

Abstract
MAPK-targeting in cancer often fails due to MAPK-reactivation. MEK inhibitor (MEKi) monotherapy provides limited clinical benefits but may serve as a foundation for combination. Here, we showed that combining a type II RAFi with an allosteric MEKi durably prevents and overcomes acquired resistance among cancers with KRAS, NRAS, NF1, BRAFnon-V600 and BRAFV600 mutations. Tumor cell-intrinsically, type II RAFi plus MEKi sequester MEK in RAF complexes, reduce MEK/MEK dimerization, and uncouple MEK from ERK in acquired-resistant tumor subpopulations. Immunologically, this combination expands memory and activated/exhausted CD8+ T-cells, and durable tumor regression elicited by this combination requires CD8+ T-cells, which can be reinvigorated by anti-PD-L1 therapy. Whereas MEKi reduces dominant intra-tumoral T-cell clones, type II RAFi co-treatment reverses this effect and promotes T-cell clonotypic expansion. These findings rationalize the clinical development of type II RAFi plus MEKi and their further combination with PD-1/L1-targeted therapy.
INTRODUCTION
RAS-RAF-MEK-ERK signaling is hyperactivated in more than 30-40% of human cancers (1), including ~70% of advanced melanoma driven by BRAFV600, non-BRAFV600 (atypical BRAF), NRAS and NF1 mutants. Type I RAF inhibitor (RAFi), such as vemurafenib, dabrafenib, encorafenib, were first developed successfully against advanced BRAFV600MUT melanoma, but acquired resistance is almost universal. As a single-agent, allosteric MEK1/2 inhibitors (MEKi), such as cobimetinib, trametinib, binimetinib and selumetinib elicits limited clinical response rates in a wide range of malignancies that harbor RAF and RAS mutations, e.g., BRAFV600MUT and NRASMUT melanoma (2,3). However, the combination of type I RAFi+MEKi suppresses acquired resistance in tumors driven by BRAFV600 mutants and increases the therapeutic window in patients (4,5), although acquired resistance caused by MAPK pathway reactivation is still commonplace (6–8). In human cancers where RAS-RAF-MEK-ERK signaling is not hyperactivated by BRAFV600 mutants, a MEKi-based combination that can sustainably block the MAPK pathway and resistance-associated reactivation has not been developed successfully.
A durably effective MEKi-based combination should prevent MAPK reactivation (9,10) and preserve and/or promote anti-tumor T-cell immunity. MAPK-targeted therapy combined with immune checkpoint blockade is undergoing clinical testing (11,12), and anti-tumor T-cell immunity has been proposed to contribute to the durability of MAPK-targeted therapy (13). MEKi may be deleterious to tumor antigen-specific T-cells (14)(15), which may be ameliorated by pulsatile MEKi dosing (16).
Next-generation MAPK pathway inhibitors may help overcome MEKi resistance commonly caused by MAPK reactivation, which is in turn caused by up-regulation of “back-to-back” RAF/RAF and/or “face-to-face” RAF/MEK dimerization. For instance, BRAFV600 mutants, which signal as monomers but form dimers due to resistance-associated alterations, and non-BRAFV600-activating mutants, which signal as RAS-independent dimers, can both be targeted by a novel dimer breaker, PLX8394 (17). This type of compounds “breaks” the paradox of type I RAFi, which is BRAF monomer-specific but has the liability of inducing BRAFV600MUT-containing dimers and causing paradoxical MAPK hyperactivation in RAS-activated cancer cells. However, paradox breakers are not effective against CRAFWT or BRAFWT homo- or hetero-dimers, which drive MAPK hyperactivation in the majority of cancers. Type II RAFi (aka dimeric or omni-RAFi) has anti-tumor activity in RASMUT or BRAFMUT cancers (18–21) (22) but, as a single-agent, does not appear to be highly active clinically (23) (24).
To evaluate the clinical potential and mechanisms underlying the combination of type II RAFi and allosteric MEKi, we investigated: (i) durability in preventing as well as overcoming acquired MEKi resistance across potentially MAPK-addicted cancer lineages, (ii) combined mechanistic action consistent with preventing MAPK-reactivation, and (iii) in vivo T-cell impacts.
RESULTS
Type II RAFi Combination Prevents and Overcomes Acquired MEKi Resistance
We tested type II RAFi (BGB-283, RAF-709) at a sub-micromolar (0.5 μM) concentration and/or allosteric MEKi (trametinib, binimetinib) at a nanomolar (20 nM) concentration against a panel (n=22) of human melanoma, colorectal carcinoma (CRC), pancreatic ductal carcinoma (PDAC), and non-small cell lung cancer (NSCLC) cell lines driven by BRAFV600E, NF1−/−, NRASMUT or KRASMUT in short-term (14-day or 14d) clonogenic assays (Fig. 1A; Supplementary Fig. S1A; Supplementary Table S1 for a list of cell lines and in vivo models used in this study). In general, while type II RAFi or MEKi individually was ineffective at preventing short-term growth, type II RAFi+MEKi prevented macroscopic growth over 14d. Moreover, using a panel of BRAFV600E melanoma sub-lines (n=4) with acquired resistance to type I RAFi+MEKi (vemurafenib+selumetinib/AZD-6244) (7), we tested whether switching from type I RAFi to type II RAFi, would overcome resistance (Fig. 1B). In one set of cultures, we maintained these lines with both type I RAFi and MEKi or withdrew one or both inhibitors. As expected, withdrawal of both reduced growth fitness due to drug addiction (7,25). In two additional sets of cultures, we introduced (or switched to) a type II RAFi (BGB-283 or RAF-709). Importantly, switching from type I to type II RAFi (over 10-15d) overcame growth of resistant sub-lines, but this occurred only when an allosteric MEKi was present.
Figure 1.
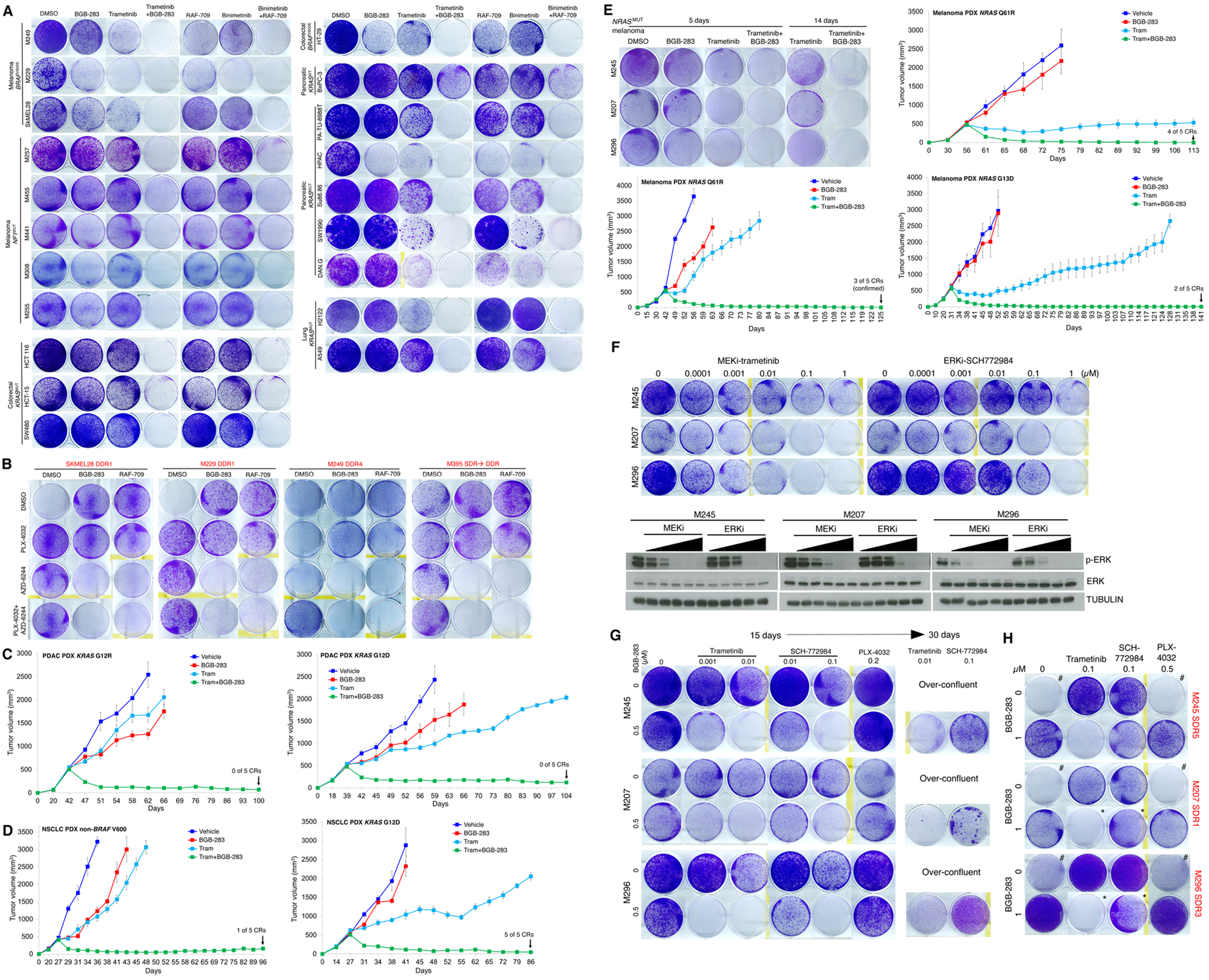
Type II RAFi+MEKi forestalls and overcomes acquired resistance in BRAFMUT, NF1MUT, KRAS MUT and NRAS MUT cancers.
A, Clonogenic growth (14 days) of indicated inhibitor-naïve BRAFV600 or NF1 mutant melanoma, BRAFV600MUT or KRASMUT colorectal carcinoma (CRC), KRASMUT non-small cell lung carcinoma (NSCLC), and KRASMUT (vs. KRASWT) pancreatic ductal adenocarcinoma (PDAC) cell lines after treatment with vehicle (DMSO), BGB-283 (0.5 μM), trametinib (0.02 μM), or trametinib (0.02 μM) plus BGB-283 (0.5 μM) or alternatively with RAF709 (0.5 μM), binimetinib (0.02 μM), or binimetinib (0.02 μM) plus RAF709 (0.5 μM). Data representative of two replicates.
B, Clonogenic growth (15 days, left two sub-lines; 10 days, right two sub-lines) of indicated type I RAFi+MEKi double-drug resistant (DDR; hereafter all names of sub-lines with acquired drug resistance shown in red text) BRAFV600E melanoma sub-lines. DDR sub-lines were maintained on both PLX4032 (1 μM) plus AZD6244 (1 μM), either PLX4032 or AZD6244, or withdrawn from both inhibitors. On top of these four conditions, a type II RAFi (BGB-283 at 1 μM or RAF-709 at 1 μM) was added. Data representative of two replicates.
C, D, Tumor volumes of PDAC (n=2; XWR6, left; XWR7, right, c) and NSCLC (n=2; LBM013-P, left; TM00302, right, d) PDXs in mice that were treated with vehicle, trametinib (3 mg/kg/day PO), BGB-283 (20 mg/kg/day PO), or the combination of trametinib and BGB-283. Vehicle or inhibitor treatments were started at tumor volumes of ~500 mm3. N=5 tumors per group; means ± SEMs.
E, Clonogenic growth (5 or 14 days, upper left panel) of indicated inhibitor-naïve NRASMUT melanoma cell lines after treatment with vehicle (DMSO), BGB-283 (0.5 μM), trametinib (0.02 μM), or trametinib (0.02 μM) plus BGB-283 (0.5 μM). Data representative of two replicates. Tumor volumes of NRASMUT (n=3; NRAS_PDX3, top; NRAS_PDX4, middle; NRAS_PDX1, bottom) PDXs in mice that were treated with vehicle, trametinib (3 mg/kg/day PO), BGB-283 (20 mg/kg/day PO), or the combination of trametinib and BGB-283. Vehicle or inhibitor treatments were started at tumor volumes of ~500 mm3. N=5 tumors per group; means ± SEMs.
F, (Top) Clonogenic growth (10 days) of indicated inhibitor-naïve NRASMUT melanoma cell lines after treatment with vehicle (DMSO) or indicated concentration of MEKi (trametinib) or ERKi (SCH772984). Data representative of two replicates. (Bottom) Western blots of lysates from NRASMUT melanoma cell lines treated (2 hours) with vehicle (DMSO) or the same concentrations of MEKi or ERKi (Top) with indicated antibodies.
G, Clonogenic growth (15 or 30 days) of indicated inhibitor-naïve NRASMUT melanoma cell lines after treatment with vehicle (DMSO) or trametinib, SCH772984, or the type I RAFi PLX-4032 (15 day only) at the indicated concentrations, ± BGB-283. Over-confluent cultures without BGB-283 co-treatment were terminated early and not shown for day 30. Data representative of two replicates.
H, As in G, except NRASMUT melanoma sub-lines (single-drug resistant or SDR, indicated in red) with acquired MEKi (trametinib) resistance were used. Day 15 cultures are shown except those marked with * (which indicates day 30 cultures). Cultures marked with # display the drug addiction phenotype.
In patient-derived xenograft (PDX) models of PDAC (n=2, KRASMUT) or of NSCLC (n=1, non-BRAFV600MUT; n=1, KRASMUT), daily treatment with BGB-283 (20 mg/kg PO) or trametinib (3 mg/kg PO) alone led to minimal or transient tumor growth-deceleration (Fig. 1C and Fig. 1D). However, in all four PDXs, combination treatment achieved durable tumor regression (86d to 104d follow-ups), with complete responses (CRs) noted in both NSCLC PDXs. Since the combination of type II RAFi+MEKi was highly effective in preventing MEKi resistance in short-term cultures of NRASMUT melanoma cell lines (Fig. 1E; Supplementary Fig. S1A), we also tested them against PDXs of NRASMUT melanoma (n=3). In vivo, responses to daily treatment with BGB-283 (20 mg/kg PO) or trametinib (3 mg/kg PO) alone ranged from none to tumor stabilization (without regression) (Fig. 1E). In all three NRASMUT melanoma PDXs, the combination achieved highly durable tumor regression in all tumors, resulting in 2/5 to 4/5 CRs (113d to 141d follow-ups) (Fig. 1E). CRs were confirmed in one PDX model by stopping treatment with both type II RAFi+MEKi at d125, with no relapse during a 109d follow-up. Tumors that relapsed in two mice were sensitive to retreatment on d162 with both inhibitors (secondary CRs) (Supplementary Fig. S1B).
