

Abstract
Cranial neural crest cells (CNCCs) are a population of multipotent stem cells that give rise to craniofacial bone and cartilage during development. Bone morphogenetic protein (BMP) signaling and autophagy have been individually implicated in stem cell homeostasis. Mutations that cause constitutive activation of the BMP type I receptor ACVR1 cause the congenital disorder fibrodysplasia ossificans progressiva (FOP), which is characterized by ectopic cartilage and bone in connective tissues in the trunk and sometimes includes ectopic craniofacial bones. Here, we showed that enhanced BMP signaling through the constitutively activated ACVR1 (ca-ACVR1) in CNCCs in mice induced ectopic cartilage formation in the craniofacial region through an autophagy-dependent mechanism. Enhanced BMP signaling suppressed autophagy by activating mTORC1, thus blocking the autophagic degradation of β-catenin, which, in turn, caused CNCCs to adopt a chondrogenic identity. Transient blockade of mTORC1, reactivation of autophagy, or suppression of Wnt–β-catenin signaling reduced ectopic cartilages in ca-Acvr1 mutants. Our results suggest that BMP signaling and autophagy coordinately regulate β-catenin activity to direct the fate of CNCCs during craniofacial development. These findings may also explain why some patients with FOP develop ectopic bones through endochondral ossification in craniofacial regions.
INTRODUCTION
Multipotent cranial neural crest cells (CNCCs) are the largest contributor to the developing face (1, 2). During craniofacial development, CNCCs delaminate from the neural tube and migrate into branchial arches (BAs), where they differentiate into various distinct cell types, such as osteocytes, chondrocytes, and glia, and give rise to most of the anterior craniofacial tissues (1–3). Defects in the delamination, migration, or differentiation of CNCCs lead to a variety of craniofacial abnormalities (4). CNCCs have multipotency before, during, and after their active migration (5–9); however, questions concerning the molecular mechanisms underlying the fine control of differential cell fate specification from CNCCs during craniofacial development are far from resolved.
The craniofacial skeleton has unique characters compared to the bones of the appendicular skeleton. The anterior cranial bones and cartilages are derived from CNCCs, whereas the posterior part is derived from the paraxial mesoderm, which is the same origin for axial bones (9, 10). Most of the elements of the craniofacial skeleton are formed through intramembranous ossification, in which CNCC-derived progenitors proliferate, condense, and differentiate directly into osteoblasts without generating a cartilage intermediate. Bones in the skull base and parts of the mandible, such as the condyle process, are formed through endochondral ossification by CNCC-derived chondrocytes. Most of the appendicular, spine, and thoracic skeletons are derived from mesodermal tissues and formed through endochondral ossification. Neural crest cells that developed in the trunk region do not participate in appendicular skeletogenesis (11).
Bone morphogenetic protein (BMP) signaling, which is mediated by intracellular Smad proteins, plays important roles in craniofacial development by balancing migration, self-renewal, cell fate specification, survival, and differentiation of CNCCs, thus contributing to both shape and functionality of normal craniofacial features (12, 13). The appropriate amount of BMP signaling is required for proper craniofacial morphogenesis (14). We and others (15–17) have reported that constitutively activated or loss-of-function mutation of Bmpr1a, which encodes a BMP type I receptor, in neural crest cells differentially affects orthotopic craniofacial tissue morphogenesis. ACVR1, another BMP type I receptor, has received much clinical attention because an activating mutation in ACVR1 is responsible for fibrodysplasia ossificans progressiva (FOP), a rare disorder characterized by heterotopic bone formation through endochondral ossification in connective tissues (18, 19). Some patients with FOP develop mandible hypoplasia and ectopic chondrogenesis and bone in the craniofacial region involving the temporomandibular joint, muscles, and associated fascia of the head and neck (20).
Although mice lacking Acvr1 in CNCCs display multiple craniofacial defects, overall cell fate specification and the formation of major cartilage structures are not affected (21). Mice lacking Acvr1 in chondrocytes display subtle changes in cartilage development (22), suggesting that BMP signaling mediated by ACVR1 plays a role in cartilage formation and homeostasis. ACVR1 may have different functions from BMPR1A in craniofacial development, because we previously found that heterozygous null mutations in Acvr1 fail to rescue the craniosynostosis caused by constitutively active BMPR1A, whereas heterozygous null mutations in Bmpr1a rescue it (15, 23). Here, we report an unexpected role for constitutively activated ACVR1 (ca-ACVR1), such as that occurs in patients with FOP, in fine-tuning BMP signaling to promote CNCC fate specification toward a chondrogenic lineage, resulting in ectopic cartilage formation within the craniofacial region.
Autophagy, a highly coordinated and evolutionarily conserved catabolic process, plays a crucial role during early embryonic development and in maintaining stem cell homeostasis (24). Dysregulation of autophagy is associated with a variety of human diseases and developmental defects, such as cancer and congenital disorders of autophagy (25–27). It has been reported that autophagy induced by ciliation directs human embryonic stem cells to a neuroectoderm lineage by degrading the fate determinant (28). In neural crest cells, autophagy is known to be involved in regulating their generation, survival, and differentiation into neurons in vitro (29, 30). However, it remains unclear whether functional coordination between BMP and autophagy contributes to the regulation of stem cell fate, especially CNCCs in the context of craniofacial development.
We found that augmented BMP signaling through ca-ACVR1 in CNCCs suppressed autophagic activity, thus directing CNCCs to an aberrant chondrogenic fate. Mechanistically, augmented BMP signaling suppressed autophagy by stimulating mammalian target of rapamycin complex 1 (mTORC1) activity, thus blocking the autophagic degradation of β-catenin and increasing Wnt–β-catenin signaling activity in CNCCs, leading to chondrogenic fate specification. Together, our results identify a role for a previously unreported BMP-autophagy–β-catenin signaling axis in regulating chondrogenic cell fate specification from neural crest cells during craniofacial development.
RESULTS
ca-ACVR1 in neural crest cells causes ectopic cartilage formation during craniofacial development
To investigate the function of enhanced BMP signaling mediated by ACVR1 in CNCCs in vivo, we bred transgenic mice that express a constitutively active form of Acvr1 (ca-Acvr1flox/flox or ca-Acvr1flox/+) (31) with neural crest–specific P0-Cre mice (32). Compared with Cre-negative controls [ca-Acvr1fx/+;P0-Cre(−), designated as wild type] and the transgene negative controls (ca-Acvr1+/+;P0-Cre, designated as controls), the mutant mice (ca-Acvr1flox/+;P0-Cre, hereafter referred to as ca-Acvr1 mutants) died soon after birth with severe craniofacial abnormalities including cleft lip, cleft palate, and strongly attenuated craniofacial bone formation, including the skull, maxilla, and mandible (Fig. 1A and fig. S1, A and B). Because an internal ribosome entry site–enhanced green fluorescent protein (IRES-EGFP) cassette was placed 3′ to ca-Acvr1 in the transgene construct, EGFP fluorescence can be used to trace ca-Acvr1-expressing cells after Cre recombination (31). In the ca-Acvr1 mutants, a cluster of EGFP signal was detected in the craniofacial region (Fig. 1B). We observed higher amounts of phosphorylated Smads 1, 5, and 9 (pSmad1/5/9), which mediate canonical BMP signaling, in the facial region of the ca-Acvr1 mutants compared with wild type and controls (about twofold; Fig. 1C and fig. S10). Therefore, ca-ACVR1 in CNCCs enhanced BMP signaling in vivo.
Fig. 1. Constitutively activated ACVR1 in CNCCs causes craniofacial defects and ectopic cartilage formation during development.
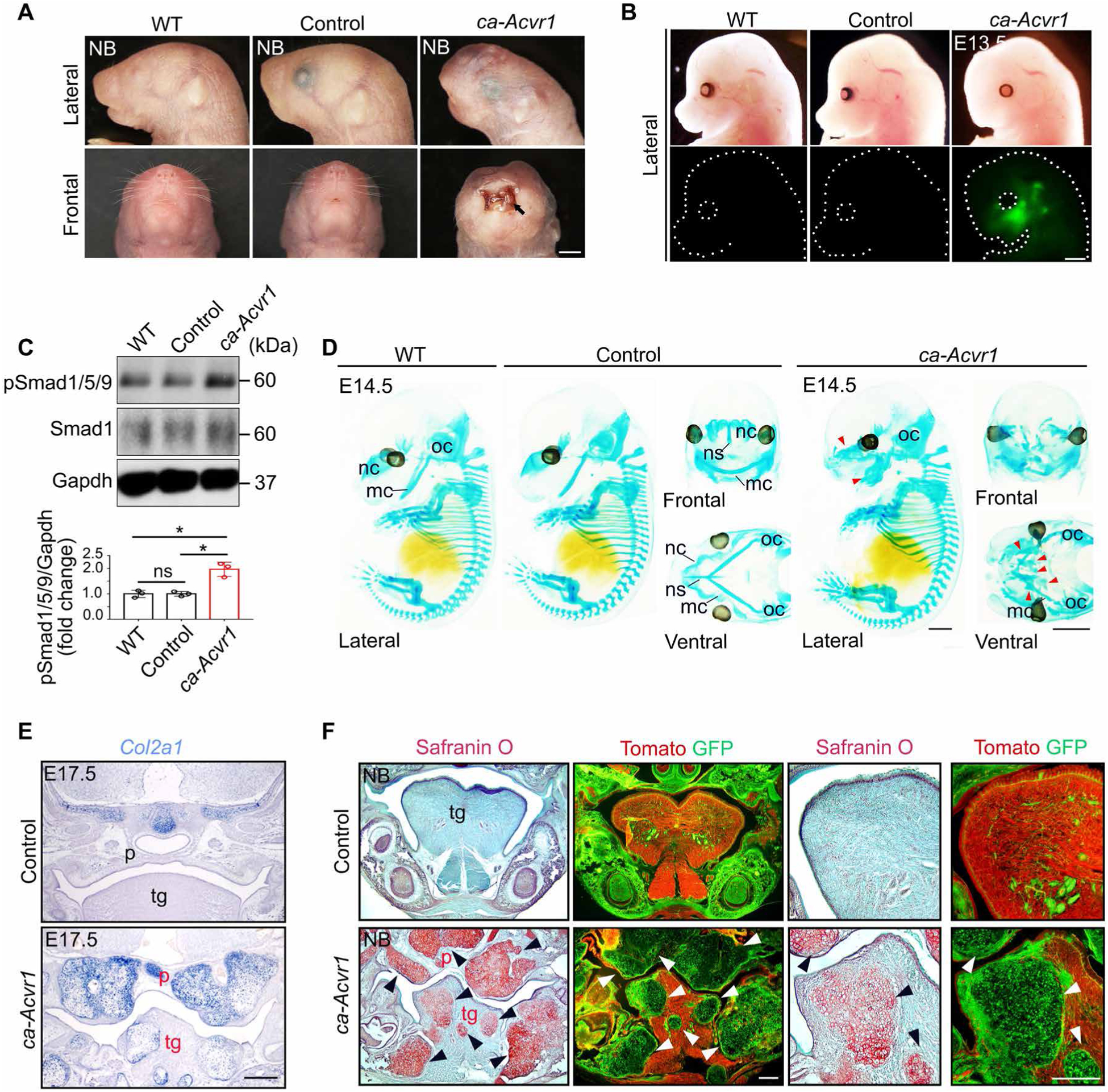
(A) Representative lateral and frontal views of the heads of wild-type (WT), control (ca-Acvr1+/+;P0-Cre), and mutant (ca-Acvr1flox/+;P0-Cre) newborn (NB) mice. Black arrow indicates the cleft lip in the mutants. Scale bar, 1 mm. n = 3 mice per group. (B) Representative lateral views to show EGFP marking the expression of the ca-Acvr1 transgene in the craniofacial region at E13.5. Scale bar, 1 mm. n = 3 mice per group. (C) Immunoblot and quantification of phosphorylated Smad1/5/9 (pSmad/1/5/9) and total Smad1 in tissue lysates from the facial regions of WT, control, and mutant embryos. Gapdh is a loading control. n = 3 independent experiments. Uncropped Western blot images are shown in fig. S10. (D) Representative cartilage structures of embryos stained with Alcian blue at E14.5. Red arrowheads indicate ectopic cartilage. Scale bars, 1 mm. n = 3 mice per group. (E) Col2a1 expression assessed by in situ hybridization in coronal sections of heads at E17.5. Scale bar, 500 μm. n = 3 mice per group. (F) The pattern of ectopic cartilages (Safranin O staining) and P0-Cre–labeled cells (GFP positive, using R26RmTmG reporter) in serial sections of heads from NB mice. Scale bars, 100 μm. n = 3 mice per group. Red labels indicate defective structures. Arrowheads indicate ectopic cartilages. mc, Meckel’s cartilage; nc, nasal capsule; ns, nasal septum; oc, otic capsule; p, palate; tg, tongue. Error bars are means ± SD. ns, not significant; P > 0.05; *P < 0.05; ANOVA.
Although ca-Acvr1 mutants showed normal cartilage structure in the trunk region, they displayed ectopic cartilages in the craniofacial region (Fig. 1D and fig. S1B). The ectopic cartilages robustly expressed Col2a1 (Fig. 1E), which encodes the alpha 1 chain of collagen type II, the principal and specific matrix protein deposited by chondrocytes (33). Safranin O staining further illustrated that ectopic cartilages were formed extensively throughout the craniofacial region in the mutants, including the skull base, palate, jaw, and tongue (Fig. 1F). Serial cranial sections of R26RmTmG reporter mice, in which ubiquitously expressed tdTomato is replaced by EGFP in Cre-expressing cells, showed that P0-Cre-driven cells (GFP positive) were abundantly present in ectopic cartilages (Fig. 1F and fig. S1C). In addition, the phenotype of ectopic cartilage formation was recapitulated in mice using a different neural crest-specific Cre driver, Wnt1-Cre (ca-Acvr1flox/+;Wnt1-Cre; fig. S1D). These results show that ca-ACVR1 in CNCCs caused extensive ectopic cartilage formation during craniofacial development.
ca-ACVR1 commits CNCCs to a chondrogenic lineage
We then determined how ectopic cartilages were formed. The first branchial arch (BA1) is derived from CNCCs and develops into several craniofacial elements, such as the mandible and tongue (1, 2). Histological analysis showed that the morphology of BA1 was comparable between controls and ca-Acvr1 mutants at embryonic day 10.5 (E10.5; fig. S1E). Abnormal structures were observed in the tongue at E11.5 (fig. S1E) and then extended and showed cartilage morphology in the mutants as development progressed (fig. S1E). We next analyzed the pattern of Sox9 and the density of Sox9-positive cells (cell number per square millimeter) during development, because Sox9 is required for chondrogenic lineage identity, and condensation of Sox9-positive cells is a prerequisite for chondrogenesis (34). Sox9 production in control embryos was restricted in the areas where mesenchymal condensation was initiated to develop orthotopic craniofacial cartilages, such as Meckel’s cartilage, at E11.5 and E12.5 (Fig. 2, A and B, and fig. S2D). However, Sox9 was ectopically distributed in the uncondensed mesenchymal areas of the tongue in ca-Acvr1 mutants at E11.5 (Fig. 2A). The density of these ectopic Sox9-positive cells, which are presumptive ectopic cartilages, increased to a similar level with that of orthotopic Sox9-positive cells in controls at E12.5 (Fig. 2B). The expression of Sox9 transcripts in the BA1 tissues of mutants was also significantly higher than that in controls at E11.5 (Fig. 2C). These data suggest that E11.5 was a critical time point for the initiation of ectopic chondrogenesis in ca-Acvr1 mutants.
Fig. 2. Activation of BMP signaling causes the specification of CNCCs into an aberrant chondrogenic lineage.
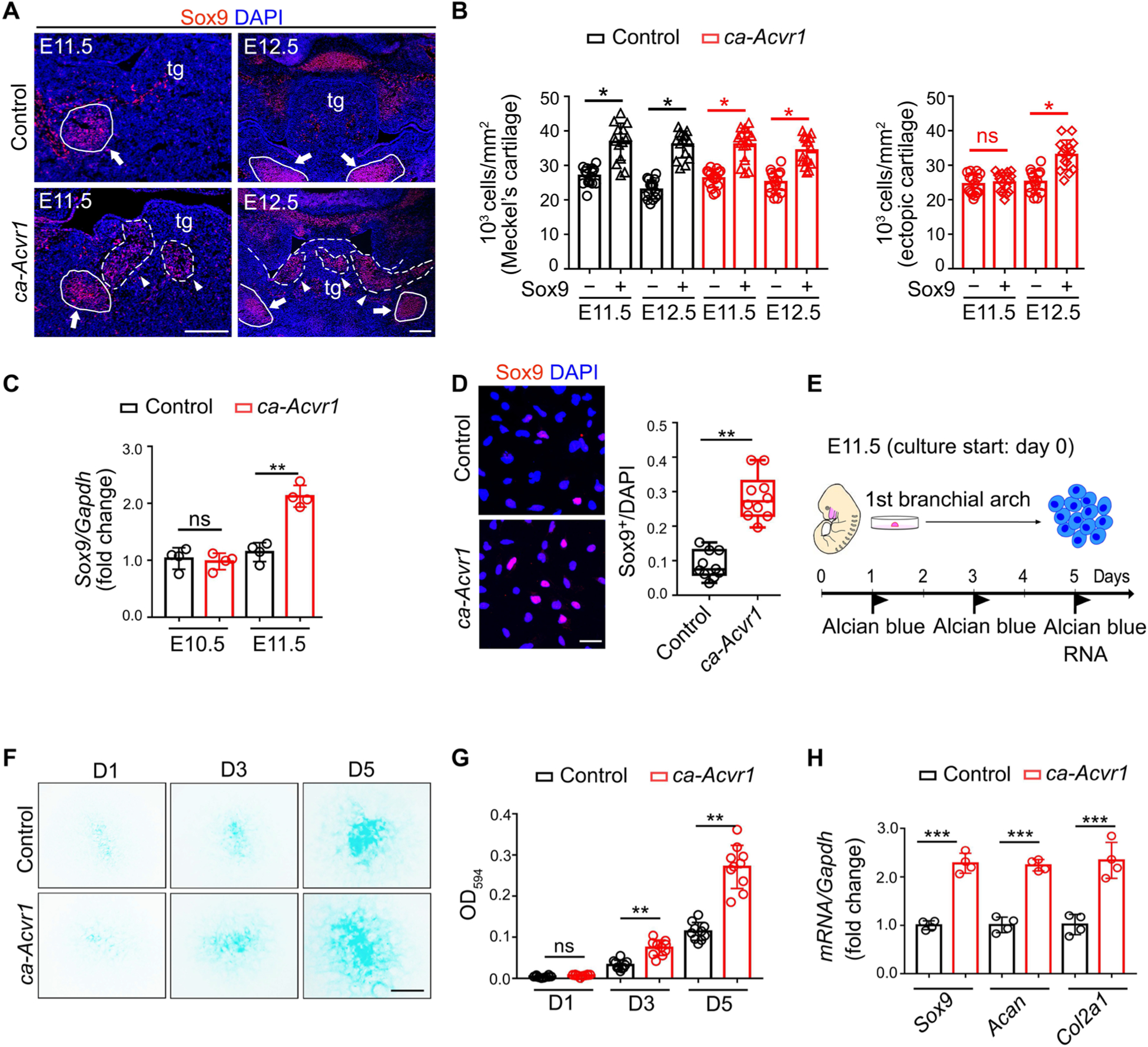
(A) Sox9 (red) immunofluorescence of coronal head sections from control (ca-Acvr1+/+;P0-Cre) and mutant (ca-Acvr1flox/+;P0-Cre) embryos at the indicated stages. Scale bars, 100 μm. n = 5 mice per group. White arrowheads indicate ectopic cartilages, and white arrows indicate Meckel’s cartilages; tg, tongue. Nuclei are stained with DAPI (4′,6-diamidino-2-phenylindole; blue). (B) Condensation of cells in orthotopic (Meckel’s cartilage) and ectopic cartilages. The density of cells in each area was calculated from 5 embryos with three images (n = 5 embryos for each group). (C) Relative expression of Sox9 in the first branchial arch (BA1) tissues from control and mutant embryos at E10.5 and E11.5. n = 4 mice per group. (D) Immunostaining and quantification of Sox9 in CNCCs isolated from BA1 tissues (BA1 cells) of control or mutant embryos. Scale bar, 20 μm. n = 5 mice per group. (E) Schematic representation of micromass cell culture model used to determine chondrogenic capacity of BA1 cells. (F) Alcian blue staining of cells during 5-day micromass culture. Scale bar, 1 mm. n = 10 independent experiments. (G) Quantification of the optical density (OD at 594 nm) of the dye in (F). n = 10 independent experiments. (H) Relative expression of Sox9, Acan, and Col2a1 in control and mutant BA1 cells after 5 days in culture. n = 3 independent experiments. Error bars are means ± SD. ns, not significant; P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; t test.
