

Abstract
Background –
Epidemiological studies have established obesity as an independent risk factor for atrial fibrillation (AF) but the underlying pathophysiological mechanisms remain unclear. Reduced cardiac sodium channel expression is a known causal mechanism in AF. We hypothesized that obesity decreases Nav1.5 expression via enhanced oxidative stress, thus reducing INa, and enhancing susceptibility to AF.
Methods –
To elucidate the underlying electrophysiologic (EP) mechanisms a diet-induced obese (DIO) mouse model was used. Weight, BP, glucose, F2-isoprostanes (F2-IsoPs), NADPH oxidase 2 (NOX2), and protein kinase C (PKC) were measured in obese mice and compared to lean controls. Invasive EP, immunohistochemistry, Western blotting and patch clamping of membrane potentials was performed to evaluate the molecular and EP phenotype of atrial myocytes.
Results –
Pacing induced AF in 100% of DIO mice versus 25% in controls (P< 0.01) with increased AF burden. Cardiac sodium channel expression, INa and atrial action potential duration (APD) were reduced and potassium channel expression (Kv1.5) and current (IKur) and F2-IsoPs, NOX2, and PKC-ɑ/δ expression and atrial fibrosis were significantly increased in DIO mice as compared to controls. A mitochondrial antioxidant reduced AF burden, restored INa, ICa,L, IKur, APD and reversed atrial fibrosis in DIO mice as compared with controls.
Conclusions –
Inducible AF in obese mice is mediated in part by a combined effect of sodium, potassium and calcium channel remodeling and atrial fibrosis. Mitochondrial antioxidant therapy abrogated the ion channel and structural remodeling and reversed the obesity-induced AF burden. Our findings have important implications for the management of obesity-mediated AF in patients.
Keywords: atrial fibrillation, obesity, ionic remodeling, oxidative stress, antiarrhythmic drug
Graphical Abstract
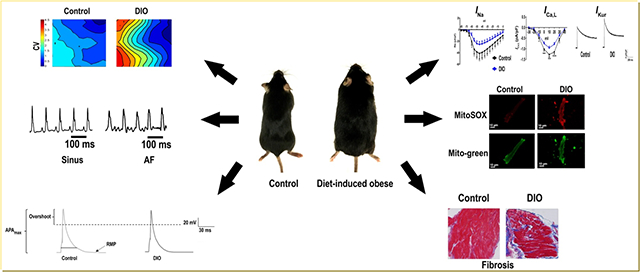
Introduction
Obesity is a global pandemic, which is steadily on the rise; between 1980–2013, the global prevalence of overweight and obese individuals rose by 27.5% for adults.1 While studies have established an association between obesity and AF, it was only recently that a causal link was confirmed.2 In sheep fed a high-calorie diet, left atrium (LA) enlargement, atrial fibrosis, increased inflammatory markers, lipid infiltration, and atrial electrophysiologic (EP) changes were observed, all of which have been associated with AF.3, 4 In spontaneously hypertensive rats, obesity is an independent risk factor for the progression of metabolic syndrome and AF.5 Whereas, weight reduction decreases AF burden.6 These results suggest that while the underlying mechanisms of AF are multifactorial, addressing a single modifiable risk factor, e.g., obesity may reduce AF burden and suppress disease progression.7, 8
Prior studies have shown that atrial fibrosis creates a substrate for AF in diet-induced obese (DIO) mice but the role of ion channel remodeling in obesity-mediated AF remains unclear.9, 10 While increased expression of delayed rectifier potassium currents (IK) leading to shortened action potential duration (APD) has been implicated in AF in obese guinea pigs, whether other ion channels are also involved in modulating the atrial AP and conduction velocity (CV) is unknown.11 Both an increase and decrease in cardiac sodium channel expression has been associated with AF suggesting a key role for this channel in disease pathogenesis.12, 13 However, it remains unclear if obesity-mediated AF is due to alterations in Nav1.5 and other ion channels by mediators such as oxidative stress, a central downstream molecular pathway modulated by obesity.14 We hypothesized that obesity decreases Nav1.5 expression via enhanced oxidative stress, reducing the sodium current (INa), and creating an EP substrate for reentrant AF. To test this hypothesis, we determined the association between obesity and Nav1.5 expression, INa, oxidative stress, and AF inducibility in DIO mice.
Methods
The data that support the findings of this study are available from the corresponding authors on reasonable request.
Mouse Model of Obesity
DIO Mice
Animal studies were performed according to protocols approved by the institutional animal care and use committee (ACC) at UIC, and as per NIH guidelines. DIO mice were bred on a C57BL/6J genetic background, and were fed a high fat diet (HFD, 60% fat; Teklad #06414) starting at 8 weeks of age. Wild-type (lean) controls were fed a regular mouse diet. Mice were considered obese when at least 33g in body mass as previously described.15 Female mice did not meet the weight threshold despite a HFD and therefore were excluded from the study. Detailed methods for invasive EP, echocardiography, BP and glucose measurements, Western blotting, and immunohistochemistry are provided in the Supplemental Methods.
Measurement of Mitochondrial Reactive Oxygen Species (ROS)
Freshly isolated atrial myocytes from control and DIO mice were incubated with 5μM Mito-Sox for 10 min at 37°C. To normalize for mitochondrial mass and assess mitochondrial structure, cells were also stained with 50 nM MitoTracker Green for 30 mins at 37°C with the fluorescence intensity measured using confocal microscopy (Zeiss LSM810) and Image J. Mito-Tracker Green was excited at 488nm and Mito-Sox at 561nm laser. The ratio of Mito-Sox fluorescence over MitoTracker fluorescence was used as a normalized indicator of mitochondrial ROS.
