

Abstract
The MYC oncoprotein activates and represses gene expression in a transcription‐dependent or transcription‐independent manner. Modification of mRNA emerges as a key gene expression regulatory nexus. We sought to determine whether MYC alters mRNA modifications and report here that MYC promotes cancer progression by down‐regulating N6‐methyladenosine (m6A) preferentially in transcripts of a subset of MYC‐repressed genes (MRGs). We find that MYC activates the expression of ALKBH5 and reduces m6A levels in the mRNA of the selected MRGs SPI1 and PHF12. We also show that MYC‐regulated m6A controls the translation of MRG mRNA via the specific m6A reader YTHDF3. Finally, we find that inhibition of ALKBH5, or overexpression of SPI1 or PHF12, effectively suppresses the growth of MYC‐deregulated B‐cell lymphomas, both in vitro and in vivo. Our findings uncover a novel mechanism by which MYC suppresses gene expression by altering m6A modifications in selected MRG transcripts promotes cancer progression.
Keywords: ALKBH5, m6A, MYC, MYC‐repressed genes, oncogenesis
Subject Categories: Cancer; Chromatin, Epigenetics, Genomics & Functional Genomics; RNA Biology
MYC suppresses gene expression by indirectly altering m6A modifications in transcripts of MYC‐repressed genes via ALKBH5 to promote cancer progression.
Introduction
MYC is critical for cell proliferation, apoptosis, differentiation, metabolism, somatic cell reprogramming, and other key processes under normal or pathological conditions, and mediates these functions by regulating numerous target genes (Fernandez et al, 2003; Dang, 2012; Carroll et al, 2018). MYC regulates up to 15% of all human genes, among which nearly one‐third of the putative target genes are repressed by MYC (O'Connell et al, 2003; Dang et al, 2006; Zeller et al, 2006; Luscher & Vervoorts, 2012). Studies suggest that instead of regulating transcription of a new gene set, deregulated MYC functions as a transcriptional signal amplifier of extant active genes in a context‐dependent manner (Lin et al, 2012; Nie et al, 2012); additional evidence also suggests that oncogenic MYC regulates gene expression selectively to promote cellular growth and cancer progression (Sabo et al, 2014; Walz et al, 2014). Despite many advances in understanding the roles of MYC, functions of MYC that are independent of canonical transcriptional regulation have not been fully evaluated.
Also, while the mechanisms whereby MYC activates transcription have been studied in considerable detail, the mode by which MYC represses gene expression is less well understood. Several mechanisms have been proposed: (i) A zinc‐finger transcription factor MIZ‐1 binds to initiator elements and activates transcription, whereas subsequent binding of MYC to MIZ‐1 inhibits MIZ‐1‐mediated activation (Herkert & Eilers, 2010; Cole, 2014). (ii) Another mechanism, which may overlap with the first, is that MYC recruits histone deacetylases (HDACs) to a set of its target genes, and directly leads to histone deacetylation and compaction of chromatin structure to inhibit gene activation (Herkert & Eilers, 2010; Cole, 2014). (iii) Overexpression of MYC activates the histone‐lysine N‐methyltransferase EZH2 or G9a, and thereby enhances the levels of H3K27me3 or H3K9me2 on certain MYC‐repressed genes, which inhibits expression of these genes (Kaur & Cole, 2013; Tu et al, 2018). And (iv) a critical indirect transcriptional regulation mode for MYC‐mediated gene repression occurs via its ability to activate non‐coding RNAs—such as microRNAs and long‐non‐coding RNAs—and thus repress gene expression at the protein level (Dang, 2012). However, these mechanisms explain only about 50% of MRGs; mechanisms affecting the remaining MRGs are still unknown.
N6‐Methyladenosine (m6A) is the most prevalent internal modification in mammalian mRNAs, and participates in various fundamental bioprocesses as well as cancer (Cao et al, 2016; Meyer & Jaffrey, 2017; Yang et al, 2018; Shi et al, 2019). Methyltransferases and demethylases, including METTL3, METTL14, ALKBH5, and FTO, are involved in the progression of certain types of cancers by regulation of m6A modification (Lin et al, 2016; Zhang et al, 2016a; Barbieri et al, 2017; Zhang et al, 2017; Li et al, 2017b; Su et al, 2018; Weng et al, 2018; Lan et al, 2019). Whether and how MYC regulates mRNA m6A modifications remain largely unexplored.
Here we use validated models of MYC‐driven B‐cell lymphoma to investigate the role of mRNA modifications in the aberrant expression of MRGs. Using integrative LC‐MS, high‐throughput sequencing, functional studies, and human cancer sample analyses, we document that MYC down‐regulates m6A preferentially in transcripts of certain MRGs, thereby reducing the expression of these MRGs, resulting in cancer progression. We thus unveil a novel mechanism by which MYC represses gene expression via RNA modifications during cancer progression.
Results
MYC down‐regulates m6A levels of mRNA in B‐cell lymphoma cells
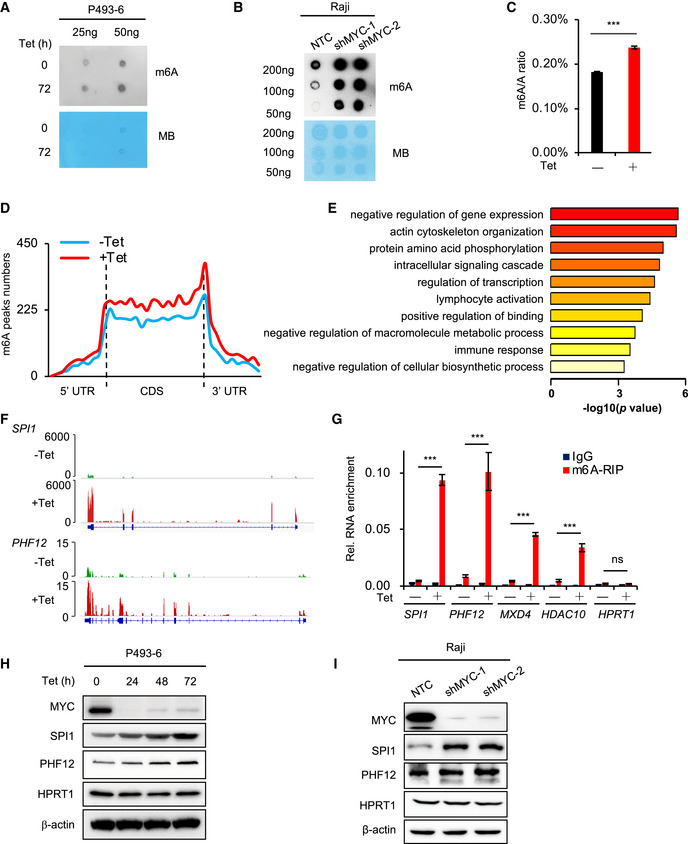
To investigate the effects of MYC on mRNA m6A modification, we used the human P493‐6 B‐cell model of Burkitt’s lymphoma (BL) (Schuhmacher et al, 1999; Pajic et al, 2000). P493‐6 cells carry a MYC tetracycline (Tet)‐off system, which enables the generation of cells with low or high MYC expression. To investigate whether MYC regulates mRNA m6A levels, we determined the levels of m6A in P493‐6 cells that were treated with Tet for 72 h or 0 h. Use of a dot blot assay with purified mRNA showed that the level of mRNA m6A modification was significantly increased when P493‐6 cells were treated with Tet (Fig 1A). Our results were confirmed in the widely used BL cell line Raji, which also exhibited significantly elevated mRNA m6A levels after knockdown of MYC with specific shRNAs (Fig 1B). The dot blot results thus indicate that MYC down‐regulates total mRNA m6A levels. This finding was verified with use of HPLC‐MS, and the same samples of P493‐6 cells, treated with Tet or not: We found that the m6A/A ratio is significantly increased in the Tet‐treated sample (Fig 1C). We thus conclude that loss of MYC in B‐cell lymphoma cells results in increased global mRNA m6A levels.
Figure 1. MYC down‐regulates mRNA m6A levels and inhibits protein expression of selected MRGs.
-
A, Bm6A dot blot of the P493‐6 cells treated with Tet for 0 h or 72 h (A) or Raji cells that expressed NTC or MYC shRNAs (B); equal mRNA loading was verified by methylene blue staining. The shown data are representative of at least three independent experiments.
-
CQuantification of m6A abundance in (A) by HPLC‐MS. ***P < 0.001 as compared to corresponding high MYC group (mean ± SD, n = 3 biological replicates, Student's t‐test).
-
DMetagene profiles of m6A peak distribution along a normalized transcript composed of three rescaled non‐overlapping segments: 5′UTR, CDS, and 3′UTR in P493‐6 cells treated with Tet for 0 h or 72 h.
-
ERepresentative GO term analysis of transcripts with significantly up‐regulated m6A peaks in P493‐6 cells treated with Tet for 72 h.
-
FIGV graph showing location of m6A peaks on representative genes.
-
Gm6A‐RIP assay in P493‐6 cells treated with Tet for 0 h or 72 h. HPRT1 serves as negative control. ***P < 0.001 as compared to corresponding high MYC group, ns, not significant (mean ± SD, n = 3 biological replicates, Student's t‐test).
-
H, IWestern blot analysis for protein levels in P493‐6 cells treated with Tet for 0, 24, 48, and 72 h (H) or in Raji cells that expressed NTC or MYC shRNAs (I). HPRT1 and β‐actin serve as negative and loading controls, respectively. Data are representative of at least three independent experiments.
Source data are available online for this figure.
To identify transcriptome‐wide methylation patterns regulated by MYC, we immunoprecipitated m6A methylated poly (A+) RNAs (MeRIP) and then sequenced and profiled mRNA m6A methylation in P493‐6 cells that were treated with Tet for 72 h or 0 h. Consistent with previous reports, mRNA m6A peaks for both high MYC and low MYC samples were abundant in coding sequences (CDS), intron sequences, and 3′ untranslated regions (3′UTR) (Figs 1D and EV1A), and the m6A peak was significantly enriched in GGACU/A, in both high MYC and low MYC samples (Fig EV1B). Analysis of the peak numbers and the density of m6A peaks in both samples revealed that down‐regulation of MYC resulted in higher m6A modifications across UTRs, CDS, and introns (Figs 1D and EV1C), consistent with a global effect of MYC on gene‐associated mRNA m6A levels.
Figure EV1. MeRIP analyses show that MYC down‐regulates m6A levels of mRNA.
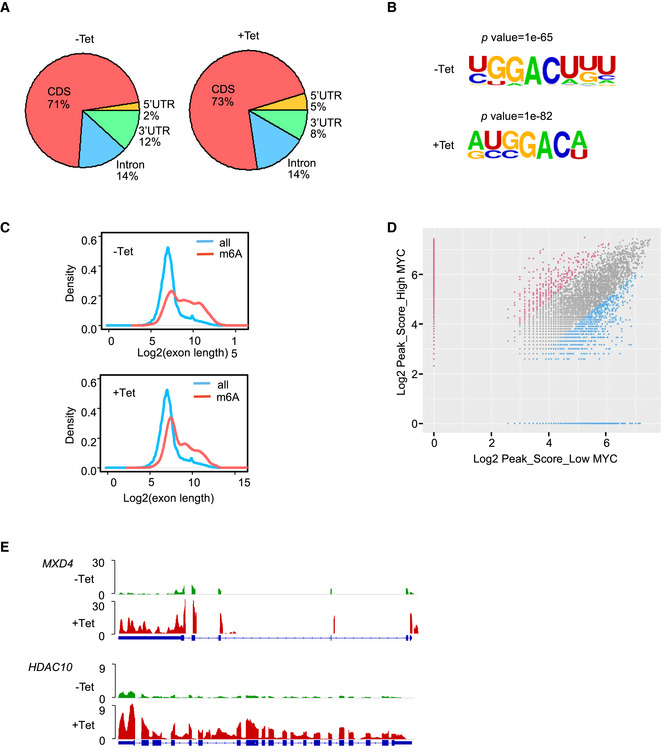
- Pie chart depicting the fraction of m6A peaks in four transcript segments in P493‐6 cells treated with Tet for 0 h or 72 h.
