

Summary
Gastric biopsies represent one of the most frequent specimens that the pathologist faces in routine activity. In the last decade or so, the landscape of gastric pathology has been changing with a significant and constant decline of H. pylori-related pathologies in Western countries coupled with the expansion of iatrogenic lesions due to the use of next-generation drugs in the oncological setting. This overview will focus on the description of the elementary lesions observed in gastric biopsies and on the most recent published recommendations, guidelines and expert opinions.
Key words: gastritis, H. pylori, secondary prevention, staging, endoscopy
Introduction
It would seem impractical to deal with the topic of gastritis without a proper definition of this disease. The term “gastritis” derived from the Greek words gastḗr gastrós and defines every flogistic process affecting the stomach, confirmed by histological evaluation. In this brief overview we will see how not only the inflammatory cells present in the biopsy sampling are fundamental, but also (and above all) the histological modifications of long-standing inflammation, possible ideal ground for the development and progression of neoplastic lesions of the stomach. For this reason, the most important goal of histology is to distinguish between non-atrophic and atrophic gastritis, in order to be informative about which patients require clinic/endoscopic surveillance.
Normal histology
Before treating the various aspects of disease, it is important to recall the normal anatomy of the gastric mucosa, which is fundamental in the understanding of how different etiologies involve different regions of the stomach. Gastric mucosa is divided in two main zones that differ both in histological and in functional aspects: the oxyntic region (fundus and corpus) and the muco-secreting region (antrum and incisura angularis). The oxyntic mucosa is composed of specialized glands containing: i) parietal (oxyntic) cells, large round or pyramidal, highly acidophilic cells, important for intrinsic factor production and hydrochloric acid (HCl) secretion, ii) the chief (zymogenic) cells, basophilic, important for pepsinogen I and gastric lipase secretion; and iii) enterochromaffin-like (ECL) cells. On the other hand, the antral mucosa is characterized by glands rich in mucous cells, also producing pepsinogen II, as well as neuroendocrine G cells (gastrin producing).
In addition to these two specialized regions, the histological transitional zones take on importance, as in other organs, mainly in neoplastic pathology. The transitional zones are defined as the junctions between the different types of mucosa: antrum-body, body-cardia, and antrum-duodenum. None of these transitions is abrupt and they all involve a gradual merging of mucosal types. Although the location of the transitional zones is easy to determine histologically by the finding of intermediate mucosa, their extent is less clear-cut. The most useful criteria to determine the crossing from body into antrum is not disappearance of the parietal cells but the absence of chief cells and the change from simple tubular glands in the body to branched glands in the antrum 1. The cardiac mucosa extends distally from the gastroesophageal junction over a variable distance, ranging from 0.5 to 4 cm. Approximately 50% of the mucosal thickness is occupied by glands lined by mucin-secreting cells. The glands have a tubular structure, in which cystic dilatation is commonly seen. Occasional chief and parietal cells can be seen, and endocrine cells are frequently present. Moving distally from the cardia, the transitional zone between body and cardia is identified as the area where chief and parietal cells start to become abundant. In the literature, the antrum-duodenal transitional zone has received little attention.
Gastritis classification
The inflammatory pathology of the stomach may be classified according to different criteria. One of the most used are temporal classifiers which divided gastritis in acute (self-limited) and chronic (non self-limited) forms. It is crucial to remember that this classification is strictly clinical and words such as “acute” and “chronic” should be avoided in the histological report. Indeed, almost all gastrites requiring histological examination are chronic forms and the word “acute” used to describe neutrophilic inflammation is confusing. At the histologic level, it is preferable to use a classification based on etiology, considering the potential role of various causes in the evolution to atrophy. In Table I the etiologic factors are summarized and below we will deal with the most important etiologies 2.
Table I.
Gastritis Classification.
| Classification of Gastritis | |
|---|---|
| Acute | Drugs, Stress induced, Uremia, Ischemia, Shock, Corrosive agents, Radiation, Certain food, Sepsis, Trauma, Certain infection, Acute alcoholism, Severe burns, Alkaline-Bile reflux, Major surgery Multiorgan failure, Portal hypertension, Congestive heart failure, Respiratory failure, Increase intracranial pressure. |
| Reactive (chemical) gastropathy | Endotossic (Alkaline Reflux-Bile Reflux, Uremic) Exotoxic (Drugs-NSAIDs, alchol, etc.) Stress induced |
| Chronic |
Helicobacter pylori (and H. Heilmannii) Autoimmune Hp-Negative Cronic Gastritis |
| Special | Lymphocytic Collagenous Eosinophilic (food induced) Radiation Graft versus host disease (GVHD) Bacterial (Syphilis, Tubercolosis, Rickettsial Infections) Viral gastritis (CMV and HSV) Fungal gastritis (Candida, Aspergillus, Mucor, Coccidioides, Histoplasma, Cryptococcus neoformans, Pneumocystis carinii and Torulopsis glabrata) Parasitic Gastritis (Anisakis, Cryptosporidium, Ascaris lumbricoides, Giardia, Toxoplasma, Schistosoma etc.) |
| Granulomatous | Idiopathic Crohn Disease Sarcoidosis Food and Barium Granulomas |
| Hypertrophic gastropathies |
Ménétrier Disease, Zollinger-Ellison Syndrome, Hypertrophic, Hypersecretory Gastropathy (with protein loss; Hp-associated) |
| Gastric vasculopathies | Ischemic, Antral Vascular Ectasia (Watermelon Stomach), Portal Hypertensive Gastropathy (Congestive Gastropathy), Varices, Angiodysplasia, Caliber-Persistent Artery (Dieulafoy Lesion); Hemodialysis-Associated Telangiectasias |
| Gastric involvement in systemic diseases | Inflammatory Bowel Disease, Amyloid, Diabetes, Mastocytosis, Sjögren Syndrome, Hypercalcemia, Siderosis |
HELICOBACTER PYLORI GASTRITIS
The discovery of Helicobacter pylori (H. pylori) in 1982 by Warren and Marshall 3 as the cause of gastritis in the vast majority of cases radically changed the epidemiology and the clinico-pathologic approach to the disease. It became evident that chronic gastritis could be cured with eradication of H. pylori, resulting in restitutio ad integrum of the mucosa in those patients in whom the morphologic pattern has not developed to atrophy.
The association between H. pylori and gastric cancer has attracted great interest worldwide when the International Agency for Research on Cancer (IARC) identified H. pylori as a “group 1 (definite carcinogen)” in 1994 on the basis of the results of epidemiologic studies 4.
