

To the Editor:
Identification of immunoglobulin (IG) and T-cell receptor (TR) gene rearrangements in acute lymphoblastic leukemia (ALL) patients at initial presentation is crucial for monitoring of minimal residual disease (MRD) during subsequent follow-up and thereby for appropriate risk-group stratification. In a diagnostic setting, IG/TR gene rearrangements are generally identified using DNA-based PCR analysis, followed by classical Sanger sequencing or next generation sequencing (NGS) [1, 2]. Nowadays, whole transcriptome RNA sequencing (RNAseq) is frequently used to identify fusion genes and to assign patients into distinct molecular subgroups according to WHO 2016 classification, or for protocol-based clinical decisions [3]. Hence, it would be beneficial if RNAseq data could also be used for the identification of IG/TR gene rearrangements pertaining to the leukemic clone.
Recently, Yeoh and colleagues reported on the use of RNAseq data for identification of IG heavy chain (IGH) gene rearrangements, which was successful in approximately 90% of B-ALL patients [4]. Almost two-thirds of clonal IGH rearrangements were unproductive, whereas the vast majority (>98%) of background rearrangements were productive. Even though these data are promising, they are also incomplete since other IG/TR loci were not evaluated. Furthermore, these data underline that caution is warranted in the analysis of RNAseq data for IG/TR marker screening in ALL (and in other lymphoproliferative disorders requiring multiple RNA/DNA analyses): [5, 6] applying computational methods that only focus on productive rearrangements (e.g., like for most repertoire analyses) will clearly result in incomplete interpretation of IG/TR data for marker identification.
Within the EuroClonality-NGS Working Group and EuroMRD, we have developed, validated, and published assays for IG/TR DNA amplicon (‘DNAamp’ hereafter) and DNA capture-based analysis of relevant samples [1, 7, 8]. In line with these recent efforts, we decided to explore the possibilities and limitations of extracting data on IG/TR gene rearrangements from RNAseq data. Here we report on a complete but preliminary analysis of all IG/TR loci from RNAseq data, using DNAamp data as benchmark.
RNAseq data from 165 ALL patients at time of diagnosis were obtained using Illumina TruSeq Library Prep or Universal RNAseq kit (Nugen, Tecan) and sequenced by Illumina Next-Seq or Novaseq (2 × 75 bp). DNAamp IG/TR data were obtained from the same patients using the aforementioned assays developed by EuroClonality-NGS, which employ separate primer sets: IGHV-IGHD-IGHJ, IGHD-IGHJ, IGK (often split in two tubes: IGKV-IGKJ/Kde and intronRSS-Kde), TRBV-TRBD-TRBJ, TRBD-TRBJ, TRG, TRD—note the absence of an IGL and TRA primer set [1, 7]. The ARResT/Interrogate bioinformatics pipeline [9], which has also been developed and validated within EuroClonality-NGS [7, 9, 10], was used to produce profiles of 22 gene IG/TR rearrangement types, or junction classes, both complete and incomplete, (potentially) productive and unproductive, across all seven IG/TR loci: IGH, IGK, IGL, TRA, TRB, TRG, and TRD (see Table 1). Rearrangements were organized (e.g., for calculating their abundance) by junction class (rearrangement type), 5′ gene, junctional segmentation and N-(D)-N region statistics (5′ gene deletions, N-(D)-N length, 3′ gene deletions), 3′ gene, and junction amino acid sequence [7]. We only allowed IG/TR rearrangements appearing uniquely across cases to minimize potential contamination and artefacts. For identifying potential MRD markers in DNAamp data, we required a minimum abundance of 10 reads and 5% of reads (amplified in separate tubes by the corresponding EuroClonality-NGS primer set) [1].
Table 1.
