

Abstract
S100 calcium-binding protein A9 (S100A9) is elevated in plasma and bronchoalveolar lavage fluid (BALF) of patients with chronic obstructive pulmonary disease (COPD), and aging enhances S100A9 expression in several tissues. Currently, the direct impact of S100A9-mediated signaling on lung function and within the aging lung is unknown. Here, we observed that elevated S100A9 levels in human BALF correlated with age. Elevated lung levels of S100A9 were higher in older mice compared with in young animals and coincided with pulmonary function changes. Both acute and chronic exposure to cigarette smoke enhanced S100A9 levels in age-matched mice. To examine the direct role of S100A9 on the development of COPD, S100a9−/− mice or mice administered paquinimod were exposed to chronic cigarette smoke. S100A9 depletion and inhibition attenuated the loss of lung function, pressure-volume loops, airway inflammation, lung compliance, and forced expiratory volume in 0.05 s/forced vital capacity, compared with age-matched wild-type or vehicle-administered animals. Loss of S100a9 signaling reduced cigarette smoke-induced airspace enlargement, alveolar remodeling, lung destruction, ERK and c-RAF phosphorylation, matrix metalloproteinase-3 (MMP-3), matrix metalloproteinase-9 (MMP-9), monocyte chemoattractant protein-1 (MCP-1), interleukin-6 (IL-6), and keratinocyte-derived chemokine (KC) release into the airways. Paquinimod administered to nonsmoked, aged animals reduced age-associated loss of lung function. Since fibroblasts play a major role in the production and maintenance of extracellular matrix in emphysema, primary lung fibroblasts were treated with the ERK inhibitor LY3214996 or the c-RAF inhibitor GW5074, resulting in less S100A9-induced MMP-3, MMP-9, MCP-1, IL-6, and IL-8. Silencing Toll-like receptor 4 (TLR4), receptor for advanced glycation endproducts (RAGE), or extracellular matrix metalloproteinase inducer (EMMPRIN) prevented S100A9-induced phosphorylation of ERK and c-RAF. Our data suggest that S100A9 signaling contributes to the progression of smoke-induced and age-related COPD.
Keywords: aging, cigarette smoke, kinase, pulmonary function, S100A9
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is the fourth leading cause of death in the United States. The Centers for Disease Control and Prevention attributes over 480,000 deaths annually or ∼1 in 5 deaths to cigarette smoke (44a). Exposure to cigarette smoke is the primary environmental factor associated with the development of COPD in the developed world. Cellular responses triggered by cigarette smoke cause the release of inflammatory and proteolytic mediators that contribute to the pathogenesis of COPD (21). There are several proposed mechanisms that lead to loss of lung function, including chronic inflammation (10), accumulation of secretions, protease responses (25), matrix remodeling, apoptosis (48), immunosenescence (43), and damage to airway mucosa (4). Importantly, COPD is regarded as an age-related disease, with the incidence rate rising with increasing age (78). During the normal aging process, pulmonary function begins to decline as a consequence of structural and physiological changes to the lung (44). There is also a strong association between smoke-induced diseases and damage-associated molecular patterns (DAMPs) (24, 49). DAMPs are endogenous molecules released from damaged or dying cells and that activate the innate immune system via pattern recognition receptors. Despite having important roles in immune responses, several DAMPs are associated with disease pathogenesis, including S100 calcium-binding protein A9 (S100A9) (24). However, apart from elevated levels of S100A9 observed in the serum of patients with COPD during an exacerbation (50) and in the bronchoalveolar lavage fluid (BALF) of exacerbation-free patients with COPD (24) and S100A9 expression in many tissues enhancing with age (68), we currently know little about the functional role of S100A9 in disease progression and the aging lung.
There are additional studies linking S100A9 to other pulmonary and cardiovascular diseases, with plasma S100A8/A9 levels associated with pediatric obstructive sleep apnea and possibly reflecting an increased risk for cardiovascular morbidity (37). S100A9 frequently forms a heterodimer with S100A8, and both play an important role in many biological processes (74), but S100A8 and S100A9 are not always coexpressed (40). S100A9 is expressed at low levels in healthy tissue but is frequently observed in diseased tissue (20) and infiltrating immune cells (6). Intracellularly, S100A9 is known to regulate NADPH oxidase activity (6), which is a major source of reactive oxygen species in neutrophils. Extracellularly, high concentrations of S100A9 are observed in tissues with elevated inflammation or in the serum of patients with inflammatory diseases (13). In cardiovascular disease, plasma S100A9 levels correlate with blood neutrophil counts and with the incidence of coronary events and cardiovascular mortality (14). Our group determined that enhanced S100A9 signaling coincides with lung damage during respiratory syncytial virus (RSV) infection in mice (24). We have also determined that S100A9 is enhanced by cigarette smoke exposure and further enhanced during viral exacerbations in mice and human primary lung cells (24).
S100A9 is highly expressed in phagocytes, is elevated in COPD samples, and triggers degranulation of neutrophils releasing inflammatory and proteolytic enzymes (64). Therefore, we tested the hypothesis that S100A9 signaling in the lung contributes to age-related changes in pulmonary function and the development of COPD. We used animals genetically deficient for S100a9 and paquinimod, a chemical inhibitor of S100A9, to explore the impact of S100a9 deficiency or inhibition on lung structure and function and signaling changes during chronic smoke exposure and in aged animals. We used BALF and human primary lung fibroblasts to correlate S100A9 levels to age and to allow modulation of S100A9 responses in vitro, respectively. Fibroblasts were used because they express S100A9-mediated cytokines and proteases, and fibroblasts play a major role in the production and maintenance of the extracellular matrix in emphysema (45, 69). DAMP-associated receptors, kinases, cytokines, and proteases were investigated in lung fibroblasts to further elucidate molecular mechanisms regulated by S100A9 in COPD.
METHODS
Human samples.
BALF was collected from healthy never smokers, smokers, and patients with COPD free from exacerbations for 6 mo (see Table 1 for demographics). Written informed consent was obtained from all study participants, and the study was approved by the Institutional Review Board of the University of Miami and conformed to the Declaration of Helsinki.
Table 1.
Patient demographics for BALF donors
| Nonsmoker | Smoker | COPD | P value | |
|---|---|---|---|---|
| Number | 11 | 28 | 76 | |
| Age, yr | 57.6 ± 13.2 | 57.1 ± 8.9 | 61.1 ± 9.3 | 0.331 |
| Sex, male/female | 6/5 | 15/13 | 42/34 | 0.483 |
| Race (Caucasian/Hispanic/African American) | 46.6%/46.6%/6.6% | 25%/61%/14% | 5.3%/92%/2.7% | 0.154 |
| Pack years | 0 ± 0 | 30.2. ± 11.4 | 48.9 ± 3.1 | >0.001 |
| FEV1% predicted | 102.1 ± 14.9 | 93.9 ± 10.3 | 59.9 ± 11.9 | 0.011 |
| FVC % predicted | 96.9 ± 10.5 | 95.2 ± 11.7 | 81.8 ± 1.8 | 0.015 |
| FEV1/FVC % | 81.2 ± 6.7 | 80.6 ± 4.7 | 52.9 ± 6.4 | >0.001 |
| DLCO % predicted | 102 ± 10.6 | 92.9 ± 10.8 | 69.5 ± 10.9 | 0.013 |
BALF, bronchoalveolar lavage fluid; COPD, chronic obstructive pulmonary disease; FEV, forced expiratory volume; FVC, forced vital capacity; DLCO, diffusing capacity for carbon monoxide.
Animal models.
S100a9−/− mice (3), on a C57BL/6J background, were maintained in a specific pathogen-free facility at SUNY Downstate Medical Center. We chose to investigate the S100a9−/− mice because lung levels of S100a9 were similar in wild-type and heterozygous animals (Supplemental Fig. S1; see https://doi.org/10.6084/m9.figshare.12272816.v1). Both male and female mice (8-wk-old) mice were used at the initiation point for the majority of experiments. Mice were whole body exposed to cigarette smoke in a chamber (Teague Enterprises, Davis, CA) for 3 h a day, 5 days per week, at a total particulate matter concentration of 80–120 mg/m3. Smoke exposure was continued for 6 mo. Marlboro cigarettes were used to generate cigarette smoke. A/J mice were used for the paquinimod studies. Oral paquinimod (Active Biotech AB, Sweden) was daily administered to animals at a final concentration of 3.75 mg/kg. Paquinimod is water-soluble, and control animals received water only as a vehicle. Mice received paquinimod 1) throughout the smoke study or 2) for 2 mo into a 4-mo smoking study or 3) daily for 2 mo in aged animals without smoke exposure. A cohort of C57BL/6J animals was aged (18 mo) to compare age-dependent changes in S100A9 levels and the impact of paquinimod on lung function. Equally, age-matched animals were exposed to acute (1-mo) or chronic (6-mo) smoke exposure. All animal experiments were performed with approval from SUNY Downstate Health Sciences University’s Institutional Animal Care and Use Committee. This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and Institutional Animal Care and Use Committee guidelines.
Expiratory measurements.
Mice were anesthetized with an intraperitoneal injection of ketamine/xylazine hydrochloride solution (100/10 mg/kg; MilliporeSigma, Burlington, MA). Animals were tracheostomized and connected via an endotracheal cannula to the SCIREQ flexiVent system (SCIREQ Inc., Montreal, Canada). After initiating mechanical ventilation, animals were paralyzed with 1 mg/kg pancuronium bromide (MilliporeSigma) via an intraperitoneal injection, and several pulmonary function measurements [pressure-volume (PV) loops, lung compliance, compliance, and forced expiration extension (forced expiratory volume or FEV) in the first 0.05 s of forced vital capacity (FVC)] were performed, as previously described (62). The area under the curve of the PV loops was calculated for each animal.
Histology and lung immune cell measurements.
Following euthanasia by cervical dislocation, the lungs underwent pressure-fixation and morphometric analysis in accordance with our previously published protocol (23) and in accordance with the American Thoracic Society/European Respiratory Society (ATS/ERS) issue statement on quantitative assessment of lung structure (35). Fixed step sections (4 μm in thickness) every 200 μm of paraffin-embedded lungs were hematoxylin-eosin (H&E) stained. The lung was imaged with a high-throughput scanner (Leica AT2). Whole slide scans of several layers were cropped into multiple images (at least 40 images per animal) before performing histology analysis based on stereological principles. Mean linear intercept analysis was performed as previously described (22). Alveolar boundary size and ductal destructive measurements were performed on images of the H&E-stained tissues, as previously described (77). Bronchoalveolar lavage fluid (BALF) and BALF cells were obtained from animals of each group. BALF cells were cytocentrifuged onto slides to determine macrophage and neutrophil numbers. Cells were stained with Diff-Quik stain and at least 200 cells were examined per slide. Goat polyclonal anti-S100A9 antibody (Santa Cruz Biotechnologies) was used to stain sections for immunohistochemistry. Briefly, deparaffinized sections underwent antigen retrieval using tri-sodium citrate at pH 6 for 30 min in a 98°C water bath. After allowing the sections to cool, they were washed in PBS containing 0.05% Tween 20 (PBST) and blocked in 3% bovine serum albumin (BSA) in PBS for 1 h at room temperature. Goat polyclonal anti-S100A9 antibody (Santa Cruz Biotechnologies) was added in 3% BSA and incubated overnight at 4°C on a rocking shaker. Sections were washed in PBS with Tween three times for 10 min. Detection was by an avidin-biotin complex (ABC)-based method (Vector Laboratories, Burlingame, CA) using a biotinylated secondary antibody, which is linked to an ABC-enzyme complex, horseradish peroxidase. 3,3′-Diaminobenzidine (Vector Laboratories) was used as a substrate for peroxidase complexes yielding a brown reaction product at the site of the target antigen. Sections were counterstained with hematoxylin (blue). Isotype control goat IgG was used as a negative control.
BALF analysis.
Mouse and human matrix metalloproteinases (MMPs), cytokines, and S100 (A8 and A9) proteins were examined in BALF using beads assays (MILLIPLEX MAP MMP Magnetic Bead and MILLIPLEX Cytokine Magnetic Bead Panels, Millipore Sigma, Billerica, MA; R&D Systems Magnetic Luminex Assays) with the Bio-Rad Bio-Plex 200 system. Mouse desmosine levels were measured in concentrated BALF using the Biorbyt Mouse Desmosine ELISA Kit, as recommended by manufacturers (Cat. No. orb409382, Biorbyt LLC, San Francisco, CA). Human BALF data were standardized to total urea concentrations, as determined by a commercially available assay as per the manufacturer’s instructions (Abnova, Walnut, CA).