H. pylori infection is mainly acquired in childhood, up to the age of 12 years, in developed countries mostly by intra-familial transmission 5. H. pylori eradication can reduce the risk for cancer, but this effect is largely confined to patients without atrophy and metaplasia; indeed, cancer could occur more than 10 years after eradication 6. For these reasons, the Kyoto global consensus report defined the search and screening for H. pylori gastritis as appropriate at an age when new infections become less likely (> 12 years) and before development of atrophic gastritis and intestinal metaplasia. This all depends on the geographical location and epidemiological context, taking into account the prevalence of infection and age-related cancer incidence 7.
At histologic exam, the bacterium may be detected on hematoxylin and eosin but is usually more easily detectable by histochemical (modified Giemsa staining) and immunohistochemical techniques (Fig. 1A). The histological signs of H. pylori gastritis include a diffuse or nodular lymphocytic inflammation in almost all cases, together with neutrophilic infiltrate. H. pylori may be difficult to detect in cases of extensive intestinal metaplasia, or during anti-secretory (PPI) therapy; in such cases, H. pylori infection is suggested by the features of inflammation. In diagnostic practice, any semi-quantitative scoring systems (+--; ++-; +++) of the bacterium’s density may be used but these have no clinically significant implications and the inter-observer reproducibility is low. Indeed, a distinction between H. pylori negative versus positive status is considered adequate.
Figure 1.
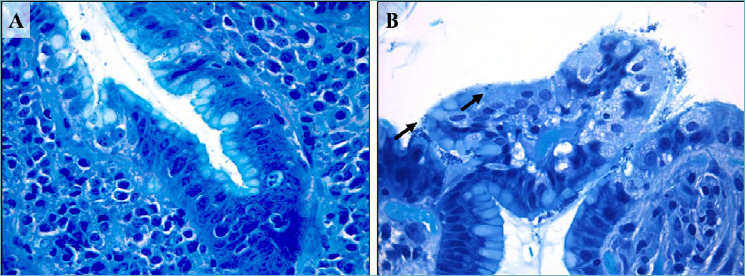
(A-B). Helicobacter pylori active gastritis (Giemsa staining). Lymphocytic inflammation and neutrophilic epithelium infiltration with conventional spiral-shaped H. pylori (A; 630x magnification). Dormant or stressed coccoid microorganisms form (arrows, B; 630 x magnification).
H. pylori is mainly present as a spiral-shaped bacterium in gastric biopsy specimens. When influenced by adverse factors (temperature or pH changes or use of antibacterial drugs), non-spore-forming microorganisms can be transformed into a latent coccoid form (Fig. 1B). The ability of H. pylori to transform from the spiral-shaped form to the coccoid form is one of its most important special adaptive mechanisms allowing it to survive extreme situations in the human organism, when cultivated, and to survive in the external environment 8.
Clinically, non-invasive diagnostic tests such as the [13C]-urea breath test, fecal antigen test and serological parameters (pepsinogen I, II, I/II, H. pylori antibody) serve as surrogate markers of H. pylori gastritis and indicators of gastritis severity 9.
REACTIVE GASTRITIS (GASTROPATHIES)
As reported in Table I, this category includes: drug-induced gastritis; alcoholic gastritis; radiation gastritis, gastritis due to duodenal reflux (etc.). Reactive gastritis shares some etiological agents (e.g. drugs) of the acute forms. Bile reflux into the stomach, due to partial gastrectomy or dysmotility and nonsteroidal anti-inflammatory drugs (NSAIDs) are the most frequent injuries and may result in a broad spectrum of histological mucosal lesions, associated with low-grade inflammation of the gastric mucosa (gastropathies). The chemical noxae increase the turnover of the gastric epithelium, resulting in foveolar hyperplasia. Pro-inflammatory cytokines produce vascular ectasia, edema and muscularis mucosae hyperplasia. These entities are frequently asymptomatic, but they may lead to multiple erosions or ulcers, even with bleeding. Atrophic changes are rare. Recently, the increasing use of immune checkpoint inhibitors for the treatment of multiple metastatic malignancies has revealed a wide spectrum of immune-related adverse events affecting the gastro-intestinal tract. While colitis has been extensively described, gastritis has only been sporadically reported. Johncilla et al. 10 characterized histological features of this kind of gastritis identifying a series of patterns which mimic other etiological aspects which include intraepithelial lymphocytosis and increased apoptotic activity, neutrophilic infiltration within glandular epithelium and foveolar hyperplasia. Checkpoint inhibitors-associated gastritis is rare but must be considered and promptly treated, as this might affect the course of the underlying oncological therapy 11.
AUTOIMMUNE GASTRITIS
Autoimmune gastritis (AG) is due to an immune-mediated aggression targeting the parietal cells and affects exclusively the oxyntic mucosa of the body-fundus; it is associated with serum anti-parietal cell and anti-intrinsic factor antibodies 12,13. Clinical signs of AG include hypo/achlorhydria, hypergastrinemia, low pepsinogen I/pepsinogen II ratio and vitamin B12-deficient macrocytic anaemia 14.
The disease may coexist with other immuno-mediated diseases, such as Hashimoto’s thyroiditis, insulin-dependent diabetes, and vitiligo and it was also reported as an association with H. pylori infection with aggravation of the gastric picture and increased risk of atrophic pangastritis. Studies using T cells from patients infected with H. pylori and patients with autoimmune atrophic gastritis have identified molecular mimicry between H. pylori and structural protein of the parietal cells as the gastric H+/K+ ATPase, suggesting that the infection might stimulate T cells targeting specialized corpus cells 15. The histopathological changes of autoimmune atrophic gastritis can be divided into three evolving phases. The early phase is characterized by a multifocal, dense lymphocytic and plasma cell infiltration of the oxyntic mucosa involving the entire thickness of the lamina propria with an accentuation in the deeper part, often mixed with eosinophils and mast cells. Patchy destruction of individual oxyntic glands by lymphocytes might occur, and the parietal cells exhibit pseudo-hypertrophic changes (PPI-like, Fig. 2A). These histologic features are non-specific, and the histologic report should only raise the possibility of early autoimmune gastritis and recommend appropriate serologic tests 16. The second phase is characterized by diffuse lymphoplasmacytic infiltration of the lamina propria, marked atrophy of oxyntic glands and increase in the thickness of the foveolar component. Pseudopyloric metaplasia is often extensive 17, and intestinal metaplasia becomes increasingly prominent. These findings are sufficiently pathognomonic for a diagnosis of autoimmune etiology, particularly if the antrum does not show inflammatory and atrophic “lesions”. Confirmation of diagnosis, however, still rests on the demonstration of antibodies directed against parietal cell and intrinsic factor antigens. The end stage is characterized by a marked replacement of oxyntic glands, which might be completely absent; foveolar hyperplasia with underlying microcystic change and the constitution of hyperplastic and inflammatory polyps. Pseudopyloric, pancreatic, and intestinal metaplasia becomes wide spread and the muscularis mucosae is thickened (Fig. 2B). Although scattered lymphoid aggregates may be seen, inflammation is usually minimal. Enterochromaffin-like (ECL) cells hyperplasia (both linear and micronodular) is detectable in increasing degrees from the initial to the second phase, while adenomatoid hyperplasia or ECL cell dysplasia may affect the late phase and progress into well-differentiated endocrine tumor (type I carcinoid).
