

Abstract
The ability of antibodies to recognize their target antigens with high specificity is fundamental to their natural function. Nevertheless, therapeutic antibodies display variable and difficult-to-predict levels of non-specific and self-interactions that can lead to various drug development challenges, including antibody aggregation, abnormally high viscosity and rapid antibody clearance. Here we report a method for predicting the overall specificity of antibodies in terms of their relative risk for displaying high levels of non-specific and/or self-interactions at physiological conditions. We find that individual and combined sets of chemical rules that limit the maximum and minimum numbers of certain solvent-exposed residues in antibody variable regions are strong predictors of specificity for large panels of preclinical and clinical-stage antibodies. We also demonstrate how the chemical rules can be used to identify sites that mediate non-specific interactions in suboptimal antibodies and guide the design of targeted sub-libraries that yield variants with high antibody specificity. These findings can be readily used to improve the selection and engineering of antibodies with drug-like specificity.
Keywords: polyspecificity, pharmacokinetics, solubility, aggregation, viscosity, developability
Graphical Abstract
INTRODUCTION
Monoclonal antibodies (mAbs) are one of the most promising classes of therapeutics because of their many attractive properties, including their high affinity and specificity for target molecules and their ability to recruit potent effector functions after target recognition.1 The generation and affinity maturation of mAbs involve introducing significant sequence variation in their six binding loops (complementarity-determining regions, CDRs) and, to a lesser extent, in their framework regions. Although maximal antibody sequence diversity is unimaginably large, the fraction of antibody sequences that give rise to mAbs with drug-like properties is expected to be dramatically lower. Natural filtering mechanisms used by the immune system eliminate many undesirable mAb sequences during antibody generation.2 However, antibodies generated by the immune system (as well as those discovered using in vitro display methods) are not optimized for the extreme requirements of many therapeutic applications.3, 4 Indeed, several examples of poor physicochemical properties of mAbs have been reported that are linked to specific antibody sequences.5–13
Recent work suggests that high specificity is a key indicator of drug-like antibodies.14 Out of twelve biophysical assays of non-specific interactions, self-association, hydrophobicity and aggregation that were used to profile 137 clinical-stage antibodies, only assays that measured antibody non-specific interactions (three assays) and self-interactions (two assays) were able to identify approved antibody drugs as having superior biophysical properties relative to antibodies in phase 2 and 3 clinical trials. Nevertheless, it remains extremely challenging to identify the molecular determinants of antibody specificity for multiple reasons. First, antibody specificity is a relative concept that is dependent on the type of methods used to measure non-specific and self-interactions. Therefore, analysis of the molecular determinants of antibody specificity based on data obtained using a single type of assay may lead to conclusions that are not generally applicable to other types of antibody specificity measurements. Second, there has not been sufficient data available until recently14,15 for detailed statistical analysis of the molecular determinants of antibody specificity. These new comprehensive data sets provide several different types of specificity measurements for diverse panels of antibodies, which could enable a more holistic analysis of the molecular determinants of antibody specificity than has been previously possible.
In this work, we have sought to develop chemical (amino acid composition) rules that are able to identify mAbs with high, drug-like specificity and reduced risk of displaying high levels of non-specific and self-interactions at physiological conditions. Our approach is to first segregate clinical-stage mAbs into two groups – namely those with low and high specificity – based on several different types of specificity measurements. Next, we have sought to develop chemical rules based on physicochemical properties of different regions in antibody variable fragments that are able to selectively identify mAbs with low specificity. This approach seeks to identify the most important chemical properties of the variable regions of antibodies linked to specificity in order to improve the identification and engineering of drug-like antibodies. Here we report individual and combined sets of chemical rules that selectively identify mAbs with high specificity based only on antibody sequences and predicted site-specific solvent accessibilities, and we apply these rules to guide the re-engineering of a suboptimal mAb to identify mutations that increase antibody specificity.
EXPERIMENTAL SECTION
Antibody sequence and biophysical data.
The amino acid sequences of the variable (VH and VL) regions of 137 clinical-stage antibodies and their corresponding measurements of non-specific and self-interactions (Table S1) were obtained from a previous publication.14 The amino acid sequences of a panel of preclinical antibodies were provided by Adimab (Table S2).15 The relative solvent accessibilities of the clinical (Table S3) and preclinical (Table S4) antibodies were calculated using a Random Forest Regression method that was trained on over 900 antibodies in the Protein Data Bank.16 The maximum and minimum counts for each type of amino acid present in the CDRs and different regions of antibody variable regions (Tables S1 and S2) weighted by their solvent accessible surface areas (Tables S3 and S4) for the clinical-stage antibodies are reported in Table S5. The similarities of mAbs relative to those in the training sets are reported as the difference between 100% and the percentage of the 286 specified amino acid limits that are violated (Table S5). The preclinical mAbs (Table S2) have greater than 99% similarity relative to the clinical-stage mAbs (Table S1). The CDRs were defined using a combination of Chothia and Kabat numbering, and heavy chain CDR3 was defined to also include two additional N-terminal residues (as reported, for example, in Tables S1 and S2). The theoretical net charges of various antibody regions were calculated at pH 7.4 by assigning charges of +1 for Lys and Arg, +0.1 for His, and −1 for Asp and Glu.
Chemical rules for identifying antibodies with low specificity.
Rules for describing antibody specificity were calculated in MATLAB using the procedure described below and in the Supplemental Methods. First, the specificities of clinical-stage mAbs were experimentally evaluated using five specificity tests that set maximum limits on the levels of non-specific interactions [>4.3 signal/background for baculovirus particle (BVP) binding, >0.27 for polyspecificity reagent (PSR) binding and >1.9 signal/background for ELISA] and self-interactions [>11.8 nm for affinity-capture self-interaction nanoparticle spectroscopy (AC-SINS) and >0.01 response units for clone self-interaction by biolayer interferometry (CSI)].14 Second, each antibody was assigned to one of two groups based on its number of physical flags, as defined by the number of times an antibody exceeded the five maximum limits for non-specific and self-interactions. One group with <2 physical flags is defined as the high specificity group and a second group with ≥2 physical flags is defined as the low specificity group. The clinical-stage mAbs were assigned to the two groups based on their value of each individual biophysical measurement (BVP, PSR, ELISA, AC-SINS, and CSI) relative to the limits described above. The preclinical mAbs15 were also assigned to two groups with high specificity (≤0.27 for PSR) or low specificity (>0.27 for PSR). Third, rules for the counts of amino acids in the CDRs and variable regions, weighted by their relative solvent exposures, were evaluated using the summed values (increments of 0.1) for various combinations of specific residues that spanned the values observed in the clinical-stage antibodies (as described in the Results section). An amino acid was considered solvent accessible if its relative solvent exposure was ≥10% (otherwise it was excluded from analysis), and glycine was assumed to be fully exposed.
The chemical rules were generated using threefold cross-validation methods and were required to meet a number of constraints (as described in detail in the Supplemental Methods). Briefly, the clinical-stage mAbs (137) were split into training (80%) and test (20%) sets in ten different ways using stratified sampling (Table S6). The training sets were further divided into three partitions (folds), two of which were used for training and the other for validation. Individual rules were required to satisfy the constraints summarized in Table S7. Finally, the rules were required to be observed in each of the ten 80/20% splits, although different values for the rules were allowed for each split.
The significance of each rule for selectively flagging antibodies with low specificity was assessed using adjusted accuracies and 2×2 contingency tables (Fisher’s exact test) for evaluating p-values. Adjusted accuracy was calculated by equally weighting the true and false positives with the true and false negatives.
Combined rules for enhancing the identification of mAbs with low specificity.
Sets of rules were generated by combining single rules together (up to six single rules per combined set were evaluated), as explained in the Results section. Each mAb was considered to have low specificity if flagged by four or five rules (as defined on a case-by-case basis). Sets of rules in the first round of analysis were only accepted if they met the constraints summarized in Table S7. Finally, the combined rules (with the same values for each rule) were required to be observed in each of the ten 80/20% splits. The best sets of combined rules in the first round of analysis were identified as those with the lowest coefficients of variation for the average validation accuracy (ten 80/20% splits). This process was repeated again for mAbs that were not flagged as polyspecific in the first round of analysis, as described in the Supplemental Methods. Briefly, single rules were first generated using similar constraints as those used in the first round of analysis (Table S7). Next, combined sets of rules using the best set of rules from the first round of analysis and up to six additional rules were required to satisfy the constraints summarized in Table S7. Finally, the combined rules (with the same values for each rule) were required to be observed in each of the ten 80/20% splits. The best sets of combined rules (from the first and second rounds) were identified as those with the lowest coefficients of variation for the average validation accuracy (ten 80/20% splits).
Measurements of antibody non-specific and colloidal interactions.
mAb variants (39 IgGs) with sequence differences in their frameworks and CDRs were expressed as IgG1 antibodies in CHO-3E7 cells (L-11992, National Research Council Canada) and purified via Protein A chromatography. Preparative size-exclusion chromatography was also performed (when necessary) to reduce the aggregate content below 10%.
The levels of antibody non-specific binding were evaluated using an ELISA method reported previously14 with minor modifications. Immulon 2HB plates (3655TS, Thermo Fisher Scientific) were coated separately with six non-antigens as reported previously except that insulin was immobilized at 0.2 mg/mL (1 h at 37 °C). The plates were washed three times (0.2 mL/well) using PBST (PBS with 0.05% Tween 20) and were not blocked. Next, each mAb (1 μM in PBST) was added to the wells for 1 h. After washing the wells three times (0.2 mL/well of PBST per wash), the secondary antibody (HRP conjugated goat anti-human IgG antibody, 10 ng/mL; 109-035-008, Jackson Immuno Research) was added (1 h). Finally, after removal of unbound secondary antibody, TMB substrate (TMBS-1000-01, Surmodics) was added and the plates were developed (5 min) before quenching with 2 M sulfuric acid. The volume of the solutions added to the wells was 50 μL/well unless otherwise specified. The absorbance values were evaluated at 450 nm using a Biotek Synergy 2 plate reader, and the signal over background was calculated using background values evaluated without mAb and with all other reagents.
The levels of antibody self-interactions were measured for the mAbs using affinity-capture self-interaction nanoparticle spectroscopy (AC-SINS), as reported previously.17 Briefly, the nanoparticle conjugates were prepared by first adsorbing goat anti-human Fc polyclonal antibody and then co-adsorbing human mAbs and human polyclonal antibodies at different ratios (fixed total concentration of 20 μg/mL of human antibody). The reported plasmon shifts are averages of those evaluated at three different percentages of human mAbs (5, 15 and 25%). The control values used to calculate the plasmon shifts were those for 0% human mAb (100% human polyclonal antibody). The absorbance spectra used to evaluate the plasmon shifts were measured using a Biotek Synergy 2 plate reader.
Antibody sub-library design and sorting.
Sites for mutation in the variable heavy (VH) region of emibetuzumab were identified using the combined chemical rules. In particular, sites in the CDRs were targeted if they were i) flagged by the maximum limits (rules 1–6 in the combined rules), ii) hydrophobic or positively charged, iii) solvent exposed (>10% SASA) and iv) relatively uncommon at a given antibody site (<50%) in tens of thousands of human antibodies.18 The last requirement that the wild-type residue must not be highly conserved aims to avoid mutations at sites that are critical to antibody folding and/or stability. The resulting antibody library was generated with mutations at eight sites in VH (Y33, R50, R54, R55, G56, A95, W97 and Y102) with the goal of reducing the number of chemical flags in the variable regions of emibetuzumab. For each mutation site, degenerate codons were designed to sample the wild-type residue as well as at least one negatively charged residue and one polar residue, as well as up to three additional residues with similar properties relative to the wild-type. For example, a degenerate codon at Y33 in VH was used to sample Tyr (wild type), Phe (aromatic and hydrophobic), Val and Ala (hydrophobic), Ser (polar) and Asp (negatively charged). The total library size was 106 variants, the library design is summarized in Figure S1, and the single-chain Fab (scFab) library was constructed and displayed on the surface of yeast, as described previously.19
The initial rounds of library sorting were conducted by incubating 109 (round 1) or 107 (round 2) surface-displaying yeast with 107 Dynabeads (Protein A, 10002D; Thermo Fisher Scientific) saturated with antigen (hepatocyte growth factor receptor as an Fc fusion protein, HGFR-Fc; MET-H5256, Acro Biosciences) in PBSB (PBS with 1 mg/mL BSA) and 1% milk for 3 h at room temperature. The final round of sorting (round 3) was completed via FACS (MoFlo Astrios, Beckman-Coulter) using 107 cells following incubation with soluble antigen or polyspecificity reagents. Ovalbumin (Sigma, A5503) and soluble membrane proteins isolated from CHO cells (polyspecificity reagent or PSR) were biotinylated using Sulfo-NHS-LC-Biotin (Pierce, P121335; Thermo Fisher Scientific) and HGFR-Fc was used as purchased. Cells were incubated with ovalbumin (260 μg/mL), PSR (130 μg/mL), or HGFR-Fc (1 nM with 1% milk) for 3 h at room temperature (ovalbumin, HGFR-Fc) or 20 min on ice (PSR) in PBSB with an anti-myc tag mouse mAb (1:1000 dilution; 2276S, Cell Signaling Technologies). After one wash with PBSB, cells were incubated with secondary reagents to detect scFab display (1:100 goat anti-mouse Alexa Fluor 488; A11001, Life Technologies) and binding (1:1000 streptavidin Alexa Fluor 647, S32357, Life Technologies for ovalbumin and PSR; 1:300 goat anti-human Fc Alexa Fluor 647, 109605098, Jackson Immuno Research Labs for HGFR-Fc). Finally, the cells were washed with PBSB and sorted for positive display and non-binding to ovalbumin and PSR or binding to HGFR-Fc.