Differences of Type II RAFi+MEKi (vs. Type II RAFi+ERKi) in Antagonizing Acquired MEKi Resistance
We then tested whether the durability of type II RAFi+MEKi in preventing acquired MEKi resistance is specific to this combination when compared to type II RAFi+ERKi. To compare the relative combinatorial potencies in preventing or overcoming acquired MEKi resistance, we first identified concentrations of MEKi (trametinib) vs. ERKi (SCH772984 or BVD-523) with functionally equivalent impacts on the short-term (10d) growth of MEKi-naïve, parental NRASMUT melanoma cell lines (M207, M245, M296) or the short-term (8-11d) growth of isogenic NRASMUT melanoma sub-lines with acquired MEKi (trametinib, 0.1 μM) resistance (M207 SDR1, M245 SDR5, M296 SDR3; SDR, Single Drug Resistant) (25). In MEKi-naïve, parental cells, 0.01 μM trametinib and 0.1 μM SCH772984 were approximately equivalent in suppressing short-term (10d) growth or short-term (2h) p-ERK levels (Fig. 1F). In isogenic sub-lines with acquired MEKi resistance, switching from trametinib at 0.1 μM to SCH772984 at 0.1 μM or BVD-523 at 1 μM had no or minimal impact on short-term (8-11d) growth, as measured by the MTT assay, viable cell counting or clonogenic assay (Supplementary Fig. S1C and Fig. S1D). We also evaluated the temporal impact of single-agent MEKi vs. ERKi doses on the MAPK pathway in NRASMUT melanoma sub-lines with acquired MEKi resistance. We measured p-ERK and p-p90RSK levels after treatment with MEKi or ERKi for 1h or 48h, with or without prior treatments with the same inhibitor for 0, 2, 4 or 6 days (Supplementary Fig. S1E to Fig. S1G). Within this time span, treatment(s) with trametinib at 0.1 μM, SCH772984 at 0.1 μM or BVD-523 at 1 μM resulted in similar p-p90RSK levels, which are consistent with similar effective ERK activities and growth rates. However, in all acquired-resistant sub-lines, the highly similar effective ERK activities over time (as measured by p-p90RSK levels and cellular growth rates) between MEKi vs. ERKi treatments contrasted glaringly with p-ERK levels. Only with ERKi (but not MEKi) treatment did we observe high rebounding levels of p-ERK 1h and 48h after the last fresh dose of treatment, regardless of the duration of prior ERKi treatment. This p-ERK rebound was stronger with BVD-523 and MK-8353 than with SCH772984. Unlike BVD-523 and MK-8353, SCH-772984 is known to inhibit both the kinase activity of ERK and MEK-mediated phosphorylation of the ERK activation loop. Rebound p-ERK levels in response to BVD-523 (1 μM) and MK-8353 (1 μM) treatment was abundant within 1h after treatment but were also readily detectable within 2d of SCH-772984 at 0.4 μM and within 4-6d of SCH-772984 at 0.1 μM. To our surprise, ERKi-induced p-ERK rebound levels did not accelerate growth rates of NRASMUT melanoma sub-lines with acquired MAPKi resistance.
Having established growth rate-equivalent concentrations of MEKi vs. ERKi, we compared the relative potencies of combination with type II RAFi in preventing acquired MEKi resistance in drug-naïve cells. In cancer cell lines, acquired resistance to targeted therapy develops through temporal stages: early slow-cycling or drug-tolerant persistence (pseudo-senescence) and later fast-cycling or proliferative growth (26). Accordingly, using parental NRASMUT melanoma cell lines (n=3) in short-term (15d) or long-term (30d) cultures (Fig. 1G), we compared the efficacies of type II RAFi (BGB-283 at 0.5 μM) in preventing growth when combined with functionally equivalent doses of MEKi (trametinib) vs. ERKi (SCH772984) (Fig. 1F). While BGB-283+trametinib and BGB-283+SCH772984 were similarly effective at preventing 15d growth, BGB-283+trametinib was far more effective than BGB-283+SCH772984 at preventing 30d growth. Since proliferative MEKi-resistant clones are expected to arise with longer-term culture, type II RAFi+MEKi may be more active than type II RAFi+ERKi in overcoming the growth of proliferative MEKi-resistant clones. To test this hypothesis, we compared the relative potencies of type II RAFi+MEKi vs. type II RAFi+ERKi in overcoming acquired MAPKi resistance in three melanoma subsets: NRASMUT melanoma sub-lines (n=3) with acquired MEKi resistance (Fig. 1H and Supplementary Fig. S1H), BRAFV600MUT melanoma sub-lines (n=4) with acquired resistance to type I RAFi+MEKi (Supplementary Fig. S1I and Fig. S1J), and NF1−/− melanoma sub-lines with acquired MEKi resistance (n=4) (Supplementary Fig. S1K and Fig. S1L). Using functionally equivalent doses of MEKi (trametinib) vs. ERKi (SCH772984, BVD-523, MK-8353) (Fig. 1H and Supplementary Fig. S1C to S1G), we observed consistently (in 11 of 11 acquired MAPKi resistant melanoma sub-lines) that BGB-283+trametinib was more effective than BGB-283+SCH772984 (or BVD-523, MK-8353) at overcoming the growth of established acquired resistant clones.
NRASMUT Melanoma Acquire MEKi Resistance via ERK Reactivation
Given the durability of type II RAFi+MEKi in preventing and overcoming MEKi resistance, we tested the hypothesis that NRASMUT melanoma acquire MEKi resistance predominantly through ERK reactivation. We treated NRASMUT PDXs (n=5) with trametinib (5 mg/kg/day) until acquired resistant tumors (n=16) emerged (Fig. 2A). The durability of response was highly variable across distinct and within each model(s). Together with aforementioned NRASMUT melanoma parental (P) (n=3) and isogenic, acquired trametinib-resistant cell lines (n=5), we extracted genomic DNAs and total RNAs from vehicle-treated tumors and isogenic resistant (R) tumors for whole exome sequencing (WES, along with patient-matched normal gDNAs) and RNA-seq. We then integrated WES/RNA-seq analysis to evaluate the recurrence of gain- or loss-of-function (GOF or LOF) gene-based events of 723 cancer-related genes (COSMIC v.88) (27) (Supplementary Table S2). We rank-ordered the recurrence of somatic, resistance-associated alterations based primarily on sample counts and secondarily on patient frequencies (Supplementary Fig. S2A). Among top 20 GOF genes were overlapping cMET and ERBB2 mRNA up-regulation (≥ 2-fold). However, the most recurrent GOF genes at the genomic level were NRAS (7/21 samples with copy number gain or mutant allele-specific loss-of-heterozygosity or LOH events) and RAF1/CRAF (5/21) (Supplementary Fig. S2A, Supplementary Tables S3 and S4). We have previously shown (4,7,13,26,28,29) that BRAFV600 mutant melanoma acquire resistance to type I RAFi+MEKi by omic alterations (e.g., BRAFV600E/K amplification-driven over-expression and RAF-regulated MEK1/2 mutants (7,30)) that enhance RAF/RAF and/or RAF/MEK interactions. Although RAF1/CRAF genomic alterations are not known to cause MAPKi resistance in BRAFV600 mutant melanoma, we observed highly recurrent (and largely mutual exclusivity) GOF alterations in NRAS, RAF1/CRAF, BRAF and MAP2K1/2 (Fig. 2B and Fig. 2C). MAP2K2 harbored resistance-causative somatic mutations (F57L, V64F and F133L) (Supplementary Fig. S2B), as equivalent positions in MAP2K1 (F53, V60 and F129) have been shown to confer MAPKi resistance in BRAFV600MUT melanoma (7). Phylogenetically, resistant tumors or sub-lines are sometimes distantly related to common ancestral clones (Fig. 2D and Supplementary Fig. S2C), indicating that tumor heterogeneity (preexisting or induced by therapy) contributes to MEKi resistance.
Figure 2.
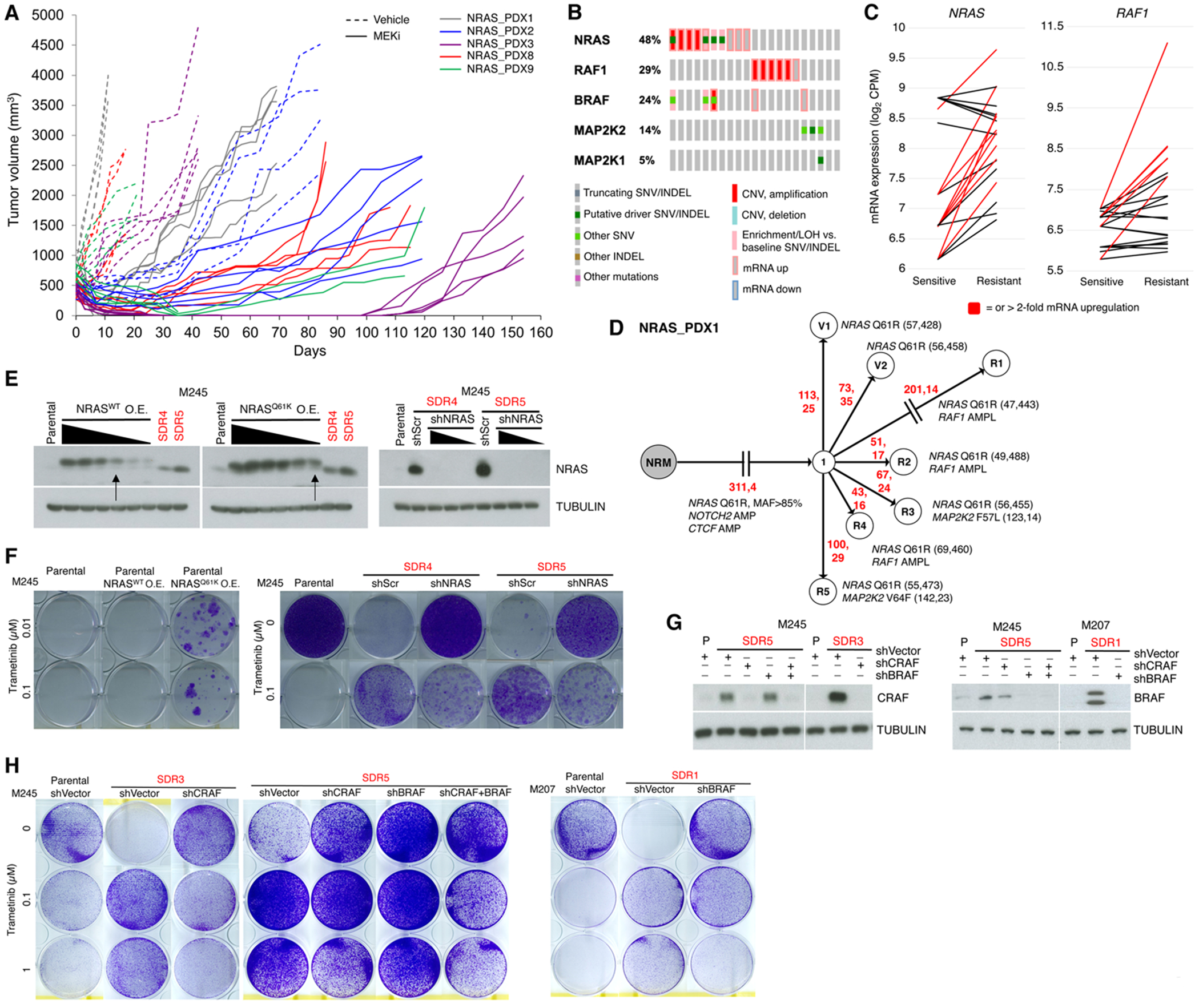
NRAS, BRAF, CRAF and MEK1/2 genomic and transcriptomic alterations drive acquired MEKi resistance in NRASMUT melanoma.
A, Volumes of all PDX tumors individually displayed over the course of daily vehicle (dotted lines) or trametinib at 5 mg/kg/day PO (solid lines) treatment (initiated when tumor volumes near 500 mm3).
B, Most recurrent ERK pathway GOF genes in trametinib-resistant NRASMUT melanoma PDX tumors or sub-lines vs. vehicle-treated PDX tumors or isogenic, parental cell lines, respectively.
C, Levels of NRAS and RAF1(CRAF) mRNA in MEKi-resistant vs. -sensitive NRASMUT melanoma.
D, Phylogenetic relationships of MEKi-resistant vs. vehicle-treated NRASMUT melanoma PDX tumors. Branch lengths proportional to the numbers (in red) of somatic SNVs and INDELs. Nearest common ancestors enumerated. Shared and private genomic alterations of CGC genes listed along with wild type and mutant allele frequencies.
E, NRAS Western blots (WBs) of indicated cell lines transduced to over-express (O.E.) NRASWT or NRASQ61K (left) or to express shSCRAMBLE (shScr) or shNRAS (right). Single (MEKi) Drug Resistant (SDR) sub-lines indicated in red. TUBULIN, loading controls. Arrows indicate engineered cell lines for assay in f.
F, Clonogenic growth (14 days) of indicated cell lines (e) at two indicated trametinib concentrations.
G, CRAF or BRAF WBs of indicated cell lines transduced to express shCRAF and/or shBRAF.
H, Clonogenic growth (14 days) of indicated cell lines in g at indicated trametinib concentrations.
To evaluate the functional roles of NRASMUT and RAF1/CRAF up-regulation, we first over-expressed NRASMUT (vs. NRASWT) in the M245 P line to a level similar to NRASMUT up-regulation observed in M245 SDR4 or SDR5 (Fig. 2E, left). We also knocked down NRAS over-expression in M245 SDR4 and SDR5 using two independent shRNAs (Fig. 2E, right). We observed in clonogenic assays that NRASMUT over-expression conferred resistance to trametinib relative to M245 P or M245 P over-expressing NRASWT (Fig. 2F, left). We also observed that NRAS knockdown in either M245 SDR4 or SDR5 sensitized these sub-lines to 0.1 μM of trametinib (Fig. 2F, right). As we have reported (25), MEKi-resistant NRASMUT melanoma sub-lines are highly addicted to MEKi withdrawal. NRAS knockdown in the SDR sub-lines abolished MEKi addiction, likely because NRASMUT over-expression drives p-ERK rebound and hence cell death after MEKi withdrawal. Moreover, M245 SDR3 up-regulates CRAF expression; M245 SDR5, both CRAF and BRAF; and M207 SDR1, BRAF (Supplementary Tables S1, S3 and S4). Thus, we knocked down CRAF alone, both CRAF/BRAF, or BRAF alone in the respective SDR sub-lines using previously validated shRNAs (7,29) (Fig. 2G). Whereas P lines were highly sensitive to 0.1 μM of trametinib and all three SDR sub-lines grew even in 1 μM of trametinib, CRAF and/or BRAF knockdown in the SDR sub-lines conferred sensitivity to 0.1 μM of trametinib in clonogenic assays (Fig. 2H).
Type II RAFi+MEKi Stabilize RAF/MEK and Uncouple MEK/ERK Complexes
We then sought to understand the mechanism underlying the durability of type II RAFi+MEKi in overcoming acquired MEKi resistance. Consistent with NRASMUT, BRAF and CRAF alterations, we detected enhanced endogenous BRAF-CRAF interaction via co-immunoprecipitation (co-IP) in all SDR sub-lines (vs. isogenic P lines) (Fig. 3A). M296 SDR3, in particular, harbors both sub-clonal MAP2K1 (MEK1 F53L) and MAP2K2 (MEK2 F133L) mutations, which disrupt negative regulation of the kinase domain by helix A and result in RAF-dependent ERK activation (30,31). The RAF-dependency of MEK1 F53L and MEK2 F133L may promote BRAF/CRAF complexes, analogous to allosteric BRAF activation by MEK binding to KSR (32). We then tested how type II RAFi dose-dependently affects growth, BRAF/CRAF interaction and activation-associated phosphorylation of MEK and ERK in MEKi-treated/resistant cells (vs. MEKi-naïve/non-treated P cells). Type II RAFi (BGB-283, BGB-3245, RAF709, LXH254) dose-dependently re-sensitized SDR sub-lines to MEKi (Fig. 3B and Supplementary Fig. S3A to S3D). Consistently, type II RAFi dose-dependently and sustainably reversed p-ERK levels or ERK reactivation in SDR sub-lines in the presence of trametinib (Fig. 3C and Fig. 3D), but, unexpectedly, did not reverse p-MEK accumulation (Fig. 3D). Thus, type II RAFi co-treatment of MEKi-treated/resistant sub-lines led to p-ERK loss but curiously p-MEK persistence.
Figure 3.
