

Abstract
Lilium lankongense Franchet is a lily species found on the Qinghai-Tibet Plateau. It is pink with deep red spots, has a high ornamental value, and is used in hybrid breeding of horticultural lily varieties. We have insufficient knowledge of the genetic resources of L. lankongense and its phylogenetic relationships with related species. Recent molecular phylogenetic studies have shown a very close phylogenetic relationship between L. lankongense and the five species L. duchartrei, L. stewartianum, L. matangense, L. lophophorum, and L. nanum. However, molecular markers still lack sufficient signals for population-level research of the genus Lilium. We sequenced and compared the complete plastid sequences of L. lankongense and its five related species. The genomes ranged from 152,307 bp to 152,611 bp. There was a slight inconsistency detected in inverted repeat and single copy boundaries and there were 53 to 63 simple sequence repeats in the six species. Two of the 12 highly variable regions (trnC-petN and rpl32-trnL) were verified in 11 individuals and are promising for population-level studies. We used the complete sequence of 33 plastid genomes, the protein-coding region sequence, and the nuclear ITS sequence to reconstruct the phylogenetic tree of Lilium species. Our results showed that the plastid gene tree and nuclear gene tree were not completely congruent, which may be caused by hybridization, insufficient information contained in the nuclear ITS, or the small number of samples. The results of phylogenetic analysis based on plastid genomes indicated that the six Lilium species were closely related. Our study provides a preliminarily rebuilt backbone phylogeny that is significant for future molecular and morphological studies of Lilium.
Keywords: Lilium, Lilium-specific primers, Lilium lankongense, Phylogeny, Plastid genome
Introduction
Plastids are semi-autonomous organelles that play an extremely important role in photosynthesis. The plastid genome is a circular double-stranded DNA ranging from 120–217 kb (Li et al., 2013; Palmer, 1985). The plastid genomes of most angiosperms exhibit a typical quadripartite structure, composed of large single copy (LSC) and small single copy (SSC) regions, which are separated by two copy regions known as the inverted repeat (IR). The most common structure follows the formulation derived from Nicotiana tabacum L. (tobacco). The IR, which is flanked by genes ycf1 and trnH-GUG, is designated as IRA and the other IR as IRB (genes rps19 and ndhF are on either side), the junctions between IR and LSC or SSC are designated as JLB(LSC/IRB), JSB(SSC/IRB), JSA (SSC/IRA) and JLA(LSC/IRA) (Shinozaki et al., 1986). The plastid genome is considered to be ideal for studying endangered species conservation (Zhao et al., 2018), relationships of lower taxonomic levels (Kang et al., 2019; Parks, Cronn & Liston, 2009), population genetics studies (Ahmed, 2014) and phylogenetics (Xie et al., 2019b; Xie et al., 2018; Yang et al., 2020) due to its compact size, maternal inheritance, absence of recombination, and low evolutionary rate (Palmer, 1985). The cost of plastid genome sequencing has been reduced by the development of sequencing technologies, providing convenience for related analyses based on the plastid genome. Scholars have developed many molecular markers applicable to population genetics and phylogeny based on the whole plastid genome (Scarcelli et al., 2011; Shaw et al., 2007).
At present, Lilium L. (Liliaceae) includes approximately 120 species (Peruzzi, 2016). Almost all of the species are distributed in the temperate and cold regions of the Northern hemisphere and are intermittently distributed in East Asia, Europe, and North America (Liang & Tamura, 2000; MacRae, 1998). Since the development of molecular biology, there have been some discrepancies in the research of classification of the genus Lilium based on molecular systematics and morphological characteristics. Most early researchers focused on the classification, origin, evolution, and phylogeny of Lilium with the nuclear ITS sequence and proposed suggestions to revise some Lilium species (Du et al., 2014; Dubouzet & Shinoda, 1999; Ikinci, Oberprieler & Güner, 2006; Lee et al., 2011; Nishikawa et al., 2001; Nishikawa et al., 1999). Plastid gene fragments have become common molecular markers in the phylogeny within Lilium. For example, Hayashi & Kawano (2000) distinguished the evolutionary relationships between Lilium and related groups with the rbcL and matK genes, and determined that the previous division of Lilium based on morphological traits should be revised. Due to the limited information contained in plastid gene fragments, researchers usually combine nuclear genes and plastid gene fragments to analyze the subgenus classification and interspecies relationships of Lilium. However, the phylogenetic location of some species was unclear if the phylogenetic trees constructed by ITS and plastid genes were incongruous (Gao et al., 2013; Gao, Harris & He, 2015; Givnish et al., 2020; Huang et al., 2018). In the latest research, Givnish et al. (2020) reconstructed phylogenetic trees with 69 Lilium whole plastomes and 440 nuclear genes loci of 67 Lilium species, and further resolved the phylogenetic relationship of Lilium. Although the phylogenetic trees constructed by nuclear genes and plastid genes initially revised the phylogenetic location of some species of Lilium, the relationship among related species still needs to be resolved using population-level analyses (Lai et al., 2016; Shen, Zhou & He, 2014). Plastid non-coding regions (intergenic regions and introns) are useful for interspecies and intraspecies analyses due to the different rates of nucleotide substitutions among different taxa. A plastid non-coding region may cause analyses to vary greatly in different taxa (Gielly & Taberlet, 1994; Shaw et al., 2005). It has been found that universal primers are not appropriate for the intraspecific level studies of Lilium, especially at the population level (Jiang, 2017; Lai et al., 2016). New Lilium-specific primers are needed to amplify suitable plastid fragments for intraspecific phylogeographic and population genetic studies within the genus Lilium.
Lilium lankongense Franchet has an ovoid-globose bulb, pink tepals with deep red spots, and is endemic to the Qinghai-Tibet Plateau. It has revolute tepals in the margin and nectaries papillose on both surfaces. L. lankongense is the parent of hybrids in lily cultivars due to its high resistance to Botrytis blight (an important and very damaging disease of Asiatic hybrids) (Liang & Tamura, 2000; MacRae, 1998; North & Wills, 1969; Proscevičius, Rančeliene & Dambrauskaite, 2007; Van Tuyl et al., 2002). In early molecular and morphological studies, L. lankongense was considered to be closely related to L. duchartrei Franchet. However, recent molecular evidence has shown that L. lankongense, L. duchartrei, L. stewartianum I.B. Balfour & W.W. Smith, L. matangense J.M. Xu, L. lophophorum (Bureau & Franchet) Franchet and L. nanum Klotzsch, have very close phylogenetic relationships (Gao, Harris & He, 2015; Huang et al., 2018). This is inconsistent with the traditional classification system based on morphological characteristics. L. lankongense, L. duchartrei, L. stewartianum, and L. matangense have similar floral features with revolute tepals, while the corolla shape of L. lophophorum and L. nanum are distinct from the four aforementioned Lilium species, which are campanulate (Fig. 1). The other five species possess ovoid to oblong bulbs with multiple scales, and are mainly found in the Qinghai-Tibet Plateau, similar to L. lankongense (Gao, Harris & He, 2015; Huang et al., 2018; Liang & Tamura, 2000). Full plastid genomes more accurately reflect interspecies relationships as opposed to single or few plastid DNA (ptDNA) fragments (Yang et al., 2019). Most clades of the plastid genome trees have higher support values in Lilium compared with the phylogenetic tree constructed by the nuclear ITS (Liu et al., 2018b). Therefore, it is essential to reconstruct the phylogenetic relationship of Lilium based on its plastid genomes. More effective molecular markers also need to be developed to better solve the interspecies relationship of the related species of Lilium.
Figure 1. Floral morphology of L. lankongense and its five related species.

(A) L. lankongense (B) L. duchartrei (C) L. matangense (D) L. stewartianum (E)L. lophophorum (F) L. nanum.
We sequenced, assembled, characterized, and compared the whole plastid genomes of L. lankongense and its five related species. We sought to: (1) explore the phylogenetic relationship of Lilium, particularly for L. lankongense, L. duchartrei, L. stewartianum, L. matangense, L. lophophorum, and L. nanum using the six plastid genomes and ITS sequences measured in this paper and the published plastid genomes of other Lilium species and their corresponding ITS sequences and discuss the possible reasons for the discrepancy between the ITS tree and the plastid gene tree in Lilium. (2) Select non-coding regions with relatively more variable loci to develop useful Lilium-specific primers for population studies by comparing the six plastid genomes and previously published plastid genomes of other Lilium species on the NCBI (https://www.ncbi.nlm.nih.gov/). We provide basic data on plastid genomes for classification, species identification, molecular breeding, biogeography, and genetic diversity in the genus Lilium.
Materials & Methods
Plant materials and DNA extraction
Leaf samples were collected from each field site (Table S1) and then immediately dried with silica gel to preserve them for DNA extraction. The total genomic DNA was extracted via the NuClean Plant Genomic DNA Kit (ComWin Biosciences, Jiangsu, China) following the manufacturer’s instructions.