Atrial Fibrosis Analysis using Masson’s Trichrome Staining
We fixed the harvested hearts with 10% neutral formalin overnight, and then embedded in paraffin; 4um thick sections were cut with microtome and stained with Masson’s trichrome stain (Sigma) after de-paraffination. Using Image J, we calculated the cardiac fibrosis ratio by dividing total cardiomyocyte area in atrium.
Optical Mapping
We harvested intact mouse hearts with cannulation via the aorta onto a Langendorff perfusion system and loaded with Rh237 (10 mM; Invitrogen) dye. The LA was then placed in the middle of the perfusion chamber with the posterior wall facing the objective of the microscope for all mapped hearts. We carefully positioned the stimulating electrode in the same area of the LA, adjacent to the aorta. Voltage-sensitive fluorescent signals (Vm; RH-237 dye) were acquired at a spatial resolution of 137.5μm and sampling rate of 2kHz/channel and analyzed as described.16, 17 The AP amplitude (APA) was defined as the difference between the maximal signal at the peak of upstroke and the baseline fluorescence signal. The activation time was defined as the time when the upstroke rose to 50% of APA. APD was defined as the time interval from activation time to 90% of repolarization (APD90). Upstroke slope was calculated as the maximal dV/dtmax of AP rise. CVs at a pacing CL of 100 msec were calculated via vector field analysis with the CV vector calculated from activation times measured at 5×5 neighboring pixels. Conduction paths were assigned according to the group of vectors spreading from the central stimulation site for every 10° (0–360°). The pathway containing the largest population of vectors was considered as the major CV pathway. We analyzed 3 beats for each mouse at 100 ms CL. The average value of the major-pathway CV from the three beats of each LA was used for statistical analysis.
Measurement of F2-Isoprostanes (F2-IsoPs)
Blood samples were centrifuged immediately after collection, and isolated plasma was stored in EDTA-containing vials at −80°C until assayed. Plasma 8-isoprostrane (8-IsoP) concentrations were measured using a competitive enzyme immunoassay kit (Cayman Chemicals). Briefly, 50μL of standard/plasma samples were placed in 96-well plate that was pre-coated with mouse monoclonal antibody after purification using C-18 solid phase extraction cartridge. Thereafter, 50μL of 8-IsoP tracer and antiserum was added into each well and incubated for 18hr at room temperature. After washing with wash buffer, 200μL of Ellman’s reagent containing acetylcholinesterase substrate was added. The plates were read at 412 nm and the values of plasma 8-IsoPs were calculated from a curve drawn using standard concentrations
Statistical Analysis
Categorical variables are expressed as counts and percentages, and continuous variables are expressed as mean±standard deviation (SD). Bivariate analyses of a continuous and categorical variable was assessed using two sample t-tests or repeated measures analysis of variance (ANOVA) as appropriate, and two categorical variables were compared using the Chi-square test for independence and also Fisher’s exact test when cell sizes were less than 5. Multivariate analysis was performed with logistic regression. Statistical significance was defined by P<0.05. Statistical software SAS 9.4 was used to conduct the analysis.
Results
DIO obese mice are prone to AF
We determined whether obesity increases AF in DIO mice. The average weight of DIO mice was 37.5±3.84g versus 24.3±4.1g controls (P ≤0.0001; Figure 1A). Mice were considered obese when at least 33g in body weight using a threshold that would require a weight gain of 30–50% (Figure 1B). The average age of DIO mice achieving the 33g threshold was 20–22 weeks. Females failed to reach this threshold and thus were not included in the study (P<0.0001; Figure 1C). To evaluate AF inducibility in DIO mice, we performed TE programmed stimulation of the atria as previously described.18 Upon atrial stimulation (Figure 1D) all DIO mice (100%) developed AF versus only 25% in control mice (P≤0.0001; Figure 1E). AF duration was 284±295 seconds in DIO mice versus 13±26 seconds in control mice (P<0.0001; Figure 1F), supporting a higher incidence and burden of pacing-induced AF in obese mice. AF duration in DIO mice on TE pacing increased with increase in weight (P ≤0.0001; Figure 1G). Echocardiography of DIO mice showed no difference in LA size (Table 1).
Figure 1:

Diet-induced obese (DIO) mice are prone to atrial fibrillation (AF). The association between obesity and pacing-induced AF was examined in wild-type (lean) control, and DIO mice. A: Photograph showing the obesity phenotype in control, and DIO mice. B: Weights in control and DIO mice. Bars represent mean weight (g) in each group. Red dashed line represents the 33g threshold for obesity. C: Average weight (g) of mice who received high fat diet (HFD) over 10 weeks duration. D: Atrial electrograms showing sinus rhythm at baseline (top), pacing-induced AF in DIO mice (middle) and sinus rhythm restoration in DIO mice (bottom). E: Incidence of AF among the 2 groups. F: Burden (duration, s) of pacing-induced AF in DIO (N=24) and control (N=16) mice. G: Increase in burden (duration, s) of pacing-induced AF with increase in weight. *P<0.05, **P<0.01; ***P<0.001, ****P<0.0001.