Western blot and RT-PCR.
Protein was collected from lung tissue by bead beater disruption (Minibeadbeater-16, BioSpec Products, Bartlesville, OK). Tissue was placed in radioimmunoprecipitation assay buffer with 50 mg of 1-mm-diameter Zirconia beads (BioSpec Products) and disrupted for 30 s in the bead beater. Fibroblasts were also lysed with the same buffer. Soluble proteins were collected following 10-min centrifugation at 13,000 g at 4°C. Immunoblots for ERK phosphorylation (Thr202/Tyr204 and total ERK1/2), c-RAF phosphorylation (Ser259 and total c-RAF), β-actin, and GAPDH (all antibodies from Cell Signaling Technologies, Beverly, MA) were performed to confirm equal levels of protein were loaded per sample. Chemiluminescence detection was performed using the Bio-Rad Laboratories Molecular Imager ChemiDoc XRS+ imaging system. Densitometry was performed and represented as a ratio of pixel intensity of the phosphorylated ERK or c-RAF compared with total ERK or c-RAF, using Bio-Rad Laboratories Image Laboratory software (version 4.0, build 16). This densitometry ratio was defined as densitometry units (DUs).
Gene expression was determined by qPCR using Taqman probes (Life Technologies/Applied Biosystems, Carlsbad, CA). RNA was isolated using the Qiagen RNeasy kit following tissue or cell homogenization, and mRNA was reverse transcribed using the Applied Biosystems high-capacity cDNA kit (Life Technologies). qPCR was performed on the Bio-Rad CFX384 real-time system. All qPCR results are represented as relative quantification compared with room air or nonsmoker controls and corrected to ACTB and GAPDH levels, using the ΔΔCt method.
Cell cultures.
Primary adult pulmonary fibroblasts expanded in cell culture from the lung tissues of healthy controls were purchased from PromoCell (Heidelberg, Germany). Fibroblasts were cultured in DMEM (Corning cellgro, Manassas, VA) supplemented with 10% bovine calf serum (BCS; HyClone Laboratories, GE Healthcare Life Sciences, Logan, UT), 2 mM l-glutamine (Gemini Bio-Products, West Sacramento, CA), 1 mM sodium pyruvate, MEM nonessential amino acid solution, and antibiotic-antimycotic (10,000 units/mL penicillin, 10,000 μg/mL streptomycin, and 25 μg/mL amphotericin B), all from Gibco Life Technologies (Thermo Fisher Scientific, Waltham, MA). Fibroblast cultures were maintained in T75 culture flasks (Nest Biotechnology, Rahway, NJ) in a humidified atmosphere with 5% CO2 at 37°C. For experiments, fibroblast cultures were incubated with DMEM medium containing the same supplements and 0.5% BCS for at least 12 h before testing. Fibroblasts were passaged by washing with filtered PBS, trypsinizing with 2–5 mL of 0.25% trypsin-EDTA (Gibco Life Technologies), and reconstituting cells in DMEM with 10% BCS before transferring to new T75 flasks, dishes, or six-well plates. In all experiments, fibroblast cell cultures were tested in passages 4-7. The ERK inhibitor LY3214996 or the c-RAF inhibitor GW5074 (both purchased from Selleckchem.com, Houston, TX) were added to the media of the cultures and incubated for 24 h at 37°C, 5% CO2. Cells were also transfected with siRNA specific for Toll-like receptor 4 (TLR4), advanced glycosylation end product-specific receptor (AGER), basigin (BSG), and scrambled control (QIAGEN) before treatment with recombinant human 1 μg/mL S100A9 protein (Novus Biologicals, Centennial, CO). Cells were collected for protein or RNA analysis.
Statistical analyses.
The majority of the data are expressed as dot plots with the means ± SE highlighted. Normality testing (D'Agostino & Pearson omnibus normality test) was performed on all data sets. A comparison of groups was performed by Student’s t test (two-tailed) when data passed the normality test and by Mann–Whitney test if data did not pass the normality test. Experiments with more than two groups were analyzed by two-way ANOVA with Bonferroni posttest analysis. P values for significance were set at 0.05, and all significant changes were noted with the exact P value. All analyses were performed using GraphPad Prism Software (Version 6.0h for Mac OS X).
RESULTS
Age and cigarette smoke exposure enhance lung S100A9 levels.
Previously, we demonstrated that S100A9 levels are elevated in COPD BALF samples (24). Here, we investigated whether age, smoking, and disease status correlate with lung S100A9 levels. Within the subject cohort, S100A9 levels were significantly elevated in subjects with COPD compared with in nonsmoker controls (Fig. 1A). S100A9 concentration within BALF from nonsmokers (R2 = 0.53 and P = 0.011), smokers (R2 = 0.19 and P = 0.020), and subjects with COPD (R2 = 0.17 and P < 0.001) correlated with age (Fig. 1B). As a binding partner and signaling modulator of S100A9, S100A8 levels were measured in the same cohort. However, no significant changes in BALF S100A8 levels were observed (Supplemental Fig. S2, A and B; see https://doi.org/10.6084/m9.figshare.12272858.v1).
Fig. 1.
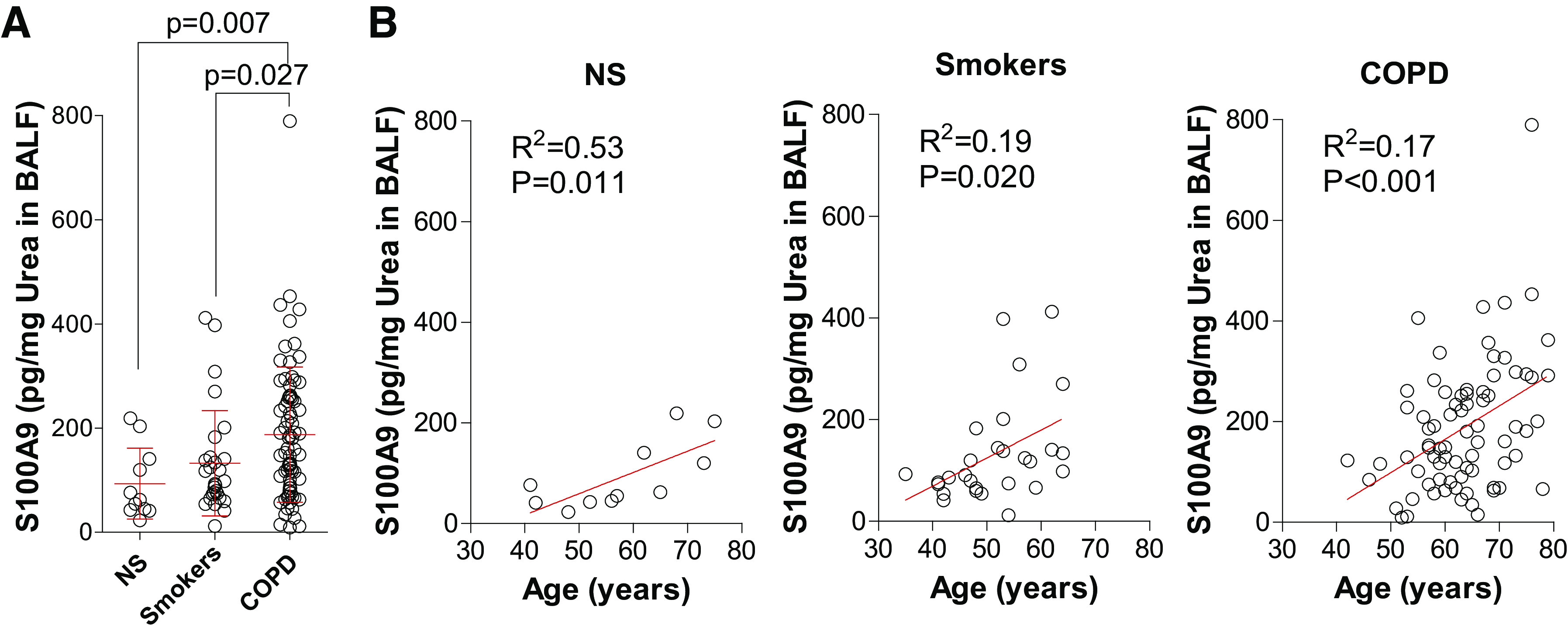
Lung levels of S100 calcium-binding protein A9 (S100A9) increase with aging and disease status in humans. A: human bronchoalveolar lavage fluid (BALF) S100A9 levels were quantified in BALF from nonsmokers, smokers, and subjects with chronic obstructive pulmonary disease (COPD). S100A9 levels were corrected to total BALF urea concentrations. B: S100A9 levels were plotted against subject age for nonsmokers, smokers, and subjects with COPD. Graphs are represented as means ± SD, where n ≥ 11 per group. P values shown when comparing both treatments connected by a line were determined by Mann–Whitney tests. Linear regression and Pearson correlation coefficient were also performed.
To understand the role for S100A9 during the processes of aging and the development of COPD, we analyzed murine models to confirm our results in the patient cohort. As the development of chronic lung diseases is associated with aging, we examined gene expression and BALF levels of S100A9 in young (7-wk-old) and old (18-mo-old) C57BL/6J mice (Fig. 2A). Older mice expressed and secreted higher levels of S100A9 than young mice (Fig. 2A). This was also true for S100A8 (Supplemental Fig. S2, B and C). To examine whether S100a9 expression correlated with age-mediated changes in lung function and morphometry, we analyzed pressure-volume loops, lung compliance, FEV0.05/FVC, and mean linear intercept (MLI) in the same young and old mice. Aged mice showed changes in both physiology and histology parameters consistent with the development of emphysema (Fig. 2B). Equally, elevated IL-6 and KC were observed in the BALF of aged animals (Fig. 2C).
Fig. 2.
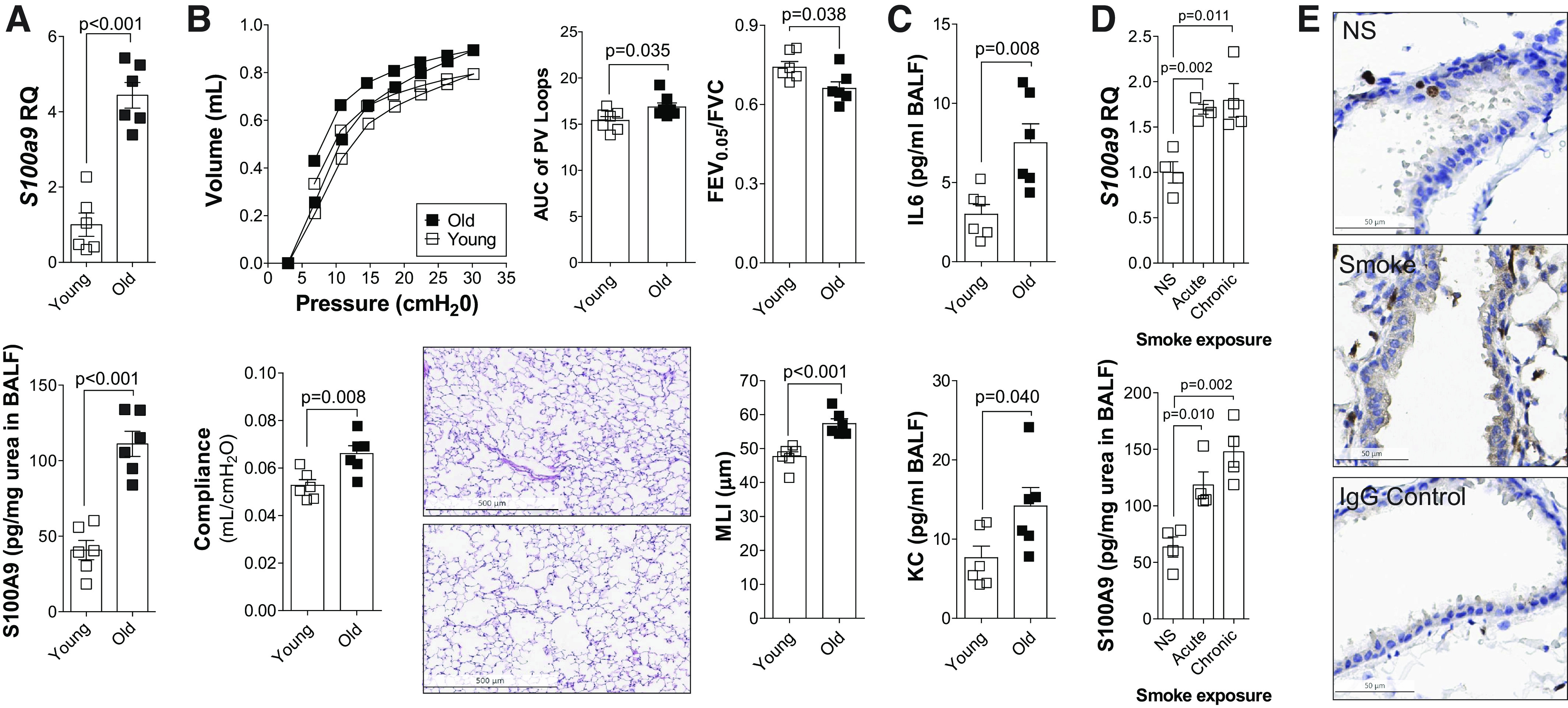
Lung levels of S100 calcium-binding protein A9 (S100A9) increase with aging and smoke exposure in mice. A: S100A9 gene expression and BALF concentration were measured in young (7-wk-old) and old (18-mo-old) C57BL/6J. B: negative pressure-driven forced expiratory and forced oscillation technique maneuvers were performed in young and old animals. Forced expiration extension (forced expiratory volume or FEV) in the first 0.05 s of forced vital capacity (FVC), compliance, and pressure-volume loops with the area under the curve analysis were determined in each animal. Mean linear intercepts (MLIs) were measured in the lungs of the mice to assess airspace size, and comparative histology images of the four mouse groups are presented here (scale bars = 500 µm). C: IL-6 and KC levels were quantified in BALF from young and old animals. D: S100A9 gene expression and bronchoalveolar lavage fluid (BALF) concentration were measured in 8-mo-old C57BL/6J following exposure to no smoke (NS), 1 mo (acute) of cigarette smoke, or 6 mo (chronic) of cigarette smoke. Graphs are represented as means ± SE, where n ≥ 4 per group. P values shown when comparing both treatments connected by a line were determined by Student’s t tests. E: immunohistochemistry (IHC) was performed for S100A9 (brown stain) in NS and chronic smoke-exposed animals. IgG negative control is also shown (scale bars = 50 µm). KC, keratinocyte-derived chemokine.
