

SUMMARY
Systemic lupus erythematosus (SLE) is a severe autoimmune disease mediated by pathogenic autoantibodies. While complement protein C4 is associated with SLE, its isoforms (C4A and C4B) are not equal in their impact. Despite being 99% homologous, genetic studies identified C4A as more protective than C4B. By generating gene-edited mouse strains expressing either human C4A or C4B and crossing these with the 564lgi lupus strain, we show that, overall, C4A-like 564Igi mice develop less humoral autoimmunity than C4B-like 564Igi mice. This includes a decrease in the number of GCs, autoreactive B cells, autoantibodies, and memory B cells. The higher efficiency of C4A in inducing self-antigen clearance is associated with the follicular exclusion of autoreactive B cells. These results explain how the C4A isoform is protective in lupus and suggest C4A as a possible replacement therapy in lupus.
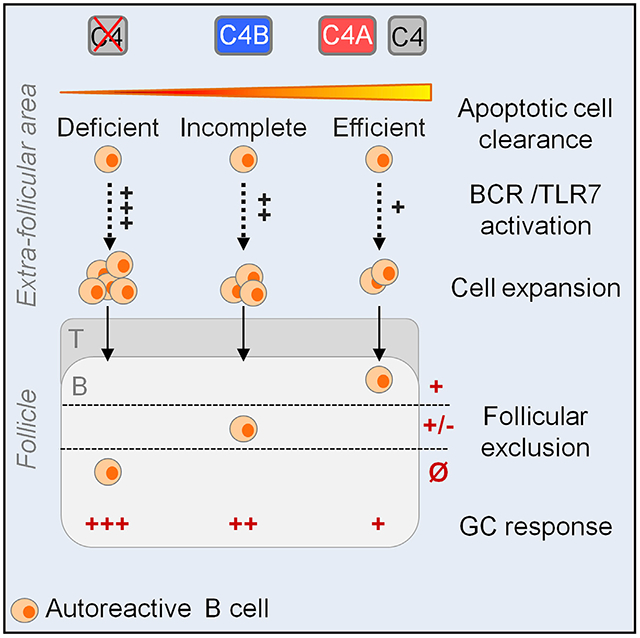
Graphical Abstract
In Brief
Simoni et al. address a long-standing question about how complement C4A and C4B isoforms differ in function in vivo in autoimmunity. They find that C4A leads to an increased protection in humoral autoimmunity relative to C4B. Autoantibody diversity is likewise dependent on the C4 protein isotype.
INTRODUCTION
Systemic lupus erythematosus (SLE) is a severe, systemic autoimmune disease characterized by the production of pathogenic autoantibodies responsible for damaging multiple organs (Omarjee et al., 2019; Tsokos, 2011). Strongly associated with the development of SLE, or lupus-like disease, is the complete deficiency of complement component C1q or C4, which increases the risk to 90% and 75%, respectively (Belot and Cimaz, 2012). The complement system facilitates targeting antigen to the B cell compartment and enhances T cell immunity during adaptive responses (Arbore et al., 2016; Carroll, 2004; Carroll and Isenman, 2012; Kolev et al., 2015; Lalli et al., 2008; Liszewski et al., 2013; Strainic et al., 2008; Walport, 2001a, 2001b; West et al., 2018). In innate immunity, complement drives the clearance of apoptotic debris and pathogens by scavenger cells and is a source of inflammatory anaphylatoxins (C3a and C5a). Similar to humans, mice deficient in C1q or C4 spontaneously develop lupus-like disease (Botto et al., 1998; Fischer et al., 1996), and this phenotype correlates with the defective clearance of apoptotic cells by peritoneal macrophages (Taylor et al., 2000). We hypothesize that this impaired clearance of apoptotic debris in the extracellular environment leads to B cell loss of tolerance (clearance hypothesis) (Carroll, 2004).
B cell tolerance mechanisms are thought to occur both in the bone marrow (checkpoint I) and in the periphery (checkpoint II) (Goodnow et al., 2005; Meffre and Wardemann, 2008). In the periphery, upon entering the spleen, immature B cells undergo negative selection based on B cell receptor (BCR) specificity and interclonal competition (Cyster et al., 1994; Goodnow et al., 1988; Harless et al., 2001). Although the mechanism behind interclonal competition is not fully understood, one hypothesis suggests that autoreactive B cell clones compete with non-self-reactive clones for B cell survival factor (BAFF). Insufficient BAFF-derived signaling leads to their exclusion from the B cell follicle, thus eliminating the self-reactive clones (Cyster et al., 1994; Lesley et al., 2004; Thien et al., 2004). This follicular exclusion mechanism was originally described in the hen egg lysozyme (HEL) transgenic model (Schmidt and Cyster, 1999), where HEL-autoantigen-binding B cells are excluded from follicles and rapidly eliminated in a T cell-independent way. This mechanism is also observed in other autoreactive B cell murine models, such as the 564Igi mouse (Chatterjee et al., 2013).
564Igi mice bear rearranged immunoglobulin (Ig) heavy- and light-chain genes inserted into their respective loci that are together specific for nucleolar antigens (Berland et al., 2006). Although the animals present high titers of IgG autoantibodies, they show minor symptoms of the disease late in life. The 564Igi mouse model constitutes a good model to study B cell tolerance. In heterozygote mice, the self-reactive transgenic B cells (Id+) are outcompeted by Id− B cells, resulting in their exclusion from the B cell follicles at the immature B cell stage. However, in heterozygous 564Igi mice deficient for C4, self-reactive B cells fully enter the follicles and form spontaneous germinal centers (GCs) (Chatterjee et al., 2013). It was proposed that defective clearance caused by C4 deficiency drives an accumulation of nucleic acid-containing self-antigens in the extracellular compartment, thus enhancing the escape of negative selection by self-reactive B cells. Internalization of nucleic acid (danger-associated patterns, or DAMPS) upon binding to the self-reactive BCR triggers TLR7 receptor pathway activation, thus inducing a second survival signal (Chatterjee et al., 2013).
The C4 locus is located in the human leukocyte antigen (HLA) class III region on chromosome 6 in humans and in the class III region of the murine major histocompatibility complex (MHC) on chromosome 17. In humans, this locus encodes 2 paralogous genes, C4A and C4B. The 2 isoforms are strongly conserved, differing only by 4 amino acids in the isotypic region, located in the C4d domain (Belt et al., 1984; Carroll et al., 1984). This region includes the internal thioester group that confers covalent binding specificity (van den Elsen et al., 2002). Functional studies comparing the 2 proteins found that C4A preferentially forms a covalent bond to amino group-containing substrates (e.g., proteins), whereas C4B preferentially binds to hydroxyl group-containing substrates (e.g., bacterial cell walls) (Law et al., 1984). This suggests that C4A is more efficient in immune clearance, whereas C4B is more responsible for microbial defense. Several studies have associated low C4A expression with SLE susceptibility in humans (Chen et al., 2016; Pereira et al., 2019; Yang et al., 2007). Notably, a recent large-scale genetic screen including >6,000 lupus patients and a similar number of healthy controls reported that, while both isoforms are protective relative to no C4, individuals deficient in the C4A isoform were at a higher relative risk of developing lupus (Kamitaki et al., 2020). The mechanism or mechanisms by which C4A protects from autoimmunity are not understood. The hypothesis of a difference between C4A and C4B affinity for a C4 receptor such as CR1 is still unclear (Reilly and Mold, 1997). More recent biochemical experiments show no difference in the binding affinity of C4A and C4B to the CR1 receptor using Biacore measurements (Clemenza and Isenman, 2004). Moreover, the crystal structure of the C4d region and positioning of the isotypic residues adjacent to the covalent binding site support a difference in chemical binding as a potential factor in protection (van den Elsen et al., 2002).
To understand how the 2 isoforms differ in function in vivo and how this relates to SLE, we used gene editing to modify the isotypic region within the murine C4 locus (positions 1,101–1,106) to express either human C4A or human C4B (mice express only 1 form of C4) (Belt et al., 1984; Blanchong et al., 2001). To compare the relative protection of the 2 isoforms in vivo, the strains were crossed with the 564Igi autoreactive strain (Berland et al., 2006). Our results show that 564Igi mice expressing the human C4A isotypic region exhibited a lower percentage of autoreactive B cells in the periphery and an overall reduction in autoimmunity relative to the C4B-expressing 564Igi mice. Improved protection correlated with more efficient clearance of apoptotic cells and an increase in follicular exclusion by B cells in the C4A 564Igi mice. Remarkably, the 2 strains varied in their autoantibody diversity, supporting the hypothesis that C4 binding efficiency to self-antigens contributes to the fate of self-reactive B cells and their expansion and secretion of autoantibodies.
RESULTS
Generation of Human C4A or C4B Mice by Genome Editing
Human C4A and C4B are distinguished by specific residues located within the isotypic region (positions 1,101–1,106), where they differ by 4 substitutions (PCPVLD for C4A versus LSPVIH for C4B) (Figure 1A). Mice express only 1 C4 gene that is functionally active in the classical pathway. Interestingly, mice express a second “C4-like” gene referred to as Slp (sex-limited protein) based on its expression only in males in certain strains of mice. Unlike mouse C4, Slp is not expressed by mice bearing the H-2b MHC such as the C57BL/6 strain, and Slp is not activated by complement C1s (Nonaka et al., 1986; Ogata and Zepf, 1991). The mouse C4 isotypic region is a hybrid of both human C4A and C4B residues (PCPVIH) (Belt et al., 1984; Chaplin et al., 1983). We generated “C4A” and “C4B” mouse models by micro-injecting C57BL/6J (B6) zygotes with a mixture of Cas9 mRNA, target-specific single guide RNA (sgRNA), and oligonucleotides coding for either the C4A or C4B human isotypic region, then transferred them to a pseudo-pregnant female (Figure 1A). To confirm (heterozygous) genome editing, litters were genotyped by PCR for C4A and C4B and then bred to homozygosity.