Plastid genome sequencing, assembling, and annotation
All complete plastid genomes were sequenced using the Illumina Novaseq 6000 Platform (Novagene, Beijing, China) with an average paired-end read length of 150 bp. In order to assess the quality of sequenced raw reads, we used the FastQC (Andrews, 2014) v0.11.7. The plastid genome related reads were filtered via mapping after they were assessed for quality and all the raw reads were associated with the previously reported plastid genome sequences in Liliaceae using Bowtie 2 (Langmead & Salzberg, 2012) v2.3.4.3. The published sequence with the highest overall similarity to the reads was used as the seed sequence for further assembly using NOVOPlasty v2.7.2 (Dierckxsens, Mardulyn & Smits, 2017) to obtain a draft sequence. The draft sequence was imported into Geneious v11.0.4 software (Kearse et al., 2012) and was compared with the optimal reference sequence and mapped reads to check and correct for mismatches. The annotation of the plastid genome was also performed in Geneious. Every annotated gene was manually edited for start/stop codons and intron/exon boundaries to correct errors and ensure the accurate annotation of the genome. Finally, circular plastid genome maps were drawn using OGDRAW (Lohse et al., 2013).
Simple sequence repeats (SSRs) and IR borders analysis
Perl script MISA (Thiel et al., 2003) was used to detect SSRs loci in the six plastid genomes of Lilium with thresholds of 10, 5, 4, 3, 3, and 3 for mono-, di-, tri-, tetra-, penta-, and hexanucleotides, respectively. The IR regions were discovered using the plugin Repeat Finder v1.0 in Geneious and were manually checked. The IR plot of the six Lilium species plastid genomes was obtained using an IRscope online program to compare the IR borders and neighboring genes of the six Lilium species plastid genomes (Amiryousefi, Hyvönen & Poczai, 2018).
Lilium-specific primers
We generated multiple sequence alignments of the six newly assembled sequences and all available plastid genome sequences in Lilium (downloaded from NCBI in October 2019) using MAFFT v1.4.0 under the automatic model selection option (Katoh & Standley, 2013). We developed primers specific for the highly mutational regions in Lilium using Geneious after alignment (Kearse et al., 2012). The primers were synthesized by Sangon Biotech, China. 12 primers pairs were designed (Table S2) through a series of experiments and two segments of gene spacers were selected with the goal of identifying those primer pairs that would produce the best results. Primer sequences and polymerase chain reaction (PCR) amplification results were summarized in Table S3. PCR of trnC-petN was conducted as follows: initial denaturation at 94 °C (3 min) followed by 33 cycles with denaturation at 94 °C (40 s), annealing at 60 °C (50 s), extension at 72 °C (1 min). PCR for rpl32-trnL was: initial denaturation at 94 °C (3 min), followed by 33 cycles of denaturation at 94 °C (40 s), annealing at 54 °C (45 s) and elongation at 72 °C (1 min). All reactions ended with a final elongation at 72 °C (10 min) followed by a holding step at 4 °C. All PCR products were sequenced in both directions by Sanger sequencing (Sanger & Coulson, 1975) (performed at Sangon Biotech, China), and then assembled and edited using Seqman v7.1.0 software (DNAstar package; DNAStar Inc., Madison, WI, USA) (Burland, 2000) for obtaining consensus sequences. PCR products of these intergenic spacer regions were obtained for 11 additional accessions belonging to three Lilium species, including those from geographically remote populations. L. lankongense was represented by five populations (from Sichuan, Yunnan, and Tibet), L. duchartrei was represented by four populations (from Sichuan), and L. lophophorum was represented by two populations (from Sichuan and Yunnan); each of these species had one individual per population.
Phylogenetic analysis
We combined the 27 published plastid genome sequences (downloaded from NCBI in October 2019) and ITS sequences from the NCBI and the six plastid genome sequences and corresponding ITS sequences measured by this study (GenBank accession number: MT260888 –MT260893, leaf samples shown in Table S1). Two sequences in Fritillaria were treated as the outgroups. We adopted the maximum likelihood (ML) and Bayesian inference (BI) methods to analyze the phylogenetic relationship of Lilium. The universal primers ITS4 and ITS5 (White et al., 1990) were used to amplify the ITS according to the standard PCR protocols of Gao et al. (2013). A total of three data sets, namely 33 complete plastid genome sequences, all shared protein-coding genes (CDS) (only containing one IR). The 33 plastid genome sequences and ITS sequences of the species corresponding to these 33 plastid genome sequences were used to reconstruct the phylogenetic tree of Lilium. We extracted the shared CDS from 33 plastid genome sequences as follows: all CDS were extracted from all 33 sequences in the software Geneious v11.0.4, and shared 71 CDS (excluding additional copies in the IR) of the 33 species were selected and manually sorted. The CDS belonging to one species were concatenated to generate 33 sequences for phylogenetic analysis. The three data sets were aligned by MAFFT v1.4.0 under the automatic model selection option, trimmed via trimAl v1.2 (Capella-Gutiérrez, Silla-Martínez & Gabaldón, 2009) with parameters Trimal -in *.fas -noallgaps -fasta -out *.fas, and were compiled into three alignment matrices. The three alignment matrices were used to search for the best-fit substitution model using ModelFinder plugin in PhyloSuite v1.2.1 software before ML and BI analyses were conducted (Zhang et al., 2020). ML analysis based on the SYM+G (ITS) and GTR + I + G (plastid genome sequences and shared concatenated 71 CDS sequences) model was conducted using RAxML v8.2.8 (Stamatakis, 2014) with 1000 bootstrap replicates. BI analysis based on the GTR + I + G model was conducted using the Mrbayes v3.2.6 plugin in PhyloSuite v1.2.1 software. The Monte Carlo Markov chains (MCMCs) were run 1 × 108 generations and the first 30% of trees were discarded as burn-in. ML tree used Bootstrap support (BS) and BI tree used posterior probability (PP) to evaluate the feasibility of each branch.
Results
Plastid features of Lilium species
The complete plastid genome of the six species in Lilium was deposited in GenBank. These plastid genomes ranged from 152,307 bp (L. stewartianum) to 152,611 bp (L. lankongense) in length, with the minimum and maximum differences being 15 and 304 bp, respectively (Table 1 and Fig. 2). All six plastid genomes displayed a typical quadripartite structure, consisting of the LSC (81, 870–82, 150 bp) and SSC (17, 382–17, 542 bp) regions separated by a pair of IRs (26, 425–26, 577 bp). In the six Lilium plastid genomes, the overall GC content was 37%. Both contained 132 genes, with 86 protein-coding genes, 38 tRNA genes, eight ribosomal RNA genes (Table 1 and Table S3).
Table 1. Summary of six complete plastid genomes of Lilium.
| Taxon | Full | LSC length (bp) | SSC length (bp) | IR length (bp) | Gene number | Protein-coding | tRNAs | rRNAs | GenBank accesion number | |
|---|---|---|---|---|---|---|---|---|---|---|
| Length (bp) | GC (%) | |||||||||
| L. lankongense | 152, 611 | 37 | 81, 995 | 17, 506 | 26, 555 | 132 | 86 | 38 | 8 | MK757466 |
| L. duchartrei | 152, 566 | 37 | 81, 870 | 17, 542 | 26, 577 | 132 | 86 | 38 | 8 | MN745200 |
| L. stewartianum | 152, 307 | 37 | 81, 921 | 17, 532 | 26, 427 | 132 | 86 | 38 | 8 | MN745202 |
| L. matangense | 152, 402 | 37 | 82, 107 | 17, 531 | 26, 427 | 132 | 86 | 38 | 8 | MN745201 |
| L. lophophorum | 152, 382 | 37 | 82, 150 | 17, 382 | 26, 425 | 132 | 86 | 38 | 8 | MK493298 |
| L. nanum | 152, 417 | 37 | 82, 056 | 17, 505 | 26, 428 | 132 | 86 | 38 | 8 | MK493300 |
Figure 2. Merged gene map of the complete plastid genomes of six Lilium species.

Genes belonging to different functional groups are denoted by different colors. The dashed area in the inner circle corresponds to the GC content of the chloroplast genome.
Simple sequence repeats (SSRs) analysis
The SSRs loci in the six plastid genome sequences of Lilium were detected using MISA perl script. We detected a total of 340 SSRs in the six Lilium plastid genomes, with different numbers of SSRs per species (Table 2). 53, 54, 57, 58, 63, and 55 SSRs were observed in Lilium lankongense, L. duchartrei, L. stewartianum, L. matangense, L. lophophorum, and L. nanum (Table 2, Table S4). L. lankongense (53 SSRs) and L. lophophorum (63 SSRs) had the lowest and highest number of SSRs, respectively. All SSRs were categorized as mononucleotide, dinucleotide, trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide (Table 2). The total number of mononucleotide repeats consisting of A or T bases was the largest compared to other types of microsatellites (Fig. 3A) and was greater than the sum of the other types. There were no hexanucleotide repeats found in the six Lilium species. In the total SSRs loci, most of the repeats were located in the LSC region, followed by the SSC region and IR regions (Fig. 3B). Additionally, 20 common SSRs were found among these six plastid genome sequences (Table S4), and the number and location of these SSRs were different.
Table 2. Types and numbers of SSRs in plastid genomes of the six Lilium species.
| Species | mononucleotide | dinucleotide | trinucleotide | tetranucleotide | pentanucleotide | hexanucleotide | Total |
|---|---|---|---|---|---|---|---|
| L. lankongense | 30 | 9 | 3 | 9 | 2 | 0 | 53 |
| L. duchartrei | 31 | 8 | 2 | 10 | 3 | 0 | 54 |
| L. stewartianum | 34 | 8 | 1 | 9 | 5 | 0 | 57 |
| L. matangense | 35 | 8 | 2 | 8 | 5 | 0 | 58 |
| L. lophophorum | 41 | 9 | 1 | 8 | 4 | 0 | 63 |
| L. nanum | 32 | 9 | 2 | 9 | 3 | 0 | 55 |
| 203 | 51 | 11 | 53 | 22 | 0 | 340 |
Figure 3. Analysis of simple sequence repeats (SSRs) in six Lilium plastid genomes.

