

Abstract
Treatments are lacking for sarcopenia, a debilitating age-related skeletal muscle wasting syndrome. Here we identify elevated 15-PGDH, the Prostaglandin E2 (PGE2) degrading enzyme, as a hallmark of aged tissues, including skeletal muscle. The resulting reduction in PGE2 signaling is a major contributor to muscle atrophy in aged mice and results from 15-PGDH-expressing myofibers and interstitial cells within muscle. Inhibition of 15-PGDH, by targeted genetic knockdown or a small molecule inhibitor, increases aged muscle mass, strength, and exercise performance. These physiological benefits arise from rejuvenated PGE2 levels which augment mitochondrial function and autophagy and decrease TGF-beta and ubiquitin-proteasome pathways. Our studies demonstrate a previously unrecognized role for PGE2 signaling in countering muscle atrophy and identify 15-PGDH as a promising therapeutic target to counter sarcopenia.
Introduction
With aging, a loss of muscle function diminishes quality of life and increases morbidity and mortality (1, 2). Skeletal muscles make up 40% of the body’s mass. After the age of 50, humans lose on average 15–30% of their muscle mass per decade (3) culminating in the drastic loss of muscle strength characteristic of sarcopenia. This disseminated muscle atrophy and loss of strength, or sarcopenia, accounts for $18 billion in annual healthcare costs in the United States alone (2). Currently, there are no approved therapies for sarcopenia (1, 2).
During aging, skeletal muscles undergo structural and functional changes. This loss of function arises from disrupted cell-cell interactions and aberrant cell signaling pathways, particularly those related to inflammation, protein turnover, and mitochondrial function (1, 4–6). Due to its multifactorial etiology, untangling causal molecular pathways in order to identify therapeutic targets to delay or reverse sarcopenia has proven challenging.
Previously, we determined that in young mice PGE2 stimulates muscle stem cells (MuSCs) and is essential to the regeneration of damaged muscles (7), in good agreement with findings regarding its function in the regeneration of bone, colon, liver, and blood (8–10). We reasoned that in aging, prostaglandin signaling might go awry. Using liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) to distinguish closely related prostaglandin family members (11), we found that PGE2 and PGD2 levels are reduced in aged skeletal muscles.
We hypothesized that the decrease in prostaglandins in aged muscles might be due to increased prostaglandin catabolism by 15-hydroxyprostaglandin dehydrogenase (15-PGDH). Here we uncover that elevated 15-PGDH is a hallmark of aged muscles and several other aged tissues. Further, we show that in aged mice inhibition of 15-PGDH augments mitochondrial function leading to increased muscle mass and strength. Genetic experiments demonstrate that the beneficial effects of 15-PGDH inhibition are specific to increased PGE2 signaling. Our findings provide fresh insights into sarcopenia and suggest a potential treatment strategy.
Increase in prostaglandin degrading enzyme (15-PGDH) in aged tissues
We previously demonstrated the importance of PGE2 signaling in stimulating stem cells to regenerate damaged tissues in young mice (7). We reasoned that PGE2 might also act on mature muscle myofibers and play a crucial role in the maintenance of muscle tissue homeostasis. We postulated that in aging a decrease in PGE2 and other endogenous prostaglandins, lipid metabolites generated from membrane fatty acids, might occur and have deleterious effects on muscle tissue function. To analyze the prostaglandin composition of aged skeletal muscle, we used LC-MS/MS. This method overcomes the cross-reactivity of antibody-based assays, such as ELISAs and exceeds other mass spectrometry methods in its resolution of closely related prostaglandins of the same mass, PGE2 and PGD2, as well as PGF2α (Fig. 1A–C, S1A–C, S2A, Table S1). We observed a significant decline in PGE2 and PGD2 levels in aged muscles (Fig. 1B,C, S2A). PGE2 and PGD2 are degraded by a multi-step process initiated by the rate-limiting enzyme 15-PGDH to yield the unstable 15-keto-PGE2 and 15-keto-PGD2 metabolites which are then converted to multiple downstream metabolites, including the 13,14-dihydro-15-keto-PGE2 metabolite (PGEM) (Fig. 1A) (12, 13). These intermediates were either not detected at all or only at low levels by LC-MS/MS due to their instability (Fig. 1B,C).
Figure 1. Elevated activity of PGE2 degrading enzyme, 15-PGDH, is associated with aging, and 15-PGDH knockdown in aged muscle tissues increases strength.
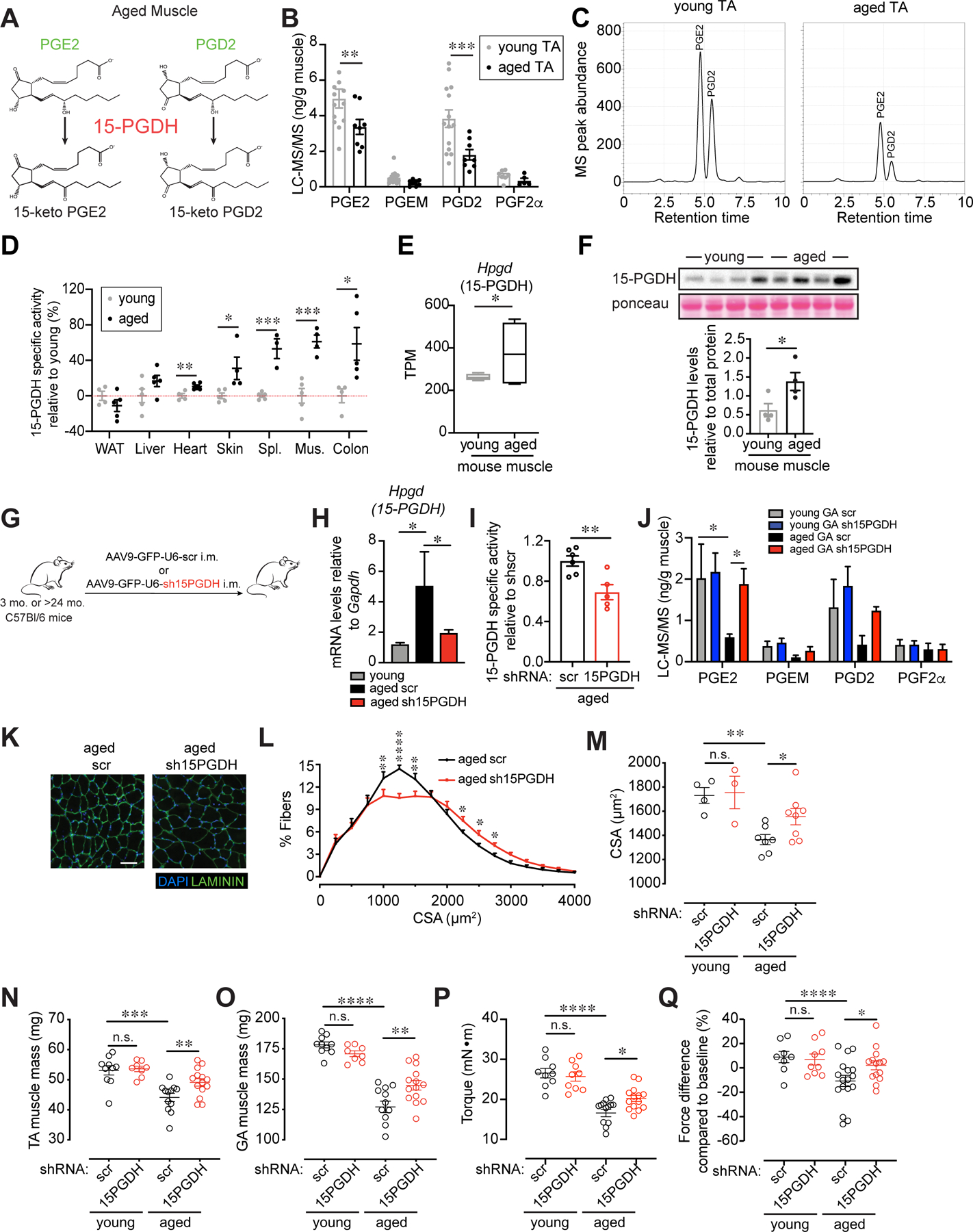
(A) PGE2 and PGD2 catabolism scheme. (B) Prostaglandin levels in Tibialis anterior (TA) muscles quantified by LC-MS/MS (n=14 young, n=8 aged). (C) Representative chromatogram of the PGE2 and PGD2 levels analyzed by LC-MS/MS in young (left) and aged (right) TA muscles. (D) 15-PGDH specific enzymatic activity assayed in tissues of young and aged mice. Activity is expressed as percent change relative to young. (n=4–5 young, n=3–5 aged). (E) 15-PGDH (Hpgd) RNAseq expression data from young (n=4) and aged (n=5) mouse muscles. TPM, transcripts per million. (F) 15-PGDH immunoblots from young and aged muscle lysates (n=4 each). (G-Q) Intramuscular (i.m.) injection of AAV9 carrying a construct of an shRNA against 15-PGDH (sh15PGDH) or scramble (scr) control into the TA and Gastrocnemius (GA) of young and aged mice. (G) Experimental scheme. (H) Expression levels of 15-PGDH in scr and sh15PGDH infected muscles and young control (n=5 young, n=4 aged scr, n=5 aged sh15PGDH). (I) 15-PGDH specific enzymatic activity assayed in muscle tissues of sh15PGDH infected aged muscles normalized to scr (n=6 aged scr, n=5 aged sh15PGDH). (J) Prostaglandin levels in GA muscles quantified by LC-MS/MS (n=5 young scr, n=4 young sh15PGDH, n=3 aged scr, n=4 aged sh15PGDH). (K) Representative GA cross-section of scr and sh15PGDH infected aged muscles. DAPI, blue; LAMININ, green. Bar=50 µm. (L) Myofiber cross-sectional areas (CSA) in scr and sh15PGDH infected aged GAs (n=7 aged scr, n=8 aged sh15PGDH). (M) Mean CSA (n=4 young scr, n=3 young sh15PGDH, n=7 aged scr, n=8 aged sh15PGDH). (N) Mass of dissected TA. (O) Mass of dissected GA. (P) Plantar flexion tetanic force (absolute values). (Q) Plantar flexion tetanic force (relative to baseline). (N-Q): n=8–10 young scr, n=8–9 young sh15PGDH, n=12–14 aged scr, n=14 aged sh15PGDH. *P<0.05, **P<0.01, ****P<0.0001. ANOVA test with Bonferroni correction (N-Q) or Fisher’s LSD (B,H,J,L,M) for multiple comparisons; unpaired t-tests (D-F,I). Means±s.e.m. Abbreviations: PGEM, 13,14-dihydro-15-keto-PGE2; Spl. Spleen; Mus. Muscle; mo. months; i.m. intramuscular. C57Bl/6 young (2–4 mo.) and aged (>24 mo.) mice were used.