- Sequence motif identified within m6A peaks by HOMER database.
- Distribution of m6A sites along the length of mRNA transcripts.
- Scatter plots showing the m6A enrichment in mRNA of high MYC and low MYC samples. m6A‐containing mRNA with significantly increased and decreased peak enrichment is highlighted in red and green, respectively (chi‐square test, P < 0.05).
- IGV graph showing location of m6A peaks on representative genes.
MYC preferentially down‐regulates m6A levels in mRNAs and inhibits the expression of selected MRGs
To study which genes have their m6A methylations regulated by MYC, we used the MeRIP‐seq data and identified 2,542 genes that showed significantly increased m6A levels in low MYC samples (Fig EV1D), suggesting that MYC rewires a global m6A modification of genes. Gene ontology (GO) term enrichment analysis revealed that this group of negative regulation of gene expression is highly enriched (Fig 1E). These genes from this enriched group are repressed by MYC, suggesting that MYC preferentially down‐regulates the levels of mRNA m6A modification of these MYC‐repressed genes (MRGs). Integrative Genomics Viewer (IGV) analysis showed that m6A peaks were increased in genes, such as B‐lymphoid cell development activation transcription factor SPI1, SIN3A‐interacting transcriptional repressor PHF12, Max‐interacting transcriptional repressor MXD4, and the histone deacetylase HDAC10 (Figs 1F and EV1E). To document whether MYC regulates m6A modification at these genes, we purified poly (A)+ mRNA and used an m6A antibody to perform a MeRIP assay in P493‐6 cells that were treated or not with Tet. MeRIP results showed that m6A enrichment was significantly higher at these MRG transcripts in cell samples with low MYC expression, relative to that in high MYC samples (Fig 1G). As a negative control, we found no effects on HPRT1 (Fig 1G). These results suggest that MYC preferentially down‐regulates the levels of mRNA m6A modification of certain MRGs.
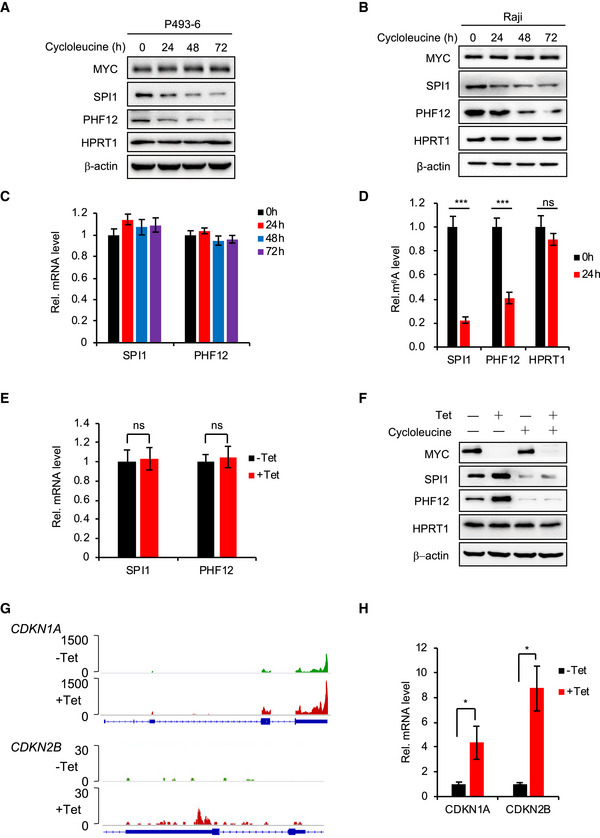
We next investigated whether MYC‐regulated mRNA m6A modification affects the expression of these MRGs. We blocked the m6A modification by treating P493‐6 cells and Raji cells with cycloleucine, a competitive inhibitor of methionine adenosyltransferase that inhibits m6A modification (Niu et al, 2013), and found no changes in MYC expression over 72 h (Fig EV2A and B). Cycloleucine treatment had little effect on the mRNA expression levels of selected MRGs SPI1 and PHF12 (Fig EV2C) but significantly decreased their protein levels in P493‐6 cells as well as Raji cells (Fig EV2A and B). The m6A RNA immunoprecipitation (RIP) assay confirmed that cycloleucine significantly reduced mRNA m6A levels of SPI1 and PHF12 (Fig EV2D). Together, these results suggest that the m6A modification affects the protein expression of MRGs. Reduced MYC expression had no effect on mRNA levels of SPI1 and PHF12 (Fig EV2E) but up‐regulated the protein levels of genes in both P493‐6 and Raji cells (Fig 1H and I).
Figure EV2. MYC and m6A down‐regulate protein expression but not mRNA expression of SPI1 and PHF12.
-
A, BWestern blot analysis for protein levels of MYC, SPI1, and PHF12 in P493‐6 cells (A) or Raji cells (B) that treated with the cycloleucine for 0, 24, 48, and 72 h. HPRT1 and β‐actin serve as negative control and loading control, respectively. Data are representative of at least three independent experiments.
-
CRT–qPCR analysis of the mRNA level of SPI1 and PHF12 in P493‐6 cells treated with cycloleucine for 0, 24, 48, and 72 h. Data were presented as mean (±SD), n = 3 biological replicates.
-
DRT–qPCR analysis of the m6A levels of MRGs (SPI1 and PHF12) in P493‐6 cells treated with cycloleucine for 0 h or 24 h. HPRT1 serves as negative control. Data are mean (±SD), n = 3 biological replicates. ***P < 0.001 relative to corresponding 0 h group (Student's t‐test).
-
ERT–qPCR analysis of the mRNA level of SPI1 and PHF12 in P493‐6 cells treated with Tet for 0 h or 24 h. Data were presented as mean (±SD), n = 3 biological replicates (Student's t‐test).
-
FWestern blot analysis for protein levels of MYC and MRGs (SPI1 and PHF12) in P493‐6 cells treated with Tet, cycloleucine, or both. HPRT1 and β‐actin serve as negative and loading controls, respectively. Data are representative of at least three independent experiments.
-
GIGV showing the locations of m6A peaks on CDKN1A and CDKN2B.
-
HRT–qPCR analysis of the mRNA levels of CDKN1A and CDKN2B in P493‐6 cells treated with Tet for 0 h or 24 h; data were presented as mean (±SD), n = 3 biological replicates, *P < 0.05 relative to corresponding ‐Tet group (Student's t‐test).
Source data are available online for this figure.
To test whether MYC regulates the protein levels of MRGs via m6A modification, we undertook a combination experiment with Tet as well as cycloleucine treatments in P493‐6 cells: Tet treatment increased the protein expression of SPI1 and PHF12, but this increase was eliminated when the cells were further treated with cycloleucine (Fig EV2F). This strongly suggests that MYC regulates the protein levels of these MRGs via m6A modification.
We also looked at both the m6A modification and gene expression of the well‐known MRGs CDKN1A and CDKN2B. IGV analysis showed that the level of m6A modification of CDKN1A mRNA was also increased when MYC was low (Fig EV2G), suggesting the possibility that MYC regulates the gene expression of CDKN1A via m6A modification. Of note, though there was a tiny peak at the 3′UTR region of CDKN2B, the m6A modification level of CDKN2B was very low and there was no enriched m6A peak for CDKN2B by our peak calling method (Fig EV2G), suggesting that CDKN2B is not an m6A modification target in this context. Our RT–qPCR results showed that MYC repressed the mRNA level of CDKN1A and CDKN2B (Fig EV2H), indicating a transcriptional regulation by MYC. These data suggest that, in addition to transcriptional regulation of CDKN1A, MYC might also regulate the mRNA m6A modification of CDKN1A, and MYC might regulate CDKN2B only by transcription.
ALKBH5 demethylates m6A‐modified mRNA and inhibits protein expression of selected MRGs
We next wanted to identify the enzymes responsible for regulating mRNA m6A modification by MYC. We assessed the levels of the mRNA m6A methyltransferases METTL3 and METTL14, and demethylases ALKBH5 and FTO in P493‐6 cells and Raji cells. Tet treatment or MYC knockdown by shRNAs reduced both mRNA and protein levels of the demethylases ALKBH5 and FTO (Fig 2A–C). In contrast, there was virtually no change in the expression of the methyltransferases METTL3 or METTL14 (Fig 2A–C). Analyzing the expression of MYC, ALKBH5, and FTO in 78 lymphocyte cell lines from Cancer Cell Line Encyclopedia (CCLE) datasets and examining in various other cell lines, we found a strong correlation of MYC and ALKBH5 and FTO (Fig EV3, EV4, EV5).
Figure 2. ALKBH5 removes m6A from mRNA and inhibits the protein expression of SPI1/PHF12.
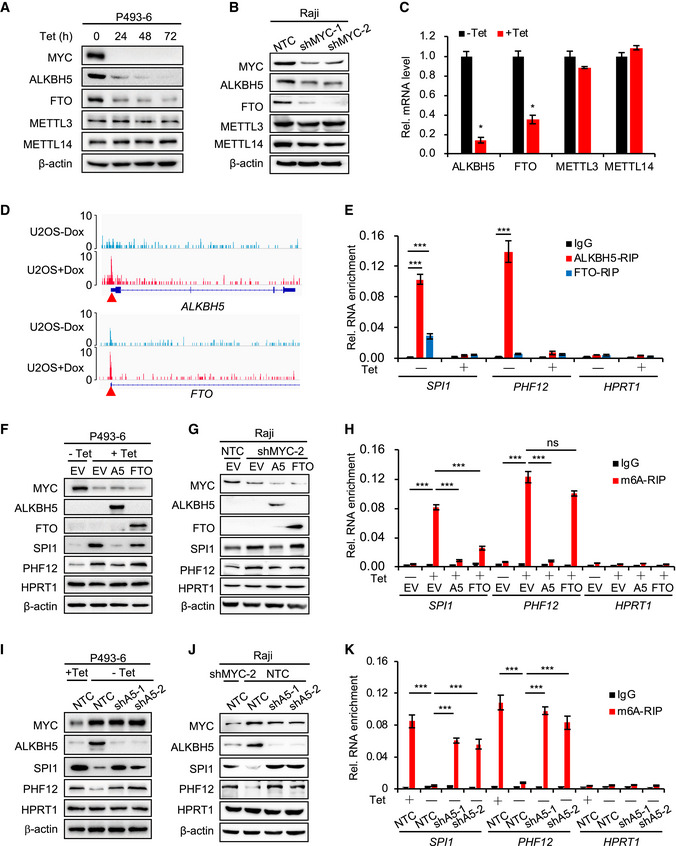
-
A, BWestern blot analysis for protein levels of m6A methyltransferases (METTL3 and METTL14) and demethylases (ALKBH5 and FTO) in P493‐6 cells treated with Tet for 0, 24, 48, and 72 h (A) or in Raji cells that expressed NTC or MYC shRNAs (B). β‐actin serves as loading controls. Data are representative of at least three independent experiments.