Deep sequencing and data analysis.
The sorted antibody library samples were evaluated using deep sequencing by extracting the scFab plasmids from yeast using the Zymoprep Yeast Plasmid Miniprep II Kit (D2004; Zymo Research). The VH region of the scFab gene was amplified via two-step PCR using Q5 polymerase (M0491; New England Biolabs). The first reaction was performed using primers that were complementary to the VH domain in addition to Illumina adapter sequences and barcodes (see Supplemental Methods for more detail). The PCR product was gel purified (1% agarose) and isolated using a QIAquick Gel Extraction Kit (28704; Qiagen, Germantown, MD). The second reaction used 2 μL of the purified PCR product with primers identical to the Illumina adapter sequences, and was also gel purified following the manufacturer’s recommendations. Concentrations of each sample were determined using a Qubit 4 Fluorometer (Q33240; Waltham, MA) and pooled together at an equimolar ratio. The pooled samples were evaluated using deep sequencing (Illumina MiSeq in a 300 bp paired-end sequencing reaction). The detailed data analysis is summarized in the Supplemental Methods.
Next, the deep sequencing data were analyzed to identify antibody variants observed in four library samples in two different biological repeats for the third round of sorting, namely the i) input library and the samples sorted for ii) negative ovalbumin binding (OVA-), iii) negative PSR binding (PSR-), and iv) positive HGFR-Fc binding. From these two repeats, 3,465 unique scFabs were identified that were present in all of the eight analyzed samples. To identify the mutations that are most strongly linked to high specificity, sets of one to four mutations were evaluated in the 3,465 scFabs that were most strongly correlated with enrichment in the samples sorted for low non-specific binding. First, all possible combinations of mutations for the eight mutated sites were evaluated. Because the statistical significance of the sets with four mutations was found to be highest, we focused on these 43,750 mutational sets. Each mutational set [e.g. Y33F, R54T, G56D, and Y102A in VH] was evaluated by first identifying clones that contain such mutations (regardless of whether they have wild-type or mutant residues at other sites), which are referred to as the four mutant (4MT) group. Similarly, the clones with wild-type residues at the same four sites (regardless of whether they have wild-type or mutant residues at the other sites) were identified, which are referred to as the four wild-type residue (4WT) group. Only the 4MT/4WT sets that contain more than ten clones in each group were further evaluated to maximize statistical significance. Next, a Spearman’s rank correlation coefficient was evaluated for each set of clones in the 4WT/4MT sets of antibodies based on whether they have the mutations (0 or 100%) relative to their enrichment ratios for PSR- and OVA- samples. Mutational sets were identified as significant if they have Spearman correlation coefficients ≥0.6 and p-values <0.05.
RESULTS
Chemical rules for identifying antibodies with high specificity
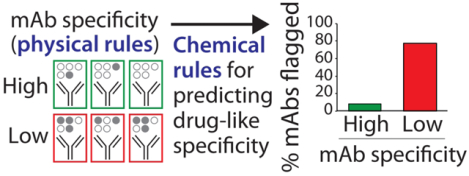
Our approach to identify the molecular determinants of antibody specificity is outlined in Fig. 1. We applied five tests of antibody specificity to 137 clinical-stage mAbs that are either approved drugs or are (or were) in phase 2 and 3 clinical trials using previously reported specificity measurements.14 The specificity measurements were obtained using different variable (VH and VL) regions for each clinical-stage mAb and the same constant regions (IgG1) regardless of the actual isotype. The five assays included three non-specific binding assays that evaluate antibody interactions with various types of proteins, DNA and virus particles [ELISA14, BVP20 and PSR21] and two assays that evaluate antibody self-association [AC-SINS22 and CSI23]. We assigned each mAb up to five physical flags if they exceeded previously reported upper limits for non-specific and self-interactions that segregate the top 90% of approved antibody drugs from the bottom 10%.14 We define antibodies with high specificity as those with few (<2) physical flags. Therefore, we segregated the clinical-stage mAbs (Table S1) into two groups, namely those with high specificity (<2 physical flags, 97 mAbs) and low specificity (≥2 physical flags, 40 mAbs), and evaluated chemical rules that selectively identify mAbs with low specificity.
Figure 1.
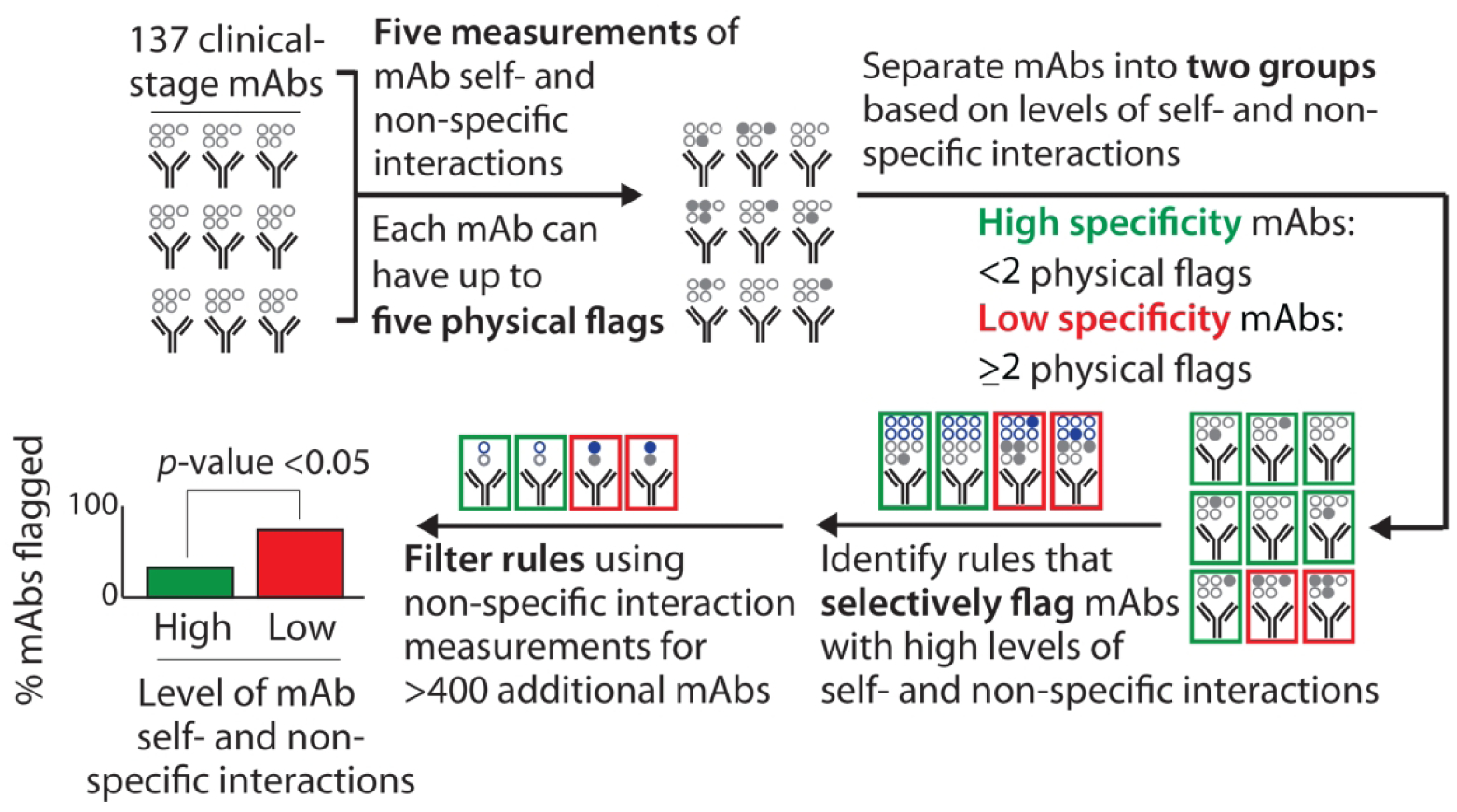
Overview of the methodology used to evaluate the molecular determinants of antibody specificity for monoclonal antibodies (mAbs). Each mAb received up to five physical flags based on exceeding limits for two self-interaction tests (AC-SINS >11.8 nm and CSI >0.01 response units) and three non-specific interaction tests (PSR >0.27, ELISA >1.9 signal/noise and BVP >4.3 signal/noise). The experimental data and limits were reported in a previous publication.14 The statistical significance was evaluated for the ability of the chemical rules to selectively flag mAbs with low specificity (≥2 physical flags) relative to mAbs with high specificity (<2 physical flags). The chemical rules were filtered using non-specific interaction measurements for an additional set of 424 preclinical mAbs to identify the most robust and general chemical rules.
Our approach to identify such chemical rules involved first evaluating maximum limits on the combined numbers of specified residues (weighted by their solvent exposure) for all possible combinations of 19 amino acids (excluding cysteine due to its rarity) for rules composed of as few as one and as many as 10 residues. This process was performed for the entire antibody variable fragment and 66 subregions of Fv, including the Fv framework (without the CDRs), VH, VL, individual CDRs (heavy chain CDRs 1, 2 and 3 and light chain CDRs 1, 2 and 3), and all possible combinations of CDRs that include as few as two and as many as six CDRs (e.g., heavy chain CDRs 1 and 3 and light chain CDR2). These rules sampled values that spanned the minimum and maximum values observed in the clinical-stage antibodies in increments of 0.1. In total, we evaluated >107 maximum rules based on the antibody Fv.
We required that the rules meet a number of constraints (see Supplemental Methods and Table S7 for full details), including that they selectively flag clinical-stage mAbs with low specificity relative to mAbs with high specificity. We also required that each rule flag mAbs with low specificity (as judged by the PSR assay) in a selective manner for a second training set of 424 human (preclinical) mAbs. The amino acid sequences and non-specific binding (PSR) values for the preclinical mAbs used are given in Table S2,15 the relative solvent accessibilities are given in Tables S3 (clinical-stage antibodies) and S4 (preclinical antibodies), and the amino acid composition limits that define the clinical-stage and preclinical mAbs are given in Table S5.
Our findings for chemical rules that identify mAbs with poor specificity based on maximum limits on the number of solvent-exposed amino acids in antibody variable regions are summarized in Figure 2 and Table S8. Despite evaluating >107 different chemical rules, only 16 ultimately met our constraints and passed our statistical analyses. Our most significant rule flags mAbs with a sum of >5.0 solvent-exposed Gln, Arg, His, Pro, Met, Leu, Tyr and Trp residues in heavy chain CDRs 1, 2 and 3 (H123) and light chain CDRs 2 and 3 (L23; Fig. 2A). This single rule flagged more than half (55%) of mAbs with low specificity while only flagging relatively few (14%) mAbs with high specificity (p-value of 2.6×10−6). While eight residues contributed to the rule, we evaluated how each residue contributed to the differences in the observed values for low specific mAbs relative to high specific mAbs for the entire panel of clinical-stage mAbs and not only for the subset of mAbs flagged by each rule. Notably, the most important residues were Gln (accounts for >30% of the difference observed between low and high specific mAbs), and Arg and His (each of which contribute 10–30%). Conversely, Pro, Met, Leu and Tyr contributed modestly (0–10%) and Trp contributed negatively (<0%). The last finding is due to the fact that mAbs with low specificity actually have less Trp (heavy chain CDRs 1, 2 and 3 and light chain CDRs 2 and 3) when considering the entire panel of clinical-stage mAbs but they have more Trp when considering the subset of mAbs flagged by this particular rule. The distribution of values for this chemical rule reveals that most mAbs with values >5.0 possess low specificity and those with values <5.0 have high specificity. Finally, the accuracy of the training (71%) and test (69%) sets of mAbs are similar, suggesting that our cross-validation procedures prevent overfitting of the training data.