Figure 1. Generation of C4A and C4B Gene-Edited Mouse Strains.

(A) Simplified representation of C4A and C4B mouse generation using Crispr-Cas9 technology. sgRNA target in green, mutations in red, isotypic residues modulating covalent binding affinities of C4 in bold, and isotypic region in capital letters.
(B and C) Assessment of C4- and C3-coated immune complexes (ICs) in vitro. WT, C4A, C4B, or C4−/− mouse serum was incubated with an artificial IC. Left panels, representative dot plot; right panels, C3 and C4 mean fluorescence intensity (MFI) on ICs evaluated by flow cytometry. Means ± SEMs. One dot represents 1 mouse. n > 6 for each strain; 1-way ANOVA with Tukey’s test.
(D and E) C4 puncta quantification on FDC networks in spleen.
(D) Representative image of spleen immunostained for FDCs (anti-CD21 Ab, 7E9 clone, blue) and mouse C4 (16D2 clone, red). Scale bar represents 200 μm.
(E) The number of C4 puncta per μm2 of FDC network was quantified using Cell Profiler software. Means ± SEMs. One dot represents 1 field of view; n > 9; 3 mice per group.
(F and G) Analysis of hemolytic activity of C4−/−, WT, C4A, and C4B serum. Mean ± SEM (F); 1 dot represents the average value of 8 mice ± SD. H2O was considered 100% of lysis. Human serum was used as a positive control. Two-way ANOVA with Tukey’s test. (G) Results for 1/30 dilution; Means ± SEMs; 1 dot represents 1 mouse; 1-way ANOVA with Tukey’s test.
****p < 0.0001. See also Figure S1.
We first assessed the level of C4 mRNA expression in liver and spleen, 2 major tissues for C4 expression and production, by digital droplet PCR (ddPCR). As expected, C4 mRNA expression is comparable among C4A, C4B, and WT mice (Figure S1A), and no differences were observed in C4 protein levels in plasma (Figure S1B). To confirm that the edited C4 proteins coat immune complexes (ICs) and recruit C3 in vitro, we incubated serum from WT, C4A, C4B, and C4−/− mice with ICs (streptavidin-coated microspheres combined with biotinylated-IgG2a antibody [Ab]) and analyzed by fluorescence-activated cell sorting (FACS). Both C4A and C4B became activated by murine C1 complex and formed a functional C3 convertase, similar to WT mouse C4 (Figures 1B and 1C). When C4 is spontaneously activated in the spleen and binds acceptor sites in vivo, the complex is taken up by follicular dendritic cells (FDCs) through the CD35/CD21 receptor. To measure the functional activation in vivo of our edited mouse C4, we assayed C4 deposition on splenic FDC using immunohistochemistry (IHC) and confocal imaging. No significant differences in terms of C4 deposition pattern (Figure 1D) and abundance (Figure 1E) were observed among C4A, C4B, or WT mice.
C4B is more efficient in promoting red cell lysis than C4A due to the increased efficiency of histidine (at 1,106) at binding carbohydrate groups on the red cell surface (Isenman and Young, 1984). To confirm this functional activity in the mutant strains, we tested sera isolated from C4A, C4B, WT, and C4−/− mice for hemolysis using the sheep red blood cell (SRBC) assay. We found that C4A is less efficient than C4B sera in promoting SRBC hemolysis (Figure 1F), a difference of ~3-fold at a 1/30 serum dilution (Figure 1G), similar to that described for human C4A and C4B (Isenman and Young, 1984).
Regulation of Autoreactive B Cells Is Impaired in C4B-Expressing Lupus Mice
In 564Igi autoimmune mice, B cells express a transgenic (Tg) BCR specific for ribonuclear complexes and spontaneously produce high titers of nuclear autoantibodies largely independent of T cell help (Berland et al., 2006). In heterozygous 564Igi mice, a major fraction of the idiotype (Id)+ autoreactive B cells are eliminated by follicular exclusion from the B cell follicles (Chatterjee et al., 2013). More recent studies confirmed that a substantial fraction of the autoreactive B cells were Id− as they expressed an endogenous Ig heavy-chain V region and outcompeted the Id+ Tg B cells from the follicles (Degn et al., 2017). Notably, this regulation of Id+ B cells is in part dependent on C4 as Tg B cells in heterozygous 564Igi mice deficient in C4 appear to escape follicular exclusion and enter the B cell follicles (Chatterjee et al., 2013). To evaluate whether the 2 C4 isoforms affect the negative selection of self-reactive B cells, we examined the Id+ B cell population among the 4 groups of lupus mice and WT controls.
Mice were bled every 2 weeks for a period of 4 months (from 8-week-old [wo] to 24 wo) to determine the percentage of self-reactive transgenic B cells in peripheral blood by flow cytometry using anti-Id Ab (Figure 2A). There was a significant difference in the evolution of circulating Id+ B cells between C4A and C4B 564Igi mice over time. Notably, this population is decreased in C4A 564Igi compared to C4B564Igi between 14 and 19 wo (Figure 2B). This decrease was not due to a general reduction in B cells, as the frequency of total B220+ B cells in peripheral blood was equivalent for both strains (Figure S2A).
Figure 2. Absence of C4A Leads to Impaired Regulation of Autoreactive B Cells in Periphery.

(A and B) Analysis of autoreactive B cell population (B220+ Id+) in peripheral blood of C4A 564Igi (n = 5) and C4B 564Igi (n = 8) by flow cytometry. (A) Representative dot plots. (B) Percentage of Id+ B cells within the total B220+ B cell population. Means ± SEMs; Mann-Whitney test; 1 dot represents the average percentage for 1 strain.
(C–E) Analysis of autoreactive Id+ B cells within the total B220+ B cell population in secondary lymphoid organs by flow cytometry.
(C) Proportion of splenic Id+ B cells within the total B220+ B cell population from 8, 15, and 20 wo C4A and C4B 564Igi mice. Means ± SEMs; Mann-Whitney test; each dot represents the average of n > 8 mice.
(D) Proportion of Id+ B cells within the total B220+ B cell population in spleen (top panel) and skin-draining LNs (bottom panel) from 8 and 15 wo C4A, C4B, C4+/+, and C4−/− 564Igi. Means ± SEMs; 1-way ANOVA with Tukey’s test; each dot represents 1 mouse; n > 4 for each strain.
(E) Gating strategy to discern immature (i, B220+ AA4.1+), transitional (t, B220+ AA4.1int), and mature (m, B220+ AA4.1−) splenic B cell subpopulations.
(F) Proportions of Id+ B cells within immature, transitional, and mature splenic B cell subpopulations of 15 wo C4A, C4B, C4+/+, and C4−/− 564Igi. Means ± SEMs; 1-way ANOVA with Tukey’s test; each dot represents 1 mouse; n > 6.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figure S2.
Although a small fraction of autoreactive B cells are not Id+ in the 564Igi het mice, the Id marker was used as a surrogate to study the self-reactive B cell population. Lymphoid tissues, spleen, skin-draining lymph nodes (LNs), and mesenteric LNs were harvested and analyzed at different time points: 8, 15, and 20 wo. We found that the frequency of Id+ B cells in the peripheral blood correlated with that in the secondary lymphoid organs such as the spleen (Figure S2B). The frequency of Id+ B cells in C4−/− 564Igi was, as previously reported (Chatterjee et al., 2013), higher in both spleen and skin-draining LNs compared to all C4-sufficient strains for all ages (Figures 2D, S2C, and S2D). Similar to what was observed in circulation, differences in the proportions of transgenic B cells in secondary lymphoid organs emerged by 15 wo (Figures 2C and 2D). The frequency of splenic Id+ B cells was greater in C4B 564Igi compared to C4A 564Igi and C4+/+ 564Igi at 15 wo (average of 34.4% versus 22.7% and 23.8%, respectively; Figure 2D, top right) and 20 wo (average of 27.4% versus 19.1% and 17.9%, respectively; Figure S2C, left). A similar pattern was observed in skin-draining LNs (Figures 2D and S2C) and mesenteric LNs (Figure S2D). We also observed reduced total B220+ B cell frequencies in secondary lymphoid tissues from 564Igi lupus strains compared to WT mice, as previously described. However, this reduction was not affected by the different C4 isoforms or C4 deficiency (Figure S2E).
To compare the negative selection of immature Id+ B cells in the periphery among strains, immature (AA4.1high [CD93]), transitional (AA4.1dim), and mature (AA4.1neg) B cell subpopulations from the spleen were analyzed by flow cytometry (Figure 2E). In contrast to C4−/− 564Igi mice, the C4-sufficient lupus strains had reduced differentiation of Id+ immature B cells into the transitional and mature stages. The high proportion of autoreactive B cells present in the immature compartment was reduced within both transitional and mature stages due to negative selection (Figures 2F and S2F). Strikingly, the frequency of Id+ B cells within all B cell subpopulations was significantly elevated in C4B 564Igi compared to C4A 564Igi and C4+/+ 564Igi mice at 15 wo (Figure 2F).
To assess whether the difference in frequency of Id+ B cells occurs in primary lymphoid tissue, bone marrow cells were analyzed by flow cytometry. No differences were observed among the groups (Figure S2G). Thus, the general increase in Id+ B cells in C4B 564Igi mice is likely due to the increased escape of tolerance at a very early stage in the periphery.