-
CRT–qPCR analysis of the mRNA levels of methyltransferases and demethylases in P493‐6 cells treated with Tet for 0 h or 24 h. Data were presented as mean (±SD), n = 3 biological replicates. *P < 0.05 relative to corresponding ‐Tet group (Student's t‐test).
- D
-
ERIP assay, using ALKBH5, FTO, or IgG antibody to detect the binding to MRGs (SPI1 and PHF12) in P493‐6 cells treated with Tet or not. HPRT1 serves as negative control. ***P < 0.001 as compared to corresponding IgG group (mean ± SD, n = 3 biological replicates, Student's t‐test).
-
F, GWestern blot analysis for protein levels in P493‐6 cells that overexpressed empty vector (EV), ALKBH5, or FTO and were then treated with Tet or not (F) or in Raji cells expressed NTC or MYC shRNAs (G). HPRT1 and β‐actin served as negative and loading controls, respectively. Data are representative of at least three independent experiments.
-
Hm6A RIP assay in (F) that P493‐6 cells that overexpressed EV, ALKBH5, or FTO and were then treated with Tet or not. HPRT1 serves as negative control. ***P < 0.001 as compared between indicated groups. ns, not significant (mean ± SD, n = 3 biological replicates, Student's t‐test).
-
I, JWestern blot analysis for protein levels in P493‐6 cells that expressed NTC or ALKBH5 shRNAs and were then treated with Tet or not (I) or in Raji cells expressed NTC or ALKBH5 shRNAs and knocked down MYC or not (J). HPRT1 and β‐actin serve as negative and loading controls, respectively. Data are representative of at least three independent experiments.
-
Km6A RIP assay in (I). HPRT1 serves as negative control. ***P < 0.001 as compared between indicated groups (mean ± SD, n = 3 biological replicates, Student's t‐test).
Source data are available online for this figure.
Figure EV3. MYC regulates demethylases ALKBH5 and FTO, and ALKBH5 inhibits the protein expression of SPI1 and PHF12.
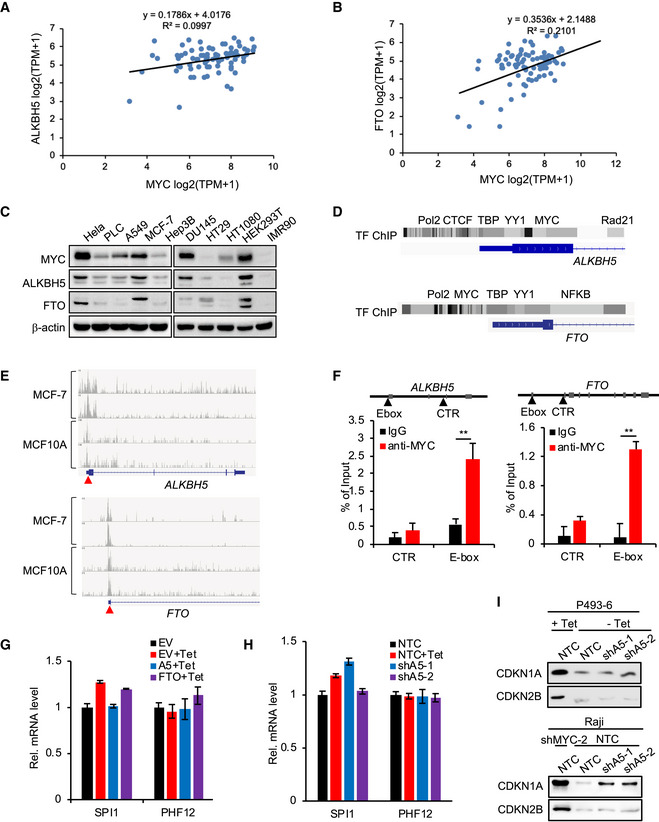
-
A, BAnalyses of the co‐expression of MYC and ALKBH5 (A) or MYC and FTO (B) in 78 lymphocyte cell lines from CCLE. Linregress P value for (A) is 4.87E‐3 and for (B) is 2.44E‐5.
-
CWestern blot analysis for protein levels of MYC, ALKBH5, and FTO in ten different human cell lines. β‐actin serves as loading controls. Western blot data are representative of at least three independent experiments.
- D
- E
-
FChIP experiment was performed in P493‐6 cells using IgG or anti‐MYC antibody. The occupancy of potential E‐Box in ALKBH5 and FTO genes by MYC was determined by RT–qPCR. Control (CTR) primer sets were also included. Data were presented as mean (±SD), n = 3 biological replicates. **P < 0.01 as compared to corresponding IgG group (Student's t‐test).
-
GRT–qPCR analysis of the mRNA level of SPI1 and PHF12 in P493‐6 cells that overexpressed EV, ALKBH5, or FTO and were then treated with Tet or not. RT–qPCR data were presented as mean (±SD), n = 3 biological replicates.
-
HRT–qPCR analysis of the mRNA level of SPI1 and PHF12 in P493‐6 cells that expressed NTC or ALKBH5 shRNAs and were then treated with Tet or not. RT–qPCR data were presented as mean (±SD), n = 3 biological replicates.
-
IWestern blot analysis for protein levels of CDKN1A and CDKN2B in Fig 2I and J samples that P493‐6 cells expressing NTC or ALKBH5 shRNAs and were then treated with or without Tet or in Raji cells expressing NTC or ALKBH5 shRNAs with or without MYC knockdown.
Source data are available online for this figure.
Figure EV4. YTHDF3 does not regulate mRNA expression but promotes mRNA translation.
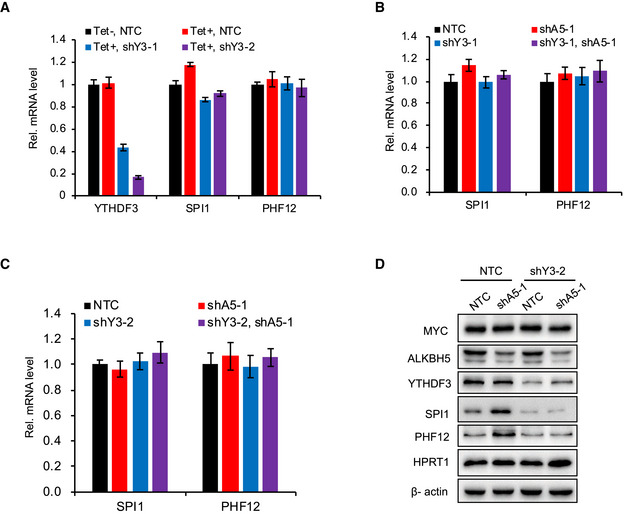
-
ART–qPCR analysis of the mRNA level of SPI1 and PHF12 in P493‐6 cells that expressed NTC or YTHDF3 shRNAs and were then treated with Tet or not. Data were presented as mean (±SD), n = 3 biological replicates.
-
B, CRT–qPCR assay for mRNA levels of SPI1 and PHF12 in P493‐6 cells that expressed NTC, ALKBH5 shRNA, or YTHDF3 shRNA, or co‐expressed ALKBH5 as well as YTHDF3 shRNAs. Data were presented as mean (±SD), n = 3 biological replicates.
-
DWestern blot analysis for protein levels in P493‐6 cells that expressed NTC, or ALKBH5 shRNA, or YTHDF3 shRNA. HPRT1 and β‐actin serve as negative and loading controls, respectively. Western blot data are representative of at least three independent experiments.
Source data are available online for this figure.
Figure EV5. The MYC‐ALKBH5‐m6A‐SPI1/PHF12 axis is critical for cancer progression.
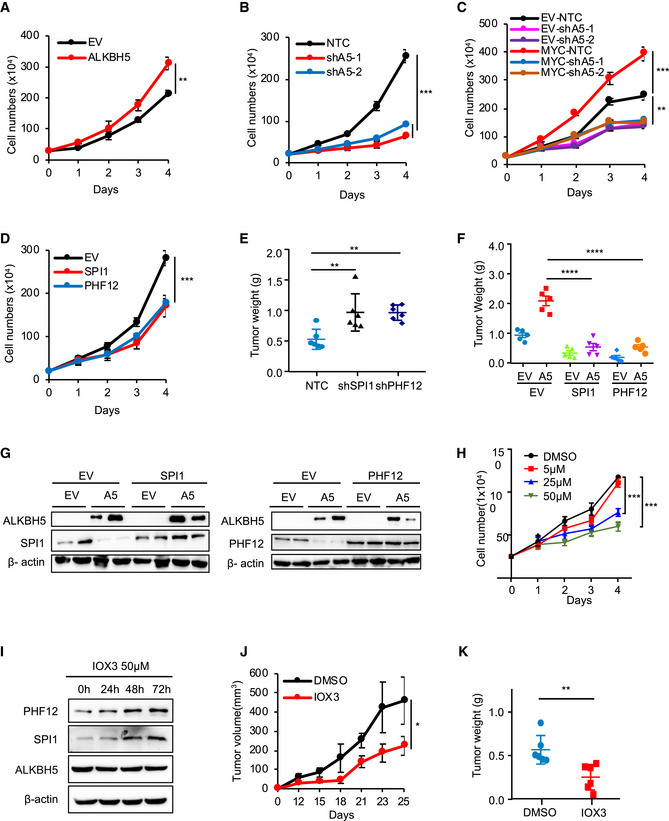
-
A, BTrypan blue counting was used to analyze growth curves for P493‐6 cells overexpressing ALKBH5 (A) and ALKBH5 shRNAs (B). **P < 0.01 and ***P < 0.001 as compared between indicated groups (mean ± SD, n = 4 biological replicates, Student's t‐test).
-
CTrypan blue counting was used to analyze growth curves for EV‐ or MYC‐expressing Raji cells that further infected with shALKBH5. **P < 0.01 and ***P < 0.001 as compared between indicated groups (mean ± SD, n = 3 biological replicates, Student's t‐test).
-
DTrypan blue counting was used to analyze growth curves for P493‐6 cells overexpressing MRGs (SPI1 or PHF12). ***P < 0.001 as compared between indicated groups (mean ± SD, n = 3 biological replicates, Student's t‐test).
- E, F
-
GWestern blot analysis for protein levels of ALKBH5, SPI1, and PHF12 from xenograft tumors. β‐actin serves as loading controls.
-
HCell growth curves for P493‐6 cells that treated with different doses of IOX3. ***P < 0.001 as compared to between indicated groups (mean ± SD, n = 3 biological replicates, Student's t‐test).
-
IWestern blot analysis for protein levels of ALKBH5, SPI1, and PHF12 in P493‐6 cells treated with the ALKBH5 inhibitor IOX3 for 0, 24, 48, and 72 h. β‐actin serves as loading control. The Western blot data are representative of at least three independent experiments.
-
J, KP493‐6 cells were injected subcutaneously into nude mice (n = 5 for each group). Mice were received every other day either IOX3 or DMSO (5% DMSO/H2O) by intragastric administration from 12 days postinjection (J). Tumor weights were measured at the end of the experiment (day 25) (K). Data are presented as mean (±SEM). *P < 0.05 and ***P < 0.001 as compared between indicated groups (Student's t‐test).
Source data are available online for this figure.
