

Abstract
Chalcone [(E)-1,3-diphenyl-2-propene-1-one], a small molecule with α, β unsaturated carbonyl group is a precursor or component of many natural flavonoids and isoflavonoids. It is one of the privileged structures in medicinal chemistry. It possesses a wide range of biological activities encouraging many medicinal chemists to study this scaffold for its usefulness to oncology, infectious diseases, virology and neurodegenerative diseases including Alzheimer’s disease (AD). Small molecular size, convenient and cost-effective synthesis, and flexibility for modifications to modulate lipophilicity suitable for blood brain barrier (BBB) permeability make chalcones a preferred candidate for their therapeutic and diagnostic potential in AD. This review summarizes and highlights the importance of chalcone and its analogs as single target small therapeutic agents, multi-target directed ligands (MTDLs) as well as molecular imaging agents for AD. The information summarized here will guide many medicinal chemist and researchers involved in drug discovery to consider chalcone as a potential scaffold for the development of anti-AD agents including theranostics.
Keywords: Chalcone, Alzheimer’s Disease, multi-target directed ligands, cholinesterase inhibitors, Amyloid beta inhibition, neuroinflammation, molecular probes, theranostic
Graphical Abstract
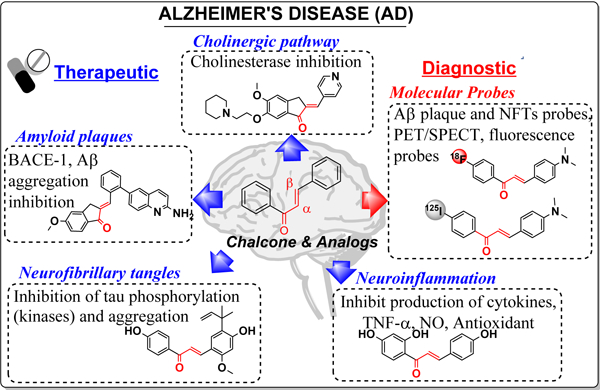
1. Introduction
Chalcone [(E)-1,3-diphenyl-2-propene-1-one] is a simple molecule with two aryl rings (A and B) separated by α, β unsaturated carbonyl group (Figure 1). Chalcone found as the precursor or component of many naturally occurring flavonoids and isoflavonoids can exist in both cis or trans forms, although the trans form is thermodynamically more stable [1, 2].The chalcone skeleton is one of the privileged scaffolds studied for several medicinal applications such as anti-cancer, anti-ulcer, anti-tuberculosis, anti-inflammatory, and anti-bacterial[3–10]. Presence of α, β unsaturated carbonyl functional group, which acts as a potential Michael acceptor, allows the chalcone molecule to interact with sulfhydryl of cysteine residue or other thiol groups. This interaction is believed to be important for their biological activities [11–13]. Given the promise of chalcone in preclinical studies, some of the chalcone-based molecules such as metochalcone (choleretic drug), hesperidin methyl chalcone (vascular protective) and sofalcone (antiulcer and mucoprotective) have been approved for clinical applications (Figure 1) [1, 14, 15].
Figure 1.
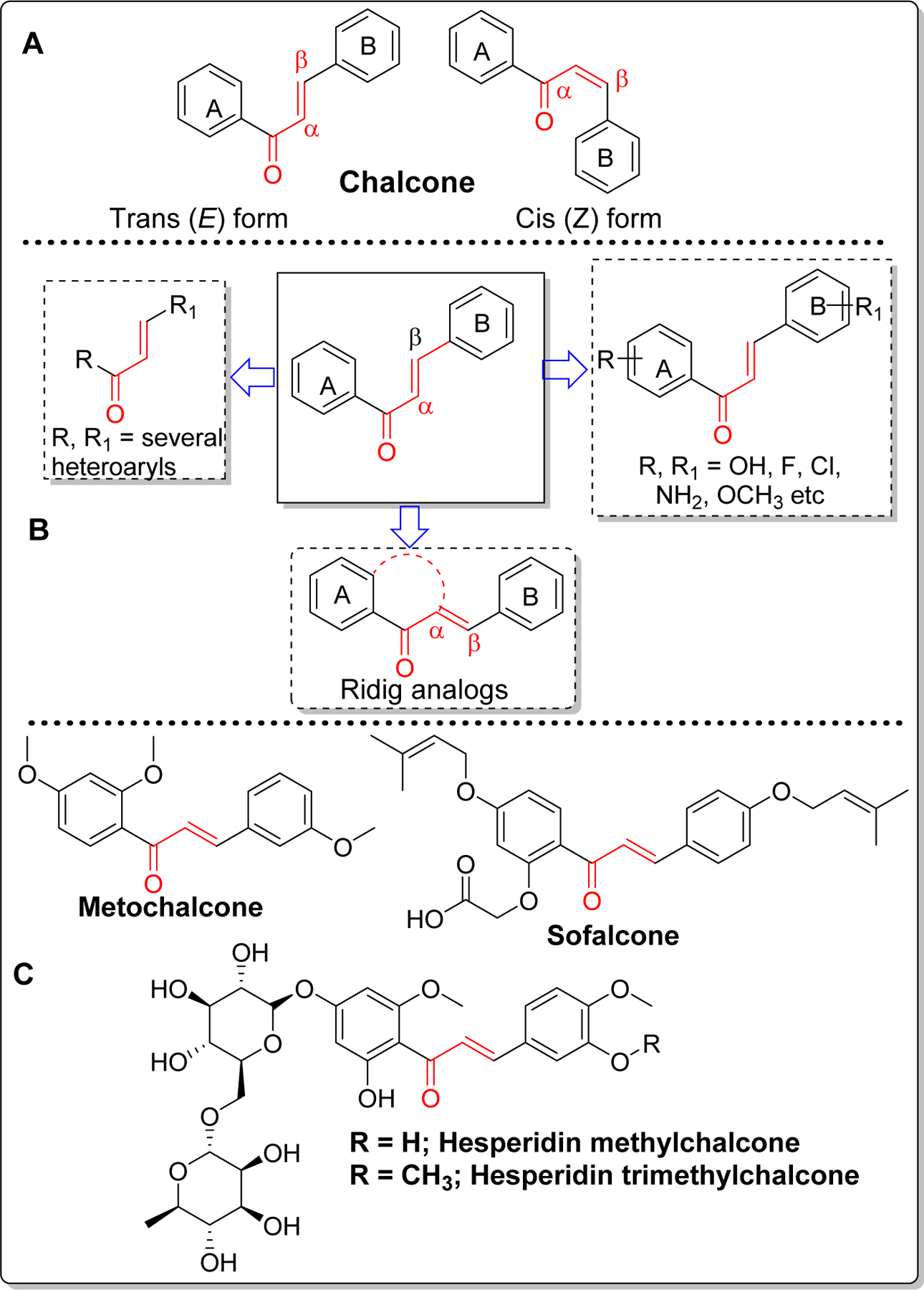
Structure of chalcone and its form (A), major possible modifications (B) and clinically used and/or tested chalcone-based drugs (C).
Because of chalcone’s low molecular weight, convenient and efficient synthesis and easy optimization of lipophilicity (logP) with appropriate substituents, this scaffold has gained significant attention in drug discovery of central nervous system (CNS) diseases including Alzheimer’s Disease (AD). This simple scaffold can be modified in several ways as shown in Figure 1. Among the possible modifications, most prominent ones are (i) substitution of ring A and B with different heteroaryls; (ii) substitution on phenyl ring with several functional groups such as hydroxyl, methoxy, halogens (Cl, F, Br), amine etc. (iii) fusion of ring A and α-carbon and various combinations of the above modifications. Synthetically, it can be prepared by acid or base-catalyzed condensation reactions. Claisen-Schmidt condensation is the most common classical reaction used for preparation of chalcone(s) [16, 17]. However, depending upon the substituents, this reaction occasionally gives a complex mixture of isomers and other side products. Several other name reactions such as Suzuki coupling reaction [18], Friedel-crafts acylation [19], Wittig reaction [20] etc have been optimized to prepare chalcones.
Naturally occurring flavonoids and isoflavonoids are derived from chalcones which are biosynthesized with the aid of chalcone synthase (CHS, EC 2.3.1.74). CHS exists as a homodimer of two 42kDa polypeptides and the key amino acid residues in active/catalytic site of CHS are Cys164, Phe215, His303, and Asn336 [21]. Chalcone biosynthesis occurs by the condensation of one molecule of p-coumaroyl-CoA and three malonyl-CoA catalyzed by CHS. First, a coumaroyl moiety from p-coumaroyl-CoA is transferred to cysteine (Cys164) at active site of CHS [22]. Thereafter, condensation of three acetates from 3 malonyl-CoA extend the polyketide intermediate to form thioester-linked tetraketide intermediate. The tetraketide subsequently undergoes regiospecific intramolecular Claisen-type condensation and forms a ring system to yield chalcone [23, 24]. Several flavonoids and isoflavonoids are produced from chalcone by the action of chalcone isomerase (CHI) [25]. Similarly, five membered cyclized products, aurones are biosynthesized from chalcone by the action of aurone synthase (AUS) (Figure2) [26].
Figure 2.
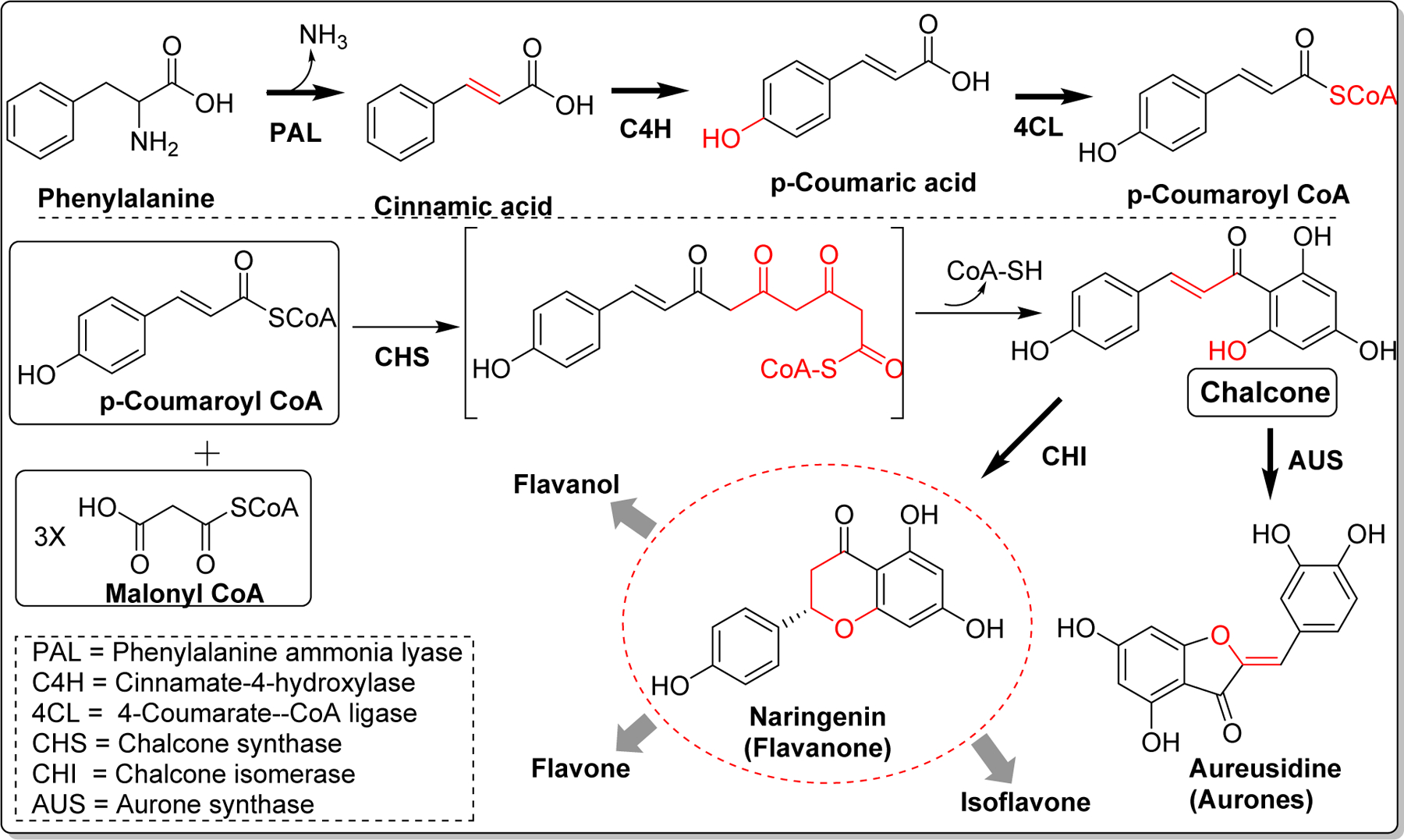
Biosynthesis of chalcone and its subsequent conversion to natural flavonoids.
This review will focus on providing comprehensive insight on recent development related to chalcone and its analogs as therapeutic or diagnostic agent(s) for treating or diagnosing AD. A brief description of AD and its significance will be followed by outline on current advances on potentially using chalcones as new clinically useful compounds.
2. Alzheimer’s Disease (AD)
Alzheimer’s disease (AD), a progressive neurodegenerative disorder is a complex multifactorial disease characterized by formation and deposition of extracellular amyloid β (Aβ) plaques and intracellular neurofibrillary tangles (NFTs), oxidative stress, mitochondrial dysfunction and neuroinflammation. The underlying mechanisms lead to neuronal loss with associated progressive decline in cognitive functions including memory loss. AD is the most common cause of dementia in elderly. Currently, estimated that 5.8 million Americans age 65 and older are living with Alzheimer’s related dementia and this number is expected to rise to 13.8 million by 2050 [27]. AD was first observed by German clinical psychiatrist and neuroanatomist, Alois Alzheimer on November 26, 1901 while examining a patient Auguste Deter, 50-year-old woman who presented symptoms of sleep disorders, memory disturbance, and progressive confusion. After her death in 1906, post-mortem of the brain showed senile plaques and neurofibrillary tangles. Later, the disease was coined as “Alzheimer’s disease” [28].
Drug development for AD so far has mainly focused on cholinergic, beta amyloid hypothesis and tauopathy. Recently, the field has expanded to other pathologies including mitochondrial dysfunction and neuroinflammation [29]. Extensive research on drug discovery and development has led to FDA-approved drugs which include cholinesterase inhibitors such as donepezil (DPZ) [30–32], rivastigmine (RIV) [33–35], galantamine [36, 37], and a partial N -methyl-D-aspartate (NMDA) antagonist, Memantine[38] (Figure 3) [39]. Unfortunately, these drugs can only manage the symptoms of AD for short-term and do not slow or stop the disease progression. Thus, a more effective therapeutic agents which can prevent progression or reverse the disease course is warranted. Chalcone has been studied as potential scaffold for the discovery of anti-AD agents.
Figure 3.
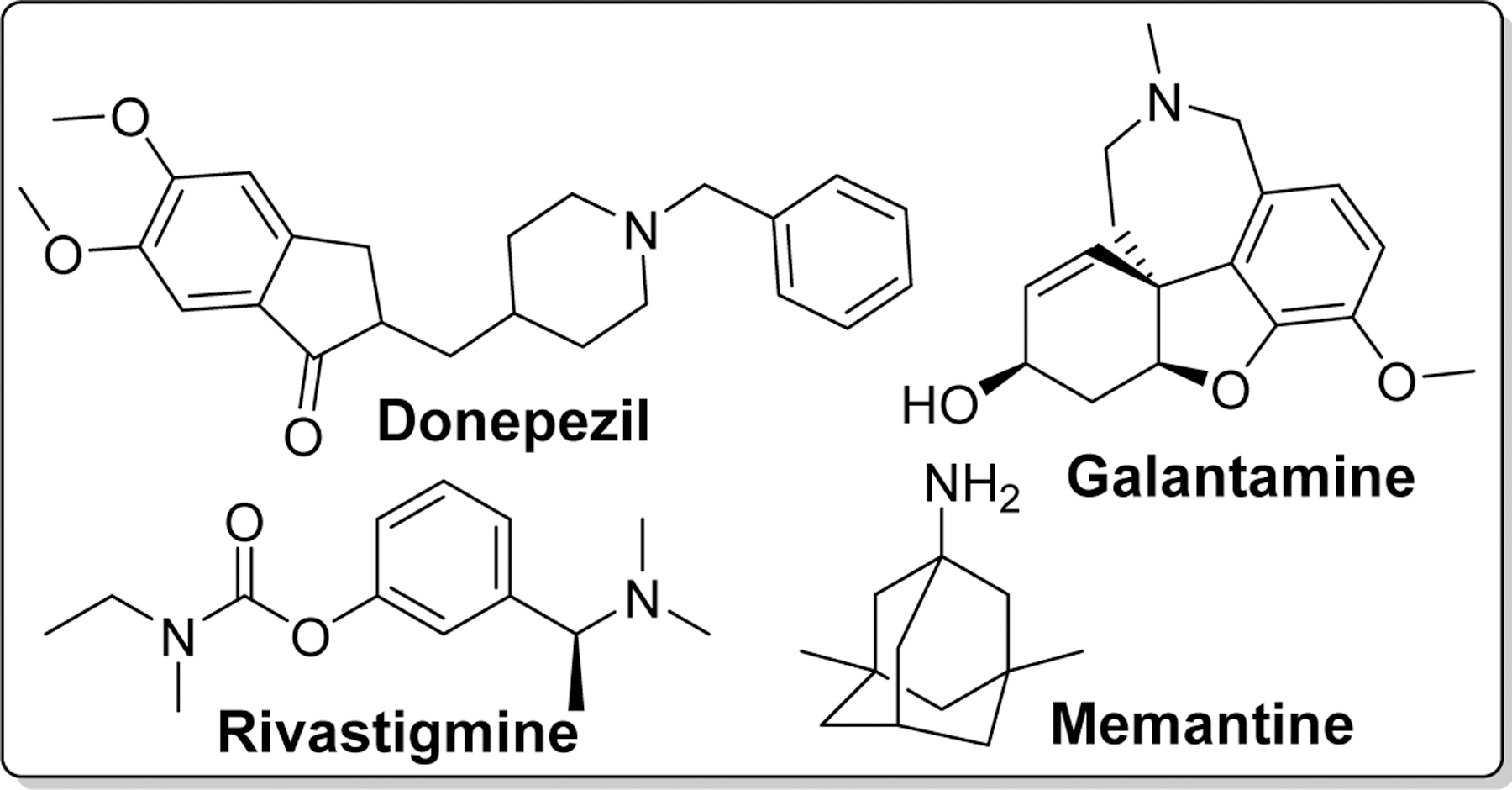
Structures of clinically approved drugs for the treatment of AD.
Several chalcone analogs have been prepared as inhibitors of either one or more enzymes/proteins involved in the pathogenesis of AD such as cholinesterase (Bu/AChE), beta secretase (BACE), tau proteins etc. Moreover, chalcone has gained an attention as a potential molecular probe for in vivo imaging of Aβ deposits in AD. Use of several chalcone derivatives for single or multiple targets involved in AD pathogenesis or diagnostic purpose for AD will be described in the following sections.
3. Chalcone and its analogs as therapeutic agent(s) for AD
3.1. Cholinesterase inhibitors (ChEIs)
Development of cholinesterase inhibitors is based on the cholinergic hypothesis [40]. According to the cholinergic hypothesis, first proposed by Peter Davies and A.J.Maloney in 1976 [41], neuronal synapses in AD patient have low levels of neurotransmitter, acetylcholine (ACh) accompanied by a decreased activity of choline acetyltransferase (ChAT). ACh is synthesized in cytoplasm of presynaptic cholinergic neurons from choline and acetyl coenzyme A (Acetyl-CoA) catalyzed by ChAT [42]. Once formed, it is transferred into synaptic vesicles. Exocytosis from presynaptic neuron upon depolarization releases ACh into synapses, which can bind with nicotinic or muscarinic receptors in postsynaptic neurons causing neurotransmission [43]. In synapses, ACh is primarily hydrolyzed by the enzyme called acetylcholinesterase into choline and acetic acid which switches off the cholinergic neurotransmission [44]. Reuptake of choline by presynaptic neurons for re-synthesis of ACh completes the cycle [45].
Inhibition of AChE to prevent the reduction of ACh level in AD patients has been a popular strategy. Butyrylcholinesterase (BuChE, pseudocholinesterase) is another abundant cholinesterase and it differs from AChE with regards to substrate specificity. AChE is highly specific to ACh whereas BuChE favors butyrylcholine (BuCh) and several other cholinesters including ACh. Structurally, AChE and BuChE share 65% amino acid sequence homology and their active site contains the peripheral anionic site (PAS), a narrow gorge (~20 Aº deep, 5 Aº wide) which gives access to the catalytic anionic site (CAS) [46, 47]. Catalytic site of AChE is responsible for the hydrolysis and is located at the bottom. The three essential amino acids, Ser200, His440 and Glu327, a catalytic triad, in AChE are involved in the transfer of acetyl group to Ser200 [48]. In BuChE, Ser198, His438 and Glu325 forms the catalytic triad [49]. AChE anionic site which comprise Trp84, Tyr130, Phe330 and Phe331 facilitates the binding of quaternary ammonium group by cation-π interactions [50]. The three amino acid residues present in the PAS of AChE are not found in the PAS of BuChE. Moreover, BuChE has wider gorge compared to AChE [51]. Initially, due to higher expression of AChE in CNS, and cholinergic neurons, compared to BuChE, AChE was preferred target of interest for AD drug development. The significance of BuChE in AD has been historically underestimated with renewed interest in BuChE as one of the key targets for AD drug discovery and development [52, 53]. In healthy brain, ACh is predominantly hydrolyzed by AChE and BuChE plays a supportive role. However, in AD, the AChE level remains unchanged or decline whereas BuChE levels increases with the disease progression [54, 55]. In concurrence, selective inhibition of BuChE increased the ACh levels, reduced Aβ levels and improved cognitive performance in vivo [56]. Furthermore, increased levels of BuChE positively correlate with the transformation of relatively inert Aβ plaques to pathogenic forms associated with neuritic tissue degeneration and dementia [57, 58]. The potential of BuChE inhibition for treatment of AD has resulted in several patent applications on BuChE inhibitors (selective or multi-targeting) [59] and discovery of several classes of compound that selectively inhibit BuChE [60].
In the quest to find novel cholinesterase inhibitors, many researchers including medicinal chemists have explored the role of chalcone and its analogs. The availability of crystal structure of AChE [61] and BuChE [62] has facilitated this process. Recently, Chandrika et al. [63] reported chalcone-donepezil hybrids with significant cholinesterase and Aβ fibril assembly inhibition. They developed a series of 1,3- (Compound 1a-f) and 1,4- (Compound 2a-f) chalcone donepezil hybrids (Figure 4). In comparison, 1,4-chalcone-donepezil hybrids (2a-2f) showed better AChE inhibition than 1,3-chalcone-donepezil hybrids. In the series, compound 2a showed better AChE inhibition than donepezil (IC50: 2a; 0.07 vs DPZ; 0.12 µM). In 1,3-chalcone series, 1b (IC50 = 0.65 µM, AChE) was the most potent AChE inhibitor. Similarly, most compounds in the 1,4-series (2a-f) showed better BuChE inhibition. In 1,3-series (1a-f), compounds 1a (IC50 = 0.020 µM) and 1b (IC50 = 0.03 µM) showed significant BuChE inhibition. It was observed that smaller linker between chalcone and donepezil favors cholinesterase inhibitory effects.
Figure 4.
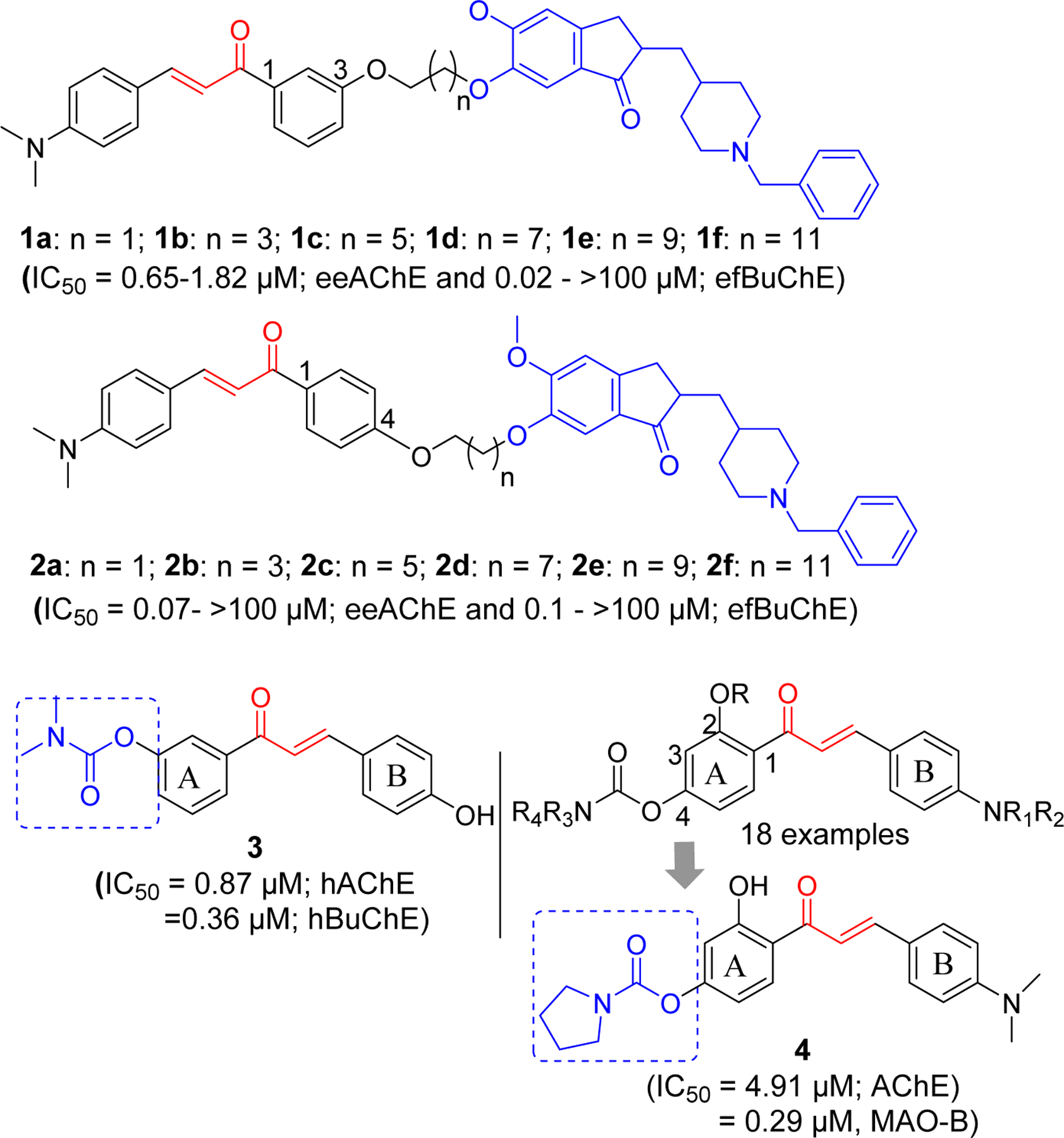
Chalcone-Rivastigmine/Donepezil hybrids.
Similar approach of hybridizing chalcone with clinically approved rivastigmine has been reported [64]. These investigators modified chalcone ring A and B using several functional groups including the carbamate groups present in rivastigmine and tested for AChE and BuChE inhibitory effects. Several compounds showed hBuChE inhibitory effects at micromolar to sub-micromolar concentrations. These compounds showed selectivity for BuChE. Compound 3 was the most potent with activity comparable to rivastigmine against hBuChE (IC50; 3: 0.36 µM vs Riv: 0.38 µM). In addition, this compound showed hAChE inhibition at sub-micromolar concentration (IC50 = 0.87 µM, hAChE). Molecular modeling showed that compound 3 occupied the catalytic pocket of hAChE and hBuChE. The carbamate ester of compound 3 binds with the catalytic triad of hAChE (Ser203 and His447) and hBuChE (Ser198 and His438).