Figure 2.
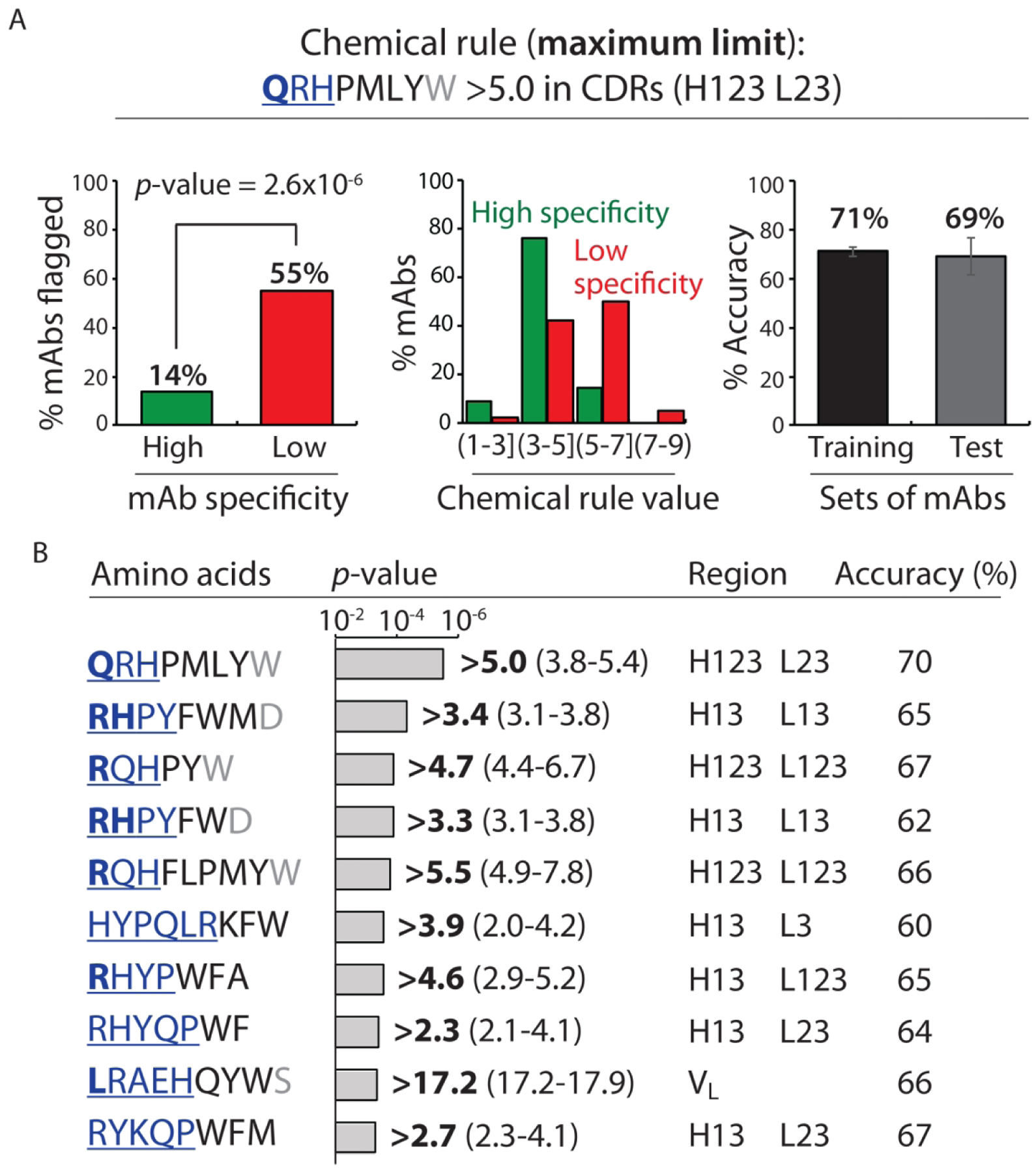
Chemical rules for selectively flagging mAbs with low specificity that limit the maximum allowable number of solvent-accessible residues in antibody variable regions. Each chemical rule is a maximum limit on the summed counts of different types of amino acids in the CDRs weighted by their relative solvent accessibilities. (A) Most selective maximum chemical rule for identifying mAbs with low specificity. mAbs with >5.0 Gln, Arg, His, Pro, Met, Leu, Tyr and Trp residues – weighted by their solvent exposures – in five CDRs (heavy chain 1, 2 and 3 and light chain 2 and 3) are flagged. On the left, the percentage of mAbs flagged with high and low specificity are reported for entire set (137 mAbs). In the middle, the distribution of the percentage of mAbs with ranges of chemical flag values are reported. On the right, the average adjusted accuracy of the chemical rule for flagging low specific antibodies relative to high specific ones is reported for the training and test sets. (B) Summary of the ten most selective chemical rules that limit the maximum sum of particular types of residues. The bolded value of each rule is the most statistically significant one when evaluated for the entire panel of clinical-stage mAbs, while the range of values reflect those that met the constraints used during cross validation. In (A) and (B), the contributions of the residues to each rule are reported in terms of their contributions to the differences in the observed rule values for mAbs with low specificity (40 clinical-stage mAbs) relative to those with high specificity (97 clinical-stage mAbs). The relative contributions of each amino acid are represented as bold and underlined blue font (most important, >30%), regular and underlined blue font (important, 10–30%), black font (minor importance, 0–10%) and grey font (least important, <0%). The negative contributions of some residues are due to the fact that the contributions are calculated for the entire set of clinical-stage mAbs (137 mAbs) and not only for those mAbs flagged by each rule. mAbs with low and high specificity are defined as described in Fig. 1. The p-values were calculated using a 2×2 contingency table (Fisher’s exact test), and the reported accuracies are adjusted to account for the different numbers of mAbs with high (97) and low (40) specificity. In (A), the average adjusted accuracies are calculated based on the training (80%) and test (20%) sets for each of the ten splits of the training and test sets. In (B), the adjusted accuracies are calculated for the entire set of 137 clinical-stage mAbs using the best flag values.
It is notable that Arg is the most important contributor to the maximum chemical rules (Fig. 2B). Of the top ten maximum rules, Arg is one of the most significant contributors (>30% contribution) in half of the rules and a significant contributor (10–30%) in all of the other rules. Moreover, His and Gln are also key contributors to the maximum rules (e.g., both contribute >30% in at least one of the rules), suggesting that certain positively charged and polar residues may be particularly important in mediating polyspecificity. Finally, although we considered many different subregions in the antibody variable regions, most (85%) of the chemical rules involve various combinations of heavy and light CDRs.
A key hypothesis in our preceding analysis is that over-enrichment of specific types of solvent-accessible residues in antibody variable regions is linked to poor specificity. We also sought to test the converse hypothesis by evaluating if underrepresentation of other types of residues may also be predictive of antibody specificity. Therefore, we evaluated minimum limits on the number of the residues weighted by their solvent exposure in antibody variable regions for all possible combinations of as many as ten residues (19 amino acids excluding cysteine; a total of >107 rules).
We identified a small subset of minimum rules (24) that met our constraints (Fig. 3 and Table S8). For example, the most significant minimum rule was a sum <11.6 Asn, Asp, Leu, Ala, Pro, Met, His, Glu and Gln residues in the variable heavy domain (VH; Fig. 3A). This single rule flagged half of the mAbs with low specificity while flagging few (13%) of mAbs with high specificity (p-value of 1.5×10−5). The most significant contributors were negatively charged (Asp) and polar (Asn) residues. Of the top ten minimum chemical rules, it is notable that Asp is the single most important contributor, and Asn and Glu are also key contributors. These findings suggest that the presence of negatively-charged and certain polar residues in antibody variable regions are linked to high specificity, which is consistent with previous work.5, 7, 8, 19, 24–30
Figure 3.
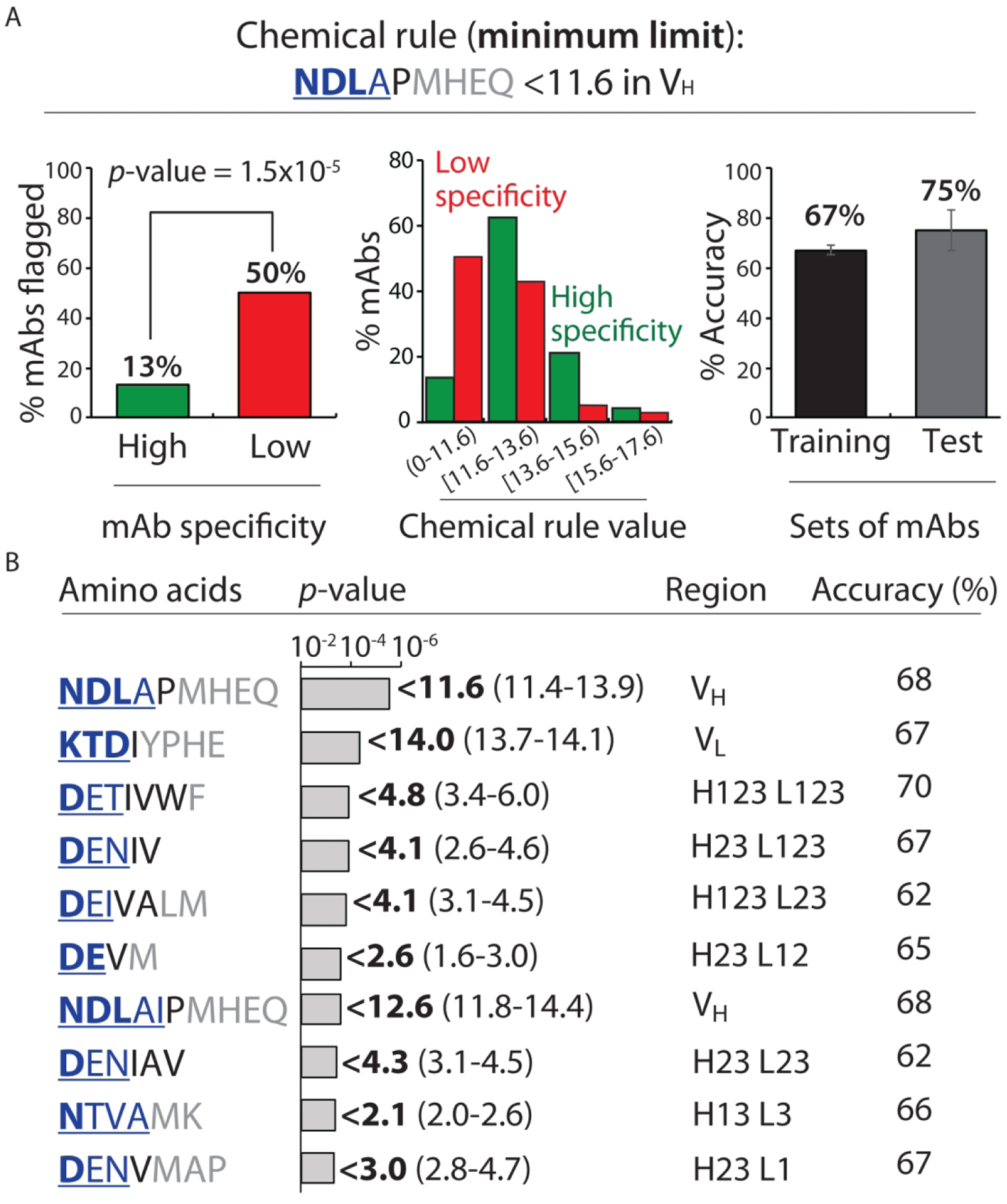
Chemical rules for selectively flagging mAbs with low specificity that limit the minimum allowable number of solvent accessible residues in antibody variable regions. Each chemical rule is a minimum limit on the summed counts of different types of amino acids in the CDRs weighted by their relative solvent accessibilities. (A) Most selective minimum chemical rule for identifying mAbs with low specificity. mAbs with <11.6 Asn, Asp, Leu, Ala, Pro, Met, His, Glu and Gln residues – weighted by their solvent exposures – in VH are flagged. The graphs are presented as described in Fig. 2. (B) Summary of the ten most selective chemical rules that limit the minimum sum of particular types of residues. In (A) and (B), the contributions of the residues to each rule are reported are described in Fig. 2 except that the differences in the observed rule values are calculated for high specific mAbs relative to low specific mAbs. mAbs with low and high specificity are defined as described in Fig. 1. The p-values and accuracies were calculated as described in Fig. 2.