We analyzed the open chromatin transcription factor binding sites by chromatin immunoprecipitation sequencing (ChIP‐seq) from Encyclopedia of DNA Elements (ENCODE) datasets and found the binding sites of MYC at both ALKBH5 promoter and FTO promoter (Fig EV3D). Specifically, MYC bound to the promoter regions of ALKBH5 and FTO in MCF‐7 and MCF10A cells (Fig EV3E). We also analyzed published ChIP‐seq datasets (Walz et al, 2014) and found high levels of MYC bound to the promoter regions of ALKBH5 and FTO in U2OS cells (Fig 2D). And ChIP‐qPCR results in P493‐6 cells showed that MYC bound to the E‐box of ALKBH5 and FTO (Fig EV3F), supporting the hypothesis that MYC transcriptionally up‐regulates these two demethylases.
To investigate whether ALKBH5 and FTO demethylate the mRNA m6A modifications of MRG transcripts, we performed a RIP assay using antibodies against ALKBH5 and FTO. We found abundant ALBKH5 bound to the mRNA of the selected MRGs SPI1 and PHF12 (Fig 2E) but notably less so FTO. In Tet‐treated cells, overexpression of ALKBH5 decreased protein levels of SPI1 and PHF12 (Fig 2F), without affecting mRNA levels (Fig EV3G). The effect of MYC on expression of MRG proteins was eliminated when ALKBH5 was overexpressed (Fig 2F). Also, overexpression of FTO had little or no effect on the expression of PHF12 and SPI1 (Fig 2F). Similar results were observed in Raji cells (Fig 2G).
Next, we performed an m6A RIP assay in the same cells as in Fig 2F and found that when MYC was low following Tet treatment, the mRNA m6A levels of MRGs SPI1 and PHF12 were high, and the effect of MYC on mRNA m6A modification of MRGs was eliminated when ALKBH5 (but not FTO) was overexpressed (Fig 2H). To further validate whether ALKBH5 regulates mRNA m6A modification and protein expression of MRGs, we performed gene knockdown experiments in P493‐6 and Raji cells using ALKBH5 shRNAs. As expected, protein but not mRNA expression of SPI1 and PHF12 was decreased in high MYC‐expressing cells compared to low MYC‐expressing cells (Figs 2I and J, and EV3H). These effects were reversed by knockdown of ALKBH5 (Fig 2I and J). Knockdown of ALKBH5 increased the mRNA m6A levels of SPI1 and PHF12 and reversed the inhibitory effect of MYC on m6A modification (Fig 2K). Collectively, our data demonstrate that ALKBH5 demethylates mRNA m6A modifications of certain MRGs, which reduces their protein expression.
Of note, we also investigated whether ALKBH5 is involved in regulating CDKN1A and CDKN2B and detected the protein level of CDKN1A and CDKN2B in the ALKBH5 knockdown P493‐6 and Raji cell samples (same to Fig 2I and J), and found that the protein level of CDKN1A was increased when knocked down ALKBH5 but not as high as in low MYC‐expressing cells and CDKN2B remained the same (Fig EV3I). These data suggest that CDKN1A protein level is regulated by MYC via both transcriptional mechanism and m6A modification. As to CDKN2B, MYC might regulate it only by transcription. Hence, these genes were not the focus of the subsequent studies.
YTHDF3 binds m6A‐modified mRNAs and facilitates translation of selected MRGs
We next asked how MYC regulates the expression of MRGs via mRNA m6A methylation. YT521‐B homology (YTH) domain family members are representative m6A‐binding proteins, and YTH m6A readers are involved in m6A methylation‐mediated RNA fate, such as RNA stability, splicing, translation, and miRNA processing (Berlivet et al, 2019; Lan et al, 2019; Shi et al, 2019). We thus performed a RIP assay by using antibodies against YTH m6A readers, to unveil the mechanisms that underlie regulation of MRG expression by m6A modification. RIP assay results indicated that only YTHDF3 can specifically bind to the SPI1 and PHF12 under Tet treatment (Fig 3A), and suggested that the reader YTHDF3 binds to the MRG transcripts SPI1 and PHF12 and serves an important role in MYC‐mediated MRG expression via m6A modification. We then examined whether YTHDF3 regulates MRG protein expression. Using shRNAs that target YTHDF3 in P493‐6 cells that were treated with Tet, YTHDF3 but not YTHDF1 or YTHDF2 was significantly decreased (Fig 3B and C), and neither MYC nor YTHDF3 regulated the mRNA levels of SPI1 and PHF12 (Fig EV4A). However, the increase in protein levels of SPI1 and PHF12 following Tet treatment of P493‐6 cells or by MYC shRNA in Raji cells was significantly reduced in the absence of YTHDF3 (Fig 3B and C), suggesting that MRG protein levels are up‐regulated by YTHDF3.
Figure 3. YTHDF3 binds m6A‐modified mRNAs and facilitates translation of selected MRG transcripts.
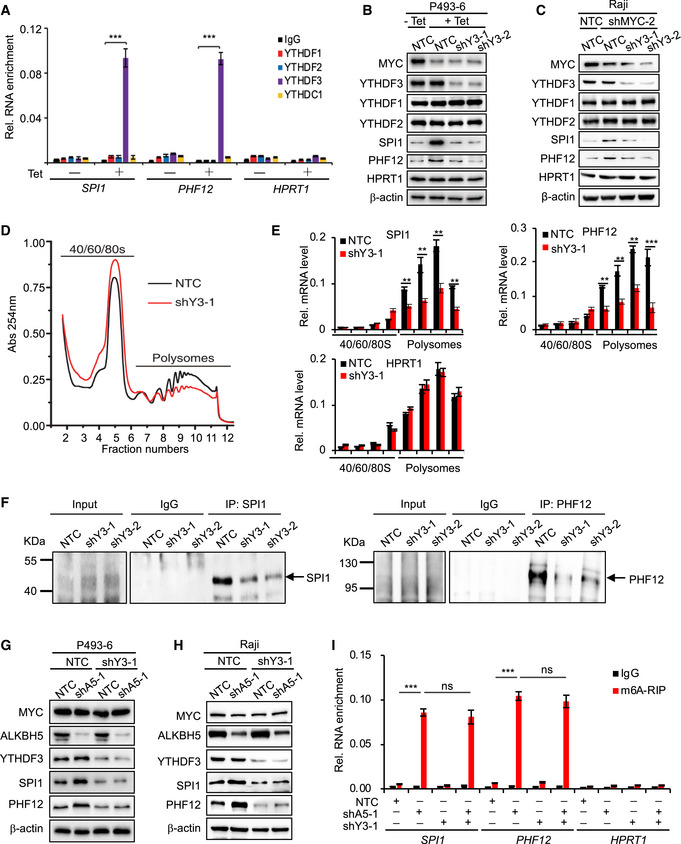
-
ARIP assay in P493‐6 cells treated with Tet for 0 h or 72 h. HPRT1 serves as negative control. ***P < 0.001 relative to indicating groups (mean ± SD, n = 3 biological replicates, Student's t‐test).
-
B, CWestern blot analysis for protein levels in P493‐6 cells that expressed NTC or YTHDF3 shRNAs and were then treated with Tet or not (B) or in Raji cells that expressed NTC or YTHDF3 shRNAs and knocked down MYC or not (C). HPRT1 and β‐actin serve as negative and loading controls, respectively. Data are representative of at least three independent experiments.
-
DPolysomes profiling assay in P493‐6 cells that expressed NTC or YTHDF3 shRNA.
-
ERT–qPCR assay for mRNA levels of MRGs (SPI1 and PHF12) in different fractions in (D). HPRT1 serves as negative control. Data were presented as mean (±SD), n = 3 biological replicates. **P < 0.01 and ***P < 0.001 as compared to NTC group (Student's t‐test).
-
FClick‐iT AHA (L‐azidohomoalanine) experiments were performed using IgG, anti‐SPI1, or anti‐PHF12 antibody. P493‐6 cells expressing NTC or YTHDF3 shRNAs were incubated for 1 h in medium containing 100 µg/ml AHA. The translated proteins were detected by Western blot. Arrow indicates translated MRGs.
-
G, HWestern blot analysis for protein levels in P493‐6 cells (G) or in Raji cells (H) that expressed NTC, or ALKBH5 shRNA, or YTHDF3 shRNA. β‐actin serves as negative and loading controls, respectively. Data are representative of at least three independent experiments.
-
Im6A RIP assay in (G). HPRT1 serves as negative control. ***P < 0.001 as compared between indicated groups, ns, not significant (mean ± SD, n = 3 biological replicates, Student's t‐test).
Source data are available online for this figure.
Given that knockdown of YTHDF3 directly regulates the protein levels of MRGs (Fig 3B and C), we hypothesized that YTHDF3 regulates the translation of m6A modified mRNA. To test this, we performed a polysome profiling analysis and found that such profiling of shYTHDF3 was much lower than in the non‐targeting control (NTC) sample (Fig 3D), demonstrating that YTHDF3 regulates the translation of mRNAs. We also collected each polysome fraction, isolated the RNA, and analyzed mRNA levels of MRGs in each fraction. Our results showed that knocking down YTHDF3 significantly impaired the amount of SPI1 and PHF12 mRNA that bound to polysomes (Fig 3E), indicating that YTHDF3 up‐regulates the translation activity of MRGs mRNA. We also performed a Click‐iT AHA (L‐azidohomoalanine) assay, which is used to detect the nascent proteins, to investigate whether YTHDF3 regulates the translation of these selected MRG transcripts, and found that translation of SPI1 and PHF12 was significantly decreased in the absence of YTHDF3 (Fig 3F). We thus conclude that YTHDF3 facilitates the protein translation of MRGs.
Our data have shown that MYC down‐regulates the protein expression of selected MRGs by down‐regulating m6A modification via ALKBH5, we next investigated whether this regulatory role of m6A in gene expression is YTHDF3‐dependent. We analyzed MRG expression and showed that neither knocking down ALKBH5 alone nor knocking down both ALKBH5 and YTHDF3 regulates the mRNA levels of SPI1 and PHF12 in P493‐6 cells (Fig EV4B and C). And knocking down ALKBH5 increased the protein levels of SPI1 and PHF12, but the increased protein levels were significantly attenuated when YTHDF3 was further knocked down in both P493‐6 cells and Raji cells (Figs 3G and H, and EV4D), indicating that ALKBH5‐mediated inhibition of MRG expression is YTHDF3‐dependent. We then performed a MeRIP assay in the same cell samples as in Fig 3G. Consistently, shRNA suppression of ALKBH5 expression significantly increased m6A enrichment levels (Fig 3I). And knocking down YTHDF3 did not affect the m6A enrichment levels of SPI1 and PHF12, eliminating the possibility that YTHDF3 regulates m6A modifications (Fig 3I). Taken together, these data demonstrate that the m6A reader YTHDF3 specifically binds m6A and facilitates the translation of MRG transcripts.
The MYC‐ALKBH5‐m6A‐SPI1/PHF12 axis is critical for cancer progression
The above data established that MYC preferentially down‐regulates the level of mRNA m6A modification in mRNA of certain MRGs by transcriptionally up‐regulating ALKBH5, which specifically demethylates the selected MRG mRNA, then impairs the translation of these genes, and inhibits their protein expression. We next explored the effect of the MYC‐ALKBH5‐m6A‐SPI1/PHF12 axis on cell proliferation and cancer progression, and found that overexpression of ALKBH5 promotes cell proliferation in P493‐6 cells (Fig EV5A) and knockdown of ALKBH5 significantly impaired cell proliferation (Fig EV5B), indicating that the MYC‐regulated m6A demethylase ALKBH5 is critical for cancer cell proliferation. Similar results were observed in Raji cells that knockdown of ALKBH5 alone significantly impaired cell proliferation, and overexpression of MYC promotes cell proliferation, which was eliminated when further knocking down ALKBH5 (Fig EV5C).
