

Abstract
Histone‐3‐lysine‐4 (H3K4) methylation is catalysed by the multiprotein complex known as the Set1/COMPASS or MLL/COMPASS‐like complex, an element that is highly evolutionarily conserved from yeast to humans. However, the components and mechanisms by which the COMPASS‐like complex targets the H3K4 methylation of plant‐pathogenic genes in fungi remain elusive. Here we present a comprehensive analysis combining biochemical, molecular, and genome‐wide approaches to characterize the roles of the COMPASS‐like family in the rice blast fungus Magnaporthe oryzae, a model plant pathogen. We purified and identified six conserved subunits of COMPASS from M. oryzae: MoBre2 (Cps60/ASH2L), MoSpp1 (Cps40/Cfp1), MoSwd2 (Cps35), MoSdc1 (Cps25/DPY30), MoSet1 (MLL/ALL), and MoRbBP5 (Cps50), using an affinity tag on MoBre2. We determined the sequence repeat in dual‐specificity kinase splA and ryanodine receptors domain of MoBre2 can interact directly with the DPY30 domain of MoSdc1 in vitro. Furthermore, we found that deletion of the genes encoding COMPASS subunits of MoBre2, MoSPP1, and MoSwd2 caused similar defects regarding invasive hyphal development and pathogenicity. Genome‐wide profiling of H3K4me3 revealed that it has remarkable co‐occupancy at the transcription start site regions of target genes. Significantly, these target genes are often involved in spore germination and pathogenesis. Decreased gene expression caused by the deletion of MoBre2, MoSwd2, or MoSpp1 was highly correlated with a decrease in H3K4me3. These results suggest that MoBre2, MoSpp1, and MoSwd2 function as a whole COMPASS complex, contributing to fungal development and pathogenesis by regulating H3K4me3‐targeted genes in M. oryzae.
Keywords: COMPASS complex, H3K4me3, Magnaporthe oryzae, pathogenesis, target gene, type III secretion system
The histone epigenetic COMPASS‐like complex modulates fungal growth, conidiation, and pathogenesis by regulating H3K4me3‐mediated targeted gene expression in TSS regions of the rice blast fungus.

1. INTRODUCTION
The eukaryotic genome is found in complexes with an equal mass of histone proteins to form nucleosomes, the basic units of chromatin (Kornberg, 1974; Smith & Shilatifard, 2010). Chromatin assembly and folding into higher structures is a dynamic process that ultimately affects the activation or repression of gene expression. Chromatin remodelling and covalent modifications of histones are two means by which variation is introduced into the chromatin polymer, thereby regulating the structure and function of chromatin and ultimately gene expression (Bhaumik et al., 2007; Lachner & Jenuwein, 2002; Luger et al., 1997; Shilatifard, 2008; Strath & Allis, 2000). Chromatin‐modifying activities are often recruited to specific gene regulatory sequences, whereupon they cause localized changes in the chromatin structure and specific transcriptional effects (Allis et al., 2007; Ng et al., 2003). Several post translational modifications are known to be present on the terminal domains of histones, such as acetylation, phosphorylation, ubiquitination, ADP ribosylation, and methylation (Berger, 2007; Kouzarides, 2007; Workman & Kingston, 2003). Lysine methylation has been well documented on histones H3 and H4 in vivo, and residues 4, 9, 27, and 36 are typical methylation sites (Wang et al., 2010; Yang et al., 2012).
Histone‐3‐lysine‐4 (H3K4) methylation is associated with active chromatin in a wide range of eukaryotic organisms. Many of the genes involved in H3K4 methylation encode proteins bearing a 130‐ to 140‐amino‐acid motif called the SET domain. This domain takes its name from the Drosophila proteins Su(var)3–9, Enhancer of zeste (E(z)), and Trithorax. Many SET domain‐containing proteins have been shown to possess histone or lysine methyltransferase activity (Shilatifard, 2012; Stassen et al., 1995; Tschiersch et al., 1994). In Saccharomyces cerevisiae, the Set1/COMPASS (Complex Proteins Associated with Set1), which consists of the seven polypeptides Set1, Cps60/Bre2, Cps50/Swd1, Cps40/Spp1, Cps35/Swd2, Cps30/Swd3, and Cps25/Sdc1, was the first H3K4 methylase to be identified (Krogan et al., 2003; Miller et al., 2001). Cps60 is the product of the BRE2/YLR015w gene, which encodes a protein similar to both Drosophila and human ASH2L proteins. ASH2L is a member of the Trx family of homeodomain DNA‐binding proteins that is thought to regulate gene expression, morphogenesis, and differentiation in humans. Cps25 is encoded by the YDR469 gene, which has weak similarity to a DPY30 protein involved in dosage compensation of expression of genes on the X chromosome. Some studies have revealed that Cps60 (Bre2) and Cps25 (Sdc1) form a globular structure sitting at the base of the Y‐shaped COMPASS structure sandwich (Chen et al., 2012; Li et al., 2016). Cps35 is the only essential subunit of the COMPASS. In human embryonic kidney 293 cells, the depletion of WDR5 (a Cps35 homologue) can result in a global loss of histone H3K4 methylation and concomitant loss of Hox gene expression (Li et al., 2016). The mammalian Wdr82 (a Cps35 homologue) is required for the targeting of Setd1A‐mediated H3K4 trimethylation near transcription start sites (TSSs) via tethering to RNA polymerase II, an event that is a consequence of transcription initiation (Lee & Skalnik, 2008; Palmer et al., 2013). Null mutants missing any one of the six COMPASS subunits grow more slowly and display greater hydroxyurea sensitivity than wild‐type cells under normal conditions, and they also exhibit a defect in silencing gene expression near chromosomal telomeres (Dover et al., 2002; Roguev et al., 2001).
Previous studies have shown that the COMPASS family of H3K4 methyltransferases is highly conserved from yeast to plants and humans (Jiang et al., 2011; Shilatifard, 2012). In Drosophila melanogaster, there are three subclasses within COMPASS‐like complexes, namely SetD1A, Trx, and trithorax‐related (Trr), all of which are homologues to yeast Set1 (Ardehali et al., 2011; Mohan et al., 2011; Shilatifard, 2012). While Set1D1A is homologous to yeast Set1, Trx and Trr are more distantly related (Czermin et al., 2002). dSet1 (Drosophila Set1), Trx, and Trr each interact with a set of core complex subunits, with additional subunits unique to each (Mohan et al., 2011). In human cells, Set1A, Set1B, MLL1, MLL2, MLL3, and MLL4 are homologous to yeast Set1 (Cho et al., 2007). A biochemical study showed that human cells bear at least six COMPASS family members, each of which is capable of methylating H3K4 with nonredundant functions (Dover et al., 2002; Roguev et al., 2001). These enzymes participate in diverse gene regulatory networks and are fully active in the context of an MLL/COMPASS‐like complex that includes the proteins WDR5, RbBP5, ASH2L, and DPY‐30 (WRAD). WRAD components function by engaging in physical interactions that recruit the SET1 family of proteins to target chromatin sites (Ali & Tyagi, 2017; Ernst & Vakoc, 2012; Hsu et al., 2018).
In Schizosaccharomyces pombe (fission yeast), Set3 was first characterized by its feature of containing a plant homeodomain finger and SET domain. Set3C (Set3/Hos2 Complex) is the functional core of a histone deacetylase complex. Set3C plays both repressive and activating roles in transcription that contributes to heterochromatin integrity by promoting transcription of subunits of Clr4‐Rik1‐Cul4 histone methyltransferase complex (Lee & Skalnik, 2008; Yu et al., 2016). The Set1/COMPASS and mediator are repurposed to promote epigenetic transcriptional memory by a highly conserved mechanism (D’Urso et al., 2016). In Arabidopsis thaliana, the COMPASS‐like complexes mediate H3K4 trimethylation (me3) to control floral transition and plant development (Jiang et al., 2011).
In fungi, the first COMPASS protein identified was SET1 in Candida albicans. SET1 was involved in H3K4 methylation‐related pathogenesis of invasive candidiasis (Raman et al., 2006). In Aspergillus spp., the CclA (a Bre2 ortholog) and SppA (an Spp1 ortholog) are two members of the conserved eukaryotic COMPASS complex that methylates H3K4, first identified to play critical roles in the regulation of several secondary metabolites (Bok et al., 2009; Palmer et al., 2013; Shinohara et al., 2015). Deletion of the COMPASS components CCL1 in Fusarium spp. also results in increased expression of several secondary metabolites in these fungi. Some quantitative PCR‐coupled chromatin immunoprecipitation (ChIP) experiments also proved that Ccl1 regulates balance between H3K4me2 and H3K4me3 independently of the transcriptional status of secondary metabolite genes (Shinohara et al., 2015; Studt et al., 2016, 2017). FgSet1 is a Set1 orthologue of Fusarium graminearum and is one subunit of the COMPASS‐like complex, which also plays an important role in regulating fungal growth and secondary metabolism (Bachleitner et al., 2019; Connolly et al., 2013; Liu et al., 2015). Magnaporthe oryzae is a model plant‐pathogenic fungus, being the causal agent of rice blast disease, which is the most devastating and persistent disease of cultivated rice (Cao et al., 2016; Liu et al., 2018; Yan & Talbot, 2016; Zhang et al., 2018). MoSet1 is the homologue of S. cerevisiae Set1. When MoSet1 is deleted (Δmoset1), H3K4 methylation becomes impaired, resulting in severe defects in infection‐related morphogenesis, including in conidiation and appressorium formation (Li et al., 2016; Pham et al., 2015). The homologues of the COMPASS complex exist in many fungi, but other than the Set1 subunit, no clear systemic functions of COMPASS have been reported in M. oryzae.
Here, we report the identification of a conserved COMPASS‐like complex that contains six subunits: MoSet1, MoBre2, MoSwd1, MoSpp1, MoSwd2, and MoSdc1. When the component genes MoBre2, MoSpp1, and MoSwd2 are deleted, H3K4me3 signals are dramatically decreased. Our results show that MoBre2, MoSpp1, and MoSwd2 not only have unique regulation roles in fungal development, but also function as a whole complex to methylate H3K4 in the TSS region of its pathogenicity genes. Moreover, most of these key target genes are involved in conidia, mycelial growth, and infectious hyphal formation in M. oryzae.
2. RESULTS
2.1. Phylogenetic analysis and affinity purification of five subunits, MoBre2, MoSwd1, MoSdc1, MoSet1, and MoJmjd2, from the COMPASS‐like complex of M. oryzae
To explore whether homologues of the core COMPASS‐like complex components are present in M. oryzae, we performed a phylogenetic analysis of COMPASS‐like homologues from several animals and plants. The results suggested that the subunits of the COMPASS‐like complex are highly conserved in M. oryzae. We searched for yeast homologues using BLAST in the M. oryzae protein database with the amino acid sequences of proteins such as Cps60/Bre2, Cps50/Swd1, Cps40/Spp1, Cps35/Swd2, Cps25/Sdc, Cps30/WDR5, and Cps30/Swd3. We identified six homologues for these proteins, namely MoSet1 (MGG_15053), MoBre2 (MGG_01427), MoSwd1 (MGG_04651), MoSpp1 (MGG_03005), MoSwd2 (MGG_06406), and MoSdc1 (MGG_00350) (Figure 1 and Table 1). Orthologues of all the components in the COMPASS‐like complex are single copies in M. oryzae, and their orthologues from representative species, including S. cerevisiae, M. oryzae, F. graminearum, Arabidopsis, Drosophila, and mammalian species (Figures 1a and S1a,b, and Tables 1 and S1).