Using CNCCs isolated from E11.5 BA1 tissues (BA1 cells), we determined whether ca-ACVR1 in CNCCs changed their in vitro chondrogenic capacity (35–37). Whereas their proliferation rate was comparable to that of controls (fig. S2A), ca-Acvr1 mutant BA1 cells produced significantly more Sox9 than did controls (Fig. 2D). Using a micromass culture system to evaluate chondrogenesis (Fig. 2E), we found that ca-Acvr1 BA1 cells developed larger cartilaginous nodules (Fig. 2, F and G) and showed higher expression of chondrogenic markers compared with controls (Fig. 2H). Because mutant embryos were characterized by the formation of ectopic cartilages rather than the expansion of orthotopic cartilages, these results indicate that ca-ACVR1 altered CNCC fate to generate an aberrant chondrogenic lineage.
We examined whether enhanced BMP signaling affects migration, proliferation, and/or survival of CNCCs. Fate mapping analysis after crossing the R26RLacZ reporter into mutant embryos (ca-Acvr1flox/+;P0-Cre; R26RLacZ) showed no overt migration abnormalities compared with controls (ca-Acvr1+/+;P0-Cre; R26RLacZ; fig. S2B). Amounts of cell proliferation and death were comparable between control and mutant BA1 tissues (fig. S2C). Together, these results suggest that enhanced BMP signaling through ca-ACVR1 altered the fate of CNCCs into an aberrant chondrogenic lineage without influencing cell migration, proliferation, or survival.
Skeletal stem cells (SSCs), the multipotent progenitors of skeletal tissues, play important roles in skeletal development (38). SSCs have been reported to be in CNCC-derived cranial sutures, which contribute to skull bone formation (39), suggesting that SSCs may be present in other CNCC derivatives. Thus, we investigated whether BA1 tissues showed SSC characters that were disturbed by enhanced BMP signaling in CNCCs. We examined Gli1, a marker of stem cells in facial development (39), and several skeletal stem markers frequently used in the study of long bone development, including CD44, Thy1, Lepr, Pdgfra, and Ctsk. The expression of these SSC markers did not change in mutant BA1 cells compared to controls (fig. S3A). In addition, the distributions of CD146, CD44, and Thy1 were comparable between control and ca-Acvr1 mutant BA1 tissues (fig. S3B). These findings suggest that SSC markers were expressed in BA1 tissues but were not affected by enhanced BMP signaling during craniofacial development.
Commitment of CNCCs to the chondrogenic lineage depends on enhanced canonical BMP-Smad signaling
BMP signaling is transduced by activating Smad-dependent (canonical signaling) and/or Smad-independent (noncanonical signaling) pathways (12). We therefore determined the downstream signaling mediated by ca-ACVR1 in CNCCs. We found that although pSmad1/5/9 was sporadically distributed in BA1 tissues of controls, including the tongue and Meckel’s cartilage, it was significantly increased and abnormally aggregated in ca-Acvr1 mutants (Fig. 3, A and B, and fig. S2D). Although P0-Cre labeled almost all CNCCs, pSmad1/5/9 was only detected in a subset of CNCCs in BA1 tissues of ca-Acvr1 mutants (fig. S4A). Furthermore, pSmad1/5/9-positive cells were aggregated and mostly colocalized with EGFP, which marks cells expressing the ca-Acvr1 transgene (Fig. 3A). In control embryos, Sox9-positive cells were aggregated in the mandible and scattered in other areas, such as the tongue. In control embryos, some Sox9-positive cells in the mandible were also positive for pSmad1/5/9, whereas Sox9-positive cells were all negative for pSmad1/5/9 in the tongue and other areas (Fig. 3B). However, in ca-Acvr1 mutant embryos, Sox9-positive cells were aggregated in the mandible and other areas (Fig. 3B). Compared with controls, the ca-Acvr1 mutant embryos showed more pSmad1/5/9-positive cells colabeled with Sox9 in the tongue and other craniofacial regions, including the mandible (Fig. 3B). Similarly, treating BA1 cells from control embryos with exogenous BMP7 significantly increased the number of Sox9-positive cells, most of which were colocalized with pSmad1/5/9 (Fig. 3C). However, the activities of Smad-independent BMP pathways, including mitogen-activated protein kinase (MAPK) pathways [phosphorylated transforming growth factor–β (TGF-β)–activated kinase 1 (pTAK1), phosphorylated p38 (pP38), phosphorylated extracellular signal–regulated kinase (pERK), and phosphorylated Jun N-terminal kinase (pJNK)], and Smad-dependent TGF-β signaling (pSmad2) were unchanged in the mutants compared to controls (fig. S4, B to D and F). These observations indicate that the MAPK and Smad-dependent TGF-β signaling pathways did not contribute to the phenotypes caused by the expression of ca-ACVR1 and were further supported by the finding that the deletion of Tak1 (ca-Acvr1flox/+;Tak1flox/flox;P0-Cre) failed to rescue the craniofacial abnormalities observed in ca-Acvr1 mutants (fig. S4E).
Fig. 3. Canonical BMP-Smad signaling is activated by ca-ACVR1 and commits CNCCs to the aberrant chondrogenic lineage.
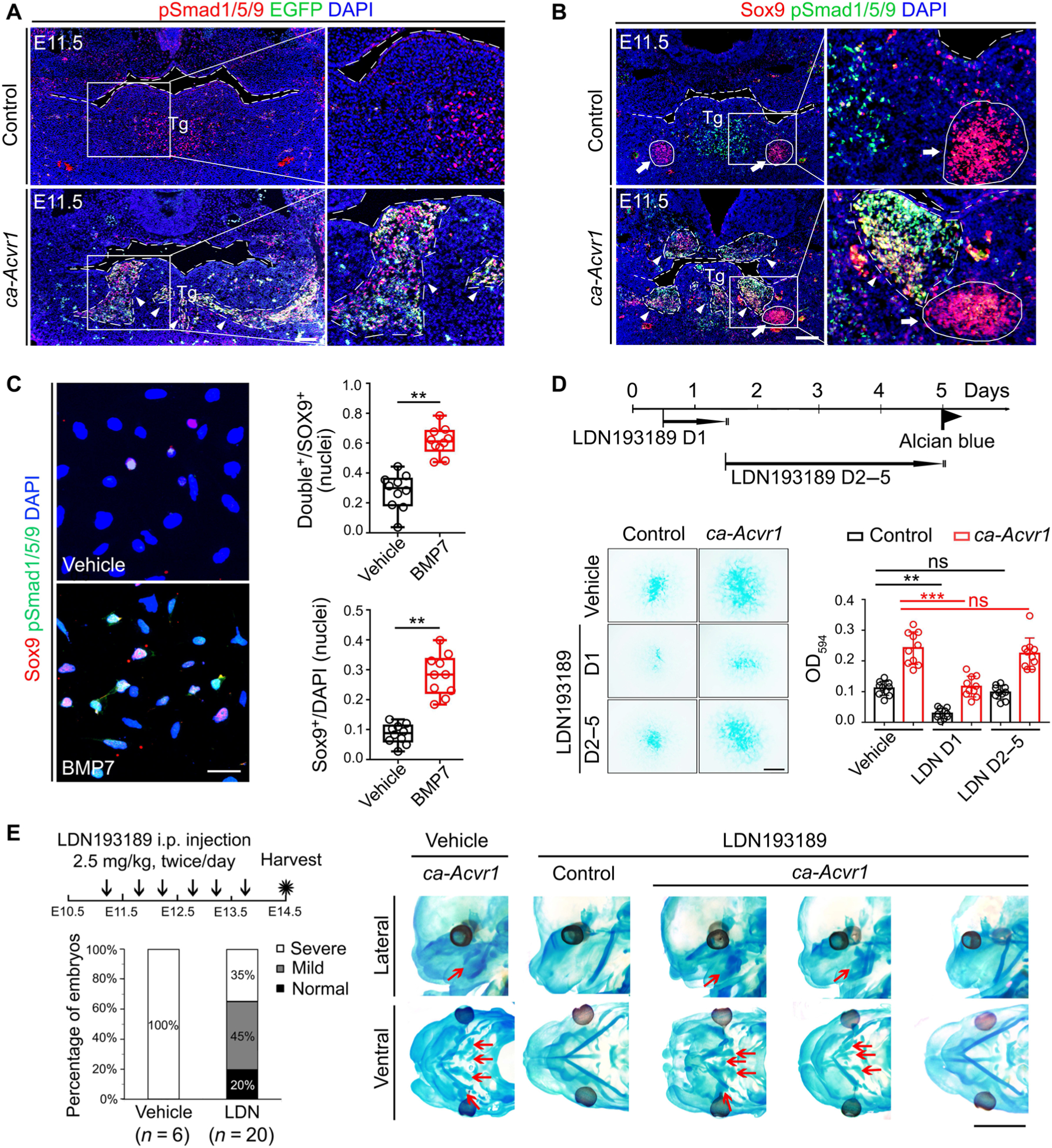
(A and B) Double immunofluorescence of phosphorylated Smad1/5/9 (pSmad1/5/9; red) and the ca-Acvr1 transgene marker EGFP (green) (A) and Sox9 (red) and pSmad1/5/9 (green) (B) in coronal head sections from control (ca-Acvr1+/+;P0-Cre) and mutant (ca-Acvr1flox/+;P0-Cre) embryos at E11.5. Green signals in control are autofluorescent blood cells. White arrows indicate Meckel’s cartilages, and white arrowheads indicate ectopic cartilages. Scale bars, 100 μm. n = 6 mice per group. (C) Immunostaining and quantification of Sox9 (red) and pSmad1/5/9 (green) in BA1 cells from control embryos treated with or without BMP7. n = 5 independent experiments. (D) Experimental scheme, representative images of Alcian blue staining, and optical density quantification to assess chondrogenesis of BA1 cells stimulated with LDN193189 (LDN) at the indicated time. Scale bar, 1 mm. n = 10 independent experiments. (E) Experimental scheme for treating embryos in utero with the BMP signaling inhibitor LDN193189 or vehicle from E11.25 to E13.5, the ratios of each cartilage phenotype (severe, mild, and normal) in treated mutant embryos, and representative images showing the cartilage phenotypes in treated control and mutant embryos by Alcian blue staining. The numbers of mice examined are shown in parentheses. Red arrows indicate ectopic cartilages. Scale bar, 2 mm. Error bars are means ± SD. ns, not significant; P > 0.05; **P < 0.01; ***P < 0.001; t test (C); ANOVA (D). i.p., intraperitoneally.
To determine the involvement of Smad-dependent signaling in ectopic cartilage formation, we suppressed phosphorylation of Smad1/5/9 with LDN193189, a selective chemical inhibitor of BMP type I receptor kinases (18). LDN193189 reduced the phosphorylation of Smad1/5/9 in BA1 cells (fig. S5, A and H) while leaving Smad-independent pathways intact (fig. S5, B and I), as expected. LDN193189 suppressed cartilage matrix deposition of BA1 cells in a dose-dependent manner (fig. S5C). LDN193189 treatment at day 1 of cell culture significantly decreased cartilage matrix deposition by BA1 cells, but treatment on days 2 to 5 did not (Fig. 3D). We further confirmed that the BA1 cells on day 2 were more committed to the chondrogenic fate compared with day 0, especially in ca-Acvr1 mutants, whereas cell proliferation and cell death were unaltered (fig. S6, A to I). Although the number of cells expressing osteogenic and myogenic markers was increased on day 2 compared with day 0, no difference was identified between the controls and the mutants for those markers (fig. S6, G and H). These findings suggest that the amount of Smad-dependent BMP signaling is critical for proper cell fate specification during the phase of chondrogenic lineage commitment.
Consistent with these observations in vitro, administration of LDN193189 (2.5 mg/kg) twice per day into pregnant females starting at E11.25 suppressed ectopic cartilage formation with decreased pSmad1/5/9 in BA1 tissues (Fig. 3E and fig. S5, D and J). However, LDN193189 treatment at later stages, starting at E12.25, did not suppress ectopic cartilage formation even though it reduced pSmad1/5/9 abundance in BA1 tissues (fig. S5, D to F). Treatment with LDN193189 (7.5 mg/kg) from E11.25 to E13.5 diminished orthotopic cartilages, such as Meckel’s cartilage and occipital arch cartilage, in both control and ca-Acvr1 mutant embryos (fig. S5G), suggesting that ectopic cartilage is formed through the same mechanisms as normal cartilage. Together, these results indicate that enhanced BMP-Smad signaling was responsible for the ectopic chondrogenic fate specification and facial cartilage formation in ca-Acvr1 mutants.
Commitment of CNCCs to the chondrogenic lineage requires activation of the BMP-mTORC1 signaling axis
Because mTORC1 is critical for chondrogenesis by CNCCs during craniofacial development (40), we investigated whether enhanced BMP signaling associated with mTORC1 signaling in CNCCs in ca-Acvr1 mutants. Phosphorylation of the S6 ribosomal protein (pS6), a surrogate marker of mTORC1 activity, was increased in the BA1 tissues of ca-Acvr1 mutants (Fig. 4A and fig. S10). pS6-positive cells, which were abnormally aggregated in the BA1 tissue of mutants, were mostly colabeled with Sox9 (Fig. 4B), indicating a potential correlation between the activation of mTORC1 and ectopic cartilage formation. LDN193189 treatment reduced pS6 in BA1 cells from mutants (Fig. 4C and fig. S10), suggesting that mTORC1 functioned downstream of BMP signaling.
Fig. 4. mTORC1 signaling is activated by enhanced BMP signaling and responsible for committing CNCCs into an aberrant chondrogenic lineage.
