

Abstract
B cell receptor (BCR) signaling is involved in the pathogenesis of B cell malignancies. Activation of BCR signaling promotes the survival and proliferation of malignant B cells. Bruton tyrosine kinase (BTK) is a key component of BCR signaling, establishing BTK as an important therapeutic target. Several covalent BTK inhibitors have shown remarkable efficacy in the treatment of B cell malignancies, especially chronic lymphocytic leukemia. However, acquired resistance to covalent BTK inhibitors is not rare in B cell malignancies. A major mechanism for the acquired resistance is the emergence of BTK cysteine 481 (C481) mutations, which disrupt the binding of covalent BTK inhibitors. Additionally, adverse events due to the off-target inhibition of kinases other than BTK by covalent inhibitors are common. Alternative therapeutic options are needed if acquired resistance or intolerable adverse events occur. Non-covalent BTK inhibitors do not bind to C481, therefore providing a potentially effective option to patients with B cell malignancies, including those who have developed resistance to covalent BTK inhibitors. Preliminary clinical studies have suggested that non-covalent BTK inhibitors are effective and well-tolerated. In this review, we discussed the rationale for the use of non-covalent BTK inhibitors and the preclinical and clinical studies of non-covalent BTK inhibitors in B cell malignancies.
Keywords: BTK, B cell malignancies, Ibrutinib, C481 mutations, Non-covalent inhibitors
Background
B cell receptor (BCR) signaling plays a crucial role in B cell development and adaptive immune response and also contributes to the pathogenesis of different types of B cell malignancies [1, 2]. The BCR signaling cascades involve several essential kinases, including spleen tyrosine kinase (SYK), Bruton tyrosine kinase (BTK), and phosphatidylinositol-3-kinase (PI3K) (Fig. 1) [1]. Briefly, BCR ligation by antigen leads to phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM) of CD79A and CD79B, thereby recruiting SYK [1]. SYK then phosphorylates and activates BTK. BTK activation initiates further downstream signaling pathways including nuclear factor-κB (NF-κB) pathway, MAPK/ERK pathway, and other pathways.
Fig. 1.

BCR signaling. The binding of antigens to the B cell receptor leads to the phosphorylation of the intracellular immunoreceptor tyrosine-based activation motifs (ITAMs) of CD79A and CD79B. The phosphorylation of CD79A/CD79B initiates SYK activation, which then results in BTK activation and subsequent PLCG2 activation. This signal cascade ultimately leads to the activation of NF-κB and MAPK/ERK pathways, contributing to the survival and proliferation of CLL cells. BTK and PLCG2 mutations are detected in BTK inhibitor-resistant CLL cases
The essential role of BTK in BCR signaling makes it an ideal target for suppressing BCR signaling. BTK was originally identified as a non-receptor protein tyrosine kinase in 1993 [3, 4]. BTK is a member of the Tec family kinases, which contain interleukin-2-inducible T cell kinase (ITK), tyrosine kinase expressed in hepatocellular carcinoma (TEC), resting lymphocyte kinase (RLK), and bone marrow expressed kinase (BMX)[5]. BTK loss-of-function mutations result in X-linked agammaglobulinemia, a type of immunodeficiency that is characterized by the lack of mature B cells and immunoglobulins and consequent opportunistic infections in young boys [3, 4], highlighting the importance of BTK in B cell development and humoral immunity. The process of B cell development and the role of BCR in this process are described in Fig. 2. BTK comprises five different protein interaction domains, which include an amino-terminal pleckstrin homology (PH) domain, a proline-rich TEC homology (TH) domain, SRC homology (SH) domains SH2 and SH3, and a kinase domain (Fig. 3) [6, 7]. BTK has two critical tyrosine phosphorylation sites, Y223 in the SH3 domain and Y551 in the kinase domain. During BCR signaling, the phosphorylation by SYK at Y551 enhances the catalytic activity of BTK and initiates subsequent Y223 autophosphorylation [7]. BTK inhibitors, including covalent and non-covalent inhibitors, bind to the BTK kinase domain and block the catalytic activity of BTK, thereby suppressing subsequent Y223 autophosphorylation [6]. Several covalent BTK inhibitors, including ibrutinib, acalabrutinib, zanubrutinib, and orelabrutinib, have been developed for targeting BTK in B cell malignancies [8–11]. These covalent inhibitors have shown remarkable efficacy in B cell malignancies, including chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL), Waldenström's macroglobulinemia (WM), and marginal zone lymphoma (MZL). However, treatment failure due to drug resistance or adverse events (AEs) is not rare in clinical practice. Overcoming drug resistance and reducing severe AEs are of vital importance to improve the outcomes of patients. Using non-covalent BTK inhibitors could be a strategy for overcoming drug resistance and reducing adverse events. In this review, we discussed the rationale of the application of non-covalent BTK inhibitors and their preclinical and clinical studies in B cell malignancies.
Fig. 2.

BCR in B cell development. In the bone marrow, progenitor B (pro-B) cells undergo the rearrangement and development of immunoglobulin heavy-chain variable (V), diversity (D), and joining (J) gene segments to form the pre-BCR. The pre-BCR is an immature form of the BCR providing signals for survival, proliferation, and cellular differentiation. After light-chain gene rearrangement occurs, immature B cells express BCR, leave the bone marrow, and mature in the periphery. Mature B cells undergo somatic hypermutation (SHM) driven by the expression of activation-induced cytidine deaminase (AID) in the germinal center (GC) to complete BCR affinity maturation and antibody diversification. The genotoxic stress induced by SHM may lead to the apoptosis of these B cells. However, the continuous BCR signaling could provide pro-survival signals for these B cells, thereby preventing them from apoptosis. Therefore, B cells deficient in Bruton tyrosine kinase tend to undergo apoptosis during the development. The B cells that have completed affinity maturation and antibody diversification then undergo class-switch recombination (CSR), after which they develop into memory B cells with high-affinity BCRs or plasma cells secreting antibodies. DZ, dark zone; LZ, light zone; FDC, follicular dendritic cell; Tfh cell, T follicular helper cell
Fig. 3.

The structural diagram of Bruton tyrosine kinase (BTK). The BTK protein is a 77 kDa protein of 659 amino acids, which contains five different protein interaction domains. There are two critical tyrosine phosphorylation sites, Y223 in the SH3 domain and Y551 in the kinase domain. BTK inhibitors bind to the BTK kinase domain and blocks the catalytic activity of BTK. Currently available covalent BTK inhibitors, including ibrutinib, acalabrutinib, zanubrutinib, and orelabrutinib, selectively bind to C481 residue in the allosteric inhibitory segment of the BTK kinase domain. The non-covalent BTK inhibitors do not bind to C481. For example, ARQ 531 binds to BTK by forming hydrogen bonds with E475 and Y476 residues [56]. Fenebrutinib forms hydrogen bonds with K430, M477, and D539 residues [50]
Covalent BTK inhibitors
Several covalent BTK inhibitors, which include ibrutinib, acalabrutinib, and zanubrutinib, have been tested in clinical trials and have been approved for treating patients with B cell malignancies. The results of phase III studies with covalent BTK inhibitors are summarized in Table 1. Ibrutinib (PCI-32765) is an irreversible, highly potent small molecule BTK inhibitor that covalently binds to cysteine481 (C481) in the active site of BTK and blocks the full activation of BTK by inhibiting its autophosphorylation at tyrosine 223, suppressing signaling downstream of BTK (Fig. 4a, c) [12]. Ibrutinib has shown remarkable efficacy in CLL, MCL, WM, and MZL [8, 10, 13–16]. The use of ibrutinib has revolutionized the treatment of B cell malignancies, especially CLL. Several phase III trials have demonstrated the superiority of ibrutinib monotherapy in both relapsed/refractory and treatment-naïve CLL patients [8, 14–18]. A recent pooled analysis of four clinical trials showed that first-line ibrutinib treatment resulted in high long-term efficacy (progression-free survival 79% and overall survival 88% at 48 months) in CLL patients with TP53 aberrations, a group of patients with historically poor prognosis [19]. Combinations of ibrutinib with other novel drugs or regimens result in more profound responses and much higher rates of minimal residual disease (MRD) negativity [20, 21]. Other covalent BTK inhibitors, including acalabrutinib and zanubrutinib [9, 22], have also shown promising efficacy in B cell malignancies. Although the covalent BTK inhibitors are effective in B cell malignancies, resistance to these BTK inhibitors, including primary and acquired resistance, is frequent in patients with B cell malignancies [23]. The major mechanisms for the resistance to the covalent BTK inhibitors are summarized in Table 2. Here, we briefly discussed the mechanisms for the acquired resistance to covalent BTK inhibitors.
Table 1.
