

Summary
Some cancers originate from a single mutation event in a single cell. Blood cancers known as myeloproliferative neoplasms (MPNs) are thought to originate when a driver mutation is acquired by a hematopoietic stem cell (HSC). However, when the mutation first occurs in individuals and how it affects the behavior of HSCs in their native context is not known. Here we quantified the effect of the JAK2-V617F mutation on the self-renewal and differentiation dynamics of HSCs in treatment-naive individuals with MPNs and reconstructed lineage histories of individual HSCs using somatic mutation patterns. We found that JAK2-V617F mutations occurred in a single HSC several decades before MPN diagnosis—at age 9 ± 2 years in a 34-year-old individual and at age 19 ± 3 years in a 63-year-old individual—and found that mutant HSCs have a selective advantage in both individuals. These results highlight the potential of harnessing somatic mutations to reconstruct cancer lineages.
Keywords: myeloproliferative neoplasm, single cell sequencing, stem cell dynamics, lineage tree, blood cancer, JAK2
Graphical abstract
Highlights
-
•
Single-cell transcriptome and whole-genome sequencing of HSPCs from individuals with MPNs
-
•
The JAK2-V617F mutation occurs in a single HSC decades before diagnosis
-
•
JAK2-V617F HSCs have increased fitness in native human hematopoiesis
-
•
JAK2 mutant fraction varies in myeloid progenitor compartments in the same individuals
Van Egeren et al. investigated the effect of the JAK2-V617F mutation in individuals with myeloproliferative neoplasms (MPNs) using single-cell profiling and found that the mutation occurs decades before MPN diagnosis and increases the fitness of HSCs. JAK2-V617F induces a megakaryocyte-erythroid differentiation bias. The JAK2-mutant fraction varies in myeloid compartments in the same individuals.
Introduction
In seminal studies, the JAK2-V617F mutation was identified to underlie the molecular pathogenesis of the majority of Philadelphia chromosome-negative myeloproliferative neoplasms (MPNs) (Baxter et al., 2005; James et al., 2005; Kralovics et al., 2005; Levine et al., 2005), a chronic blood cancer. More recently, JAK2 was identified as one of the most commonly mutated genes in clonal hematopoiesis (Genovese et al., 2014; Jaiswal et al., 2014; Xie et al., 2014). Notably, JAK2-V617F clonal hematopoiesis is associated with an increased risk of cardiovascular disease (Jaiswal et al., 2017) and venous thrombosis (Cordua et al., 2019; Wolach et al., 2018), in addition to sharing the same germline variants that predispose to the development of JAK2 mutant MPN (Hinds et al., 2016).
The JAK2-V617F mutation results in activated JAK2 signaling, leading to increased production of mature blood cells of the myeloid lineage, ultimately resulting in MPNs. Some individuals with MPN present primarily with increased numbers of red blood cells (polycythemia vera [PV]), others with increased numbers of platelets (essential thrombocythemia [ET]), and, more rarely, some with scarring (“fibrosis”) of the bone marrow (primary myelofibrosis [PMF]). Although in MPN it has been shown that the JAK2-V617F mutation is detectable in hematopoietic stem cells (HSCs) (Jamieson et al., 2006) and in all mature cell lineages (Delhommeau et al., 2007; Ishii et al., 2006), it is unclear how the mutation affects HSC differentiation and proliferation of human HSCs in their native bone marrow microenvironment to engender clonal hematopoiesis and MPN. Therefore, to understand the behavior of JAK2-V617F mutant HSCs and development of MPN, we studied unrelated individuals with newly diagnosed JAK2-V617F+ ET and PV and ascertained when the JAK2-V617F mutation first occurred in each individual, how the number of JAK2 mutant cells expanded over time, and the extent to which the differentiation trajectories of the JAK2 mutant cells deviated from those of cells without the mutation.
Although the effect of the JAK2-V617F mutation on HSCs in vivo has been modeled previously using Jak2-V617F transgenic mice and patient-derived xenograft MPN models, these experimental systems do not accurately recreate the native microenvironment or the behavior of JAK2 mutant hematopoietic stem and progenitor cells (HSPCs) in humans. The discovery that JAK2-V617F causes clonal hematopoiesis (Genovese et al., 2014; Jaiswal et al., 2014; Xie et al., 2014) and the observation that JAK2-V617F is often the sole somatic mutation detected in individuals with MPNs (Grinfeld et al., 2018; Lundberg et al., 2014) suggest that the JAK2 mutation promotes HSC self-renewal and confers a selective advantage. However, this has never been measured directly. Measurement of the self-renewal and differentiation ability of JAK2 mutant HSCs in individuals with MPNs is not feasible because direct observation of dynamic cell behaviors is not possible in human bone marrow. However, static single-cell genomic and transcriptomic measurements can be used to reconstruct the self-renewal history and differentiation behavior in unperturbed cell populations (Lee-Six et al., 2018; Tusi et al., 2018).
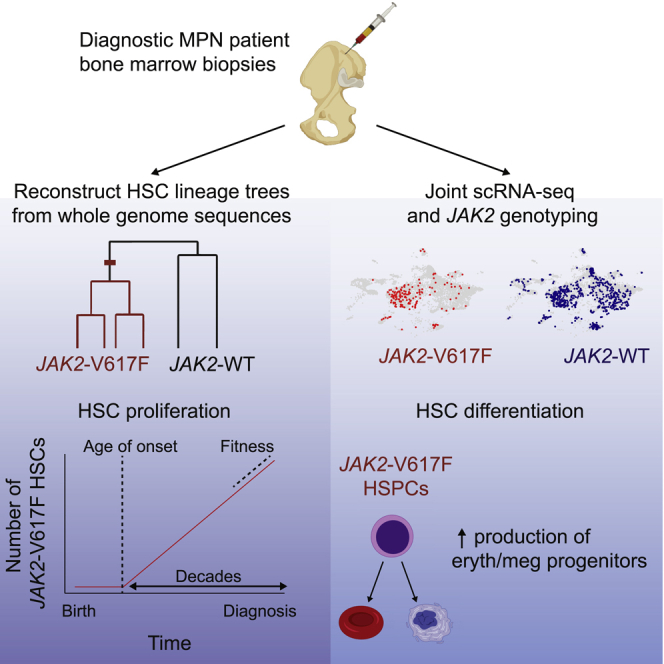
Therefore, to directly assess the consequences of the JAK2-V617F mutation on HSC differentiation and self-renewal in their native microenvironment, we reconstructed lineage trees of JAK2 mutant and wild-type HSCs obtained from individuals with MPN and inferred the history of MPN development in 2 people with ET. In addition, to determine how the JAK2 mutation affects the differentiation trajectories of the progenies of HSCs, we profiled the transcriptomes of individual cells obtained from bone marrow aspirates of 7 individuals with MPN.
Results
To investigate the effect of JAK2 mutations in individuals with ET and PV, we performed single-cell transcriptomic profiling of HSPCs from 7 newly diagnosed, untreated individuals with PV (n = 3) and ET (n = 4) as well as healthy controls (n = 2) (Figure 1). The JAK2-V617F mutation was detected in 6 individuals, whereas the remaining individuals with ET had a JAK2 variant previously unreported in humans (JAK2-V617L), with fibroblast germline testing confirming a somatic origin of the mutation. Of note, JAK2-V617L has been shown previously to induce cytokine independence and constitutive downstream signaling in Ba/F3 cells (Dusa et al., 2008). The individuals with ET did not harbor additional myeloid malignancy-associated mutations, as measured by a clinical next-generation sequencing (NGS) assay performed on whole white blood cells from peripheral blood (i.e., rapid heme panel; Kluk et al., 2016), whereas somatic truncating mutations in TET2 (2 individuals) and EZH2 (1 individual) were identified in people with PV (Figure 1B). From each individual with MPNs and healthy donor, we collected a bone marrow aspirate, isolated mononuclear cells, and then enriched for CD34 expression to isolate HSPCs (STAR Methods).
Figure 1.

Experimental Design
(A) Individual hematopoietic stem and progenitor cells (HSPCs) from bone marrow aspirates of individuals with MPNs were analyzed in two ways. First, hematopoietic stem cells (HSCs) and multipotent progenitors (MPPs) were expanded in vitro and characterized using WGS. Second, we simultaneously read out the transcriptional profiles and somatic mutations in single HSPCs.
(B) Information about the individuals with MPNs sampled in this study. “Allelic burden peripheral blood (PB)” and “secondary mutations” refer to VAFs of JAK2 mutations and other hematopoiesis-associated mutations in PB, respectively. The numbers of JAK2 WT and mutant cells identified in the HSPCs using scRNA-seq are given in the last two rows.
See also Figure S1.
JAK2-Mutant HSPCs Exhibit Fate Bias
To determine how JAK2 mutations affect HSPC differentiation dynamics in individuals with MPN, we simultaneously measured the full transcriptome and genotyped the JAK2 mutation in individual CD34+ cells obtained from each bone marrow aspirate (Figure 1A). To do so, we developed a protocol for amplifying specific transcripts from single-cell RNA sequencing (RNA-seq) libraries. Briefly, we used the 10X platform to generate barcoded single-cell cDNA libraries. Before fragmenting the libraries for sequencing, we generated amplicon libraries of the target loci for the somatic mutations of interest by performing three rounds of nested PCR with locus-specific reverse primers and generic forward primers (Figure S1; STAR Methods). The somatic mutations were mapped to the transcriptional profiles using the shared single-cell barcodes across the two libraries (Figure S1; STAR Methods).
Using this approach, we detected the JAK2 mutation site (locus encoding codon 617) in at least one transcript in 5%–15% of cells (mean 9.5%) in the 7 libraries, improving over the existing methods for detecting somatic mutations in single-cell libraries (Nam et al., 2019; Psaila et al., 2020). We designated cells in which at least one mutated JAK2 transcript was detected as JAK2 mutant cells. Importantly, cells with a heterozygous JAK2 mutation express wild-type (WT) JAK2 and mutant JAK2. We accounted for this when computing the fraction of mutated cells in specific subpopulations.
Using the expression levels of marker genes, we identified all major hematopoietic lineage progenitors in all samples (Figures 2A and 2B) and found that individuals with JAK2-mutant MPNs showed a similar hematopoietic differentiation hierarchy as healthy controls (Figure S2). JAK2-V617F cells had similar gene expression profiles compared with WT cells from the same individuals and were found to be generally intermixed with the WT cells in UMAP visualizations (Figure 2C). A significant fraction of the HSC subpopulation was mutated in all individuals (ranging from 5%–62%). Interestingly, we found that the JAK2-V617F allele fraction varied in different myeloid compartments in the same individual. The JAK2-V617F mutation frequency was higher in megakaryocyte/erythroid progenitors and lower in lymphoid progenitors and granulocyte-macrophage progenitors (GMPs) (combined p < 10−10 for erythroid versus lymphoid and erythroid progenitors versus GMPs for all individuals with V617F mutation, Fisher’s exact test with Fisher’s method) (Figures 2C and 2D). In contrast, the JAK2-V617L mutation showed no significant megakaryocyte-erythroid lineage bias (Figures 2C and 2D). TET2 mutations were amplified and identified similarly in the single-cell RNA-seq (scRNA-seq) libraries from individuals PV 2 and PV 3 and were present in JAK2-mutant cells in both individuals, suggesting that the TET2 and JAK2 mutations occurred in the same clone (Figure S2). Both individuals had a higher JAK2 allele fraction than TET2 allele fraction (Figure 1B; p < 10−10 for PV 2, p = 0.003 for PV 3, Fisher’s exact test).
Figure 2.

Erythroid and Megakaryocyte Progenitors from Individuals with MPNs Are More Likely to Have the JAK2-V617F Mutation than Other CD34+ Bone Marrow HSPCs
(A) UMAP of CD34-enriched bone marrow scRNA-seq data from ET 1, colored by cell type.