Deregulated MYC contributes human malignancies and therefore, we asked whether MYC promotes cancer cell proliferation by inhibiting the expression of these MRGs. We first studied the effect of MRGs on cell proliferation and found that overexpressing SPI1 or PHF12 alone significantly inhibited cell proliferation (Fig EV5D) and knocking down SPI1 or PHF12 significantly promoted cell proliferation (Fig 4A), indicating that MRGs SPI1 and PHF12 inhibit cancer cell proliferation and function as tumor suppressors. Also, overexpression of SPI1 or PHF12 in ALKBH5 overexpressing cells significantly suppressed the enhanced proliferation by ALKBH5 (Fig 4B and C), demonstrating that MRGs SPI1 and PHF12 are involved in ALKBH5‐regulated cancer cell proliferation. We conclude that MYC down‐regulates certain MRGs by regulating ALKBH5, and then blocks the inhibitory effect of these MRGs on cell proliferation, thereby promoting cancer cell proliferation and growth.
Figure 4. The MYC‐ALKBH5‐m6A‐SPI1/PHF12 axis is critical for cancer progression.
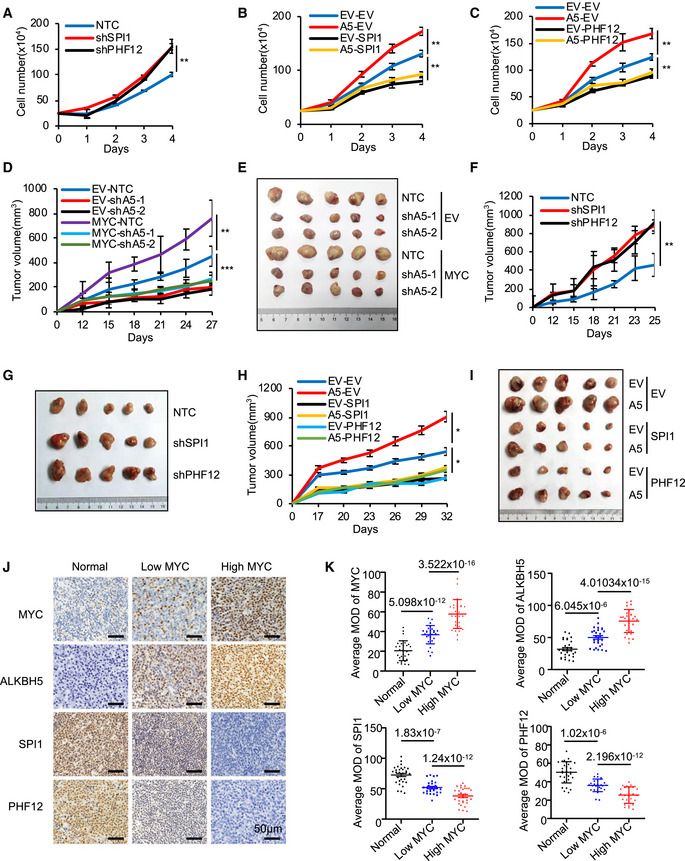
-
ATrypan blue counting was used to analyze growth curves for P493‐6 cells that expressed NTC or SPI1 shRNA or PHF12 shRNA. **P < 0.01 as compared between indicated groups (mean ± SD, n = 4 biological replicates, Student's t‐test).
-
B, CTrypan blue counting was used to analyze growth curves for P493‐6 cells overexpressing ALKBH5 and further infected with viruses expressing SPI1 (B) or PHF12 (C). **P < 0.01 as compared between indicated groups (mean ± SD, n = 4 biological replicates, Student's t‐test).
-
D, ERaji cells stably expressing EV or MYC were infected with viruses expressing shALKBH5. Cells were injected subcutaneously into nude mice (n = 5 for each group). Tumor growth curves were measured starting from 12 days postinjection (D). Photograph of tumors collected at the end of the experiment (day 27) (E). Data are presented as mean (±SEM). **P < 0.01 or ***P < 0.001 as compared between indicated groups (Student's t‐test).
-
F, GP493‐6 cells stably expressing NTC or shSPI1 or shPHF12 were injected subcutaneously into nude mice (n = 5 for each group). Tumor growth curves were measured starting from 12 days postinjection (F). Photograph (G) of tumors collected at the end of the experiment (day 25). Data are presented as mean (±SEM). **P < 0.01 as compared between indicated groups (Student's t‐test).
-
H, IP493‐6 cells stably expressing EV or ALKBH5 were infected with viruses expressing SPI1 or PHF12. Cells were injected subcutaneously into nude mice (n = 5 for each group). Tumor growth curves were measured starting from 17 days postinjection (H). Photograph (I) of tumors collected at the end of the experiment (day 32). Data are presented as mean (±SEM). *P < 0.05 as compared between indicated groups (Student's t‐test).
-
J, KRepresentative IHC images (J), and analysis (K) of MYC, ALKBH5, SPI1, and PHF12 expressions in normal lymphocyte tissue (normal) and lymphoma specimens. Scale bar: 50 µm. Data are presented as mean (±SEM), clinical samples n = 30 for normal group, n = 28 for low MYC expression group, and n = 27 for high MYC expression group; P value was presented between indicated groups (Student's t‐test).
Given that our in vitro experiments show that the MYC‐ALKBH5‐m6A‐SPI1/PHF12 axis could promote cancer cell proliferation, we evaluated the effect of this axis on cancer progression in vivo. First, mice were xenografted with Raji cells stably expressing MYC and ALKBH5 shRNAs. Our results showed that knockdown of ALKBH5 alone significantly impaired tumor growth, and overexpression of MYC promoted tumor growth, which was eliminated by further knockdown of ALKBH5 (Fig 4D and E). Second, mice were xenografted with P493‐6 cells expressing shSPI1 or shPHF12. Our data showed that knockdown of SPI1 or PHF12 markedly accelerated tumor growth in vivo (Figs 4F and G, and EV5E), indicating that SPI1/PHF12 act as tumor suppressors and play an important role in tumor growth. Third, mice received a xenograft of P493‐6 cells stably overexpressing ALKBH5 and MRGs: Forced expression of ALKBH5 led to a markedly accelerated tumor growth in vivo, and this enhanced growth was significantly suppressed by overexpression of MRGs SPI1 or PHF12 (Figs 4H and I, and EV5F). These findings are consistent with the in vitro data and underscore the critical role of this axis in tumor growth in vivo. Overexpression of ALKBH5 and MRGs was confirmed via Western blot analysis of protein lysates from xenograft tumors (Fig EV5G).
To further evaluate the physiological relevance and potential clinical significance of the MYC‐ALKBH5‐m6A‐SPI1/PHF12 regulatory axis, we used immunohistochemistry (IHC) to assess the expression of MYC, ALKBH5, SPI1, and PHF12 in samples from a retrospective cohort of 55 clinicopathologically characterized diffuse large B‐cell lymphoma (DLBCL) cases and from 30 normal lymphocyte tissue samples (Li et al, 2020). DLBCL is the most common type of non‐Hodgkin lymphoma (NHL) and is highly invasive and malignant. MYC is highly expressed in cells of this tumor and is translocated to the nucleus, which increases tumor malignancy and leads to a poor prognosis in patients with DLBCLs (Savage et al, 2009; Barrans et al, 2010; Green et al, 2012). As expected, DLBCLs display elevated MYC staining relative to normal lymph nodes (Fig 4J). HistoQuest software was used to analyze the mean optical density (MOD) of MYC staining for all clinical samples. A MOD cutoff of ≥ 50% defined high MYC expression, as reported (Kluk et al, 2012; Carey et al, 2015). Thus, the 55 DLBCL samples were stratified by MYC expression levels: 28 samples had low MYC expression and 27 samples had high MYC expression. The level of protein expression was determined in these two groups and also in the normal lymphocyte tissue samples: Compared to the normal samples and to the low MYC‐expressing DLBCL tumor group, expression of ALKBH5 was significantly higher in the high MYC‐expressing DLBCL tumors (Fig 4J). In contrast, SPI1 and PHF12 were significantly decreased in the high MYC‐expressing DLBCL tumors, compared to normal samples as well as to low MYC‐expressing tumors (Fig 4J). Quantitative analysis of IHC images revealed a strong positive correlation between MYC and ALKBH5, and a notable negative correlation between MYC and SPI1, as well as MYC and PHF12 (Fig 4K), underscoring the potential of the MYC‐ALKBH5‐SPI1/PHF12 regulatory axis as a significant prognostic pathway in DLBCL patients.
Finally, we explored the clinical and translational potential of our findings in cancers, wherein MYC is deregulated, by investigating whether a chemical inhibitor of m6A demethylase inhibits B‐cell lymphoma. We treated P493‐6 cells with the ALKBH5 inhibitor IOX3 (Aik et al, 2014) to determine whether it affects cell proliferation and MRG expression. IOX3 treatment significantly impaired cell proliferation and increased the protein levels of SPI1 and PHF12 (Fig EV5H and I), demonstrating that IOX3 is able to inhibit MYC‐deregulated cell proliferation and the regulation of ALKBH5 on MRGs. To determine whether IOX3 affects cancer progression in vivo, mice were xenografted with P493‐6 cells and treated with or without IOX3. Our data showed that IOX3 treatment significantly impaired tumor growth in vivo (Fig EV5J and K), indicating that IOX3 is able to inhibit MYC‐deregulated tumor growth. The results also confirm that blocking the MYC‐ALKBH5‐m6A‐SPI1/PHF12 regulatory axis with ALKBH5 inhibitor suppresses tumor growth in vivo and represents a promising clinical target.
Discussion
MYC‐mediated gene regulation and cancer progression have been the focus of attention by many scientists and clinicians. MYC regulates up to 15% of human genes, many of which, particularly those genes which MYC represses, are not regulated via classical transcriptional regulation nor in a transcription‐dependent manner (Dang et al, 2006; Dang, 2012; Cole, 2014; Baluapuri et al, 2020). We document here that MYC suppression of certain MRGs is exerted preferentially by reducing the m6A modification of their transcripts and uncover a novel mechanism whereby MYC facilitates cancer progression, mainly through epigenetic modification of RNA (Fig 5). We show that treatment with IOX3, which inhibits the activity of the m6A demethylase ALKBH5 and blocks the MYC‐ALKBH5‐m6A‐SPI1/PHF12 regulatory pathway, represses tumor growth in vitro as well as in vivo, unveiling a therapeutic pathway for MYC‐dependent cancers.
Figure 5. Working model: MYC suppression of gene expression via m6A is critical for cancer progression.
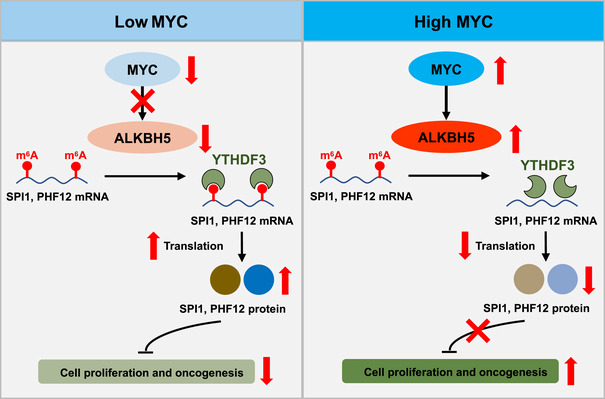
MYC down‐regulates the m6A modification preferentially in certain MRGs, by up‐regulating the demethylase ALKBH5. The m6A reader YTHDF3‐mediated translation of MRGs SPI1 and PHF12 is attenuated as decreased m6A modification, and thus releases the inhibitory effect of MRGs on cell proliferation, thereby promoting cancer progression. The up‐direction red arrows indicate high, and down‐direction red arrows indicate low.