Published phase III studies of covalent BTK inhibitors in B cell malignancies
| Drug | Number | Patients | Regimen | Response rates | Survival |
|---|---|---|---|---|---|
| Ibrutinib | NCT02165397 | TN or R/R WM (n = 150) | IR versus placebo + RTX | IR: MR 72%, CR 3%, VGPR 23%; Placebo plus RTX: MR 32%, CR 1%, VGPR 4% | IR: PFS 68% and OS 86% at 54 mon; placebo plus RTX: PFS 25% and OS 84% at 54 mon [72, 73] |
| Ibrutinib | NCT01611090 | R/R CLL/SLL (n = 578) | Ibrutinib + BR versus placebo + BR | Ibrutinib + BR: ORR 87.2%, CR/CRi 38.1%; placebo + BR: ORR 66.4%, CR/CRi: 8.0% | Ibrutinib + BR: PFS 68.0% and OS 81.6% at 3 yr; Placebo + BR: PFS 13.9% and OS 72.9% at 3 yr [74] |
| Ibrutinib | NCT01578707 | R/R CLL/SLL (n = 391) | Ibrutinib versus ofatumumab | Ibrutinib: ORR 91%, CR/CRi 11%; ofatumumab: ORR 4.1%, CR/CRi 1%; | Ibrutinib: median PFS 44.1 mon and median OS 67.7 mon; ofatumumab: median PFS 8.0 mon and median OS 65.1 mon [17, 18] |
| Ibrutinib | TN CLL/SLL, age ≥ 65 yr (n = 269) | Ibrutinib versus chlorambucil | Ibrutinib: ORR 92%, CR/CRi 30%; chlorambucil: ORR: 37%, CR/CRi 2% | Ibrutinib: PFS 70% and OS 83% at 5 yr; chlorambucil: PFS 12% and OS 68% at 5 yr [8] | |
| Ibrutinib | NCT01886872 | TN CLL, age ≥ 65 yr (n = 547) | Ibrutinib versus IR versus BR |
Ibrutinib: ORR 93%, CR 7% IR: ORR: 94%; CR:12% BR: ORR: 81%; CR:26% |
Ibrutinib: PFS 87% and 90% at 2 yr; IR: PFS 88% and OS 94% at 2 yr; BR: PFS 74% and OS 95% at 2 yr [16] |
| Ibrutinib | NCT02048813 | TN CLL/SLL, age ≤ 70 yr (n = 529) | IR versus FCR |
IR: ORR 95.8%, CR/CRi 17.2% FCR: ORR 81.1%, CR/CRi 30.3% |
IR: PFS 89.4% and OS 98.8% at 3 yr FCR: PFS 72.9% and OS: 91.5% at 3 yr [15] |
| Ibrutinib | NCT02264574 | TN CLL/SLL (n = 229) | Ibrutinib + GA-101 versus chlorambucil + GA-101 | Ibrutinib + GA-101: ORR 88%, CR/CRi 19%; chlorambucil + GA-101: ORR 73%, CR/CRi 8% | Ibrutinib + GA-101: PFS 79% and OS 86% at 30 mon; chlorambucil + GA-101: PFS: 31% and OS 85% at 30 mon [14] |
| Ibrutinib | NCT01646021 | R/R MCL (n = 280) | Ibrutinib versus temsirolimus | Ibrutinib: ORR 72%, CR 19%; temsirolimus: ORR 40% CR 1% | Ibrutinib: median PFS 15.6 mon and median OS 30.3 mon; temsirolimus: median PFS 6.2 mon and median OS 23.5 mon [13] |
| Acalabrutinib | NCT02475681 | TN CLL (n = 535) | Acalabrutinib versus acalabrutinib + GA-101 versus chlorambucil + GA-101 | Acalabrutinib: ORR 94%, CR/CRi 24%; acalabrutinib + GA-101: ORR 85%, CR/CRi 1%; chlorambucil + GA-101: ORR 79%, CR/CRi 5%; | Acalabrutinib: PFS 87% at OS 95% at 24 mon; acalabrutinib + GA-101: PFS 93% and OS 95% at 24 mon; chlorambucil + GA-101: PFS 47% and OS 92% at 24 mon [9] |
| Ibrutinib zanubrutinib | NCT03053440 | TN or R/R WM with MYD88L265P (n = 201) | Ibrutinib versus zanubrutinib |
Ibrutinib: MR 78%, VGPR: 19% zanubrutinib: MR 77%, VGPR 28% |
Ibrutinib: PFS 84% and OS 93% at 18 mon; zanubrutinib: PFS 85% and OS 97% at 18 mon [10] |
| Ibrutinib | NCT01855750 | TN non-GCB DLBCL (n = 838) | Ibrutinib + R-CHOP versus R-CHOP | Ibrutinib + R-CHOP: ORR 87.3%, CR 67.3%; R-CHOP: ORR 93.1%, CR 68.0% |
For age < 60 yr: Ibrutinib + R-CHOP: PFS 77.4% and OS 93.2% at 3 yr; R-CHOP: PFS 66.3% and OS 80.9% at 3 yr For age ≥ 60 yr: Ibrutinib + R-CHOP: PFS 66.8% and OS 76.6% at 3 yr; R-CHOP: PFS 69.6% and OS 81.7% at 3 yr [75] |
TN treatment naïve, R/R relapsed/refractory, WM Waldenstrom's macroglobulinemia, IR ibrutinib and rituximab, RTX rituximab, MR major response, CR complete response, CRi complete response with incomplete hematologic recovery, VGPR very good partial response, PFS progression free survival, OS overall survival, mon months, CLL/SLL chronic lymphocytic leukemia/small lymphocytic lymphoma, BR bendamustine and rituximab, ORR overall response rate, yr years, FCR fludarabine, cyclophosphamide, and rituximab, MCL mantle cell lymphoma, non-GCB Non-germinal center B cell, R-CHOP rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone
Fig. 4.

Mechanisms for the action of representative BTK inhibitors. a Chemical structure of the covalent BTK inhibitor ibrutinib. b Chemical structure of the non-covalent BTK inhibitor ARQ 531. c Ibrutinib covalently binds to BTK cysteine 481 (C481), competes with ATP in the ATP binding pocket, and inhibits autophosphorylation of BTK. The action of ibrutinib requires its binding to BTK C481, and BTK C481 mutations abrogate the binding of ibrutinib and lead to the resistance to ibrutinib. d ARQ 531 non-covalently interacts with BTK, occupies the ATP binding pocket, and inhibits BTK autophosphorylation. The effect of ARQ 531 does not require its binding to BTK C481. Therefore, ARQ 531 remains active in patients with BTK C481 mutations. c, d were made by using PyMOL 0.99
Table 2.
Resistance mechanisms to BTK inhibitors in B cell malignancies
| B cell malignancy subtype | Covalent BTK inhibitors | Mechanisms underlying resistance |
|---|---|---|
| Primary resistance | ||
| MCL | Ibrutinib |
Mutations involving NF-κB pathway: A20 mutations, TRAF2 mutations, BIRC3 mutations or BIRC2 mutations, RELA E39Q mutation, and others [76, 77] Sustained PI3K/AKT/mTOR activation [28, 78] Tumor microenvironment [79] Metabolic reprogramming toward oxidative phosphorylation and glutaminolysis [80] CCND1 mutation [81] |
| WM | Ibrutinib | CXCR4 WHIM-like mutations [82] |
| DLBCL | Ibrutinib |
PIM1 mutation [83] PI3K/AKT activation [84] MAPK activation [84] Aberrations activating NF-κB pathway: CARD11 mutation, A20 aberrations [85] High expression of PDGFD [86] |
| Acquired resistance | ||
| CLL/SLL | Ibrutinib |
BTK C481 and T474 mutations [24, 87, 88] PLCG2 mutations (R665W, S707, L845F, and others) [87] del(8p) [89] |
| CLL/SLL | Acalabrutinib | BTK C481 mutations and T474I mutation, PLCG2 mutations [29] |
| CLL/SLL | Zanubrutinib | BTK Leu528Trp mutation and C481 mutation [31] |
| MCL | Ibrutinib |
BTK C481S mutation [28] PLCG2 mutations [76] CARD11 mutation [76] Tumor microenvironment [79] |
| WM | Ibrutinib |
BTK C481 mutations [26] PLCG2 Tyr495His mutation [26] |
| MZL | Ibrutinib |
BTK C481S mutation [90] PLCG2 R665W [90] |
| DLBCL | Ibrutinib | BTK C481S mutation [91] |
BTK Bruton tyrosine kinase, MCL mantle cell lymphoma, WM Waldenstrom's macroglobulinemia, DLBCL diffuse large B cell lymphoma, CLL/SLL chronic lymphocytic leukemia/small lymphocytic lymphoma, MZL marginal zone lymphoma
Acquired resistance to BTK inhibitors
Despite the clinical success ibrutinib has achieved, it should be noted that CLL progression or Richter transformation (RT) occurs in a subset of CLL patients administrated with ibrutinib [24]. Most of the patients who experience CLL relapse have BTK C481 mutations and less commonly PLCG2 mutations [24]. The BTK C481 mutations, most of which are BTK C481S, involve the cysteine where ibrutinib binding occurs, rendering ibrutinib unable to inhibit BTK and downstream pathways. BTK C481 mutations are also detected in approximately 30% of patients who underwent RT on ibrutinib, suggesting BTK C481 mutations could be involved in mediating RT on ibrutinib [25]. Ibrutinib resistance has also been observed in WM patients with active ibrutinib therapy. BTK C481 mutations are commonly observed in WM patients who experienced progression, suggesting BTK C481 mutations are also responsible for ibrutinib resistance in WM [26]. Both primary resistance (10.2–35%) and acquired resistance (17.5–54%) to ibrutinib are common in ibrutinib-treated patients with MCL [27]. Acquired BTK C481S mutation is detected in a small proportion of MCL patients who relapsed after ibrutinib therapy [28].