Xiao et al. [65] has investigated the 4’-Aminochalcone-rivastigmine hybrids. All prepared compounds (18 examples) were assayed for AChE and BuChE inhibitory effects. Compound 4 showed the most potent AChE inhibition with IC50 = 4.91 µM. Incorporation of carbamates on both 2 and 4 positions resulted in weaker AChE inhibitory activity. Molecular modeling of compound 4 with TcAChE (PDB: 1EVE) showed that it occupied CAS, mid-gorge sites and PAS of the enzyme. It (ring B) forms π-π interactions with Phe288, Phe331 and Phe290 in the PAS site along with hydrophobic interactions with Ile287, Arg289, Leu282. In the gorge of enzyme, hydrophobic interaction of α, β-unsaturated ketone group with Tyr334, π-π interaction of ring A with Phe330 and hydrogen bond of hydroxyl group in ring A with Tyr121 were observed. The carbamate moiety of compound 4 forms the hydrophobic interactions with Acn85, Asp72 and Gln69 in catalytic site. In addition, this compound showed good antioxidant property compared to Trolox (2.83 equivalent) and selective monoamine oxidase-B inhibitory effect (IC50 = 0.29 µM). Artificial membrane permeation assay [66, 67] to determine blood brain barrier permeability showed that compound 4 could penetrate the blood-brain barrier making it a promising candidate for AD drug development.
The Liu group has investigated a series of non-halogenated [68] as well as halogenated (F and Cl) [69, 70] chalcone derivatives for cholinesterase inhibitory activity. First, they prepared a series of non-halogenated chalcones with alkyl amine substituents at the hydroxyl group with variable carbon spacer (Figure 5). It was observed that compound 5 with the two-carbon spacer was the most potent selective AChE inhibitor. The group later developed the two-carbon spacer alkyl amine derivatives with fluorine or chlorine group at 2-, 3-, or 4- position of the phenyl ring on the other side. Among fluorinated derivatives, compound 6 was the most potent compound against AChE with IC50 of 0.21 µM. In series of 12 compounds, most of them showed selectivity to AChE compared to BuChE. Similarly, among chlorinated derivatives, compound 7 was the most potent compound. It showed higher selectivity to AChE over BuChE as compare to compound 6. They prepared the pyrazoline derivatives at α, β carbon position. All these derivatives showed weaker AChE inhibition. Authors remarked that α, β carbon position possibly plays an important role in determining the AChE inhibition. To determine the reason for specificity to AChE, they performed the molecular modeling of compound 5 with both AChE and BuChE. Interestingly, they found that compound 5 retains the multipoint interaction with AChE (π-π stacking with Trp279 and Ph330 at PAS and cation-π interaction with Trp84 in the CAS) whereas it retains only cation-π interaction with Trp82 in BuChE. They also prepared a series of compounds with alkyl amine substituents at different position (meta) of phenyl ring (Figure 5) [71]. Compound 8 was the most potent selective AChE inhibitor. Molecular modeling showed similar binding pattern with compound 5.
Figure 5.
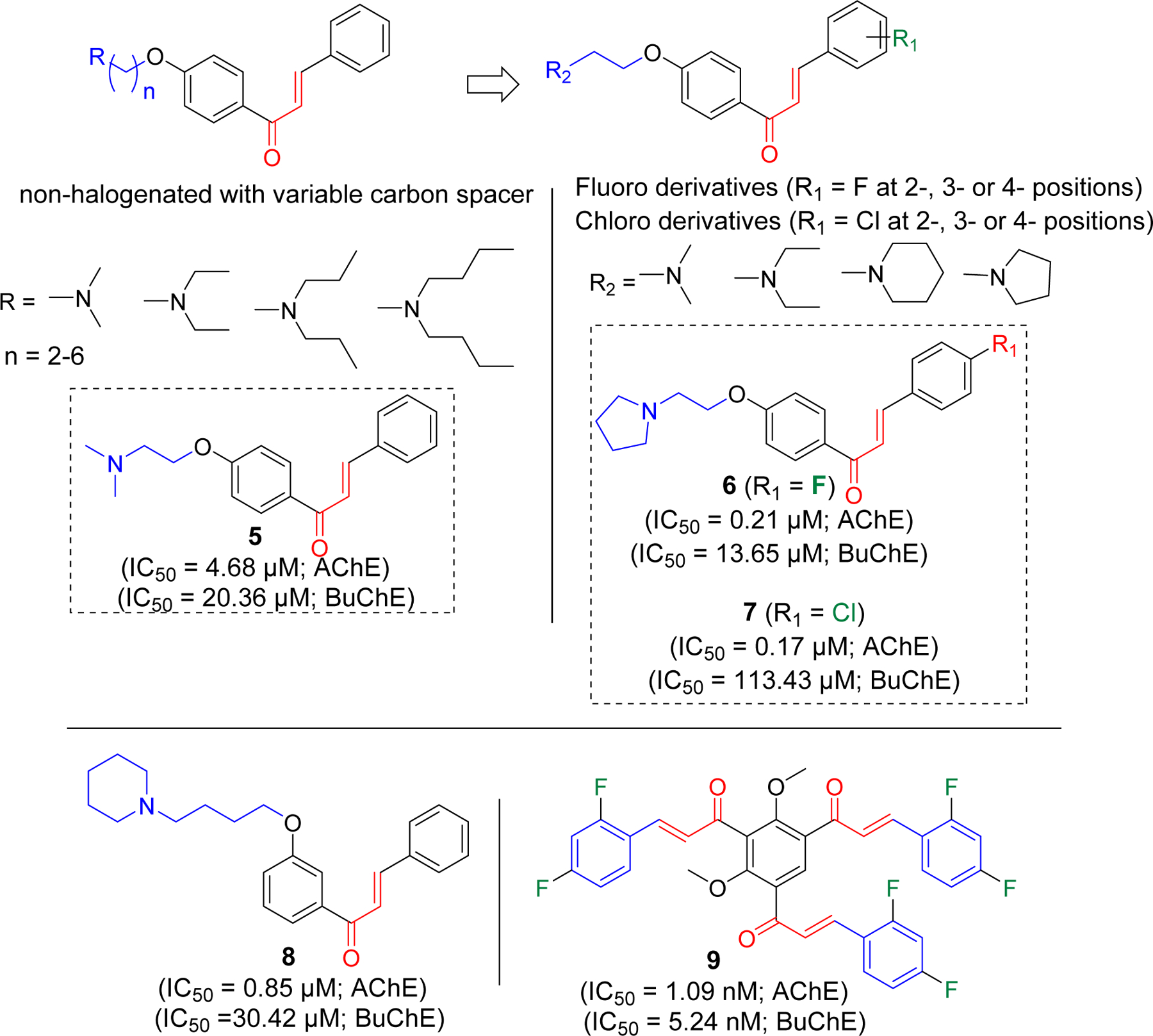
Halogenated and non-halogenated alkyl amine derivatives of chalcones.
Burmaoglu et al. [72] has reported tris-chalcones with fluorine(s) group on different position of phenyl ring. Nine derivatives were prepared and evaluated for their cholinesterase inhibitory activity. All evaluated compounds showed both AChE and BuChE inhibition at nanomolar concentrations. Compound 9 was the most potent among the series with IC50 of 1.09 nM (AChE) and 5.24 nM (BuChE).
Rampa et al. [73] incorporated both alkylamine and carbamate groups in their chalcone derivatives as shown in compounds 10a-f. All derivatives showed the cholinesterase activity at nanomolar concentrations with specificity to AChE. The most potent compound for AChE inhibition was 10d with IC50 of 0.81 nM. For BuChE it showed IC50 at 106 nM concentration. Compounds 10a-10f showed the AChE inhibition in the range of 0.81 to 51.8 nM. Compound 10f (IC50 = 51.8 nM) with longest chain length showed weaker AChE inhibition in comparison to other compounds. Several derivatives with piperidine spacer were developed by Zhao et al. [74]. Among 8 examples, compound 11 showed the most potent AChE and BuChE inhibition (IC50 at 1.12 and 3.23 µM, respectively). From structure activity relationship (SAR) study, authors observed that electron donating groups (e.g. Methyl as in compound 11, methoxy) showed better activity compared to electron withdrawing (e.g. nitro, halogens) substituents in this series of compounds (Figure 6).
Figure 6.
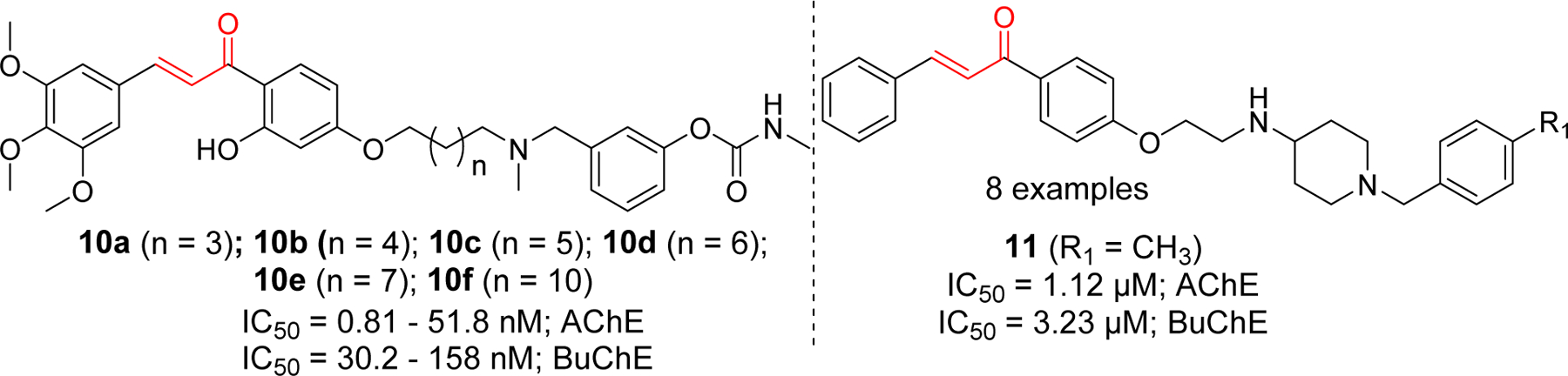
Chalcone derivative with alkylamine functional groups.
Importance of alkylamine functional group for cholinesterase inhibition led researchers to incorporate various alkylamine groups such as benzyl piperidine, piperidine, pyrrole and diethylamine on two side of chalcones (Figure 7) [75]. Compound 12 showed the most potent eeAChE inhibitory effect. Compound 13 showed significant hAChE and hBuChE inhibition. Similarly, compound 14 has shown the most potent eqBuChE inhibitory activity. Molecular docking study of compound 13 showed that it occupied the entire CAS, mid-gorge and PAS of AChE (PDB: 1EVE). Chalcone benzene ring form the π-π interaction with Phe330, two s-p interactions with Trp84 and one hydrogen bond with Phe288. In BuChE (PDB: 4TPK), compound 13 showed π-π and sp interaction with key amino acid Tyr332. The hydroxyl and carbonyl group form the H-bond with Pro285. Similarly, pyrrole ring forms the H-bond with Thr120. Other hydrophobic interactions were observed between 13 and Thr120, Asp70, Gly116, Phe329, Ala328, Tyr332, Trp82, His438.
Figure 7.
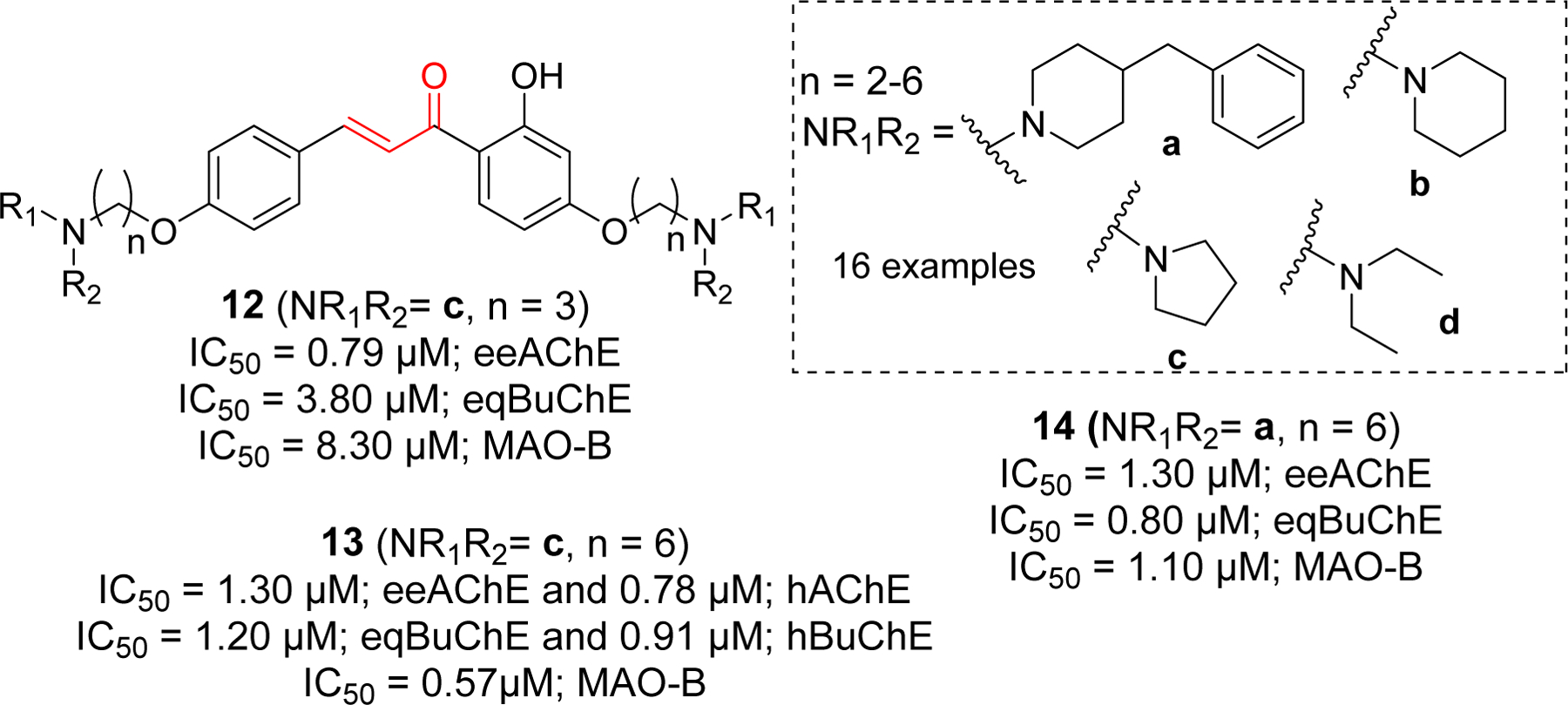
Chalcone derivative with di O-alkylamine substituents.
Studies on tetramethyl pyrazine derivatives of chalcone has yielded few compounds with potent AChE inhibitory activity [76]. Compound 15 (IC50 = 0.025 µM) showed better AChE inhibition than standard donepezil (IC50 = 0.055 µM). In comparison, it showed weaker BuChE inhibition with IC50 of 2.7 µM. Similarly, compounds 16 and 17 showed significant AChE inhibitory effects with IC50 of 0.062 and 0.10 µM, respectively (Figure 8).
Figure 8.
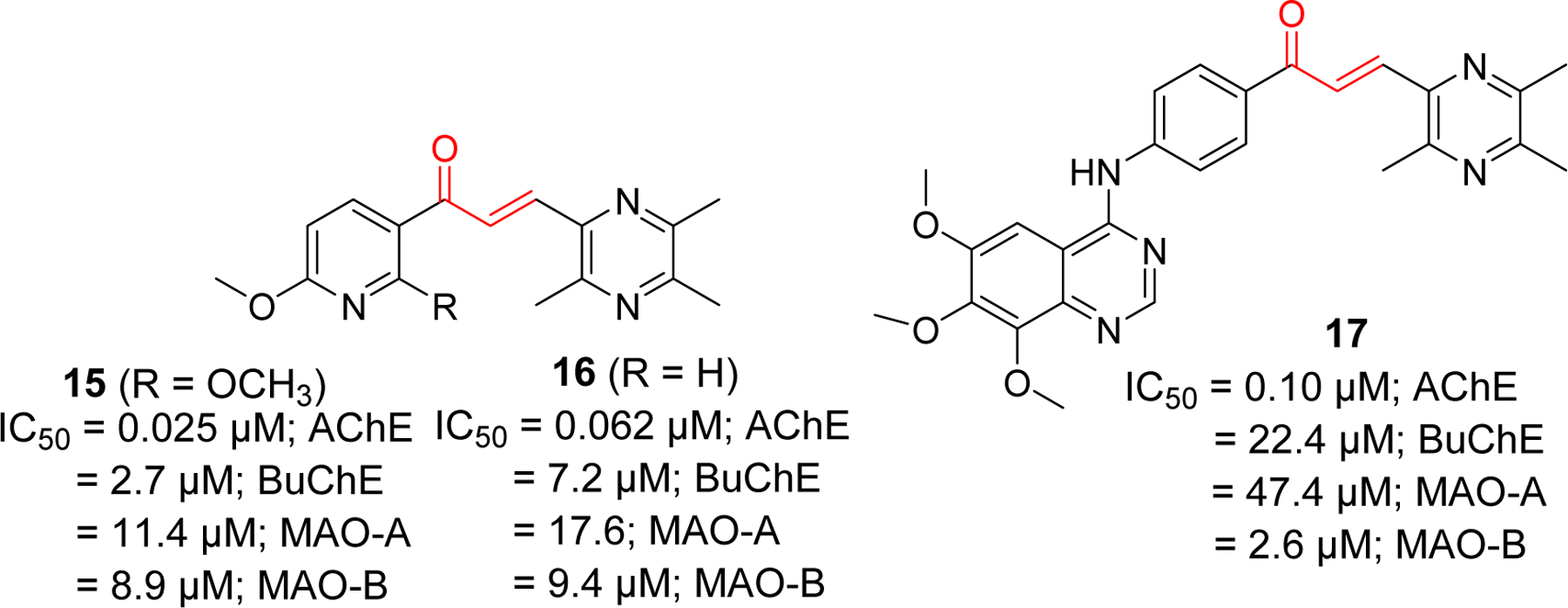
Tetramethyl pyrazine derivatives of chalcone.
Several quinoline and piperidyl derivatives of chalcone were reported as cholinesterase inhibitors by Shah et al. (Figure 9) [77, 78] In the quinoline series with methyl or methoxy group [77], most compounds showed better BuChE inhibition except compound 18, which significantly showed both AChE and BuChE inhibitory effects at IC50 of 0.32 µM and 0.90 µM, respectively. Similarly, compound 19 showed significant BuChE inhibition but weaker AChE inhibition. Among methoxy derivatives, compound 20 and 21 showed significant BuChE inhibitory effect. Docking study of compound 18 with AChE (PDB: 1EVE) showed that quinoline ring forms π-π T-shaped interaction with Trp84 and π-π stacking with His440. Compound 19 was docked with BuChE (PDB: 4BDS). The dioxo benzene ring forms the π-π stacking with Trp82, Phe329 and His438. Similarly, the quinoline ring form the π-π stacking Tyr332 and Asp70. It also forms the π-sigma interactions with Trp82 and Asp70. From SAR and molecular docking study, authors believed that presence of bulky groups in the compounds might be the reason for better BuChE inhibitory effect as BuChE has larger active site gorge and can better accommodate the larger compounds than AChE. Previously, same group reported piperidyl derivatives of chalcones as cholinesterase inhibitors [78]. Interestingly, these derivates were found to be selective AChE inhibitors. Compound 22 was the most potent among the piperidyl derivatives with IC50 of 0.16 µM. Compound 23 in the same class also showed significant AChE inhibition in sub micromolar concentration. Molecular docking study of compound 22 with AChE showed that it occupies both PAS and CAS of enzyme. Piperidine ring in compound 22 form one π-π stacking with Phe330, and two π-π T-shaped interaction with Tyr121 and Phe331. Another π-π interaction was observed between thiophene ring and Trp29.
Figure 9.
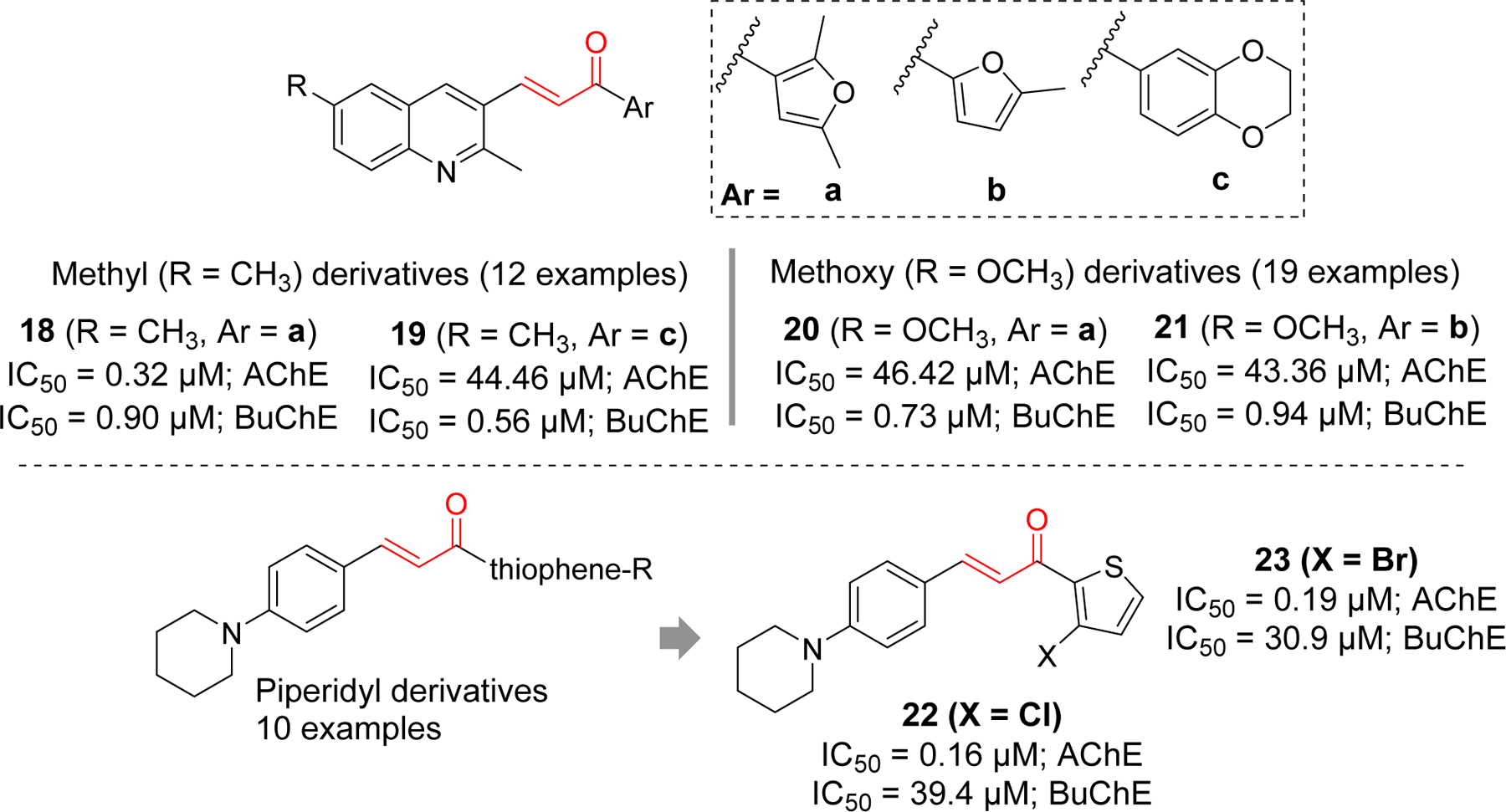
Quinoline and piperidyl derivatives as cholinesterase inhibitors.
Several chalcone derivates containing imides (compound 24 and 25) [79], coumarin (compound 26) [80], and 2-acyl aniline (compound 27) [81] were reported to inhibit AChE at sub micromolar concentrations (Figure 10). Most of the reported imide containing chalcones showed AChE inhibition in nanomolar range. Compound 24 and 25 showed the most significant activity. Similarly, among a series of coumarin containing compounds, 26 showed the most potent AChE inhibitory effect. In addition, compound 26 also inhibited BuChE at IC50 = 4.11 μM. Molecular modeling of compound 26 with AChE showed that it forms multiple interactions occupying both PAS and CAS. In PAS, the coumarin ring form the π-π interaction with Trp279. Similarly, phenyl ring forms π-π interaction with Tyr334. In CAS, cation-π interaction was observed between protonated nitrogen of pyrrolidine ring and Trp84. Molecular docking of compound 26 with BuChE showed only cation-π interaction with Trp84.
Figure 10.
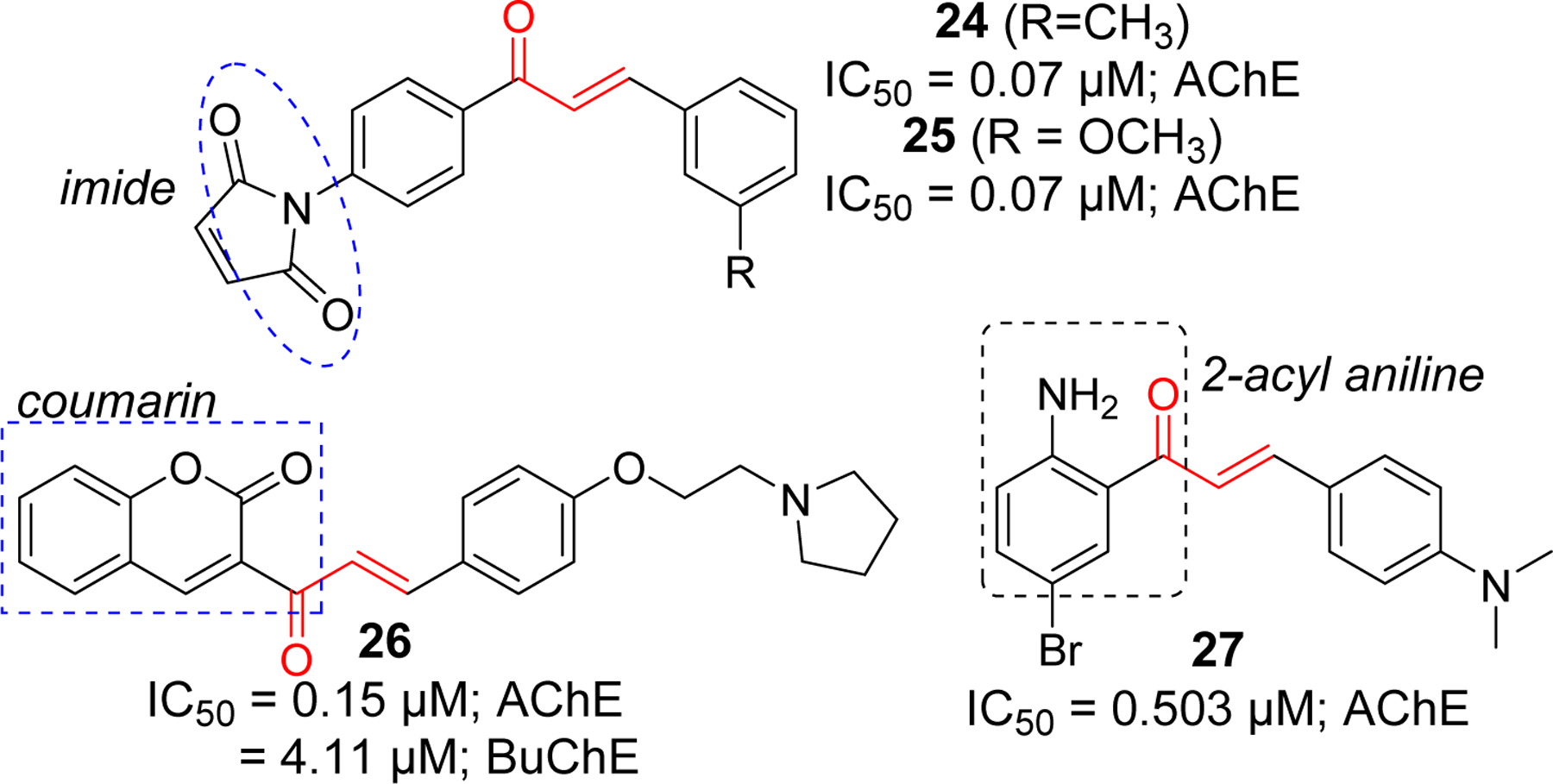
Chalcones derivatives with imide, coumarin and aniline functional groups as cholinesterase inhibitors.
Arylidene indanone, a rigid homolog of chalcone, has been investigated extensively for cholinesterase inhibitory effects due to its resemblance to clinically approved AChE inhibitor donepezil. Seng et al. [82] have reported the preparation of a series of indanone (16 examples) and aurone (16 examples) derivatives. All prepared compounds were tested for AChE and BuChE inhibitory effects. The arylidene derivative, compound 28 was the most potent AChE inhibitor (IC50 = 0.035 μM). Almost all of the reported compounds showed selectivity towards AChE. From SAR, they deduced that double bond (CH=CH) was important for AChE inhibition. However, there was not much differences in activities while comparing indanone and aurone derivatives. Molecular docking study of compound 28 with AChE showed that it occupies both PAS and CAS. In CAS, the charged diethylamine forms cation-π interaction with Trp84 and a hydrophobic interaction with Asp72 and Phe331. In the gorge, π-π stacking between phenyl ring and Tyr334 and Phe330 was observed. In the PAS, the indanone ring forms π-π interaction with Trp279 and the carbonyl group interacts with Phe288 via H-bonding.