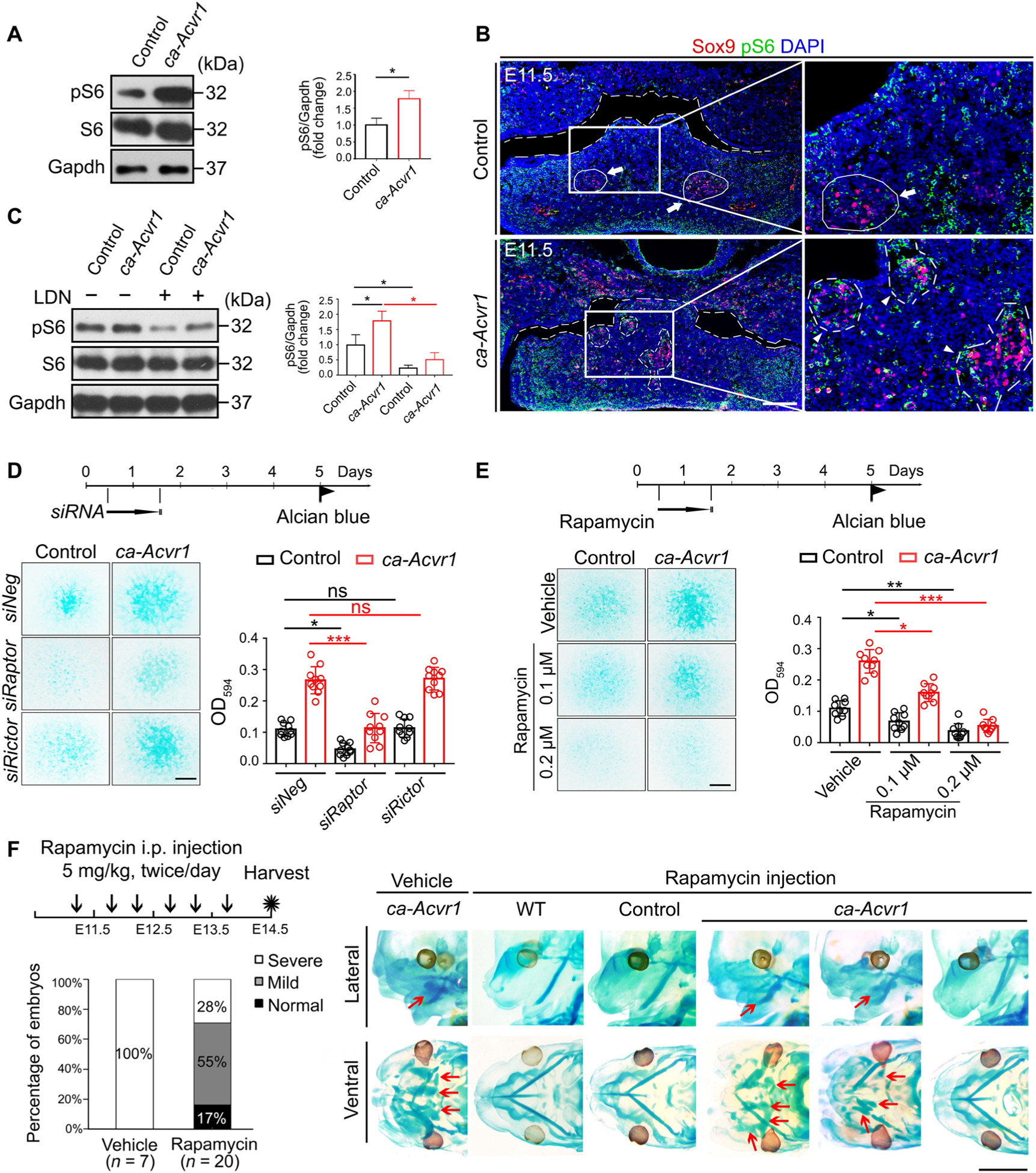
(A) Western blot analysis and quantification of phosphorylated S6 (pS6) in BA1 tissues of control and ca-Acvr1 mutant embryos. Gapdh is a loading control. n = 4 mice per group. Uncropped Western blot images are shown in fig. S10. (B) Representative images of Sox9 (red) and pS6 (green) double immunofluorescence of coronal head sections from control and mutant embryos at E11.5. White arrows indicate Meckel’s cartilages, and white arrowheads indicate ectopic cartilages. Scale bar, 100 μm. n = 4 embryos per genotype. (C) Western blot analysis of pS6 in control and mutant BA1 cells treated with LDN193189 for 24 hours. n = 3 independent experiments. (D) Experimental scheme, representative images of Alcian blue staining, and optical density quantification to assess the chondrogenesis of BA1 cells stimulated with control siRNA (siNeg) or siRNAs targeting Raptor (siRaptor) or Rictor (siRictor) at day 1. Scale bar, 1 mm. n = 10 independent experiments. (E) Scheme, representative images of Alcian blue staining, and optical density quantification of BA1 cells treated with the indicated concentration of rapamycin at day 1. Scale bar, 1 mm. n = 10 independent experiments. (F) Experimental scheme for treating embryos in utero with vehicle or rapamycin from E11.25 to E13.5, the ratios of each cartilage phenotype (severe, mild, and normal) in mutant embryos, and representative images showing cartilage phenotypes with whole-mount Alcian blue staining of embryos. The numbers of mice examined are shown in parentheses. Red arrows indicate ectopic cartilages. Scale bar, 2 mm. Error bars are means ± SD. ns, not significant; P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; t test (A); ANOVA [(C) to (E)].
To determine the role of mTORC1 signaling in chondrogenic fate specification, we suppressed mTOR signaling in BA1 cells using small interfering RNA (siRNA) against Raptor and Rictor, major components of mTORC1 and mTORC2, respectively. Knocking down Raptor, but not Rictor, decreased the amount of pS6 (fig. S7, A and G) and efficiently suppressed cartilage matrix deposition by ca-Acvr1 mutant BA1 cells (Fig. 4D). The mTORC1 inhibitor rapamycin also reduced the amount of pS6 and suppressed chondrogenesis in ca-Acvr1 mutant BA1 cells (Fig. 4E and fig. S7, B and H). About 70% of the ca-Acvr1 mutant embryos from mothers treated with rapamycin from E11.25 to E13.5 developed less ectopic cartilages compared to mutant embryos from vehicle-treated mothers (Fig. 4F and fig. S7, C and I). Rapamycin treatment starting at later stages (E12.25 to E13.5) reduced pS6 but failed to suppress ectopic cartilage formation in ca-Acvr1 mutants (fig. S7, C, D, and I), suggesting that reduction of mTOR signaling during chondrogenic commitment was critical. These results indicate that enhanced mTORC1 signaling by ca-ACVR1 played a pivotal role in committing CNCCs toward a chondrogenic lineage.
Wnt–β-catenin signaling activated by mTORC1 alters the fate of CNCCs
Wnt–β-catenin signaling is involved in BMP2-induced ectopic cartilage formation (41). We found that β-catenin-positive cells were abnormally aggregated and colabeled with EGFP in the BA1 tissue of ca-Acvr1 mutants (Fig. 5A). The amounts of total and active β-catenin in mutant BA1 tissues were higher than those in controls (Fig. 5B and fig. S10). In addition, the expression of Wnt–β-catenin target genes, including Cnx43, Ccnd1, and Lef1, was also significantly higher in the BA1 tissues of mutants (Fig. 5C). BMP7 treatment significantly increased the number of BA1 cells colabeled by Sox9 and β-catenin (Fig. 5D). We examined the expression of key regulators of Wnt–β-catenin signaling, such as Dkk1, Dkk2, Sost, and Lrp5. Although the amounts of Dkk2, Sost, and Lrp5 transcripts were unaltered, Dkk1 mRNA increased (fig. S7E). We previously reported that osteoblast-specific Acvr1 knockout mice activate Wnt–β-catenin signaling through the suppression of the Wnt signaling inhibitor DKK1 (Dickkopf-related protein 1) (42). In agreement with that finding, we found an activation of Wnt signaling and increased expression of Dkk1 in ca-Acvr1 mutants. These findings suggest that the up-regulation of Wnt–β-catenin signaling in ca-Acvr1 mutants may be caused by a DKK1-independent mechanism. Knockdown of Raptor or treatment with rapamycin significantly attenuated the accumulation of β-catenin and active β-catenin (Fig. 5E and fig. S10), as well as the expression of Cnx43, Ccnd1, and Lef1 (fig. S7F) in BA1 cells from ca-Acvr1 mutants. These results indicate that Wnt–β-catenin signaling activity may be increased through mTORC1 signaling in ca-Acvr1 mutants.
Fig. 5. Wnt–β-catenin signaling is activated by mTORC1 signaling and responsible for committing CNCCs into an aberrant chondrogenic lineage.
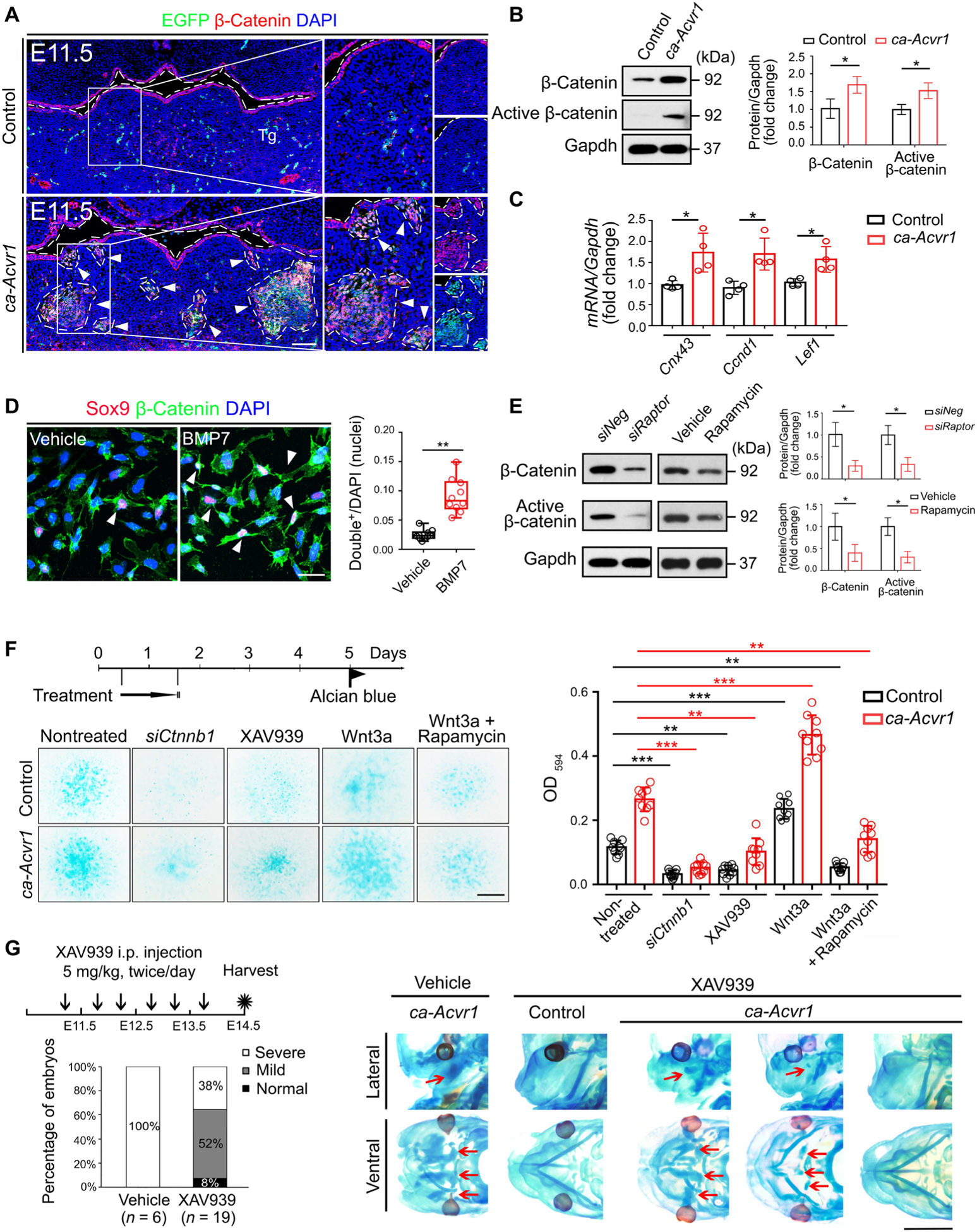
(A) Double immunofluorescence of β-catenin (red) and the ca-Acvr1 transgene marker EGFP (green) in coronal head sections from control and ca-Acvr1 mutant embryos at E11.5. Green signals in control are autofluorescent blood cells. White arrowheads indicate ectopic cartilages. Scale bar, 100 μm. n = 4 mice per group. (B) Western blot analysis and quantification of β-catenin and active β-catenin in control and mutant BA1 tissues. Gapdh is a loading control. n = 4 independent experiments. (C) Relative expression of Cnx43, Ccnd1, and Lef1 in BA1 tissues at E11.5. n = 4 independent experiments. (D) Immunostaining and quantification of Sox9 (red) and β-catenin (green) in the nuclei of BA1 cells from control embryos treated with vehicle or BMP7. White arrowheads indicate Sox9 and β-catenin double-positive nuclei. Scale bar, 20 μm. n = 5 independent experiments. (E) Western blot analysis and quantification of β-catenin and active β-catenin in control or mutant BA1 cells treated with siRaptor or rapamycin for 24 hours. Uncropped Western blot images are shown in fig. S10. n = 3 independent experiments. (F) Experimental scheme for treating control and mutant BA1 cells with siRNA targeting β-catenin (siCtnnb1), the tankyrase inhibitor XAV939, Wnt3a alone, or Wnt3a plus rapamycin for 24 hours; representative images of Alcian blue staining of treated cells; and optical density quantification to assess chondrogenesis. Scale bar, 1 mm. n = 10 independent experiments. (G) Schematic representation, the ratios of each cartilage phenotype (severe, mild, and normal), and representative images of Alcian blue staining of mutant embryos treated in utero with rapamycin from E11.25 to E13.5. The numbers of mutant mice examined are shown in parentheses. Red arrows indicate ectopic cartilages. Scale bar, 2 mm. Error bars are means ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; t test [(B) to (E)]; ANOVA (F).
Functionally, we found that cartilage matrix deposition of ca-Acvr1 mutant BA1 cells was suppressed by knocking down β-catenin (encoded by Ctnnb1) or XAV939, a tankyrase inhibitor that suppresses Wnt–β-catenin signaling (Fig. 5F) (43) but enhanced by exogenous Wnt3a (Fig. 5F). We confirmed that the amount of β-catenin was reduced by the treatment with either siCtnnb1 or XAV939 in control and ca-Acvr1 mutant BA1 cells (fig. S8, A, B, K, and L) and increased by treatment with exogenous Wnt3a (fig. S8, C and M). Rapamycin efficiently mitigated the effect of Wnt3a on chondrogenic fate specification from BA1 cells (Fig. 5F), indicating that the chondrogenic effect of Wnt–β-catenin signaling depended on mTORC1 signaling. XAV939 efficiently suppressed cartilage matrix deposition when applied at day 1 but not when applied on days 2 to 5 (fig. S8, D to F), suggesting that the mTOR-Wnt–β-catenin signaling axis is responsible for committing CNCCs into the chondrogenic lineage. Maternal administration of XAV939 starting at E11.25 efficiently suppressed ectopic cartilage formation in most mutant embryos (Fig. 5G and fig. S8, G and N). However, XAV939 treatment starting at later stages (from E12.25 to E13.5) failed to rescue ectopic cartilage formation, although it reduced the amount of β-catenin in the BA1 tissues of ca-Acvr1 mutant embryos (fig. S8, G and H). It has been reported that Wnt–β-catenin signaling suppresses chondrogenesis in mesenchymal progenitor cells (44, 45). Consistent with this notion, we found that suppression of Wnt–β-catenin signaling using siCtnnb1 or XAV939 enhanced chondrogenesis in primary cells from limb buds (fig. S8, I, J, and O). These findings demonstrate that the function of Wnt–β-catenin signaling in promoting or inhibiting chondrogenesis is context dependent.
Augmented BMP signaling suppresses mTORC1-dependent autophagic degradation of β-catenin
In the canonical Wnt–β-catenin signaling pathway, the amount of β-catenin is regulated by a destruction complex that depends on the activity of glycogen synthase kinase-3 β (GSK3β), which phosphosphorylates β-catenin, thus targeting it for degradation. Wnt stimulation inhibits the destruction of β-catenin by promoting the phosphorylation of GSK3β at Ser9 (46). The amounts of both GSK3β phosphorylated at Ser9 and total GSK3β were unaltered in BA1 tissues from ca-Acvr1 mutants (Fig. 6A and fig. S11), suggesting that the increased activity of β-catenin in ca-Acvr1 mutants could not be explained simply by the inhibition of the canonical β-catenin destruction complex.
Fig. 6. Autophagy is suppressed in ca-Acvr1 mutants through mTORC1 signaling, and β-catenin is an autophagy target.
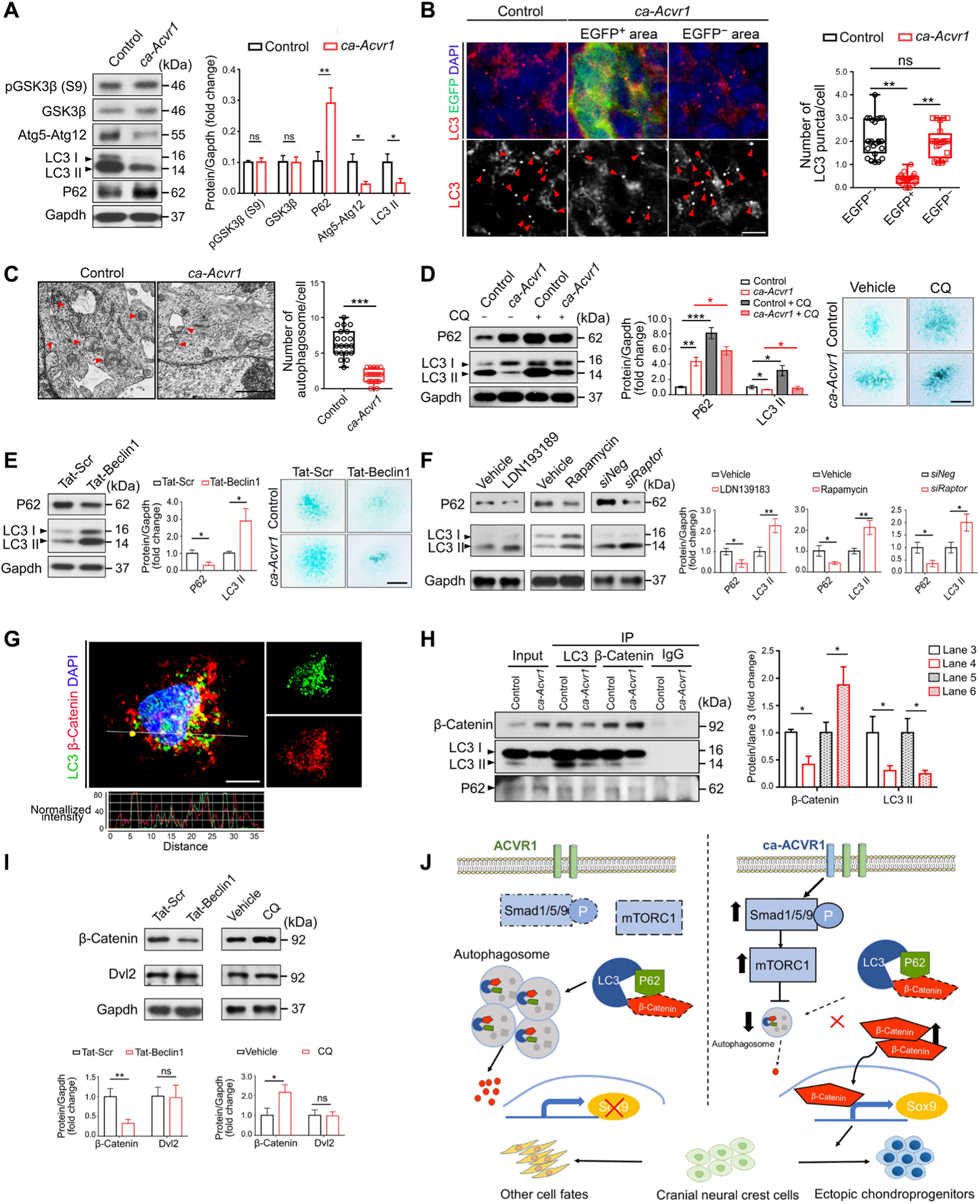
(A) Western blot analysis and quantification of GSK3β phosphorylated at Ser9 [pGSK3β (S9)], total GSK3β, the Atg5-Atg12 conjugate, LC3, and P62 in BA1 tissues. Gapdh is a loading control. n = 4 mice per group. (B) Double immunofluorescence for LC3 (red) and the ca-Acvr1 transgene marker EGFP (green) in coronal head sections from control and ca-Acvr1 mutant embryos at E11.5. Numbers of LC3 puncta (red arrowheads) per cell were quantified in EGFP-negative and EGFP-positive cells. Scale bar, 10 μm. n = 25 cells from four embryos per group. Schematic drawings illustrating the sources of the magnified regions are shown in fig. S9A. (C) Representative images and quantification of autophagic vacuoles (red arrowheads) per cell in the condensed area of the tongue from mutant embryos and its corresponding area from control embryos by transmission electron microscopy (TEM). Scale bar, 1 μm. n = 20 cells from three embryos per group. (D and E) Western blot analysis and quantification of P62 and LC3 in control and ca-Acvr1 BA1 cells treated with chloroquine (CQ) (D) and in control BA1 cells treated with Tat-Beclin 1 peptide (E) for 24 hours. n = 3 independent experiments. Representative images of Alcian blue staining to assess chondrogenesis of BA1 cells in each set of experiments are shown. Scale bars, 1 mm. n = 10 independent experiments. The optical density quantification of the dye is shown in fig. S9B. (F) Western blot and quantification of P62 and LC3 in BA1 cells treated with LDN193189, rapamycin, or siRaptor for 24 hours. n = 3 independent experiments. (G) Representative double immunofluorescence for LC3 (green) and β-catenin (red) in control BA1 cells with line scan analyses showing staining intensity of the indicated puncta. Scale bar, 10 μm. n = 5 independent experiments. (H) Immunoblotting and quantification of β-catenin, LC3, and P62 in LC3 and β-catenin immunoprecipitates (IP) from lysates from control and mutant BA1 cells. Immunoglobulin G (IgG) IP is a negative control. n = 3 independent experiments. (I) Western blot analysis and quantification of β-catenin and Dvl2 in mutant BA1 cells after stimulation with Tat-Beclin 1 peptide or CQ for 24 hours. n = 3 independent experiments. (J) Proposed model of enhanced BMP-Smad signaling in regulating CNCC fate. During normal development, there is a low level of pSmad1/5/9 and mTORC1 in CNCCs, and β-catenin is degraded by autophagy. When ca-ACVR1 is expressed in CNCCs, pSmad1/5/9-mTORC1 activity increases, which, in turn, inhibits autophagosome formation and autophagy-mediated β-catenin degradation. This increase in β-catenin signaling causes CNCCs to preferentially acquire ectopic chondrogenic fates. Uncropped Western blot images are shown in fig. S11. Error bars are means ± SD. ns, not significant P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; t test [(A), (C), (E), (F), (H), and (I)]; ANOVA [(B) and (D)].