Disease progression due to drug resistance has also been reported in patients treated with other covalent inhibitors. According to the study by Woyach et al., with a median follow-up of 47.5 months, CLL relapse occurred in 17 of 105 CLL patients on acalabrutinib therapy [29]. BTK C481 mutations were identified in 11 of these 16 patients, indicating that the mechanism for acalabrutinib resistance is similar to that for ibrutinib resistance [29]. Thirty-one percent of R/R MCL patients on acalabrutinib had disease progression with a median follow-up of only 15.2 months. The mechanisms responsible for acalabrutinib resistance in MCL have not been characterized yet [30]. The acquired resistance also occurs in CLL patients treated with zanubrutinib, although at a relatively low rate [22]. CLL patients progressing on zanubrutinib had concurrent BTK Leu528Trp and BTK C481 mutations [31]. The co-occurrence of BTK Leu528Trp and BTK C481 mutations suggests these mutations may cooperate in mediating resistance to zanubrutinib.
Adverse events of covalent BTK inhibitors
In addition to ibrutinib resistance, ibrutinib intolerance is also a concern in patients treated with ibrutinib, frequently leading to ibrutinib discontinuation [32, 33]. Some kinases also harbor a modifiable cysteine residue that is homologous to C481 in BTK; therefore, these kinases could also be inhibited by ibrutinib [34]. A variety of receptor tyrosine kinases and non-receptor tyrosine kinases, including EGFR, ITK, TEC, ERBB4, BMK, JAK3, and HER2 [35], are covalently inhibited by ibrutinib. The inhibition of these non-BTK kinases accounts for part of the AEs related to ibrutinib. The common AEs in patients treated with ibrutinib include diarrhea, bleeding, atrial fibrillation, infection, and others, and serious AEs may cause discontinuation. According to a pooled analysis, diarrhea occurred in approximately half of the CLL patients treated with ibrutinib and 5% of patients had grade 3 diarrhea [36]. Ibrutinib-related diarrhea could be attributed to the inhibition of EGFR [37]; however, the exact mechanism remains to be determined. Bleeding/bruising events occurred in 55% of CLL patients on ibrutinib therapy, and 7.6% of patients experienced major hemorrhage events [36]. The inhibition of BTK and other Tec family kinases impairs glycoprotein VI signalings, suppressing platelet aggregation, and thereby contributing to bleeding events [38–40]. Approximately 11% of CLL patients developed atrial fibrillation during ibrutinib monotherapy, and 5% of patients developed grade 3 atrial fibrillation [36]. It should be noted that the frequency of atrial fibrillation is various in different studies, with younger patients having a lower rate of atrial fibrillation (7.4% in the E1912 study) [15]. Ibrutinib inhibits BTK and TEC in cardiac tissue, resulting in the downregulation of the cardiac PI3K/AKT pathway. It has been reported that reduced PI3K/AKT activity increased the susceptibility to atrial fibrillation [41]. Therefore, ibrutinib may lead to atrial fibrillation by inhibiting the PI3K/AKT pathway in cardiac tissue [42]. Infection was prevalent (83%) in CLL patients on ibrutinib therapy, and grade 3 or 4 infection occurred in 29% of patients [36]. The significantly increased risk of infection could be caused by immune impairment, which is attributed to the inhibition of ITK in T cells and BTK in macrophages [43]. Other common AEs include arthralgia, fatigue, hypertension, and rash. Consistently, the second-generation covalent BTK inhibitors, including acalabrutinib, zanubrutinib, and orelabrutinib, bind to C481 residue. Current studies have shown that the second-generation covalent BTK inhibitors have higher specificity and fewer off-target toxicities, while still cause adverse events. For instance, headache (43%) and diarrhea (39%) were very common in acalabrutinib-treated relapsed CLL patients [44]. Further, the follow-up of patients treated with these second-generation covalent BTK inhibitors is relatively short compared to those treated with ibrutinib, so that longer follow-up is warranted to observe potential adverse events.
Non-covalent BTK inhibitors in B cell malignancies
Non-covalent BTK inhibitors do not bind to C481 (Figs. 3, 4d), therefore providing a potentially effective option to patients with B cell malignancies, including those who are resistant to covalent BTK inhibitors due to BTK C481 mutations. These non-covalent BTK inhibitors could administrate to patients who have not received covalent BTK inhibitors previously to reduce the risk of acquired resistance; meanwhile, they may also have fewer side effects caused by the off-target inhibition of kinases other than BTK. Several non-covalent BTK inhibitors have been studied and have shown promising efficacy and manageable safety profiles. The clinical trials involving non-covalent BTK inhibitors are summarized in Table 3.
Table 3.
Clinical trials with non-covalent BTK inhibitors
| Non-covalent BTK inhibitor | Phase | Patient population | Strategy | ClinicalTrials.gov ID | Status |
|---|---|---|---|---|---|
| Vecabrutinib | I/II dose escalation and expansion trial | CLL/SLL or NHL (including LPL/WM, MCL, MZL, ABC-DLBCL, FL) patients who have failed prior standard of care therapies including a BTK inhibitor where one is approved for the indication | Monotherapy | NCT03037645 | Terminated |
| ARQ 531 | I/II dose escalation and expansion trial | Dose escalation cohorts: R/R CLL/SLL, FL, MCL, MZL, and WM patients who have received ≥ 2 prior systemic therapies; expansion cohorts include R/R CLL/SLL after ≥ 2 prior systemic therapies including a covalent BTKi, with or without a C481 mutation | Monotherapy | NCT03162536 | Recruiting |
| Fenebrutinib | Ia dose escalation trial | R/R NHL (including FL, DLBCL, MCL, PLL/WM) or CLL | Monotherapy | NCT01991184 | Active, not recruiting |
| LOXO-305 | I/II dose escalation and expansion trial | CLL/SLL or NHL patients who have failed or are intolerant to standard of care | Monotherapy; combination therapy | NCT03740529 | Recruiting |
| LOXO-305 | III | MCL patients who have been previously treated with at least one prior line of systemic therapy and were BTK inhibitor naive | Monotherapy (versus investigator choice of BTK Inhibitor) | NCT04662255 | Not yet recruiting |
| LOXO-305 | III | CLL/SLL patients who were previously treated with a covalent BTK inhibitor | Monotherapy (versus idelalisib plus rituximab or bendamustine plus rituximab) | NCT04666038 | Not yet recruiting |
CLL/SLL chronic lymphocytic leukemia/small lymphocytic lymphoma, NHL non-Hodgkin's lymphoma, LPL lymphoplasmacytic lymphoma, WM Waldenstrom's macroglobulinemia, MCL mantle cell lymphoma, MZL marginal zone lymphoma, DLBCL diffuse large B cell lymphoma, ABC-DLBCL the activated B cell subtype of DLBCL, FL follicular lymphoma, BTK Bruton tyrosine kinase, R/R relapsed and/or refractory, PLL prolymphocytic leukemia
Vecabrutinib (SNS-062)
Vecabrutinib is a selective, reversible, non-covalent BTK inhibitor with nanomolar potency. In vitro studies have demonstrated vecabrutinib shows activity against both wild-type and C481S-mutated BTK. Vecabrutinib decreases surface expression of B cell activation markers and viability of primary cells from CLL patients, resulting in a phenotypic alteration that is comparable to ibrutinib [45]. Vecabrutinib also inhibits ITK but not EGFR; therefore, it may have less EGFR-mediated toxicities including rash and diarrhea [46].
A phase I study of vecabrutinib that enrolled 32 healthy participants was completed [47]. All AEs were grade 1, including headache (n = 5) and nausea, constipation, bronchitis, fatigue, orthostatic hypotension, and supraventricular tachycardia (n = 1 each), except for 1 subject who received 300 mg vecabrutinib experiencing grade 2 headache and fatigue [47]. The occupation produced by vecabrutinib and duration of BTK inhibition is encouraging [47]. A phase Ib/II dose-escalation and cohort-expansion study is ongoing in patients with relapsed/refractory advanced B cell malignancies who progressed on covalent BTK inhibitor therapy. According to the reported data, 27 patients (CLL, n = 21; MCL, n = 2; WM, n = 3; MZL = 1) have been treated with doses ranging from 25 to 300 mg, twice daily [48]. The maximum tolerated dose (MTD) has not been reached. Data regarding safety are available for 24 patients; the most frequent AEs included anemia (37.5%), neutropenia (25%), night sweats (25%), and headache (25%) [48]. Grade 3 drug-related AEs included alanine aminotransferase (ALT) elevation, neutropenia and worsening anemia (all in 1 patient), and leukocytosis (2 patients). Regarding efficacy, no response was observed, and 4 CLL patients, including three with BTK C481S mutation, showed stable disease. Preliminary results revealed that vecabrutinib in dose levels from 25 to 200 mg twice daily was safe in patients with B cell malignancies. The 300 mg twice daily dose level was being evaluated when the study was presented at the 2019 American Society of Hematology (ASH) meeting [48]. Vecabrutinib shows manageable safety profiles, while its efficacy in patients with B cell malignancies remains to be explored.
