

Abstract
Spastic cerebral palsy (CP), despite the name, is not consistently identifiable by specific brain lesions. CP animal models focus on risk factors for development of CP, yet few reproduce the diagnostic symptoms. Animal models of CP must advance beyond risk factors to etiologies, including both the brain and spinal cord.
Introduction
Cerebral palsy (CP) is the most common motor disability in childhood with a prevalence of 2.1 per 1,000 live births in developed countries, although a higher prevalence of CP is likely in lower income countries (152, 155). CP began as a description of physical symptoms and impairments by Dr. William John Little in 1862 (118, 119). Although Little originally termed this a “cerebro-spinal disorder,” the nomenclature quickly transitioned to the term cerebral palsy (118, 119). With the CP terminology, the brain and cerebral cortex became the focus of the etiology of the symptoms in both research and clinical interventions.
Since 1862, medical and scientific advances have been made in the prevention and medical management of CP. However, the physical impairments in individuals with CP remain incurable, and the prevalence has remained largely unchanged (155). Potential contributors to the lack of curative therapies for CP include the focus on the brain and cerebral cortex as the driver of the symptoms. Furthermore, CP is an inherently complex disorder due to the heterogeneity of etiologies and physical impairments.
The heterogeneity of CP and long-held clinical and scientific beliefs that CP is only a developmental brain injury has hampered animal models of this disorder. Specifically, the focus of animal models has been on inflicting a brain injury and subsequent description of the neuropathological changes in the brain. Yet, CP is a disorder of movement and postural control, symptoms absent in most animal models.
The following review will critically assess the current state of CP animal models in research, with a goal of promoting animal models that display the CP phenotype. We will briefly discuss the history of CP, symptoms and diagnosis of CP, risk factors for CP, the contribution of animal models to our understanding of CP pathophysiology, and gaps in our understanding of CP. The gaps in our understanding of the mechanistic underpinnings of CP are largely due to the sole, laser-like focus on a brain injury only. This limitation is further compounded by a lack of animal models with the physical symptoms of CP.
Is Cerebral Palsy an Accurate Term?
History of Cerebral Palsy
In 1862, Dr. William John Little, an orthopedic surgeon with a particular interest in tenotomy procedures, described children who had “congenital distortions” of their limbs (118, 119). Little described this disorder based on observations of muscle stiffness and limb deformity in children who had difficult births including prematurity, prolonged labor, breech presentations, and even nuchal umbilical cords (118, 119). In his original manuscript, Little referred to the symptoms as a “cerebro-spinal disorder” (118, 119) because of postmortem evidence of brain and spinal cord involvement. The abnormalities included venous and capillary congestion on the surface of the brain and spinal cord, and blood in the cerebrospinal fluid in the brain ventricles and within the subarachnoid space surrounding the brain and spinal cord (118, 119). Despite this early recognition of both brain and spinal involvement, CP quickly became referred to as “cerebral paresis” (49). This loss of the “spinal” component of this descriptive name may be due to the commonly held belief at that time that the spinal cord was more completely formed than the brain at birth (118, 119). Little hypothesized that this disorder was due to anoxia (asphyxia neonatorum) secondary to birth difficulties, recognizing a central nervous system disorder as the driver of the physical symptoms (118, 178). This was a novel hypothesis since the prevailing belief was that the limb deformities were from direct mechanical injury (118, 178). However, Little had sparse evidence to support this hypothesis, although he recognized the work of others who noted abnormal engorgement of the meninges and blood surrounding the brain and spinal cord in infants who died from suspected asphyxia neonatorum (118, 119). Over time, the spastic diplegic form of CP became known as Little’s Disease (49, 159). Although Little’s original concept of a “cerebro-spinal disorder” may be more accurate than the subsequent term of “cerebral paresis,” this long-held concept of an injury to the brain due to anoxia at birth has persisted despite evidence suggesting that CP may not be the result of an isolated injury at a single time or a single insult (147, 194).
The actual term “cerebral palsy” (CP), which continues to be used today, was coined in 1889 by Sir William Osler from a series of five lectures titled “The Cerebral Palsies of Children” (156, 178). In this series of lectures, Osler described numerous patients with symptoms of CP who had difficult deliveries (156, 178). Shortly thereafter, Dr. Sigmund Freud developed a classification system for CP and described other conditions associated with CP (65). His classification system was based on hypothesized timing of the injury occurrence, including antepartum, intrapartum, and postnatal (65, 178). However, Freud strayed from this research when he was unable to definitively describe unique neuropathological brain abnormalities to set CP apart from other conditions along with confusion of CP with other conditions resulting in similar impairments, including spinal disorders like poliomyelitis (178). The reasons for abandoning additional work in this area, in retrospect, were a herald for the possibility that CP is more than an injury to the brain.
Cerebral Palsy in Modern Medicine
Eventually, CP and its definition were revisited, although without a change in nomenclature. In 1959, the definition of CP was updated in a consensus report to a “persisting qualitative motor disorder due to non-progressive interference with development of the brain occurring before the growth of the central nervous system (CNS) is complete” (125). Although these authors recognized the incomplete development of the entire CNS at the time of the insult or injury, they unfortunately only included an insult to the brain as part of the definition of CP, excluding the role of the spinal cord. This definition of CP was further refined in 1964 in another consensus report by Bax et al. as primarily a “disorder of movement and posture due to a defect or lesion of the immature brain,” again without mentioning the spinal cord involvement (11). In 2004, the definition of CP was revisited yet again by a group of international experts. This definition, which continues to be used today, describes CP as a “group of permanent disorders of the development of movement and posture, causing activity limitation, that are attributed to non-progressive disturbances that occurred in the developing fetal or infant brain,” with further description of other potential associated conditions (169). Surprisingly, although the length of these descriptions of CP progressively increased with time, they exclude mention that CP symptoms also relate to spinal cord dysfunction (i.e., hyperexcitability/spasticity and delays in limb and trunk motor development). The involvement of abnormal spinal cord development is unmentioned in the current clinically accepted definition of CP, and the focus remains on the brain despite the severity or even presence of physical impairments being poorly predicted by brain imaging (10, 114, 203). However, despite the “progress” in the clinical consensus of CP, the definition of CP continues to focus on the brain without acknowledgment of developmental spinal cord dysfunction.
Is a Brain Injury Required for Diagnosis and Symptoms of Cerebral Palsy?
Diagnosis of Cerebral Palsy
CP is a clinical diagnosis, typically made before 2 years of age, with most being diagnosed in the first year of life (77, 90, 92). It is diagnosed based on both clinical symptoms and neurological signs, although not all signs or symptoms are present in an individual (Table 1). Typically, it is the delay in reaching normal developmental milestones that prompt a clinical evaluation for CP, especially in infants with risk factors for developing CP. Abnormal brain imaging is not a diagnostic requirement, nor is the presence of abnormal brain imaging diagnostic of CP if there are no motor symptoms.
Table 1.
Signs and symptoms of cerebral palsy
| Neurological | Orthopedic | Cognitive | Vision/Hearing | Aerodigestive | Other |
|---|---|---|---|---|---|
| Ataxia | Femoral anteversion | Autism | Conductive hearing impairment | Aspiration pneumonia | Disorder sleep initiation |
| Athetosis | Hip dislocation | Epilepsy | Cortical visual impairment | Constipation | Disordered sleep maintenance |
| Dystonia | Hip dysplasia | Intellectual impairment | Dyskinetic strabismus | Dysarthria | Failure to thrive/ poor growth |
| Gait disorder | Hip subluxation | Learning disabilities | High myopia | Dysphagia | Low bone mineral density |
| Hyperreflexia | Joint contracture | Retinopathy of Prematurity | Gastresophageal reflux | Neurogenic bladder | |
| Hypotonia | Patella alta | Sensorineural hearing impairment | Obstructive sleep apnea | Muscle spasms | |
| Impaired gross motor coordination | Scoliosis | Respiratory disorders (reduced forced vital capacity, forced expiratory volume, peak expiratory flow) | Pain | ||
| Impaired fine motor coordination | Tibial torsion | Sialorrhea | |||
| Spasticity |
Symptoms of Cerebral Palsy
The primary symptoms of CP include spasticity (most common), dyskinesias (such as dystonia, athetosis, chorea), ataxia, hyper-reflexia, and in rare cases hypotonia (89, 152, 181). Spasticity is detected clinically as a velocity-dependent resistance of a muscle to stretch (112, 173). It is thought to be due to loss of descending inhibition of the motor neuron (102, 132). It is measured clinically by subjective assessment on physical exam and is typically scored in severity using the Modified Ashworth Scale and Modified Tardieu Scale (74, 135, 141). Dyskinesias are unwanted, often sustained, involuntary movements commonly due to an injury or pathology of deep brain structures (172). These can be evaluated clinically using the Barry Albright Dystonia Scale, Dyskinesia Impairment Scale, and Hypertonia Assessment Tool (1, 9, 98, 104, 139). Ataxia involves incoordination of movement, typically due cerebellar injury or maldevelopment (105). Hyper-reflexia is an increase in muscle stretch reflex, often elicited on physical exam via a tendon tap (i.e., patellar tendon, Achilles tendon, and biceps brachii tendons), with subjective grading of robustness of response (197). Hyper-reflexia, similar to spasticity, is thought to be due to a loss of descending inhibition of the motor neuron (197). Hypotonia, or decreased muscle tone, is most commonly associated with muscle or peripheral nerve disorders (117). However, it can occur in central nervous system, genetic, and metabolic disorders, although the mechanism underlying hypotonia is poorly understood (117). Hypotonia is graded clinically on presence and subjectively on severity, although no formal grading scales have been agreed upon.
These varied symptoms point to varied CP etiologies involving brain and spinal cord pathophysiology. Furthermore, the complexity and heterogeneity of CP is reflected by the secondary conditions associated with CP. These include epilepsy, musculoskeletal disorders (hip dysplasia, boney torsional deformities, scoliosis), autism, intellectual disability, chewing and swallowing disorders, respiratory dysfunction, sleep disorders, chronic pain, cortical visual impairment, hearing impairment, and bladder and bowel disorders (Table 1; FIGURE 1) (22, 44, 55, 111, 140, 148, 152, 154, 162, 169, 185). The most common form of CP is spastic CP (>80% of all diagnosed cases) (89), which almost certainly involves dysfunction of the spinal cord since spasticity is related to disinhibition of the motor neuron (102, 132). Spastic CP is diagnosed based on clinical symptoms, as noted above, with the presence of spasticity, which is also determined clinically. To advance our understanding of CP, it may be best to separate CP into those who predominantly display symptoms of spasticity and those who do not, since the spastic component is likely indicating significant involvement of the spinal cord. Furthermore, >80% of individuals with CP have spasticity (4). In this review, we will narrow our focus to spastic CP.
FIGURE 1.
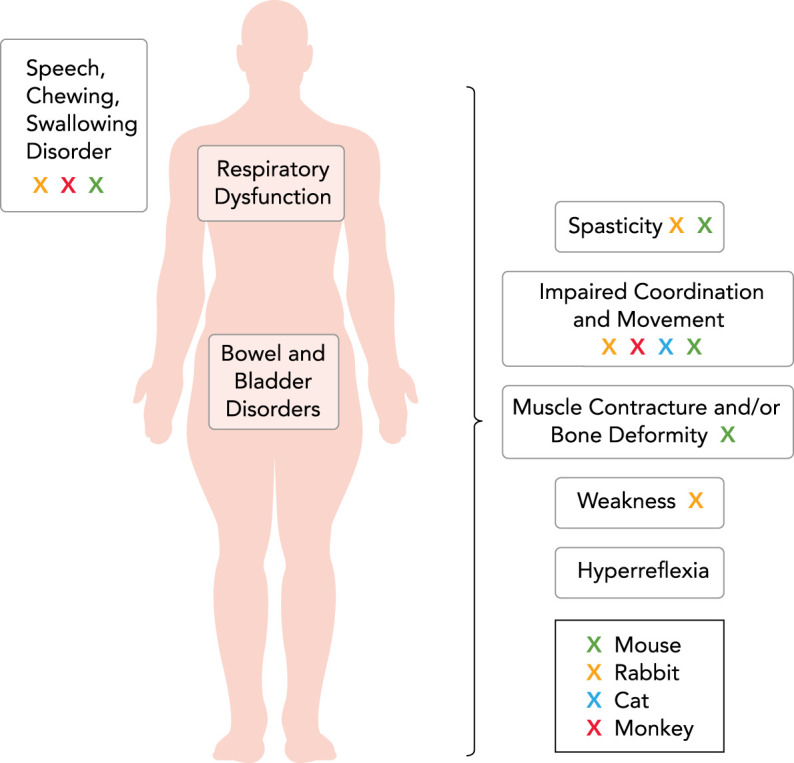
Physical impairments and secondary disorders associated with spastic cerebral palsy in humans
In addition to spasticity, the essential diagnostic criterion for spastic CP includes impaired movement and/or coordination, which are determined clinically based on developmental norms for acquiring motor skills. In humans, impaired movement includes difficulty achieving motor milestones, crawling, standing, or walking, or difficulty with sitting or postural control. Note that although impaired movement is observed in monkey (red), cat (blue), rabbit (yellow), and mouse (green) models, spasticity is exhibited only in the mouse and rabbit. Neither cardinal symptoms nor signs of spastic CP are observed in any of the hamster, rat, or sheep models. Clearly, animal models that more closely resemble clinical phenotypes are needed for rational approaches to the understanding of the disease mechanisms and eventual translation to treatment.