Table 1:
Echocardiographic parameters of control and diet-induced obese (DIO) mice.
| Control | DIO | P-value | |
|---|---|---|---|
| Heart rate (bpm) | 515 ± 28 | 570 ± 252 | 0.68 |
| Left atrial diameter (mm) | 1.59 ± 0.20 | 1.56 ± 0.06 | 0.78 |
| Left ventricular ejection fraction (%) | 60.42 ± 2.61 | 72.74 ± 7.61 | 0.02 |
| Fractional shortening (%) | 31.44 ± 1.96 | 41.45 ± 6.74 | 0.03 |
| End systolic diameter (mm) | 2.33 ± 0.09 | 2.05 ± 0.42 | 0.25 |
| End diastolic diameter (mm) | 3.40 ± 0.23 | 3.49 ± 0.38 | 0.72 |
| Intraventricular septum in systole (mm) | 0.65 ± 0.10 | 0.72 ± 0.08 | 0.28 |
| Intraventricular septum in diastole (mm) | 0.45 ± 0.02 | 0.56 ± 0.10 | 0.08 |
| Left ventricular posterior wall in systole (mm) | 0.83 ± 0.14 | 0.75 ± 0.18 | 0.5 |
| Left ventricular wall in diastole (mm) | 0.52 ± .12 | 0.52 ± 0.11 | 0.92 |
APD shortening and reduced CV
To investigate the underlying EP mechanism of AF in DIO mice, we measured the APD20, APD50 and APD90 using whole cell patch clamping (Figure 2A–D). DIO mice showed shortened APD20, APD50, APD90 and APAmax but the resting membrane potential (RMP) remained unchanged (P<0.01, Figure 2B–F). The maximal upstroke velocity of the AP, dV/dTmax was significantly reduced in DIO mice as compared to controls (P<0.001, Figure 2G). Furthermore, optical mapping confirmed delayed upstroke of the AP with reduced atrial CV strongly supporting a reentrant mechanism for obesity-induced AF (P<0.001; Figure 2H–I)
Figure 2:

Atrial action potential duration (APD) is altered in DIO mice. Patch-clamped atrial myocytes displayed reduced APD, a condition associated with AF induction. A: Representative AP tracings in atrial myocytes in DIO and control mice. Measured parameters include: B: APD at 20% repolarization (APD20; n=6 cells); C: APD at 50% repolarization (APD50; n=6 cells); D: APD at 90% repolarization (APD90; n=6 cells). E: Maximum AP amplitude (APAmax; n=6 cells). F: Resting membrane potential (RMP; n=6 cells). G: Instantaneous rate of voltage change over time (dV/dTmax), an indicator of atrial conduction velocity (CV; n=6 cells). H: Representative images of atrial optical mapping experiments in DIO and control mice. I: Quantification of mean atrial CV with optical mapping (N=6 hearts). *P<0.05, **P<0.01; ***P < 0.001, ****P< 0.0001.
Downregulation of Nav1.5 in obese mice with AF
Yanni et al19 previously reported a reduction of Nav1.5 in the sinoatrial nodes of aged obese rats. In humans, genetic variations in SCN5A are prone to alterations in cardiac repolarization, reduction of APD, and susceptibility to AF. Given this association, we sought to determine the role of Nav1.5 expression in the obesity-AF phenotype. Western blotting of Nav1.5 in control and DIO mouse atria revealed a significant reduction of Nav1.5 protein in obese mice versus age-matched controls (P<0.0001; Figure 3A–B), thus establishing that obesity reduced Nav1.5 expression independent of the age. Immunohistochemical staining of Nav1.5 in the 2 groups also revealed a significant reduction in Nav1.5 protein expression versus controls (P < 0.05, respectively, Figure 3C–D).
Figure 3:

The cardiac sodium current (INa) is reduced in DIO mice. Obese mice have reduced sodium channel current (INa) versus controls. A: Western blot showing Nav1.5 expression in atria of control, and DIO mice (N=7 hearts). B: Quantification of Nav1.5 expression among the 2 groups (N=7 hearts). C: Atrial sections immunostained for Nav1.5 (left), DAPI (center), and merged (right). D: Quantification of corrected total cell fluorescence (CTCF) of Nav1.5 protein in immunostained samples (n=14 cells). E: Sodium current and voltage relationship (I-V curves) in control and DIO mice (n=6 atrial cells). F: Comparisons of peak INa at −50mV in control (n=6 cells) and DIO (n=6 cells) mice. G: Steady-state inactivation and activation of the sodium currents in control (n=6 cells) and DIO mice (n=6 cells). H: DIO mice did not significantly alter sodium windows currents compared to control. *P<0.05, **P<0.01; ***P<0.001, ****P<0.0001.
INa is reduced in obese mice
We performed patch clamping of atrial cells isolated from the 2 groups of mice. We found that INa was significantly reduced at all test potentials between −65 mV and −5 mV (P<0.01 and P=0.03; Figure 3E) and the peak INa was reduced by −37% in DIO mice (P<0.0001, Figure 3F). INa kinetics and voltage dependence were not significantly changed in DIO mice (Figure 3G–H). Thus, in mice with acquired obesity, reduced Nav1.5 expression is associated with reduced INa, shortened APD and reduced dV/dTmax and CV, recognized risk factors for AF initiation.
L-type calcium and potassium channel expression
To fully elucidate the EP phenotype, we evaluated the role of the ultra-rapid potassium current (Ikur), and calcium current (ICa,L) as well as calcium handling proteins as potential regulators of APD in obesity-induced AF. First, there was significant reduction in CaV1.2 expression and ICa,L density in DIO mice (P<0.05, Figure 4A–B). ICa,L was significantly reduced at all test potentials between −10 mV and +40 mV (P<0.01 and P<0.001; Figure 4C) and the peak ICa,L was reduced by −23% in DIO mice (P<0.05, Figure 4D). ICa,L kinetics and voltage dependence however were unchanged in DIO mice (Supplementary Figure 1G). Second, we observed increased Kv1.5 expression in DIO mice (P<0.001; Figure 4E–F). Third, there was no change in Kv4.2/3 expression (Supplementary Figure 1E–F). Fourth, there was a significant increase in IKur in DIO mice (P<0.01, Figure 4G–H). Although connexin expression is important in atrial CV changes,20, 21 there were no differences in connexin-40/43 expression in DIO mice versus controls (Supplementary Figure 1).