(B) Marker gene expression in ET 1 CD34-enriched bone marrow.
(C) Cell type classifications and JAK2 WT/mutant transcript calls in scRNA-seq data from individuals with MPNs (columns).
(D) Fraction of JAK2 mutant cells (colors) in different bone marrow cell types from individuals with MPNs.
(E) Relationship between the PB VAF and JAK2 V617F mutant transcript fraction in bone marrow HSCs (blue) and erythroid progenitors (red). Error bars indicate 95% confidence intervals.
(F) Mean expression of selected marker genes in CD14+ cells that are upregulated in monocyte subsets or were differentially expressed in CD14+ cells between individuals with ET and PV or between individuals with MPNs and healthy controls.
See also Figure S2.
In clinical practice, MPN clone size can be approximated by the peripheral blood JAK2-V617F variant allele frequency (VAF), which reflects the fraction of nucleated blood cells that harbor the mutation but does not measure the contribution from anucleated mature red blood cells and platelets (Steensma, 2006). Our single-cell analysis demonstrates that the peripheral blood VAF consistently underestimates the degree to which JAK2 mutant cells contribute to steady-state erythropoiesis (Figure 2E). Indeed, the large fraction of JAK2-V617F mutant erythroid progenitor cells in individuals with PV (79% to more than 95%) suggests that nearly all erythropoiesis arises from the JAK2-V617F clone. Additionally, most of the erythroid progenitors in ET 1 and ET 2 are JAK2 mutant, suggesting that the JAK2-V617F mutation induces an erythroid fate bias even in individuals diagnosed with ET. Furthermore, the peripheral blood VAF does not accurately reflect the extent of disease in HSCs (Figure 2E).
We identified a CD14+ population in our scRNA-seq data that showed enhanced expression of type I-II interferon-regulated genes in individuals with MPNs relative to healthy controls (Figure 2F; Figure S2). These cells did not express CD34 mRNA and therefore likely represent a contaminating CD34− monocyte-like bone marrow population. Interestingly, this CD14+ bone marrow population showed increased expression of SLAMF7 compared with healthy controls (particularly in individuals with PV). SLAMF7 is a cell surface protein reported recently to be highly expressed on monocytes from individuals with established JAK2 mutant myelofibrosis (Maekawa et al., 2019).
Our observations show that JAK2-V617F HSPCs have gene expression profiles similar to those of WT HSPCs but show a clear bias toward the megakaryocyte-erythroid fate. In addition, a significant fraction of HSCs was mutated in each individual. To understand how this population of mutated stem cells emerged, we set out to determine when the JAK2-V617F mutation first occurred in each individual and how the population of mutated stem cells subsequently expanded.
Lineage Trees of Individual Mutated and WT Stem Cells
To infer the disease history prior to clinical presentation with MPN, we reconstructed the lineage trees of the JAK2 mutant HSCs of two individuals with ET, ET 1 and ET 2, using the pattern of somatic mutations accrued by individual cells. Somatic mutations occur at random and are passed to a cell’s descendants and can be used to establish lineage relationships. To read out the somatic mutations in each cell, we isolated individual HSCs and multipotent progenitor (MPP) cells using established cell surface markers (STAR Methods) from the CD34+-enriched bone marrow cells of ET 1 and ET 2. We then expanded each HSC or MPP cell ex vivo by culturing them for ∼8 weeks and performed whole-genome sequencing (WGS) on the single-cell colonies (STAR Methods). We selected colonies to balance the number of JAK2 mutant and WT cells sequenced: 22 JAK2 mutant colonies and 20 WT colonies for ET 1 and 13 JAK2 mutant colonies and 21 WT colonies for ET 2. We observed that JAK2 mutant HSCs likely had a proliferative advantage under our culture conditions because the fraction of JAK2 mutant HSCs and MPPs after culturing was higher than the fraction of JAK2 mutant HSCs found in the scRNA-seq data (10%/2% of cultured MPPs and 73%/32% of cultured HSCs versus 29%/8% of HSCs identified by scRNA-seq for ET 1/ET 2, respectively; see STAR Methods for additional information).
We found that the younger individual (ET 1, 34 years old) had, on average, 713 ± 45 somatic point mutations in individual HSCs/MPPs, whereas the older individual (ET 2, 63 years old) had 1,185 ± 75 mutations in each cell. Using the number of point mutations found in each cell and the age of each individual, we estimated a constant somatic point mutation rate of 19 ± 1 per year, consistent with previous observations in healthy donors (Lee-Six et al., 2018; Osorio et al., 2018; Figure S3). In both individuals, the number of somatic mutations in JAK2 mutant (ET 1, 732 ± 26; ET 2, 1,209 ± 35) and JAK2 WT cells (ET 1, 690 ± 52; ET 2, 1,170 ± 89) was comparable, and after accounting for the shared ancestry of cancer cells, the difference in mutation rate was not significant (ET 1, p = 0.06; ET 2, 0.21; Figure S3; STAR Methods). Although the number of HSPCs analyzed in this study is limited, our results suggest that the somatic mutation rate was not altered by the JAK2-V617F mutation. However, the fraction of C > T mutations at NpCpG trinucleotides was increased significantly in JAK2 mutant HSPCs in ET 2 (p < 0.01; Wilcoxon rank sum test), and the average telomere length was shorter in JAK2 mutant HSPCs in both individuals (p < 0.01, Wilcoxon rank-sum test; Figure S3; STAR Methods), suggesting that JAK2 mutant cells might have undergone more cell divisions than JAK2 WT cells (Alexandrov et al., 2015). The number of somatic mutations was similar in HSCs and MPPs in both individuals (p = 0.07 for ET 1, p = 0.21 for ET 2; STAR Methods). No somatic structural variants or copy number aberrations were detected, except for loss of one copy of chromosome X in one colony from individual ET 2.
Analysis of the single-base substitution (SBS) mutation signatures revealed that spontaneous aging-associated clock mutations (COSMIC signatures 1 and 5) predominated in WT and JAK2-V617F HSCs/MPPs (Alexandrov et al., 2020; Figures 3A and 3B), consistent with previous analyses of somatic mutations in healthy HSPCs (Lee-Six et al., 2018; Machado et al., 2019; Osorio et al., 2018). Other than JAK2-V617F, no deleterious somatic mutations were detected in JAK2 mutant cells. The other mutations we identified that occurred in genes that could potentially affect stem cell function, such as ASH1L and FAT1, were not predicted to affect protein function nor have they been reported previously to be pathogenic (STAR Methods). Therefore, the JAK2-V617F mutation is likely the disease-initiating MPN driver mutation in these two individuals.
Figure 3.

Somatic Mutations Can Be Used to Reconstruct the Lineage Trees of WT and Mutant HSCs
(A and B) Lineage trees constructed using somatic SNVs for ET 1 (A) and ET 2 (B). The heatmap below the lineage trees shows the relative contribution of the SBS mutational signatures SBS1, SBS2, SBS5, SBS19, SBS23, and SBS32 (STAR Methods) to the mutational spectrum defined by the private mutations detected in each HSC-derived colony.
(C) Number of mutant stem cells as a function of time inferred from the ET 1 lineage tree, assuming one generation per year. The dashed lines on the bottom show the times of the coalescent events in the tree.
(D) Same as (C) but for individual ET 2
Next we used Wagner parsimony to reconstruct the phylogenies of the stem cells from the pattern of somatic mutations (Figures 3A and 3B; Table S2; STAR Methods). Two distinct clades were found in each individual that were defined by the presence or absence of the heterozygous JAK2-V617F mutation. These phylogenies suggest that, in both individuals, a single JAK2 mutation event initiated the disease, followed by expansion of the mutated stem cells. No mutations were shared across all JAK2 WT stem cells in ET 1 or in ET 2, suggesting that the common ancestor of JAK2 WT stem cells dates back to embryonic development, before most somatic mutations occurred. However, there were many mutations shared across JAK2 mutant stem cells (220 in ET 1 and 398 in ET 2), indicating that all mutated cells descended from a single common ancestor in which the JAK2 mutation occurred. Using the inferred somatic mutation rate, we estimated that the disease-initiating mutation occurred ∼25 years prior to sampling in ET 1 (the 34-year-old individual) and ∼40 years prior to sampling in ET 2 (the 63-year-old individual).
Reconstructing the History of Disease Progression in Individuals
To reconstruct the history of disease development, we inferred the number of mutated stem cells in each individual from the time of the initial JAK2 mutation event to the time of sampling by applying a phylogenetic dynamics inference algorithm (Karcher et al., 2017; Lee-Six et al., 2018) to the reconstructed lineage trees. This algorithm assumes that all mutated HSCs in the population are equivalent and that the population size over time can be modeled as a Gaussian process. Additionally, we assumed that HSCs divide symmetrically once per year (Abkowitz et al., 1996; Catlin et al., 2011; Lee-Six et al., 2018; Osorio et al., 2018); this assumption is required to infer the absolute population size but not to infer the rate of expansion of the population of mutated cells. No additional assumptions are made; e.g., whether the population of mutated cells expands or shrinks or at what rate. We found, in both individuals, that fewer than 100 mutated stem cells were present in the first decade after the JAK2 mutation occurred. The number of mutated stem cells in both individuals grew exponentially for decades (Figures 3C and 3D).
To quantitatively estimate the difference in growth rates between JAK2 WT and JAK2 mutant HSCs in vivo, we constructed a mathematical model of stem cell self-renewal based on the Wright-Fisher model (Figure 4A; STAR Methods). Importantly, the Wright-Fisher model can be simulated efficiently to infer its parameter from the observed lineage trees (STAR Methods). Our model contains three parameters: the maximum number of mutated stem cells, the age at which the disease-initiating JAK2 mutation occurred, and the fitness s of the mutant stem cells, corresponding to the proliferative advantage of the mutated stem cells over WT stem cells. This model assumes that all mutated stem cells have the same constant fitness value. If the mutant stem cell population survives stochastic extinction early, when its size is small, it will grow exponentially as (1+s)t, where t is time in years, until the mutant cells take over the majority of the population (Methods S1). Critically, unlike population size (Kimura, 1983; Lee-Six et al., 2018), fitness s and the time of occurrence of disease can be inferred without any knowledge of the HSC division rate (Figures 4A–4C; Methods S1). This is because changing the division rate scales the inferred population size and the minimum population size required to evade stochastic extinction in the same way. These two effects cancel each other, and s can be inferred directly from the observed lineage trees without knowledge of the division rate.
Figure 4.

The History of JAK2-Mutant HSC Expansion Is Reconstructed from the Lineage Trees
(A) Schematics showing the effect of scaling the number of generations by a factor of 2 while keeping the onset of the disease and fitness the same. As a result, the number of mutant cells doubles because early on the number of mutant cells (shown in white) fluctuates to increase by a factor of 2 to escape stochastic extinction. Increasing the number of generations increases the coalescent rate. Increasing the number of mutant cells decreases the coalescent rate. These effects cancel each other, and the trees are indistinguishable.
(B) Green curves (c = 1) represent 50 simulated mutant HSC trajectories that survived extinction with fitness s = 0.8 and a maximum population size of 50,000. Blue curves are similar, except the number of generations and maximum population size are scaled by a factor of 1,000 (c = 1,000). This scaling results in 1,000 times as many mutant HSCs through time (blue) because a larger initial population is needed to escape stochastic extinction.
(C) Trees corresponding to the blue and green trajectories in (B) are statistically indistinguishable (STAR Methods).
(D) Inference on data from ET 1 and ET 2. For both individuals, we show 50 inferred trajectories of the number of mutant stem cells as a function of time. Heatmaps show the inferred joint distribution of the fitness of the cancer cells and the age when the disease initiating mutation occurred. The marginal distributions are shown as histograms.