(A) Number of SSRs in different repeat types; (B) frequency of identified SSRs in different regions.
IR expansion and contraction
The IR boundaries of the six Lilium species were shown in Fig. 4. The rps19, ndhF, ycf1, and psbA genes were distributed in the JLB, JSB, JSA, and JLA regions, respectively (Fig. 4). The order and types of genes were very conservative. Despite the similar lengths of these six species IR regions (ranging from 26,425 to 26, 577 bp) there is a slight difference in the IR expansion and contraction. The rps19 gene crosses the LSC and IRB regions (JLB), with 140–145 bp extension into the IRB region. The JSA line intersects the ycf1 gene and the SSC and IRA regions are the same in Lilium stewartianum and L. matangense (4,327 bp in SSC and 1,226 bp in IRB) but are different in other species (4,323 to 4,333 bp in SSC and 1,226 to 1,244 bp in IRB) (Fig. 4). The psbA gene in the JLA regions remained completely within the LSC region, 78 to 83 bp away from JLA line. The changes in the boundary transformations between the LSC/IR and JSA regions were relatively stable, whereas the JSB regions varied among the six species to some extent. The JSB line is located between ycf1 and ndhF. Compared to the six Lilium species, the JSB region showed that ndh F expanded in three species (L. stewartianum, L. matangense, L. lophophorum), but contracted in two species (L. duchartrei, L. nanum) (Fig. 4).
Figure 4. Comparison of the junction positions of large single-copy (LSC), small single-copy (SSC), and inverted repeat (IR) regions among six Lilium plastid genomes.

Colored boxes for genes indicate the gene position.
Development of Lilium-specific primers
Two of the 12 primer pairs designed to sequence the plastid genome of the genus Lilium and tested on three species of Lilium, amplified consistently and easily produced good quality sequences. The two primer pairs, listed in Table 3 and Fig. 5, were tested on 11 individuals belonging to Lilium lankongense, L. duchartrei, L. lophophorum, including those from geographically remote populations (Table S5). Comparisons of intergenic spacer regions among L. lankongense, L. duchartrei, L. lophophorum plastomes provided evidence of high variability within trnC-petN, rpl32-trnL regions. The 620 bp trnC-petN and 793 bp rpl32-trnL regions were aligned for 11 individuals (Table S6, Supplementary Nucleotide alignment trnC-petN and Nucleotide alignment rpl32-trnL). We found nine, four, and one mutation sites in the trnC-petN region in L. lankongense, L. duchartrei, and L. lophophorum, respectively. Sequences in the rpl32-trnL region aligned data set were observed in the different number of variable sites in L. lankongense (10), L. duchartrei (4), L. lophophorum (1) (Table 4 and Table S6).
Table 3. Sequences of primers used for PCR amplification and sequencing.
| Region | Primer name | Sequence (5′-3′) | GC (%) |
|---|---|---|---|
| trnC-petN | trnC | CCTTTATCCCCAGTTCAAATCTG | 43.48 |
| petN | CCCAAGCGAGACKTACTATATCCAT | 46.00 | |
| rpl32-trnL | rpl32 | TGTTTTTGAAWGGCGGTTCC | 45.00 |
| trnL | CAGCGTGTCTACCAATTTCAC | 47.62 |
Figure 5. Positions and directions of two new primers used to amplify.

Tips of arrows indicate the 3′ ends of the primers. The length of the amplified regions in the figure corresponds to the aligned length of all published chloroplast genome sequences in Lilium using MAFFT. (A) trnC-petN; (B) rpl32-trnL.
Table 4. Numbers of variable sites and Genbank Accession Number for the amplified sequences in this study.
| Species | Numbers of variable sites | Genbank Accession Number | ||
|---|---|---|---|---|
| trnC-petN | rpl32-trnL | trnC-petN | rpl32-trnL | |
| L. lophophorum | 1 | 1 | MN764136 | MN764115 |
| MN764135 | MN764120 | |||
| L. duchartrei | 4 | 4 | MN764128 | MN764122 |
| MN764124 | MN764112 | |||
| MN764127 | MN764113 | |||
| MN953785 | MN953787 | |||
| L. lankongense | 9 | 10 | MN953784 | MN953786 |
| MN764132 | MN764116 | |||
| MN764134 | MN764111 | |||
| MN764125 | MN764123 | |||
| MN764131 | MN764114 | |||
| 13 | 15 | |||
Phylogenetic analysis
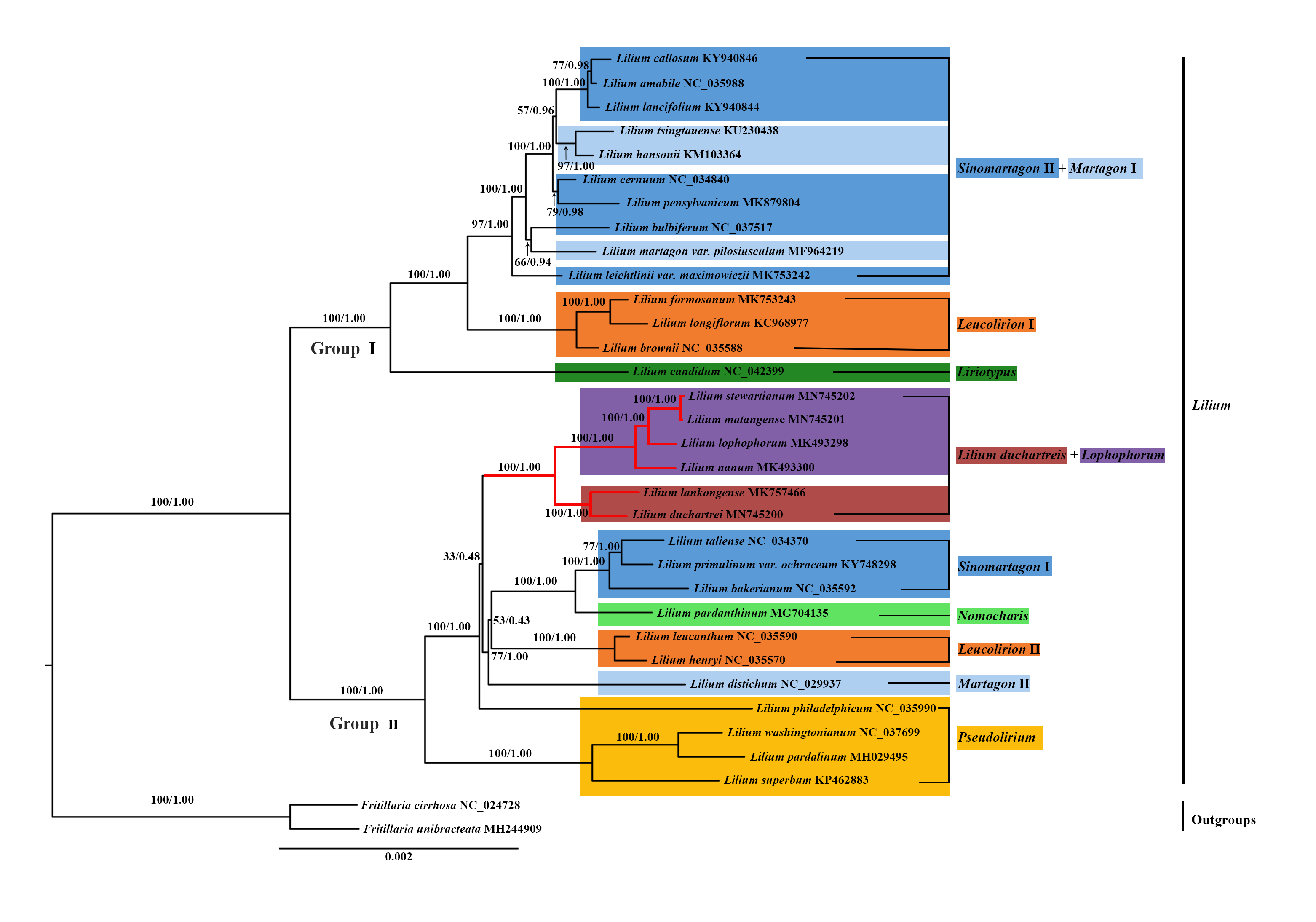
We constructed the phylogenetic tree (containing 31 Lilium species, two Fritillaria as the outgroups) from the three data sets of the complete plastid genome sequences, the shared 71 CDS sequences, and ITS sequences using the maximum likelihood and Bayesian inference methods. The results are shown in Figs. 6 and 7, and Fig. S1. The phylogenetic trees obtained by ML and the BI methods had identical topologies. The topologies of the phylogenetic tree constructed by ML and BI methods were largely the same based on the complete plastid genome sequences and the shared 71 CDS sequences (Fig. 6 and Fig. S1). The trees strongly supported that Lilium was a monophyletic group. The support values of phylogenetic trees constructed by the whole plastid genome sequences were slightly improved when compared with the phylogenetic trees constructed by shared CDS sequences. The phylogenetic relationships of most species were relatively clear and had high support values. Two groups were identified among the 31 Lilium species. We considered traditional sections (Comber, 1949; Wang & Tang, 1980) and used Nomocharis or Lilium duchartreis (including L. duchartrei and L. lankongense) as a clade, respectively, for clarification. Group I was composed of 14 species and was divided into the Sinomartagon II + Martagon I, Leucolirion I, and Liriotypus clades. Group II was made up of the remaining 17 species, belonging to the Lilium duchartreis + Lophophorum, Sinomartagon I, Nomocharis, Martagon II, Leucolirion II, and Pseudolirium clades. The ML tree and the BI tree also indicated that Sect. Sinomartagon, Sect. Martagon, Sect. Leucolirion and Sect. Pseudolirium did not form monophyletic groups. Sect. Sinomartagon, Sect. Martagon and Sect. Leucolirion each formed two clades. L. lankongense, L. duchartrei, L. stewartianum, L. matangense, L. lophophorum, and L. nanum were clustered into the Lilium duchartreis + Lophophorum branch with strong support (BS = 100%, PP = 1.00). The phylogenetic trees (ML tree and BI tree) reconstructed by ITS sequencing also strongly suggested that Lilium was a monophyletic group (Fig. 7). The main structure of Lilium was divided into four groups, Group A included the Leucolirion I, Sinomartagon II, Sinomartagon I, and Martagon clades, Group B contained the the Lophophorum, Pseudolirium, and Leucolirion II clades, Nomocharis and Liriotypus clades made up Group C, and the Lilium duchartreis clade alone formed Group D. The phylogenetic tree, established by the 33 plastid genomes or the shared 71 CDS, was not completely consistent with the phylogenetic tree constructed by ITS. In the ITS phylogenetic tree, four species in Sect. Martagon and four species in Sect. Pseudolirium were respectively monophyletic clades. L. lankongense and L. duchartrei belonged to Lilium duchartreis clade, while L. stewartianum, L. matangense, L. lophophorum, and L. nanum belonged to another clade (Lophophorum clade), and the Lilium duchartreis clade and the Lophophorum clade were in two different groups. Compared with the phylogenetic trees formed by complete plastid genome sequences and its shared 71 CDS sequences, the ITS phylogenetic tree was more in line with the previous classification based on morphology.