Fenebrutinib (GDC-0853)
Fenebrutinib is a highly selective, reversible, non-covalent BTK inhibitor that does not bind to the C481 residue for its action and does not inhibit EGFR or ITK [49, 50]. Fenebrutinib potently inhibits BCR signaling through BTK inhibition. In vitro studies showed that fenebrutinib decreased the activation of BTK and its downstream targets upon stimulation with αIgM and reduced viability, NF-κB gene transcription, activation, and migration in CLL cells [49]. Fenebrutinib inhibits C481S BTK mutant that mediates ibrutinib resistance and is toxic to CLL cells with BTK C481S mutation. Unlike ibrutinib, which antagonizes rituximab-mediated NK cell–mediated cytotoxicity through ITK inhibition, fenebrutinib does not inhibit ITK and preserves NK cell-mediated cytotoxicity that is dependent on anti-CD20 antibodies [51]. Thus, exploration of fenebrutinib as monotherapy and in combination with anti-CD20 antibodies is promising, especially in patients with acquired resistance to ibrutinib [49, 52].
Fenebrutinib was well-tolerated with favorable safety, selectivity, and pharmacokinetic/pharmacodynamic (PK/PD) profiles in healthy volunteers [53]. A phase I study has evaluated fenebrutinib in 24 patients with relapsed/refractory B cell malignancies (CLL, n = 14; follicular lymphoma [FL], n = 4; diffuse large B cell lymphoma [DLBCL], n = 3; MCL, n = 2; prolymphocytic leukemia plus WM, n = 1) [54]. The enrolled patients were treated at 100, 200, or 400 mg once daily, orally. There was no dose-limiting toxicity. This phase I trial of fenebrutinib was prematurely terminated during dose escalation and the MTD was not reached. The most common AEs included fatigue (37%), nausea (33%), diarrhea (29%), thrombocytopenia (25%), and headache (20%). Eight of 24 patients had a response to fenebrutinib. Of these 8 patients who responded, an MCL patient achieved complete response (CR) and 7 CLL patients achieved partial response (PR) or PR with lymphocytosis (PR-L), including 1 of 5 heavily pretreated patients with BTK C481S mutant CLL [54]. Two additional patients with BTK C481S mutation showed a decrease in the size of target tumors (− 23% and − 44%). The median duration of response in all responding patients and patients with CLL is 3.8 months and 2.5 months, respectively. As the MTD that may provide greater BTK inhibition was not reached, the short duration of response should be interpreted cautiously. Despite this, this study provides evidence of clinical activities of reversible non-covalent BTK inhibitors in B cell malignancies [54].
ARQ 531
ARQ 531 is a potent, reversible inhibitor of both wild-type (WT) and mutant BTK (WT, IC50 = 0.85 nM; C481S, IC50 = 0.39 nM) with additional activities against SRC family kinases, ERK, and AKT (Fig. 4b, d). In vitro, ARQ 531 suppresses BTK-dependent functions including BCR signaling and transcription of NF-κB genes, thereby suppressing viability, cell activation, and migration of primary CLL cells [55]. ARQ 531 potently inhibits C481S-mutated BTK and downstream signaling and is also toxic to BTK C481S-mutated CLL cells [55]. In ibrutinib-resistant CLL cells with mutant PLCG2, PLCG2 can be directly activated by LYN and SYK, thereby bypassing the activation by BTK. ARQ 531 effectively inhibits signaling downstream of mutant PLCG2 and is toxic to ibrutinib-resistant CLL cells with PLCG2 mutations. In vivo studies have demonstrated the superiority of ARQ 531 over ibrutinib in mouse models of CLL and RT [56].
A phase I dose-escalation study of ARQ 531 has been completed in patients with relapsed or refractory B cell malignancies [57]. Totally, 40 patients (CLL/SLL, n = 26; RT, n = 6; DLBCL, n = 3; FL, n = 4; MCL, n = 1) were enrolled [57]. The enrolled patients were treated with a median of 4 prior therapies and were all previously treated with an irreversible BTK inhibitor. Most patients with CLL (22/26, 85%) had BTK C481S mutation. Doses of 5, 10, 15, 20, 30, 45, 65, and 75 mg daily were used. ARQ 531 was well tolerated, and most of the drug-related treatment emergent AEs (TEAEs) were grade 1 or 2. Partial response was achieved in 10 patients, including patients with CLL (n = 7), RT (n = 1), DLBCL (n = 1), and FL (n = 1) [57]. Patients that responded to ARQ 531 were mainly from the higher dose cohorts [65 mg daily]. ARQ 531 at 65 mg daily showed manageable safety profiles and significant anti-tumor efficacy; therefore, 65 mg daily was determined as the recommended phase 2 dose in patients with B cell malignancies [57]. This trial suggests ARQ531 could be an effective therapeutic option for patients with relapsed or refractory B cell malignancies, including BTK C481S mutated CLL cases that are resistant to covalent BTK inhibitors. The phase I b expansion portion at 65 mg daily of this study is ongoing, and updated data are pending [57].
ARQ 531 may be also effective in other types of hematological malignancies. ARQ 531 could target multiple pathways including BTK, MYB, AKT, ERK, and other pathways in acute myeloid leukemia (AML) [58]. In preclinical models of AML, ARQ 531 showed in vitro and in vivo activities against AML [59]. ARQ 531 was demonstrated to be synergistic with venetoclax in AML xenograft model [59], suggesting the combination of ARQ 531 with venetoclax could be a potential therapeutic option in the treatment of AML.
LOXO-305
LOXO-305 is a next-generation, reversible BTK inhibitor, which potently inhibits both WT and C481S mutant BTK with nanomolar potency and shows high selectivity with minimal off-target inhibition [60]. LOXO-305 potently inhibits Y223 autophosphorylation of all active BTK mutants (BTK C481S, C481T, and C481R). In vitro studies showed that LOXO-305 potently led to inhibition of BCR signaling and cell survival in both treatment-naive and BTK C481 mutant CLL primary cells, indicating that LOXO-305 could be used for treating treatment-naïve and ibrutinib-resistant CLL patients [60–62]. In high proliferating tumors, high rates of BTK turnover may result in incomplete target inhibition by covalent inhibitors, which ultimately lead to resistance to these covalent BTK inhibitors. LOXO-305 achieves remarkable target coverage even in the presence of high rates of BTK turnover, providing a rationale for using LOXO-305 in aggressive B cell lymphomas including DLBCL.
A multicenter phase I/II BRUIN trial evaluating oral LOXO-305 in patients with previously treated B cell malignancies is currently ongoing [63, 64]. Preliminary results were reported at the 2020 ASH meeting [63, 64]. A total of 186 patients include 94 patients with CLL/SLL, 38 with MCL, 19 with DLBCL, 17 with WM, 6 with FL, 5 with MZL, and 7 patients with other (B-PLL and Richter’s transformation) [63]. A 3 + 3 dose-escalation design was used, and patients were treated on 7 dose levels (25 mg to 300 mg QD) [15]. The enrolled patients were heavily treated, and the median number of prior therapies was 4 for CLL/SLL (range 1–10), 2 for MCL (range 2–8), and 3 for other NHLs (range 2–11). In total, 84% of CLL/SLL patients were previously treated with a BTK inhibitor and 31% venetoclax. And 92% of MCL patients had received a prior BTK inhibitor [64]. LOXO-305 demonstrated high oral bioavailability, with doses ≥ 100 mg QD resulting in higher than 90% of the maximum inhibition for the entire dosing interval [63]. There was no dose-limiting toxicity or dose reductions. The only emergent AEs related to LOXO-305 were fatigue (n = 29, 16%) and diarrhea (n = 28, 15%). A recommended phase 2 dose of 200 mg QD was selected as the recommended dose for future studies. The clinical activity of LOXO-305 was demonstrated within the first cycle of therapy and at the first dose level. Among the 94 CLL/SLL patients, 88 patients remained on therapy. And among the 65 CLL/SLL patients that were efficacy-evaluable (58 BTK inhibitor-treated, 7 BTK inhibitor-naïve), the overall response rate (ORR) was 57% with 23 PRs and 14 PR-Ls [63]. The therapeutic efficacy of LOXO-305 in MCL patients was also remarkable. Among 35 efficacy-evaluable MCL, the ORR was 51% with 9 CRs and 9 PRs [64]. For other NHL patients, 15 DLBCL patients (ORR: 20%, with 3 CRs), 5 FL patients (ORR: 60%, with 3 PRs), 3 MZL patients (ORR: 67%, with 2 PRs), and 6 other patients (ORR: 33%, with 2 PRs) were efficacy-evaluable [64]. This study suggested that LOXO-305 was well-tolerated and was effective in patients with heavily pretreated CLL/SLL and NHLs, including those who had developed resistance to ibrutinib and venetoclax. However, longer follow-up and a larger number of patients are needed to determine its efficacy and safety.