FIGURE 1.
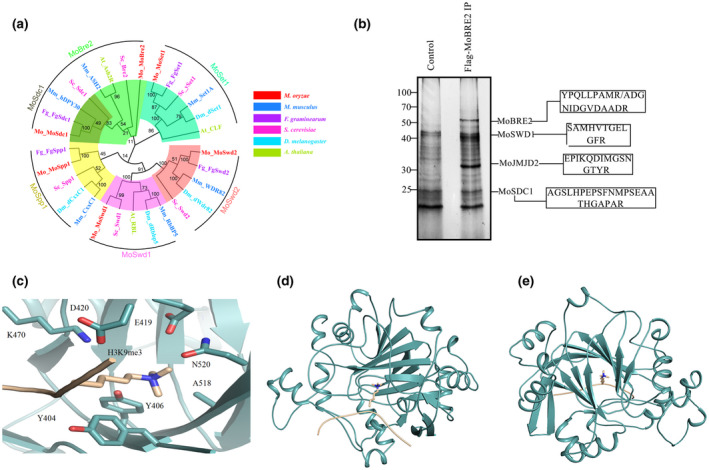
(a) Neighbour‐joining tree of candidate COMPASS or COMPASS‐like orthologues from Magnaporthe oryzae, Saccharomyces cerevisiae, Fusarium graminearum, Arabidopsis thaliana, Drosophila melanogaster, and Mus musculus. (b) Coimmunoprecipitation and mass spectrometry analysis of the COMPASS‐like complex from M. oryzae. The boxes contain the peptide sequences identified using tandem mass spectrometry. (c–e) Modelled structure of the core Tudor domain of MoJmjd2 in complex with histone H3K9me3. The front view (c), side view (d), and top view (e) of a modelled MoJmjd2(cyan)–H3K9me3(gold) complex are shown
TABLE 1.
Subunit composition of COMPASS‐like family from yeast to human
| Species | Conserved subunits of COMPASS‐like complex | |||||
|---|---|---|---|---|---|---|
| Saccharomyces cerevisiae | ySet1 | Cps60(Bre2) | cps40(Spp1) | cps35(Swd2) | cps25(Sdc1) | cps50(Swd1) |
| Drosophila | SetD1A/Trithorax (trx)/Trithorax‐related (Trr) | Ash2 | dCxxC1 | dWdr82 | dDpy30 | dRbbp5 |
| Mammalian | Set1A/Set1B/MLL1/MLL2/MLL3/MLL4 | ASH2 | CxxC1 | Wdr82 | hDPY30 | RbBP5 |
| Fusarium graminearum | FgSet1 (FGSG_07445) | FgCcl1 (FGSG_09564) | FgSpp1 (FGSG_09543) | FgSwd2 (FGSG_05413) | FgSdc1 (FGSG_07047) | – |
| Aspergillus | – | CclA | SppA | – | – | – |
| Magnaporthe oryzae | MoSet1 (MGG_015053) | MoBRE2 (MGG_01427) | MoSPP1 (MGG_03005) | MoSWD2 (MGG_06406) | MoSDC1 (MGG_00350) | MoSWD1 (MGG_04651) |
To purify the Set1/COMPASS‐like complex in M. oryzae, we first generated transformation strains stably expressing FLAG‐MoBre2, and performed an affinity purification using the total nuclear extracts from the FLAG‐MoBre2 strains (Figures 1b and S2). Immunoprecipitated complexes were subjected to silver staining and protein identification using multidimensional protein identification technology (MudPIT). Tandem mass spectrometry analysis was used to identify the five proteins, MoBre2, MoSwd1, MoSdc1, MoSet1, and a novel JMJC domain‐containing protein (MGG_09186) (Table S2). A sequence alignment of MGG_09186 revealed that it contains Jumonji (JmjC) catalytic domains, which can demethylate histone trimethylated lysine, suggesting that this protein may act as a histone demethylase. We named MGG_09186 as MoJmjd2.
The MoJmjd2 has a conserved Tudor domain, which is highly conserved with the Tudor domain of human JMJD2A. We identified the Tudor domain in Mojmjd2 (residues 90–590) that possesses an extra uncharacterized motif (residues 194–341), but the residues that recognize trimethylated lysine in the core Tudor domain are highly conserved (Figures 1c and S3) (Chen et al., 2007; Ozboyaci et al., 2017; Ulucan et al., 2017). Hence, the core Tudor domain of Mojmjd2 without the aforementioned extra motif that is complexed with H3K9me3 was modelled using the crystal structures of the JMJD2A Tudor domain and the H3K9me3 complex (PDB code 2006; Ng et al., 2007) using docking methods. The modelled structure showed that the trimethylated H3K9me3 inserts into a pocket composed of residues Y404, Y406, E419, D420, K470, A518, and N520 (Figure 1c). The predicted binding model indicates that trimethylated H3K9me3 interacts with residues Y404 and Y406 by cation–π interactions and with E419, D420, and N520 through hydrogen bonds (Figure 1c).
In addition, although we did not detect an obvious MoSet1 band in the sodium dodecyl sulphate polyacrylamide electrophoresis (SDS‐PAGE) gels, we did find that our MoBre2‐FLAG preparations contained significant amounts of MoSet1 (Table S2). However, the MudPIT did not identify peptides unique to other homologous subunits, such as MoSpp1, MoSwd2, and MoSdc1. The results clearly indicate that MoBre2, MoSwd1, MoSdc1, MoSet1, and MoJmjd2 have stable associations with COMPASS‐like complexes of M. oryzae in vivo.
2.2. MoBre2 physically interacts with MoSdc1 via its sequence repeat in dual‐specificity kinase splA and ryanodine receptors (SPRY) domain401–613 in vitro and is subcellularly colocalized in vivo
In M. oryzae, MoBre2 shares 17.36% and 12.70% identity with yeast Bre2 and human ASH2L, respectively (Figure S1c–e). In addition, MoSdc1 and yeast Cps25 have a conserved DPY30 domain of approximately 37 amino acids with 68.57% shared identity (Figure S1d). The MoBre2 protein contains two SPRY domains from the N‐ to the C‐terminus (Figure 2a). Recently, the ASH2L–DPY30 interaction was reported to be important for the regulation of H3K4 methylation (Chen et al., 2012; Li et al., 2016; Tremblay et al., 2014).
FIGURE 2.
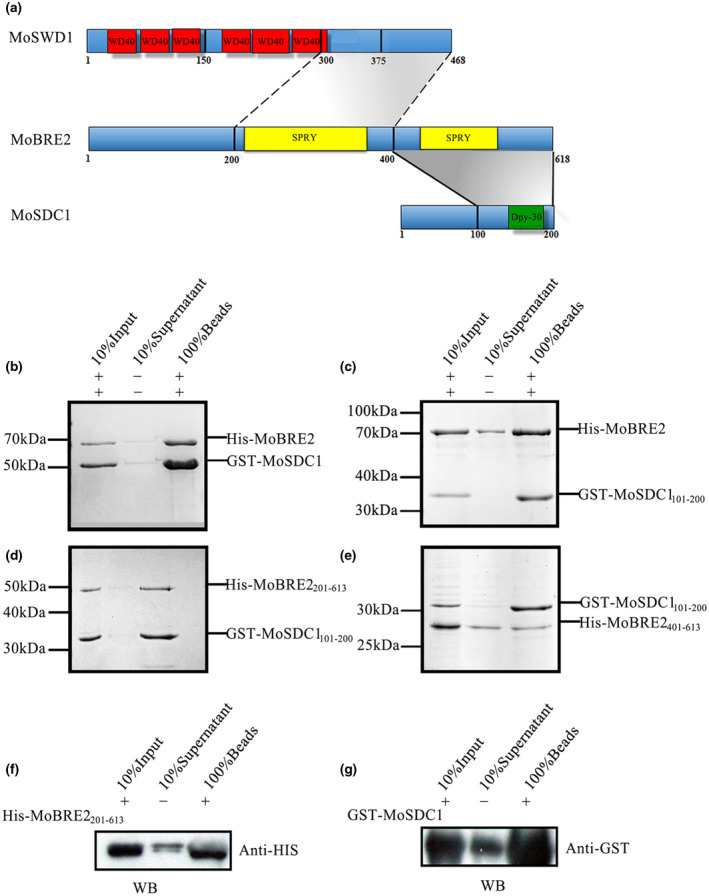
MoBre2 interacts directly with MoSdc1 in vitro. (a) Domain organization of MoBre2, MoSdc1, and MoSwd1. In MoSwd1, the WD40‐repeat domain is shown in red; in MoBre2, the SPRY domain (SPLA and RY anodine receptor domains) is shown in yellow; in MoSdc1, the DPY30 domain is shown in green. Numbers indicate residue numbers at the boundaries of various subdivisions. Protein interactions are indicated with grey areas. (b) Glutathione‐S‐transferase (GST) pull‐down assay with MoBre2 and MoSdc1. GST‐tagged MoSdc1 was incubated with His‐tagged MoBre2. The eluates were subjected to SDS‐polyacrylamide gel electrophoresis. (c, d) Full‐length MoBre2 interacts with the C‐terminal region of MoSdc1 (residues 101–200) but not with the N‐terminal region of MoSdc1 (residues 1–100). (e) The fragments of MoSdc1 (residues 101–200) interact with the two deletion mutants (MoBre2201–613, MoBre2401–613). (f, g) Western blot analysis shows that His‐tagged MoBre22 01–613 interacts with GST‐tagged full‐length MoSdc1
To investigate whether MoBre2 interacts with MoSdc1 or MoSwd1, we expressed and purified recombinant proteins corresponding to the full length, N‐terminal, and C‐terminal regions of MoBre2, MoSdc1, and MoSwd1. Our immunoprecipitation experiments showed that MoBre2 can directly interact with MoSdc1 (Figure 2a–g and Table S3), but not with MoSwd1 (Figure S4a,b). To understand which residues are required for interactions between MoBre2 and MoSdc1, we constructed three deletion mutants of MoBre2 (amino acids 1–200, termed MoBre21–200; amino acids 201–613, termed MoBre2201–613; amino acids 401–613, termed MoBre2401–613) and two deletion mutants of MoSdc1 (amino acids 1–100, termed MoSdc11–100; amino acids 101–200, termed MoSdc1101–200) (Figure 2c–e). As shown in Figure 2, a pull‐down assay showed that MoBre2 retained the binding activity with MoSdc1101–200 (Figure 2c), but not MoSdc11‐100 (Figure S4a).