Spontaneous GC Response Is Enhanced in the C4B 564Igi Strain
GCs are important sites within the lymphoid follicles, where activated B cells receiving T cell help undergo proliferation, somatic hypermutation, and clonal selection (Victora and Nussenzweig, 2012). 564Igi het mice have T cell-dependent spontaneous GCs harboring autoreactive B cells specific for a broad range of selective reactivities. These autoreactive B cells can undergo clonal evolution and compete for T cell help and antigen, resulting in the emergence of dominant clones in many GCs (Degn et al., 2017). Interestingly, C4 deficiency promotes an increase in spontaneous GC formation, possibly due to impaired follicular exclusion (Chatterjee et al., 2013).
To evaluate whether the C4 isoforms affect GC responses, we measured B220+ CD38dim/− GL7+ GC B cell frequencies in spleen and skin-draining LNs by flow cytometry. In 15 wo mice, splenic GC B cell frequencies were increased in C4B 564Igi mice compared to C4A 564Igi mice and C4+/+ 564Igi mice (average of 3.0% versus 1.9% and 1.5%, respectively) (Figure 3A). Similar results were observed in skin-draining LNs (Figure S3A). In parallel, we observed an increase in the frequency of splenic T follicular helper (Tfh) cells in the C4B 564Igi strain (Figure S3B). GC activity may be reflected by their size and frequency. Using IHC, we observed more GCs in the spleens of C4B 564Igi mice than in C4A 564Igi or C4+/+ 564Igi mice (Figures 3B and 3C). In addition, GCs in C4B 564Igi animals were larger, as evidenced by the increased GC area (Figures 3D and S3C). As the BCR encoded by the 564 heavy and light chains is reactive against nucleolar antigens, we assessed the level of anti-nucleolar autoantibodies in the different 564Igi strains by using purified nucleoli (Das et al., 2017; Degn et al., 2017). Consistent with an increase in GCs in the C4B 564Igi mice, there was a trend toward higher levels of nucleoli-reactive IgG autoantibodies in C4B 564Igi mice (Figure 3E). However, the measurement by ELISA of anti-single stranded DNA (ssDNA) and anti-double stranded DNA (dsDNA) IgG autoantibodies, both typical lupus autoantibodies, did not reveal striking differences among the strains tested (Figure S3D). These results show that spontaneous formation and expansion of GCs is reduced in C4A mice compared to C4B 564Igi mice, suggesting that the C4A and C4B isotypic sequences confer functional differences that affect the GC response.
Figure 3. GC Structures Are More Frequent and Larger in C4B 564Igi Mice Than in C4A 564Igi Mice.

(A) Splenic GC B cell population (B220+ CD38dim/− GL7high) analyzed by flow cytometry in 15 wo C4A, C4B, and C4+/+ 564Igi. Means ± SEMs; 1-way ANOVA with Tukey’s test; 1 dot represents 1 mouse; n > 6.
(B–D) Analysis of GC structures by immunohistochemistry.
(B) Representative images of spleen follicles. Marginal zone macrophages: CD169 (red), B cell zone: B220 (blue), T cell zone: CD3 (green) and GC: GL7 (white); the scale bar represents 100 μm.
(C) Frequency of GL7+ follicles. Means ± SEMs; 1-way ANOVA with Tukey’s test; 1 dot represents 1 mouse; n > 8 for each 564Igi strain.
(D) Quantification of individual GL7+ area using ImageJ software. Means ± SEMs; 1-way ANOVA with Tukey’s test; 1 dot represents 1 GL7+ area; n > 7 animals for each 564Igi strain.
(E) Serum anti-nucleoli IgG titers measured by ELISA. Means ± SEMs; 1 dot represents 1 mouse; n > 8.
*p < 0.05, **p < 0.01, ****p < 0.0001). See also Figure S3.
We next assessed B cell differentiation toward Ab-secreting cells among the 3 groups of autoreactive mice. By flow cytometry, no difference in the percentage of plasma cells (B220− CD138+) was observed in secondary lymphoid organs at both 8 wo (not shown) and 15 wo (Figure S3E). Similarly, no significant difference in the serum level of total IgG was observed (Figure S3F). This result was not unexpected, as plasma cells are not only derived from the GC reaction in this model but are also independent of T cell help and can form spontaneously due to the rearranged BCR transgene in Id+ B cells (Berland et al., 2006).
C4A Expression Leads to a Reduced Autoreactive Memory B Cell Population
To compare the population of memory B cells among the C4 564Igi strains, we crossed the lupus mice with the Aicda-CreER-T2EYFP fate-mapping reporter, which marks GC-experienced B cells (Dogan et al., 2009). The 564Igi strains were treated with tamoxifen at 6 wo and analyzed at 15 wo (Figure 4A). We refer to the B220+ CD138neg GL7neg EYFP+ cells as “memory B cells,” which are cells that turned on the EYFP reporter (because they were GC derived at the time of tamoxifen treatment), but were negative for GL7 (GC) or CD138 (plasma blasts) (Figure 4B). Consistent with the differences observed in the GC stage, the frequency of memory B cells was reduced in the spleens of C4A 564Igi mice compared to C4B 564Igi mice (Figure 4C). To a lesser extent, a similar pattern was observed in skin-draining and mesenteric LNs (Figure S4).
Figure 4. Reduction of Autoreactive Memory-like B Cells in C4A 564Igi.

(A) Experiment timeline.
(B) Gating strategy to identify memory-like B cells (B220+ EYFP+ GL7− CD138−).
(C) Analysis of autoreactive memory-like B cell population (B220+ YFP+ CD138− GL7−) by flow cytometry. Means ± SEMs; 1-way ANOVA with Tukey’s test; 1 dot represents 1 mouse; n > 3.
*p < 0.05.
See also Figure S4.
C4A Helps Maintain B Cell Tolerance by Inducing Clearance of Self-Antigens
Previous studies demonstrated the importance of complement C1q and C4 in clearing apoptotic cells in vivo (Taylor et al., 2000). They reported that mice deficient in C1q or C4 were less efficient in the uptake and clearance of labeled thymocytes by thioglycolate-activated peritoneal macrophages. To investigate whether the differences between C4A and C4B affect clearance, we injected mice intraperitoneally with pH-Rhodo-labeled apoptotic murine thymocytes 4 days after inducing sterile peritonitis with thioglycolate (Taylor et al., 2000) (Figure 5A). The apoptotic state of the murine thymocytes was confirmed by flow cytometry using propidium iodide and annexinV staining prior to injection (Figures 5A and 5B). A short period (10 min) was selected to assess the clearance efficiency based on the earlier study (Taylor et al., 2000). After collection of peritoneal exudate, cells were kept on ice to avoid ex vivo phagocytosis (Figure S5A). The uptake of apoptotic cells by elicited macrophages was analyzed by measuring the proportion of pH-Rhodo+ F4/80+ cells by flow cytometry (Figure 5C). As expected, based on earlier observations, C4−/− mice cleared labeled cells less efficiently than WT mice (Figure 5D) (Taylor et al., 2000). Strikingly, macrophages in C4A mice cleared apoptotic cells with higher efficiency in vivo compared to C4B and C4−/− mice (Figure 5D). Flow cytometry showed no differences in macrophage recruitment (Figures S5B and S5C) or the expression of complement receptor 3 (CD11b) or activation marker CD206 (Figures S5D and S5E).
Figure 5. C4A Is More Efficient Than C4B in Inducing Clearance of Apoptotic Cells In Vivo and Driving Effective Follicular Exclusion of Id+ B Cells.

(A) Experiment timeline.
(B) Representative dot plot of apoptotic stages analyzed by flow cytometry using propidium iodide (PI) and annexinV (AnnV) staining (a [live cells]: AnnV− PI−; b [early apoptosis]: AnnV+ PI−; c [late apoptosis]: AnnV+ PI+).
(C and D) Analysis of apoptotic cell uptake by F4/80+ macrophages. (C) Representative dot plots of apoptotic cell uptake by macrophages (pH-Rhodo+) for all of the strains. (D) Frequency of macrophages ingesting apoptotic bodies, analyzed by flow cytometry and relative to WT mice. Means ± SEMs; each dot represents 1 mouse; n > 10 over 6 independent experiments; 1-way ANOVA with Tukey’s test; *p < 0.05; ***p < 0.005.
(E and F) Analysis of Id+ B cell follicular exclusion at 20 wo. (E) Representative image of splenic follicles. Follicular B cells: IgD (blue), T cell zone: CD3 (white), Id+ B cells: anti-idiotype (9D11 clone) (red). The scale bar represents 100 μm. The dashed lines determine B cell follicles. (F) Follicular exclusion, expressed as percentage of follicular B cell zone occupied by Id+ B cells, was quantified using Cell Profiler software. Means ± SEMs; each dot represents 1 mouse; n > 5; 1-way ANOVA with Tukey’s test; *p < 0.05; ****p < 0.001.
See also Figure S5.
The association between the impaired clearance of apoptotic cells and reduced follicular exclusion in C4−/− relative to C4+/+ heterozygous 564Igi mice supports the “clearance hypothesis” (Carroll, 2004). Thus, reduced clearance of apoptotic cells would increase the amount of apoptotic material in the extracellular compartment that is available for binding by cognate B cells. Accordingly, BCR uptake of nucleic acid-containing material and triggering of TLR7 pathway would provide a second signal fostering the escape of tolerance by self-reactive B cells at the transitional stage or checkpoint II (Botto et al., 1998; Wardemann and Nussenzweig, 2007; Green et al., 2012; Leadbetter et al., 2002). Our finding that C4A 564Igi mice clear apoptotic cells more efficiently than C4B and C4−/− strains suggests that follicular exclusion may be similarly affected. To test this possibility, spleens were isolated from the 4 strains at 15–20 wo. Localization of the Id+ B cells to the follicular area was determined by IHC (Figure 5E). Follicular exclusion was evaluated by quantifying the percentage of the follicular B cell zone, defined as IgD+, occupied by Id+ B cells (Figure 5F). Interestingly, the values we obtained were in concordance with the percentage of Id+ B cells present in the spleen (Figure 2C, top right). Thus, while Id+ B cells occupy a mean of ~70% of the B cell follicle in C4-deficient 564Igi animals, exclusion was significantly greater in C4+/+ 564Igi mice (mean 30%) (Figure 5F). A significant reduction in Id+ B cells in splenic follicles was observed in C4A mice (mean 30%) relative to C4B 564Igi mice (mean 43%) (Figures 5F and S5F). As expected, no difference between C4-sufficient strains was observed at 8 wo (Figures S5G and S5H).