What Is Known About the Etiology of Spastic Cerebral Palsy?
Symptoms of spastic CP give us insight into etiology. Spastic CP is classified topographically as unilateral (hemiplegia) or bilateral (diplegia or quadriplegia) (152, 177). Individuals with diplegic spastic CP tend to have greater involvement of the legs than the arms (i.e., disproportionately impaired spasticity and motor control of the legs compared with the arms), suggesting a rostral-caudal pattern of injury for individuals with this type of spastic CP (158). Some known risk factors for developing spastic CP include asphyxia, hemorrhagic or ischemic stroke (prenatal or neonatal), infection (prenatal or neonatal), brain malformation, trauma (inflicted or accidental), non-progressive genetic disorders/differences, and dysmaturation (Table 2) (5–7, 171). The mechanisms by which these risk factors cause or contribute to a neurodevelopmental injury to the brain is discussed in many reviews (54, 57, 99, 171, 182, 194–196, 205). Yet, there is scant information about the impact these risk factors have on spinal cord development. The spinal cord should not be ignored, since it is vulnerable to a developmental injury during this same period, and in particular the motor neurons (23, 189).
Table 2.
Prevalence of cerebral palsy by live birth based on risk factors and percentage of individuals with cerebral palsy diagnosis who had risk factors
| Risk Factor | Prevalence of CP | Percentage (%) of Individuals with CP | |
|---|---|---|---|
| Birth prematurity | <28-wk gestation | 82 per 1,000 live births (155) | 73% (90) |
| 28- to 31-wk gestation | 43 per 1,000 live births (155) | ||
| 32- to 36-wk gestation | 7 per 1,000 live births (155) | ||
| In utero insults (i.e., uterine ischemia, intrauterine infections, uterine inflammation) | Odds ratio of developing CP if maternal infection during pregnancy 1.55, with greater risk if infection at 21–40 wk of gestation (153) | 12% of those with spastic CP (79) | |
| Brain malformations | 8.6–11.1% (70, 107, 108) | ||
| Stroke | 1 per 4,000 live births (122) | 1% (from postnatal stroke) (72) | |
| Hypoxic-ischemic encephalopathy | 1.5 per 1,000 live births (110) | 14.5% of term infants (76) | |
| Asphyxia | 2–10% (53, 165) | ||
| Twin gestation | Odds ratio of CP diagnosis if twin gestation 6.62 (153); 7–12 per 1,000 live births (20, 113, 160, 209) | ||
The second column shows the prevalence of cerebral palsy per live births according to risk factors (first column). The last column displays percentage of individuals living with cerebral palsy who had a particular risk factor exposure. Note that some individuals with cerebral palsy may have been exposed to more than one risk factor.
Therapeutic interventions for spasticity in CP may also provide insight into etiology. Interventions to reduce spasticity symptoms generally target motor neuron hyperexcitability, although the targeting depends on the intervention (i.e., ranging from a specific muscle to the entire spinal cord). These spasticity-reducing interventions act on the motor neuron either through direct modulation of neurotransmitter input or indirectly through various mechanisms (FIGURE 2). For example, baclofen is a GABAB receptor agonist commonly used for spasticity treatment in individuals with spastic CP (25, 134). GABA is one of two inhibitory (hyperpolarizing) neurotransmitters in the central nervous system, with the metabotropic GABAB receptors located in both the brain and spinal cord (136). Baclofen is often administered intrathecally, targeting the spinal cord (18). An intervention that indirectly mitigates motor neuron hyperexcitability is a selective dorsal rhizotomy (FIGURE 2) (27, 133). This is a procedure that removes some afferent input to the motor neuron, some of which occurs directly via reduction of Ia afferent inputs through selectively sectioning portions (rootlets) of the lumbar sensory nerves (usually L2–S1 with generally 25–50% of rootlets sectioned), and some of which may occur indirectly. The end result is a reduction or elimination of spasticity in the legs via interruption of the sensory excitatory input to the motor neuron (133). Both of these interventions target leg spasticity, which typically involves all muscles of the lower limb at varying degrees. Symptoms of spastic CP always involve the legs, with variable involvement of the trunk and arms, supporting a differential rostro-caudal burden of disease, suggestive of spinal cord involvement.
FIGURE 2.
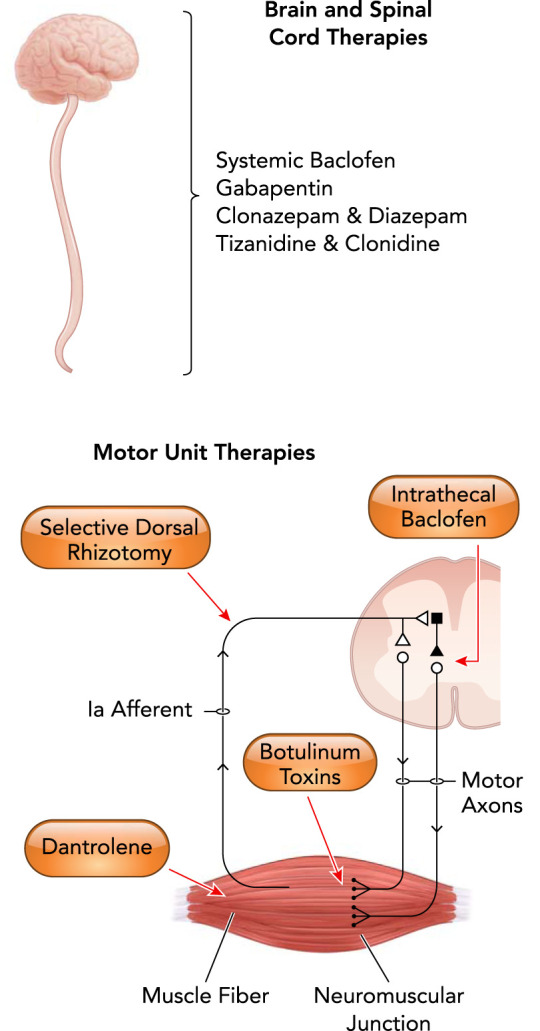
Medical and surgical interventions in the treatment of spastic cerebral palsy
All of the current clinical treatments for spastic CP involve both the brain and the spinal cord. Indeed, most treatments exclusively affect the spinal cord, motor neuron and/or motor unit alone. Treatment with baclofen, a presynaptic GABA agonist, may be orally (systemic) or intrathecally (targeting spinal cord neurons) administered. Gabapentin is a mild GABAmimetic and binds the α2δ subunit of voltage-gated Ca2+ channels. Clonazapam and diazepam are similar GABA receptor agonists. Tizanidine and clonidine primarily reduce interneuron activity within the spinal cord via agony of α2 adrenergic receptors. Selective dorsal rhizotomy selectively removes Ia afferent inputs to motor neurons, reducing their reflex excitability. Dantrolene works at the level of skeletal muscle, inhibiting the release of Ca2+ from the sarcoplasmic reticulum.
It is likely that spasticity is mediated by spinal cord circuits, since spasticity results from disinhibition of the motor neuron (102, 132). This is in direct conflict with the long-held conceptual framework that spastic CP is exclusively a developmental brain disorder. We will critically evaluate the premise of spastic CP as a brain disorder, assess preclinical animal models of this disorder, and propose a shift in the paradigm to consideration of spastic CP as a central nervous system disorder affecting both the brain and the spinal cord.
How Is Spastic Cerebral Palsy Modeled?
To study the mechanisms underlying known, suspected, and yet to be identified etiologies of spastic CP, animal models are essential. Animal models of spastic CP must display motor symptoms. Yet, most animal models of CP have focused on the risk factors for developing spastic CP rather than the mechanisms causing the spasticity and motor symptoms. Furthermore, these insults or risk factors impact not only the brain but also spinal cord development and specifically motor neuron abnormalities. The pathognomonic symptoms of spastic CP imply that motor neuron abnormalities are the mechanistic driver of the spasticity and motor impairment. Animal models of spastic CP must move beyond the prevailing research paradigm of modeling risk factors. Progressing beyond descriptions of risk factors for spastic CP to defining the mechanisms causing the symptoms facilitates development of targeted curative interventions, as opposed to merely modulating the symptoms. To move the research paradigm forward, it is imperative to understand the state of animal models for spastic CP.
Animal Models of Spastic Cerebral Palsy Risk Factors
Traditionally, animal models of spastic CP have focused on a brain injury as the driver of symptoms. Yet, few of the commonly accepted animal models actually display the diagnostic symptoms of spasticity and motor impairment (FIGURE 1). Most animal models of spastic CP are derived based on known risk factors for developing this permanent disorder.
The most common risk factor for development of spastic CP is birth prematurity (4, 155, 158, 181). Birth prematurity has been modeled via various methods for inducing preterm birth in mice, rats, rabbits, and sheep (15, 26, 37, 47, 48, 52, 69, 71, 109, 130, 151, 166, 168, 210). Due to the preterm birth or the research protocols, these animals do not survive, thus do not develop CP symptoms. Furthermore, a notable issue with animal models using birth prematurity is that most species are born at different stages of developmental maturity (34, 51, 204). These differences in developmental stages may not be reflective of the developmental timing of preterm birth in humans.
In utero insults are known risk factors but are far less common causes of spastic CP than birth prematurity (176, 207). In utero insults that have been modeled as risk factors for spastic CP include reduced uterine blood flow (uterine ischemia) and intrauterine infections or inflammation (51, 73, 147). Uterine ischemia has been modeled in the rabbit, sheep, and monkey, with physical symptoms observed only in monkeys and rabbits (40–42, 50, 91, 130, 142, 166, 168). In monkeys, feeding difficulties, weakness, hypertonia, and abnormal movement lead to them perishing due to the feeding difficulty (142). Rabbits demonstrate physical impairments including difficulty with head control, locomotion, swimming, and feeding (40–42, 45, 46). The husbandry for these animals is arduous, since the feeding impairment necessitates orogastric delivery of rabbit milk for survival (40–42). Uterine inflammation via introduction of an endotoxin or live bacteria has been extensively studied in mice, rats, rabbits, and sheep, with many of these animals displaying brain abnormalities. However, none survived a sufficient length of time to display motor symptoms (14, 15, 26, 28, 37, 38, 43, 48, 52, 58, 71, 120, 151, 170, 210).
Brain malformations, although rare, are also a risk factor for developing spastic CP (158, 184). Brain malformations have been modeled through administration of a glutamate analog, which results in neuronal brain migration abnormalities in hamsters (78, 129). Despite the presences of these neuronal migration abnormalities, these animals do not appear to develop motor symptoms (78, 129).
Stroke is a well-known risk factor for development of unilateral spastic CP (89, 181). Unilateral spastic CP is a type of CP that is increasing in frequency (89, 181). Stroke has been induced in animals via unilateral carotid artery occlusion, although the animals do not develop motor symptoms (17, 115).
Hypoxic-ischemic encephalopathy is a risk factor for severe forms of CP, including spastic quadriplegic or dyskinetic CP (81). Spastic quadriplegic CP is a form of CP in which an individual has difficulty with movement of arms and legs, and typically also has difficulty with postural or trunk control. Dyskinetic CP may also be associated with impaired movement of all four limbs and the trunk, typically due to other movement difficulties, including dystonia, chorea, and athetosis. These are the most common types of CP to occur in infants born at term gestation (19, 80, 89, 181). Hypoxic-ischemic encephalopathy is modeled in a variety of animal species via bilateral carotid artery occlusion (29, 137, 150, 193) or unilateral carotid artery occlusion/ligation and exposure to hypoxia (21, 95, 97, 145, 146, 167, 183, 188, 191). Despite the extreme reduction in perfusion to the brain in these animal models, motor symptoms were infrequently seen (137, 150). In cats with bilateral carotid artery ligation, delays in visual and auditory orientation accompany delays in standing, walking, and running (137, 150). In a monkey, hypoxic-ischemic encephalopathy was modeled via placental abruption and delayed uterine membrane opening, with the animal exhibiting lethargy, decreased activity, easy startle, and difficulty with maintaining upright posture (164).