In addition, MoSdc1101–200 physically interacted with MoBre2201–613 and MoBre2401–613 (Figure 2d,e), whereas this construct failed to bind to MoBre21–200 (Figure S4b), which suggests that the SPRY domain located between residues 401 and 613 of MoBre2 is required for recognition with MoSdc1. Furthermore, a western blot analysis showed that MoSdc1 interacted with MoBre2401–613 (Figure 2f,g). Taken together, these results clearly indicate that a 212‐residue SPRY‐C‐terminal domain of MoBre2 (residues 401–613) is required for interactions with MoSdc1 and that the conserved DPY30‐C‐terminal regions of MoSdc1 are required for the binding activity with MoBre2 (Figure 2d,e). However, direct interactions between MoSwd11–375, MoSwd1150–375, MoSwd1300–468, and MoBre2 were not detected in pulldowns of these protein segments (Figure S4d–f), suggesting that MoBre2 cannot interact with MoSwd1 directly in vitro.
To further explore whether MoBre2 can interact with MoSdc1 in vivo, the entire coding regions of MoBre2 and MoSdc1 were fused with green fluorescent protein (GFP) and red fluorescent protein (RFP), respectively. These two constructs were cotransformed into the wild‐type strain P131. As shown in Figure 3, the GFP‐MoBre2 was colocalized with the RFP‐MoSdc1, clearly showing that MoBre2 can interact with MoSdc1 in vivo (Figure 3). Moreover, these two proteins were localized to the nucleus in the mycelium (Figure 3a–d), conidia (Figure 3e–h), appressoria (Figure 3i–l), and infectious hyphae (Figure 3m–p). Taken together, these results indicate that MoBre2 can directly interact with MoSdc1 in vitro and in vivo.
FIGURE 3.
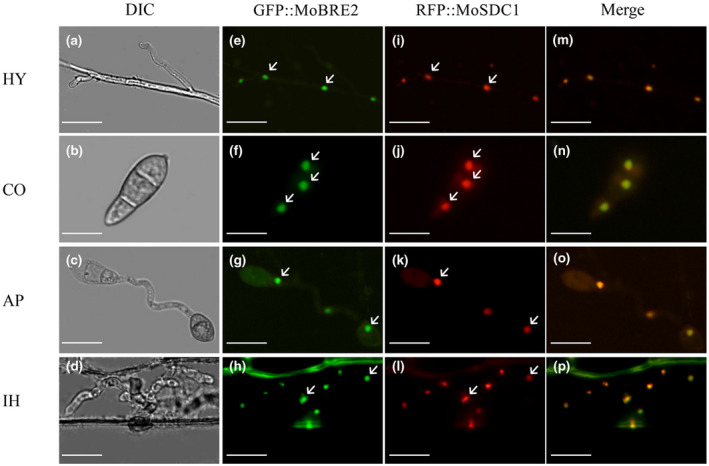
Dual‐colour imaging of GFP‐MoBre2 and RFP‐MoSdc1 in Magnaporthe oryzae by confocal laser scanning microscopy. MoSdc1 was cloned into pKS, and the RFP‐MoSdc1 fusion construct was generated. The GFP‐MoBre2 and RFP‐MoSdc1 fusion constructs were digested with NotI and transformed into the protoplasts of wild‐type P131. The GFP‐MoBre2 and RFP‐MoSdc1 signals were merged. HY, vegetative hyphae; CO, conidia; AP, appressoria; IH, infection hyphae. DIC, differential interference contrast. Scale bars = 20 μm. Arrows indicate the nuclei localized in cells
2.3. MoBre2, MoSwd2, or MoSpp1 are critical for fungal development and invasive hyphae formation during pathogenesis
Next, we used a gene replacement approach to generate null mutants of M. oryzae COMPASS‐like complex genes. We successfully obtained three null mutant strains for the MoBre2, MoSpp1, and MoSwd2 genes (Figure S5). These deletion mutants were confirmed by Southern blots (Figure S5b,e,i) and PCR analysis (Figure S5a,b). We found that these three gene deletion mutants had defects in colony growth on oatmeal tomato agar (OTA) plates (Figure 4a). Although the Δmobre2 deletion mutant displayed a reduction in colony growth, severe defects were apparent during conidium formation (Figure 4a,c,d). The Δmobre2 mutant produced approximately 30% of the conidia and Δmoswd2 produced approximately 10% of the conidia compared with the wild‐type strain (Figure 4c). In contrast, the Δmospp1 null mutants showed a small reduction in colony growth without conidium formation defects (Figure 4c and Table 2).
FIGURE 4.
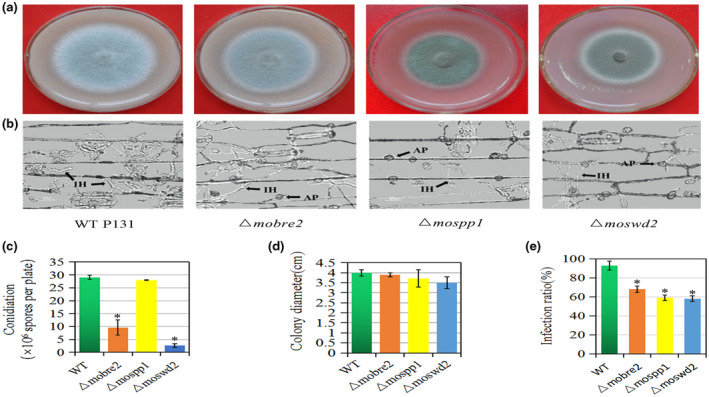
Deletion of MoBre2, MoSpp1, and MoSwd2 results in defects in hyphal growth, and pathogenic infection. (a) Five‐day‐old cultures of the wild type (WT) P131, Δmobre2, Δmospp1, and Δmoswd2 strains grown on oatmeal tomato agar (OTA). (b) Barley leaves sprayed with conidial suspensions (105 spores/ml) of P131 and the Δmobre2, Δmospp1, and Δmoswd2 deletion mutants 36 hr after inoculation. (c–e) Microscopic observation of conidium formation in the wild‐type strain P131 and the Δmobre2, Δmospp1, and Δmoswd2 deletion mutants. IH, infection hypha; AP, appresorium. Means and SDs were calculated from three independent experiments. *p < .01, n > 100
TABLE 2.
List of the downstream target genes involved in conidiation, mycelium development, or pathogenesis in fungal development and infection in the Δmobre2, Δmospp1, and Δmoswd2 deletion mutants
| Gene locus | Δmobre2 | Δmospp1 | Δmoswd2 | Protein | Function |
|---|---|---|---|---|---|
| MGG_06888 (MoGln1) | + | − | − | Glutamine synthetase‐encoding orthologue | Appressorium formation; mycelium development |
| MGG_04855 (MoAbc7) | + | + | − | ABC transporters | Conidiation |
| MGG_09639 (MoAgs1) | + | − | + | α−1,3‐glucan synthase gene | Infection |
| MGG_01176 (XDH1) | + | + | + | Xylitol dehydrogenase | Mycelium growth |
| MGG_09299 (MoPex1) | + | + | − | Putative homologue to peroxin 1 | Infection |
| MGG_11764 (FZC55) | + | − | − | Zn2Cys6 transcription factors | Appressorium formation |
| MGG_01127 (CONx7) | + | − | + | Cys2‐His2 (C2H2) zinc finger protein | Conidial differentiation |
| MGG_05871 (PTH11) | + | + | + | G‐protein‐coupled receptor | Appressoria‐like structures; infection |
| MGG_14719 (MoVelC) | + | + | + | Velvet | Pathogenesis; spore germination |
| MGG_03593 | + | + | + | Unknown | Hypothetical protein |
| MGG_09875 | + | + | + | Unknown | Hypothetical protein |
| MGG_00450 | + | − | + | Unknown | Pathogenesis |
| MGG_11261 | + | + | − | Unknown | Pathogenesis |
| MGG_00501 (TDG2) | + | − | − | Unknown | Pathogenesis |
| MGG_03451 (GCF6) | + | + | + | Unknown | Pathogenesis; sporulation |
| MGG_07833; MGG_10570; MGG_07975 | + | − | + | Unknown | Mycelium development |
| MGG_09640; MGG_08710; MGG_04206 | + | − | − | Unknown | Mycelium development |
| MGG_12574 | + | + | − | Unknown | Mycelium development |
| MGG_04676 | − | + | − | Unknown | Spore germination; pathogenesis |
| MGG_04495; MGG_07259 | − | + | − | Unknown | Spore germination |
| MGG_11236; MGG_04527 | − | + | − | Unknown | Fungal‐type vacuole |
| MGG_12462 (MoLPP5) | − | + | − | Unknown | Fungal‐type vacuole |
| MGG_07478 | + | + | + | Unknown | Fungal‐type vacuole |
| MGG_00993 | + | + | − | Unknown | Fungal‐type vacuole |
| MGG_00450 | + | + | Unknown | Sexual reproduction; sporulation | |
| MGG_07535; MGG_01818 (magA); MGG_02240 | − | − | + | Unknown | Sexual reproduction |
| MGG_07535; MGG_09499 | − | − | + | Unknown | Sporulation |
| MGG_00450 | + | − | + | Unknown | Sporulation |
| MGG_07930 | + | − | + | Unknown | Developmental process |
| MGG_02961 | + | + | + | Unknown | Hypothetical protein |
| MGG_02775 | + | + | + | Unknown | Hypothetical protein |
| MGG_00184 | + | + | + | Unknown | Regulation of biosynthetic process |
| MGG_06148 | + | + | + | Unknown | Hypothetical protein |
| MGG_12574; MGG_04855; MGG_13691 | + | + | − | Unknown | Mycelium development |
| MGG_09548; MGG_04689; MGG_00990; MGG_05631; MGG_03039 | − | + | + | Unknown | Mycelium development |
| MGG_02835; MGG_03929; MGG_04966; MGG_01782; MGG_10681; MGG_00341; MGG_12944 | − | + | − | Unknown | Mycelium development |
To evaluate the virulence of Δmobre2, Δmoswd2, and Δmospp1 null mutants, conidial suspensions of these three null mutants and from wild‐type P131 were inoculated onto the surface of barley. These three mutants showed severe defects in their invasive hyphae formation at 36 hr after inoculation (Figure 4b). At 36 hr after inoculation, wild‐type P131 formed bulbous infection hyphae and could extend to neighbouring host cells on the barley epidermis according to our microscopic observation (Figure 4b,e). The infection ratio was 68%, 59%, and 58.1% in the Δmobre2, Δmospp1, and Δmoswd2 deletion mutants, respectively, compared with wild‐type strains (Figure 4e). However, at 5 days after conidial suspension inoculation onto the seedlings of rice cultivar LTH, the typical robust lesions of rice blast were observed on all three null mutants and wild‐type P131 (Figure S6a,b), which suggests that MoBre2, MoSwd2, and MoSpp1 are required for fungal–host interaction in the early stages of pathogenesis. These results suggest that MoBre2, MoSwd2, and MoSpp1 are required for infection during invasive hyphae formation of the pathogen during fungal development in the early stages of pathogenesis.