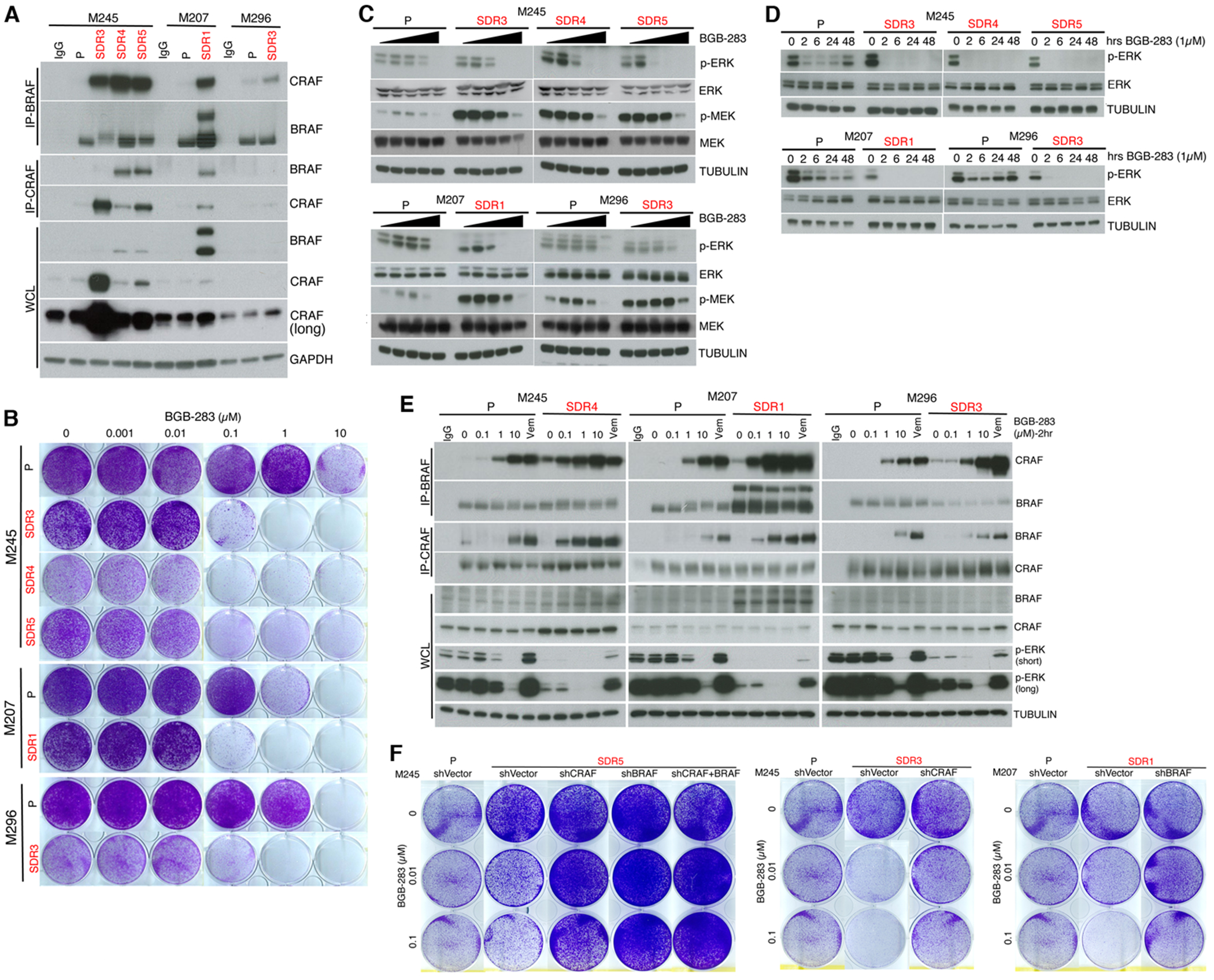
Type II RAFi+MEKi overcomes ERK re-activation and growth in acquired resistant melanoma by inducing BRAF/CRAF complex without reducing p-MEK.
A, Immunoprecipitation (IP) of indicated cell lines (parental or P without trametinib and SDR sub-lines with 0.1 μM of trametinib; this convention followed in all experiments, unless otherwise indicated) followed by WBs or direct WBs of whole cell lysates (WCLs) of indicated cell lines. IgG, isotype control for IP.
B, Clonogenic growth (15 days) of indicated cell lines treated with the indicated concentrations of BGB-283. Data representative of two replicates.
C, Total and phospho-MEK or -ERK WBs of lysates from indicated cell lines treated with increasing concentrations (0, 0.01, 0.1, 1 or 10 μM) of BGB-283 (2 hours).
D, Total and phospho-ERK WBs of lysates from indicated cell lines treated with 1 μM of BGB-283 for indicated durations (hours or hrs).
E, IP-WBs or direct WBs of WCL after treatment of indicated cell lines with increasing concentrations of BGB-283 or 0.5 μM of vemurafenib (2 hours). IgG, isotype control for IP. Last trametinib (0.1 μM) dose (only for SDR sub-lines) added 16 hours prior to cell lysis.
F, Clonogenic growth (14 days) of indicated P lines and SDR sub-lines engineered as in Figure 2g and either untreated or treated with two concentrations of BGB-283.
Type II RAFi RAF709 as a single-agent has been reported to induce RAF dimerization (19). In MEKi-naïve/non-treated P cells, type I RAFi (vemurafenib, 0.5 μM) as a single-agent induced both BRAF/CRAF interaction and p-ERK levels. In contrast, type II RAFi (BGB-283 or RAF709) as a single-agent at 10 μM induced equivalent levels of BRAF/CRAF interaction but suppressed p-ERK levels (Fig. 3E and Supplementary Fig. S3E). In MEKi-treated/resistant SDR cells, type I RAFi co-treatment also induced both BRAF/CRAF interaction and p-ERK levels. Importantly, type II RAFi co-treatment induced BRAF/CRAF interaction (on top of higher preexisting levels compared to P cells) at 0.1 μM, leading to p-ERK suppression at sub-micromolar concentrations of type II RAFi. These findings suggest that the effective concentration of type II RAFi to induce signal-incompetent (p-ERK suppressed) BRAF/CRAF complexes depends on MEKi co-treatment and/or up-regulated levels of BRAF/CRAF complexes (both conditions present in SDR sub-lines compared to P lines). The up-regulated levels of BRAF/CRAF complexes are apparently still capable of phosphorylating MEK (Fig. 3C) but somehow incapable of productively signaling to ERK.
We then investigated whether the high abundance of BRAF/CRAF complexes, which confers MEKi resistance (Fig. 2), also confers sensitivity of SDR sub-lines to type II RAFi co-treatment. In support of this hypothesis, individual or combined CRAF and/or BRAF knockdown abolished the efficacy of BGB-283 combination treatment (Fig. 3F). Using SDR sub-lines, we then investigated whether MEKi-cotreatment enhances the ability of type II RAFi to induce signal-incompetent BRAF/CRAF complexes, resulting in p-ERK suppression. In this hypothetical model, type II RAFi+MEKi (i) foster BRAF/CRAF interaction (since BRAF and/or CRAF protein upregulation fosters the combinatorial efficacy of type II RAFi+MEKi, Fig. 3F); (ii) sequester or stabilize MEK/p-MEK within the BRAF/CRAF scaffold (which promotes p-MEK levels, Fig. 3C); (iii) retard p-MEK release from BRAF/CRAF (which would in turn reduce MEK dimerization and thereby reduce the pool of active MEK accessible to ERK); and (iv) retard MEK/ERK binding (which is required for ERK activation). To test this model, we used MEKi-resistant NRASMUT melanoma sub-lines and measured the effects of single (type II RAFi or MEKi) vs. double (type II RAFi+MEKi) inhibitor treatments on the in situ levels of protein/protein complexes within the MAPK pathway by proximity ligation assay (PLA). Consistent with prediction (i) of the model, acute (2h) treatment with type II RAFi+MEKi (BGB-283+trametinib, LXH-254+trametinib, or RAF-709+binimetinib) upregulated BRAF/CRAF complexes compared to single-agent treatment (Supplementary Fig. S4A to S4C). After 12d of treatments with inhibitor(s) (treatment refreshed every two days with additional last dose 12h prior to analysis), we observed that type II RAFi alone had little effect on CRAF/MEK or MEK/ERK levels. In contrast, type II RAFi+MEKi significantly induced CRAF/MEK and reduced MEK/ERK levels (Fig. 4A and 4B), consistent with predictions (ii) and (iv). Induction of CRAF/MEK and reduction of MEK/ERK were also observed as early as 2h after treatment with the aforementioned three type II RAFi+MEKi plus two additional combinations (BGB-283+binimetinib, BGB-283+cobimetinib) (Supplementary Fig. S4A to S4D).
Figure 4.
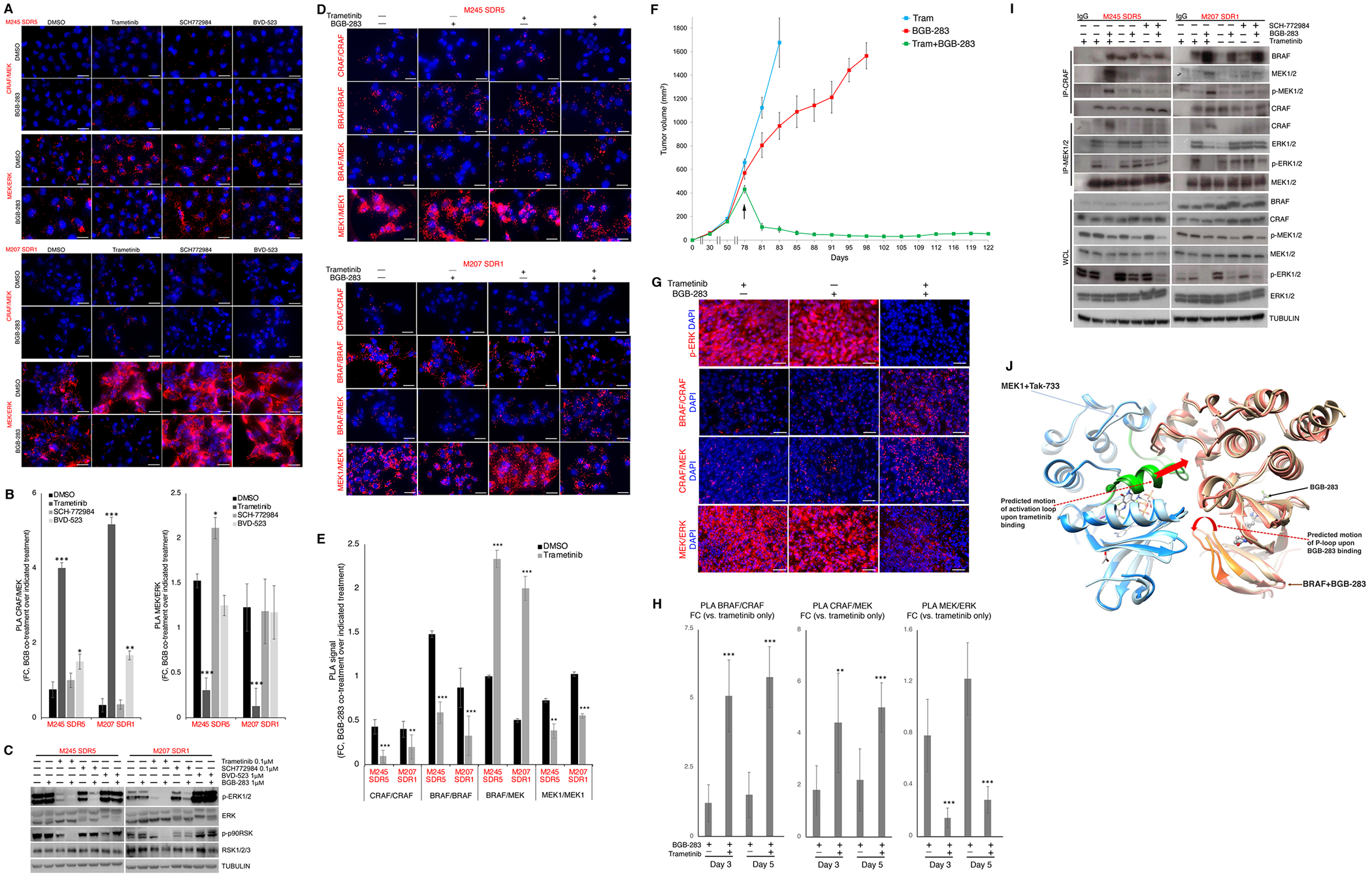
Type II RAFi and MEKi coordinate RAF/MEK stabilization, sequestering MEK from ERK.
A, Proximity ligation assays (PLAs) detecting CRAF/MEK and MEK/ERK proximity complexes in NRASMUT melanoma SDR sub-lines withdrawn from trametinib for 12 days or treated for 12 days with trametinib (0.1 μM), SCH-772984 (0.1 μM), or BVD-523 (1.0 μM), ± BGB-283 co-treatment (1 μM). Last fresh dose of inhibitor(s), 12 hours before analysis. DAPI, nuclear stain. Ruler, 20 μm.
B, Quantifications of PLA signals in (a) expressed as the fold change (FC) of PLA dots (per nucleus) of BGB-283 co-treatment over DMSO or single inhibitor treatment. N=5 fields; mean ± SDs; all comparisons with respect to FC (BGB-283+DMSO/DMSO); *p<0.1, **p<0.05 and ***p <0.01 based on t-test.
C, Western blots (Wbs) of lysates from culture conditions in A using indicated antibodies.
D, PLA detecting CRAF/CRAF, BRAF/BRAF, BRAF/MEK and MEK1/MEK1 proximity complexes in two NRASMUT melanoma SDR sub-lines treated with vehicle or trametinib (0.1 μM) for 48 hours, ± BGB-283 (1 μM, last 2 hours). DAPI, nuclear stain. Ruler, 20 μm.
E, Quantification of PLA data in c. N=5 fields; mean ± SDs. FCs: black, BGB-283+DMSO/DMSO; grey, BGB-283+tram/tram. Latter FC is compared to the former FC; **p<0.05 and ***p <0.01 based on t-test.
F, Tumor volumes of NRAS_PDX1 R2 (with acquired trametinib resistance) in mice that were (starting on day 78, indicated by an arrow) maintained on trametinib (5 mg/kg/day PO), switched to BGB-283 (20 mg/kg/day PO), or treated with BGB-283 (20 mg/kg/day PO) on top of trametinib (5 mg/kg/day PO). N=5 tumors per group; means ± SEMs.
G, Immunofluorescence (IF) of p-ERK levels (top row) and PLA of indicated protein/protein proximity complexes (bottom three rows) for three groups of PDX tumors in e. Tumors were collected on days 3 or 5 (from day 78). Images representative of five tumors per group.
H, Quantifications of PLA signals of CRAF/BRAF, CRAF/MEK and MEK/ERK complexes expressed as FC of PLA dots per nucleus of each indicated treatment condition vs. continuous trametinib monotherapy. Mean ± SDs; **p<0.05 and ***p <0.01 based on pairwise t-test.
I, IP-WBs or direct WBs of WCLs after treatment of indicated trametinib-resistant sub-lines with vehicle (DMSO) or indicated inhibitor(s) as in a. IgG, isotype control for IP.
J, Predicted conformational rearrangements of MEK1 and BRAF in complex upon binding to trametinib and BGB-283, respectively. Light brown, BRAF in its MEK1-bound tetrameric conformation; dark brown, BRAF in its BGB-283-bound conformation (BRAF P-loop in orange). Dark blue, MEK1 in BRAF-bound tetrameric conformation; light blue, MEK1 in its Tak-733-bound conformation (MEK1 activation loop in green). Red arrows, predicted and actual rearrangements of MEK1 and BRAF upon trametinib and BGB-283 binding, respectively.