The Rampa group reported a series of indanone derivatives based upon hybrids of donepezil and their previously reported potent AChE inhibitor AP2238 [83]. They tested all prepared compounds against both human recombinant AChE and BuChE from human serum. Compound 29 was the most potent AChE (IC50 = 0.19 µM) inhibitor in the indanone series with significant hBuChE (IC50 = 2.70 µM) inhibitory effect. Previously, Belluti et al. [84] prepared several indanone derivatives that showed significant AChE inhibitory effect in nanomolar ranges. Among the arylidene indanone derivatives, compound 30 was the most potent AChE inhibitor with IC50 = 1.80 nM. Similarly, Huang et al. [85] reported several arylidene derivatives with significant AChE and Aβ aggregation inhibitory effects. Compounds 31 and 32 showed both significant AChE (31; IC50 = 14.8 nM and 32; IC50 = 18.6 nM) and Aβ aggregation inhibition (31; 85.5% and 32; 83.8% at 20 μM). Importantly, compounds 31 and 32 exhibited good BBB permeability in vitro. Molecular docking study in AChE (PDB: 2CMF), showed that compounds 31 and 32 has similar interactions and occupied both CAS and PAS. In PAS, benzyl ring of docked compounds forms π-π stacking with Trp279. In the bottom of the gorge, cation-π interaction between protonated piperidine (31 and 32) and Phe330 and Trp84 was observed. Indanone ring of compounds 31 and 32 forms hydrophobic and π-π interaction with Val71, Ser122, Phe331, Tyr121 and Tyr334 along the gorge.
Mishra et al. [86] also reported several arylidene derivatives that exhibit considerable inhibition of AChE and Aβ aggregation. Antioxidant and Aβ aggregation inhibitory properties of curcumin inspired authors to hybridize donepezil with α, β carbonyl group of curcumin to obtain several arylidene indanones. Most of the compounds showed potent AChE inhibition at sub-micromolar concentrations and greater inhibition of Aβ aggregation than curcumin. Particularly, compound 33, 34 and 35 (IC50 = 0.025 – 0.045 μM) showed AChE inhibition at similar or lower concentration than standard Donepezil (IC50 = 0.039 μM).
Chierrito et al. [87] reported several hybrids of donepezil and tacrine with α, β unsaturated carbonyl group as arylidene indanones. They evaluated all prepared compounds for AChE and BuChE inhibition and observed that compound 36 with α, β unsaturated carbonyl group and quinoline amine moiety was the most effective at inhibiting AChE (IC50 = 0.014 μM). It also possesses moderate BuChE inhibition (IC50 = 3.69 μM). Besides AChE and Aβ aggregation inhibition, Meng et al. [88] reported several arylidene indanones with significant AChE and good metal chelating effects. These compounds were prepared by modifying the hydroxyl group at 6-position of indanone with several amine functional groups linked with different carbon chains. In this series, compound 37 with piperidine and 2 carbon linkers at 6-position of indanone and pyridine on β-carbon showed the most potent AChE inhibition (IC50 = 0.0018 μM).
Rampa group [83] has also reported a series of tetralone derivatives. Among the series, compound 38 (IC50 = 0.056 µM) and 39 (IC50 = 0.052 µM) showed the strong AChE inhibition at nanomolar concentrations. Similarly, Belluti et al. [84], in addition to the indanones, has prepared a series of aurone derivatives and tested their cholinesterase activity. Aurone derivative, 40 showed potent AChE inhibition (IC50 = 0.52 nM). Additionally, compound 41 possessed significant AChE inhibitory activity (IC50 = 2.76 nM). Both compound 40 and 41 exhibited considerable BuChE inhibitory activity. Compound 41 was noted by investigators as it also showed a potential for inhibiting Aβ aggregation (67% at 100 μM) induced by AChE (2.3 μM).
The Shafiee group has extensively studied other conformationally constrained rigid groups such as indolinone [89], chromanone [90], and aurones [91, 92] for their cholinesterase inhibitory activities. They reported several potent AChE inhibitors with IC50 in sub-micromolar range. Compounds 42 (indolinone), 43 (chromanone), and 44 (aurone) were found to be the most potent compounds in their classes reported by the group. Molecular modeling of 43 with AChE showed the cation-π interaction between the charged pyridinium moiety with Tyr334 in CAS. Other possible interactions observed at CAS were π-π stacking of benzyl and pyridinium ring with Trp84 and Phe330, respectively. In PAS, hydrophobic interaction between chromanone Trp279 was observed. In addition, there was formation of H-bonding between carbonyl group of chromanone and Tyr121.
Pourshojaei et al. [93] have reported a series of 18 chromanone derivatives that were tested for inhibitory effects on AChE and BuChE. Most derivatives with piperidinyl ethoxy residue showed the potency against AChE at sub-micromolar level. Compound 45 with IC50 of 0.122 μM (AChE) was the most potent among the group. This compound also showed significant BuChE inhibition (IC50 = 0.854 μM). Recently, Wang et al. [94] have investigated isochromanone containing compounds as a fused analog of chalcones for their cholinesterase activities. Most of the compounds showed significant AChE inhibition in sub micromolar range. Compound 46 with 1-(4-fluorobenzyl) substituent displayed the most potent AChE inhibition (IC50 = 8.93 nM). Molecular modeling study of compound 46 with AChE showed that charged pyridinium moiety forms cation-π interaction and π-π stacking with Phe330 in CAS. Moreover, phenyl ring forms the π-π interaction with Trp84. In the PAS, isochromanone functional group forms the π-π stacking with Trp279 and H-bond with Tyr121. The Lee group [95] has also reported the AChE inhibitory effects of aurone derivatives. They carried out modification based upon the sulfuretin, an aurone flavonoid, and found compound 47 as the most potent AChE inhibitor with IC50 of 0.45 µM (Figure 12).
Figure 12.
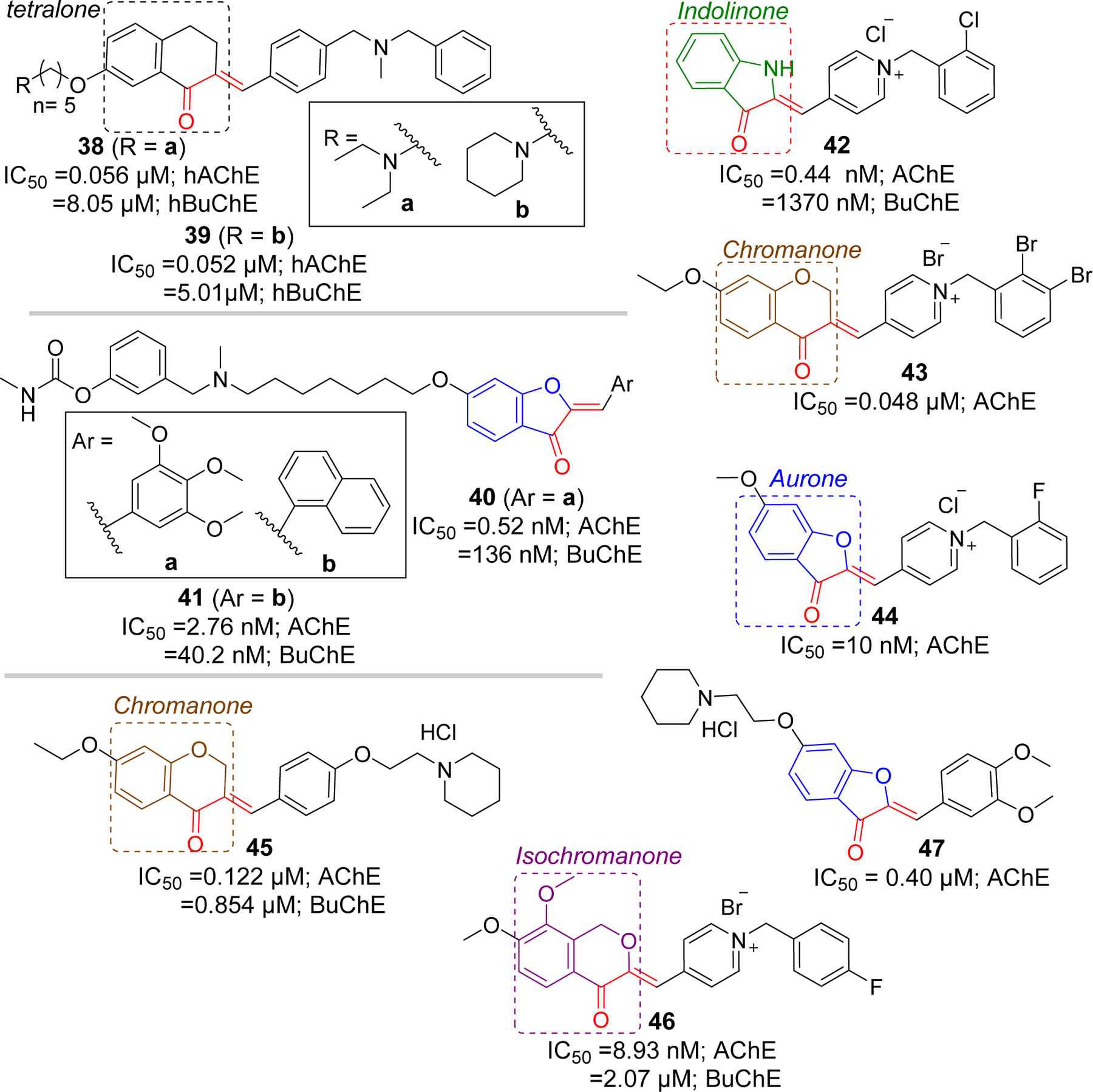
Other rigid analogs of chalcones as cholinesterase inhibitors.
3.2. Amyloid inhibition
The amyloid cascade hypothesis was proposed in the early 1990s [96, 97]. According to this hypothesis, deposition of Aβ due to imbalance in production and clearance results in Alzheimer’s disease. Formation of insoluble amyloid plaques along with neurofibrillary tangles are believed to promote neuronal degeneration and cognitive decline. Aβ was produced from integral membrane protein amyloid precursor protein (APP) by the action of two proteases β- and γ- secretase [98]. APP follows two metabolic pathways, non-amyloidogenic or amyloidogenic, depending on its processing by α- or β- secretase, respectively.
In non-amyloidogenic pathway, APP undergoes α-secretase mediated proteolysis (step 1), which produces a soluble sAPPα and membrane bound COOH-terminus fragment (αCTF or C83). C83 is subsequently cleaved by γ- secretase (step 2) to produce non-amyloidogenic soluble peptides p3 and amyloid intracellular domain (AICD).
In amyloidogenic pathway, APP is first cleaved by β-secretase [99] near the N-terminus at extracellular β-site of the APP (step 1) to release sAPPβ, and membrane bound COOH-terminus fragment (βCTF or C99). C99 fragment is then subsequently cleaved by transmembrane γ-secretase at different γ-sites to produce Aβ of different lengths (step 2). The major species are Aβ40 and Aβ42, however, Aβ42 has shown to have the strongest aggregating property among the species [100, 101].
Inhibition of amyloid plaque formation has been one of the main strategies for developing the disease modifying AD therapies. Investigators have persistently tried to develop inhibitors of α-and β-secretase as these enzyme activities play key roles in Aβ production. However, due to involvement of α- secretase in Notch signaling and severe toxicity associated, β-secretase (beta-site amyloid precursor protein cleaving enzyme 1; BACE-1) has gained much attention as an optimum therapeutic target for AD.
3.2.1. BACE-1 inhibitors
BACE-1, primarily present in the neurons, is an aspartyl protease bound to the membrane which is responsible for cleavage of APP at the beta site and considered the rate limiting step in the generation of Aβ. Its predominant location in CNS, role in Aβ production, and proof of no significant phenotypic alterations in BACE-1 knockout mice [102] encouraged many researchers to develop its inhibitor as a disease modifying agents for AD. Development of crystal structure of BACE-1 with substrate inhibitor OM99–2 [103] further support the concept of exploiting BACE-1 as a potential therapeutic target. Initially, BACE-1 inhibitors were peptidomimetics based upon the transition analog of APP. However, these compounds lacked good pharmacokinetic (PK) profile including poor BBB permeability. Therefore, researchers searched for non-peptidomimetic(s) large enough to fit the larger cavity of BACE-1 active site and display favorable PK profile and BBB permeability. Moreover, compounds selectivity to BACE-1 as compared to other aspartyl protease such as its homolog BACE-2, cathepsin D was an important factor to consider while developing BACE-1 inhibitors to avoid adverse side effects. Several large companies were involved in the discovery and development of BACE-1 inhibitors. Thus several potent non-peptidomimetic BACE-1 inhibitors such as verubecestat (MK-8931), lanabecestat (AZD3293; LY3314814), elenbecestat (E2609), atabecestat (JNJ-54861911), Umibecestat (CNP-520) (Figure 13) were developed and tested in clinical trials for treatment of AD [104–108]. Unfortunately, most of these compounds were unsuccessful and the studies has been discontinued due to low efficacy and/or adverse side effects [109]. Failure of most BACE-1 inhibitors necessitates a reconsideration of understanding the target enzyme or development of multitargeting drugs.
Figure 13.
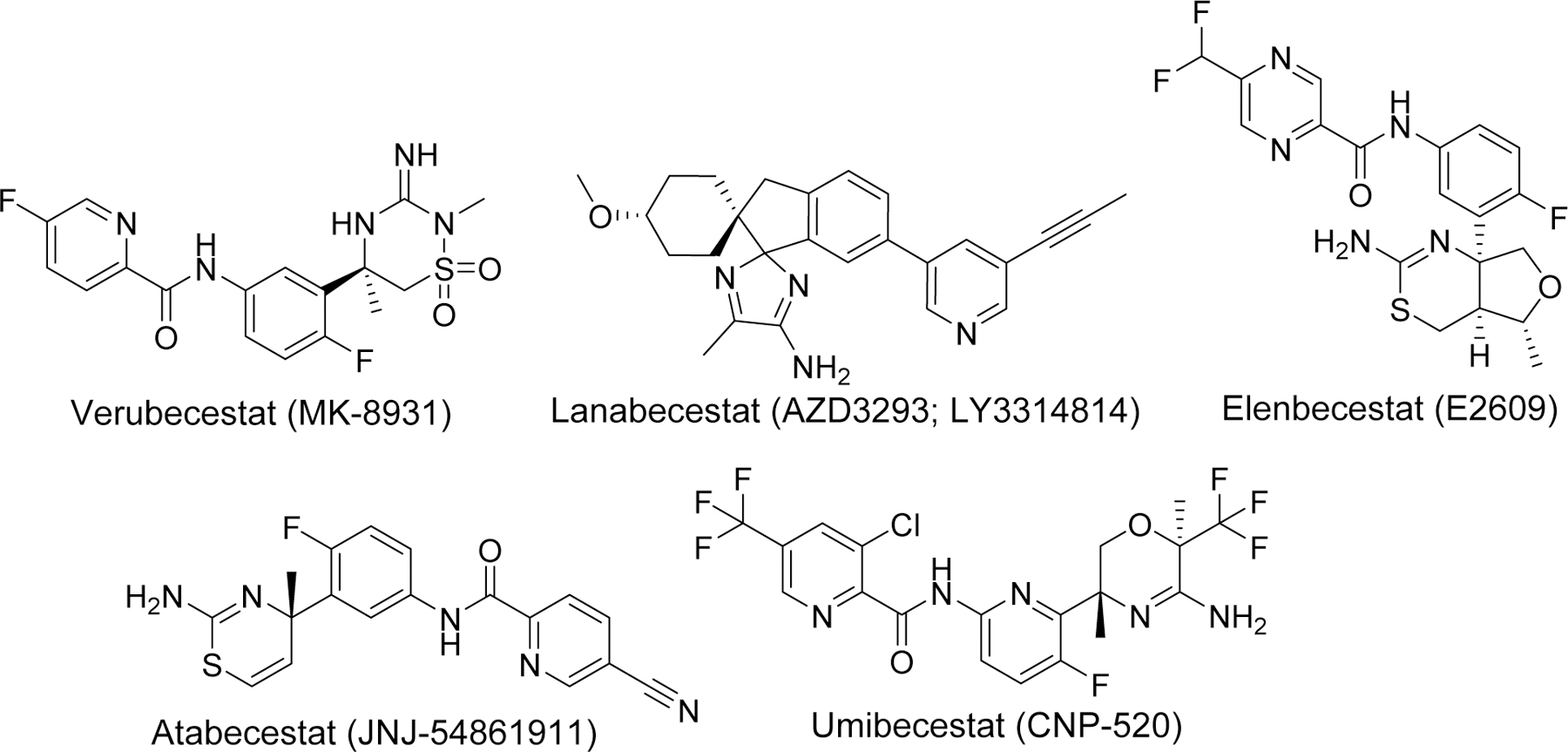
Examples of potent BACE-1 inhibitors that enter clinical trials.
Several natural and synthetic chalcone derivatives have also been explored as BACE-1 inhibitors. Youn et al. [110] reported the BACE-1 inhibitory activities of three flavonoids from Boesenbergia Rotunda including a chalcone, cardamonin (compound 48). Among three flavonoids, compound 48 (IC50 = 4.35 μM) showed the most potent noncompetitive BACE-1 inhibitory activity. Docking study revealed compound 48 having hydrophobic interaction rather than H-bonds with the enzyme. Another natural chalcone, 2, 2′,4′-Trihydroxychalcone acid (TDC) (compound 49) from Glycyrrhiza glabra L (licorice) reportedly displayed specific non-competitive BACE-1 inhibitory activity with IC50 of 2.45 μM [111]. After enzyme assay, in vitro (HEK293-APPswe cells) and in vivo (B6C3-Tg mice) studies showed that compound 49 effectively decreased the Aβ in cells without any toxicity. Furthermore, there was no effect on other secretases (α- and γ). In a mouse model, at a dose of 9 mg/kg per day, compound 49 significantly decreased the Aβ40 and Aβ42. Morris water maze (MWM) test, after 100 days of compound 49 administration, clearly indicated improved memory function.
Rampa et al. [112] designed several chalcone derivatives based on naturally inspired small molecules. After screening in house collection of small molecules, two hit compounds which includes a chalcone derivative (compound 50) and a benzophenone derivative was identified (Figure 14). Computational modeling showed that N, N’-benzyl methylamine groups of benzophenone derivative interact with the catalytic dyad of the BACE-1. Thus, investigators fused compound 50 (along with other various aryl groups) with benzophenone derivative to give new chalcone derivatives including 51 and 52. The most potent BACE-1 inhibitor in the series was compound 51 (IC50 = 1.06 μM). In comparison, 3,4,5-trimethoxyphenyl ring was more favorable than naphthyl ring for enzyme inhibition. In addition, compound 51 showed good Cu (II) chelating activity and significantly reduce the formation of Cu (II) mediated ROS.
Figure 14.
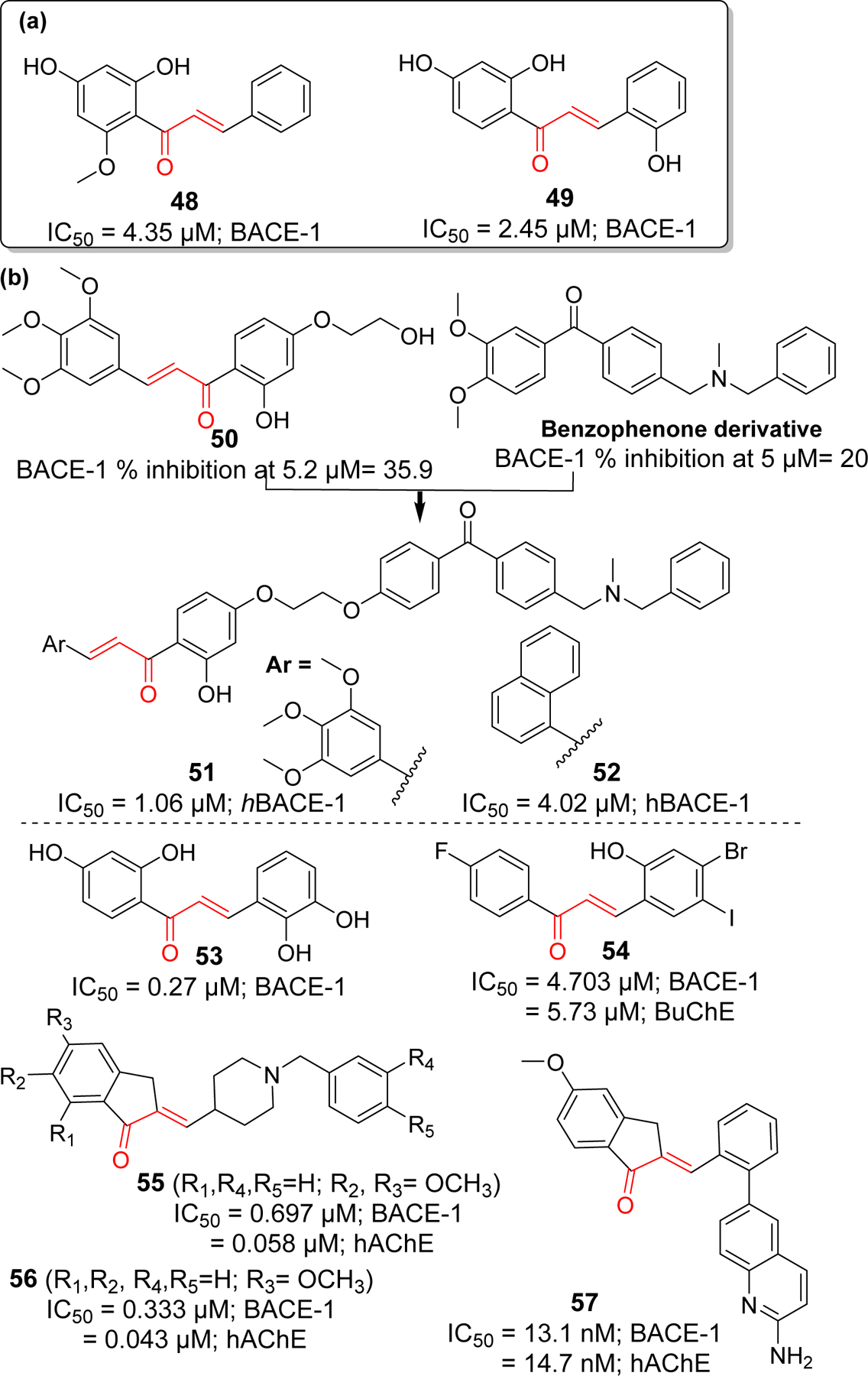
BACE-1 inhibitors: (a) natural chalcone and (b) synthetic derivatives.
On the basis of natural chalcone isoliquiritigenin (IC50 = 33.0 μM) with moderate BACE-1 inhibitory activity, Ma et al. [113] prepared a series of 21 hydroxyl substituted chalcone derivatives and tested for BACE-1 inhibitory activity. They observed several compounds with better BACE-1 inhibitory effects than isoliquiritigenin, compound 53 (IC50 = 0.27 μM) being the most potent. Mphahlele et al. [114] studied several halogenated chalcone derivatives for BACE-1 inhibition along with cholinesterase inhibitory effects. They found compound 54 (IC50 = 4.7 μM) was the most potent BACE-1 inhibitor in the series. It also possessed moderate BuChE inhibitory activity. In one study [115], donepezil was modified to yield several chalcone derivatives. Prepared compounds possessed the donepezil scaffold with α, β-unsaturated carbonyl functionality like chalcone. All the prepared compounds were tested for cholinesterase (AChE and BuChE) and BACE-1 inhibitory activity. Compound 55 and 56 displayed the potent dual functionality against AChE and BACE-1. Compound 56 showed the most potent BACE-1 and AChE inhibition in the series with IC50 = 0.333 and 0.043 μM, respectively. Inspired with compound 56, Gabr et al. [116] synthesized a new series of arylidene indanone (chalcones) as rigid analogs of donepezil. All prepared compounds showed better dual inhibitory effects on AChE and BACE-1 in nanomolar ranges. Compound 57 showed the most potent inhibition of BACE-1 (IC50 = 13.1 nM) and AChE (IC50 = 14.7 nM). From docking study, it was observed that 2-aminoquinoline moiety of 57 is oriented towards the catalytic Asp32 and Asp228 residues of S3 pocket of BACE-1. The hydrophobic phenyl ring occupied the S1 pocket and its π-π interaction with Tyr198 facilitates the favorable orientation of 2-aminoquinoline.
3.2.2. Amyloid beta aggregation inhibitors
Another approach in impeding the formation of amyloid plaque is to prevent the aggregation of Aβ into pathogenic oligomers and fibrils. This approach is believed to be more beneficial than inhibition of physiologically relevant Aβ production as it may avoid the mechanism-based toxicity. Therefore, development of disease modifying Aβ aggregation inhibitors has emerged as an attractive area of AD drug discovery [117, 118]. Development of small molecules as inhibitors of Aβ aggregation is challenging due to several reasons including larger size and geometry of protein-protein interaction surface, and absence of grooves or binding pockets for small molecules to fit [119]. However, discovery of specific amino acids responsible for Aβ fibril formation [120] has provided hope to the idea of developing small molecule inhibitors of Aβ aggregation. All subregions of Aβ such as N-terminus, hydrophobic core, hinge or turn regions and C-terminus contribute differently and are critical for Aβ aggregation. His13-Lys16 (HHQK), central hydrophobic core, and hydrophobic C-terminus are the key sites important for nucleation of β-sheet rich conformation [121, 122]. The formation of hinge or turn regions helps to bring two hydrophobic segments to initiate the β-strand formation and subsequently assembly of β-sheet. Electrostatic and hydrophobic interaction plays and important role in Aβ aggregation. Moreover, it has been identified that the C-terminus of Aβ has significant contribution in aggregation. Two additional hydrophobic residues at C-terminus of Aβ42 is responsible for its higher tendency for aggregation and neurotoxicity as compared to Aβ40 [123]. Development of Aβ aggregation inhibitors has focused on targeting these subsections. Several compounds such as Tramiprosate, RS-0406, and Scyllo-inositol (Figure 15) which binds specifically to HHQK region (N-terminus), central hydrophobic region and C-terminus, respectively, have entered the clinical trials [124–126]. However, none of the inhibitors have made it to clinic yet. Another strategy to inhibit the Aβ aggregation is by disruption of interaction between metal and Aβ. It is evident that excess metals such as zinc, copper worsened the Aβ mediated oxidative stress and act as a catalyst for Aβ aggregation [127, 128]. PBT2 (Figure 15) is an example of copper/zinc ionophore and metal mediated Aβ aggregation inhibitor [129].
Figure 15.