Figure 2.
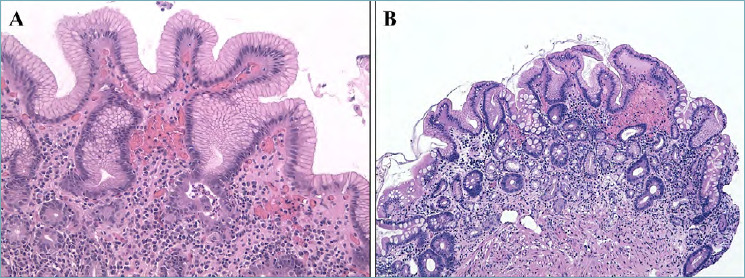
(A-B). Autoimmune gastritis. Early phase of autoimmune gastritis with hypertrophic glandular changes and mild lymphocytic and granulocytic infiltrate in the lamina propria (A; 200x magnification). The end stage is characterized by a marked replacement of oxyntic glands with pseudopyloric and intestinal metaplasia with mild inflammation of the lamina propria (B; 100x magnification).
“SPECIAL-TYPE GASTRITIS” (FIG. 3)
Figure 3.
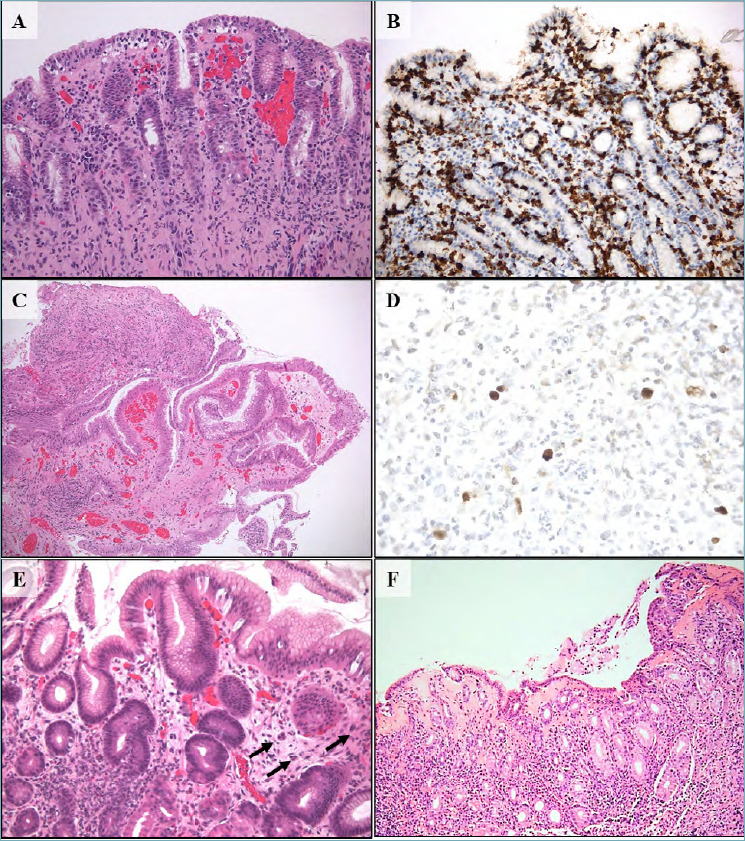
(A-F). Special-type gastrites. Lymphocytic gastritis is defined by the presence of at least 25 intraepithelial lymphocytes CD3+ per 100 gastric epithelial cells (A; hematoxylin-eosin, 200x magnification) (B; CD 3 immunostaining, 200x magnification). Cytomegalovirus (CMV) gastritis is characterized abundant granulation tissue with important inflammatory reaction (C; hematoxylin-eosin, 200x magnification) and CMV inclusions are visible in endothelial cells and also in macrophages (D; CMV immunostaining, 630x magnification). Graft versus host disease (GVHD) gastric mucosa shows apoptotic bodies (arrows) and gland abscess (E; hematoxylin-eosin, 200x magnification). Collagenous gastritis is typically defined by the subepithelial deposition of collagen bands thicker than 10 μm and the intense inflammation response in the lamina propria (F; hematoxylin-eosin, 200x magnification).
Lymphocytic gastritis
About thirty years ago, Haot et al. 18 described endoscopic and microscopic features of the condition, which they called lymphocytic gastritis (LG), characterized by accumulation of lymphocytes in the gastric epithelium. In fact, the lesion probably corresponds to previously reported one (in 1945) called varioliform gastritis which is characterized by nodular and eroded lesions running along the gastric rugae in the corpus. The diagnosis of LG is based on microscopic examination of the gastric mucosa and according to current definition the main feature is a presence of at least 25 intraepithelial lymphocytes (IEL) per 100 gastric epithelial cells (Fig. 3A). Most of these lymphocytes present CD3+ and CD8+ phenotype (Fig. 3B). H. pylori infection is known to be associated with LG, reaching from 0 up to 27% of cases. An association between LG and celiac disease is well-established, varying from 10 to 38% 19.
Eosinophilic gastritis
Eosinophilic gastritis (EG) is part of the eosinophilic gastrointestinal disorders family which includes eosinophilic esophagitis, eosinophilic gastroenteritis, and eosinophilic enteritis/colitis. These disorders are considered immune-mediated chronic inflammatory processes with strong associations to food allergen triggers. Indeed, many patients have a history of atopic conditions including asthma, allergic rhinitis or atopic dermatitis, and allergy to medicine food or pollen 20. Recent studies strongly support that Th-2 cytokines (e.g. IL-4, IL-5 and IL-13) and chemokines such as eotaxin play a critical role in the pathogenesis of EG 21. The exact incidence of EG is difficult to estimate; the current literature suggests that EG occurs most commonly between 30 and 40 years of age, with a slightly male predominance and the prevalence data varies from 2.5 to 28/100,000 22. Although symptoms of EG are non-specific, abdominal pain and nausea/vomiting result the most frequent presenting symptoms in children and adults. Young patients may also show growth retardation and delayed puberty or amenorrhea. Unlike esophageal mucosa, the lamina propria of healthy stomach contains a quote of eosinophils and the threshold number of gastric eosinophils required in EG diagnosis varies among authors. Recently, Lwin et al. 23 recommended two main criteria for EG diagnosis: i) gastric biopsies with an average density > 127 eosinophils/mm2 (or > 30 eosinophils/ high power field (HPF) on microscopes equipped with wide-lens oculars (FN 22) in at least five separate HPFs; ii) no known associated causes of eosinophilia (e.g. H. pylori infection, Crohn’s disease, parasitic infections, and hematological or lymphoid disorders). Considering clinical manifestations and depth of eosinophilic infiltration into the gastric wall, Klein et al. 24 proposed a classification of EG into three different patterns, including predominantly mucosal pattern, predominantly muscular pattern, and predominantly serosal pattern. Currently, treatment strategies consist in medical agents able to limit the inflammation (corticosteroids) and in dietary therapy.