It has been reported that autophagy negatively controls β-catenin activity in cancer cells and human embryonic kidney 293 cells (47–49). Because mTORC1 is a master regulator of autophagy (24), together with our data showing that β-catenin activity was suppressed by Raptor knockdown or rapamycin treatment (Fig. 5E), we hypothesized that β-catenin activity in CNCCs could be regulated through autophagy. To test this possibility, first, we examined the expression of several autophagy markers. The amounts of the autophagy proteins Atg5 and LC3-II were reduced, whereas P62, a known substrate for autophagic degradation (24), was increased in BA1 tissues in ca-Acvr1 mutants (Fig. 6A and fig. S11). The number of LC3 puncta in EGFP-positive cells in mutants was lower than that in EGFP-negative cells in these tissues in both control and ca-Acvr1 mutants, including the tongue, maxilla, and mandible (Fig. 6B and fig. S9A). Transmission electron microscopy (TEM) analyses confirmed that the number of membranous vacuoles resembling autophagic vacuoles was significantly lower in the condensed cells of the ca-Acvr1 mutants compared with controls (Fig. 6C), consistent with a reduction in autophagy in the mutant cells. Treatment with chloroquine (CQ), an inhibitor of lysosomal acidification and autophagosome degradation (24), reduced the accumulation of P62 and LC3-II in ca-Avcr1 mutant cells compared to control cells (Fig. 6D and fig. S11), suggesting that the autophagic machinery was functional in the mutant cells but that there was reduced autophagic flux. In addition, CQ increased cartilage matrix deposition by both control and BA1 cells (Fig. 6D and fig. S9B). In comparison, transactivating regulatory protein-Beclin 1 (Tat-Beclin1), an autophagy-inducing peptide (50), increased autophagic flux and suppressed the cartilage matrix deposition of control BA1 cells (Fig. 6E and figs. S9B and S11). These findings indicate that the suppression of autophagy is involved in the ectopic specification of chondrogenic fate in ca-Acvr1 mutants.
Next, we determined whether autophagy acted downstream of the BMP-mTORC1 axis. Treatment with LDN193189, rapamycin, or siRaptor reactivated autophagy in ca-Acvr1 mutant BA1 cells, as indicated by an increase in LC3-II and a reduction in p62 (Fig. 6F and fig. S11). On the other hand, in the presence of CQ, treatment with either LDN193189 or rapamycin did not suppress the deposition of cartilage matrix by ca-Acvr1 mutant BA1 cells (fig. S9C). These findings were further supported by superimposing null alleles of Atg5 (51) into ca-Acvr1 mutants to genetically block autophagic activity (fig. S9, D and J). In addition, treatment with LDN193189 or rapamycin failed to suppress the ectopic cartilage formation in the compound Atg5, ca-Acvr1 mutant embryos (ca-Acvr1flox/+;Atg5flox/flox;P0-Cre; fig. S9E). These results indicate that autophagy acted downstream of BMP-mTORC1 signaling during ectopic cartilage formation.
To evaluate a potential direct interaction between autophagy and β-catenin abundance in CNCCs (47–49), we examined the sub-cellular distribution of β-catenin and LC3-II. Confocal microscopy revealed the colocalization of β-catenin and LC3 puncta in control BA1 cells (Fig. 6G). Molecular association among β-catenin, LC3, and P62 was confirmed by coimmunoprecipitation in both control and ca-Acvr1 mutant BA1 cells (Fig. 6H and fig. S11). Although the amount of β-catenin was increased in ca-Acvr1 mutant cells, the amount of β-catenin that associated with LC3 in the cells was decreased compared with control cells (Fig. 6H and fig. S11). We obtained similar results using lysates from BA1 tissues from control and ca-Acvr1 mutant embryos (fig. S9, F and K). Furthermore, the amounts of β-catenin, as well as expression of Wnt–β-catenin target genes, were reduced in control BA1 cells upon Tat-Beclin 1 treatment (Fig. 6I and figs. S9G and S11) but increased upon CQ treatment (Fig. 6I and figs. S9H and S11), suggesting that autophagy inhibits β-catenin activity in BA1 cells.
It has been reported that autophagy indirectly promotes the degradation of β-catenin by stimulating the degradation of Dishevelled 2 (Dvl2) (52), thus reducing Dvl2-mediated inhibition of the β-catenin destruction complex downstream of Wnt receptors. However, treatment with either Tat-Beclin 1 or CQ did not change the abundance of Dvl2 in BA1 cells from ca-Acvr1 mutants (Fig. 6I and fig. S11). These results are consistent with β-catenin being directly targeted for autophagic degradation, possibly through an interaction with LC3, in CNCCs. β-catenin has also been reported to be degraded through the ubiquitin-proteasome pathway (53). Treatment of ca-Acvr1 BA1 cells with MG-132 (N-carbobenzyloxy-l-leucyl-l-leucyl-l-leucinal), an inhibitor of the ubiquitin-proteasome system (54), slightly increased the abundance of β-catenin (fig. S9, I and L), suggesting that the ubiquitin-proteasome system may play a minor role in degrading β-catenin in these cells. The amount of β-catenin was higher in CQ-treated cells than that in MG-132–treated cells (fig. S9, I and L). These results indicate that autophagy rather than the ubiquitin-proteasome system plays a major role in degrading β-catenin in ca-Acvr1 mutants. Together, these data suggest that autophagy was suppressed through the activation of mTORC1 signaling, thus reducing autophagic degradation of β-catenin and leading to the chondrogenic specification of CNCCs and ectopic cartilage formation in ca-Acvr1 mutants (Fig. 6J).
DISCUSSION
During craniofacial development, the fate of multipotent neural crest cells is regulated by the tissue context-dependent integration of extrinsic and intrinsic signals that drive their differentiation to produce appropriate cell types at the proper time and location (55). However, it remains elusive how the skeletogenic fate is determined in CNCCs (56). Here, using ca-ACVR1 mouse models, we uncovered previously unrecognized coordination among BMP, mTOR, Wnt–β-catenin signaling, and autophagy that play pivotal roles to regulate chondrogenic fate during craniofacial development.
BMP signaling controls skeletal development and regeneration (9, 12, 16, 57). Mice lacking a type I BMP receptor, Acvr1, in either chondrocytes or neural crest cells display subtle changes in cartilage development (21, 22). Double deficiency of the genes encoding the type I BMP receptors BMPR1A and BMPR1B in cartilage causes severe generalized chondrodysplasia in mice (58). These loss-of-function studies demonstrate that an appropriate range of BMP signaling through BMP type I receptors is required for normal cartilage development. In humans, gain-of-function mutation of ACVR1 causes FOP, an aggressive form of endochondral ossification (18, 19). It has been reported that some patients with FOP develop ectopic bones in the craniofacial region; however, the molecular pathogenesis of this phenotype is still unclear (20). Here, we show that enhanced BMP signaling through ACVR1 in CNCCs induced ectopic cartilages. This molecular mechanism could give rise to ectopic bones, as observed in some patients with FOP, if the ectopic cartilages progressed to endochondral ossification.
In the CNCC ca-Acvr1 mouse model, we found a mosaic pattern of enhanced BMP-Smad signaling in the facial region (Fig. 3, A and B, and fig. S4A). Because P0-Cre labels almost all CNCCs, the activation of BMP-Smad signaling in a mosaic rather than ubiquitous pattern may be attributable to heterogeneous activation of the CAG promoter driving expression of the ca-Acvr1 transgene, which we reported previously (31). Another possibility is that some subpopulations of CNCCs are more sensitive to ca-ACVR1. This would be consistent with our finding that a mere twofold increase in BMP-Smad signaling was associated with the ca-ACVR1-driven development of ectopic cartilages (Fig. 1C). We hypothesize that a small increase of BMP signaling in CNCCs would be sufficient to induce ectopic cartilage. This is well supported by our chemical inhibitor experiments, because relatively low doses of these inhibitors, below that required for complete BMP signaling inhibition, suppressed ectopic cartilage formation in ca-Acvr1 mutants but did not cause any facial abnormalities in control mice (Figs. 3E, 4F, and 5G). Therefore, it may be reasonable to conclude that a relatively small increase of BMP signaling induced by ca-ACVR1 could be sufficient to induce ectopic chondrogenic specification of CNCCs in a mosaic manner in ca-Acvr1 mutants.
We previously reported that enhanced BMP signaling induced by ca-Bmpr1a in CNCCs leads to craniosynostosis, failure of nasal septum fusion, and secondary palate fusion (15, 57). The ca-Acvr1;P0-Cre mutants, like ca-Bmpr1a;P0-Cre mutants, had defects in skull development, but the development of ectopic cartilage in ca-Acvr1;P0-Cre mice was not observed in ca-Bmpr1a;P0-Cre mice. These discrepancies could be explained by a difference in the BMP signaling levels induced in ca-Acvr1;P0-Cre and ca-Bmpr1a;P0-Cre mutants. In support of this hypothesis, the amount of pSmad1/5/9 is similar between controls and ca-Bmpr1a;P0-Cre mutants during early stages of craniofacial development and increased mid to late gestations (59), but the amount of pSmad1/5/9 was already increased during the early stages in ca-Acvr1;P0-Cre mutants (Figs. 1C and 2, A and B). Alternatively, one might speculate that these BMP type I receptors may have distinct roles in CNCCs, with the activation of BMPR1A or ACVR1 mediating different transcriptional responses. For example, if activation of BMP signaling through ACVR1 but not BMPR1A stimulates the expression of chondrogenesis-related genes such as Sox9, then only ca-Acvr1;P0-Cre mutants would develop ectopic cartilage in the face even though both mutants develop craniofacial abnormalities. Therefore, while both mutants showed craniofacial abnormalities, ca-Acvr1;P0-Cre mutants developed ectopic cartilage in the face, whereas ca-Bmpr1a;P0-Cre mutants did not. However, mutants expressing constitutively activated Bmpr1a using a different neural crest-Cre driver (ca-Bmpr1a;Wnt1-Cre) also display ectopic cartilage formation in the craniofacial region (16). Because these different ca-Bmpr1a mutant mouse models were generated using different transgenic strategies, it is difficult to conclude whether these phenotypes are due to differences in pSmad1/5/9 signaling amounts attained in ca-Bmpr1a;P0-Cre mice with that in ca-Bmpr1a;Wnt1-Cre mice. One potential future direction is to address these points using ca-Acvr1;P0-Cre, ca-Bmpr1a;P0-Cre, and ca-Bmpr1a;Wnt1-Cre mice.
mTOR integrates both intracellular and extracellular signals and serves as a central regulator of cell proliferation, survival, and differentiation (60). Neural crest–specific deletion of the mTORC1 component Raptor results in the absence of facial cartilage (40). We have reported that ca-Bmpr1a in neural crest cells activates mTORC1 signaling during skull development (61), suggesting the potential for cross-talk between BMP and mTORC1 signaling. mTORC1 signaling has been identified as one of the downstream pathways that is critical for the aberrant chondrogenesis in FOP mouse models (62). Therefore, our study further supports the notion that activation of the BMP-mTORC1 signaling axis induces ectopic cartilage during craniofacial development.
Canonical Wnt–β-catenin signaling functions as either a positive or a negative regulator in many developmental contexts (44, 45). During craniofacial development, inactivation of β-catenin in CNCCs deletes most craniofacial cartilage (63). In support of this notion, we found that enhanced BMP signaling through ca-ACVR1 activated Wnt–β-catenin signaling and induced ectopic cartilage in the face (Fig. 5, A to D, F, and G). These findings are further supported by the fact that Wnt–β-catenin signaling is involved in ectopic cartilage formation induced by exogenous BMPs (41, 64–66), demonstrating that BMP–Wnt–β-catenin signaling pathway positively controls craniofacial cartilage formation.
Both BMP and Wnt–β-catenin signaling were previously known to be required for craniofacial development (12). Here, we identified autophagy downstream of BMP signaling as important for regulating the activity of β-catenin in CNCC-derived chondrocytes. This mechanism is distinct from the mechanism by which BMP controls Wnt–β-catenin in chondrogenesis of mesodermal cells in the trunk. Conditional knockout of Acvr1 in osteoblasts or mesenchymal stem cells activates canonical Wnt–β-catenin signaling through suppression of the Wnt inhibitors Dkk1, Sost, or Gsk3β (67, 68). On the other hand, we found that β-catenin activity increased in ca-Acvr1 BA1 cells despite an increase in Dkk1, suggesting that the mechanism downstream of BMP regulating Wnt–β-catenin signaling depends on cell context.
Autophagy is a self-degradative process that is important for stem cell homeostasis and cell differentiation (24, 69), yet little is known about how the activity of the autophagy machinery is coordinated with growth factor signaling. We found that the stabilization of β-catenin in ca-Acvr1;P0-Cre mutants was not due to effects on the β-catenin destruction complex but rather the dysfunction of the autophagy machinery that is involved in the degradation of β-catenin. Several groups, including ours, have previously reported that BMP and Wnt–β-catenin signaling cross-talk regulates skeletal development (41, 42, 64–67, 70), but the intracellular mechanism of how enhanced BMP signaling leads to the stabilization of β-catenin has not been reported. During chondrogenic fate determination in craniofacial development, we identified that β-catenin stabilization is regulated by the autophagy machinery in a BMP signaling–dependent manner. Consistent with our findings, other studies have demonstrated the involvement of the BMP-autophagy pathway in the chondrogenic differentiation of mesenchymal stem cells (71) and chondrosarcoma (72, 73). Therefore, our study may provide a molecular explanation, at least in part, for why patients with FOP develop ectopic bone in the face, possibly through the suppression of autophagic β-catenin degradation. In addition to FOP, BMP signaling is associated with many pathologies, including obesity, diabetes, and vascular diseases (74). Our study highlights the importance of considering autophagy in developing therapeutic strategies to treat BMP-related diseases.
In summary, we uncovered a previously unrecognized mechanism by which a BMP-autophagy-β-catenin axis regulates cell fate specification during craniofacial development. We propose a model in which, during the cell fate specification in CNCCs, β-catenin interacts with LC3 and is directly targeted for autophagic degradation, thus favoring a nonchondrogenic fate by inhibiting Wnt–β-catenin signaling (Fig. 6J). We hope that our findings will contribute to developing therapeutic methods for preventing ectopic facial chondrogenesis in patients with FOP by using not only BMP blockers but also autophagy activators or Wnt inhibitors.
MATERIALS AND METHODS
Mice
ca-Acvr1flox/flox mice (31) and Tak1flox/flox mice (75) were generated in our laboratory. P0-Cre mice, C57BL/6J-Tg(P0-Cre)94Imeg (ID 148), were provided by the Center for Animal Resources and Development, Kumamoto University, Japan. Atg5flox/flox mice were obtained from N. Mizushima, Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan (51). Wnt1-Cre, R26RLacZ, R26RtdTomato, and R26RmTmG mice were obtained from the Jackson laboratory. ca-Acvr1flox/flox or ca-Acvr1flox/+ mice were crossed with P0-Cre transgenic mice to obtain wild-type, control (ca-Acvr1+//+;P0-Cre), and ca-Acvr1 mutant (ca-Acvr1flox//+;P0-Cre) mice. ca-Acvr1flox/flox mice were crossed with Wnt1-Cre mice to obtain ca-Acvr1;Wnt1-Cre (ca-Acvr1flox/+; Wnt1-Cre) mice. Embryos with compound mutation of ca-Acvr1 and Tak1 (ca-Acvr1flox/+;Tak1flox/flox;P0-Cre) were generated by crossing ca-Acvr1flox/flox;Tak1flox/+ mice and Tak1flox/+;P0-Cre mice. Embryos with compound mutation of ca-Acvr1 and Atg5 (ca-Acvr1flox/+; Atg5flox/flox;P0-Cre) were generated by crossing ca-Acvr1flox/flox;Atg5flox/+ mice and Atg5flox/+;P0-Cre mice. In vivo fate mapping of neural crest–derived cells was performed on mice additionally carrying the R26RLacZ, R26RmTmG, or R26RtdTomato allele.