Combinations of rules are highly selective for identifying mAbs with high specificity
The selectivity of these rules led us to evaluate whether greater discrimination between antibodies with high and low specificity could be achieved using combinations of these rules (Fig. 4 and Table S9). Therefore, we tested all possible combinations of 40 individual rules (Table S8) that passed our constraints to generate the best sets of rules that selectively identify mAbs with low specificity. We evaluated sets of rules with as few as four and as many as six (a total of >106 sets of rules), and identified antibodies with low specificity as those with ≥4–6 chemical flags (as defined on a case-by-case basis). We eliminated the vast majority of the sets of rules by requiring that they satisfy a number of constraints (see Supplemental Methods and Table S7 for details), resulting in only 16 sets of rules that met these constraints.
Figure 4.
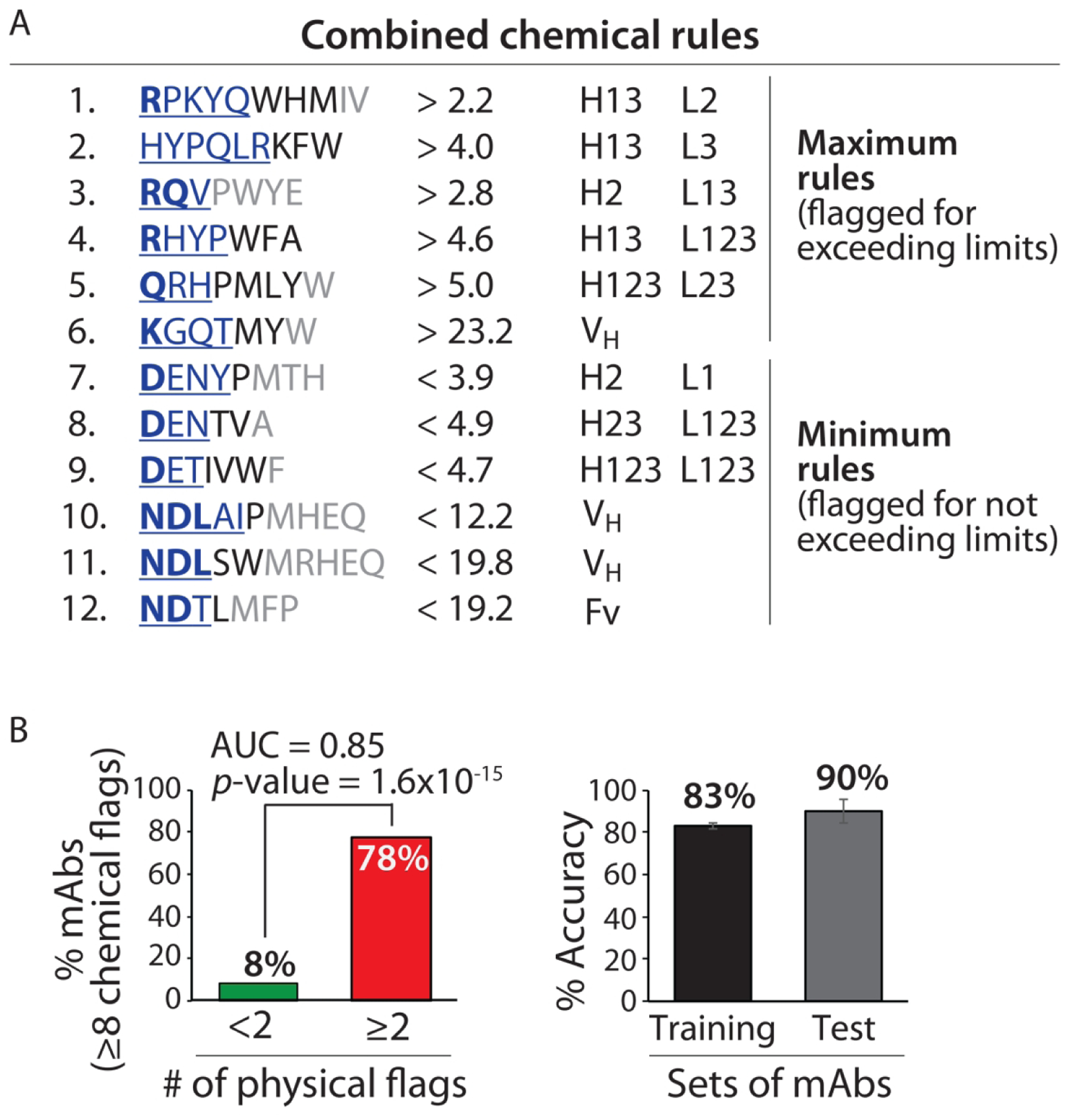
Combined chemical rules display high selectivity for identifying clinical-stage mAbs with low specificity. (A) Antibodies with predicted high specificity are required to be flagged by <8 of 12 rules. The contributions of the residues to each rule are reported as described in Figs. 2 and 3. (B) The combined rules selectively flag mAbs with low specificity (⩾2 physical flags) and display similar average adjusted accuracies for the training and test sets. The experimentally determined antibody specificities – as judged by five measurements of non-specific and self-interactions – are defined as described in Fig. 1. The p-values and adjusted accuracies were calculated as described in Fig. 2, and the area under the curve (AUC) is also reported.
The best set of chemical rules we identified comprised six chemical rules and displayed a significant improvement in performance relative to the individual rules (Table S9). This set of rules (Set A in Table S9) includes three maximum limits and three minimum limits, five of which are CDR-specific and the other is VH-specific. This set of rules was able to flag 35% of clinical-stage mAbs with low specificity while flagging only 2% of mAbs with high specificity (≥4 chemical flags corresponds to low antibody specificity). Similar to the individual rules, this set of rules displays similar average validation (66%) and test (67%) accuracies.
We reasoned that the specificity predictions could be further improved by eliminating mAbs flagged by the first set of six rules (Set A in Table S9) and generating additional rules for selectively flagging liabilities that were not identified in the first round of analysis (Fig. 4 and Tables S10–S12). Therefore, we eliminated mAbs from our training sets that were flagged by the first set of rules (Table S10), and identified individual maximum and minimum chemical rules that were best at selectively identifying the remaining mAbs with low specificity in our training sets (Table S11). We applied similar constraints and statistical methods in generating the individual rules for the second specificity test as we used for the first test (see Supplemental Methods for details).
Interestingly, we identified several (45) chemical rules (Table S11) that were markedly different than those generated in the first round of analysis (Figs. 2, 3 and Table S8). For example, Lys was the most significant contributor (>30% contribution) in most (61%) of the maximum rules in the second round of analysis, while Arg was rarely observed as one the most significant contributors (19% of the maximum rules). Moreover, most (73%) of the maximum and minimum rules in the second round of analysis were specific for one of the variable regions (VH or VL), the entire Fv or the variable framework (Fv without the CDRs), which was markedly different than the findings in the first round of analysis (27%).
We next evaluated whether the best set of rules in the first round of analysis could be combined with rules generated in the second round of analysis to further improve the selectivity of identifying mAbs with low specificity (Fig. 4 and Table S12). Therefore, we tested all possible combinations of the 45 individual rules (Table S11) with the six rules generated in the first round of analysis (Set A in Table S9) for a total of 8 to 14 chemical rules per set. As for the first round of analysis, we required that the sets of rules meet a number of constraints and statistical measures (see Supplemental Methods and Table S7 for details), and we identified mAbs with low antibody specificity as those with ≥4–12 chemical flags (as defined on a case-by-case basis).
Our best combined set of chemical rules is reported in Fig. 4A, and additional details are given in Table S9 (Set A) and Table S12 (Set F). The expanded set of 12 rules significantly improves the overall identification of clinical-stage mAbs with high specificity, as defined as those with <8 of 12 chemical flags. This set of rules flags most (78%) of clinical-stage mAbs with low specificity while flagging few (8%) mAbs with high specificity (p-value of 1.6×10−15 and area under curve of 0.85; Fig. 4B). Importantly, the average accuracy for our training (83%) and test (90%) sets of antibodies are similar.
The distribution of the number of chemical flags for the mAbs with high and low specificity reveals that most mAbs with <8 chemical flags have high specificity, while those with ≥8 chemical flags have low specificity (Fig. 5). It is also notable that the predictions of antibody specificity can be further refined. Antibodies with <4 chemical flags are all predicted correctly to have high specificity (accuracy of 100%). Likewise, antibodies with ≥8 flags are mostly predicted correctly to have low specificity (accuracy of 90%). Antibodies with 4–7 flags – which were considered in our original analysis as those with high specificity (<8 of 12 chemical flags) – are predicted correctly with more modest accuracy (75%). This suggests that a useful application of our chemical rules is to define three regions of specificity predictions, two with higher confidence (0–3 chemical flags for high specificity and 8–12 flags for low specificity), and a third with modest confidence in predicting high antibody specificity (4–7 flags).
Figure 5.
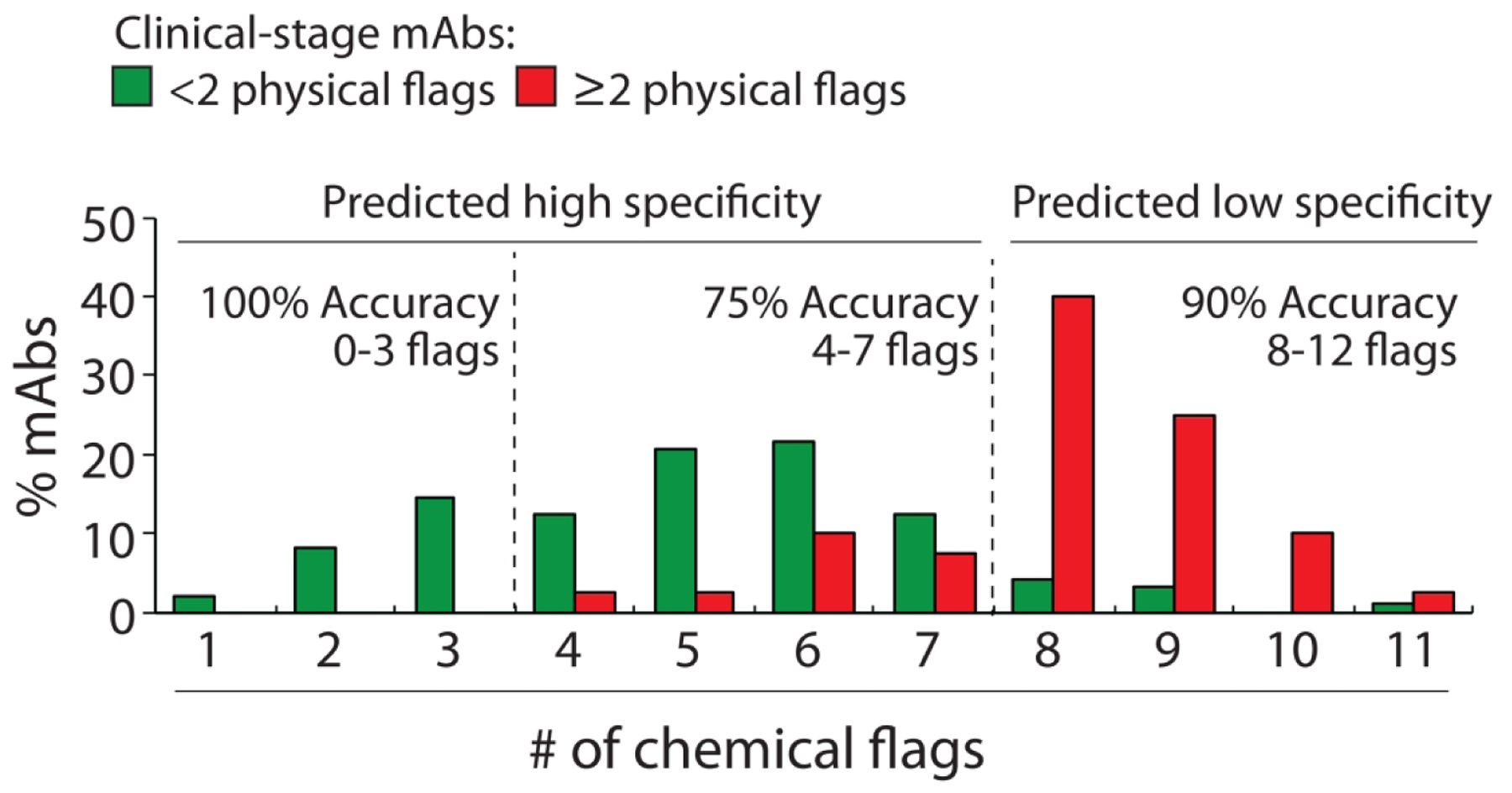
Distributions of the number of chemical flags for clinical-stage mAbs with high and low specificity. The chemical flags are defined in Fig. 4A. The experimentally determined antibody specificities – as judged by five measurements of non-specific and self-interactions – are defined as described in Fig. 1. mAbs with high specificity are those with <2 physical flags and mAbs with low specificity are those with ≥2 physical flags. The adjusted accuracies are calculated as described in Fig. 2.