Asphyxia is another risk factor of CP that is typically associated with the development of spastic and dyskinetic quadriplegic CP (19, 80). Despite the severity of the motor symptoms in humans with CP, animal models using asphyxia have had minimal, temporary motor symptoms that improve with time (88, 115, 143, 144, 189, 192).
Thus the glaring flaw in most animal models based on recapitulating risk factors for spastic CP is the absence of symptoms that characterize spastic CP.
Animal Models of Spastic Cerebral Palsy Symptoms
As noted previously, impairments in movement or posture are essential for diagnosis of CP, with spasticity being the most common symptom (89, 152, 169, 181). Accordingly, these symptoms should also be critical for modeling this condition successfully in animals.
The spa mouse, originally described in 1961, has symptoms inducible with handling or startle consisting of tip-toe walking, arched back, and difficulty righting (32, 33). In addition, these mice also are smaller than their littermates at maturity and display muscle rigidity, myoclonic jerks, exaggerated startle response, and spasticity (12, 23, 84, 138), a phenotype strikingly similar to symptoms in humans with spastic CP (FIGURE 1). Although not exhaustively chronicled, the phenotypical abnormalities in the spa mice predominantly affect the trunk and limb muscles, particularly the hind limb (35, 212). The spa mouse is not a genetic model of spastic CP but rather a phenotypic model of spastic CP via motor neuron disinhibition. Moreover, although not commonly recognized as an animal model for spastic CP, these mice have been used to model symptoms and interventions in spastic CP (35, 205, 212). In 1994, the genetic basis for these motor symptoms was found to be due to a homozygous insertion of LINE-1 in the beta subunit of the glycine receptor gene, resulting in a splicing error of this subunit (103). We have recently shown that this abnormal neurotransmitter signaling in spa mice results in excessive motor neuron loss, with these mice having fewer motor neurons, which are smaller in size (23). Specifically, spa mice had over 50% reduction in the tibialis anterior motor neurons compared with wild-type mice, with motor neuron somal size reduced. This motor neuron loss disproportionately affected the larger motor neurons compared with wild-type mice (23). Corroborating evidence of reduced numbers of motor neurons with suggestion of increased motor unit size in adult humans with spastic CP has been reported in one small study (128). However, correlation of these findings to impact on physical function is not clear due to the clinical measures used and the large variability in physical abilities of the participants with CP (128).
This early developmental difference in the CNS neurotransmission may underlie the spastic CP phenotype, which notably manifests on clinical exam as muscles with velocity-dependent resistance to stretch, hyperreflexia, motor incoordination, and gait disorder. The embryonic and perinatal periods are a critical time for spinal cord development, specifically developmental motor neuron loss. By the beginning of the third trimester, the motor neuron pool reaches its maximum number of motor neurons (82, 161, 180). Thereafter, the number of motor neurons is reduced into the neonatal period, a highly regulated process dependent on many factors, including motor neuron excitability (101, 116, 127).
Excessive motor neuron excitability, also known as hyperexcitability, is manifest clinically as spasticity due to the motor neuron activity stimulating excessive muscle activation, such as with stretch and reflex testing, a predominant symptom of spastic CP. Motor neuron hyperexcitability is multifactorial (59) and can be seen with changes in motor neuron size (i.e., smaller motor neurons) (85–87), motor neuron morphology (i.e., dendritic branching) (189), and imbalances in neurotransmitter influences (39, 64, 67, 96, 100). Imbalances in neurotransmission include excessive glutamate neurotransmission (depolarizing) and/or loss of GABA or glycine neurotransmission (both hyperpolarizing), both of which cause hyperexcitability. These neurotransmitter perturbations can occur at differing time points in development and impact both the brain and the spinal cord. For example, CNS excitotoxicity via glutamate agonists and alteration of glutamate receptor expression may contribute to susceptibility to in utero brain injury during fetal development (39, 64, 100). CNS excitotoxicity could also occur postnatally via disruption of descending (i.e., brain or corticospinal tract injury) or local (interneuronal) glycine and GABA pathways, resulting in the disinhibition of motor neurons (67, 96). Regardless of their source, reduced inhibitory inputs contribute to motor neuron hyperexcitability, likely due to a net gain in depolarizing inputs and the continued presence of persistent inward calcium and sodium currents (60, 61, 83).
Animal models with developmental neurotransmitter imbalances exist. Spa, spasmodic, and oscillator mice all have glycine receptor mutations, which reduce glycinergic neurotransmission (23, 84, 138, 212). Various glycine receptor subunits are expressed at high levels in discrete brain stem nuclei and more diffusely in both the gray and white matter of the spinal cord, with lower level regions within the forebrain and cerebellum (66, 123, 126, 131, 211). Within the brain stem, motor and auditory neurons have abundant glycine receptors, linked to audition and motor control (123). Within the spinal cord, motor neurons and interneurons involved with motor control (30, 36, 199, 200) and dorsal horn and sensory interneurons (including those associated with nocioception) (75, 94, 124, 190) express glycine receptors (123). Outside the central nervous system, glycine receptors are expressed on spermatozoa, with chloride channel activity associated with the acrosome reaction and fertilization process (24, 174). Indeed, in cases of glycine receptor mutation, fertility is markedly reduced, including in spa mice (13, 175). Homozygous knockout mutant mice display a phenotype similar to cerebral palsy and survive into maturity (23, 84, 138, 212). Gephyrin mice have both glycine and GABA receptor mutations via a deletion of exon 1 of the gephyrin gene and deletion of the upstream sequences responsible for initiating transcription and translation of gephyrin (56). Gephyrin mice have early symptoms suggestive of a severe motor impairment (8, 56, 62, 63, 93, 201). However, they do not survive past P1 due to inability to suck, progressive hyperextended posturing, and progressive apnea (8, 56, 62, 63, 93, 201).
These animal models demonstrate that abnormalities in glycine and GABA synapses impact motor control and physical function. Notably, these animal models have germline mutations, meaning that glycine receptor mutations are present in all cell types, not just motor neurons. In this respect, they are imperfect models; however, since glycine is so highly expressed in the spinal cord and the brain stem, the behavioral abnormalities inherent in mice that have GABA receptor mutations are largely avoided (2, 3, 31, 208). Glycine and GABA are particularly critical neurotransmitters since they are excitatory (depolarizing) in embryonic and early postnatal development, with a transition to their inhibitory (hyperpolarizing) influence later in life (16, 68, 106). Regulation of chloride transport is responsible for this transition from depolarizing to hyperpolarizing actions. Driving this transition is the developmental downregulation of the sodium-potassium-chloride cotransporter (NKCC1) and simultaneous upregulation of the neuron-specific potassium-chloride cotransporter (KCC2) (93, 96, 121, 187), resulting in changes in the intracellular chloride concentration (FIGURE 3). For all motor neurons, extracellular chloride concentration is ~110 mM (16, 157, 179). However intracellular chloride concentration differs based on maturity, with immature neurons being ~30 mM vs. ~5 mM in mature neurons (179, 198). This change in intracellular chloride concentration results in changes in the equilibrium potential of chloride. Specifically, in the embryonic and early postnatal period, the equilibrium potential for chloride is ~20 mV, with maturity being ~75 mV (16, 157, 202). However, the resting membrane potential of the cell is ~65 mV, with the action potential threshold of ~45 mV in development and maturity. Thus, during early developmental periods, activation of GABA and glycine receptors results in chloride efflux, depolarizing the motor neuron (FIGURE 3) (16, 206). As postnatal maturity progresses, the equilibrium potential for chloride becomes more negative than the resting membrane potential, due to upregulation of the KCC2 channel, resulting in chloride contributing to hyperpolarization (inhibition) of the motor neuron when GABA and glycine receptors are activated (FIGURE 3) (16).
FIGURE 3.

Maturation of inhibitory neurotransmission is dependent on chloride channel expression
During development, the opening of ionotropic chloride channels due to GABAergic or glycinergic receptor activation (glycine receptor pentamers with gephyrin clustering shown in orange) leads to chloride efflux (blue arrows) from the neuron, depolarizing the membrane (black bilayer). This is due to high intracellular chloride concentrations (~30 mM) engendered by the abundant expression of NKCC1 chloride transporter (teal) and low expression of the KCC2 chloride transporter (green). The relative expression of these transporters reverses during later postnatal neuronal maturation, such that KCC2 becomes much more abundant and NKCC1 expression is negligible. As a result, internal chloride concentrations in mature neurons is much lower (~5 mM), such that GABAergic or glycinergic receptor activation leads to influx of chloride through ionotropic channels and the hyperpolarization of the membrane. Note that potassium (and sodium in NKCC1) is also co-transported through these channels (yellow arrows).
Animal models with KCC2 mutations have been developed, with rodents homozygous for the KCC2 mutation displaying abnormal in utero and neonatal motor impairments and the inability to breathe (93, 186). Similar to gephyrin mice, mice with KCC2 mutations do not survive (8, 56, 62, 63, 93, 186, 201). Due to such a severe motor phenotype and lethality, the KCC2 and gephyrin mice may be suitable for modeling CP via studying interventions to reduce the severity of the phenotype, as long as this promotes survival. Importantly, these types of studies may provide insight into potential treatments for individuals with spastic CP.
How Do We Target the Gaps in Cerebral Palsy Research?
As discussed, critical gaps in the concept of CP and in animal modeling of CP persist. To truly reflect CP, the model must have an abnormality of movement or posture, such as a change in locomotion, difficulty feeding, or impairment in righting (152, 169). Many of the current animal models are focused on the risk factors for the developmental brain injury due to the long-held dogma about the etiology of CP. Animal models have largely ignored the impaired posture and movement that is essential to the diagnosis of CP or the mechanisms driving the impaired movement and posture.
Animal models of CP must overcome the gaps in CP research, including the long-held misconceptions regarding an isolated brain injury as a driver of the symptoms. Animals for modeling CP must survive without spontaneous neurological recovery to facilitate study of the CNS mechanistic drivers of the phenotype over a lifespan. Animal models that focus on inflicting a developmental brain injury without regard for a postural or movement abnormality are not models of CP, since many infants at risk of CP also have brain abnormalities, although most do not develop CP (90, 203). White matter injury of the brain is a common feature of animal models, although it is neither predictive of CP development nor prognostic of movement disorder severity in CP (114). Brain lesions are neither sensitive nor specific to CP, since not all individuals with CP have abnormal brain imaging (10, 203). Furthermore, the vast majority of our clinical interventions to therapeutically modulate spastic CP symptoms act on the spinal cord, motor neuron, and/or motor unit (133, 163) (FIGURE 2). Thus animal models with a developmental difference of movement regardless of whether they display gross anatomical evidence of a brain injury should be embraced as models of CP and closely evaluated with regard to mechanistic causes of the movement disorder (23, 205, 212).
Recently, the NIH published a strategic plan for CP research (149). One recognized area of focus was the need for multiple animal models to address the complexities of the biology of CP (149). Also noted was that the focus is not only on the brain but also should include the impact of any potential developmental disruption, including genetic/genomic differences, on the neurodevelopment of the spinal cord and musculoskeletal system (149). However, these complex developmental disruptions cannot be studied if appropriate and varied animal models are not recognized, encouraged, available, and used. To study the basic science of CP, a variety of animal models with differing neurodevelopmental insults are needed. It is critical that all models display a phenotype of CP to identify whether the neurodevelopmental insult results in motor impairments. After all, the motor impairment is both essential for diagnosing CP (169) and a critical marker for tracking response to therapeutic interventions.
Conclusions
CP, although being defined as a developmental brain injury, is not clearly or consistently traceable to specific brain lesions. The single commonality of CP is the presence of a developmental disturbance resulting in motor or postural disabilities. Although many animal models of CP have focused on inflicting a brain injury, few have consistently reproduced the motor symptoms of CP, an essential component of the diagnosis. Animal models of CP must display disordered movement or posture. In addition, animal models of CP should be heterogeneous in etiology to include developmental disruptions that affect the entire CNS. Since scientific advancements are identifying new etiologies of CP, including genetic disorders and neurotransmitter imbalances, animal models of CP also need to advance. Animal models of CP also need to go beyond risk factors that are known or presumed to continue to advance the science of not only the etiology of CP but also CP prevention and cure. Critically, the field needs to embrace the original conception of CP as a “cerebro-spinal” disorder, both in our clinical work and in animal models.