Next, we sought to determine whether cigarette smoke exposure affected the expression of S100A9. Age-matched mice were exposed to room air, 1 mo (acute) of cigarette smoke, or 6 mo (chronic) of cigarette smoke and analyzed at 8 mo of age (Fig. 2D). Both acute and chronic exposure to cigarette smoke enhanced S100A9 expression and secretion in C57BL/6J mice (Fig. 2D). BALF S100A8 was also increased (Supplemental Fig. S2, C and D). Immunohistochemistry demonstrated that S100A9 is localized to infiltrating immune cells as well as airway epithelial cells (Fig. 2E). We also identified that airway epithelium and immune cells are a source of S100A9. Therefore, both age and smoke exposure influence S100A9 levels in the murine models, recapitulating our patient cohort data.
S100a9 deficiency reduces cigarette smoke-induced loss of lung function.
Since increased levels of S100A9 expression and release into the BALF were correlated to loss of lung function, the significance of S100A9 signaling was assessed by genetic knockout. We used the SCIREQ flexiVent system to determine pulmonary function in wild-type and S100a9−/− mice following room air or 6-mo exposure to cigarette smoke. Knockout of S100A9 was confirmed by analysis of lung and BALF (Supplemental Fig. S1). As expected, chronic smoke exposure altered lung compliance, FEV0.05/FVC, and pressure-volume loops in wild-type animals (Fig. 3, A and B). However, S100a9-deficient animals were less susceptible to functional changes as a result of smoke exposure (Fig. 3, A and B). Similarly, histological analysis of the lungs demonstrated that S100a9 deficiency significantly attenuated airspace enlargements, the number of alveolar airspaces, and ductal/destructive fraction compared with in wild-type smoke-exposed animals (Fig. 3, C and D). These data demonstrate that S100A9 contributes to structural and physiological changes to the lung.
Fig. 3.

S100a9−/− mice have reduced disease characteristics in a smoke exposure model. Wild-type and S100a9−/− mice were exposed to smoke exposure daily for 6 mo. A and B: negative pressure-driven forced expiratory and forced oscillation technique maneuvers were performed in all animal groups. Forced expiration extension (forced expiratory volume or FEV) in the first 0.05 s of forced vital capacity (FVC), compliance, and pressure-volume loops with the area under the curve analysis were determined in each animal. C: mean linear intercepts (MLIs) were measured in the lungs of the mice to assess airspace size, and comparative histology images of the four mouse groups are presented here (scale bars = 200 µm). D: alveolar count and ductal/destructive fractions were quantified in each animal by parenchymal airspace profiling. Measurements are means ± SE. P values shown when comparing both treatments connected by a line were determined by two-way ANOVA with Tukey’s post hoc test.
S100a9-deficient animals have reduced immune cell infiltration and associated cytokine and protease signaling following smoke exposure.
Inhibition of S100A9 in an arthritis animal model preserved the structural integrity of bone and collagen and reduced inflammation (11). Therefore, we examined the impact of S100A9 on inflammation and protease production in S100a9−/− mice. S100a9 deficiency reduced cigarette smoke-induced immune cell infiltration (Fig. 4A) and MCP-1, IL-6, and KC secretion into the airways (Fig. 4B). No S100A9-mediated changes were observed for IL-5, IL-9, IL-13, IL-17, and eotaxin (Supplemental Fig. S3A; see https://doi.org/10.6084/m9.figshare.12272864.v1). In human osteoarthritis synovium, the S100A9 inhibitor paquinimod blocks MMP-1 and MMP-3 secretion (58), which could impact COPD (15). Therefore, we examined the expression of several MMPs in all animal groups. Loss of S100a9 expression resulted in reduced MMP-3 and MMP-9 in the BALF of smoke-exposed mice (Fig. 4C). No S100A9-mediated changes were observed for MMP-2, MMP-8, and MMP-12 (Supplemental Fig. S3B). Compared with wild-type mice, smoke-exposed S100a9−/− mice had reduced desmosine in their BALF (Fig. 4C), suggesting less elastin degradation within the lungs. We evaluated possible pathways that could be responsible for these S100A9-mediated signaling downstream effects and determined that loss of S100A9 influenced ERK and c-RAF phosphorylation (Fig. 4D). No S100A9-mediated changes were observed for Src, JNK, and p38 (Supplemental Fig. S4; see https://doi.org/10.6084/m9.figshare.12272876.v1). Therefore, S100A9 mediates several cytokines, proteases, and kinases during smoke exposure possibly via ERK and c-RAF.
Fig. 4.
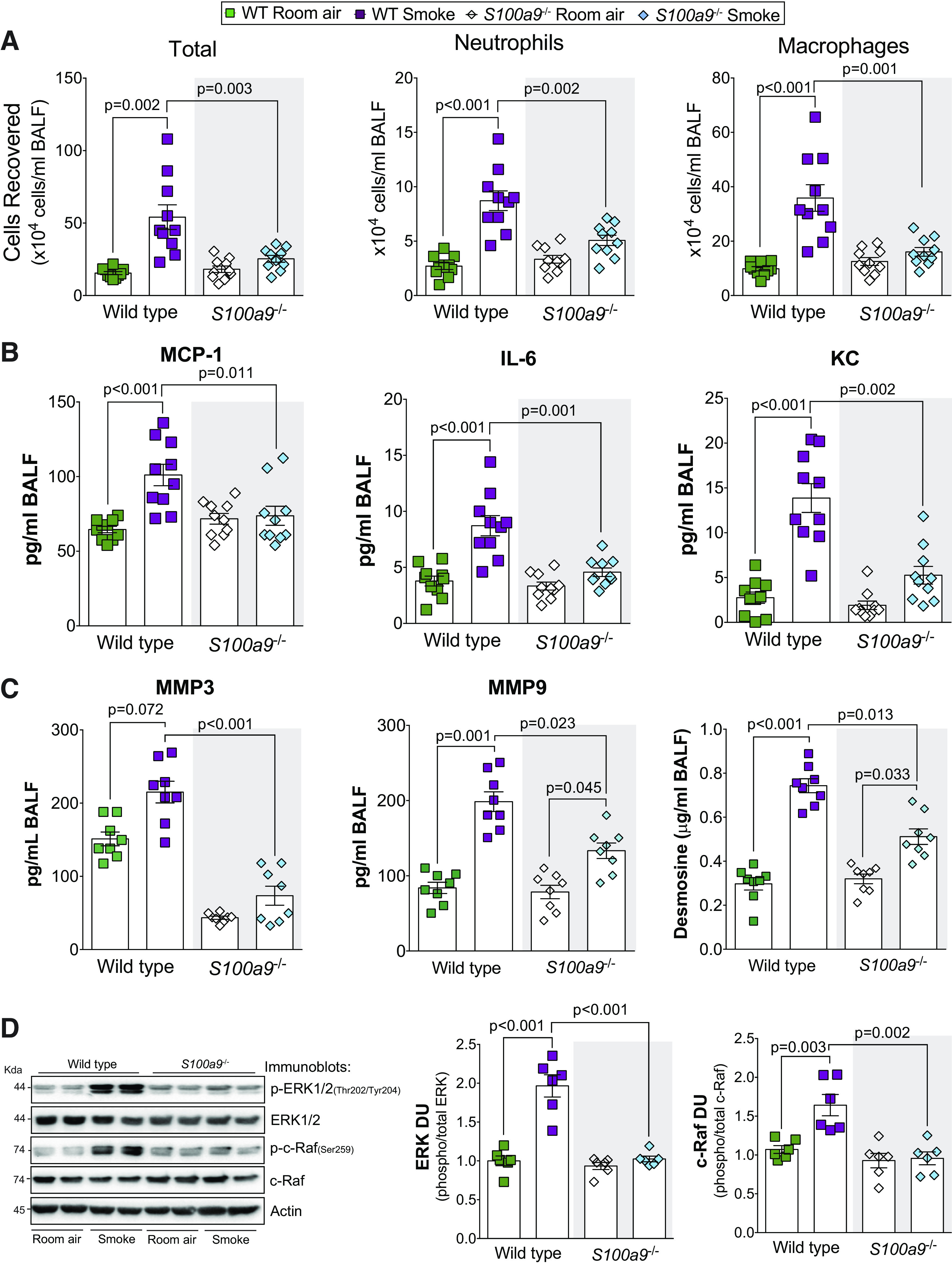
S100a9 deficiency reduced inflammation and protease responses during smoke exposure. A: bronchoalveolar lavage fluid (BALF) cellularity levels were examined in wild-type and S100a9−/− animals after 6 mo of smoke exposure. BALF concentrations for MCP-1, IL-6, and KC (B) and matrix metalloproteinase-3 (MMP-3), matrix metalloproteinase-9 (MMP-9), and desmosine (C) were quantified using Luminex bead assays. D: ERK and c-RAF phosphorylation were examined in lung tissue of smoke-exposed animals by Western blot and densitometry analysis. Graphs are represented as means ± SE, where n ≥ 3 per group. P values shown when comparing both treatments connected by a line were determined by two-way ANOVA with Tukey’s post hoc test.
Early administration of paquinimod prevents smoke-induced COPD in mice.
To determine whether the S100A9 inhibitor paquinimod could prevent the loss of lung function observed in subjects with COPD, we performed a series of exposure studies in mice. First, to determine whether long-term administration of paquinimod exhibited any notable toxicity, animal weight and liver-to-body weight ratios were determined in mice receiving daily doses of paquinimod for 2 mo. No significant changes in animal weight or liver-to-body weight ratios were observed (Fig. 5A). Since long-term paquinimod treatment had little to no negative effects in mice, paquinimod was first administered to mice at the same time as smoke exposure. This approach determined whether extracellular S100A9 was contributing to lung disease initiation. Similar to S100a9 deficiency, treatment with paquinimod prevented loss of lung function in wild-type mice, determined by flexiVent measurements of FEV0.05/FVC, lung compliance, and pressure-volume loops (Fig. 5, B and C). Equally, when quantifying histology changes to lung tissue, paquinimod reduced smoke-induced airspace enlargement, alveolar remodeling, and destruction (Fig. 5, D and E). Early administration of paquinimod also reduced smoke-induced inflammation in the airways (Fig. 6A) and the release of MCP-1, IL-6, KC (Fig. 6B), MMP-3, MMP-9, and desmosine into the airways (Fig. 6C). Paquinimod also prevented phosphorylation of ERK and c-RAF (Fig. 6D). Similar to S100a9 deficiency, paquinimod had no impact on eotaxin, IL-5, IL-9, IL-13, IL-17, MMP-2, MMP-8, MMP-12, p38, JNK, and c-Src (Supplemental Fig. S5; see https://doi.org/10.6084/m9.figshare.12272888.v1). Therefore, paquinimod treatment mimics the profile of S100a9 deficiency by modulating kinase, cytokine, and protease responses.