We hypothesized that an increase in the degrading enzyme 15-PGDH could account for the observed reduction in PGE2 and PGD2 in muscle and might be a general characteristic of aged tissues. In agreement, we found that the specific activity of the enzyme was elevated not only in aged skeletal muscles, but also in aged cardiac, skin, spleen and colon tissues (Fig. 1D, S3). Accordingly, 15-PGDH mRNA and protein levels are significantly increased in aged muscles (Fig. 1E,F and S4A,B). To determine the relevance of this finding to human aging, we analyzed publicly available microarray data for young and aged human muscle samples (14) and found that expression of 15-PGDH was significantly increased in aged human (78 ± 6 yr) biopsies from the vastus lateralis muscle compared to those from young populations (25 ± 3 yr) (Fig. S5A). Together, these data identify 15-PGDH as a potential driver of the decline in prostaglandin levels seen in aged muscles.
Increase in aged muscle mass and strength following inhibition of 15-PGDH
We postulated that inhibition of 15-PGDH could lead to increased levels of PGE2 and PGD2 which in turn could ameliorate muscle wasting in aged mice. Like humans, aged mice exhibit sarcopenia, a general loss of muscle strength (1). We first used a genetic approach to reduce enzyme levels that entailed adeno-associated virus (AAV9) mediated intramuscular (i.m.) delivery of either GFP and shRNA to 15-PGDH or control AAV9 encoding GFP and a scrambled (scr) shRNA under the control of a ubiquitous promoter U6 (Fig. 1G). The resulting localized intramuscular gene therapy delivery strategy led to a significant reduction in 15-PGDH mRNA levels and specific activity and an increase in PGE2 and PGD2 levels assessed by LC-MS/MS (Fig. 1H–J). Efficiency of knockdown was confirmed by immunofluorescence analysis of the GFP reporter and reduced levels of 15-PGDH protein in shRNA transduced Tibialis anterior (TA) and Gastrocnemius (GA) muscles (Fig. S6A,B). Genetic knockdown of 15-PGDH in aged, but not young, muscles was accompanied by a marked increase in cross-sectional myofiber area in 15-PGDH shRNA treated aged muscles compared to controls (Fig. 1K–M). Furthermore, knockdown of 15-PGDH in aged muscles resulted in a significant increase in both muscle mass and muscle force one month after treatment (Fig. 1N–Q).
To test if the disseminated muscle wasting seen in sarcopenia could be overcome by systemic delivery of a small molecule inhibitor of 15-PGDH, we treated aged mice and young control mice intraperitoneally with SW033291 (SW) or vehicle (10) (Fig. 2A). SW was previously extensively characterized as a specific inhibitor of 15-PGDH in vitro (ki of 0.1 nM) (10). In vivo, SW was shown to increase PGE2 levels 2-fold, and to a lesser extent PGD2 levels, in bone marrow, colon, lung and liver, which augmented regeneration following injury of these tissues in young mice (10). We found that after one month of daily intraperitoneal SW treatment, 15-PGDH specific activity was significantly reduced in aged muscles and a concomitant increase in the levels of PGE2 and PGD2 was detected by LC-MS/MS that was on par with young muscles (Fig. 2B,C and S7A,B). Histological analysis revealed that myofiber cross-sectional area was significantly augmented in SW-treated aged mice, indicating that muscle atrophy in the aged was attenuated (Fig. 2D–F). Fiber type analysis revealed that SW treatment promoted an increase in the cross-sectional area of both oxidative (type IIa) and glycolytic (type IIb) fast twitch fibers (Fig. 2G–J). Young mice treated with SW exhibited a trend toward increased muscle mass and absolute strength which reached statistical significance for plantar flexor force difference compared to baseline (Fig. 2K, L). SW-treated aged mice were more profoundly impacted than young mice and exhibited a significant increase in mass of TA, GA and soleus muscles (Fig. 2K) as well as in both plantar flexor absolute and relative to baseline muscle force (Fig. 2L). The reduced response of young relative to aged mice is in keeping with the levels of 15-PGDH and the degree of increase in PGE2 levels induced in muscle tissue by SW at these ages (Fig. 1D–F, 2C and S2A, S3). Notably, for both young and aged mice endurance (time to exhaustion on a treadmill) was significantly increased, suggestive of an overall systemic beneficial effect in addition to muscle strength (Fig. 2M). Taken together, our studies using the small molecule inhibitor, SW, corroborate our findings using a genetic loss of function via a localized shRNA. Notably, SW appears to be more efficacious than AAV9-mediated knockdown, presumably because the small molecule gains access to and inhibits enzyme activity in both non-myogenic and myogenic cell types, whereas AAV9 is well known to exhibit myofiber tropism (15–17). Together, these findings show that a systemic decrease in 15-PGDH activity for a period of one month suffices to counter skeletal muscle atrophy and augment muscle function in aged mice.
FIG. 2. Increase in muscle mass and strength is induced in aged muscles by small molecule inhibition of 15-PGDH.
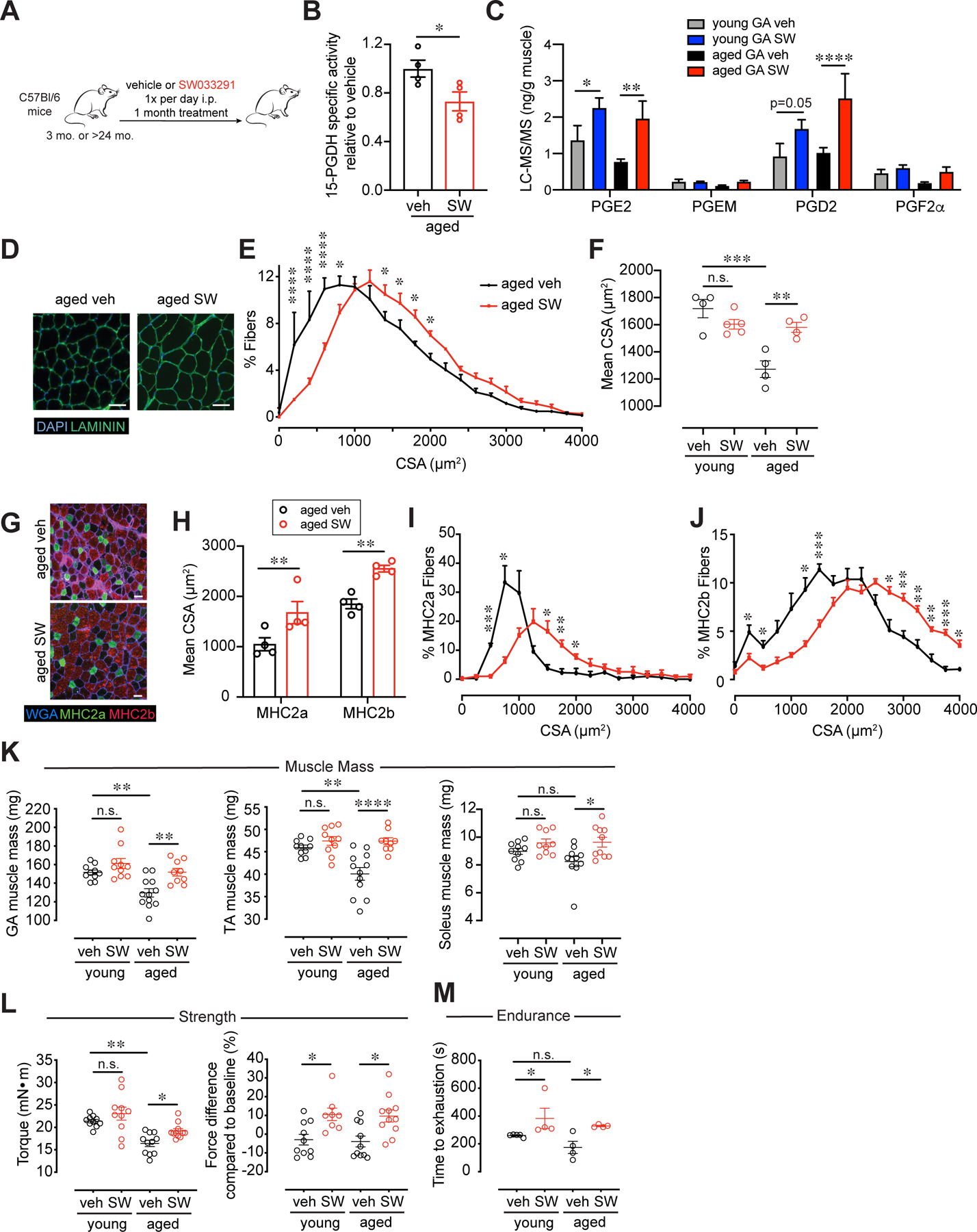
(A) Experimental scheme. Young and aged mice were treated daily with 15-PGDH inhibitor, SW033291 (SW) or vehicle for 1 mo. (B) 15-PGDH specific enzymatic activity assayed in muscle tissues of SW treated aged muscles normalized to vehicle treated (n=4 mice per age group). (C) Prostaglandin levels in Gastrocnemius (GA) muscles quantified by LC-MS/MS (n=8 young veh, n=9 young SW, n=8 aged veh, n=6 aged SW). Muscles were analyzed 3 hours post vehicle or SW injection. (D) Representative Tibialis anterior (TA) cross-section of 1 mo. treated vehicle or SW treated aged muscles. DAPI, blue; LAMININ, green. Bar=50 µm. (E) Myofiber cross-sectional areas (CSA) in vehicle and SW treated aged TA (n=4 per group). (F) Mean CSA (n=5 young veh, n=4 young SW, n=4 aged veh, n=4 aged SW). (G) Representative TA cross-section of 1 mo. vehicle or SW treated aged muscles stained for oxidative (MHC2a) and glycolytic fibers (MHC2b). WGA, blue; MHC2a, green; MHC2b, red. Bar=50 µm. (H) Mean CSA. n=4 per group (I) Cross-sectional area of MHC2a. n=4 per group (J) Cross-sectional area of MHC2b. n=4 per group (K) Weight of dissected GA, TA and Soleus muscles. (L) Plantar flexion tetanic force (absolute values) and plantar flexion tetanic force (relative to baseline). (K-L): n=10 young veh, n=8–10 young SW, n=8–12 aged veh, n=7–11 aged SW. (M) Time to exhaustion (n=5 young veh, n=4 young SW, n=4 aged veh, n=4 aged SW). *P<0.05, **P<0.01, ****P<0.0001. Unpaired t-test (B,H); ANOVA test with Bonferroni correction (F,K,L) or Fisher’s LSD (C,E,I,J,M) for multiple comparisons. Means±s.e.m. Abbreviation: veh., vehicle, PGEM, 13,14-dihydro-15-keto-PGE2; mo. months; i.p. intraperitoneal. C57Bl/6 young (2–4 mo.) and aged (>24 mo.) mice were used.