Many efforts have been made to understand the biology of MYC oncoproteins recently. For instance, Posternak and colleagues have proposed a global role for MYC in promoting mRNA cap methylation of Wnt/β‐catenin signaling genes, and in increasing their translational capacity (Posternak et al, 2017), while Baluapuri et al (2019) showed that high levels of MYC sequester SPT5 into non‐functional complexes, and thereby decrease gene expression. However, the mechanism by which MYC directly or indirectly represses genes is not well understood (Cole & Cowling, 2008; Baluapuri et al, 2020). In this context, our findings elucidate a mechanism by which MYC represses genes indirectly via m6A. We show that MYC down‐regulates m6A levels of mRNAs by activating the demethylases ALKBH5 and FTO, and thereby down‐regulates mRNA m6A levels of selected MRGs and inhibits their protein expression. Among these MRGs that were enriched by the GO analyses as shown in Fig 1E, considering the complexity of the genes that could be regulated by MYC at both mRNA level and m6A modification level, such as CDKN1A, we chose those genes that are less studied and only could be regulated by MYC at m6A modification level. We thus focused on SPI1 and PHF12 that are important in lymphomagenesis in this study. Mechanistically, by using integrative RIP and MeRIP assays, YTHDF3 knockdown experiments, polysome profiling analysis, and Click‐IT AHA assay, we find that YTHDF3 specifically binds m6A and facilitates the translation of SPI1 and PHF12 mRNA. This mechanism provides new insight into how MYC governs mRNA translation via the mRNA m6A modification, in addition to the known mechanisms that involve stimulation of ribosome biogenesis, tRNA synthesis, transcription of genes that encode translation factors, and interacting with RNA‐binding proteins (Cole & Cowling, 2008; Cargnello & Topisirovic, 2019; Singh et al, 2019).
Analyses of mouse models and clinical samples reveal that MRGs play an essential role in MYC‐deregulated cancers. The sample analyses document that SPI1 and PHF12 are highly expressed in normal tissue, relative to tumor tissue in which cells exhibit high MYC expression (Fig 4J and K). Our in vitro and in vivo experiments demonstrate that overexpression of SPI1 or PHF12 impairs cancer progression (Figs 4B, C, H, I, and EV5D and F) and that knocking down SPI1 or PHF12 significantly accelerates cancer progression (Figs 4A, F, G, and EV5E). These observations indicate not only that MYC activated genes, but also these MRGs—which are not very well studied—make important contributions to the normal development as well as to many oncogenic functions of MYC. Based on these findings, we suggest that when MYC is low, m6A levels of certain MRG transcripts are high, which facilitates their and leads to cell proliferation and growth inhibition under the control of the MRGs. Conversely, when MYC is overexpressed, m6A levels of MRG transcripts are reduced, which reduces protein expressions of MRGs, and in turn triggers uncontrolled cell growth. Additional studies of various MYC‐deregulated tumor mouse models and of clinical cancer samples will enable us to more fully elucidate the importance of MYC, m6A, and MRGs, and how they are associated with the development of cancers, by tracking m6A levels and MRG expression.
Since the first m6A modification enzyme FTO was discovered in 2011, interest in mRNA m6A modifications has increased greatly: Many m6A modification‐related enzymes have been reported, and m6A modifications are now known to participate in a number of fundamental biological processes. Emerging evidence suggests m6A also plays an important role in malignancy (Bansal et al, 2014; Li et al, 2017b; Nishizawa et al, 2018; Su et al, 2018; Weng et al, 2018; Cheng et al, 2019; Lan et al, 2019). Several reports claim that MYC is a target of the m6A methyltransferases METTL3 and METTL14, and that the mRNA m6A modification of MYC is regulated by METTL3 or METTL14 in some cancers (Lin et al, 2016; Su et al, 2018; Weng et al, 2018; Cheng et al, 2019), implicating MYC and m6A in cancer. Here, we have examined whether MYC regulates mRNA m6A modifications, investigated the underlying mechanisms, and reported on the consequences for cancer progression. We conclude that MYC down‐regulates mRNA m6A levels. Unlike the methyltransferases METTL3 and METTL14, which regulate the mRNA m6A level of MYC, MYC itself regulates the expression of the demethylases ALKBH5 and FTO. Further, RIP and MeRIP assays show that ALKBH5 specifically demethylates the mRNA m6A levels of certain MRG transcripts. Moreover, consistent with previous findings, the m6A reader YTHDF3 facilitates mRNA translation (Shi et al, 2017; Li et al, 2017a), thus promoting protein expression of MRGs. Finally, the in vitro and in vivo experiments show that the MYC‐ALKBH5‐m6A‐SPI1/PHF12 regulatory axis is critical for MYC‐deregulated cancer progression, and that inhibition of ALKBH5 with IOX3 or knockdown of ALKBH5 with shRNAs significantly impairs cancer progression. This is consistent with reports from several other groups that knocking down ALKBH5 with shRNAs decreases cancer progression (Zhang et al, 2016a; Zhang et al, 2016b; Zhang et al, 2017). Of note, there is no ALKBH5‐specific inhibitor currently available and IOX3 that we used here might have other potential targets. Another limitation of this study is that we were able to explore only a small subset of MRGs in detail; thus, it is possible that the other MRG transcripts are regulated by FTO, which is reportedly related to obesity and cancers (Lin et al, 2016; Li et al, 2017b; Su et al, 2018; Huang et al, 2019; Yang et al, 2019; Melstrom & Chen, 2020). Targeting FTO with the specific inhibitor FB23‐2 significantly inhibits AML progression (Huang et al, 2019), suggesting that FTO might be another potential target for MYC‐deregulated cancer. Together, we show that targeting ALKBH5 and blocking the MYC‐ALKBH5‐m6A‐SPI1/PHF12 regulatory pathway represses tumor growth in vitro as well as in vivo, unveiling a therapeutic potential of m6A demethylases for MYC‐dependent cancers.
Overall, our work reveals a novel regulatory pathway downstream of MYC regulating gene expression by showing global alterations of m6A levels. Specifically, we show that the oncogene MYC down‐regulates the m6A level of mRNA by activating demethylases and preferentially down‐regulates the expression of certain MRGs via mRNA m6A modifications. We document that not only does MYC down‐regulate the m6A levels of mRNAs in cancer cells, but we also unveil a novel mechanism where MYC suppresses of gene expression via m6A modifications, providing new insights into how MRGs are regulated. The MYC‐ALKBH5‐m6A‐SPI1/PHF12 regulatory axis that we have identified here is critical for cancer progression and may represent a promising clinical target for human malignancies with aberrant MYC expression.
Materials and Methods
Cell culture and reagents
HEK293T cells (from ATCC) were cultured in Dulbecco’s modified Eagle medium DMEM (Gibco) with 10% FBS (Gibco). P493‐6 B cells (gift of Dr. Chi V. Dang at Ludwig Institute for Cancer Research) were cultured in RPMI‐1640 (Gibco) with 10% Tet system approved FBS (Takara Bio). Raji cells (from the Type Culture Collection of Chinese Academy of Sciences, Shanghai, China) were cultured in RPMI‐1640 medium (Gibco) with 10% FBS (Gibco). All cell lines were tested for and found to be free of mycoplasma contamination. DMEM and RPMI‐1640 were supplemented with 1% penicillin–streptomycin (Gibco). Cells were maintained in 5% CO2 at 37°C. To repress MYC expression in P493‐6 B cells, 0.1 µg/ml tetracycline (Sigma‐Aldrich, T7660) was added to the culture medium. Chemical m6A inhibition was achieved with use of 20 mM cycloleucine (Sigma‐Aldrich, A48105). ALKBH5 inhibitor IOX3 was purchased from Selleckchem (Catalog No. S7979).
Plasmids and established stable cells
pCDH‐EV empty vector or pCDH‐SPI1, pCDH‐PHF12/pSin‐3XFlag‐EV, pSin‐3XFlag‐ALKBH5, pSin‐3XFlag‐FTO vectors were co‐transfected with plasmids encoding Δ8.9 and VSVG, into HEK293T packaging cells, using lipofectamine 2000 (Invitrogen). P493‐6 B cells were infected with produced lentivirus in the presence of polybrene, and selected with 0.5 µg/ml puromycin, to establish stable cells. Lentiviral shRNAs targeting human MYC, ALKBH5, YTHDF3, SPI1, or PHF12 were purchased from Sigma (targeting sequences listed in Table EV1). Virus‐expressing shRNAs were produced in HEK293T cells and transduced into P493‐6 B cells and Raji cells in the presence of polybrene.
RNA extraction and quantitative real‐time PCR
Total RNA was isolated using TRIzol reagent (Life technologies) and treated with DNase (Ambion). One microgram of RNA was used to synthesize cDNA, employing the iScriptcDNA Synthesis Kit (Bio‐Rad) (sequences of used primers shown in Table EV2). Quantitative real‐time PCR was performed using iQ SYBR Green Supermix and the iCycler Real‐time PCR Detection System (Bio‐Rad). mRNA levels were compared to 18S rRNA or RPL13A, and the fold change of target mRNA expression was calculated based on a threshold cycle (Ct), where ΔCt = Cttarget − Ct18S and Δ(ΔCt) = ΔCtControl − ΔCtIndicated condition.
Western blotting
Proteins were extracted using RIPA buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.1% SDS, 1% NP‐40), supplemented with a protease inhibitor cocktail, and quantified with a Bradford assay kit (Sangon Bio). Equal amounts of protein were fractionated by 5–12% SDS–PAGE. The following primary antibodies were used: METTL14 (1:1,000, HPA038002), ALKBH5 (1:1,000, HPA007196) (Sigma‐Aldrich, St Louis, MO, USA); YTHDC1 (1:1,000, ab122340), FTO (1:1,000, ab124892) (Abcam, Cambridge, USA); MYC (1:2,000, 9402s), SPI1 (1:1,000, 2258s) (Cell Signaling Technology, Beverly, MA, USA); METTL3 (1:1,000, 15073‐1‐AP), YTHDF1 (1:1,000, 17479‐1‐AP), YTHDF2 (1:1,000, 24744‐1‐AP), YTHDF3 (1:1,000, 25537‐1‐AP), PHF12 (1:1,000, 24485‐1‐AP), HPRT1 (1:1,000, 15059‐1‐AP), CDKN1A (1:1,000, 10355‐1‐AP), CDKN2B (1:1,000, 12877‐1‐AP), β‐actin (1:5,000, 66009‐1‐Ig) (Proteintech, Rosemont, IL, USA). HRP‐conjugated anti‐rabbit and anti‐mouse (1:10,000, Bio‐Rad) secondary antibodies were used. Signaling was detected by Western ECL Substrate (Bio‐Rad).