Fig. 5.
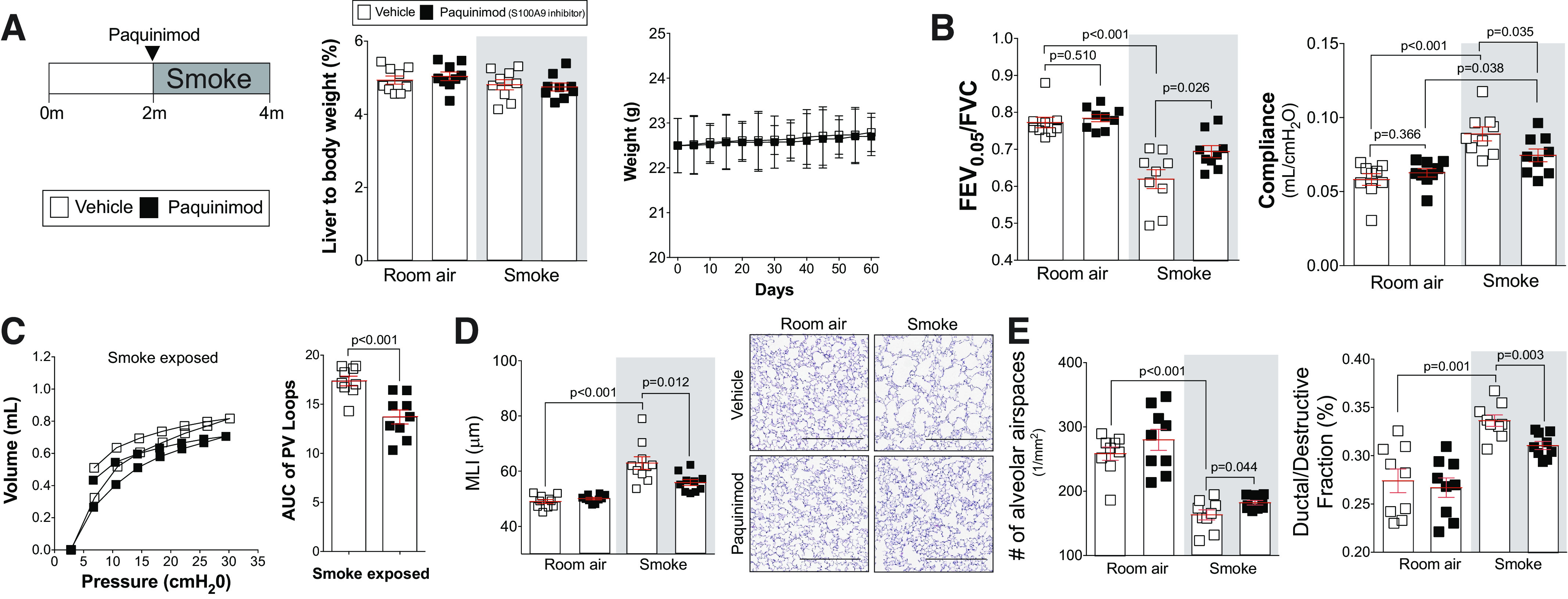
Administration of the S100 calcium-binding protein A9 (S100A9) inhibitor paquinimod shows no toxicity in mice and prevents the development of smoke-induced chronic obstructive pulmonary disease (COPD). A/J mice received paquinimod orally each day before smoke exposure. A: at the end of the study, liver-to-body weight ratios were determined. B: animal body weight was calculated throughout the study. Forced expiratory volume (FEV) in the first 0.05 s of forced vital capacity (FVC), compliance, and pressure-volume loops (C) with the area under the curve analysis were determined in each animal. D: mean linear intercept (MLI) was measured in the lungs of the mice to assess airspace size, and comparative histology images of the four mouse groups are presented here (scale bars = 500 µm). E: alveolar count and ductal/destructive fractions were quantified in each animal by parenchymal airspace profiling. Measurements are means ± SE. P values shown when comparing both treatments connected by a line were determined by two-way ANOVA with Tukey’s post hoc test. Graphs with two groups were analyzed by Student’s t tests.
Fig. 6.
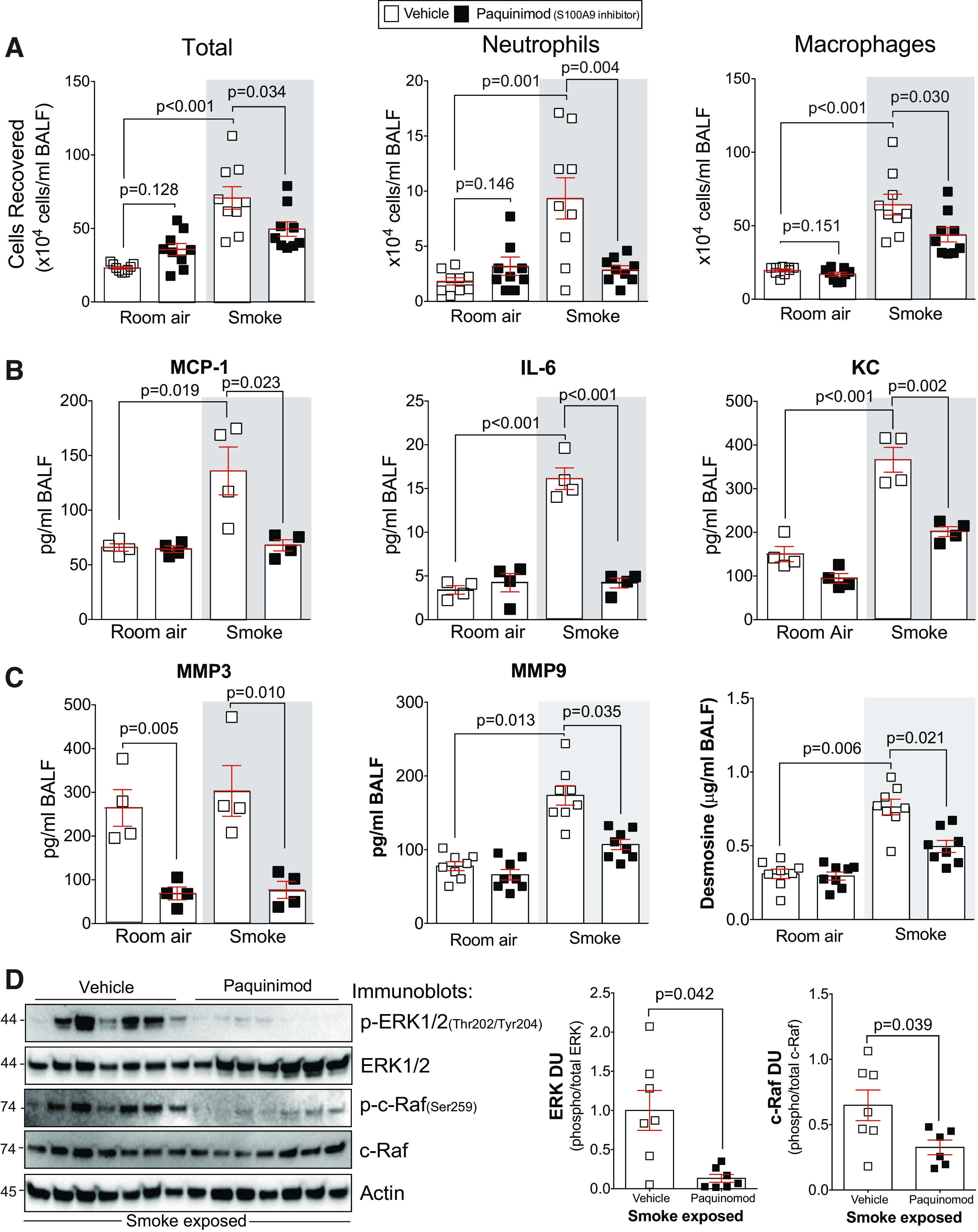
Paquinimod prevents inflammation and protease responses during smoke exposure. A: bronchoalveolar lavage fluid (BALF) cellularity levels were examined in A/J animals when paquinimod was administered daily before daily smoke exposure. BALF concentrations for MCP-1, IL-6, and keratinocyte-derived chemokine (KC) (B) and matrix metalloproteinase-3 (MMP-3), matrix metalloproteinase-9 (MMP-9), and desmosine (C) were quantified using Luminex bead assays. D: ERK and c-RAF phosphorylation were examined in lung tissue of smoke-exposed animals by Western blot and densitometry analysis. The representative blot shows data from seven animals per group. Graphs are represented as means ± SE, where n ≥ 5 per group. P values shown when comparing both treatments connected by a line were determined by two-way ANOVA with Tukey’s post hoc test. Graphs with two groups were analyzed by Student’s t tests.
Paquinimod treatment slows the progression of already established smoke-induced and age-related COPD in mice.
To confirm that paquinimod could be used to treat already established COPD, paquinimod was administered to mice following 2 mo of cigarette smoke exposure. The animals were exposed for an additional 2 mo with daily paquinimod intervention (Fig. 7). Similar to the S100a9−/− mouse and early treatment with paquinimod, delayed paquinimod treatment prevented smoke-induced ERK and c-RAF phosphorylation (Fig. 7A), loss of lung function, airspace enlargements and tissue destruction (Fig. 7, B–C), immune cell infiltration (Fig. 7D), and MCP-1, IL-6, and KC (Fig. 7E) and MMP-3, MMP-9, and desmosine release (Fig. 7F). Therefore, paquinimod treatment successfully attenuated several key factors associated with COPD progression.
Fig. 7.