We had shown earlier (Fig. 1H and Supplementary Fig. S1C to S1G) that that BGB-283+trametinib was more effective than BGB-283+SCH772984 (or BVD-523, MK-8353) at overcoming the growth of established acquired MEKi-resistant melanoma sub-clones. SCH772984, compared with trametinib, acutely (2h) and minimally induced CRAF/MEK and reduced MEK/ERK levels in conjunction with BGB-283 (Supplementary Fig. S4A and S4B). With prolonged (12d) treatment(s), Type II RAFi+ERKi (BGB-283+SCH772984 or BGB-283+BVD-523) failed to induce CRAF/MEK or reduce MEK/ERK levels (Fig. 4A and 4B). Consistently, after 12d of treatment(s), BGB-283 co-treatment was able to reduce the p-p90RSK level only in trametinib- but not SCH772984- or BVD-523-cultured NRASMUT melanoma sub-lines with acquired resistance (Fig. 4C). As observed previously, single-agent ERKi treatment led to rebound p-ERK levels (BVD-523 > SCH772984), which curiously failed to increase the growth rate (Supplementary Fig. S1C to Fig. S1G). BGB-283 co-treatment with BVD-523 did not reduce the rebounding p-ERK level and with SCH772984, which unlike BVD-253 inhibits ERK phosphorylation by MEK, did reduce the p-ERK rebound level but only to a level comparable to that with trametinib alone (Fig. 4C).
To validate our model further, we performed additional PLA assays in NRASMUT melanoma sub-lines with acquired MEKi-resistance. While BGB-283+trametinb induced BRAF/CRAF complexes, this combination reduced BRAF or CRAF homo-complexes (Fig. 4D and 4E). BRAF/MEK, just like CRAF/MEK, complexes were induced by BGB-283+trametinib. Importantly, BGB-283+trametinib reduced MEK1 homo-complexes, suggesting that RAF-phosphorylated/sequestered MEK could not be released from BRAF/CRAF scaffolds and thereby could not homo-dimerize and become activated. We also performed additional PLA assays in KRASMUT non-melanoma tumor cell lines. First, we derived sub-lines (HCT116-R, Su86.86-R, H2122-R) from KRASMUT CRC, PDAC, NSCLC cell lines (Fig. 1A) that had adapted to increasing doses of trametinib (up to 0.05 μM) with proliferative resistance. In short-term (7d) clonogenic assays, adding type II RAFi (BGB-283 or RAF-709 at 0.5 μM) to MEKi (trametinib or binimetinb, respectively, at 0.05 μM) strongly reduced clonogenic growth (Supplementary Fig. S4E). With 6h of treatment with inhibitor(s), we found that BGB-283 addition to trametinib (vs. BGB-283 alone) induced BRAF/CRAF and CRAF/MEK protein complex levels and reduced MEK/ERK and p-ERK levels (Supplementary Fig. S4F and S4G) in all three non-melanoma cancer cell lines with acquired MEKi resistance. To validate key mechanistic features of our model in vivo, we re-transplanted the NRAS_PDX1 R2 (Fig. 2) acquired trametinib-resistant tumor into mice that were treated daily with trametinib. Trametinib-resistant tumors (≅ 500 mm3) were assigned into three groups: trametinib (5 mg/kg/day PO), BGB-283 (20 mg/kg/day PO), or trametinib (5 mg/kg/day PO) plus BGB-283 (20 mg/kg/day PO). Switching from trametinib to BGB-283 did not induce tumor regression, while the combination of trametinib and BGB-283 rapidly induced tumor regression (CRs in 2 of 5 mice or tumors) (Fig. 4F). We then analyzed NRAS_PDX1 R2 tumors early (days 3 and 5) on each of the three treatments for CRAF/BRAF, CRAF/MEK and MEK/ERK in situ complexes by PLA and for p-ERK levels by immunofluorescence (Fig. 4G and 4H). Consistent with cell line observations, type II RAFi+MEKi induced BRAF/CRAF and CRAF/MEK levels concomitant with loss of MEK/ERK and p-ERK levels. To corroborate PLA findings, we performed co-IP and observed that type II RAFi+MEKi co-treatment preferentially (vs. type II RAFi+ERKi) induced endogenous CRAF/MEK and CRAF/p-MEK complexes and reduced MEK/ERK and especially MEK/p-ERK complexes (Fig. 4I and Supplementary Fig. S4H and S4I).
To predict how RAF binding to BGB-283 and MEK binding to trametinib might enhance RAF/MEK complexes, we modeled the structure of the BRAF/MEK1 dimer bound to these inhibitors and calculated the change in buried solvent accessible surface area (SASA) as a result of dual inhibitor binding (See details in Methods). We superimposed the individual experimental or predicted structures of trametinib-bound MEK1 and BGB-283-bound BRAF (PDB ID 3PP1 and 4R5Y) on a dimer of the BRAF/MEK1 tetramer (PDB 4MNE) (Supplementary Fig. S5). It is thought that BRAF and MEK1 form a face-to-face dimer (Supplementary Fig. S5A), with contribution to binding from the activation loop of both kinases (Supplementary Fig. S5B to S5F). When this model is compared to the BRAF and MEK1 conformations in the BRAF/MEK1 tetramer (Fig. 4J), the conformational changes of the activation loop of MEK1 upon trametinib binding and of the P-loop of BRAF upon BGB-283 binding could increase the contact surface between the two proteins and thereby the binding affinity. To estimate the effect of the latter conformational change, we calculated the variation of the buried SASA upon binding of BRAF and MEK1, for both the apo conformation of BRAF (PDB ID 4MNE) and its BGB-283-bound conformation. Since the P-loop of BRAF was not resolved in the X-ray crystal structure of the apo form, we modeled it ab initio. For the residues of the BRAF P-loop (465-469) and for the MEK1 residues in the vicinity (residues 73-82 and 97-101), we calculated that the total buried SASA increased from 78±5 Å2 for the apo form of BRAF to 98 Å2 for the BGB-283-bound BRAF, which suggests stronger binding.
Type II RAFi+MEKi Elicit CD8+ T-cell-Mediated Tumor Regression
Beyond tumor cell-intrinsic mechanisms, we evaluated the contribution of CD8 T-cells to tumor regression. First, we introduced mutational burden into a syngeneic model of murine NrasQ61R melanoma (called NIL) we recently reported (25) by exposing it to radiation (UV), thereby deriving a sub-line called NILER1-4 and generating a relative increase of 25.3 mutations per megabase of gDNA. As subcutaneous allografts, NILER1-4 tumors displayed increased durability of MEKi (trametinib 3 mg/kg/d) response (vs. NIL tumors; starting volume at ~100 mm3) (Fig. 5A). However, systemic CD8+ T-cell neutralization abolished this gain in durability, while having no effect on MEKi resistance development in NIL tumors. These findings suggest that CD8+ T-cells suppress acquired MEKi resistance by recognizing neoantigen(s). Using larger (~200 mm3) NILER tumors, we tested a non-tumor regressive dose of trametinib (1 mg/kg/d), two doses of BGB-283 (10 or 20 mg/kg/d) and their combinations. Comparable to the studies using PDXs, BGB-283 at these doses did not induce tumor regression. Combination with trametinib at the higher dose (20 mg/kg/d) of BGB-283 induced durable tumor regression beyond 35d and 7 of 7 CRs on 52d (with 3 of 7 confirmed CRs after treatment withdrawal on d52) (Fig. 5B). The average weekly body weights of mice in all treatment groups increased commensurate with the Jackson laboratory reference weights. However, NILER tumor-bearing mice treated with trametinib (1 mg/kg/d)+BGB-283 (20 mg/kg/d) began to lose weight after 40d of continuous treatments.
Figure 5.
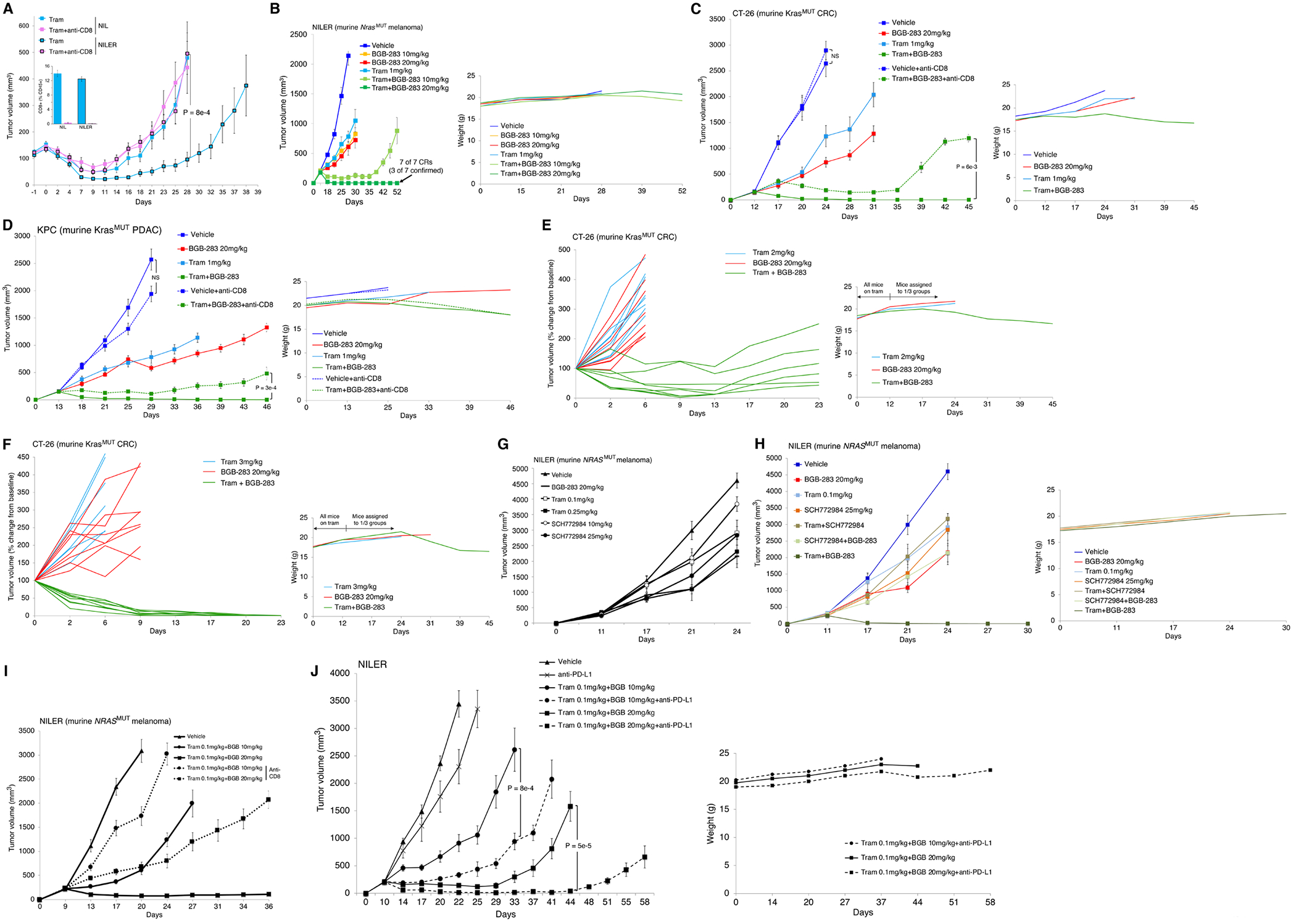
Tumor regression in response to Type II RAFi plus MEKi in syngenic models requires CD8+ T-cells.
A, Volumes of NIL vs. NILER1-4 tumors in response to trametinib (Tram) treatment (3 mg/kg/day PO) (starting day 0), ± anti-CD8 neutralization (starting -1 day). Inset, CD8+ T-cell levels as a % of CD45+ splenocytes at the end of experiments. N=8 tumors/group; means ± SEMs. P-value, Student t test.
B, Volumes of NILER1-4 tumors with indicated treatments starting at 200 mm3. Trametinib at 1 mg/kg/day PO. N=7 tumors/group; means ± SEMs. Complete responses (CRs) confirmed by withdrawal of tram+BGB-283 (20 mg/kg/day PO) without tumor recurrence for 35 days of observation. Right, average body weights of mice in each group measured twice a week.
C, D, Volumes of CT-26 (C) or KPC (D) tumors with indicated treatments starting on day 12 and 13, respectively (at 200 mm3 tumor volume). Trametinib at 1 mg/kg/day PO. Anti-CD8 treatment started on day 11 and 12, respectively. N=8 tumors/group; means ± SEMs. Unconfirmed CRs, 6/8 (C) or 2/8 (D) in the tram+BGB-283 (20 mg/kg/day PO) groups on day 45 or 46. P-value, Student t test. Right, average body weights of mice in each group measured twice a week.
E, F, Volumes of individual CT-26 tumors relative to tumor-matched baseline volumes (400-450 mm3) on day 0, when tumor-bearing mice were assigned to each of three indicated groups. Trametinib 2 (E) or 3 (F) mg/kg/day PO. Tumors/group (tram, BGB-283, tram+BGB-283): 8, 8, 6 (E) or 8, 8, 8 (F). Unconfirmed CRs, 0/6 (E) or 1/8 (F) in the tram+BGB-283 (20 mg/kg/day PO) groups on day 23. Right, average body weights of mice in each group measured twice a week.
G, Volumes of NILER1-4 tumors with indicated daily single-agent treatments starting at 200 mm3. N=8 tumors/group; means ± SEMs. Functionally equivalent doses of trametinib vs. SCH772984 were selected for use in H.
H, Volumes of NILER1-4 tumors with indicated treatment starting at 200 mm3. Trametinib at 0.1 mg/kg/day PO, SCH772984 at 25 mg/kg/day IP. N=8 tumors/group; means ± SEMs. Right, average body weights of mice in each group measured twice a week.
I, Volumes of NILER1-4 tumors with indicated treatment starting at 200 mm3. Trametinib at 0.1 mg/kg/day plus BGB-283 at 10 or 20 mg/kg/day ± anti-CD8 neutralization (starting day 8). N=8 tumors/group; means ± SEMs.
J, Volumes of NILER1-4 tumors with indicated treatment starting at 200 mm3 and day 10. Trametinib at 0.1 mg/kg/day plus BGB-283 at 10 or 20 mg/kg/day ± anti-PD-L1 treatment. N=8 tumors/group; means ± SEMs. P-value, Student t test. Right, average body weights of mice in each group measured twice a week.
We also tested trametinib and BGB-283 at these dosages in syngeneic tumor models of other cancer lineages. In KrasMUT CRC (CT-26) and PDAC (KPC) syngeneic models, neither trametinib (1 mg/kg/d) nor BGB-283 (20 mg/kg/d) alone induced regression of established (~200 mm3) tumors, but the combination elicited durable tumor regression in both CRC and PDAC models, resulting in 6/8 and 2/8 CRs respectively on d42. Body weight loss was noted only in mice treated with combination, beginning around d24 (Fig. 5C and 5D). To determine whether the addition of BGB-283 can overcome well-established acquired trametinib-resistant tumors (~ 450 mm3), we began treating mice bearing ~200 mm3 CT-26 tumors with higher doses (2 or 3 mg/kg/d) of trametinib to achieve tumor volume stabilization. When tumors resumed growth and reached an average tumor volume of 450 mm3, we assigned mice into three groups: (i) continued trametinib at the prior dosage, (ii) BGB-283 (20 mg/kg/d), or (iii) trametinib+BGB-283. Only in the third group did we observe tumor regression, which was associated with body weight loss (Fig. 5E and 5F).