In summary, we identified an association between the efficiency of apoptotic cell clearance and exclusion of self-reactive B cells. Thus, mice expressing C4A were relatively more efficient than those with C4B in clearing apoptotic cells and showed less escape of tolerance by self-reactive B cells.
C4A and C4B Expression Is Associated with Different Ab Profiles in 564Igi Mice
Given the differences between C4A and C4B lupus mice in their follicular exclusion efficiency and spontaneous GC formation, we hypothesized that the strains may also differ in autoantibody reactivities. To understand the diversity of autoantibodies produced by the lupus mice, serum IgG was isolated from the 2 groups of lupus mice (8 C4A 564Igi and 10 C4B 564Igi) and assayed for binding in a self-antigen array assay (Table S6). The self-antigens selected were common autoimmune targets (e.g., Ro60, ssDNA) or epitope-spread antigens identified in the 564Igi model (e.g., Smd2, BPI) (Degn et al., 2017). Self-antigens not thought to be recognized by the 564Igi Tg BCR are referred to as “epitope spread” (Degn et al., 2017). Overall, IgG antibodies from both mutant 564Igi strains were reactive against a broad panel of self-antigens, as expected. Although the differences were not statistically significant, sera from C4B 564Igi showed a relative increase in reactivity to 70% of the self-antigens tested when compared to C4A 564Igi sera (Figure 6A).
Figure 6. C4A Lessens Autoantibody Production in the 564Igi Lupus Model.

(A) Volcano plot comparison of median IgG signal intensities in C4A 564Igi (n = 8) relative to C4B 564Igi (n = 10) mice. Log2(fold change) appears on the x axis. Significance (presented as −10 × log10[p value]) is on the y axis. The p value was obtained running multiple unpaired t tests corrected with the Holm-Sidak method. One dot represents 1 self-Ag.
(B–D) Dosage of anti-Ro60, -BPI, and -CTGF IgG titers in serum of 15–20 wo C4−/−, C4A, C4B, and C4+/+ 564Igi by ELISA. Means ± SEMs; 1 dot represents 1 mouse; n > 4; 1-way ANOVA with Tukey’s test; *p < 0.05.
ELISA was used to validate the reactivities identified in the self-antigen array. Despite similar anti-ssDNA titers (Figure S3D), a higher production of IgG antibodies targeting Ro60/SSA by C4B 564Igi mice compared to C4A 564Igi mice and, to a lesser extent, the other 564Igi strains, was confirmed by ELISA (Figure 6B). Four of the self-antigens tested by ELISA were considered epitope-spread antigens in 564Igi mice, in other words, BPI (bactericidal/permeability-increasing protein), CTGF (connective tissue growth factor), Smd2, and C1q. A trend of higher IgG titers was observed in C4B compared to C4A 564Igi mice for 3 of the 4 self-antigens tested, with a statistical difference for BPI (Figures 6C, S6A, and S6B). Remarkably, anti-CTGF autoantibody, identified in the self-antigen array, was more abundant in C4A than in C4B 564Igi animals and was produced largely by 2 C4A 564Igi mice (Figure 6D).
These results reveal that only a few amino acid differences in the C4 isotypic region can affect autoantibody secretion and diversity.
DISCUSSION
A current enigma in autoimmune disease is that a deficiency in C4, despite its importance in mediating inflammation, almost always results in lupus. This observation has been referred to as the “lupus paradox,” and it supports the importance of a protective role for complement in SLE (Carroll, 2004). Although complete deficiency is rare, partial deficiency (i.e., the absence of C4A or C4B) is quite common, with a gene frequency of ~1%–2%. Whether the 2 isoforms are equally protective in autoimmunity has been a long-standing question. Early studies proposed that, given the chemical binding differences between the 2 isotypes of C4, individuals deficient in either form may have an impaired response to specific pathogens or increased susceptibility to certain diseases (Law et al., 1984; Porter, 1983). Genetic studies attempting to address this question have been difficult to interpret, due to small cohorts of patients and linkage disequilibrium in the HLA region (i.e., limited recombination over long stretches of this region, which covers >5 Mbp). Despite the complications of linkage disequilibrium, a large genetic screen identified C4A as a major factor in susceptibility for schizophrenia (Sekar et al., 2016). Using a similar approach, Kamitaki et al. (2020) screened >6,000 SLE patients for a number of recombinant haplotypes in which deficiency in C4A or C4B was distinct from the HLA DR region. Importantly, they found that the increased copy number of C4A was relatively more protective than C4B.
To develop an animal model to test the relative importance of the 2 isotypes in autoimmunity, we used a gene editing approach to introduce nucleotide substitutions to modify the mouse locus to encode either a C4A- or C4B-like isoform. One advantage of this approach is that the other genes within the MHC remain constant, thereby circumventing the problem of linkage disequilibrium. Thus, an altered immune response between the 2 strains can be interpreted as due to C4 isotype and not neighboring MHC class I or class II genes.
Characterization of the 2 mutant strains confirmed that the C4A and C4B mice express similar levels of complement C4, as predicted. Both strains showed a similar generation of C3 convertase and activation of C3. More important, functional differences in covalent binding to target antigens were distinct – in other words, sera from the C4B mice were ~3-fold more active in the SRBC hemolysis assay than sera from the C4A mice. These results are in line with previous studies comparing human C4A and C4B (Isenman and Young, 1984).
Using a lupus-like strain, we found that C4A 564Igi mice developed less humoral autoimmunity relative to C4B 564Igi mice beginning at 12 wo. C4A 564Igi mice had fewer circulating self-reactive B cells based on the frequency of Id+ B cells, a reduced frequency of spontaneous GC B cells, a reduced number and size of spontaneous GC, and a reduced memory B cell population. Despite a significant reduction of these autoimmune parameters in the C4A 564Igi mice, anti-nucleolar autoantibody titers and the frequency of plasma cells were not significantly reduced relative to C4B 564Igi mice. This could be explained by the high background of anti-nucleolar antibodies secreted spontaneously by the 564Igi transgenic B cells. Berland et al. (2006) reported that, even on a RAG-2-deficient background, the Tg B cells in homozygous 564Igi mice secrete a relative high titer of anti-nucleolar antibody that is TLR7 dependent. Notably, after an initial screen using an autoantigen array, we observed differential reactivity to specific self-antigens between the 2 strains. For example, the serum titers for Ro60 and BPI in C4A 564Igi mice were significantly reduced relative to that observed in C4B 564Igi mice. By contrast, the titer of anti-CTGF autoantibody was increased in C4A mice relative to C4B lupus mice. We attribute these isotype-specific differences in response to self-antigens as due to the chemical binding difference between C4A and C4B, much like that observed in the SRBC lysis assay. In future studies, it will be important to test this hypothesis by immunizing the C4A and C4B 564Igi mice with the relative self-antigens. Given our findings in these murine models, it would be interesting to compare autoantibody reactivity in lupus patients according to their C4 isotype.
A current paradigm in lupus is that impaired clearance of apoptotic cells can lead to the escape of B cell tolerance via a TLR7-dependent pathway (Green et al., 2012; Leadbetter et al., 2002). Given the importance of C1q and C4 in apoptotic cell clearance, we propose that complement is also a factor in the paradigm. Accordingly, the rapidity of complement binding to apoptotic cells can influence their rate of clearance and availability for binding by cognate B cells. The early studies by Law and Dodds (1997) quantitated the time required for covalent binding of C4A and C4B to small molecule ligands (i.e., glycine and glycerol, respectively). Although apoptotic cells are more complex and less well defined, it is likely that the isotypic residues that are located adjacent to the thioester site affect C4 contact and the rate of clearance by phagocytic cells. Thus, we propose that chemical differences within the isotypic region confer to C4A a more rapid interaction and covalent binding and clearance of apoptotic material than C4B. Therefore, according to the clearance hypothesis, less contact with apoptotic material by self-reactive B cells in C4A mice would limit their escape from follicular exclusion relative to C4B. This interpretation is supported by our finding that peritoneal macrophages are more efficient in the uptake of apoptotic cells in C4A relative to C4B mice. Moreover, a gradient of the follicular exclusion of B cells was observed among the 4 groups, with limited exclusion in C4−/− and near-complete exclusion in C4A mice. Thus, a correlation was observed in the level of follicular exclusion and formation of spontaneous GC. It will be important in future studies to use the C4A and C4B 564 Igi mice to test this hypothesis by tracking the clearance of apoptotic cells within the secondary lymphoid compartment and uptake of self-antigens on FDCs.
Additional factors that may play a role in the differential uptake of apoptotic cells in C4A or C4B lupus mice are C4 receptors. While an early study comparing the binding of C4A and C4B protein to the C4b receptor CR1 (CD35) suggested the isotypic region affected receptor binding (Reilly and Mold, 1997), more recent biochemical studies reported a similar affinity of binding for both isoforms (Clemenza and Isenman, 2004). Most recently, Neuropilin-1 (NRP-1) was identified as a receptor for the C4d region, which includes the isotypic sites (Battin et al., 2019). NRP-1 is expressed by many immune cells, including macrophages (Fantin et al., 2013; Aung et al., 2016). Whether C4d binding to NRP-1 is affected by the isotypic region was not reported.
