

Abstract
Inducible conditional knockout mice are important tools for studying gene function and disease therapy, but their generation is costly and time-consuming. We introduced clustered regularly interspaced short palindromic repeats (CRISPR) and Cre into an LSL-Cas9 transgene-carrying mouse line by using adeno-associated virus (AAV)-PHP.eB to rapidly knockout gene(s) specifically in central nervous system (CNS) cells of adult mice. NeuN in neurons and GFAP in astrocytes were knocked out 2 weeks after an intravenous injection of vector, with an efficiency comparable to that of inducible Cre-loxP conditional knockout. For functional testing, we generated astrocyte-specific Act1 knockout mice, which exhibited a phenotype similar to mice with Cre-loxP-mediated Act1 knockout, in an animal model of multiple sclerosis (MS), an autoimmune disorder of the CNS. With this novel technique, neural cell-specific knockout can be induced rapidly (few weeks) and cost-effectively. Our study provides a new approach to building inducible conditional knockout mice, which would greatly facilitate research on CNS biology and disease.
Keywords: CRISPR, Conditional knockout, AAV PHP.eB, Neural-specific knockout, Multiple sclerosis, Knockout mouse model, Rapid in vivo knockout
Graphical abstract
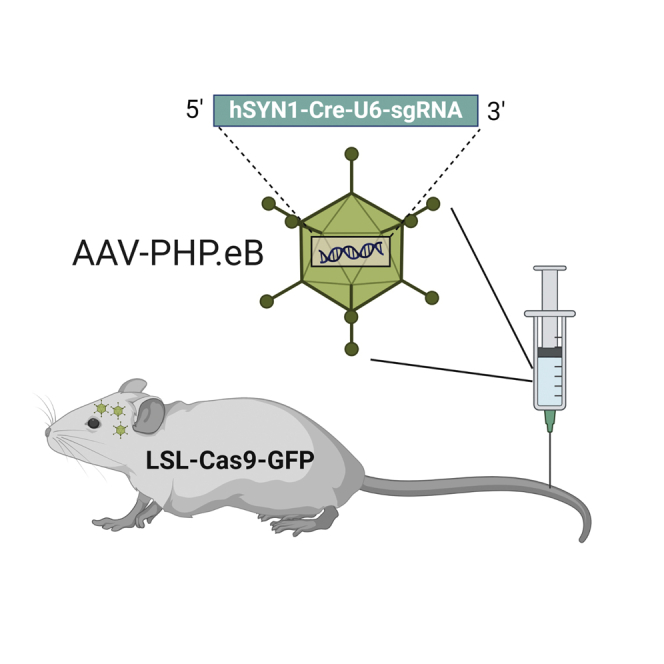
We built a method based on CRISPR to generate neural cell-specific conditional knockout mice. Through i.v. injection of CRISPR carrying AAV into LSL-Cas9 transgenic mice, knockout mice can be built in a couple of weeks. Our method generates knockout mice with efficiency comparable with the Cre-loxP system but with much less time cost.
Introduction
Development of inducible conditional gene knockout mouse lines through breeding of CreER-donor and loxP-carrying mice, with typically 70%–80% knockout efficiency of a target gene, allows for time-controlled and cell-specific gene modification.1,2 While this technique is a cornerstone in basic scientific studies on disease mechanisms and pre-clinical studies of potential therapies, it is time- and labor-consuming to generate CreER and loxP mice, to obtain certain specific mouse lines from non-commercial resources, and then to breed and genotype them to generate homozygous CreER-loxP mice. The clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated (Cas)-based genome-editing technique has a proven capacity for efficient genome editing.3, 4, 5, 6, 7 CRISPR-mediated gene knockout in vivo has the advantage of being time-, labor-, and cost-saving compared with the Cre-loxP breeding method. Highly efficient CRISPR-mediated gene knockout has been achieved to develop disease models in peripheral tissues such as lung and liver.5,7, 8, 9, 10, 11 However, efficient gene knockout in the central nervous system (CNS) using CRISPR is limited by the blood-brain barrier (BBB). Some attempts have been made to edit genes by stereotaxic intracerebral injections into the CNS of adult mice,12, 13, 14 or to take advantage of the underdeveloped BBB of neonatal mice to knockout genes in neurons.15 However, we still do not have a method to knock out genes of a specific cell type in the entire adult CNS. The development of adeno-associated virus (AAV)-PHP.eB, a new variant of AAV9 that can transduce neurons, astrocytes, and oligodendrocytes, but not microglia, with high efficiency through intravenous (i.v.) injection,16 makes this possible.