15-PGDH expression by myofibers and interstitial cells in the aged muscle microenvironment
We sought to identify the source of 15-PGDH in aged muscle tissue. We capitalized on multiplex tissue imaging (CODEX, CO-Detection by indEXing) to visualize the localization of multiple markers together with 15-PGDH in young and aged muscle tissue sections. CODEX uses antibodies conjugated with unique DNA barcodes that are iteratively rendered visible by cycles of hybridization and chemical denaturation with fluorophore-conjugated complementary DNA probes (18, 19). This method enables simultaneous visualization of multiple antibody stainings in a single section, circumvents spectral overlap, and allows for the analysis of co-localization of 15-PGDH with markers of major cell types in muscle. It also overcomes non-specific and autofluorescence signals in aged muscle. 15-PGDH staining was detected at significant levels in a subset of myofibers in the aged TA and GA, but present at reduced levels in those muscles in young mice (Fig. 3A,B and S6B). CODEX also revealed that interstitial cells, in particular macrophages, are a source of 15-PGDH in muscle. 15-PGDH was detected in cells co-stained with macrophage markers CD11b and CD45 in the aged GA (Fig. 3A,B). This was confirmed by a striking increase in 15-PGDH transcript levels in FACS purified macrophages, relative to endothelial or myogenic stem and progenitor cells isolated from aged muscles (Fig. 3C and S8A,B). To confirm that myofibers in aged mice are a source of 15-PGDH, we isolated the Flexor digitorum brevis (FDB) muscles and digested them with collagenase to obtain isolated single myofibers. This collagenase digestion depleted interstitial cell types such as macrophages and fibroadipocytes (FAPs) (Fig. S8C). Both whole FDB muscles and isolated single myofibers exhibited increased Hpgd (15-PGDH) transcript levels in the aged compared to young (Fig. 3D). These results suggest that in aged muscles myofibers and macrophages are the primary sites of the prostaglandin degradation that drives the dysfunction of the aged myogenic microenvironment, or niche.
FIG. 3. Myofibers and interstitial macrophages comprise cellular sources of 15-PGDH in the aged muscle microenvironment.
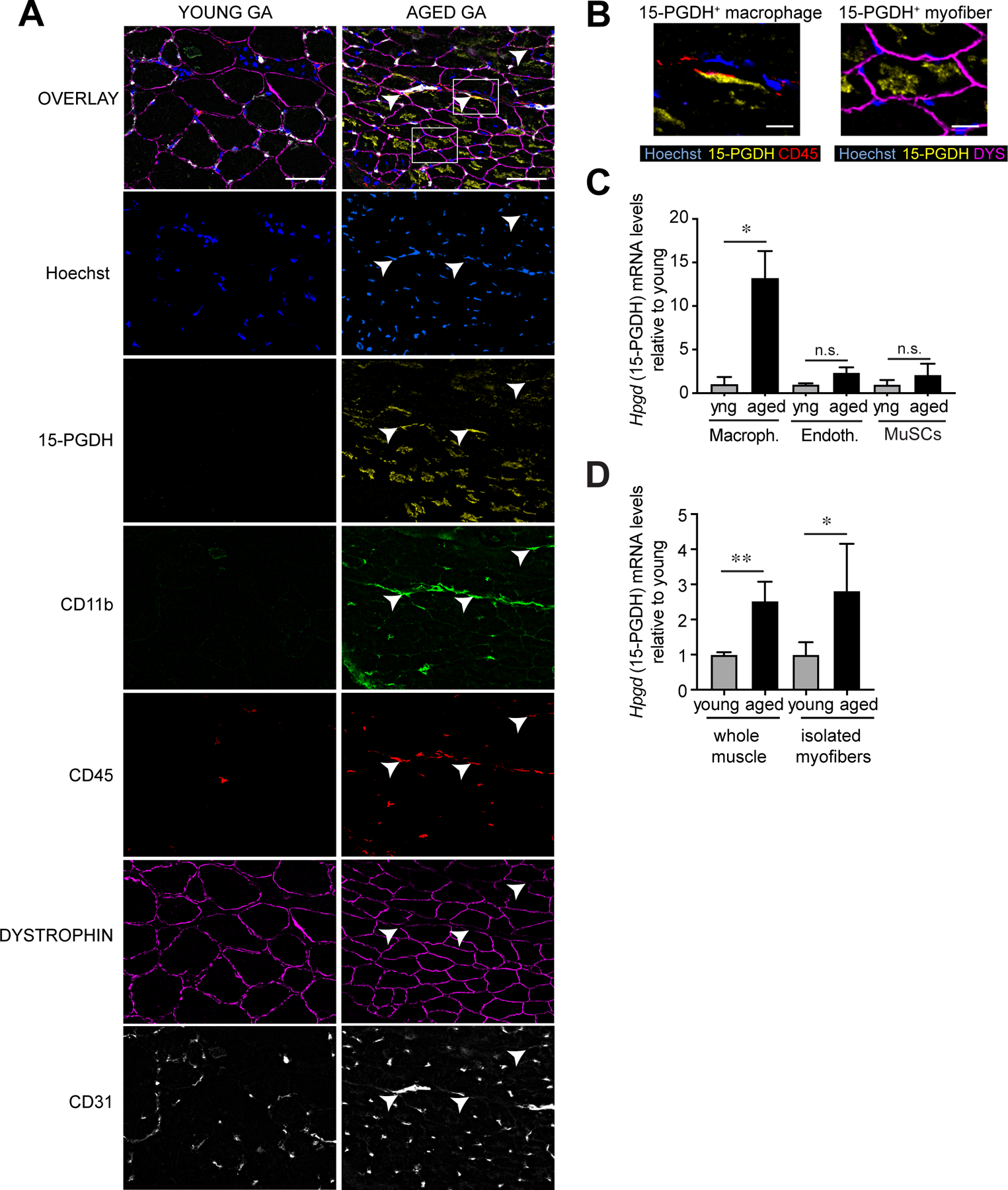
(A) Multiplexed immunofluorescence co-detection of 15-PGDH (yellow) in major cell types in tissue sections of GA muscles from young and aged. CD45 (red) stains all immune cells; CD11b (green) stains myeloid cells including macrophages; DYSTROPHIN (magenta) stains the sarcolemma of myofibers; CD31 (PECAM; grays) stains capillary and vascular endothelial cells. n=4 per group; tissues were imaged in a 5×7 grid with 26 z-slices for each stain; best focused Z-slice shown. Arrows indicate 15-PGDH+ macrophages. Bar=50 µm. (B) Magnified regions from (A) of co-localized 15-PGDH staining in CD45+CD11b+ macrophages (left) and dystrophin+ myofibers (right) in aged GA muscles. Bar=10 µm. (C) Expression of 15-PGDH (Hpgd) in sorted macrophages (CD11b+/CD11c−/F4/80+/CD31−) (n=3 young, n=8 aged), endothelial (CD31+/CD11b−/CD11c−/F4/80−) (n=3 young, n=5 aged) and muscle stem cells (MuSCs) (α7+/CD11b−/CD45−/CD31−/Sca1−) (n=5 young, n=5 aged) from young and aged hindlimb muscles. (D) Expression of 15-PGDH (Hpgd) in whole EDL muscle lysate and collagenase dissociated single isolated myofibers (n=6 young whole muscle, n=6 aged whole muscle, n=4 young isolated myofibers, n=3 aged isolated myofibers). *P<0.05, **P<0.01. Unpaired t-tests (C,D). Means±s.e.m. Abbreviation: yng, young. C57Bl/6 young (2–4 mo.) and aged (>24 mo.) mice were used.
Reduced muscle strength after ectopic expression of 15-PGDH in young muscles
We reasoned that if 15-PGDH plays a major role in the loss of muscle function seen with aging, ectopic expression of the PGE2 degrading enzyme in muscles of young mice would have a deleterious effect on muscle function. To test this hypothesis, we used AAV9 to deliver and overexpress the 15-PGDH gene (Hpgd) under the control of the ubiquitous cytomegalovirus (CMV) promoter (Fig. 4A). We confirmed that upon intramuscular injection of AAV9-CMV-15-PGDH, expression of 15-PGDH was increased by qRT-PCR (Fig. 4B). Additionally, analysis by LC-MS/MS revealed a marked decrease in prostaglandins PGE2 and PGD2 in young muscles expressing 15-PGDH (Fig. 4C), similar to the decline in these prostaglandins seen in aged muscles. The reduction in these prostaglandins for a period of only one month resulted in a significant decrease in the average cross-sectional area of individual myofibers, loss of muscle mass and force in young adult mice (Fig. 4D–H). We analyzed markers of muscle atrophy by qRT-PCR and found that the atrogenes, ubiquitin ligases Trim63 (MuRF1) and Fbxo32 (Atrogin-1) (Fig. 4I) were upregulated in muscles overexpressing 15-PGDH, in accordance with findings by others in acute models of atrophy (20). These data provide strong evidence that 15-PGDH overexpression plays a causal role in decreasing PGE2 and PGD2 levels in muscle, which in turn, leads to a decrease in muscle mass and strength. Moreover, they show that elevated 15-PGDH activity alone has a profound effect on young muscle homeostasis and is capable of inducing an atrophy phenotype.
FIG. 4. Muscle atrophy and weakness are induced in young mice by overexpression of 15-PGDH.