2.4. MoBre2, MoSwd2, and MoSpp1 fine‐regulate H3K4me3 distribution near TSS overlapped target regions
Considering that the COMPASS complex is highly conserved in its H3K4me3 methylation function and activates its target genes expression, we first performed western blot analysis using H3K4 mono‐ (me1), di‐ (me2), and trimethylation‐specific antibodies in Δmobre2, Δmospp1, and Δmoswd2 gene deletion mutants (Figure 5). In these three null mutants, there was a robust decrease in H3K4 trimethylation levels throughout the entire genome, whereas those of H3K4 di‐ and monomethylation were unaltered (Figure 5a). To determine H3K4me3’s chromatin distribution pattern at the genome‐wide level, we performed chromatin IP‐sequencing (ChIP‐seq) experiments using antibodies against H3K4me3 in the strains that were devoid of MoBre2, MoSpp1, and MoSwd2 genes.
FIGURE 5.
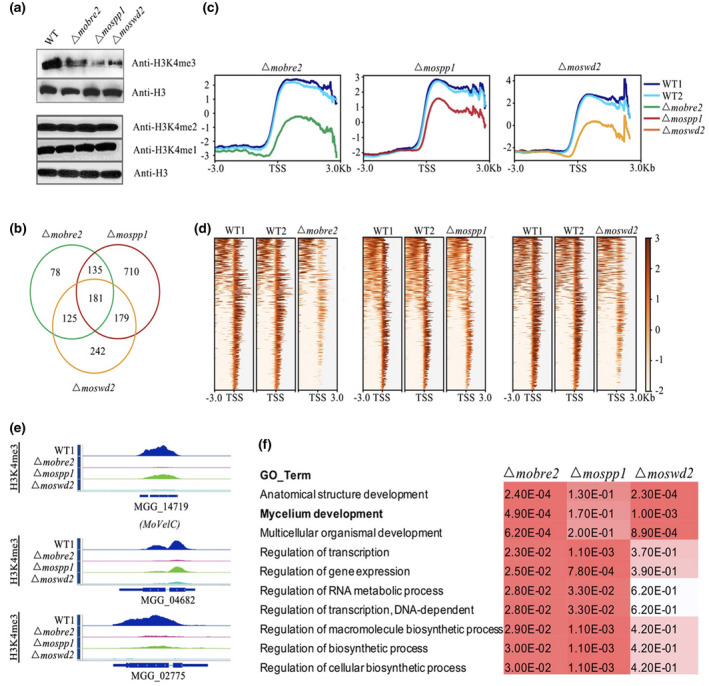
The MoBre2, MoSwd2, and MoSpp1 deletion mutants display significantly decreased H3K4me3 profiles in transcription start site (TSS) overlapped target regions. (a) Western blot analysis of histone H3K4me3, H3K4me2, and H3K4me1 modifications in the wild‐type (WT) P131 and Δmobre2, Δmospp1, and Δmoswd2 strains. (b) The number in the Venn diagram shows the H3K4me3 decreased peaks in the deletion mutants compared with the WT. (c) The H3K4me3‐ChIP‐seq distribution of enriched peaks around the TSSs of the overlapped genes in MoBre2, MoSwd2, and MoSpp1 deletion mutants. (d) The heatmaps show the distribution of H3K4me3‐ChIP‐seq peak signal density in different samples. (e) The read coverage shows that the peaks in these three deletion mutants were significantly decreased compared with the WT strain in some sample genes. (f) Gene ontology (GO) term comparisons of the down‐regulated genes of deletion mutants, especially in spore germination and pathogenesis in the Δmobre2, Δmospp1, and Δmoswd2 mutants
In total, we identified 519, 1,205, and 727 H3K4me3‐decreased peaks in the Δmobre2, Δmospp1, and Δmoswd2 deletion mutants, respectively, compared with wild‐type strains (Figure 5b and Table S4). Remarkably, close to 60% (316) of the H3K4me3 decreased peaks in the Δmobre2 deletion mutant overlapped with the Δmospp1 deletion mutant and close to 59% (306) of the H3K4me3 decreased peaks in the Δmobre2 deletion mutant overlapped with the Δmoswd2 deletion mutant (Figure 5b). Such striking similarity at the genome‐wide level strongly suggests a functional connection among MoBre2, MoSpp1, and MoSwd2.
Significantly, when we sorted all the genes according to their H3K4me3‐ChIP‐seq reads at the TSS regions (−3,000 bp to +3,000 bp) in wild‐type strains and then analysed their H3K4me3‐ChIP‐seq reads in the Δmobre2, Δmospp1, and Δmoswd2 deletion mutants (Figure 5c,d) we found that the Δmobre2, Δmospp1, and Δmoswd2 deletion mutants displayed a highly similar profile in the TSS regions (Figure 5c,d). The signals of enriched H3K4me3‐ChIP‐seq reads in wild types were largely decreased when compared with deletion mutants in the TSS region (Figure 5c). In the Δmobre2 mutant, the H3K4me3‐ChIP‐seq reads were reduced 3.5‐fold compared to the wild‐type strain (Figure 6b, left), while in the Δmospp1 and Δmoswd2 mutants, the signal displayed 2.6‐ and 2.2‐fold changes, respectively (Figure 5c).
FIGURE 6.
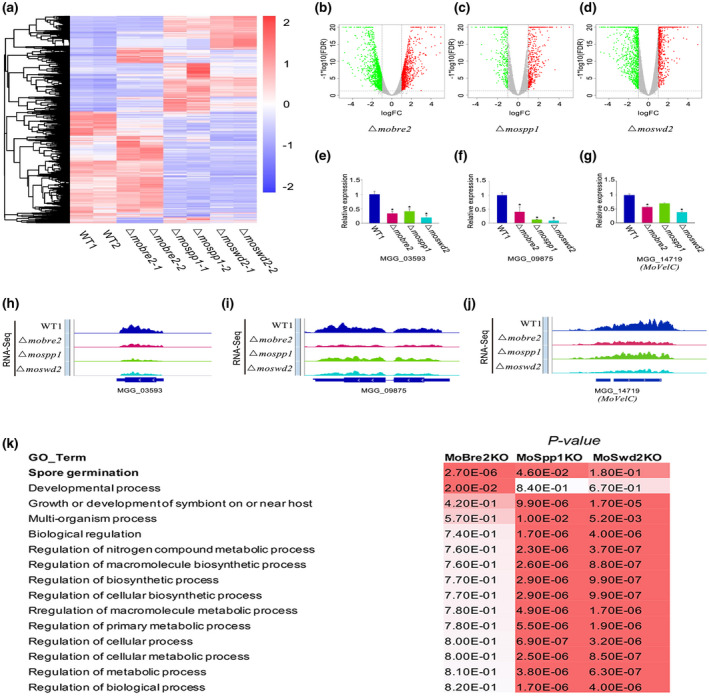
The MoBre2, MoSwd2, or MoSpp1 gene deletion mutants exhibit gene expression decrease. (a) The heat map shows all the differentially expressed genes in wild types (WT) compared with three deletion mutants. (b–d) The differentially expressed genes are shown in volcano plots, the x axis shows the fold‐changes transformed by log2, and the y axis shows the false discovery ratio (FDR) transformed by − log10. (e–g) The read coverage shows that the expression signals of selected targets in spore germination in these three deletion mutants were significantly decreased compared with the wild‐type strain in some sample genes. (h–j) Quantitative reverse transcription PCR for spore germination genes shows the expression in WT decreased compared with knockouts (right panel). (k) Gene ontology (GO) term analysis of the candidate genes involved in mycelium development with H3K4me3‐ChIP‐seq peaks decreased in Δmobre2, Δmospp1, and Δmoswd2 mutants. The value shown on the table represents the frequency of the GO term in the GO annotation database. The colour represents the enrichment p value (Fisher's exact test) of each GO term. The scaled relative colour is shown in the heatmap
Gene ontology term comparisons showed that these 2,451 genes (519, 1,205, and 727 H3K4me3‐decreased peaks) are especially involved in mycelium development, multicellular organismal development, the regulation of gene expression, the RNA metabolic process, and other regulation of cellular biosynthetic process (Tables S4 and S5, and Figure 5f). More than half of the transcriptional changes (1,367/2,451) were hypothetical genes (Table S4). We observed that MoBre2, MoSwd2, and MoSpp1 were required for conidiation or infection defects during the pathogenesis stage (Figure 4), and thus we reasoned that MoBre2, MoSwd2, and MoSpp1 might specifically target fungal development genes. We then performed the comparisons for enriched biological processes in the deletion mutants of Δmobre2, Δmospp1, and Δmoswd2 versus the wild‐type strain that showed that their target genes are especially involved in mycelium development (Tables S4 and S5, and Figure 5f). In these target genes, we identified 69/519, 128/1205, and 103/727 genes involved in mycelium development and pathogenesis in the MoBre2, MoSpp1, and MoSwd2 gene deletion mutants, respectively (Table S5). Among them, there were 64 candidate genes that are more likely to be involved in mycelium development in these three deletion mutants (Figure 5f). Moreover, we selected seven candidate genes from the 181 overlapped genes, MoGln1 (MGG_06888), MoAbc7 (MGG_04855), MoAgs1(MGG_09639), XDH1(MGG_01176), MoPex1(MGG_09299), FZC55(MGG_11764), and CONx7(MGG_01127). All seven genes are involved in spore germination, fungal growth, or pathogenesis (Figure 5b and Table S5). MoABC7 has been previously revealed to be involved in virulence, conidiation, and abiotic stress tolerance, and MoPEX1 has been shown to be involved in infection‐related morphogenesis and pathogenicity; these genes were previously demonstrated to be involved in mycelium development (Deng et al., 2016; Kim et al., 2013; Kou et al., 2016). We also selected three overlapped target genes, MGG_04682, MGG_14719, and MGG_02775, and confirmed that the H3K4me3 coverages are significantly decreased in Δmobre2, Δmospp1, and Δmoswd2 deletion mutants (Figure 5e). Such a striking similarity of phenotypes among the Δmobre2, Δmospp1, and Δmoswd2 knockouts at the genome level strongly suggests a functional connection among MoBre2, MoSpp1, and MoSwd2.
2.5. Decreased target gene expression caused by deletion of MoBre2, MoSwd2, or MoSpp1 highly correlates with decreased H3K4me3
We further investigated whether the deletion of MoBre2, MoSwd2, or MoSpp1 would cause changes in gene expression. We then performed RNA‐seq in both wild‐type strains and each deletion mutant. Notably, the depletion of MoBre2 showed relatively moderate transcriptional changes, while the depletion of MoSpp1 or MoSwd2 caused more obvious and consistent alterations with the thresholds above 2‐fold changes (deletion mutants vs. wild‐types) and a false discovery ratio less than 0.1, including about 42.47% and 55.83% consistently up‐ and down‐regulated genes in Δmospp1 or Δmoswd2 vs. wild‐types (Figure 6a–d and Table S6). We detailed gene expression patterns of three genes, MGG_03593, MoVelC (MGG_14719), and MGG_09875. The read coverage of these genes indicated a significant drop in expression compared with the wild‐type strain (Figure 6e–g). Furthermore, quantitative reverse transcription PCR (RT‐qPCR) experiments for these genes selected indicate that expression in the wild type was increased compared with knockouts (Figure 6h–j).