Figure 6. Phylogenetic tree reconstruction including 33 species using Bayesian inference (BI) method based on the plastid genomes.

Species of the L. lankongense and five related species are highlighted in red branches. The color patches highlight species in each of the traditional sections and Nomocharis or Lilium duchartreis clade. Bootstrap support values in the maximum likelihood (ML) trees and posterior probabilities in the BI trees are shown at the corresponding nodes. Nodes that do not occur in the ML tree are indicated by #.
Figure 7. Phylogenetic tree reconstruction including 33 species using Bayesian inference (BI) method based on nuclear ITS.

Species of the L. lankongense and five related species are highlighted in red branches. The color patches highlight species in each of the traditional sections and Nomocharis or Lilium duchartreis clade. Bootstrap support values in the the maximum likelihood (ML) trees and posterior probabilities in the BI trees are shown at the corresponding nodes.
Discussion
Comparative analysis of Lilium plastid genomes
The plastid genomes of the six Lilium species were compared and analyzed. Similar to other Lilium species (Du et al., 2017; Kim & Kim, 2013; Liu et al., 2018a; Liu et al., 2018b), the plastid genomes of these six species all have LSC and SSC regions, and a pair of IRS (Table 1). Moreover, these six plastid genomes have the same protein-coding genes, tRNAs, and rRNAs (Table 1), and have the typical structure of land plant plastid genomes (Wicke et al., 2011). Their lengths varied from 152,307 to 152,611 bp (Table 1), indicating a slight variation. The LSC and IR boundaries and the JSA region of the six plastid genomes were relatively stable with small differences, while the JSB region was relatively obvious. The ndh F gene of the JSB region showed signs of contraction and expansion. Typically, the IR (IRA and IRB) length differs among plant species. In angiosperms, the size variations in the plastid genome are attributed to the expansion and contraction of the IR region and single-copy (SC) border regions (Chumley et al., 2006) and play a vital role in its evolution (Wang et al., 2008). According to our results, the IR expansion and contraction may act as a primary mechanism for the variation in the length of the six plastid genomes. Similar results have been shown in most land plants (Gao et al., 2019; Hansen et al., 2007; Huang et al., 2014).
Microsatellites (SSRs) also known as short tandem repeats (STRs), are usually composed of one to six nucleotide repeat units (Chen et al., 2006; Powell, Machray & Provan, 1996), and are found in the majority of eukaryotic genomes (Tautz & Renz, 1984). SSRs are suitable molecular markers because of their high rate of polymorphism, and they are mostly co-dominant, and highly transferable (Scott et al., 2000). The SSRs in the plastid genome, which were viewed as complements to the nuclear microsatellite markers, were useful in research of phylogeography, population genetics, systematics, and breeding (Gaudeul et al., 2011; Hashimoto et al., 2004; Sánchez-Pérez et al., 2005; Xue, Wang & Zhou, 2012). The repeat units in plastid SSRs of these Lilium genomes were mostly comprised of adenine (A) or thymine (T) repeats, but were rarely guanine (G) or cytosine (C) repeats (Fig. 3A), which resulted from the AT-richness of the plastid genome (Zhang et al., 2016). These results are identical to other Lilium species (Du et al., 2017; Liu et al., 2018b). We identified a total of 340 SSR information sites. These SSR information sites can provide a reference for the subsequent development and application of SSRs molecular markers within Lilium.
Lilium-specific primers
Noncoding regions of the plastid genome play an important molecular role in systematic, phylogeographic, and population genetic studies of angiosperms (Perret et al., 2003; Xie et al., 2019a; Yang et al., 2016). Previous researchers, including Lai et al. (2016) tried to amplify the trnV-ndhC and petL-petE regions of the plastid genome but failed to amplify the corresponding fragments of all individuals. Jiang (2017) was unable to find suitable ptDNA fragments by screening 33 published universal primers for their phylogeographic study. No suitable ptDNA fragments from the genus Lilium have been found for use in population studies to date. In this study, 12 primers were designed according to the conserved regions of published Lilium plastid genomes to amplify a wide range of Lilium species. Two of the 12 pairs of primers (trnC-petN and rpl32-trnL) could amplify the products in all experimental individuals. The trnC-petN and rpl32-trnL regions proved to be effective in phylogeny and phylogeography in other genera (Liu, 2017; Lo Presti & Oberprieler, 2011; Yu et al., 2011) and the two non-coding regions of trnC-petN and rpl32-trnL may be used for intraspecies research of Lilium. The other 10 primer pairs (Table S2) successfully amplified some individuals, such as trnQ-rps16, trnF-ndhJ, and trnE-trnT, which had similar proportions of variant sites in amplified sequences and were applied to population-level studies in other genera (Chen et al., 2013; Dane & Liu, 2007; Qiu et al., 2009). We suggest that trnC-petN and rpl32-trnL with the two new primer pairs can be used in subsequent population studies (Table 3). Future studies should focus on discovering better primers for other potential regions to amplify their products. The two pairs of primers (trnC-petN and rpl32-trnL) were selected based on their plastid genomes and provide basic data for the genetic diversity and molecular breeding of this important wild flower of Lilium.
Phylogenetic analysis
We constructed a phylogenetic tree based on 3 data sets (the plastid genome sequences, the shared 71 CDS sequences, and ITS sequences). The tree supported Lilium as a monophyletic group with subgroups that include Sect. Sinomartagon and Sect. Leucolirion, which were both polyphyletic (Figs. 6, 7, and Fig. S1). Sect. Martagon and Sect. Pseudolirium were both monophyletic in the ITS tree, but none of them formed a monophyletic group in the plastid genome and the shared 71 CDS trees (Figs. 6, 7, and Fig. S1). Givnish et al. (2020) also sequenced the plastomes and 440 nuclear loci of 67 species of Lilium, and the results are similar to ours. Previous studies have also shown a conflict between the nuclear gene tree and the plastid gene tree in Lilium. This discordance may be due to hybridization in Lilium (Gao et al., 2013; Gao, Harris & He, 2015; Givnish et al., 2020; Gong et al., 2017; Huang et al., 2018). Due to the limited dispersion of seeds and the pollination by wind or pollinators, hybridization in Lilium is likely to occur among neighboring species. Hybridization may play an important role in Lilium flower morphology (Gao, Harris & He, 2015; Givnish et al., 2020). Furthermore, there are paralogous homologs in the ITS sequences of Liliaceae that may not reflect the true interspecific relationships in the phylogenetic tree (Day et al., 2014; Gao et al., 2012b; Huang et al., 2018). The 31 species of Lilium studied could not exclude the paralogous homology of the ITS sequences. Finally, L. lankongense, L. duchartrei, L. stewartianum, L. matangense, L. lophophorum, and L. nanum in the ITS tree were split into two different clades. The phylogenetic trees constructed by Huang et al. (2018) based on the ITS of 140 accessions in Liliaceae showed that the six species occur together in a clade. Aside from those results, the phylogenetic trees constructed with 440 nuclear genes and their spacers by Givnish et al. (2020) supported five species (except L. matangense), including L. lankongense, L. duchartrei, L. stewartianum, L. lophophorum, and L. nanum as a monophyletic group. The nuclear gene phylogenetic tree in this study only involved 33 ITS sequences (the number of samples was small), the information reflected by the ITS sequences was limited, and some clades (such as the Pseudolirium and Sinomartagon I clades) had low support values. The aforementioned three points are also one of the reasons for discordance between the ITS tree and the plastid gene tree.