We also sought to test the performance of our chemical rules if we eliminated the use of specific experimental limits to define antibody specificity (e.g., mAbs with PSR values >0.27 have low specificity) and instead simply ranked the antibodies from the most specific to the least specific based on experimental measurements (Fig. 6). To do this, we ranked the 137 clinical-stage mAbs from best (lowest levels of non-specific or self-interactions) to worst (highest levels of non-specific interactions or self-interactions) for each of the five biophysical assays and used the average rank percentile of the five assays to define the most specific mAbs (lowest rank). Given that there are 97 of 137 mAbs with <2 physical flags in our original definition of high specificity, we would expect that these antibodies would be ranked mostly in the top 71% (97 of 137 mAbs). Indeed, we find that most (94%, 91 of 97 mAbs) of the antibodies ranked in the top 71% of the mAbs have <2 physical flags (Fig. 6A), suggesting that our original definition of antibody specificity is weakly influenced by the use of experimental limits to determine poor specificity. Moreover, we find that our chemical rules segregate the best and worst antibodies in a similar manner as the physical rules (Fig. 6B). We also observe that mAbs with 0–3 chemical flags are mostly (79%, 19 of 24 mAbs) ranked in the top half of the antibodies, while mAbs with 8–12 chemical flags are all (100%) ranked in the bottom half of the antibodies and those with 4–7 chemical flags show intermediate average ranks (Fig. 6C). These findings further suggest that our chemical rules provide the strongest predictions of high specificity for mAbs with 0–3 chemical flags and low specificity for mAbs with 8–12 chemical flags.
Figure 6.
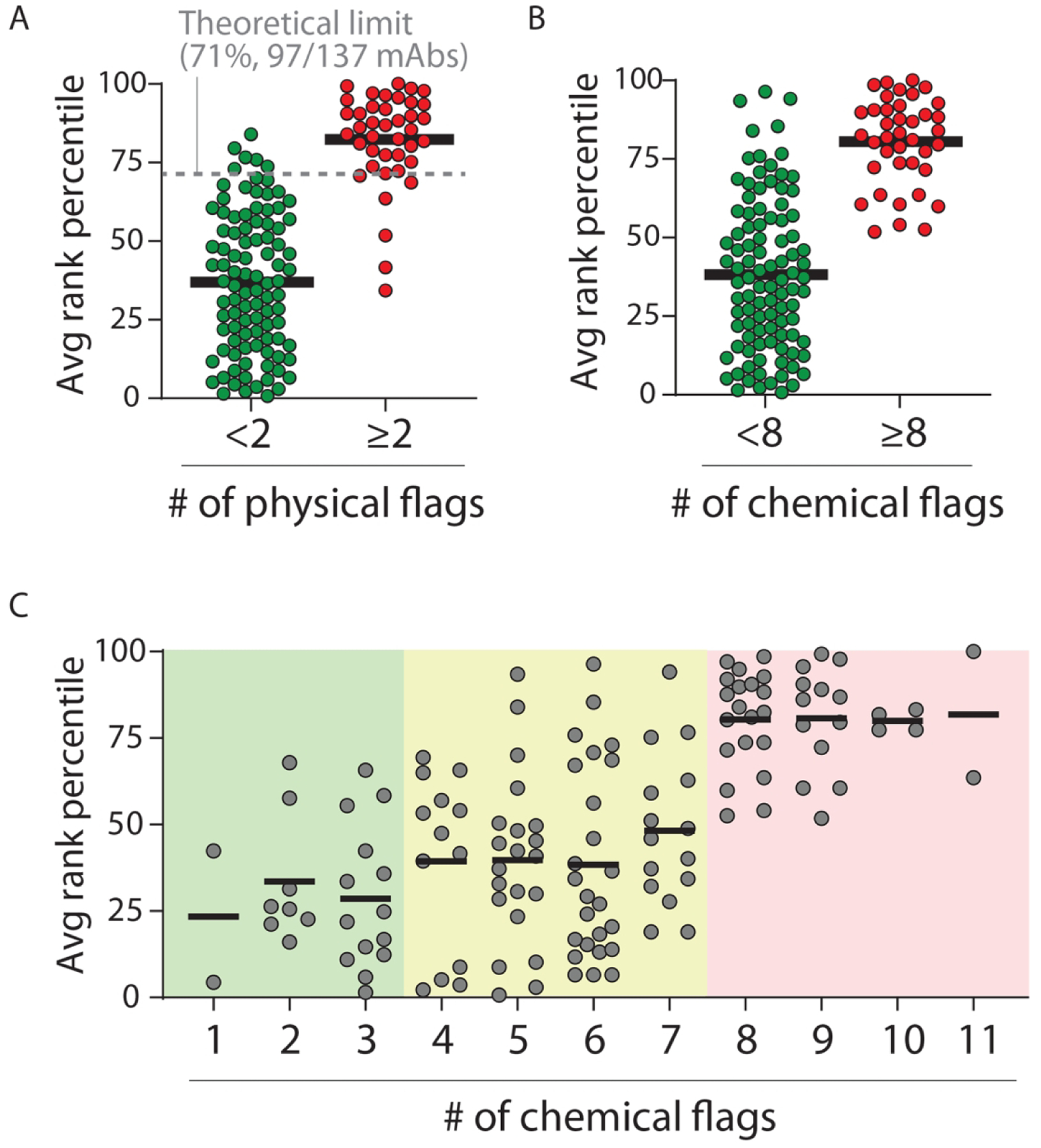
Comparison of the average rank for clinical-stage mAbs based on five measures of non-specific and self-interactions and the corresponding number of physical and chemical flags. (A) The average rank of mAbs with <2 physical flags (97 of 137 mAbs, 71% of mAbs) and ⩾2 physical flags (40 of 137 mAbs, 29% of mAbs) were calculated based on their ranks in five assays of self- and non-specific interactions. (B, C) The average experimental rank of mAbs compared to (B) <8 versus ⩾8 chemical flags and (C) the number of chemical flags. In (C), three regions are shown, one with predicted high specificity (0–3 chemical flags), a second one with intermediate specificity (4–7 chemical flags) and a third one with low specificity (8–12 chemical flags).
Our goal in developing the chemical rules in this work was to broadly describe antibody specificity and not rely on any single type of experimental specificity measurement. Nevertheless, we next evaluated how the rules that emerged from our analysis would perform in the context of each individual specificity assay (three non-specific binding assays and two self-interaction assays). For each biophysical assay, the antibodies were segregated into two groups based on previously established limits for high levels of non-specific and self-interactions.14 Next, the specificity test in Fig. 4 was applied to each group of antibodies defined by single non-specific and self-interaction measurements (Figs. S2–S6). Encouragingly, the performance of the chemical rules was both strong and relatively similar for the five individual assays (p-values <10−5 and accuracy of ≥75% for each assay). Moreover, we observed that the accuracies for the PSR (Fig. S2) and AC-SINS (Fig. S3) assays were particularly strong for predicting high specificity of mAbs with <4 chemical flags (100% accuracy). More generally, we also observed strong performance for the PSR (Fig. S2), AC-SINS (Fig. S3), ELISA (Fig. S5) and BVP (Fig. S6) assays for predicting mAbs with high specificity (>80% accuracy, <4 chemical flags) and low specificity (>80% accuracy, 8–12 chemical flags).
Evaluation of combinations of chemical rules using independent sets of antibodies
We also evaluated our chemical rules using independent sets of antibodies not included in our training or test sets. We first evaluated an independent set of non-specific interaction (PSR) measurements for an additional 359 preclinical mAbs (Tables S13 and S14) that largely fall within the amino acid compositions that we evaluated previously in this work (≥98% and <99% similarity based on the maximum and minimum limits in Table S5). Importantly, the combined specificity rules correctly identified more than half (55%) of these preclinical mAbs with low specificity while flagging few mAbs (16%) with high specificity (p-value of 1.6×10−4 and accuracy of 69%; Fig. S7). It is also notable that the accuracies for the training (70%) and test (69%) sets of preclinical antibodies were similar.
We also tested our predictions using a second independent set of mAbs (39 IgGs) that we generated and characterized using non-specific binding (ELISA14) and self-association (AC-SINS17) assays (Fig. 7). Importantly, the mAbs predicted by our specificity rules to have poor specificity (≥8 chemical flags) displayed significantly higher levels of non-specific binding than those that pass our test (<8 flags, p-value of 6.3×10−4; Fig. 7A). Moreover, we find that the same specificity test is also able to identify mAbs with high levels of self-association (Fig. 7B). The mAbs identified by our specificity test (≥8 flags) displayed markedly higher levels of self-association relative to mAbs that were not flagged (<8 flags, p-value of 1.2×10−9). Moreover, we observed that mAbs with <4 chemical flags generally had lower levels of non-specific binding and self-association than those with 4–7 flags, although the difference for non-specific binding is not significant. These results demonstrate the generality of our methodology for identifying antibodies with drug-like specificity.
Figure 7.
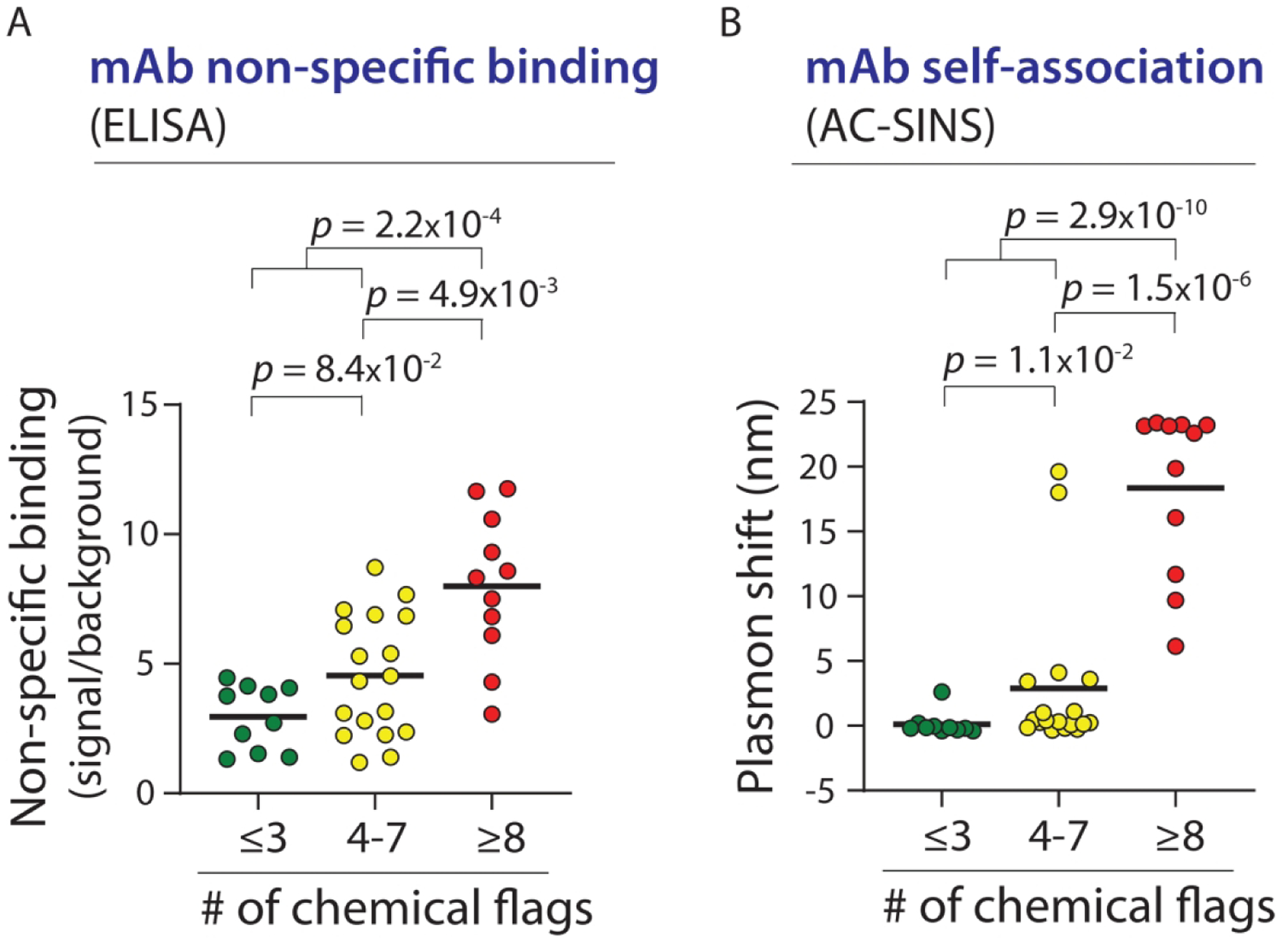
Combined chemical rules strongly differentiate between mAbs with different levels of non-specific and self-interactions for an independent set of antibodies. (A) Non-specific interactions (ELISA) and (B) self-interactions (AC-SINS) were measured for 39 mAbs that were not included in the training and test sets used to generate the combined chemical rules. The p-values were calculated using a two sample Anderson-Darling test. In (A), the difference between ⩽3 chemical flags and 4–7 chemical flags is not significant.