To investigate which biological processes are affected by the MoBre2, MoSpp1, and MoSwd2 genes, we further performed a gene ontology analysis for the down‐regulated genes in each knockout strain (Figure 6k and Table S6). Some selected candidate genes such as PTH11 (MGG_05871), MoVelC (MGG_14719), and MGG_03593 are all involved in spore germination and pathogenesis (Tables [Link], [Link], [Link]; Deng et al., 2016; Kim et al., 2013; Kou et al., 2016). These genes affected in Δmobre2, Δmospp1, and Δmoswd2 were more likely to be involved in developmental processes such as spore germination, transcriptional regulation (transcription factor activities), multiorganism processes, and pathogenesis (Figure 6k and Tables [Link], [Link], [Link]). In these target genes, more than half of target genes were hypothetical genes (Table S6). More importantly, the genes showing decreased expression in the MoSpp1 or MoSwd2 deletion mutants are also involved in growth or development of symbionts on or near host multiorganism processes or other aspects of pathogenesis (Figure 6). The functions of these selected genes are clearly consistent with the phenotypes we have observed in Δmobre2, Δmospp1, or Δmoswd2 deletion mutants, which have defects in colony growth, infection, and other related aspects of early pathogenesis (Figure 4b).
To further observe the relationship between gene expression changes and H3K4me3 changes, we compared the down‐regulated genes in Δmobre2, Δmospp1, and Δmoswd2 deletion mutants. For each comparison between the deletion mutant and the wild type (Figure 7a), we observed that the down‐regulated genes were likely to have decreased the H3K4me3 peaks, which suggests that the down‐regulated genes that are targets of MoBre2, MoSpp1, and MoSwd2 overlap. In total, we identified 38, 141, and 106 decreased peaks both in ChIP‐seq and RNA‐seq experiments in the Δmobre2, Δmospp1, and Δmoswd2 deletion mutants, respectively, compared with wild‐type strains (Figures 7a and S6). In Δmobre2 deletion mutants, there were 8 (3.5%) and 12 (5.3%) genes in common with the Δmospp1 and Δmoswd2 deletion mutants, respectively (Figure 7a). Notably, five genes that had decreased expression, MGG_02961, MGG_02775, MGG_00184, MoVelC (MGG_14719), and MGG_06148, followed the same trend of expression in each of the Δmobre2, Δmospp1, and Δmoswd2 knockout lines (Figure 7a,b and Tables [Link], [Link], [Link]). We identified expression levels through RNA‐seq for those genes with decreased H3K4me3 peaks and found that these genes were also consistently down‐regulated in the knockout strains (Figure 7b). We further confirmed that a gene we obtained (Figure 7a) with reduced gene expression in RNA‐seq had a similar H3K4me3 reduction pattern in ChIP‐seq (Figure 7a,c). For MGG_02775, MoVelC (MGG_14719), and MGG_04682, the read coverage of these genes indicated a significant drop in both gene expression and H3K4me3 occupancy (Figure 7c–h). Significantly, the majority of H3K4me3‐decreased peak target genes had significant expression changes, which correlates with the well‐known effects of histone modification on gene expression.
FIGURE 7.
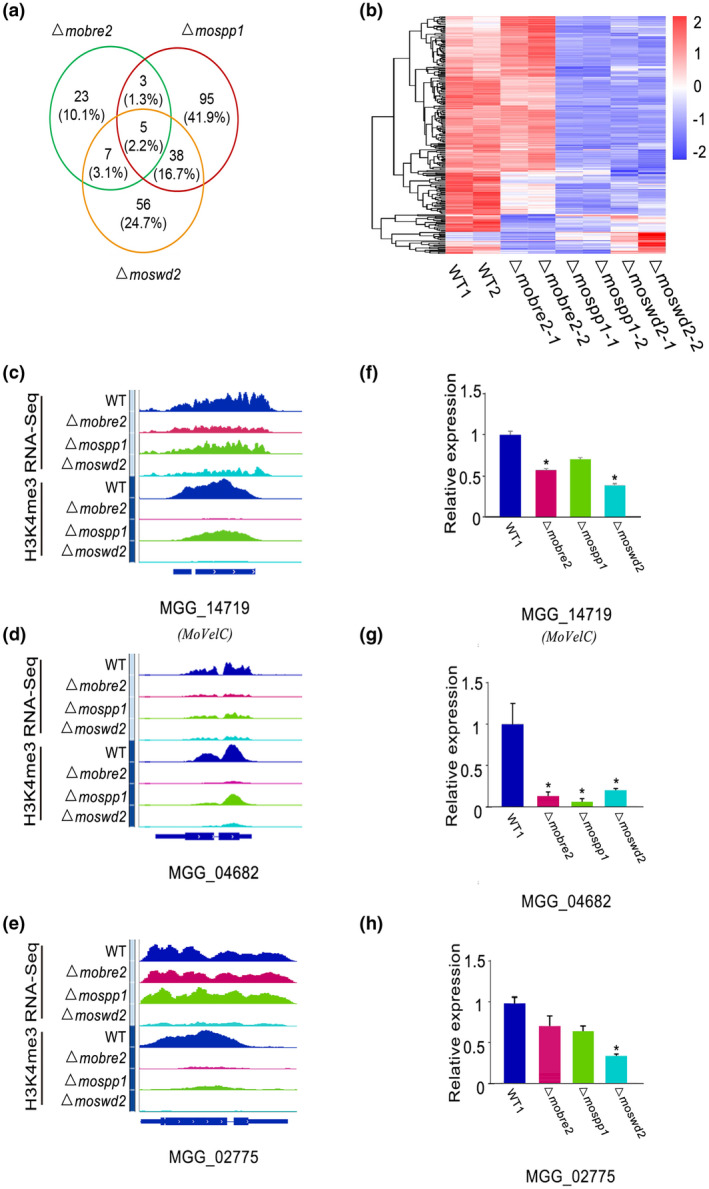
Decreased gene expression in the MoBre2, MoSwd2, or MoSpp1 gene deletion mutants is highly correlated with decreased H3K4me3. (a) Volcano plots show changes in down‐regulated gene expression in knockouts compared to the wild type (WT) strains. The numbers of down‐regulated genes are indicated. (b) Heat map of differentially expressed genes between knockouts and the WT strains. (c–e) Example genomic tracks showing genes that exhibit decreased H3K4me3‐ChIP‐seq signal levels and down‐regulated RNA‐seq target genes. (f–h) Quantitative reverse transcription PCR for selected genes shows the expression in WT decreased compared with deletion mutants. Identified genes show decreased signals and the expression level is reduced in deletion mutants
Together, these results suggest that MoBre2, MoSpp1, and MoSwd2 may assemble into a functional COMPASS complex and mediate H3K4me3‐regulated gene expression.
3. DISCUSSION
3.1. Highly conserved roles of COMPASS‐like H3K4 methyltransferase complexes in M. oryzae
This study is the first to uncover the molecular mechanism of the COMPASS‐like complex in a plant‐pathogenic fungus, providing new insight into the epigenetic regulatory mechanism of early stage target gene expression during pathogenesis.
In F. graminearum, FgSet1 physically interacts with multiple proteins, including FgBre2, FgSpp1, and FgSwd2. FgBre2 further interacts with FgSdc1 (Liu et al., 2015). Our biochemical studies have demonstrated that the M. oryzae complexes are very similar to their mammalian counterparts in terms of subunit composition. We found that all the COMPASS family members are also highly conserved in M. oryzae. There are several common subunits, namely MoBre2, MoSwd1, MoSdc1, MoSet1, MoSpp1, and MoSwd2, that are homologues of Cps60, Cps50, Cps25, ySet1, Cps40, and Cps35, respectively (Figure 1 and Table 1). A study showed that MoSet1, a component of COMPASS, is required for global gene expression during infection‐related morphogenesis, which is consistent with the findings presented here (Pham et al., 2015). However, we were unable to identify the homologues of yeast Cps30/Wdr5 in M. oryzae. Moreover, we did not purify any Cps30‐like protein from the immunoprecipitation assay. Further study is required to determine whether there are some Cps30‐like proteins in the M. oryzae genome.
Previously published crystal structures of COMPASS or COMPASS‐like in yeast and humans revealed that a Y‐shaped architecture with Cps50 and Cps30 localizes on the top of two adjacent lobes and Cps60‐Cps25 forming the base at the bottom (Takahashi et al., 2011; Zhang et al., 2014). In M. oryzae, our results also showed that MoBre2/Cps60 can interact directly with MoSdc1 in vitro, but not with MoSwd2. Although we did not find that MoSwd1 can interact with MoBre2 in a pull‐down assay, we did find that FLAG‐MoBre2 can be copurified with MoSwd1 in an immunoprecipitation assay in vivo (Table S4), which suggests that MoSwd1 does indeed function in the COMPASS complex of M. oryzae.
Interestingly, we also found that our FLAG‐MoBre2 preparations contained significant MoJmjd2 enrichment (Figure 1 and Table S2). In mammalian cells, some previous studies showed that UTX, a histone H3K27me3 demethylase, can copurify with the MLL3/MLL4 complexes (Shilatifard, 2012). The Trr/MLL3/MLL4‐containing COMPASS complexes function as the histone H3K4 methylase of Trx, while UTX is thought to be involved in counteracting PcG‐mediated gene silencing (Agger et al., 2007; Mohan et al., 2011). Here, MoJMJD2 may serve as an H3K9me3 histone demethylase in the COMPASS‐like complexes and their associated bivalent H3K9/27 demethylase and H3K4me3 methyltransferase activities may be involved in dynamic target gene activation in M. oryzae. This finding suggests that these demethylase proteins may first demethylate the repressive histone markers, such as H3K9me3 or H3K27me3, and then recruit the histone methylase enzymes for gene activation. However, identifying which regions and mechanisms underlie how MoJMJD2 interacts with other components of COMPASS‐like complex is under consideration for future research.
3.2. Contribution by COMPASS‐like complex components provides novel insights into the regulation of target genes in TSS regions during fungal development and infection‐related pathogenesis in fungi
The COMPASS complex and its product, H3K4me3, are basic epigenetic regulators and are involved in a variety of biological processes in many plants and animals. Previously, Δmoset1 mutants exhibited the most severe reduction in vegetative growth, conidiation, and pathogenicity. MoSet1 was reported to affect the expression of genes related to infection‐related morphogenesis, especially the genes involved in the cAMP‐signalling pathway leading to appressorium formation (Pham et al., 2015).
In addition, Pham et al. also found that MoKMT1, MoKMT3, MoKMT6, and MoKMT2H played significant roles in infection‐related morphogenesis and/or pathogenicity to varying degrees (Pham et al., 2015). The Δmokmt3 and Δmokmt2h mutants showed moderate defects in all phenotypic traits. The Δmokmt6 mutants also showed severe reduction in conidiation and slight defects in appressorium formation.