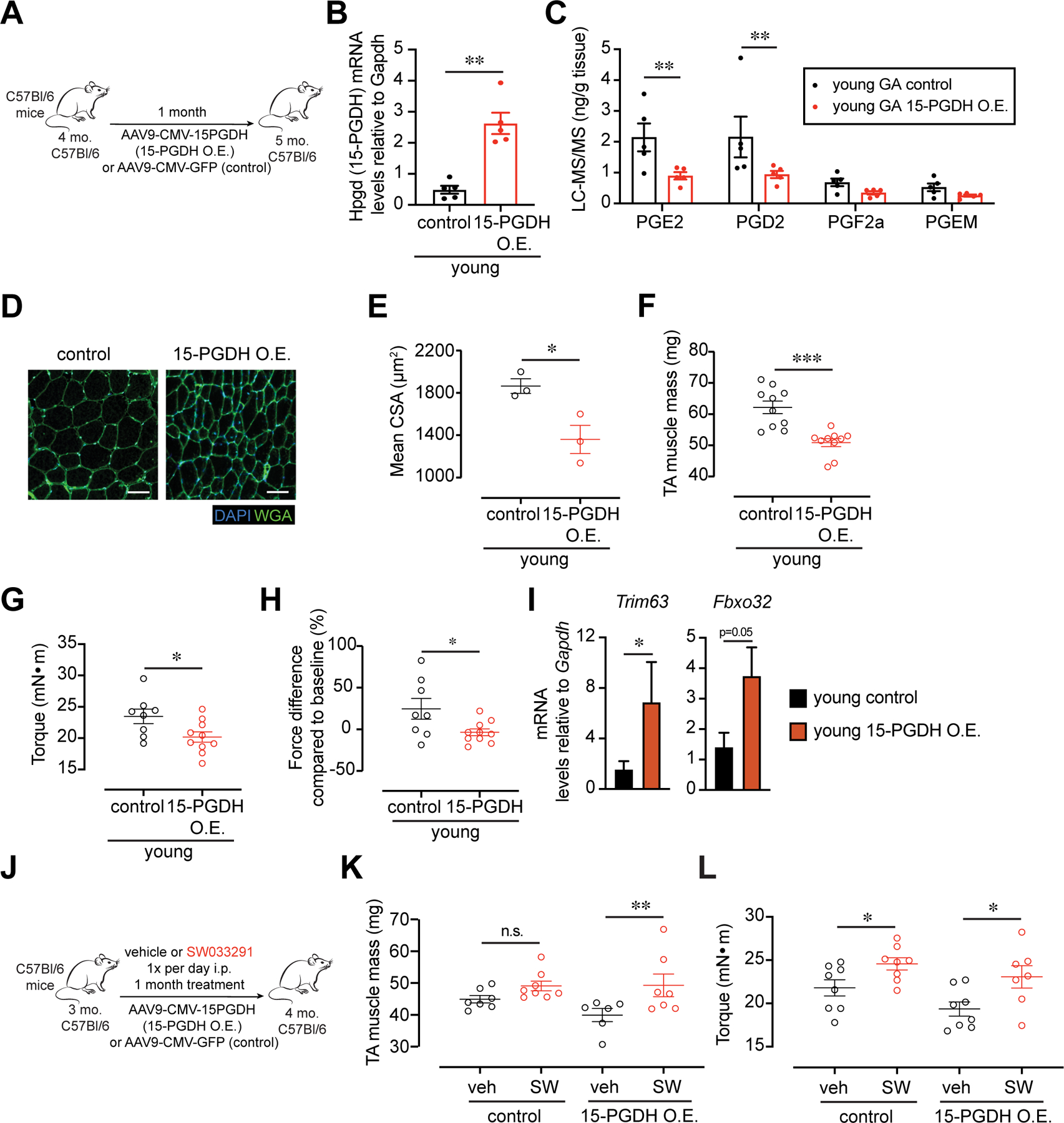
(A-H) Intramuscular (i.m.) injection of AAV9 carrying a construct of CMV driving 15-PGDH expression or control into the Tibialis anterior (TA) or Gastrocnemius (GA) of young mice. (A) Experimental scheme. (B) Expression of 15-PGDH (Hpgd) in control and 15-PGDH O.E. infected young muscles (n=5 per group). (C) Prostaglandin levels in GA muscles quantified by LC-MS/MS (n=5 per group). (D) Representative TA cross-section 1-mo. post i.m. injection. DAPI, blue; WGA, green. Bar=50 µm. (E) Myofiber cross sectional area of muscles injected with 15-PGDH overexpression vector and control (n=3 per group). (F) Mass of dissected Tibialis anterior (TA) muscles. (n=10 per group) (G) Plantar flexion tetanic force (absolute values) (n=8 control, n=10 15-PGDH O.E.). (H) Plantar flexion tetanic force (relative values) (n=7 control, n=9 15-PGDH O.E.). (I) Expression level of MuRF1 (Trim63), Atrogin-1 (Fbxo32) measured by qPCR (n=5 per group). (J-L) I.m. injection of AAV9 carrying a construct of CMV driving 15-PGDH expression or control into the TA of young mice together with daily intraperitoneal (i.p.) treatment with 15-PGDH inhibitor, SW033291 (SW) or vehicle. (J) Experimental scheme. (K) Mass of dissected TA muscles. (L) Plantar flexion tetanic force (absolute values). (J-L): n=7–8 control veh, n=8 control SW, n=6–8 15-PGDH O.E. veh, n=7 15-PGDH O.E. SW. *P<0.05, **P<0.01, ***P<0.001 ****P<0.0001. Unpaired t-tests (B,C,E-I), ANOVA with Fisher’s LSD (K,L) for multiple comparisons. Means±s.e.m. Abbreviation: PGEM, 13,14-dihydro-15-keto-PGE2. C57Bl/6 young (2–4 mo.) mice were used.
To determine the specificity of SW for its target 15-PGDH, we performed a rescue experiment in young mice overexpressing the enzyme following intramuscular AAV9-mediated gene delivery. We reasoned that inhibition of the over-expressed enzyme by SW should overcome the deleterious effects seen upon 15-PGDH overexpression. Accordingly, we treated control and 15-PGDH overexpressing young mice systemically with vehicle or SW (Fig. 4J) and found that muscle mass (Fig. 4K) and strength (Fig. 4L) were restored. These data demonstrate that 15-PGDH inhibition using the small molecule SW specifically targets 15-PGDH and improves muscle function.
Increase in strength in aged mice mediated by PGE2 but not PGD2
15-PGDH degrades both PGE2 and PGD2 in aged muscles. Notably, the two prostaglandins differ in their receptors and in their downstream signaling cascades (21). To determine which prostaglandin was responsible for driving the improvement in aged muscle function, we increased their levels by 15-PGDH inhibition using SW while inhibiting the expression of the PGD2 synthesizing enzyme, Ptgds. This was achieved by intramuscular injection of aged muscles with an AAV9 virus encoding either an shRNA that targets PTGDS (shPTGDS) or a scrambled control shRNA and treating the aged mice for one month with the 15-PGDH inhibitor, SW or vehicle (Fig. 5A). We validated knockdown of PTGDS in transduced aged muscles by confirming reduced Ptgds mRNA levels by qRT-PCR and decreased levels of PGD2 by LC-MS/MS (Fig. 5B,C). Upon knockdown of PTGDS, an increase in muscle mass, force and endurance was seen after SW treatment (Fig. 5D–G). These results identify PGE2, not PGD2, as the mediator of the increased muscle function seen in aged muscles upon 15-PGDH inhibition.
FIG. 5. Beneficial effects of 15-PGDH inhibition on muscle strength are specific to PGE2 and the EP4 receptor on myofibers.
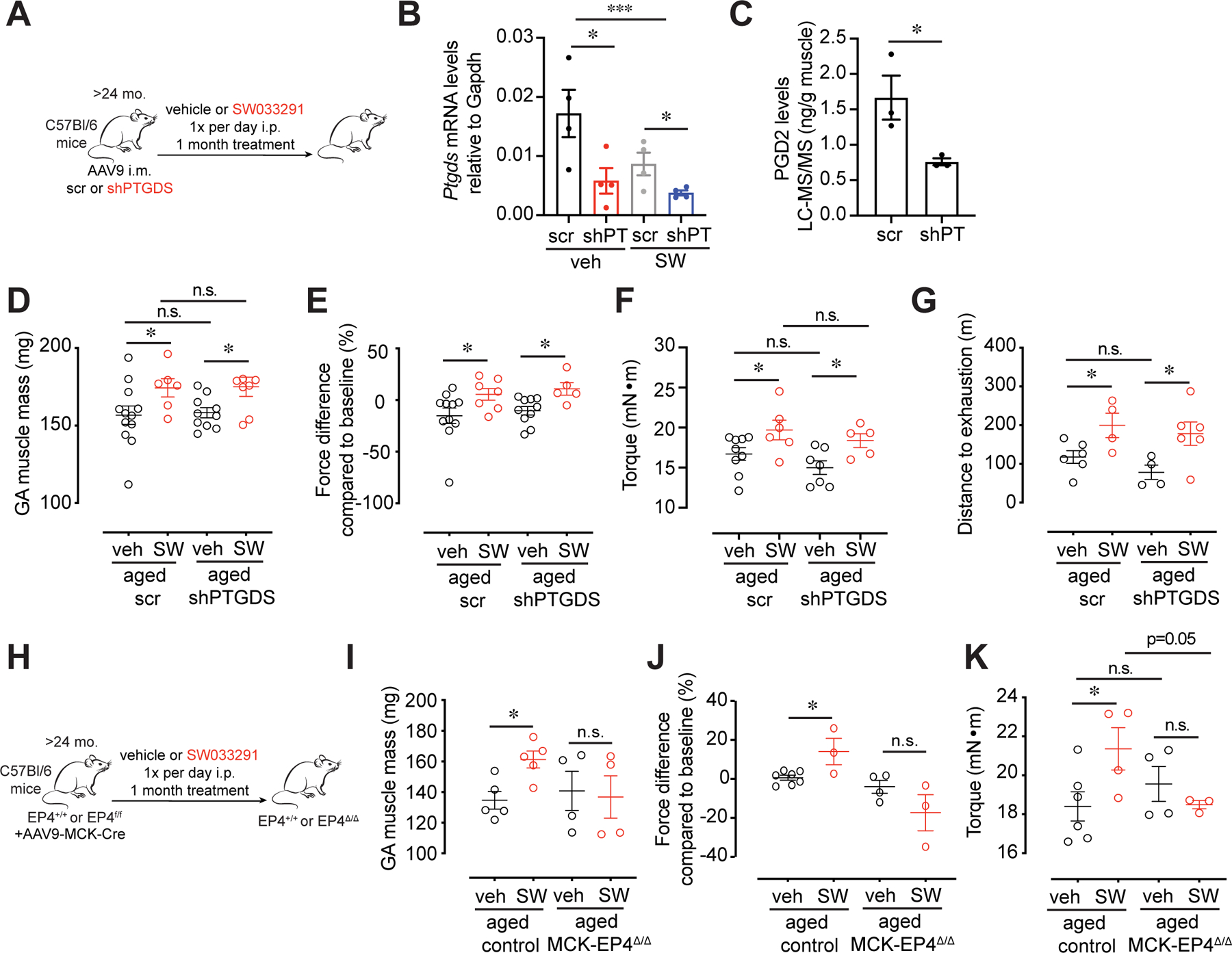
(A-G) Intramuscular (i.m.) injection of AAV9 carrying a construct of an shRNA against Prostaglandin D2 Synthase, PTGDS (shPTGDS) or scramble (scr) control into the Gastrocnemius (GA) of aged mice. (A) Experimental scheme. (B) Expression of Ptgds measured by qPCR (n= 4 per group). (C) PGD2 level in GA muscle tissues quantified by LC-MS/MS (n=3 per group). (D) Mass of dissected GA. (E) Plantar flexion tetanic force (values normalized to baseline) (F) Plantar flexion tetanic force (absolute values). (D-F): n=9–12 scr veh, n=6–7 scr SW, n=7–10 shPTGDS veh, n=5–8 shPTGDS SW. (G) Distance to exhaustion on treadmill (n=6 scr veh, n=4 scr SW n=4 shPTGDS veh, n=6 shPTGDS SW). (H-K) I.m. injection of AAV9 carrying a construct of MCK promoter driving Cre expression into the GA of EP4f/f mice or littermate controls (EP4+/+, control). Mice were then treated daily with 15-PGDH inhibitor, SW033291 (SW) or vehicle and muscle function was measured at 1 mo. (H) Experimental scheme. (I) Mass of dissected GA. (J) Plantar flexion tetanic force (values normalized to baseline). (K) Plantar flexion tetanic force (absolute values). (I-K): n=5–7 control veh, n=3–5 control SW, n=4 EP4 KO veh, n=3–4 EP4 KO SW. *P<0.05, **P<0.01, ***P<0.001 ****P<0.0001. ANOVA with Fisher’s LSD (B,D-K) for multiple comparisons; Unpaired t-test (C). Means±s.e.m. Abbreviation: mo. months; i.p. intraperitoneal; i.m. intramuscular; shPT, shPTGDS. C57Bl/6 aged (>24 mo.) mice were used.