CMV gastritis
Cytomegalovirus (CMV) gastritis is the only viral infection with a distinct pathologic pattern in the stomach. It predominantly affects children and immunocompromised patients. Usually, different sites of the gastrointestinal tract are involved. Endoscopically, the gastric mucosa may appear completely normal or show erosions and ulcers 25. Rarely, it may occur as a mass called “pseudotumor”. Histologically, numerous CMV inclusions may be seen less in epithelial than in endothelial cells and also in macrophages, with little or absent inflammation. In this case, the immunological status of the patients is usually very compromised. In other cases, abundant granulation tissue with important inflammatory reaction can be observed (Fig. 3C), while the CMV inclusions are difficult to see without immunohistochemical or in situ hybridization techniques (Fig. 3D). Eosinophilic infiltrates can also be an important component of the CMV gastritis inflammation.
GVHD gastritis
Graft versus host disease (GVHD) follows allogeneic bone marrow transplant or transfusions, especially in immunocompromised patients. Acute GVHD occurs within the first 100 days post-transplant and mainly targets the liver, gastrointestinal tract and skin. Among cases of GVHD of the gastrointestinal tract, GVHD rarely affects the upper tract 26. Patients with isolated gastric GVHD present with nausea, vomiting, and upper abdominal pain without diarrhea. In the early stages, the stomach appears endoscopically normal (30% to 80%) and some patients may show gastric mucosal exfoliation. In full-blown disease, the stomach appears variably congested and atrophic. Histologically, the mucosa shows apoptotic cells predominantly in the mucous neck regions (in both antral and oxyntic mucosa) and gland abscesses may also present (Fig. 3E). Different degrees of inflammation and granular eosinophilic material may occur in the lamina propria and in the glands. Even though apoptotic bodies may be present in small numbers, their presence is diagnostic of GVHD in the appropriate setting 27.
Collagenous gastritis
Collagenous gastritis (CG) is a rare disease characterized by the subepithelial deposition of collagen bands thicker than 10 μm and the infiltration of inflammatory mononuclear cells in the lamina propria (Fig. 3F). Collagenous colitis seems to be part of the same disease entity, although the colonic form occurs more frequently. Since the disease was first reported in 1989, only 60 cases have been documented 28. Because of the small number of cases, no standard therapy has been found. Based on case reports, two phenotypes of the disease (pediatric and adult) have been defined 29. The clinical symptoms of the pediatric type are mainly of the upper gastrointestinal tract, including abdominal pain and anemia secondary to the stomach specific inflammation and collagen deposition. In contrast, the adult type is characterized by the simultaneous occurrence of collagenous colitis, which may be related to autoimmune processes and celiac disease.
Morphological features: gastritis “grading”
The Sydney System 30 and its Update in Houston Gastritis Workshop 1994 31 introduce a common terminology for the standardization of the histological report, providing the histopathological and morphological features characterizing every nosologic type of gastritis. In order to describe the intensity and severity of the histological signs of gastritis, the use of a scale for grading was also proposed. Below we analyze the main elementary lesions that histologically define the gastritis landscape.
GASTRITIS INTENSITY: MONONUCLEAR LEUKOCYTE INFILTRATE
In the normal gastric mucosa, only scattered chronic inflammatory cells as lymphocytes and plasma cells are present in the lamina propria. Thus, every increase in their count defines a chronic gastritis. Although it is not easy to define the exact count of mononucleate cells in the normal gastric mucosa, a number of 2 to 5 lymphocytes, plasma cells and macrophages per high power microscope field or 2-3 of them between foveolae is acceptable.
The increase in these numbers defines gastritis as mild (+--); moderate (++-) and marked (+++).
Density of mononuclear cells in the lamina propria should be graded in areas far from the lymphoid follicles that typically occur in H. pylori infection.
Lymphocytes may also be found in the glandular epithelium, usually in the surface. The finding of a number greater than 25/100 epithelial cells defines the picture of lymphocytic gastritis as previously described.
GASTRITIS ACTIVITY: NEUTROPHILIC INFILTRATE
The presence of neutrophils (not eosinophils) in the gastric mucosa defines the “activity” of the gastritis which typically characterizes some etiological forms as H. pylori infection. Inflammation in the gastric mucosa (within the lamina propria and/or the glandular lumen) is as defined in the previous paragraph. The localization of the neutrophils defines the grade: neutrophils in the lamina propria as mild (+--) activity; neutrophils in the epithelium as moderate (++-) activity and neutrophils in the glandular lumen as marked (+++) activity.
H. PYLORI DENSITY
As previously reported, for clinical management it is important to report if H. pylori is present or not. Since the absolute number of the bacteria may have a role in the degree of the inflammation, in reactive glandular hyperplasia and in lesions inducing lymphoproliferative disorders, the density of H. pylori, if mentioned, should be graded as mild (+--), moderate (++-), and marked (+++).
FOVEOLAR HYPERPLASIA
Expansion of the proliferative compartment of the gastric glands results in foveolar hyperplasia. All inflammatory conditions and direct acute noxae of the gastric mucosa induce regenerative epithelial changes, especially in association with erosions and peptic ulcers. Chemicals or infectious noxae increase the cell turnover and lead to hyperplastic foveolae. The most important differential diagnosis of these changes is with dysplasia; indeed, in some contexts where the inflammatory component is severe, it is not easy to distinguish an atypical regeneration of the glandular neck from dysplastic lesions. For these reasons, the term “indefinite for dysplasia, atypical foveolar hyperproliferation” type was coined to identify this histological frame and pay attention to the follow-up of the patient 32.
OXYNTIC EPITHELIUM HYPERTROPHY
Hypertrophic changes in the oxyntic gland, usually as a result of treatment with proton pump inhibitors (PPI) and in the early phase of autoimmune gastritis as “vicariant” reaction, represent a remodeling of the epithelial structure due to cytoskeletal rearrangements.
The clinical significance is not yet well known, but it is recommended to report these changes in the histological report.