Acknowledgments
No conflicts of interest, financial or otherwise, are declared by the author(s).
J.E.B. and M.J.F. prepared figures; J.E.B. drafted manuscript; J.E.B., M.J.F., and G.C.S. edited and revised manuscript; J.E.B., M.J.F., and G.C.S. approved final version of manuscript.
References
- 1.Albright L, Andrews M. Development of the Hypertonia Assessment Tool (HAT). Dev Med Child Neurol 52: 411–412, 2010. doi: 10.1111/j.1469-8749.2009.03477.x. [DOI] [PubMed] [Google Scholar]
- 2.Anstee QM, Knapp S, Maguire EP, Hosie AM, Thomas P, Mortensen M, Bhome R, Martinez A, Walker SE, Dixon CI, Ruparelia K, Montagnese S, Kuo YT, Herlihy A, Bell JD, Robinson I, Guerrini I, McQuillin A, Fisher EM, Ungless MA, Gurling HM, Morgan MY, Brown SD, Stephens DN, Belelli D, Lambert JJ, Smart TG, Thomas HC. Mutations in the Gabrb1 gene promote alcohol consumption through increased tonic inhibition. Nat Commun 4: 2816, 2013. doi: 10.1038/ncomms3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arain FM, Boyd KL, Gallagher MJ. Decreased viability and absence-like epilepsy in mice lacking or deficient in the GABAA receptor α1 subunit. Epilepsia 53: e161–e165, 2012. doi: 10.1111/j.1528-1167.2012.03596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arneson CL, Durkin MS, Benedict RE, Kirby RS, Yeargin-Allsopp M, Van Naarden Braun K, Doernberg NS. Prevalence of cerebral palsy: Autism and Developmental Disabilities Monitoring Network, three sites, United States, 2004. Disabil Health J 2: 45–48, 2009. doi: 10.1016/j.dhjo.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Back SA. Brain injury in the preterm infant: new horizons for pathogenesis and prevention. Pediatr Neurol 53: 185–192, 2015. doi: 10.1016/j.pediatrneurol.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Back SA. Cerebral white and gray matter injury in newborns: new insights into pathophysiology and management. Clin Perinatol 41: 1–24, 2014. doi: 10.1016/j.clp.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Back SA, Miller SP. Brain injury in premature neonates: A primary cerebral dysmaturation disorder? Ann Neurol 75: 469–486, 2014. doi: 10.1002/ana.24132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banks GB, Kanjhan R, Wiese S, Kneussel M, Wong LM, O’Sullivan G, Sendtner M, Bellingham MC, Betz H, Noakes PG. Glycinergic and GABAergic synaptic activity differentially regulate motoneuron survival and skeletal muscle innervation. J Neurosci 25: 1249–1259, 2005. doi: 10.1523/JNEUROSCI.1786-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barry MJ, VanSwearingen JM, Albright AL. Reliability and responsiveness of the Barry-Albright Dystonia Scale. Dev Med Child Neurol 41: 404–411, 1999. doi: 10.1017/S0012162299000870. [DOI] [PubMed] [Google Scholar]
- 10.Bax M, Tydeman C, Flodmark O. Clinical and MRI correlates of cerebral palsy: the European Cerebral Palsy Study. JAMA 296: 1602–1608, 2006. doi: 10.1001/jama.296.13.1602. [DOI] [PubMed] [Google Scholar]
- 11.Bax MC. Terminology and classification of cerebral palsy. Dev Med Child Neurol 6: 295–297, 1964. doi: 10.1111/j.1469-8749.1964.tb10791.x. [DOI] [PubMed] [Google Scholar]
- 12.Becker CM, Hermans-Borgmeyer I, Schmitt B, Betz H. The glycine receptor deficiency of the mutant mouse spastic: evidence for normal glycine receptor structure and localization. J Neurosci 6: 1358–1364, 1986. doi: 10.1523/JNEUROSCI.06-05-01358.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Becker L, Hartenstein B, Schenkel J, Kuhse J, Betz H, Weiher H. Transient neuromotor phenotype in transgenic spastic mice expressing low levels of glycine receptor beta-subunit: an animal model of startle disease. Eur J Neurosci 12: 27–32, 2000. doi: 10.1046/j.1460-9568.2000.00877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bell MJ, Hallenbeck JM. Effects of intrauterine inflammation on developing rat brain. J Neurosci Res 70: 570–579, 2002. doi: 10.1002/jnr.10423. [DOI] [PubMed] [Google Scholar]
- 15.Beloosesky R, Weiner Z, Ginsberg Y, Ross MG. Maternal N-acetyl-cysteine (NAC) protects the rat fetal brain from inflammatory cytokine responses to lipopolysaccharide (LPS). J Matern Fetal Neonatal Med 25: 1324–1328, 2012. doi: 10.3109/14767058.2011.632793. [DOI] [PubMed] [Google Scholar]
- 16.Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci 3: 728–739, 2002. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- 17.Benjelloun N, Renolleau S, Represa A, Ben-Ari Y, and Charriaut-Marlangue C. Inflammatory responses in the cerebral cortex after ischemia in the P7 neonatal rat. Stroke 30: 1916–1923; discussion 1923–1914, 1999. [DOI] [PubMed] [Google Scholar]
- 18.Bernards CM. Cerebrospinal fluid and spinal cord distribution of baclofen and bupivacaine during slow intrathecal infusion in pigs. Anesthesiology 105: 169–178, 2006. doi: 10.1097/00000542-200607000-00027. [DOI] [PubMed] [Google Scholar]
- 19.Blair E. Trends in cerebral palsy. Indian J Pediatr 68: 433–438, 2001. doi: 10.1007/BF02723024. [DOI] [PubMed] [Google Scholar]
- 20.Blickstein I. Cerebral palsy in multifoetal pregnancies. Dev Med Child Neurol 44: 352–355, 2002. doi: 10.1111/j.1469-8749.2002.tb00823.x. [DOI] [PubMed] [Google Scholar]
- 21.Bona E, Andersson AL, Blomgren K, Gilland E, Puka-Sundvall M, Gustafson K, Hagberg H. Chemokine and inflammatory cell response to hypoxia-ischemia in immature rats. Pediatr Res 45: 500–509, 1999. doi: 10.1203/00006450-199904010-00008. [DOI] [PubMed] [Google Scholar]
- 22.Bottcher L. Children with spastic cerebral palsy, their cognitive functioning, and social participation: a review. Child Neuropsychol 16: 209–228, 2010. doi: 10.1080/09297040903559630. [DOI] [PubMed] [Google Scholar]
- 23.Brandenburg JE, Gransee HM, Fogarty MJ, Sieck GC. Differences in lumbar motor neuron pruning in an animal model of early onset spasticity. J Neurophysiol 120: 601–609, 2018. doi: 10.1152/jn.00186.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bray C, Son JH, Kumar P, Harris JD, Meizel S. A role for the human sperm glycine receptor/Cl(-) channel in the acrosome reaction initiated by recombinant ZP3. Biol Reprod 66: 91–97, 2002. doi: 10.1095/biolreprod66.1.91. [DOI] [PubMed] [Google Scholar]
- 25.Brogden RN, Speight TM, Avery GS. Baclofen: a preliminary report of its pharmacological properties and therapeutic efficacy in spasticity. Drugs 8: 1–14, 1974. doi: 10.2165/00003495-197408010-00001. [DOI] [PubMed] [Google Scholar]
- 26.Burd I, Bentz AI, Chai J, Gonzalez J, Monnerie H, Le Roux PD, Cohen AS, Yudkoff M, Elovitz MA. Inflammation-induced preterm birth alters neuronal morphology in the mouse fetal brain. J Neurosci Res 88: 1872–1881, 2010. doi: 10.1002/jnr.22368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cahan LD, Kundi MS, McPherson D, Starr A, Peacock W. Electrophysiologic studies in selective dorsal rhizotomy for spasticity in children with cerebral palsy. Appl Neurophysiol 50: 459–462, 1987. [DOI] [PubMed] [Google Scholar]
- 28.Cai Z, Pan Z-L, Pang Y, Evans OB, Rhodes PG. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr Res 47: 64–72, 2000. doi: 10.1203/00006450-200001000-00013. [DOI] [PubMed] [Google Scholar]
- 29.Cai Z, Pang Y, Xiao F, Rhodes PG. Chronic ischemia preferentially causes white matter injury in the neonatal rat brain. Brain Res 898: 126–135, 2001. doi: 10.1016/S0006-8993(01)02180-1. [DOI] [PubMed] [Google Scholar]
- 30.Callister RJ, Graham BA. Early history of glycine receptor biology in Mammalian spinal cord circuits. Front Mol Neurosci 3: 13, 2010. doi: 10.3389/fnmol.2010.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cellot G, Cherubini E. GABAergic signaling as therapeutic target for autism spectrum disorders. Front Pediatr 2: 70, 2014. doi: 10.3389/fped.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chai CK. Hereditary spasticity In mice. J Hered 52: 241–243, 1961. doi: 10.1093/oxfordjournals.jhered.a107083. [DOI] [Google Scholar]
- 33.Chai CK, Roberts E, Sidman RL. Influence of aminooxyacetic acid, a γ-aminobutyrate transaminase inhibitor, on hereditary spastic defect in the mouse. Proc Soc Exp Biol Med 109: 491–495, 1962. doi: 10.3181/00379727-109-27245. [DOI] [PubMed] [Google Scholar]
- 34.Clancy B, Finlay BL, Darlington RB, Anand KJ. Extrapolating brain development from experimental species to humans. Neurotoxicology 28: 931–937, 2007. doi: 10.1016/j.neuro.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cosgrove AP, Graham HK. Botulinum toxin A prevents the development of contractures in the hereditary spastic mouse. Dev Med Child Neurol 36: 379–385, 1994. doi: 10.1111/j.1469-8749.1994.tb11863.x. [DOI] [PubMed] [Google Scholar]
- 36.Curtis DR, Hösli L, Johnston GA. Inhibition of spinal neurons by glycine. Nature 215: 1502–1503, 1967. doi: 10.1038/2151502a0. [DOI] [PubMed] [Google Scholar]
- 37.Dalitz P, Harding R, Rees SM, Cock ML. Prolonged reductions in placental blood flow and cerebral oxygen delivery in preterm fetal sheep exposed to endotoxin: possible factors in white matter injury after acute infection. J Soc Gynecol Investig 10: 283–290, 2003. [DOI] [PubMed] [Google Scholar]
- 38.Debillon T, Gras-Leguen C, Vérielle V, Winer N, Caillon J, Rozé JC, Gressens P. Intrauterine infection induces programmed cell death in rabbit periventricular white matter. Pediatr Res 47: 736–742, 2000. doi: 10.1203/00006450-200006000-00009. [DOI] [PubMed] [Google Scholar]
- 39.Degos V, Peineau S, Nijboer C, Kaindl AM, Sigaut S, Favrais G, Plaisant F, Teissier N, Gouadon E, Lombet A, Saliba E, Collingridge GL, Maze M, Nicoletti F, Heijnen C, Mantz J, Kavelaars A, Gressens P. G protein-coupled receptor kinase 2 and group I metabotropic glutamate receptors mediate inflammation-induced sensitization to excitotoxic neurodegeneration. Ann Neurol 73: 667–678, 2013. doi: 10.1002/ana.23868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Derrick M, Drobyshevsky A, Ji X, Chen L, Yang Y, Ji H, Silverman RB, Tan S. Hypoxia-ischemia causes persistent movement deficits in a perinatal rabbit model of cerebral palsy: assessed by a new swim test. Int J Dev Neurosci 27: 549–557, 2009. doi: 10.1016/j.ijdevneu.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Derrick M, Drobyshevsky A, Ji X, Tan S. A model of cerebral palsy from fetal hypoxia-ischemia. Stroke 38, Suppl: 731–735, 2007. doi: 10.1161/01.STR.0000251445.94697.64. [DOI] [PubMed] [Google Scholar]