We performed additional experiments to substantiate the specific role of PGE2 in attenuating muscle atrophy in aged mice. As an alternative approach to targeting the three enzymes responsible for PGE2 synthesis, we focused on the PGE2 receptors in muscle. qRT-PCR revealed that the PGE2 receptor, EP4 (Ptger4), is the most highly expressed prostaglandin receptor in differentiated myotubes (Fig. S9A). To determine conclusively if the observed muscle hypertrophy was due to PGE2-mediated EP4 signaling in mature muscle myofibers in vivo, we used intramuscular AAV9-mediated delivery of muscle creatine kinase (MCK)-promoter driven Cre to the GA myofibers of aged EP4f/f mice (MCK-EP4Δ/Δ) to genetically ablate EP4 in myofibers. Strikingly, loss of EP4 expression in myofibers of aged mice abrogated the beneficial effect on muscle mass and strength induced by SW-mediated 15-PGDH inhibition (Fig 5H–K, S9B). These data demonstrate that the observed effects of SW treatment result primarily from PGE2 signaling through the EP4 receptor on aged myofibers.
Decreased proteolysis and TGF-beta signaling in aged muscles following 15-PGDH inhibition
To identify downstream signaling pathways through which 15-PGDH inhibition exerts its effects on aged muscles, we performed transcriptomic analysis of vehicle and SW-treated aged muscles. Gene expression analysis revealed a decline in signaling pathways linked to age-related muscle atrophy. Among the top downregulated genes upon SW treatment of aged muscles are members of ubiquitin pathways (Fig 6A,B). PGE2 signaling has previously been implicated in the activation of the AKT pathway in non-muscle cells (12, 22, 23). We therefore sought to determine if this pathway might function in muscle to regulate the expression of E3 ubiquitin ligases that are known to play a role in muscle atrophy (24). To this end, muscle cells, in the absence of other cell types, were subjected to an acute exposure to PGE2. As shown by western blot analysis, differentiated myotubes derived from human donor myoblasts treated with PGE2 for 15 or 30 minutes exhibited increased levels of pAKT which inactivated FOXO (pFOXO3a) (Fig. S10A). Additionally, PGE2 treated myotubes activated the downstream target phospho-S6 ribosomal protein (pS6rp), indicative of increased protein synthesis (Fig. S10A) and exhibited a marked increase in diameter, not seen in the presence of the PGE2 antagonist (ONO-AE3–208) (Fig. S10B,C). In corroboration, we observed an increase in protein synthesis quantified by puromycin incorporation after PGE2 treatment of myotubes (Fig. S10D). These in vitro data show that PGE2 can act directly on myotubes to activate AKT signaling and enhance myotube growth and protein synthesis, providing evidence for a previously understudied role for PGE2 in countering muscle atrophy.
FIG. 6. 15-PGDH inhibition alters multiple pathways to improve aged muscle function.
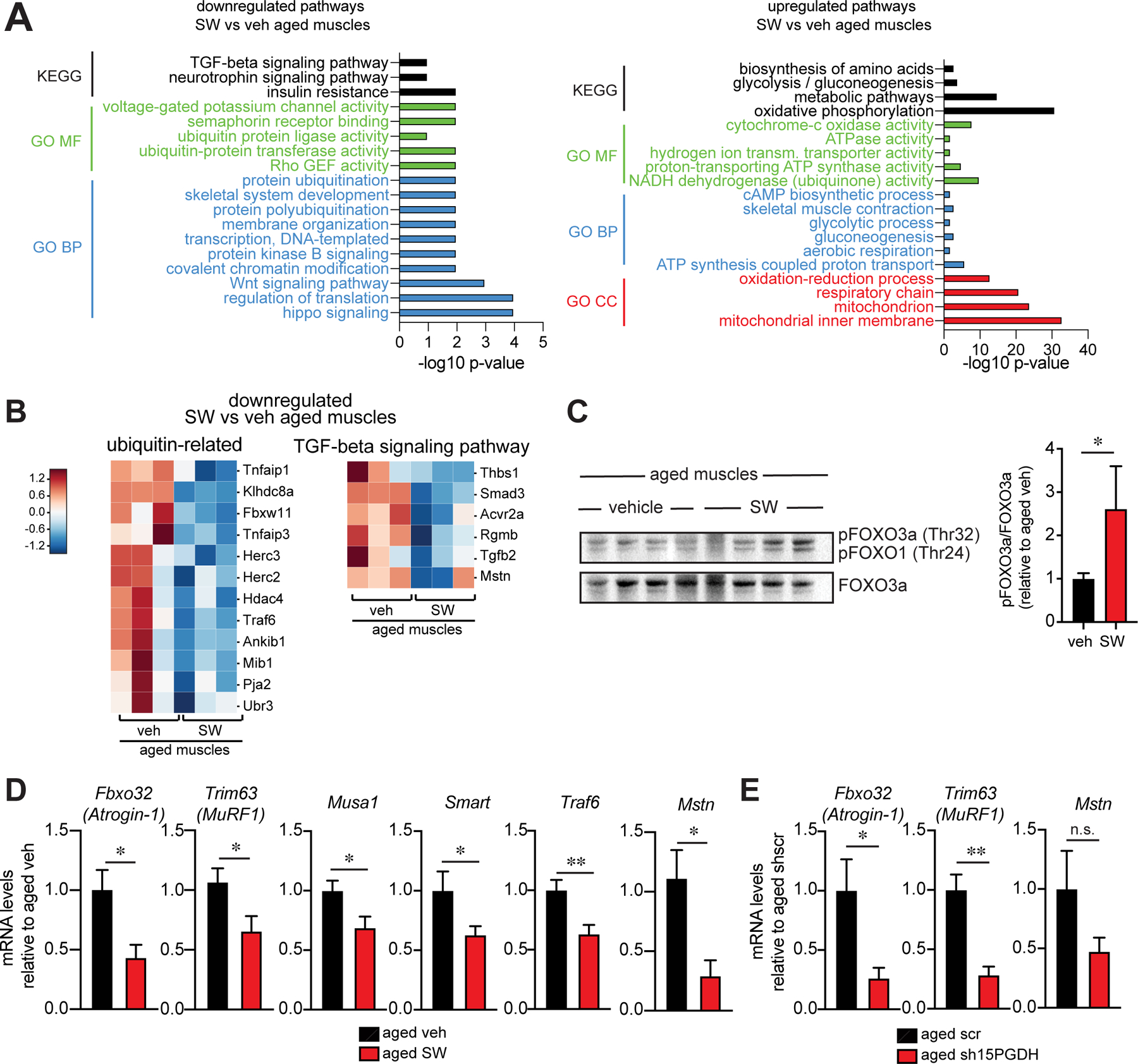
(A-B) RNA sequencing analysis of muscles from aged muscle mice that were treated daily with 15-PGDH inhibitor, SW033291 (SW) or vehicle for 1 mo. (n=3 each). (A) KEGG and GO Term analysis of downregulated (left) and upregulated (right) genes. (B) Heatmap of protein ubiquitin related genes (left) and TGF-beta signaling pathway (right) identified in (A). (C) Immunoblots of muscle lysates from aged vehicle and SW treated mice (left) and quantification (right) (n=3 aged veh, n=4 aged SW). (D) Expression level of MuRF1 (Trim63), Atrogin-1 (Fbxo32) and Myostatin (Mstn) (n=15 aged veh, n=8 aged SW) and Musa, Smart and Traf6 (n=7 aged veh, n=6 aged SW). (E) Expression level of MuRF1 (Trim63), Atrogin-1 (Fbxo32) and Myostatin (Mstn) measured by qPCR (n=5 aged scr, n=4 aged sh15PGDH). *P<0.05, **P<0.01. Unpaired t-test (C-E). Means±s.e.m. Abbreviation: KEGG: Kyoto Encyclopedia of Genes and Genomes; GO: Gene Ontology; BP: Biological Process; MF: Molecular Function; CC: Cellular Component. C57Bl/6 aged (>24 mo.) mice were used.
We sought to determine in vivo in aged muscle tissue if elevation of PGE2 due to 15-PGDH inhibition leads to signaling via the AKT pathway, as seen in vitro in myotubes. We found that inactive pFOXO was increased in SW treated aged muscles compared to vehicle treated controls (Fig. 6C). FOXO has previously been shown by others to play a role in increasing expression of the muscle-specific atrophy-related E3 ubiquitin ligases Atrogin-1 (Fbxo32), MuRF1 (Trim63), Musa1 and Smart (25–27). Analysis by RT-qPCR revealed that expression of all of these atrogenes, as well as the E3 ubiquitin ligase Traf6 (28), was diminished in SW treated aged muscles compared to vehicle treated controls (Fig. 6D), suggesting that a modulation of proteolysis contributes to the attenuation of muscle atrophy. This finding fits with our transcriptome analysis of aged muscles compared to young muscles which shows that the genes in the ubiquitin ligase pathway are among the top enriched upregulated genes in aged muscles (Fig. S4A,B,D) and is in agreement with reports that atrogene expression is increased with aging (29–31). We observed a similar decrease in E3 ubiquitin ligase expression following a genetic inhibition of the 15-PGDH enzyme in aged muscles mediated by intramuscular delivery of an shRNA to 15-PGDH compared to scr shRNA control (Fig. 6E). Of interest, the histone deacetylase Hdac4, another mediator of muscle atrophy that deacetylates proteins such as MyHC and PGC1α leading to their ubiquitination, was diminished in SW-treated muscles (Fig. 6B). Reduced Hdac4 could also contribute to the lower levels of atrogenes Atrogin-1 and MuRF1 (32, 33). These results show that PGE2 leads to a modulation of atrogene expression that tempers the increased protein degradation seen in aged muscles and contributes to the observed amelioration of muscle atrophy in aged muscles.