IG/TR rearrangements detected by DNA amplicon-based methods (DNAamp) and by RNAseq.
| Rearrangements/casea | RNAseq MRD markers according to DNAamp benchmarkb | |||||
|---|---|---|---|---|---|---|
| DNAamp | RNAseq (“damaged”) | RNAseq Productive | DNAamp | RNAseq | RNAseq (%) | |
| IGH complete VJ:Vh-(Dh)-Jh | 2241 | 643 (215) | 364 | 223 | 188 | 84.3 |
| IGH incomplete DJ:Dh-Jh | 1407 | 21 (9) | na | 89 | 69 | 77.5 |
| IGK complete VJ:Vk-Jk | 762 | 436 (61) | 323 | 63 | 23 | 36.5 |
| IGK other Vk-Kde | 285 | <1 | na | 57 | 2 | 3.5 |
| IGK other intron-Kde | 780 | <1 | na | 45 | 1 | 2.2 |
| IGL complete VJ:Vl-Jl | nac | 329 (53) | 233 | na | ||
| TRA complete VJ:Va-Ja | 11 | 62 (20) | 25 | 0 | ||
| TRA + D complete VJ:Va-Jd | 10 | <1 | <1 | 0 | ||
| TRA + D complete VJ:Vd-(Dd)-Ja | 133 | 2 (1) | <1 | 33 | 14 | 42.4 |
| TRA + D incomplete DJ:Dd-Ja | 71 | 37 (25) | na | 27 | 1 | 3.7 |
| TRA + D incomplete VD:Va-Dd3 | na | <1 | na | na | ||
| TRB complete VJ:Vb-(Db)-Jb | 1921 | 128 (34) | 72 | 101 | 53 | 52.5 |
| TRB incomplete DJ:Db-Jb | 1555 | 13 (6) | na | 71 | 19 | 26.8 |
| TRB incomplete VD:Vb-Db | na | 1 (<1) | na | na | ||
| TRB other DD:Db-Db | na | <1 | na | na | ||
| TRD complete VJ:Vd-(Dd)-Jd | 184 | 2 (<1) | 1 | 17 | 4 | 23.5 |
| TRD incomplete DJ:Dd2-Jd | 139 | 1 (1) | na | 11 | 1 | 9.1 |
| TRD incomplete DJ:Dd3-Jd | na | <1 | na | na | ||
| TRD incomplete VD:Vd-Dd3 | 188 | <1 | na | 82 | 1 | 1.2 |
| TRD other DD:Dd2-Dd3 | 74 | 0 | na | 36 | 0 | 0 |
| TRD other DD:Dd3-Dd2 | na | <1 | na | na | ||
| TRG complete VJ:Vg-Jg | 2163 | 12 (2) | 3 | 198 | 91 | 46 |
aAverage number of rearrangements per case as detected by DNAamp and RNAseq. The RNAseq data refer to total number of rearrangements (with in between brackets the number of “damaged” sequences) and to the number of productive rearrangements. A total of 165 ALL patients were evaluated.
bNumber of potential MRD markers identified by DNAamp (as per abundance thresholds), and number and percentage of those markers identified by RNAseq at any abundance. A total of 165 ALL patients were evaluated.
cna: not analyzed/not applicable.
Comparison of DNAamp and RNAseq IG/TR data revealed clear differences in the number of identified IG/TR rearrangements (summarized in Fig. 1 and Table 1). Importantly, using an RNAseq read length of ~150 (2 × 75bp), around 25% of RNAseq rearrangements are missing part of the junctional residues (herein “damaged”) when the fragment is longer than the read coverage. Apart from the expected absence of TRA and IGL events from the DNAamp data, we observed additional differences in the average absolute number of rearrangements per case for each locus (Fig. 1A): the DNAamp data provide an average of 1703 rearrangements per case per locus (range 11–3648) compared to 211 (range 3–664) for RNAseq. When “damaged” rearrangements were ignored, RNAseq numbers fell to 157 (range 2–440). The most noticeable difference in relative incidence is the remarkable under-representation of TRB, TRD and TRG gene rearrangements in the RNAseq data. Furthermore, of the rearrangements identified by RNAseq, the vast majority is complete (96%) and the absolute majority is productive (>60%; Fig. 1B). In contrast, by DNAamp the complete rearrangements are relatively less frequent (~60%) and nonproductive rearrangements are found in about 40% of patients.
Fig. 1. Comparison between IG/TR rearrangements detected by amplicon-based assays (‘DNAamp’) and RNAseq in 165 ALL patients.