Click‐iT AHA (L‐azidohomoalanine)
For the Click‐iT AHA analysis, P493‐6 cells were washed with PBS twice and then incubated in DMEM supplemented with or without methionine, cysteine for 1 h, followed by culturing in DMEM supplied with 100 μg/ml AHA (C10102; Invitrogen) plus 10% FBS, for 1 h in 5% CO2 at 37°C. Cells were lysed with IP buffer (1% NP‐40, 20 mM HEPES (pH 7.4), 150 mM NaCl, 2 mM EDTA, 1.5 mM MaCl2) supplemented with a protease inhibitor cocktail for 45 min on ice, followed by centrifugation at 16,000 g for 10 min at 4°C. Supernatants were incubated with the indicated antibodies for 4 h at 4°C and then with protein A/G‐conjugated beads for 2 h. Beads were washed five times with IP buffer. Eluted samples were incubated with 40 µM Biotin‐PEG4‐Alkyne for 30 min in Click‐iT Protein Reaction Buffer (C10276; Invitrogen) following the manufacturer’s protocol. The proteins were extracted with methanol and chloroform and analyzed by Western blot using streptavidin‐conjugated horseradish peroxidase.
Polysome profiling analysis
P493‐6 cells were treated with a final concentration of 100 μg/ml cycloheximide (Sigma‐Aldrich) for 5 min. Cells were washed 2× in 10 ml of ice‐cold PBS containing 100 μg/ml cycloheximide and lysed in lysis buffer (5 mM Tris–HCl (pH 7.5), 2.5 mM MgCl2, 1.5 mM KCl, 0.1 mg/ml cycloheximide, 10 mM DTT, 0.5% Triton X‐100, 0.5% sodium deoxycholate) supplemented with a protease inhibitor cocktail (EDTA‐free) plus the RNase inhibitor RNasin (Promega), and centrifuged at 21,000 g for 5 min at 4°C. Supernatants were quantified by NanoDrop, and same OD amount of lysate was loaded onto each gradient. Each gradient was centrifuged at 164,000 g for 2 h at 4°C using the SW40Ti rotor in a Beckman Coulter ultracentrifuge. Each tube was screwed onto the ISCO UV detector, and the chasing solution was run through the gradient. Data were collected with use of the TracerDAQ program and fractions collected over time. RNA was isolated from each fraction and detected by RT–qPCR.
Chromatin immunoprecipitation
The ChIP assay was performed as described (Wu et al, 2017) with use of an EZ‐ChIP kit (Millipuro), according to the manufacturer’s instructions. Briefly, cells were fixed with 1% formaldehyde, quenched in 0.125 M glycine, and sonicated in a Bioruptor Sonication System UCD‐300. DNA was immunoprecipitated with use of control IgG or a primary antibody against MYC (Cell Signaling Technology, 9402s), followed by RT–qPCR analysis (Bio‐Rad). Oligos used for this analysis are listed in Table EV2.
m6A dot blot assay
Immediately after harvesting the cells, total RNA was isolated using Trizol (Invitrogen, 15596‐018, as per the manufacturer’s instructions) and purified with the Dynabeads® mRNA purification kit (Ambion, 61006) for two rounds. Isolated mRNA was first denatured by heating at 95°C for 3 min, followed by chilling on ice. RNA samples were quantified using NanoDrop. Twofold serial dilutions were spotted on an Amersham Hybond‐N + membrane, which was optimized for nucleic acid transfer (GE Healthcare). After UV crosslinking in a Stratagene Stratalinker 2400 UV Crosslinker, the membrane was washed with PBST buffer, blocked in 5% non‐fat milk in PBST, incubated with anti‐m6A antibody (1:2,000; Synaptic Systems) overnight at 4°C, and incubated with HRP‐conjugated anti‐rabbit IgG secondary antibody and detected by an ECL Western Blotting Detection Kit (Bio‐Rad). To verify that equal amounts of mRNA were spotted on the membrane, the blot was stained with 0.02% methylene blue in 0.3 M sodium acetate (pH 5.2).
RNA immunoprecipitation
Cells were collected, pelleted by centrifuge for 5 min at 600 g, and washed twice with cold PBS. The cell pellet was re‐suspended with 1 ml of lysis buffer (150 mM KCl, 10 mM HEPES pH 7.6, 2 mM EDTA, 0.5% NP‐40, 0.5 mM DTT, 1:100 protease inhibitor cocktail, 400 U/ml RNase inhibitor), and then the messenger ribonucleoprotein (mRNP) lysate was incubated on ice for 30 min and shock‐frozen at −80°C with liquid nitrogen. The mRNP lysate was thawed on ice and centrifuged at 15,000 g for 15 min to clear the lysate. Cell lysate was pre‐cleared with 40 µl protein A/G beads. 50 µl of pre‐cleared lysate was saved as input, mixed with 50 µl TRIzol, and stored at −80°C. Protein A/G beads were coated with indicated antibody (2 μg) or IgG control antibody for 2 h at 4°C. Then, the antibody‐coated beads were mixed with pre‐cleared cell lysate and incubated overnight at 4°C. mRNP bound to protein A/G beads was washed 6 times with washing buffer (200 mM NaCl, 50 mM HEPES pH 7.6, 2 mM EDTA, 0.05% NP‐40, 0.5 mM DTT, 200 U/ml RNase inhibitor), and RNA was extracted with 500 µl TRIzol.
m6A immunoprecipitation
Total RNAs were extracted with TRNzol (Invitrogen, 15596‐018) and then purified with the Dynabeads® mRNA purification kit (Ambion, 61006) for two rounds. For detecting levels of m6A, the purified mRNAs were analyzed through UHPLC‐MS/MS as described (Jia et al, 2011).
To immunoprecipitated m6A, purified mRNAs were digested by DNase I and then incubated at 94°C for 30 s in fragmentation buffer (10 mM ZnCl2, 10 mM Tris–HCl, pH 7.0), to cut them into pieces of ~ 300‐nt. The reaction was stopped with 0.05 M EDTA (Ambion, AM8740) and was followed by standard ethanol precipitation and collection. Anti‐m6A polyclonal antibody (12 µg antibody for 6 µg mRNAs; Synaptic Systems, 202003) was incubated with 50 µl protein A beads (Sigma, P9424) in IPP buffer (150 mM NaCl, 0.1% NP‐40, 10 mM Tris–HCl, pH 7.4) for 1 h at room temperature. The mRNAs (6 µg) were incubated with the prepared antibody‐beads mixture for 4 h at 4°C. After washing, bound RNAs were extracted by TRNzol and reverse‐transcribed, and then amplified by PCR. Enrichment of m6A was quantified using RT–qPCR. Sequences of the qPCR primers are listed in Table EV2.
MeRIP‐seq and data analysis
MeRIP was performed as above, except that RNA was cut into fragments of ~ 100‐nt. An Illumina HiSeq 2000 platform was used for the MeRIP sequencing; reads were mapped to the UCSC human genome hg19 using TopHat (version 2.0.9). Only unique mapped reads with mapping quality more than or equal to 20 were kept for the subsequent analysis for each sample. The m6A‐enriched regions in each MeRIP sample were extracted using MACS2 software (version 2.0.10), with the corresponding input sample serving as control. Motifs were analyzed by HOMER (Hypergeometric Optimization of Motif EnRichment) (version 3.12). Gene traces were visualized using the Integrative Genomics Viewer. Original data are available in NCBI GEO (accession number GSE150892).
Animal studies
All animal studies were approved by the Animal Research Ethics Committee of the South China University of Technology. For xenograft experiments, 2 × 107 P493‐6 cells or 1 × 107 Raji cells were injected subcutaneously into 6‐week‐old male BALb/c nude mice (n = 5 for each group) (SJA Laboratory Animal Company, China); starting at 10 days postinjection, tumors were measured every 2 or 3 days with a caliper and volumes were calculated using the equation: volume = width*depth*length*0.5. IOX3 was dissolved first in dimethylsulfoxide (DMSO) and then in sterile H2O (pH 7.0) (5% DMSO/H2O). A single dose of IOX3 (15 mg/kg) or vehicle (5% DMSO/H2O) was given to mice through intragastric administration every other day.
Clinical human tissue specimen and immunohistochemistry
Formalin‐fixed, paraffin‐embedded primary DLBCLs and normal lymph node specimens obtained from 85 patients were randomly selected from the archives of the First Affiliated Hospital of Anhui Medical University. To use these clinical materials for research purposes, written informed consent from patients and approval from the Institutional Research Ethics Committee of the First Affiliated Hospital of Anhui Medical University were obtained. Immunohistochemistry (IHC) was performed as described (Li et al, 2020). Images were acquired with a Zeiss AxioImager Z1 and quantified with HistoQuest (Tissue Gnostics GmbH, Vienna, Austria, www.tissuegnostics.com). Images of ten zones (X200 objective) in each sample were analyzed, to verify the mean optical density (MOD); data were statistically analyzed by a t‐test. Exposure time, signal amplification, and objectives were the same for all samples when obtaining images. Primary antibodies against the following proteins were used for IHC: MYC (ZA0555, ZSGB‐BIO), ALKBH5 (HPA007196, Sigma‐Aldrich), SPI1 (2258s, Cell Signaling Technology), PHF12 (PA5‐48127, Thermo Fisher Scientific).
Statistical analyses
Clinicopathological characteristics were analyzed by the chi‐square test. Statistical analysis was determined using Student’s t‐test for other experiments. Differences were considered to be statistically significant at the P < 0.05 level (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s. not significant). Error bars represent SD or SEM.
Author contributions
PG, GW, Y‐GY, and JC conceived this study. GW, CS, LS, HZ, and PG designed the experiments. GW, CS, YY, YZ, S‐TL, DY, YW, YC, and NW executed the experiments. SS and YY analyzed MeRIP sequencing data. GW, CS, and PG wrote the manuscript. All the authors analyzed the results, read, and approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Acknowledgements
We thank Drs. David M Weinstock and Sonal Jhaveri at Dana‐Farber Cancer Institute for critical reading of this manuscript and valuable suggestions. This work is supported in part by National Key R&D Program of China (2018YFA0800300, 2018YFA0107103), National Natural Science Foundation of China (91957203, 81525022, 81930083, 31571472, 81530076, 81821001, 31625016, 81874060), the Chinese Academy of Sciences (XDB39020100), the Program for Guangdong Introducing Innovative and Entrepreneurial Teams (2017ZT07S054), and Outstanding Scholar Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory (2018GZR110102001), The K. C. Wong Education Foundation, Shanghai Municipal Science and Technology Major Project (2017SHZDZX01).
EMBO reports (2021) 22: e51519
Contributor Information
Gongwei Wu, Email: gongwei_wu@dfci.harvard.edu.
Yun‐Gui Yang, Email: ygyang@big.ac.cn.
Jie Cao, Email: eycaojie@scut.edu.cn.
Ping Gao, Email: pgao2@ustc.edu.cn.
Data availability
The data that support the findings of this study are available from the corresponding author on reasonable request. The MeRIP‐seq datasets reported in this study are available in the NCBI Gene Expression Omnibus GSE150892 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150892).