All mice were maintained in a mixed background and were used and genotyped as previously described (31). Embryos were collected from timed-pregnant mice. Embryonic ages were determined by the day when the vaginal plug was found, which was designated as E0.5. Embryonic tails were subjected to DNA extraction for genotyping as previously described (31). For genotyping primers, see table S1. All mouse procedures used in this study were approved by the Institutional Animal Care and Use Committee at the University of Michigan (#PRO00007715) and the University of Texas (AWC-18–0137).
Whole-mount Alcian blue staining
Embryos collected from timed-pregnant females at indicated time points were fixed in Bouin’s solution for 2 hours at room temperature, rinsed with a solution of 1% NH4OH diluted in 70% ethanol for 24 hours until embryos appeared white, equilibrated with 5% acetic acid, and stained with 0.05% Alcian blue solution for 2 hours. Samples were then rinsed with 5% acetic acid, dehydrated with 100% methanol, cleared, and stored in benzyl alcohol benzyl benzoate solution before taking pictures under a stereomicroscope.
Whole-mount skeletal preparation
After removing skins and adipose tissue, embryo heads were fixed in 95% ethanol overnight, stained with Alcian blue solution [0.015% (w/v)], destained by 70 and 95% ethanol, and precleared with 2% potassium hydroxide solution. After staining with Alizarin red [0.015% (w/v)] for 2 hours, samples were destained in 20% glycerol/1% KOH solution and stored in 50% glycerol/47.5% ethanol solution before taking pictures under a stereomicroscope.
Histological and morphometric analyses
Embryos collected from timed-pregnant females at indicated time points were fixed with 4% paraformaldehyde (PFA), followed by optimal cutting temperature (OCT) compound embedding. Consecutive serial sections (10 μm) were collected for hematoxylin and eosin staining, Safranin O and Fast Green staining, immunofluorescence staining, or in situ hybridization according to standard protocols. For analysis of cell ultrastructure, samples were cut from the BA1 tissues and immersed immediately in a mixture of PFA and glutaraldehyde. Samples were processed for TEM as described (24).
Immunostaining
Immunohistochemistry was performed according to standard protocols. The antibodies used are listed in table S3. Experiments and analyses were performed in a blinded manner. For statistical analysis, the density of Sox9-positive or Sox9-negative cells (n per square millimeter) was defined as cell count number divided by the region of interest area for different regions. The number of LC3 puncta in E11.5 BA1 tissues was counted randomly in 25 EGFP-positive or EGFP-negative ectomesenchyme cells from the sections. The number of pH3-positive cells was counted randomly in a minimum of 300 mesenchyme cells from the sections. Results were presented as percentages of positive cells against total cells.
In situ hybridization
E17.5 embryos’ heads were fixed with 4% PFA for 2 hours and saturated with 30% sucrose in diethyl pyrocarbonate–treated phosphate-buffered saline (PBS) overnight at 4°C, embedded in OCT compound, and sectioned at 10 μm. Antisense RNA probe of Col2a1 labeled with digoxigenin-UTP (uridine 5′-triphosphate) was transcribed from a complementary DNA (cDNA) clone. After overnight hybridization with RNA probe, sections were detected with anti-digoxigenin-alkaline phosphatase (11093274910, Roche) and then developed by nitro blue tetrazolium.
Isolation and culture of CNCCs from BA1 tissues
The isolation of CNCCs from BA1 tissues (BA1 cells) was performed as previously described (35–37). In brief, BA1 tissues from E11.5 embryos were isolated, dissociated into a single-cell suspension by digesting with TrypLE enzyme (12605028, Thermo Fisher Scientific) for 10 min at 37°C, and filtered to remove undigested tissues. For micromass culture, cells were pelleted by centrifugation at 1000 rpm for 5 min, then reconstituted 1 × 107 cells/ml in Dulbecco’s modified Eagle’s medium (DMEM) media supplemented with 10% fetal bovine serum (FBS), antibiotics, and fibronectin (100 μg/ml; 33016–015, Thermo Fisher Scientific), and spotted as 20-μl drops on culture dishes. After a 2-hour attachment period, the DMEM media was supplemented with 10% FBS and antibiotics, fibronectin (10 μg/ml) was gently added, and cultures were left resting for a further 12 hours. Then, fresh medium [DMEM/F12 (2:3) medium supplemented with 4% FBS and antibiotics] was added to let cells differentiate for 5 days. Chondrogenesis was determined by examining the gene expression of chondrogenic markers Sox9, Aggrecan (Acan), and Col2a1 by qRT-PCR (quantitative reverse transcription polymerase chain reaction) and accumulation of sulfated proteoglycans using Alcian blue staining. For semiquantification of Alcian blue staining, after taking pictures, we extracted the dye by incubating cultures with 500 μl of 6 M guanidine hydrochloride (SRE0066, Sigma-Aldrich) overnight at room temperature. The absorption of the extracted dye was measured at 595 nm in a microplate reader. Graphs were generated, and statistical analyses were performed using Prism. Cells at 12 hours (day 0, before changing to fresh medium) and day 2 (24 hours after changing to fresh medium) were harvested and processed for qRT-PCR analysis to determine the expression of differentiation markers for osteogenic, neurogenic, myogenic, and chondrogenic lineages.
For siRNA transfection, BA1 cells were maintained as micromass culture for 12 hours, washed twice with PBS, and changed to the culture medium without antibiotics 1 hour before transfection. Negative control siRNA (scrambled, 20 pmol per reaction; Thermo Fisher Scientific), siRictor (s95670, 20 pmol per reaction; Thermo Fisher Scientific), siRptor (s92711, 20 pmol per reaction; Thermo Fisher Scientific), or siCtnnb1 (s63417, 20 pmol per reaction; Thermo Fisher Scientific) was transfected into cells using Lipofectamine RNAiMAX (13778150, Thermo Fisher Scientific) following the manufacturer’s standard protocol. After 24 hours of transfection, cells were cultured for up to 5 days in fresh medium or harvested for Western blot or qRT-PCR analysis.
For inhibitors or ligands treatment, BA1 cells were maintained as micromass culture for 12 hours, stimulated with inhibitors or ligands at indicated time and concentrations. Inhibitors or ligands used in this study were as follows: LDN193189 (100 or 200 nmol final concentration), rapamycin (0.1 or 0.2 μmol final concentration; 9904S, Cell Signaling Technology), CQ (10 μmol final concentration; C6628, Sigma-Aldrich), XAV939 (10 μmol final concentration; X3004, Sigma-Aldrich), Tat-Beclin 1 peptide (10 μmol final concentration; NBP2–49888, Novus Biologicals), Tat-inactive scrambled peptide (10 μmol final concentration; NBP2–49887, Novus Biologicals), MG-132 (10 μmol final concentration; M7449, Sigma-Aldrich), and Wnt3a (50 ng/ml final concentration; P27467, R&D Systems).
For immunocytochemistry, cells were seeded onto eight-well chambers with DMEM medium supplemented with 10% FBS. Cells were harvested at the indicated time points after seeding or stimulated with BMP7 (100 ng/ml final concentration; NP_031583, R&D Systems) for 12 hours. Cells were fixed with 4% PFA. Immunostaining was performed as standard protocol with indicated primary antibodies.
In vivo inhibitor administration
LDN193189, rapamycin, or XAV939 was used for the treatment of animals. LDN-193189 was dissolved in sterile endotoxin-free water (18). Rapamycin (R-5000, LC Laboratories) was reconstituted in absolute ethanol at 10 mg/ml and diluted in 5% Tween 80 (Sigma-Aldrich) and 5% polyethylene glycol 400 (Hampton Research) (62). XAV939 was dissolved in dimethyl sulfoxide to prepare stock solution (10 mg/ml) and further diluted in PBS before injection (43). After optimizing the dosage and injection schedule, 2.5 mg of the LDN193189, 5 mg of the rapamycin, or 5 mg of the XAV939 per kilogram body weight twice per day was used. Each chemical was intraperitoneally injected into pregnant mice starting on days E11.25 or E12.25 through E13.5. BA1-derived tissues, including the maxilla, mandible, and tongue, were dissected at E13.5, homogenized in immunoprecipitation lysis buffer containing protease and phosphatase inhibitors, and processed for Western blot analysis. Embryos at E14.5 were collected for whole-mount Alcian blue staining.
RNA extraction and gene expression analysis
Total RNAs were isolated using the TRIzol reagent following the manufacturer’s protocol. First-strand cDNA was synthesized with 1 mg of denatured RNA using the SuperScript First-Strand Synthesis System (11904–018, Thermo Fisher Scientific). The real-time qRT-PCR analysis was performed with ABI PRISM 7500 (Applied Biosystems) to measure the relative mRNA levels using a SYBR Green kit (4367659, Thermo Fisher Scientific). Experiments were replicated with six samples for each genotype. Each reaction was performed in triplicate. The quantity of each experimental sample was first determined using a standard curve based on the cycle threshold (Ct) values and then expressed relative to the internal control. Data were normalized to Gapdh mRNA levels using the 2−ΔΔCt method. The DNA sequences of primers used for qRT-PCR are summarized in table S2.
Western blot analysis
For protein extraction, harvested BA1 tissues in each embryo or BA1 cells were washed with PBS containing a protease inhibitor cocktail (Roche). Tissues were homogenized with Precellys Tissue Homogenizer (Bertin Instruments) in radioimmunoprecipitation assay buffer supplemented with deoxyribonuclease I and protease inhibitor cocktail. SDS–polyacrylamide gel electrophoresis (PAGE) and Western blot were carried out according to standard protocol. Antigen detection was performed using antibodies listed in table S3. Bound primary antibodies were detected with horseradish peroxidase-conjugated species-specific secondary antibodies (Cell Signaling Technology) using the Super Signal Pico system (34079, Thermo Fisher Scientific). The immunoreactive bands were quantified using ImageJ, and the mean ratios of the indicated protein from three independent experiments are shown at the bottom of the figures.
Immunoprecipitation and Western blot analysis
The BA1 tissues or cells isolated from BA1 tissues of E11.5 embryos were used for this experiment. BA1 cells were harvested and seeded on a 10-cm dish. Tissues or confluent cells were homogenized in 200 μl of immunoprecipitation lysis buffer containing protease and phosphatase inhibitors (05892970001, Sigma-Aldrich). The homogenate was incubated on ice for 30 min and centrifuged at 14,000g at 4°C for 15 min. Total lysates were subjected to immunoprecipitation with the indicated antibodies in protein G magnetic beads (LSKMAGG02, Sigma-Aldrich). Beads with immunoprecipitates were washed three times with cold lysis buffer, heated in SDS buffer at 95°C for 5 min, and subjected to SDS-PAGE on 4 to 20% Mini-PROTEAN TGX Precast Gels (4561093, Bio-Rad Laboratories). Protein expression was detected by indicated primary antibodies. Bound primary antibodies were detected with horseradish peroxidase–conjugated species-specific secondary antibodies (Cell Signaling Technology) using the Super Signal Pico system (34079, Thermo Fisher Scientific).
TUNEL assays
E11.5 heads were fixed in 4% PFA at 4°C overnight, incubated in 30% sucrose in PBS at 4°C overnight, embedded in OCT compound, and sectioned at 10 μm. BA1 cells were fixed in 4% PFA at 4°C for 30 min. Levels of cell death were measured by using the terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) method using the In Situ Cell Death Detection Kit (11684795910, Roche). DNA strand breaks were labeled with fluorescein and visualized with fluorescein isothiocyanate.
β-Galactosidase staining
For whole-mount staining, E10.5 embryos with R26RLacZ reporter were fixed in 2.5% glutaraldehyde at 4°C for 2 hours. After rinsing with LacZ rinse [0.1% Na deoxycholate, 0.2% NP-40, and 2 mM MgCl2 in 0.1 M sodium phosphate buffer (pH 7.3)], embryos were stained with X-gal staining solution (1 mg/ml) for 8 hours at 37°C protected from light. The LacZ staining solution was made using X-gal (1 mg/ml), 5 mM potassium ferricyanide, and 5 mM potassium ferrocyanide in LacZ rinse. Embryos were then washed with PBS and postfixed/stored in 4% PFA in PBS at 4°C before taking pictures under a stereomicroscope.
For staining of sections, after fixation with 2.5% glutaraldehyde for 2 hours at 4°C, E10.5 embryos with R26RLacZ reporter were cryoprotected in 30% sucrose in PBS overnight at 4°C and embedded in OCT compound. Consecutive serial sections (10 μm) were collected for histological analysis of β-galactosidase by X-gal staining and counterstained with fast red.
Proliferation assay
BA1 cells from control or ca-Acvr1 embryos were seeded 10,000 cells per well in quadruplicate in 96-well flat-bottom tissue culture plates. WST-1 analysis (5015944001, Sigma-Aldrich) was performed according to the manufacturer’s instructions. Absorption at 450 nm was measured using a microplate reader (Bio-Rad Laboratories). Cell viability was evaluated at days 0, 1, 2, 3, and 5 after plating.
Statistical analysis and quantification
All results were replicated in at least three different animals. Results were presented as percentages of positive cells against total cells. Graphs were generated, and statistical analyses were performed using Prism. All values were expressed as means ± SD. Unpaired Student’s t test or analysis of variance (ANOVA) was used to analyze the differences between or among groups. A P value of less than 0.05 was considered statistically significant. All representative experiments shown were repeated three or more times. Experiments and analyses were performed in a blinded manner.
Supplementary Material
Acknowledgments:
We thank K. Yamamura (Kumamoto University) for providing P0-Cre mice, J. Harrison (University of Michigan Microscopy Core) for assistance in TEM imaging, and Y. Ohsumi for suggestions on autophagy experiments.
Funding: This work was supported by grants from the NIH (R01DE020843 to Y.M. and R01DE025897 to Y.K.), International Fibrodysplasia Ossificans Progressiva Association (to Y.M.), and the grant-in-aid from the National Natural Science Foundation of China (31500788 to J.Y.).
Footnotes
SUPPLEMENTARY MATERIALS
stke.sciencemag.org/cgi/content/full/14/665/eaaz9368/DC1
Fig. S1. Phenotypes due to expression of ca-ACVR1 in CNCCs during craniofacial development.
Fig. S2. Cell migration, proliferation, and survival are unaltered in ca-Acvr1 mutants.
Fig. S3. Skeletal stem cell populations are unaltered in ca-Acvr1 mutants.
Fig. S4. Noncanonical BMP signaling and TGF-β signaling are unaffected in ca-Acvr1 mutants.
Fig. S5. LDN193189 suppresses Smad1/5/9 phosphorylation and chondrogenesis of BA1 cells in a concentration- and time-dependent manner.
Fig. S6. Increased commitment of BA1 cells to the chondrocyte fate in ca-Acvr1 mutants.
Fig. S7. siRaptor or rapamycin suppresses mTORC1 signaling and Wnt–β-catenin signaling.
Fig. S8. Blocking Wnt–β-catenin signaling suppresses chondrogenesis of BA1 cells but enhances chondrogenic differentiation of limb bud cells.
Fig. S9. Autophagy inhibits β-catenin signaling and is suppressed by BMP-mTORC1 signaling in BA1 cells.
Fig. S10. Uncropped Western blots for Figs. 1, 4, and 5.
Fig. S11. Uncropped Western blots for Fig. 6.
Table S1. Genotyping PCR primers.
Table S2. Quantitative real-time PCR primers.
Table S3. Antibodies used in this study.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The ca-Acvr1 mouse line is available from Y.M. under a material transfer agreement with the NIH. Raw data files are available from the corresponding author upon reasonable request.