In sections delimited by Comber (1949), which were based on morphology, L. lankongense, L. duchartrei, L. stewartianum, L. lophophorum, and L. nanum belong to Sect. Sinomartagon. L. lankongense, L. duchartrei belong to Sect. Sinomartagon 5a, while L. stewartianum, L. lophophorum, and L. nanum belong to Sect. Sinomartagon 5c (Comber, 1949). Sect. Sinomartagon Comber, which is the largest group of the genus Lilium, includes rather disparate species, and various researchers had different treatments for the division of its subsection (Comber, 1949; Du et al., 2014; Gao, Harris & He, 2015; Givnish et al., 2020; Nishikawa et al., 2001). Sect. Sinomartagon Comber was divided into the Sinomartagon, Lophophorum, and Lilium duchartreis clades based on the ITS tree (Fig. 7). The species in the Lophophorum clade contained L. stewartianum, L. matangense, L. lophophorum, and L. nanum; the Lilium duchartreis clade was composed of L. lankongense and L. duchartrei. The plastid gene trees (the plastid genome sequences, the shared 71CDS sequences) and the ITS tree both supported that Sect. Sinomartagon Comber was not monophyletic (Figs. 6, 7, and Fig. S1). In the plastid and ITS trees, L. lankongense was sister to L. duchartrei, L. stewartianum was evolutionarily close to L. matangense. Different phylogenetic relationships were shown among L. lankongense, L. duchartrei, L. stewartianum, L. matangense, L. lophophorum, and L. nanum. These six species were a monophyletic group in the plastid gene trees, and two clades were formed in the ITS tree. Previous studies have shown that L. lankongense and L. duchartrei was a monophyletic group, while L. stewartianum, L. matangense, L. lophophorum, and L. nanum were clustered into another clade in the ITS tree (Gao et al., 2013; Gao, Harris & He, 2015). Huang et al. (2018) constructed a phylogenetic tree based on the ITS sequence, three ptDNA fragments, and the combined (ITS + ptDNA) data sets. All trees supported that the six species were a monophyletic group (Sect. Sinomartagon I). Phylogenetic trees constructed with 440 nuclear genes and their spacers by Givnish et al. (2020) showed that the five species (L. lankongense, L. duchartrei, L. stewartianum, L. lophophorum, and L. nanum) aggregated into a single clade. Hence, L. lankongense, L. duchartrei, L. stewartianum, L. matangense, L. lophophorum, and L. nanum in the phylogenetic trees of ITS did not form a clade, which may be due to the insufficient number of ITS samples and the limited information contained in the ITS sequence. A close relationship has been shown among the six species (L. lankongense, L. duchartrei, L. stewartianum, L. matangense, L. lophophorum, and L. nanum) reflected in the plastid genome datum.
The revolute tepals of L. lankongense, L. duchartrei, L. stewartianum, L. matangense are different from those of the campanulate flowers of L. lophophorum and L. nanum (Fig. 1). However, the morphological characteristics of Lilium do not reflect the actual interspecies relationships. Convergent evolution, divergent evolution, retention and loss of ancestral characteristics, hybridization, and non-one-to-one correspondence between genes and morphological traits may affect plant’s morphology. The results of these five factors may be different morphologies of closely related species and similar morphologies of distantly related species, which confuses the judgment of interspecies relationships (Givnish et al., 2020). The habitats of L. lophophorum and L. nanum are mainly alpine grasslands and the adjacent niche at higher altitudes. These habitats often experience torrential downpours and have strong ultraviolet rays. The nodding and campanulate flowers can protect the reproductive structures against heavy rainfall and ultraviolet rays (Gao, Harris & He, 2015). The other four species found in the regions of lower altitudes have flowers with a larger opening, due to the revolute tepals, which may be more beneficial to pollination. The common floral characteristics of these six Lilium species are the inner tepals with projections on both surfaces of the nectaries, however the shape of the projections is not exactly the same. The projections of L. lankongense and L. duchartrei are papillose, L. stewartianum has cristate projections, and L. matangense, L. lophophorum, and L. nanum have fimbriate projections (Fig. S2). L. lankongense and L. duchartrei have habitats in lower elevations than the other four species and their inner and outer tepals have papillose projections. The other four species have habitats at higher elevations and only have projections in the inner tepals. L. stewartianum grows in open and rocky places on limestone mountains or along valleys and forest margins. Its habitat is distinct from those of the other three species, and the cristate projections are also different. L. matangense grows on grassy slopes, L. lophophorum and L. nanum grow mainly on bushy slopes and alpine grasslands. These three species have similar habitats and their projections are fimbriate. We speculate that the adaptation to different habitats may cause the dissimilarities of floral morphology among L. lankongense, L. duchartrei, L. stewartianum, L. matangense, L. lophophorum, and L. nanum.
Although the flower morphology of L. lophophorum and L. nanum was different from the other four species, they shared similar karyotypes that were measured using MAML (Altinordu et al., 2016). Gao, Zhou & He (2011) found that L. lophophorum, L. nanum, and L. matangense held a 3A type and relative asymmetry index and there were multiple, approximative parameters of the karyotype between L. stewartianum and L. lophophorum (Wan et al., 2011). The phylogenetic trees in this study demonstrated that the four species were more closely related (Figs. 6, 7, and Fig. S1). L. matangense, L. lophophorum, and L. nanum had karyotypes that resembled a clade formed by L. duchartrei and L. lankongense (Gao et al., 2012a). Therefore, it is reasonable to conclude that these six species constituted a clade. L. duchartrei and L. lankongense, which are closely related (Jefferson-Brown & Howland, 1995; MacRae, 1998), were thought to be one species (Wang & Tang, 1980), but were later separated due to the different colors of the tepal (L. lankongense: pink tepal, L. duchartrei: white tepal), the obvious geography variations (within the Daxue Mountains), and the significant genetic differentiation of the 22 populations (Gao et al., 2013; Liang & Tamura, 2000; Shen, Zhou & He, 2014). There are obvious micromolecular differences in the leaf dermis such as the inner margin and the waxy ornamentation of the outer stomatal rim, and the characteristics of the cuticular membrane under the electron microscope (L. lankongense: the pattern of anticlinal wall of leaf epidermal cells are all straight and the guard cells have no T pieces; L. duchartrei: the pattern of anticlinal wall of leaf epidermal cells are wavy and the guard cells have T pieces at the two poles) (Zhou, 2008). Our research supports the classification of L. lankongense as an independent species and agrees with the classification of Flora of China (Liang & Tamura, 2000). The two species have been clustered in the same clade based on the internal transcribed spacer region or plastid genes (Du et al., 2014; Gao et al., 2012a; Givnish et al., 2020; Lee et al., 2011; Nishikawa et al., 2001) (Figs. 6, 7, and Fig. S1). L. duchartrei also showed a sister relationship with L. lankongense in the phylogenetic trees of ITS and plastid genomes, which is in agreement with results from previous studies (Figs. 6, 7, and Fig. S1). We reconstructed the phylogenetic relationship of Lilium with 33 plastid whole genomes. However, we only conducted a preliminary analysis of the relationship of some species in Lilium due to the limited plastid genome datum. More plastid genomes should be obtained to better clarify the classification at the section level of Lilium and the relationship among species.
Conclusions
The plastid genomes of the six Lilium species shared a high similarity in genome organization, order, and content, however the IR and single copy boundaries were slightly inconsistent. The total number of SSRs ranged from 53 to 63. We designed a series of new primers by the conservative regions learned from all available Lilium plastid genomes alignments. Two Lilium-specific primer pairs were verified to be effective. Two intergenic spacer regions (trnC-petN and rpl32-trnL) can be well-amplified with the two new primer pairs and may be applied in future population-level studies of Lilium. The phylogenetic inference supported that Lilium is a monophyletic group. The phylogenetic tree of ITS is not completely consistent with the phylogenetic tree based on two data sets of the plastid genome (the plastid genome sequences, the shared 71 CDS sequences). The inconsistencies may be due to hybridization, paralogous homology of ITS sequences, the limited information contained in ITS sequences, and the small number of Lilium species involved in this study. The results of phylogenetic studies on plastid genomes have shown that the six species, namely L. lankongense, L. duchartrei, L. stewartianum, L. matangense, L. lophophorum, have close phylogenetic relationships.
Supplemental Information
Species of the L. lankongense and five related species are highlighted in red branches. The color patches highlight species in each of the traditional sections and Nomocharis or Lilium duchartreis clade. Bootstrap support values in the ML trees and posterior probabilities in the BI trees are shown at the corresponding nodes.
{kind=link}
Scale bar indicating two mm.
{kind=link}
Primers were designed using genes alignment of all published plastid genome sequences in Lilium. For each primer pair, forward (F) and reverse (R) primers are indicated. Amplifications were tested on different Lilium species. Ok = good amplification; No = partial or no amplification.
Marked red repeats are common SSR to the six Lilium species.
Acknowledgments
The authors appreciate the open genome data from the NCBI an wish to thank Junpei Chen, Xin Yang, Hongyi Zheng for their assistance in the use of the software.