In the present study, we combined an AAV-PHP.eB, CRISPR, and LSL-Cas9 mouse line to build a method that knocks out genes rapidly, efficiently, and cell specifically in the entire CNS. We knocked out NeuN specifically in neurons and GFAP specifically in astrocytes with an efficiency comparable with that of the CreER-loxP method. Finally, we generated astrocyte-specific Act1 knockout mice, which showed a phenotype similar to mice generated using the Cre-loxP method when tested in experimental autoimmune encephalomyelitis (EAE), a mouse model for MS. Our data demonstrate that our approach is a powerful method for neurophysiological and pathological research.
Results
Neuron-specific knockout of NeuN using AAV PHP.eB-CRISPR
Owing to the size limitations of AAV vectors,17 there is not enough space for Cas9, single guide RNA (sgRNA), and a tissue-specific promoter to be included in the same vector. We thus generated the AAV PHP.eB vector carrying sgRNA and Cre gene that is expressed under the control of a neural cell-specific promoter (Figure 1A). This vector was i.v. injected into LSL-Cas9 transgenic mice on a C57BL/6 background, carrying a Cas9-P2A-GFP cassette driven by the inactive CAG promoter due to a floxed stop signal18 (Figure 1B). In this manner, CNS-specific knockout mice can be generated in less than 1 month, which is much more rapid and easier than the traditional Cre-loxP-based conditional gene knockout method (Figure 1C; Table S1). Cell-specific gene knockout was first tested using a neuron-specific promoter, hSYN1, to knock out NeuN, a neuron marker. Three target sites for sgRNAs were selected (Figure 1D), sgRNA carrying plasmids were transfected into the N2A-C9 cell line, and sgRNA activity was analyzed by a T7 endonuclease 1 (T7E1) assay (Figure 1E). To avoid sense mutation induced by single sgRNA, two sgRNAs (#1 and #2) with the highest activity were cloned in series into the AAV transfer plasmid to further improve knockout efficiency; the plasmid was named pAAV-sgNeuN-hSYN1-Cre. Cleaving efficiency of plasmid carrying two sgRNAs was also analyzed in the N2A-C9 cell line (Figure 1F). The corresponding AAVs were produced and purified as reported,16 and named PHP.eB-sgNeuN-hSYN1-Cre. Control plasmid named pAAV-sgRNA scramble (sgScram)-hSYN1-Cre was generated by replacing NeuN sgRNA with a scramble sgRNA, and the resulting virus was named PHP.eB-sgScram-hSYN1-Cre. To knock out NeuN in vivo, 5 × 1011 vector genomes (vg) of AAV were injected per mouse through the tail vein, with three mice in each group. The knockout efficiency was analyzed by immunostaining and flow cytometry 2 weeks after injection (Figure 2). In the spinal cord of PHP.eB-sgScram-hSYN1-Cre-injected mice, about 72% of NeuN+ cells were GFP+, which indicates Cas9 expression in neurons, while only 2% of NeuN− cells expressed GFP (Figure 2A). This demonstrates highly efficient and specific gene expression in the target cells. Injection of PHP.eB-sgNeuN-hSYN1-Cre knocked out NeuN in 65% of total neurons (from 9.35% down to 3.32%; Figure 2A). This result was confirmed by immunostaining showing a 66% reduction in NeuN+ cells (from 26.05% down to 8.83%) (Figures 2B and 2C). NeuN was knocked out in 98% of GFP+ neurons, i.e., from 93% in the control group down to 2% in the NeuN sgRNA-carrying AAV group (Figure 2D). NeuN knockout in the cerebrum was also analyzed, as exemplified by immunofluorescence images of the cortex (Figures 3A and 3B), hippocampus (Figure 3C), and thalamus (Figure S1). Statistical analysis of the immunofluorescence data showed that NeuN was knocked out in 82.2% of the total NeuN+ cells (from 38.96% down to 6.94%; Figure 3D) and in 99% of transduced (GFP+) neurons (Figure 3E) after treatment with NeuN-sgRNA-carrying AAV. It has been reported that the liver and heart were heavily transduced by AAV PHP.eB in rats.19 We thus tested whether our virus induced GFP expression in liver and heart, which may lead to undesirable expression of Cas9 in the peripheral tissue. Immunostaining of these organs from AAV-Scram sgRNA-transduced LSL-Cas9 mice showed no GFP+ cells (Figure S2), indicating that our neuron-specific targeting AAV virus is not active in peripheral tissues, most likely because of the high specificity of the hSYN1 (neuron-specific) promoter.