REFERENCES AND NOTES
- 1.Santagati F, Rijli FM, Cranial neural crest and the building of the vertebrate head. Nat. Rev. Neurosci 4, 806–818 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Kaucka M, Ivashkin E, Gyllborg D, Zikmund T, Tesarova M, Kaiser J, Xie M, Petersen J, Pachnis V, Nicolis SK, Yu T, Sharpe P, Arenas E, Brismar H, Blom H, Clevers H, Suter U, Chagin AS, Fried K, Hellander A, Adameyko I, Analysis of neural crest-derived clones reveals novel aspects of facial development. Sci. Adv 2, e1600060 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalcheim C, Neural crest emigration: From start to stop. Genesis 56, e23090 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Takahashi Y, Sipp D, Enomoto H, Tissue interactions in neural crest cell development and disease. Science 341, 860–863 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Baker CV, Bronner-Fraser M, Le Douarin NM, Teillet MA, Early- and late-migrating cranial neural crest cell populations have equivalent developmental potential in vivo. Development 124, 3077–3087 (1997). [DOI] [PubMed] [Google Scholar]
- 6.Baroffio A, Dupin E, Le Douarin NM, Clone-forming ability and differentiation potential of migratory neural crest cells. Proc. Natl. Acad. Sci. U.S.A 85, 5325–5329 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung IH, Yamaza T, Zhao H, Choung PH, Shi S, Chai Y, Stem cell property of postmigratory cranial neural crest cells and their utility in alveolar bone regeneration and tooth development. Stem Cells 27, 866–877 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bronner-Fraser M, Fraser SE, Cell lineage analysis reveals multipotency of some avian neural crest cells. Nature 335, 161–164 (1988). [DOI] [PubMed] [Google Scholar]
- 9.Mishina Y, Snider TN, Neural crest cell signaling pathways critical to cranial bone development and pathology. Exp. Cell Res 325, 138–147 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noden DM, Trainor PA, Relations and interactions between cranial mesoderm and neural crest populations. J. Anat 207, 575–601 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Achilleos A, Trainor PA, Neural crest stem cells: Discovery, properties and potential for therapy. Cell Res. 22, 288–304 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Graf D, Malik Z, Hayano S, Mishina Y, Common mechanisms in development and disease: BMP signaling in craniofacial development. Cytokine Growth Factor Rev. 27, 129–139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grafe I, Alexander S, Peterson JR, Snider TN, Levi B, Lee B, Mishina Y, TGF-β family signaling in mesenchymal differentiation. Cold Spring Harb. Perspect. Biol 10, a022202 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bellacosa A, Developmental disease and cancer: Biological and clinical overlaps. Am. J. Med. Genet. A 161A, 2788–2796 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Komatsu Y, Yu PB, Kamiya N, Pan H, Fukuda T, Scott GJ, Ray MK, Yamamura K, Mishina Y, Augmentation of Smad-dependent BMP signaling in neural crest cells causes craniosynostosis in mice. J. Bone Miner. Res 28, 1422–1433 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li L, Wang Y, Lin M, Yuan G, Yang G, Zheng Y, Chen Y, Augmented BMPRIA-mediated BMP signaling in cranial neural crest lineage leads to cleft palate formation and delayed tooth differentiation. PLOS ONE 8, e66107 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu S, Wu W, Liu C, Yang L, Sun C, Ye W, Li X, Chen J, Long F, Chen Y, BMPRIA mediated signaling is essential for temporomandibular joint development in mice. PLOS ONE 9, e101000 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML, Hong DW, McManus PM, Katagiri T, Sachidanandan C, Kamiya N, Fukuda T, Mishina Y, Peterson RT, Bloch KD, BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat. Med 14, 1363–1369 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS, A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet 38, 525–527 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Carvalho DR, Farage L, Martins BJ, Speck-Martins CE, Craniofacial findings in fibrodysplasia ossificans progressiva: Computerized tomography evaluation. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod 111, 499–502 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Dudas M, Sridurongrit S, Nagy A, Okazaki K, Kaartinen V, Craniofacial defects in mice lacking BMP type I receptor Alk2 in neural crest cells. Mech. Dev 121, 173–182 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Rigueur D, Brugger S, Anbarchian T, Kim JK, Lee Y, Lyons KM, The type I BMP receptor ACVR1/ALK2 is required for chondrogenesis during development. J. Bone Miner. Res 30, 733–741 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan H, Zhang H, Abraham P, Komatsu Y, Lyons K, Kaartinen V, Mishina Y, BmpR1A is a major type 1 BMP receptor for BMP-Smad signaling during skull development. Dev. Biol 429, 260–270 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ, Adler SG, Agam G, Agarwal R, Aghi MK, Agnello M, Agostinis P, Aguilar PV, Aguirre-Ghiso J, Airoldi EM, Ait-Si-Ali S, Akematsu T, Akporiaye ET, Al-Rubeai M, Albaiceta GM, Albanese C, Albani D, Albert ML, Aldudo J, Algül H, Alirezaei M, Alloza I, Almasan A, Almonte-Beceril M, Alnemri ES, Alonso C, Altan-Bonnet N, Altieri DC, Alvarez S, Alvarez-Erviti L, Alves S, Amadoro G, Amano A, Amantini C, Ambrosio S, Amelio I, Amer AO, Amessou M, Amon A, An Z, Anania FA, Andersen SU, Andley UP, Andreadi CK, Andrieu-Abadie N, Anel A, Ann DK, Anoopkumar-Dukie S, Antonioli M, Aoki H, Apostolova N, Aquila S, Aquilano K, Araki K, Arama E, Aranda A, Araya J, Arcaro A, Arias E, Arimoto H, Ariosa AR, Armstrong JL, Arnould T, Arsov I, Asanuma K, Askanas V, Asselin E, Atarashi R, Atherton SS, Atkin JD, Attardi LD, Auberger P, Auburger G, Aurelian L, Autelli R, Avagliano L, Avantaggiati ML, Avrahami L, Awale S, Azad N, Bachetti T, Backer JM, Bae D-H, Bae J-S, Bae O-N, Bae SH, Baehrecke EH, Baek S-H, Baghdiguian S, Bagniewska-Zadworna A, Bai H, Bai J, Bai X-Y, Bailly Y, Balaji KN, Balduini W, Ballabio A, Balzan R, Banerjee R, Bánhegyi G, Bao H, Barbeau B, Barrachina MD, Barreiro E, Bartel B, Bartolomé A, Bassham DC, Bassi MT, Bast RC Jr, Basu A, Batista MT, Batoko H, Battino M, Bauckman K, Baumgarner BL, Ulrich Bayer K, Beale R, Beaulieu J-F, Beck GR Jr, Becker C, David Beckham J, Bédard P-A, Bednarski PJ, Begley TJ, Behl C, Behrends C, Behrens GM, Behrns KE, Bejarano E, Belaid A, Belleudi F, Bénard G, Berchem G, Bergamaschi D, Bergami M, Berkhout B, Berliocchi L, Bernard A, Bernard M, Bernassola F, Bertolotti A, Bess AS, Besteiro S, Bettuzzi S, Bhalla S, Bhattacharyya S, Bhutia SK, Biagosch C, Bianchi MW, Biard-Piechaczyk M, Billes V, Bincoletto C, Bingol B, Bird SW, Bitoun M, Bjedov I, Blackstone C, Blanc L, Blanco GA, Blomhoff HK, Boada-Romero E, Böckler S, Boes M, Boesze-Battaglia K, Boise LH, Bolino A, Boman A, Bonaldo P, Bordi M, Bosch J, Botana LM, Botti J, Bou G, Bouché M, Bouchecareilh M, Boucher M-J, Boulton ME, Bouret SG, Boya P, Boyer-Guittaut M, Bozhkov PV, Brady N, Braga VM, Brancolini C, Braus GH, Bravo-San Pedro JM, Brennan LA, Bresnick EH, Brest P, Bridges D, Bringer M-A, Brini M, Brito GC, Brodin B, Brookes PS, Brown EJ, Brown K, Broxmeyer HE, Bruhat A, Brum PC, Brumell JH, Brunetti-Pierri N, Bryson-Richardson RJ, Buch S, Buchan AM, Budak H, Bulavin DV, Bultman SJ, Bultynck G, Bumbasirevic V, Burelle Y, Burke RE, Burmeister M, Bütikofer P, Caberlotto L, Cadwell K, Cahova M, Cai D, Cai J, Cai Q, Calatayud S, Camougrand N, Campanella M, Campbell GR, Campbell M, Campello S, Candau R, Caniggia I, Cantoni L, Cao L, Caplan AB, Caraglia M, Cardinali C, Cardoso SM, Carew JS, Carleton LA, Carlin CR, Carloni S, Carlsson SR, Carmona-Gutierrez D, Carneiro LA, Carnevali O, Carra S, Carrier A, Carroll B, Casas C, Casas J, Cassinelli G, Castets P, Castro-Obregon S, Cavallini G, Ceccherini I, Cecconi F, Cederbaum AI, Ceña V, Cenci S, Cerella C, Cervia D, Cetrullo S, Chaachouay H, Chae H-J, Chagin AS, Chai C-Y, Chakrabarti G, Chamilos G, Chan EY, Chan MT, Chandra D, Chandra P, Chang C-P, Chuen-Chung Chang R, Chang TY, Chatham JC, Chatterjee S, Chauhan S, Che Y, Cheetham ME, Cheluvappa R, Chen C-J, Chen G, Chen G-C, Chen G, Chen H, Chen JW, Chen J-K, Chen M, Chen M, Chen P, Chen Q, Chen Q, Chen S-D, Chen S, Chen SS-L, Chen W, Chen W-J, Chen WQ, Chen W, Chen X, Chen Y-H, Chen Y-G, Chen Y, Chen Y, Chen Y, Chen Y-J, Chen Y-Q, Chen Y, Chen Z, Chen Z, Cheng A, Cheng CH, Cheng H, Cheong H, Cherry S, Chesney J, Cheung CHA, Chevet E, Chi HC, Chi S-G, Chiacchiera F, Chiang H-L, Chiarelli R, Chiariello M, Chieppa M, Chin L-S, Chiong M, Chiu GN, Cho D-H, Cho S-G, Cho WC, Cho Y-Y, Cho Y-S, Choi AM, Choi E-J, Choi E-K, Choi J, Choi ME, Choi S-I, Chou T-F, Chouaib S, Choubey D, Choubey V, Chow K-C, Chowdhury K, Chu CT, Chuang T-H, Chun T, Chung H, Chung T, Chung Y-L, Chwae Y-J, Cianfanelli V, Ciarcia R, Ciechomska IA, Ciriolo MR, Cirone M, Claerhout S, Clague MJ, Clària J, Clarke PG, Clarke R, Clementi E, Cleyrat C, Cnop M, Coccia EM, Cocco T, Codogno P, Coers J, Cohen EE, Colecchia D, Coletto L, Coll NS, Colucci-Guyon E, Comincini S, Condello M, Cook KL, Coombs GH, Cooper CD, Mark Cooper J, Coppens I, Corasaniti MT, Corazzari M, Corbalan R, Corcelle-Termeau E, Cordero MD, Corral-Ramos C, Corti O, Cossarizza A, Costelli P, Costes S, Cotman SL, Coto-Montes A, Cottet S, Couve E, Covey LR, Cowart LA, Cox JS, Coxon FP, Coyne CB, Cragg MS, Craven RJ, Crepaldi T, Crespo JL, Criollo A, Crippa V, Cruz MT, Cuervo AM, Cuezva JM, Cui T, Cutillas PR, Czaja MJ, Czyzyk-Krzeska MF, Dagda RK, Dahmen U, Dai C, Dai W, Dai Y, Dalby KN, Valle LD, Dalmasso G, D’Amelio M, Damme M, Darfeuille-Michaud A, Dargemont C, Darley-Usmar VM, Dasarathy S, Dasgupta B, Dash S, Dass CR, Davey HM, Davids LM, Dávila D, Davis RJ, Dawson TM, Dawson VL, Daza P, de Belleroche J, de Figueiredo P, de Figueiredo RCBQ, de la Fuente J, De Martino L, De Matteis A, De Meyer GR, De Milito A, De Santi M, de Souza W, De Tata V, De Zio D, Debnath J, Dechant R, Decuypere J-P, Deegan S, Dehay B, Bello BD, Del Re DP, Delage-Mourroux R, Delbridge LM, Deldicque L, Delorme-Axford E, Deng Y, Dengjel J, Denizot M, Dent P, Der CJ, Deretic V, Derrien B, Deutsch E, Devarenne TP, Devenish RJ, Bartolomeo SD, Daniele ND, Domenico FD, Nardo AD, Paola SD, Pietro AD, Renzo LD, Antonio AD, Díaz-Araya G, Díaz-Laviada I, Diaz-Meco MT, Diaz-Nido J, Dickey CA, Dickson RC, Diederich M, Digard P, Dikic I, Dinesh-Kumar SP, Ding C, Ding W-X, Ding Z, Dini L, Distler JH, Diwan A, Djavaheri-Mergny M, Dmytruk K, Dobson RC, Doetsch V, Dokladny K, Dokudovskaya S, Donadelli M, Dong XC, Dong X, Dong Z, Donohue TM Jr, Doran KS, D’Orazi G, Dorn GW II, Dosenko V, Dridi S, Drucker L, Du J, Du L-L, Du L, du Toit A, Dua P, Duan L, Duann P, Dubey VK, Duchen MR, Duchosal MA, Duez H, Dugail I, Dumit VI, Duncan MC, Dunlop EA, Dunn WA Jr, Dupont N, Dupuis L, Durán RV, Durcan TM, Duvezin-Caubet S, Duvvuri U, Eapen V, Ebrahimi-Fakhari D, Echard A, Eckhart L, Edelstein CL, Edinger AL, Eichinger L, Eisenberg T, Eisenberg-Lerner A, Tony Eissa N, El-Deiry WS, El-Khoury V, Elazar Z, Eldar-Finkelman H, Elliott CJ, Emanuele E, Emmenegger U, Engedal N, Engelbrecht A-M, Engelender S, Enserink JM, Erdmann R, Erenpreisa J, Eri R, Eriksen JL, Erman A, Escalante R, Eskelinen E-L, Espert L, Esteban-Martínez L, Evans TJ, Fabri M, Fabrias G, Fabrizi C, Facchiano A, Færgeman NJ, Faggioni A, Fairlie WD, Fan C, Fan D, Fan J, Fang S, Fanto M, Fanzani A, Farkas T, Faure M, Favier FB, Fearnhead H, Federici M, Fei E, Felizardo TC, Feng H, Feng Y, Feng Y, Ferguson TA, Fernández ÁF, Fernandez-Barrena MG, Fernandez-Checa JC, Fernández-López A, Fernandez-Zapico ME, Feron O, Ferraro E, Veríssima Ferreira-Halder C, Fesus L, Feuer R, Fiesel FC, Filippi-Chiela EC, Filomeni G, Fimia GM, Fingert JH, Finkbeiner S, Finkel T, Fiorito F, Fisher PB, Flajolet M, Flamigni F, Florey O, Florio S, Andres Floto R, Folini M, Follo C, Fon EA, Fornai F, Fortunato F, Fraldi A, Franco R, Francois A, François A, Frankel LB, Fraser ID, Frey N, Freyssenet DG, Frezza C, Friedman SL, Frigo DE, Fu D, Fuentes JM, Fueyo J, Fujitani Y, Fujiwara Y, Fujiya M, Fukuda M, Fulda S, Fusco C, Gabryel B, Gaestel M, Gailly P, Gajewska M, Galadari S, Galili G, Galindo I, Galindo MF, Galliciotti G, Galluzzi L, Galluzzi L, Galy V, Gammoh N, Gandy S, Ganesan AK, Ganesan S, Ganley IG, Gannagé M, Gao F-B, Gao F, Gao J-X, Nannig LG, Véscovi EG, Garcia-Macía M, Garcia-Ruiz C, Garg AD, Garg PK, Gargini R, Gassen NC, Gatica D, Gatti E, Gavard J, Gavathiotis E, Ge L, Ge P, Ge S, Gean P-W, Gelmetti V, Genazzani AA, Geng J, Genschik P, Gerner L, Gestwicki JE, Gewirtz DA, Ghavami S, Ghigo E, Ghosh D, Giammarioli AM, Giampieri F, Giampietri C, Giatromanolaki A, Gibbings DJ, Gibellini L, Gibson SB, Ginet V, Giordano A, Giorgini F, Giovannetti E, Girardin SE, Gispert S, Giuliano S, Gladson CL, Glavic A, Gleave M, Godefroy N, Gogal RM Jr, Gokulan K, Goldman GH, Goletti D, Goligorsky MS, Gomes AV, Gomes LC, Gomez H, Gomez-Manzano C, Gómez-Sánchez R, Gonçalves DA, Goncu E, Gong Q, Gongora C, Gonzalez CB, Gonzalez-Alegre P, Gonzalez-Cabo P, González-Polo RA, Goping IS, Gorbea C, Gorbunov NV, Goring DR, Gorman AM, Gorski SM, Goruppi S, Goto-Yamada S, Gotor C, Gottlieb RA, Gozes I, Gozuacik D, Graba Y, Graef M, Granato GE, Grant GD, Grant S, Gravina GL, Green DR, Greenhough A, Greenwood MT, Grimaldi B, Gros F, Grose C, Groulx J-F, Gruber F, Grumati P, Grune T, Guan J-L, Guan K-L, Guerra B, Guillen C, Gulshan K, Gunst J, Guo C, Guo L, Guo M, Guo W, Guo X-G, Gust AA, Gustafsson ÅB, Gutierrez E, Gutierrez MG, Gwak H-S, Haas A, Haber JE, Hadano S, Hagedorn M, Hahn DR, Halayko AJ, Hamacher-Brady A, Hamada K, Hamai A, Hamann A, Hamasaki M, Hamer I, Hamid Q, Hammond EM, Han F, Han W, Handa JT, Hanover JA, Hansen M, Harada M, Harhaji-Trajkovic L, Wade Harper J, Harrath AH, Harris AL, Harris J, Hasler U, Hasselblatt P, Hasui K, Hawley RG, Hawley TS, He C, He CY, He F, He G, He R-R, He X-H, He Y-W, He Y-Y, Heath JK, Hébert M-J, Heinzen RA, Helgason GV, Hensel M, Henske EP, Her C, Herman PK, Hernández A, Hernandez C, Hernández-Tiedra S, Hetz C, Hiesinger PR, Higaki K, Hilfiker S, Hill BG, Hill JA, Hill WD, Hino K, Hofius D, Hofman P, Höglinger GU, Höhfeld J, Holz MK, Hong Y, Hood DA, Hoozemans JJ, Hoppe T, Hsu C, Hsu C-Y, Hsu L-C, Hu D, Hu G, Hu H-M, Hu H, Hu MC, Hu Y-C, Hu Z-W, Hua F, Hua Y, Huang C, Huang H-L, Huang K-H, Huang K-Y, Huang S, Huang S, Huang W-P, Huang Y-R, Huang Y, Huang Y, Huber TB, Huebbe P, Huh W-K, Hulmi JJ, Hur