Funding Statement
This research was funded by the National Natural Science Foundation of China [Grant nos. 31570198], and the Chinese Ministry of Science and Technology through the “National Science and Technology Infrastructure Platform” project [Grant no. 2005DKA21403-JK]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Additional Information and Declarations
Competing Interests
The authors declare there are no competing interests.
Author Contributions
Danmei Su conceived and designed the experiments, performed the experiments, analyzed the data, prepared figures and/or tables, authored or reviewed drafts of the paper, and approved the final draft.
Fumin Xie performed the experiments, analyzed the data, prepared figures and/or tables, and approved the final draft.
Haiying Liu performed the experiments, analyzed the data, authored or reviewed drafts of the paper, and approved the final draft.
Dengfeng Xie, Juan Li and Xianlin Guo analyzed the data, authored or reviewed drafts of the paper, and approved the final draft.
Xingjin He and Songdong Zhou conceived and designed the experiments, authored or reviewed drafts of the paper, and approved the final draft.
DNA Deposition
Data Availability
The following information was supplied regarding data availability:
The raw measurements are available in the Supplemental Files.
All Voucher specimens are deposited in SZ (Sichuan University Herbarium). The herbarium number of L. lankongense, L. duchartrei, L. matangense, L. stewartianum, L. lophophorum, and L. nanum are respectively KS2018071601, KS20180704, LS20180622, KS2018070203, LJ2017062301, YM20140828-3.
References
- Ahmed (2014).Ahmed I. D. Phil. Thesis. 2014. Evolutionary dynamics in taro (Colocasia esculenta L.) [Google Scholar]
- Altinordu et al. (2016).Altinordu F, Peruzzi L, Yu Y, He X. A tool for the analysis of chromosomes: KaryoType. Taxon. 2016;65(3):586–592. doi: 10.12705/653.9. [DOI] [Google Scholar]
- Amiryousefi, Hyvönen & Poczai (2018).Amiryousefi A, Hyvönen J, Poczai P. IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics. 2018;34(17):3030–3031. doi: 10.1093/bioinformatics/bty220. [DOI] [PubMed] [Google Scholar]
- Andrews (2014).Andrews S. FastQC, A quality control tool for high throughput sequence data. 2014. http://www.bioinformatics.babrahamacuk/projects/fastqc/ [6 October 2011]. http://www.bioinformatics.babrahamacuk/projects/fastqc/
- Burland (2000).Burland TG. DNASTAR’s lasergene sequence analysis software. Methods in Molecular Biology. 2000;132:71–91. doi: 10.1385/1-59259-192-2:71. [DOI] [PubMed] [Google Scholar]
- Capella-Gutiérrez, Silla-Martínez & Gabaldón (2009).Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. TrimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25(15):1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen et al. (2013).Chen T, Wang X, Tang H, Chen Q, Huang X, Chen J. Genetic diversity and population structure of Chinese Cherry revealed by chloroplast DNA trnQ-rps16 intergenic spacers variation. Genetic Resources and Crop Evolution. 2013;60(6):1859–1871. doi: 10.1007/s10722-013-9960-9. [DOI] [Google Scholar]
- Chen et al. (2006).Chen C, Zhou P, Choi YA, Huang S, Gmitter FG. Mining and characterizing microsatellites from citrus ESTs. Theoretical and Applied Genetics. 2006;112(7):1248–1257. doi: 10.1007/s00122-006-0226-1. [DOI] [PubMed] [Google Scholar]
- Chumley et al. (2006).Chumley TW, Palmer JD, Mower JP, Fourcade HM, Calie PJ, Boore JL, Jansen RK. The complete chloroplast genome sequence of Pelargonium× hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Molecular Biology and Evolution. 2006;23(11):2175–2190. doi: 10.1093/molbev/msl089. [DOI] [PubMed] [Google Scholar]
- Comber (1949).Comber HF. A new classification of the genus Lilium. London: Royal Horticultural Scoeity; 1949. [Google Scholar]
- Dane & Liu (2007).Dane F, Liu J. Diversity and origin of cultivated and citron type watermelon (Citrullus lanatus) Genetic Resources and Crop Evolution. 2007;54(6):1255–1265. doi: 10.1007/s10722-006-9107-3. [DOI] [Google Scholar]
- Day et al. (2014).Day P, Berger M, Hill L, Fay M, Leitch A, Leitch I, Kelly L. Evolutionary relationships in the medicinally important genus Fritillaria L. (Liliaceae) Molecular Phylogenetics and Evolution. 2014;80:11–19. doi: 10.1016/j.ympev.2014.07.024. [DOI] [PubMed] [Google Scholar]
- Dierckxsens, Mardulyn & Smits (2017).Dierckxsens N, Mardulyn P, Smits G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Research. 2017;45(4):e18C5. doi: 10.1093/nar/gkw955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du et al. (2017).Du Y, Bi Y, Yang F, Zhang M, Chen X, Xue J, Zhang X. Complete chloroplast genome sequences of Lilium: insights into evolutionary dynamics and phylogenetic analyses. Scientific Reports. 2017;7:5751. doi: 10.1038/s41598-017-06210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du et al. (2014).Du Y, He H, Wang ZX, Li S, Wei C, Yuan XN, Cui Q, Jia GX. Molecular phylogeny and genetic variation in the genus Lilium native to China based on the internal transcribed spacer sequences of nuclear ribosomal DNA. Journal of Plant Research. 2014;127(2):249–263. doi: 10.1007/s10265-013-0600-4. [DOI] [PubMed] [Google Scholar]
- Dubouzet & Shinoda (1999).Dubouzet J, Shinoda K. Phylogenetic analysis of the internal transcribed spacer region of Japanese Lilium species. Theoretical and Applied Genetics. 1999;98:954–960. doi: 10.1007/s001220051155. [DOI] [Google Scholar]
- Gao et al. (2019).Gao K, Li J, Khan WU, Zhao T, Yang X, Yang X, Guo B, An X. Comparative genomic and phylogenetic analyses of Populus section Leuce using complete chloroplast genome sequences. Tree Genetics & Genomes. 2019;15(3):32. doi: 10.1007/s11295-019-1342-9. [DOI] [Google Scholar]
- Gao, Harris & He (2015).Gao YD, Harris AJ, He XJ. Morphological and ecological divergence of Lilium and Nomocharis within the Hengduan Mountains and Qinghai-Tibetan Plateau may result from habitat specialization and hybridization. BMC Evolutionary Biology. 2015;15:147. doi: 10.1186/s12862-015-0405-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao et al. (2013).Gao YD, Harris A, Zhou SD, He XJ. Evolutionary events in Lilium (including Nomocharis, Liliaceae) are temporally correlated with orogenies of the Q-T plateau and the Hengduan Mountains. Molecular Phylogenetics and Evolution. 2013;68(3):443–460. doi: 10.1016/j.ympev.2013.04.026. [DOI] [PubMed] [Google Scholar]
- Gao et al. (2012b).Gao YD, Hohenegger M, Harris A, Zhou SD, He XJ, Wan J. A new species in the genus Nomocharis Franchet (Liliaceae): evidence that brings the genus Nomocharis into Lilium. Plant Systematics and Evolution. 2012b;298(1):69–85. doi: 10.1007/s00606-011-0524-1. [DOI] [Google Scholar]
- Gao, Zhou & He (2011).Gao YD, Zhou SD, He XJ. Karyotype studies in thirty-two species of Lilium (Liliaceae) from China. Nordic Journal of Botany. 2011;29(6):746–761. doi: 10.1111/j.1756-1051.2011.01069.x. [DOI] [Google Scholar]
- Gao et al. (2012a).Gao YD, Zhou SD, He XJ, Wan J. Chromosome diversity and evolution in tribe Lilieae (Liliaceae) with emphasis on Chinese species. Journal of Plant Research. 2012a;125(1):55–69. doi: 10.1007/s10265-011-0422-1. [DOI] [PubMed] [Google Scholar]
- Gaudeul et al. (2011).Gaudeul M, Giraud T, Kiss L, Shykoff JA. Nuclear and chloroplast microsatellites show multiple introductions in the worldwide invasion history of common ragweed, Ambrosia artemisiifolia. PLOS ONE. 2011;6(3):e17658. doi: 10.1371/journal.pone.0017658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gielly & Taberlet (1994).Gielly L, Taberlet P. Chloroplast DNA polymorphism at the intrageneric level and plant phylogenies. Comptes rendus de l’Academie des sciences Série III, Sciences de la vie. 1994;317(7):685–692. doi: 10.1016/1367-8280(94)90114-7. [DOI] [Google Scholar]
- Givnish et al. (2020).Givnish TJ, Skinner MW, Rešetnik I, Ikinci N, Kriebel R, Lemmon AR, Lemmon EM, Gao YD. Evolution, geographic spread and floral diversification of the Genus Lilium with special reference to the lilies of North America. North American Lily Society Year Book. 2020;74:26–44. [Google Scholar]