Figure 4:

Obesity affects potassium and L-type calcium channels. A: Western blot comparing atrial CaV1.2 expression in DIO and control mice (N=6 hearts). B: Quantification of atrial CaV1.2 expression among the 2 groups (N=6 hearts). C: I/V curve with ICa,L recordings in mouse atrial cells in control (n=6 cells) and DIO mice (n=6 cells) at different test potentials. D: Comparisons of peak ICa,L at 10 mV in control (n=6 cells) and DIO (n=6 cells) mice. E: Western blot comparing Kv1.5 expression in DIO and control mice (N=6 hearts). F: Quantification of Kv1.5 expression among the 2 groups (N=6 hearts). G: Representative IKur recordings in control and DIO mice; H: Comparisons of peak IKur density among the 2 groups (n=6, 6 cells respectively). *P<0.05, **P<0.01; ***P<0.001, ****P<0.0001.
DIO mice are prone to atrial oxidation
Obesity and AF are both independently associated with oxidative stress and compromised redox balance.22, 23 However, the association between obesity and atrial oxidation is not firmly established. Obese mice were phenotyped to determine the atrial substrate for the induction and maintenance of AF. First, serum glucose was significantly higher in DIO mice (120±12 mg/dL) than controls (100±11 mg/dL; P<0.05, Supplementary Figure 2A), consistent with prior reports in obese patients with metabolic syndrome. Second, BP readings of mean arterial pressure (MAP) showed significant reduction in DIO mice (P<0.05; Supplementary Figure 2B). Third, Masson’s Trichrome staining of atrial samples from the DIO and Control groups showed that there was significant increase in fibrosis in DIO mice (P <0.01; Supplementary Figure 2C–D). Fourth, there was no difference in expression of calcium handling proteins such as Ca2+/calmodulin-dependent protein kinase II (CaMKII), the cardiac sodium-calcium exchanger (NCX1) and ryanodine receptor type 2 (RyR2) in DIO mice (Supplementary Figure 3C–H). However, SERCA2a expression was significantly increased in DIO mice (Supplementary Figure 3A–B). Collectively our findings not only suggest that obesity directly increases atrial oxidation in DIO mice but that ion channel remodeling especially downregulation of the cardiac INa is the predominant plasma membrane current contributing to the cellular AF phenotype in an acquired mouse model of obesity.
To measure mitochondrial ROS production, freshly isolated atrial myocytes from DIO and control mice were incubated with the mitochondrial-specific ROS fluorescence dye Mito-Sox. We established that Mito-Sox fluorescence signals were abolished by the mitochondrial antioxidant MitoTEMPO (MT), thus confirming that Mito-Sox was measuring mitochondrial ROS. The cells were stained with Mito-Tracker Green and the fluorescence intensity was measured using confocal microscopy. We showed that the mitochondrial ROS levels were increased 3-fold and 5-fold in the atrial myocytes of DIO mice when compared to control mice (Figure 5A–B). Furthermore, protein expression of NOX2, one of the major enzymes involved in superoxide and ROS production in the atria, was increased (Figure 5C–D). We also observed increased protein expression of PKC isoforms PKC-α and PKC-δ which modulate INa ion channel expression and function (Figure 5E–H).24 We next assessed whether acute exposure to a mitochondrial ROS generating agent (antimycin, AMA) would alter INa density in vitro by patch-clamping healthy control murine atrial myocytes in the presence of AMA (20 uM) loaded in each patching pipette.25 Here, there was significant reduction in INa (P<0.05, Supplementary Figure 2E–F). Collectively these data support the observation that obesity-mediated atrial oxidation specific to the mitochondria is associated with reduced Nav1.5 expression and INa.
Figure 5:

The atrial substrate in obesity favors AF. Characterization of the atrial substrate in DIO mice shows atrial fibrosis and oxidation versus controls. A: Atrial sections immunostained for MitoSOX, a marker of mitochondrial oxidative stress in Control and DIO mice. B: Quantification of MitoSOX staining in DIO mice and controls (n=9, 7 cells respectively). C: Western blot comparing NOX2 expression among the 2 groups. D: Quantification of NOX2 expression (N=6 hearts); E: Western blot comparing PKC-δ expression among two groups. F: Quantification of PKC-δ expression. G: Western blot comparing PKC-α expression among two groups. H: Quantification of PKCα expression (N=6 hearts). I: Masson’s trichrome staining of atrial myocytes in Control and DIO mice. J: Fold change in fibrosis (%) in the 2 groups of mice showing a significant increase in atrial fibrosis in DIO mice (n=10, 11 cells respectively). *P<0.05, **P<0.01; ***P<0.001, ****P<0.0001.
Treatment of DIO mice with MT reduces oxidative stress and AF inducibility
Currently there are no FDA-approved antiarrhythmic drugs (AADs) that reliably increase INa. Furthermore, Class I AADs such as flecainide or propafenone, have the potential to further reduce INa, and potentially cause a profibrillatory substrate in patients with obesity. As our data suggested that mitochondrial oxidant stress impairs Nav1.5 function, we tested the therapeutic efficacy of a mitochondrial-targeted superoxide scavenger, MT to reduce AF burden and improve INa in DIO mice.