In summary, we find that only a few amino acid differences between the 2 complement isoforms, C4A and C4B, can lead to changes in B cell tolerance and autoimmunity. We suggest that an increased rate of binding and clearance of apoptotic cells in C4A mice is more effective than C4B in regulating self-reactive B cells. Our results suggest how C4 isotype can shape adaptive immunity in autoimmune disease and provide support for recent genetic studies identifying C4A as more protective than C4B as a risk factor for SLE (Chen et al., 2016; Kamitaki et al., 2020; Pereira et al., 2019; Yang et al., 2007).
Based on this differential role for the 2 isoforms of C4 and the increased susceptibility of individuals that express C4B only, C4A protein replacement may provide a therapy for lupus patients with clearance defect.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Michael C. Carroll (michael.carroll@childrens.harvard.edu).
Materials Availability
C57BL/6.mC4A/A and C57BL/6.mC4B/B mouse lines generated in this study will be deposited to Jackson Laboratory.
Data and Code Availability
The published article includes all datasets/code generated or analyzed during this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal models
C57BL/6J mice were obtained from Jackson Laboratories. 564Igi mice (Berland et al., 2006) were initially provided by Theresa Imanishi-Kari (Tufts University) and were maintained in-house. AicdaCreERT2 flox-STOP-flox-EYFP mice (Dogan et al., 2009) were from Claude-Agnès Reynaud and Jean-Claude Weill (Institut Necker, Paris) then maintained in-house. 564Igi mice were bred with AicdaCreERT2 flox-STOP-flox-EYFP mice to obtain 564Igi- AicdaCreERT2 flox-STOP-flox-EYFP mice. C4-deficient mice (Fischer et al., 1996), backcrossed more than 18 times with C57BL/6J,were bred with 564Igi mice to obtain C4−/− 564Igi mice. For C4A and C4B mice new strains, C57BL/6J zygotes were microinjected with a mixture of Cas9 mRNA, target-specific sgRNA, and oligonucleotides coding for either C4A or C4B human isotypic region (Table S1). Oligonucleotides had ~60-bp homology arms and included synonymous mutations to introduce a unique restriction site and additional silent mutations to remove the targeted PAM site. The design of the sgRNA guide is further explained below. After injection, zygotes were grown to the 2-cell stage and transferred to the oviducts of pseudo pregnant females. Resulting litters may contain C4A or C4B mice through homology directed repair (HDR) or small insertions or deletions due to error prone nonhomologous end joining (NHEJ). To confirm successful (heterozygous) genome editing, litters were genotyped by PCR for C4A or C4B (Table S2). After breeding to homozygosity, C4A and C4B mice were then crossed with either 564Igi or 564Igi AicdaCreERT2 flox-STOP-flox-EYFP mice. All the experiments were performed on 8–25 wo mice. As no phenotype differences were observed between genders, data from both males and females were analyzed together. C57BL/6J mice were used as non-auto-immune controls for experiments and are named WT. All mice were bred and maintained in the AAALAC-accredited facility at Harvard Medical School. All animal experiments were conducted in accordance with the guidelines of the Laboratory Animal Center of the National Institutes of Health. The Institutional Animal Care and Use Committee of Harvard Medical School approved all animal protocols (protocol number IS00000111).
sgRNA guide design for the generation of C4A and C4B edited strains
sgRNA guides were designed using https://zlab.bio/guide-design-resources. Because of the 100% homology with Slp around the C4 isotypic regions, all guides annealed to both C4B and Slp. sgRNA was generated as follows: complementary oligonucleotides coding for the guide RNA were ordered from IDT and cloned into px330 vector. Insertion of the guide RNA into the plasmid was confirmed using Sanger sequencing (DFCI/Harvard core). sgRNA DNA templates were generated using a forward primer containing a T7 promoter sequence and a reverse primer annealing at the end of the tracrRNA sequence. Purified sgRNA templates were then used in an in vitro transcription reaction to generate the final sgRNAs (Mega Shortscript, Thermo Fisher Scientific). Cas9 template DNA (Table S1) was amplified from px330 and transcribed to capped, poly-A tailed mRNA using the mMessage mMachine kit (Thermo fisher Scientific). Several guides were first tested by piezo-injected assisted injection of Cas9 mRNA and sgRNA in the cytoplasm of B6D2F1 zygotes. These were then grown to blastocysts, which were subsequently tested for the presence of NHEJ by nested PCR amplification followed by Sanger sequencing. Efficiency was calculated as the percent of blastocysts showing NHEJ in one or two alleles at the targeted region. The guide “C4B-3” (GGATGACTGGACATGGGTCG) was selected despite it displaying moderate efficiency (33%), since it anneals directly within the isotypic region.
Additional procedures
Recombination in AicdaCreERT2 flox-STOP-flox-EYFP mice was induced by a single gavage of 15 mg of tamoxifen (Sigma) dissolved in sunflower seed oil (Spectrum chemical) at 30 mg/mL.
METHOD DETAILS
Genotyping, FACS typing
Genotyping was performed on tail DNA using the primers and reaction conditions indicated in Table S2. For C4A and C4B, PCR products were digested for 2 hours with restriction enzymes: BAMHI-HF at 37°C for C4A and BsAWI at 60°C for C4B (Table S3). For FACStyping, 564Igi mice were bled retroorbitally using heparinized capillary tubes (Drummond). For PBMC isolation, about 70 μL of blood was added into Eppendorf tubes containing 30 μL of acid-citrate-dextrose solution (Sigma). After a brief centrifugation, blood was underlayered with 1 mL of Lymphocyte Separation Medium (Corning) and spun for 30 min, at 1800 rpm at RT, without brake. The mononuclear cell layer was aspirated and transferred into 1 mL of ice-cold FACSbuffer (PBS1x, 2mM EDTA, 2% FCS, 8mM Na Azide), mixed, then pelleted at 1400 rpm for 5 min. Cells were resuspended in FACSbuffer and processed for flow cytometry analysis using anti-B220, -IgMa, -IgMb (Biolegend), and –idiotype (9D11 clone, in-house generated, conjugated to biotin) antibodies.
Analysis of C4 mRNA expression by ddPCR
RNA from spleen and liver was isolated with TRIzol Reagent (Ambion, Life technology) according to manufacturer’s protocols. cDNA was obtained with iScript gDNA clear cDNA Synthesis kit (BioRad). To measure C4 mRNA expression, droplet digital PCR (ddPCR) was performed. The primer sequences for C4 and reference gene product eif4h are presented in Table S4. 20 to 50 ng of cDNA was mixed with 11 μL of 2 × ddPCR mix and 1 μL of each 20 × probe. For each sample, the reaction mixture was emulsified into approximately 20,000 droplets in an oil/aqueous emulsion, using a microfluidic droplet generator (BioRad). The droplets containing the reaction mixture underwent PCR amplification using the following cycling conditions: 95°C for 10 min, 40 cycles of 94°C for 30 s, 60°C for 1 min, followed by 98°C for 10 min. After PCR, the fluorescence (both colors) in each droplet was read using a QX200 droplet reader (BioRad). Data were then analyzed using the QuantaSoft software (BioRad), which provides absolute quantification of cDNA target abundance.
Antibody detection and C4 protein quantification by ELISA
Antibodies and proteins used for coating (Table S5) were diluted in PBS and subsequently bound to a high-binding microplate (Greiner). After O/N incubation at 4°C, the plates were washed 3 times with PBS and 0.1% (v/v) Tween20 (PBS-T). The wells were then blocked by incubation with PBS and 1% BSA at 37°C for 45 minutes. Then, standard, blank (buffer only), and samples (serum or plasma) diluted in PBS-T as specified in Table S5 were added for 1.5 h at 37°C. Of note, for C4 titration, prior to serum preparation, 1 mM EDTA was added to blood to avoid complement activation and C4 spontaneous hydrolysis. After 3 more washes with PBS-T, secondary AP- or HRP-conjugated antibodies diluted in PBS-T were added to the wells for 30 min at 37°C. Phosphatase substrate, 1 mg/mL in AP substrate buffer (1 M diethanolamine, pH 9.8, with 0.5 mM MgCl2) or TMB substrate (ThermoScientific) was used to develop the reaction, and it was read out on a SPECTRAmax 340 Microplate Reader at λ = 405nm for AP substrate or λ = 450nm for TMB, running SoftMax Pro V.5.4 software.
To measure the levels of anti-nucleoli IgG, nucleoli were purified from Raji cells (ATCC). The cells were grown to a density of _1 * 106 cells per ml. For the isolation, 108 cells were spun down at_200 g (1000 rpm, Beckman GS-6 centrifuge, GH-3.8 rotor) for 5 min and washed twice with ice-cold PBS. After another centrifugation at the same speed, the supernatant was decanted and the cells were resuspended in 5 mL fresh ice-cold Buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT). After incubation on ice for 5 min, the cells were transferred to a pre-cooled 7 mL dounce homogenizer (Wheaton Scientific) and subjected to 10 strokes using a tight pestle (“A” specification: 0.0010” - 0.0030” clearance), while keeping the homogenizer on ice. An aliquot of the homogenate was checked under a phase contrast microscope, and the procedure repeated as necessary, until > 90% of the cells were burst, leaving intact nuclei, with various amounts of cytoplasmic material attached. The homogenate was then centrifuged 200 g for 5 min at 4°C. Following aspiration of the supernatant, the pellet, containing enriched but not highly pure nuclei, was resuspended in 3 mL S1 solution (0.25 M Sucrose, 10 mM MgCl2). The resuspended pellet was layered over 3 mL of S2 solution (0.35 M Sucrose, 0.5 mM MgCl2) and centrifuged at 1,400 g (2,500 rpm) for 5 min at 4°C. The pellet was resuspended in 3 mL of S2 solution and the resulting nuclear suspension was sonicated for 6 × 10 s bursts (with 10 s rest intervals between each burst) using a Misonix XL 2020 sonicator fitted with a microtip probe, and set at power setting 5. The sonicated nuclei were periodically checked under a phase contrast microscope to ensure the virtual absence of intact cells and nuclei, and confirm the presence of free nucleoli observable as dense refractile bodies. The sonicated sample was layered over 3 mL of S3 solution (0.88 M Sucrose, 0.5 mM MgCl2) and centrifuged at 3,000 g (3,500 rpm) for 10 min at 4°C. The nucleoplasmic fraction was aspirated and the nucleolar pellet was resuspended in 0.5 mL of S2 solution, followed by centrifugation at 1,430 g (2,500 rpm) for 5 min at 4°C and resuspension in 0.5 mL of S2 solution. Each batch was checked carefully under a phase contrast microscope to ensure presence of highly purified nucleoli without any other material, and stored at −80°C until use. A volume of 100 μL of purified nucleoli diluted in PBS was used to coat one microtiter plate. To allow an effective coating, plates were spun down for 10 min at 2500 rpm (1300 g) at 4°C. After 3 washes and a blocking step as described above, blank and diluted samples were added to the wells for 1.5 h at 37°C. The final steps are similar as those described above.