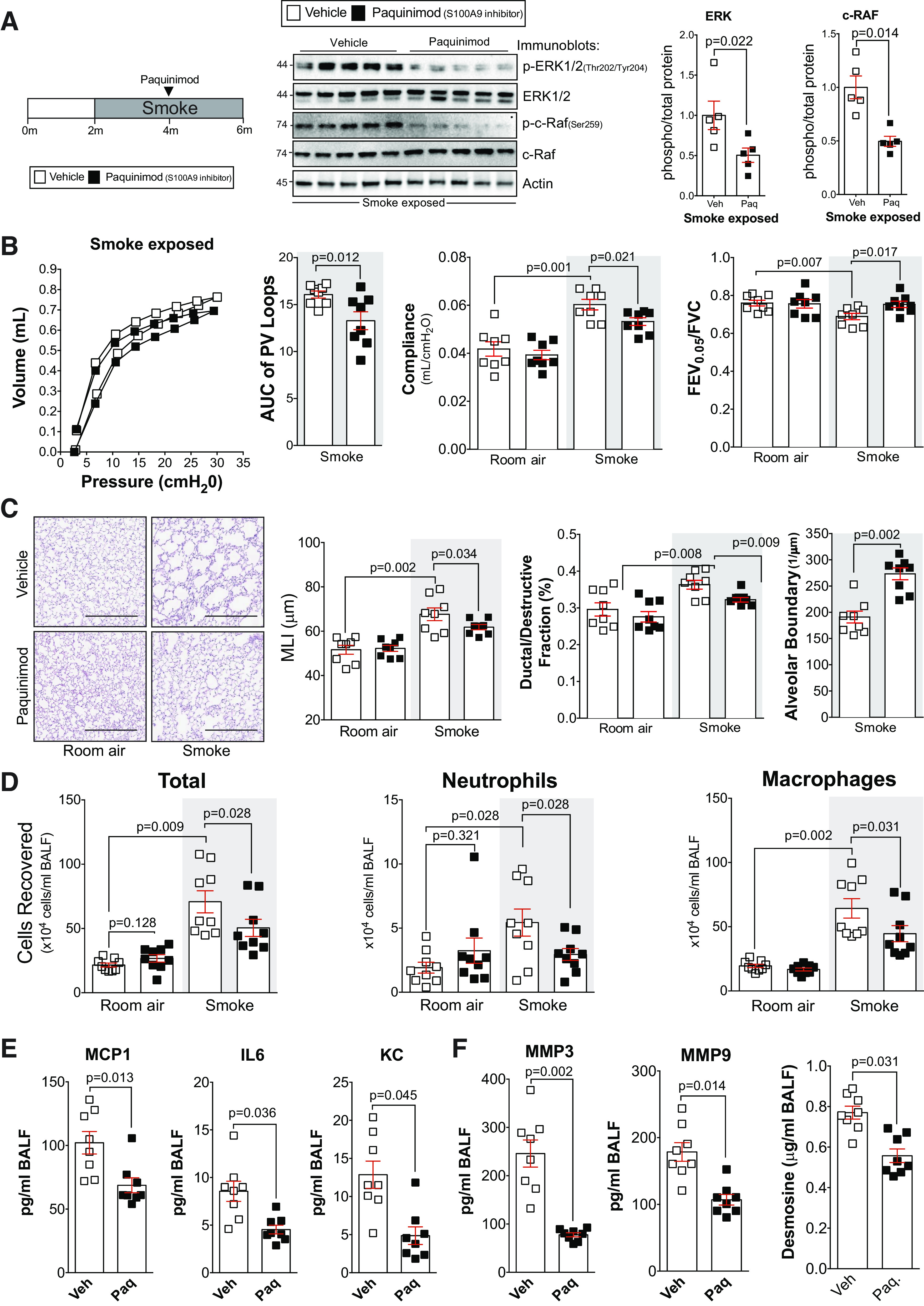
Delayed administration of paquinimod reduces the loss of lung function in smoke-exposed mice. A: A/J mice were smoke-exposed for 4 mo and began receiving paquinimod orally each day at the 2-mo mark after initiation of smoke exposure. ERK and c-RAF phosphorylation were examined in lung tissue of smoke-exposed animals by Western blot and densitometry analysis. B: forced expiratory volume (FEV) in the first 0.05 s of forced vital capacity (FVC), compliance, and pressure-volume loops with the area under the curve analysis were determined in each animal. C: mean linear intercept (MLI) was measured in the lungs of the mice to assess airspace size, and comparative histology images of the four mouse groups are presented here (scale bars = 500 µm). Alveolar count and ductal/destructive fractions were quantified in each animal by parenchymal airspace profiling. D: bronchoalveolar lavage fluid (BALF) cellularity levels were examined in A/J animals when paquinimod was administered daily before daily smoke exposure. BALF concentrations for monocyte chemoattractant protein-1 (MCP-1), IL-6, and keratinocyte-derived chemokine (KC) (E) and matrix metalloproteinase-3 (MMP-3), matrix metalloproteinase-9 (MMP-9), and desmosine (F) were quantified using Luminex bead assays. Graphs are represented as means ± SE, where n ≥ 5 per group. P values shown when comparing both treatments connected by a line were determined by two-way ANOVA with Tukey’s post hoc test. Graphs with two groups were analyzed by Student’s t tests.
Lung function declines with advancing age (60) and S100A9 levels increase with age (68). Therefore, we administered paquinimod to older animals and examined whether inhibition of S100A9 influenced age-dependent loss of lung function in the absence of cigarette smoke exposures. Paquinimod was administered to 18-mo-old mice for 2 mo (Fig. 8). Treatment reduced loss of lung function, airspace enlargements (Fig. 8, A–B), and IL-6, KC, and MMP-3 release (Fig. 8C). Therefore, age-dependent S100A9 signaling contributes to loss of lung function and can be modulated by treatment with paquinimod.
Fig. 8.
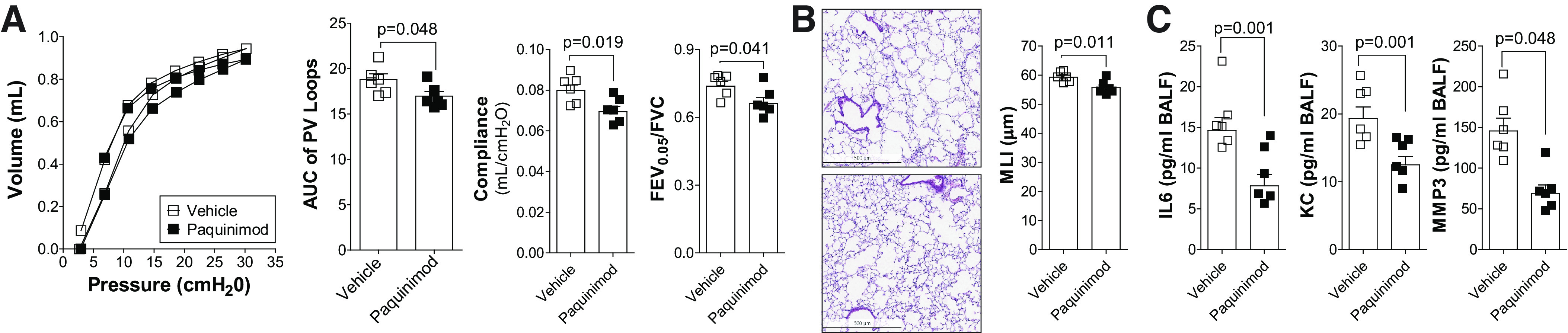
Paquinimod reduces the loss of lung function in aged mice. 18-mo-old C57BL/6J mice were administered paquinimod or vehicle orally each day for 2 mo. A: pressure-volume loops with the area under the curve, forced expiratory volume (FEV) in the first 0.05 s of forced vital capacity (FVC), and compliance analysis were determined in each animal. B: mean linear intercept (MLI) was measured in the lungs of the mice to assess airspace size, and comparative histology images of the four mouse groups are presented here (scale bars = 500 µm). C: bronchoalveolar lavage fluid (BALF) concentrations for IL-6, keratinocyte-derived chemokine (KC), and matrix metalloproteinase-3 (MMP-3) were quantified using Luminex bead assays. Graphs are represented as means ± SE, where n = 6 animals per group. P values shown when comparing both treatments connected by a line were determined by Student t tests.
ERK/c-RAF regulates S100A9-mediated cytokine and protease responses in human pulmonary fibroblasts.
We previously demonstrated that S100A9 protein induces TLR4 signaling, ERK phosphorylation, and secretion of MCP-1 and IL-8 in human primary small airway epithelial cells (24). However, to investigate MMP-3 changes, we used human primary fibroblasts because they appear to be the only pulmonary cells expressing MMP-3 when using the single-cell sequencing tool outlined by Reyfman et al. (52). We treated fibroblasts with ERK (LY3214996) and c-RAF (GW5074) inhibitors before S100A9 stimulation. Fibroblasts were treated with combined concentrations that induced no cell death, determined by lactate dehydrogenase (LDH) release assays (Fig. 9A). Inhibition of ERK or c-RAF prevented S100A9 induction of MCP-1, IL-6, IL-8, MMP-3, and MMP-9 gene expression (Fig. 9A).
Fig. 9.
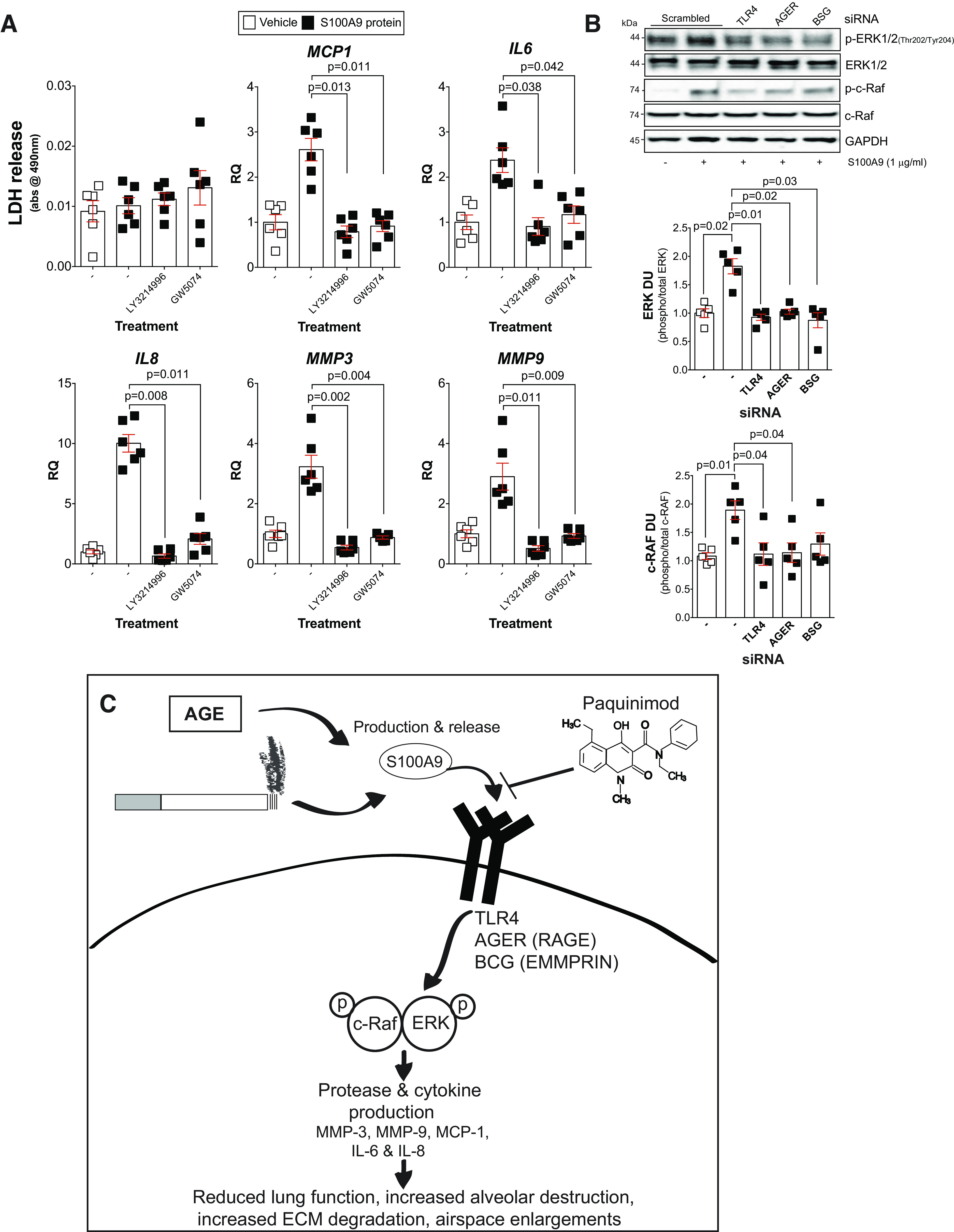
ERK and c-RAF phosphorylation are required for S100 calcium-binding protein A9 (S100A9) induction of cytokines and proteases in lung fibroblasts. A: fibroblasts from nonsmoker (NS) individuals were grown in media supplemented with the ERK inhibitor LY3214996 or the c-RAF inhibitor GW5074, and toxicity assays were performed by measuring lactate dehydrogenase (LDH) release. Real-time PCR analysis was performed on these cells to examine levels of monocyte chemoattractant protein-1 (MCP-1), IL-6, IL-8, matrix metalloproteinase-3 (MMP-3), and matrix metalloproteinase-9 (MMP-9). B: immunoblots subsequent to densitometry analysis were performed in fibroblasts following silencing of Toll-like receptor 4 (TLR4), advanced glycosylation end product-specific receptor (AGER), and sasigin (BSG), and ERK and c-RAF phosphorylation were determined. Data are represented as means ± SE, where each measurement was performed with n ≥ 4 subjects per group. P values shown when comparing both treatments connected by a line were determined by two-way ANOVA with Bonferroni posttests or by Student’s t tests. C: possible signaling mechanism following age- and cigarette smoke-induced S100A9 signaling.
S100A9 promotes fibroblast growth and activation to secrete cytokines and collagen production, via RAGE, ERK, MAPK, and NF-κB pathways (79). Therefore, we examine whether TLR4, RAGE (AGER), or EMMPRIN (BSG) were required to regulate S100A9-dependent ERK and c-RAF phosphorylation. Using siRNA (Supplemental Fig. S6; see https://doi.org/10.6084/m9.figshare.12863831.v1), we observed that S100A9 can induce ERK and c-RAF phosphorylation via all three receptors (Fig. 9B). Therefore, we cannot rule out any of these receptors in the regulation of ERK and c-RAF in S100A9-mediated signaling in fibroblasts.