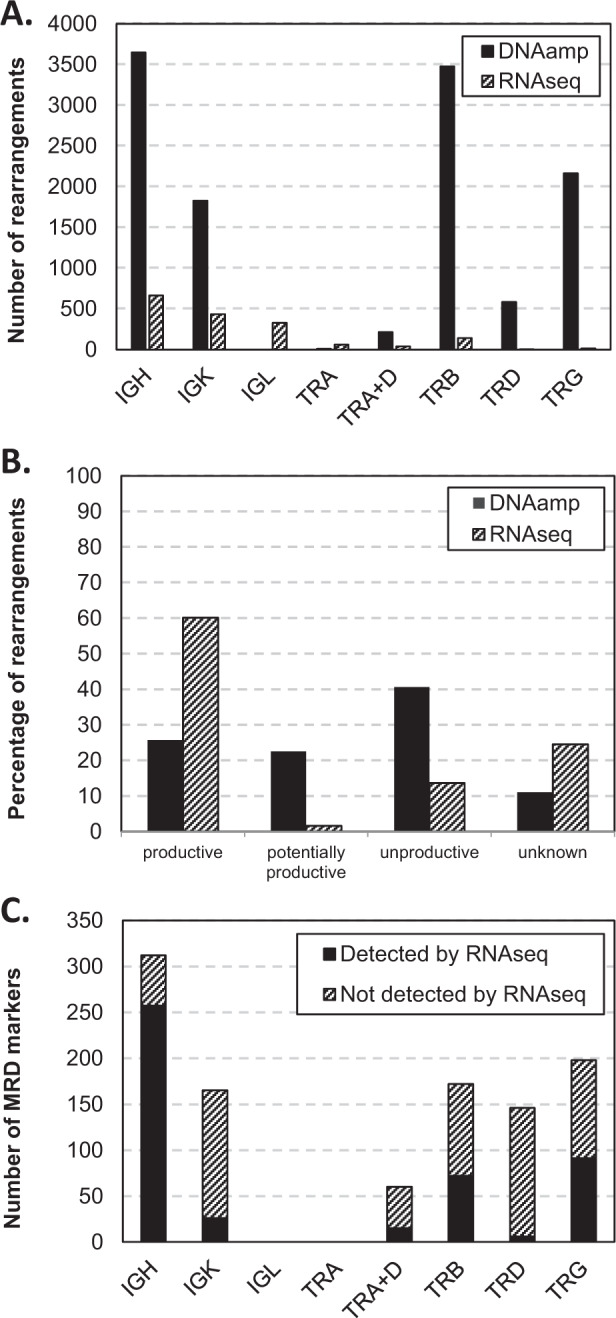
A Average number of rearrangements detected for the various IG/TR loci per case. B Frequencies of productive, potentially productive (incomplete sequences that have the potential to be productive when they would be complete), and unproductive rearrangements. Unknown: non-VJ and non-DJ classes, and “damaged” rearrangements from RNAseq. C Number of RNAseq re-identifiable MRD markers per target according to DNAamp benchmark.
The lower number of total IG/TR rearrangements detected by RNAseq as compared to DNAamp also limited the identification of potential MRD markers. Figure 1C and Table 1 show the absolute number of potential MRD markers above the aforementioned abundance thresholds in the DNAamp data, and results from using DNAamp as benchmark for the sensitivity of RNAseq to identify MRD markers. At present, the non-trivial question of appropriate thresholds for marker screening in RNAseq has not been answered. Therefore, we decided to search for the respective MRD marker obtained by the DNAamp approach in the RNAseq data at any abundance. This probably means that our results are overestimating the ability of RNAseq to recover DNAamp markers. Overall, less than half of the DNAamp markers were found in the RNAseq data (467/1053; 44%). Best concordance was observed for IGH (complete and incomplete) markers (82% of DNAamp markers also identified by RNAseq); other complete markers (IGK, TRB, TRA + D, TRD and TRG) were partially detected by RNAseq (24–52%), while most incomplete markers and virtually all IGK-Kde markers were only limitedly identified by RNAseq (<10%). Evaluation of the individual ALL cases showed that in eight out of 165 cases (5%) none of the DNAamp markers were found in the RNAseq results (again, at any abundance), while identical marker profiles were seen in only 19/165 cases (12%). In the remaining 138 patients, at least some of the potential markers were missed. When the same abundance criteria were used for both DNAamp and RNAseq, by RNAseq, 38 cases (23%) had one potential marker, 50 cases (30%) two, and 37 cases (22%) three (and seven cases had none); for DNAamp these numbers were six (4%), three (2%), and 14 (8%), respectively. The remaining cases (i.e., 33 (20%) in RNAseq, 142 (86%) in DNAamp) had four or more potential markers available. Thus, at least 2 IG/TR markers (required in most current clinical protocols) were identified by DNAamp in 159/165 (96%) cases and by RNAseq in 120/165 cases (73%).