FIBROSIS OF THE LAMINA PROPRIA AND MUSCULARIS MUCOSAE HYPERPLASIA
Focal fibrosis of the lamina propria may occur as scarring after ulcer and inflammatory process, but it should be distinguished from mucosa atrophy. Hyperplasia of the muscularis mucosae is a mesenchymal response to many noxae as duodenal reflux, but could be an antrum remark in association with autoimmune gastritis.
ENDOCRINE CELL HYPERPLASIA
Endocrine cell hyperplasia is secondary to corpus oxyntic atrophy which, by inducing hypo/achlorhydria status, causes a proliferative stimulus towards enterochromaffin-like (ECL) cells. These cells are the precursor lesions of the neuroendocrine tumor type I of the stomach. ECL hyperplasia is graded as: i) linear hyperplasia; characterized by a chain of at least 5 cells, linearly growing within the gastric gland; ii) micronodular hyperplasia; defined as nodular clusters of at least 5 cells not exceeding the diameter of gastric glands (< 150 μm in diameter), usually located within the lamina propria at the deep of the oxyntic glands (Fig. 4A). Recently, in the cascade towards neuroendocrine tumor, other two lesions have been described: adenomatoid ECL hyperplasia; defined as compact collections of ECL micronodules (150-500 μm in diameter) in the deep part of the lamina propria (Fig. 4B); and ECL dysplasia, described as large confluent ECL micronodules, ranging from 150 to 500 μm in diameter and characterized by the exceedance of the basement membrane with microinfiltration of the lamina propria and formation of new stroma 13,33 (Tab. II).
Figure 4.
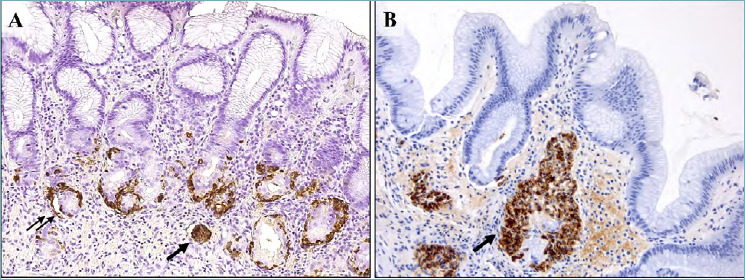
(A-B). Enterochromaffin-like cells disorders in autoimmune gastritis. Linear hyperplasia of endocrine cells growing within the gastric gland (double arrow) and micronodular hyperplasia in the lamina propria (single arrow) (A; Chromogranin A immunostaining, 200x magnification). Adenomatoid endocrine hyperplasia (arrow) is defined as a nodule of 150-500 mμ in diameter in the deep of the lamina propria (B; Chromogranin A immunostaining, 200x magnification).
Table II.
Enterochromaffin-like cells hyperplasia.
| Linear hyperplasia | Chain of at least 5 cells, linearly growing within the gastric gland |
| Micronodular hyperplasia | Nodular clusters of at least 5 cells; < 150 μm in diameter; Located within the lamina propria |
| Adenomatoid hyperplasia | Compact collection of micronodules; 150-500 μm in diameter; Located in the deep of lamina propria |
| ECL cells dysplasia | Large confluent ECL cells micronodules; 150-500 μm in diameter; Microinfiltration of the lamina propria; Formation of new stroma |
MUCOSAL ATROPHY
The accepted definition of gastric mucosa atrophy is “loss of appropriate glands” for the sampling site. Indeed, normal gastric biopsy samples show different types of glands (muco-secreting or oxyntic), appropriate for the function of the anatomic site (antrum or corpus) from which the specimen is obtained; the discrepancy between the expected glands and what is actually observed at the histologic exam represents the concept of atrophy 34-37. This definition is derived from an international group of pathologists (Atrophy Club 2000) who established new diagnostic criteria for the two main phenotypes of the chronic gastritis (non-atrophic and atrophic) and proposed a classification of atrophy, which includes a non-metaplasic and a metaplasic category (Tab. III) 34-37. The first framework is characterized by the decrease or complete loss of glandular units, replaced by collagen deposition in lamina propria, like a fibrotic scar. This situation causes the reduction of glandular mass, but does not imply any change of the original cell phenotype. Sometimes, severe inflammation may hide the normal glands, mimicking atrophy and making difficult to assess. These cases can be temporarily labeled as “indefinite for atrophy” and the analysis can be delayed until the inflammation has regressed (e.g. after H. pylori eradication). Diversely, in the metaplasic cathegory the native glands are replaced by cells featuring a new commitment (intestinal and/or pseudopyloric). Two main forms of metaplasia are recognized: pseudopyloric metaplasia (or spasmolytic polypeptide-expressing metaplasia, SPEM), corpus-restricted; and intestinal metaplasia (IM), potentially located in all gastric regions. Other metaplasic changes as pseudo-pancreatic metaplasia and clear-cell metaplasia are not considered in this definition because they are not related (to date) to any carcinogenic process.
Table III.
Mucosal atrophy classification.
| Gastric atrophy - Loss of glands appropriate for the sampling site - | ||
|---|---|---|
| Type | Histological lesions | Scoore |
| Non-metaplasic | Glandular disapperance
|
1 = 1%-30% 2 = 31%-60% 3 = >60% |
| Metaplasic | Glandular replacement
|
|
SPEM is characterized by the substitution of oxyntic epithelia with antral-like mucosa. SPEM (restricted to the oxyntic mucosa by definition) can be more easily assessed by exploiting its positive immunostaining for Trefoil Factor 2 (TFF2) 38.
Intestinal metaplasia (IM) may arise in native muco-secreting (antral) epithelia or in previously antralized oxyntic glands (SPEM). Different subtypes of intestinal metaplasia have been classified, based on whether the metaplastic epithelium phenotype resembles large bowel epithelia or the small intestinal mucosa 39,40.