Figure 1.

Schematic illustration of AAV-CRISPR-mediated CNS-specific knockout and NeuN targeting sgRNA selection in vitro
(A) Diagram of the structure of AAV transfer plasmid used. TSP, tissue-specific promoter. (B) Diagram of the structure of transgene in LSL-Cas9 mice. (C) Cartoon shows the timeline of the Cre-loxP or CRISPR-based conditional gene knockout method. (D) Location of sgRNAs targeting NeuN and detection primers on the genome. (E) T7E1 assay of single NeuN sgRNA activity. (F) T7E1 assay of the cleavage efficiency of two NeuN sgRNAs.
Figure 2.

Neuron-specific NeuN knockout in spinal cord
PHP.eB-sgNeuN-hSYN1-Cre or PHP.eB-sgScram-hSYN1-Cre was i.v. injected into naive adult LSL-Cas9 mice, 8–10 weeks of age, at 5 × 1011 vg per mouse. Spinal cords were harvested 2 weeks later for flow cytometry analysis or immunostaining. (A) Flow cytometry analysis of NeuN+ cells in the spinal cord of PHP.eB-sgNeuN-hSYN1-Cre versus control groups, and GFP+ cells in the NeuN+ cells and NeuN− cells. One representative result of two independent experiments is shown. (B) Representative confocal images of GFP and NEUN staining in the transverse spinal cord sections. Scale bars, 50 μm. (C and D) Data shown in (B) were quantified for the knockout efficiency in total neurons (C) or vector-transduced (GFP+) neurons (D). Data in (C) and (D) are shown as mean ± SD (n = 3 mice per group, five to six images for each mouse). ∗∗∗∗p < 0.0001, by an unpaired two-tailed t test.
Figure 3.

Neuron-specific NeuN knockout in cerebrum
Brains from PHP.eB-sgNeuN-hSYN1-Cre- or PHP.eB-sgScram-hSYN1-Cre-injected mice were harvested 2 weeks after injection and analyzed by immunostaining. (A–C) Representative confocal images of GFP and NEUN staining in the cortex (A and B) and hippocampus (C). Arrows in (B) depict GFP and NEUN double-positive cells in the PHP.eB-sgNeuN-hSYN1-Cre-injected group. For (A) and (B): scale bars, 100 μm, scale bars of zoomed images, 20 μm; for (C), scale bars, 50 μm. (D and E) Quantification of knockout efficiency in total neurons (D) or vector-infected transduced (GFP+) neurons (E) in the cerebrum. Data in (D) and (E) are shown as mean ± SD (n = 3 mice per group, five to six images for each mouse). ∗∗∗∗p < 0.0001, by an unpaired two-tailed t test.
