality of Redox-Active Cysteines Is Required for Restriction of Retroviral Replication by SAMHD1. Cell Rep, 2018. 24(4): p. 815–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi A, et al. , Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol, 2004. 24(16): p. 7130–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang DD and Hannink M, Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol, 2003. 23(22): p. 8137–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moi P, et al. , Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proceedings of the National Academy of Sciences, 1994. 91(21): p. 9926–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Venugopal R and Jaiswal AK, Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci U S A, 1996. 93(25): p. 14960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chorley BN, et al. , Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res, 2012. 40(15): p. 7416–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bannai S, Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem, 1986. 261(5): p. 2256–63. [PubMed] [Google Scholar]
- 23.Sato H, et al. , Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem, 1999. 274(17): p. 11455–8. [DOI] [PubMed] [Google Scholar]
- 24.Sasaki H, et al. , Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem, 2002. 277(47): p. 44765–71. [DOI] [PubMed] [Google Scholar]
- 25.Habib E, et al. , Expression of xCT and activity of system xc(−) are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol, 2015. 5: p. 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lo M, Wang YZ, and Gout PW, The x(c)- cystine/glutamate antiporter: a potential target for therapy of cancer and other diseases. J Cell Physiol, 2008. 215(3): p. 593–602. [DOI] [PubMed] [Google Scholar]
- 27.Sugano K, et al. , Expression of xCT as a predictor of disease recurrence in patients with colorectal cancer. Anticancer Res, 2015. 35(2): p. 677–82. [PubMed] [Google Scholar]
- 28.Ji X, et al. , xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene, 2018. 37(36): p. 5007–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arensman MD, et al. , Cystine-glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity. Proc Natl Acad Sci U S A, 2019. 116(19): p. 9533–9542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaleeba JA and Berger EA, Kaposi’s sarcoma-associated herpesvirus fusion-entry receptor: cystine transporter xCT. Science, 2006. 311(5769): p. 1921–4. [DOI] [PubMed] [Google Scholar]
- 31.Veettil MV, et al. , Kaposi’s sarcoma-associated herpesvirus forms a multimolecular complex of integrins (alphaVbeta5, alphaVbeta3, and alpha3beta1) and CD98-xCT during infection of human dermal microvascular endothelial cells, and CD98-xCT is essential for the postentry stage of infection. J Virol, 2008. 82(24): p. 12126–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chakraborty S, Veettil MV, and Chandran B, Kaposi’s Sarcoma Associated Herpesvirus Entry into Target Cells. Front Microbiol, 2012. 3: p. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qin Z, et al. , Upregulation of xCT by KSHV-encoded microRNAs facilitates KSHV dissemination and persistence in an environment of oxidative stress. PLoS Pathog, 2010. 6(1): p. e1000742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kandasamy RK, et al. , A time-resolved molecular map of the macrophage response to VSV infection. NPJ Syst Biol Appl, 2016. 2: p. 16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barger CJ, et al. , Pan-Cancer Analyses Reveal Genomic Features of FOXM1 Overexpression in Cancer. Cancers (Basel), 2019. 11(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jimenez-Vidal M, et al. , Thiol modification of cysteine 327 in the eighth transmembrane domain of the light subunit xCT of the heteromeric cystine/glutamate antiporter suggests close proximity to the substrate binding site/permeation pathway. J Biol Chem, 2004. 279(12): p. 11214–21. [DOI] [PubMed] [Google Scholar]
- 37.Fogal B, et al. , System x(c)- activity and astrocytes are necessary for interleukin-1 beta-mediated hypoxic neuronal injury. J Neurosci, 2007. 27(38): p. 10094–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gout PW, et al. , Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia, 2001. 15(10): p. 1633–40. [DOI] [PubMed] [Google Scholar]
- 39.Plosker GL and Croom KF, Sulfasalazine: a review of its use in the management of rheumatoid arthritis. Drugs, 2005. 65(13): p. 1825–49. [DOI] [PubMed] [Google Scholar]
- 40.Yang WS, et al. , Regulation of ferroptotic cancer cell death by GPX4. Cell, 2014. 156(1–2): p. 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu M, et al. , Targeted exosome-encapsulated erastin induced ferroptosis in triple negative breast cancer cells. Cancer Sci, 2019. 110(10): p. 3173–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonifati S, et al. , SAMHD1 controls cell cycle status, apoptosis and HIV-1 infection in monocytic THP-1 cells. Virology, 2016. 495: p. 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hrecka K, et al. , Lentiviral Vpr usurps Cul4-DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc Natl Acad Sci U S A, 2007. 104(28): p. 11778–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laguette N, et al. , SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature, 2011. 474(7353): p. 654–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wen X, et al. , The HIV1 protein Vpr acts to promote G2 cell cycle arrest by engaging a DDB1 and Cullin4A-containing ubiquitin ligase complex using VprBP/DCAF1 as an adaptor. J Biol Chem, 2007. 282(37): p. 27046–57. [DOI] [PubMed] [Google Scholar]