Other non-covalent BTK inhibitors
In addition to the several non-covalent BTK inhibitors that have shown promising efficacy in clinical trials, there are also some other non-covalent BTK inhibitors with significant antitumor effects in preclinical studies (Table 4). For instance, XMU-MP-3, a non-covalent inhibitor with potent BTK inhibitory activity, inhibited B cell lymphoma cells with or without BTK C481S mutation in vitro and in vivo [65], suggesting it could be effective in treating B cell lymphomas including those resistant to ibrutinib. Several other non-covalent BTK inhibitors including CB1763, GNE-431, and CGI-1746 have shown potent inhibitory effects on both wildtype and C481S mutant BTK [66, 67]. Further clinical trials are warranted to investigate the safety and efficacy of these novel agents.
Table 4.
Representative non-covalent BTK inhibitors in preclinical stage
| Novel non-covalent BTK Inhibitor | Chemical structure | Molecular Formula | In vitro | In vivo | References |
|---|---|---|---|---|---|
| XMU-MP-3 |
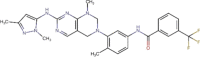
|
C27H27F3N8O | XMU-MP-3 is a potent BTK inhibitor with IC50 of 10.7 nM for WT BTK. It also effectively inhibits C481S mutant BTK in vitro. XMU-MP-3 suppresses the proliferation of BTK-transformed Ba/F3 cell with an IC50 of 11.4 nM. It remains active against C481S mutant BTK-transformed Ba/F3 cells with an IC50 of 182.3 nM | XMU-MP-3 remarkably inhibits tumor growth in BTK-transformed Ba/F3 and Ramos in mouse xenograft models without affecting animal weights | [65] |
| CB1763 (also known as AS-1763) | NA | NA | CB1763 potently, reversibly inhibits both WT and C481S mutant BTKs (IC50 = 0.85 and 0.99 nM for WT and C481S, respectively). CB1763 substantially reduces BTK Tyr223 autophosphorylation at nanomolar concentration in HEK293 cells that are transfected with C481S mutant BTK | CB1763 shows excellent antitumor activity in the BTK-driven OCI-Ly10 xenograft model | [92] |
| GNE-431 |
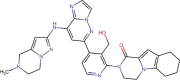
|
C30H32N10O2 | GNE-431 potently inhibits WT BTK and C481S mutant BTK (IC50 = 3.2 and 2.5 nM for WT and C481S mutant, respectively). Additionally, GNE-431 shows high potency against several other BTK mutants including C481R, T474I, and T474M mutants (IC50 = 7.5–10 nM). GNE-431 potently suppresses BTK autophosphorylation in C481S BTK mutant-transfected cells | No in vivo data regarding the activity of GNE-431 in B cell malignancies to date | [66] |
| CGI-1746 |
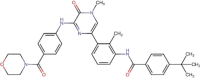
|
C34H37N5O4 | By occupying an H3 binding pocket, CGI-1746 stabilizes an inactive conformation of BTK. CGI-1746 reversibly and selectively inhibits BTK with IC50 of 8.9 nM and 16 nM for WT BTK and C481S mutant, respectively. CGI-1746 induces significant cell cycle arrest of Ramos cells in vitro | No in vivo data regarding the activity of CGI-1746 in B cell malignancies to date | [67, 93] |
BTK Bruton tyrosine kinase, IC50 half-maximal inhibitory concentration, nM nmol/l, WT wildtype, NA not available
Future directions
Although some non-covalent BTK inhibitors have shown promising efficacy in the treatment of relapsed or refractory B cell malignancies, the currently available data are all from phase I or II studies with a small number of participants. Phase II studies that have larger enrollment are needed to verify the safety and efficacy of non-covalent BTK inhibitors. In addition to patients who have developed resistance to covalent BTK inhibitors, patients who are unable to tolerate covalent BTK inhibitors may also benefit from non-covalent BTK inhibitors, which remains to be verified. It is also important to conduct phase III trials to compare non-covalent BTK inhibitors with covalent BTK inhibitors in the treatment of B cell malignancies. As the combinations of ibrutinib with immunochemotherapy or venetoclax have resulted in high rates of MRD negativity in patients with untreated CLL [20, 21], it would be interesting to see whether combining non-covalent BTK inhibitors with immunochemotherapy or venetoclax produces similar effects in patients with CLL.
In addition to non-covalent BTK inhibitors, proteolysis targeting chimeras (PROTACs) are also designed to target BTK and BTK mutants [68]. PROTACs simultaneously bind to an E3 ligase and a target protein, thereby resulting in ubiquitination and subsequent degradation of the target protein. MT-802 is a PROTAC that recruits BTK to the cereblon E3 ligase complex and triggers ubiquitination and subsequent degradation of both wild-type and C481S mutant BTK, thereby providing a rationale for using PROTACs to treat B cell malignancies including those resistant to ibrutinib due to BTK C481S mutation [69]. Another PROTAC P13I could remarkably inhibit the proliferation of BTK C481S mutant DLBCL cell line HBL-1, which is resistant to ibrutinib [70], providing a potential treatment for ibrutinib-resistant B cell malignancies. Several other PROTACs have also shown potential therapeutic efficacy for BTK C481S mutant B cell malignancies in preclinical models [71]. Clinical trials are required to explore the safety and efficacy of PROTACs in patients with B cell malignancies including those resistant to ibrutinib.
Conclusions
BTK, the essential component of BCR signaling, plays a vital role in the origin and development of B cell malignancies. The covalent BTK inhibitors, especially ibrutinib, acalabrutinib, and zanubrutinib, bring benefits for patients with CLL and other B cell malignancies. With resistance and off-target toxicities caused by covalent inhibitors, preclinical and clinical studies of non-covalent BTK inhibitors are ongoing. To date, several non-covalent BTK inhibitors studied in B cell malignancies have shown great specificity and mild AEs in clinical trials, while long-term efficacy and safety remain uncertain; and the others in preclinical studies need further evaluation before clinical trials. Compared to covalent inhibitors, non-covalent inhibitors display prominent advantages when treating patients with BTK C481 mutations. Nonetheless, further clinical and preclinical studies of non-covalent BTK inhibitors are essential to evaluate the specificity, efficacy, and safety.
Acknowledgements
Not applicable.
Abbreviations
- BCR
B cell receptor
- BTK
Bruton tyrosine kinase
- SYK
Spleen tyrosine kinase
- PI3K
Phosphatidylinositol-3-kinase
- ITAM
Immunoreceptor tyrosine-based activation motif
- NF-κB
Nuclear factor κB
- ITK
Interleukin-2-inducible T cell kinase
- TEC
Tyrosine kinase expressed in hepatocellular carcinoma
- RLK
Resting lymphocyte kinase
- BMX
Bone marrow expressed kinase
- CLL
Chronic lymphocytic leukemia
- SLL
Small lymphocytic lymphoma
- MCL
Mantle cell lymphoma
- WM
Waldenström's macroglobulinemia
- MZL
Marginal zone lymphoma
- FL
Follicular lymphoma
- DLBCL
Diffuse large B cell lymphoma
- RT
Richter transformation
- AE
Adverse event
- MRD
Minimal residual disease
- PLCG2
Phospholipase C-gamma-2
- MTD
Maximum tolerated dose
- ASH
American Society of Hematology
- CR
Complete response
- PR
Partial response
- PR-L
PR with lymphocytosis
- WT
Wild type
- ORR
Overall response rate
- PROTACs
Proteolysis targeting chimeras
Authors’ contributions
DG and HT were major contributors in writing the original draft. JW revised the manuscript. JL and YM are the corresponding authors who were in charge of supervision and editing the manuscript. All authors have read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81720108002), National Major Science and Technology Projects of China (Grant No. 2018ZX09734-007), Jiangsu Natural Science Foundation (Grant No. BK20171079).
Availability of data and materials
Not applicable.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Danling Gu and Hanning Tang have contributed equally to this work
Contributor Information
Jianyong Li, Email: lijianyonglm@126.com.
Yi Miao, Email: miaoyi1992@hotmail.com.