- Gong et al. (2017).Gong X, Hung KH, Ting YW, Hsu TW, Malikova L, Tran HT, Huang CW, Liu SH, Chiang TY. Frequent gene flow blurred taxonomic boundaries of sections in Lilium L.(Liliaceae) PLOS ONE. 2017;12(7):e0183209. doi: 10.1371/journal.pone.0183209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen et al. (2007).Hansen DR, Dastidar SG, Cai Z, Penaflor C, Kuehl JV, Boore JL, Jansen RK. Phylogenetic and evolutionary implications of complete chloroplast genome sequences of four early-diverging angiosperms: Buxus (Buxaceae), Chloranthus (Chloranthaceae), Dioscorea (Dioscoreaceae), and Illicium (Schisandraceae) Molecular Phylogenetics and Evolution. 2007;45(2):547–563. doi: 10.1016/j.ympev.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Hashimoto et al. (2004).Hashimoto Z, Mori N, Kawamura M, Ishii T, Yoshida S, Ikegami M, Takumi S, Nakamura C. Genetic diversity and phylogeny of Japanese sake-brewing rice as revealed by AFLP and nuclear and chloroplast SSR markers. Theoretical and applied genetics. 2004;109(8):1586–1596. doi: 10.1007/s00122-004-1794-6. [DOI] [PubMed] [Google Scholar]
- Hayashi & Kawano (2000).Hayashi K, Kawano S. Molecular systematics of Lilium and allied genera (Liliaceae): phylogenetic relationships among Lilium and related genera based on the rbcL and matK gene sequence data. Plant Species Biology. 2000;15(1):73–93. doi: 10.1046/j.1442-1984.2000.00025.x. [DOI] [Google Scholar]
- Huang et al. (2014).Huang H, Shi C, Liu Y, Mao S, Gao L. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evolutionary Biology. 2014;14:151. doi: 10.1186/1471-2148-14-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang et al. (2018).Huang J, Yang LQ, Yu Y, Liu YM, Xie DF, Li J, He XJ, Zhou SD. Molecular phylogenetics and historical biogeography of the tribe Lilieae (Liliaceae): bi-directional dispersal between biodiversity hotspots in Eurasia. Annals of Botany. 2018;122(7):1245–1262. doi: 10.1093/aob/mcy138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikinci, Oberprieler & Güner (2006).Ikinci N, Oberprieler C, Güner A. On the origin of European lilies: phylogenetic analysis of Lilium section Liriotypus (Liliaceae) using sequences of the nuclear ribosomal transcribed spacers. Willdenowia. 2006;36:647–657. doi: 10.3372/wi.36.36201. [DOI] [Google Scholar]
- Jefferson-Brown & Howland (1995).Jefferson-Brown MJ, Howland H. The gardener’s guide to growing lilies. Timber Press; Portlan: 1995. [Google Scholar]
- Jiang (2017).Jiang F. M. Phil. Thesis. 2017. Phylogeography of Lilium pumilum Redouté in southeast of Qinghai-Tibetan Plateau. [Google Scholar]
- Kang et al. (2019).Kang L, Xie D, Xiao Q, Peng C, Yu Y, He X. Sequencing and analyses on chloroplast genomes of Tetrataenium candicans and two allies give new insights on structural variants, DNA barcoding and phylogeny in Apiaceae subfamily Apioideae. Peerj. 2019;7:e8063. doi: 10.7717/peerj.8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh & Standley (2013).Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse et al. (2012).Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim & Kim (2013).Kim JS, Kim JH. Comparative genome analysis and phylogenetic relationship of order Liliales insight from the complete plastid genome sequences of two Lilies (Lilium longiflorum and Alstroemeria aurea) PLOS ONE. 2013;8(6):e68180. doi: 10.1371/journal.pone.0068180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai et al. (2016).Lai S, Shen C, Zhou S, XJ HE. Phylogeny and interspecific relationship of Lilium section Leucoliron based on three gene sequences. Acta Botanica Boreali-Occidentalia Sinica. 2016;36(8):1541–1550. doi: 10.7606/j.issn.1000-4025.2016.08.1541. [DOI] [Google Scholar]
- Langmead & Salzberg (2012).Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee et al. (2011).Lee CS, Kim SC, Yeau SH, Lee NS. Major lineages of the genus Lilium (Liliaceae) based on nrDNA ITS sequences, with special emphasis on the Korean species. Journal of Plant Biology. 2011;54(3):159–171. doi: 10.1007/s12374-011-9152-0. [DOI] [Google Scholar]
- Li et al. (2013).Li XW, Gao HH, Wang YT, Song JY, Henry R, Wu HZ, Hu ZG, Yao H, Luo HM, Luo K. Complete chloroplast genome sequence of Magnolia grandiflora and comparative analysis with related species. Science China Life Sciences. 2013;56(2):189–198. doi: 10.1007/s11427-012-4430-8. [DOI] [PubMed] [Google Scholar]
- Liang & Tamura (2000).Liang S, Tamura M. Lilium. In: Wu ZY, Raven PH, editors. Flora of China. Vol. 24. Beijing: Science Press; 2000. pp. 135–149. [Google Scholar]
- Liu et al. (2018a).Liu H, Li J, Xie D, He X, Yu Y, Zhou S. The complete chloroplast genome of Nomocharis pardanthina. Mitochondrial DNA Part B. 2018a;3(1):103–104. doi: 10.1080/23802359.2018.1424581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu et al. (2018b).Liu H, Yu Y, Deng Y, Li J, Huang Z, Zhou S. The Chloroplast Genome of Lilium henrici: Genome Structure and Comparative Analysis. Molecules. 2018b;23(6):1276. doi: 10.3390/molecules23061276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu (2017).Liu PL. D. Phil. Thesis. 2017. Phylogeny And Biogeography Of The Genus Hedysarum L. (FABACEAE) [Google Scholar]
- Lohse et al. (2013).Lohse M, Drechsel O, Kahlau S, Bock R. OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Research. 2013;41:W575–W581. doi: 10.1093/nar/gkt289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Presti & Oberprieler (2011).Lo Presti RM, Oberprieler C. The central Mediterranean as a phytodiversity hotchpotch: phylogeographical patterns of the Anthemis secundiramea group (Compositae, Anthemideae) across the Sicilian Channel. Journal of Biogeography. 2011;38(6):1109–1124. doi: 10.1111/j.1365-2699.2010.02464.x. [DOI] [Google Scholar]
- MacRae (1998).MacRae EA. Lilies: a guide for growers and collectors. Timber Press; Portland: 1998. [Google Scholar]
- Nishikawa et al. (2001).Nishikawa T, Okazaki K, Arakawa K, Nagamine T. Phylogenetic analysis of section Sinomartagon in genus Lilium using sequences of the internal transcribed spacer region in nuclear ribosomal DNA. Breeding Science. 2001;51(1):39–46. doi: 10.1270/jsbbs.51.39. [DOI] [Google Scholar]
- Nishikawa et al. (1999).Nishikawa T, Okazaki K, Uchino T, Arakawa K, Nagamine T. A molecular phylogeny of Lilium in the internal transcribed spacer region of nuclear ribosomal DNA. Journal of Molecular Evolution. 1999;49(2):238–249. doi: 10.1007/PL00006546. [DOI] [PubMed] [Google Scholar]
- North & Wills (1969).North C, Wills AB. Inter-specific pecific hybrids of Lilium lankongense Franchet produced by embryo-culture. Euphytica. 1969;18(3):430–434. doi: 10.1007/BF00397793. [DOI] [Google Scholar]
- Palmer (1985).Palmer JD. Comparative organization of chloroplast genomes. Annual Review of Genetics. 1985;19(1):325–354. doi: 10.1146/annurev.ge.19.120185.001545. [DOI] [PubMed] [Google Scholar]
- Parks, Cronn & Liston (2009).Parks M, Cronn R, Liston A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biology. 2009;7:84. doi: 10.1186/1741-7007-7-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perret et al. (2003).Perret M, Chautems A, Spichiger R, Kite G, Savolainen V. Systematics and evolution of tribe Sinningieae (Gesneriaceae): evidence from phylogenetic analyses of six plastid DNA regions and nuclear ncpGS. American Journal of Botany. 2003;90(3):445–460. doi: 10.3732/ajb.90.3.445. [DOI] [PubMed] [Google Scholar]
- Peruzzi (2016).Peruzzi L. A new infrafamilial taxonomic setting for Liliaceae, with a key to genera and tribes. Plant Biosystems. 2016;150(6):1341–1347. doi: 10.1080/11263504.2015.1115435. [DOI] [Google Scholar]
- Powell, Machray & Provan (1996).Powell W, Machray GC, Provan J. Polymorphism revealed by simple sequence repeats. Trends in Plant Science. 1996;1:215–222. doi: 10.1016/1360-1385(96)86898-1. [DOI] [Google Scholar]
- Proscevičius, Rančeliene & Dambrauskaite (2007).Proscevičius J, Rančeliene V, Dambrauskaite D. Cytogenetic analysis of progeny derived from allotriploid inter-specific hybrids of Lilium. Biologija. 2007;53(2):8–12. [Google Scholar]
- Qiu et al. (2009).Qiu Y, Guan B, Fu C, Comes HP. Did glacials and/or interglacials promote allopatric incipient speciation in East Asian temperate plants? Phylogeographic and coalescent analyses on refugial isolation and divergence in Dysosma versipellis. Molecular Phylogenetics and Evolution. 2009;51(2):281–293. doi: 10.1016/j.ympev.2009.01.016. [DOI] [PubMed] [Google Scholar]
- Sánchez-Pérez et al. (2005).Sánchez-Pérez R, Ruiz D, Dicenta F, Egea J. Application of simple sequence repeat (SSR) markers in apricot breeding: molecular characterization, protection, and genetic relationships. Scientia Horticulturae. 2005;103(3):305–315. doi: 10.1016/j.scienta.2004.06.009. [DOI] [Google Scholar]
- Sanger & Coulson (1975).Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. Journal of Molecular Biology. 1975;94(3):441–448. doi: 10.1016/0022-2836(75)90213-2. [DOI] [PubMed] [Google Scholar]