DISCUSSION
Here, we demonstrate that age and cigarette smoke exposure enhance lung levels of S100A9, and using animal models of cigarette smoke-induced COPD, we confirm that S100A9 contributes to smoke-induced loss of lung function. Our findings demonstrate that targeting S100A9 signaling with an inhibitor, such as paquinimod, could slow the progression of cigarette smoke-associated COPD and age-associated loss of tissue function. Knockout or inhibition of S100A9 attenuated loss of lung function, airspace enlargement, protease and cytokine release into the airways, elastin degradation, and ERK/c-RAF phosphorylation. Changes in inflammation, kinase, and protease responses could contribute to the protection of the lung against smoke inhalation in mice. Our data also confirm that S100A9 triggers ERK and c-RAF phosphorylation through its receptors TLR4, RAGE, and EMMPRIN. Equally, inhibition of ERK and c-RAF activity with chemical inhibitors prevents S100A9-induced expression of MCP-1, IL-6, IL-8, MMP-3, and MMP-9 in pulmonary fibroblasts. There is a robust change in S100A9 expression during aging, which coincided with changes in lung structure and physiology. Since COPD is regarded as an age-related disease, targeting S100A9 signaling may counter several elements of lung aging. Therefore, smoke and age-related induction of S100A9 contributes to elevated kinases, protease, and cytokine responses that could collectively alter lung function (see Fig. 9C for the proposed mechanism scheme).
S100A9 expression is associated with aging, particularly in the central nervous system (68). Interestingly, C57BL/6 mice do not have changes in S100A9 expression in their lungs due to aging (68). However, this was reported to be strain dependent because the same group showed significant age-dependent changes in S100A9 expression in CB6F1 mice (68). Here, we observe changes in S100A9 expression in both C57BL/6J and A/J mice when comparing young and old animals, both in lung expression and secretion into the airways. We also see a correlation with age and S100A9 BALF concentration in human samples. Elevated age-related S100A9 expression in mice is partially due to the E26 transformation-specific (ETS) transcription factor SPI1/PU.1 (68). PU.1 is upregulated in fibroblasts of various fibrotic diseases and regulates fibrosis (76). There is evidence to suggest that PU.1 plays a role in COPD, with increased transcriptional activity in mice susceptible to cigarette smoke-induced COPD (9). Interestingly, vitamin D receptor (VDR)-deficient mice develop emphysema possibly due to the interaction of the VDR with PU.1 (36). PU.1 can also modulate T-cell receptor expression levels in CD4+ T-cells via regulating the DNA-binding activity of GATA-3 and subsequentially regulating T helper type 2 (Th2) cell development (12). PU.1 also helps macrophages maintain identity through controlling the miR-424-dependent translational repression of the nuclear factor 1 A-type protein (54). S100A9 expression is also sensitive to the Src kinase inhibitor PP2 (65) and STAT3 expression (39). We have previously identified that protein tyrosine phosphatase 1B (PTP1B) is a major negative regulator of S100A9 expression (24). Long-term exposure to cigarette smoke desensitizes PTP1B, resulting in persistent inflammatory signaling (24, 30), and exposing Ptp1b−/− mice to cigarette smoke exaggerates immune cell infiltration in the BALF and increased airspace enlargement (24). The DEAD-box RNA helicase DDX21 can also interact with TRIF to mediate S100A9 signaling (70). Therefore, evidence suggests that S100A9 and signaling that regulates its expression are altered due to aging or exposure to cigarette smoke. S100A9 induces senescence in mesenchymal stromal cells, which triggers inflammasome responses (63). Further studies are required to assess whether S100A9 is inducing senescence in COPD. It is also noteworthy to highlight that there are posttranslational changes that occur to S100A9 that could influence its responses. Relatively few proteins are targets of S-nitrosylation, but both S100A8 and S100A9 can undergo S-nitrosylation by nitric oxide (NO) donors, which alters their immune modulation responses, including leukocyte-endothelial cell interactions (41). Equally, the phosphorylated form of S100A9 is a potent inducer of cytokines (59), but little is known about the impact of cigarette smoke on S100A9 phosphorylation and subsequent binding to S100A8. Finally, S100A9 directly interacts with S100A8 and forms heterotetramers and dimers. Therefore, the relationship of S100A9 with S100A8 also requires addressing in COPD.
S100A9 is highly expressed in early infiltrating phagocytes (2), but S100A9 also influences leukocyte recruitment (56). We observe strong staining for S100A9 in immune cells within the lung but also positive staining in the epithelium. Profiling S100A9-positive cells within the lung would be of interest. Equally, examining the significance of neutrophil S100A9 in cell-specific animal models would further increase our knowledge of S100A9 in COPD. S100A9 stimulates the shedding of l-selectin, upregulates and activates Mac-1/CD11b, and induces neutrophil adhesion to fibrinogen (56). This is important in COPD, as l-selectin plays a major role in leukocyte recruitment (31), and leukocytes are elevated in COPD airways and their intracellular components, notably proteases, modulate extracellular matrix remodeling (75). S100A8 increases protease expression and activation in vivo, including MMP-2, MMP-3, MMP-9, MMP-13, A disintegrin and metalloproteinases (ADAMS)-4 and ADAMS-5 (71). However, little is known about S100A9 induction of proteases in the lungs. S100A9 binds to EMMPRIN, an inducer of matrix metalloproteinase synthesis, to regulate MMP-1 expression in a melanoma cell model (34). In human osteoarthritis synovium, paquinimod treatment blocked MMP-1 and MMP-3 secretion (58). Here, we observed that MMP-3 and MMP-9 are sensitive to S100A9 signaling during smoke exposure. MMP-1 is not expressed by mice but could also be mediated by S100A9 as TLR4 regulates its expression (28). MMP-9 is well documented to potentially play a role in COPD development (22). However, less is known about MMP-3 in COPD. MMP-3 polymorphisms are associated with cancer development in patients with COPD (8). We observed reduced desmosine in the BALF of S100a9−/− and paquinimod-treated animals. Changes in MMP-9 levels could contribute to this, but other proteases may also contribute to the degradation of elastin. S100 proteins also activates neutrophils (18), and S100A9 directly binds p67phox and p47phox (18) to potentiate NADPH oxidase activation in neutrophils. Therefore, inhibition of S100A9 could also prevent neutrophil-associated inflammation in COPD lungs in addition to preventing immune cell recruitment and expression of tissue-damaging cytokines and proteases.
S100A9 signaling is associated with multiple diseases, but it has a high affinity for lung localization, as B16F10 melanoma cells preferentially metastasize to the lungs and overexpress S100A8 and S100A9 in uteroglobin-knockout mice, and S100A9 is expressed highest in the lungs of uteroglobin-knockout mice, which are sensitized to RAGE signaling (57). Importantly, we previously demonstrated that RSV-infected human bronchial epithelial (HBE) cells isolated from COPD donors and fully differentiated and cultured in air-liquid interface secreted more S100A9 into the apical surface compared with cells from nonsmokers (24). Equally, exposing HBE cells isolated from COPD donors to S100A9 protein resulted in greater G-CSF and MCP-1 secretion than with cells from smokers and nonsmokers (24). Equally, cigarette smoke extract causes nitric oxide (NO) synthesis changes, with cigarette smoke extract causing an irreversible inhibition of endothelial nitric oxide synthase (eNOS) activity observed in pulmonary artery endothelial cells (67). S100A9 also promotes inflammation via the activation of TLR4 (72), RAGE (33), and CD147/ EMMPRIN (46). Cigarette smoke is known to modulate signaling of TLR4 (28), RAGE (53), and EMMPRIN (7). Therefore, smoke not onl increases S100A9 expression but also enhances the responses of known S100A9 receptors. S100A9 mediates ERK and c-RAF signaling that could influence many downstream responses, including ribosomal S6 kinases (27), cell cycle proteins, and mRNA translation-mediated proteins (55). ERK1/2 is closely associated with cell aging due to its regulation of proliferation, senescence, and mitochondria fate (80). Uniquely, here we observe that S100A9 did not influence c-Src, p38, or JNK responses. Most stimuli that activate p38 also activate JNK (38). We previously observed that smoke induced c-Src via PKC-α responses (29), which appear to be independent of S100A9 despite ERK being sensitive to c-Src activity.
Paquinimod, the S100A9 inhibitor used in this study, effectively treats experimental lupus and encephalomyelitis (32) and is well tolerated in patients with systemic lupus erythematosus (5). Paquinimod can influence macrophage populations (66) and T-cell proliferation (32) and can prevent S100A9 from being a chemoattractant (56). Paquinimod prevents S100A9 binding to these receptors (73). Paquinimod also reduces activation of disease-promoting transgenic natural killer T-II cells and CD115+ Ly6Chi monocytes and CD11b+ F4/80+ CD206+ macrophages, which coincided with reduced liver fibrosis in an animal model (26). Our paquinimod data suggest that paquinimod may be a good candidate for treating COPD and due to its role in fibrosis, it could possibly be used in pulmonary fibrosis (79). However, it should also be noted that the role of S100A9 in bacterial pathogen clearance needs to be investigated in the context of COPD exacerbations. S100A9 is linked to disease severity in sepsis shock (1, 19), effect on neutrophil recruitment in Streptococcus pneumoniae (51), and Salmonella infection (17) without impacting the bacterial load. However, S100a9−/− mice infected intranasally with pneumococci rapidly have elevated mortality (16). Equally, S100A9 protects from CD4+ T-helper type 2 cell hyperinflammation in response to Alternaria alternata (47). Therefore, additional studies on paquinimod during infection are required. Finally, S100A8 was recently shown to protect type II pneumonocytes from smoke-induced cell death (42). Whether S100A9 plays a similar role is unknown, and despite both proteins frequently interacting, both proteins also have independent signaling. Further studies on the interactions of both proteins in COPD and intracellular S100A9 signaling are required. Therefore, S100A9 or the proteins it interacts with could have multiple roles in the lungs of patients with COPD and require further studies to observe any plausible complications from long-term inhibition of S100A9. Finally, the correlation analysis in our study for human S100A9 BALF levels and age were not altered for possible confounding factors such as race, BMI, FEV1, and diffusing capacity for carbon monoxide (DLCO). Whether these or other factors contribute to S100A9 levels needs to be explored further.
Together, our data identify that cigarette smoke-induced S100A9 contributes to loss of lung function, airspace enlargements, elastin degradation, enhanced phosphorylation of ERK and c-RAF, and altered expression of MMP-3, MMP-9, MCP-1, IL-6, and KC/IL-8. Smoke- and age-dependent modulation of this pathway contribute to cigarette smoke-induced COPD.
GRANTS
This work was supported by grants made available to P. Geraghty by Flight Attendant Medical Research Institute (YCSA113380 and CIA160005) and the Alpha-1 Foundation (493373 and 614218).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.G. conceived and designed research; C.R., A.L., K. Z., H.G., M.C., A.W., B.J., M.P., M.R., A.D., S.M.M., M.E., and P.G. performed experiments; C.R., A.L., K.Z., H.G., M.R., and P.G. analyzed data; C.R., B.J., S.M.M., and P.G. interpreted results of experiments; C.R. and P.G. prepared figures; C.R. and P.G. drafted manuscript; C.R., A.L., B.J., S.M.M., R.F., M.E., and P.G. edited and revised manuscript; C.R., A.L., K.Z., H.G., M.C., A.W., B.J., M.P., M.R., A.D., S.M.M., R.F., M.E., and P.G. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank the Pulmonary Division of SUNY Downstate Medical Centre for the support. Paquinimod was provided by Active Biotech AB, Sweden.