Measuring the hemolytic complement activity of serum
Mice were bled retroorbitally and blood was placed into a tube containing 5 μL of EDTA (0.1 M, pH 8) then kept on ice. Blood was spun twice at 3000 rpm and 9000 rpm for 8 min at 4°C in order to collect plasma. 1 mL of EA cells (sheep red blood cells coated with hemolysin, Complement Technology) was washed with GVB (Complement Technology) and spun at 1500 rpm for 5 min at RT until supernatant was clear. The pellet was resuspended in 400 μL of GVB. The cell concentration was evaluated by mixing 10 μL of cell suspension to 140 μL of H2O and absorbance was measured at λ415nm. The volume needed to be at 1 × 109 cells/mL and was calculated as: 400 × (OD415/0.62). The cell suspension was diluted 10 times to be at a working concentration of 1 × 108 cells/mL. A series of two-fold dilutions of control and test plasma (1:30 to 1:120 for mouse samples, 1:30 to 1:100000 for human serum) was prepared in GVB. 50 μL of diluted plasma, 50 μL of 2% C4-deficient guinea pig serum (Complement technology), and 30 μL of EA cells at 1 × 108 cells/mL were mixed together in a 96-well plate and incubated for 30 min at 37°C. A complete lysis control (100 μL of H20 + 30 μL EA cells) and a negative control (100 μL of GVB + 30 μL EA cells) were also included. After 5 min of centrifugation at 1500 rpm at RT to sediment the RBCs, the supernatant was collected. The absorbance of the samples was read at 541 nm using a spectrophotometer (Shimadzu, UV-1700).
Immunohistochemistry
Spleens were frozen at −80°C in Tissue-Tek OCT embedding media (Fisher Healthcare). Cryosections of 12 μm were prepared, fixed with cold acetone, blocked with PBS, 0.01% Tween20, 2% BSA, and 5% FCS for 1 hour at RT, and then stained for 1 hour at RT. Antibodies (including PB, FITC, PE and APC anti-CD169, -B220, -CD3, -GL7, -IgM, -IgD, -CD21/35 (All from Biolegend), rat anti-mouse C4 (clone 16D2, gift from E. Kremmer, Munich, Germany), and mouse anti-idiotype (clone 9D11, in-house generated) were diluted in PBS containing 0.01% (v/v) Tween20. When needed, slides were washed and secondary antibodies, donkey anti-goat IgG or goat anti-rat IgG diluted in PBS containing 0.01% Tween20, were added for 1 hour at RT. The stained slides were then mounted using mounting medium (FluoroGel, Electron Microscopy Sciences). Images were acquired with confocal microscope (Olympus, Fluoview FV1000), using Fluoview software. The images were processed with ImageJ software. GC frequency and size were analyzed using ImageJ software. Follicular exclusion of 9D11+ B cells and C4 and C3 deposition in spleen follicles were quantified using Cell Profiler software (Lamprecht et al., 2007).
Flow cytometry
Analysis of cell phenotype and apoptotic body uptake by macrophages
Immune cell phenotypes of spleen, lymph nodes (LNs), BM, isolated PBMC (see “FACS typing”), or peritoneal exudate were studied by eight-color fluorescence analysis according to standard protocol. The following antibodies were used: Pacific Blue, FITC, PE, PercpCy5.5, APC, PECy7, or biotin-conjugated anti-mouse B220, GL7, CD38, CD93, CD138, IgMa, IgMb, 9D11, CD4, CD3, CXCR5 (CD185), PD-1 (CD279), F4/80, CD11b, CD206 (all from Biolegend). Fixable viability Dye efluor780 (eBioscience) was used as a viability marker. To measure the level of apoptosis, 1 × 105 cells were resuspended in a mix of 100 μL of AnnexinV specific buffer (10mM HEPES pH7.4, 0.14M NaCl, 2.5mM CaCl2), 5 μL of FITC-labeled AnnexinV (Biolegend), and 0.5 μg/mL of propidium iodide (PI, Sigma). After an incubation of 15 min in the dark at RT, apoptosis stages were analyzed by flow cytometry.
C3 and C4 binding on immune complexes
Streptavidin-coated polystyrene microspheres (Bangs lab) were incubated with 1.5 μg/mL of IgG2a-biotinylated antibody for 1 h on ice to form immune complexes (IC). IC were spun for 10 min at 1500 rpm and resuspended in GVB buffer (Complement Technology). 30 μL of IC was incubated with 50 μL of 2% C4-deficient guinea pig serum (Complement technology) and 50 μL of serum diluted in GVB (10%, 1%, 0.1% final concentration) for 30 min at 37°C. The reaction was stopped by adding 100 μL of PBS with 5 mM EDTA, 2% FCS. The beads were washed and stained for C3 and C4 using rabbit anti-human C4c (Dako) and goat anti-mouse C3 (MP Biomedicals) antibodies according to standard protocols.
For all staining, flow cytometric analyses were performed on a standard 3 lasers-configuration (405 nm, 488 nm, 633 nm) FACS-Canto2 (BD Biosciences) with 8-colors. The data were analyzed with FlowJo software.
Apoptotic cells preparation
Mouse thymocytes were obtained by mechanical dissociation of thymus from 3–5 week-old B6 mice and were induced to undergo apoptosis by exposure to UVC (30W) for 2 hours followed by an O/N culture in RPMI without any source of serum. This resulted in about 50%–75% apoptotic cells (Taylor et al., 2000). Apoptosis was confirmed by annexinV binding by flow cytometry, as described above (see “flow cytometry”). Apoptotic cells were then labeled with 4 μg of pH-Rhodo, a pH-sensitive fluorophore (Life technology) for 30 min on ice.
In vivo clearance of apoptotic cells by peritoneal macrophages
To induce sterile peritonitis, 1 mL of sterile 4% thioglycollate was injected intraperitoneally into transgenic and control mice. 4 days later, 10–15 × 106 labeled apoptotic murine thymocytes in a volume of 200 μL were injected intraperitoneally into mice. The mice were euthanized 10 min after injection, and the peritoneal cells were recovered by lavages with 4.5 mL of ice-cold PBS. To avoid phagocytosis activity ex vivo, the peritoneal cells were kept on ice until their analysis by flow cytometry; and a control condition comprising the incubation of peritoneal cells with apoptotic cells on ice was performed for each experiment (Figure S2E).
Autoantigen array
Autoantigen arrays were generated and assays on the arrays were performed as described previously (Ayoglu et al., 2016). Self-antigens and controls tested are listed in Table S6. 8 ug of each Ag, control mouse and anti-mouse antibodies were diluted in PBS and transferred into 96-well plates. Analytes were coupled to 1 × 106 carboxylated magnetic beads per ID (MagPlex-C, Luminex Corp.). Beads were distributed into 96-well plates (Greiner BioOne), washed, and re-suspended in phosphate buffer (0.1 M NaH2PO4, pH 6.2) using a plate washer (Biotek). Bead surface was activated by addition of 100 μL of phosphate buffer containing 0.5 mg of 1-ethyl-3(3-dimethylamino-propyl) carbodiimide (Pierce) and 0.5 mg N-hydroxysuccinimide (Pierce). After 20 min incubation on a shaker (Grant Bio), beads were washed and re-suspended in activation buffer (0.05 M MES, pH 5.0). Diluted Ags and control antibodies were incubated with beads for 2 hr at RT. The beads were washed 3 × in 100 μL PBS-T, re-suspended in 60 μL storage buffer (Blocking reagent for ELISA, Roche), and stored in plates at 4°C overnight. Immobilization of the Ags was confirmed, and the assay conditions were optimized by analysis of several mouse monoclonal antibodies, such as anti-La/SSB (SantaCruz), anti-Ro52 (Santa Cruz), anti-IL17A (eBiosciences), and a rabbit polyclonal Ab, all at 1 mg/ml (data not shown).