To test the hypothesis that reduction of atrial oxidation reduces pacing-induced AF, we administered intra-peritoneal MT (0.7 mg/kg) to DIO and control mice daily for 2 weeks. We then evaluated whether the antioxidant alters the AF substrate, and incidence. First, DIO mice treated with MT showed a significant increase in Nav1.5 protein expression and peak INa density when compared to DIO mice not treated with the antioxidant (Figure 6A–B). Also, the INa was significantly restored at all test potentials between −60 mV and −20 mV and the peak INa was increased by −40% in DIO mice (P<0.01 and P=0.03; Figure 5G–H). Second, Kv1.5 expression and IKur were significantly reduced in DIO mice treated with MT as compared with control DIO mice (P<0.05; Figure 6E–F, 6I-J). Third, atrial fibrosis and plasma 8-IsoPs were significantly reduced in MT treated DIO treated mice versus untreated DIO mice (Figure 7). Fourth, APD20 and APD90 were normalized while there was no significant difference in APD50 (P<0.05; Figure 8A–D) and the dV/dTmax was completely reversed in MT treated DIO mice (P<0.001; Figure 8E). Finally, AF burden was significantly reduced in DIO mice treated with MT as compared to DIO controls (P<0.05; Figure 8F). Collectively, our data suggests that the obesity-mediated EP substrate for AF can be abrogated by targeting oxidative stress with reversal of ion channel remodeling, normalized APD and maximum upstroke velocity.
Figure 6:

The antioxidant MitoTEMPO (MT) normalizes cardiac sodium channel and potassium channel expression in obese mice. A: Western blot comparing Nav1.5 expression in DIO and DIO-MT treated mice. B: Quantification of Nav1.5 expression among the 2 groups (N=6 hearts); C: Western blot comparing Cav1.2 expression in DIO and DIO-MT treated mice. D: Quantification of CaV1.2 expression among the 2 groups (N=6 hearts). E: Western blot comparing Kv1.5 expression in DIO and DIO-MT treated mice. F: Quantification of Kv1.5 expression among the 2 groups (N=6 hearts). G: I/V curve of INa at different test voltages for DIO and DIO MT mice. H: Comparisons of peak INa among control, control-MT, DIO and DIO-MT mice; n=6 cells each respectively. I: Representative IKur current images in control, DIO and DIO-MT mice; J: Comparisons of peak IKur among Control, Control-MT, DIO and DIO-MT mice; n = 6 respectively each. **P<0.01; ***P<0.001, ****P<0.0001.
Figure 7:

The antioxidant MitoTEMPO (MT) reverses atrial fibrosis and reduces oxidative stress in DIO mice. A: Western blot showing NOX2 expression in DIO and DIO-MT mice (N=3 hearts). B: Quantification of reduced NOX2 expression in DIO MT compared to DIO mice (n=3 cells). C: Mitosox staining image in DIO and DIO-MT mice. D: Isoprostane levels in the control and DIO mice pre- and post-MT treatment; n=4 mice; *P<0.05. E: Masson’s trichrome staining of atrial myocytes from DIO mice before and chronic MT treatment. F: Fold change in fibrosis (%) in the 2 groups of mice showing a significant reduction in fibrosis after MT (n=6, 7 cells each respectively). *P<0.05; **P<0.01.
Figure 8:

The antioxidant MT reduces AF Burden in DIO obese mice through increased APD90 and maximum upstroke velocity, dV/dTmax. A: Representative AP tracings in atrial myocytes among DIO and DIO-MT mice; B: APD20 obtained in atrial myocytes pre- and post-MT; C: APD50 obtained in atrial myocytes pre- and post-MT; D: APD90 obtained in atrial myocytes pre- and post-MT (n=6 cells); E: dV/dTmax in atrial myocytes pre- and post-MT (n=6 cells); F: AF burden in mice pre- and post-MT. **P < 0.01; ***P < 0.001, ****P<0.0001.
Discussion
Obesity has long been described as a significant clinical risk factor for the development of AF but the underlying EP mechanisms are poorly understood. We showed that DIO mice were not only more prone to develop AF but that this risk is mediated in part by a combined effect of sodium, potassium and calcium channel remodeling and atrial fibrosis. Mitochondrial antioxidant therapy reduced AF burden, restored INa, ICa,L, IKur, APD and reversed atrial fibrosis. Our findings may have important implications for the management of obesity-mediated AF in patients.
Although an association between obesity and AF is established, the underlying causal mechanisms remain unclear.2 One reason for this is that obesity is a complex phenotype with acquired, genetic, and metabolic components.49 To elucidate the underlying EP mechanisms of obesity-mediated AF, we studied an acquired mouse model. DIO mice showed a high susceptibility to pacing induced AF and had shortened atrial APD, decreased atrial CV and maximum upstroke velocity. Our findings that there is significant downregulation of Nav1.5 and reduced INa and ICa,L in DIO mice and increase in IKur may explain the reduction in APD, CV and dV/dTmax. Collectively, our data supports obesity-mediated AF is due to a combined effect of sodium, potassium and calcium channel remodeling and atrial fibrosis. Our data showing an increase in PKC-ɑ and PKC-δ and NOX2 expression in DIO mice may provide an explanation for the observed alterations in Nav1.5 and INa as previously reported.24
Recent studies have demonstrated that a HFD in mice is associated with atrial EP changes such as shortened PR interval, prolonged sinoatrial recovery time, and reduced atrial CV; however the underlying mechanisms contributing to this EP phenotype have remained unclear.21, 26 In obese rats with advanced age, expression of Nav1.5 is markedly decreased in sinoatrial nodal cells and associated with bradycardia.39 We showed that DIO mice are more prone to AF in part by modulation of the cardiac sodium channel with shortening of the atrial APD (Figure 3). However, shortening of the atrial APD may also relate to modulation of potassium repolarizing currents through the up-regulation of atrial Kv4.2/3 and/or atrial specific Kv1.5.11, 26 While we found no change in the expression of atrial Kv4.2/3 in DIO mice (Supplementary Figure 1E–F), there was significant up-regulation of Kv1.5 and IKur which along with Nav1.5 and INa downregulation, may explain the shortening of the atrial APD. DIO mice also displayed reduced CaV1.2 and Ica,L expression when compared to controls. Inhibition of the L-type calcium channel shortens the APD in human induced pluripotent stem cell-derived atrial cardiomyocytes and can potentially be pro-arrhythmic.27 Collectively our findings support our hypothesis that obesity mediates AF in mice in part through shortened APD and reduced CV by creating a reentrant substrate for AF.