References
- 1.Burger JA, Wiestner A. Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat Rev Cancer. 2018;18(3):148–167. doi: 10.1038/nrc.2017.121. [DOI] [PubMed] [Google Scholar]
- 2.Miao Y, Medeiros LJ, Li Y, Li J, Young KH. Genetic alterations and their clinical implications in DLBCL. Nat Rev Clin Oncol. 2019;16(10):634–652. doi: 10.1038/s41571-019-0225-1. [DOI] [PubMed] [Google Scholar]
- 3.Vetrie D, Vořechovský I, Sideras P, Holland J, Davies A, Flinter F, et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature. 1993;361(6409):226–233. doi: 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- 4.Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. 1993;72(2):279–290. doi: 10.1016/0092-8674(93)90667-F. [DOI] [PubMed] [Google Scholar]
- 5.Bradshaw JM. The Src, Syk, and Tec family kinases: distinct types of molecular switches. Cell Signal. 2010;22(8):1175–1184. doi: 10.1016/j.cellsig.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Wu J, Liu C, Tsui ST, Liu D. Second-generation inhibitors of Bruton tyrosine kinase. J Hematol Oncol. 2016;9(1):80. doi: 10.1186/s13045-016-0313-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber ANR, Bittner Z, Liu X, Dang TM, Radsak MP, Brunner C. Bruton's tyrosine kinase: an emerging key player in innate immunity. Front Immunol. 2017;8:1454. doi: 10.3389/fimmu.2017.01454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burger JA, Barr PM, Robak T, Owen C, Ghia P, Tedeschi A, et al. Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia. 2020;34(3):787–798. doi: 10.1038/s41375-019-0602-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharman JP, Egyed M, Jurczak W, Skarbnik A, Pagel JM, Flinn IW, et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment-naive chronic lymphocytic leukaemia (ELEVATE TN): a randomised, controlled, phase 3 trial. Lancet. 2020;395(10232):1278–1291. doi: 10.1016/S0140-6736(20)30262-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tam CS, Opat S, D'Sa S, Jurczak W, Lee HP, Cull G, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenstrom macroglobulinemia: the ASPEN study. Blood. 2020;136(18):2038–2050. doi: 10.1182/blood.2020006844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu W, Song Y, Li Z, Yang S, Liu L, Hu Y, et al. Safety, tolerability and efficacy of orelabrutinib, once a day, to treat Chinese patients with relapsed or refractory chronic lymphocytic leukemia/small cell leukemia. Blood. 2019;134(Supplement_1):4319. doi: 10.1182/blood-2019-123331. [DOI] [Google Scholar]
- 12.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci. 2010;107(29):13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dreyling M, Jurczak W, Jerkeman M, Silva RS, Rusconi C, Trneny M, et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet. 2016;387(10020):770–778. doi: 10.1016/S0140-6736(15)00667-4. [DOI] [PubMed] [Google Scholar]
- 14.Moreno C, Greil R, Demirkan F, Tedeschi A, Anz B, Larratt L, et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20(1):43–56. doi: 10.1016/S1470-2045(18)30788-5. [DOI] [PubMed] [Google Scholar]
- 15.Shanafelt TD, Wang XV, Kay NE, Hanson CA, O'Brien S, Barrientos J, et al. Ibrutinib-rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N Engl J Med. 2019;381(5):432–443. doi: 10.1056/NEJMoa1817073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woyach JA, Ruppert AS, Heerema NA, Zhao W, Booth AM, Ding W, et al. Ibrutinib regimens versus chemoimmunotherapy in older patients with untreated CLL. N Engl J Med. 2018;379(26):2517–2528. doi: 10.1056/NEJMoa1812836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byrd JC, Brown JR, O'Brien S, Barrientos JC, Kay NE, Reddy NM, et al. Ibrutinib versus of atumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213–223. doi: 10.1056/NEJMoa1400376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Munir T, Brown JR, O'Brien S, Barrientos JC, Barr PM, Reddy NM, et al. Final analysis from RESONATE: Up to six years of follow-up on ibrutinib in patients with previously treated chronic lymphocytic leukemia or small lymphocytic lymphoma. Am J Hematol. 2019;94(12):1353–1363. doi: 10.1002/ajh.25638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Allan JN, Shanafelt T, Wiestner A, Moreno C, O'Brien SM, Braggio E, et al. Long-term efficacy of first-line ibrutinib treatment for chronic lymphocytic leukemia (CLL) with 4 years of follow-up in patients with TP53 aberrations (del(17p) or TP53 mutation): a pooled analysis from 4 clinical trials. Blood. 2020;136(Supplement 1):23–24. doi: 10.1182/blood-2020-134431. [DOI] [Google Scholar]
- 20.Jain N, Keating M, Thompson P, Ferrajoli A, Burger J, Borthakur G, et al. Ibrutinib and venetoclax for first-line treatment of CLL. N Engl J Med. 2019;380(22):2095–2103. doi: 10.1056/NEJMoa1900574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davids MS, Brander DM, Kim HT, Tyekucheva S, Bsat J, Savell A, et al. Ibrutinib plus fludarabine, cyclophosphamide, and rituximab as initial treatment for younger patients with chronic lymphocytic leukaemia: a single-arm, multicentre, phase 2 trial. Lancet Haematol. 2019;6(8):e419–e428. doi: 10.1016/S2352-3026(19)30104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu W, Yang S, Zhou K, Pan L, Li Z, Zhou J, et al. Treatment of relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma with the BTK inhibitor zanubrutinib: phase 2, single-arm, multicenter study. J Hematol Oncol. 2020;13(1):48. doi: 10.1186/s13045-020-00884-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou H, Hu P, Yan X, Zhang Y, Shi W. Ibrutinib in Chronic lymphocytic leukemia: clinical applications, drug resistance, and prospects. Onco Targets Ther. 2020;13:4877–4892. doi: 10.2147/OTT.S249586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woyach JA, Ruppert AS, Guinn D, Lehman A, Blachly JS, Lozanski A, et al. BTK(C481S)-mediated resistance to ibrutinib in chronic lymphocytic leukemia. J Clin Oncol. 2017;35(13):1437–1443. doi: 10.1200/JCO.2016.70.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kadri S, Lee J, Fitzpatrick C, Galanina N, Sukhanova M, Venkataraman G, et al. Clonal evolution underlying leukemia progression and Richter transformation in patients with ibrutinib-relapsed CLL. Blood Adv. 2017;1(12):715–727. doi: 10.1182/bloodadvances.2016003632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu L, Tsakmaklis N, Yang G, Chen JG, Liu X, Demos M, et al. Acquired mutations associated with ibrutinib resistance in Waldenstrom macroglobulinemia. Blood. 2017;129(18):2519–2525. doi: 10.1182/blood-2017-01-761726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hershkovitz-Rokah O, Pulver D, Lenz G, Shpilberg O. Ibrutinib resistance in mantle cell lymphoma: clinical, molecular and treatment aspects. Br J Haematol. 2018;181(3):306–319. doi: 10.1111/bjh.15108. [DOI] [PubMed] [Google Scholar]
- 28.Chiron D, Di Liberto M, Martin P, Huang X, Sharman J, Blecua P, et al. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 2014;4(9):1022–1035. doi: 10.1158/2159-8290.CD-14-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woyach J, Huang Y, Rogers K, Bhat SA, Grever MR, Lozanski A, et al. Resistance to acalabrutinib in CLL is mediated primarily by BTK mutations. Blood. 2019;134(Supplement_1):504. doi: 10.1182/blood-2019-127674. [DOI] [Google Scholar]
- 30.Wang M, Rule S, Zinzani PL, Goy A, Casasnovas O, Smith SD, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet. 2018;391(10121):659–667. doi: 10.1016/S0140-6736(17)33108-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Handunnetti SM, Tang CPS, Nguyen T, Zhou X, Thompson E, Sun H, et al. BTK Leu528Trp-a potential secondary resistance mechanism specific for patients with chronic lymphocytic leukemia treated with the next generation BTK inhibitor zanubrutinib. Blood. 2019;134(Supplement_1):170. doi: 10.1182/blood-2019-125488. [DOI] [Google Scholar]