- 46.Tan L, Ehrlich E, and Yu X-F, DDB1 and Cul4A Are Required for Human Immunodeficiency Virus Type 1 Vpr-Induced G2 Arrest. 2007. 81(19): p. 10822–10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Israel N, et al. , Redox status of cells influences constitutive or induced NF-kappa B translocation and HIV long terminal repeat activity in human T and monocytic cell lines. J Immunol, 1992. 149(10): p. 3386–93. [PubMed] [Google Scholar]
- 48.Stroud JC, et al. , Structural basis of HIV-1 activation by NF-kappaB--a higher-order complex of p50:RelA bound to the HIV-1 LTR. J Mol Biol, 2009. 393(1): p. 98–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Griffin GE, et al. , Activation of HIV gene expression during monocyte differentiation by induction of NF-kappa B. Nature, 1989. 339(6219): p. 70–3. [DOI] [PubMed] [Google Scholar]
- 50.Bachelerie F, et al. , HIV enhancer activity perpetuated by NF-kappa B induction on infection of monocytes. Nature, 1991. 350(6320): p. 709–12. [DOI] [PubMed] [Google Scholar]
- 51.Campbell LA, et al. , In vitro modeling of HIV proviral activity in microglia. Febs j, 2017. 284(23): p. 4096–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Graf L, et al. , Analysis of Early Phase HIV-1 Replication and Integration Events by Using Real-time PCR. Bio-protocol, 2019. 9(4): p. e3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kane M, et al. , MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature, 2013. 502(7472): p. 563–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu CC, et al. , Inhibition of Epstein-Barr virus reactivation in nasopharyngeal carcinoma cells by dietary sulforaphane. Mol Carcinog, 2013. 52(12): p. 946–58. [DOI] [PubMed] [Google Scholar]
- 55.Li Z, et al. , Natural Sulforaphane From Broccoli Seeds Against Influenza A Virus Replication in MDCK Cells. Natural Product Communications, 2019. 14(6): p. 1934578X19858221. [Google Scholar]
- 56.Kesic MJ, et al. , Nrf2 expression modifies influenza A entry and replication in nasal epithelial cells. Free Radic Biol Med, 2011. 51(2): p. 444–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wyler E, et al. , Single-cell RNA-sequencing of herpes simplex virus 1-infected cells connects NRF2 activation to an antiviral program. Nat Commun, 2019. 10(1): p. 4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilen CB, Tilton JC, and Doms RW, HIV: cell binding and entry. Cold Spring Harb Perspect Med, 2012. 2(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Finkelshtein D, et al. , LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci U S A, 2013. 110(18): p. 7306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gažová I, et al. , The Transcriptional Network That Controls Growth Arrest and Macrophage Differentiation in the Human Myeloid Leukemia Cell Line THP-1. 2020. 8(498). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goujon C, et al. , Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature, 2013. 502(7472): p. 559–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kabe Y, et al. , Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal, 2005. 7(3–4): p. 395–403. [DOI] [PubMed] [Google Scholar]
- 63.Mihm S, et al. , Inhibition of HIV-1 replication and NF-kappa B activity by cysteine and cysteine derivatives. Aids, 1991. 5(5): p. 497–503. [DOI] [PubMed] [Google Scholar]
- 64.Makropoulos V, Bruning T, and Schulze-Osthoff K, Selenium-mediated inhibition of transcription factor NF-kappa B and HIV-1 LTR promoter activity. Arch Toxicol, 1996. 70(5): p. 277–83. [DOI] [PubMed] [Google Scholar]
- 65.Weber CK, et al. , Suppression of NF-kappaB activity by sulfasalazine is mediated by direct inhibition of IkappaB kinases alpha and beta. Gastroenterology, 2000. 119(5): p. 1209–18. [DOI] [PubMed] [Google Scholar]
- 66.Okamoto K, et al. , Redox-dependent regulation of nuclear import of the glucocorticoid receptor. J Biol Chem, 1999. 274(15): p. 10363–71. [DOI] [PubMed] [Google Scholar]
- 67.Le Rouzic E, et al. , HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the Cul4-DDB1 ubiquitin ligase. Cell Cycle, 2007. 6(2): p. 182–8. [DOI] [PubMed] [Google Scholar]
- 68.Wahl C, et al. , Sulfasalazine: a potent and specific inhibitor of nuclear factor kappa B. J Clin Invest, 1998. 101(5): p. 1163–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cronstein BN and Sitkovsky M, Adenosine and adenosine receptors in the pathogenesis and treatment of rheumatic diseases. Nat Rev Rheumatol, 2017. 13(1): p. 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Norton TD, et al. , Vpx-containing dendritic cell vaccine induces CTLs and reactivates latent HIV-1 in vitro. Gene Ther, 2015. 22(3): p. 227–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Siva C and Brasington RD, Worsening of arthritis with antiretroviral therapy: the coexistence of rheumatoid arthritis and human immunodeficiency virus infection revisited. J Clin Rheumatol, 2001. 7(1): p. 42–6. [DOI] [PubMed] [Google Scholar]
- 72.Hernandez-Ramirez RU, et al. , Cancer risk in HIV-infected people in the USA from 1996 to 2012: a population-based, registry-linkage study. Lancet HIV, 2017. 4(11): p. e495–e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S3: L-[14C] cystine uptake is linear up to 40 minutes. PMA-differentiated WT THP-1 cells were washed and incubated in prewarmed Basal media for twenty minutes at 37°C followed by a five, ten, twenty or forty-minute incubation in [14C] cystine-containing transport media. The reaction was stopped by adding ice-cold basal media and DPMs were measured. Representative samples were lysed in 2% SDS solution and protein concentrations were measured by BCA assay. The graph includes data from one experiment. Error bars represent ± one SD from the mean.