See also Figure S4.
The model parameters were inferred from the reconstructed single-cell WGS lineage trees using approximate Bayesian computation (ABC). Briefly, we chose the parameter values from a prior distribution, simulated the model, randomly sampled a subset of cells, and obtained their lineage tree (Methods S1). We then compared the simulated tree with the observed tree (as measured by the lineage-through-time metric). If the simulated and observed trees were sufficiently similar, then we retained the parameter values. Otherwise, they were discarded, obtaining the posterior distribution for the parameter values (STAR Methods). We validated the inference procedure using simulated data, particularly in cases in which the observed tree was generated using slightly different dynamics than those used for ABC (Methods S1). In all cases, we were still able to accurately infer the effective fitness and age of onset. Therefore, our inference framework is robust to expected deviations in the actual dynamics of stem cell proliferation from the simplified Wright-Fisher model.
We then applied the inference procedure to the observed lineage trees of the JAK2 mutant stem cells from ET 1 and ET 2 (Figure 4D; Figure S4). We inferred that in ET 1, the JAK2-V617F mutation first occurred at age 9 ± 2 and had a fitness effect of 63% ± 15%. Similarly, in ET 2, the JAK2-V617F mutation first occurred at age 19 ± 3 and had a fitness effect of 44% ± 13%. Our analyses show that JAK2 mutant HSCs have a selective advantage over WT HSCs and increase in number over the decades before MPN diagnosis.
Discussion
To determine the precise effect of the JAK2-V617F mutation on the behavior of human HSCs in their native bone marrow microenvironment, we performed WGS and single-cell profiling on HSPCs isolated from the bone marrow of newly diagnosed individuals with MPNs. Although it has long been known that MPN are clonal disorders (Adamson et al., 1976; Gilliland et al., 1991) and it has been shown previously that the JAK2-V617F mutation is detectable in HSPCs in MPNs (Jamieson et al., 2006) and that JAK2-V617F cells are clonal in MPNs (Beer et al., 2009), in this study, we trace acquisition of the JAK2-V617F mutation to a single HSC decades before MPN diagnosis. Subsequently, the population of JAK2 mutant stem cells grows exponentially but may exhibit large fluctuations and even stochastic extinction when its size is small in the first few years after occurrence of the mutation.
Using the WGS data, we estimated the time interval between JAK2-V617F acquisition and MPN development in individuals. Our findings that the JAK2-V617F mutation occurred in the first decade of life (9 ± 2 years) in a man who developed ET at age 34 and in the second decade of life (19 ± 3 years) in a woman who developed ET at age 63 are striking in terms of the young age at the time of JAK2-V617F acquisition and the decades-long interval to MPN development in both cases. While our paper was under review, similar findings were reported in an independent study (Williams et al., 2020).
We found that, at the time of MPN diagnosis, a significant fraction of HSCs (5% or more) are descendants of the original JAK2 mutant HSC. In addition, we inferred the fitness advantage of JAK2 mutant HSCs in 2 individuals with MPNs during the pre-diagnosis period to be approximately 63% ± 15% and 44% ± 13% in individuals ET 1 and ET 2, respectively. We emphasize that this fitness is inferred from the coalescent structure of the HSC lineage trees. Because most of the observed coalescent events occur close to the root of the tree (when the number of mutated stem cells is low), the inferred fitness value reflects the growth rate in the first decade after occurrence of the mutation. We did not sample enough HSCs to determine whether or when the growth in the population of mutated stem cells saturates. Our inferred fitness advantage is larger than that found in a population-level study of clonal hematopoiesis of indeterminate potential (CHIP), which analyzed peripheral blood variant allele fractions in large cohorts of healthy individuals (Watson et al., 2020). This discrepancy suggests that development of full-blown MPNs may require a faster-growing JAK2 mutant clone than that observed in clonal hematopoiesis.
Our study focused on newly diagnosed, treatment-naive individuals with JAK2 mutant ET and PV. We found that, in addition to modifying HSC proliferation dynamics, the JAK2-V617F mutation also affects the differentiation dynamics of their progenies. In contrast to a recent study of myelofibrosis that found marked aberrant megakaryopoiesis (Psaila et al., 2020), our study indicates that the hematopoietic differentiation hierarchy is largely preserved in individuals with ET and PV at diagnosis. However, JAK2 mutant HSPCs showed a lineage bias toward the megakaryocyte-erythroid fate, and, strikingly, we found that the fraction of JAK2 mutant cells varied significantly across different progenitor cell populations in the same patient. This suggests that peripheral blood monitoring may not accurately assess JAK2 mutant allele burden, particularly in megakaryocyte-erythroid lineage cells and HSCs. Finally, the JAK2-V617F mutation has been shown to have cell-intrinsic effects not only in leukocytes (Rampal et al., 2014; Wolach et al., 2018) but also in erythroid cells (Chen et al., 2010; De Grandis et al., 2013) and platelets (Gangaraju et al., 2020; Guo et al., 2020). Our observation of high JAK2-V617F allele fractions in megakaryocyte-erythroid lineage cells in individuals with low peripheral blood JAK2-V617F mutational burden may help explain the development of thrombosis in these people.
Many cancers start when a genetic alteration arises in a single cell and confers a fitness advantage over other cells. By the time the disease manifests clinically, this cell has expanded to millions of cells or more. Naturally occurring somatic mutations provide a glimpse into the history of cancer in each individual, revealing when the driver mutations first occurred, how the population of cancer cells expanded, and how their proliferation and differentiation dynamics differ from healthy cells. The framework we developed to harness somatic mutations as a clock to reconstruct the lineage tree of cancer cells and follow the differentiation trajectories of their progenies is broadly applicable in oncology.
Limitations of Study
Although our study traced the acquisition of the JAK2-V617F mutation in newly diagnosed individuals with MPNs, it has some clear limitations. First, we studied a total of seven individuals with ET/PV and performed WGS to reconstruct lineage trees for two of these individuals with ET. It would be informative to expand the study to a larger cohort of individuals with MPNs and study sequential samples from the same person (including during JAK2 mutant clonal hematopoiesis before development of MPNs). Second, we sequenced the whole genome of a limited number of HSCs from each individual. Although a sufficient number of coalescent events were observed to infer the behavior of mutated cells early in their history, extending the number of cells analyzed will provide further insights into the behavior of these cells closer to the time of diagnosis. More broadly, we inferred the history of disease expansion from final time point measurements. Any inference framework is only valid up to its assumptions. With larger lineage trees, we may be able to relax some of our assumptions and identify variations in fitness across the mutated stem cells over time.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD34-PB | BioLegend | Cat# 343512; RRID:AB_1877197 |
| CD38-PE-Cy7 | Thermo Fisher Scientific | Cat# 25-0388-42; RRID:AB_2573346 |
| CD45RA-FITC | BioLegend | Cat# 304106; RRID:AB_314410 |
| CD49f-APC-Cy7 | BioLegend | Cat# 313628; RRID:AB_2616784 |
| Thy1/CD90-PE | BioLegend | Cat# 328110; RRID:AB_893433 |
| EasySep Human CD34 Positive Selection Kit II | STEMCELL Technologies | Cat# 17856 |
| Biological Samples | ||
| Whole bone marrow samples | Massachusetts General Hospital; Dana-Farber Cancer Institute | N/A |
| Peripheral blood samples | Dana-Farber Cancer Institute | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Human SCF | PeproTech | Cat#300-07 |
| Recombinant Human TPO | PeproTech | Cat#300-18 |
| Recombinant Human FLT3-L | PeproTech | Cat#300-19 |
| Recombinant Human IL-6 | PeproTech | Cat#200-06 |
| Recombinant Human IL-3 | PeproTech | Cat#160-01 |
| Chromium Single Cell 3′ GEM, Library & Gel Bead Kit v3, 16 rxns | 10x Genomics | Cat#1000075 |
| Chromium Chip B Single Cell Kit, 48 rxns | 10x Genomics | Cat#1000073 |
| Chromium i7 Multiplex Kit, 96 rxns | 10x Genomics | Cat#120262 |
| Buffer EB | QIAGEN | Cat#19086 |
| High Sensitivity D5000 ScreenTape | Agilent | Cat#5067-5592 |
| High Sensitivity D5000 Reagents | Agilent | Cat#5067-5593 |
| MiSeq Reagent Kit v2 (500 cycles) | Illumina | Cat#MS-102-2003 |
| NovaSeq 6000 SP Reagent Kit (200 cycles) | Illumina | Cat#20040326 |
| Lymphoprep | STEMCELL Technologies | Cat#07801 |
| EasySep Buffer | STEMCELL Technologies | Cat#20144 |
| SPRIselect | Beckman Coulter | Cat#B23318 |
| QIAmp UCP DNA Micro Kit | QIAGEN | Cat#56204 |
| Qubit dsDNA HS Assay Kit | Invitrogen | Cat#32854 |
| Monarch DNA Gel Extraction Kit | New England Biolabs | Cat#T1020L |
| Dulbecco’s phosphate-buffered saline | Thermo Scientific | Cat#14040133 |
| SFEM medium | STEMCELL Technologies | Cat#09650 |
| DMEM-F12 medium | GIBCO | Cat#11320082 |
| Iscove Modified Dulbecco Medium (IMDM) | GIBCO | Cat#31980030 |
| Roswell Park Memorial Institute (RPMI) | GIBCO | Cat#12633012 |
| Horse Serum | Thermo Scientific | Cat#16050130 |
| Hydrocortisone Solution | Sigma-Aldrich | Cat#H6909-10ML |
| Fetal Bovine Serum | VWR | Cat#89510-186 |
| Deposited Data | ||
| scRNA-seq and WGS data | dbGAP | phs002308.v1.p1 |
| Experimental Models: Cell Lines | ||
| UKE-1 | Ann Mullally | N/A |
| Molt4 | Sahand Hormoz | N/A |
| Oligonucleotides | ||
| Primers | This paper | Table S1 |
| Software and Algorithms | ||
| StemCellSim | This paper | https://gitlab.com/hormozlab/stemcellsim |
| Scripts used for scRNA-seq analysis | This paper | https://gitlab.com/hormozlab/mpn-scrnaseq-analysis |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sahand Hormoz (sahand_hormoz@hms.harvard.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
We developed a C++ object called StemCellSim for simulating clonal expansions and inferring the parameters of our model. StemCellSim has the ability to generate simulated data under various models, and to infer model parameters from either simulated or real data with ABC. The StemCellSim code and the Python scripts used to analyze and plot the scRNA-seq data can be found on GitLab (https://gitlab.com/hormozlab). Raw scRNA-seq and whole-genome sequencing data have been deposited in dbGAP:phs002308.v1.p1.
Experimental Model and Subject Details
Human bone marrow biopsies
Prospective MPN patients were identified through the outpatient clinic. Patients were required to have a confirmed diagnosis of PV or ET according to WHO criteria and next-generation sequencing (RapidHEME panel or SnapShot) documenting the presence of JAK2-V617F. Use of anti-platelet agents was permitted but disease-modifying treatments (e.g., hydroxyurea, interferon-alpha, ruxolitinib) were an exclusion criterion for the study. Consequently, our cohort consisted of newly diagnosed, treatment-naive JAK2 mutant MPN patients. Bone marrow aspirate samples were uniformly collected at the time of diagnostic bone marrow biopsy under tissue banking protocols at the participating centers. The study was approved by and conducted in accordance with the Declaration of Helsinki protocol (Dana-Farber Cancer Institute IRB protocol no. 01-206 and Massachusetts General Hospital protocol 13-583). All patients provided informed consent. The healthy donor samples were purchased as de-identified samples from the Boston Children’s Hospital. Healthy donor 1 was a 22-year-old female and healthy donor 2 was a 29-year-old female. The age and sex of all other study subjects can be found in Figure 1B. Bone marrow biopsies were performed on all donors. RapidHEME panel screen on peripheral blood from the same patients was performed as a part of clinical diagnostic testing. Approximately 10-20mL of bone marrow aspirate from each donor was collected in EDTA-coated tubes. The syringes and tubes used were sterile with no preservative-free heparin coating. The bone marrow aspirates (BMA) were kept at room temperature until use.