Obese mice accumulate pericardial fat, and atrial and systemic oxidative stress, as evidenced by histological analysis of the atrial substrate. DIO mice had increased atrial fibrosis, which may in part be due to the HFD. Mitochondrial dysfunction and fibrosis are known contributing factors to the development of AF and our data suggests that alleviation of atrial mitochondrial oxidative stress with MT abrogated both ion channel remodeling, and atrial fibrosis and reversed the burden of pacing-induced AF in obese mice.
Our results have important clinical implications for ‘personalizing’ therapy for AF in patients with obesity. First, reduced atrial INa resulting from obesity may not be treatable with Class I (sodium channel blocker) AADs such as flecainide/propafenone. Support for this comes from our recent report where we showed that obese patients had greater recurrence of AF than non-obese patients when treated with sodium channel blocker AADs.28 Furthermore, this clinical finding was confirmed in DIO mice which showed reduced efficacy of flecainide as compared with sotalol in suppressing pacing-induced AF. Second, obesity-mediated AF may have a reversible component. Hohl et al.5 showed that reducing obesity can reverse AF; our results complement these clinical observations by suggesting that reducing oxidative stress in obesity may also be an effective means by which AF may be suppressed by reversing the pathologic obesity-induced atrial substrate for AF. Third, our studies suggest that the EP substrate for AF in acquired obesity is dependent in part upon the content of the diet. DIO mice fed a HFD had significantly more atrial fibrosis than control mice; thus this diet may directly contribute to a reentrant mechanism for AF.
Our study has some limitations that should be acknowledged. First, we performed detailed phenotyping of DIO mice to uncover the underlying EP substrate for obesity-mediated AF. Similar to obese patients, DIO mice have elevated fasting serum glucose. However, in these young (4–6 month old) mice baseline MAPs were similar, thus indicating that these mice do not fully recapitulate the human phenotype of advanced obesity and metabolic syndrome. Second, our focus was on identifying obesity-induced molecular pathways which may promote AF but we did not perform in silico computer simulations that could be useful to ascertain the relative contributions of the identified obesity-induced pathways in promoting AF. Our finding of a 7-fold increase in both PKC-ɑ and PKC-δ and a 3-fold increase in NOX2 expression in DIO mice as compared to controls (Figure 5C–H) is consistent with earlier reports29, 30 and strongly suggests a cause-effect relationship between ion channel remodeling and mitochondrial ROS production. Future in silico modeling studies could be useful to determine the relative contributions of these and other molecular pathways in promoting AF. Third, our studies were carried out only in mice which can only approximate human AF. However, challenges associated with procuring left atrial tissue, in-vivo imaging of thin-walled atria, and heterogeneity of the underlying substrate render animals an attractive model to study AF mechanisms. While models of AF have generally been developed in dogs and sheep, the elegance of performing definitive genetic and mechanistic studies in transgenic mouse models has resulted in the development of important murine AF models which elucidate the underlying mechanisms; permit access to tissue samples from different regions of the heart; and conduct in-vivo, ex-vivo and in-vitro studies with whole hearts and isolated myocytes. Furthermore, we and others have shown that transgenic AF mouse models expressing cardiac ion channel mutations have identified shortening of the APD, creation of a substrate for reentry, and generation of early afterdepolarization and delayed afterdepolarization triggered activity associated with cardiac RyR2 channel dysfunction as important mechanisms underlying AF.31–35 It has been recently reported that obese patients had greater recurrence of AF than non-obese patients when treated with sodium channel blocker antiarrhythmic drugs.28 This clinical finding can be explained by the mechanistic studies in DIO mice presented in the current work which showed reduced efficacy of flecainide versus sotalol in suppressing pacing-induced AF. Fourth, we did not assess the temporal relationship between ion channel remodeling and atrial fibrosis. While our data suggests that obesity-induced AF is mediated by both ion channel remodeling and atrial fibrosis, determining which one comes first would be challenging and necessitate the use of transgenic mouse models of obesity and is thus beyond the scope of this study. Nonetheless, this is an important question and one which we plan on addressing in future studies.
Conclusions
Obesity is a significant risk factor for AF in susceptible patients. We showed that obesity-mediated AF is due in part to a combined effect of ion channel and structural remodeling. Mitochondrial antioxidant therapy reversed the obesity-induced AF burden. Our findings may not only impact the management of obesity-mediated AF in patients but also provide important insights into the underlying mechanisms for the differential response to antiarrhythmic therapy and the potential role of targeted antioxidant therapy.
Supplementary Material
What is known?
Epidemiological studies have established obesity as an independent risk factor for atrial fibrillation (AF)
Other studies have shown that reduced cardiac sodium channel expression is a known causal mechanism of AF.
The pathophysiological mechanisms underlying obesity-mediated AF are not understood.
What the study adds?
Mice fed a high fat diet were more prone to develop pacing-induced AF with reduced expression of the cardiac sodium and calcium channel, currents (INa; ICa,L) and atrial action potential duration (APD) and increased potassium channel (Kv1.5), current (IKur), markers of oxidative stress, NOX2 (NADPH oxidase 2) and PKC (protein kinase C) isoforms.
A mitochondrial antioxidant reduced the AF burden, restored INa, ICa,L, IKur, APD and reversed atrial fibrosis in obese mice.
Inducible AF in obese mice is mediated by a combined effect of ion channel and structural remodeling with a mitochondrial antioxidant abrogating the ion channel and structural remodeling. Our findings may have important implications for the treatment of obesity-mediated AF.