Secondary prevention: gastritis staging
Gastric cancer (GC) is one of the most lethal epithelial malignancies, and its mortality rate prompts a global prevention strategy 41,42. Gastric mucosal inflammation, mainly caused by H. pylori infection, is a preneoplastic condition, which may promote the transition of stem cells into cancer stem cells involved in both intestinal- and diffuse-type GCs. Along the progression into atrophic phenotypes, coincidental pathogenetic processes, even carcinogenic ones, are potentially triggered, concurring in the well-known “Correa cascade”, and they may contribute to processes that link the atrophic gastritis with intestinal type GC 43. Knowing this carcinogenic process, a window of opportunity to prevent progression in carcinoma is available. While inflammation coexists with mucosal atrophy in the majority of patients with intestinal GC, diffuse-type GCs do not show any coexisting atrophic changes and its “field of cancerization” is still not well-known. Discontinuing the inflammatory cascade triggered by H. pylori is crucial to preventing GC. For patients who have already developed gastric atrophy, the severity and the topography (antrum, corpus or both) of the atrophic changes correlates with cancer risk. Based on this assumption, the Operative Link Gastritis Assessment (OLGA) staging system defines five stages of gastritis with increasing cancer risk (stages 0-IV) based on atrophy score (Tab. IV). At each mucosal compartment, the atrophy score is calculated as average of the atrophic changes as detected in antral vs oxyntic mucosa. At the single biopsy level, atrophy is graded as a percentage of atrophic transformations (both metaplasic and non-metaplasic). To obtain this type of staging, it is crucial that multiple biopsy samples have been obtained to explore the different mucosa compartments. The OLGA approach recommends at least 5 biopsy samples from: i) the greater and lesser curvatures of the distal antrum (A1-A2 = mucus-secreting mucosa); ii) the lesser curvature at the incisura angularis (A3), where the earliest atrophic-metaplastic changes tend to occur; and iii) the anterior and posterior walls of the proximal corpus/fundus (C1-C2 = oxyntic mucosa) (Fig. 5).
Table IV.
Gastritis staging.
| Olga Staging System | |||||
|---|---|---|---|---|---|
| Overall Atrophy score 0: No atrophy 1: 1-30% of biopsy samples 2: 31-60% of biopsy samples 3: >60% of biopsy samples |
Oxintic (Corpus and Fundus) overall atrophy score | ||||
| Score 0 | Score 1 | Score 2 | Score 3 | ||
|
Mucosecreting (Antrum and Angularis Incisura) overall atrophy score |
Score 0 | No Stage | Stage I | Stage II | Stage II |
| Score 1 | Stage I | Stage I | Stage II | Stage III | |
| Score 2 | Stage II | Stage II | Stage III | Stage IV | |
| Score 3 | Stage III | Stage III | Stage IV | Stage IV | |
Figure 5.
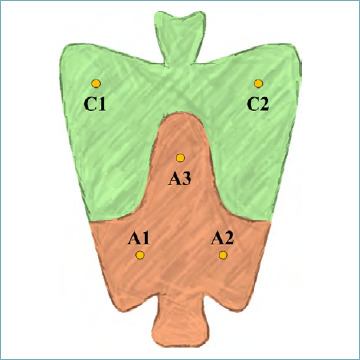
OLGA sample protocol for the gastritis staging.
International guidelines for the secondary prevention of GC use the OLGA staging system to distinguish between low-risk (stage 0-I-II) and high-risk (stage III and IV) gastritis 44.
An alternative staging system, the Operative Link on Gastric Intestinal Metaplasia Assessment (OLGIM) proposes to consider only intestinal metaplasia for the atrophy score 45. This approach could have better inter-observer reproducibility, but in some studies OLGIM seemed to have less sensitivity in identifying patients at high risk of gastric cancer 46. Indeed, IM change is considered just one of the phenotypes of atrophy involved in the cancer-prone atrophic microenvironment 47. Indeed, the clinic-pathological correlation between SPEM and dysplastic lesions, have been less extensively explored though recent findings suggest an important role in “non-classic gastric” carcinogenesis 38,48.
Histology report
The diagnosis of gastritis is both clinical and histologic: clinical information should always be given to the pathologist for the interpretation of the endoscopic and histological findings. The Sydney System protocol, which requires biopsy sampling from the antral, incisura angularis and oxyntic areas, is the most widely applied standardized biopsy protocol. Additional specimens from any focal lesions should be added. Antral and incisura angularis specimens can be placed in the same container and the corpus/fundus biopsies in another; this is essential to differentiate special forms of gastritis such ad as AG. Biopsy material should be handled with care. Stains which should be used in routine practice are H&E and modified Giemsa for H. pylori detection.
The pathology request form should include essential information such as the patient’s clinical history, endoscopic features, and the biopsy sampling map. Serological tests (e.g. pepsinogen I/II and Gastrin 17 levels, autoantibody against parietal cell and intrinsic factor) should be reported, if available.
The histology report should include 4 main chapters (Tab. V): i) sample description, including the number of samples for each biopsy site; ii) clinical and endoscopic information; iii) histological description, according to the Houston modified-Sydney System gradation of the elementary lesions; iv) the OLGA staging system; and finally v) information about the etiology of gastritis (i.e. H. pylori infection; autoimmune disease, etc.) where possible.
Table V.
Histology report.
| Histology report |
|---|
| Samples description |
|
| Clinical and endoscopic information |
|
| Histological description |
|
| OLGA staging and etiological information |
| A (antrum and incisura angularis overall atrophy score) 0-1-2-3; C (corpus and fundus overall atrophy score) 0-1-2-3; Olga staging: 0, I, II, III, IV- Hp-positive / Hp-negative / Morphological features suggestive for Hp / Autoimmune / Chemical Gastritis (Gastropathy) |
Conclusion
Biopsy examination of the stomach represents one of the most frequent pathology samples that the pathologist faces in his/her routine activity. The sheer number of samples and the time-related factor can lead to the perception that the diagnostic activity is monotonous and possibly poorly informative (this may be in part due to the few clinical reports). As pathologists, we should formulate reproducible histopathologic reports that will help the clinicians in the tailoring of secondary prevention strategies for gastric cancer or allow a better definition of the etiology of gastric disease.