Our transcriptome analysis revealed a reduction in a second signaling pathway, the TGF-beta pathway, after one month of SW treatment, providing evidence of another synergistic beneficial effect of 15-PGDH inhibition in aged muscles. The expression of key TGF-beta pathway genes, such as myostatin, that are known to be detrimental to muscle function and associated with muscle atrophy in aging (Mstn, Tgfb2, Acrv2a, Smad3) (24), was decreased (Fig. 6B,D,E), which likely contributed to the observed attenuation of muscle atrophy. Notably, no significant changes were observed in other aging and inflammatory markers assayed in muscles of SW treated aged mice (Fig. S11A,B). Together, these results show that a one month 15-PGDH inhibition and consequent elevation of PGE2 in aged muscles stimulates several synergistic signaling pathways leading to the observed improvement in muscle function in aged mice.
Elevation of mitochondrial function and biogenesis and increase in autophagy following 15-PGDH inhibition
Among the most striking changes in the aged muscle tissue transcriptome after SW treatment was a strong enrichment for mitochondrial pathways, including mitochondrial oxidative phosphorylation, ATP synthesis and other metabolic and energy generating functions (Fig 6A, 7A). Numerous components of the mitochondria complexes I, II, IV and V of the electron transport chain were markedly increased in SW-treated aged muscles (Fig 7A). PGE2 signaling through the G-coupled protein receptor, EP4, is known to be mediated by cyclic AMP (cAMP) (12, 21, 34). We confirmed in skeletal muscles that PGE2 activates the cyclic AMP response element binding protein (CREB) (Fig S12A). Importantly, when we assayed mRNA levels of the master regulator for mitochondrial biogenesis peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Pgc1α), which has a CREB binding motif in its promoter (35), we found that its level was restored in aged muscles to that seen in young (Fig 7B). Overall mitochondrial content was increased, as reflected by the increased mitochondrial DNA content relative to nuclear DNA content following SW treatment of aged muscles (Fig 7C).
FIG. 7. 15-PGDH inhibition boosts mitochondrial biogenesis and function in aged muscles.
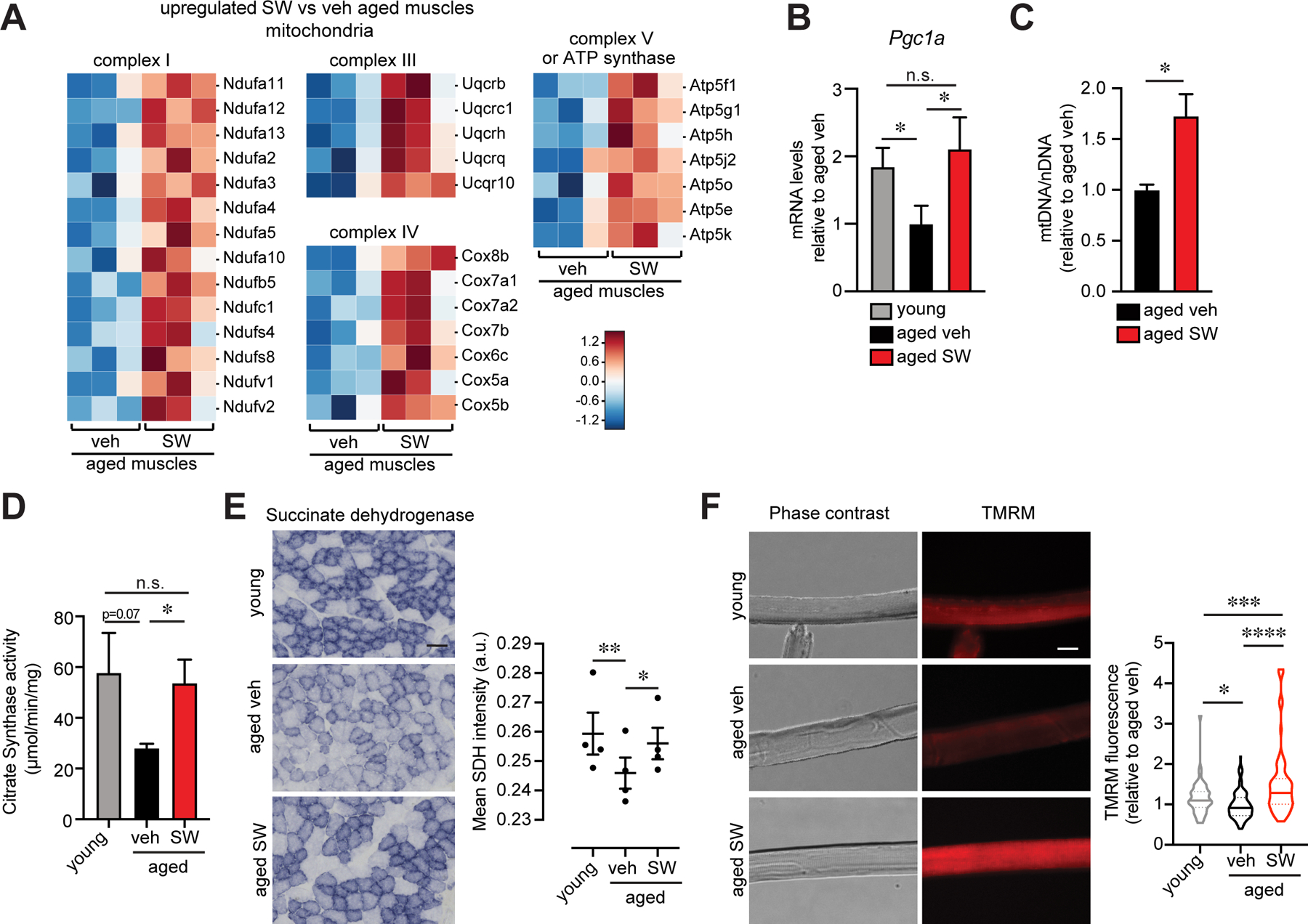
(A) Heatmap of mitochondrial genes identified in (6A). (B) Expression level of Pgc1a by qPCR (n=4 young, n=4 aged veh, n=3 aged SW033291 (SW)). (C) Relative quantification of mitochondrial DNA to nuclear DNA (n=4 per group). (D) Citrate synthase activity of Gastrocnemius muscles (n=3 young, n=4 aged veh, n=4 aged SW). (E) Representative Tibilais anterior (TA) cross-section stained for succinate dehydrogenase (SDH) (left). Quantification of SDH mean average intensity per fiber (n=4 mice per condition). Bar=50 µm. (F) Representative images of mitochondrial membrane potential (TMRM staining) in isolated Extensor digitorum longus (EDL) myofibers from young and aged vehicle and SW treated mice (n=4 mice per condition; total number of myofibers: n=52 young, n=139 aged veh, n=89 aged SW). Bar=10 µm. *P<0.05, **P<0.01, ***P<0.001 ****P<0.0001. ANOVA with Fisher’s LSD for multiple comparisons (B,D-F); Unpaired t-test (C). Means±s.e.m. C57Bl/6 young (2–4 mo.) and aged (>24 mo.) mice were used.
To assess mitochondrial function, we analyzed citrate synthase activity, the first enzyme of the Krebs cycle (36), in isolated mitochondria and found that it was significantly increased in SW-treated aged muscles compared to controls, reaching levels comparable to young (Fig 7D and S12B). SW treated aged muscle mitochondria also showed increased succinate dehydrogenase staining which reflects enzymatic activity, a key component of both the Krebs cycle and the electron transport chain (36) (Fig 7E). Additionally, we observed a marked increase in mitochondrial membrane potential in myofibers isolated from EDL muscles of SW treated aged mice compared to controls (Fig 7F).
A morphological assessment by transmission electron microscopy (TEM) of Extensor digitorum longus (EDL) muscles after SW treatment revealed an increase in the number of intermyofibrillar (IMF) mitochondria (Fig 8A,B and S13A). In addition, the morphology of the mitochondria improved. The shift in IMF mitochondrial area from compact and round in young to a larger size with disorganized morphology was evident in aged tissues, in agreement with reports by others (37, 38) (Fig 8A,B and S13A). Following one month of SW treatment, aged mitochondrial morphology was restored to a compact circular mitochondrial morphology resembling that seen in young (Fig 8A,B and S13A). TEM analysis also revealed an increase in myofibril width consistent with the increased cross-sectional area and muscle mass seen in the aged treated with SW compared to vehicle controls (Fig. 2D–L, 8A,B and S13A,B). A decrease in autophagy and an accumulation of dysfunctional organelles such as mitochondria has been associated with sarcopenia (38). In accordance with the increase in mitochondrial function, we observed an increase in autophagy flux. An increase in autophagy was evident by western blot of p62 upon colchicine treatment of muscles of aged mice treated with SW compared to controls (Fig. S13C). Together, these data provide strong evidence that a one month 15-PGDH inhibition and consequent elevation of PGE2 in aged muscles triggers mitochondrial biogenesis and increased mitochondrial function.
FIG. 8. 15-PGDH inhibition in aged mice increases mitochondrial number and improves mitochondrial morphology.