Acknowledgments
Sources of Funding: Dr. McCauley is supported by the NIH K08 HL130587; Dr. Hong is supported by AHA 19CDA34630041; Dr. Bonini is supported by the NIH RO1HL125356; Dr. Rehman is supported by the P01 HL060678, R01 HL126516 and R01 HL060678; Dr. Darbar is supported by T32 HL139439 and VA Merit Award I01 BX004268; Dr. McCauley is supported by R01 HL151508; and Dr. Ai by NIH HL113640 and R01 AA024769. Dr. Silva is supported by FAPESP 2014/21464-8.
Nonstandard Abbreviations and Acronyms
- AF
atrial fibrillation
- APA
action potential amplitude
- APD
AP duration
- CaMKII
calcium calmodulin-dependent protein kinase II
- CL
cycle length
- CV
conduction velocity
- DIO
diet-induced obesity
- dV/dTmax
maximal upstroke of AP
- EP
electrophysiology
- F2-IsoPs
F2-isoprostanes
- 8-IsoP
8-isoprostane
- HFD
high fat diet
- ICa,L
L-type calcium current
- IKur
ultra-rapid potassium current
- INa
sodium current
- LA
left atrium
- MAP
mean arterial pressure
- ROS
reactive oxygen species
- PKC
protein kinase C
- NOX2
NADPH oxidase 2
- SD
standard deviation
Footnotes
Disclosures: None
References:
- 1.Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, Mullany EC, Biryukov S, Abbafati C, Abera SF, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014;384:766–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chatterjee NA, Giulianini F, Geelhoed B, Lunetta KL, Misialek JR, Niemeijer MN, Rienstra M, Rose LM, Smith AV, Arking DE, et al. Genetic Obesity and the Risk of Atrial Fibrillation: Causal Estimates from Mendelian Randomization. Circulation. 2017;135:741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abed HS, Wittert GA, Leong DP, Shirazi MG, Bahrami B, Middeldorp ME, Lorimer MF, Lau DH, Antic NA, Brooks AG, et al. Effect of weight reduction and cardiometabolic risk factor management on symptom burden and severity in patients with atrial fibrillation: a randomized clinical trial. JAMA. 2013;310:2050–60. [DOI] [PubMed] [Google Scholar]
- 4.Miller JD, Aronis KN, Chrispin J, Patil KD, Marine JE, Martin SS, Blaha MJ, Blumenthal RS, Calkins H. Obesity, Exercise, Obstructive Sleep Apnea, and Modifiable Atherosclerotic Cardiovascular Disease Risk Factors in Atrial Fibrillation. J Am Coll Cardiol. 2015;66:2899–2906. [DOI] [PubMed] [Google Scholar]
- 5.Hohl M, Lau DH, Muller A, Elliott AD, Linz B, Mahajan R, Hendriks JML, Bohm M, Schotten U, Sanders P, Linz D. Concomitant Obesity and Metabolic Syndrome Add to the Atrial Arrhythmogenic Phenotype in Male Hypertensive Rats. J Am Heart Assoc. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jamaly S, Carlsson L, Peltonen M, Jacobson P, Sjostrom L, Karason K. Bariatric Surgery and the Risk of New-Onset Atrial Fibrillation in Swedish Obese Subjects. J Am Coll Cardiol. 2016;68:2497–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lau DH, Nattel S, Kalman JM, Sanders P. Modifiable Risk Factors and Atrial Fibrillation. Circulation. 2017;136:583–596. [DOI] [PubMed] [Google Scholar]
- 8.McCabe PJ, Darbar D. Is Achieving the American Heart Association’s Life Simple 7 Goals Sufficient to Reduce the Burden of Atrial Fibrillation? No Simple Answers. J Am Heart Assoc. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kondo H, Abe I, Gotoh K, Fukui A, Takanari H, Ishii Y, Ikebe Y, Kira S, Oniki T, Saito S, et al. Interleukin 10 Treatment Ameliorates High-Fat Diet-Induced Inflammatory Atrial Remodeling and Fibrillation. Circ Arrhythm Electrophysiol. 2018;11:e006040. [DOI] [PubMed] [Google Scholar]
- 10.Shuai W, Kong B, Fu H, Shen C, Jiang X, Huang H. MD1 Deficiency Promotes Inflammatory Atrial Remodelling Induced by High-Fat Diets. Can J Cardiol. 2019;35:208–216. [DOI] [PubMed] [Google Scholar]
- 11.Aromolaran AS, Colecraft HM, Boutjdir M. High-fat diet-dependent modulation of the delayed rectifier K(+) current in adult guinea pig atrial myocytes. Biochem Biophys Res Commun. 2016;474:554–559. [DOI] [PubMed] [Google Scholar]
- 12.Musa H, Kline CF, Sturm AC, Murphy N, Adelman S, Wang C, Yan H, Johnson BL, Csepe TA, Kilic A, et al. SCN5A variant that blocks fibroblast growth factor homologous factor regulation causes human arrhythmia. Proc Natl Acad Sci U S A. 2015;112:12528–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayashi K, Konno T, Tada H, Tani S, Liu L, Fujino N, Nohara A, Hodatsu A, Tsuda T, Tanaka Y, et al. Functional Characterization of Rare Variants Implicated in Susceptibility to Lone Atrial Fibrillation. Circ Arrhythm Electrophysiol. 2015;8:1095–104. [DOI] [PubMed] [Google Scholar]