Figures and tables
References
- 1.Van Zanten SJ, Dixon MF, Lee A. The gastric transitional zones: neglected links between gastroduodenal pathology and helicobacter ecology. Gastroenterology 1999;116:1217-29. https://doi.org/10.1016/s0016-5085(99)70025-9 10.1016/s0016-5085(99)70025-9 [DOI] [PubMed] [Google Scholar]
- 2.Hunt RH, Camilleri M, Crowe SE, et al. The stomach in health and disease. Gut 2015;64:1650-68. https://doi.org/10.1136/gutjnl-2014-307595 10.1136/gutjnl-2014-307595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1(8390):1311-5. https://doi.org/10.1016/s0140-6736(84)91816-6 10.1016/s0140-6736(84)91816-6 [DOI] [PubMed] [Google Scholar]
- 4.Schistosomes liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7-14 June 1994. IARC Monogr Eval Carcinog Risks Hum 1994;61:1-241. [PMC free article] [PubMed] [Google Scholar]
- 5.Okuda M, Sugiyama T, Fukunaga K, et al. A strain-specific antigen in Japanese Helicobacter pylori recognized in sera of Japanese children. Clin Diagn Lab Immunol 2005;12:1280-4. https://doi.org/10.1128/CDLI.12.11.1280-1284.2005 10.1128/CDLI.12.11.1280-1284.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ford AC, Forman D, Hunt RH, et al. Helicobacter pylori eradication therapy to prevent gastric cancer in healthy asymptomatic infected individuals: systematic review and meta-analysis of randomised controlled trials. BMJ 2014;348:g3174. https://doi.org/10.1136/bmj.g3174 10.1136/bmj.g3174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sugano K, Tack J, Kuipers EJ, et al. Kyoto global consensus report on Helicobacter pylori gastritis. Gut 2015;64:1353-67. https://doi.org/10.1136/gutjnl-2015-309252 10.1136/gutjnl-2015-309252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bode G, Mauch F, Malfertheiner P. The coccoid forms of Helicobacter pylori. Criteria for their viability. Epidemiol Infect 1993;111:483-90. https://doi.org/10.1017/s0950268800057216 10.1017/s0950268800057216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malfertheiner P, Megraud F, O’Morain CA, et al. Management of Helicobacter pylori infection--the Maastricht IV/ Florence Consensus Report. Gut 2012;61:646-64. https://doi.org/10.1136/gutjnl-2012-302084 10.1136/gutjnl-2012-302084 [DOI] [PubMed] [Google Scholar]
- 10.Johncilla M, Grover S, Zhang X, et al. Morphological spectrum of immune check-point inhibitor therapy-associated gastritis. Histopathology 2020;76:531-9. https://doi.org/10.1111/his.14029 10.1111/his.14029 [DOI] [PubMed] [Google Scholar]
- 11.Rovedatti L, Lenti MV, Vanoli A, et al. Nivolumab-associated active neutrophilic gastritis. J Clin Pathol 2020. Online ahead of print. https://doi.org/10.1136/jclinpath-2020-206540 10.1136/jclinpath-2020-206540 [DOI] [PubMed] [Google Scholar]
- 12.Rugge M, Fassan M, Pizzi M, et al. Autoimmune gastritis: histology phenotype and OLGA staging. Aliment Pharmacol Ther 2012;35:1460-6. https://doi.org/10.1111/j.1365-2036.2012.05101.x 10.1111/j.1365-2036.2012.05101.x [DOI] [PubMed] [Google Scholar]
- 13.Coati I, Fassan M, Farinati F, et al. Autoimmune gastritis: Pathologist’s viewpoint. World J Gastroenterol 2015;21:12179-89. https://doi.org/10.3748/wjg.v21.i42.12179 10.3748/wjg.v21.i42.12179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neumann WL, Coss E, Rugge M, Genta RM. Autoimmune atrophic gastritis--pathogenesis, pathology and management. Nat Rev Gastroenterol Hepatol 2013;10:529-41. https://doi.org/10.1038/nrgastro.2013.101 10.1038/nrgastro.2013.101 [DOI] [PubMed] [Google Scholar]
- 15.Bergman MP, D’Elios MM. Cytotoxic T cells in H. pylori-related gastric autoimmunity and gastric lymphoma. J Biomed Biotechnol. 2010;2010:104918. https://doi.org/10.1155/2010/104918 10.1155/2010/104918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Junca J, de Soria PL, Granada ML, et al. Detection of early abnormalities in gastric function in first-degree relatives of patients with pernicious anemia. Eur J Haematol 2006;77:518-22. https://doi.org/10.1111/j.0902-4441.2006.t01-1-EJH2913.x 10.1111/j.0902-4441.2006.t01-1-EJH2913.x [DOI] [PubMed] [Google Scholar]
- 17.Rugge M. Biologic profiles meet clinical priorities: incorporating pseudopyloric, and spasmolytic-expressing metaplasia in the assessment of gastric atrophy. Virchows Arch 2020. Online ahead of print. https://doi.org/10.1007/s00428-020-02814-8 10.1007/s00428-020-02814-8 [DOI] [PubMed] [Google Scholar]
- 18.Haot J, Hamichi L, Wallez L, Mainguet P. Lymphocytic gastritis: a newly described entity: a retrospective endoscopic and histological study. Gut 1988;29:1258-64. https://doi.org/10.1136/gut.29.9.1258 10.1136/gut.29.9.1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feeley KM, Heneghan MA, Stevens FM, McCarthy CF. Lymphocytic gastritis and coeliac disease: evidence of a positive association. J Clin Pathol 1998;51:207-10. https://doi.org/10.1136/jcp.51.3.207 10.1136/jcp.51.3.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonsalves N. Eosinophilic Gastrointestinal Disorders. Clin Rev Allergy Immunol. 2019;57:272-85. https://doi.org/10.1007/s12016-019-08732-1 10.1007/s12016-019-08732-1 [DOI] [PubMed] [Google Scholar]
- 21.Caldwell JM, Collins MH, Stucke EM, et al. Histologic eosinophilic gastritis is a systemic disorder associated with blood and extragastric eosinophilia, TH2 immunity, and a unique gastric transcriptome. J Allergy Clin Immunol. 2014;134:1114-24. https://doi.org/10.1016/j.jaci.2014.07.026 10.1016/j.jaci.2014.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang M, Li Y. Eosinophilic gastroenteritis: A state-of-the-art review. J Gastroenterol Hepatol 2017;32:64-72. https://doi.org/10.1111/jgh.13463 10.1111/jgh.13463 [DOI] [PubMed] [Google Scholar]
- 23.Lwin T, Melton SD, Genta RM. Eosinophilic gastritis: histopathological characterization and quantification of the normal gastric eosinophil content. Mod Pathol 2011;24:556-63. https://doi.org/10.1038/modpathol.2010.221 10.1038/modpathol.2010.221 [DOI] [PubMed] [Google Scholar]