Furthermore, by reducing the trametinib dose 10-fold to 0.1 mg/kg/d, we evaluated the superiority of type II RAFi plus MEKi (vs. +ERKi) in vivo, as suggested by cell line studies (Fig. 1), and the contribution of CD8 T-cells to tumor regression elicited by type II RAFi+MEKi. Using NILER tumor-bearing mice, we first identified trametinib at 0.1 mg/kg/d and SCH772984 at 25 mg/kg/d as having similar sub-optimal anti-tumor activities (Fig. 5G). We then compared the relative anti-tumor activities of trametinib+BGB-283 vs. SCH772984+BGB-283 (vs. trametinb+SCH772984). It was clear that only trametinib+BGB-283 has tumor-regressive activity, which was achieved with only 0.1 mg/kg/d of trametinib and associated with improved tolerability (Fig. 5H). Moreover, NILER tumor regression (trametinib 0.1 mg/kg/d+BGB-283 20 mg/kg/d) or transient stabilization (trametinib 0.1 mg/kg/d+BGB-283 10 mg/kg/d) was strongly dependent on CD8 T-cells, as their systemic neutralization strongly undercut combinatorial efficacies (Fig. 5I). Similar observations were made in CT26 and KPC models, although the contribution of CD8 T-cells to combo-elicited tumor regression was less (Fig. 5C and 5D). Instead of neutralizing CD8 T-cells (Fig. 5I), we invigorated CD8 T-cells by treatment of NILER tumor-bearing mice with anti-PD-L1 (Fig. 5J). Anti-PD-L1 treatment by itself has minimal, if any, impact on vehicle-treated tumors. However, triplet treatments incorporating anti-PD-L1 improved the efficacy over that induced by doublet (trametinib+BGB-283) treatments, which is consistent with systemic and/or intratumoral CD8 T-cells being an important effector of type II RAFi+MEKi in immune competent hosts.
Type II RAFi+MEKi Expand TN/TCM and Shrink TREG Compartments Systemically
We evaluated NILER tumor histology and T-cell infiltration levels and pattern, on d4 and d11, in response to treatment with vehicle, BGB-283 (20 mg/kg/d), trametinib (1 mg/kg/d) or both (Fig. 5B). Vehicle-treated tumors displayed sheets of spindled tumor cells, immature blood vessels and scattered necrosis (Fig. 6A and 6B) as well as tumor-infiltrating lymphocytes (TILs) (Supplementary Fig. S6A), consistent with findings of CD4+ and CD8+ immunohistochemistry (IHC) (Fig. 6B). Tumors treated with BGB-283 or trametinib alone, which only displayed growth deceleration but not regression (Fig. 5B), showed small foci of tumor regression on d4 and d11 and large clefts on d11 and increased TILs in both the invasive margins and tumor centers (Fig. 6A; Supplementary Fig. S6A). In contrast, durable tumor regression induced by BGB-283+trametinib was associated with a marked histologic response characterized by large foci of tumor regression and necrosis, tumor cell balloon degeneration, apoptosis and melanosis (Fig. 6B). Maximal TIL scores was reached by d4 in both tumor centers/margins, along with extensive infiltration by histiocytes and melanophages. In vehicle- or trametinib-treated tumors, the CD8+:CD4+ ratios were 1 or less. In BGB-283-treated tumors, this ratio was consistently 1. However, by d11 on BGB-283+trametinib treatment, this ratio was 2-3 (Supplementary Fig. S6A), consistent with CD8+ T-cell expansion.
Figure 6.
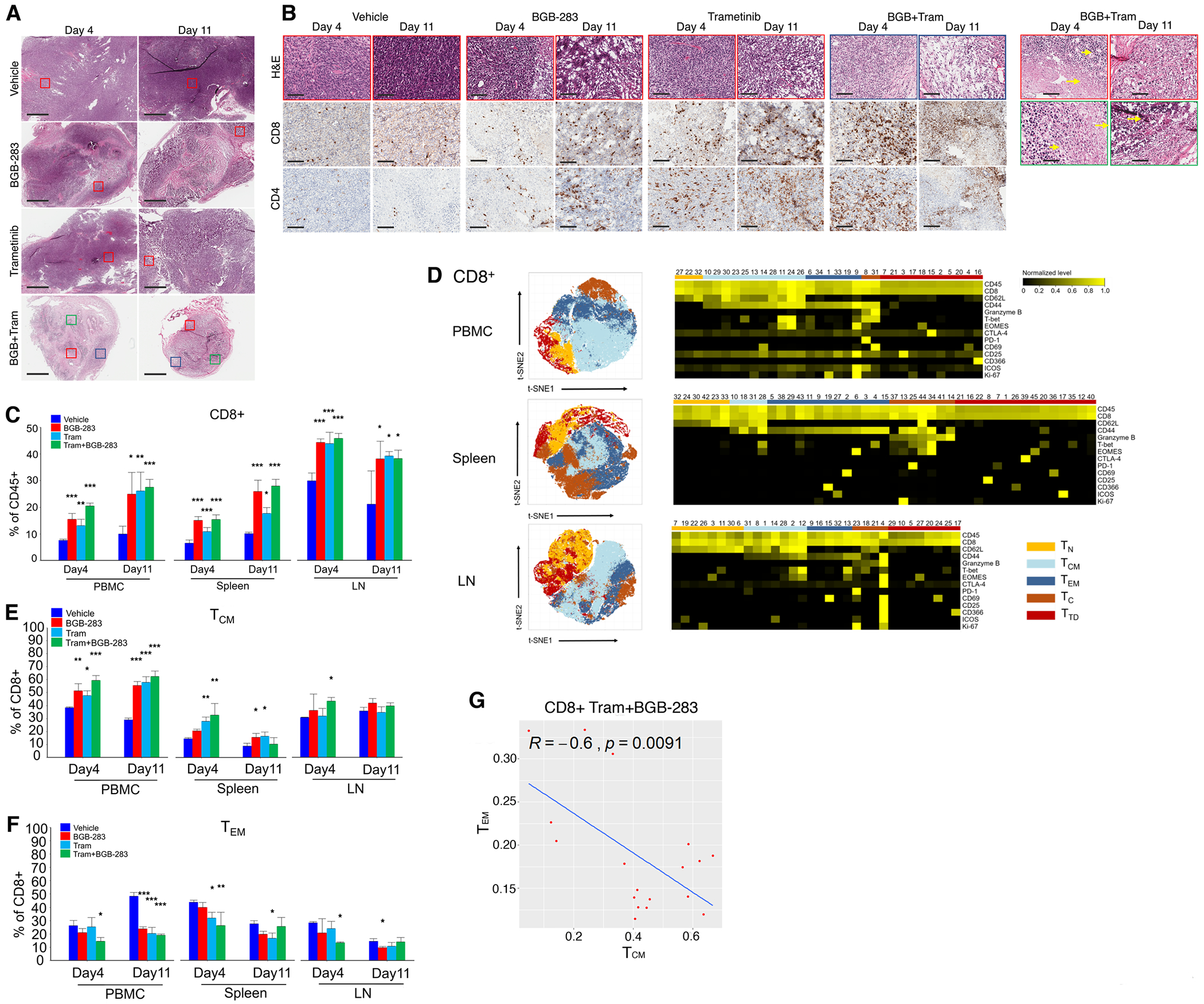
Type II RAFi+MEKi elicits systemic expansion of central memory CD8+ T-cells in mice-bearing NrasMUT melanoma.
A, Hematoxylin and eosin (H&E) stains of NILER1–4 tumors (days 4 and 11) on vehicle, BGB-283 (20 mg/kg/d), trametinib (1 mg/kg/d), or both. Representative of three tumors per group. Ruler=1 mm. Colored boxes delineate magnified areas in e.
B, Magnified areas of NILER1–4 tumors analyzed by H&E and representative CD8 and CD4 immnuohistochemistry. Ruler=100 μm. Highlighted H&E images of BGB+tram-treated tumors show areas of inflammation (short arrows), necrosis (long arrows), apoptotic bodies (d11, top) and tumor regression with melanosis (d11, bottom).
C, Indicated tissue samples (PBMC, spleen, draining lymph nodes or LNs; n=3 per site) were obtained for CyTOF analysis on days 4 and 11 after starting each treatment group. Frequencies of CD4+ or CD8+ T-cells. Mean ± SEMs. Pairwise group comparisons with respect to the vehicle-treated group. P-value, paired Student’s t test. *p<0.05, **p<0.01 and ***p <0.001.
D, t-SNE maps (left) of CD8+ T-cell subsets (within CD45+ cells) in the PMBC, spleen and draining LNs analyzed by CyTOF (pooled data from days 4 and 11). Heatmaps (right) showing the expression values of immune phenotypic protein markers normalized to the maximum mean value across subsets.
E, F, As in C, except CyTOF analysis is for indicated CD8+ TCM (h) or TEM (i) subsets.
G, Pearson correlation between CD8+ TCM vs. TEM subset frequencies in BGB-283+tram treated mice on (pooled data from days 4 and 11).
To assess systemic T-cell impacts of non-tumor-regressive (single-agent) vs. tumor-regressive (combination) vs. vehicle treatments (Fig. 5B) in NILER tumor-bearing mice, we performed mass cytometry (CyTOF) on peripheral blood mononuclear cells (PBMCs) and dissociated secondary lymphoid organs (tumor-draining lymph nodes and spleens) at d4 and d11 (n=3 mice/group). In all T-cell compartments, single or combined inhibitor treatment, short- and long-term, induced the percentages of CD4+ and CD8+ T-cells as percentages of CD45+ cells (Fig. 6C; Supplementary Fig. S6B). We analyzed further T-cell subpopulations by t-SNE (Fig. 6D; Supplementary Fig. S6C and S6D). Short- and long-term, single and combined inhibitor treatments tended to induce the proportion of CD4+ and CD8+ TN (T naïve, CD62L+CD44−) and TCM (T central memory, CD62L+CD44+) but reduce CD8+ TEM (T effector memory, CD62L−CD44+) cells (Fig. 6E and 6F; Supplementary Fig. S6E to S6H). This pattern was more consistent on d4 (during maximal tumor volume reduction by combination treatment) in response to type II RAFi+MEKi, but not to type II RAFi or MEKi alone. Consistently, only among mice co-treated with type II RAFi+MEKi, the proportions of CD8+ TCM and TEM cells were significantly anti-correlated (Fig. 6G) (vehicle, R=−0.35, p=0.14; type II RAFi, R=−0.43, p=0.078; MEKi, R=−0.058. p=0.82; type II RAFi+MEKi, R=−0.6, p=0.0091). Moreover, combination type II RAFi+MEKi treatment reduced the levels of CD4+ TREG (T regulatory, CD4+FOXP3+) cells and their proliferation (Supplementary Fig. S6I and S6J). We also analyzed the profiles of splenic T-cells (collected on d4) after ex vivo cultures for 3 or 4 days with or without anti-CD3+anti-CD28 stimulation. We found that CD4+ and CD8+ T-cells alike from the spleens of mice that were treated in vivo with single-agent or combined inhibitors were more capable of activation (Supplementary Fig. S7A and S7B). Moreover, splenic CD4+ TREG cells from inhibitor-treated mice (especially type II RAFi+MEKi-treated mice on ex vivo day 3) were less capable of ex vivo expansion (Supplementary Fig. S7C). Ex vivo stimulation-induced T-cell proliferation as well as activation and/or effector marker expression were more robust among mice treated with inhibitor(s) (Supplementary Fig. S7D and S7E). Consistent with prior analysis (Fig. 6), CD4+ and CD8+ T-cell compartments from the spleens of inhibitor-treated tumor-bearing mice, with and without ex vivo stimulation, expanded the TN and TCM subpopulations at the expense of the TEM subpopulation (Supplementary Fig. S7F).
Type II RAFi+MEKi Expand Intratumoral CD8+ TEM and Activated T-cells
To assess intratumoral T-cell impacts, we performed CyTOF on dissociated vehicle- vs. type II RAFi+MEKi-treated tumors (n=3 tumors/group) on d5 and observed that combination treatment increased CD8+ T-cells (% of CD45+ cells) by ~7-fold, based on t-SNE analysis (Fig. 7A; Supplementary Fig. S8A). This finding indicates an even higher increase in the CD8+ T-cell to tumor cell ratio, given induction of the CD45+ and reduction of the tumor cell compartments elicited by combination therapy. We analyzed further CD8+ T-cell subpopulations by t-SNE (Fig. 7B; Supplementary Fig. S8B). Importantly, type II RAFi+MEKi elevated the levels of intratumoral CD8+ TEM (~3-fold), TC (~3-fold), and TTD (~5-fold) cells (% CD45+ cells) (Fig. 7C). PD-1+ expression expanded from <0.5% (vehicle) to ~5% and ~7% (type II RAFi+MEKi) in respectively the CD8+ TEM and TTD subpopulations (Fig. 7D). This was accompanied by an increase in Ki-67 positivity among the CD8+ TTD cells. We corroborated these CyTOF findings with scRNA-seq analysis of combined CD4+ and CD8+ T-cells sorted from vehicle- vs. type II RAFi+MEKi-treated tumors on d5 (sorted cells from four independent tumors were combined per condition). T-cell subpopulations visualized by t-SNE were analyzed for differential gene expression (Fig. 7E). Importantly, CD8+ T-cells (as a % of all CD8+ T-cells) that co-express activation- and exhaustion-associated genes increased from ~20% to ~65% in vehicle- to type II RAFi+MEKi-treated tumors (Fig. 7F). An increase in the activated/exhausted CD8+ T-cell to tumor cell ratio is expected to be even greater, given expansion in both the CD45+ and CD8+ T-cell compartments elicited by combination therapy. Consistently, type II RAFi+MEKi-treated tumors harbored CD8+ T-cells with higher activation/exhaustion score and expression of the master regulator of exhaustion, Tox (Fig. 7G and 7H) (33,34).
Figure. 7.
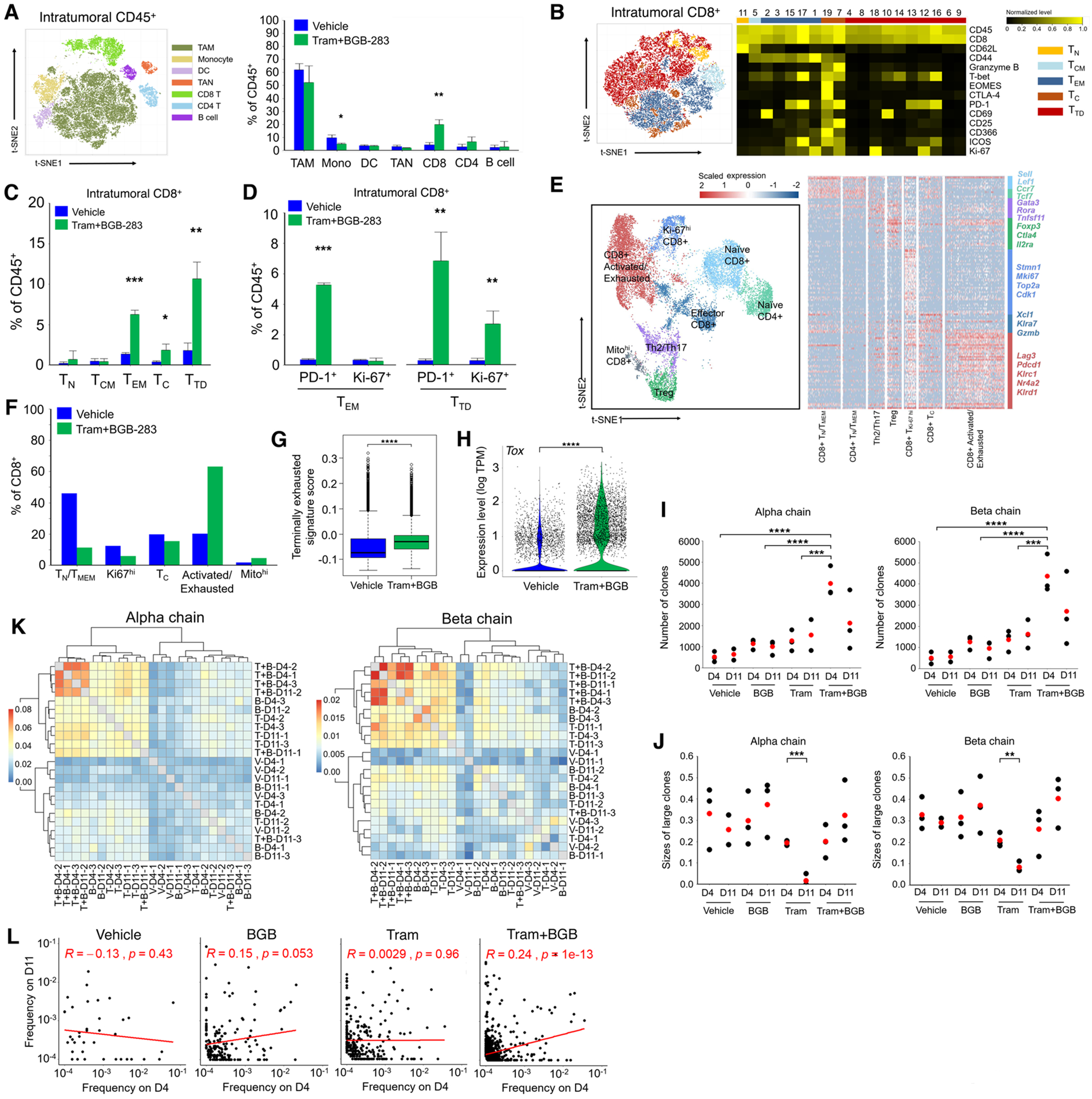
Co-treatment with type II RAFi+MEKi heightens tumor infiltration by effector memory and activated/exhausted CD8+ T-cells and T-cell clonal expansion.