In this study, we found that MoBre2, MoSpp1, and MoSwd2 were essential for colony growth, conidiation, and invasive hyphae formation in M. oryzae (Figure 6a–e). These three gene deletion mutants exhibited pleiotropic defects as described for the Δmoset1 mutants (Li et al., 2016; Pham et al., 2015). According to the ChIP‐seq and RNA‐seq results, several previously characterized target genes, such as MoABC7, MoPEX1, FZC55, MoAGS1, and GLN1, were among the COMPASS‐like complex down‐regulated genes with H3K4me3‐decreased peaks. In Δmoswd2 strains, the expression of some target genes involved in conidiation and pathogenesis was decreased, and this finding is consistent with the mutant defects in invasive hyphae formation (Figure 6b,e and Table S8).
More interestingly, we also found down‐regulated expression of MgHTF1 (MGG_00184), a gene known to regulate conidial production. The Δmghtf1 mutant failed to form conidia and lost pathogenicity (Liu et al., 2010). The expression of MgCONx2 (MGG_02775), a gene that encodes a zinc finger C2H2‐like protein, also decreased. The MgCONx2 mutant was previously deemed essential for conidiogenesis and pathogenesis in M. oryzae (Zhang et al., 2014) (Figure 7 and Table S8). The ChIP‐seq results showed that MgHTF1 and MgCONx2 both had H3K4me3‐decreased peaks in the Δmoswd2 strains, which suggests that MoSwd2 activates H3K4me3 to regulate the transcription of target genes (Figures 6 and 7, and Table S4). The expression levels of MgAnd1 (MGG_02961) and MgFox2 (MGG_06148) were also significantly down‐regulated in the Δmospp1 mutant. MgAnd1 encodes a cell cortex protein involved in nuclear migration and positioning. Deletion of MgAnd1 from M. oryzae leads to a delay in the penetration of the host plants (Rahman & De Sousa, 2014). Mg Fox2 encodes a multifunctional enzyme that catalyses the second and third enzymatic reactions of the β‐oxidation pathway, and its disruption also leads to a significant reduction in virulence (Patkar et al., 2012).
Notably, among the genes affected by the COMPASS‐like complex, the gene MgVelC (MGG_14719) was reported to be a positive regulator of conidiation and pathogenicity in M. oryzae (Kim et al., 2014). The expression of MgVelC was down‐regulated in both Δmobre2 and Δmoswd2 gene deletion strains. Furthermore, the ChIP‐seq results also showed that MgVelC had H3K4me3‐negative peaks in both gene deletion mutants. These results suggest that MgVelC is a potential target gene that is regulated by the COMPASS‐like complex. The transcriptional deficiency of these genes may be the cause of severe defects in terms of colony growth, conidiation, and pathogenicity in different mutants (Dang & Jones, 2001; Wang et al., 2012). Moreover, in agreement with our findings, deletion of COMPASS components cclA and FgSet1 in Aspergillus spp. and Fusarium spp., respectively, resulted in hyphal growth defects, cell wall stress, and virulence reduction. The Δfgset1 strains were restricted to the inoculated spikelets after point inoculation (Liu et al., 2015; Studt et al., 2017). Collectively, these findings suggest that the COMPASS functions as a global regulator to control target genes for fungal pathogenicity and development in these filamentous fungi. However, the details of how these target genes regulate pathogenesis remain unknown.
In conclusion, our biological, biochemical, and proteomic studies identified novel components of the COMPASS‐like complex and elaborated on its contribution to H3K4me3‐mediated target gene expression in M. oryzae. This work serves as a functional resource for studying plant‐pathogenic processes in fungi and provides a greater breadth of knowledge in epigenetics regarding gene regulatory mechanisms.
4. EXPERIMENTAL PROCEDURES
4.1. Strains and culture conditions
The M. oryzae wild‐type strain P131 was used in this study. The wild‐type strain and corresponding transformants were grown on OTA and cultured at 25 °C under light conditions. For extraction of genomic DNA, RNA, and protein, as well as the isolation of protoplasts, fresh mycelia were broken and cultured in liquid complete medium (0.6% yeast extract, 0.3% enzymatic casein hydrolysate, 0.3% acidic casein hydrolysate, and 1% glucose) and shaken at 150 rpm at 25 °C for 36 hr. For measuring colony sizes, mycelial plugs of 5 mm were inoculated into the centre of OTA plates. Colony diameter on each plate was measured for 3 days. Each experiment was repeated three times.
4.2. Conidiation experiments and virulence tests of M. oryzae
The wild‐type, deletion mutant, and complementation strains were cultured on OTA plates at 25 °C for 4 days. Then, 2 ml double‐distilled water was added to each plate and mycelia were collected with an inoculation loop and transferred to a new OTA plate and dried. The plates were covered with three layers of gauze and cultured at 25 °C for 12–16 hr. Two millilitres of double‐distilled water was added to each dish and conidia were scraped with sterile cotton swabs. Conidial suspensions were transferred into a new 50 ml tube and centrifuged for 5 min at a minimum of 5,000 × g at 25 °C. The supernatant was removed and pellets were resuspended to obtain 2 × 104 conidia per millilitre in 0.025% (vol/vol) Tween 20 solution. A haemocytometer was used to count conidia under the optical microscope. For the infection assay, 4‐week‐old seedlings of rice (Oryza sativa 'LTH') and 8‐day‐old seedlings of barley (Hordeum vulgare 'E9') were used for spray infection assays. Plant incubation was performed as described previously (Cao et al., 2016). All inoculation and conidiation experiments were repeated three times independently.
4.3. Western blot analysis
For all tested strains, total proteins were extracted as described previously (Cao et al., 2016) then separated by 13% denaturing SDS‐PAGE, transferred to an Immobilon‐P transfer membrane (Millipore) with a Bio‐Rad electroblotting apparatus, and evaluated using the antibodies anti‐histone H3 (Abcam, ab1791), anti‐H3K4me3 (Abcam, ab8580), anti‐H3K4me2 (Abcam, ab7766), and anti‐H3K4me1 (Abcam, ab8895). Samples were incubated with a secondary antibody (Santa Cruz, sc‐2313) for chemiluminescent detection.
4.4. Co‐IP and silver staining
Immunoprecipitation experiments were performed using a 3 × MoBre2‐FLAG transgenic strain. About 0.5 g of 7‐day‐old mycelia from the FLAG‐MoBre2‐expressing strain was harvested and ground in liquid nitrogen. Subsequently, total proteins were extracted with protein extraction buffer (50 mM Tris.HCl [pH 7.5], 100 mM NaCl, 5 mM EDTA, 1% Triton X‐100, 2 mM PMSF). Then, 15 ml protein extract was incubated in a 50 μl slurry of anti‐FLAG M2 affinity gel (Sigma, A2220) and rotated at 4 °C overnight. The beads were washed with 150 ml BC100 (20 mM Tris.HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, 0.1% NP‐40). Subsequently, target proteins were eluted with the FLAG peptide (DYKDDDDK). The samples were then boiled in the SDS‐PAGE loading buffer and separated in an SDS‐PAGE gel. Silver staining experiments were performed using the ProteoSilver plus silver stain kit (Sigma). The silver‐stained bands were analysed by mass spectrometry.
4.5. Protein expression in Escherichia coli and purification
The recombinant deletion vectors of MoBre2, MoSdc1, and MoSwd2 were transformed into Rosetta (DE3) E. coli bacteria (Biomed). After sequencing to determine positive clones, the bacteria were incubated at 37 °C at 220 rpm to OD600 = 0.6 with Luria‐Bertani (LB) medium, then induced by the addition of 1 mM β‐d‐1‐thiogalactopyranoside (IPTG) and incubated at 16 °C for 14 hr to express glutathione‐S‐transferase (GST)‐tagged fusion proteins. The fusion proteins were purified using glutathione‐sepharose 4B (GE Healthcare) following the manufacturer's instructions.
4.6. In vitro pull‐down assay
The fusion proteins were purified as described previously (Mao et al., 2014). Then, equivalent protein of His‐MoBre2FL and GST‐MoSdc1FL fusion proteins were added to a 1.5 ml centrifuge tube with BC100 (20 mM Tris.HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, 0.1% NP‐40) to 400 μl. Next, 40 μl glutathione‐sepharose 4B (GE Healthcare) was added to the solution. The tubes were then rotated and incubated for 4 hr at 4 °C to pull down GST‐tagged fusion proteins. This was followed by four washes with BC500 (20 mM Tris.HCl [pH 8.0], 500 mM NaCl, 1 mM EDTA, 0.1% NP‐40). The beads were boiled in SDS loading buffer for 10 min, centrifuged at 1,575 × g at 4 °C for 5 min, and then the supernatant was separated by 13% denaturing SDS‐PAGE. Finally, the gel was stained with Coomassie Brilliant Blue R‐250 (AMRESCO) and western blotting using anti‐His (Transgen Biotech, HT501) and anti‐GST antibodies (Transgen Biotech, HT601) (Mao et al., 2017) for observation.
4.7. Subcellular localization analysis
For colocalization analysis of MoBre2 and MoSdc1, the full‐length of MoBre2 was amplified by PCR from wild‐type P131 cDNA with primer pairs MoBre2‐GFP‐F and MoBre2‐GFP‐R. The resulting fragment and pKNTGRP27 vector were digested with EcoRI and BamHI, and MoBre2 was cloned into pKNTGRP27, which was generated by cloning the strong, constitutive RP27 promoter into pKNTG. The full‐length of MoSdc1 was amplified by PCR from wild‐type P131 cDNA with primer pairs MoSdc1‐RFP‐F and MoSdc1‐RFP‐R. The resulting fragment and pKS vector were digested with EcoRI and BamHI, MoSdc1 was cloned into pKS, and the RFP‐MoSdc1 fusion construct was generated. The GFP‐MoBre2 and RFP‐MoSdc1 fusion constructs were digested with NotI and cotransformed into wild‐type P131. Colocalization of GFP‐MoBre2 and RFP‐MoSdc1 fusion proteins was performed with a Nikon A1 laser scanning confocal microscope.
4.8. ChIP‐seq
Protoplasts for ChIP sequencing were prepared as described previously (Cao et al., 2016). Briefly, the protoplasts were crosslinked with 37% formaldehyde for 10 min, followed by termination of the crosslinking reaction with 10 × glycine for 5 min. Protoplasts were collected by centrifugation at 1,800 × g for 15 min, washed with 0.7 M NaCl, and diluted to 108/ml. Centrifugation and suspension of protoplasts occurred with 750 μl RIPA buffer (50 mM Tris.HCl [pH 8.0], 150 mM NaCl, 2 mM EDTA, 1% NP‐40, 0.5% sodium dexycholate, 0.1% SDS). The chromatin was sheared by sonication with JY 92‐IIDN (SCIENTZJ) for 8 min (25% W, output 3S, stop 5S). Chromatin immunopurification with H3K4me3 antibody (Abcam ab8580) and Dynabeads Protein A (Invitrogen 10002D) were incubated overnight at 4 °C as previously described (Mao et al., 2014). Subsequently, DNA fragments were extracted using the phenol‐chloroform method for constructing an Illumina sequencing library and sequenced with single‐ends on a Hiseq 2000. All the ChIP‐seq experiments had three independent replicates.