GM, Hurley JH, Husak Z, Hussain SN, Hussain S, Hwang JJ, Hwang S, Hwang TI, Ichihara A, Imai Y, Imbriano C, Inomata M, Into T, Iovane V, Iovanna JL, Iozzo RV, Ip NY, Irazoqui JE, Iribarren P, Isaka Y, Isakovic AJ, Ischiropoulos H, Isenberg JS, Ishaq M, Ishida H, Ishii I, Ishmael JE, Isidoro C, Isobe K-I, Isono E, Issazadeh-Navikas S, Itahana K, Itakura E, Ivanov AI, Iyer AKV, Izquierdo JM, Izumi Y, Izzo V, Jäättelä M, Jaber N, Jackson DJ, Jackson WT, Jacob TG, Jacques TS, Jagannath C, Jain A, Jana NR, Jang BK, Jani A, Janji B, Jannig PR, Jansson PJ, Jean S, Jendrach M, Jeon J-H, Jessen N, Jeung E-B, Jia K, Jia L, Jiang H, Jiang H, Jiang L, Jiang T, Jiang X, Jiang X, Jiang X, Jiang Y, Jiang Y, Jiménez A, Jin C, Jin H, Jin L, Jin M, Jin S, Jinwal UK, Jo E-K, Johansen T, Johnson DE, Johnson GV, Johnson JD, Jonasch E, Jones C, Joosten LA, Jordan J, Joseph A-M, Joseph B, Joubert AM, Ju D, Ju J, Juan H-F, Juenemann K, Juhász G, Jung HS, Jung JU, Jung Y-K, Jungbluth H, Justice MJ, Jutten B, Kaakoush NO, Kaarniranta K, Kaasik A, Kabuta T, Kaeffer B, Kågedal K, Kahana A, Kajimura S, Kakhlon O, Kalia M, Kalvakolanu DV, Kamada Y, Kambas K, Kaminskyy VO, Kampinga HH, Kandouz M, Kang C, Kang R, Kang T-C, Kanki T, Kanneganti T-D, Kanno H, Kanthasamy AG, Kantorow M, Kaparakis-Liaskos M, Kapuy O, Karantza V, Karim MR, Karmakar P, Kaser A, Kaushik S, Kawula T, Murat Kaynar A, Ke P-Y, Ke Z-J, Kehrl JH, Keller KE, Kemper JK, Kenworthy AK, Kepp O, Kern A, Kesari S, Kessel D, Ketteler R, do Carmo Kettelhut I, Khambu B, Khan MM, Khandelwal VK, Khare S, Kiang JG, Kiger AA, Kihara A, Kim AL, Kim CH, Kim DR, Kim D-H, Kim EK, Kim HY, Kim H-R, Kim J-S, Kim JH, Kim JC, Kim JH, Kim KW, Kim MD, Kim M-M, Kim PK, Kim SW, Kim S-Y, Kim Y-S, Kim Y, Kimchi A, Kimmelman AC, Kimura T, King JS, Kirkegaard K, Kirkin V, Kirshenbaum LA, Kishi S, Kitajima Y, Kitamoto K, Kitaoka Y, Kitazato K, Kley RA, Klimecki WT, Klinkenberg M, Klucken J, Knævelsrud H, Knecht E, Knuppertz L, Ko J-L, Kobayashi S, Koch JC, Koechlin-Ramonatxo C, Koenig U, Koh YH, Köhler K, Kohlwein SD, Koike M, Komatsu M, Kominami E, Kong D, Kong HJ, Konstantakou EG, Kopp BT, Korcsmaros T, Korhonen L, Korolchuk VI, Koshkina NV, Kou Y, Koukourakis MI, Koumenis C, Kovács AL, Kovács T, Kovacs WJ, Koya D, Kraft C, Krainc D, Kramer H, Kravic-Stevovic T, Krek W, Kretz-Remy C, Krick R, Krishnamurthy M, Kriston-Vizi J, Kroemer G, Kruer MC, Kruger R, Ktistakis NT, Kuchitsu K, Kuhn C, Kumar AP, Kumar A, Kumar A, Kumar D, Kumar D, Kumar R, Kumar S, Kundu M, Kung H-J, Kuno A, Kuo S-H, Kuret J, Kurz T, Kwok T, Kwon TK, Kwon YT, Kyrmizi I, La Spada AR, Lafont F, Lahm T, Lakkaraju A, Lam T, Lamark T, Lancel S, Landowski TH, Lane DJR, Lane JD, Lanzi C, Lapaquette P, Lapierre LR, Laporte J, Laukkarinen J, Laurie GW, Lavandero S, Lavie L, La Voie MJ, Kwan Law BY, Law HK-W, Law KB, Layfield R, Lazo PA, Cam LL, Le Roch KG, Stunff HL, Leardkamolkarn V, Lecuit M, Lee B-H, Lee C-H, Lee EF, Lee GM, Lee H-J, Lee H, Lee JK, Lee J, Lee J-H, Lee JH, Lee M, Lee M-S, Lee PJ, Lee SW, Lee S-J, Lee S-J, Lee SY, Lee SH, Lee SS, Lee S-J, Lee S, Lee Y-R, Lee YJ, Lee YH, Leeuwenburgh C, Lefort S, Legouis R, Lei J, Lei Q-Y, Leib DA, Leibowitz G, Lekli I, Lemaire SD, Lemasters JJ, Lemberg MK, Lemoine A, Leng S, Lenz G, Lenzi P, Lerman LO, Barbato DL, Leu JI-J, Leung HY, Levine B, Lewis PA, Lezoualc’h F, Li C, Li F, Li F-J, Li J, Li K, Li L, Li M, Li M, Li Q, Li R, Li S, Li W, Li X, Li Y, Lian J, Liang C, Liang Q, Liao Y, Liberal J, Liberski PP, Lie P, Lieberman AP, Lim HJ, Lim K-L, Lim K, Lima RT, Lin C-S, Lin C-F, Lin F, Lin F, Lin F-C, Lin K, Lin K-H, Lin P-H, Lin T, Lin W-W, Lin Y-S, Lin Y, Linden R, Lindholm D, Lindqvist LM, Lingor P, Linkermann A, Liotta LA, Lipinski MM, Lira VA, Lisanti MP, Liton PB, Liu B, Liu C, Liu C-F, Liu F, Liu H-J, Liu J, Liu J-J, Liu J-L, Liu K, Liu L, Liu L, Liu Q, Liu R-Y, Liu S, Liu S, Liu W, Liu X-D, Liu X, Liu X-H, Liu X, Liu X, Liu X, Liu Y, Liu Y, Liu Z, Liu Z, Liuzzi JP, Lizard G, Ljujic M, Lodhi IJ, Logue SE, Lokeshwar BL, Long YC, Lonial S, Loos B, López-Otín C, López-Vicario C, Lorente M, Lorenzi PL, Lõrincz P, Los M, Lotze MT, Lovat PE, Lu B, Lu B, Lu J, Lu Q, Lu S-M, Lu S, Lu Y, Luciano F, Luckhart S, Lucocq JM, Ludovico P, Lugea A, Lukacs NW, Lum JJ, Lund AH, Luo H, Luo J, Luo S, Luparello C, Lyons T, Ma J, Ma Y, Ma Y, Ma Z, Machado J, Machado-Santelli GM, Macian F, MacIntosh GC, MacKeigan JP, Macleod KF, MacMicking JD, MacMillan-Crow LA, Madeo F, Madesh M, Madrigal-Matute J, Maeda A, Maeda T, Maegawa G, Maellaro E, Maes H, Magariños M, Maiese K, Maiti TK, Maiuri L, Maiuri MC, Maki CG, Malli R, Malorni W, Maloyan A, Mami-Chouaib F, Man N, Mancias JD, Mandelkow E-M, Mandell MA, Manfredi AA, Manié SN, Manzoni C, Mao K, Mao Z, Mao Z-W, Marambaud P, Marconi AM, Marelja Z, Marfe G, Margeta M, Margittai E, Mari M, Mariani FV, Marin C, Marinelli S, Mariño G, Markovic I, Marquez R, Martelli AM, Martens S, Martin KR, Martin SJ, Martin S, Martin-Acebes MA, Martín-Sanz P, Martinand-Mari C, Martinet W, Martinez J, Martinez-Lopez N, Martinez-Outschoorn U, Martínez-Velázquez M, Martinez-Vicente M, Martins WK, Mashima H, Mastrianni JA, Matarese G, Matarrese P, Mateo R, Matoba S, Matsumoto N, Matsushita T, Matsuura A, Matsuzawa T, Mattson MP, Matus S, Maugeri N, Mauvezin C, Mayer A, Maysinger D, Mazzolini GD, McBrayer MK, Call KM, Cormick CM, McInerney GM, McIver SC, Kenna SM, McMahon JJ, McNeish IA, Mechta-Grigoriou F, Medema JP, Medina DL, Megyeri K, Mehrpour M, Mehta JL, Mei Y, Meier U-C, Meijer AJ, Meléndez A, Melino G, Melino S, de Melo EJT, Mena MA, Meneghini MD, Menendez JA, Menezes R, Meng L, Meng L-H, Meng S, Menghini R, Menko AS, Menna-Barreto RF, Menon MB, Meraz-Ríos MA, Merla G, Merlini L, Merlot AM, Meryk A, Meschini S, Meyer JN, Mi M-T, Miao C-Y, Micale L, Michaeli S, Michiels C, Migliaccio AR, Mihailidou AS, Mijaljica D, Mikoshiba K, Milan E, Miller-Fleming L, Mills GB, Mills IG, Minakaki G, Minassian BA, Ming X-F, Minibayeva F, Minina EA, Mintern JD, Minucci S, Miranda-Vizuete A, Mitchell CH, Miyamoto S, Miyazawa K, Mizushima N, Mnich K, Mograbi B, Mohseni S, Moita LF, Molinari M, Molinari M, Møller AB, Mollereau B, Mollinedo F, Mongillo M, Monick MM, Montagnaro S, Montell C, Moore DJ, Moore MN, Mora-Rodriguez R, Moreira PI, Morel E, Morelli MB, Moreno S, Morgan MJ, Moris A, Moriyasu Y, Morrison JL, Morrison LA, Morselli E, Moscat J, Moseley PL, Mostowy S, Motori E, Mottet D, Mottram JC, Moussa CE-H, Mpakou VE, Mukhtar H, Levy JMM, Muller S, Muñoz-Moreno R, Muñoz-Pinedo C, Münz C, Murphy ME, Murray JT, Murthy A, Mysorekar IU, Nabi IR, Nabissi M, Nader GA, Nagahara Y, Nagai Y, Nagata K, Nagelkerke A, Nagy P, Nanjundan M, Napolitano G, Naqvi NI, Nardacci R, Narendra DP, Narita M, Nascimbeni AC, Natarajan R, Navegantes LC, Nawrocki ST, Nazarko TY, Nazarko VY, Neill T, Neri LM, Netea MG, Netea-Maier RT, Neves BM, Ney PA, Nezis IP, Nguyen HT, Nguyen HP, Nicot A-S, Nilsen H, Nilsson P, Nishimura M, Nishino I, Niso-Santano M, Niu H, Nixon RA, Njar VC, Noda T, Noegel AA, Nolte EM, Norberg E, Norga KK, Noureini SK, Notomi S, Notterpek L, Nowikovsky K, Nukina N, Nürnberger T, O’Donnell VB, O’Donovan T, O’Dwyer PJ, Oehme I, Oeste CL, Ogawa M, Ogretmen B, Ogura Y, Oh YJ, Ohmuraya M, Ohshima T, Ojha R, Okamoto K, Okazaki T, Oliver FJ, Ollinger K, Olsson S, Orban DP, Ordonez P, Orhon I, Orosz L, O’Rourke EJ, Orozco H, Ortega AL, Ortona E, Osellame LD, Oshima J, Oshima S, Osiewacz HD, Otomo T, Otsu K, Ou J-HJ, Outeiro TF, Ouyang D-Y, Ouyang H, Overholtzer M, Ozbun MA, Hande Ozdinler P, Ozpolat B, Pacelli C, Paganetti P, Page G, Pages G, Pagnini U, Pajak B, Pak SC, Pakos-Zebrucka K, Pakpour N, Palková Z, Palladino F, Pallauf K, Pallet N, Palmieri M, Paludan SR, Palumbo C, Palumbo S, Pampliega O, Pan H, Pan W, Panaretakis T, Pandey A, Pantazopoulou A, Papackova Z, Papademetrio DL, Papassideri I, Papini A, Parajuli N, Pardo J, Parekh VV, Parenti G, Park J-I, Park J, Park OK, Parker R, Parlato R, Parys JB, Parzych KR, Pasquet J-M, Pasquier B, Pasumarthi KB, Patschan D, Patterson C, Pattingre S, Pattison S, Pause A, Pavenstädt H, Pavone F, Pedrozo Z, Peña FJ, Peñalva MA, Pende M, Peng J, Penna F, Penninger JM, Pensalfini A, Pepe S, Pereira GJ, Pereira PC, Pérez-de la Cruz V, Pérez-Pérez ME, Pérez-Rodríguez D, Pérez-Sala D, Perier C, Perl A, Perlmutter DH, Perrotta I, Pervaiz S, Pesonen M, Pessin JE, Peters GJ, Petersen M, Petrache I, Petrof BJ, Petrovski G, Phang JM, Piacentini M, Pierdominici M, Pierre P, Pierrefite-Carle V, Pietrocola F, Pimentel-Muiños FX, Pinar M, Pineda B, Pinkas-Kramarski R, Pinti M, Pinton P, Piperdi B, Piret JM, Platanias LC, Platta HW, Plowey ED, Pöggeler S, Poirot M, Polčic P, Poletti A, Poon AH, Popelka H, Popova B, Poprawa I, Poulose SM, Poulton J, Powers SK, Powers T, Pozuelo-Rubio M, Prak K, Prange R, Prescott M, Priault M, Prince S, Proia RL, Proikas-Cezanne T, Prokisch H, Promponas VJ, Przyklenk K, Puertollano R, Pugazhenthi S, Puglielli L, Pujol A, Puyal J, Pyeon D, Qi X, Qian W-B, Qin Z-H, Qiu Y, Qu Z, Quadrilatero J, Quinn F, Raben N, Rabinowich H, Radogna F, Ragusa MJ, Rahmani M, Raina K, Ramanadham S, Ramesh R, Rami A, Randall-Demllo S, Randow F, Rao H, Ashutosh Rao V, Rasmussen BB, Rasse TM, Ratovitski EA, Rautou P-E, Ray SK, Razani B, Reed BH, Reggiori F, Rehm M, Reichert AS, Rein T, Reiner DJ, Reits E, Ren J, Ren X, Renna M, Reusch JE, Revuelta JL, Reyes L, Rezaie AR, Richards RI, Richardson DR, Richetta C, Riehle MA, Rihn BH, Rikihisa Y, Riley BE, Rimbach G, Rippo MR, Ritis K, Rizzi F, Rizzo E, Roach PJ, Robbins J, Roberge M, Roca G, Roccheri MC, Rocha S, Rodrigues CM, Rodríguez CI, de Cordoba SR, Rodriguez-Muela N, Roelofs J, Rogov VV, Rohn TT, Rohrer B, Romanelli D, Romani L, Romano PS, Roncero MIG, Rosa JL, Rosello A, Rosen KV, Rosenstiel P, Rost-Roszkowska M, Roth KA, Roué G, Rouis M, Rouschop KM, Ruan DT, Ruano D, Rubinsztein DC, Rucker III EB, Rudich A, Rudolf E, Rudolf R, Ruegg MA, Ruiz-Roldan C, Ruparelia AA, Rusmini P, Russ DW, Russo GL, Russo G, Russo R, Rusten TE, Ryabovol V, Ryan KM, Ryter SW, Sabatini DM, Sacher M, Sachse C, Sack MN, Sadoshima J, Saftig P, Sagi-Eisenberg R, Sahni S, Saikumar P, Saito T, Saitoh T, Sakakura K, Sakoh-Nakatogawa M, Sakuraba Y, Salazar-Roa M, Salomoni P, Saluja AK, Salvaterra PM, Salvioli R, Samali A, Sanchez AM, Sánchez-Alcázar JA, Sanchez-Prieto R, Sandri M, Sanjuan MA, Santaguida S, Santambrogio L, Santoni G, Dos Santos CN, Saran S, Sardiello M, Sargent G, Sarkar P, Sarkar S, Sarrias MR, Sarwal MM, Sasakawa C, Sasaki M, Sass M, Sato K, Sato M, Satriano J, Savaraj N, Saveljeva S, Schaefer L, Schaible UE, Scharl M, Schatzl HM, Schekman R, Scheper W, Schiavi A, Schipper HM, Schmeisser H, Schmidt J, Schmitz I, Schneider BE, Marion Schneider E, Schneider JL, Schon EA, Schönenberger MJ, Schönthal AH, Schorderet DF, Schröder B, Schuck S, Schulze RJ, Schwarten M, Schwarz TL, Sciarretta S, Scotto K, Ivana Scovassi A, Screaton RA, Screen M, Seca H, Sedej S, Segatori L, Segev N, Seglen PO, Seguí-Simarro JM, Segura-Aguilar J, Seki E, Sell C, Seiliez I, Semenkovich CF, Semenza GL, Sen U, Serra AL, Serrano-Puebla A, Sesaki H, Setoguchi T, Settembre C, Shacka JJ, Shajahan-Haq AN, Shapiro IM, Sharma S, She H, James Shen C-K, Shen C-C, Shen H-M, Shen S, Shen W, Sheng R, Sheng X, Sheng Z-H, Shepherd TG, Shi J, Shi Q, Shi Q, Shi Y, Shibutani S, Shibuya K, Shidoji Y, Shieh J-J, Shih C-M, Shimada Y, Shimizu S, Shin DW, Shinohara ML, Shintani M, Shintani T, Shioi T, Shirabe K, Shiri-Sverdlov R, Shirihai O, Shore GC, Shu C-W, Shukla D, Sibirny AA, Sica V, Sigurdson CJ, Sigurdsson EM, Sijwali PS, Sikorska B, Silveira WA, Silvente-Poirot S, Silverman GA, Simak J, Simmet T, Simon AK, Simon H-U, Simone C, Simons M, Simonsen A, Singh R, Singh SV, Singh SK, Sinha D, Sinha S, Sinicrope FA, Sirko A, Sirohi K, Sishi BJ, Sittler A, Siu PM, Sivridis E, Skwarska A, Slack R, Slaninová I, Slavov N, Smaili SS, Smalley KS, Smith DR, Soenen SJ, Soleimanpour SA, Solhaug A, Somasundaram K, Son JH, Sonawane A, Song C, Song F, Song HK, Song J-X, Song W, Soo KY, Sood AK, Soong TW, Soontornniyomkij V, Sorice M, Sotgia F, Soto-Pantoja DR, Sotthibundhu A, Sousa MJ, Spaink HP, Span PN, Spang A, Sparks JD, Speck PG, Spector SA, Spies CD, Springer W, Clair DS, Stacchiotti A, Staels B, Stang MT, Starczynowski DT, Starokadomskyy P, Steegborn C, Steele JW, Stefanis L, Steffan J, Stellrecht CM, Stenmark H, Stepkowski TM, Stern ST, Stevens C, Stockwell BR, Stoka V, Storchova Z, Stork B, Stratoulias V, Stravopodis DJ, Strnad P, Strohecker AM, Ström A-L, Stromhaug P, Stulik J, Su Y-X, Su Z, Subauste CS, Subramaniam S, Sue CM, Suh SW, Sui X, Sukseree S, Sulzer D, Sun F-L, Sun J, Sun J, Sun S-Y, Sun Y, Sun Y, Sun Y, Sundaramoorthy V, Sung J, Suzuki H, Suzuki K, Suzuki N, Suzuki T, Suzuki YJ, Swanson MS, Swanton C, Swärd K, Swarup G, Sweeney ST, Sylvester PW, Szatmari Z, Szegezdi E, Szlosarek PW, Taegtmeyer H, Tafani M, Taillebourg E, Tait SW, Takacs-Vellai K, Takahashi Y, Takáts S, Takemura G, Takigawa N, Talbot NJ, Tamagno E, Tamburini J, Tan C-P, Tan L, Tan ML, Tan M, Tan Y-J, Tanaka K, Tanaka M, Tang D, Tang D, Tang G, Tanida I, Tanji K, Tannous BA, Tapia JA, Tasset-Cuevas I, Tatar M, Tavassoly I, Tavernarakis N, Taylor A, Taylor GS, Taylor GA, Paul Taylor J, Taylor MJ, Tchetina EV, Tee AR, Teixeira-Clerc F, Telang S, Tencomnao T, Teng B-B, Teng R-J, Terro F, Tettamanti G, Theiss AL, Theron AE, Thomas KJ, Thomé MP, Thomes PG, Thorburn A, Thorner J, Thum T, Thumm M, Thurston TL, Tian L, Till A, Ting JP-Y, Titorenko VI, Toker L, Toldo S, Tooze SA, Topisirovic I, Torgersen ML, Torosantucci L, Torriglia A, Torrisi MR, Tournier C, Towns R, Trajkovic V, Travassos LH, Triola G, Tripathi DN, Trisciuoglio D, Troncoso R, Trougakos IP, Truttmann AC, Tsai K-J, Tschan MP, Tseng Y-H, Tsukuba T, Tsung A, Tsvetkov AS, Tu S, Tuan H-Y, Tucci M, Tumbarello DA, Turk B, Turk V, Turner RF, Tveita AA, Tyagi SC, Ubukata M, Uchiyama Y, Udelnow A, Ueno T, Umekawa M, Umemiya-Shirafuji R, Underwood BR, Ungermann C, Ureshino RP, Ushioda R, Uversky VN, Uzcátegui NL, Vaccari T, Vaccaro MI, Váchová L, Vakifahmetoglu-Norberg H, Valdor R, Valente EM, Vallette F, Valverde AM, Van den Berghe G, Van Den Bosch L, van den Brink GR, van der Goot FG, van der Klei IJ, van der Laan LJ, van Doorn WG, van Egmond M, van Golen KL, Van Kaer L, van Lookeren Campagne M, Vandenabeele P, Vandenberghe W, Vanhorebeek I, Varela-Nieto I, Helena Vasconcelos M, Vasko R, Vavvas DG, Vega-Naredo I, Velasco G, Velentzas AD, Velentzas PD, Vellai T, Vellenga E, Vendelbo MH, Venkatachalam K, Ventura N, Ventura S, Veras