- Scarcelli et al. (2011).Scarcelli N, Barnaud A, Eiserhardt W, Treier UA, Seveno M, d’Anfray A, Vigouroux Y, Pintaud J-C. A set of 100 chloroplast DNA primer pairs to study population genetics and phylogeny in monocotyledons. PLOS ONE. 2011;6(5):e19954. doi: 10.1371/journal.pone.0019954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott et al. (2000).Scott KD, Eggler P, Seaton G, Rossetto M, Ablett EM, Lee LS, Henry RJ. Analysis of SSRs derived from grape ESTs. Theoretical and Applied Genetics. 2000;100:723–726. doi: 10.1007/s001220051344. [DOI] [Google Scholar]
- Shaw et al. (2005).Shaw J, Lickey EB, Beck JT, Farmer SB, Liu W, Miller J, Siripun KC, Winder CT, Schilling EE, Small RL. The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. American Journal of Botany. 2005;92(1):142–166. doi: 10.3732/ajb.92.1.142. [DOI] [PubMed] [Google Scholar]
- Shaw et al. (2007).Shaw J, Lickey EB, Schilling EE, Small RL. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. American Journal of Botany. 2007;94(3):275–288. doi: 10.3732/ajb.94.3.275. [DOI] [PubMed] [Google Scholar]
- Shen, Zhou & He (2014).Shen C, Zhou S, He X. Genetic diversity of Lilium duchartrei and Lilium lankongense revealed by ISSR markers. Acta Botanica Boreali-Occidentalia Sinica. 2014;34:1331–1338. doi: 10.7606/j.issn.1000-4025.2014.07.1331. [DOI] [Google Scholar]
- Shinozaki et al. (1986).Shinozaki K, Ohme M, Tanaka M, Wakasugi T, Hayashida N, Matsubayashi T, Zaita N, Chunwongse J, Obokata J, Yamaguchi-Shinozaki K. The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. The EMBO Journal. 1986;5(9):2043–2049. doi: 10.1002/j.1460-2075.1986.tb04464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis (2014).Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz & Renz (1984).Tautz D, Renz M. Simple sequences are ubiquitous repetitive components of eukaryotic genomes. Nucleic Acids Research. 1984;12(10):4127–4138. doi: 10.1093/nar/12.10.4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel et al. (2003).Thiel T, Michalek W, Varshney R, Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.) Theoretical and Applied Genetics. 2003;106(3):411–422. doi: 10.1007/s00122-002-1031-0. [DOI] [PubMed] [Google Scholar]
- Van Tuyl et al. (2002).Van Tuyl JM, Chung MY, Chung JD, Lim KB. Introgression studies using GISH in interspecific Lilium hybrids of L. longiflorum x Asiatic, L. longiflorum x L. rubellum and L. auratum x L. henryi. North American Lily Yearbook. 2002;55:1–7. [Google Scholar]
- Wan et al. (2011).Wan J, Zhou S, Gao Y, He X. Karyotypes of twenty-five populations of thirteen species in Nomocharis and Lilium. Plant Diversity and Resources. 2011;33:477–494. doi: 10.3724/SP.J.1143.2011.11017. [DOI] [Google Scholar]
- Wang et al. (2008).Wang R, Cheng C, Chang C, Wu C, Su T, Chaw SM. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evolutionary Biology. 2008;8(1):36. doi: 10.1186/1471-2148-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang & Tang (1980).Wang FZ, Tang J. Flora Reipublicae Popularis Sinicae. vol. 14. Beijing: Science Press; 1980. Lilium L; pp. 116–157. [Google Scholar]
- White et al. (1990).White TJ, Bruns T, Lee S, Taylor J. Amplification and direct seqencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, editors. PCR protocols: a guide to methods and applications. Vol. 38. USA: Academic Press; 1990. pp. 315–322. [Google Scholar]
- Wicke et al. (2011).Wicke S, Schneeweiss GM, Depamphilis CW, Müller KF, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Molecular Biology. 2011;76:273–297. doi: 10.1007/s11103-011-9762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie et al. (2019a).Xie C, Xie D, Zhong Y, Guo X, Liu Q, Zhou S, He X. The effect of Hengduan Mountains Region (HMR) uplift to environmental changes in the HMR and its eastern adjacent area: Tracing the evolutionary history of Allium section Sikkimensia (Amaryllidaceae) Molecular Phylogenetics and Evolution. 2019a;130:380–396. doi: 10.1016/j.ympev.2018.09.011. [DOI] [PubMed] [Google Scholar]
- Xie et al. (2018).Xie DF, Yu Y, Deng YQ, Li J, Liu HY, Zhou SD, He XJ. Comparative analysis of the chloroplast genomes of the Chinese endemic genus Urophysa and their contribution to chloroplast phylogeny and adaptive evolution. International Journal of Molecular Sciences. 2018;19(7):1847. doi: 10.3390/ijms19071847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie et al. (2019b).Xie DF, Yu HX, Xie C, Deng YQ, Chen J, Yu Y, Zhou S, He X. Phylogeny of Chinese Allium species in Section Daghestanica and adaptive evolution of Allium (Amaryllidacea, Allioideae) species revealed by the chloroplast complete genomee. Frontiers in Plant Science. 2019b;10:460. doi: 10.3389/fpls.2019.00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, Wang & Zhou (2012).Xue J, Wang S, Zhou SL. Polymorphic chloroplast microsatellite loci in Nelumbo (Nelumbonaceae) American Journal of Botany. 2012;99(6):e240–e244. doi: 10.3732/ajb.1100547. [DOI] [PubMed] [Google Scholar]
- Yang et al. (2016).Yang LQ, Hu HY, Xie C, Lai SP, Yang M, He XJ, Zhou SD. Molecular phylogeny, biogeography and ecological niche modelling of Cardiocrinum (Liliaceae): insights into the evolutionary history of endemic genera distributed across the Sino-Japanese floristic region. Annals of Botany. 2016;119(1):59–72. doi: 10.1093/aob/mcw210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang et al. (2019).Yang Z, Wang G, Ma Q, Ma W, Liang L, Zhao T. The complete chloroplast genomes of three Betulaceae species: implications for molecular phylogeny and historical biogeography. Peerj. 2019;7:e6320. doi: 10.7717/peerj.6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang et al. (2020).Yang X, Xie DF, Chen JP, Zhou SD, Yu Y, He XJ. Comparative analysis of the complete chloroplast genomes in allium subgenus Cyathophora (Amaryllidaceae): phylogenetic relationship and adaptive evolution. BioMed Research International. 2020;2020:1–17. doi: 10.1155/2020/1732586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu et al. (2011).Yu Y, Downie SR, He X, Deng X, Yan L. Phylogeny and biogeography of Chinese Heracleum (Apiaceae tribe Tordylieae) with comments on their fruit morphology. Plant Systematics and Evolution. 2011;296:179–203. doi: 10.1007/s00606-011-0486-3. [DOI] [Google Scholar]
- Zhang et al. (2020).Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, Wang GT. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Molecular Ecology Resources. 2020;20:348–355. doi: 10.1111/1755-0998.13096. [DOI] [PubMed] [Google Scholar]
- Zhang et al. (2016).Zhang Y, Du L, Liu A, Chen J, Wu L, Hu W, Zhang W, Kim K, Lee SC, Yang TJ. The complete chloroplast genome sequences of five Epimedium species: lights into phylogenetic and taxonomic analyses. Frontiers in Plant Science. 2016;7:306. doi: 10.3389/fpls.2016.00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao et al. (2018).Zhao K, Wang J, Cai Y, Zhu Z, López-Pujol J, Wang H. Complete chloroplast genome sequence of Heritiera angustata (Malvaceae): an endangered plant species. Mitochondrial DNA Part B. 2018;3(1):141–142. doi: 10.1080/23802359.2017.1422398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou (2008).Zhou SD. D. Phil. Thesis. 2008. The phylogenetic classification and evolution of Trib. Lilieae (Liliaceae s.str.) in China. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Species of the L. lankongense and five related species are highlighted in red branches. The color patches highlight species in each of the traditional sections and Nomocharis or Lilium duchartreis clade. Bootstrap support values in the ML trees and posterior probabilities in the BI trees are shown at the corresponding nodes.
Scale bar indicating two mm.
Primers were designed using genes alignment of all published plastid genome sequences in Lilium. For each primer pair, forward (F) and reverse (R) primers are indicated. Amplifications were tested on different Lilium species. Ok = good amplification; No = partial or no amplification.
Marked red repeats are common SSR to the six Lilium species.
Data Availability Statement
The following information was supplied regarding data availability:
The raw measurements are available in the Supplemental Files.
All Voucher specimens are deposited in SZ (Sichuan University Herbarium). The herbarium number of L. lankongense, L. duchartrei, L. matangense, L. stewartianum, L. lophophorum, and L. nanum are respectively KS2018071601, KS20180704, LS20180622, KS2018070203, LJ2017062301, YM20140828-3.