- 14.Nattel S, Dobrev D. Deciphering the fundamental mechanisms of atrial fibrillation: a quest for over a century. Cardiovasc Res. 2016;109:465–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li XF, Lytton J. An essential role for the K+-dependent Na+/Ca2+-exchanger, NCKX4, in melanocortin-4-receptor-dependent satiety. J Biol Chem. 2014;289:25445–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan J, Kong W, Zhang Q, Beyer EC, Walcott G, Fast VG, Ai X. c-Jun N-terminal kinase activation contributes to reduced connexin43 and development of atrial arrhythmias. Cardiovasc Res. 2013;97:589–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan J, Thomson JK, Zhao W, Wu X, Gao X, DeMarco D, Kong W, Tong M, Sun J, Bakhos M, et al. The stress kinase JNK regulates gap junction Cx43 gene expression and promotes atrial fibrillation in the aged heart. J Mol Cell Cardiol. 2018;114:105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li N, Wang T, Wang W, Cutler MJ, Wang Q, Voigt N, Rosenbaum DS, Dobrev D, Wehrens XH. Inhibition of CaMKII phosphorylation of RyR2 prevents induction of atrial fibrillation in FKBP12.6 knockout mice. Circ Res. 2012;110:465–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yanni J, Tellez JO, Sutyagin PV, Boyett MR, Dobrzynski H. Structural remodelling of the sinoatrial node in obese old rats. J Mol Cell Cardiol. 2010;48:653–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi K, Sasano T, Sugiyama K, Kurokawa J, Tamura N, Soejima Y, Sawabe M, Isobe M, Furukawa T. High-fat diet increases vulnerability to atrial arrhythmia by conduction disturbance via miR-27b. J Mol Cell Cardiol. 2016;90:38–46. [DOI] [PubMed] [Google Scholar]
- 21.Meng T, Cheng G, Wei Y, Ma S, Jiang Y, Wu J, Zhou X, Sun C. Exposure to a chronic high-fat diet promotes atrial structure and gap junction remodeling in rats. Int J Mol Med. 2017;40:217–225. [DOI] [PubMed] [Google Scholar]
- 22.Simon JN, Ziberna K, Casadei B. Compromised redox homeostasis, altered nitroso-redox balance, and therapeutic possibilities in atrial fibrillation. Cardiovasc Res. 2016;109:510–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mauro C, Smith J, Cucchi D, Coe D, Fu H, Bonacina F, Baragetti A, Cermenati G, Caruso D, Mitro N, et al. Obesity-Induced Metabolic Stress Leads to Biased Effector Memory CD4(+) T Cell Differentiation via PI3K p110delta-Akt-Mediated Signals. Cell Metab. 2017;25:593–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joseph LC, Avula UMR, Wan EY, Reyes MV, Lakkadi KR, Subramanyam P, Nakanishi K, Homma S, Muchir A, Pajvani UB, et al. Dietary Saturated Fat Promotes Arrhythmia by Activating NOX2 (NADPH Oxidase 2). Circ Arrhythm Electrophysiol. 2019;12:e007573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu M, Liu H, Dudley SC, Jr. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ Res. 2010;107:967–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang F, Hartnett S, Sample A, Schnack S, Li Y. High fat diet induced alterations of atrial electrical activities in mice. Am J Cardiovasc Dis. 2016;6:1–9. [PMC free article] [PubMed] [Google Scholar]
- 27.Wu J, Wang X, Chung YY, Koh CH, Liu Z, Guo H, Yuan Q, Wang C, Su S, Wei H. L-Type Calcium Channel Inhibition Contributes to the Proarrhythmic Effects of Aconitine in Human Cardiomyocytes. PLoS One. 2017;12:e0168435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ornelas-Loredo A, Kany S, Abraham V, Alzahrani Z, Darbar FA, Sridhar A, Ahmed M, Alamar I, Menon A, Zhang M, et al. Association Between Obesity-Mediated Atrial Fibrillation and Therapy With Sodium Channel Blocker Antiarrhythmic Drugs. JAMA Cardiol. 2019; 5: 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joseph LC, Barca E, Subramanyam P, Komrowski M, Pajvani U, Colecraft HM, Hirano M, Morrow JP. Inhibition of NAPDH Oxidase 2 (NOX2) Prevents Oxidative Stress and Mitochondrial Abnormalities Caused by Saturated Fat in Cardiomyocytes. PLoS One. 2016;11:e0145750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu M, Shi G, Yang KC, Gu L, Kanthasamy AG, Anantharam V, Dudley SC Jr. Role of protein kinase C in metabolic regulation of the cardiac Na(+) channel. Heart Rhythm. 2017;14:440–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Faggioni M, Savio-Galimberti E, Venkataraman R, Hwang HS, Kannankeril PJ, Darbar D, Knollmann BC. Suppression of spontaneous ca elevations prevents atrial fibrillation in calsequestrin 2-null hearts. Circ Arrhythm Electrophysiol. 2014;7:313–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishida K, Michael G, Dobrev D, Nattel S. Animal models for atrial fibrillation: clinical insights and scientific opportunities. Europace. 2010;12:160–72. [DOI] [PubMed] [Google Scholar]
- 33.Riley G, Syeda F, Kirchhof P and Fabritz L. An introduction to murine models of atrial fibrillation. Front Physiol. 2012;3:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirchhof P, Kahr PC, Kaese S, Piccini I, Vokshi I, Scheld HH, Rotering H, Fortmueller L, Laakmann S, Verheule S, et al. PITX2c is expressed in the adult left atrium, and reducing Pitx2c expression promotes atrial fibrillation inducibility and complex changes in gene expression. Circ Cardiovasc Genet. 2011;4:123–33. [DOI] [PubMed] [Google Scholar]
- 35.Purohit A, Rokita AG, Guan X, Chen B, Koval OM, Voigt N, Neef S, Sowa T, Gao Z, Luczak ED, et al. Oxidized Ca(2+)/calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation. 2013;128:1748–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.