The strong performance of our chemical rules for identifying antibodies with high specificity using multiple independent sets of data (Figs. 7 and S7) suggest that our rules identify some of the most important determinants of antibody specificity. However, we sought to evaluate our predictions using much larger data sets to better evaluate their utility in identifying antibodies with high specificity. Therefore, we sought to mutagenize a clinical-stage antibody (emibetuzumab) that is flagged by all 5 biophysical assays (ELISA, PSR, BVP, AC-SINS and CSI)14 and 8 out of 12 of the chemical rules. Our strategy was to identify sites in the variable regions that were flagged by our maximum rules and mutate them to residues that reduced the number of chemical flags, including those that are most important in the minimum rules (e.g., D and T). We identified eight sites in the heavy chain CDRs to mutagenize, and sampled five mutations per site in addition to the wild-type residue using degenerate codons (Fig. S1), which resulted in >106 variants. This library was then displayed on yeast as single-chain Fab fragments, sorted for low non-specific binding against two reagents (PSR and ovalbumin) or high (specific) binding to the target antigen, and the selected antibody variants with high specificity were identified using deep sequencing (see Methods for more details).
Our findings are summarized in Figure 8. Of the 3,465 antibodies that we identified in two independent experiments, we first sought to identify sets of mutations that were most strongly correlated with significant enrichment for high specificity during selection. Our initial analysis led us to focus on sets of four mutations to maximize statistical significance. For example, we identified a set of four mutations (Y33F, R54T, G56D and Y102A in VH) that shows strong correlation between antibody variants with such mutations and their enrichment ratios during selection for low polyspecificity (Spearman’s correlation coefficients of 0.81 for PSR and 0.83 for ovalbumin; Fig. 8A). Of the top ten sets of four mutations we identified, all of them included a mutation that introduces negative charge, further suggesting that negative charge is linked to increased antibody specificity (Fig. 8B and Table S15). It is also notable that most (80%) of top sets of mutations involved eliminating at least one Arg in the CDRs. In total, we identified 13 sets of four mutations and 161 antibodies with such mutations that are expected to possess high specificity (see Methods for more details).
Figure 8.
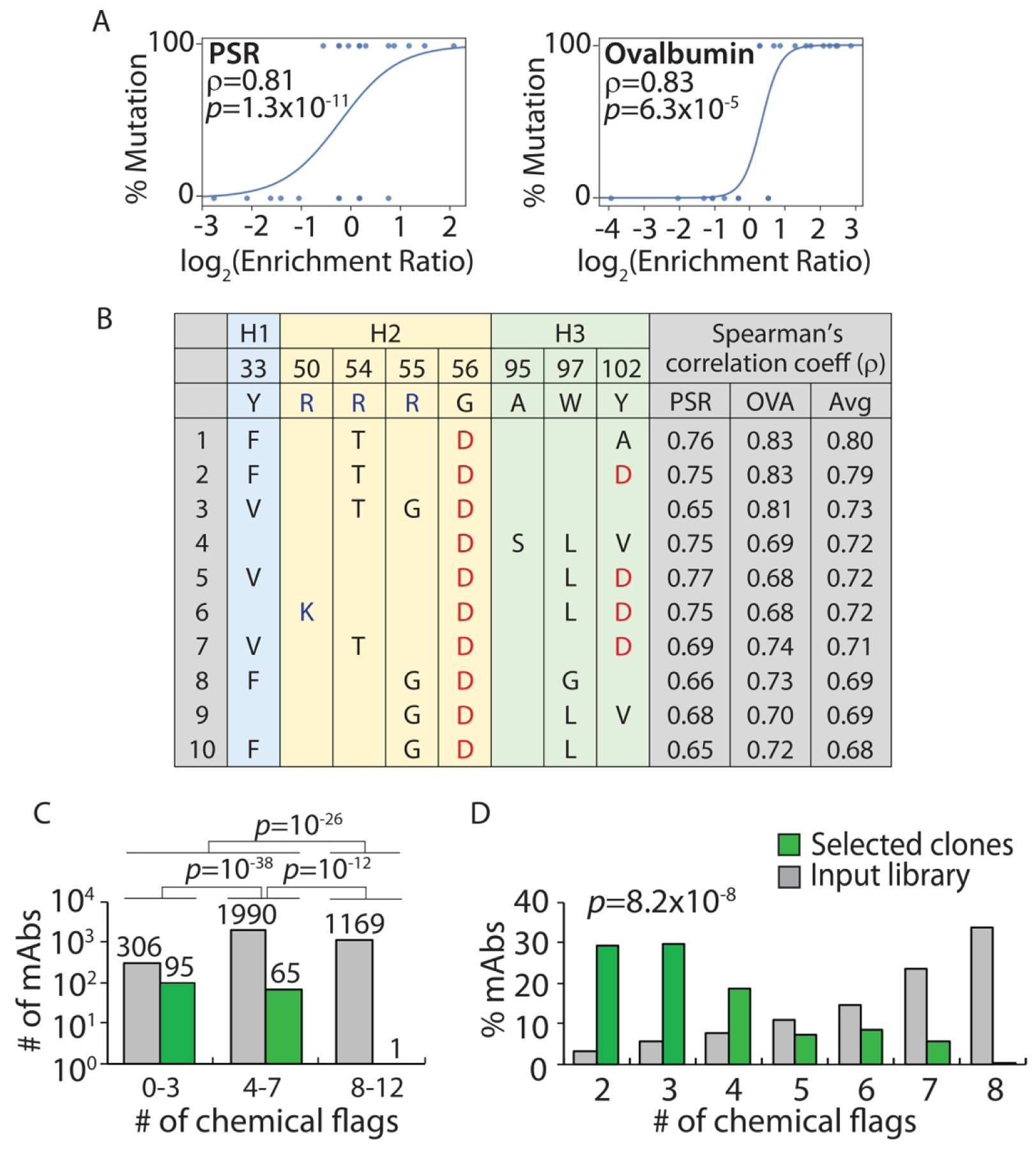
Design of Fab sub-libraries of emibetuzumab guided by the combined chemical rules and evaluation of selected mutants with improved antibody specificity. The VH domain of emibetuzumab was mutated at eight solvent-exposed sites (Y33, R50, R54, R55, G56, A95, W97 and Y102) in the three heavy chain CDRs that were flagged by the maximum chemical rules. The mutations sampled the wild-type residue as well as five mutations that are predicted to reduce the number of chemical flags. The libraries were constructed as single-chain Fab fragments (scFabs) on yeast, sorted for non-binding to two polyspecificity reagents [PSR and ovalbumin (OVA)], and evaluated via deep sequencing. (A) Enrichment ratios for antibody variants with a set of four mutations (F33, T54, D56 and A102 in VH) relative to antibody variants with wild-type residues at the same positions (Y33, R54, G56 and Y102 in VH) for two different polyspecificity reagents. The curves (logistic regressions) are guides to the eye. (B) Top ten sets of four mutation combinations that are most strongly correlated with reduced binding to polyspecificity reagents and increased specificity. (C, D) The (C) number and (D) percentage of mAb variants selected with high specificity as a function of the number of chemical flags relative to the corresponding values for the input library. In (A), the mAbs included in the wild-type or mutant groups are only required to have wild-type or mutant residues at the four evaluated sites and can have either wild-type or mutant residues at the other four sites. Moreover, the p-values are for the Spearman’s correlation coefficients (ρ). In (C), the p-values for the comparisons of the number of mAbs were calculated using a 2×2 contingency table (Fisher’s exact test). In (D), the p-value for comparing the distributions of mAbs was calculated using paired sample t-test (two tailed).
We next sought to test the ability of our chemical rules to correctly predict antibodies with high specificity (Fig. 8C). Strikingly, despite observing thousands of antibodies with 0–7 chemical flags (2,370 antibodies) and 8–12 flags (1,095 antibodies with 8 chemical flags) in our input library, our rules correctly identified almost all selected antibodies with high specificity (160 of 161 antibodies with <8 chemical flags, p-value of 10−26). Moreover, we find that antibodies with 0–3 flags were much more strongly enriched than even those with 4–7 flags (p-value <10−38), which further suggests that our predictions of specificity are strongest for antibodies with 0–3 chemical flags. More generally, we find that the distributions of the number chemical flags for the selected mAbs with high specificity relative to the input library are significantly different (p-value of 10−8; Fig. 8D). These findings demonstrate how our chemical rules can be used to guide the design of antibody libraries that target sites involved in polyspecificity and facilitate the identification of antibody mutants with significant improvements in specificity.
DISCUSSION
Our findings provide a relatively simple yet powerful description of the overall specificity of monoclonal antibodies at physiological conditions. To the best of our knowledge, there are no prior methods for predicting the overall propensity of antibodies to interact non-specifically with various types of molecules or themselves based simply on their primary structures and their corresponding sequence-based solvent accessibilities. Importantly, the conceptual framework that we have developed is able to explain many previous disparate findings and observations. Our finding that antibodies with high specificity have low levels of positively charged CDR residues is consistent with previous findings that increased levels of positive charge in the CDRs and variable (VH and VL) regions are linked to poor antibody specificity2, 10, 19, 31–35 and pharmacokinetics10, 12, 34–38 as well as high viscosity in concentrated antibody solutions.39 Conversely, our finding that antibodies with high specificity have increased levels of negative charge in the CDRs is consistent with previous findings that increased levels of negative charge in antibody CDRs and variable regions are linked to high solubility and low aggregation propensity,5, 7, 8, 24, 25, 30, 40–42 low self-association17 (although exceptions have been noted for mAbs with abnormal levels of negative charge41, 42) and favorable pharmacokinetics.12, 37, 38, 43 Moreover, our finding that antibodies with high specificity also have increased levels of polar CDR residues (e.g., Asn) is consistent with the fact that increased levels of polar residues in the CDRs are linked to increased antibody specificity31, 32 and improved pharmacokinetics.9 Our specificity rules capture multiple factors that govern the physicochemical properties of antibodies and simplify them into powerful guidelines for rapidly identifying mAbs that are expected to display favorable specificity, solubility and biodistribution.
What are implications of our chemical rules for generating and engineering antibodies with high affinity? Interestingly, the theoretical net charge of the CDRs (pH 7.4) of the mAbs with high specificity is near zero (−0.1±2.4) and more than half of them (61%) have positively charged CDRs. While mAbs with low specificity do have more positively charged CDRs on average than those with high specificity (1.8±2.7 for mAbs with low specificity vs. −0.1±2.4 for mAbs with high specificity), we do not expect that antigen-binding sites that are strongly negatively charged are likely to be generally compatible with high affinity binding. It is plausible that antibodies with CDR net charges near zero (pH 7.4) – and with appropriate amounts of non-charged hydrophilic and hydrophobic residues as predicted by our rules – may be most attractive for achieving both high affinity and specificity. Nevertheless, these speculative ideas await additional experimental and computational analysis.