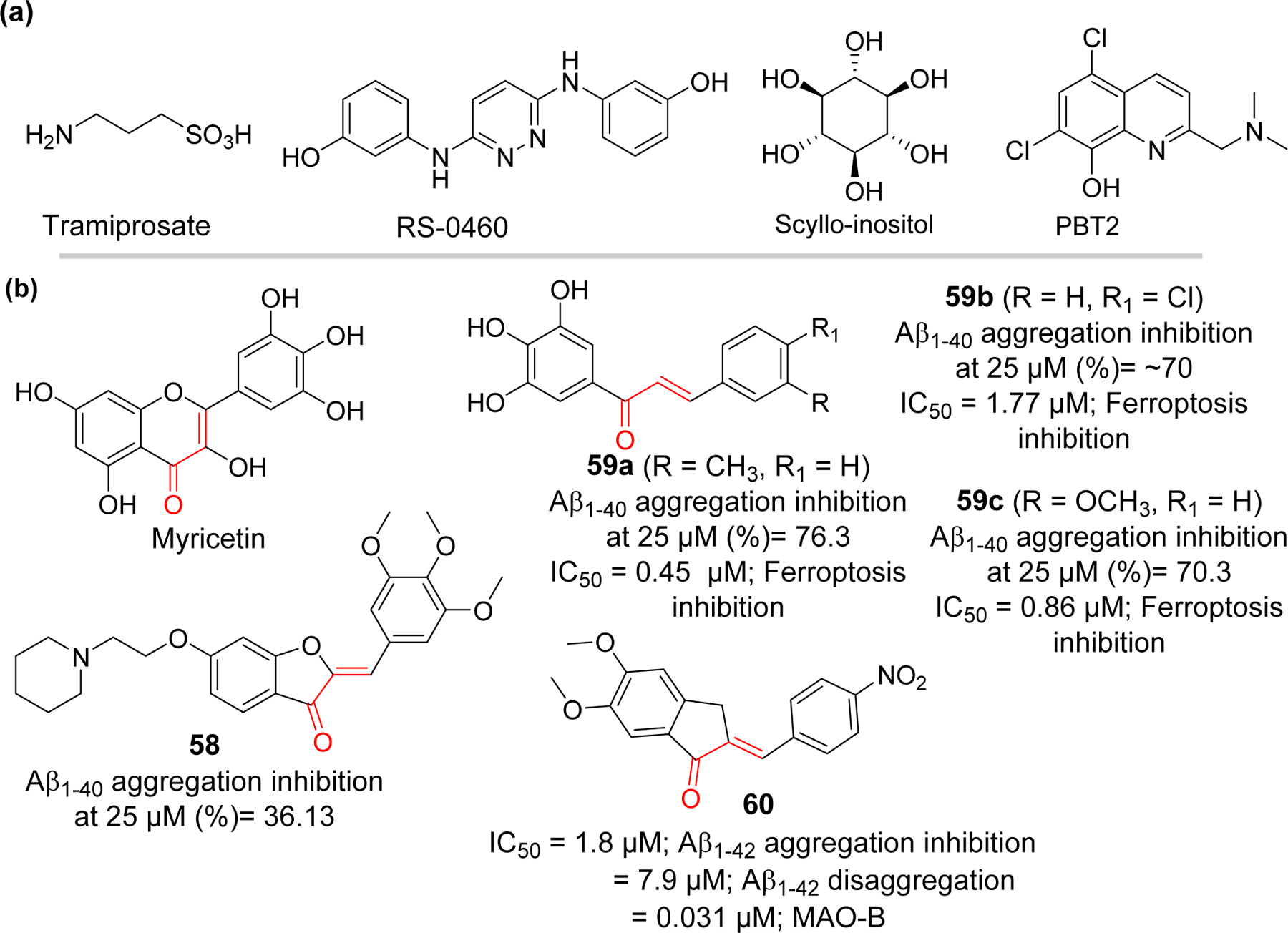
Aβ aggregation inhibitors: (a) compounds that entered clinical trials and (b) natural and synthetic chalcone-based compounds.
Natural product curcumin which is closely related to chalcone has been reported to prevent oligomerization of Aβ and disaggregate preformed Aβ fibrils [130]. Hirohata et al.[131] have studied the mechanism for anti-amyloidogenic effects of five flavonoids. It was observed that Myricetin bound reversibly to the Aβ fibrils rather than the monomers.
Liew et al. [132] reported a synthesis of several aurone derivatives with amine and carbamate functional groups. They evaluated these compounds for their Aβ aggregation activity along with cholinesterase and monoamine oxidase inhibitory effects. Compound 58 with piperidine side chain was the most potent to inhibit Aβ aggregation (36.1% at 25 μM) as compared to curcumin (30.5% at 25 μM). Compound 58 demonstrated neuroprotective effect in Caenorhabditis elegans neurodegeneration model against Aβ and 6-hydroxydopamine induced toxicities.
Cong et al. [133] prepared a series of hydroxylated chalcones (10 examples) and tested for their dual inhibitory effects on Aβ aggregation and ferroptosis. They observed that trihydroxy compounds 59a-c showed better Aβ aggregation inhibition (76.3% to 70.3% at 25 μM) than other compounds. In vitro study showed that trihydroxy substituted chalcones showed better neuroprotection against Aβ1–42 induced neurotoxicity as compared to EGCG and curcumin. In addition, these compounds were evaluated for inhibition of ferroptotic cell death induced by RSL3 and erastin in vitro. Compound 59a was the most potent inhibitor of ferroptosis with IC50 = 0.45 μM (RSL3 induced) via inhibition of lipid peroxidation.
A series of indanone derivatives were reported by Wang et al. [134] and evaluated for their Aβ aggregation and monoamine oxidase B inhibitory effects. Among twelve evaluated derivatives, compound 60 showed the most potent inhibition of Aβ aggregation (IC50 = 1.8 μM). It disaggregated the preformed Aβ fibrils with IC50 = 7.9 μM. Compound 60 exhibited the selective MAO-B inhibition (IC50 = 0.031 μM) as well as neuroprotective effects against Aβ1–42 induced toxicity in SH-SY5Y cells.
3.3. Tau-based therapeutics
Neurofibrillary tangles (NFTs), aggregates of hyperphosphorylated tau protein are one of the hallmarks of several neurodegenerative diseases (tauopathies) including AD. Tau, a microtubule-associated protein, is thought to be responsible for stabilization of neuronal microtubules and help in intracellular transport [135]. Hyperphosphorylation of tau is believed to be one of the earliest events in pathogenesis of AD [136]. Tau has multiple phosphorylation sites with the process of phosphorylation and dephosphorylation being tightly regulated by protein kinases and phosphatases, respectively. Examples of such kinases are cyclin-dependent-like kinase 5 (CDK5), [137–139] glycogen synthase kinase-3β (GSK3β), [140–142] tyrosine kinase Fyn [143, 144] and c-JUN N-terminal kinase (JNK) [145, 146]. Besides hyperphosphorylation, other post-translational modifications such as acetylation, [147, 148] and N-glycosylation [149] are thought to be associated with abnormal tau proteins. In AD, these underlying mechanisms lead to tau protein conformational changes and misfolding resulting in loss of tau binding to microtubules, subsequently dissociation, and aggregation to form fibrillary tangles inside neurons [150].
There has been an increased focus on tau-based agents subsequent to failure of drug development based on Aβ related targets in clinical trials. Inhibitors of kinases responsible for tau hyper phosphorylation, modification of other post translational events, microtubule stabilizers, tau-aggregation inhibitors and anti-tau immunotherapy are the major current strategies for developing tau-based therapeutics [151, 152]. This has led to several molecules such as tideglusib [153, 154], BMS-986168 (BIIB092) [155] methylene blue, Salsalate [148], curcumin and other drug candidates that has entered clinical trials (Figure 16).
Figure 16.
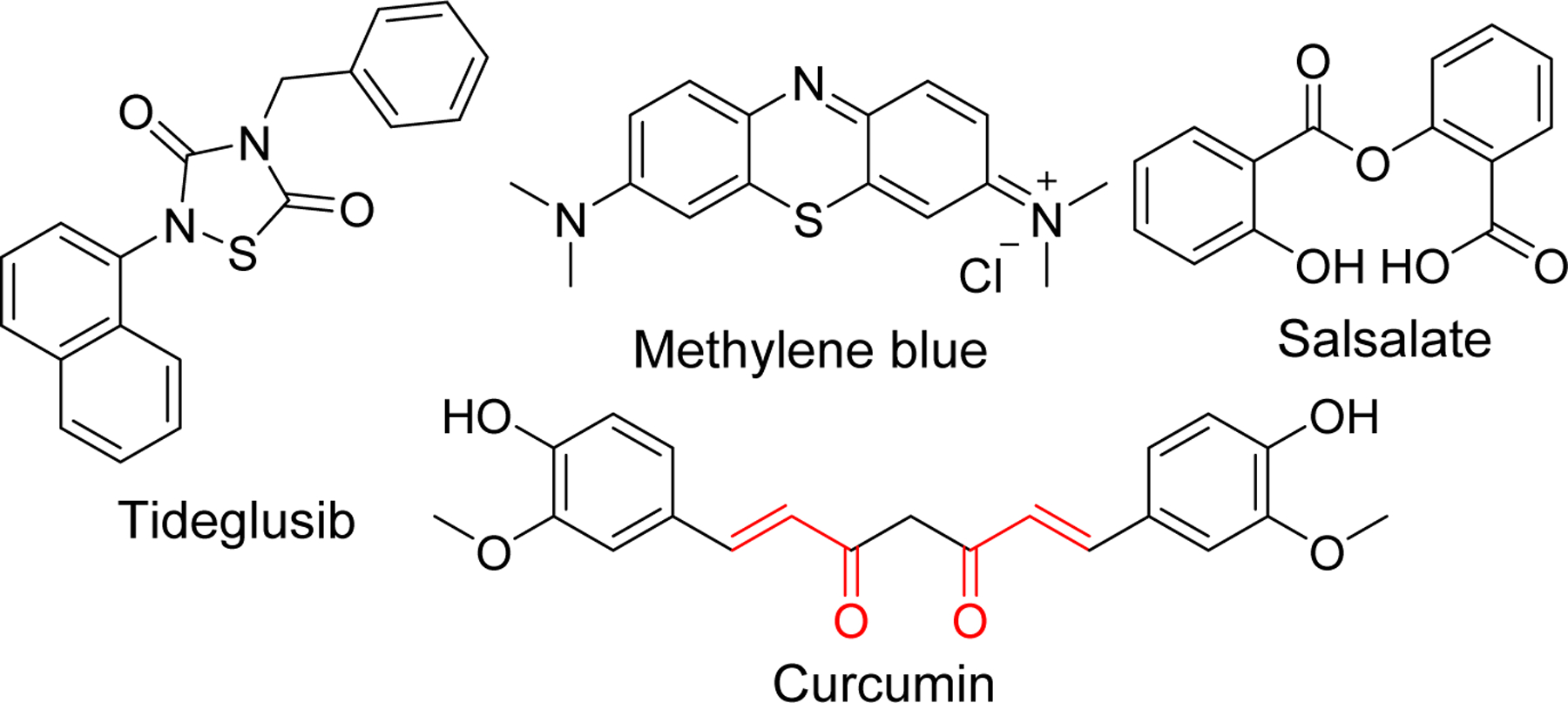
Structure of tau-based inhibitors in clinical trials
Several researchers have reported the chalcone-based compounds that could prevent the formation of neurofibrillary tangles. These reported compounds exert their activity by either directly inhibiting the a) tau protein oligomerization or b) kinases involved in the hyperphosphorylation of tau proteins responsible for conformational changes, dissociation from microtubule, and subsequently aggregation to form tangles.
3.3.1. Tau aggregation inhibition
It is noteworthy that covalent inhibitors of tau aggregation contain α, β unsaturated carbonyl group which act as an electrophilic Michael acceptor similar to chalcones (Figure 17). For example, Cinnamaldehyde has been reported to inhibit the tau aggregation in vitro [156]. From the study, it was observed that the compound interacted with two cysteine residues in tau protein undergoing nucleophilic attack at Michael acceptor by cysteine. To confirm that α, β unsaturated carbonyl group was involved in interaction, investigators tested the 2-Phenylpropionaldehyde (2-PA), a reduced form of cinnamaldehyde (without α, β unsaturated carbonyl group). It did not show inhibitory effects on tau aggregation confirming β-carbon in cinnamaldehyde is essential for its activity. Another well-known natural compound that shares this electrophilic pharmacophore is curcumin. Presence of Michael acceptor, with similar mechanism, makes chalcone (and its analogs) a potential scaffold to develop as tau aggregation inhibitors. Xanthohumol, a natural chalcone based compound has been reported to inhibit tau protein aggregation and even disaggregate tau fibrils by directly interacting with tau proteins [157]. In vitro study showed that it reduces the apoptosis induced by tau oligomers. Similarly, Sonawane et al. [158] reported the anti-tau aggregation activity of another chalcone based natural product Baicalein (IC50 = 35.8 μM). It was able to disaggregate the preformed tau fibrils as well as oligomers. Authors demonstrated that the Baicalein inhibit the tau aggregation by covalent modification as adduct of tau and baicalein was observed in mass analysis.
Figure 17.
Structures of (a) tau aggregation and (b) kinase inhibitors containing α,-β unsaturated carbonyl group
Lin et al. [159] studied licochalcone A and five derivatives for their ability to prevent tau misfolding, ROS scavenging and neuroprotection in human cells expressing proaggregant ΔK280 TauRD-DsRed. Among tested compounds, licochalcone A and compound 61 showed reduced tau misfolding and associated ROS. Increase in expression of protein HSPB1 (heat shock protein B1), involved in prevention of tau misfolding [160], by these compounds was attributed for their tau misfolding inhibitory activity. In streptozocin-induced hyperglycemic 3 × Tg-AD mice, compound 61 reduced the Aβ and tau levels and ameliorated cognitive dysfunction.
3.3.2. Kinase inhibition
The Bellluti group [161] developed a series of β-keto-enol curcumin analogs and tested their dual inhibitory effects in BACE-1 and GSK-3β. Compound 62 and 63 were the most potent BACE-1 and GSK-3β inhibitors, respectively. Compound 64 showed balanced inhibitory activity against BACE-1 and GSK-3β. Docking simulation showed that Cys199 of GSK-3β interact with α, β unsaturated carbonyl of compound 63. Experimentally, they performed the thiol trapping assay using 1H NMR. It yields thia-Michael adduct in a very short time as evident by the disappearance of olefin proton signals.
Jeon et al. [162] reported chalcone derivatives that reduce tau phosphorylation and insoluble Aβ. They prepared 11 analogs and tested for the µ-calpain and cathepsin B inhibitory effects. In AD, hyperactivated µ-calpain are observed that cleaves p35 to p25 [163]. This causes hyperactivation of CDK5 which is associated with hyperphosphorylation of tau leading to formation of NFT. Among the tested compounds, investigators selected compound 65 (IC50 = 18.83 µM for µ-Calpain and 6.34 µM for Cathepsin B) and 66 (IC50 = 16.81 µM for µ-Calpain and 11.05 µM for Cathepsin B) for further studies based upon their potency against cathepsin B as well as µ-calpain. Compound 65 and 66 reduced the phosphorylated tau protein via inhibition of MAPK phosphorylation and cleavage of p35 to p25 associated with µ-calpain inhibition.
3.4. Neuroinflammation Inhibition
Several hypotheses exist to explain the causes and progression of AD, including those based on the cholinergic system, Aβ aggregate, NFTs, glucose hypometabolism and mitochondrial dysfunction, deficiency of autophagy, calcium homeostasis, neurovascular dysfunction, inflammatory hypothesis, metal ion hypothesis, and the lymphatic system hypothesis [164]. Neuroinflammation has emerged as a common feature of AD and a central link that corroborates how innate immune system mediate many of the pro-inflammatory processes. The significance of inflammation in AD was initially noted due to accumulation of immune-related proteins and cells within close proximity to amyloid plaques, protection against AD by anti-inflammatory drugs (e.g. NSAIDs), and reduction in AD pathology by NSAIDs in transgenic animals [165]. Acute inflammation is considered to be neuroprotective while chronic inflammation can be injurious. Sustained activation of immune cells in the brain is now considered to be the third core pathology in AD besides amyloid plaques and phosphorylated tau protein [166, 167]. Thus, acute-phase activation of immune cells to protect neurons may start contributing to injury through persistent generation of pro-inflammatory molecules.
3.4.1. Microglial activation and production of inflammatory mediators
Activation of glial cells (astrocytes, microglia) results in accumulation of proinflammatory molecules exacerbating Aβ and tau pathology among other factors and causes progressive neuronal damage [165]. Microglia are also considered as resident immune cells of central nervous system. They have been recognized for their diverse role in surveying the microenvironment in CNS, phagocytosis of plaques, cellular debris or molecules. Microglia can initiate the immune response with the help of pattern recognitions receptors (PRRs) including TLRs that are present on their surface. TLRs 1 to 9 are expressed in microglia [168]. TLRs can recognize both pathogen-associated molecular patterns (PAMP) and endogenous danger signals known as damage associated molecular patterns (DAMP). In AD, DAMPs such as Aβ or tau aggregates can activate microglia by binding with TLRs, receptors for advanced glycation end products (RAGE), TREM2 and scavenger receptors [169, 170]. This results in production of proinflammatory cytokines such as IL-1β, IL-18, IL-10, IL-6, and TNF. Expression of these cytokines in acutely activated microglia drive enhanced phagocytosis, uptake and clearance of Aβ. However, if the activation persists, the phagocytic nature of microglia is compromised and lead to continuous generation of cytokines that causes neuroinflammation and ultimately neurotoxicity and neuronal cell death. Ion channels involved in potassium (THIK-1, Kv), chloride and purinergic signaling aid in microglial function through regulation of mobility and surveillance [165, 171–173].
Inflammasome plays a key role in inflammatory pathway. These are the protein complexes of sensor molecules (NLRs), apoptosis associated spike like protein (ASC) and pro-caspase that are formed in the cytosol in response to damage or pathogen- associated molecular patterns. NLRP1, NLRP3, NLRC4, AIM2 and PYRIN are the major cytoplasmic sensors that have been reported to form the inflammasome. NLRP3 inflammasome is one of the best studied among all the inflammasomes [174]. Assembly of these protein complex facilitates the conversion pro-caspase-1 to mature caspase-1 via proximity-induced autocatalysis. Caspase-1 is then involved in processing of precursor of inflammatory cytokines pro-1β and pro-IL8 into mature IL-1β and IL-18. Once secreted extracellularly, these matured cytokines can exhibit proinflammatory role. Moreover, casaspe-1 can trigger the cleavage of pore-forming Gasdermin D (GSDMD) which in turn induces the pro-inflammatory cell death called pyroptosis [175, 176].
Endogenous Aβ plaques can initiate the NLRP3 inflammasome mediated inflammatory responses which produces several proinflammatory and neurotoxic cytokines and chemokines [177]. Direct association of NLRP3 inflammasome in the development of AD has been demonstrated in vivo [178]. The transgenic mice (APP/PS1) with NLRP3 and caspase-1 deficiency have reduced AD-related pathogenesis. These mice have reduced neuroinflammation, and cognitive impairment as well as reduced Aβ secretion [178]. Recently, work by Ising et al. [179] demonstrated that microglia and NLRP3 inflammasome plays an important role in promoting Aβ and tau pathology. Their work supports the hypothesis that NFTs are the downstream product of amyloid-beta-induced microglial activation. Association of NLRP3 inflammasome in AD pathogenesis and demonstration that pharmacological intervention of NLRP3 inflammasome improves cognitive impairment in AD mouse model has attracted many researchers on developing NLRP3 inflammasome inhibitors for AD [180, 181].
With increasing evidence supporting a central role of inflammatory processes in AD and other neurodegenerative disease, it is imperative to develop drugs to modulate targets involved in neuroinflammation. Currently used drugs mostly act as cholinesterase inhibitors [182]. A recent report showed 121 total drugs in clinical trials including drugs for cognitive function enhancement (12 agents) and for treating behavioral and neuropsychiatric symptoms (12 agents). In addition, they include several disease-modifying agents (97 agent) that target Aβ, tau, inflammation, oxidative stress and mitochondrial function. This report indicates an increasing number of disease-modifying agents that targets pathways other than amyloid or tau in 2020 compared to 2019 [183, 184]. There is an unmet need for developing new drugs that target pro-inflammatory molecules/pathways.
Chalcones and several flavonoids have been studied for their anti-inflammatory properties. Presence of Michael acceptors in chalcones makes it easier to target many proteins involved in immune responses and inflammation. They can undergo Michael addition with the cysteine present in these proteins. Inhibitory kappa B kinases (IKKs) are one such group whose inhibition causes the suppression of nuclear translocation of NF-ƙB [185] which is a key mediator of inflammatory immune response [186, 187]. Some examples of chalcones that inhibit the IKKβ and NF-ƙB mediated inflammatory responses include butein [188], isoliquiritigenin, and licochalcone A [189, 190]. Isoliquiritigenin, a natural flavonoid from licorice root has been demonstrated to reverse LPS induced cognitive impairment due to its antioxidant and anti-inflammatory properties [191].Moreover, Isoliquiritigenin was found to inhibit NLRP3 inflammasome independent of its inhibitory potency on TLR4 [192]. Prenylated chalcones, Bavachalcone and isobavachalcone were demonstrated to have anti-inflammatory activity in BV-2 microglial cells [193]. Similarly, plant derived 2,2’,5’-trihydroxychalcone (225THC), a potent antioxidant, exerts neuroprotective activity against the TLR4 mediated inflammation in microglia. It inhibited the LPS stimulated TNF-α and IL-6 secretion [194].
Lee et al. [195] reported a synthetic chalcone derivative (compound 67) and its anti-inflammatory effects in microglial cells (BV2) by blocking (TLR4)-mediated inflammatory responses. Inhibition of Akt by compound 67 decreases the IkB phosphorylation which ultimately downregulate the NF-ƙB. Downregulation of NF-kB signaling pathway alleviates the expression of iNOS, COX-2, as well as the production of pro-inflammatory cytokines IL-1β and IL-6. Mateeva et al. [196] synthesized a series of 24 chalcone and flavone derivatives and tested their anti-inflammatory activity on LPS stimulated microglial cells (BV2). Compound 68 and 69 caused a potent inhibition of NO production and iNOS protein expression. Compound 68 reduced the production and release of pro-inflammatory cytokines IL-1α, IL-6 and IL-10.
However, an unsuccessful clinical trial using the anti-inflammatory properties of tetracycline antibiotic minocycline to treat the progression of AD in individuals with mild disease [197] points to explore drugs with ability to reach more than one target.
3.5. Multi-target directed ligands (MTDLs)
Drug discovery and development has mainly focused on selectively targeting a single protein to avoid side effects involving off-targets. However, polypharmacology [198], single agent targeting two or more proteins, is emerging as a new approach for treating many complex and chronic diseases including neurodegenerative diseases such as AD [199–201]. Extensive studies on this multi-targeting approach have already provided several drugs with dual and/or multiple target effects. Data from 2015–2017 show that 21% of drugs approved were multi-targeting drugs [199]. Some of the examples are brexpiprazole, a partial agonist of dopamine (D2) and serotonin (5-HT1A) receptors, cariprazine, partial agonist of D2 and D3 receptors, and midostaurin, a multi-kinase inhibitor. Partial inhibition of multiple targets, instead of complete inhibition of one target, is considered advantageous as it would maintain the balance between the normal physiological functions of proteins targets and prevention of the disease progression [202]. Moreover, it has an advantage of easier prediction and control of pharmacokinetic profile (PK) of single drug compare to combination therapy along with lower or no risk of drug interactions as in combination therapy [203, 204].
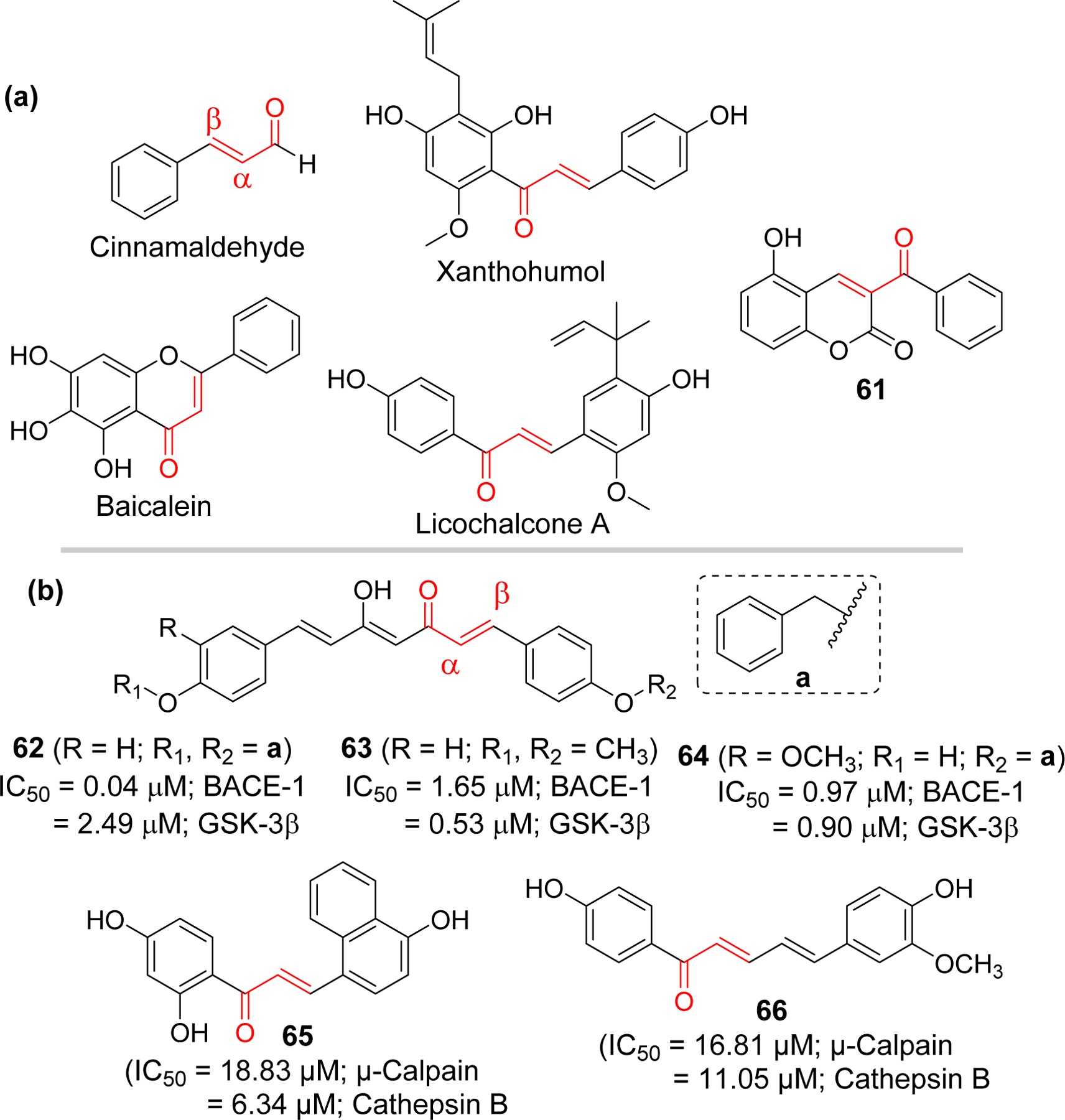
In multifactorial diseases such as AD, multiple-target directed ligands (MTDLs) have gained attention for drug development [205, 206] as approaches targeting single mechanism (proteins) or pathology has not led to a successful disease modifying drugs. Several chalcone derivatives targeting multiple pathologies of AD are being investigated due to flexibility of chalcone for modifications (Figure 19). Sun et al. [207] reported the multitargeted tacrine-homoisoflavonoid (rigid chalcone analogs) as an inhibitors of cholinesterases and monoamine oxidase B (MAO-B). They connected tacrine and homoisoflavonoid with different chain length carbon spacer. Among the tested compounds, compound 70 showed the most promising results with IC50 at sub-micromolar range for all three targets (IC50 = 67.9 nM, eeAChE; 33.0 nM, eqBuChE; and 0.401 µM, hMAO-B). Li et al. [208] have also prepared a series of homoisoflavonoid as a multitargeting agents. They tested the prepared compound for cholinesterases, MAO, Aβ1–42 aggregation and antioxidant properties. Compound 71 showed the most potent AChE inhibition at nanomolar concentration (IC50 = 2.49 nM). In addition, it showed MAO-B inhibitory activity with IC50 of 1.74 µM. The compound showed good self or Cu2+ induced Aβ1–42 aggregation inhibitory and metal chelating activity. Molecular modeling showed that compound 71 occupied the CAS, and PAS of AChE.
Figure 19.
Rigid analogs (chromanone, aurone) of chalcone and flurbiprofen-chalcone hybrids as multifunctional agents in AD.
Li et al. [209] also used the aurone functional group and prepared several aurone Mannich bases as rigid analogs of chalcones. Compound 72, along with good metal chelating ability, Aβ-aggregation inhibition, and antioxidant activity, showed the most potent inhibitory effect on AChE from different species (IC50 = 8.78 nM, ratAChE; 21.2 nM, eeAChE and 37.1 nM, hAChE). This compound displayed the significant neuroprotective effect against H2O2 induced injury in PC-12 cell. Previously, they reported a series of 4-hydroxyl aurone derivatives and evaluated their effects on MAOs, Aβ-aggregation, metal chelation and antioxidant properties [210]. Based upon balanced activities, they selected 73 as a candidate compound for further investigations.
Other chalcone derivatives reported by the Deng group as multifunctional ligands for AD include the chalcone-flurbiprofen hybrids 74–76 [211, 212]. Cao et al. [211] prepared a series of 18 4’-OH flurbiprofen-chalcone hybrids and evaluated for MAO inhibitory effects, Aβ-aggregation inhibition, metal chelating effects and antioxidant activity. Compound 74 and 75 displayed a significant inhibition of Aβ aggregation and antioxidant activity. Anti-inflammatory activity of 74 and 75 were performed in BV-2 cells by analyzing inhibition of LPS-induced NO and TNF-α production. Both compounds showed better TNF-α and NO inhibitory activity as compared to flurbiprofen. Both compounds exhibited 3-fold higher antioxidant effect compared to efficient antioxidant Trolox as determined by in vitro oxygen radical absorbance capacity-fluorescein (ORAC-FL) assay.