4.9. Mass spectrometry (LC‐MS/MS) and data analysis
Samples were destained in Proteo Silver Destainer A and Proteo Silver Destainer B, and then rinsed with water several times. Proteins were digested with 500 ng of sequencing‐grade modified trypsin (Promega), and the resulting peptides were resolved on a nano‐capillary reverse phase column. Using a water–acetonitrile gradient system, at 300 nl/min, sample was directly introduced into an ion‐trap mass spectrometer in the data‐dependent mode to collect MS/MS spectra on the five most intense ions from each full MS scan. MS/MS spectra were matched to peptides in a database containing M. oryzae protein sequences. All proteins with a Protein Prophet probability score of 0.9 were considered positive identifications.
4.10. RNA‐seq
Total RNA was isolated from the tissue of wild‐type strain P131, Δmobre2, Δmospp1, and Δmoswd2 deletion mutants, and mRNA was then isolated using a Poly(A) Purist MAG kit (Ambion). Each sample was repeated three times, and three experiments were replicated. DNA was removed by RNase‐free DNase (Qiagen) followed by column clean‐up according to the manufacturer's instructions. Illumina TruSeq RNA Sample Preparation kits were used to construct RNA‐seq libraries; cDNA was sequenced on an Illumina Hiseq 2000.
4.11. Bioinformatics
ChIP‐seq read quality was assessed using FASTQC (0.11.7) and aligned to M. oryzae genome assembly (MG8) using Bowtie2 v. 2.3.4.3 software. Only uniquely mapped reads were kept for analysis. H3K4me3 peaks were called by MACS2 v. 2.1.1.20160309 with read extension to average fragment size and the threshold of FDR < 10−6. Based on the MACS2‐called peaks and aligned read files, DiffBind v. 2.6.6 was used to identify differential binding peaks of H3K4me3 with FDR < 0.01 and binding fold change ≥2.
RNA‐seq reads were aligned to M. oryzae transcriptome annotation (MG8) with the STAR v. 2.6.0c aligner. Differentially expressed genes and quantification were done using the edgeR v. 3.20.9 package. Plots and charts were drawn with R software and MS Excel.
4.12. Sequence alignment and structural modeling
The sequences of Unknown and JMJD2A were aligned in ClustalX2 and then coloured by ESPript 3.0 (http.//espript.ibcp.fr/ESPript/ESPript/; Robert & Gouet, 2014). Using the online phyre2 modelling machine (http.//www.sbg.bio.ic.ac.uk/phyre2/html/ page.cgiid = index; Kelley et al., 2015), we constructed and selected the complex of the core Tudor domain of Unknown and H3K9me3 with the highest score, based on the template of the JMJD2A Tudor domain–H3K4me3 complex (PDB code 2006; Ng et al., 2007).
AUTHOR CONTRIBUTIONS
W.X.W. designed the experiments. S.D.Z., W.Y.S., M.Y.Z., S.P., Y.Y., D.H., M.S., J.Y., L.E.C., and J.Y.W. performed the biological experiments. X.Y.L. and Q.Z. performed the bioinformatics analysis. W.X.W. wrote the manuscript.
Supporting information
ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China (grant no. 31871638), the Special Scientific Research Project of Beijing Agriculture University (YQ201603), the Scientific Project of Beijing Educational Committee (KM201610020005), the Research Fund for Academic Degree & Graduate Education of Beijing University of Agriculture (2019YJS037), the National Laboratory of Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, and the National Natural Science Foundation of China (2018kf06).
Zhou S, Liu X, Sun W, et al. The COMPASS‐like complex modulates fungal development and pathogenesis by regulating H3K4me3‐mediated targeted gene expression in Magnaporthe oryzae . Mol Plant Pathol. 2021;22:422–439. 10.1111/mpp.13035
DATA AVAILABILITY STATEMENT
The raw data of RNA‐seq are available at NCBI BioProject, url http://www.ncbi.nlm.nih.gov/bioproject/ accession 649063. The raw data of ChIP‐seq are available at NCBI BioProject, http://www.ncbi.nlm.nih.gov/bioproject/ accession 649321.
REFERENCES
- Agger, K. , Cloos, P.A.C. , Christensen, J. , Pasini, D. , Rose, S. , Rappsilber, J. et al. (2007) UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature, 449, 731–734. [DOI] [PubMed] [Google Scholar]
- Ali, A. & Tyagi, S. (2017) Diverse roles of Wdr5‐RbBP5‐ASH2L‐DPY30 (wrad) complex in the functions of the Set1 histone methyltransferase family. Journal of Biosciences, 42, 155–159. [DOI] [PubMed] [Google Scholar]
- Allis, C.D. , Berger, S.L. , Cote, J. , Dent, S. , Jenuwien, T. , Kouzarides, T. et al. (2007) New nomenclature for chromatin‐modifying enzymes. Cell, 131, 131–633. [DOI] [PubMed] [Google Scholar]
- Ardehali, M.B. , Mei, A. , Zobeck, K.L. , Caron, M. , Lis, J.T. & Kusch, T. (2011) Drosophila Set1 is the major histone H3 lysine 4 trimethyltransferase with role in transcription. The EMBO Journal, 30, 2817–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachleitner, S. , Sørensen, J.L. , Gacek‐Matthews, A. , Sulyok, M. , Studt, L. & Strauss, J. (2019) Evidence of a demethylase independent role for the H3K4‐specific histone demethylases in Aspergillus nidulans and Fusarium graminearum secondary metabolism. Frontiers in Microbiology, 10, 1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger, S.L. (2007) The complex language of chromatin regulation during transcription. Nature, 447, 407–412. [DOI] [PubMed] [Google Scholar]
- Bhaumik, S.R. , Smith, E. & Shilatifard, A. (2007) Covalent modifications of histones during development and disease pathogenesis. Nature Structural and Molecular Biology, 14, 1008–1016. [DOI] [PubMed] [Google Scholar]
- Bok, J.W. , Chiang, Y.‐M. , Szewczyk, E. , Reyes‐Dominguez, Y. , Davidson, A.D. , Sanchez, J.F. et al. (2009) Chromatin‐level regulation of biosynthetic gene clusters. Nature Chemical Biology, 5, 462–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, Z.J. , Yin, Y. , Sun, X. , Han, J. , Sun, Q.P. , Lu, M. et al. (2016) An ash1‐like protein MoKMT2H null mutantis delayed for conidium germination and pathogenesis in Magnaporthe oryzae . BioMed Research International, 10, 1575–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Cao, F. , Wan, B. , Dou, Y. & Lei, M. (2012) Structure of the SPRY domain of human ASH2L and its interactions with RbBP5 and DPY30. Cell Research, 22, 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. , Zang, J. , Kappler, J. , Hong, X. , Crawford, F. , Wang, Q. et al. (2007). Structural basis of the recognition of a methylated histone tail by JMJD2A. Proceedings of the National Academy of Sciences of the United States of America, 104, 10818–10823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, Y.‐W. , Hong, T. , Hong, SunHwa , Guo, H. , Yu, H. , Kim, D. et al. (2007) PTIP associates with MLL3‐and‐MLL4‐containing histone H3 lysine 4 methyltransferase complex. The Journal of Biological Chemistry, 282, 20395–20406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly, L.R. , Smith, K.M. & Freitag, M. (2013) The Fusarium graminearum histone H3K27 methyltransferase KMT6 regulates development and expression of secondary metabolite gene clusters. PLoS Genetics, 9, e1003916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czermin, B. , Melfi, R. , McCabe, D. , Seitz, V. , Imhof, A. & Pirrotta, V. (2002) Drosophila enhancer of zeste/esc complexes have a histone H3 methyltransferase activity that marks chromosomal polycomb sites. Cell, 111, 185–196. [DOI] [PubMed] [Google Scholar]
- D’Urso, A. , Takahashi, Y. , Xiong, B. , Marone, J. , Coukos, R. , Hinchliff, C.R. , et al. (2016) Set1/COMPASS and mediator are repurposed to promote epigenetic transcriptional memory. eLife, 5, e16691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang, J.L. & Jones, J.D.G. (2001) Plant pathogens and integrated defence response to infection. Nature, 411, 826–833. [DOI] [PubMed] [Google Scholar]
- Deng, S. , Gu, Z. , Yang, N. , Li, L. , Yue, X. , Que, Y. et al. (2016) Identification and characterization of the peroxin 1 gene MoPEX1 required for infection‐related morphogenesis and pathogenicity in Magnaporthe oryzae . Scientific Reports, 10, 36292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dover, J. , Schneider, J. , Tawiah‐Boateng, M.A. , Wood, A. , Dean, K. , Johnston, M. et al. (2002) Methylation of histone H3 by compass requires ubiquitination of histone H2B by Rad6. Journal of Biological Chemistry, 277, 28368–28371. [DOI] [PubMed] [Google Scholar]
- Ernst, P. & Vakoc, C.R. (2012) WRAD: Enabler of the Set1‐family of H3K4 methyltransferases. Briefings in Functional Genomics, 11, 217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, P.L. , Li, H. , Lau, H.‐T. , Leonen, C. , Dhall, A. , Ong, S.‐E. et al. (2018) Crystal structure of the compass H3K4 methyltransferase catalytic module. Cell, 174, 1106–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, D.H. , Kong, N.C. , Gu, X.F. , Li, Z.C. & He, Y.H. (2011) Arabidopsis compass‐like complexes mediate histone H3 lysine‐4 trimethylation to control floral transition and plant development. PLoS Genetics, 7, e1001330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley, L.A. , Mezulis, S. , Yates, C.M. , Wass, M.N. & Sternberg, M.J. (2015) The phyre2web portal for protein modeling, prediction and analysis. Nature Protocols, 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H.J. , Han, J.H. , Kim, K.S. & Lee, Y.H. (2014) Comparative functional analysis of the velvet gene family reveals unique roles in fungal development and pathogenicity in Magnaporthe oryzae . Fungal Genetics and Biology, 66, 33–43. [DOI] [PubMed] [Google Scholar]
- Kim, Y. , Park, S.‐Y. , Kim, D. , Choi, J. , Lee, Y.‐H. , Lee, J.‐H. et al. (2013) Genome‐scale analysis of ABC transporter genes and characterization of the ABCC type transporter genes in Magnaporthe oryzae . Genomics, 10, 354–361. [DOI] [PubMed] [Google Scholar]
- Kornberg, R.D. (1974) Chromatin structure: A repeating unit of histones and DNA. Science, 184, 868–871. [DOI] [PubMed] [Google Scholar]
- Kou, Y.J. , Tan, Y.H. , Ramanujam, R.I. & Naqvi, N. (2016) Structure‐function analyses of the Pth11 receptor reveal an important role for CFEM motif and redox regulation in rice blast. New Phytologist, 214, 330–342. [DOI] [PubMed] [Google Scholar]
- Kouzarides, T. (2007) Chromatin modifications and their function. Cell, 128, 693–705. [DOI] [PubMed] [Google Scholar]
- Krogan, N.J. , Dover, J. , Wood, A. , Schneider, J. , Heidt, J. , Boateng, M.A. et al. (2003) The paf1 complex is required for histone H3 methylation by COMPASS and dot1p: Linking transcriptional elongation to histone methylation. Molecular Cell, 11, 721–729. [DOI] [PubMed] [Google Scholar]