Cell lines
The UKE-1 and MOLT4 cell lines used in the control experiments were collected from a 59-year-old female and a 19-year-old male. UKE-1 cells were maintained in Iscove modified Dulbecco medium (IMDM) supplemented with 10% fetal bovine serum, 10% horse serum and 1 μM hydrocortisone. MOLT4 cells were maintained in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% fetal bovine serum. All cultures were maintained in standard tissue culture conditions of 37°C and 5% CO2.
Method Details
Mononuclear cell isolation
Mononuclear cells (MNCs) were isolated from the BMA via a density gradient centrifugation protocol using StemCell Technologies, Inc.’s SepMate system. BMA, phosphate-buffered saline (PBS) with 2% fetal bovine serum (PBS + 2% FBS; StemCell Technologies, Inc. #07905), Lymphoprep (StemCell Technologies, Inc. #07801), and the centrifuge (Eppendorf #5810R) were all acclimated at room temperature. Approximately 20-22mL Lymphoprep was added to the 50mL SepMate tube (StemCell Technologies, Inc. #15450) by carefully pipetting it through the central hole of the SepMate insert, ensuring that as few air bubbles as possible were present. BMA was diluted with an equal volume of PBS + 2% FBS and mixed gently with wide-bore pipette. Keeping the SepMate tube vertical, the diluted sample was added by slowly pipetting it down the side of the tube. The diluted BMW was centrifuged at 1200 x g for 20 minutes at room temperature, with the brake off. For BMA rich in platelets/plasma, the top layer of platelets/plasma was pipetted off. The remaining volume down to the SepMate central hole (containing all the enriched MNCs) was poured into a new 50mL tube. The MNCs were washed by topping up until 45mL with PBS + 2% FBS and mixing well with a wide-bore pipette. The MNCs were then centrifuged at 300 x g for 12 minutes at room temperature, with the brake low, and the supernatant was removed. The MNC pellet was topped up again with PBS + 2% FBS, and the volume was mixed well with a wide-bore pipette. Centrifuge at 120 x g for 12 minutes at room temperature, with the brake off. The supernatant was then removed. For BMA rich in platelets, an additional wash and centrifugation at 12 minutes at room temperature, with the break off was performed. The MNC pellet was resuspended in 1mL of EasySep buffer (StemCell Technologies, Inc. #20144) and mixed with a wide-bore pipette. Cell concentration was counted with a hemocytometer (Reichert) and a Tali Image Cytometer system (ThermoFisher #T10796) using Tali Image Analysis Slides (ThermoFisher #T10794). Cells were placed on ice until further use.
CD34+ enrichment
CD34+ MNCs were isolated using the protocol for EasySep Human CD34 Positive Selection Kit II (StemCell Technologies, Inc. #17856). MNCs (at concentration of > 108 cells/mL EasySep buffer) were added to 5mL (12 × 75 mm) polystyrene round-bottom tube (StemCell Technologies, Inc. #38007). EasySep Human CD34 Positive Selection Cocktail (StemCell Technologies, Inc. #17856C) was added at a concentration of 100 μL per 1mL of sample. The sample was then mixed and incubated at room temperature for 10 minutes. EasySep Dextran RapidSpheres (StemCell Technologies, Inc. #50100) were vortexed for 30 s. RapidSpheres were added at a concentration of 75 μL per 1 mL of sample. The sample was mixed and incubated at room temperature for 5 minutes. The tube was topped up to 2.5mL with EasySep buffer and gently mixed. The tube was placed in EasySep magnet (StemCell Technologies, Inc. #18000) and incubated at room temperature for 3 minutes. The supernatant was discarded by inverting the magnet with the tube inside. This process was repeated 4 more times for a total of 5 rounds of enrichment. Cells were resuspended in PBS+2% FBS after the last round of enrichment. Cell concentration was counted with a hemocytometer (Reichert #1492) and Tali Image Cytometer. Cells were placed on ice until further use. CD34+ MNCs were used to create single-cell cDNA, single-cell RNA-Seq (scRNA-seq) and JAK2 amplicon libraries as described below.
Single-cell cDNA libraries
The isolated CD34+ MNC suspensions were used to generate single-cell gel bead emulsions (GEMs) using a 10x Genomics Chromium controller (10x Genomics #120223). Following steps 1 and 2 of the protocol “Chromium Single Cell 3′ Reagent Kit v3” (10x Genomics, CG000183 Rev A), single-cell cDNA libraries were constructed using the Chromium Single Cell 3′ Library & Gel Bead Kit v3 (10x Genomics #1000075). The protocol yields 40 μL of cDNA per sample after step 2.4. scRNA-Seq libraries were generated from 10 μL of single-cell cDNA libraries using Step 3 of the Chromium Single Cell 3′ Reagent Kit v3 user guide (10x Genomics, CG000183 Rev A).
Locus-specific single-cell amplicon libraries
We developed the following protocol to preferentially amplify transcripts containing loci-of-interest from single-cell cDNA libraries (ex. JAK2-V617F), thereby generating locus-specific single-cell amplicon libraries (Figures S1A–S1C).
A triple-nested PCR approach was used to amplify the transcripts carrying the loci-of-interest from single-cell cDNA libraries with high sensitivity and specificity. The approach used locus-specific reverse primers that flank the mutation site combined with generic forward primers that preserves the single-cell barcoding structure. In total, there were 5 total PCR steps (3 nested steps that increasingly filtered for a specific transcript, 1 step that added a Read2 sequence, and 1 step that added an Illumina P7 adaptor sequence). In Step 1, a PCR was conducted using a forward primer containing both the Illumina P5 sequence and part of the Read 1 sequence and a reverse primer containing a locus-specific sequence approximately ∼300bp upstream of the mutation site. In Step 2, a PCR was conducted using a shortened version of the forward primer in Step 1 and a reverse primer containing a locus-specific sequence approximately ∼150bp upstream of the mutation site. In Step 3, a PCR was conducted using the forward primer from Step 2 and a reverse primer containing a locus-specific sequence approximately ∼50bp upstream of the mutation site. In Step 4, a PCR was conducted using the forward primer from Step 2 and a reverse primer containing part of the locus-specific sequence used in Step 3 combined with a Read2 sequence. In Step 5, a PCR was conducted using the forward primer used in Step 1 and a reverse primer containing the Read2 sequence combined with the Illumina P7 sequence.
The following was the specific protocol used for constructing the JAK2-V617F amplicon libraries. All PCR’s were conducted in TempAssure PCR tubes (USA Scientific #14024700) on a Bio-Rad C1000 Touch Thermal Cycler.
In Step 1, a 25uL PCR mixture was made containing 12.5uL of Amplification Master Mix (10x Genomics #220125), 1.25uL of cDNA additive (10x Genomics #220067), 1.25uL of forward primer (P5-Partial Read 1, AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTC) at 20uM, 1.25ul of reverse primer (Reverse Ext 1, ACCAACCTCACCAACATTACAGAGGCCT) at 10uM, 3ng of cDNA library material, and remaining volume with nuclease-free water. With the thermal cycler lid set to 105°C, the following thermal cycling protocol is used: initial denaturation at 98°C for 45 s, 10 cycles of denaturing at 98°C for 20 s, annealing at 67°C for 30 s, extension at 72°C for 180 s, and a final extension at 72°C for 60 s.
The PCR reaction mixture was purified using SPRIselect (Beckman Coulter #B23318) as follows. 20uL (i.e., 0.8x) of SPRIselect reagent was added to the reaction mixture, pipette mixed, and incubated at room temperature for 5 minutes. The PCR tube was placed on the 10x Magnetic Separator (10x Genomics #230003) on High until solution clears (usually about a minute). Supernatant was removed and discarded. 200uL of 80% ethanol in nuclease-free water was added to the pellet and allowed to sit for 30 s. The ethanol wash was removed and repeated once more. The PCR tube was briefly centrifuged and put back into the magnet at Low setting. Any remaining ethanol wash was removed, and the pellet was allowed to air dry for 1 minute. The DNA was then eluted by removing the PCR tube from the magnet, pipetting 20uL Buffer EB (QIAGEN #19086) onto the pellet, pipette mixing, allowing the mixture to equilibrate for 2 minutes, placing the tube on the magnet on Low, and then eluting the liquid into a new tube.
In Step 2, a 25uL PCR mixture was made containing 12.5uL of Amplification Master Mix (10x Genomics #220125), 1.25uL of cDNA additive (10x Genomics #220067), 1.25uL of forward primer (Partial P5, AATGATACGGCGACCACCGAGATCT) at 20uM, 1.25ul of reverse primer (Reverse Ext 2, AGGAGACTACGGTCAACTGCATGAAACAGA) at 10uM, 5uL of the DNA product from Step 1, and 3.75uL of nuclease-free water. With the thermal cycler lid set to 105°C, the following thermal cycling protocol is used: initial denaturation at 98°C for 45 s, 10 cycles of denaturing at 98°C for 20 s, annealing at 67°C for 30 s, extension at 72°C for 180 s, and a final extension at 72°C for 60 s. The PCR reaction mixture was purified using SPRIselect (0.8x) as described previously.
In Step 3, a 25uL PCR mixture was made containing 12.5uL of Hot Start Taq 2x Master Mix (NEB #M0496S), 1.25uL of cDNA additive (10x Genomics #220067), 1.25uL of forward primer (Partial P5, AATGATACGGCGACCACCGAGATCT) at 20uM, 1.25ul of reverse primer (Reverse Ext 3, GCAGCAAGTATGATGAGCAAGCTTTCTCACA) at 10uM, 5uL of the DNA product from Step 2, and 3.75uL of nuclease-free water. With the thermal cycler lid set to 105°C, the following thermal cycling protocol is used: initial denaturation at 95°C for 30 s, 10 cycles of denaturing at 95°C for 30 s, annealing at 60°C for 60 s, extension at 68°C for 210 s, and a final extension at 68°C for 300 s. The PCR reaction mixture was purified using SPRIselect (0.8x) as described previously.
In Step 4, a 25uL PCR mixture was made containing 12.5uL of Amplification Master Mix (10x Genomics #220125), 1.25uL of cDNA additive (10x Genomics #220067), 1.25uL of forward primer (Partial P5, AATGATACGGCGACCACCGAGATCT) at 20uM, 1.25ul of reverse primer (Partial Reverse Ext 3-Read 2, GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTAGCAGCAAGTATGATGAGCA) at 10uM, 2uL of the DNA product from Step 3, and 6.75uL of nuclease-free water. With the thermal cycler lid set to 105°C, the following thermal cycling protocol is used: initial denaturation at 98°C for 45 s, 10 cycles of denaturing at 98°C for 20 s, annealing at 67°C for 30 s, extension at 72°C for 195 s, and a final extension at 72°C for 60 s. The PCR reaction mixture was purified using SPRIselect (0.8x) as described previously.
In Step 5, a 25uL PCR mixture was made containing 12.5uL of Amplification Master Mix (10x Genomics #220125), 0.5uL of SI-PCR primer (10x Genomics #220111), 2.5ul of from one well (recorded for future reference) from the Chromium i7 Sample Index Plate (10x Genomics #220103), 2uL of the DNA product from Step 4, and 7.5uL of nuclease-free water. With the thermal cycler lid set to 105°C, the following thermal cycling protocol is used: initial denaturation at 98°C for 45 s, 10 cycles of denaturing at 98°C for 20 s, annealing at 54°C for 30 s, extension at 72°C for 195 s, and a final extension at 72°C for 60 s. The PCR reaction mixture was purified using SPRIselect (0.8x) as described previously.