Astrocyte-specific knockout of GFAP using AAV PHP.eB-CRISPR
To test the efficiency of gene knockout in astrocytes, we replaced the hSYN1 promoter by a GFAP promoter. The GFAP gene, a marker for astrocytes, was chosen as the target gene to knock out. Four sgRNAs targeting the GFAP gene were selected (Figure 4A), and sgRNA activities were tested in the N2A-C9 cell line and analyzed by a T7E1 assay (Figure 4B). Two sgRNAs with the highest efficiency (#3 and #4) were selected and cloned successively into an AAV-transfer plasmid that carries the GFAP promoter and Cre gene, and cleavage efficiency was analyzed in the N2A-C9 cell line (Figure 4C). AAV was i.v. injected into adult mice as described in Figure 2, with three mice in each group. The transduction efficiency and specificity in CNS cells were analyzed 2 weeks later by flow cytometry. In mice injected with PHP.eB-sgScram-GFAP-Cre, up to 86% of astrocytes in the spinal cord (Figure 4D) and 54% in the cerebrum (Figure 4E) were transduced by AAV, as indicated by GFP expression. The transduction specificity in the spinal cord was high, since only 6.8% of GFAP− cells expressed GFP (Figure 4D), whereas transduction specificity in the cerebrum (Figure 4E) was lower than in the spinal cord. Knockout efficiencies were also analyzed by western blot. Injection of GFAP-targeting AAV reduced GFAP expression by 79% in spinal cord and 46% in cerebrum (Figures 4F and 4G). These results show that CRISPR-carrying AAV knocked out the GFAP gene in astrocytes of the spinal cord with high efficiency and specificity, but the knockout efficiency and specificity in the cerebrum were somewhat low. This could be because the GFAP promoter activity in some cerebrum astrocytes is not high enough to induce the expression of Cre.20 Nonetheless, our method shows efficiency comparable with reports that GFAP-Cre/loxP conditional knockout mice have a 70%–80% knockout efficiency.21,22
Figure 4.

Astrocyte-specific GFAP knockout in vivo
(A) Location of sgRNAs targeting GFAP and detection primers on the genome. (B) T7E1 assay of single GFAP sgRNA activity. (C) T7E1 assay of the cleavage efficiency of two GFAP sgRNAs. PHP.eB-sgGFAP-GFAP-Cre or PHP.eB-sgScram-GFAP-Cre was i.v. injected into naive adult LSL-Cas9 mice, 8–10 weeks of age, at 5 × 1011 vg per mouse. (D and E) GFAP expression in spinal cord (D) and cerebrum (E) of mice that received PHP.eB-sgScram-GFAP-Cre was determined at 2 weeks after injection by flow cytometry. n = 2 mice each group. (F) Western blot analysis of the knockout efficiency of GFAP in spinal cord (top panel) and cerebrum (bottom panel). AAV-injected mice were sacrificed 2 weeks after injection and astrocytes were purified using an ACSA-2 MicroBead kit. The isolated astrocytes were lysed by RIPA buffer and GFAP expression was determined by western blot. (G) Quantification of GFAP knockout efficiency was determined by the density of the western blot bands. The results are presented as the ratio of density between GFAP and GAPDH. The density of bands was analyzed using ImageJ. Data are shown as mean ± SD (n = 3 mice per group). ∗∗p < 0.01, ∗∗∗∗p < 0.0001, by a one-way ANOVA.
Functional test in vivo
For in vivo functional assessment, we compared the conditional knockout mice generated using our method with Cre-loxP mice. Act1 is an essential intracellular adaptor for interleukin (IL)-17A signaling, which is important for the development of EAE.23,24 Significantly reduced EAE severity was seen in mice with Cre-loxP-mediated knockout of Act1 in neuroectoderm-derived cells, in which astrocytes are a major part, in addition to neurons and oligodendrocytes.25 To knock out Act1 in astrocytes in adult mice, four sgRNAs targeting mouse Act1 were designed and screened (Figures 5A and 5B). Two sgRNAs with the highest efficiency (# 2 and #3) were successively cloned into GFAP-Cre-carrying AAV transfer plasmid, and activity was tested in vitro (Figure 5C). Mice were injected through the tail vein with Act1-targeting AAV (five mice) and control AAV (seven mice). Ten days later, mice were immunized for EAE induction and were sacrificed 30 days post-immunization (p.i.), after which transduction efficiency in the spinal cord, where the main EAE lesions are located, was analyzed by immunostaining. Astrocytes in both white and gray matter were transduced by AAV with high efficiency, as shown by a large number of GFP+GFAP+ cells (Figure 6A and 6B), likely owing to the activation of astrocytes during EAE. This was in contrast to naive mice, in which mainly astrocytes in white matter were transduced (as shown in Figure S3). Act1 in astrocytes was knocked out with high efficiency, as shown by immunostaining (Figures 6C and 6D). Mice injected with AAV that targets Act1 had delayed and ameliorated EAE development compared to control mice (Figure 6E), with significantly reduced numbers of CNS-infiltrating mononuclear cells (Figure 6F). The size and number of GFAP+ cells in the Act1 knockout group were also greatly reduced, most likely owing to reduced astrocyte activation (Figure 6C). These results show that CRISPR-mediated conditional knockout mice have similar characteristics as Cre-loxP conditional knockout mice, thus validating the usefulness of this approach in studies that involve the CNS. AAV can also be injected at the onset or peak of disease for therapeutic studies.