References
- Aik W, Scotti JS, Choi H, Gong L, Demetriades M, Schofield CJ, McDonough MA (2014) Structure of human RNA N(6)‐methyladenine demethylase ALKBH5 provides insights into its mechanisms of nucleic acid recognition and demethylation. Nucleic Acids Res 42: 4741–4754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baluapuri A, Hofstetter J, Dudvarski Stankovic N, Endres T, Bhandare P, Vos SM, Adhikari B, Schwarz JD, Narain A, Vogt M et al (2019) MYC Recruits SPT5 to RNA polymerase II to promote processive transcription elongation. Mol Cell 74: 674–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baluapuri A, Wolf E, Eilers M (2020) Target gene‐independent functions of MYC oncoproteins. Nat Rev Mol Cell Biol 21: 255–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Penalva LO, Uren PJ, Suresh U, Carew JS, Karnad AB et al (2014) WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia 28: 1171–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan‐Zambrano G, Robson SC, Aspris D, Migliori V, Bannister AJ, Han N et al (2017) Promoter‐bound METTL3 maintains myeloid leukaemia by m(6)A‐dependent translation control. Nature 552: 126–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrans S, Crouch S, Smith A, Turner K, Owen R, Patmore R, Roman E, Jack A (2010) Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B‐cell lymphoma treated in the era of rituximab. J Clin Oncol 28: 3360–3365 [DOI] [PubMed] [Google Scholar]
- Berlivet S, Scutenaire J, Deragon JM, Bousquet‐Antonelli C (2019) Readers of the m(6)A epitranscriptomic code. Biochim Biophys Acta Gene Regul Mech 1862: 329–342 [DOI] [PubMed] [Google Scholar]
- Cao G, Li HB, Yin Z, Flavell RA (2016) Recent advances in dynamic m6A RNA modification. Open Biol 6: 160003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey CD, Gusenleitner D, Chapuy B, Kovach AE, Kluk MJ, Sun HH, Crossland RE, Bacon CM, Rand V, Dal Cin P et al (2015) Molecular classification of MYC‐driven B‐cell lymphomas by targeted gene expression profiling of fixed biopsy specimens. J Mol Diagn 17: 19–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cargnello M, Topisirovic I (2019) c‐Myc steers translation in lymphoma. J Exp Med 216: 1471–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll PA, Freie BW, Mathsyaraja H, Eisenman RN (2018) The MYC transcription factor network: balancing metabolism, proliferation and oncogenesis. Front Med 12: 412–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M, Sheng L, Gao Q, Xiong Q, Zhang H, Wu M, Liang Y, Zhu F, Zhang Y, Zhang X et al (2019) The m(6)A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF‐kappaB/MYC signaling network. Oncogene 38: 3667–3680 [DOI] [PubMed] [Google Scholar]
- Cole MD, Cowling VH (2008) Transcription‐independent functions of MYC: regulation of translation and DNA replication. Nat Rev Mol Cell Biol 9: 810–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole MD (2014) MYC association with cancer risk and a new model of MYC‐mediated repression. Cold Spring Harb Perspect Med 4: a014316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F (2006) The c‐Myc target gene network. Semin Cancer Biol 16: 253–264 [DOI] [PubMed] [Google Scholar]
- Dang CV (2012) MYC on the path to cancer. Cell 149: 22–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers M (2014) Gene Expression Omnibus GSM1231597 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM1231597). [DATASET]
- Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B (2003) Genomic targets of the human c‐Myc protein. Genes Dev 17: 1115–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey T, Zhang Z, Song L, Crawford G, Giresi P, Lieb J, Liu Z, McDaniell R, Lee B, Iyer V et al (2011) Gene Expression Omnibus GSE33213 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE33213) [DATASET]
- Green TM, Nielsen O, de Stricker K, Xu‐Monette ZY, Young KH, Moller MB (2012) High levels of nuclear MYC protein predict the presence of MYC rearrangement in diffuse large B‐cell lymphoma. Am J Surg Pathol 36: 612–619 [DOI] [PubMed] [Google Scholar]
- Herkert B, Eilers M (2010) Transcriptional repression: the dark side of myc. Genes Cancer 1: 580–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, Ni T, Zhang ZS, Zhang T, Li C et al (2019) Small‐molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell 35: 677–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG et al (2011) N6‐methyladenosine in nuclear RNA is a major substrate of the obesity‐associated FTO. Nat Chem Biol 7: 885–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur M, Cole MD (2013) MYC acts via the PTEN tumor suppressor to elicit autoregulation and genome‐wide gene repression by activation of the Ezh2 methyltransferase. Cancer Res 73: 695–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluk MJ, Chapuy B, Sinha P, Roy A, Dal Cin P, Neuberg DS, Monti S, Pinkus GS, Shipp MA, Rodig SJ (2012) Immunohistochemical detection of MYC‐driven diffuse large B‐cell lymphomas. PLoS One 7: e33813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Q, Liu PY, Haase J, Bell JL, Huttelmaier S, Liu T (2019) The critical role of RNA m(6)A methylation in cancer. Cancer Res 79: 1285–1292 [DOI] [PubMed] [Google Scholar]
- Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y, Sun HY, Zhu Q, Baidya P, Wang X et al (2017a) Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation. Cell Res 27: 444–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, Huang H, Nachtergaele S, Dong L, Hu C et al (2017b) FTO plays an oncogenic role in acute myeloid leukemia as a N(6)‐methyladenosine RNA demethylase. Cancer Cell 31: 127–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Huang D, Shen S, Cai Y, Xing S, Wu G, Jiang Z, Hao Y, Yuan M, Wang N et al (2020) Myc‐mediated SDHA acetylation triggers epigenetic regulation of gene expression and tumorigenesis. Nat Metab 2: 256–269 [DOI] [PubMed] [Google Scholar]
- Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA (2012) Transcriptional amplification in tumor cells with elevated c‐Myc. Cell 151: 56–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Choe J, Du P, Triboulet R, Gregory RI (2016) The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell 62: 335–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher B, Vervoorts J (2012) Regulation of gene transcription by the oncoprotein MYC. Gene 494: 145–160 [DOI] [PubMed] [Google Scholar]
- Melstrom L, Chen J (2020) RNA N(6)‐methyladenosine modification in solid tumors: new therapeutic frontiers. Cancer Gene Ther 27: 625–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Jaffrey SR (2017) Rethinking m(6)A readers, writers, and erasers. Annu Rev Cell Dev Biol 33: 319–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R et al (2012) c‐Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151: 68–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa Y, Konno M, Asai A, Koseki J, Kawamoto K, Miyoshi N, Takahashi H, Nishida N, Haraguchi N, Sakai D et al (2018) Oncogene c‐Myc promotes epitranscriptome m(6)A reader YTHDF1 expression in colorectal cancer. Oncotarget 9: 7476–7486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu Y, Zhao X, Wu YS, Li MM, Wang XJ, Yang YG (2013) N6‐methyl‐adenosine (m6A) in RNA: an old modification with a novel epigenetic function. Genomics Proteomics Bioinformatics 11: 8–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell BC, Cheung AF, Simkevich CP, Tam W, Ren X, Mateyak MK, Sedivy JM (2003) A large scale genetic analysis of c‐Myc‐regulated gene expression patterns. J Biol Chem 278: 12563–12573 [DOI] [PubMed] [Google Scholar]
- Pajic A, Spitkovsky D, Christoph B, Kempkes B, Schuhmacher M, Staege MS, Brielmeier M, Ellwart J, Kohlhuber F, Bornkamm GW et al (2000) Cell cycle activation by c‐myc in a burkitt lymphoma model cell line. Int J Cancer 87: 787–793 [DOI] [PubMed] [Google Scholar]
- Posternak V, Ung MH, Cheng C, Cole MD (2017) MYC mediates mRNA Cap methylation of canonical Wnt/beta‐catenin signaling transcripts by recruiting CDK7 and RNA methyltransferase. Mol Cancer Res 15: 213–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, Morelli MJ, Bora P, Doni M, Verrecchia A et al (2014) Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 511: 488–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage KJ, Johnson NA, Ben‐Neriah S, Connors JM, Sehn LH, Farinha P, Horsman DE, Gascoyne RD (2009) MYC gene rearrangements are associated with a poor prognosis in diffuse large B‐cell lymphoma patients treated with R‐CHOP chemotherapy. Blood 114: 3533–3537 [DOI] [PubMed] [Google Scholar]
- Schuhmacher M, Staege MS, Pajic A, Polack A, Weidle UH, Bornkamm GW, Eick D, Kohlhuber F (1999) Control of cell growth by c‐Myc in the absence of cell division. Curr Biol 9: 1255–1258 [DOI] [PubMed] [Google Scholar]
- Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, Liu C, He C (2017) YTHDF3 facilitates translation and decay of N(6)‐methyladenosine‐modified RNA. Cell Res 27: 315–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Wei J, He C (2019) Where, when, and how: context‐dependent functions of rna methylation writers, readers, and erasers. Mol Cell 74: 640–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K, Lin J, Zhong Y, Burcul A, Mohan P, Jiang M, Sun L, Yong‐Gonzalez V, Viale A, Cross JR et al (2019) c‐MYC regulates mRNA translation efficiency and start‐site selection in lymphoma. J Exp Med 216: 1509–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder M, Gerstein M, Weissman S, Farnham P, Struhl K (2011) Gene Expression Omnibus GSE31477 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE31477) [DATASET]
- Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, Deng X, Wang Y, Weng X, Hu C et al (2018) R‐2HG exhibits anti‐tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell 172: 90–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu WB, Shiah YJ, Lourenco C, Mullen PJ, Dingar D, Redel C, Tamachi A, Ba‐Alawi W, Aman A, Al‐Awar R et al (2018) MYC interacts with the G9a histone methyltransferase to drive transcriptional repression and tumorigenesis. Cancer Cell 34: 579–595 [DOI] [PubMed] [Google Scholar]
- Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, Rycak L, Dumay‐Odelot H, Karim S, Bartkuhn M et al (2014) Activation and repression by oncogenic MYC shape tumour‐specific gene expression profiles. Nature 511: 483–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, Shi H, Skibbe J, Shen C, Hu C et al (2018) METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell 22: 191–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Yuan M, Shen S, Ma X, Fang J, Zhu L, Sun L, Liu Z, He X, Huang D et al (2017) Menin enhances c‐Myc‐mediated transcription to promote cancer progression. Nat Commun 8: 15278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Hsu PJ, Chen YS, Yang YG (2018) Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res 28: 616–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wei J, Cui YH, Park G, Shah P, Deng Y, Aplin AE, Lu Z, Hwang S, He C et al (2019) m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti‐PD‐1 blockade. Nat Commun 10: 2782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller KI, Zhao X, Lee CW, Chiu KP, Yao F, Yustein JT, Ooi HS, Orlov YL, Shahab A, Yong HC et al (2006) Global mapping of c‐Myc binding sites and target gene networks in human B cells. Proc Natl Acad Sci USA 103: 17834–17839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, He X, Semenza GL (2016a) Hypoxia induces the breast cancer stem cell phenotype by HIF‐dependent and ALKBH5‐mediated m(6)A‐demethylation of NANOG mRNA. Proc Natl Acad Sci USA 113: E2047–E2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Zhi WI, Lu H, Samanta D, Chen I, Gabrielson E, Semenza GL (2016b) Hypoxia‐inducible factors regulate pluripotency factor expression by ZNF217‐ and ALKBH5‐mediated modulation of RNA methylation in breast cancer cells. Oncotarget 7: 64527–64542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, Chen Y, Sulman EP, Xie K, Bogler O et al (2017) m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem‐like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell 31: 591–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Eilers M (2014) Gene Expression Omnibus GSM1231597 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM1231597). [DATASET]
- Furey T, Zhang Z, Song L, Crawford G, Giresi P, Lieb J, Liu Z, McDaniell R, Lee B, Iyer V et al (2011) Gene Expression Omnibus GSE33213 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE33213) [DATASET]
- Snyder M, Gerstein M, Weissman S, Farnham P, Struhl K (2011) Gene Expression Omnibus GSE31477 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE31477) [DATASET]
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Data Availability Statement
The data that support the findings of this study are available from the corresponding author on reasonable request. The MeRIP‐seq datasets reported in this study are available in the NCBI Gene Expression Omnibus GSE150892 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150892).