- 24.Klein NC, Hargrove RL, Sleisenger MH, Jeffries GH. Eosinophilic gastroenteritis. Medicine (Baltimore) 1970;49:299-319. https://doi.org/10.1097/00005792-197007000-00003 10.1097/00005792-197007000-00003 [DOI] [PubMed] [Google Scholar]
- 25.El-Zimaity H, Choi WT, Lauwers GY, Riddell R. The differential diagnosis of Helicobacter pylori negative gastritis. Virchows Arch 2018;473:533-50. https://doi.org/10.1007/s00428-018-2454-6 10.1007/s00428-018-2454-6 [DOI] [PubMed] [Google Scholar]
- 26.Nomura K, Iizuka T, Kaji D, Yamamoto H, Kuribayashi Y, Kimura R, et al. Clinicopathological features of patients with acute graft-versus-host disease of the upper digestive tract. J Gastroenterol Hepatol 2014;29:1867-72. https://doi.org/10.1111/jgh.12651 10.1111/jgh.12651 [DOI] [PubMed] [Google Scholar]
- 27.Tobin JM, Sinha B, Ramani P, et al. Upper gastrointestinal mucosal disease in pediatric Crohn disease and ulcerative colitis: a blinded, controlled study. J Pediatr Gastroenterol Nutr 2001;32:443-8. https://doi.org/10.1097/00005176-200104000-00010 10.1097/00005176-200104000-00010 [DOI] [PubMed] [Google Scholar]
- 28.Kamimura K, Kobayashi M, Sato Y, et al. Collagenous gastritis: Review. World J Gastrointest Endosc 2015;7:265-73. https://doi.org/10.4253/wjge.v7.i3.265 10.4253/wjge.v7.i3.265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lagorce-Pages C, Fabiani B, Bouvier R, et al. Collagenous gastritis: a report of six cases. Am J Surg Pathol 2001;25:1174-9. https://doi.org/10.1097/00000478-200109000-00008 10.1097/00000478-200109000-00008 [DOI] [PubMed] [Google Scholar]
- 30.Price AB. The Sydney System: histological division. J Gastroenterol Hepatol 1991;6:209-22. https://doi.org/10.1111/j.1440-1746.1991.tb01468.x 10.1111/j.1440-1746.1991.tb01468.x [DOI] [PubMed] [Google Scholar]
- 31.Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol 1996;20:1161-81. https://doi.org/10.1097/00000478-199610000-00001 10.1097/00000478-199610000-00001 [DOI] [PubMed] [Google Scholar]
- 32.Fassan M, Pizzi M, Farinati F, et al. Lesions indefinite for intraepithelial neoplasia and OLGA staging for gastric atrophy. Am J Clin Pathol 2012;137:727-32. https://doi.org/10.1309/AJCPEU41HTGXSJDQ 10.1309/AJCPEU41HTGXSJDQ [DOI] [PubMed] [Google Scholar]
- 33.Vanoli A, La Rosa S, Luinetti O, et al. Histologic changes in type A chronic atrophic gastritis indicating increased risk of neuroendocrine tumor development: the predictive role of dysplastic and severely hyperplastic enterochromaffin-like cell lesions. Hum Pathol 2013;44:1827-37. https://doi.org/10.1016/j.humpath.2013.02.005 10.1016/j.humpath.2013.02.005 [DOI] [PubMed] [Google Scholar]
- 34.Rugge M, Correa P, Dixon MF, et al. Gastric mucosal atrophy: interobserver consistency using new criteria for classification and grading. Aliment Pharmacol Ther. 2002;16:1249-59. https://doi.org/10.1046/j.1365-2036.2002.01301.x 10.1046/j.1365-2036.2002.01301.x [DOI] [PubMed] [Google Scholar]
- 35.Rugge M, Pennelli G, Pilozzi E, et al. Gastritis: the histology report. Dig Liver Dis. 2011;43 Suppl 4:S373-84. https://doi.org/10.1016/S1590-8658(11)60593-8 10.1016/S1590-8658(11)60593-8 [DOI] [PubMed] [Google Scholar]
- 36.Rugge M, Correa P, Di Mario F, et al. OLGA staging for gastritis: a tutorial. Dig Liver Dis 2008;40:650-8. https://doi.org/10.1016/j.dld.2008.02.030 10.1016/j.dld.2008.02.030 [DOI] [PubMed] [Google Scholar]
- 37.Rugge M, Meggio A, Pennelli G, et al. Gastritis staging in clinical practice: the OLGA staging system. Gut 2007;56:631-6. https://doi.org/10.1136/gut.2006.106666 10.1136/gut.2006.106666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rugge M, Sacchi D, Graham DY, Genta RM. Secondary prevention of gastric cancer: merging the endoscopic atrophic border with OLGA staging. Gut 2020;69:1151-2. https://doi.org/10.1136/gutjnl-2019-319107 10.1136/gutjnl-2019-319107 [DOI] [PubMed] [Google Scholar]
- 39.Niwa T, Tsukamoto T, Toyoda T, et al. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res 2010;70:1430-40. https://doi.org/10.1158/0008-5472.CAN-09-2755 10.1158/0008-5472.CAN-09-2755 [DOI] [PubMed] [Google Scholar]
- 40.Correa P. Chronic gastritis: a clinico-pathological classification. Am J Gastroenterol 1988;83:504-9. [PubMed] [Google Scholar]
- 41.Rugge M, Fassan M, Graham DY. Clinical guidelines: Secondary prevention of gastric cancer. Nat Rev Gastroenterol Hepatol 2012;9:128-9. https://doi.org/10.1038/nrgastro.2012.19 10.1038/nrgastro.2012.19 [DOI] [PubMed] [Google Scholar]
- 42.Rugge M, Genta RM, Di Mario F, et al. Gastric cancer as preventable disease. Clin Gastroenterol Hepatol 2017;15:1833-43. https://doi.org/10.1016/j.cgh.2017.05.023 10.1016/j.cgh.2017.05.023 [DOI] [PubMed] [Google Scholar]
- 43.Correa P, Piazuelo MB. The gastric precancerous cascade. J Dig Dis 2012;13:2-9. https://doi.org/10.1111/j.1751-2980.2011.00550.x 10.1111/j.1751-2980.2011.00550.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.WHO Classification of Tumours - Digestive system tumours. 5th Ed. Lyon: International Agency for Research on Cancer; 2019. [Google Scholar]
- 45.Capelle LG, de Vries AC, Haringsma J, et al. The staging of gastritis with the OLGA system by using intestinal metaplasia as an accurate alternative for atrophic gastritis. Gastrointest Endosc 2010;71:1150-8. https://doi.org/10.1016/j.gie.2009.12.029 10.1016/j.gie.2009.12.029 [DOI] [PubMed] [Google Scholar]
- 46.Rugge M, Fassan M, Pizzi M, et al. Operative link for gastritis assessment vs operative link on intestinal metaplasia assessment. World J Gastroenterol. 2011;17(41):4596-601. https://doi.org/10.3748/wjg.v17.i41.4596 10.3748/wjg.v17.i41.4596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rugge M, Sugano K, Scarpignato C, et al. Gastric cancer prevention targeted on risk assessment: Gastritis OLGA staging. Helicobacter 2019;24:e12571. https://doi.org/10.1111/hel.12571 10.1111/hel.12571 [DOI] [PubMed] [Google Scholar]
- 48.Rugge M, Genta RM, Fassan M, et al. OLGA Gastritis Staging for the Prediction of Gastric Cancer Risk: A Long-term Follow-up Study of 7436 Patients. Am J Gastroenterol 2018;113:1621-8. https://doi.org/10.1038/s41395-018-0353-8 10.1038/s41395-018-0353-8 [DOI] [PubMed] [Google Scholar]