In a latter study, Tian et al. [212] prepared 7 flurbiprofen-chalcone hybrids. They tested all prepared compounds for inhibition of cholinesterase, MAO, Aβ-aggregation inhibition as well as for metal chelation and antioxidant properties. Compound 76 showed the most potent inhibitory effect on MAO-B (IC50 = 0.43 µM) and Aβ-aggregation (70.6%, self-induced and 54.9, Cu2+ induced, at 25 µM). TNF-α and nitric oxide (NO) inhibitory activity was measured to assess its anti-inflammatory properties. It inhibits NO (52.5% and 77.5% at 2.5 and 10 µM, respectively) and TNF-α (56.5% and 77.1% at 2.5 and 10.0 μM, respectively) expression better than flurbiprofen (23.0%; NO and 30.3%; TNF-α, at 10 μM).
4. Chalcone as molecular imaging agent in AD
Aβ plaques and neurofibrillary tangles (NFTs) in the brain are the pathological hallmarks of AD [213]. In vivo detection of these pathological markers using molecular probes facilitates the diagnosis as well as monitoring of the disease progression and treatment [214]. Molecular imaging methods such as positron emission tomography (PET) and single-photon emission computed tomography (SPECT) are noninvasive, sensitive and powerful diagnostic techniques that use radiolabeled molecular probe to visualize, characterize and quantify the physiological alterations in vivo and in situ [215, 216]. Several radiolabeled PET imaging agents to image Aβ or tau proteins (NFTs) have been investigated. The ideal PET imaging agent needs to be a) easily BBB permeable b) selectively bind with the target of interest e.g amyloid plaque or NFTs with high affinity and c) easily cleared from non-target brain regions. Physicochemical requirements of the probe include low molecular weight (<600), neutral species with logP value in the range of 1.0 to 3.5 for easy BBB permeation through passive diffusion [217, 218].
Several fluorinated Aβ PET tracers (florbetapir [219], florbetaben, and flutemetamol) for in vivo imaging of Aβ plaques have been approved. However, use of these probes is limited to support other clinical assessment for AD diagnosis and cannot be used to measure the extent of cognitive impairment or diagnose AD on its own as amyloid burden does not necessarily correlate with AD severity. Recently, first tau PET imaging agent, flortaucipir F18 (Tauvid) [220] has been approved by FDA as intravenous injection to determine tau pathology, precise distribution and density of tau tangles, in AD.
For imaging Aβ by PET/SPECT in vivo, the most commonly used radiolabels are [11C] (t1/2 = 20 min) and [18F] (t1/2 = 110 min) [221]. [99mTc] and [125I] are other radiolabels used for imaging Aβ. [99mTc], due to easy production using the 99Mo/99mTc generator is favored than the latter.
Small molecular size, and feasible optimization of lipophilicity (LogP) to enhance BBB permeability makes chalcone a good candidate scaffold for the development of chemical probes or imaging agents in AD. Ono and Nakayama group have reported the use of chalcones as a template for development of several radiolabeled molecular probes e.g radio iodinated chalcones, fluoro-pegylated chalcones, 99mTc labeled chalcones [222–226]. Compound 77, iodinated chalcone with N, N-dimethyl [225], showed significant Aβ1–42 binding affinity (Ki = 3.9 nM). Authors suggest that the binding site of compound 77 is different than known Aβ imaging probes Congo red and thioflavin T. In mouse model brain sections, 77 showed strong staining of Aβ plaque. In vivo biodistribution of [125I]-77 displayed good uptake (2.56% ID/g, 2 min post injection) and rapid clearance from brain (0.21%ID/g, 60 min post injection). Similarly, another iodinated chalcone 78 [222] also showed high binding affinity with Aβ1–42 (Ki = 2.9 nM) and like 77 has non-competitive binding with congo red and thiovalvin T. Radioiodinated [125I-78] was evaluated for the biodistribution study in normal mice. It was observed to have good brain uptake (2.04% ID/g, 2 min post injection) and rapid clearance from brain (0.49% ID/g, 30 min post injection). O-Substituted derivatives 79 and 80 with iodinated radio label [226], also displayed good binding affinity with Aβ1–42. Compound 79 showed better affinity than 80, however 80 has better pharmacokinetic profile as observed in in vivo biodistribution study. Uptake and clearance percentage for 80 was 4.82% ID/g at 2 min and 0.45% ID/g at 60 min, post injection, respectively). Cui et al. [227] prepared a series of indole moiety containing chalcones and evaluated them for Aβ imaging. Most of the compounds showed good binding affinity to Aβ1–42. In particular, compound 81 with 4-iodophenyl substituents, showed the most significant binding affinity (IC50 = 8.22 nM) with specific binding to Aβ plaque in vivo. However, biodistribution study in normal mice showed poor uptake into the brain (0.41% ID/g at 2 min). Therefore, authors concluded that more modifications of the indole containing chalcone derivatives is warranted. Several fluoro-pegylated chalcones were developed [223] as molecular probes for PET imaging of Aβ plaques. In this series, compound 82 exhibited significant affinity to Aβ1–42 (Ki = 38.9 nM). From SAR, it was found that dimethylamino group was important for binding activity as derivatives with this group have higher Ki values than monosubstituted or free amine group. Recently, Kaide et al. [228] has shown that substitution of iodinated chalcone with fluorine-18, compound 83 and 84, has improved its binding affinity to Aβ. Both compounds showed high binding affinity to Aβ1–42 with Kd values in nM range (4.47 and 6.50 nM for 83 and 84, respectively). Biodistribution study in normal mice showed a higher uptake in brain (4.43 and 5.47% ID/g at 2 min post injection for compound 83 and 84, respectively). It was rapidly cleared from the brain (0.52 and 0.66% ID/g at 30 min post injection for 83 and 84, respectively.
The Ono group [224] has also prepared the (99m) Tc-labeled chalcone derivatives and their corresponding rhenium analogs as probes for Aβ aggregates. Compound 85 displayed the desirable characteristics with the potential for being developed as Aβ imaging probe. In normal mice, it showed high uptake in brain (1.48% ID/g) and rapid clearance from brain (0.17% ID/g) at 2 min and 60 min post injection, respectively. These developments have inspired other group to develop chalcone based 11C PET imaging agents [229]. Authors reported the preparation of homodimeric chalcone as a ligand for Aβ. They used the ligand to image the Aβ plaques after labeling it with 11C (compound 86) and performed the comparison with monomeric 11C radiolabeled ligand (compound 87). The bivalent compound, 86, showed ~1.6-fold higher binding affinity to Aβ1–42 as compare to its monomeric congener, 87. PET images after administration of these probes indicated that the uptake of 86 [standard uptake value (SUV) ~3.6] is higher than 87 (SUV ~2.1). Studies on its molecular modeling to show the binding mode with Aβ fibril (PDB: 2BEG) and other basic chemical properties beneficial for the CNS drugs such as molecular weight, BBB permeability were also considered while preparing the core chalcone ligand.
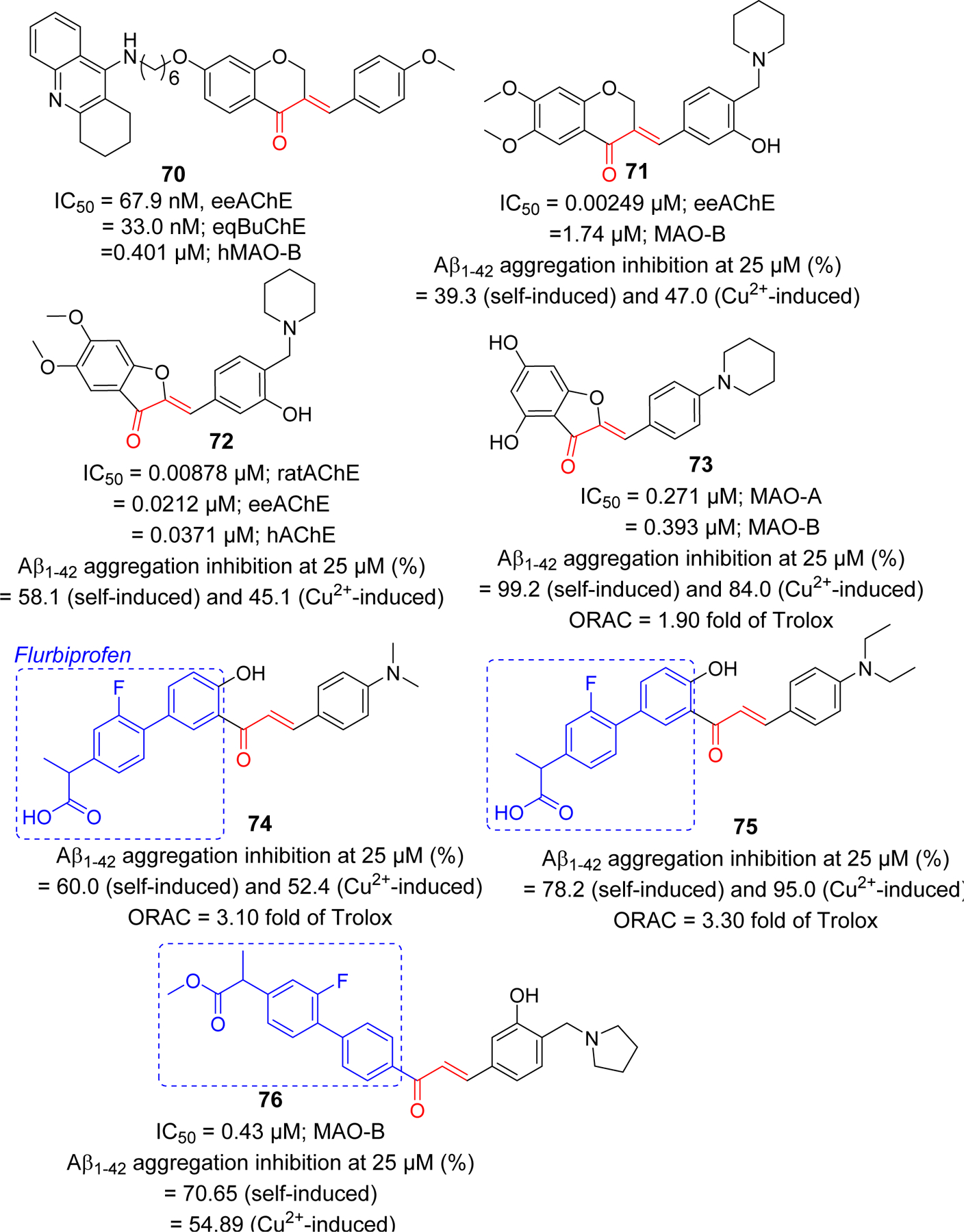
Conformationally constrained analogs of chalcone such as aurone and indanone have been explored for Aβ plaque imaging. Ono et al. [230] reported iodinated aurones with primary (88), secondary (89) and tertiary amine (90) groups in the phenyl ring. All compounds were tested for the Aβ1–42 binding affinity and showed the binding affinity in nanomolar range (Ki = 1.24–6.82 nM). The radio iodinated 88 also showed good binding affinity with Kd = 7.9 nM. The aurone derivative clearly stained Aβ plaques in AD mouse model. From biodistribution study in normal mouse, they showed good uptake to brain and clearance from the brain as well. For e.g. 125I-88 showed uptake of 4.57% ID/g at 2 min post injection and clearance of 0.49% ID/g at 30 min post injection. Later the amine moiety was replaced with ethylene oxide to give compound 91 [231]. It showed significant Aβ1–42 binding affinity with Ki of 1.05 nM. Biodistribution study clearly indicated significant brain uptake (4.5% ID/g at 2 min post injection) of 91 and its clearance from brain (0.09% ID/g at 60 min post injection).
Later, Watanabe et al. [232] introduced dual fluorinated and iodinated chalcone derivatives as a radio labeled probe for PET and SPECT imaging of Aβ. The fluorinated and iodinated chalcone (Figure 22) was evaluated for Aβ1–42 binding affinity which gives Ki value at 6.81 nM. In AD mouse model, it showed intense staining of Aβ plaques. The radio labeled derivatives 92 (125I) and 93 (18F) were prepared and tested for their biodistribution in normal mice. Both showed good brain uptake and clearance. Compound 92 showed initial uptake of 2.34% of ID/g at 2 min post injection. It was rapidly eliminated in 60 min post-injection (0.19% of ID/g). For compound 93, the radioactivity reading was 3.66% of ID/g and 1.75% of ID/g at 2 min and 60 min post injection, respectively. Iodinated indanone derivatives with methyl substituted amines were reported as potential Aβ plaques imaging probes [233]. Particularly, compound 94 and 95 with methyl amine or dimethyl amine showed significant Aβ1–40 binding affinity with Ki = 5.8 and 11.8 nM, respectively. Both compounds showed great affinity in AD patient brain homogenates with Ki values in nanomolar range. Compound 95 was radio labeled with 125I and further tested for biodistribution in mice, partition coefficient measurement, in vitro stability test etc. It was observed that compound 95 exhibited significant brain uptake of 5.29% ID/g at 2 min post-injection. Also, it showed fast clearance from brain (0.84% ID/g at 60 min post injection).
Figure 22.
Radiolabeled conformationally constrained chalcone derivatives (aurone and indanone) as a molecular probe for detection of amyloid beta plaques
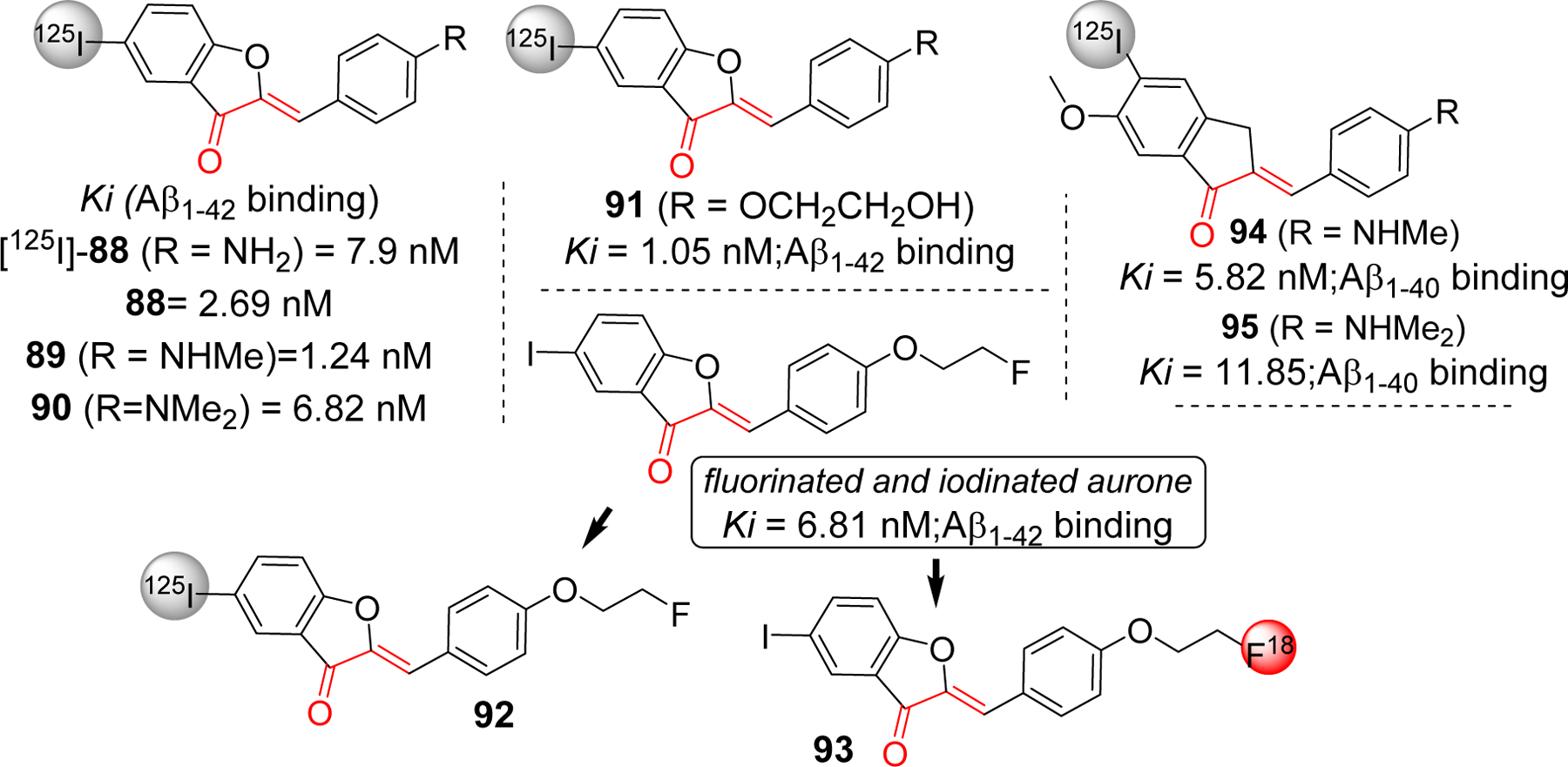
In addition to radiolabeled imaging agent, chalcone derivatives have been investigated as fluorescent probes to image the Aβ aggregates. In vivo use of these agents in humans is limited because of a lack of deep penetration. However, they are good candidates for pre-clinical investigations because they are easy to synthesize, handle and less expensive compared to radiolabeled agents. In contrast to PET/SPECT, where expensive and specialized instrument is required to generate short lived radionuclides, fluorescent probes are nonradioactive and gives real-time high-resolution imaging [234]. Chalcones conjugated system (Figure 1) with proper electron withdrawing or electron donating functional groups on the benzene ring(s) can pertain fluorescence. Jung et al. [235] have reported chalcone based fluorescent probes, 96 and 97 (Figure 23). Both probes showed increased fluorescence when bound with Aβ aggregates. The apparent binding constants (Kd) for 96 and 97 was observed at 1.59 and 2.30 µM, respectively. Probe 96 was chosen for the ex-vivo imaging because of its better LogP value. Its emission wavelength of 532 nm after binding to Aβ aggregates limits the use for in vivo experimentation. Later they developed compound 98 [236] boronic acid-based chalcones as fluorescent probes that exhibit a significant increase in binding affinity (Kd = 0.79 µM) for Aβ aggregates. The Ono group, in addition to their work on radio labeled chalcone derivatives, reported the fluoresence probes to stain β-amyloid in vitro [237]. All four probes showed moderate binding affinity for Aβ (1–42) aggregates (Ki = 72–114nM). However, only molecules 99 and 100 showed stronger fluroescence (6.7 and 14.2 fold, respectively) in the presence of Aβ (1–42) aggregates. In Tg2576 mouse brain sections, these two probes clearly visualized Aβ plaques in contrast to two other probes they developed in the study. Thus author concluded that these two probes are suitable for in vitro staining of Aβ plaques.
Figure 23.
Chalcone based fluorescent probe for imaging Aβ aggregates
Recently, some investigators are focused on developing several near infrared (NIR, 650–900 nm) fluorescence probes for Alzheimer’s disease as it has advantages of deep tissue penetration and high sensitivity. Several probes such as NIAD-4 (bisthiophene derivatives), benzophenoxazine dyes, curcumin derivatives, BODIPY based probes have been explored as NIR fluorescence probes for AD [238].
5. Future perspectives: Potential theranostic agent for AD
Theranostics defines the approach of using a single molecule or agent that possess both therapeutic and diagnostic properties [239]. In contrast to conventional strategy of using two different agents, this approach provides the advantage of avoiding differences in the biodistribution and selectivity of multiple ligands. This idea has been explored in the drug discovery and development for treating several diseases such as cancers, HIV infection, multiple sclerosis and atherosclerosis [240] and found to be beneficial. Undeniably, most of such work has been done in the field of oncology [241–243]. This approach is attainable in Alzheimer’s disease as well, however, the field of developing theranostic for AD is in its infancy.
Evidently, chalcone and its analogs have been widely investigated for their therapeutic application as cholinesterase inhibitor, Aβ aggregation inhibitor, tau protein related kinase inhibitors, anti-inflammatory agents to prevent neuroinflammation and several others. On the other hand, several radiolabeled chalcone derivatives have been studied for their possibility as PET/SPECT imaging agents to detect amyloid plaques in vitro and in vivo. Similarly, several chalcone based fluorescent probes with significant binding affinity for Aβ are currently being investigated. These studies justify an idea of incorporating both therapeutic and imaging capability in one chalcone molecule to obtain theranostic chalcone derivatives. For instance, developing a chalcone compound that can give intense fluorescence upon binding to Aβ aggregates or NFTs and inhibit the aggregation can be a plausible approach to yield theranostic chalcones as supported by a few studies on other classes of compounds. Such examples include NIR fluorescence probe phenothiazine [244–246], curcumin derivatives (18F labeled curcumin, NIR fluorescent probes such as CRANAD derivatives [244]. Phenothiazine derivative 101 [244], showed significant binding affinity to Aβ aggregates (Kd = 7.5 nM) and effectively inhibited the formation of Aβ fibrils. It also showed ability to disaggregate already formed Aβ fibrils. This compound showed maximum emission in PBS at >650 nm. Taken together, it has displayed a potential to evolve as theranostic agent for AD. Later same group developed another phenothiazine based NIR probe, 102 [245], that has protective effects on human neuroblastoma cells. It has high binding affinity to Aβ aggregates (Kd = 24.5 nM) and metal-chelating property. This probe has the ability to image Aβ plaques in AD mouse model. It prevents the Aβ aggregation and protect human neuroblastoma SH-SY5Y cells from Aβ induced toxicity and oxidative stress. One of the best examples of compounds with theranostic potential in AD is curcumin which is structurally related to chalcone. Several studies suggest that it has very high binding affinity to Aβ [248, 249]. Several curcumin derivatives have been developed to evaluate Aβ plaques in vivo e.g 18F curcumin derivatives, CRANAD derivatives etc [250–252]. In addition, it has been demonstrated to possess Aβ aggregation inhibitory activity,[130, 253, 254] metal chelating effect [255], antioxidant, and anti-inflammatory [256, 257]. The Ran group has developed several curcumin based NIR fluorescence probes (CRANAD derivatives) for Aβ plaque imaging. CRANAD-17 was designed to possess both imaging and therapeutic potential [251]. Imidazole moiety was incorporated into CRANAD-17 to compete Aβ peptide (Histidine-H13 and H14) for copper binding, thereby preventing crosslinking of Aβ. Collectively, curcumin and its derivatives show great potential to develop a theranostic agent for AD.
On the foundation of the above examples and the fact that chalcone shares structural similarity with curcumin, chalcone has a potential to be explored as theranostic agents in AD. Currently, there are no clinically approved disease modifying agents for AD. Several chalcone derivatives have been reported to have high binding affinity to Aβ, increased fluorescent after binding to Aβ, and inhibitory effects on Aβ aggregation as well as other therapeutic targets of AD. Therefore, vigorous and extensive studies are warranted to develop chalcone as MTDL which possess higher binding affinity to Aβ or NFTs and fluorescence properties, particularly at NIR range to enable molecular imaging of AD in vivo.
6. Conclusion
Limited efficacy of currently available FDA approved drugs for AD treatment and lack of any disease modifying agents has attracted many researchers including medicinal chemists to explore different chemical scaffolds in search of a potential therapeutics for AD. Chalcone is one of the privileged scaffolds with diverse biological functions that is being extensively studied for Alzheimer’s disease, both as therapeutics and molecular probe. For the development of CNS drugs, BBB permeability is one of the key factors to be considered in addition to potency of the drugs. As mentioned earlier, chalcone has low molecular weight, and easy to optimize its lipophilicity (logP) with appropriate substituents which makes chalcone scaffold interesting with a potential CNS therapeutic agent. Currently, several amyloid and tau-based agents for AD in clinical trials have failed. Thus, researchers have extended their focus on development of novel agents targeting other measures such as neuroinflammation, mitochondrial dysfunctions for the AD treatment. As evident, several natural chalcone and its analogs (e.g. butein, licochalcone, Isoliquiritigenin) has shown considerable anti-neuroinflammatory activities in vitro and in vivo. Moreover, AD is a complex multifactorial disease. Inhibition of single protein target/mechanism involved in the pathogenesis of AD, would not be sufficient to treat AD. Therefore, development of multi-target directed ligands would be an essential approach to succeed in finding novel therapeutic agents for AD. In this quest, chalcone and its analogs has shown a great potential as many derivatives has been reported to show multi-targeted functions. Furthermore, early diagnosis of disease is a critical step in determining the efficacy of treatment. In AD, several biomarkers such as Aβ42, Aβ42/40 ratio, phosphorylated tau proteins (pTau181) are specified as core neurochemical biomarkers [258]. Several Aβ and NFTs molecular probes were discovered. However, they are radiolabeled agents associated with high cost, and toxicities. Researchers are in search of developing NIR fluorescence probes to image Aβ and NFTs. One of the most studied compounds for developing molecular probe that is closely related to chalcone is a natural product curcumin. Some of the NIR fluorescence probes (related to curcumin) has theranostic potential in AD as they have ability to inhibit the protein (Aβ) aggregation as well. Chalcone share structural similarity with curcumin, and studies showed that several chalcone analogs have significant binding affinity to Aβ and inhibition of Aβ aggregation. Exploration and optimization of chalcone and its analogs is applicable for the development of NIR fluorescence theranostic agents (for AD). In summary, this review highlights different pathological mechanisms of AD and current development of chalcone and its analogs in drug discovery and molecular imaging for AD treatment and diagnosis, respectively. We hope the information provided in this review will be adequate and encourage medicinal chemists and many other researchers to consider exploration of chalcone as therapeutic or diagnostic or even beyond as a theranostic agent in their future AD drug discovery endeavors.
Figure 11.
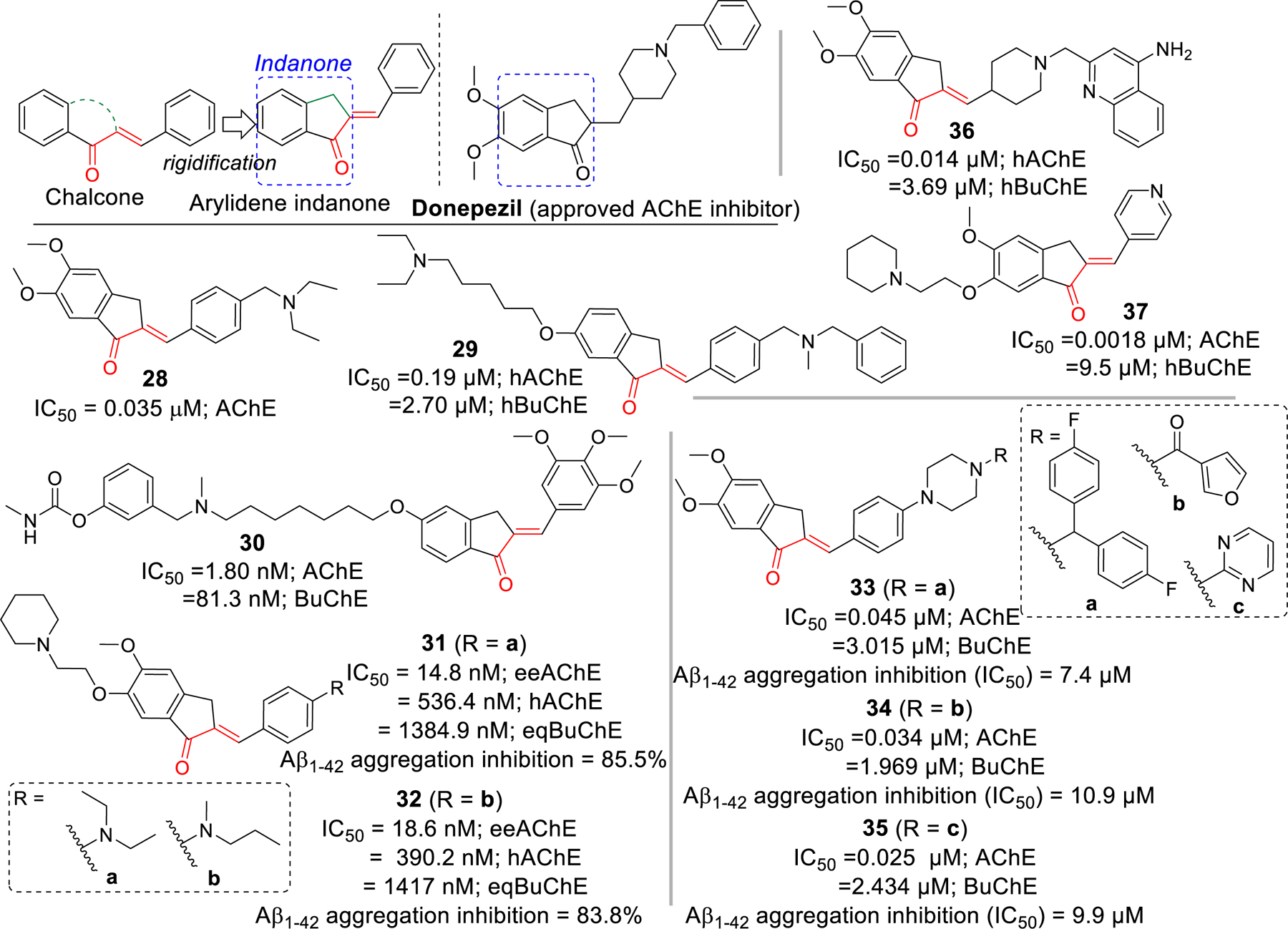
Arylidene indanones (rigid analogs of chalcones) as cholinesterase inhibitors.