Mouse sera were diluted 1:100 in an assay buffer of 0.05% PBS-T supplemented with 3% (w/v) BSA (Sigma) and transferred into a 96-well plate. The bead array was distributed into a 384-well plate (Greiner BioOne) by transfer of 5 μL bead array per well. 45 μL of the 1:100 diluted sera were aliquoted and transferred into the 384-well plate. Samples were incubated for 90 min on a shaker (Grant Bio) at RT. The beads were washed with 3 × with 60 μL PBS-T on a plate washer (EL406, Biotek). 50 μL of R-PE conjugated Fcg fragment specific goat anti-mouse IgG (Jackson) secondary antibody was diluted 1:750 in 0.05% PBS-T. After incubation with the secondary antibody for 45 min, the plate was washed with 3 × with 60 μL PBS-T and re-suspended in 60 mL PBS-T for readout in a FlexMap3D instrument (Luminex Corp.).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
All statistical analyses were performed in GraphPad Prism version 6, as defined throughout the text and figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal Rat anti-Mouse C4 (clone 16D2) | (Gift of E. Kremmer, Munich, Germany) | N/A |
| Polyclonal Rabbit anti-Human C4c | Dako | F0169 |
| Goat IgG fraction to mouse complement C3 | MP Biomedicals | Cat# ICN55463, RRID:AB_2334481 |
| Polyclonal Donkey anti-Goat IgG Ax488 | Life technology | Cat# A-11055, RRID:AB_2534102 |
| Polyclonal Goat anti-Rat IgG Ax488 | Life technology | Cat# A-11006, RRID:AB_2534074 |
| Polyclonal Goat anti-Rabbit IgG (H+L), Human/Mouse absorbed-AP | Southern biotech | Cat# 4050-04, RRID:AB_2795954 |
| Polyclonal Goat anti-Mouse IgG (H+L), Human absorbed-HRP | Southern biotech | Cat# 1031-05, RRID:AB_2794307 |
| Polyclonal Goat anti-Mouse IgG, Human absorbed-AP | Southern biotech | Cat# 1030-04, RRID:AB_2794293 |
| Polyclonal Goat anti-Mouse IgG2a, Human-absorbed-AP | Southern Biotech | Cat# 1080-04, RRID:AB_2692322 |
| IgG from murine serum | Sigma | Cat# I5381, RRID:AB_1163670 |
| Purified Mouse Ig2a | Southern biotech | Cat# 0103-01, RRID:AB_2736850 |
| Monoclonal Rat anti-Mouse/Human B220 APC | Biolegend | Cat# 103212, RRID:AB_312997 |
| Monoclonal Rat anti-Mouse/Human B220 PerCP/Cy5.5 | Biolegend | Cat# 103236, RRID:AB_893354 |
| Monoclonal Rat anti-Mouse/Human B220 Pacblue | Biolegend | Cat# 103227, RRID:AB_492876 |
| Monoclonal Rat anti-Mouse CD3 FITC | Biolegend | Cat# 100203, RRID:AB_312660 |
| Monoclonal Rat anti-Mouse CD3 PE | Biolegend | Cat100308, RRID:AB_312673 |
| Monoclonal Rat anti-Mouse CD4 FITC | Biolegend | Cat# 100405, RRID:AB_312690 |
| Monoclonal Rat anti-Mouse CD4 PB | Biolegend | Cat# 100428, RRID:AB_493647 |
| Monoclonal Rat anti-Mouse CD11b APC | Biolegend | Cat# 101212, RRID:AB_312795 |
| Monoclonal Rat anti-Mouse CD21/35 (clone 7E9) PB | Biolegend | Cat# 123414, RRID:AB_2085158 |
| Monoclonal Rat anti-Mouse CD38 PeCy7 | Biolegend | Cat# 102718, RRID:AB_2275531 |
| Monoclonal Rat anti-Mouse CD93 (AA4.1) PerCP/Cy5.5 | Biolegend | Cat# 136511, RRID:AB_10645333 |
| Monoclonal Rat anti-Mouse CD138 PE | Biolegend | Cat# 142503, RRID:AB_10915989 |
| Monoclonal Rat anti-Mouse CD169 FITC | Biolegend | Cat# 142406, RRID:AB_2563107 |
| Monoclonal Rat anti-Mouse CD185 (CXCR5) Biotin | Biolegend | Cat# 145510, RRID:AB_2562126 |
| Monoclonal Rat anti-Mouse CD206 PeCy7 | Biolegend | Cat# 141719, RRID:AB_2562247 |
| Monoclonal Rat anti-Mouse CD279 (PD-1) APC | Biolegend | Cat# 135210, RRID:AB_2159183 |
| Monoclonal Rat anti-Mouse CD357 (GITR) PeCy7 | Biolegend | Cat# 126317, RRID:AB_2563385 |
| Monoclonal Rat anti-Mouse F4/80 FITC | Biolegend | Cat# 123108, RRID:AB_893502 |
| Monoclonal Rat anti-Mouse GL7 PB | Biolegend | Cat# 144614, RRID:AB_2563292 |
| Monoclonal Rat anti-Mouse IgD PB | Biolegend | Cat# 405712, RRID:AB_1937244 |
| Monoclonal Rat anti-Mouse IgM PE | Biolegend | Cat# 406507, RRID:AB_315057 |
| Monoclonal Mouse anti-Mouse IgMa PE | Biolegend | Cat# 408608, RRID:AB_940545 |
| Monoclonal Mouse anti-Mouse IgMb FITC | Biolegend | Cat# 406206, RRID:AB_315039 |
| Monoclonal Mouse anti-idiotype (clone 9D11) | Chatterjee et al., 2013 | N/A |
| Biological Samples | ||
| EA cells | Complement technology | B202 |
| C4−/− guinea pig serum | Complement technology | A305 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ro60 Protein | Diarect | 17400 |
| BPI Protein | Diarect | 19200 |
| CTGF Protein | R&D system | 9190-CC |
| Smd2 protein | Diarect | 11900 |
| C1q protein | Meridian Life Science | A01670H |
| APRIL protein | PEPROTECH | 310-10C |
| Ebioscience™ Fixable viability Dye eFluor™ 780 | Invitrogen | 65-0865-14 |
| AnnexinV FITC | Biolegend | 640905 |
| Propidium Iodide | Sigma | P4170 |
| Streptavidin-APC | Biolegend | 405207 |
| Streptavidin-PE | Biolegend | 405204 |
| pH-Rhodo™ Red, SE | Life technology | P36600 |
| Thioglycollate | BD | 211716 |
| Tamoxifen | Sigma | T5648 |
| Sunflower seed oil | Spectrum chemical | 51929 |
| Streptavidin-coated microspheres | Bangs Laboratories | CP01N |
| Experimental Models: Organisms/Strains | ||
| Mouse: 564Igi: 564 HiKi | Berland et al., 2006 | N/A |
| Mouse: AIDCre.ERT2.eYFP: AicdaCreERT2 flox-STOP-flox-EYFP | Dogan et al., 2009 | N/A |
| Mouse: C4−/−: C57BL/6.C4−/− | Fischer et al., 1996 | N/A |
| Mouse: mC4A: C57BL/6.mC4A/A | This paper | N/A |
| Mouse: mC4B: C57BL/6.mC4B/B | This paper | N/A |
| Oligonucleotides | ||
| Primers for genotyping | This paper | Table S1 |
| Primers for ddPCR | This paper | Table S4 |
| Software and Algorithms | ||
| ImageJ | Rasband WS, ImageJ, NIH, Bethesda, Maryland, USA | https://imagej.nih.gov/ij/ |
| CellProfiler | Lamprecht et al., 2007 | https://cellprofiler.org/ |
| Other | ||
| Antigens used in autoantigen array | This paper | Table S6 |
Highlights.
Autoreactive B cell regulation is impaired in absence of complement C4A isoform
C4A helps maintain negative selection of B cells
Relative to C4B, C4A promotes better clearance of apoptotic cells
ACKNOWLEDGMENTS
We thank all of the members of the M.C.C. lab for suggestions and help with experiments and feedback on the manuscript; E.M. Carroll for valuable assistance; C. Usher for editorial assistance; K. Holscher for technical support; J.-C. Weill and Agnes Reynaud (Institut Necker Enfants Malades, Université Paris Descartes) for the generous gift of the aicda-YFP reporter mice; and the Flow Cytometry core and Harry Leung of the Optical Microscopy core at the PCMM for technical assistance. This research was supported by NIH grants 5R01AI130307 and 5R01AR074105, to M.C.C.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108330.