Figure 5.

Act1 sgRNA design and activity assay
(A) Location of sgRNAs targeting Act1 and detection primers on the genome. (B) T7E1 assay of single Act1 sgRNA activity. (C) T7E1 assay of the cleavage efficiency of two Act1 sgRNAs.
Figure 6.

Astrocyte-specific Act1 knockout in vivo and EAE induction
PHP.eB-sgAct1-GFAP-Cre or control virus was i.v. injected into naive adult LSL-Cas9 mice, 8–10 weeks old, at 5 × 1011 vg per mouse 10 days before immunization. Mice were sacrificed at day 30 p.i. and transduction/knockout efficiencies were analyzed by immunostaining. (A) Representative confocal image of GFP and GFAP staining in the transverse spinal cord sections. Scale bar, 50 μm. (B) Representative confocal image of GFP and GFAP staining in the white matter of spinal cord with a prolonged GFP exposure time. Scale bar, 100 μm; scale bar of zoomed images, 20 μm. (C) Immunostaining analysis of Act1 knockout in the spinal cord of EAE mice. Scale bar, 100 μm; zoomed images show representative cells, scale bar, 20 μm. (D) Quantification of Act1 expression in astrocytes, as calculated by the intensity of GFAP+Act1+ cells divided by the intensity of GFAP+ cells in the spinal cord. (E) EAE scores. (F) Statistical analysis of numbers of immune cells infiltrated into the CNS. Mononuclear cells from the CNS of mice in (E) were isolated by Percoll gradient centrifugation and counted using a cell counter. All data are shown as mean ± SD (n = 7 for scramble sgRNA, n = 5 for Act1 sgRNA). ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, by an unpaired two-tailed t test. One representative of two independent experiments is shown.
Discussion
Our present study provides a novel technique to generate inducible conditional knockout animals, with several significant advantages over currently used approaches. (1) The current technique to generate inducible, conditional gene knockout mouse lines is cost- and time-consuming, requiring months or even years to establish, while our approach requires a few weeks in vitro, and 1–2 weeks in vivo to knock out the gene(s) of interest in a specific CNS cell type. (2) The mouse lines (e.g., flox/flox and Cre-ER) necessary to generate inducible conditional knockout of a particular gene in a specific cell type are often unavailable, precluding relevant in vivo experiments, while any gene(s) of interest can be readily knocked out using our technique. (3) Compared with intra-CNS injections that typically knock out genes only in an area proximal to the injection site, i.v. injection of PHP.eB serotype AAV, an ideal tool for CNS delivery,16 knocks out genes throughout the CNS. Also, i.v. injection is easy to perform and causes less stress to the mice, which is important in some disease models affected by stress, e.g., EAE.26,27 (4) To save enough space for a tissue-specific promoter in AAV transfer plasmid and to further improve knockout efficiency, we used an LSL-Cas9 transgene mouse line with a C57 background, available from The Jackson Laboratory and suitable for most disease models. The GFP reporter gene in the transgene of LSL-Cas9 mice is a convenient marker for identification of knockout cells. (5) Replacement of Cas9 from Streptococcus pyogenes (spCas9) with Staphylococcus aureus Cas9 (saCas9) could allow cloning of a tissue-specific promoter, Cas9 and sgRNA, in a single plasmid, given that saCas9 is smaller than spCas9. This would extend the applicability of our method to more animal types and backgrounds. (6) Our approach can also be used to knock out multiple genes simultaneously in multiple CNS cell types with a combination of different viruses.