A, t-SNE map (left) of tumor infiltrating CD45+ cells analyzed by CyTOF (pooled data from vehicle- and Tram+BGB-283-treated groups). Frequencies of each cluster (right). N=3 tumors per group; mean ± SEMs. Pairwise group comparisons with respect to the vehicle-treated group. P-value, paired Student’s t test. *p<0.05, **p<0.01 and ***p <0.001.
B, t-SNE map (left) of tumor infiltrating CD8+ T-cell subsets analyzed by CyTOF (pooled data from vehicle- and Tram+BGB-283-treated groups). Heatmap (right) showing the expression values of immune phenotypic protein markers normalized to the maximum mean value across meta-clusters.
C, D, As in A, except CyTOF analysis is shown for indicated CD8+ T-cell subsets (c) or the percentages of PD-1 or Ki-67 positivity within indicated subsets (d).
E, UMAP of tumor-infiltrating CD4+ and CD8+T-cells (n=14,199) analyzed by scRNA-seq (pooled data from vehicle- and Tram+BGB-283-treated groups, n=4 tumors per group by combining FACS-sorted CD4+ and CD8+T-cells). Clusters denoted by distinct colors are labeled with inferred cell types (left). Heatmap (right) showing differentially expressed genes (rows) among different T-cell subsets (columns). Specific genes that are associated with different T-cell clusters are highlighted along the right.
F, Frequencies of indicated CD8+ T-cell subsets analyzed by scRNA-seq based on treatment status.
G, Box plot of terminally exhausted gene signature scores (scRNA-seq) for all CD8+ T-cells in each treatment group. P-value, Wilcoxon rank-sum test, ****p <0.0001.
H, Violin plot showing Tox expression levels (scRNA-seq) for all CD8+ T-cells in each treatment group. P-value, Wilcoxon rank-sum test, ****p <0.0001.
I, Vehicle-, BGB-283-, Tram- and Tram+BGB-283-treated tumors on days 4 and 11 (n=3 per group, except one outlier in vehicle-treated tumor, d11) were analyzed by TCR-seq. Dot plot showing the number of TCR clones for α or β chain (red dots, average values). P-value, paired Student’s t test, ****p <0.0001.
J, As in I except TCR-seq analysis is shown for sizes of large TCR clones (frequency >= 0.05). Pairwise comparison between d4 and d11 was performed in each treatment group with paired Student’s t test. *p<0.05, **p<0.01 and ***p <0.001.
K, Clustering of TCR repertoires of tumor-infiltrating T-cells by the levels of shared CDR3 sequences (using the Jaccard index).
L, Spearman correlations between frequencies on d4 and d11 of shared α chain CDR3 sequences in each condition. Mean frequency on d4 or d11 of each shared CDR3 sequence (at least in two samples of each condition) was calculated.
We then evaluated whether intratumoral expansion of activated CD8+ T-cells induced by type II RAFi+MEKi is clonal by performing TCR-seq analysis of vehicle-, BGB-283-, trametinib-, or trametinib+BGB-283-treated NILER1-4 tumors (n=3/group) on d4 and d11. By analyzing the CDR3 clonotypes of both TCRα and β chains, we observed that trametinib+BGB-283 increased the number of T-cell clones (Fig. 7I). Moreover, whereas trametinib temporally reduced the sizes of the top or all dominant (>5%) T-cell clones, BGB-283 co-treatment erased and enhanced the sizes of top or dominant clones (Fig. 7J and Supplementary Fig. S8C and S8D). We also calculated the diversity and Gini (clonality) indices, which showed that trametinib increased but trametinib+BGB-283 reduced the diversity of T-cell clonotypes. Accordingly, trametinib reduced, whereas trametinib+BGB-283 maintained, T-cell clonality (Supplementary Fig. S8E and S8F). To evaluate the overlap of T-cell clones across treatment conditions and time points, we calculated the Jaccard indices and overlap coefficients (Fig. 7K; Supplementary Fig. S8G). Importantly, sample-to-sample comparisons amongst combination-treated tumors generally exhibited the highest percentages of intersecting clones (up to 8%). Group-to-group comparisons consistently showed the highest levels of overlap when combination-treated tumors as a group was compared against any other group, including itself. We observed within the combination treatment group the most significant positive correlation between d4 and d11 TCR clonotypes’ frequencies (Fig. 7L; Supplementary Fig. S8H), suggesting that type II RAFi+MEKi elicit persistent expansion of tumor antigen-specific TCR clones. These findings suggest that type II RAFi+MEKi elicited the most robust tumor-specific T-cell reactivity, as trametinib+BGB-283 co-treatment increased TCR clonality as well as the number of distinct clones and converged T-cell clonotypes. Taken together with prior findings (Fig. 5 and 6), we conclude that type II RAFi+MEKi induce durable tumor regression by recruiting PD-L1 therapy-responsive, tumor antigen-specific CD8+ T-cells.
DISCUSSION
MAPK-targeted therapy has clinically meaningful activity in only a handful of BRAFV600MUT cancers. From recent BRAFV600MUT-focused basket trials, it remains unknown whether lack of addiction to the MAPK pathway explains poor clinical efficacy in some BRAFV600MUT cancer histologies. In RASMUT or non-BRAFV600MUT cancers, addiction to the MAPK pathway also has been cast into doubt by trials testing single-agent MEK or ERK inhibitors. Our study showing the highly durable efficacy of a specific pair of MAPK-targeted agents (type II RAFi+allosteric MEKi) across multiple driver mutations and cancer histologies (BRAFV600, non-BRAFV600, KRAS, NRAS and NF1 mutant melanoma, colorectal, pancreatic and lung cancers) strongly supports MAPK pathway addiction.
The first combinatorial therapy developed successfully against the MAPK pathway in the clinic (for patients with advanced BRAFV600 melanoma) consists of type I RAFi plus an allosteric MEKi. However, despite high response rates, acquired resistance is commonplace and frequently due to MAPK pathway reactivation. Our data suggest that a specific combination of type II RAFi+MEKi could help overcome acquired resistance to the current standard-of-care (type I RAFi+MEKi).
Clinical development of MAPK-targeted combinatorial agents in oncology has centered on direct impacts on tumor cells (vs. immune cells) via inhibition of the activity of kinase(s). Insights from this study lend credence to the importance of anti-tumor impacts arising from durable allosteric modulation of the MAPK pathway and persistent recruitment of CD8+ T-cells, which are most evident when both type II RAFi and MEKi are deployed together. This study also provides foundational knowledge for clinical development vis-à-vis identifying susceptible cancer histologies, biomarkers, and rational combinations with immunotherapies.
Previously, the allosteric action of dual MAPK inhibitors has not been examined at the level of protein/protein interactions in the MAPK pathway. Oncogenic RAS (which forms dimers) recruits cytosolic RAF/MEK heterodimers to the cell surface through the RAS-binding domain of RAFs. Face-to-face RAF/MEK heterodimers are then brought together by the back-to-back dimerization of RAFs, which facilitates cis-autophosphorylation of the RAF activation loop. Loosening of the RAF/MEK heterodimers is thought to facilitate the assembly of MEK homodimers on the RAF surface. Phosphorylation of both protomers of MEK permits release of MEK dimers from RAF dimers and ERK interaction and ERK phosphorylation. Here we provided evidence that type II RAFi and allosteric MEKi act in concert to stabilize and sequester p-MEK in RAF complexes (specifically BRAF/CRAF complexes, which is considered the most active complex among all RAF dimers). This action, which is likely facilitated by the high abundance of RAF/RAF and/or RAF/MEK complexes in MEKi-resistant tumor clones, may therefore be selectively robust against acquired resistance. The end result of MEK sequestration by RAF is reduced MEK dimerization and uncoupling of MEK or p-MEK interaction with ERK.
ERKi has been proposed to overcome MEKi resistance (35). Since MEKi or ERKi each can strongly suppress the MAPK pathway in normal cells, untoward toxicities are likely to arise with their combination. Type II RAFi as a single-agent does not appear to be highly active in suppressing oncogenic MAPK signaling, which is consistent with low single-agent anti-tumor activity from early-phase clinical trials. Here, we showed that type II RAFi+MEKi is superior to type II RAFi+ERKi (SCH772984) in preventing and overcoming acquired MEKi resistance. This correlates with the superior ability of type II RAFi+MEKi to physically stabilize RAF/MEK and uncouple MEK/ERK. The synergy derived from this allosteric mechanism may afford MEKi dose reduction and improve the therapeutic index, permitting triplet combination with immune checkpoint blockade therapy.
We also presented evidence that type II RAFi+MEKi may have favorable T-cell impacts that directly rationalize combination with anti-PD-1/L1 therapy. Prior studies (14,16) have analyzed the T-cell impacts of MEKi monotherapy. Here, we observed that type II RAFi+MEKi induce systemic levels of CD8+ TCM cells and reduce CD4+ TREG cells. Studies have implicated the importance of CD8+ TCM cells to adoptive immunotherapy or anti-PD-1 therapy (36,37). Tumor antigen-specific CD8+ TCM (vs. TEM) cells are thought to exhibit more potent in vivo anti-tumor (i.e., eradication of large established tumors) recall responses (38,39). Consistent with this, homing to secondary lymphoid tissues (e.g., spleen, LN) appeared to be required for optimal tumor eradication. That is, highly effective anti-tumor T cells were those that initially targeted secondary lymphoid tissues rather than peripheral/tumor sites (38). Intratumorally, we observed that type II RAFi+MEKi not only induced sustained immune infiltration but also the relative abundance of CD8+ TEM and TTD cells, their expression of PD-1/Ki-67 expression, and activation/exhaustion genes and signatures. Inside the tumor, CD8+ TEM cells acquire effector functions more rapidly than TCM cells. This superior tumor cytotoxicity in situ is thought to be a key mechanism through which preexisting CD8+ TEM cells mediate secondary or de novo priming of effector T-cells in the tumor-draining lymph nodes. Intratumorally, type II RAFi+MEKi also promoted expansion and convergence of T-cell clonotypes, in contrast to MEKi monotherapy. Collectively, these findings rationalize triplet combination trials of type II RAFi+MEKi+anti-PD-1/L1 therapy in RAS/MAPK-hyperactivated cancers.
METHODS
Cell Lines
All cell lines and drug-resistant sub-lines were routinely tested for mycoplasma and profiled and identified by RNA-seq and the GenePrint 10 system (Promega) at periodic intervals during the course of this study for banking and experimental studies. All cell lines were maintained in DMEM high glucose with 10% heat-inactivated FBS (Omega Scientific) and 2 mM glutamine in humidified, 5% CO2 incubator. To derive resistant sub-lines, parental human BRAFV600MUT, NRASMUT or NF1−/− melanoma cells seeded at low-density were treated with BRAFi (PLX4032)+MEKi (AZD6244) (for BRAFV600MUT lines) or MEKi (trametinib) (for NRASMUT or NF1−/− lines) every 2-3 days for 12-15 weeks, and proliferative colonies were ring-isolated and expanded. To derive resistant polyclonal sub-lines of KRASMUT PDAC, NSCLC and colorectal lines, cells seeded at low-density were treated MEKi (trametinib) every 2-3 days for 25 days and expanded. The NILER1-4 murine melaoma cell line was derived from NIL by exposure to 1 round of high-dose UVB radiation followed by ring-clonal selection and expansion.
Mice
C57BL/6 and NSG (NOD scid gamma) were obtained from the Radiation Oncology breeding colony at UCLA (Los Angeles, CA). Male or female mice were used at 4-6 weeks of age. All animal experiments were conducted according to the guidelines approved by the UCLA Animal Research Committee.
Constructs and Inhibitors
shNRAS, shCRAF, shBRAF were sub-cloned into the lentiviral vector pLL3.7 as described (4,7,29,40). NRAS over-expression and all knockdown constructs were packaged into lentiviral particles for infection. Experiments were carried out 3 days post-transduction. Inhibitors were obtained from the following sources: PLX4032 (Plexxikon), trametinib in vitro and in vivo (LC Laboratories), BGB-283 and BGB-3245 (Beigene), RAF709 (Cayman Chemical), LXH254 (Selleckchem), cobimetinib (Selleckchem), binimetinib (LC Laboratories), and SCH772984 (Chemietek).
Cell Growth Assays
For clonogenic assays, cells were plated at single-cell density in six-well plates. Data presented are representative of at least two independent replicates. Inhibitor and media were replenished every 2 days for the number of days noted. Colonies were fixed in 4% paraformaldehyde and stained with 0.05% crystal violet. For temporal measurements of cell growth, cells were plated at single-cell density in 96-well plates and treated with indicated treatments every 2-3 days for 8-11 days. Cell viability in relative light units (RLU) was measured using Celltiter-Glo every 2-3 days. Experiments were performed in triplicates. For cell counting, cells were plated at single-cell density in six-well plates, and experiments were performed in triplicates. Indicated treatment and media were replenished every 2-3 days and, after trypsinization, viable (trypan blue negative) cells were counted on the days noted. Additional plates for the last timepoint (11 days) were fixed in 4% paraformaldehyde and stained with 0.05% crystal violet.