- 42.Derrick M, Luo NL, Bregman JC, Jilling T, Ji X, Fisher K, Gladson CL, Beardsley DJ, Murdoch G, Back SA, Tan S. Preterm fetal hypoxia-ischemia causes hypertonia and motor deficits in the neonatal rabbit: a model for human cerebral palsy? J Neurosci 24: 24–34, 2004. doi: 10.1523/JNEUROSCI.2816-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dombroski RA, Woodard DS, Harper MJ, Gibbs RS. A rabbit model for bacteria-induced preterm pregnancy loss. Am J Obstet Gynecol 163: 1938–1943, 1990. doi: 10.1016/0002-9378(90)90777-5. [DOI] [PubMed] [Google Scholar]
- 44.Driscoll SW, Skinner J. Musculoskeletal complications of neuromuscular disease in children. Phys Med Rehabil Clin N Am 19: 163–194, 2008. doi: 10.1016/j.pmr.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 45.Drobyshevsky A, Cotten CM, Shi Z, Luo K, Jiang R, Derrick M, Tracy ET, Gentry T, Goldberg RN, Kurtzberg J, Tan S. Human umbilical cord blood cells ameliorate motor deficits in rabbits in a cerebral palsy model. Dev Neurosci 37: 349–362, 2015. doi: 10.1159/000374107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drobyshevsky A, Luo K, Derrick M, Yu L, Du H, Prasad PV, Vasquez-Vivar J, Batinic-Haberle I, Tan S. Motor deficits are triggered by reperfusion-reoxygenation injury as diagnosed by MRI and by a mechanism involving oxidants. J Neurosci 32: 5500–5509, 2012. doi: 10.1523/JNEUROSCI.5986-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dudley DJ, Branch DW, Edwin SS, Mitchell MD. Induction of preterm birth in mice by RU486. Biol Reprod 55: 992–995, 1996. doi: 10.1095/biolreprod55.5.992. [DOI] [PubMed] [Google Scholar]
- 48.Duncan JR, Cock ML, Scheerlinck J-PY, Westcott KT, McLean C, Harding R, Rees SM. White matter injury after repeated endotoxin exposure in the preterm ovine fetus. Pediatr Res 52: 941–949, 2002. doi: 10.1203/00006450-200212000-00021. [DOI] [PubMed] [Google Scholar]
- 49.Dunn PM. Dr William Little (1810-1894) of London and cerebral palsy. Arch Dis Child Fetal Neonatal Ed 72: F209–F210, 1995. doi: 10.1136/fn.72.3.F209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ebrahimi S, Esfahani SA, Kohkiloezadeh M, Moghaddam BH, Askarian S, Tanideh N, Tamadon A. A model of cerebral ischemia induction in neonatal rabbits. J Appl Anim Res 40: 37–42, 2012. doi: 10.1080/09712119.2011.627134. [DOI] [Google Scholar]
- 51.Elovitz MA, Mrinalini C. Animal models of preterm birth. Trends Endocrinol Metab 15: 479–487, 2004. doi: 10.1016/j.tem.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 52.Elovitz MA, Wang Z, Chien EK, Rychlik DF, Phillippe M. A new model for inflammation-induced preterm birth: the role of platelet-activating factor and Toll-like receptor-4. Am J Pathol 163: 2103–2111, 2003. doi: 10.1016/S0002-9440(10)63567-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eunson P. Aetiology and epidemiology of cerebral palsy. Paediatr Child Health 26: 367–372, 2016. doi: 10.1016/j.paed.2016.04.011. [DOI] [Google Scholar]
- 54.Fatemi A, Wilson MA, Johnston MV. Hypoxic-ischemic encephalopathy in the term infant. Clin Perinatol 36: 835–858, 2009. doi: 10.1016/j.clp.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fehlings D, Switzer L, Agarwal P, Wong C, Sochett E, Stevenson R, Sonnenberg L, Smile S, Young E, Huber J, Milo-Manson G, Kuwaik GA, Gaebler D. Informing evidence-based clinical practice guidelines for children with cerebral palsy at risk of osteoporosis: a systematic review. Dev Med Child Neurol 54: 106–116, 2012. doi: 10.1111/j.1469-8749.2011.04091.x. [DOI] [PubMed] [Google Scholar]
- 56.Feng G, Tintrup H, Kirsch J, Nichol MC, Kuhse J, Betz H, Sanes JR. Dual requirement for gephyrin in glycine receptor clustering and molybdoenzyme activity. Science 282: 1321–1324, 1998. doi: 10.1126/science.282.5392.1321. [DOI] [PubMed] [Google Scholar]
- 57.Ferriero DM. Neonatal brain injury. N Engl J Med 351: 1985–1995, 2004. doi: 10.1056/NEJMra041996. [DOI] [PubMed] [Google Scholar]
- 58.Field NT, Newton ER, Kagan-Hallet K, Peairs WA. Perinatal effects of Gardnerella vaginalis deciduitis in the rabbit. Am J Obstet Gynecol 168: 988–994, 1993. doi: 10.1016/S0002-9378(12)90858-3. [DOI] [PubMed] [Google Scholar]
- 59.Fogarty MJ. Driven to decay: Excitability and synaptic abnormalities in amyotrophic lateral sclerosis. Brain Res Bull 140: 318–333, 2018. doi: 10.1016/j.brainresbull.2018.05.023 . A corrigendum for this article is available at http://dx.doi.org/10.1016/j.brainresbull.2019.02.007. [DOI] [PubMed] [Google Scholar]
- 60.Fogarty MJ, Kanjhan R, Bellingham MC, Noakes PG. Glycinergic Neurotransmission: A Potent Regulator of Embryonic Motor Neuron Dendritic Morphology and Synaptic Plasticity. J Neurosci 36: 80–87, 2016. doi: 10.1523/JNEUROSCI.1576-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fogarty MJ, Kanjhan R, Yanagawa Y, Noakes PG, Bellingham MC. Alterations in hypoglossal motor neurons due to GAD67 and VGAT deficiency in mice. Exp Neurol 289: 117–127, 2017. doi: 10.1016/j.expneurol.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 62.Fogarty MJ, Smallcombe KL, Yanagawa Y, Obata K, Bellingham MC, Noakes PG. Genetic deficiency of GABA differentially regulates respiratory and non-respiratory motor neuron development. PLoS One 8: e56257, 2013. doi: 10.1371/journal.pone.0056257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fogarty MJ, Yanagawa Y, Obata K, Bellingham MC, Noakes PG. Genetic absence of the vesicular inhibitory amino acid transporter differentially regulates respiratory and locomotor motor neuron development. Brain Struct Funct 220: 525–540, 2015. doi: 10.1007/s00429-013-0673-9. [DOI] [PubMed] [Google Scholar]
- 64.Fontaine RH, Olivier P, Massonneau V, Leroux P, Degos V, Lebon S, El Ghouzzi V, Lelièvre V, Gressens P, Baud O. . Vulnerability of white matter towards antenatal hypoxia is linked to a species-dependent regulation of glutamate receptor subunits. Proc Natl Acad Sci USA 105: 16779–16784, 2008. doi: 10.1073/pnas.0803004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Freud S. Les diplegies cerebrales infantiles. Rev Neurol (Paris) 1: 178–183, 1893. [Google Scholar]
- 66.Frostholm A, Rotter A. Glycine receptor distribution in mouse CNS: autoradiographic localization of [3H]strychnine binding sites. Brain Res Bull 15: 473–486, 1985. doi: 10.1016/0361-9230(85)90038-3. [DOI] [PubMed] [Google Scholar]
- 67.Gackière F, Vinay L. Contribution of the potassium-chloride cotransporter KCC2 to the strength of inhibition in the neonatal rodent spinal cord in vitro. J Neurosci 35: 5307–5316, 2015. doi: 10.1523/JNEUROSCI.1674-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao BX, Ziskind-Conhaim L. Development of glycine- and GABA-gated currents in rat spinal motoneurons. J Neurophysiol 74: 113–121, 1995. doi: 10.1152/jn.1995.74.1.113. [DOI] [PubMed] [Google Scholar]
- 69.Garfield RE, Gasc JM, Baulieu EE. Effects of the antiprogesterone RU 486 on preterm birth in the rat. Am J Obstet Gynecol 157: 1281–1285, 1987. doi: 10.1016/S0002-9378(87)80315-0. [DOI] [PubMed] [Google Scholar]
- 70.Garne E, Dolk H, Krägeloh-Mann I, Holst Ravn S, Cans C; SCPE Collaborative Group . Cerebral palsy and congenital malformations. Eur J Paediatr Neurol 12: 82–88, 2008. doi: 10.1016/j.ejpn.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 71.Gayle DA, Beloosesky R, Desai M, Amidi F, Nuñez SE, Ross MG. Maternal LPS induces cytokines in the amniotic fluid and corticotropin releasing hormone in the fetal rat brain. Am J Physiol Regul Integr Comp Physiol 286: R1024–R1029, 2004. doi: 10.1152/ajpregu.00664.2003. [DOI] [PubMed] [Google Scholar]
- 72.Germany L, Ehlinger V, Klapouszczak D, Delobel M, Hollódy K, Sellier E, De La Cruz J, Alberge C, Genolini C, Arnaud C. Trends in prevalence and characteristics of post-neonatal cerebral palsy cases: a European registry-based study. Res Dev Disabil 34: 1669–1677, 2013. doi: 10.1016/j.ridd.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 73.Ginsberg Y, Khatib N, Weiner Z, Beloosesky R. Maternal Inflammation, Fetal Brain Implications and Suggested Neuroprotection: A Summary of 10 Years of Research in Animal Models. Rambam Maimonides Med J 8: e0028, 2017. doi: 10.5041/RMMJ.10305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gracies JM, Burke K, Clegg NJ, Browne R, Rushing C, Fehlings D, Matthews D, Tilton A, Delgado MR. Reliability of the Tardieu Scale for assessing spasticity in children with cerebral palsy. Arch Phys Med Rehabil 91: 421–428, 2010. doi: 10.1016/j.apmr.2009.11.017. [DOI] [PubMed] [Google Scholar]
- 75.Gradwell MA, Boyle KA, Callister RJ, Hughes DI, Graham BA. Heteromeric α/β glycine receptors regulate excitability in parvalbumin-expressing dorsal horn neurons through phasic and tonic glycinergic inhibition. J Physiol 595: 7185–7202, 2017. doi: 10.1113/JP274926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Graham EM, Ruis KA, Hartman AL, Northington FJ, Fox HE. A systematic review of the role of intrapartum hypoxia-ischemia in the causation of neonatal encephalopathy. Am J Obstet Gynecol 199: 587–595, 2008. doi: 10.1016/j.ajog.2008.06.094. [DOI] [PubMed] [Google Scholar]
- 77.Granild-Jensen JB, Rackauskaite G, Flachs EM, Uldall P. Predictors for early diagnosis of cerebral palsy from national registry data. Dev Med Child Neurol 57: 931–935, 2015. doi: 10.1111/dmcn.12760. [DOI] [PubMed] [Google Scholar]
- 78.Gressens P, Arquié C, Hill JM, Marret S, Sahir N, Robberecht P, Evrard P. VIP and PACAP 38 modulate ibotenate-induced neuronal heterotopias in the newborn hamster neocortex. J Neuropathol Exp Neurol 59: 1051–1062, 2000. doi: 10.1093/jnen/59.12.1051. [DOI] [PubMed] [Google Scholar]
- 79.Grether JK, Nelson KB. Maternal infection and cerebral palsy in infants of normal birth weight. JAMA 278: 207–211, 1997. doi: 10.1001/jama.1997.03550030047032. [DOI] [PubMed] [Google Scholar]
- 80.Hagberg B, Hagberg G, Beckung E, Uvebrant P. Changing panorama of cerebral palsy in Sweden. VIII. Prevalence and origin in the birth year period 1991-94. Acta Paediatr 90: 271–277, 2001. doi: 10.1080/080352501300067532. [DOI] [PubMed] [Google Scholar]
- 81.Hankins GDV, Speer M. Defining the pathogenesis and pathophysiology of neonatal encephalopathy and cerebral palsy. Obstet Gynecol 102: 628–636, 2003. [DOI] [PubMed] [Google Scholar]
- 82.Harris AJ, McCaig CD. Motoneuron death and motor unit size during embryonic development of the rat. J Neurosci 4: 13–24, 1984. doi: 10.1523/JNEUROSCI.04-01-00013.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Heckmann CJ, Gorassini MA, Bennett DJ. Persistent inward currents in motoneuron dendrites: implications for motor output. Muscle Nerve 31: 135–156, 2005. doi: 10.1002/mus.20261. [DOI] [PubMed] [Google Scholar]
- 84.Heller AH, Hallett M. Electrophysiological studies with the spastic mutant mouse. Brain Res 234: 299–308, 1982. doi: 10.1016/0006-8993(82)90870-8. [DOI] [PubMed] [Google Scholar]