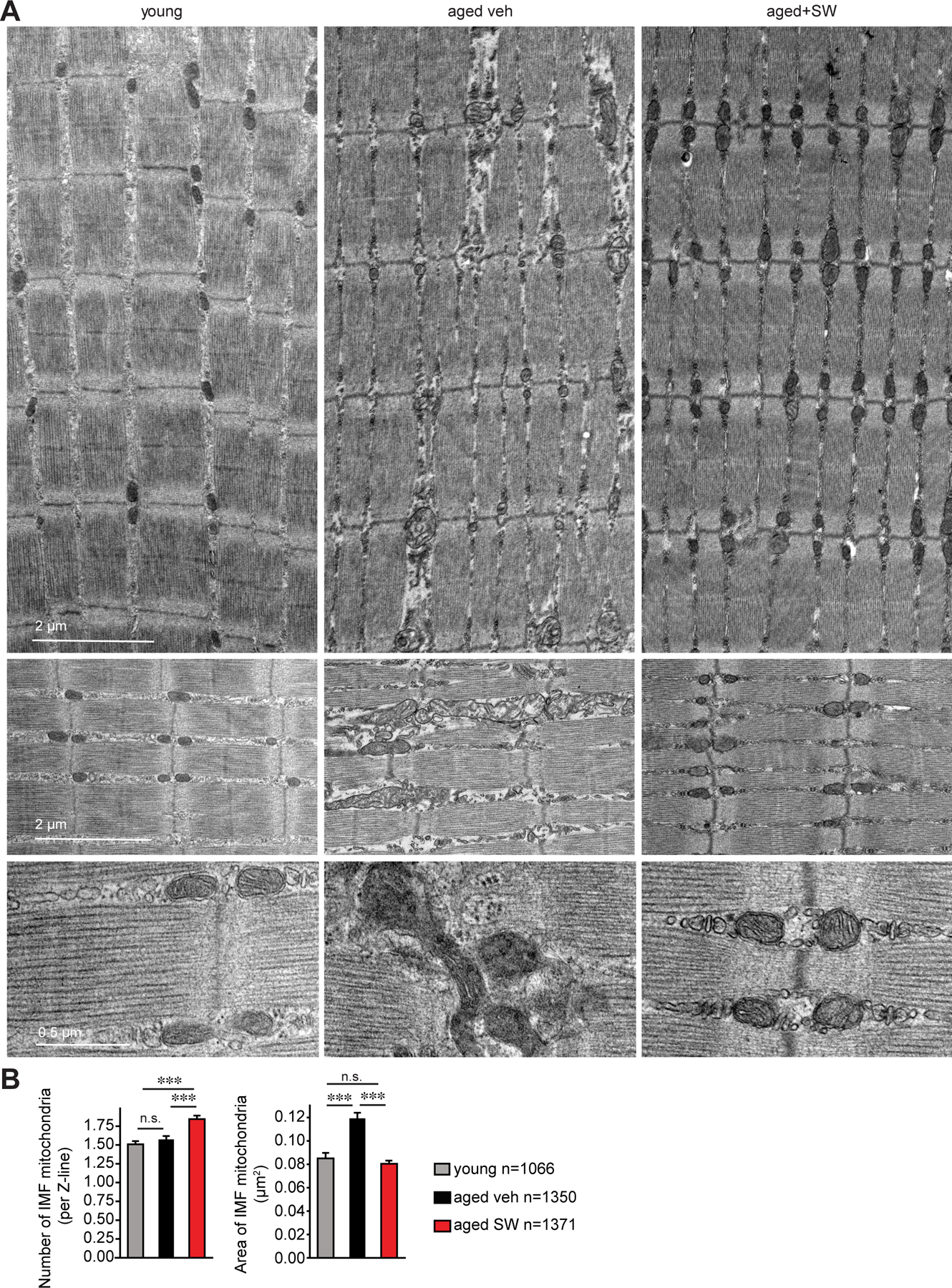
(A) Representative images of intermyofibrillar (IMF) mitochondria from transmission electron micrographs (TEM) of longitudinal sections of Extensor digitorum longus (EDL) muscles of young and aged vehicle and SW treated mice. (B) Quantifications of IMF mitochondria number and size from TEM images as in (A). (n=3 mice per condition; total mitochondria quantified: n=1,066 young, n=1,350 aged vehicle, n=1,371 aged SW). ***P<0.001. ANOVA test with Tukey’s test (B). Means±s.e.m. C57Bl/6 young (2–4 mo.) and aged (>24 mo.) mice were used.
Discussion
A drastic loss of muscle strength accompanies aging. The lower body muscles decline by 50–80% in aged humans, a loss that is manifested by a reduction in muscle mass and strength and is associated with a marked increase in mortality (1, 2, 39). There are currently no therapies for age-related muscle wasting, or sarcopenia, and its healthcare burden is high (2). Here we identify elevated expression of the prostaglandin degrading enzyme, 15-PGDH, as a new marker of aged muscles, both in mouse and human. We find that increased 15-PGDH activity is not limited to muscle, but is increased in many aged tissues, for example aged heart, skin, colon and spleen. In aged mice, inhibition of 15-PGDH, either by genetic knockdown or a small molecule, counters muscle atrophy and markedly increases muscle mass, strength and endurance. Using genetic loss of function experiments and LC-MS/MS mass spectrometry, which is capable of definitive resolution and quantification of highly similar prostaglandin family members, we show that the amelioration of muscle function is due to increased levels of PGE2 and signaling via its receptor, EP4. The striking loss of muscle mass and strength within one month of overexpressing the enzyme in young mice, highlights its causal role in muscle atrophy. We and others previously demonstrated the importance of PGE2 signaling in stimulating stem cells to regenerate damaged tissues in young mice (7–10). Here we show that PGE2 also acts on mature muscle myofibers and is crucial to the maintenance of muscle tissue homeostasis. Importantly, our data suggest that 15-PGDH is a potential therapeutic target to counter the debilitating muscle atrophy characteristic of sarcopenia.
Our data suggest that the aged muscle milieu plays a key role in the decrease in PGE2 levels in aged muscles. In addition to myofibers, tissue resident macrophages in aged muscles express elevated 15-PGDH. Future studies are warranted to investigate if 15-PGDH expression differs among muscles and among fiber types within muscles, and how it functions in an autocrine loop to induce muscle atrophy. Additionally, the mechanism by which 15-PGDH is increased with aging remains unknown. We postulate that tissue-resident macrophages expressing 15-PGDH play a critical role in fostering dysfunction by reducing PGE2 levels in a multiplicity of aged tissues via a paracrine mechanism.
Here we provide in vivo evidence that restoring endogenous PGE2 levels within a physiological range suffices to overcome muscle atrophy and protein catabolism in aged mice. In contrast, previous studies that implicated PGE2 in protein degradation used denervated excised muscles undergoing rapid muscle protein catabolism (40, 41). Our data fit well with prior studies in which perturbation of COX enzyme levels revealed a role for prostaglandins in muscle hypertrophy or recovery from muscle atrophy (21, 42, 43). However, COX2 is a problematic therapeutic target, as it is critical to the synthesis of disparate prostaglandins with antagonistic effects.
Sarcopenia is a multifactorial disease, a compendium of dysregulated signaling pathways that culminate in chronic inflammation, muscle denervation, defective mitochondria due to diminished autophagy, and disrupted proteostasis (4, 44, 45). In particular, mitochondrial function is impaired (46). Our transcriptome analysis revealed that mitochondrial functions, including Complexes I-V, are among the top upregulated genes upon 15-PGDH inhibition. We show PGE2 signaling through the EP4 receptor via cAMP/CREB in agreement with others (23). This could account for the observed increase in mitochondrial number and function by activating downstream transcriptional regulators with cAMP response elements (CREB binding motifs) including the master mitochondrial regulator Pgc1α and other oxidative genes (47, 48), as reported for other cAMP inducing agents, such as β-adrenergic receptor (β-AR) agonists or corticotropin releasing factor receptor 2 (CRFR2) agonists (49). In addition, activity of mitochondrial enzymes and membrane potential were restored upon SW treatment reaching levels similar to young muscles. Perhaps most striking was the rejuvenation of mitochondrial and tissue morphology apparent by TEM. The large distended vacuous organelles typical of aged muscle were replaced by round compact organelles in orderly doublets after one month of SW treatment. We postulate that this extensive tissue and mitochondrial remodeling is mediated by autophagy. These data demonstrate that 15-PGDH inhibition triggers a signaling cascade that culminates in a dramatic rejuvenation of mitochondrial biogenesis, form and function that is likely to be central to the marked reversal of aged muscle atrophy and increase in strength.
Ubiquitin-proteasome pathway and TGF-beta signaling genes were markedly downregulated after one month of 15-PGDH inhibition. Whether ubiquitin ligase expression plays a causal role in sarcopenia remains a matter of debate. Others have reported elevated levels of the E3 ubiquitin ligases Atrogin-1 and MuRF1 in aged rat (29, 30) and human muscles (31), similar to our findings in aged murine muscles. On the other hand, constitutive knockout of certain E3 ubiquitin ligases, including Atrogin-1 that had a deleterious effect on muscle function (50), but was protective in the context of acute denervation atrophy (51). Notably, short-term interventions that ameliorated sarcopenia, such as rapalogs, sestrin, or Apelin, reduced atrogene expression (Atrogin-1 and MuRF1) (52–54) in accordance with our results with 15-PGDH inhibition. We also observed a decline in several of the TGF-beta signaling components, including Myostatin, a major focus of therapeutic efforts to promote hypertrophy (1, 55). The reduction in atrogene expression and TGFβ signaling likely play a synergistic role in the beneficial effect of SW in attenuating muscle atrophy.
In summary, here we uncover 15-PGDH as a previously unrecognized marker and therapeutic target for strategies that aim to augment muscle mass and strength and overcome sarcopenia, the muscle wasting associated with aging. Our intervention is advantageous, as it entails a physiological restoration of homeostatic levels of PGE2 in aged mice to those found in young mice. The resulting moderate increase in PGE2 levels modulates several signaling pathways to promote mitochondrial biogenesis and function while inhibiting TGF-beta and ubiquitin proteosome pathways, leading to an increase in muscle function. Since 15-PGDH activity is elevated in a range of tissues, we postulate that its inhibition will have pleiotropic beneficial effects that extend beyond skeletal muscle to numerous tissues during aging.
Supplementary Material
Acknowledgments:
We apologize to those investigators whose important work we were unable to cite or describe owing to space constraints. We thank K. Koleckar, D.M. Burns, G. Markov for critical discussions of the manuscript and for technical assistance, W. Zhou and P. Nallagatla from Stanford Genetics Bioinformatics Service Center for analysis of transcriptomics data, G.P. Nolan for sharing CODEX methodology in advance of publication and for CODEX reagents, J. Perrino from the Cell Sciences Imaging Facility (CSIF) at Stanford for expert TEM processing and imaging, the FACS Core Facility in Stanford Lokey Stem Cell Research Building, the Vincent Coates Foundation Mass Spectrometry Laboratory (http://mass-spec.stanford.edu) and the Stanford Veterinary Service Center (VSC) for technical support.
Funding:
This study was supported by the Baxter Foundation, the Li Ka Shing Foundation, California Institute for Regenerative Medicine (CIRM) grant DISC2-10604 and US National Institutes of Health (NIH) grant 5R01AG02096115, 1R01AG069858-01 and RHG009674A (H.M.B.). This work utilized the Xevo TQ-XS mass spectrometer system that was purchased with funding from National Institutes of Health Shared Instrumentation grant S10OD026962. Y.X.W. was supported by the Canadian Institutes of Health Research and NIH K99 award K99NS120278, M.R by an SNF Early Postdoc Mobility Fellowship P2EZP3_184231, and C.M.S. by an SNF Postdoc Mobility Fellowship. Work using TEM described here was supported, in part, by ARRA Award Number 1S10RR026780-01 from the National Center for Research Resources (NCRR).