- Lachner, M. & Jenuwein, T. (2002) The many faces of histone lysine methylation. Current Opinion in Cell Biology, 14, 286–298. [DOI] [PubMed] [Google Scholar]
- Lee, J.H. & Skalnik, D.G. (2008) Wdr82 is a C‐terminal domain‐binding protein that recruits the Setd1A histone H3‐Lys4 methyltransferase complex to transcription start sites of transcribed human genes. Molecular and Cellular Biology, 28, 609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Han, J. , Zhang, Y. , Cao, F. , Liu, Z. , Li, S. et al. (2016) Structural basis for activity regulation of MLL family methyltransferases. Nature, 530, 447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C. , Li, Z. , Xing, J. , Yang, J. , Wang, Z. , Zhang, H. et al. (2018) Global analysis of sumoylation function reveals novel insightsinto development and appressorium‐mediated infection of the rice blast fungus. New Phytologist, 219, 1031–1047. [DOI] [PubMed] [Google Scholar]
- Liu, W. , Xie, S. , Zhao, X. , Chen, X. , Zheng, W. , Lu, G. et al. (2010) A homeobox gene is essential for conidiogenesis of the rice blast fungus Magnaporthe oryzae . Molecular Plant‐Microbe Interactions, 23, 366–375. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Liu, N. , Yin, Y.N. , Chen, Y. , Jiang, J.H. & Ma, Z.H. (2015) Histone H3K4 methylation regulates hyphal growth, secondary metabolism and multiple stress responses in Fusarium graminearum . Environmental Microbiology, 17, 4615–4630. [DOI] [PubMed] [Google Scholar]
- Luger, K. , Maeder, A.W. , Richmond, R.K. , Sargent, D.F. & Richmond, T.J. (1997) Crystal structure of the nucleosomecore particle at 2.8 A resolution. Nature, 389, 251–260. [DOI] [PubMed] [Google Scholar]
- Mao, Z. , Pan, L.U. , Wang, W. , Sun, J. , Shan, S. , Dong, Q. et al. (2014) Anp32e, a higher eukaryotic histone chaperone directs preferential recognition for H2A.Z. Cell Research, 24, 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, T. , Krogan, N.J. , Dover, J. , Erdjument‐Bromage, H. , Tempst, P. , Johnston, M. et al. (2001) COMPASS: A complex of proteins associatedwith a trithorax‐related SET domain protein. Proceedings of the National Academy of Sciences of the United States of America, 98, 12902–12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan, M. , Herz, H.‐M. , Smith, E.R. , Zhang, Y. , Jackson, J. , Washburn, M.P. et al. (2011) The COMPASS family of H3K4 methylases in Drosophila . Molecular and Cellular Biology, 31, 4310–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, H.H. , Robert, F. , Young, R.A. & Struhl, K. (2003) Targeted recruitment of set1 histone methylase by elongating Pol II provides a localized markand memory of recent transcriptional activity. Molecular Cell, 11, 709–719. [DOI] [PubMed] [Google Scholar]
- Ng, S.S. , Kavanagh, K.L. , McDonough, M.A. , Butler, D. , Pilka, E.S. , Lienard, B.M.R. et al. (2007) Crystal structures of histone demethylase jmjd2a reveal basis for substrate specificity. Nature, 448, 87–91. [DOI] [PubMed] [Google Scholar]
- Ozboyaci, M. , Gursoy, A. , Erman, B. & Keskin, O. (2017) Molecular recognition of H3/H4 histone tails by the tudor domains of JMJD2A: A comparative molecular dynamics simulations study. PLoS One, 6, e14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer, J.M. , Bok, J.W. , Lee, S. , Dagenais, T.R.T. , Andes, D.R. , Kontoyiannis, D.P. et al. (2013) Loss of CclA, required for histone 3 lysine 4 methylation, decreases growth but increases secondary metabolite production in Aspergillus fumigatus . PeerJ, 1, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patkar, R.N. , Ramospamplona, M. , Gupta, A.P. , Fan, Y. & Naqvi, N.I. (2012) Mitochondrial β‐oxidation regulates organellar integrity and is necessary for conidial germination and invasive growth in Magnaporthe oryzae . Molecular Microbiology, 86, 1345–1363. [DOI] [PubMed] [Google Scholar]
- Pham, K.T.M. , Inoue, Y. , Van Vu, B. , Nguyen, H.H. , Nakayashiki, T. , Ikeda, K.‐I. et al. (2015) MoSet1 (histone H3K4 methyltransferase in Magnaporthe oryzae) regulates global gene expression during infection‐related morphogenesis. PLoS Genetics, 11, e1005752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman, H. & De Sousa, R.D. (2014) Role of MoAnd1‐mediated nuclear positioning in morphogenesis and pathogenicity in the rice blast fungus, Magnaporthe oryzae . Fungal Genetics & Biology, 69, 43–51. [DOI] [PubMed] [Google Scholar]
- Raman, S.B. , Nguyen, M.H. , Zhang, Z. , Cheng, S. , Jia, H.Y. , Weisner, N. et al. (2006) Candida albicans SET1 encodes a histone 3 lysine 4 methyltransferase that contributes to the pathogenesis of invasive candidiasis. Molecular Microbiology, 60, 697–709. [DOI] [PubMed] [Google Scholar]
- Robert, X. & Gouet, P. (2014) Deciphering key features in protein structures with the new endscript server. Nucleic Acids Research, 42, W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roguev, A. , Schaft, D. , Shevchenko, A. , Pijnappel, W.W. , Wilm, M. , Aasland, R. et al. (2001) The Saccharomyces cerevisiae Set1 complex includes an ash2 homologue and methylates histone 3 lysine 4. EMBO Journal, 20, 7137–7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard, A. (2008) Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Current Opinion in Cell Biology, 20, 341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard, A. (2012) The COMPASS family of histone H3K4 methylases: Mechanisms of regulation in development and disease pathogenesis. Annual Review of Biochemistry, 81, 65–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara, Y. , Kawatani, M. , Futamura, Y. , Osada, H. & Koyama, Y. (2015) An overproduction of astellolides induced by genetic disruption of chromatin remodeling factors in Aspergillus oryzae . Journal of Antibiotics, 69, 4–8. [DOI] [PubMed] [Google Scholar]
- Smith, E. & Shilatifard, A. (2010) The chromatin signaling pathway: Diverse mechanisms of recruitment of histone‐modifying enzymes and varied biological outcomes. Molecular Cell, 40, 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stassen, M.J. , Bailey, D. , Nelson, S. , Chinwalla, V. & Harte, P.J. (1995) The Drosophila trithorax proteins contain a novel variant of the nuclear receptor type DNA binding domain and an ancient conserved motif found in other chromosomal proteins. Mechanisms of Development, 52, 209–223. [DOI] [PubMed] [Google Scholar]
- Strath, B.D. & Allis, C.D. (2000) The language of covalent histone modifications. Nature, 403, 41–45. [DOI] [PubMed] [Google Scholar]
- Studt, L. , Janevska, S. , Arndt, B. , Boedi, S. , Sulyok, M. , Humpf, H.‐U. et al. (2017) Lack of the COMPASS component Ccl1 reduces H3K4 trimethylation levels and affects transcription of secondary metabolite genes in two plant pathogenic Fusarium species. Frontiers in Microbiology, 7, 2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studt, L. , Rösler, S.M. , Burkhardt, I. , Arndt, B. , Freitag, M. , Humpf, H.U. et al. (2016) Knock‐down of the methyltransferase Kmt6 relieves H3K27me3 and results in induction of cryptic and otherwise silent secondary metabolite gene clusters in Fusarium fujikuroi . Environmental Microbiology, 18, 4037–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, Y.H. , Westfield, G.H. , Oleskie, A.N. , Trievel, R.C. , Shilatifard, A. & Skiniotis, G. (2011) Structural analysis of the core COMPASS family of histone H3K4 methylases from yeast to human. Proceedings of the National Academy of Sciences of the United States of America, 108, 20526–20531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay, V. , Zhang, P. , Chaturvedi, C.‐P. , Thornton, J. , Brunzelle, J. , Skiniotis, G. et al. (2014) Molecular basis for DPY‐30 association to COMPASS‐like and NURF complexes. Structure, 22, 1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschiersch, B. , Hofmann, A. , Krauss, V. , Dorn, R. , Korge, G. & Reuter, G. (1994) The protein encoded by the drosophila position‐effect variegation suppressor gene Su(var)3–9 combines domains of antagonistic regulators of homeotic gene complexes. EMBO Journal, 13, 3822–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulucan, O. , Keskin, O. , Erman, B. & Gursoy, A. (2017) A comparative molecular dynamics study of methylation state specificity of JMJD2A. PLoS One, 6, e24664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, G. , Wang, C. , Hou, R. , Zhou, X. , Li, G. , Zhang, S. et al. (2012) The AMT1 arginine methyltransferase gene is important for plant infection and normal hyphal growth in Fusarium graminearum . PLoS One, 7, e38324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J.K. , Tsai, M.‐C. , Poulin, G. , Adler, A.S. , Chen, S. , Liu, H. et al. (2010) The histone demethylase UTX enables RB‐dependent cell fate control. Genes & Development, 24, 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman, J.L. & Kingston, R.E. (2003) Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annual Review of Biochemistry, 67, 545–579. [DOI] [PubMed] [Google Scholar]
- Yan, X. & Talbot, N.J. (2016) Investigating the cell biology of plant infection by the rice blast fungus Magnaporthe oryzae . Current Opinion in Microbiology, 34, 147–153. [DOI] [PubMed] [Google Scholar]
- Yang, N. , Wang, W. , Wang, Y. , Wang, M. , Zhao, Q. , Rao, Z. et al. (2012) Distinct mode of methylated lysine‐4 of histone H3 recognition by tandem tudor‐like domains of spindlin1. Proceedings of the National Academy of Sciences of the United States of America, 109, 17954–17959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Y. , Zhou, H. , Deng, X.L. , Wang, W.C. & Lu, H. (2016) Set3 contributes to heterochromatin integrity by promoting transcription of subunits of Clr4‐Rik1‐Cul4 histone methyltransferase complex in fission yeast. Scientific Reports, 6, 31752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Zhao, Q. , Guo, X. , Guo, M. , Qi, Z. , Tang, W. et al. (2014) Pleiotropic function of the putative zinc‐finger protein MoMsn2 in Magnaporthe oryzae . Molecular Plant‐Microbe Interactions, 27, 446–460. [DOI] [PubMed] [Google Scholar]
- Zhang, M.Y. , Sun, X. , Yin, Y. , Cui, L.E. , Wang, W.X. et al. (2018) The plant infection test: Spray and wound‐mediated inoculation with the plant pathogen Magnaporthe grisea . Journal of Visualized Experiments, 138, e57675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data of RNA‐seq are available at NCBI BioProject, url http://www.ncbi.nlm.nih.gov/bioproject/ accession 649063. The raw data of ChIP‐seq are available at NCBI BioProject, http://www.ncbi.nlm.nih.gov/bioproject/ accession 649321.