Although our findings generally suggest that increased negative charge in antibody variable regions is linked to reduced non-specific and self-interactions, it is well established that over-enrichment in negative charge in antibody variable regions is also linked to increased self-association and viscous solution behavior at high antibody concentrations.41, 44–48 One obvious difference between this work and previous studies related to viscous antibodies are the solution conditions, as we analyzed antibody self-interaction measurements at physiological conditions (PBS at pH 7.4) and previous studies have evaluated antibody viscosity measurements in typical formulation conditions (e.g., pH 5–6 without salt or with low salt concentrations). Another notable difference is that the antibodies with high specificity in our analysis have modest amounts of negative charge – based on their theoretical net charge calculated at pH 7.4 – in their VH (0.8±1.9), VL (0.8±2.0), Fv (1.5±2.5), heavy chain CDRs (−0.6±1.7), light chain CDRs (0.6±1.9) and overall CDRs (−0.1±2.4; Table S16). Moreover, the isoelectric points of the variable regions of the antibodies with high specificity in our analysis are relatively typical of antibodies in general, as evidenced by the values for VH (7.4±1.4), VL (7.5±1.4) and Fv (7.7±1.3; Table S16).
Caution should be used when interpreting our predictions of specificity for antibodies with variable region charges and isoelectric points that fall beyond the range of values represented in our study. For example, omalizumab is viscous at high antibody concentrations (e.g., ~40 cP at ~120 mg/mL in histidine buffer at pH 6.0).41, 42, 49 Notably, this mAb has abnormal charge properties relative to those for the high specific antibodies in this study, including a more negatively charged VL (−3.9 relative to 0.8±2.0 at pH 7.4) and light chain CDRs (−4.9 relative to 0.6±1.9 at pH 7.4) as well as a lower VL isoelectric point (pI of 4.7 relative to 7.5±1.4). Moreover, mutations in the light chain CDRs of omalizumab (D28A, D30N, H92Y, E93T, D94T and Y96P in VL) that reduce the amount of negative charge (−1.0 relative to −4.9 for wild type at pH 7.4) and increase the isoelectric point of VL (pI of 6.3 relative to 4.7 for wild type) to be within the range of the high specific antibodies in this study significantly reduce the viscosity (<10 cP at ~120 mg/mL).41, 42, 50 Future work is needed to better define limits on the amount of negative charge in antibody variable regions that is favorable for specificity without promoting attractive electrostatic interactions that are unfavorable for high concentration viscoelastic behavior.
There are also multiple factors to consider when interpreting and applying our findings. We defined the limits of sequence space for our analysis in Table S5 based on the maximum and minimum numbers of each type of amino acid (weighted by their relative solvent accessibilities) in the CDRs and variable regions of preclinical and clinical-stage antibodies in our training sets. It is expected that the performance of our rules will be reduced for antibodies whose amino acid compositions and site-specific solvent accessibilities are outside the range of chemical and structural diversity that we explored in this analysis. Encouragingly, we find that the accuracy of our specificity predictions for non-specific binding (PSR assay) is weakly impacted by reducing the similarity of antibodies in our test set relative to the those in our training set (Fig. S7). However, the accuracy of such predictions is expected to decrease as the antibodies to be analyzed become more dissimilar relative to those in our training sets. Second, the antibody specificity measurements considered in this work were obtained using common antibody constant regions (IgG1 isotype with corresponding kappa and lambda light chains) regardless of the actual antibody isotype.14, 15 This is notable because it is well known that isotype differences between antibodies can lead to differences in self-association and related properties such as solubility and viscosity.14, 48, 51–54 The effects of different antibody isotypes on the physicochemical properties of mAbs have not been addressed in our specificity analysis and will need to be addressed in future work.
It is also important to consider the impact of the methods we used to calculate solvent-accessible surface areas on our findings. We employed a published machine learning method that only requires antibody variable region sequences,16 which is particularly convenient for antibodies of unknown structure. Nevertheless, it is important to note that this machine learning method yields slightly different models each time it is trained, which leads to small differences in the predicted solvent accessibility values and the resulting chemical rule values. For example, we compared two different models generated from this machine learning method and observed minor differences in the number of chemical flags for the clinical-stage antibodies (Table S17). This results in minor changes in the accuracy of the predictions for the specificity of the clinical-stage antibodies (e.g., 82–85% accuracy). We recommend not only using the same machine learning method, but also the same compiled model that we used in this work to calculate solvent accessibilities for evaluating the number of chemical flags for additional antibodies. It is also notable that the machine learning method is mostly trained on antibodies with kappa light chains, and caution should be used when applying this method to antibodies with lambda light chains. More generally, our training sets of non-specific and self-interaction measurements primarily contained antibodies with kappa light chains given that most clinical-stage antibodies have kappa light chains (91% of the clinical-stage antibodies in this study), and our chemical rules are expected to be most useful for predicting the specificity of antibodies with kappa light chains.
We expect that our findings will immediately impact therapeutic antibody development in multiple ways. First, our specificity rules will serve as valuable design guidelines for generating antibody libraries with drug-like specificity. This is particularly important for both in vitro antibody discovery and affinity maturation given that it is only possible to sample an extremely small fraction of maximum CDR chemical diversity, and it is critical to focus the CDR diversity on combinations of residues that give rise to drug-like properties. We also expect that our specificity rules will provide powerful guidelines for both identifying antibody candidates with high specificity during early antibody discovery and re-engineering existing antibodies with drug-like properties later in the optimization and development process. More generally, we expect that our novel conceptual framework – which can be readily expanded in the future to include additional structural information and incorporate additional biophysical data sets – will accelerate the generation of potent antibody therapeutics with drug-like properties.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Laura Walker for providing antibody sequences and PSR measurements of non-specific antibody binding. We thank Tushar Jain for providing support and guidance in using his machine learning methods for calculating antibody solvent accessible surface areas. We also thank Laura Walker, Tushar Jain, Mark Julian and Charles Starr for providing valuable feedback on multiple versions of this manuscript. This work was supported by the National Institutes of Health [R01GM104130, R01AG050598, RF1AG059723 and R35GM136300 to P.M.T., T32 fellowship to L.W. (T32-GM008353)], National Science Foundation [CBET 1813963, CBET 1605266 and CBET 1804313 to P.M.T., Graduate Research Fellowships to L.A.R. and M.D.S (DGE 1256260)], and the Albert M. Mattocks Chair (to P.M.T).
Footnotes
COMPETING FINANCIAL INTERESTS
None.
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS Publications website.
Methods:
Summary of methods for generating individual and combined chemical rules for identifying antibodies with low specificity, and methods for deep sequencing data generation and analysis.
Tables:
Sequences and specificity measurements for clinical-stage antibodies; Sequences and specificity (PSR) measurements for a large set of preclinical antibodies used as a training set to generate the chemical rules; Predicted relative solvent accessibilities of amino acids in the variable regions of clinical-stage antibodies; Predicted relative solvent accessibilities of amino acids in the variable regions of preclinical antibodies; Maximum and minimum values for the observed counts of amino acids (weighted by their solvent accessibilities) and net charges (pH 7.4) of different regions within the variable domains of clinical-stage and preclinical mAbs in the training sets; Summary of segregation of clinical-stage mAbs into training and test sets; Summary of the constraints used to generate the single and combined sets of rules for flagging mAbs with low specificity; Summary of the most selective rules that flag mAbs with low specificity in the first round of analysis; Summary of the best combined sets of rules generated in the first round of analysis that flag mAbs with low specificity; Segregation of mAbs that pass the combined specificity rules in the first round of analysis into training and test sets; Summary of the most selective chemical rules that identify mAbs with low specificity in the second round of analysis; Summary of the best combined sets of rules generated after the second round of analysis that identify mAbs with low specificity; Sequences and specificity measurements for an independent set of preclinical antibodies used to test the combined specificity rules; Predicted relative solvent accessibilities of amino acids in the variable regions of an independent set of preclinical antibodies; Sets of four mutations in Fab sub-libraries of emibetuzumab that are most strongly correlated with reduced binding to polyspecificity reagents [PSR and ovalbumin (OVA)]; Theoretical net charges and isoelectric points for the high specific clinical-stage antibodies in this study; Number of chemical flags for the clinical-stage antibodies calculated using two different SASA machine learning models.
Figures:
Design of a mutant VH library for single-chain Fab fragments of emibetuzumab aimed at reducing the number of chemical flags; Performance of combined chemical rules for identifying clinical-stage mAbs with high levels of non-specific interactions detected using the PSR assay; Performance of combined chemical rules for identifying clinical-stage mAbs with high levels of self-interactions detected using the AC-SINS assay; Performance of combined chemical rules for identifying clinical-stage mAbs with high levels of self-interactions detected using the CSI assay; Performance of combined chemical rules for identifying clinical-stage mAbs with high levels of non-specific interactions detected using the ELISA non-specific binding assay; Performance of combined chemical rules for identifying clinical-stage mAbs with high levels of non-specific interactions detected using the BVP assay; Performance of combined chemical rules for selectively identifying preclinical mAbs with high levels of non-specific interactions.