- 85.Henneman E. Relation between size of neurons and their susceptibility to discharge. Science 126: 1345–1347, 1957. doi: 10.1126/science.126.3287.1345. [DOI] [PubMed] [Google Scholar]
- 86.Henneman E, Somjen G, Carpenter DO. Excitability and inhibitability of motoneurons of different sizes. J Neurophysiol 28: 599–620, 1965. doi: 10.1152/jn.1965.28.3.599. [DOI] [PubMed] [Google Scholar]
- 87.Henneman E, Somjen G, Carpenter DO. Functional significance of cell size in spinal motoneurons. J Neurophysiol 28: 560–580, 1965. doi: 10.1152/jn.1965.28.3.560. [DOI] [PubMed] [Google Scholar]
- 88.Hicks SP, Cavanaugh MC, O’Brien ED. Effects of anoxia on the developing cerebral cortex in the rat. Am J Pathol 40: 615–635, 1962. [PMC free article] [PubMed] [Google Scholar]
- 89.Himmelmann K, Hagberg G, Beckung E, Hagberg B, Uvebrant P. The changing panorama of cerebral palsy in Sweden. IX. Prevalence and origin in the birth-year period 1995-1998. Acta Paediatr 94: 287–294, 2005. doi: 10.1111/j.1651-2227.2005.tb03071.x. [DOI] [PubMed] [Google Scholar]
- 90.Himpens E, Oostra A, Franki I, Vansteelandt S, Vanhaesebrouck P, den Broeck CV. Predictability of cerebral palsy in a high-risk NICU population. Early Hum Dev 86: 413–417, 2010. doi: 10.1016/j.earlhumdev.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 91.Hohimer AR, Chao CR, Bissonnette JM. The effect of combined hypoxemia and cephalic hypotension on fetal cerebral blood flow and metabolism. J Cereb Blood Flow Metab 11: 99–105, 1991. doi: 10.1038/jcbfm.1991.11. [DOI] [PubMed] [Google Scholar]
- 92.Hubermann L, Boychuck Z, Shevell M, Majnemer A. Age at referral of children for initial diagnosis of cerebral palsy and rehabilitation: current practices. J Child Neurol 31: 364–369, 2016. doi: 10.1177/0883073815596610. [DOI] [PubMed] [Google Scholar]
- 93.Hübner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 30: 515–524, 2001. doi: 10.1016/S0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- 94.Imlach WL, Bhola RF, Mohammadi SA, Christie MJ. Glycinergic dysfunction in a subpopulation of dorsal horn interneurons in a rat model of neuropathic pain. Sci Rep 6: 37104, 2016. doi: 10.1038/srep37104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ivacko JA, Sun R, Silverstein FS. Hypoxic-ischemic brain injury induces an acute microglial reaction in perinatal rats. Pediatr Res 39: 39–47, 1996. doi: 10.1203/00006450-199601000-00006. [DOI] [PubMed] [Google Scholar]
- 96.Jean-Xavier C, Pflieger J-F, Liabeuf S, Vinay L. Inhibitory postsynaptic potentials in lumbar motoneurons remain depolarizing after neonatal spinal cord transection in the rat. J Neurophysiol 96: 2274–2281, 2006. doi: 10.1152/jn.00328.2006. [DOI] [PubMed] [Google Scholar]
- 97.Jelinski SE, Yager JY, Juurlink BH. Preferential injury of oligodendroblasts by a short hypoxic-ischemic insult. Brain Res 815: 150–153, 1999. doi: 10.1016/S0006-8993(98)01053-1. [DOI] [PubMed] [Google Scholar]
- 98.Jethwa A, Mink J, Macarthur C, Knights S, Fehlings T, Fehlings D. Development of the Hypertonia Assessment Tool (HAT): a discriminative tool for hypertonia in children. Dev Med Child Neurol 52: e83–e87, 2010. doi: 10.1111/j.1469-8749.2009.03483.x. [DOI] [PubMed] [Google Scholar]
- 99.Johnston MV, Ferriero DM, Vannucci SJ, Hagberg H. Models of cerebral palsy: which ones are best? J Child Neurol 20: 984–987, 2005. doi: 10.1177/08830738050200121001. [DOI] [PubMed] [Google Scholar]
- 100.Kaindl AM, Degos V, Peineau S, Gouadon E, Chhor V, Loron G, Le Charpentier T, Josserand J, Ali C, Vivien D, Collingridge GL, Lombet A, Issa L, Rene F, Loeffler J-P, Kavelaars A, Verney C, Mantz J, Gressens P. Activation of microglial N-methyl-D-aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann Neurol 72: 536–549, 2012. doi: 10.1002/ana.23626. [DOI] [PubMed] [Google Scholar]
- 101.Kanning KC, Kaplan A, Henderson CE. Motor neuron diversity in development and disease. Annu Rev Neurosci 33: 409–440, 2010. doi: 10.1146/annurev.neuro.051508.135722. [DOI] [PubMed] [Google Scholar]
- 102.Katz RT, Rymer WZ. Spastic hypertonia: mechanisms and measurement. Arch Phys Med Rehabil 70: 144–155, 1989. [PubMed] [Google Scholar]
- 103.Kingsmore SF, Giros B, Suh D, Bieniarz M, Caron MG, Seldin MF. Glycine receptor β-subunit gene mutation in spastic mouse associated with LINE-1 element insertion. Nat Genet 7: 136–141, 1994. doi: 10.1038/ng0694-136. [DOI] [PubMed] [Google Scholar]
- 104.Knights S, Datoo N, Kawamura A, Switzer L, Fehlings D. Further evaluation of the scoring, reliability, and validity of the Hypertonia Assessment Tool (HAT). J Child Neurol 29: 500–504, 2014. doi: 10.1177/0883073813483903. [DOI] [PubMed] [Google Scholar]
- 105.Krägeloh-Mann I, Horber V. The role of magnetic resonance imaging in elucidating the pathogenesis of cerebral palsy: a systematic review. Dev Med Child Neurol 49: 144–151, 2007. doi: 10.1111/j.1469-8749.2007.00144.x. [DOI] [PubMed] [Google Scholar]
- 106.Kriegstein AR, Owens DF. GABA may act as a self-limiting trophic factor at developing synapses. Sci STKE 2001: pe1, 2001. [DOI] [PubMed] [Google Scholar]
- 107.Kułak W, Okurowska-Zawada B, Gościk E, Sienkiewicz D, Paszko-Patej G, Kubas B. Schizencephaly as a cause of spastic cerebral palsy. Adv Med Sci 56: 64–70, 2011. doi: 10.2478/v10039-011-0006-2. [DOI] [PubMed] [Google Scholar]
- 108.Kułak W, Sobaniec W, Gościk M, Oleński J, Okurowska-Zawada B. Clinical and neuroimaging profile of congenital brain malformations in children with spastic cerebral palsy. Adv Med Sci 53: 42–48, 2008. doi: 10.2478/v10039-008-0006-z. [DOI] [PubMed] [Google Scholar]
- 109.Kumarasamy V, Mitchell MD, Bloomfield FH, Oliver MH, Campbell ME, Challis JR, Harding JE. Effects of periconceptional undernutrition on the initiation of parturition in sheep. Am J Physiol Regul Integr Comp Physiol 288: R67–R72, 2005. doi: 10.1152/ajpregu.00357.2004. [DOI] [PubMed] [Google Scholar]
- 110.Kurinczuk JJ, White-Koning M, Badawi N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum Dev 86: 329–338, 2010. doi: 10.1016/j.earlhumdev.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 111.Kwon YH, Lee HY. Differences of respiratory function in children with spastic diplegic and hemiplegic cerebral palsy, compared with normally developed children. J Pediatr Rehabil Med 6: 113–117, 2013. [DOI] [PubMed] [Google Scholar]
- 112.Lance JW. Pathophysiology of spasticity and clinical experience with baclofen. In: Spasticity:Disordered Motor Control, edited by Feldman RG, Young RR, Koella WP. Chicago, IL: Year Book Medical Publishers, 1980, p. 185–220. [Google Scholar]
- 113.Laplaza FJ, Root L, Tassanawipas A, Cervera P. Cerebral palsy in twins. Dev Med Child Neurol 34: 1053–1063, 1992. doi: 10.1111/j.1469-8749.1992.tb11417.x. [DOI] [PubMed] [Google Scholar]
- 114.Lee JD, Park HJ, Park ES, Oh MK, Park B, Rha DW, Cho SR, Kim EY, Park JY, Kim CH, Kim DG, Park CI. Motor pathway injury in patients with periventricular leucomalacia and spastic diplegia. Brain 134: 1199–1210, 2011. doi: 10.1093/brain/awr021. [DOI] [PubMed] [Google Scholar]
- 115.Levine S. Anoxic-ischemic encephalopathy in rats. Am J Pathol 36: 1–17, 1960. [PMC free article] [PubMed] [Google Scholar]
- 116.Lin S, Landmann L, Ruegg MA, Brenner HR. The role of nerve- versus muscle-derived factors in mammalian neuromuscular junction formation. J Neurosci 28: 3333–3340, 2008. doi: 10.1523/JNEUROSCI.5590-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lisi EC, Cohn RD. Genetic evaluation of the pediatric patient with hypotonia: perspective from a hypotonia specialty clinic and review of the literature. Dev Med Child Neurol 53: 586–599, 2011. doi: 10.1111/j.1469-8749.2011.03918.x. [DOI] [PubMed] [Google Scholar]
- 118.Little WJ. On the influence of abnormal parturition, difficult labor, premature birth and asphyxia neonatorum on the mental and physical condition of the child, especially in relation to deformities. Trans Obstetrical Soc Lond 3: 243–344, 1862. [PubMed] [Google Scholar]
- 119.Little WJ. On the influence of abnormal parturition, difficult labours, premature birth, and asphyxia neonatorum, on the mental and physical condition of the child, especially in relation to deformities. Clin Orthop Relat Res 46: 7–22, 1966. [PubMed] [Google Scholar]
- 120.Liverman CS, Kaftan HA, Cui L, Hersperger SG, Taboada E, Klein RM, Berman NEJ. Altered expression of pro-inflammatory and developmental genes in the fetal brain in a mouse model of maternal infection. Neurosci Lett 399: 220–225, 2006. doi: 10.1016/j.neulet.2006.01.064. [DOI] [PubMed] [Google Scholar]
- 121.Lu J, Karadsheh M, Delpire E. Developmental regulation of the neuronal-specific isoform of K-Cl cotransporter KCC2 in postnatal rat brains. J Neurobiol 39: 558–568, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 122.Lynch JK, Hirtz DG, DeVeber G, Nelson KB. Report of the National Institute of Neurological Disorders and Stroke workshop on perinatal and childhood stroke. Pediatrics 109: 116–123, 2002. doi: 10.1542/peds.109.1.116. [DOI] [PubMed] [Google Scholar]
- 123.Lynch JW. Molecular structure and function of the glycine receptor chloride channel. Physiol Rev 84: 1051–1095, 2004. doi: 10.1152/physrev.00042.2003. [DOI] [PubMed] [Google Scholar]
- 124.Lynch JW, Callister RJ. Glycine receptors: a new therapeutic target in pain pathways. Curr Opin Investig Drugs 7: 48–53, 2006. [PubMed] [Google Scholar]
- 125.MacKeith R, Polani P. The Little Club: Memorandum on terminology and classification of cerebral palsy. Cerebral Palsy Bull 5: 27–35, 1959. [Google Scholar]
- 126.Malosio ML, Marquèze-Pouey B, Kuhse J, Betz H. Widespread expression of glycine receptor subunit mRNAs in the adult and developing rat brain. EMBO J 10: 2401–2409, 1991. doi: 10.1002/j.1460-2075.1991.tb07779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mantilla CB, Sieck GC. Trophic factor expression in phrenic motor neurons. Respir Physiol Neurobiol 164: 252–262, 2008. doi: 10.1016/j.resp.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Marciniak C, Li X, Zhou P. An examination of motor unit number index in adults with cerebral palsy. J Electromyogr Kinesiol 25: 444–450, 2015. doi: 10.1016/j.jelekin.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 129.Marret S, Gressens P, Evrard P. Arrest of neuronal migration by excitatory amino acids in hamster developing brain. Proc Natl Acad Sci USA 93: 15463–15468, 1996. doi: 10.1073/pnas.93.26.15463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.McClure MM, Riddle A, Manese M, Luo NL, Rorvik DA, Kelly KA, Barlow CH, Kelly JJ, Vinecore K, Roberts CT, Hohimer AR, Back SA. Cerebral blood flow heterogeneity in preterm sheep: lack of physiologic support for vascular boundary zones in fetal cerebral white matter. J Cereb Blood Flow Metab 28: 995–1008, 2008. doi: 10.1038/sj.jcbfm.9600597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.McCracken LM, Lowes DC, Salling MC, Carreau-Vollmer C, Odean NN, Blednov YA, Betz H, Harris RA, Harrison NL. Glycine receptor α3 and α2 subunits mediate tonic and exogenous agonist-induced currents in forebrain. Proc Natl Acad Sci USA 114: E7179–E7186, 2017. doi: 10.1073/pnas.1703839114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.McGuire J, Rymer WZ. Spasticity: mechanisms and management. In: Medical Management of Long-Term Disability, edited by Green D. Oxford, UK: Butterworth-Heinemann, 1996. [Google Scholar]