Quality control of each step was verified using High Sensitivity D5000 ScreenTape (Agilent #5067-5592) and High Sensitivity D5000 ScreenTape (Agilent #5067-5593) on an Agilent 2200 Tapestation system (Figure S1E). Amplicon libraries display multiple peaks in the TapeStation trace (Figure S1E), which is believed to be due to promiscuous binding of primers to poly-A like regions. Successful enrichment with correct DNA product were typically associated with “sawtooth”-like traces. After using different indexing primers in Step 5 for different libraries, several libraries were pooled and subsequently sequenced together on an Illumina Novaseq 6000 sequencing machine. The sequencing cycle settings were as follows: 28 cycles for Read 1, 8 cycles for the i7 index, and 91 cycles for Read 2.
cDNA material generated in the single-cell cDNA library construction step described in the previous section was generally adequate for making the amplicon libraries described here. If additional cDNA was needed, the single-cell cDNA library was amplified by repeating Step 2.2 of the Chromium Single Cell 3′ Reagent Kit v3 user guide (10x Genomics, CG000183 Rev A).
Depending on the mutation being targeted, optimization of the location of the locus-specific primers and their biochemical properties were needed to minimize non-specific primer binding. For the JAK2-V617F mutation, the three locus-specific primers used were ∼30bp and have melting temperatures of around 68°C. Since the PCR cycle number is dependent on the expression level of the gene harboring the targeted mutations, the number of cycles was optimized experimentally for other mutation targets (ex. TET2).
Stem cell genotyping and preparation for WGS
Several types of cells were genotyped and prepared for WGS: (1) HSCs, MPPs, and stromal cells from bone marrow biopsies, and (2) fibroblasts from a skin biopsy.
1. HSCs, MPPs, and stromal cells
10mL of bone marrow aspirates were used to isolate HSCs, MPPs, and stromal cells. Erythrocytes were removed from 9mL of bone marrow aspirate samples using red blood cell lysis buffer. CD34-enrichment was performed using magnetic-assisted cell sorting with anti-CD34 magnetic beads (Miltenyi Biotech #130-046-703). Different cell populations were purified through using FACSAria (Becton Dickinson). The following combinations of cell surface markers were used to define cell populations. HSC: CD34+CD38-CD45RA-CD90+CD49f+; MPP: CD34+CD38-CD45RA-CD90-CD49f-.
Cells were first sorted into a collection tube, and a second index sorting step was performed to seed single-cells into round bottom 384-well plates, with ∼90 μL of growth medium. Colonies were grown in StemSpan SFEM medium (StemCell Technologies, Inc. #09650) supplemented with: SCF (100 ng/ml), Flt3-L (100 ng/mL), TPO (50 ng/mL), IL3 (10 ng/mL), Tpo (50 ng/mL), Epo (10 ng/mL), and GM-CSF (10 ng/mL). Colonies were grown at 37°C in 5% CO2 for 4-6 weeks before collection, with partial media exchange and fresh cytokine repletion every 2 weeks.
Polyclonal mesenchymal stem cells (MSCs) cultures were established from 1mL of whole bone marrow aspirate samples after red blood cell lysis, cells were plated in tissue culture treated dishes in DMEM-F12 medium (GIBCO #11320082), supplemented with 10% fetal bovine serum (VWR # 89510-186). MSCs were kept in culture for a week and medium was replaced each day to remove non-adherent cells. Stromal cells were ready for collection after reaching 80%–100% confluency.
2. Skin biopsy
One skin biopsy sample was obtained from the JAK2 V617L mutant patient. The tissue was dissociated using collagenase I (StemCell Technologies, Inc. #07415) and genomic DNA was extracted as described below.
For both samples, genomic DNA was extracted from cells using QIAmp UCP DNA Micro Kits (QIAGEN #56204) and eluted into a final volume of 30 μL. The DNA concentration was quantified using a Qubit fluorometer (Invitrogen #Q32866) and Qubit dsDNA HS Assay Kit (Invitrogen #32854). Some of the genomic DNA (1 ng) was amplified using JAK2-V617F specific primers and screened for the mutation using Sanger sequencing.
To generate PCR products for the JAK2 target loci for Sanger sequencing, we performed three rounds of nested PCR with locus-specific reverse primers and generic forward primers. First, a 611bp fragment of JAK2 was amplified to obtain a sufficient amount of DNA containing the mutation site. PCR components included 1ng of gDNA template, Phusion Hot Start Flex 2x Master Mix (M0536L), forward and reverse primers ACTCTTGCTCTCTCTCACTTTG and ACCTGCCATAATCTCTTTTGCT (DNA oligos synthesized by IDT), respectively, and nuclease-free water. The amplification protocol was as follows: (1) initial denaturation at 98°C for 30 s; (2) 40 cycles of denaturation at 98°C for 10 s; (3) annealing at 63°C for 30 s; (4) extension at 72°C for 30 s; and (5) final extension at 72°C for 10 minutes. PCR reaction mixtures were slowly run on a 1.5% agarose gel in EDTA. PCR products were extracted from the gel using a Monarch DNA Gel Extraction Kit (NEB T1020L).
Second, the PCR products were Sanger sequenced in four separate reactions (each with one of four primers) through the Psomagen’s gDNA sequencing service. The four primers used were flanking both directions of the mutation site (DNA oligos synthesized by IDT):
-
1.
TGGCAGAGAGAATTTTCTGAAC (147bp upstream of the mutation)
-
2.
ACTCTTGCTCTCTCTCACTTTG (304bp upstream of the mutation)
-
3.
GTCCTACAGTGTTTTCAGTTTCA (166bp downstream of the mutation)
-
4.
ACCTGCCATAATCTCTTTTGCT (306bp downstream of the mutation)
Electropherograms from Sanger sequencing were annotated. A sample of cells was marked as likely having the mutation if all of the corresponding electropherograms for that sample contained the mutation. Samples were then selected and their gDNA submitted for WGS sequencing (Broad Genomics). The WGS sequencing results were always consistent with the Sanger sequencing, that is the JAK2 mutation was detected in the whole-genome sequencing data for the colonies that were designated as mutated using Sanger sequencing.
Of the ∼600 MPPs and ∼600 HSCs cultured from ET 1, we recovered sufficient genomic DNA from 62 MPP colonies and 22 HSC colonies after expansion. Of these, 6 MPP colonies and 16 HSC colonies had the JAK2-V617F mutation, indicating that mutated HSCs proliferated more in our culture conditions compared with WT HSCs. A similar bias was also observed in cells cultured from the patient ET 2. 384 HSC and 384 MPP colonies were cultured from the ET 2 patient. Of these, we extracted sufficient genomic DNA from 44 HSC colonies and 84 MPP colonies. Sanger genotyping revealed that 14 out of the 44 HSC colonies and 2 out of the 84 MPP colonies had a heterozygous JAK2-V617F mutation. The total sequencing depth was 1.6 billion reads for ET 1 and 480 million reads for ET 2.
Phylodynamic inference
To infer the clonal expansion of mutant HSCs, we first used BNPR (Karcher et al., 2016), an algorithm that infers population size multiplied by a constant factor from lineage trees, where the constant factor is the time between generations. BNPR assumes a Gaussian process prior on the clonal expansion and infers the marginal posterior distributions of the population size (multiplied by a constant factor) at different time points from the coalescent times of a tree. To infer population dynamics with BNPR, we used the Phylodyn package (Karcher et al., 2017).
For the 34-year-old patient tree, we used an averaging algorithm (the averaging algorithm is described in the ABC sections) to make the length from any leaf to the root of the tree the same. We then converted the branch lengths from mutations to generations by assuming 1 cell division per year (Abkowitz et al., 1996; Catlin et al., 2011), so that we could infer population size without the constant factor. The coalescent times of the tree were given to BNPR as input. The only parameter we set was lengthout = 28, which determines the number of time slices at which the population size is estimated, and the remaining were default parameters. The BNPR inference on the 63-year-old patient tree was done in an identical manner.
The BNPR inference was not sensitive to the priors we chose. In particular, changing the covariance associated with the Gaussian process did not change the interpretation of the results. We also tested BNPR on simulated clonal expansions under various scenarios, including simple exponential growth and population bottlenecks, and reliably inferred the population size over time.
Inference of JAK2 mutant HSC fitness
To more precisely infer the clonal expansion of mutated HSCs, we carried out ABC (Approximate Bayesian Computation) using the Wright-Fisher model with selection (see Methods S1 for a detailed description of ABC, the modeling, and for simulations that show that our inference is robust to model assumptions).
On each patient tree, we ran our ABC algorithm and inferred the model parameters.
For the patient with age 34, we used the following specifications for ABC:
-
1.
s was drawn from a uniform distribution on (0, 2).
-
2.
N was drawn from 10X, where X is uniformly distributed on (1, 9).
-
3.
L was drawn from round(Y), where Y is a Gaussian with mean 35 and std 5. If L < 2, we redrew L until L > = 2 since at least 2 generations are necessary to produce a tree.
-
4.
g was drawn uniformly on 2, …, L.
-
5.
k = 22 cells were sampled
-
6.
the mutation rate was (total length of patient tree in mutations)/(L - 1). In this case, the total length of the patient tree in mutations was 723.
-
7.
An epsilon threshold of 0.0225 was used.
For the patient with age 63, we used the following specifications for ABC:
-
1.
s was drawn from a uniform distribution on (0, 2).
-
2.
N was drawn from 10X, where X is uniformly distributed on (1, 9).
-
3.
L was drawn from round(Y), where Y is a Gaussian with mean 65 and std 10. If L < 2, we redrew L until L > = 2 since at least 2 generations are necessary to produce a tree.
-
4.
g was drawn uniformly on 2, …, L.
-
5.
k = 13 cells were sampled
-
6.
The mutation rate was (total length of patient tree in mutations)/(L - 1). In this case, the total length of the patient tree in mutations was 1205.
-
7.
An epsilon threshold of 0.0125 was used.
Note that we drew s from (0, 2) instead of from (0, 5) as done on simulated data in Methods S1. This was done to speed up the simulations and is justified because preliminary runs showed the distribution of s converging to a much smaller value.
For each patient we ran 400 parallel simulations. For the 34-year-old we collected 1,038,712 data points from the posterior, and for the 63-year-old we collected 8,816,199 data points from the posterior. The posterior joint distributions are plotted in Figure 4D and Figure S4.
As indicated by our analysis (see Methods S1), fitness s could be inferred from the patient trees. Our analysis also suggests that if we assume a division rate of one per year (Lee-Six et al., 2018), n can be inferred for the 34-year-old patient as 4.74 ± 0.68 of the posterior distribution. n, however, cannot be inferred for the 63-year-old patient, and the inferred distribution of n is just the prior information. This is due to the fact that the coalescent events occur in the very early history of the disease, and the information about the population size is lost. We can, however, put bounds on the point of saturation if we assume one division per year. As seen in simulation results presented in Methods S1, when the number of mutant cells approaches N, the growth of the mutant population slows down and starts to exhibit neutral dynamics. If N is sufficiently small, it changes the coalescent structure, and ABC then assumes the saturation point is n = N. Trajectories generated by ABC if N is below a certain threshold value produced coalescent structures that did not match that of the patient data. Any value of N larger than this threshold had no effect on the coalescent structure and therefore was retained as a possible inferred value. Therefore, N could not be precisely determined.