References
- 1.Tiller KE; Tessier PM Advances in antibody design. Annu Rev Biomed Eng 2015, 17, 191–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wardemann H; Yurasov S; Schaefer A; Young JW; Meffre E; Nussenzweig MC Predominant autoantibody production by early human B cell precursors. Science 2003, 301, (5638), 1374–1377. [DOI] [PubMed] [Google Scholar]
- 3.Maynard J; Georgiou G Antibody engineering. Annu Rev Biomed Eng 2000, 2, 339–76. [DOI] [PubMed] [Google Scholar]
- 4.Perchiacca JM; Tessier PM Engineering aggregation-resistant antibodies. Annu Rev Chem Biomol Eng 2012, 3, 263–86. [DOI] [PubMed] [Google Scholar]
- 5.Perchiacca JM; Ladiwala AR; Bhattacharya M; Tessier PM Aggregation-resistant domain antibodies engineered with charged mutations near the edges of the complementarity-determining regions. Protein Eng Des Sel 2012, 25, (10), 591–601. [DOI] [PubMed] [Google Scholar]
- 6.Wu SJ; Luo J; O’Neil KT; Kang J; Lacy ER; Canziani G; Baker A; Huang M; Tang QM; Raju TS; Jacobs SA; Teplyakov A; Gilliland GL; Feng Y Structurebased engineering of a monoclonal antibody for improved solubility. Protein Eng Des Sel 2010, 23, (8), 643–51. [DOI] [PubMed] [Google Scholar]
- 7.Perchiacca JM; Bhattacharya M; Tessier PM Mutational analysis of domain antibodies reveals aggregation hotspots within and near the complementarity determining regions. Proteins 2011, 79, (9), 2637–47. [DOI] [PubMed] [Google Scholar]
- 8.Dudgeon K; Rouet R; Kokmeijer I; Schofield P; Stolp J; Langley D; Stock D; Christ D General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc Natl Acad Sci U S A 2012, 109, (27), 10879–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dobson CL; Devine PW; Phillips JJ; Higazi DR; Lloyd C; Popovic B; Arnold J; Buchanan A; Lewis A; Goodman J; van der Walle CF; Thornton P; Vinall L; Lowne D; Aagaard A; Olsson LL; Ridderstad Wollberg A; Welsh F; Karamanos TK; Pashley CL; Iadanza MG; Ranson NA; Ashcroft AE; Kippen AD; Vaughan TJ; Radford SE; Lowe DC Engineering the surface properties of a human monoclonal antibody prevents self-association and rapid clearance in vivo. Sci Rep 2016, 6, 38644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Datta-Mannan A; Thangaraju A; Leung D; Tang Y; Witcher DR; Lu J; Wroblewski VJ Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pI reduces non-specific binding and improves the pharmacokinetics. mAbs 2015, 7, (3), 483–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kelly RL; Zhao J; Le D; Wittrup KD Nonspecificity in a nonimmune human scFv repertoire. Mabs-Austin 2017, 9, (7), 1029–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bumbaca Yadav D; Sharma VK; Boswell CA; Hotzel I; Tesar D; Shang Y; Ying Y; Fischer SK; Grogan JL; Chiang EY; Urban K; Ulufatu S; Khawli LA; Prabhu S; Joseph S; Kelley RF Evaluating the use of antibody variable region (Fv) charge as a risk assessment tool for predicting typical cynomolgus monkey pharmacokinetics. J Biol Chem 2015, 290, (50), 29732–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Starr CG; Tessier PM Selecting and engineering monoclonal antibodies with drug-like specificity. Curr Opin Biotech 2019, 60, 119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jain T; Sun T; Durand S; Hall A; Houston NR; Nett JH; Sharkey B; Bobrowicz B; Caffry I; Yu Y; Cao Y; Lynaugh H; Brown M; Baruah H; Gray LT; Krauland EM; Xu Y; Vasquez M; Wittrup KD Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci U S A 2017, 114, (5), 944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shehata L; Maurer DP; Wec AZ; Lilov A; Champney E; Sun TW; Archambault K; Burnina I; Lynaugh H; Zhi XY; Xu YD; Walker LM Affinity Maturation Enhances Antibody Specificity but Compromises Conformational Stability. Cell Rep 2019, 28, (13), 3300–3308. [DOI] [PubMed] [Google Scholar]
- 16.Jain T; Boland T; Lilov A; Burnina I; Brown M; Xu YD; Vasquez M Prediction of delayed retention of antibodies in hydrophobic interaction chromatography from sequence using machine learning. Bioinformatics 2017, 33, (23), 3758–3766. [DOI] [PubMed] [Google Scholar]
- 17.Alam ME; Geng SB; Bender C; Ludwig SD; Linden L; Hoet R; Tessier PM Biophysical and sequence-based methods for identifying monovalent and bivalent antibodies with high colloidal stability. Mol Pharm 2018, 15, (1), 150–163. [DOI] [PubMed] [Google Scholar]
- 18.Swindells MB; Porter CT; Couch M; Hurst J; Abhinandan KR; Nielsen JH; Macindoe G; Hetherington J; Martin ACR abYsis: Integrated Antibody Sequence and Structure-Management, Analysis, and Prediction. J Mol Biol 2017, 429, (3), 356–364. [DOI] [PubMed] [Google Scholar]
- 19.Julian MC; Rabia LA; Desai AA; Arsiwala A; Gerson JE; Paulson HL; Kane RS; Tessier PM Nature-inspired design and evolution of anti-amyloid antibodies. J Biol Chem 2019, 294, (21), 8438–8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hotzel I; Theil FP; Bernstein LJ; Prabhu S; Deng R; Quintana L; Lutman J; Sibia R; Chan P; Bumbaca D; Fielder P; Carter PJ; Kelley RF A strategy for risk mitigation of antibodies with fast clearance. mAbs 2012, 4, (6), 753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Y; Roach W; Sun T; Jain T; Prinz B; Yu TY; Torrey J; Thomas J; Bobrowicz P; Vasquez M; Wittrup KD; Krauland E Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: a FACS-based, high-throughput selection and analytical tool. Protein Eng Des Sel 2013, 26, (10), 663–70. [DOI] [PubMed] [Google Scholar]
- 22.Sule SV; Dickinson CD; Lu J; Chow CK; Tessier PM Rapid analysis of antibody self-association in complex mixtures using immunogold conjugates. Mol Pharm 2013, 10, (4), 1322–31. [DOI] [PubMed] [Google Scholar]
- 23.Sun T; Reid F; Liu Y; Cao Y; Estep P; Nauman C; Xu Y High throughput detection of antibody self-interaction by bio-layer interferometry. mAbs 2013, 5, (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jespers L; Schon O; Famm K; Winter G Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nat Biotechnol 2004, 22, (9), 1161–5. [DOI] [PubMed] [Google Scholar]
- 25.Arbabi-Ghahroudi M; To R; Gaudette N; Hirama T; Ding W; MacKenzie R; Tanha J Aggregation-resistant VHs selected by in vitro evolution tend to have disulfide-bonded loops and acidic isoelectric points. Protein Eng Des Sel 2009, 22, (2), 59–66. [DOI] [PubMed] [Google Scholar]
- 26.Rabia LA; Zhang YL; Ludwig SD; Julian MC; Tessier PM Net charge of antibody complementarity-determining regions is a key predictor of specificity. Protein Eng Des Sel 2018, 31, (11), 409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narula J; Petrov A; Bianchi C; Ditlow CC; Lister BC; Dilley J; Pieslak I; Chen FW; Torchilin VP; Khaw BA Noninvasive Localization of Experimental Atherosclerotic Lesions with Mouse-Human Chimeric Z2d3 F(Ab’)(2) Specific for the Proliferating Smooth-Muscle Cells of Human Atheroma - Imaging with Conventional and Negative Charge-Modified Antibody Fragments. Circulation 1995, 92, (3), 474–484. [DOI] [PubMed] [Google Scholar]
- 28.McCarthy BJ; Hill AS Altering the fine specificity of an anti-Legionella single chain antibody by a single amino acid insertion. J Immunol Methods 2001, 251, (1–2), 137–149. [DOI] [PubMed] [Google Scholar]
- 29.Schaefer ZP; Bailey LJ; Kossiakoff AA A polar ring endows improved specificity to an antibody fragment. Protein Sci 2016, 25, (7), 1290–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salchnini LI; Greisen PJ; Wiberg C; Bozoky Z; Lund S; Perez AMW; Karkov HS; Huus K; Hansen JJ; Bulow L; Lorenzen N; Dainiak MB; Pedersen AK Improving the Developability of an Antigen Binding Fragment by Aspartate Substitutions. Biochemistry 2019, 58, (24), 2750–2759. [DOI] [PubMed] [Google Scholar]
- 31.Birtalan S; Zhang Y; Fellouse FA; Shao L; Schaefer G; Sidhu SS The intrinsic contributions of tyrosine, serine, glycine and arginine to the affinity and specificity of antibodies. J Mol Biol 2008, 377, (5), 1518–28. [DOI] [PubMed] [Google Scholar]
- 32.Birtalan S; Fisher RD; Sidhu SS The functional capacity of the natural amino acids for molecular recognition. Mol Biosyst 2010, 6, (7), 1186–94. [DOI] [PubMed] [Google Scholar]
- 33.Tiller KE; Li L; Kumar S; Julian MC; Garde S; Tessier PM Arginine mutations in antibody complementarity-determining regions display context-dependent affinity/specificity trade-offs. J Biol Chem 2017, 292, (40), 16638–16652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Datta-Mannan A; Lu J; Witcher DR; Leung D; Tang Y; Wroblewski VJ The interplay of non-specific binding, target-mediated clearance and FcRn interactions on the pharmacokinetics of humanized antibodies. mAbs 2015, 7, (6), 1084–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hong G; Chappey O; Niel E; Scherrmann JM Enhanced cellular uptake and transport of polyclonal immunoglobulin G and Fab after their cationization. J Drug Target 2000, 8, (2), 67–77. [DOI] [PubMed] [Google Scholar]
- 36.Sharma VK; Patapoff TW; Kabakoff B; Pai S; Hilario E; Zhang B; Li C; Borisov O; Kelley RF; Chorny I; Zhou JZ; Dill KA; Swartz TE In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc Natl Acad Sci U S A 2014, 111, (52), 18601–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Igawa T; Tsunoda H; Tachibana T; Maeda A; Mimoto F; Moriyama C; Nanami M; Sekimori Y; Nabuchi Y; Aso Y; Hattori K Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel 2010, 23, (5), 385–92. [DOI] [PubMed] [Google Scholar]
- 38.Boswell CA; Tesar DB; Mukhyala K; Theil FP; Fielder PJ; Khawli LA Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjug Chem 2010, 21, (12), 2153–63. [DOI] [PubMed] [Google Scholar]
- 39.Arora J; Hu Y; Esfandiary R; Sathish HA; Bishop SM; Joshi SB; Middaugh CR; Volkin DB; Weis DD Charge-mediated Fab-Fc interactions in an IgG1 antibody induce reversible self-association, cluster formation, and elevated viscosity. mAbs 2016, 8, (8), 1561–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perchiacca JM; Lee CC; Tessier PM Optimal charged mutations in the complementarity-determining regions that prevent domain antibody aggregation are dependent on the antibody scaffold. Protein Eng Des Sel 2014, 27, (2), 29–39. [DOI] [PubMed] [Google Scholar]
- 41.Yadav S; Laue TM; Kalonia DS; Singh SN; Shire SJ The Influence of Charge Distribution on Self-Association and Viscosity Behavior of Monoclonal Antibody Solutions. Mol Pharmaceut 2012, 9, (4), 791–802. [DOI] [PubMed] [Google Scholar]
- 42.Yadav S; Sreedhara A; Kanai S; Liu J; Lien S; Lowman H; Kalonia DS; Shire SJ Establishing a Link Between Amino Acid Sequences and Self-Associating and Viscoelastic Behavior of Two Closely Related Monoclonal Antibodies. Pharm Res-Dordr 2011, 28, (7), 1750–1764. [DOI] [PubMed] [Google Scholar]
- 43.Li B; Tesar D; Boswell CA; Cahaya HS; Wong A; Zhang JH; Meng YG; Eigenbrot C; Pantua H; Diao JY; Kapadia SB; Deng R; Kelley RF Framework selection can influence pharmacokinetics of a humanized therapeutic antibody through differences in molecule charge. mAbs 2014, 6, (5), 1255–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nichols P; Li L; Kumar S; Buck PM; Singh SK; Goswami S; Balthazor B; Conley TR; Sek D; Allen MJ Rational design of viscosity reducing mutants of a monoclonal antibody: Hydrophobic versus electrostatic inter-molecular interactions. mAbs 2015, 7, (1), 212–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tomar DS; Kumar S; Singh SK; Goswami S; Li L Molecular basis of high viscosity in concentrated antibody solutions: Strategies for high concentration drug product development. Mabs-Austin 2016, 8, (2), 216–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li L; Kumar S; Buck PM; Burns C; Lavoie J; Singh SK; Warne NW; Nichols P; Luksha N; Boardman D Concentration Dependent Viscosity of Monoclonal Antibody Solutions: Explaining Experimental Behavior in Terms of Molecular Properties. Pharm Res-Dordr 2014, 31, (11), 3161–3178. [DOI] [PubMed] [Google Scholar]
- 47.Yadav S; Shire SJ; Kalonia DS Viscosity behavior of high-concentration monoclonal antibody solutions: Correlation with interaction parameter and electroviscous effects. J Pharm Sci 2012, 101, (3), 998–1011. [DOI] [PubMed] [Google Scholar]
- 48.Buck PM; Chaudhri A; Kumar S; Singh SK Highly Viscous Antibody Solutions Are a Consequence of Network Formation Caused by Domain-Domain Electrostatic Complementarities: Insights from Coarse-Grained Simulations. Mol Pharmaceut 2015, 12, (1), 127–139. [DOI] [PubMed] [Google Scholar]
- 49.Yadav S; Liu J; Shire SJ; Kalonia DS Specific Interactions in High Concentration Antibody Solutions Resulting in High Viscosity. J Pharm Sci-Us 2010, 99, (3), 1152–1168. [DOI] [PubMed] [Google Scholar]
- 50.Yadav DB; Sharma VK; Boswell CA; Hotzel I; Tesar D; Shang YL; Ying Y; Fischer SK; Grogan JL; Chiang EY; Urban K; Ulufatu S; Khawli LA; Prabhu S; Joseph S; Kelley RF Evaluating the Use of Antibody Variable Region (Fv) Charge as a Risk Assessment Tool for Predicting Typical Cynomolgus Monkey Pharmacokinetics. J Biol Chem 2015, 290, (50), 29732–29741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bethea D; Wu SJ; Luo JQ; Hyun L; Lacy ER; Teplyakov A; Jacobs SA; O’Neil KT; Gilliland GL; Feng YQ Mechanisms of self-association of a human monoclonal antibody CNTO607. Protein Eng Des Sel 2012, 25, (10), 531–537. [DOI] [PubMed] [Google Scholar]
- 52.Pepinsky RB; Silvian L; Berkowitz SA; Farrington G; Lugovskoy A; Walus L; Eldredge J; Capili A; Mi S; Graff C; Garber E Improving the solubility of anti-LINGO1 monoclonal antibody Li33 by isotype switching and targeted mutagenesis. Protein Sci 2010, 19, (5), 954–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neergaard MS; Nielsen AD; Parshad H; Van de Weert M Stability of Monoclonal Antibodies at High-Concentration: Head-to-Head Comparison of the IgG(1) and IgG(4) Subclass. J Pharm Sci 2014, 103, (1), 115–127. [DOI] [PubMed] [Google Scholar]
- 54.Tian XS; Langkilde AE; Thorolfsson M; Rasmussen HB; Vestergaard B SmallAngle X-ray Scattering Screening Complements Conventional Biophysical Analysis: Comparative Structural and Biophysical Analysis of Monoclonal Antibodies IgG1, IgG2, and IgG4. J Pharm Sci 2014, 103, (6), 1701–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.