Figure 18.
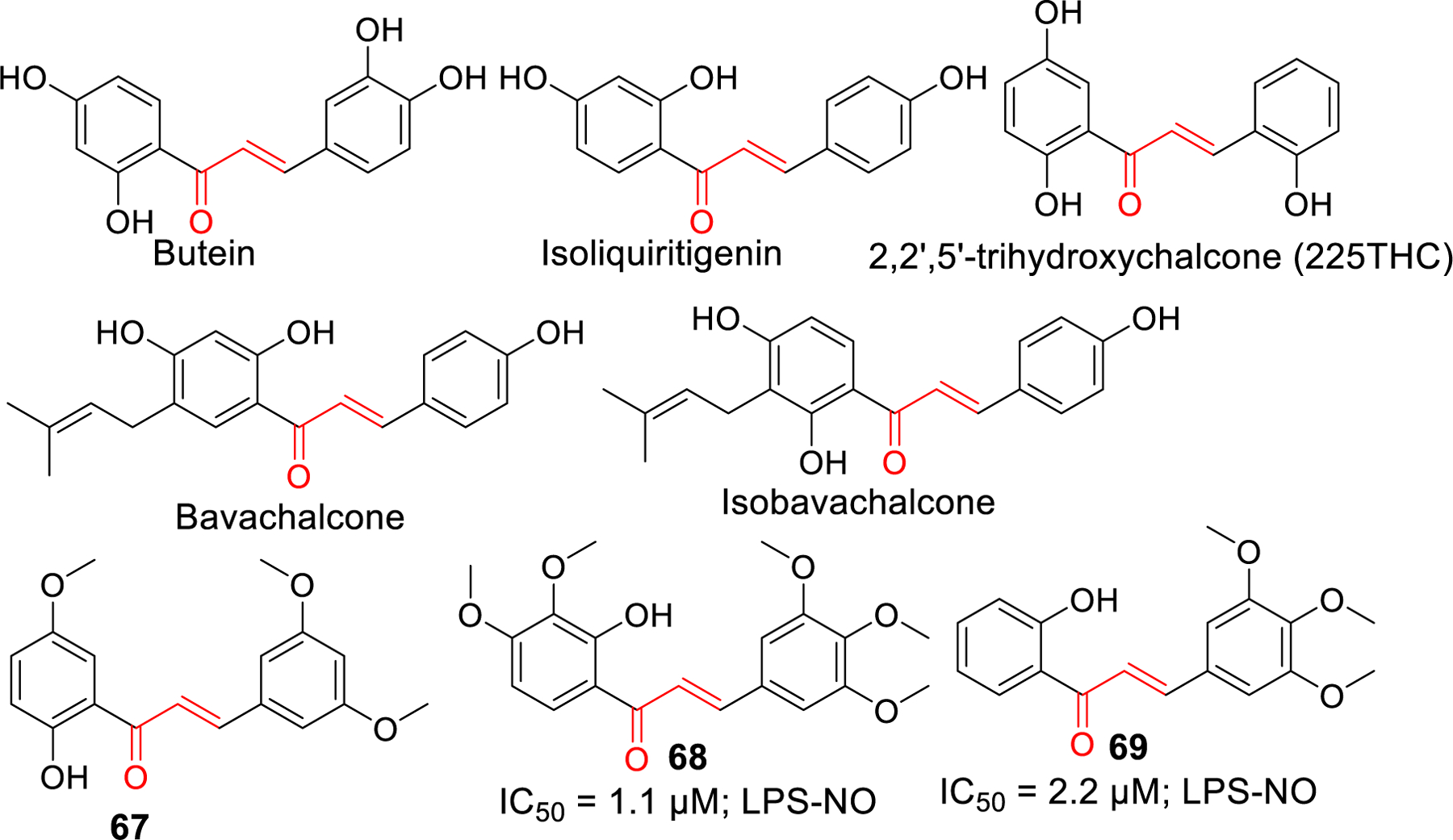
Natural and synthetic chalcone derivatives as anti-neuroinflammation agents
Figure 20.
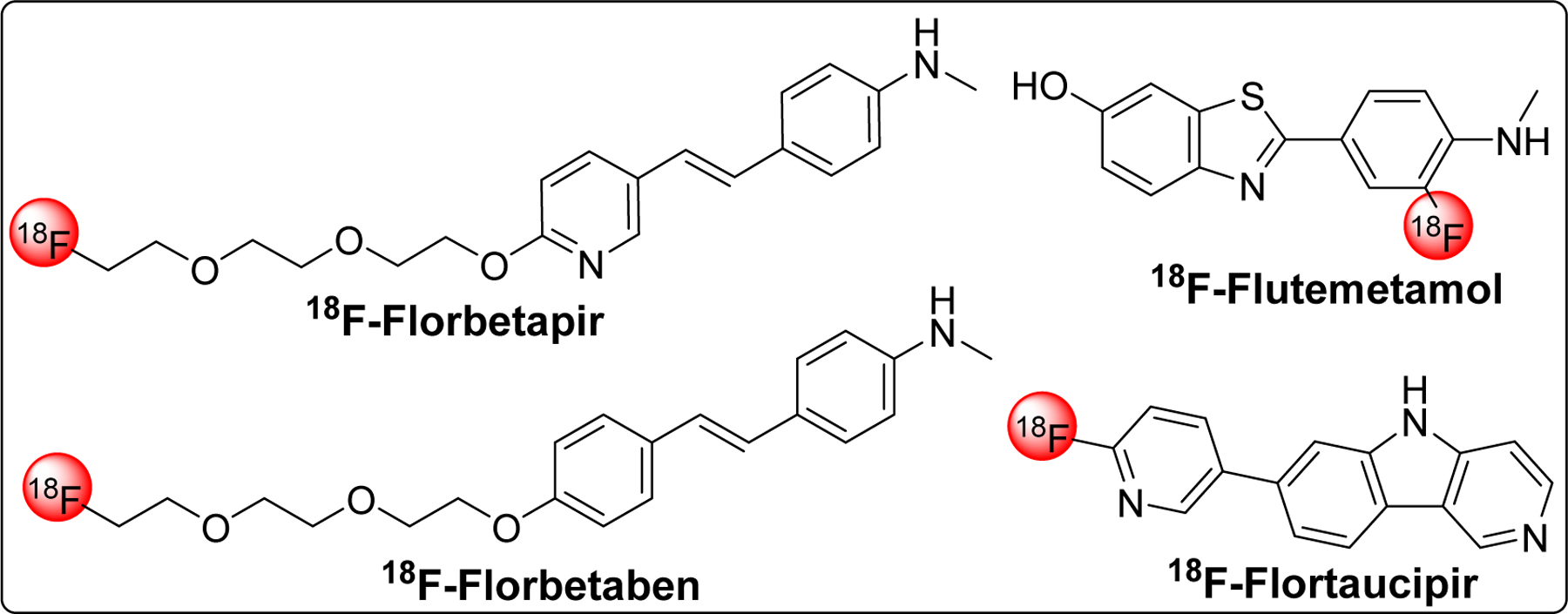
FDA approved PET imaging probe for amyloid (Aβ) and tau proteins (NFTs)
Figure 21.
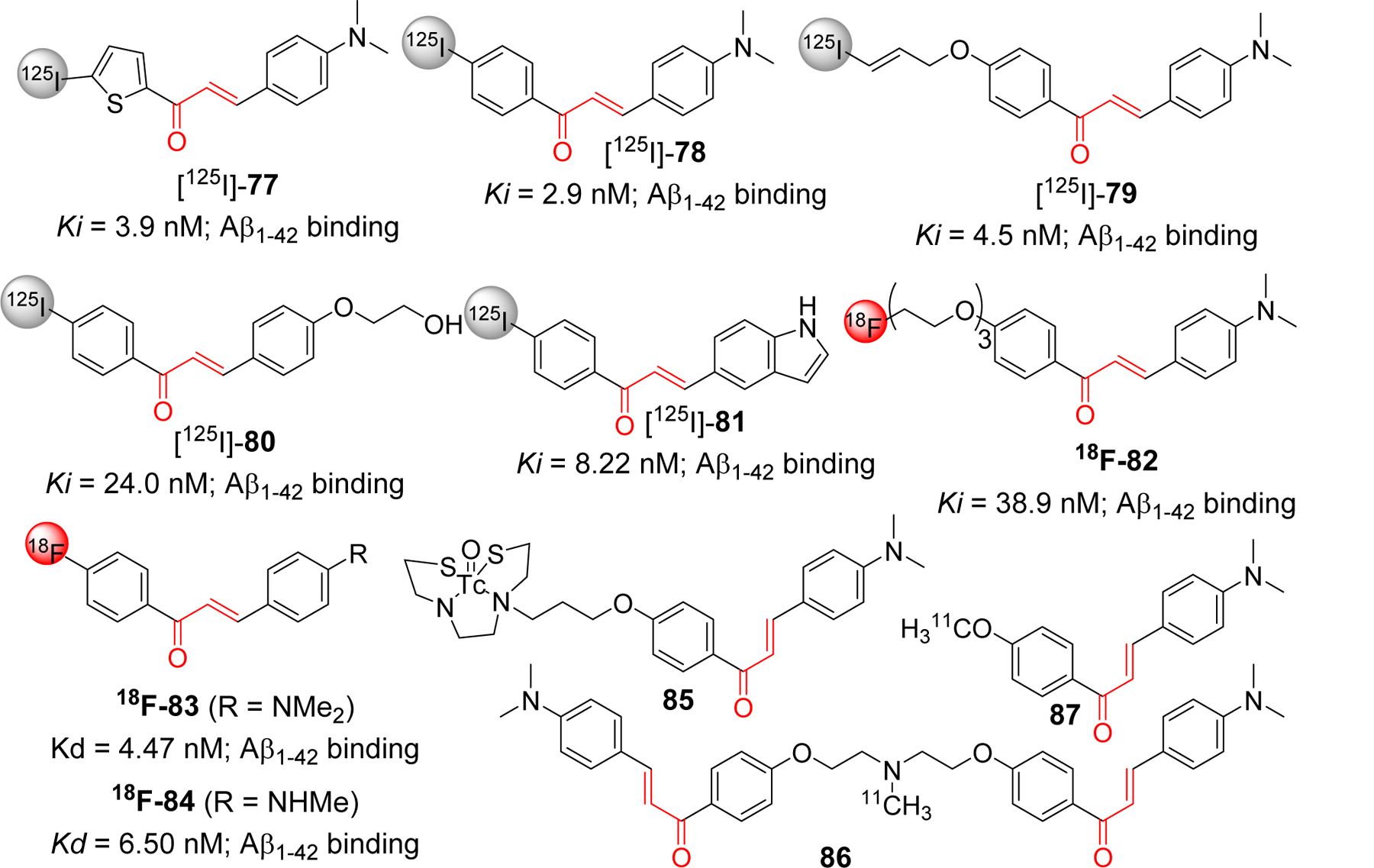
Radiolabeled chalcone derivatives (125I, 18F, 11C and 99mTc) as a molecular probe for detection of amyloid beta plaques.
Figure 24.
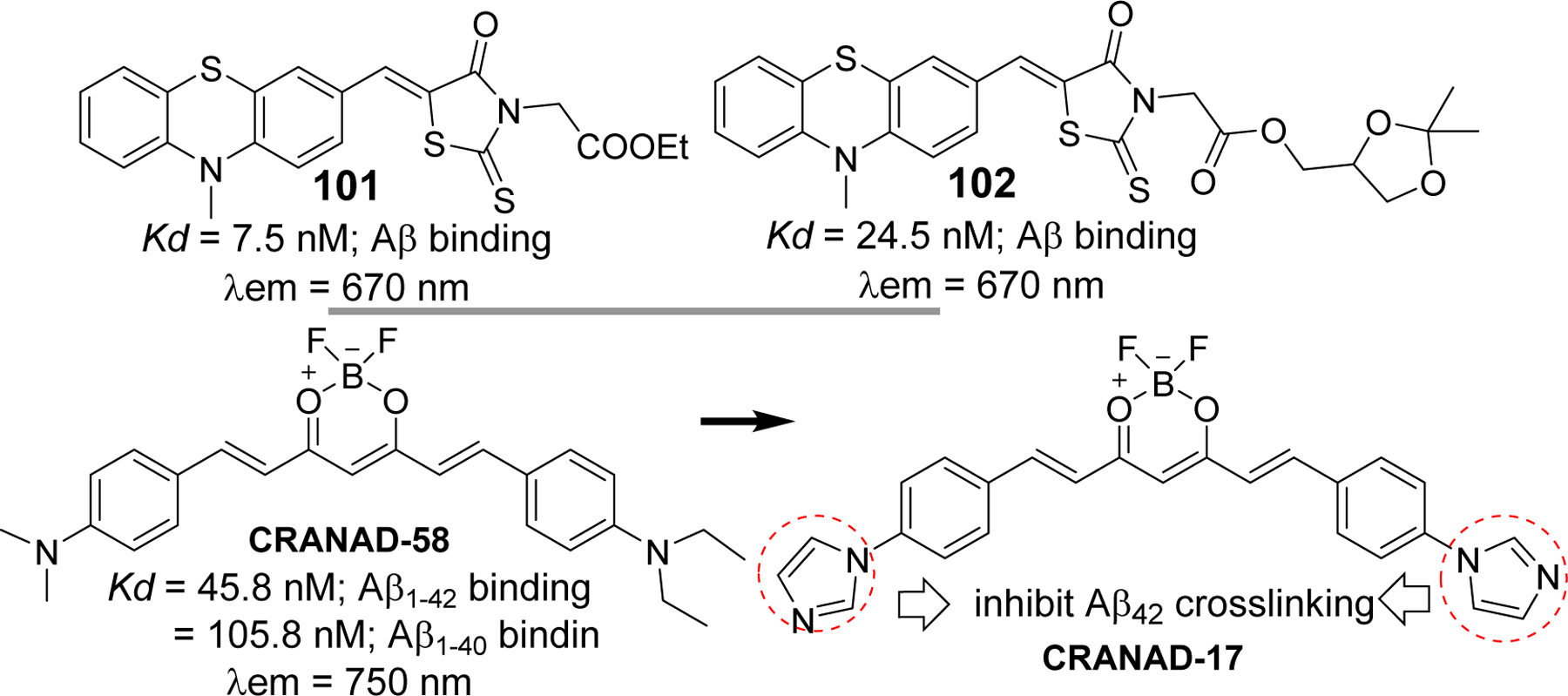
Representative phenothiazine based (101 and 102), and curcumin based (CRANAD-58 and 17) NIR fluorescence probe with theranostic potential.
Highlights.
Comprehensive overview of current developments related to chalcone and its analogs as therapeutic or diagnostic agents in Alzheimer’s disease (AD).
Chalcones as multi-target directed ligands (MTDL) in AD.
Potential use of chalcones as theranostic agents for AD.
Acknowledgements
The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States government. We would like to thank Matthew S Yorek for thorough proofreading.
Funding
This work was supported by Midwest Veterans Biomedical Research Foundation (MVBRF) at KCVA Medical Center, Kansas City, MO, 64128. MS received support, in part, from National Institutes of Health /NIDDK R01DK107490.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare no conflict of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
References
- [1].Gomes MN, Muratov EN, Pereira M, Peixoto JC, Rosseto LP, Cravo PVL, Andrade CH, Neves BJ, Chalcone Derivatives: Promising Starting Points for Drug Design, Molecules, 22 (2017) 1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Simmler C, Lankin DC, Nikolic D, van Breemen RB, Pauli GF, Isolation and structural characterization of dihydrobenzofuran congeners of licochalcone A, Fitoterapia, 121 (2017) 6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Leon-Gonzalez AJ, Acero N, Munoz-Mingarro D, Navarro I, Martin-Cordero C, Chalcones as Promising Lead Compounds on Cancer Therapy, Curr Med Chem, 22 (2015) 3407–3425. [DOI] [PubMed] [Google Scholar]
- [4].Singh P, Anand A, Kumar V, Recent developments in biological activities of chalcones: a mini review, Eur J Med Chem, 85 (2014) 758–777. [DOI] [PubMed] [Google Scholar]
- [5].Yadav VR, Prasad S, Sung B, Aggarwal BB, The role of chalcones in suppression of NF-kappaB-mediated inflammation and cancer, Int Immunopharmacol, 11 (2011) 295–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Karthikeyan C, Moorthy NS, Ramasamy S, Vanam U, Manivannan E, Karunagaran D, Trivedi P, Advances in chalcones with anticancer activities, Recent Pat Anticancer Drug Discov, 10 (2015) 97–115. [DOI] [PubMed] [Google Scholar]
- [7].Takahashi M, Maeda S, Ogura K, Terano A, Omata M, The possible role of vascular endothelial growth factor (VEGF) in gastric ulcer healing: effect of sofalcone on VEGF release in vitro, J Clin Gastroenterol, 27 Suppl 1 (1998) S178–182. [DOI] [PubMed] [Google Scholar]
- [8].Bukhari SN, Franzblau SG, Jantan I, Jasamai M, Current prospects of synthetic curcumin analogs and chalcone derivatives against mycobacterium tuberculosis, Med Chem, 9 (2013) 897–903. [DOI] [PubMed] [Google Scholar]
- [9].Dan W, Dai J, Recent developments of chalcones as potential antibacterial agents in medicinal chemistry, Eur J Med Chem, 187 (2020) 111980. [DOI] [PubMed] [Google Scholar]
- [10].Mahapatra DK, Bharti SK, Asati V, Chalcone Derivatives: Anti-inflammatory Potential and Molecular Targets Perspectives, Curr Top Med Chem, 17 (2017) 3146–3169. [DOI] [PubMed] [Google Scholar]
- [11].Dinkova-Kostova AT, Massiah MA, Bozak RE, Hicks RJ, Talalay P, Potency of Michael reaction acceptors as inducers of enzymes that protect against carcinogenesis depends on their reactivity with sulfhydryl groups, Proc Natl Acad Sci U S A, 98 (2001) 3404–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Maydt D, De Spirt S, Muschelknautz C, Stahl W, Muller TJ, Chemical reactivity and biological activity of chalcones and other alpha,beta-unsaturated carbonyl compounds, Xenobiotica, 43 (2013) 711–718. [DOI] [PubMed] [Google Scholar]
- [13].Zhuang C, Zhang W, Sheng C, Zhang W, Xing C, Miao Z, Chalcone: A Privileged Structure in Medicinal Chemistry, Chem Rev, 117 (2017) 7762–7810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Higuchi K, Watanabe T, Tanigawa T, Tominaga K, Fujiwara Y, Arakawa T, Sofalcone, a gastroprotective drug, promotes gastric ulcer healing following eradication therapy for Helicobacter pylori: a randomized controlled comparative trial with cimetidine, an H2-receptor antagonist, J Gastroenterol Hepatol, 25 Suppl 1 (2010) S155–160. [DOI] [PubMed] [Google Scholar]
- [15].Sahu NK, Balbhadra SS, Choudhary J, Kohli DV, Exploring pharmacological significance of chalcone scaffold: a review, Curr Med Chem, 19 (2012) 209–225. [DOI] [PubMed] [Google Scholar]
- [16].Jahng Y, Zhao LX, Moon YS, Basnet A, Kim EK, Chang HW, Ju HK, Jeong TC, Lee ES, Simple aromatic compounds containing propenone moiety show considerable dual COX/5-LOX inhibitory activities, Bioorg Med Chem Lett, 14 (2004) 2559–2562. [DOI] [PubMed] [Google Scholar]
- [17].Nielsen AT, Houlihan WJ, The Aldol Condensation, Org React, 16 (2011) 1–438. [Google Scholar]
- [18].Eddarir S, Cotelle N, Bakkour Y, Rolando C, An efficient synthesis of chalcones based on the Suzuki reaction, Tetrahedron Lett, 44 (2003) 5359–5363. [Google Scholar]
- [19].Shotter RG, Johnston KM, Jones JF, Reactions of unsaturated acid halides-IV1: Competitive friedel-crafts acylations and alkylations of monohalogenobenzenes by the bifunctional cinnamoyl chloride, Tetrahedron, 34 (1978) 741–746. [Google Scholar]
- [20].Xu C, Chen G, Huang X, Chalcones by the wittig reaction of a stable ylide with aldehydes under microwave irradiation, Org Prep Proced Int, 27 (1995) 559–561. [Google Scholar]
- [21].Ferrer JL, Jez JM, Bowman ME, Dixon RA, Noel JP, Structure of chalcone synthase and the molecular basis of plant polyketide biosynthesis, Nat Struct Biol, 6 (1999) 775–784. [DOI] [PubMed] [Google Scholar]
- [22].Lanz T, Tropf S, Marner FJ, Schroder J, Schroder G, The role of cysteines in polyketide synthases. Site-directed mutagenesis of resveratrol and chalcone synthases, two key enzymes in different plant-specific pathways, J Biol Chem, 266 (1991) 9971–9976. [PubMed] [Google Scholar]
- [23].Dao TT, Linthorst HJ, Verpoorte R, Chalcone synthase and its functions in plant resistance, Phytochem Rev, 10 (2011) 397–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Abe I, Morita H, Structure and function of the chalcone synthase superfamily of plant type III polyketide synthases, Nat Prod Rep, 27 (2010) 809–838. [DOI] [PubMed] [Google Scholar]
- [25].Austin MB, Noel JP, The chalcone synthase superfamily of type III polyketide synthases, Nat Prod Rep, 20 (2003) 79–110. [DOI] [PubMed] [Google Scholar]
- [26].Molitor C, Mauracher SG, Rompel A, Aurone synthase is a catechol oxidase with hydroxylase activity and provides insights into the mechanism of plant polyphenol oxidases, Proc Natl Acad Sci U S A, 113 (2016) E1806–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].A.s. Association, 2020 Alzheimer’s disease facts and figures, Alzheimers Dement, (2020). [DOI] [PubMed] [Google Scholar]
- [28].Hippius H, Neundorfer G, The discovery of Alzheimer’s disease, Dialogues Clin Neurosci, 5 (2003) 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Graham WV, Bonito-Oliva A, Sakmar TP, Update on Alzheimer’s Disease Therapy and Prevention Strategies, Annu Rev Med, 68 (2017) 413–430. [DOI] [PubMed] [Google Scholar]
- [30].Rogers SL, Doody RS, Mohs RC, Friedhoff LT, Donepezil improves cognition and global function in Alzheimer disease: a 15-week, double-blind, placebo-controlled study. Donepezil Study Group, Arch Intern Med, 158 (1998) 1021–1031. [DOI] [PubMed] [Google Scholar]
- [31].Sugimoto H, Donepezil hydrochloride: a treatment drug for Alzheimer’s disease, Chem Rec, 1 (2001) 63–73. [DOI] [PubMed] [Google Scholar]
- [32].Sugimoto H, Ogura H, Arai Y, Limura Y, Yamanishi Y, Research and development of donepezil hydrochloride, a new type of acetylcholinesterase inhibitor, Jpn J Pharmacol, 89 (2002) 7–20. [DOI] [PubMed] [Google Scholar]
- [33].Grossberg GT, Sadowsky C, Olin JT, Rivastigmine transdermal system for the treatment of mild to moderate Alzheimer’s disease, Int J Clin Pract, 64 (2010) 651–660. [DOI] [PubMed] [Google Scholar]
- [34].Articus K, Baier M, Tracik F, Kuhn F, Preuss UW, Kurz A, A 24-week, multicentre, open evaluation of the clinical effectiveness of the rivastigmine patch in patients with probable Alzheimer’s disease, Int J Clin Pract, 65 (2011) 790–796. [DOI] [PubMed] [Google Scholar]
- [35].Khoury R, Rajamanickam J, Grossberg GT, An update on the safety of current therapies for Alzheimer’s disease: focus on rivastigmine, Ther Adv Drug Saf, 9 (2018) 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Prvulovic D, Hampel H, Pantel J, Galantamine for Alzheimer’s disease, Expert Opin Drug Metab Toxicol, 6 (2010) 345–354. [DOI] [PubMed] [Google Scholar]
- [37].Raskind MA, Update on Alzheimer drugs (galantamine), Neurologist, 9 (2003) 235–240. [DOI] [PubMed] [Google Scholar]
- [38].Matsunaga S, Kishi T, Nomura I, Sakuma K, Okuya M, Ikuta T, Iwata N, The efficacy and safety of memantine for the treatment of Alzheimer’s disease, Expert Opin Drug Saf, 17 (2018) 1053–1061. [DOI] [PubMed] [Google Scholar]
- [39].Galimberti D, Scarpini E, Old and new acetylcholinesterase inhibitors for Alzheimer’s disease, Expert Opin Investig Drugs, 25 (2016) 1181–1187. [DOI] [PubMed] [Google Scholar]
- [40].Bartus RT, Dean RL, Beer B, Lippa AS, The cholinergic hypothesis of geriatric memory dysfunction, Science, 217 (1982) 408–414. [DOI] [PubMed] [Google Scholar]
- [41].Davies P, Maloney AJ, Selective loss of central cholinergic neurons in Alzheimer’s disease, Lancet, 2 (1976) 1403. [DOI] [PubMed] [Google Scholar]
- [42].Wu D, Hersh LB, Choline acetyltransferase: celebrating its fiftieth year, J Neurochem, 62 (1994) 1653–1663. [DOI] [PubMed] [Google Scholar]
- [43].Ferreira-Vieira TH, Guimaraes IM, Silva FR, Ribeiro FM, Alzheimer’s disease: Targeting the Cholinergic System, Curr Neuropharmacol, 14 (2016) 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Taylor P, Radic Z, The cholinesterases: from genes to proteins, Annu Rev Pharmacol Toxicol, 34 (1994) 281–320. [DOI] [PubMed] [Google Scholar]
- [45].Guyenet P, Lefresne P, Rossier J, Beaujouan JC, Glowinski J, Inhibition by hemicholinium-3 of (14C)acetylcholine synthesis and (3H)choline high-affinity uptake in rat striatal synaptosomes, Mol Pharmacol, 9 (1973) 630–639. [PubMed] [Google Scholar]
- [46].Greig NH, Reale M, Tata AM, New advances in pharmacological approaches to the cholinergic system: an overview on muscarinic receptor ligands and cholinesterase inhibitors, Recent Pat CNS Drug Discov, 8 (2013) 123–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sussman JL, Harel M, Frolow F, Oefner C, Goldman A, Toker L, Silman I, Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein, Science, 253 (1991) 872–879. [DOI] [PubMed] [Google Scholar]
- [48].Dvir H, Silman I, Harel M, Rosenberry TL, Sussman JL, Acetylcholinesterase: From 3D Structure to Function, Chem Biol Interact, 187 (2010) 10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Nachon F, Asojo OA, Borgstahl GEO, Masson P, Lockridge O, Role of Water in Aging of Human Butyrylcholinesterase Inhibited by Echothiophate: The Crystal Structure Suggests Two Alternative Mechanisms of Aging, Biochemistry, 44 (2005) 1154–1162. [DOI] [PubMed] [Google Scholar]
- [50].Bajda M, Wieckowska A, Hebda M, G. N., Sotriffer CA, Malawska B, Structure-based search for new inhibitors of cholinesterases, Int J Mol Sci, 14 (2013) 5608–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Saxena A, Redman AM, Jiang X, Lockridge O, Doctor BP, Differences in active site gorge dimensions of cholinesterase revealed by binding of inhibitors to human butyrylcholinesterase, Biochemistry, 36 (1997) 14642–14651. [DOI] [PubMed] [Google Scholar]
- [52].Greig NH, Lahiri DK, Sambamurti K, Butyrylcholinesterase: an important new target in Alzheimer’s disease therapy, Int Psychogeriatr, 14 (2002) 77–91. [DOI] [PubMed] [Google Scholar]
- [53].Darvesh S, Hopkins DA, Geula C, Neurobiology of butyrylcholinesterase, Nat Rev Neurosci, 4 (2003) 131–138. [DOI] [PubMed] [Google Scholar]
- [54].Perry EK, Perry RH, Blessed G, Tomlinson BE, Changes in brain cholinesterases in senile dementia of Alzhemeir type, Neuropathol Appl Neurobiol, 4 (1978) 273–277. [DOI] [PubMed] [Google Scholar]
- [55].Arendt T, Bruckner MK, Lange M, Bigl V, Changes in acetylcholinesterase and butyrylcholinesterase in Alzheimer’s disease resemble embryonic development--a study of molecular forms, Neurochem Int, 21 (1992) 381–396. [DOI] [PubMed] [Google Scholar]
- [56].Greig NH, Utsuki T, Ingram DK, Wang Y, Pepeu G, Scali C, Yu QS, Mamczarz J, Holloway HW, Giordano T, Chen D, Furukawa K, Sambamurti K, Brossi A, Lahiri DK, Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer β-amyloid peptide in rodent Proc. Natl. Acad. Sci. USA, 102 (2005) 17213–17218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Guillozet AL, Smiley JF, Mash DC, Mesulam MM, Butyrylcholinesterase in the life cycle of amyloid plaques, Ann Neurol, 42 (1997) 909–918. [DOI] [PubMed] [Google Scholar]
- [58].Mesulam MM, Geula C, Butyrylcholinesterase reactivity differentiates the amyloid plaques of aging from those of dementia, Ann Neurol, 36 (1994) 722–727. [DOI] [PubMed] [Google Scholar]
- [59].Andrisano v., Naldi M, Simone AD, Bartolini M, A patent review of butyrylcholinesterase inhibitors and reactivators 2010–2017, Expert Opin Ther Pat, 28 (2018) 455–465. [DOI] [PubMed] [Google Scholar]
- [60].Li Q, Yang H, Chen Y, Sun H, Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer’s disease, Eur J Med Chem, 132 (2017) 294–309. [DOI] [PubMed] [Google Scholar]
- [61].Cheung J, Rudolph MJ, Burshteyn F, Cassidy MS, Gary EN, Love J, Franklin MC, Height JJ, Structures of Human Acetylcholinesterase in Complex with Pharmacologically Important Ligands, J Med Chem, 55 (2012) 10282–10286. [DOI] [PubMed] [Google Scholar]
- [62].Nicolet Y, Lockridge O, Masson P, Fontecilla-Camps JC, Nachon F, Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products, J Biol Chem, 278 (2003) 41141–41147. [DOI] [PubMed] [Google Scholar]
- [63].Thamban Chandrika N, Fosso MY, Tsodikov OV, LeVine Iii H, Garneau-Tsodikova S, Combining Chalcones with Donepezil to Inhibit Both Cholinesterases and Abeta Fibril Assembly, Molecules, 25 (2019) 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Wang L, Wang Y, Tian Y, Shang J, Sun X, Chen H, Wang H, Tan W, Design, synthesis, biological evaluation, and molecular modeling studies of chalcone-rivastigmine hybrids as cholinesterase inhibitors, Bioorg Med Chem, 25 (2017) 360–371. [DOI] [PubMed] [Google Scholar]
- [65].Xiao G, Li Y, Qiang X, Xu R, Zheng Y, Cao Z, Luo L, Yang X, Sang Z, Su F, Deng Y, Design, synthesis and biological evaluation of 4’-aminochalcone-rivastigmine hybrids as multifunctional agents for the treatment of Alzheimer’s disease, Bioorg Med Chem, 25 (2017) 1030–1041. [DOI] [PubMed] [Google Scholar]
- [66].Di L, Kerns EH, Fan K, McConnell OJ, Carter GT, High throughput artificial membrane permeability assay for blood-brain barrier, Eur J Med Chem, 38 (2003) 223–232. [DOI] [PubMed] [Google Scholar]
- [67].Geldenhuys WJ, Allen DD, Bloomquist JR, Novel models for assessing blood-brain barrier drug permeation, Expert Opin Drug Metab Toxicol, 8 (2012) 647–653. [DOI] [PubMed] [Google Scholar]
- [68].Liu HR, Liu XJ, Fan HQ, Tang JJ, Gao XH, Liu WK, Design, synthesis and pharmacological evaluation of chalcone derivatives as acetylcholinesterase inhibitors, Bioorg Med Chem, 22 (2014) 6124–6133. [DOI] [PubMed] [Google Scholar]
- [69].Liu HR, Zhou C, Fan HQ, Tang JJ, Liu LB, Gao XH, Wang QA, Liu WK, Novel Potent and Selective Acetylcholinesterase Inhibitors as Potential Drugs for the Treatment of Alzheimer’s Disease: Synthesis, Pharmacological Evaluation, and Molecular Modeling of Amino-Alkyl-Substituted Fluoro-Chalcones Derivatives, Chem Biol Drug Des, 86 (2015) 517–522. [DOI] [PubMed] [Google Scholar]
- [70].Gao XH, Zhou C, Liu HR, Liu LB, Tang JJ, Xia XH, Tertiary amine derivatives of chlorochalcone as acetylcholinesterase (AChE) and buthylcholinesterase (BuChE) inhibitors: the influence of chlorine, alkyl amine side chain and alpha,beta-unsaturated ketone group, J Enzyme Inhib Med Chem, 32 (2017) 146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Liu H, Fan H, Gao X, Huang X, Liu X, Liu L, Zhou C, Tang J, Wang Q, Liu W, Design, synthesis and preliminary structure-activity relationship investigation of nitrogen-containing chalcone derivatives as acetylcholinesterase and butyrylcholinesterase inhibitors: a further study based on Flavokawain B Mannich base derivatives, J Enzyme Inhib Med Chem, 31 (2016) 580–589. [DOI] [PubMed] [Google Scholar]
- [72].Burmaoglu S, Yilmaz AO, Polat MF, Kaya R, Gulcin I, Algul O, Synthesis and biological evaluation of novel tris-chalcones as potent carbonic anhydrase, acetylcholinesterase, butyrylcholinesterase and alpha-glycosidase inhibitors, Bioorg Chem, 85 (2019) 191–197. [DOI] [PubMed] [Google Scholar]
- [73].Rampa A, Montanari S, Pruccoli L, Bartolini M, Falchi F, Feoli A, Cavalli A, Belluti F, Gobbi S, Tarozzi A, Bisi A, Chalcone-based carbamates for Alzheimer’s disease treatment, Future Med Chem, 9 (2017) 749–764. [DOI] [PubMed] [Google Scholar]
- [74].Zhao FC, Wu Y, Song XJ, Design and Development of a Novel Chalcone Derivative as an Anticholinesterase Inhibitor for Possible Treatment of Dementia, Med Sci Monit, 23 (2017) 3311–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Bai P, Wang K, Zhang P, Shi J, Cheng X, Zhang Q, Zheng C, Cheng Y, Yang J, Lu X, Sang Z, Development of chalcone-O-alkylamine derivatives as multifunctional agents against Alzheimer’s disease, Eur J Med Chem, 183 (2019) 111737. [DOI] [PubMed] [Google Scholar]
- [76].Wang M, Qin HL, Leng J, Ameeduzzafar, Amjad MW, Raja MAG, Hussain MA, Bukhari SNA, Synthesis and biological evaluation of new tetramethylpyrazine-based chalcone derivatives as potential anti-Alzheimer agents, Chem Biol Drug Des, 92 (2018) 1859–1866. [DOI] [PubMed] [Google Scholar]
- [77].Shah MS, Najam-Ul-Haq M, Shah HS, Farooq Rizvi SU, Iqbal J, Quinoline containing chalcone derivatives as cholinesterase inhibitors and their in silico modeling studies, Comput Biol Chem, 76 (2018) 310–317. [DOI] [PubMed] [Google Scholar]
- [78].Shah MS, Khan SU, Ejaz SA, Afridi S, Rizvi SUF, Najam-Ul-Haq M, Iqbal J, Cholinesterases inhibition and molecular modeling studies of piperidyl-thienyl and 2-pyrazoline derivatives of chalcones, Biochem Biophys Res Commun, 482 (2017) 615–624. [DOI] [PubMed] [Google Scholar]
- [79].Kocyigit UM, Budak Y, Gurdere MB, Erturk F, Yencilek B, Taslimi P, Gulcin I, Ceylan M, Synthesis of chalcone-imide derivatives and investigation of their anticancer and antimicrobial activities, carbonic anhydrase and acetylcholinesterase enzymes inhibition profiles, Arch Physiol Biochem, 124 (2018) 61–68. [DOI] [PubMed] [Google Scholar]
- [80].Kang L, Gao XH, Liu HR, Men X, Wu HN, Cui PW, Oldfield E, Yan JY, Structure-activity relationship investigation of coumarin-chalcone hybrids with diverse side-chains as acetylcholinesterase and butyrylcholinesterase inhibitors, Mol Divers, 22 (2018) 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Fosso MY, LeVine H 3rd, Green KD, Tsodikov OV, Garneau-Tsodikova S, Effects of structural modifications on the metal binding, anti-amyloid activity, and cholinesterase inhibitory activity of chalcones, Org Biomol Chem, 13 (2015) 9418–9426. [DOI] [PubMed] [Google Scholar]
- [82].Sheng R, Xu Y, Hu C, Zhang J, Lin X, Li J, Yang B, He Q, Hu Y, Design, synthesis and AChE inhibitory activity of indanone and aurone derivatives, Eur J Med Chem, 44 (2009) 7–17. [DOI] [PubMed] [Google Scholar]
- [83].Rizzo S, Bartolini M, Ceccarini L, Piazzi L, Gobbi S, Cavalli A, Recanatini M, Andrisano V, Rampa A, Targeting Alzheimer’s disease: Novel indanone hybrids bearing a pharmacophoric fragment of AP2238, Bioorg Med Chem, 18 (2010) 1749–1760. [DOI] [PubMed] [Google Scholar]
- [84].Belluti F, Rampa A, Piazzi L, Bisi A, Gobbi S, Bartolini M, Andrisano V, Cavalli A, Recanatini M, Valenti P, Cholinesterase inhibitors: xanthostigmine derivatives blocking the acetylcholinesterase-induced beta-amyloid aggregation, J Med Chem, 48 (2005) 4444–4456. [DOI] [PubMed] [Google Scholar]
- [85].Huang L, Miao H, Sun Y, Meng F, Li X, Discovery of indanone derivatives as multi-target-directed ligands against Alzheimer’s disease, Eur J Med Chem, 87 (2014) 429–439. [DOI] [PubMed] [Google Scholar]