Footnotes
Competing interests: A.R.P., M.R., A.T.V.H. and H.M.B. are named inventors on patent applications regarding 15-PGDH inhibition licensed to Myoforte Therapeutics. Y.X.W., C.A.H. and H.M.B. are named inventors on a patent application for processing of multiplex microscopy images. A.R.P., A.T.V.H. and H.M.B. receive consulting fees and have equity and stock options from Myoforte Therapeutics. H.M.B. is a cofounder of Myoforte Therapeutics. C.M.S. is a scientific advisor to Enable Medicine, LLC.
Data and materials availability: All data are available in the main text or the supplementary materials. The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database GSE149924. Correspondence and requests for materials should be addressed to H.M.B.
Publisher's Disclaimer: “This manuscript has been accepted for publication in Science. This version has not undergone final editing. Please refer to the complete version of record at http://www.sciencemag.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.”
References and Notes:
- 1.Cohen S, Nathan JA, Goldberg AL, Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov 14, 58–74 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Rolland Y et al. , Sarcopenia: its assessment, etiology, pathogenesis, consequences and future perspectives. J Nutr Health Aging 12, 433–450 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.von Haehling S, Morley JE, Anker SD, An overview of sarcopenia: facts and numbers on prevalence and clinical impact. J Cachexia Sarcopenia Muscle 1, 129–133 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larsson L et al. , Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol Rev 99, 427–511 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee CE, McArdle A, Griffiths RD, The role of hormones, cytokines and heat shock proteins during age-related muscle loss. Clin Nutr 26, 524–534 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Lenk K, Schuler G, Adams V, Skeletal muscle wasting in cachexia and sarcopenia: molecular pathophysiology and impact of exercise training. J Cachexia Sarcopenia Muscle 1, 9–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ho ATV et al. , Prostaglandin E2 is essential for efficacious skeletal muscle stem-cell function, augmenting regeneration and strength. Proc Natl Acad Sci U S A 114, 6675–6684 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen H et al. , Prostaglandin E2 mediates sensory nerve regulation of bone homeostasis. Nat Commun 10, 181 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.North TE et al. , Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature 447, 1007–1011 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y et al. , Inhibition of the prostaglandin-degrading enzyme 15-PGDH potentiates tissue regeneration. Science 348, aaa2340 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prasain JK, Hoang HD, Edmonds JW, Miller MA, Prostaglandin extraction and analysis in Caenorhabditis elegans. J Vis Exp, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang D, Dubois RN, Eicosanoids and cancer. Nat Rev Cancer 10, 181–193 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu YH et al. , Structural basis for catalytic and inhibitory mechanisms of human prostaglandin reductase PTGR2. Structure 16, 1714–1723 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Raue U et al. , Transcriptome signature of resistance exercise adaptations: mixed muscle and fiber type specific profiles in young and old adults. J Appl Physiol (1985) 112, 1625–1636 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kessler PD et al. , Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein. Proc Natl Acad Sci U S A 93, 14082–14087 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shin JH et al. , Microdystrophin ameliorates muscular dystrophy in the canine model of duchenne muscular dystrophy. Mol Ther 21, 750–757 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE, Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther 16, 1073–1080 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Goltsev Y et al. , Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell 174, 968–981 e915 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schurch CM et al. , Coordinated Cellular Neighborhoods Orchestrate Antitumoral Immunity at the Colorectal Cancer Invasive Front. Cell 182, 1341–1359 e1319 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sacheck JM et al. , Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J 21, 140–155 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Korotkova M, Lundberg IE, The skeletal muscle arachidonic acid cascade in health and inflammatory disease. Nat Rev Rheumatol 10, 295–303 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Fujino H, Regan JW, EP(4) prostanoid receptor coupling to a pertussis toxin-sensitive inhibitory G protein. Mol Pharmacol 69, 5–10 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Konya V, Marsche G, Schuligoi R, Heinemann A, E-type prostanoid receptor 4 (EP4) in disease and therapy. Pharmacol Ther 138, 485–502 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonaldo P, Sandri M, Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 6, 25–39 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Milan G et al. , Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun 6, 6670 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandri M et al. , Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117, 399–412 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stitt TN et al. , The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 14, 395–403 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Paul PK et al. , Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J Cell Biol 191, 1395–1411 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Altun M et al. , Muscle wasting in aged, sarcopenic rats is associated with enhanced activity of the ubiquitin proteasome pathway. J Biol Chem 285, 39597–39608 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clavel S et al. , Atrophy-related ubiquitin ligases, atrogin-1 and MuRF1 are up-regulated in aged rat Tibialis Anterior muscle. Mech Ageing Dev 127, 794–801 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Welle S, Brooks AI, Delehanty JM, Needler N, Thornton CA, Gene expression profile of aging in human muscle. Physiol Genomics 14, 149–159 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Luo L et al. , HDAC4 Controls Muscle Homeostasis through Deacetylation of Myosin Heavy Chain, PGC-1alpha, and Hsc70. Cell Rep 29, 749–763 e712 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Moresi V et al. , Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell 143, 35–45 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ricciotti E, FitzGerald GA, Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol 31, 986–1000 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandez-Marcos PJ, Auwerx J, Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 93, 884S–890 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berg JM, Tymoczko JL, Stryer L, Biochemistry. (Freeman WH, 2002).
- 37.Leduc-Gaudet JP et al. , Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 6, 17923–17937 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Romanello V, Sandri M, Mitochondrial Quality Control and Muscle Mass Maintenance. Front Physiol 6, 422 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fielding RA et al. , Sarcopenia: an undiagnosed condition in older adults. Current consensus definition: prevalence, etiology, and consequences. International working group on sarcopenia. J Am Med Dir Assoc 12, 249–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rodemann HP, Goldberg AL, Arachidonic acid, prostaglandin E2 and F2 alpha influence rates of protein turnover in skeletal and cardiac muscle. J Biol Chem 257, 1632–1638 (1982). [PubMed] [Google Scholar]
- 41.Rodemann HP, Waxman L, Goldberg AL, The stimulation of protein degradation in muscle by Ca2+ is mediated by prostaglandin E2 and does not require the calcium-activated protease. J Biol Chem 257, 8716–8723 (1982). [PubMed] [Google Scholar]
- 42.Bondesen BA, Mills ST, Pavlath GK, The COX-2 pathway regulates growth of atrophied muscle via multiple mechanisms. Am J Physiol Cell Physiol 290, C1651–1659 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Trappe TA, Liu SZ, Effects of prostaglandins and COX-inhibiting drugs on skeletal muscle adaptations to exercise. J Appl Physiol (1985) 115, 909–919 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ibebunjo C et al. , Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol Cell Biol 33, 194–212 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ubaida-Mohien C et al. , Discovery proteomics in aging human skeletal muscle finds change in spliceosome, immunity, proteostasis and mitochondria. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Calvani R et al. , Mitochondrial pathways in sarcopenia of aging and disuse muscle atrophy. Biol Chem 394, 393–414 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Austin S, St-Pierre J, PGC1alpha and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci 125, 4963–4971 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Herzig S et al. , CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413, 179–183 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Berdeaux R, Stewart R, cAMP signaling in skeletal muscle adaptation: hypertrophy, metabolism, and regeneration. Am J Physiol Endocrinol Metab 303, E1–17 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zaglia T et al. , Atrogin-1 deficiency promotes cardiomyopathy and premature death via impaired autophagy. J Clin Invest 124, 2410–2424 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bodine SC et al. , Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294, 1704–1708 (2001). [DOI] [PubMed] [Google Scholar]
- 52.Joseph GA et al. , Partial Inhibition of mTORC1 in Aged Rats Counteracts the Decline in Muscle Mass and Reverses Molecular Signaling Associated with Sarcopenia. Mol Cell Biol 39, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Segales J et al. , Sestrin prevents atrophy of disused and aging muscles by integrating anabolic and catabolic signals. Nat Commun 11, 189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vinel C et al. , The exerkine apelin reverses age-associated sarcopenia. Nat Med 24, 1360–1371 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Elkina Y, von Haehling S, Anker SD, Springer J, The role of myostatin in muscle wasting: an overview. J Cachexia Sarcopenia Muscle 2, 143–151 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cosgrove BD et al. , Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med 20, 255–264 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sacco A, Doyonnas R, Kraft P, Vitorovic S, Blau HM, Self-renewal and expansion of single transplanted muscle stem cells. Nature 456, 502–506 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gilbert PM et al. , Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science 329, 1078–1081 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brun CE, Wang YX, Rudnicki MA, Single EDL Myofiber Isolation for Analyses of Quiescent and Activated Muscle Stem Cells. Methods Mol Biol 1686, 149–159 (2018). [DOI] [PubMed] [Google Scholar]
- 60.Azzarello G et al. , Myosin isoform expression in rat rhabdomyosarcoma induced by Moloney murine sarcoma virus. J Cancer Res Clin Oncol 113, 417–429 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schiaffino S et al. , Three myosin heavy chain isoforms in type 2 skeletal muscle fibres. J Muscle Res Cell Motil 10, 197–205 (1989). [DOI] [PubMed] [Google Scholar]
- 62.Smith LR, Barton ER, SMASH - semi-automatic muscle analysis using segmentation of histology: a MATLAB application. Skelet Muscle 4, 21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rando TA, Blau HM, Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. The Journal of cell biology 125, 1275–1287 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu X et al. , Human Satellite Cell Transplantation and Regeneration from Diverse Skeletal Muscles. Stem Cell Reports 5, 419–434 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baker DJ et al. , Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Quiros PM, Goyal A, Jha P, Auwerx J, Analysis of mtDNA/nDNA Ratio in Mice. Curr Protoc Mouse Biol 7, 47–54 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dobin A et al. , STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li B, Dewey CN, RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang da W, Sherman BT, Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 71.Ju JS, Varadhachary AS, Miller SE, Weihl CC, Quantitation of “autophagic flux” in mature skeletal muscle. Autophagy 6, 929–935 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dubowitz V, Sewry C, Oldfors A, Muscle Biopsy: a Practical Approach. (Elsevier Health Sciences, ed. 5th, 2020). [Google Scholar]
- 73.Mintz EL, Passipieri JA, Lovell DY, Christ GJ, Applications of In Vivo Functional Testing of the Rat Tibialis Anterior for Evaluating Tissue Engineered Skeletal Muscle Repair. J Vis Exp, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sheth KA et al. , Muscle strength and size are associated with motor unit connectivity in aged mice. Neurobiol Aging 67, 128–136 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.