- 32.Stephens DM, Byrd JC. How I manage ibrutinib intolerance and complications in patients with chronic lymphocytic leukemia. Blood. 2019;133(12):1298–1307. doi: 10.1182/blood-2018-11-846808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mato AR, Nabhan C, Thompson MC, Lamanna N, Brander DM, Hill B, et al. Toxicities and outcomes of 616 ibrutinib-treated patients in the United States: a real-world analysis. Haematologica. 2018;103(5):874–879. doi: 10.3324/haematol.2017.182907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, et al. Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. ChemMedChem. 2007;2(1):58–61. doi: 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- 35.Burger JA, Buggy JJ. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) Leuk Lymphoma. 2013;54(11):2385–2391. doi: 10.3109/10428194.2013.777837. [DOI] [PubMed] [Google Scholar]
- 36.Coutre SE, Byrd JC, Hillmen P, Barrientos JC, Barr PM, Devereux S, et al. Long-term safety of single-agent ibrutinib in patients with chronic lymphocytic leukemia in 3 pivotal studies. Blood Adv. 2019;3(12):1799–1807. doi: 10.1182/bloodadvances.2018028761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bond DA, Woyach JA. Targeting BTK in CLL: Beyond Ibrutinib. Curr Hematol Malig Rep. 2019;14(3):197–205. doi: 10.1007/s11899-019-00512-0. [DOI] [PubMed] [Google Scholar]
- 38.Chen J, Kinoshita T, Gururaja T, Sukbuntherng J, James D, Lu D, et al. The effect of Bruton's tyrosine kinase (BTK) inhibitors on collagen-induced platelet aggregation, BTK, and tyrosine kinase expressed in hepatocellular carcinoma (TEC) Eur J Haematol. 2018;101:62–63. doi: 10.1111/ejh.13148. [DOI] [PubMed] [Google Scholar]
- 39.Rigg RA, Aslan JE, Healy LD, Wallisch M, Thierheimer ML, Loren CP, et al. Oral administration of Bruton's tyrosine kinase inhibitors impairs GPVI-mediated platelet function. Am J Physiol Cell Physiol. 2016;310(5):C373–C380. doi: 10.1152/ajpcell.00325.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Atkinson BT, Ellmeier W, Watson SP. Tec regulates platelet activation by GPVI in the absence of Btk. Blood. 2003;102(10):3592–3599. doi: 10.1182/blood-2003-04-1142. [DOI] [PubMed] [Google Scholar]
- 41.Pretorius L, Du XJ, Woodcock EA, Kiriazis H, Lin RC, Marasco S, et al. Reduced phosphoinositide 3-kinase (p110alpha) activation increases the susceptibility to atrial fibrillation. Am J Pathol. 2009;175(3):998–1009. doi: 10.2353/ajpath.2009.090126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McMullen JR, Boey EJ, Ooi JY, Seymour JF, Keating MJ, Tam CS. Ibrutinib increases the risk of atrial fibrillation, potentially through inhibition of cardiac PI3K-Akt signaling. Blood. 2014;124(25):3829–3830. doi: 10.1182/blood-2014-10-604272. [DOI] [PubMed] [Google Scholar]
- 43.Colado A, Genoula M, Cougoule C, Marin Franco JL, Almejun MB, Risnik D, et al. Effect of the BTK inhibitor ibrutinib on macrophage- and gammadelta T cell-mediated response against Mycobacterium tuberculosis. Blood Cancer J. 2018;8(11):100. doi: 10.1038/s41408-018-0136-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Byrd JC, Harrington B, O'Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):323–332. doi: 10.1056/NEJMoa1509981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fabian CA, Reiff SD, Guinn D, Neuman L, Fox JA, Wilson W, et al. SNS-062 demonstrates efficacy in chronic lymphocytic leukemia in vitro and inhibits C481S mutated Bruton tyrosine kinase. Cancer Res. 2017;77(13 supplement):1207. [Google Scholar]
- 46.Binnerts ME, Otipoby KL, Hopkins BT, Bohnert T, Hansen S, Jamieson G, et al. SNS-062 is a potent noncovalent BTK inhibitor with comparable activity against wild type BTK and BTK with an acquired resistance mutation. Mol Cancer Ther. 2015;14(12 Suppl 2):Abstract nr C186. [Google Scholar]
- 47.Neuman LL, Ward R, Arnold D, Combs DL, Gruver D, Hill W, et al. First-in-human phase 1a study of the safety, pharmacokinetics, and pharmacodynamics of the noncovalent bruton tyrosine kinase (BTK) inhibitor SNS-062 in healthy subjects. Blood. 2016;128(22):2032. doi: 10.1182/blood.V128.22.2032.2032. [DOI] [Google Scholar]
- 48.Allan JN, Patel K, Mato AR, Wierda WG, Ibarz JP, Choi MY, et al. Ongoing results of a phase 1B/2 dose-escalation and cohort-expansion study of the selective, noncovalent, reversible Bruton's tyrosine kinase inhibitor, vecabrutinib, in B-cell malignancies. Blood. 2019;134(Supplement_1):3041. doi: 10.1182/blood-2019-126286. [DOI] [Google Scholar]
- 49.Reiff SD, Muhowski EM, Guinn D, Lehman A, Fabian CA, Cheney C, et al. Noncovalent inhibition of C481S Bruton tyrosine kinase by GDC-0853: a new treatment strategy for ibrutinib-resistant CLL. Blood. 2018;132(10):1039–1049. doi: 10.1182/blood-2017-10-809020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crawford JJ, Johnson AR, Misner DL, Belmont LD, Castanedo G, Choy R, et al. Discovery of GDC-0853: a potent, selective, and noncovalent Bruton's tyrosine kinase inhibitor in early clinical development. J Med Chem. 2018;61(6):2227–2245. doi: 10.1021/acs.jmedchem.7b01712. [DOI] [PubMed] [Google Scholar]
- 51.Kohrt HE, Sagiv-Barfi I, Rafiq S, Herman SE, Butchar JP, Cheney C, et al. Ibrutinib antagonizes rituximab-dependent NK cell-mediated cytotoxicity. Blood. 2014;123(12):1957–1960. doi: 10.1182/blood-2014-01-547869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reiff SD, Guinn D, Mantel R, Smith L, Cheney C, Johnson AJ, et al. Evaluation of the novel Bruton′s tyrosine kinase (BTK) inhibitor GDC-0853 in chronic lymphocytic leukemia (CLL) with wild type or C481S mutated BTK. J Clin Oncol. 2016;34(15_suppl):7530. doi: 10.1200/JCO.2016.34.15_suppl.7530. [DOI] [Google Scholar]
- 53.Herman AE, Chinn LW, Kotwal SG, Murray ER, Zhao R, Florero M, et al. Safety, pharmacokinetics, and pharmacodynamics in healthy volunteers treated with GDC-0853, a selective reversible Bruton's tyrosine kinase inhibitor. Clin Pharmacol Ther. 2018;103(6):1020–1028. doi: 10.1002/cpt.1056. [DOI] [PubMed] [Google Scholar]
- 54.Byrd JC, Smith S, Wagner-Johnston N, Sharman J, Chen AI, Advani R, et al. First-in-human phase 1 study of the BTK inhibitor GDC-0853 in relapsed or refractory B-cell NHL and CLL. Oncotarget. 2018;9(16):13023–13035. doi: 10.18632/oncotarget.24310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reiff SD, Mantel R, Smith LL, McWhorter S, Goettl VM, Johnson AJ, et al. The Bruton's tyrosine kinase (BTK) inhibitor ARQ 531 effectively inhibits wild type and C481S mutant BTK and is superior to ibrutinib in a mouse model of chronic lymphocytic leukemia. Blood. 2016;2016(128):3232. doi: 10.1182/blood.V128.22.3232.3232. [DOI] [Google Scholar]
- 56.Reiff SD, Mantel R, Smith LL, Greene JT, Muhowski EM, Fabian CA, et al. The BTK inhibitor ARQ 531 targets ibrutinib-resistant CLL and Richter transformation. Cancer Discov. 2018;8(10):1300–1315. doi: 10.1158/2159-8290.CD-17-1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Woyach J, Stephens DM, Flinn IW, Bhat SA, Savage RE, Chai F, et al. Final results of phase 1, dose escalation study evaluating ARQ 531 in patients with relapsed or refractory B-cell lymphoid malignancies. Blood. 2019;134(Supplement_1):4298. doi: 10.1182/blood-2019-127260. [DOI] [Google Scholar]
- 58.Soncini D, Orecchioni S, Ruberti S, Minetto P, Martinuzzi C, Agnelli L, et al. The new small tyrosine kinase inhibitor ARQ531 targets acute myeloid leukemia cells by disrupting multiple tumor-addicted programs. Haematologica. 2020;105(10):2420–2431. doi: 10.3324/haematol.2019.224956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Elgamal OA, Mehmood A, Jeon JY, Carmichael B, Lehman A, Orwick SJ, et al. Preclinical efficacy for a novel tyrosine kinase inhibitor, ArQule 531 against acute myeloid leukemia. J Hematol Oncol. 2020;13(1):8. doi: 10.1186/s13045-019-0821-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brandhuber B, Gomez E, Smith S, Eary T, Spencer S, Rothenberg SM, et al. LOXO-305, a next generation reversible BTK inhibitor, for overcoming acquired resistance to irreversible BTK inhibitors. Clin Lymphoma Myeloma Leuk. 2018;18:S216. doi: 10.1016/j.clml.2018.07.081. [DOI] [Google Scholar]
- 61.Gomez EB, Isabel L, Rosendahal MS, Rothenberg SM, Andrews SW, Brandhuber BJ. Loxo-305, a highly selective and non-covalent next generation BTK inhibitor, inhibits diverse BTK C481 substitution mutations. Blood. 2019;134(Supplement_1):4644. doi: 10.1182/blood-2019-126114. [DOI] [Google Scholar]