Quantification and Statistical Analysis
scRNA-seq preprocessing and cell type identification
scRNaseq libraries and the amplicon libraries were sequenced on the NovaSeq platform. The resulting bcl files were run through the Cell Ranger 4.0.0 pipeline to generate the fastq files and the count matrices. The fastq files for the amplicon libraries were analyzed as described below. Count matrices from each patient were loaded into Scanpy (Wolf et al., 2018). Genes expressed in < 3 cells and cells with < 2,000 total UMIs or > 20% mitochondrial transcripts were excluded from further analysis. Total count normalization was performed so that each cell had 100,000 total transcripts. Log-transformed expression values were used for UMAP visualization and clustering after regressing out % of mitochondrial transcripts and total counts. UMAP coordinates were calculated using Scanpy default parameter values. To assign an HSPC cell type to each cell, the scRNA-seq data from all patients were merged and batch corrected using Seurat’s data integration workflow with the default parameter values (Stuart et al., 2019). Louvain clustering was performed on the merged and batch-corrected dataset in Scanpy and each cluster was assigned an HSPC cell type identity by manually reviewing the expression levels of marker genes in that cluster. Identification of monocyte subsets was performed similarly on CD14+ cells from all donors.
Differential gene expression analysis between CD14+ cells from different patient groups
Scanpy’s implementation of the Student’s t test was used to compare gene expression between ET, PV, and healthy CD14+ cells using total count normalized gene expression values without batch correction. To limit the impact of batch effects, we performed all pairwise comparisons between each patient in both groups (e.g., for the ET versus PV comparison, we separately compared ET 1 and PV 1, ET 1 and PV 2, etc) and identified genes that were differentially expressed in all comparisons for gene set enrichment analysis using GSEApy (https://pypi.org/project/gseapy/).
Identification of JAK2 mutant cells in the scRNA-seq data
To identify individual cells in the scRNaseq library as either WT or JAK2 mutant, we separately analyzed the fastq files of the amplicon libraries derived from the same cells. First, the reads in the fastq files were discarded if the average Illumina base quality value was less than 30. Next, only reads were retained whose single-cell barcode uniquely matched (up to at most 2 bps differences) a barcode from the list of single-cell barcodes in the scRNaseq library of the same cells. A threshold for the number of reads was determined by inspecting the plot of the number of reads from each molecule (unique cell-barcode and UMI) after rank ordering the molecules by their number of reads, corresponding to the knee in the plot, usually around 100 to 1,000 reads depending on the sequencing depth (Figure S1F). Molecules that had fewer reads than the threshold were discarded. To correct for sequencing errors in the UMIs, those molecules that shared the same barcode but had the same UMI sequence up to 2 mismatches were merged. Next, the mutation site was inspected in the remaining reads. Only reads were retained that had the expected WT nucleotide, or the expected mutated version of the nucleotide, and the where the 10 bps upstream and downstream of the mutation site matched the reference genome. A molecule was designated as “mutated” if more than half of its reads carried the mutated nucleotide, and WT otherwise. The above analysis pipeline was implemented in MATLAB R2018. JAK2 genotyping results for all cells sequenced can be found in Table S3.
Cells with at least one WT amplicon call were marked as “WT.” It is important to note that in cells with a heterozygous JAK2 mutation, the presence of a WT transcript does not guarantee that the cell is homozygous WT. However, cells with at least one JAK2-mutant transcript were definitively classified as JAK2-mutant cells. Therefore, to correct for JAK2 mutation heterozygosity in patients with < 50% peripheral blood JAK2 mutation VAF, the fraction of JAK2 mutant cells in a cell population was estimated as the fraction of JAK2 mutant molecules in the cell population multiplied by 2. This correction factor comes from the observation that, since most cells only have one JAK2 transcript call, cells with a heterozygous JAK2 mutation have approximately a 50% chance of having a JAK2 mutant transcript sequenced so approximately half of true JAK2 mutant cells have a mutant JAK2 transcript sequenced.
Whole-genome sequencing data analysis
Raw sequencing reads were mapped to the GRCh38 build of the human reference genome using BWA-MEM (Li, 2013) version 0.7.17-r1188. Aligned reads in BAM format were processed following the Genome Analysis Toolkit (GATK, version 4.1.2.0) Best Practices workflow to remove duplicates and recalibrate base quality scores (DePristo et al., 2011).
Detection of somatic single-nucleotide variants and INDELs
The germline short variant discovery workflow from GATK version 4.1.2.0 was used to detect somatic single-nucleotide variants (SNVs) and small insertions and deletions (INDELs) in the single-cell-derived WGS data. In brief, intermediate GVCF files were generated for each colony and chromosome using HaplotypeCaller in GVCF mode. Default parameter values were used except for the output-mode argument, which was set to “EMIT_ALL_SITES.” Next, GVCF files for all colonies from each patient were consolidated into a single GVCF file using the GATK functionality CombineGVCFs using default options. Finally, colonies were jointly genotyped across all sites using GenotypeGVCFs with the “–include-non-variant-site” parameter set to true.
In order to identify somatically acquired point mutations and indels in the colonies the following steps were followed.
All sites with a genotype quality of at least 50 and showing variation in at least one colony were selected.
Variants mapping less than 10bp upstream or downstream of a simple repeat reported in the RepeatMasker track from the UCSC Genome Browser were discarded.
Variants mapping less than 100bp apart from each other were removed, as in our experience these are likely artifacts.
Variants that could not be genotyped in 10 or more colonies in each patient were discarded.
To remove subclonal mutations acquired during in vitro culture the mean variant allele frequency (VAF) value across all mutated colonies was required to be between 0.3 and 0.7 for patient ET 2 (female) and for the autosomes in the case of patient ET 1 (male). Additionally, we required a minimum coverage of at least 6 sequencing reads. Variants mapping to chromosomes X and Y in the case of patient ET 1 and chromosome X in colony MPP-73 from patient ET 2, which harbors only one copy of this chromosome, were required to show a VAF value of at least 0.9 and the coverage threshold was set to 3 sequencing reads.
Sites supporting more than 4 genotypes across all colonies were removed, as after manual inspection of a number of such cases we concluded that these were likely artifacts.
We required the genotype quality in the bulk sequencing data from stromal cells to be at least 80 in order to remove variants in low-quality mapping regions.
Only variants with a homozygous reference genotype in the bulk sample were kept in order to filter out germline heterozygous polymorphisms.
Given that all cancer cells share 220 and 398 mutations in patients ET 1 and ET 2, respectively, we reasoned that any mutations occurring early in development and giving rise to both the cancer and wild-type cells should be present in all cancer colonies and in a subset of the normal colonies, but not in normal colonies and just a subset of the cancer colonies. Therefore, all mutations detected in just a subset of the cancer cells and one or more wild-type colonies were discarded, as these are likely germline polymorphisms or artifacts. We did not find any mutation present in all cancer cells and one or more normal cells. In this analysis we only focused on the discovery of heterozygous variants given that all colonies, with the exception of the loss of chromosome X in colony MPP-73 from ET 2, show diploid karyotypes with no copy number alterations.
All variants remaining after applying the filters described above were visually inspected using BAMsnap (https://github.com/parklab/bamsnap), and those deemed to be false positives were removed. The remaining variants were deemed to be somatic and were considered for further analysis. Annovar (version 2018Apr16) was used to annotate variants. Missense variants predicted to be deleterious by MetaLR and MetaSVM were considered pathogenic (Dong et al., 2015).
Detection of microsatellite mutations
Somatic mutations at microsatellite loci were detected using HipSTR version 0.6.2 (Willems et al., 2017) using de novo stutter estimation and allele generation, and the reference set of microsatellite loci provided by the authors. Subsequently, microsatellite calls were filtered and only calls satisfying the following criteria were considered for further analysis: (1) Posterior probability for the genotype higher than 0.95; (2) the fraction of indels in the reads mapping to the flanking regions of the microsatellite under consideration smaller than 0.15; (3) the fraction of reads estimated to contain a stutter artifact smaller than 0.15; (4) at least 3 sequencing reads spanning each of the supported alleles for the microsatellite under consideration; (5) log10 P value for the allele bias test implemented in HipSTR higher than 2; (6) log10 P value for the Fisher strand bias test higher than 2; (7) the ratio of the number of reads supporting each allele higher than 0.7. This filter served to remove low-VAF mutations likely arising during in vitro culture or PCR noise; and (8) a depth of at least 10 sequencing reads. Finally, only microsatellite loci with a reliable call in at least 30 samples and with at least 2 different genotypes across all colonies were considered for further analysis. All mutations satisfying the criteria listed above were further validated through visual inspection of raw sequencing reads.
Detection of somatic structural variants
Structural variants were called in each colony using Manta (version 1.6.0), LUMPY (version 0.2.13), SvABA (version 1.1.3), and Delly (version 0.8.3) (Chen et al., 2016; Layer et al., 2014; Rausch et al., 2012; Wala et al., 2018). Each algorithm was run independently on each colony using the bulk sequencing data for stromal cells from the corresponding patient as control, and in a second run using a randomly selected JAK2-WT colony as control. The calls generated by each algorithm were merged using the Python library mergevcf. (https://github.com/ljdursi/mergevcf) and only calls generated by at least two algorithms were kept for further analysis.
Somatic copy number calling
The software package ascatNGS (Raine et al., 2016) was used to detect somatic copy number alterations in each colony and to estimate their purity and ploidy. The bulk sequencing data from bone marrow stromal cells from the same patient was used as the normal sample in all cases.
Mutational signature analysis
Mutational signature analysis was performed using the R package MutationalPatterns (Blokzijl et al., 2018). To quantify the contribution of mutational processes known to be operative in MPNs (Alexandrov et al., 2020) (namely SBS1, SBS2, SBS5, SBS19, SBS23, and SBS32) to the observed spectrum of somatic point mutations in each colony, we used the function fit_to_signatures using default parameter options. The goodness of fit was determined by computing the cosine similarity between the observed mutational pattern and the reconstructed one using the estimated signature contributions. In all cases we obtained cosine similarity values > 0.95, suggesting that our analysis explained most of the variance related to the contribution of different mutational processes to the observed mutational spectra.
Telomere length estimation
The length of telomeres was estimated for all colonies from the same patient jointly using Telomerecat version 3.4.0 (Farmery et al., 2018) and the default options except for batch correction. The average telomere length across 100 runs was considered for further analysis.
Comparing the mutation rate between JAK2 mutant and JAK2-WT colonies
To assess whether the mutation rate in JAK2 mutant and JAK2-WT colonies is statistically significant, we had to account for the fact that JAK2 mutant colonies are clonally related, as they share hundreds of mutations in both ET 1 and ET 2. To account for this shared ancestry, we computed the difference between the mean number of mutations in JAK2 mutant and JAK2-WT colonies. Next, we computed the expected variance by accounting for the clonal relatedness of JAK2 mutant colonies. Specifically, we scaled the variance of the number of mutations in JAK2-WT colonies by the number of years at which the clonal expansion started (that is, 9/34 and 19/63 in the case of ET 1 and ET 2, respectively), and computed the square root. We scaled the number of mutations by the variance rather than by the standard deviation given that we assume that the accumulation of mutations in HSPCs can be modeled as a Poisson process. If we then consider the distribution of mean differences to be Gaussian with mean zero, we can compute a z score by computing the mean difference divided by the estimated standard deviation, and then estimate the corresponding one-sided P value.