REFERENCES
- 1.Alkhateeb T, Kumbhare A, Bah I, Youssef D, Yao ZQ, McCall CE, El Gazzar M. S100A9 maintains myeloid-derived suppressor cells in chronic sepsis by inducing miR-21 and miR-181b. Mol Immunol 112: 72–81, 2019. doi: 10.1016/j.molimm.2019.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Averill MM, Kerkhoff C, Bornfeldt KE. S100A8 and S100A9 in cardiovascular biology and disease. Arterioscler Thromb Vasc Biol 32: 223–229, 2012. doi: 10.1161/ATVBAHA.111.236927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bah I, Kumbhare A, Nguyen L, McCall CE, El Gazzar M. IL-10 induces an immune repressor pathway in sepsis by promoting S100A9 nuclear localization and MDSC development. Cell Immunol 332: 32–38, 2018. doi: 10.1016/j.cellimm.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 138: 16–27, 2016. doi: 10.1016/j.jaci.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 5.Bengtsson AA, Sturfelt G, Lood C, Rönnblom L, van Vollenhoven RF, Axelsson B, Sparre B, Tuvesson H, Ohman MW, Leanderson T. Pharmacokinetics, tolerability, and preliminary efficacy of paquinimod (ABR-215757), a new quinoline-3-carboxamide derivative: studies in lupus-prone mice and a multicenter, randomized, double-blind, placebo-controlled, repeat-dose, dose-ranging study in patients with systemic lupus erythematosus. Arthritis Rheum 64: 1579–1588, 2012. doi: 10.1002/art.33493. [DOI] [PubMed] [Google Scholar]
- 6.Berthier S, Paclet MH, Lerouge S, Roux F, Vergnaud S, Coleman AW, Morel F. Changing the conformation state of cytochrome b558 initiates NADPH oxidase activation: MRP8/MRP14 regulation. J Biol Chem 278: 25499–25508, 2003. doi: 10.1074/jbc.M209755200. [DOI] [PubMed] [Google Scholar]
- 7.Betsuyaku T, Tanino M, Nagai K, Nasuhara Y, Nishimura M, Senior RM. Extracellular matrix metalloproteinase inducer is increased in smokers’ bronchoalveolar lavage fluid. Am J Respir Crit Care Med 168: 222–227, 2003. doi: 10.1164/rccm.200301-103OC. [DOI] [PubMed] [Google Scholar]
- 8.Brzóska K, Bartłomiejczyk T, Sochanowicz B, Cymerman M, Grudny J, Kołakowski J, Kapka-Skrzypczak L, Kruszewski M, Sliwiński P, Roszkowski-Śliż K. Matrix metalloproteinase 3 polymorphisms as a potential marker of enhanced susceptibility to lung cancer in chronic obstructive pulmonary disease subjects. Ann Agric Environ Med 21: 546–551, 2014. doi: 10.5604/12321966.1120599. [DOI] [PubMed] [Google Scholar]
- 9.Cabanski M, Fields B, Boue S, Boukharov N, DeLeon H, Dror N, Geertz M, Guedj E, Iskandar A, Kogel U, Merg C, Peck MJ, Poussin C, Schlage WK, Talikka M, Ivanov NV, Hoeng J, Peitsch MC. Transcriptional profiling and targeted proteomics reveals common molecular changes associated with cigarette smoke-induced lung emphysema development in five susceptible mouse strains. Inflamm Res 64: 471–486, 2015. doi: 10.1007/s00011-015-0820-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cazzola M, Page CP, Calzetta L, Matera MG. Emerging anti-inflammatory strategies for COPD. Eur Respir J 40: 724–741, 2012. doi: 10.1183/09031936.00213711. [DOI] [PubMed] [Google Scholar]
- 11.Cesaro A, Anceriz N, Plante A, Pagé N, Tardif MR, Tessier PA. An inflammation loop orchestrated by S100A9 and calprotectin is critical for development of arthritis. PLoS One 7: e45478, 2012. doi: 10.1371/journal.pone.0045478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang HC, Han L, Jabeen R, Carotta S, Nutt SL, Kaplan MH. PU.1 regulates TCR expression by modulating GATA-3 activity. J Immunol 183: 4887–4894, 2009. doi: 10.4049/jimmunol.0900363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi IY, Gerlag DM, Herenius MJ, Thurlings RM, Wijbrandts CA, Foell D, Vogl T, Roth J, Tak PP, Holzinger D. MRP8/14 serum levels as a strong predictor of response to biological treatments in patients with rheumatoid arthritis. Ann Rheum Dis 74: 499–505, 2015. doi: 10.1136/annrheumdis-2013-203923. [DOI] [PubMed] [Google Scholar]
- 14.Cotoi OS, Dunér P, Ko N, Hedblad B, Nilsson J, Björkbacka H, Schiopu A. Plasma S100A8/A9 correlates with blood neutrophil counts, traditional risk factors, and cardiovascular disease in middle-aged healthy individuals. Arterioscler Thromb Vasc Biol 34: 202–210, 2014. doi: 10.1161/ATVBAHA.113.302432. [DOI] [PubMed] [Google Scholar]
- 15.D’Armiento J, Dalal SS, Okada Y, Berg RA, Chada K. Collagenase expression in the lungs of transgenic mice causes pulmonary emphysema. Cell 71: 955–961, 1992. doi: 10.1016/0092-8674(92)90391-O. [DOI] [PubMed] [Google Scholar]
- 16.De Filippo K, Neill DR, Mathies M, Bangert M, McNeill E, Kadioglu A, Hogg N. A new protective role for S100A9 in regulation of neutrophil recruitment during invasive pneumococcal pneumonia. FASEB J 28: 3600–3608, 2014. doi: 10.1096/fj.13-247460. [DOI] [PubMed] [Google Scholar]
- 17.De Jong HK, Achouiti A, Koh GC, Parry CM, Baker S, Faiz MA, van Dissel JT, Vollaard AM, van Leeuwen EM, Roelofs JJ, de Vos AF, Roth J, van der Poll T, Vogl T, Wiersinga WJ. Expression and function of S100A8/A9 (calprotectin) in human typhoid fever and the murine Salmonella model. PLoS Negl Trop Dis 9: e0003663, 2015. doi: 10.1371/journal.pntd.0003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doussiere J, Bouzidi F, Vignais PV. The S100A8/A9 protein as a partner for the cytosolic factors of NADPH oxidase activation in neutrophils. Eur J Biochem 269: 3246–3255, 2002. doi: 10.1046/j.1432-1033.2002.03002.x. [DOI] [PubMed] [Google Scholar]
- 19.Dubois C, Marcé D, Faivre V, Lukaszewicz AC, Junot C, Fenaille F, Simon S, Becher F, Morel N, Payen D. High plasma level of S100A8/S100A9 and S100A12 at admission indicates a higher risk of death in septic shock patients. Sci Rep 9: 15660, 2019. doi: 10.1038/s41598-019-52184-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ehrchen JM, Sunderkötter C, Foell D, Vogl T, Roth J. The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J Leukoc Biol 86: 557–566, 2009. doi: 10.1189/jlb.1008647. [DOI] [PubMed] [Google Scholar]
- 21.Fischer BM, Pavlisko E, Voynow JA. Pathogenic triad in COPD: oxidative stress, protease-antiprotease imbalance, and inflammation. Int J Chron Obstruct Pulmon Dis 6: 413–421, 2011. doi: 10.2147/COPD.S10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foronjy R, Nkyimbeng T, Wallace A, Thankachen J, Okada Y, Lemaitre V, D’Armiento J. Transgenic expression of matrix metalloproteinase-9 causes adult-onset emphysema in mice associated with the loss of alveolar elastin. Am J Physiol Lung Cell Mol Physiol 294: L1149–L1157, 2008. doi: 10.1152/ajplung.00481.2007. [DOI] [PubMed] [Google Scholar]
- 23.Foronjy RF, Dabo AJ, Taggart CC, Weldon S, Geraghty P. Respiratory syncytial virus infections enhance cigarette smoke induced COPD in mice. PLoS One 9: e90567, 2014. doi: 10.1371/journal.pone.0090567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foronjy RF, Ochieng PO, Salathe MA, Dabo AJ, Eden E, Baumlin N, Cummins N, Barik S, Campos M, Thorp EB, Geraghty P. Protein tyrosine phosphatase 1B negatively regulates S100A9-mediated lung damage during respiratory syncytial virus exacerbations. Mucosal Immunol 9: 1317–1329, 2016. doi: 10.1038/mi.2015.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Foronjy RF, Taggart CC, Dabo AJ, Weldon S, Cummins N, Geraghty P. Type-I interferons induce lung protease responses following respiratory syncytial virus infection via RIG-I-like receptors. Mucosal Immunol 8: 161–175, 2015. doi: 10.1038/mi.2014.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fransén Pettersson N, Deronic A, Nilsson J, Hannibal TD, Hansen L, Schmidt-Christensen A, Ivars F, Holmberg D. The immunomodulatory quinoline-3-carboxamide paquinimod reverses established fibrosis in a novel mouse model for liver fibrosis. PLoS One 13: e0203228, 2018. doi: 10.1371/journal.pone.0203228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frödin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol 151: 65–77, 1999. doi: 10.1016/S0303-7207(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 28.Geraghty P, Dabo AJ, D’Armiento J. TLR4 protein contributes to cigarette smoke-induced matrix metalloproteinase-1 (MMP-1) expression in chronic obstructive pulmonary disease. J Biol Chem 286: 30211–30218, 2011. doi: 10.1074/jbc.M111.238824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geraghty P, Hardigan A, Foronjy RF. Cigarette smoke activates the proto-oncogene c-src to promote airway inflammation and lung tissue destruction. Am J Respir Cell Mol Biol 50: 559–570, 2014. doi: 10.1165/rcmb.2013-0258OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geraghty P, Hardigan AA, Wallace AM, Mirochnitchenko O, Thankachen J, Arellanos L, Thompson V, D’Armiento JM, Foronjy RF. The glutathione peroxidase 1-protein tyrosine phosphatase 1B-protein phosphatase 2A axis. A key determinant of airway inflammation and alveolar destruction. Am J Respir Cell Mol Biol 49: 721–730, 2013. doi: 10.1165/rcmb.2013-0026OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hafezi-Moghadam A, Thomas KL, Prorock AJ, Huo Y, Ley K. L-selectin shedding regulates leukocyte recruitment. J Exp Med 193: 863–872, 2001. doi: 10.1084/jem.193.7.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Helmersson S, Sundstedt A, Deronic A, Leanderson T, Ivars F. Amelioration of experimental autoimmune encephalomyelitis by the quinoline-3-carboxamide paquinimod: reduced priming of proinflammatory effector CD4(+) T cells. Am J Pathol 182: 1671–1680, 2013. doi: 10.1016/j.ajpath.2013.01.032. [DOI] [PubMed] [Google Scholar]
- 33.Hermani A, De Servi B, Medunjanin S, Tessier PA, Mayer D. S100A8 and S100A9 activate MAP kinase and NF-kappaB signaling pathways and trigger translocation of RAGE in human prostate cancer cells. Exp Cell Res 312: 184–197, 2006. doi: 10.1016/j.yexcr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 34.Hibino T, Sakaguchi M, Miyamoto S, Yamamoto M, Motoyama A, Hosoi J, Shimokata T, Ito T, Tsuboi R, Huh NH. S100A9 is a novel ligand of EMMPRIN that promotes melanoma metastasis. Cancer Res 73: 172–183, 2013. doi: 10.1158/0008-5472.CAN-11-3843. [DOI] [PubMed] [Google Scholar]
- 35.Hsia CCW, Hyde DM, Ochs M, Weibel ER; ATS/ERS Joint Task Force on Quantitative Assessment of Lung Structure . An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am J Respir Crit Care Med 181: 394–418, 2010. doi: 10.1164/rccm.200809-1522ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu G, Dong T, Wang S, Jing H, Chen J. Vitamin D3-vitamin D receptor axis suppresses pulmonary emphysema by maintaining alveolar macrophage homeostasis and function. EBioMedicine 45: 563–577, 2019. doi: 10.1016/j.ebiom.2019.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim J, Bhattacharjee R, Snow AB, Capdevila OS, Kheirandish-Gozal L, Gozal D. Myeloid-related protein 8/14 levels in children with obstructive sleep apnoea. Eur Respir J 35: 843–850, 2010. doi: 10.1183/09031936.00075409. [DOI] [PubMed] [Google Scholar]
- 38.Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Keys JR, Landvatter SW, Strickler JE, McLaughlin MM, Siemens IR, Fisher SM, Livi GP, White JR, Adams JL, Young PR. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372: 739–746, 1994. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 39.Li C, Zhang F, Lin M, Liu J. Induction of S100A9 gene expression by cytokine oncostatin M in breast cancer cells through the STAT3 signaling cascade. Breast Cancer Res Treat 87: 123–134, 2004. doi: 10.1023/B:BREA.0000041594.36418.f6. [DOI] [PubMed] [Google Scholar]