Our preliminary data thus show that IG/TR rearrangements detected by DNA amplicon-based methods are clearly distinct from RNAseq-identified rearrangements. Obviously, this is to a large extent explained by the underlying (immuno)biology of IG/TR gene rearrangements, which will mainly be transcribed if complete and productive, thus allowing production of a functional IG/TR chain. In contrast, non-transcribed unproductive and incomplete rearrangements are hardly or not detectable by RNAseq. Of importance, about 70% of IGH rearrangements in ALL patients are unproductive, whereas non-leukemic rearrangements generally are productive [4, 6]. While it is appropriate to filter out out-of-frame immune rearrangements in analysis of functional repertoires, this is certainly not appropriate for ALL IG/TR marker analysis [5]. In our analysis, we did not yet take full transcription levels into account (i.e., we counted in de-duplicated reads, or unique fragments, as in DNA capture analyses), but it will be interesting to evaluate whether for example TR rearrangements in reactive T-cell clones show differences in transcript levels compared to cross-lineage TR rearrangements in B-cell precursor type ALL. From a technical perspective, it should be noted that we applied 2 × 75 bp sequencing and it may be expected that IG/TR rearrangement detection will be improved if longer reads are used. From a clinical perspective, RNAseq data may be used for MRD marker identification in those cases where DNAamp methods are unsuccessful or unavailable, if appropriate bioinformatic strategies are used.
Of note, IG/TR rearrangements may also be derived from whole-exome sequencing (WES) or whole-genome sequencing (WGS) datasets that, in contrast to RNAseq data, do not depend on the transcriptional level of rearrangements. This creates a clear advantage as was recently showcased in work introducing IgCaller for WGS-derived IGH data [11].
We therefore plan to continue and expand on the evaluation of not only RNAseq, but also WES and WGS data, mainly in ALL but also in NHL, in comparison to results obtained from a large set of DNAamp and targeted DNA capture data from the EuroClonality-NGS/EuroMRD groups. Challenges will include compiling appropriate rules and thresholds for marker identification, preparing specific protocols and suggesting limits for the safe use for each technology, for example and especially for MRD monitoring.
Acknowledgements
GC was supported by a grant from the Italian Association for Cancer Research (AIRC), grant IG2015-17593. BS was supported by a grant from the Dutch Cancer Society (2017/11137).
EuroMRD
Vincent H. J. van der Velden1, Monika Brüggemann2, Giovanni Cazzaniga3, Simona Songia3, Jan Trka6
EuroClonality-NGS Working Group
Monika Brüggemann2, Giovanni Cazzaniga3, Blanca Scheijen4, Jan Trka6, Karol Pal2,7, Sonja Hänzelmann2, Grazia Fazio3, Simona Songia3, Anton W. Langerak1, Nikos Darzentas2,7
Compliance with ethical standards
Conflict of interest
The EuroClonality-NGS Working Group is an independent scientific subdivision of EuroClonality that aims at innovation, standardization and education in the field of diagnostic clonality analysis. The revenues of the previously obtained patent (PCT/NL2003/000690), which is collectively owned by the EuroClonality Foundation and licensed to InVivoScribe, are exclusively used for EuroClonality activities, such as for covering costs of the Working Group meetings, collective WorkPackages and the EuroClonality Educational Workshops. The EuroClonality consortium operates under an umbrella of ESLHO, which is an official EHA Scientific Working Group. VHJvdV: contract research for Pfizer and Janssen, Service Level Agreements with BD Biosciences and Agilent. MB reports personal fees from Incyte (advisory board), financial support for reference diagnostics from Affimed, Amgen and Regeneron, grants and personal fees from Amgen (advisory board, speakers bureau, travel support), personal fees from Janssen (speakers bureau), personal fees from Molecular Health (advisory board), all outside the submitted work. AWL: contract research for Roche-Genentech, research support from Gilead, advisory board for AbbVie, speaker for Gilead, Janssen. The other authors declare no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Lists of authors and their affiliations of EuroMRD and EuroClonality-NGS Working Group appear at the below Acknowledgements.