- [86].Mishra CB, Kumari S, Manral A, Prakash A, Saini V, Lynn AM, Tiwari M, Design, synthesis, in-silico and biological evaluation of novel donepezil derivatives as multi-target-directed ligands for the treatment of Alzheimer’s disease, Eur J Med Chem, 125 (2017) 736–750. [DOI] [PubMed] [Google Scholar]
- [87].Chierrito TPC, Pedersoli-Mantoani S, Roca C, Requena C, Sebastian-Perez V, Castillo WO, Moreira NCS, Perez C, Sakamoto-Hojo ET, Takahashi CS, Jimenez-Barbero J, Canada FJ, Campillo NE, Martinez A, Carvalho I, From dual binding site acetylcholinesterase inhibitors to allosteric modulators: A new avenue for disease-modifying drugs in Alzheimer’s disease, Eur J Med Chem, 139 (2017) 773–791. [DOI] [PubMed] [Google Scholar]
- [88].Meng FC, Mao F, Shan WJ, Qin F, Huang L, Li XS, Design, synthesis, and evaluation of indanone derivatives as acetylcholinesterase inhibitors and metal-chelating agents, Bioorg Med Chem Lett, 22 (2012) 4462–4466. [DOI] [PubMed] [Google Scholar]
- [89].Akrami H, Mirjalili BF, Khoobi M, Nadri H, Moradi A, Sakhteman A, Emami S, Foroumadi A, Shafiee A, Indolinone-based acetylcholinesterase inhibitors: synthesis, biological activity and molecular modeling, Eur J Med Chem, 84 (2014) 375–381. [DOI] [PubMed] [Google Scholar]
- [90].Arab S, Sadat-Ebrahimi SE, Mohammadi-Khanaposhtani M, Moradi A, Nadri H, Mahdavi M, Moghimi S, Asadi M, Firoozpour L, Pirali-Hamedani M, Shafiee A, Foroumadi A, Synthesis and Evaluation of Chroman-4-One Linked to N-Benzyl Pyridinium Derivatives as New Acetylcholinesterase Inhibitors, Arch Pharm (Weinheim), 348 (2015) 643–649. [DOI] [PubMed] [Google Scholar]
- [91].Nadri H, Pirali-Hamedani M, Shekarchi M, Abdollahi M, Sheibani V, Amanlou M, Shafiee A, Foroumadi A, Design, synthesis and anticholinesterase activity of a novel series of 1-benzyl-4-((6-alkoxy-3-oxobenzofuran-2(3H)-ylidene) methyl) pyridinium derivatives, Bioorg Med Chem, 18 (2010) 6360–6366. [DOI] [PubMed] [Google Scholar]
- [92].Nadri H, Pirali-Hamedani M, Moradi A, Sakhteman A, Vahidi A, Sheibani V, Asadipour A, Hosseinzadeh N, Abdollahi M, Shafiee A, Foroumadi A, 5,6-Dimethoxybenzofuran-3-one derivatives: a novel series of dual Acetylcholinesterase/Butyrylcholinesterase inhibitors bearing benzyl pyridinium moiety, Daru, 21 (2013) 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Pourshojaei Y, Gouranourimi A, Hekmat S, Asadipour A, Rahmani-Nezhad S, Moradi A, Nadri H, Moghadam FH, Emami S, Foroumadi A, Shafiee A, Design, synthesis and anticholinesterase activity of novel benzylidenechroman-4-ones bearing cyclic amine side chain, Eur J Med Chem, 97 (2015) 181–189. [DOI] [PubMed] [Google Scholar]
- [94].Wang C, Wu Z, Cai H, Xu S, Liu J, Jiang J, Yao H, Wu X, Xu J, Design, synthesis, biological evaluation and docking study of 4-isochromanone hybrids bearing N-benzyl pyridinium moiety as dual binding site acetylcholinesterase inhibitors, Bioorg Med Chem Lett, 25 (2015) 5212–5216. [DOI] [PubMed] [Google Scholar]
- [95].Lee YH, Shin MC, Yun YD, Shin SY, Kim JM, Seo JM, Kim NJ, Ryu JH, Lee YS, Synthesis of aminoalkyl-substituted aurone derivatives as acetylcholinesterase inhibitors, Bioorg Med Chem, 23 (2015) 231–240. [DOI] [PubMed] [Google Scholar]
- [96].Hardy J, Allsop D, Amyloid deposition as the central event in the aetiology of Alzheimer’s disease, Trends Pharmacol Sci, 12 (1991) 383–388. [DOI] [PubMed] [Google Scholar]
- [97].Hardy J, Selkoe DJ, The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics, Science, 297 (2002) 353–356. [DOI] [PubMed] [Google Scholar]
- [98].Selkoe DJ, Hardy J, The amyloid hypothesis of Alzheimer’s disease at 25 years, EMBO Mol Med, 8 (2016) 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M, Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE, Science, 286 (1999) 735–741. [DOI] [PubMed] [Google Scholar]
- [100].Golde TE, The pathogenesis of Alzheimer’s disease and the role of Abeta42, CNS Spectr, 12 (2007) 4–6. [DOI] [PubMed] [Google Scholar]
- [101].Haass C, Kaether C, Thinakaran G, Sisodia S, Trafficking and proteolytic processing of APP, Cold Spring Harb Perspect Med, 2 (2012) a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, Vassar R, Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation, Nat Neurosci, 4 (2001) 231–232. [DOI] [PubMed] [Google Scholar]
- [103].Hong L, Koelsch G, Lin X, Wu S, Terzyan S, Ghosh AK, Zhang XC, Tang J, Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor, Science, 290 (2000) 150–153. [DOI] [PubMed] [Google Scholar]
- [104].Scott JD, Li SW, Brunskill APJ, Chen X, Cox K, Cumming JN, Forman M, Gilbert EJ, Hodgson RA, Hyde LA, Jiang Q, Iserloh U, Kazakevich I, Kuvelkar R, Mei H, Meredith J, Misiaszek J, Orth P, Rossiter LM, Slater M, Stone J, Strickland CO, Voigt JH, Wang G, Wang H, Wu Y, Greenlee WJ, Parker EM, Kennedy ME, Stamford AW, Discovery of the 3-Imino-1,2,4-thiadiazinane 1,1-Dioxide Derivative Verubecestat (MK-8931)-A beta-Site Amyloid Precursor Protein Cleaving Enzyme 1 Inhibitor for the Treatment of Alzheimer’s Disease, J Med Chem, 59 (2016) 10435–10450. [DOI] [PubMed] [Google Scholar]
- [105].Kennedy ME, Stamford AW, Chen X, Cox K, Cumming JN, Dockendorf MF, Egan M, Ereshefsky L, Hodgson RA, Hyde LA, Jhee S, Kleijn HJ, Kuvelkar R, Li W, Mattson BA, Mei H, Palcza J, Scott JD, Tanen M, Troyer MD, Tseng JL, Stone JA, Parker EM, Forman MS, The BACE1 inhibitor verubecestat (MK-8931) reduces CNS beta-amyloid in animal models and in Alzheimer’s disease patients, Sci Transl Med, 8 (2016) 363ra150. [DOI] [PubMed] [Google Scholar]
- [106].Neumann U, Ufer M, Jacobson LH, Rouzade-Dominguez ML, Huledal G, Kolly C, Luond RM, Machauer R, Veenstra SJ, Hurth K, Rueeger H, Tintelnot-Blomley M, Staufenbiel M, Shimshek DR, Perrot L, Frieauff W, Dubost V, Schiller H, Vogg B, Beltz K, Avrameas A, Kretz S, Pezous N, Rondeau JM, Beckmann N, Hartmann A, Vormfelde S, David OJ, Galli B, Ramos R, Graf A, Lopez Lopez C, The BACE-1 inhibitor CNP520 for prevention trials in Alzheimer’s disease, EMBO Mol Med, 10 (2018) e9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Ye N, Monk SA, Daga P, Bender DM, Rosen LB, Mullen J, Minkwitz MC, Kugler AR, Clinical Bioavailability of the Novel BACE1 Inhibitor Lanabecestat (AZD3293): Assessment of Tablet Formulations Versus an Oral Solution and the Impact of Gastric pH on Pharmacokinetics, Clin Pharmacol Drug Dev, 7 (2018) 233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Timmers M, Streffer JR, Russu A, Tominaga Y, Shimizu H, Shiraishi A, Tatikola K, Smekens P, Borjesson-Hanson A, Andreasen N, Matias-Guiu J, Baquero M, Boada M, Tesseur I, Tritsmans L, Nueten LV, Engelborghs S, Pharmacodynamics of atabecestat (JNJ-54861911), an oral BACE1 inhibitor in patients with early Alzheimer’s disease: randomized, double-blind, placebo-controlled study, Alzheimers Res Ther, 10 (2018) 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Das B, Yan R, A Close Look at BACE1 Inhibitors for Alzheimer’s Disease Treatment, CNS Drugs, 33 (2019) 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Youn K, Jun M, Biological Evaluation and Docking Analysis of Potent BACE1 Inhibitors from Boesenbergia rotunda, Nutrients, 11 (2019) 662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Zhu Z, Li C, Wang X, Yang Z, Chen J, Hu L, Jiang H, Shen X, 2,2’,4’-trihydroxychalcone from Glycyrrhiza glabra as a new specific BACE1 inhibitor efficiently ameliorates memory impairment in mice, J Neurochem, 114 (2010) 374–385. [DOI] [PubMed] [Google Scholar]
- [112].Rampa A, Tarozzi A, Mancini F, Pruccoli L, Di Martino RM, Gobbi S, Bisi A, De Simone A, Palomba F, Zaccheroni N, Belluti F, Naturally Inspired Molecules as Multifunctional Agents for Alzheimer’s Disease Treatment, Molecules, 21 (2016) 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Ma L, Yang Z, Li C, Zhu Z, Shen X, Hu L, Design, synthesis and SAR study of hydroxychalcone inhibitors of human beta-secretase (BACE1), J Enzyme Inhib Med Chem, 26 (2011) 643–648. [DOI] [PubMed] [Google Scholar]
- [114].Mphahlele MJ, Agbo EN, Gildenhuys S, Synthesis and Evaluation of the 4-Substituted 2-Hydroxy-5-Iodochalcones and Their 7-Substituted 6-Iodoflavonol Derivatives for Inhibitory Effect on Cholinesterases and beta-Secretase, Int J Mol Sci, 19 (2018) 4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Costanzo P, Cariati L, Desiderio D, Sgammato R, Lamberti A, Arcone R, Salerno R, Nardi M, Masullo M, Oliverio M, Design, Synthesis, and Evaluation of Donepezil-Like Compounds as AChE and BACE-1 Inhibitors, ACS Med Chem Lett, 7 (2016) 470–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Gabr MT, Abdel-Raziq MS, Structure-based design, synthesis, and evaluation of structurally rigid donepezil analogues as dual AChE and BACE-1 inhibitors, Bioorg Med Chem Lett, 28 (2018) 2910–2913. [DOI] [PubMed] [Google Scholar]
- [117].Estrada LD, Soto C, Disrupting beta-amyloid aggregation for Alzheimer disease treatment, Curr Top Med Chem, 7 (2007) 115–126. [DOI] [PubMed] [Google Scholar]
- [118].Nie Q, Du XG, Geng MY, Small molecule inhibitors of amyloid beta peptide aggregation as a potential therapeutic strategy for Alzheimer’s disease, Acta Pharmacol Sin, 32 (2011) 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Whitty A, Kumaravel G, Between a rock and a hard place?, Nat Chem Biol, 2 (2006) 112–118. [DOI] [PubMed] [Google Scholar]
- [120].Maji SK, Ogorzalek Loo RR, Inayathullah M, Spring SM, Vollers SS, Condron MM, Bitan G, Loo JA, Teplow DB, Amino acid position-specific contributions to amyloid beta-protein oligomerization, J Biol Chem, 284 (2009) 23580–23591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Hilbich C, Kisters-Woike B, Reed J, Masters CL, Beyreuther K, Substitutions of hydrophobic amino acids reduce the amyloidogenicity of Alzheimer’s disease beta A4 peptides, J Mol Biol, 228 (1992) 460–473. [DOI] [PubMed] [Google Scholar]
- [122].Serpell LC, Alzheimer’s amyloid fibrils: structure and assembly, Biochim Biophys Acta, 1502 (2000) 16–30. [DOI] [PubMed] [Google Scholar]
- [123].Jarrett JT, Berger EP, Lansbury PT Jr., The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease, Biochemistry, 32 (1993) 4693–4697. [DOI] [PubMed] [Google Scholar]
- [124].Liu Y, Xu LP, Wang Q, Yang B, Zhang X, Synergistic Inhibitory Effect of GQDs-Tramiprosate Covalent Binding on Amyloid Aggregation, ACS Chem Neurosci, 9 (2018) 817–823. [DOI] [PubMed] [Google Scholar]
- [125].Nakagami Y, Nishimura S, Murasugi T, Kaneko I, Meguro M, Marumoto S, Kogen H, Koyama K, Oda T, A novel beta-sheet breaker, RS-0406, reverses amyloid beta-induced cytotoxicity and impairment of long-term potentiation in vitro, Br J Pharmacol, 137 (2002) 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Ma K, Thomason LA, McLaurin J, scyllo-Inositol, preclinical, and clinical data for Alzheimer’s disease, Adv Pharmacol, 64 (2012) 177–212. [DOI] [PubMed] [Google Scholar]
- [127].Dong J, Canfield JM, Mehta AK, Shokes JE, Tian B, Childers WS, Simmons JA, Mao Z, Scott RA, Warncke K, Lynn DG, Engineering metal ion coordination to regulate amyloid fibril assembly and toxicity, Proc Natl Acad Sci U S A, 104 (2007) 13313–13318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Hane F, Leonenko Z, Effect of metals on kinetic pathways of amyloid-beta aggregation, Biomolecules, 4 (2014) 101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A, Bedo J, Harrison J, Lannfelt L, Blennow K, Zetterberg H, Ingelsson M, Masters CL, Tanzi RE, Cummings JL, Herd CM, Bush AI, PBT2 rapidly improves cognition in Alzheimer’s Disease: additional phase II analyses, J Alzheimers Dis, 20 (2010) 509–516. [DOI] [PubMed] [Google Scholar]
- [130].Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM, Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo, J Biol Chem, 280 (2005) 5892–5901. [DOI] [PubMed] [Google Scholar]
- [131].Hirohata M, Hasegawa K, Tsutsumi-Yasuhara S, Ohhashi Y, Ookoshi T, Ono K, Yamada M, Naiki H, The anti-amyloidogenic effect is exterted against alzheimer’s β-amyloid fibrils in vitro by preferential and reversible binding of flavonoids to the amyloid fibril structure, Biochemistry, 46 (2007) 1888–1899. [DOI] [PubMed] [Google Scholar]
- [132].Liew KF, Lee EHC, Chan KL, Lee CY, Multi-targeting aurones with monoamine oxidase and amyloid-beta inhibitory activities: Structure-activity relationship and translating multi-potency to neuroprotection, Biomed Pharmacother, 110 (2019). [DOI] [PubMed] [Google Scholar]
- [133].Cong L, Dong X, Wang Y, Deng Y, Li B, Dai R, On the role of synthesized hydroxylated chalcones as dual functional amyloid-β aggregation and ferroptosis inhibitors for potential treatment of Alzheimer’s disease, Eur J Med Chem, 166 (2019) 11–21. [DOI] [PubMed] [Google Scholar]
- [134].Wang K, Yu L, Shi J, Liu W, Sang Z, Multifuntional indanone-chalcone hybrid compounds with anti-β-amyloid (Aβ) aggregation, monoamine oxidase B (MAO-B) inhibition and neuroprotective properties against Alzheimer’s disease, Med Chem Res, 28 (2019) 1912–1922. [Google Scholar]
- [135].Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW, A protein factor essential for microtubule assembly, Proc Natl Acad Sci U S A, 72 (1975) 1858–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI, Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology, Proc Natl Acad Sci U S A, 83 (1986) 4913–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Wen Y, Yang SH, Liu R, Perez EJ, Brun-Zinkernagel AM, Koulen P, Simpkins JW, Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats, Biochim. Biophys. Acta, 1772 (2007) 473–483. [DOI] [PubMed] [Google Scholar]
- [138].Kimura T, Ishiguro K, Hisanaga S, Physiological and pathological phosphorylation of tau by Cdk5, Front Mol Neurosci, 7 (2014) 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Shukla V, Skuntz S, Pant HC, Deregulated Cdk5 activity is involved in inducing Alzheimer’s disease, Arch Med Res, 43 (2012) 655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Rankin CA, Sun Q, Gamblin TC, Tau phosphorylation by GSK-3beta promotes tangle-like filament morphology, Mol Neurodegener, 2 (2007) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Jin N, Yin X, Yu D, Cao M, Gong CX, Iqbal K, Ding F, Gu X, Liu F, Truncation and activation of GSK-3beta by calpain I: a molecular mechanism links to tau hyperphosphorylation in Alzheimer’s disease, Sci Rep, 5 (2015) 8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Hanger DP, Noble W, Functional implications of glycogen synthase kinase-3-mediated tau phosphorylation, Int J Alzheimers Dis, 2011 (2011) 352805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Bekris LM, Millard S, Lutz F, Li G, Galasko DR, Farlow MR, Quinn JF, Kaye JA, Leverenz JB, Tsuang DW, Yu CE, Peskind ER, Tau phosphorylation pathway genes and cerebrospinal fluid tau levels in Alzheimer’s disease, Am J Med Genet B Neuropsychiatr Genet, 159B (2012) 874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Nygaard HB, van Dyck CH, Strittmatter SM, Fyn kinase inhibition as a novel therapy for Alzheimer’s disease, Alzheimers Res Ther, 6 (2014) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Ploia C, Antoniou X, Sclip A, Grande V, Cardinetti D, Colombo A, Canu N, Benussi L, Ghidoni R, Forloni G, Borsello T, JNK plays a key role in tau hyperphosphorylation in Alzheimer’s disease models, J Alzheimers Dis, 26 (2011) 315–329. [DOI] [PubMed] [Google Scholar]
- [146].Vogel J, Anand VS, Ludwig B, Nawoschik S, Dunlop J, Braithwaite SP, The JNK pathway amplifies and drives subcellular changes in tau phosphorylation, Neuropharmacology, 57 (2009) 539–550. [DOI] [PubMed] [Google Scholar]
- [147].Choi H, Kim HJ, Yang J, Chae S, Lee W, Chung S, Kim J, Choi H, Song H, Lee CK, Jun JH, Lee YJ, Lee K, Kim S, Sim HR, Choi YI, Ryu KH, Park JC, Lee D, Han SH, Hwang D, Kyung J, Mook-Jung I, Acetylation changes tau interactome to degrade tau in Alzheimer’s disease animal and organoid models, Aging Cell, 19 (2020) e13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, Meyers D, Cole PA, Ott M, Gan L, Acetylation of tau inhibits its degradation and contributes to tauopathy, Neuron, 67 (2010) 953–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Wang JZ, Grundke-Iqbal I, Iqbal K, Glycosylation of microtubule-associated protein tau: an abnormal posttranslational modification in Alzheimer’s disease, Nat Med, 2 (1996) 871–875. [DOI] [PubMed] [Google Scholar]
- [150].Hyman BT, Augustinack JC, Ingelsson M, Transcriptional and conformational changes of the tau molecule in Alzheimer’s disease, Biochim. Biophys. Acta, 1739 (2005) 150–157. [DOI] [PubMed] [Google Scholar]
- [151].Medina M, An Overview on the Clinical Development of Tau-Based Therapeutics, Int J Mol Sci, 19 (2018) 1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Congdon EE, Sigurdsson EM, Tau-targeting therapies for Alzheimer disease, Nat Rev Neurol, 14 (2018) 399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Dominguez JM, Fuertes A, Orozco L, del Monte-Millan M, Delgado E, Medina M, Evidence for irreversible inhibition of glycogen synthase kinase-3beta by tideglusib, J Biol Chem, 287 (2012) 893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Martinez A, Alonso M, Castro A, Perez C, Moreno FJ, First non-ATP competitive glycogen synthase kinase 3 beta (GSK-3beta) inhibitors: thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer’s disease, J Med Chem, 45 (2002) 1292–1299. [DOI] [PubMed] [Google Scholar]
- [155].Qureshi IA, Tirucherai G, Ahlijanian MK, Kolaitis G, Bechtold C, Grundman M, A randomized, single ascending dose study of intravenous BIIB092 in healthy participants, Alzheimers Dement (N Y), 4 (2018) 746–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].George RC, Lew J, Graves DJ, Interaction of cinnamaldehyde and epicatechin with tau: implications of beneficial effects in modulating Alzheimer’s disease pathogenesis, J Alzheimers Dis, 36 (2013) 21–40. [DOI] [PubMed] [Google Scholar]
- [157].Zhang M, Wu Q, Yao X, Zhao J, Zhong W, Liu Q, Xiao S, Xanthohumol inhibits tau protein aggregation and protects cells against tau aggregates, Food Funct, 10 (2019) 7865–7874. [DOI] [PubMed] [Google Scholar]
- [158].Sonawane SK, Balmik AA, Boral D, Ramasamy S, Chinnathambi S, Baicalein suppresses repeat tau fibrillization by sequestering oligomers, Arch Biochem Biophys, 675 (2019) 108119. [DOI] [PubMed] [Google Scholar]
- [159].Lin TH, Chiu YJ, Lin CH, Lin CY, Chao CY, Chen YC, Yang SM, Lin W, Mei Hsieh-Li H, Wu YR, Chang KH, Lee-Chen GJ, Chen CM, Exploration of multi-target effects of 3-benzoyl-5-hydroxychromen-2-one in Alzheimer’s disease cell and mouse models, Aging Cell, 19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Shimura H, Miura-Shimura Y, Kosik KS, Binding of tau to heat shock protein 27 leads to decreased concentration of hyperphosphorylated tau and enhanced cell survival, J Biol Chem, 279 (2004) 17957–17962. [DOI] [PubMed] [Google Scholar]
- [161].Di Martino RM, De Simone A, Andrisano V, Bisignano P, Bisi A, Gobbi S, Rampa A, Fato R, Bergamini C, Perez DI, Martinez A, Bottegoni G, Cavalli A, Belluti F, Versatility of the Curcumin Scaffold: Discovery of Potent and Balanced Dual BACE-1 and GSK-3beta Inhibitors, J Med Chem, 59 (2016) 531–544. [DOI] [PubMed] [Google Scholar]
- [162].Jeon KH, Lee E, Jun KY, Eom JE, Kwak SY, Na Y, Kwon Y, Neuroprotective effect of synthetic chalcone derivatives as competitive dual inhibitors against mu-calpain and cathepsin B through the downregulation of tau phosphorylation and insoluble Abeta peptide formation, Eur J Med Chem, 121 (2016) 433–444. [DOI] [PubMed] [Google Scholar]
- [163].Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S, Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25, J Biol Chem, 275 (2000) 17166–17172. [DOI] [PubMed] [Google Scholar]
- [164].Liu PP, Xie Y, Meng XY, Kang JS, History and progress of hypotheses and clinical trials for Alzheimer’s disease, Signal Transduct Target Ther, 4 (2019) 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT, Inflammation as a central mechanism in Alzheimer’s disease, Alzheimers Dement (N Y), 4 (2018) 575–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Gomez-Nicola D, Boche D, Post-mortem analysis of neuroinflammatory changes in human Alzheimer’s disease, Alzheimers Res Ther, 7 (2015) 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Walters A, Phillips E, Zhen R, Biju M, Kuruvilla T, Evidence for neuroinflammation in Alzheimer’s disease, Prog Neurol Psychiatry, 20 (2016) 25–31. [Google Scholar]
- [168].Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, Shapiro A, Antel JP, TLR signaling tailors innate immune response in human microglia and astrocytes, J Immunol, 175 (2005) 4320–4330. [DOI] [PubMed] [Google Scholar]