- 133.McLaughlin J, Bjornson K, Temkin N, Steinbok P, Wright V, Reiner A, Roberts T, Drake J, O’Donnell M, Rosenbaum P, Barber J, Ferrel A. Selective dorsal rhizotomy: meta-analysis of three randomized controlled trials. Dev Med Child Neurol 44: 17–25, 2002. doi: 10.1017/S0012162201001608. [DOI] [PubMed] [Google Scholar]
- 134.McLaughlin MJ, He Y, Brunstrom-Hernandez J, Thio LL, Carleton BC, Ross CJD, Gaedigk A, Lewandowski A, Dai H, Jusko WJ, Leeder JS. Pharmacogenomic variability of oral baclofen clearance and clinical response in children with cerebral palsy. PM R 10: 235–243, 2018. doi: 10.1016/j.pmrj.2017.08.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mehrholz J, Wagner K, Meissner D, Grundmann K, Zange C, Koch R, Pohl M. Reliability of the modified Tardieu Scale and the modified Ashworth Scale in adult patients with severe brain injury: a comparison study. Clin Rehabil 19: 751–759, 2005. doi: 10.1191/0269215505cr889oa. [DOI] [PubMed] [Google Scholar]
- 136.Malcangio M, Bowery NG. GABA and its receptors in the spinal cord. Trends Pharmacol Sci 17: 457–462, 1996. doi: 10.1016/S0165-6147(96)01013-9. [DOI] [PubMed] [Google Scholar]
- 137.Miller B, Nagy D, Finlay BL, Chance B, Kobayashi A, Nioka S. Consequences of reduced cerebral blood flow in brain development. I. Gross morphology, histology, and callosal connectivity. Exp Neurol 124: 326–342, 1993. doi: 10.1006/exnr.1993.1203. [DOI] [PubMed] [Google Scholar]
- 138.Molon A, Di Giovanni S, Hathout Y, Natale J, Hoffman EP. Functional recovery of glycine receptors in spastic murine model of startle disease. Neurobiol Dis 21: 291–304, 2006. doi: 10.1016/j.nbd.2005.05.030. [DOI] [PubMed] [Google Scholar]
- 139.Monbaliu E, Ortibus E, De Cat J, Dan B, Heyrman L, Prinzie P, De Cock P, Feys H. The Dyskinesia Impairment Scale: a new instrument to measure dystonia and choreoathetosis in dyskinetic cerebral palsy. Dev Med Child Neurol 54: 278–283, 2012. doi: 10.1111/j.1469-8749.2011.04209.x. [DOI] [PubMed] [Google Scholar]
- 140.Murphy KP, Boutin SA, Ide KR. Cerebral palsy, neurogenic bladder, and outcomes of lifetime care. Dev Med Child Neurol 54: 945–950, 2012. doi: 10.1111/j.1469-8749.2012.04360.x. [DOI] [PubMed] [Google Scholar]
- 141.Mutlu A, Livanelioglu A, Gunel MK. Reliability of Ashworth and Modified Ashworth scales in children with spastic cerebral palsy. BMC Musculoskelet Disord 9: 44, 2008. doi: 10.1186/1471-2474-9-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Myers RE. Fetal asphyxia due to umbilical cord compression. Metabolic and brain pathologic consequences. Biol Neonate 26: 21–43, 1975. doi: 10.1159/000240714. [DOI] [PubMed] [Google Scholar]
- 143.Myers RE. Maternal psychological stress and fetal asphyxia: a study in the monkey. Am J Obstet Gynecol 122: 47–59, 1975. doi: 10.1016/0002-9378(75)90614-6. [DOI] [PubMed] [Google Scholar]
- 144.Myers RE. Two patterns of perinatal brain damage and their conditions of occurrence. Am J Obstet Gynecol 112: 246–276, 1972. doi: 10.1016/0002-9378(72)90124-X. [DOI] [PubMed] [Google Scholar]
- 145.Nagata N, Saji M, Ito T, Ikeno S, Takahashi H, Terakawa N. Repetitive intermittent hypoxia-ischemia and brain damage in neonatal rats. Brain Dev 22: 315–320, 2000. doi: 10.1016/S0387-7604(00)00123-6. [DOI] [PubMed] [Google Scholar]
- 146.Nelson C, Silverstein FS. Acute disruption of cytochrome oxidase activity in brain in a perinatal rat stroke model. Pediatr Res 36: 12–19, 1994. doi: 10.1203/00006450-199407001-00003. [DOI] [PubMed] [Google Scholar]
- 147.Nelson KB. Causative factors in cerebral palsy. Clin Obstet Gynecol 51: 749–762, 2008. doi: 10.1097/GRF.0b013e318187087c. [DOI] [PubMed] [Google Scholar]
- 148.Newman CJ, O’Regan M, Hensey O. Sleep disorders in children with cerebral palsy. Dev Med Child Neurol 48: 564–568, 2006. doi: 10.1017/S0012162206001198. [DOI] [PubMed] [Google Scholar]
- 149.NINDS, NICHD . NINDS/NICHD Strategic Plan for Cerebral Palsy Research (Online). Bethesda, MD: National Institute of Neurological Disorders and Stroke, 2017. https://www.ninds.nih.gov/About-NINDS/Strategic-Plans-Evaluations/Strategic-Plans/2017-NINDS-NICHD-Strategic-Plan-Cerebral-Palsy [Google Scholar]
- 150.Nioka S, Zaman A, Nagy D, Miller B, Finlay BL, Chance B. Consequences of reduced cerebral blood flow in brain development. II. Retardation of neurological outcome and phosphorus metabolism. Exp Neurol 124: 343–350, 1993. doi: 10.1006/exnr.1993.1204. [DOI] [PubMed] [Google Scholar]
- 151.Nitsos I, Rees SM, Duncan J, Kramer BW, Harding R, Newnham JP, Moss TJM. Chronic exposure to intra-amniotic lipopolysaccharide affects the ovine fetal brain. J Soc Gynecol Investig 13: 239–247, 2006. doi: 10.1016/j.jsgi.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 152.Novak I, Morgan C, Adde L, Blackman J, Boyd RN, Brunstrom-Hernandez J, Cioni G, Damiano D, Darrah J, Eliasson AC, de Vries LS, Einspieler C, Fahey M, Fehlings D, Ferriero DM, Fetters L, Fiori S, Forssberg H, Gordon AM, Greaves S, Guzzetta A, Hadders-Algra M, Harbourne R, Kakooza-Mwesige A, Karlsson P, Krumlinde-Sundholm L, Latal B, Loughran-Fowlds A, Maitre N, McIntyre S, Noritz G, Pennington L, Romeo DM, Shepherd R, Spittle AJ, Thornton M, Valentine J, Walker K, White R, Badawi N. Early, accurate diagnosis and early intervention in cerebral palsy: advances in diagnosis and treatment. JAMA Pediatr 171: 897–907, 2017. doi: 10.1001/jamapediatrics.2017.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.O’Callaghan ME, MacLennan AH, Gibson CS, McMichael GL, Haan EA, Broadbent JL, Goldwater PN, Dekker GA; Australian Collaborative Cerebral Palsy Research Group . Epidemiologic associations with cerebral palsy. Obstet Gynecol 118: 576–582, 2011. doi: 10.1097/AOG.0b013e31822ad2dc. [DOI] [PubMed] [Google Scholar]
- 154.Odding E, Roebroeck ME, Stam HJ. The epidemiology of cerebral palsy: incidence, impairments and risk factors. Disabil Rehabil 28: 183–191, 2006. doi: 10.1080/09638280500158422. [DOI] [PubMed] [Google Scholar]
- 155.Oskoui M, Coutinho F, Dykeman J, Jetté N, Pringsheim T. An update on the prevalence of cerebral palsy: a systematic review and meta-analysis. Dev Med Child Neurol 55: 509–519, 2013. doi: 10.1111/dmcn.12080. [DOI] [PubMed] [Google Scholar]
- 156.Osler W. The Cerebral Palsies of Childhood. London: HK Lewis, 1889. [Google Scholar]
- 157.Owens DF, Kriegstein AR. Is there more to GABA than synaptic inhibition? Nat Rev Neurosci 3: 715–727, 2002. doi: 10.1038/nrn919. [DOI] [PubMed] [Google Scholar]
- 158.Pakula AT, Van Naarden Braun K, Yeargin-Allsopp M. Cerebral palsy: classification and epidemiology. Phys Med Rehabil Clin N Am 20: 425–452, 2009. doi: 10.1016/j.pmr.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 159.Panteliadis C, Panteliadis P, Vassilyadi F. Hallmarks in the history of cerebral palsy: from antiquity to mid-20th century. Brain Dev 35: 285–292, 2013. doi: 10.1016/j.braindev.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 160.Pharoah PO, Cooke T. Cerebral palsy and multiple births. Arch Dis Child Fetal Neonatal Ed 75: F174–F177, 1996. doi: 10.1136/fn.75.3.F174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pittman RH, Oppenheim RW. Neuromuscular blockade increases motoneurone survival during normal cell death in the chick embryo. Nature 271: 364–366, 1978. doi: 10.1038/271364a0. [DOI] [PubMed] [Google Scholar]
- 162.Pruitt DW, Tsai T. Common medical comorbidities associated with cerebral palsy. Phys Med Rehabil Clin N Am 20: 453–467, 2009. doi: 10.1016/j.pmr.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 163.Delgado MR, Hirtz D, Aisen M, Ashwal S, Fehlings DL, McLaughlin J, Morrison LA, Shrader MW, Tilton A, Vargus-Adams J; Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society . Practice parameter: pharmacologic treatment of spasticity in children and adolescents with cerebral palsy (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 74: 336–343, 2010. doi: 10.1212/WNL.0b013e3181cbcd2f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ranck JB Jr, Windle WF. Brain damage in the monkey, macaca mulatta, by asphyxia neonatorum. Exp Neurol 1: 130–154, 1959. doi: 10.1016/0014-4886(59)90032-9. [DOI] [PubMed] [Google Scholar]
- 165.Reddihough DS, Collins KJ. The epidemiology and causes of cerebral palsy. Aust J Physiother 49: 7–12, 2003. doi: 10.1016/S0004-9514(14)60183-5. [DOI] [PubMed] [Google Scholar]
- 166.Reddy K, Mallard C, Guan J, Marks K, Bennet L, Gunning M, Gunn A, Gluckman P, Williams C. Maturational change in the cortical response to hypoperfusion injury in the fetal sheep. Pediatr Res 43: 674–682, 1998. doi: 10.1203/00006450-199805000-00017. [DOI] [PubMed] [Google Scholar]
- 167.Rice JE III, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 9: 131–141, 1981. doi: 10.1002/ana.410090206. [DOI] [PubMed] [Google Scholar]
- 168.Riddle A, Luo NL, Manese M, Beardsley DJ, Green L, Rorvik DA, Kelly KA, Barlow CH, Kelly JJ, Hohimer AR, Back SA. Spatial heterogeneity in oligodendrocyte lineage maturation and not cerebral blood flow predicts fetal ovine periventricular white matter injury. J Neurosci 26: 3045–3055, 2006. doi: 10.1523/JNEUROSCI.5200-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Rosenbaum P, Paneth N, Leviton A, Goldstein M, Bax M, Damiano D, Dan B, Jacobsson B. A report: the definition and classification of cerebral palsy April 2006. Dev Med Child Neurol Suppl 109: 8–14, 2007. [PubMed] [Google Scholar]
- 170.Rousset CI, Chalon S, Cantagrel S, Bodard S, Andres C, Gressens P, Saliba E. Maternal exposure to LPS induces hypomyelination in the internal capsule and programmed cell death in the deep gray matter in newborn rats. Pediatr Res 59: 428–433, 2006. doi: 10.1203/01.pdr.0000199905.08848.55. [DOI] [PubMed] [Google Scholar]