REFERENCES
- Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, Dutow P, Woodruff TM, Yu ZX, O’Neill LA, et al. (2016). T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science 352, aad1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aung NY, Ohe R, Meng H, Kabasawa T, Yang S, Kato T, and Yamakawa M (2016). Specific neuropilins expression in alveolar macrophages among tissue-specific macrophages. PLOS ONE 11, e0147358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoglu B, Schwenk JM, and Nilsson P (2016). Antigen arrays for profiling autoantibody repertoires. Bioanalysis 8, 1105–1126. [DOI] [PubMed] [Google Scholar]
- Battin C, De Sousa Linhares A, Paster W, Isenman DE, Wahrmann M, Leitner J, Zlabinger GJ, Steinberger P, and Hofer J (2019). Neuropilin-1 acts as a receptor for complement split products. Front. Immunol 10, 2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belot A, and Cimaz R (2012). Monogenic forms of systemic lupus erythematosus: new insights into SLE pathogenesis. Pediatr. Rheumatol. Online J 10, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belt KT, Carroll MC, and Porter RR (1984). The structural basis of the multiple forms of human complement component C4. Cell 86, 907–914. [DOI] [PubMed] [Google Scholar]
- Berland R, Fernandez L, Kari E, Han JH, Lomakin I, Akira S, Wortis HH, Kearney JF, Ucci AA, and Imanishi-Kari T (2006). Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity 25, 429–440. [DOI] [PubMed] [Google Scholar]
- Blanchong CA, Chung EK, Rupert KL, Yang Y, Yang Z, Zhou B, Moulds JM, and Yu CY (2001). Genetic, structural and functional diversities of human complement components C4A and C4B and their mouse homologues, Slp and C4. Int. Immunopharmacol 1, 365–392. [DOI] [PubMed] [Google Scholar]
- Botto M, Dell’Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, Loos M, Pandolfi PP, and Walport MJ (1998). Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat. Genet 19, 56–59. [DOI] [PubMed] [Google Scholar]
- Carroll MC (2004). A protective role for innate immunity in systemic lupus erythematosus. Nat. Rev. Immunol 4, 825–831. [DOI] [PubMed] [Google Scholar]
- Carroll MC, and Isenman DE (2012). Regulation of humoral immunity by complement. Immunity 37, 199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll MC, Belt T, Palsdottir A, and Porter RR (1984). Structure and organization of the C4 genes. Philos. Trans. R. Soc. Lond. B Biol. Sci 306, 379–388. [DOI] [PubMed] [Google Scholar]
- Chaplin DD, Woods DE, Whitehead AS, Goldberger G, Colten HR, and Seidman JG (1983). Molecular map of the murine S region. Proc. Natl. Acad. Sci. USA 80, 6947–6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee P, Agyemang AF, Alimzhanov MB, Degn S, Tsiftsoglou SA, Alicot E, Jones SA, Ma M, and Carroll MC (2013). Complement C4 maintains peripheral B-cell tolerance in a myeloid cell dependent manner. Eur. J. Immunol 43, 2441–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, Wu YL, Mok MY, Wu YJ, Lintner KE, Wang CM, Chung EK, Yang Y, Zhou B, Wang H, et al. (2016). Effects of Complement C4 Gene Copy Number Variations, Size Dichotomy, and C4A Deficiency on Genetic Risk and Clinical Presentation of Systemic Lupus Erythematosus in East Asian Populations. Arthritis Rheumatol. 68, 1442–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemenza L, and Isenman DE (2004). The C4A and C4B isotypic forms of human complement fragment C4b have the same intrinsic affinity for complement receptor 1 (CR1/CD35). J. Immunol 172, 1670–1680. [DOI] [PubMed] [Google Scholar]
- Cyster JG, Hartley SB, and Goodnow CC (1994). Competition for follicular niches excludes self-reactive cells from the recirculating B-cell repertoire. Nature 371, 389–395. [DOI] [PubMed] [Google Scholar]
- Das A, Heesters BA, Bialas A, O’Flynn J, Rifkin IR, Ochando J, Mittereder N, Carlesso G, Herbst R, and Carroll MC (2017). Follicular Dendritic Cell Activation by TLR Ligands Promotes Autoreactive B Cell Responses. Immunity 46, 106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degn SE, van der Poel CE, Firl DJ, Ayoglu B, Al Qureshah FA, Bajic G, Mesin L, Reynaud CA, Weill JC, Utz PJ, et al. (2017). Clonal Evolution of Autoreactive Germinal Centers. Cell 170, 913–926.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan I, Bertocci B, Vilmont V, Delbos F, Mégret J, Storck S, Reynaud CA, and Weill JC (2009). Multiple layers of B cell memory with different effector functions. Nat. Immunol 10, 1292–1299. [DOI] [PubMed] [Google Scholar]
- Fantin A, Vieira JM, Plein A, Denti L, Fruttiger M, Pollard JW, and Ruhrberg C (2013). NRP1 acts cell autonomously in endothelium to promote tip cell function during sprouting angiogenesis. Blood 121, 2352–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer MB, Ma M, Goerg S, Zhou X, Xia J, Finco O, Han S, Kelsoe G, Howard RG, Rothstein TL, et al. (1996). Regulation of the B cell response to T-dependent antigens by classical pathway complement. J. Immunol 157, 549–556. [PubMed] [Google Scholar]
- Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K, et al. (1988). Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 334, 676–682. [DOI] [PubMed] [Google Scholar]
- Goodnow CC, Sprent J, Fazekas de St Groth B, and Vinuesa CG (2005). Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature 435, 590–597. [DOI] [PubMed] [Google Scholar]
- Green NM, Moody KS, Debatis M, and Marshak-Rothstein A (2012). Activation of autoreactive B cells by endogenous TLR7 and TLR3 RNA ligands. J. Biol. Chem 287, 39789–39799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harless SM, Lentz VM, Sah AP, Hsu BL, Clise-Dwyer K, Hilbert DM, Hayes CE, and Cancro MP (2001). Competition for BLyS-mediated signaling through Bcmd/BR3 regulates peripheral B lymphocyte numbers. Curr. Biol 11, 1986–1989. [DOI] [PubMed] [Google Scholar]
- Isenman DE, and Young JR (1984). The molecular basis for the difference in immune hemolysis activity of the Chido and Rodgers isotypes of human complement component C4. J. Immunol 132, 3019–3027. [PubMed] [Google Scholar]
- Kamitaki N, Sekar A, Handsaker RE, de Rivera H, Tooley K, Morris DL, Taylor KE, Whelan CW, Tombleson P, Loohuis LMO, et al. ; Schizophrenia Working Group of the Psychiatric Genomics Consortium (2020). Complement genes contribute sex-biased vulnerability in diverse disorders. Nature 582, 577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolev M, Dimeloe S, Le Friec G, Navarini A, Arbore G, Povoleri GA, Fischer M, Belle R, Loeliger J, Develioglu L, et al. (2015). Complement Regulates Nutrient Influx and Metabolic Reprogramming during Th1 Cell Responses. Immunity 42, 1033–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, and Heeger PS (2008). Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood 112, 1759–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht MR, Sabatini DM, and Carpenter AE (2007). CellProfiler: free, versatile software for automated biological image analysis. Biotechniques 42, 71–75. [DOI] [PubMed] [Google Scholar]
- Law SKA, and Dodds AW (1997). The internal thioester and the covalent binding properties of the complement proteins C3 and C4. Protein Sci. 6, 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law SK, Dodds AW, and Porter RR (1984). A comparison of the properties of two classes, C4A and C4B, of the human complement component C4. EMBOJ. 3, 1819–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, and Marshak-Rothstein A (2002). Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 416, 603–607. [DOI] [PubMed] [Google Scholar]
- Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, and Cyster JG (2004). Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity 20, 441–453. [DOI] [PubMed] [Google Scholar]
- Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, Subias M, Pickering MC, Drouet C, Meri S, et al. (2013). Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 39, 1143–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meffre E, and Wardemann H (2008). B-cell tolerance checkpoints in health and autoimmunity. Curr. Opin. Immunol 20, 632–638. [DOI] [PubMed] [Google Scholar]
- Nonaka M, Nakayama K, Yeul YD, and Takahashi M (1986). Complete nucleotide and derived amino acid sequences of sex-limited protein (Slp), nonfunctional isotype of the fourth component of mouse complement (C4). J. Immunol 136, 2989–2993. [PubMed] [Google Scholar]
- Ogata RT, and Zepf NE (1991). The murine Slp gene. Additional evidence that sex-limited protein has no biologic function. J. Immunol 147, 2756–2763. [PubMed] [Google Scholar]
- Omarjee O, Picard C, Frachette C, Moreews M, Rieux-Laucat F, Soulas-Sprauel P, Viel S, Lega JC, Bader-Meunier B, Walzer T, et al. (2019). Monogenic lupus: dissecting heterogeneity. Autoimmun. Rev 18, 102361. [DOI] [PubMed] [Google Scholar]
- Pereira KMC, Perazzio S, Faria AGA, Moreira ES, Santos VC, Grecco M, da Silva NP, and Andrade LEC (2019). Impact of C4, C4A and C4B gene copy number variation in the susceptibility, phenotype and progression of systemic lupus erythematosus. Adv. Rheumatol 59, 36. [DOI] [PubMed] [Google Scholar]
- Porter RR (1983). Complement polymorphism, the major histocompatibility complex and associated diseases: a speculation. Mol. Biol. Med 1, 161–168. [PubMed] [Google Scholar]
- Reilly BD, and Mold C (1997). Quantitative analysis of C4Ab and C4Bb binding to the C3b/C4b receptor (CR1, CD35). Clin. Exp. Immunol 110, 310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt KN, and Cyster JG (1999). Follicular exclusion and rapid elimination of hen egg lysozyme autoantigen-binding B cells are dependent on competitor B cells, but not on T cells. J. Immunol 162, 284–291. [PubMed] [Google Scholar]
- Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J, Baum M, Van Doren V, et al. ; Schizophrenia Working Group of the Psychiatric Genomics Consortium (2016). Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, Shapiro VS, Dubyak GR, Heeger PS, and Medof ME (2008). Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity 28, 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, Savill JS, Henson PM, Botto M, and Walport MJ (2000). A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J. Exp. Med 192, 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F, and Brink R (2004). Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity 20, 785–798. [DOI] [PubMed] [Google Scholar]
- Tsokos GC (2011). Systemic lupus erythematosus. N. Engl. J. Med 365, 2110–2121. [DOI] [PubMed] [Google Scholar]
- van den Elsen JM, Martin A, Wong V, Clemenza L, Rose DR, and Isenman DE (2002). X-ray crystal structure of the C4d fragment of human complement component C4. J. Mol. Biol 322, 1103–1115. [DOI] [PubMed] [Google Scholar]
- Victora GD, and Nussenzweig MC (2012). Germinal centers. Annu. Rev. Immunol 30, 429–457. [DOI] [PubMed] [Google Scholar]
- Walport MJ (2001a). Complement. First of two parts. N. Engl. J. Med 344, 1058–1066. [DOI] [PubMed] [Google Scholar]
- Walport MJ (2001b). Complement. Second of two parts. N. Engl. J. Med 344, 1140–1144. [DOI] [PubMed] [Google Scholar]
- Wardemann H, and Nussenzweig MC (2007). B-cell self-tolerance in humans. Adv. Immunol 95, 83–110. [DOI] [PubMed] [Google Scholar]
- West EE, Kolev M, and Kemper C (2018). Complement and the Regulation of T Cell Responses. Annu. Rev. Immunol 36, 309–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, Zhou B, Hebert M, Jones KN, Shu Y, Kitzmiller K, et al. (2007). Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am. J. Hum. Genet 80, 1037–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets/code generated or analyzed during this study.