Fig S1: SFN treatment causes upregulation of xCT in hMDM but not T cells. Primary CD4+ T cells and hMDMs were treated with 10 μM SFN or the equivalent amount of DMSO for twenty-four hours before lysis and characterization by western blot with antibodies for Nrf2, xCT or tubulin
Fig S2: CRISPR/Cas9-mediated frame-shifts in both alleles of xCT gene. WT THP-1 monocytes were transduced with a lentiviral vector that expresses CRISPR/Cas9 and xCT-specific gRNA. (A) PMA-differentiated WT and xCT KO THP-1 cells were lysed and characterized by western blotting with antibodies for xCT or tubulin. (B) The genomic DNA that includes the sequence targeted by the guide RNA was amplified using PCR and sequenced with the Sanger method. The upper chromatogram shows the mutant alleles and the lower shows the chromatogram from wild-type THP-1 cells for comparison. KO allele 1 (taller peaks) provided a stronger signal than KO allele 2 (minor peaks). KO allele 1 shows a single-residue deletion of a C (labeled Δ1) while KO allele 2 shows a 7-residue deletion, GATTTGG, (labeled Δ2). (C) The protein sequences, predicted from the sequencing data, for the xCT KO THP-1 line is shown.
Fig S4: xCT-mediated restriction is independent of reporter activity. PMA-differentiated WT THP-1 were treated with 5 μM AZT, 0.3125 mM, 0.625 mM or 1.25 mM SSZ or DMSO alone for twenty-four hours before being infected with VSV-G pseudotyped HIV-1 GFP reporter virus. Twenty-four hours post infection, viral supernatant was replaced with complete RPMI-1640 containing PMA. Twenty-four hours later, cells were fixed in 4% paraformaldehyde and GFP positive cells were measured by flow cytometry. The graphs represent data from one experiment. Error bars represent ± one SD from the mean.
Fig S5: SAMHD1 KO confirmation. WT and SAMHD1 KO THP-1 cells were lysed in Laemmli buffer and run on an SDS-PAGE gel for characterization by western blot with antibodies specific for SAMHD1, xCT or tubulin.
Fig S6: SSZ treatment decreases glutathione levels in all cell types tested. (A) THP-1 monocytes were treated with 0.625 mM or 1.25 mM SSZ or DMSO (solvent) alone in complete RPMI for twenty-four hours before being lysed and measured for glutathione concentrations according to manufacturer’s protocol. (B) Similar to (A) but HeLa treated in complete DMEM. (C) Similar to (B) but HEK293T. (D) Similar to (A) but SupT1. The graphs include data from one experiment. Error bars represent ± one SD from the mean.
Fig S7: SSZ treatment results in an increase in 2-LTR circle formation in HEK293T cells. HEK293T cells were treated with 0.625 mM SSZ, the equivalent amount of DMSO, or 10 μM AZT 24 hours before being infected with VSV-G pseudotyped HIV-1 GFP reporter virus. 8 and 24 hours post infection, cells were lysed and total DNA was extracted. Samples lysed at 8 hours post infection were probed with primers targeting sequence markers for late reverse transcription or 2-LTR circle formation for samples lysed 24 hours post infection by qPCR.
File S1: xCT is the highest upregulated transcript in hMDMs treated with SFN. Primary hMDMs were treated with 10, 20 or 30 μM SFN or the equivalent amount of DMSO prior to performing a microarray expression analysis. We compared the level of expression in SFN-treated samples versus the DMSO-treated controls and filtered for mRNAs that changed expression by at least 1.5-fold. xCT was expressed over 7-fold more than in DMSO-treated cells.