- 171.Rumajogee P, Bregman T, Miller SP, Yager JY, Fehlings MG. Rodent hypoxia-ischemia models for cerebral palsy research: a systematic review. Front Neurol 7: 57, 2016. doi: 10.3389/fneur.2016.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sanger TD, Chen D, Fehlings DL, Hallett M, Lang AE, Mink JW, Singer HS, Alter K, Ben-Pazi H, Butler EE, Chen R, Collins A, Dayanidhi S, Forssberg H, Fowler E, Gilbert DL, Gorman SL, Gormley ME Jr, Jinnah HA, Kornblau B, Krosschell KJ, Lehman RK, MacKinnon C, Malanga CJ, Mesterman R, Michaels MB, Pearson TS, Rose J, Russman BS, Sternad D, Swoboda KJ, Valero-Cuevas F. Definition and classification of hyperkinetic movements in childhood. Mov Disord 25: 1538–1549, 2010. doi: 10.1002/mds.23088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sanger TD, Delgado MR, Gaebler-Spira D, Hallett M, Mink JW; Task Force on Childhood Motor Disorders . Classification and definition of disorders causing hypertonia in childhood. Pediatrics 111: e89–e97, 2003. doi: 10.1542/peds.111.1.e89. [DOI] [PubMed] [Google Scholar]
- 174.Santi CM, Orta G, Salkoff L, Visconti PE, Darszon A, Treviño CL. K+ and Cl- channels and transporters in sperm function. Curr Top Dev Biol 102: 385–421, 2013. doi: 10.1016/B978-0-12-416024-8.00014-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sato Y, Son JH, Tucker RP, Meizel S. The zona pellucida-initiated acrosome reaction: defect due to mutations in the sperm glycine receptor/Cl(-) channel. Dev Biol 227: 211–218, 2000. doi: 10.1006/dbio.2000.9882. [DOI] [PubMed] [Google Scholar]
- 176.Schendel DE, Schuchat A, Thorsen P. Public health issues related to infection in pregnancy and cerebral palsy. Ment Retard Dev Disabil Res Rev 8: 39–45, 2002. doi: 10.1002/mrdd.10011. [DOI] [PubMed] [Google Scholar]
- 177.Schiariti V, Fowler E, Brandenburg JE, Levey E, Mcintyre S, Sukal-Moulton T, Ramey SL, Rose J, Sienko S, Stashinko E, Vogtle L, Feldman RS, Koenig JI. A common data language for clinical research studies: the National Institute of Neurological Disorders and Stroke and American Academy for Cerebral Palsy and Developmental Medicine Cerebral Palsy Common Data Elements Version 1.0 recommendations. Dev Med Child Neurol 60: 976–986, 2018. doi: 10.1111/dmcn.13723. [DOI] [PubMed] [Google Scholar]
- 178.Schifrin BS, Longo LD. William John Little and cerebral palsy. A reappraisal. Eur J Obstet Gynecol Reprod Biol 90: 139–144, 2000. doi: 10.1016/S0301-2115(00)00261-X. [DOI] [PubMed] [Google Scholar]
- 179.Schulte JT, Wierenga CJ, Bruining H. Chloride transporters and GABA polarity in developmental, neurological and psychiatric conditions. Neurosci Biobehav Rev 90: 260–271, 2018. doi: 10.1016/j.neubiorev.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 180.Sheard P, McCaig CD, Harris AJ. Critical periods in rat motoneuron development. Dev Biol 102: 21–31, 1984. doi: 10.1016/0012-1606(84)90171-4. [DOI] [PubMed] [Google Scholar]
- 181.Shevell M, Dagenais L, Oskoui M. The epidemiology of cerebral palsy: new perspectives from a Canadian registry. Semin Pediatr Neurol 20: 60–64, 2013. doi: 10.1016/j.spen.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 182.Silbereis JC, Huang EJ, Back SA, Rowitch DH. Towards improved animal models of neonatal white matter injury associated with cerebral palsy. Dis Model Mech 3: 678–688, 2010. doi: 10.1242/dmm.002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Silverstein F, Johnston MV. Effects of hypoxia-ischemia on monoamine metabolism in the immature brain. Ann Neurol 15: 342–347, 1984. doi: 10.1002/ana.410150407. [DOI] [PubMed] [Google Scholar]
- 184.Smithers-Sheedy H, Badawi N, Blair E, Cans C, Himmelmann K, Krägeloh-Mann I, McIntyre S, Slee J, Uldall P, Watson L, Wilson M. What constitutes cerebral palsy in the twenty-first century? Dev Med Child Neurol 56: 323–328, 2014. doi: 10.1111/dmcn.12262. [DOI] [PubMed] [Google Scholar]
- 185.Stevenson RD, Conaway M, Chumlea WC, Rosenbaum P, Fung EB, Henderson RC, Worley G, Liptak G, O’Donnell M, Samson-Fang L, Stallings VA; North American Growth in Cerebral Palsy Study . Growth and health in children with moderate-to-severe cerebral palsy. Pediatrics 118: 1010–1018, 2006. doi: 10.1542/peds.2006-0298. [DOI] [PubMed] [Google Scholar]
- 186.Stil A, Jean-Xavier C, Liabeuf S, Brocard C, Delpire E, Vinay L, Viemari JC. Contribution of the potassium-chloride co-transporter KCC2 to the modulation of lumbar spinal networks in mice. Eur J Neurosci 33: 1212–1222, 2011. doi: 10.1111/j.1460-9568.2010.07592.x. [DOI] [PubMed] [Google Scholar]
- 187.Stil A, Liabeuf S, Jean-Xavier C, Brocard C, Viemari JC, Vinay L. Developmental up-regulation of the potassium-chloride cotransporter type 2 in the rat lumbar spinal cord. Neuroscience 164: 809–821, 2009. doi: 10.1016/j.neuroscience.2009.08.035. [DOI] [PubMed] [Google Scholar]
- 188.Szaflarski J, Burtrum D, Silverstein FS. Cerebral hypoxia-ischemia stimulates cytokine gene expression in perinatal rats. Stroke 26: 1093–1100, 1995. doi: 10.1161/01.STR.26.6.1093. [DOI] [PubMed] [Google Scholar]
- 189.Takahashi S, Tanaka H, Oki J. Development of spinal motoneurons in rats after a neonatal hypoxic insult. Pediatr Neurol 21: 715–720, 1999. doi: 10.1016/S0887-8994(99)00080-6. [DOI] [PubMed] [Google Scholar]
- 190.Takazawa T, Choudhury P, Tong CK, Conway CM, Scherrer G, Flood PD, Mukai J, MacDermott AB. Inhibition mediated by glycinergic and GABAergic receptors on excitatory neurons in mouse superficial dorsal horn is location-specific but modified by inflammation. J Neurosci 37: 2336–2348, 2017. doi: 10.1523/JNEUROSCI.2354-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Towfighi J, Mauger D, Vannucci RC, Vannucci SJ. Influence of age on the cerebral lesions in an immature rat model of cerebral hypoxia-ischemia: a light microscopic study. Brain Res Dev Brain Res 100: 149–160, 1997. doi: 10.1016/S0165-3806(97)00036-9. [DOI] [PubMed] [Google Scholar]
- 192.Turner CP, Seli M, Ment L, Stewart W, Yan H, Johansson B, Fredholm BB, Blackburn M, Rivkees SA. A1 adenosine receptors mediate hypoxia-induced ventriculomegaly. Proc Natl Acad Sci USA 100: 11718–11722, 2003. doi: 10.1073/pnas.1931975100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Uehara H, Yoshioka H, Kawase S, Nagai H, Ohmae T, Hasegawa K, Sawada T. A new model of white matter injury in neonatal rats with bilateral carotid artery occlusion. Brain Res 837: 213–220, 1999. doi: 10.1016/S0006-8993(99)01675-3. [DOI] [PubMed] [Google Scholar]
- 194.Van Steenwinckel J, Schang AL, Sigaut S, Chhor V, Degos V, Hagberg H, Baud O, Fleiss B, Gressens P. Brain damage of the preterm infant: new insights into the role of inflammation. Biochem Soc Trans 42: 557–563, 2014. doi: 10.1042/BST20130284. [DOI] [PubMed] [Google Scholar]
- 195.Vannucci RC, Vannucci SJ. Perinatal hypoxic-ischemic brain damage: evolution of an animal model. Dev Neurosci 27: 81–86, 2005. doi: 10.1159/000085978. [DOI] [PubMed] [Google Scholar]
- 196.Vannucci SJ, Hagberg H. Hypoxia-ischemia in the immature brain. J Exp Biol 207: 3149–3154, 2004. doi: 10.1242/jeb.01064. [DOI] [PubMed] [Google Scholar]
- 197.Walker HK. Deep Tendon Reflexes. In: The History, Physical, and Laboratory Examinations, edited by Walker HK, Hall WD, Hurst JW. Boston: Butterworths, 1990. [PubMed] [Google Scholar]
- 198.Watanabe M, Fukuda A. Development and regulation of chloride homeostasis in the central nervous system. Front Cell Neurosci 9: 371, 2015. doi: 10.3389/fncel.2015.00371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Werman R, Davidoff RA, Aprison MH. Inhibition of motoneurones by iontophoresis of glycine. Nature 214: 681–683, 1967. doi: 10.1038/214681a0. [DOI] [PubMed] [Google Scholar]
- 200.Werman R, Davidoff RA, Aprison MH. Inhibitory of glycine on spinal neurons in the cat. J Neurophysiol 31: 81–95, 1968. doi: 10.1152/jn.1968.31.1.81. [DOI] [PubMed] [Google Scholar]
- 201.Wojcik SM, Katsurabayashi S, Guillemin I, Friauf E, Rosenmund C, Brose N, Rhee JS. A shared vesicular carrier allows synaptic corelease of GABA and glycine. Neuron 50: 575–587, 2006. doi: 10.1016/j.neuron.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 202.Wong-Riley MT, Liu Q. Neurochemical and physiological correlates of a critical period of respiratory development in the rat. Respir Physiol Neurobiol 164: 28–37, 2008. doi: 10.1016/j.resp.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Woodward LJ, Anderson PJ, Austin NC, Howard K, Inder TE. Neonatal MRI to predict neurodevelopmental outcomes in preterm infants. N Engl J Med 355: 685–694, 2006. doi: 10.1056/NEJMoa053792. [DOI] [PubMed] [Google Scholar]
- 204.Workman AD, Charvet CJ, Clancy B, Darlington RB, Finlay BL. Modeling transformations of neurodevelopmental sequences across mammalian species. J Neurosci 33: 7368–7383, 2013. doi: 10.1523/JNEUROSCI.5746-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Wright J, Rang M. The spastic mouse. And the search for an animal model of spasticity in human beings. Clin Orthop Relat Res 253: 12–19, 1990. [PubMed] [Google Scholar]
- 206.Wu WL, Ziskind-Conhaim L, Sweet MA. Early development of glycine- and GABA-mediated synapses in rat spinal cord. J Neurosci 12: 3935–3945, 1992. doi: 10.1523/JNEUROSCI.12-10-03935.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wu YW, Colford JM Jr. Chorioamnionitis as a risk factor for cerebral palsy: a meta-analysis. JAMA 284: 1417–1424, 2000. doi: 10.1001/jama.284.11.1417. [DOI] [PubMed] [Google Scholar]
- 208.Yee BK, Keist R, von Boehmer L, Studer R, Benke D, Hagenbuch N, Dong Y, Malenka RC, Fritschy JM, Bluethmann H, Feldon J, Möhler H, Rudolph U. A schizophrenia-related sensorimotor deficit links alpha 3-containing GABAA receptors to a dopamine hyperfunction. Proc Natl Acad Sci USA 102: 17154–17159, 2005. doi: 10.1073/pnas.0508752102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Yokoyama Y, Shimizu T, Hayakawa K. Prevalence of cerebral palsy in twins, triplets and quadruplets. Int J Epidemiol 24: 943–948, 1995. doi: 10.1093/ije/24.5.943. [DOI] [PubMed] [Google Scholar]
- 210.Yoon BH, Kim CJ, Romero R, Jun JK, Park KH, Choi ST, Chi JG. Experimentally induced intrauterine infection causes fetal brain white matter lesions in rabbits. Am J Obstet Gynecol 177: 797–802, 1997. doi: 10.1016/S0002-9378(97)70271-0. [DOI] [PubMed] [Google Scholar]
- 211.Zarbin MA, Wamsley JK, Kuhar MJ. Glycine receptor: light microscopic autoradiographic localization with [3H]strychnine. J Neurosci 1: 532–547, 1981. doi: 10.1523/JNEUROSCI.01-05-00532.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ziv I, Blackburn N, Rang M, Koreska J. Muscle growth in normal and spastic mice. Dev Med Child Neurol 26: 94–99, 1984. doi: 10.1111/j.1469-8749.1984.tb04412.x. [DOI] [PubMed] [Google Scholar]