In summary, we have established a rapid, simple, and economic approach to cell-specific gene knockout in CNS cells, whereby any gene(s) of interest can be knocked out in a short time (about 2 weeks). This technique has the capacity to significantly simplify the inducible conditional knockout mouse system for CNS studies in vivo.
Materials and methods
Cell culture
The N2A-Cas9 cell line was purchased from Genecopoeia (Rockville, MD, USA) and grown in DMEM containing 10% fetal bovine serum (FBS). HEK293T cells were also grown in DMEM containing 10% FBS. Cells were maintained at 37°C in 5% CO2 atmosphere.
Mice
LSL-Cas9 (stock no. 026175) mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Mice, 8–10 weeks old, with a body weight of approximately 20 g were used in all experiments in this study. Mice were kept in a specific pathogen-free animal facility at Thomas Jefferson University. All experiments were carried out in accordance with guidelines by the Institutional Animal Care and Use Committee (IACUC) of Thomas Jefferson University.
Plasmid
lentiCRISPR v2 (Addgene plasmid #52961) and AAV:inverted terminal repeat (ITR)-U6-sgRNA (backbone)-pCBh-Cre-WPRE-hGHpA-ITR (Addgene plasmid #60229) were provided by Dr. Feng Zhang. pAAV-hSyn1-mRuby2 was provided by Viviana Gradinaru (Addgene plasmid #99126). lentiCRISPR v2 was used as a template to amplify U6, using U6 KpnI forward and U6 SfuI reverse primers. The acquired fragment was used to replace the U6 promoter in lentiCRISPR v2 through KpnI and SfuI to introduce an XbaI before U6, and the obtained plasmid was named lentiCRISPR v3 (Figure S4). sgRNAs targeting NeuN, GFAP, and Act1 were designed using https://www.benchling.com/crispr/; corresponding primers were synthesized by Integrated DNA Technologies (IDT, Coralville, IA, USA). Primers were annealed and ligated into lentiCRISPR v3 through BsmBI to test cleaving efficiency in the N2A-C9 cell. To clone two sgRNAs in one plasmid, the U6-sgRNA fragment was cleaved from the first plasmid through KpnI and NheI and inserted into the second plasmid through KpnI and XbaI. Scramble sgRNA was also cloned into lentiCRISPR v3 as control.
Primers for the MPAA linker were synthesized and annealed at room temperature. The MPAA linker was cloned into AAV:ITR-U6-sgRNA (backbone)-pCBh-Cre-WPRE-hGHpA-ITR to replace U6-sgRNA-pCBH cassette through MluI and AgeI; the resulting plasmid was named pAAV-MCS-Cre. For neuron-specific gene knockout, the hSYN1 promoter was amplified from pAAV-hSyn1-mRuby2 and cloned into pAAV-MCS-Cre through NheI and AscI; the resulting plasmid was named pAAV-MCS-hSYN1-Cre (Figure S5A). U6-sgRNA expression cassettes carrying two NeuN sgRNAs or scrambled sgRNAs were cleaved from lentiCRISPR v3 and cloned into pAAV-MCS-hSYN1-Cre separately through KpnI and NheI; the resulting plasmid was named pAAV-sgNeuN-hSYN1-Cre or pAAV-sgScram-hSYN1-Cre.
For astrocyte-specific gene knockout, the GFAP promoter was amplified from the pLenti-Gfap-eGFP-mir30-shAct1 vector28 and cloned into pAAV-MCS-Cre through NheI and AscI; the resulting plasmid was named pAAV-MCS-GFAP-Cre (Figure S5B). For U6-sgRNA expression cassettes carrying two GFAP sgRNAs, two Act1 sgRNAs or scrambled sgRNAs were cleaved from lentiCRISPR v3 and ligated into pAAV-MCS-GFAP-Cre separately, through KpnI and NheI; the resulting plasmid was named pAAV-sgGFAP-GFAP-Cre, pAAV-sgAct1-GFAP-Cre, or pAAV-sgScram-GFAP-Cre.