- [169].Doens D, Fernandez PL, Microglia receptors and their implications in the response to amyloid beta for Alzheimer’s disease pathogenesis, J Neuroinflammation, 11 (2014) 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Zhong L, Wang Z, Wang D, Wang Z, Martens YA, Wu L, Xu Y, Wang K, Li J, Huang R, Can D, Xu H, Bu G, Chen XF, Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2), Mol Neurodegener, 13 (2018) 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Hanisch UK, Microglia as a source and target of cytokines, Glia, 40 (2002) 140–155. [DOI] [PubMed] [Google Scholar]
- [172].Izquierdo P, Attwell D, Madry C, Ion Channels and Receptors as Determinants of Microglial Function, Trends Neurosci, 42 (2019) 278–292. [DOI] [PubMed] [Google Scholar]
- [173].Wang WY, Tan MS, Yu JT, Tan L, Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease, Ann Transl Med, 3 (2015) 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Poudel B, Gurung P, An update on cell intrinsic negative regulators of the NLRP3 inflammasome, J Leukoc Biol, 26 (2018) 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Place DE, Kanneganti TD, Recent Advances In Inflammasome Biology, Curr Opin Immunol, 50 (2018) 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Yang Y, Wang H, Kouadir M, Song H, Shi F, Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors, Cell Death Dis, 10 (2019) 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT, The NALP3 inflammasome is involved in the innate immune response to amyloid-beta, Nat Immunol, 9 (2008) 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT, NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice, Nature, 493 (2013) 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM, Tejera D, Griep A, Santarelli F, Brosseron F, Opitz S, Stunden J, Merten M, Kayed R, Golenbock DT, Blum D, Latz E, Buee L, Heneka MT, NLRP3 inflammasome activation drives tau pathology, Nature, 575 (2019) 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Lonnemann N, Hosseini S, Marchetti C, Skouras DB, Stefanoni D, D’Alessandro A, Dinarello CA, Korte M, The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease, Proc Natl Acad Sci U S A, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Dempsey C, Rubio Araiz A, Bryson KJ, Finucane O, Larkin C, Mills EL, Robertson AAB, Cooper MA, O’Neill LAJ, Lynch MA, Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-beta and cognitive function in APP/PS1 mice, Brain Behav Immun, 61 (2017) 306–316. [DOI] [PubMed] [Google Scholar]
- [182].Sharma K, Cholinesterase inhibitors as Alzheimer’s therapeutics (Review), Mol Med Rep, 20 (2019) 1479–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K, Alzheimer’s disease drug development pipeline: 2019, Alzheimers Dement (N Y), 5 (2019) 272–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K, Alzheimer’s disease drug development pipeline: 2020, Alzheimers Dement (N Y), 6 (2020) e12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Yamamoto Y, Gaynor RB, IkappaB kinases: key regulators of the NF-kappaB pathway, Trends Biochem Sci, 29 (2004) 72–79. [DOI] [PubMed] [Google Scholar]
- [186].Liu T, Zhang L, Joo D, Sun SC, NF-kappaB signaling in inflammation, Signal Transduct Target Ther, 2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Hayden MS, Ghosh S, NF-kappaB in immunobiology, Cell Res, 21 (2011) 223–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Pandey MK, Sandur SK, Sung B, Sethi G, Kunnumakkara AB, Aggarwal BB, Butein, a tetrahydroxychalcone, inhibits nuclear factor (NF)-kappaB and NF-kappaB-regulated gene expression through direct inhibition of IkappaBalpha kinase beta on cysteine 179 residue, J Biol Chem, 282 (2007) 17340–17350. [DOI] [PubMed] [Google Scholar]
- [189].Chu X, Ci X, Wei M, Yang X, Cao Q, Guan M, Li H, Deng Y, Feng H, Deng X, Licochalcone a inhibits lipopolysaccharide-induced inflammatory response in vitro and in vivo, J Agric Food Chem, 60 (2012) 3947–3954. [DOI] [PubMed] [Google Scholar]
- [190].Hu J, Liu J, Licochalcone A Attenuates Lipopolysaccharide-Induced Acute Kidney Injury by Inhibiting NF-kappaB Activation, Inflammation, 39 (2016) 569–574. [DOI] [PubMed] [Google Scholar]
- [191].Zhu X, Liu J, Chen S, Xue J, Huang S, Wang Y, Chen O, Isoliquiritigenin attenuates lipopolysaccharide-induced cognitive impairment through antioxidant and anti-inflammatory activity, BMC Neurosci, 20 (2019) 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Honda H, Nagai Y, Matsunaga T, Okamoto N, Watanabe Y, Tsuneyama K, Hayashi H, Fujii I, Ikutani M, Hirai Y, Muraguchi A, Takatsu K, Isoliquiritigenin is a potent inhibitor of NLRP3 inflammasome activation and diet-induced adipose tissue inflammation, J Leukoc Biol, 96 (2014) 1087–1100. [DOI] [PubMed] [Google Scholar]
- [193].Xu QX, Hu Y, Li GY, Xu W, Zhang YT, Yang XW, Multi-Target Anti-Alzheimer Activities of Four Prenylated Compounds from Psoralea Fructus, Molecules, 23 (2018) 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Jiwrajka M, Phillips A, Butler M, Rossi M, Pocock JM, The Plant-Derived Chalcone 2,2’,5’-Trihydroxychalcone Provides Neuroprotection against Toll-Like Receptor 4 Triggered Inflammation in Microglia, Oxid Med Cell Longev, 2016 (2016) 6301712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Lee YH, Jeon SH, Kim SH, Kim C, Lee SJ, Koh D, Lim Y, Ha K, Shin SY, A new synthetic chalcone derivative, 2-hydroxy-3’,5,5’-trimethoxychalcone (DK-139), suppresses the Toll-like receptor 4-mediated inflammatory response through inhibition of the Akt/NF-kappaB pathway in BV2 microglial cells, Exp Mol Med, 44 (2012) 369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Mateeva N, Gangapuram M, Mazzio E, Eyunni S, Soliman KF, Redda KK, Biological evaluation of synthetic chalcone and flavone derivatives as anti-inflammatory agents, Med Chem Res, 24 (2015) 1672–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Gyengesi E, Munch G, In search of an anti-inflammatory drug for Alzheimer disease, Nat Rev Neurol, 16 (2020) 131–132. [DOI] [PubMed] [Google Scholar]
- [198].Zhang W, Bai Y, Wang Y, Xiao W, Polypharmacology in Drug Discovery: A Review from Systems Pharmacology Perspective, Curr Pharm Des, 22 (2016) 3171–3181. [DOI] [PubMed] [Google Scholar]
- [199].Ramsay RR, Popovic-Nikolic MR, Nikolic K, Uliassi E, Bolognesi ML, A perspective on multi-target drug discovery and design for complex diseases, Clin Transl Med, 7 (2018) 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Rosini M, Simoni E, Caporaso R, Minarini A, Multitarget strategies in Alzheimer’s disease: benefits and challenges on the road to therapeutics, Future Med Chem, 8 (2016) 697–711. [DOI] [PubMed] [Google Scholar]
- [201].Albertini C, Salerno A, de P Pinheiro Sena Murteira, Bolognesi ML, From combinations to multitarget-directed ligands: A continuum in Alzheimer’s disease polypharmacology, Med Res Rev, (2020). [DOI] [PubMed] [Google Scholar]
- [202].Csermely P, Agoston V, Pongor S, The efficiency of multi-target drugs: the network approach might help drug design, Trends Pharmacol Sci, 26 (2005) 178–182. [DOI] [PubMed] [Google Scholar]
- [203].Berube G, An overview of molecular hybrids in drug discovery, Expert Opin Drug Discov, 11 (2016) 281–305. [DOI] [PubMed] [Google Scholar]
- [204].Ament PW, Bertolino JG, Liszewski JL, Clinically significant drug interactions, Am Fam Physician, 61 (2000) 1745–1754. [PubMed] [Google Scholar]
- [205].Wang Y, Wang H, Chen HZ, AChE Inhibition-based Multi-target-directed Ligands, a Novel Pharmacological Approach for the Symptomatic and Disease-modifying Therapy of Alzheimer’s Disease, Curr Neuropharmacol, 14 (2016) 364–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Leon R, Garcia AG, Marco-Contelles J, Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease, Med Res Rev, 33 (2013) 139–189. [DOI] [PubMed] [Google Scholar]
- [207].Sun Y, Chen J, Chen X, Huang L, Li X, Inhibition of cholinesterase and monoamine oxidase-B activity by Tacrine-Homoisoflavonoid hybrids, Bioorg Med Chem, 21 (2013) 7406–7417. [DOI] [PubMed] [Google Scholar]
- [208].Li Y, Qiang X, Luo L, Yang X, Xiao G, Zheng Y, Cao Z, Sang Z, Su F, Deng Y, Multitarget drug design strategy against Alzheimer’s disease: Homoisoflavonoid Mannich base derivatives serve as acetylcholinesterase and monoamine oxidase B dual inhibitors with multifunctional properties, Bioorg Med Chem, 25 (2017) 714–726. [DOI] [PubMed] [Google Scholar]
- [209].Li Y, Qiang X, Luo L, Yang X, Xiao G, Liu Q, Ai J, Tan Z, Deng Y, Aurone Mannich base derivatives as promising multifunctional agents with acetylcholinesterase inhibition, anti-beta-amyloid aggragation and neuroprotective properties for the treatment of Alzheimer’s disease, Eur J Med Chem, 126 (2017) 762–775. [DOI] [PubMed] [Google Scholar]
- [210].Li Y, Qiang X, Luo L, Li Y, Xiao G, Tan Z, Deng Y, Synthesis and evaluation of 4-hydroxyl aurone derivatives as multifunctional agents for the treatment of Alzheimer’s disease, Bioorg Med Chem, 24 (2016) 2342–2351. [DOI] [PubMed] [Google Scholar]
- [211].Cao Z, Yang J, Xu R, Song Q, Zhang X, Liu H, Qiang X, Li Y, Tan Z, Deng Y, Design, synthesis and evaluation of 4’-OH-flurbiprofen-chalcone hybrids as potential multifunctional agents for Alzheimer’s disease treatment, Bioorg Med Chem, 26 (2018) 1102–1115. [DOI] [PubMed] [Google Scholar]
- [212].Tian C, Qiang X, Song Q, Cao Z, Ye C, He Y, Deng Y, Zhang L, Flurbiprofen-chalcone hybrid Mannich base derivatives as balanced multifunctional agents against Alzheimer’s disease: Design, synthesis and biological evaluation, Bioorg Chem, 94 (2020) 103477. [DOI] [PubMed] [Google Scholar]
- [213].Goedert M, Spillantini MG, A century of Alzheimer’s disease, Science, 314 (2006) 777–781. [DOI] [PubMed] [Google Scholar]
- [214].Rowe CC, Villemagne VL, Amyloid imaging with PET in early Alzheimer disease diagnosis, Med Clin North Am, 97 (2013) 377–398. [DOI] [PubMed] [Google Scholar]
- [215].Wu M, Shu J, Multimodal Molecular Imaging: Current Status and Future Directions, Contrast Media Mol Imaging, 2018 (2018) 1382183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Piel M, Vernaleken I, Rosch F, Positron emission tomography in CNS drug discovery and drug monitoring, J Med Chem, 57 (2014) 9232–9258. [DOI] [PubMed] [Google Scholar]
- [217].Dishino DD, Welch MJ, Kilbourn MR, Raichle ME, Relationship between lipophilicity and brain extraction of C-11-labeled radiopharmaceuticals, J Nucl Med, 24 (1983) 1030–1038. [PubMed] [Google Scholar]
- [218].Waterhouse RN, Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents, Mol Imaging Biol, 5 (2003) 376–389. [DOI] [PubMed] [Google Scholar]
- [219].Clark CM, Pontecorvo MJ, Beach TG, Bedell BJ, Coleman RE, Doraiswamy PM, Fleisher AS, Reiman EM, Sabbagh MN, Sadowsky CH, Schneider JA, Arora A, Carpenter AP, Flitter ML, Joshi AD, Krautkramer MJ, Lu M, Mintun MA, Skovronsky DM, Group A-AS, Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-beta plaques: a prospective cohort study, Lancet Neurol, 11 (2012) 669–678. [DOI] [PubMed] [Google Scholar]
- [220].Fleisher AS, Pontecorvo MJ, Devous MD Sr., Lu M, Arora AK, Truocchio SP, Aldea P, Flitter M, Locascio T, Devine M, Siderowf A, Beach TG, Montine TJ, Serrano GE, Curtis C, Perrin A, Salloway S, Daniel M, Wellman C, Joshi AD, Irwin DJ, Lowe VJ, Seeley WW, Ikonomovic MD, Masdeu JC, Kennedy I, Harris T, Navitsky M, Southekal S, Mintun MA, Positron Emission Tomography Imaging With [18F]flortaucipir and Postmortem Assessment of Alzheimer Disease Neuropathologic Changes, JAMA Neurol, 77 (2020) 829–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Rowe CC, Ackerman U, Browne W, Mulligan R, Pike KL, O’Keefe G, Tochon-Danguy H, Chan G, Berlangieri SU, Jones G, Dickinson-Rowe KL, Kung HP, Zhang W, Kung MP, Skovronsky D, Dyrks T, Holl G, Krause S, Friebe M, Lehman L, Lindemann S, Dinkelborg LM, Masters CL, Villemagne VL, Imaging of amyloid beta in Alzheimer’s disease with 18F-BAY94–9172, a novel PET tracer: proof of mechanism, Lancet Neurol, 7 (2008) 129–135. [DOI] [PubMed] [Google Scholar]
- [222].Ono M, Haratake M, Mori H, Nakayama M, Novel chalcones as probes for in vivo imaging of beta-amyloid plaques in Alzheimer’s brains, Bioorg Med Chem, 15 (2007) 6802–6809. [DOI] [PubMed] [Google Scholar]
- [223].Ono M, Watanabe R, Kawashima H, Cheng Y, Kimura H, Watanabe H, Haratake M, Saji H, Nakayama M, Fluoro-pegylated chalcones as positron emission tomography probes for in vivo imaging of beta-amyloid plaques in Alzheimer’s disease, J Med Chem, 52 (2009) 6394–6401. [DOI] [PubMed] [Google Scholar]
- [224].Ono M, Ikeoka R, Watanabe H, Kimura H, Fuchigami T, Haratake M, Saji H, Nakayama M, Synthesis and evaluation of novel chalcone derivatives with (99m)Tc/Re complexes as potential probes for detection of beta-amyloid plaques, ACS Chem Neurosci, 1 (2010) 598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Ono M, Hori M, Haratake M, Tomiyama T, Mori H, Nakayama M, Structure-activity relationship of chalcones and related derivatives as ligands for detecting of beta-amyloid plaques in the brain, Bioorg Med Chem, 15 (2007) 6388–6396. [DOI] [PubMed] [Google Scholar]
- [226].Fuchigami T, Yamashita Y, Haratake M, Ono M, Yoshida S, Nakayama M, Synthesis and evaluation of ethyleneoxylated and allyloxylated chalcone derivatives for imaging of amyloid beta plaques by SPECT, Bioorg Med Chem, 22 (2014) 2622–2628. [DOI] [PubMed] [Google Scholar]
- [227].Cui M, Ono M, Kimura H, Liu BL, Saji H, Synthesis and biological evaluation of indole-chalcone derivatives as beta-amyloid imaging probe, Bioorg Med Chem Lett, 21 (2011) 980–982. [DOI] [PubMed] [Google Scholar]
- [228].Kaide S, Ono M, Watanabe H, Shimizu Y, Nakamoto Y, Togashi K, Yamaguchi A, Hanaoka H, Saji H, Conversion of iodine to fluorine-18 based on iodinated chalcone and evaluation for beta-amyloid PET imaging, Bioorg Med Chem, 26 (2018) 3352–3358. [DOI] [PubMed] [Google Scholar]
- [229].Chauhan K, Tiwari AK, Chadha N, Kaul A, Singh AK, Datta A, Chalcone Based Homodimeric PET Agent, (11)C-(Chal)2DEA-Me, for Beta Amyloid Imaging: Synthesis and Bioevaluation, Mol Pharm, 15 (2018) 1515–1525. [DOI] [PubMed] [Google Scholar]
- [230].Ono M, Maya Y, Haratake M, Ito K, Mori H, Nakayama M, Aurones serve as probes of beta-amyloid plaques in Alzheimer’s disease, Biochem Biophys Res Commun, 361 (2007) 116–121. [DOI] [PubMed] [Google Scholar]
- [231].Maya Y, Ono M, Watanabe H, Haratake M, Saji H, Nakayama M, Novel radioiodinated aurones as probes for SPECT imaging of beta-amyloid plaques in the brain, Bioconjug Chem, 20 (2009) 95–101. [DOI] [PubMed] [Google Scholar]
- [232].Watanabe H, Ono M, Kimura H, Kagawa S, Nishii R, Fuchigami T, Haratake M, Nakayama M, Saji H, A dual fluorinated and iodinated radiotracer for PET and SPECT imaging of beta-amyloid plaques in the brain, Bioorg Med Chem Lett, 21 (2011) 6519–6522. [DOI] [PubMed] [Google Scholar]
- [233].Nan DD, Gan CS, Wang CW, Qiao JP, Wang XM, Zhou JN, 6-Methoxy-indanone derivatives as potential probes for beta-amyloid plaques in Alzheimer’s disease, Eur J Med Chem, 124 (2016) 117–128. [DOI] [PubMed] [Google Scholar]
- [234].Ntziachristos V, Fluorescence molecular imaging, Annu Rev Biomed Eng, 8 (2006) 1–33. [DOI] [PubMed] [Google Scholar]
- [235].Jung SJ, Park SH, Lee EJ, Park JH, Kong YB, Rho JK, Hur MG, Yang SD, Park YD, Development of fluorescent probes that bind and stain amyloid plaques in Alzheimer’s disease, Arch Pharm Res, 38 (2015) 1992–1998. [DOI] [PubMed] [Google Scholar]
- [236].Jung SJ, Lee JY, Kim TH, Lee DE, Jeon J, Yang SD, Hur MG, Min JJ, Park YD, Discovery of boronic acid-based fluorescent probes targeting amyloid-beta plaques in Alzheimer’s disease, Bioorg Med Chem Lett, 26 (2016) 1784–1788. [DOI] [PubMed] [Google Scholar]
- [237].Watanabe H, Saji H, Ono M, Novel fluorescence probes based on the chalcone scaffold for in vitro staining of beta-amyloid plaques, Bioorg Med Chem Lett, 28 (2018) 3242–3246. [DOI] [PubMed] [Google Scholar]
- [238].Tong H, Lou K, Wang W, Near-infrared fluorescent probes for imaging of amyloid plaques in Alzheimers disease, Acta Pharm Sin B, 5 (2015) 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Kelkar SS, Reineke TM, Theranostics: combining imaging and therapy, Bioconjug Chem, 22 (2011) 1879–1903. [DOI] [PubMed] [Google Scholar]
- [240].Lobatto ME, Fuster V, Fayad ZA, Mulder WJ, Perspectives and opportunities for nanomedicine in the management of atherosclerosis, Nat Rev Drug Discov, 10 (2011) 835–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Fernandez-Fernandez A, Manchanda R, McGoron AJ, Theranostic applications of nanomaterials in cancer: drug delivery, image-guided therapy, and multifunctional platforms, Appl Biochem Biotechnol, 165 (2011) 1628–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Cheng P, Pu K, Activatable Phototheranostic Materials for Imaging-Guided Cancer Therapy, ACS Appl Mater Interfaces, 12 (2020) 5286–5299. [DOI] [PubMed] [Google Scholar]
- [243].Li J, Van Valkenburgh J, Hong X, Conti PS, Zhang X, Chen K, Small molecules as theranostic agents in cancer immunology, Theranostics, 9 (2019) 7849–7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Dao P, Ye F, Liu Y, Du ZY, Zhang K, Dong CZ, Meunier B, Chen H, Development of Phenothiazine-Based Theranostic Compounds That Act Both as Inhibitors of beta-Amyloid Aggregation and as Imaging Probes for Amyloid Plaques in Alzheimer’s Disease, ACS Chem Neurosci, 8 (2017) 798–806. [DOI] [PubMed] [Google Scholar]
- [245].Li Y, Yan L, Cai J, Zhang W, Li L, Du Z, Dong C, Meunier B, Chen H, Development of novel theranostic agents for in vivo amyloid imaging and protective effects on human neuroblastoma cells, Eur J Med Chem, 181 (2019) 111585. [DOI] [PubMed] [Google Scholar]
- [246].Li YL, Cai J, Yan L, Zhang W, Li L, Du Z, Fang YX, Dong CZ, Meunier B, Chen H, Phenothiazine-based theranostic compounds for in vivo near-infared fluorescence imaging of β-amyloid plaques and inhibition of Aβ aggregation, Dyes and Pigments, 171 (2019) 107744. [Google Scholar]
- [247].Chen M, Du ZY, Zheng X, Li DL, Zhou RP, Zhang K, Use of curcumin in diagnosis, prevention, and treatment of Alzheimer’s disease, Neural Regen Res, 13 (2018) 742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Jakubowski JM, Orr AA, Le DA, Tamamis P, Interactions between Curcumin Derivatives and Amyloid-beta Fibrils: Insights from Molecular Dynamics Simulations, J Chem Inf Model, 60 (2020) 289–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].den Haan J, Morrema THJ, Rozemuller AJ, Bouwman FH, Hoozemans JJM, Different curcumin forms selectively bind fibrillar amyloid beta in post mortem Alzheimer’s disease brains: Implications for in-vivo diagnostics, Acta Neuropathol Commun, 6 (2018) 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Kim H, Im YH, Ahn J, Yang J, Choi JY, Lee KH, Kim BT, Choe YS, Synthesis and in vivo characterization of (18)F-labeled difluoroboron-curcumin derivative for beta-amyloid plaque imaging, Sci Rep, 9 (2019) 6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Zhang X, Tian Y, Li Z, Tian X, Sun H, Liu H, Moore A, Ran C, Design and synthesis of curcumin analogues for in vivo fluorescence imaging and inhibiting copper-induced cross-linking of amyloid beta species in Alzheimer’s disease, J Am Chem Soc, 135 (2013) 16397–16409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Zhang X, Tian Y, Yuan P, Li Y, Yaseen MA, Grutzendler J, Moore A, Ran C, A bifunctional curcumin analogue for two-photon imaging and inhibiting crosslinking of amyloid beta in Alzheimer’s disease, Chem Commun (Camb), 50 (2014) 11550–11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Yanagisawa D, Taguchi H, Morikawa S, Kato T, Hirao K, Shirai N, Tooyama I, Nov el curcumin derivatives as potent inhibitors of amyloid beta aggregation, Biochem Biophys Rep, 4 (2015) 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Orlando RA, Gonzales AM, Royer RE, Deck LM, Vander Jagt DL, A chemical analog of curcumin as an improved inhibitor of amyloid Abeta oligomerization, PLoS One, 7 (2012) e31869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Ferrari E, Benassi R, Sacchi S, Pignedoli F, Asti M, Saladini M, Curcumin derivatives as metal-chelating agents with potential multifunctional activity for pharmaceutical applications, J Inorg Biochem, 139 (2014) 38–48. [DOI] [PubMed] [Google Scholar]
- [256].Hamaguchi T, Ono K, Yamada M, REVIEW: Curcumin and Alzheimer’s disease, CNS Neurosci Ther, 16 (2010) 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Mishra S, Palanivelu K, The effect of curcumin (turmeric) on Alzheimer’s disease: An overview, Ann Indian Acad Neurol, 11 (2008) 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Lewczuk P, Lukaszewicz-Zajac M, Mroczko P, Kornhuber J, Clinical significance of fluid biomarkers in Alzheimer’s Disease, Pharmacol Rep, 72 (2020) 528–542. [DOI] [PMC free article] [PubMed] [Google Scholar]