- 40.Lian Z, Wang L, Yamaga S, Bonds W, Beazer-Barclay Y, Kluger Y, Gerstein M, Newburger PE, Berliner N, Weissman SM. Genomic and proteomic analysis of the myeloid differentiation program. Blood 98: 513–524, 2001. doi: 10.1182/blood.V98.3.513. [DOI] [PubMed] [Google Scholar]
- 41.Lim SY, Raftery M, Cai H, Hsu K, Yan WX, Hseih HL, Watts RN, Richardson D, Thomas S, Perry M, Geczy CL. S-nitrosylated S100A8: novel anti-inflammatory properties. J Immunol 181: 5627–5636, 2008. doi: 10.4049/jimmunol.181.8.5627. [DOI] [PubMed] [Google Scholar]
- 42.Lin CR, Bahmed K, Criner GJ, Marchetti N, Tuder RM, Kelsen S, Bolla S, Mandapati C, Kosmider B. S100A8 protects human primary alveolar type II cells against injury and emphysema. Am J Respir Cell Mol Biol 60: 299–307, 2019. doi: 10.1165/rcmb.2018-0144OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer KC. The role of immunity and inflammation in lung senescence and susceptibility to infection in the elderly. Semin Respir Crit Care Med 31: 561–574, 2010. doi: 10.1055/s-0030-1265897. [DOI] [PubMed] [Google Scholar]
- 44.Miller MR. Structural and physiological age-associated changes in aging lungs. Semin Respir Crit Care Med 31: 521–527, 2010. doi: 10.1055/s-0030-1265893. [DOI] [PubMed] [Google Scholar]
- 44a.National Center for Chronic Disease Prevention and Health Promotion (US) Office on Smoking and Health . The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General. Atlanta: US Department of Health and Human Services, Centers for Disease Control and Prevention (US), 2014. [PubMed] [Google Scholar]
- 45.Ohnishi K, Takagi M, Kurokawa Y, Satomi S, Konttinen YT. Matrix metalloproteinase-mediated extracellular matrix protein degradation in human pulmonary emphysema. Lab Invest 78: 1077–1087, 1998. [PubMed] [Google Scholar]
- 46.Olsson A, Nakhlé J, Sundstedt A, Plas P, Bauchet AL, Pierron V, Bruetschy L, Deronic A, Törngren M, Liberg D, Schmidlin F, Leanderson T. Tasquinimod triggers an early change in the polarization of tumor associated macrophages in the tumor microenvironment. J Immunother Cancer 3: 53, 2015. doi: 10.1186/s40425-015-0098-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Palmer LD, Maloney KN, Boyd KL, Goleniewska AK, Toki S, Maxwell CN, Chazin WJ, Peebles RS Jr, Newcomb DC, Skaar EP. The innate immune protein S100A9 protects from T-helper cell type 2-mediated allergic airway inflammation. Am J Respir Cell Mol Biol 61: 459–468, 2019. doi: 10.1165/rcmb.2018-0217OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petrache I, Natarajan V, Zhen L, Medler TR, Richter AT, Cho C, Hubbard WC, Berdyshev EV, Tuder RM. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med 11: 491–498, 2005. doi: 10.1038/nm1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pouwels SD, Heijink IH, ten Hacken NH, Vandenabeele P, Krysko DV, Nawijn MC, van Oosterhout AJ. DAMPs activating innate and adaptive immune responses in COPD. Mucosal Immunol 7: 215–226, 2014. doi: 10.1038/mi.2013.77. [DOI] [PubMed] [Google Scholar]
- 50.Pouwels SD, Nawijn MC, Bathoorn E, Riezebos-Brilman A, van Oosterhout AJ, Kerstjens HA, Heijink IH. Increased serum levels of LL37, HMGB1 and S100A9 during exacerbation in COPD patients. Eur Respir J 45: 1482–1485, 2015. doi: 10.1183/09031936.00158414. [DOI] [PubMed] [Google Scholar]
- 51.Raquil MA, Anceriz N, Rouleau P, Tessier PA. Blockade of antimicrobial proteins S100A8 and S100A9 inhibits phagocyte migration to the alveoli in streptococcal pneumonia. J Immunol 180: 3366–3374, 2008. doi: 10.4049/jimmunol.180.5.3366. [DOI] [PubMed] [Google Scholar]
- 52.Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, Fernandez R, Akbarpour M, Chen CI, Ren Z, , et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med 199: 1517–1536, 2019. doi: 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robinson AB, Stogsdill JA, Lewis JB, Wood TT, Reynolds PR. RAGE and tobacco smoke: insights into modeling chronic obstructive pulmonary disease. Front Physiol 3: 301, 2012. doi: 10.3389/fphys.2012.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosa A, Ballarino M, Sorrentino A, Sthandier O, De Angelis FG, Marchioni M, Masella B, Guarini A, Fatica A, Peschle C, Bozzoni I. The interplay between the master transcription factor PU.1 and miR-424 regulates human monocyte/macrophage differentiation. Proc Natl Acad Sci USA 104: 19849–19854, 2007. doi: 10.1073/pnas.0706963104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 68: 320–344, 2004. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA. Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol 170: 3233–3242, 2003. doi: 10.4049/jimmunol.170.6.3233. [DOI] [PubMed] [Google Scholar]
- 57.Saha A, Lee YC, Zhang Z, Chandra G, Su SB, Mukherjee AB. Lack of an endogenous anti-inflammatory protein in mice enhances colonization of B16F10 melanoma cells in the lungs. J Biol Chem 285: 10822–10831, 2010. doi: 10.1074/jbc.M109.083550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schelbergen RF, Geven EJ, van den Bosch MH, Eriksson H, Leanderson T, Vogl T, Roth J, van de Loo FA, Koenders MI, van der Kraan PM, van den Berg WB, Blom AB, van Lent PL. Prophylactic treatment with S100A9 inhibitor paquinimod reduces pathology in experimental collagenase-induced osteoarthritis. Ann Rheum Dis 74: 2254–2258, 2015. doi: 10.1136/annrheumdis-2014-206517. [DOI] [PubMed] [Google Scholar]
- 59.Schenten V, Plançon S, Jung N, Hann J, Bueb JL, Bréchard S, Tschirhart EJ, Tolle F. Secretion of the phosphorylated form of S100A9 from neutrophils is essential for the proinflammatory functions of extracellular S100A8/A9. Front Immunol 9: 447, 2018. doi: 10.3389/fimmu.2018.00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schulte H, Mühlfeld C, Brandenberger C. Age-related structural and functional changes in the mouse lung. Front Physiol 10: 1466, 2019. doi: 10.3389/fphys.2019.01466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shalaby KH, Gold LG, Schuessler TF, Martin JG, Robichaud A. Combined forced oscillation and forced expiration measurements in mice for the assessment of airway hyperresponsiveness. Respir Res 11: 82, 2010. doi: 10.1186/1465-9921-11-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shi L, Zhao Y, Fei C, Guo J, Jia Y, Wu D, Wu L, Chang C. Cellular senescence induced by S100A9 in mesenchymal stromal cells through NLRP3 inflammasome activation. Aging (Albany NY) 11: 9626–9642, 2019. doi: 10.18632/aging.102409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Simard JC, Girard D, Tessier PA. Induction of neutrophil degranulation by S100A9 via a MAPK-dependent mechanism. J Leukoc Biol 87: 905–914, 2010. doi: 10.1189/jlb.1009676. [DOI] [PubMed] [Google Scholar]
- 65.Spijkers-Hagelstein JA, Schneider P, Hulleman E, de Boer J, Williams O, Pieters R, Stam RW. Elevated S100A8/S100A9 expression causes glucocorticoid resistance in MLL-rearranged infant acute lymphoblastic leukemia. Leukemia 26: 1255–1265, 2012. doi: 10.1038/leu.2011.388. [DOI] [PubMed] [Google Scholar]
- 66.Stenström M, Nyhlén HC, Törngren M, Liberg D, Sparre B, Tuvesson H, Eriksson H, Leanderson T. Paquinimod reduces skin fibrosis in tight skin 1 mice, an experimental model of systemic sclerosis. J Dermatol Sci 83: 52–59, 2016. doi: 10.1016/j.jdermsci.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 67.Su Y, Han W, Giraldo C, De Li Y, Block ER. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am J Respir Cell Mol Biol 19: 819–825, 1998. doi: 10.1165/ajrcmb.19.5.3091. [DOI] [PubMed] [Google Scholar]
- 68.Swindell WR, Johnston A, Xing X, Little A, Robichaud P, Voorhees JJ, Fisher G, Gudjonsson JE. Robust shifts in S100a9 expression with aging: a novel mechanism for chronic inflammation. Sci Rep 3: 1215, 2013. doi: 10.1038/srep01215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Togo S, Holz O, Liu X, Sugiura H, Kamio K, Wang X, Kawasaki S, Ahn Y, Fredriksson K, Skold CM, Mueller KC, Branscheid D, Welker L, Watz H, Magnussen H, Rennard SI. Lung fibroblast repair functions in patients with chronic obstructive pulmonary disease are altered by multiple mechanisms. Am J Respir Crit Care Med 178: 248–260, 2008. doi: 10.1164/rccm.200706-929OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsai SY, Segovia JA, Chang TH, Morris IR, Berton MT, Tessier PA, Tardif MR, Cesaro A, Bose S. DAMP molecule S100A9 acts as a molecular pattern to enhance inflammation during influenza A virus infection: role of DDX21-TRIF-TLR4-MyD88 pathway. PLoS Pathog 10: e1003848, 2014. doi: 10.1371/journal.ppat.1003848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van Lent PL, Grevers LC, Blom AB, Arntz OJ, van de Loo FA, van der Kraan P, Abdollahi-Roodsaz S, Srikrishna G, Freeze H, Sloetjes A, Nacken W, Vogl T, Roth J, van den Berg WB. Stimulation of chondrocyte-mediated cartilage destruction by S100A8 in experimental murine arthritis. Arthritis Rheum 58: 3776–3787, 2008. doi: 10.1002/art.24074. [DOI] [PubMed] [Google Scholar]
- 72.Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, Nacken W, Foell D, van der Poll T, Sorg C, Roth J. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med 13: 1042–1049, 2007. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 73.Wache C, Klein M, Ostergaard C, Angele B, Häcker H, Pfister HW, Pruenster M, Sperandio M, Leanderson T, Roth J, Vogl T, Koedel U. Myeloid-related protein 14 promotes inflammation and injury in meningitis. J Infect Dis 212: 247–257, 2015. doi: 10.1093/infdis/jiv028. [DOI] [PubMed] [Google Scholar]
- 74.Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100A8/A9 in Inflammation. Front Immunol 9: 1298, 2018. doi: 10.3389/fimmu.2018.01298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wight TN, Frevert CW, Debley JS, Reeves SR, Parks WC, Ziegler SF. Interplay of extracellular matrix and leukocytes in lung inflammation. Cell Immunol 312: 1–14, 2017. doi: 10.1016/j.cellimm.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wohlfahrt T, Rauber S, Uebe S, Luber M, Soare A, Ekici A, Weber S, Matei AE, Chen CW, Maier C, Karouzakis E, Kiener HP, Pachera E, Dees C, Beyer C, Daniel C, Gelse K, Kremer AE, Naschberger E, Stürzl M, Butter F, Sticherling M, Finotto S, Kreuter A, Kaplan MH, Jüngel A, Gay S, Nutt SL, Boykin DW, Poon GMK, Distler O, Schett G, Distler JHW, Ramming A. PU.1 controls fibroblast polarization and tissue fibrosis. Nature 566: 344–349, 2019. doi: 10.1038/s41586-019-0896-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xiao R, Goldklang MP, D’Armiento JM. Parenchymal airspace profiling: sensitive quantification and characterization of lung structure evaluating parenchymal destruction. Am J Respir Cell Mol Biol 55: 708–715, 2016. doi: 10.1165/rcmb.2016-0143OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xie M, Liu X, Cao X, Guo M, Li X. Trends in prevalence and incidence of chronic respiratory diseases from 1990 to 2017. Respir Res 21: 49, 2020. doi: 10.1186/s12931-020-1291-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xu X, Chen H, Zhu X, Ma Y, Liu Q, Xue Y, Chu H, Wu W, Wang J, Zou H. S100A9 promotes human lung fibroblast cells activation through receptor for advanced glycation end-product-mediated extracellular-regulated kinase 1/2, mitogen-activated protein-kinase and nuclear factor-κB-dependent pathways. Clin Exp Immunol 173: 523–535, 2013. doi: 10.1111/cei.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zou J, Lei T, Guo P, Yu J, Xu Q, Luo Y, Ke R, Huang D. Mechanisms shaping the role of ERK1/2 in cellular senescence Mol Med Rep 19: 759–770, 2019. doi: 10.3892/mmr.2018.9712. [DOI] [PMC free article] [PubMed] [Google Scholar]