Inference and validation of phylogenetic trees
The somatic mutations detected across all colonies in a given patient were used to reconstruct phylogenetic trees using the software package PHYLIP version 3.695 (https://evolution.genetics.washington.edu/phylip.html). For each patient and mutation type, namely, SNVs, INDELs, and microsatellite mutations, as well as for these three combined, we constructed a binary matrix with rows indexed by somatic mutations and columns by colonies such that the i,j entry in each matrix was set to one if mutation i is present in colony j, and to zero otherwise. Only mutations detected in at least two colonies were considered to build lineage trees, as private mutations are uninformative to establish the phylogeny of the colonies. We detected a total of 21,699 SNVs (935 present in at least two colonies), 1,396 (60) indels, and 482 (31) microsatellite mutations across the single-cell-colonies derived from patient ET 1 (Table S2). In the case of ET 2, we detected a total of 33,994 SNVs (1,245), 2,464 (94) indels, and 891 (70) microsatellite mutations (Table S2).
For each input mutation matrix, we generated 100 bootstrap replicates by sampling with replacement using the Seqboot method. Lineage trees were then estimated for each resample using the Wagner parsimony algorithm as implemented in the Mix method using the bulk data from stromal cells as the outgroup. The consensus tree across all bootstrap samples was generated using the extended majority rule method as implemented in the program Consense. Once the consensus tree was determined, we assigned to each branch those mutations that were present in all the descendant colonies of that branch and in none of the other colonies. Lineage tree representations were generated using the R package ggtree (Yu, 2020).
As expected given the high number of mutations shared across cancer colonies, the clonal architecture of cancer cells was largely consistent across bootstrap resamples irrespective of the type of mutations considered for lineage tree inference. In fact, the clonal architecture for the cancer colonies was the same across all resamples when using somatic SNVs as input. More variability was observed when the lineage trees were constructed using indels or microsatellite mutations as input, although the majority of splits in the tree were consistent across more than 90% of resamples. This is expected given that variant callers generally show lower sensitivity and specificity for the detection of small insertions and deletions as compared to point mutations (Campbell et al., 2020). This is also consistent with the fact that the highest rates of private INDELs and microsatellite are detected for those colonies with the lowest sequencing quality in our cohort (e.g., HSC-49 from ET 1). The clonal architecture of WT colonies varied across resamples, as indicated by the low bootstrap values we obtained for nodes splitting clades of WT colonies. The low concordance observed for node splits across resamples is likely due to the low number of somatic mutations detected in more than one WT colony, consistent with previous lineage tree analyses of human HSPCs using somatic mutations (Lee-Six et al., 2018). Overall, the reliability of the consensus trees we have generated is supported by the following: (1) the clonal architecture, in particular for cancer colonies, observed across lineage trees inferred using different types of somatic mutations is overall consistent, (2) the nodes in the trees are largely concordant across bootstrapping resamples for cancer colonies, and (3) 96% and 99% of the SNVs detected in at least 2 colonies from ET 1 and ET 2, respectively, could be unambiguously assigned to the consensus lineage tree generated using SNVs.
Acknowledgments
We thank the individuals who participated in our study. We thank Drs. David Weinstock and Julie-Aurore Losman for valuable feedback on the manuscript. Portions of this research were conducted on the O2 High Performance Compute Cluster, supported by the Research Computing Group, at Harvard Medical School (https://it.hms.harvard.edu/our-services/research-computing/). S.H. acknowledges funding from NIH NIGMS R00GM118910 and NIH NHLBI R01HL158269, the DFCI BCB Fund Award, the Jayne Koskinas Ted Giovanis Foundation, The William F. Milton Fund at Harvard University, an AACR-MPM Oncology Charitable Foundation Transformative Cancer Research grant, and Gabrielle’s Angel Foundation for Cancer Research. S.H. and A.M. acknowledge funding from the Claudia Adams Barr Program in Cancer Research. A.M. acknowledges funding from NIH NHLBI (R01HL131835) and the MPN Research Foundation. A.M. is a Scholar of The Leukemia & Lymphoma Society. C.R.R acknowledges funding from NIH NHLBI T32HL116324. D.V.E. acknowledges funding from the NSF-Simons Center for Mathematical and Statistical Analysis of Biology at Harvard, award number 1764269, and the Harvard Quantitative Biology Initiative. D.V.E and F.M. acknowledge support from the Ludwig Center at Harvard and the Dana-Farber Cancer Institute Physical Science-Oncology Center (NIH U54CA193461 to F.M.). I.C.-C. and M.K. acknowledge funding from EMBL. J.E. acknowledges funding from NIH NIGMS R25GM109436. F.D.C. is supported by a grant from the Edward Evans MDS Foundation and NIH NHLBI P01HL131477. We thank Dr. Francesc Mutas Remolar for fruitful discussions.
Author Contributions
R.M.S., A.M., and S.H. conceived the project. M.N., S.L., and S.H. designed the experiments. M.N. devised and optimized the single-cell amplicon sequencing protocol with help from S.L. and supervision from S.H. M.N. and S.L. processed the samples and generated all sequencing libraries with help from B.K. C.R.R., G.S.H., and A.M. devised the individual selection criteria. D.J.D., I.G., M.W., E.S.W., M.R.L., R.M.S., J.S.G., G.S.H., and A.M. helped obtain samples, coordinated by C.R.R. D.V.E. analyzed all the single-cell data with help from C.R.R. and B.K., supervised by F.M. and S.H. M.K. and I.C.-C. analyzed the whole-genome sequencing data and reconstructed the lineage trees. S.P. isolated and cultured individual HSCs, supervised by F.D.C. J.E. devised and implemented the algorithms for inference of growth dynamics from lineage trees, supervised by S.H. D.V.E., J.E., M.N., S.L., C.R.R., F.M., A.M., I.C.-C., and S.H. wrote the manuscript with input from all authors. A.M., I.C.-C., and S.H. supervised the project.
Declaration of Interests
A.M. has consulted for Janssen, PharmaEssentia, Constellation, and Relay Therapeutics and receives research support from Janssen and Actuate Therapeutics. E.S.W. reports personal fees from Jazz Pharmaceuticals, Takeda Pharmaceutical Company, Novartis, and Pfizer. F.M. is the co-founder of an oncology company. J.S.G. has consulted for AbbVie, Takeda, and Astellas and receives research support from AbbVie, Genentech, Prelude, AstraZeneca, and Eli Lilly. D.J.D. receives research support from Glycomimetics, Novartis, AbbVie, and Blueprint Medicines and has consulted for Incyte, Jazz, Novartis, Pfizer, Shire, Takeda, Amgen, Forty-Seven, Agios, Autolos, and Blueprint Medicines. G.S.H. has received research support from Bayer, Merck, Incyte, and Constellation and has received honoraria from Constellation, Jazz, Novartis, and Celgene/BMS. R.M.S. has advisory board, DSMB, and/or steering committee membership at Syntrix/ACI Clinical, Takeda, Elevate Bio, Syndax Pharma, AbbVie, Syros, Gemoab, BerGenBio, Foghorn Thera, GSK, Aprea, Innate, Actinium, and OncoNova.
Published: February 22, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stem.2021.02.001.
Contributor Information
Ann Mullally, Email: ann_mullally@dfci.harvard.edu.
Isidro Cortes-Ciriano, Email: icortes@ebi.ac.uk.
Sahand Hormoz, Email: sahand_hormoz@hms.harvard.edu.
Supporting Citations
The following references appear in the supplemental information: Fisher (1923); Kimura (1962); Kingman (1982a); Kingman (1982b); Kingman (1982c); Wright, (1931).
Supplemental Information
References
- Abkowitz J.L., Catlin S.N., Guttorp P. Evidence that hematopoiesis may be a stochastic process in vivo. Nat. Med. 1996;2:190–197. doi: 10.1038/nm0296-190. [DOI] [PubMed] [Google Scholar]
- Adamson J.W., Fialkow P.J., Murphy S., Prchal J.F., Steinmann L. Polycythemia vera: stem-cell and probable clonal origin of the disease. N. Engl. J. Med. 1976;295:913–916. doi: 10.1056/NEJM197610212951702. [DOI] [PubMed] [Google Scholar]
- Alexandrov L.B., Jones P.H., Wedge D.C., Sale J.E., Campbell P.J., Nik-Zainal S., Stratton M.R. Clock-like mutational processes in human somatic cells. Nat. Genet. 2015;47:1402–1407. doi: 10.1038/ng.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov L.B., Kim J., Haradhvala N.J., Huang M.N., Tian Ng A.W., Wu Y., Boot A., Covington K.R., Gordenin D.A., Bergstrom E.N., PCAWG Mutational Signatures Working Group. PCAWG Consortium The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101. doi: 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter E.J., Scott L.M., Campbell P.J., East C., Fourouclas N., Swanton S., Vassiliou G.S., Bench A.J., Boyd E.M., Curtin N., Cancer Genome Project Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- Beer P.A., Jones A.V., Bench A.J., Goday-Fernandez A., Boyd E.M., Vaghela K.J., Erber W.N., Odeh B., Wright C., McMullin M.F. Clonal diversity in the myeloproliferative neoplasms: independent origins of genetically distinct clones. Br. J. Haematol. 2009;144:904–908. doi: 10.1111/j.1365-2141.2008.07560.x. [DOI] [PubMed] [Google Scholar]
- Blokzijl F., Janssen R., van Boxtel R., Cuppen E. MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med. 2018;10:33. doi: 10.1186/s13073-018-0539-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell P.J., Getz G., Korbel J.O., Stuart J.M., Jennings J.L., Stein L.D., Perry M.D., Nahal-Bose H.K., Ouellette B.F.F., Li C.H., ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93. [Google Scholar]
- Catlin S.N., Busque L., Gale R.E., Guttorp P., Abkowitz J.L. The replication rate of human hematopoietic stem cells in vivo. Blood. 2011;117:4460–4466. doi: 10.1182/blood-2010-08-303537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen E., Beer P.A., Godfrey A.L., Ortmann C.A., Li J., Costa-Pereira A.P., Ingle C.E., Dermitzakis E.T., Campbell P.J., Green A.R. Distinct clinical phenotypes associated with JAK2V617F reflect differential STAT1 signaling. Cancer Cell. 2010;18:524–535. doi: 10.1016/j.ccr.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Schulz-Trieglaff O., Shaw R., Barnes B., Schlesinger F., Källberg M., Cox A.J., Kruglyak S., Saunders C.T. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–1222. doi: 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- Cordua S., Kjaer L., Skov V., Pallisgaard N., Hasselbalch H.C., Ellervik C. Prevalence and phenotypes of JAK2 V617F and calreticulin mutations in a Danish general population. Blood. 2019;134:469–479. doi: 10.1182/blood.2019001113. [DOI] [PubMed] [Google Scholar]
- De Grandis M., Cambot M., Wautier M.-P., Cassinat B., Chomienne C., Colin Y., Wautier J.-L., Le Van Kim C., El Nemer W. JAK2V617F activates Lu/BCAM-mediated red cell adhesion in polycythemia vera through an EpoR-independent Rap1/Akt pathway. Blood. 2013;121:658–665. doi: 10.1182/blood-2012-07-440487. [DOI] [PubMed] [Google Scholar]
- Delhommeau F., Dupont S., Tonetti C., Massé A., Godin I., Le Couedic J.P., Debili N., Saulnier P., Casadevall N., Vainchenker W., Giraudier S. Evidence that the JAK2 G1849T (V617F) mutation occurs in a lymphomyeloid progenitor in polycythemia vera and idiopathic myelofibrosis. Blood. 2007;109:71–77. doi: 10.1182/blood-2006-03-007146. [DOI] [PubMed] [Google Scholar]
- DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C., Wei P., Jian X., Gibbs R., Boerwinkle E., Wang K., Liu X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015;24:2125–2137. doi: 10.1093/hmg/ddu733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusa A., Staerk J., Elliott J., Pecquet C., Poirel H.A., Johnston J.A., Constantinescu S.N. Substitution of pseudokinase domain residue Val-617 by large non-polar amino acids causes activation of JAK2. J. Biol. Chem. 2008;283:12941–12948. doi: 10.1074/jbc.M709302200. [DOI] [PubMed] [Google Scholar]
- Farmery J.H.R., Smith M.L., Lynch A.G., NIHR BioResource - Rare Diseases Telomerecat: A ploidy-agnostic method for estimating telomere length from whole genome sequencing data. Sci. Rep. 2018;8:1300. doi: 10.1038/s41598-017-14403-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher R.A. XXI.—On the Dominance Ratio. Proc. R. Soc. Edinb. 1923;42:321–341. [Google Scholar]
- Gangaraju R., Song J., Kim S.J., Tashi T., Reeves B.N., Sundar K.M., Thiagarajan P., Prchal J.T. Thrombotic, inflammatory, and HIF-regulated genes and thrombosis risk in polycythemia vera and essential thrombocythemia. Blood Adv. 2020;4:1115–1130. doi: 10.1182/bloodadvances.2019001379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G., Kähler A.K., Handsaker R.E., Lindberg J., Rose S.A., Bakhoum S.F., Chambert K., Mick E., Neale B.M., Fromer M. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliland D.G., Blanchard K.L., Levy J., Perrin S., Bunn H.F. Clonality in myeloproliferative disorders: analysis by means of the polymerase chain reaction. Proc. Natl. Acad. Sci. USA. 1991;88:6848–6852. doi: 10.1073/pnas.88.15.6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinfeld J., Nangalia J., Baxter E.J., Wedge D.C., Angelopoulos N., Cantrill R., Godfrey A.L., Papaemmanuil E., Gundem G., MacLean C. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018;379:1416–1430. doi: 10.1056/NEJMoa1716614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B.B., Linden M.D., Fuller K.A., Phillips M., Mirzai B., Wilson L., Chuah H., Liang J., Howman R., Grove C.S. Platelets in myeloproliferative neoplasms have a distinct transcript signature in the presence of marrow fibrosis. Br. J. Haematol. 2020;188:272–282. doi: 10.1111/bjh.16152. [DOI] [PubMed] [Google Scholar]
- Hinds D.A., Barnholt K.E., Mesa R.A., Kiefer A.K., Do C.B., Eriksson N., Mountain J.L., Francke U., Tung J.Y., Nguyen H.M. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood. 2016;128:1121–1128. doi: 10.1182/blood-2015-06-652941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii T., Bruno E., Hoffman R., Xu M. Involvement of various hematopoietic-cell lineages by the JAK2V617F mutation in polycythemia vera. Blood. 2006;108:3128–3134. doi: 10.1182/blood-2006-04-017392. [DOI] [PubMed] [Google Scholar]
- Jaiswal S., Fontanillas P., Flannick J., Manning A., Grauman P.V., Mar B.G., Lindsley R.C., Mermel C.H., Burtt N., Chavez A. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S., Natarajan P., Silver A.J., Gibson C.J., Bick A.G., Shvartz E., McConkey M., Gupta N., Gabriel S., Ardissino D. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017;377:111–121. doi: 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James C., Ugo V., Le Couédic J.-P., Staerk J., Delhommeau F., Lacout C., Garçon L., Raslova H., Berger R., Bennaceur-Griscelli A. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- Jamieson C.H.M., Gotlib J., Durocher J.A., Chao M.P., Mariappan M.R., Lay M., Jones C., Zehnder J.L., Lilleberg S.L., Weissman I.L. The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc. Natl. Acad. Sci. USA. 2006;103:6224–6229. doi: 10.1073/pnas.0601462103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karcher M.D., Palacios J.A., Bedford T., Suchard M.A., Minin V.N. Quantifying and Mitigating the Effect of Preferential Sampling on Phylodynamic Inference. PLoS Comput. Biol. 2016;12:e1004789. doi: 10.1371/journal.pcbi.1004789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karcher M.D., Palacios J.A., Lan S., Minin V.N. phylodyn: an R package for phylodynamic simulation and inference. Mol. Ecol. Resour. 2017;17:96–100. doi: 10.1111/1755-0998.12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. On the probability of fixation of mutant genes in a population. Genetics. 1962;47:713–719. doi: 10.1093/genetics/47.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. Cambridge University Press; 1983. The Neutral Theory of Molecular Evolution. [Google Scholar]
- Kingman J.F.C. Exchangeability and the evolution of large populations. In: Finetti B.D., Koch G., Spizzichino F., editors. Proceedings of the International Conference on Exchangeability in Probability and Statistics. North-Holland Publishing Company; 1982. pp. 97–112. [Google Scholar]
- Kingman J.F.C. On the genealogy of large populations. In: Moran A.P., Gani J.M., Hannan E.J., editors. Essays in Statistical Science. Applied Probability Trust; 1982. pp. 27–43. [Google Scholar]
- Kingman J.F.C. The coalescent. Stochastic Process. Appl. 1982;13:235–248. [Google Scholar]
- Kluk M.J., Lindsley R.C., Aster J.C., Lindeman N.I., Szeto D., Hall D., Kuo F.C. Validation and Implementation of a Custom Next-Generation Sequencing Clinical Assay for Hematologic Malignancies. J. Mol. Diagn. 2016;18:507–515. doi: 10.1016/j.jmoldx.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralovics R., Passamonti F., Buser A.S., Teo S.-S., Tiedt R., Passweg J.R., Tichelli A., Cazzola M., Skoda R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- Layer R.M., Chiang C., Quinlan A.R., Hall I.M. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15:R84. doi: 10.1186/gb-2014-15-6-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Six H., Øbro N.F., Shepherd M.S., Grossmann S., Dawson K., Belmonte M., Osborne R.J., Huntly B.J.P., Martincorena I., Anderson E. Population dynamics of normal human blood inferred from somatic mutations. Nature. 2018;561:473–478. doi: 10.1038/s41586-018-0497-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine R.L., Wadleigh M., Cools J., Ebert B.L., Wernig G., Huntly B.J.P., Boggon T.J., Wlodarska I., Clark J.J., Moore S. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013 https://arxiv.org/abs/1303.3997 arXiv:1303.3997. [Google Scholar]
- Lundberg P., Karow A., Nienhold R., Looser R., Hao-Shen H., Nissen I., Girsberger S., Lehmann T., Passweg J., Stern M. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood. 2014;123:2220–2228. doi: 10.1182/blood-2013-11-537167. [DOI] [PubMed] [Google Scholar]
- Machado H.E., Øbro N.F., Mitchell E., Davies M., Green A.R., Saeb-Parsy K., Hodson D.J., Kent D., Campbell P.J. Life History of Normal Human Lymphocytes Revealed By Somatic Mutations. Blood. 2019;134 1045–1045. [Google Scholar]
- Maekawa T., Kato S., Kawamura T., Takada K., Sone T., Ogata H., Saito K., Izumi T., Nagao S., Takano K. Increased SLAMF7high monocytes in myelofibrosis patients harboring JAK2V617F provide a therapeutic target of elotuzumab. Blood. 2019;134:814–825. doi: 10.1182/blood.2019000051. [DOI] [PubMed] [Google Scholar]
- Nam A.S., Kim K.-T., Chaligne R., Izzo F., Ang C., Taylor J., Myers R.M., Abu-Zeinah G., Brand R., Omans N.D. Somatic mutations and cell identity linked by Genotyping of Transcriptomes. Nature. 2019;571:355–360. doi: 10.1038/s41586-019-1367-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio F.G., Rosendahl Huber A., Oka R., Verheul M., Patel S.H., Hasaart K., de la Fonteijne L., Varela I., Camargo F.D., van Boxtel R. Somatic Mutations Reveal Lineage Relationships and Age-Related Mutagenesis in Human Hematopoiesis. Cell Rep. 2018;25:2308–2316.e4. doi: 10.1016/j.celrep.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psaila B., Wang G., Rodriguez-Meira A., Li R., Heuston E.F., Murphy L., Yee D., Hitchcock I.S., Sousos N., O’Sullivan J., NIH Intramural Sequencing Center Single-Cell Analyses Reveal Megakaryocyte-Biased Hematopoiesis in Myelofibrosis and Identify Mutant Clone-Specific Targets. Mol. Cell. 2020;78:477–492.e8. doi: 10.1016/j.molcel.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raine K.M., Van Loo P., Wedge D.C., Jones D., Menzies A., Butler A.P., Teague J.W., Tarpey P., Nik-Zainal S., Campbell P.J. ascatNgs: Identifying Somatically Acquired Copy-Number Alterations from Whole-Genome Sequencing Data. Curr. Protoc. Bioinformatics. 2016;56:15.9.1–15.9.17. doi: 10.1002/cpbi.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampal R., Al-Shahrour F., Abdel-Wahab O., Patel J.P., Brunel J.-P., Mermel C.H., Bass A.J., Pretz J., Ahn J., Hricik T. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood. 2014;123:e123–e133. doi: 10.1182/blood-2014-02-554634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T., Zichner T., Schlattl A., Stütz A.M., Benes V., Korbel J.O. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–i339. doi: 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steensma D.P. JAK2 V617F in myeloid disorders: molecular diagnostic techniques and their clinical utility: a paper from the 2005 William Beaumont Hospital Symposium on Molecular Pathology. J. Mol. Diagn. 2006;8:397–411, quiz 526. doi: 10.2353/jmoldx.2006.060007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusi B.K., Wolock S.L., Weinreb C., Hwang Y., Hidalgo D., Zilionis R., Waisman A., Huh J.R., Klein A.M., Socolovsky M. Population snapshots predict early haematopoietic and erythroid hierarchies. Nature. 2018;555:54–60. doi: 10.1038/nature25741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wala J.A., Bandopadhayay P., Greenwald N.F., O’Rourke R., Sharpe T., Stewart C., Schumacher S., Li Y., Weischenfeldt J., Yao X. SvABA: genome-wide detection of structural variants and indels by local assembly. Genome Res. 2018;28:581–591. doi: 10.1101/gr.221028.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson C.J., Papula A.L., Poon G.Y.P., Wong W.H., Young A.L., Druley T.E., Fisher D.S., Blundell J.R. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science. 2020;367:1449–1454. doi: 10.1126/science.aay9333. [DOI] [PubMed] [Google Scholar]
- Willems T., Zielinski D., Yuan J., Gordon A., Gymrek M., Erlich Y. Genome-wide profiling of heritable and de novo STR variations. Nat. Methods. 2017;14:590–592. doi: 10.1038/nmeth.4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams N., Lee J., Moore L., Baxter E.J., Hewinson J., Dawson K.J., Menzies A., Godfrey A.L., Green A.R., Campbell P.J. Phylogenetic reconstruction of myeloproliferative neoplasm reveals very early origins and lifelong evolution. bioRxiv. 2020 doi: 10.1101/2020.11.09.374710. [DOI] [Google Scholar]
- Wolach O., Sellar R.S., Martinod K., Cherpokova D., McConkey M., Chappell R.J., Silver A.J., Adams D., Castellano C.A., Schneider R.K. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018;10:eaan8292. doi: 10.1126/scitranslmed.aan8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S. Evolution in Mendelian Populations. Genetics. 1931;16:97–159. doi: 10.1093/genetics/16.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M., Lu C., Wang J., McLellan M.D., Johnson K.J., Wendl M.C., McMichael J.F., Schmidt H.K., Yellapantula V., Miller C.A. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014;20:1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G. Using ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinformatics. 2020;69:e96. doi: 10.1002/cpbi.96. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We developed a C++ object called StemCellSim for simulating clonal expansions and inferring the parameters of our model. StemCellSim has the ability to generate simulated data under various models, and to infer model parameters from either simulated or real data with ABC. The StemCellSim code and the Python scripts used to analyze and plot the scRNA-seq data can be found on GitLab (https://gitlab.com/hormozlab). Raw scRNA-seq and whole-genome sequencing data have been deposited in dbGAP:phs002308.v1.p1.