- 62.Naeem AS, Nguy WI, Tyekucheva S, Fernandes SM, Rai V, Ebata K, et al. LOXO-305: targeting C481S Bruton tyrosine kinase in patients with Ibrutinib-resistant CLL. Blood. 2019;134:478. doi: 10.1182/blood-2019-124362. [DOI] [Google Scholar]
- 63.Mato AR, Pagel JM, Coombs CC, Shah NN, Lamanna N, Lech-Marańda E, et al. LOXO-305, a next generation, highly selective, non-covalent BTK inhibitor in previously treated CLL/SLL: results from the phase 1/2 BRUIN study. Blood. 2020;136(Supplement 1):35–37. doi: 10.1182/blood-2020-134970. [DOI] [Google Scholar]
- 64.Wang M, Shah NN, Alencar AJ, Gerson JN, Patel MR, Fakhri B, et al. LOXO-305, a next generation, highly selective, non-covalent BTK inhibitor in previously treated mantle cell lymphoma, Waldenström's macroglobulinemia, and other non-hodgkin lymphomas: results from the phase 1/2 BRUIN study. Blood. 2020;136(Supplement 1):8–10. [Google Scholar]
- 65.Gui F, Jiang J, He Z, Li L, Li Y, Deng Z, et al. A non-covalent inhibitor XMU-MP-3 overrides ibrutinib-resistant Btk(C481S) mutation in B-cell malignancies. Br J Pharmacol. 2019;176(23):4491–4509. doi: 10.1111/bph.14809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Johnson AR, Kohli PB, Katewa A, Gogol E, Belmont LD, Choy R, et al. Battling Btk mutants with noncovalent inhibitors that overcome Cys481 and Thr474 mutations. ACS Chem Biol. 2016;11(10):2897–2907. doi: 10.1021/acschembio.6b00480. [DOI] [PubMed] [Google Scholar]
- 67.Guo X, Yang D, Fan Z, Zhang N, Zhao B, Huang C, et al. Discovery and structure-activity relationship of novel diphenylthiazole derivatives as BTK inhibitor with potent activity against B cell lymphoma cell lines. Eur J Med Chem. 2019;178:767–781. doi: 10.1016/j.ejmech.2019.06.035. [DOI] [PubMed] [Google Scholar]
- 68.Sun X, Rao Y. PROTACs as potential therapeutic agents for cancer drug resistance. Biochemistry. 2020;59(3):240–249. doi: 10.1021/acs.biochem.9b00848. [DOI] [PubMed] [Google Scholar]
- 69.Buhimschi AD, Armstrong HA, Toure M, Jaime-Figueroa S, Chen TL, Lehman AM, et al. Targeting the C481S Ibrutinib-resistance mutation in Bruton's tyrosine kinase using PROTAC-mediated degradation. Biochemistry. 2018;57(26):3564–3575. doi: 10.1021/acs.biochem.8b00391. [DOI] [PubMed] [Google Scholar]
- 70.Dobrovolsky D, Wang ES, Morrow S, Leahy C, Faust T, Nowak RP, et al. Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood. 2019;133(9):952–961. doi: 10.1182/blood-2018-07-862953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun Y, Ding N, Song Y, Yang Z, Liu W, Zhu J, et al. Degradation of Bruton's tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas. Leukemia. 2019;33(8):2105–2110. doi: 10.1038/s41375-019-0440-x. [DOI] [PubMed] [Google Scholar]
- 72.Dimopoulos MA, Tedeschi A, Trotman J, Garcia-Sanz R, Macdonald D, Leblond V, et al. Phase 3 trial of Ibrutinib plus rituximab in Waldenstrom's macroglobulinemia. N Engl J Med. 2018;378(25):2399–2410. doi: 10.1056/NEJMoa1802917. [DOI] [PubMed] [Google Scholar]
- 73.Buske C, Tedeschi A, Trotman J, García-Sanz R, MacDonald D, Leblond V, et al. Five-year follow-up of ibrutinib plus rituximab vs placebo plus rituximab for Waldenstrom's macroglobulinemia: final analysis from the randomized phase 3 iNNOVATETM study. Blood. 2020;136(Supplement 1):24–26. doi: 10.1182/blood-2020-134460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fraser G, Cramer P, Demirkan F, Silva RS, Grosicki S, Pristupa A, et al. Updated results from the phase 3 HELIOS study of ibrutinib, bendamustine, and rituximab in relapsed chronic lymphocytic leukemia/small lymphocytic lymphoma. Leukemia. 2019;33(4):969–980. doi: 10.1038/s41375-018-0276-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Younes A, Sehn LH, Johnson P, Zinzani PL, Hong X, Zhu J, et al. Randomized phase III trial of ibrutinib and rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in non-germinal center B-cell diffuse large B-cell lymphoma. J Clin Oncol. 2019;37(15):1285–1295. doi: 10.1200/JCO.18.02403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lenz G, Balasubramanian S, Goldberg J, Rizo A, Schaffer M, Phelps C, et al. Sequence variants in patients with primary and acquired resistance to ibrutinib in the phase 3 MCL3001 (RAY) trial. J Clin Oncol. 2016;34(15_suppl):7570. doi: 10.1200/JCO.2016.34.15_suppl.7570. [DOI] [Google Scholar]
- 77.Saba NS, Liu D, Herman SE, Underbayev C, Tian X, Behrend D, et al. Pathogenic role of B-cell receptor signaling and canonical NF-kappaB activation in mantle cell lymphoma. Blood. 2016;128(1):82–92. doi: 10.1182/blood-2015-11-681460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ma J, Lu P, Guo A, Cheng S, Zong H, Martin P, et al. Characterization of ibrutinib-sensitive and -resistant mantle lymphoma cells. Br J Haematol. 2014;166(6):849–861. doi: 10.1111/bjh.12974. [DOI] [PubMed] [Google Scholar]
- 79.Zhao X, Lwin T, Silva A, Shah B, Tao J, Fang B, et al. Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma. Nat Commun. 2017;8:14920. doi: 10.1038/ncomms14920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang L, Yao Y, Zhang S, Liu Y, Guo H, Ahmed M, et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci Transl Med. 2019;11(491):1167. doi: 10.1126/scitranslmed.aau1167. [DOI] [PubMed] [Google Scholar]
- 81.Mohanty A, Sandoval N, Das M, Pillai R, Chen L, Chen RW, et al. CCND1 mutations increase protein stability and promote ibrutinib resistance in mantle cell lymphoma. Oncotarget. 2016;7(45):73558–73572. doi: 10.18632/oncotarget.12434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cao Y, Hunter ZR, Liu X, Xu L, Yang G, Chen J, et al. The WHIM-like CXCR4(S338X) somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom's Macroglobulinemia. Leukemia. 2015;29(1):169–176. doi: 10.1038/leu.2014.187. [DOI] [PubMed] [Google Scholar]
- 83.Kuo HP, Ezell SA, Hsieh S, Schweighofer KJ, Cheung LW, Wu S, et al. The role of PIM1 in the ibrutinib-resistant ABC subtype of diffuse large B-cell lymphoma. Am J Cancer Res. 2016;6(11):2489–2501. [PMC free article] [PubMed] [Google Scholar]
- 84.Kim JH, Kim WS, Ryu K, Kim SJ, Park C. CD79B limits response of diffuse large B cell lymphoma to ibrutinib. Leuk Lymphoma. 2016;57(6):1413–1422. doi: 10.3109/10428194.2015.1113276. [DOI] [PubMed] [Google Scholar]
- 85.Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. 2015;21(8):922–926. doi: 10.1038/nm.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jin J, Wang L, Tao Z, Zhang J, Lv F, Cao J, et al. PDGFD induces ibrutinib resistance of diffuse large Bcell lymphoma through activation of EGFR. Mol Med Rep. 2020;21(5):2209–2219. doi: 10.3892/mmr.2020.11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–2294. doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Maddocks KJ, Ruppert AS, Lozanski G, Heerema NA, Zhao W, Abruzzo L, et al. Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol. 2015;1(1):80–87. doi: 10.1001/jamaoncol.2014.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Burger JA, Landau DA, Taylor-Weiner A, Bozic I, Zhang H, Sarosiek K, et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun. 2016;7:11589. doi: 10.1038/ncomms11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Epperla N, Shana'ah AY, Jones D, Christian BA, Ayyappan S, Maddocks K, et al. Resistance mechanism for ibrutinib in marginal zone lymphoma. Blood Adv. 2019;3(4):500–502. doi: 10.1182/bloodadvances.2018029058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Scherer F, Kurtz DM, Newman AM, Stehr H, Craig AF, Esfahani MS, et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci Transl Med. 2016;8(364):364ra155. doi: 10.1126/scitranslmed.aai8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Asami T, Kawahata W, Kashimoto S, Sawa M. CB1763, a highly selective, novel non-covalent BTK inhibitor, targeting ibrutinib-resistant BTK C481S mutant. Mol Cancer Ther. 2018;17(1 Suppl):Abstract nr B152. [Google Scholar]
- 93.Di Paolo JA, Huang T, Balazs M, Barbosa J, Barck KH, Bravo BJ, et al. Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat Chem Biol. 2011;7(1):41–50. doi: 10.1038/nchembio.481. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.