Protein Detection
Cells were lysed in IP lysis buffer (Thermo Fisher Scientific) with Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific) for immunoprecipitation (IP) and Western blotting. Dynabeads (Thermo Fisher Scientific) were used to immunoprecipitate proteins of interest based on the manufacturer’s protocol. For immunocytochemistry (ICC), tissues were fixed in 4% paraformaldehyde (PFA) and sucrose and cryoprotected in OCT or in formalin followed by embedding in paraffin (formalin-fixed and paraffin-embedded or FFPE). For FFPE tissues, after de-paraffinization and re-hydration, tissue sections were subjected to heat for antigen-retrieval. PFA/OCT sections were not subjected to antigen retrieval. Cell lines were fixed with 4% PFA. ICC of both tissue and cell lines were performed with Alexa Fluor-conjugated secondary antibodies (Life Technologies). Nuclei were counterstained by DAPI. Duolink Proximity Ligation Assay (PLA) (Sigma Aldrich) was used to detect in situ protein-protein proximity interactions by following the manufacturer’s protocol. Fluorophore signals were captured with a Zeiss microscope (AXIO Imager A1) mounted with a charge-coupled device camera (Retiga EXi QImaging), and the images captured by Image-pro plus 6.0. PLA signals were quantified by counting signals per cell in each field (n=5 fields per condition). IP, Western blots, immunofluorescence, immunohistochemistry, and PLA assays were performed using the following antibodies: CD4 (#183685 from Abcam), CD8 (14-0808-80 from Invitrogen/ThermoFisher), BRAF (sc-5284 from Santa Cruz), CRAF (#53745, #12552 from Cell Signaling), ERK (#9101, #4696 from Cell Signaling), p-ERK1/2 (#5726, #4695 from Cell Signaling), p-MEK1/2 (#9154 from Cell Signaling), MEK1/2 (#9126 from Cell Signaling, sc-81504 from Santa Cruz), MEK1 (sc-6250 from Santa Cruz, #9146 from Cell Signaling), p-90RSK (#11989 from Cell Signaling), RSK1/2/3 (#9355 from Cell Signaling), NRAS (sc-519, Santa Cruz), and TUBULIN (T9026 from Sigma Aldrich).
PDX, Syngeneic Mouse Models, Treatments, and Tissue Collection
To develop PDX models, tumor fragments derived from NRASMUT metastatic melanoma, pancreatic ductal adenocarcinoma (PDAC) or non-small cell lung cancer (NSCLC), with approval by the local Institutional Review Boards, were transplanted subcutaneously in sex-matched NSG mice (4-6 week old). One tumor fragment was implanted in each mouse. Tumors were measured with a calliper every 2 days, and tumor volumes were calculated using the formula (length x width2)/2. Tumors with tumor volumes around 500mm3 were randomly assigned into experimental groups. For syngeneic melanoma models, C57BL/6 mice were subcutaneously injected on both flanks with either one million NIL or NILER1-4 cells. Once tumors reached a size of 150-200 mm3, mice were assigned randomly into experimental groups. αCD8a and isotype control antibodies were administrated intraperitoneally (200 μg/mouse) on day −1, day 0 and then twice a week. Special mice diets (for NSG and C57BL/6) were generated by incorporating trametinib at 1, 3 or 5 mg/kg to facilitate daily drug dosing and to reduce animal stress (Test Diet, Richmond, IN, USA). BGB-283 (10 or 20 mg/kg/day) was administered to mice via oral gavage and SCH772984 (10 or 25 mg/kg/day) intraperitoneally. Tumors were excised from mice, minced and digested to single-cell suspensions using the tumor dissociation kit and gentleMACSTM Octo Dissociator (Miltenyi Biotech). Spleens were manually homogenized, mashed through 45 μm filters into RPMI-1640 supplemented with 10% FBS. Red blood cells in single cell suspensions were lysed using ACK lysis buffer (Lonza). Intracardiac blood samples were collected in the presence of heparin, and PBMC were obtained through density centrifugation (1500 g at 22°C for 30 min) using Lympholyte M (Cedarlane). Draining lymph nodes were collected and single cell suspensions were obtain using a spleen dissociation kit according to manufacturer’s protocol (Milteny Biotec).
Histologic Evaluation
Following tissue fixation, paraffin-embedding and sectioning, histologic evaluation was performed by a dermatopathologist (P.O.S.) blinded to the identity of the samples. TIL scoring was performed using a slight modification of standard methodology (41,42). After identifying the tumor invasive margin and tumor core, lymphocyte counts were performed from 3 HPFs. TIL scores were defined as: 1= 0-10% of total cells; 2= 20-40% of total cells; 3= 50-70% of total cells; 4= 80-100% of total cells. These counts were confirmed using immunohistochemistry for CD4 and CD8. The ratio of CD8:CD4 cells was performed in 3 distinct tumor regions with the highest TIL infiltration by counting CD4+ and CD8+ cells in the same area.
Ex Vivo T-cells Activation
Splenocytes from NILER1-4 tumor-bearing mice with or without in vivo MAPKi treatment were collected and then seeded at 1 million cells/ml in 24-well plates, with or without anti-CD3 (145-2C11, 2 μg/ml) and anti-CD28 (37.51, 2 μg/ml). After 48 hours, media were refreshed using RPMI-1640 + 7.5% FBS + 0.1% β-mercaptoethanol+ 30U/ml mIL-2, without antibodies. At 72 and 96 hours, cells were collected and analyzed by mass cytometry.
Mass Cytometry of Murine Tissues
2x106 or fewer cells were incubated with 2% of FBS in PBS with 25 μg/mL of 2.4G2 antibody at 4°C for 10 min prior to surface staining with an antibody cocktail at 4°C for 30 min in a 50 μL volume. Cells were incubated with 2.5 μM 194Pt monoisotopic cisplatin (Fluidigm) at 4°C for 1 min. Cells were then washed twice with FACS buffer and barcoded using palladium metal barcoding reagents according to manufacturer’s protocol (Fluidigm). Subsequently, fixation and permeabilization were performed using the Foxp3 fix and permeabilization kit according to the manufacturer’s protocol (eBioscience). Cells were then stained with an intracellular stain antibody cocktail (Foxp3, Ki67, granzyme B, T-bet, iNos, Eomes) for 30 min at room temperature. Cells were then washed twice with Foxp3 permeabilization buffer, twice with FACS buffer, and incubated overnight in 1.6% PFA PBS with 100 nM iridium nucleic acid intercalator (Fluidigm). Cells were then washed twice with PBS with 0.5% BSA, filtered, and washed twice with water with 0.1% BSA prior to analysis. Samples were analyzed using a Helios mass cytometer based on the Helios 6.5.358 acquisition software (Fluidigm).
CyTOF Data Analysis
All the samples were pre-processed by CATALYST, including normalization, debarcoding and compensation. The normalized fcs files were then uploaded into Cytobank (43), and data were gated to exclude beads and to only include live, single cells. The CD8+ and CD4+ T cells were gated from the CD45+CD3+ populations, and respectively data were downloaded separately into individual files for each sample. We applied Cytofkit(44) to perform the t-Distribution Stochastic Neighbor Embedding (t-SNE) analysis separately on the manually gated CD4+ and CD8+ populations from PBMC, spleen and lymph node samples. We selected 5,000 events/sample to ensure equal representation of cells across samples. For CD4+ T cells, 12 markers, including CD44, CD62L, CD25, CD69, CD366, FoxP3, PD1, CTLA-4, ICOS, EOMES, T-bet and Ki67, were used to cluster the cell populations. For CD8+ T cells, CD44, CD62L, CD25, CD69, CD366, granzyme B, PD-1, CTLA-4, ICOS, EOMES, T-bet and Ki67 were used. We chose the 3,000 iterations, perplexity of 30 and theta of 0.5, as the standard t-SNE parameters. Mean intensity values of markers in each cluster were calculated and visualized via heatmaps. Cells were assigned to different populations on the basis of the local gradient expression of known markers, e.g., CD44, CD62L, Granzyme B and FOXP3. Numbers of cells and percentages of different immune cell subsets were calculated for each sample. Ex vivo cultures stained with a reduced antibody panel (Cd45, Cd4, Cd8, Cd44, Cd62L, Foxp3 ICOS, T-bet, Pd-1, Ctla-4, Ki67) were analyzed using the FlowJo software.
WES and RNA-Seq Data and Analysis
Thirty-six PDX tumors and cell lines and matched normal (or surrogate normal) tissue specimens were subjected to WES and RNA-seq. Sequencing was performed using paired-end sequencing with read length of 2x150 bps based on either the Illumina HiSeq3000 or the NovaSeq V4 platform. We called SNVs and small INDELs as we reported previously(7,28). Mutations were annotated for coding/non-coding alterations using the stand-alone version of Oncotator (45). Copy numbers were called using the intersection of copy number calls derived from Sequenza (46) and VarScan2 (47). Recurrent gene alteration events were visualized using the OncoPrint tool while the MEKi resistance specific mutations on the MAP2K2 gene were visualized using MutationMapper (48,49). GOF alterations include known oncogenic missense mutations (from COSMIC v88), copy number amplification, and/or mRNA over-expression (≥ 2-fold up). LOF alterations include truncating mutations (nonsense, splice site, frameshift), copy number loss and/or mRNA down-expression (≥ 2-fold down). Missense/in-frame INDELS of unknown significance were counted as both GOF and LOF events. CNV-related differential gene expression events were defined as concurrent copy number gain and mRNA over-expression (at least 1.5-fold of genomic copy number gain and 2-fold mRNA over-expression). We applied the same cutoffs for copy number loss (at least 1.5-fold of copy number loss and 2-fold mRNA down-expression). Paired-end, 2x150bp RNA-seq reads were mapped to the Genome Reference Consortium Human Build 38 (GRCh38) reference genome using HISAT2(50). Gene level counts are generated by the htseqcount (51) program, and we took log2 counts per million (CPM) as normalized gene expression values. We added a pseudo CPM count of 0.1 to avoid taking the log of zero. The phylogenetic analysis was performed using the PHYLIP program as in our previous studies(7,28).
For the syngeneic murine model NILER1-4, the paired-end reads were aligned to the mouse reference sequence (GRCm38) using the bwa-mem algorithm. The aligned reads were then processed using Picard tools and GATK (version 4.0) for deduplication and base quality recalibration prior to mutation detection. SNVs and INDELs were identified with MuTect2 using wild-type C57BL/6J mouse as the germline control. Variants were filtered by FilterMutectCalls using GATK default thresholds. Accumulated mutations in NILER1-4, presumably caused by UV irradiation, were detected as novel variants compared with the parental line NIL.
Generation and Analysis of scRNA-Seq Data
To sort tumor-infiltrating lymphocytes (TILs), single-cell suspensions of digested tumor were stained for 5 minutes at room temperature with Fc block (anti-CD16/32) and then stained with primary antibodies in staining buffer (PBS with 20% FCS) for 30 minutes. Sorting of live TILs (TER119−CD45+CD8+CD4− and TER119−CD45+CD4+CD8−) was performed on a BD FACSAria II (BD Biosciences). For each treatment condition, TILs from 4 different tumors were pooled together. Droplet-based 3’ end massively parallel single-cell RNA sequencing (scRNA-seq) was performed by encapsulating sorted, live TILs into droplets, and libraries were prepared using Chromium Single Cell 3’ Reagent Kits v3 according to the manufacturer’s protocol (10x Genomics). The generated scRNAseq libraries were sequenced using Illumina NovaSeq.
Alignment to GRCm38 reference genome, barcode and unique molecular identifier (UMI) counting were performed by using Cell Ranger v2.1.0 (10x Genomics). Seurat package (52) was used for downstream analysis. In brief, cells with less than 500 genes detected or greater than 10% mitochondrial RNA content were excluded from analysis. Raw UMI counts were normalized to UMI count per million total counts and log-transformed. Variable genes were detected based on average expression and dispersion for each dataset independently. We then use CellCycleScoring function to calculate scores of S and G2/M cell cycle phases for each cell. Single cells from vehicle- and Tram+BGB-283-treated samples were integrated into a single assay based on variable genes identified from each sample. We then use the ScaleData function to calculate scaled z-scores of each variable gene in the integrated assay and regress out the effect of number of genes per cell, mitochondrial RNA content, and cell cycle score differences (between S phase score and G2/M phase score). This scaled data set was then used for principle component analysis (PCA) for cells. Clusters and UMAP plots were generated based on top 30 PCA dimensions. Cell clusters expressing markers of both T-cells (i.e., Cd3d, Cd3e, Cd8a and Cd4) and other cell types (i.e., Cd19, B cells; Klrcs1, NK cells; Csf1r, macrophages; Flt3, dendritic cells) were defined as doublets and excluded from further analysis. T-cell clusters were annotated by identifying differentially expressed marker genes with log-fold change higher than 0.4 using MAST in FindAllMarkers function. Finally, scores of T cell’s terminal exhaustion were assigned to each cell by using Seurat’s AddModuleScore function based on the gene sets previously reported(34).
Generation and Analysis of TCR-Seq Data
Total RNA was extracted from frozen tissue stored in RNALater using QIAGEN All Prep DNA/RNA Mini Kit and Ambion mirVana miRNA Isolation Kit. 600 ng of RNA was used as input to construct libraries with the QIAGEN QIAseq Immune Repertoire RNA Library Kit – T-cell Receptor Panel. Briefly, RNA was reverse transcribed using a pool of TCR gene-specific primers against the constant region for the T cell receptor alpha, beta, gamma, and delta genes. The resulting cDNA was then ligated to an oligo containing one side of sample index and unique molecular index (UMI). After reaction cleanup, a single primer extension was used to capture the T-cell receptor using a pool of gene-specific primers. The resulting captured sequences were amplified and purified using QIAseq beads. Libraries were then sample indexed on the other side by using an unique sample index primer and an universal primer to amplify the library and introduce platform-specific adapter sequences. The dual indexed sample PCR fragment was purified, then quantified for absolute quantification of amplifiable libraries (DNA with adaptors at both ends) in triplicate by real-time qPCR using QIAGEN QIAseq Library Quant Array Kit. For sequencing, each library was diluted to 4 nM, pooled, and denatured. 1.2 pM of denatured library pool was run with QIAseq A Read1 Primer on Illumina NextSeq 500 Mid Output Kit using v2.5 chemistry for 300 cycles with an asymmetrical paired end 261/41 bp read for CDR3 region.
Raw reads were analyzed from the QIAGEN GeneGlobe Data Analysis Center (https://www.qiagen.com/us/shop/genes-and-pathways/data-analysis-center-overview-page/), which estimates the abundance of reads of unique CDR3 sequence and generate TCR clonotype calls. Briefly, raw reads were trimmed and randomly down-sampled to control the over-sequencing error in UMI and CDR3 sequences. Paired R1 and R2 were then merged into one read with trimmed V-regions. Clonotypes were called by IMSEQ(53), which clustered highly similar CDR3 sequences. CDR3 calls that did not have at least one UMI supported by three reads were excluded from downstream analysis. R package tcR (54) was used to perform all the statistical analysis for TCR repertoires, including: 1) size of the largest clone, top 3 clones and large clones with frequency higher than 5%; 2) diversity estimation using ecological diversity and Gini-Simpson index, 3) similarities of TCR repertories by calculating Jaccard index and overlap coefficient between every pair of samples based on their unique alpha or beta chains’ CDR3 sequences. The Jaccard index was calculated using jaccard.index function in tcR package, and heatmaps were generated using R pheatmap package. The comparison of overlap coefficients intra- and across treatment groups were performed using Wilcoxon rank-sum test.
Molecular Modeling
Solvent Accessible Surface Areas (SASAs) were calculated using the analytical module for surface calculation of the CHARMM molecular modelling package (55), with water molecules described as spheres with a radius of 1.4 Å. The value of SASA buried upon the binding of MEK1 to BRAF was computed for the residues belonging to the BRAF P-loop (residues 465-469) and for MEK1 residues situated in the vicinity (i.e. residues 73-82 and 97-101) by the difference in the SASA of these residues between the complex and the isolated MEK1 and BRAF proteins, in the same conformation. The buried SASA was calculated for the apo and BGB-283 bound forms of BRAF, to estimate its variation as a result of BGB-283 binding.
The conformation of the MEK1/BRAF heterodimeric complex with the apo form of BRAF was taken from the experimental 3D structure of the BRAF/MEK1 hetero-tetramer complex, involving the apo form of BRAF and the G-573/ACP-bound MEK1 (PDB ID 4MNE) (56). Since the P-loop residues were not resolved in this structure, they were modelled using the MODELLER program (57). For this, 5000 conformations of the P-loop were modelled with DOPE-based loop modeling classes of MODELLER. The 50 top-ranked conformations according to MODELLER were used to calculate the average SASA buried upon the binding of MEK1 to BRAF.