Contributor Information
Vincent H. J. van der Velden, Email: v.h.j.vandervelden@erasmusmc.nl
EuroMRD:
References
- 1.Bruggemann M, Kotrova M, Knecht H, Bartram J, Boudjogrha M, Bystry V, et al. Standardized next-generation sequencing of immunoglobulin and T-cell receptor gene recombinations for MRD marker identification in acute lymphoblastic leukaemia; a EuroClonality-NGS validation study. Leukemia. 2019;33:2241–53. doi: 10.1038/s41375-019-0496-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Velden VH, Cazzaniga G, Schrauder A, Hancock J, Bader P, Panzer-Grumayer ER, et al. Analysis of minimal residual disease by Ig/TCR gene rearrangements: guidelines for interpretation of real-time quantitative PCR data. Leukemia. 2007;21:604–11. doi: 10.1038/sj.leu.2404586. [DOI] [PubMed] [Google Scholar]
- 3.Grioni A, Fazio G, Rigamonti S, Bystry V, Daniele G, Dostalova Z, et al. A Simple RNA target capture NGS strategy for fusion genes assessment in the diagnostics of pediatric B-cell acute lymphoblastic. Leukemia. Hemasphere. 2019;3:e250. doi: 10.1097/HS9.0000000000000250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Z, Jiang N, Lim EH, Chin WHN, Lu Y, Chiew KH, et al. Identifying IGH disease clones for MRD monitoring in childhood B-cell acute lymphoblastic leukemia using RNA-Seq. Leukemia. 2020;34:2418–29. doi: 10.1038/s41375-020-0774-4. [DOI] [PubMed] [Google Scholar]
- 5.Bueno C, Ballerini P, Varela I, Menendez P, Bashford-Rogers R. Shared D-J rearrangements reveal cell of origin of TCF3-ZNF384 and PTPN11 mutations in monozygotic twins with concordant BCP-ALL. Blood. 2020;136:1108–11. doi: 10.1182/blood.2020006604. [DOI] [PubMed] [Google Scholar]
- 6.Abdo C, Thonier F, Simonin M, Kaltenbach S, Valduga J, Petit A, et al. Caution encouraged in next-generation sequencing immunogenetic analyses in acute lymphoblastic leukemia. Blood. 2020;136:1105–7. doi: 10.1182/blood.2020005613. [DOI] [PubMed] [Google Scholar]
- 7.Knecht H, Reigl T, Kotrova M, Appelt F, Stewart P, Bystry V, et al. Quality control and quantification in IG/TR next-generation sequencing marker identification: protocols and bioinformatic functionalities by EuroClonality-NGS. Leukemia. 2019;33:2254–65. doi: 10.1038/s41375-019-0499-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stewart P, Gazdova J, Darzentas N, Wren D, Proszek P, Fazio G, et al. Euroclonality-NGS DNA capture panel for integrated analysis of IG/TR rearrangements, translocations, copy number and sequence variation in lymphoproliferative disorders. Blood. 2019;134(Supplement 1):S888. doi: 10.1182/blood-2019-127822. [DOI] [Google Scholar]
- 9.Bystry V, Reigl T, Krejci A, Demko M, Hanakova B, Grioni A, et al. ARResT/Interrogate: an interactive immunoprofiler for IG/TR NGS data. Bioinformatics. 2017;33:435–7. doi: 10.1093/bioinformatics/btw634. [DOI] [PubMed] [Google Scholar]
- 10.Scheijen B, Meijers RWJ, Rijntjes J, van der Klift MY, Mobs M, Steinhilber J, et al. Next-generation sequencing of immunoglobulin gene rearrangements for clonality assessment: a technical feasibility study by EuroClonality-NGS. Leukemia. 2019;33:2227–40. doi: 10.1038/s41375-019-0508-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nadeu F, Mas-de-Les-Valls R, Navarro A, Royo R, Martin S, Villamor N, et al. IgCaller for reconstructing immunoglobulin gene rearrangements and oncogenic translocations from whole-genome sequencing in lymphoid neoplasms. Nat Commun. 2020;11:3390. doi: 10.1038/s41467-020-17095-7. [DOI] [PMC free article] [PubMed] [Google Scholar]