PS, Verdier M, Vertessy BG, Viale A, Vidal M, Vieira HLA, Vierstra RD, Vigneswaran N, Vij N, Vila M, Villar M, Villar VH, Villarroya J, Vindis C, Viola G, Viscomi MT, Vitale G, Vogl DT, Voitsekhovskaja OV, von Haefen C, von Schwarzenberg K, Voth DE, Vouret-Craviari V, Vuori K, Vyas JM, Waeber C, Walker CL, Walker MJ, Walter J, Wan L, Wan X, Wang B, Wang C, Wang C-Y, Wang C, Wang C, Wang C, Wang D, Wang F, Wang F, Wang G, Wang H-J, Wang H, Wang H-G, Wang H, Wang H-D, Wang J, Wang J, Wang M, Wang M-Q, Wang P-Y, Wang P, Wang RC, Wang S, Wang T-F, Wang X, Wang X-J, Wang X-W, Wang X, Wang X, Wang Y, Wang Y, Wang Y, Wang Y-J, Wang Y, Wang Y, Wang YT, Wang Y, Wang Z-N, Wappner P, Ward C, McVey Ward D, Warnes G, Watada H, Watanabe Y, Watase K, Weaver TE, Weekes CD, Wei J, Weide T, Weihl CC, Weindl G, Weis SN, Wen L, Wen X, Wen Y, Westermann B, Weyand CM, White AR, White E, Lindsay Whitton J, Whitworth AJ, Wiels J, Wild F, Wildenberg ME, Wileman T, Wilkinson DS, Wilkinson S, Willbold D, Williams C, Williams K, Williamson PR, Winklhofer KF, Witkin SS, Wohlgemuth SE, Wollert T, Wolvetang EJ, Wong E, Wong GW, Wong RW, Wong VKW, Woodcock EA, Wright KL, Wu C, Wu D, Wu GS, Wu J, Wu J, Wu M, Wu M, Wu S, Wu WK, Wu Y, Wu Z, Xavier CP, Xavier RJ, Xia G-X, Xia T, Xia W, Xia Y, Xiao H, Xiao J, Xiao S, Xiao W, Xie C-M, Xie Z, Xie Z, Xilouri M, Xiong Y, Xu C, Xu C, Xu F, Xu H, Xu H, Xu J, Xu J, Xu J, Xu L, Xu X, Xu Y, Xu Y, Xu Z-X, Xu Z, Xue Y, Yamada T, Yamamoto A, Yamanaka K, Yamashina S, Yamashiro S, Yan B, Yan B, Yan X, Yan Z, Yanagi Y, Yang D-S, Yang J-M, Yang L, Yang M, Yang P-M, Yang P, Yang Q, Yang W, Yang WY, Yang X, Yang Y, Yang Y, Yang Z, Yang Z, Yao M-C, Yao PJ, Yao X, Yao Z, Yao Z, Yasui LS, Ye M, Yedvobnick B, Yeganeh B, Yeh ES, Yeyati PL, Yi F, Yi L, Yin X-M, Yip CK, Yoo Y-M, Yoo YH, Yoon S-Y, Yoshida K-I, Yoshimori T, Young KH, Yu H, Yu JJ, Yu J-T, Yu J, Yu L, Yu WH, Yu X-F, Yu Z, Yuan J, Yuan Z-M, Yue BY, Yue J, Yue Z, Zacks DN, Zacksenhaus E, Zaffaroni N, Zaglia T, Zakeri Z, Zecchini V, Zeng J, Zeng M, Zeng Q, Zervos AS, Zhang DD, Zhang F, Zhang G, Zhang G-C, Zhang H, Zhang H, Zhang H, Zhang H, Zhang J, Zhang J, Zhang J, Zhang J, Zhang J-P, Zhang L, Zhang L, Zhang L, Zhang L, Zhang M-Y, Zhang X, Zhang XD, Zhang Y, Zhang Y, Zhang Y, Zhang Y, Zhang Y, Zhao M, Zhao W-L, Zhao X, Zhao YG, Zhao Y, Zhao Y, Zhao Y-X, Zhao Z, Zhao ZJ, Zheng D, Zheng X-L, Zheng X, Zhivotovsky B, Zhong Q, Zhou G-Z, Zhou G, Zhou H, Zhou S-F, Zhou X-J, Zhu H, Zhu H, Zhu W-G, Zhu W, Zhu X-F, Zhu Y, Zhuang S-M, Zhuang X, Ziparo E, Zois CE, Zoladek T, Zong W-X, Zorzano A, Zughaier SM, Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levine B, Kroemer G, Biological functions of autophagy genes: A disease perspective. Cell 176, 11–42 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi AM, Ryter SW, Levine B, Autophagy in human health and disease. N. Engl. J. Med 368, 651–662 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Ebrahimi-Fakhari D, Saffari A, Wahlster L, Lu J, Byrne S, Hoffmann GF, Jungbluth H, Sahin M, Congenital disorders of autophagy: An emerging novel class of inborn errors of neuro-metabolism. Brain 139, 317–337 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jang J, Wang Y, Lalli MA, Guzman E, Godshalk SE, Zhou H, Kosik KS, Primary cilium-autophagy-Nrf2 (PAN) Axis activation commits human embryonic stem cells to a neuroectoderm fate. Cell 165, 410–420 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Wang G, Chen EN, Liang C, Liang J, Gao LR, Chuai M, Munsterberg A, Bao Y, Cao L, Yang X, Atg7-mediated autophagy is involved in the neural crest cell generation in chick embryo. Mol. Neurobiol 55, 3523–3536 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Wang XY, Li S, Wang G, Ma ZL, Chuai M, Cao L, Yang X, High glucose environment inhibits cranial neural crest survival by activating excessive autophagy in the chick embryo. Sci. Rep 5, 18321 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fukuda T, Scott G, Komatsu Y, Araya R, Kawano M, Ray MK, Yamada M, Mishina Y, Generation of a mouse with conditionally activated signaling through the BMP receptor, ALK2. Genesis 44, 159–167 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Yamauchi Y, Abe K, Mantani A, Hitoshi Y, Suzuki M, Osuzu F, Kuratani S, Yamamura K, A novel transgenic technique that allows specific marking of the neural crest cell lineage in mice. Dev. Biol 212, 191–203 (1999). [DOI] [PubMed] [Google Scholar]
- 33.de Crombrugghe B, Lefebvre V, Behringer RR, Bi W, Murakami S, Huang W, Transcriptional mechanisms of chondrocyte differentiation. Matrix Biol. 19, 389–394 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Mori-Akiyama Y, Akiyama H, Rowitch DH, de Crombrugghe B, Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc. Natl. Acad. Sci. U.S.A 100, 9360–9365 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zalc A, Rattenbach R, Aurade F, Cadot B, Relaix F, Pax3 and Pax7 play essential safeguard functions against environmental stress-induced birth defects. Dev. Cell 33, 56–66 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Zhao H, Bringas P Jr., Chai Y, An in vitro model for characterizing the post-migratory cranial neural crest cells of the first branchial arch. Dev. Dyn 235, 1433–1440 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.John N, Cinelli P, Wegner M, Sommer L, Transforming growth factor β-mediated Sox10 suppression controls mesenchymal progenitor generation in neural crest stem cells. Stem Cells 29, 689–699 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Bianco P, Robey PG, Skeletal stem cells. Development 142, 1023–1027 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao H, Feng J, Ho TV, Grimes W, Urata M, Chai Y, The suture provides a niche for mesenchymal stem cells of craniofacial bones. Nat. Cell Biol 17, s386–s396 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nie X, Zheng J, Ricupero CL, He L, Jiao K, Mao JJ, mTOR acts as a pivotal signaling hub for neural crest cells during craniofacial development. PLOS Genet. 14, e1007491 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y, Whetstone HC, Youn A, Nadesan P, Chow EC, Lin AC, Alman BA, β-Catenin signaling pathway is crucial for bone morphogenetic protein 2 to induce new bone formation. J. Biol. Chem 282, 526–533 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Kamiya N, Kaartinen VM, Mishina Y, Loss-of-function of ACVR1 in osteoblasts increases bone mass and activates canonical Wnt signaling through suppression of Wnt inhibitors SOST and DKK1. Biochem. Biophys. Res. Commun 414, 326–330 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, Hild M, Shi X, Wilson CJ, Mickanin C, Myer V, Fazal A, Tomlinson R, Serluca F, Shao W, Cheng H, Shultz M, Rau C, Schirle M, Schlegl J, Ghidelli S, Fawell S, Lu C, Curtis D, Kirschner MW, Lengauer C, Finan PM, Tallarico JA, Bouwmeester T, Porter JA, Bauer A, Cong F, Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461, 614–620 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Day TF, Guo X, Garrett-Beal L, Yang Y, Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 8, 739–750 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Hill TP, Spater D, Taketo MM, Birchmeier W, Hartmann C, Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 8, 727–738 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Stamos JL, Weis WI, The β-catenin destruction complex. Cold Spring Harb. Perspect. Biol 5, a007898 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jia Z, Wang J, Wang W, Tian Y, XiangWei W, Chen P, Ma K, Zhou C, Autophagy eliminates cytoplasmic β-catenin and NICD to promote the cardiac differentiation of P19CL6 cells. Cell. Signal 26, 2299–2305 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Wang BS, Yang Y, Lu HZ, Shang L, Zhang Y, Hao JJ, Shi ZZ, Wang XM, Liu YZ, Zhan QM, Jia XM, Wang MR, Inhibition of atypical protein kinase Cι induces apoptosis through autophagic degradation of β-catenin in esophageal cancer cells. Mol. Carcinog 53, 514–525 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Petherick KJ, Williams AC, Lane JD, Ordonez-Moran P, Huelsken J, Collard TJ, Smartt HJ, Batson J, Malik K, Paraskeva C, Greenhough A, Autolysosomal β-catenin degradation regulates Wnt-autophagy-p62 crosstalk. EMBO J. 32, 1903–1916 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, Wilkins AD, Sun Q, Pallauf K, MacDuff D, Huerta C, Virgin HW, Helms JB, Eerland R, Tooze SA, Xavier R, Lenschow DJ, Yamamoto A, King D, Lichtarge O, Grishin NV, Spector SA, Kaloyanova DV, Levine B, Identification of a candidate therapeutic autophagy-inducing peptide. Nature 494, 201–206 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N, Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Gao C, Cao W, Bao L, Zuo W, Xie G, Cai T, Fu W, Zhang J, Wu W, Zhang X, Chen YG, Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat. Cell Biol 12, 781–790 (2010). [DOI] [PubMed] [Google Scholar]
- 53.Salomon D, Sacco PA, Roy SG, Simcha I, Johnson KR, Wheelock MJ, Ben-Ze’ev A, Regulation of beta-catenin levels and localization by overexpression of plakoglobin and inhibition of the ubiquitin-proteasome system. J. Cell Biol 139, 1325–1335 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshida T, Shiraishi T, Nakata S, Horinaka M, Wakada M, Mizutani Y, Miki T, Sakai T, Proteasome inhibitor MG132 induces death receptor 5 through CCAAT/enhancer-binding protein homologous protein. Cancer Res. 65, 5662–5667 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Simoes-Costa M, Bronner ME, Reprogramming of avian neural crest axial identity and cell fate. Science 352, 1570–1573 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Soldatov R, Kaucka M, Kastriti ME, Petersen J, Chontorotzea T, Englmaier L, Akkuratova N, Yang Y, Haring M, Dyachuk V, Bock C, Farlik M, Piacentino ML, Boismoreau F, Hilscher MM, Yokota C, Qian X, Nilsson M, Bronner ME, Croci L, Hsiao WY, Guertin DA, Brunet JF, Consalez GG, Ernfors P, Fried K, Kharchenko PV, Adameyko I, Spatiotemporal structure of cell fate decisions in murine neural crest. Science 364, eaas9536 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Hayano S, Komatsu Y, Pan H, Mishina Y, Augmented BMP signaling in the neural crest inhibits nasal cartilage morphogenesis by inducing p53-mediated apoptosis. Development 142, 1357–1367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM, Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc. Natl. Acad. Sci. U.S.A 102, 5062–5067 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu X, Hayano S, Pan H, Inagaki M, Ninomiya-Tsuji J, Sun H, Mishina Y, Compound mutations in Bmpr1a and Tak1 synergize facial deformities via increased cell death. Genesis 56, e23093 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Laplante M, Sabatini DM, mTOR signaling in growth control and disease. Cell 149, 274–293 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kramer K, Yang J, Swanson WB, Hayano S, Toda M, Pan H, Kim JK, Krebsbach PH, Mishina Y, Rapamycin rescues BMP mediated midline craniosynostosis phenotype through reduction of mTOR signaling in a mouse model. Genesis 56, e23220 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hino K, Horigome K, Nishio M, Komura S, Nagata S, Zhao C, Jin Y, Kawakami K, Yamada Y, Ohta A, Toguchida J, Ikeya M, Activin-A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J. Clin. Invest 127, 3339–3352 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R, Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128, 1253–1264 (2001). [DOI] [PubMed] [Google Scholar]
- 64.Fischer L, Boland G, Tuan RS, Wnt-3A enhances bone morphogenetic protein-2-mediated chondrogenesis of murine C3H10T1/2 mesenchymal cells. J. Biol. Chem 277, 30870–30878 (2002). [DOI] [PubMed] [Google Scholar]
- 65.Papathanasiou I, Malizos KN, Tsezou A, Bone morphogenetic protein-2-induced Wnt/beta-catenin signaling pathway activation through enhanced low-density-lipoprotein receptor-related protein 5 catabolic activity contributes to hypertrophy in osteoarthritic chondrocytes. Arthritis Res. Ther 14, R82 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fu HD, Wang HR, Li DH, BMP-7 accelerates the differentiation of rabbit mesenchymal stem cells into cartilage through the Wnt/β-catenin pathway. Exp. Ther. Med 14, 5424–5428 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kamiya N, Kobayashi T, Mochida Y, Yu PB, Yamauchi M, Kronenberg HM, Mishina Y, Wnt inhibitors Dkk1 and Sost are downstream targets of BMP signaling through the type IA receptor (BMPRIA) in osteoblasts. J. Bone Miner. Res 25, 200–210 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Y, Sun JC, Wang HB, Xu XM, Kong QJ, Wang YJ, Zheng B, Shi GD, Shi JG, ACVR1-knockout promotes osteogenic differentiation by activating the Wnt signaling pathway in mice. J. Cell. Biochem 120, 8185–8194 (2018). [DOI] [PubMed] [Google Scholar]
- 69.Boya P, Codogno P, Rodriguez-Muela N, Autophagy in stem cells: Repair, remodelling and metabolic reprogramming. Development 145, dev146506 (2018). [DOI] [PubMed] [Google Scholar]
- 70.Kamiya N, Ye L, Kobayashi T, Mochida Y, Yamauchi M, Kronenberg HM, Feng JQ, Mishina Y, BMP signaling negatively regulates bone mass through sclerostin by inhibiting the canonical Wnt pathway. Development 135, 3801–3811 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang X, Lin Q, Zhang T, Wang X, Cheng K, Gao M, Xia P, Li X, Low-intensity pulsed ultrasound promotes chondrogenesis of mesenchymal stem cells via regulation of autophagy. Stem Cell Res. Ther 10, 41 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Niu J, Yan T, Guo W, Wang W, Zhao Z, Insight into the role of autophagy in osteosarcoma and its therapeutic implication. Front. Oncol 9, 1232 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Boeuf S, Bovee JV, Lehner B, van den Akker B, van Ruler M, Cleton-Jansen AM, Richter W, BMP and TGFbeta pathways in human central chondrosarcoma: Enhanced endoglin and Smad 1 signaling in high grade tumors. BMC Cancer 12, 488 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim M, Choe S, BMPs and their clinical potentials. BMB Rep. 44, 619–634 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yumoto K, Thomas PS, Lane J, Matsuzaki K, Inagaki M, Ninomiya-Tsuji J, Scott GJ, Ray MK, Ishii M, Maxson R, Mishina Y, Kaartinen V, TGF-β-activated kinase 1 (Tak1) mediates agonist-induced Smad activation and linker region phosphorylation in embryonic craniofacial neural crest-derived cells. J. Biol. Chem 288, 13467–13480 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.