The conformation of the MEK1/BRAF heterodimeric complex with the BGB-283-bound form of BRAF was obtained by superimposing the experimental structure of the complex between BGB-283 and BRAF (PDB ID 4R5Y)(20) on the experimental structure of the MEK1/BRAF heterodimeric complex (PDB ID 4MNE) using UCSF Chimera (58).
Statistical Analysis
No statistical methods were used to predetermine sample size. The paired t-test was performed to determine the statistical significance of differences between two variables. All statistical analyses were carried out using R and GraphPad Prism 7.
Data Availability
Raw sequencing files of RNA-seq, scRNA-seq and TCR-seq data are available at the Gene Expression Omnibus (GEO158610). Mass cytometry data are deposited at FlowRepository (http://flowrepository.org/) using the experiment ID FR-FCM-Z34M. WES data have been made available through the Sequence Read Archive (SRA) at the accession number PRJNA666070.
Supplementary Material
SIGNIFICANCE.
Type I RAFi+MEKi are indicated only in certain BRAFV600MUT cancers. In contrast, type II RAFi+MEKi are durably active against acquired MEKi-resistance across broad cancer indications, which reveals exquisite MAPK addiction. Allosteric modulation of MAPK protein/protein interactions and temporal preservation of intra-tumoral CD8+ T-cells are mechanisms that may be further exploited.
Acknowledgements
We thank Lusong Luo (BeiGene Inc.) for providing BGB-283 and −3245 and Robert H. Vonderheide for providing the KPC syngeneic PDAC cell line. We would like to acknowledge the support of the National Institutes of Health (NIH) (1P01CA168585 and 1P01CA244118-01A1) (to R.S. Lo and A. Ribas). This research was also supported by grants (to R.S. Lo) from: National Institutes of Health (NIH) (1R01CA176111A1; 1R21CA215910-01), Melanoma Research Alliance (MRA) (Team Science Award), and V Foundation for Cancer Research (Translational Award). Additional funding was provided by: Department of Defense Horizon Award (to A. Hong), MRA Dermatology Fellows Award (to S. Liu), National Cancer Center Postdoctoral Fellowship (to Z. Yang), JCCC Postdoctoral Fellowships (to S. Liu and Z. Yang), JCCC Postdoctoral Seed Grant (to Z. Yang), Dermatology Foundation Career Development Award (to G. Moriceau), NIGMS P20GM121293 (to A.J. Tackett), Huntsman Cancer Foundation (to S.L. Holmen), and Steven C. Gordon Family Foundation (to R.S. Lo). We acknowledge use of the HCI Biorepository and Molecular Pathology (BMP) Shared Resource supported by P30CA042014 awarded to HCI from the National Cancer Institute. We also thank the Huntsman Cancer Institute (HCI) Preclinical Research Resource (PRR) for assistance with PDX models. Flow cytometry and CyTOF were performed in the UCLA Jonsson Comprehensive Cancer Center (JCCC) Flow Cytometry Core Facility that is supported by National Institutes of Health award P30 CA016042, the JCCC, the David Geffen School of Medicine at UCLA, the UCLA Chancellor’s Office, and the UCLA Vice Chancellor’s Office of Research. We would like to thank the Technology Center for Genomics and Bioinformatics at UCLA for excellent technical support. R.S. Lo and A. Ribas are especially grateful to the Ressler Family Foundation for its long-term support. R.S. Lo dedicates this study to the memory of Waun Ki Hong, M.D.
Disclosure of Potential Conflicts of Interest
R.S.L. receives research funding from Merck, Array BioPharma, and OncoSec Medical and consulted in the last three years with Amgen, Shire, Novartis, Array BioPharma and Merck. S.L.H. receives research funding from Agios. A.R. consulted with Amgen, Bristol-Myers Squibb, Chugai, Genentech, Merck, Novartis and Roche, and is or has been a member of the scientific advisory board and holds stock in Advaxis, Arcus Biosciences, Bioncotech Therapeutics, Compugen, CytomX, Five Prime, FLX-Bio, ImaginAb, Isoplexis, Kite-Gilead, Lutris Pharma, Merus, PACT Pharma, Rgenix and Tango Therapeutics. S.D. serves on the scientific advisory boards of J&J Lung Cancer Initiative, AstraZeneca, LungLifeAI, Early Diagnostics, Inc., and T-Cure Biosciences and receives research funding from J&J and Novartis.
References
- 1.Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov 2014;13:928–42 [DOI] [PubMed] [Google Scholar]
- 2.Dummer R, Schadendorf D, Ascierto PA, Arance A, Dutriaux C, Di Giacomo AM, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2017;18:435–45 [DOI] [PubMed] [Google Scholar]
- 3.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14 [DOI] [PubMed] [Google Scholar]
- 4.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468:973–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 2012;366:207–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Long GV, Fung C, Menzies AM, Pupo GM, Carlino MS, Hyman J, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun 2014;5:5694. [DOI] [PubMed] [Google Scholar]
- 7.Moriceau G, Hugo W, Hong A, Shi H, Kong X, Yu CC, et al. Tunable-Combinatorial Mechanisms of Acquired Resistance Limit the Efficacy of BRAF/MEK Cotargeting but Result in Melanoma Drug Addiction. Cancer Cell 2015;27:240–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, et al. MAP Kinase Pathway Alterations in BRAF-Mutant Melanoma Patients with Acquired Resistance to Combined RAF/MEK Inhibition. Cancer Discov 2014;4:61–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamba S, Russo M, Sun C, Lazzari L, Cancelliere C, Grernrum W, et al. RAF suppression synergizes with MEK inhibition in KRAS mutant cancer cells. Cell Rep 2014;8:1475–83 [DOI] [PubMed] [Google Scholar]
- 10.Lito P, Saborowski A, Yue J, Solomon M, Joseph E, Gadal S, et al. Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell 2014;25:697–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ascierto PA, Ferrucci PF, Fisher R, Del Vecchio M, Atkinson V, Schmidt H, et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat Med 2019;25:941–6 [DOI] [PubMed] [Google Scholar]
- 12.Ribas A, Lawrence D, Atkinson V, Agarwal S, Miller WH, Jr., Carlino MS, et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat Med 2019;25:936–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015;162:1271–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ebert PJ, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016;44:609–21 [DOI] [PubMed] [Google Scholar]
- 15.Dushyanthen S, Teo ZL, Caramia F, Savas P, Mintoff CP, Virassamy B, et al. Agonist immunotherapy restores T cell function following MEK inhibition improving efficacy in breast cancer. Nat Commun 2017;8:606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi H, Deng J, Li S, Silk T, Dong L, Brea EJ, et al. Pulsatile MEK Inhibition Improves Anti-tumor Immunity and T Cell Function in Murine Kras Mutant Lung Cancer. Cell Rep 2019;27:806–19 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao Z, Gao Y, Su W, Yaeger R, Tao J, Na N, et al. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat Med 2019;25:284–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peng SB, Henry JR, Kaufman MD, Lu WP, Smith BD, Vogeti S, et al. Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell 2015;28:384–98 [DOI] [PubMed] [Google Scholar]
- 19.Shao W, Mishina YM, Feng Y, Caponigro G, Cooke VG, Rivera S, et al. Antitumor Properties of RAF709, a Highly Selective and Potent Inhibitor of RAF Kinase Dimers, in Tumors Driven by Mutant RAS or BRAF. Cancer Res 2018;78:1537–48 [DOI] [PubMed] [Google Scholar]
- 20.Tang Z, Yuan X, Du R, Cheung SH, Zhang G, Wei J, et al. BGB-283, a Novel RAF Kinase and EGFR Inhibitor, Displays Potent Antitumor Activity in BRAF-Mutated Colorectal Cancers. Mol Cancer Ther 2015;14:2187–97 [DOI] [PubMed] [Google Scholar]
- 21.Vakana E, Pratt S, Blosser W, Dowless M, Simpson N, Yuan XJ, et al. LY3009120, a panRAF inhibitor, has significant anti-tumor activity in BRAF and KRAS mutant preclinical models of colorectal cancer. Oncotarget 2017;8:9251–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramurthy S, Taft BR, Aversa RJ, Barsanti PA, Burger MT, Lou Y, et al. Design and Discovery of N-(3-(2-(2-Hydroxyethoxy)-6-morpholinopyridin-4-yl)-4-methylphenyl)-2-(trifluorom ethyl)isonicotinamide, a Selective, Efficacious, and Well-Tolerated RAF Inhibitor Targeting RAS Mutant Cancers: The Path to the Clinic. J Med Chem 2019;63:2013–27 [DOI] [PubMed] [Google Scholar]
- 23.Sullivan RJ, Hollebecque A, Flaherty KT, Shapiro GI, Rodon Ahnert J, Millward MJ, et al. A Phase 1 Study of LY3009120, a Pan-RAF Inhibitor, in Patients with Advanced or Metastatic Cancer. Mol Cancer Ther 2019;19:460–7 [DOI] [PubMed] [Google Scholar]
- 24.Yen I, Shanahan F, Merchant M, Orr C, Hunsaker T, Durk M, et al. Pharmacological Induction of RAS-GTP Confers RAF Inhibitor Sensitivity in KRAS Mutant Tumors. Cancer Cell 2018;34:611–25 e7 [DOI] [PubMed] [Google Scholar]
- 25.Hong A, Moriceau G, Sun L, Lomeli S, Piva M, Damoiseaux R, et al. Exploiting drug addiction mechanisms to select against MPAKi-resistant melanoma. Cancer Discov 2018;8:1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song C, Piva M, Sun L, Hong A, Moriceau G, Kong X, et al. Recurrent Tumor Cell-Intrinsic and -Extrinsic Alterations during MAPKi-Induced Melanoma Regression and Early Adaptation. Cancer Discov 2017;7:1248–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 2019;47:D941–D7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, et al. Acquired Resistance and Clonal Evolution in Melanoma during BRAF Inhibitor Therapy. Cancer Discov 2014;4:80–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi H, Moriceau G, Kong X, Lee MK, Lee H, Koya RC, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun 2012;3:724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao Y, Chang MT, McKay D, Na N, Zhou B, Yaeger R, et al. Allele-Specific Mechanisms of Activation of MEK1 Mutants Determine Their Properties. Cancer Discov 2018;8:648–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan J, Ng WH, Tian Z, Yap J, Baccarini M, Chen Z, et al. Activating mutations in MEK1 enhance homodimerization and promote tumorigenesis. Sci Signal 2018;11:eaar6795. [DOI] [PubMed] [Google Scholar]
- 32.Lavoie H, Sahmi M, Maisonneuve P, Marullo SA, Thevakumaran N, Jin T, et al. MEK drives BRAF activation through allosteric control of KSR proteins. Nature 2018;554:549–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim K, Park S, Park SY, Kim G, Park SM, Cho JW, et al. Single-cell transcriptome analysis reveals TOX as a promoting factor for T cell exhaustion and a predictor for anti-PD-1 responses in human cancer. Genome Med 2020;12:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao C, Sun HW, Lacey NE, Ji Y, Moseman EA, Shih HY, et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8(+) T cell persistence in chronic infection. Nat Immunol 2019;20:890–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hatzivassiliou G, Liu B, O’Brien C, Spoerke JM, Hoeflich KP, Haverty PM, et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol Cancer Ther 2012;11:1143–54 [DOI] [PubMed] [Google Scholar]
- 36.Takeuchi Y, Tanemura A, Tada Y, Katayama I, Kumanogoh A, Nishikawa H. Clinical response to PD-1 blockade correlates with a sub-fraction of peripheral central memory CD4+ T cells in patients with malignant melanoma. Int Immunol 2018;30:13–22 [DOI] [PubMed] [Google Scholar]
- 37.Wu F, Zhang W, Shao H, Bo H, Shen H, Li J, et al. Human effector T cells derived from central memory cells rather than CD8(+)T cells modified by tumor-specific TCR gene transfer possess superior traits for adoptive immunotherapy. Cancer Lett 2013;339:195–207 [DOI] [PubMed] [Google Scholar]
- 38.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A 2005;102:9571–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberts AD, Ely KH, Woodland DL. Differential contributions of central and effector memory T cells to recall responses. J Exp Med 2005;202:123–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi H, Moriceau G, Kong X, Koya RC, Nazarian R, Pupo GM, et al. Preexisting MEK1 exon 3 mutations in V600E/KBRAF melanomas do not confer resistance to BRAF inhibitors. Cancer Discov 2012;2:414–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hendry S, Salgado R, Gevaert T, Russell PA, John T, Thapa B, et al. Assessing Tumor-Infiltrating Lymphocytes in Solid Tumors: A Practical Review for Pathologists and Proposal for a Standardized Method from the International Immuno-Oncology Biomarkers Working Group: Part 2: TILs in Melanoma, Gastrointestinal Tract Carcinomas, Non-Small Cell Lung Carcinoma and Mesothelioma, Endometrial and Ovarian Carcinomas, Squamous Cell Carcinoma of the Head and Neck, Genitourinary Carcinomas, and Primary Brain Tumors. Adv Anat Pathol 2017;24:311–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saldanha G, Flatman K, Teo KW, Bamford M. A Novel Numerical Scoring System for Melanoma Tumor-infiltrating Lymphocytes Has Better Prognostic Value Than Standard Scoring. Am J Surg Pathol 2017;41:906–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kotecha N, Krutzik PO, Irish JM. Web-based analysis and publication of flow cytometry experiments. Curr Protoc Cytom 2010;Chapter 10:Unit10 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen H, Lau MC, Wong MT, Newell EW, Poidinger M, Chen J. Cytofkit: A Bioconductor Package for an Integrated Mass Cytometry Data Analysis Pipeline. PLoS Comput Biol 2016;12:e1005112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, et al. Oncotator: cancer variant annotation tool. Hum Mutat 2015;36:E2423–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, et al. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann Oncol 2015;26:64–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012;22:568–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 2015;12:357–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015;31:166–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature biotechnology 2018;36:411–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuchenbecker L, Nienen M, Hecht J, Neumann AU, Babel N, Reinert K, et al. IMSEQ--a fast and error aware approach to immunogenetic sequence analysis. Bioinformatics 2015;31:2963–71 [DOI] [PubMed] [Google Scholar]
- 54.Nazarov VI, Pogorelyy MV, Komech EA, Zvyagin IV, Bolotin DA, Shugay M, et al. tcR: an R package for T cell receptor repertoire advanced data analysis. BMC bioinformatics 2015;16:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brooks BR, Brooks CL 3rd, Mackerell AD Jr., Nilsson L, Petrella RJ, Roux B, et al. CHARMM: the biomolecular simulation program. J Comput Chem 2009;30:1545–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haling JR, Sudhamsu J, Yen I, Sideris S, Sandoval W, Phung W, et al. Structure of the BRAF-MEK Complex Reveals a Kinase Activity Independent Role for BRAF in MAPK Signaling. Cancer Cell 2014;26:402–13 [DOI] [PubMed] [Google Scholar]
- 57.Webb B, Sali A. Protein Structure Modeling with MODELLER. Methods Mol Biol 2017;1654:39–54 [DOI] [PubMed] [Google Scholar]
- 58.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–12 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing files of RNA-seq, scRNA-seq and TCR-seq data are available at the Gene Expression Omnibus (GEO158610). Mass cytometry data are deposited at FlowRepository (http://flowrepository.org/) using the experiment ID FR-FCM-Z34M. WES data have been made available through the Sequence Read Archive (SRA) at the accession number PRJNA666070.