All of the primers used are listed in Table S2.
pAdDeltaF6 was provided by James M. Wilson (Addgene plasmid #112867); pUCmini-iCAP-PHP.eB was provided by Viviana Gradinaru16 (Addgene plasmid #103005).
AAV packaging and purification
The AAV particles were generated as reported by Chan et al.16 Briefly, low-passaged 293T cells were transfected with transfer plasmid, pUCmini-iCAP-PHP.eB and pAdDeltaF6 using PEI-MAX (Polysciences, Warrington, PA, USA). Viral particles were collected at 72 h after transfection from the medium, and at 120 h after transfection from the cells and medium. The viruses were then purified by iodixanol (Sigma, St. Louis, MO, USA) step gradients (15%, 25%, 40%, and 60%),29 concentrated using Amicon filters (EMD Millipore, Burlington, MA, USA), and formulated in sterile PBS with 0.001% Pluronic F-68 (Gibco, Gaithersburg, MD, USA). Virus titers were measured by determining the number of DNase I-resistant vg using qPCR, with a linearized genome plasmid as the standard.30
Isolation of cells from adult mouse CNS
Mice were perfused with 1× PBS and the isolated brain or spinal cord was chopped with a razor blade. Tissues were digested into single-cell suspensions, and myelin was removed using an adult brain dissociation kit (Miltenyi Biotec, Gaithersburg, MD, USA). Cells were re-suspended in PBS for flow cytometry analysis or specific cell isolation.
For western blot, astrocytes were isolated from single-cell suspension using an anti-ACSA-2 MicroBead kit (Miltenyi Biotec) following the manufacturer’s instructions. The purified astrocytes were re-suspended in PBS for western blot analysis.
Immunofluorescent labeling and imaging
Frozen tissues were cut into 10-μm sections. Frozen sections were air-dried, rehydrated in Tris-buffered saline (TBS), permeabilized by TBS with 0.2% Triton X-100, and blocked in TBS with 10% horse serum and 1% BSA for 30 min. The primary antibodies were then incubated in TBS with 1% BSA at 4°C overnight. The following day, the slides were washed three times in TBS with 0.025% Triton X-100 and incubated with secondary antibody (Jackson ImmunoResearch, West Grove, PA, USA) in TBS with 1% BSA at room temperature for 1 h. Antibodies used were as follows: anti-NeuN clone A60 (EMD Millipore), anti-GFP (Abcam, Branford, CT, USA), anti-GFAP clone D1F4Q (Cell Signaling Technology [CST], Danvers, MA, USA), and anti-Act1 clone WW-18 (Santa Cruz Biotechnology, Dallas, TX, USA).
Finally, all of the sections were washed and mounted in Prolong Gold antifade reagent with DAPI (Invitrogen, Carlsbad, CA, USA). Imaging was performed using a Nikon A1R microscope and Nikon NIS Elements acquisition and analysis software. Images were processed and analyzed by ImageJ.
Flow cytometry
Cells were first stained with surface antibodies at 4°C for 20 min, fixed by medium A (Invitrogen), washed, and then incubated with intracellular antibodies in medium B (Invitrogen) at 4°C overnight. Antibodies used in these experiments were as follows: anti-NeuN-phycoerythrin (PE) clone A60 (EMD Millipore), anti-GFP-AF488 (Invitrogen), and anti-GFAP-AF647 clone 2.2B10 (Invitrogen). Compensation was performed using UltraComp eBeads (Invitrogen).
Western blot
1 × 106 astrocytes were lysed in 100 μL of radioimmunoprecipitation assay (RIPA) lysis buffer (Thermo Scientific, Waltham, MA, USA) containing protease inhibitors (Sigma). Cells were incubated on ice for 30 min and sonicated on ice for 20 s. Cell lysate was centrifuged at 10,000 rpm for 10 min at 4°C and supernatant was collected. Protein concentration in the supernatant was determined using the bicinchroninic acid (BCA) protein assay kit (Pierce Biotechnology, Waltham, MA, USA). Protein lysate was diluted in SDS-PAGE sample buffer, separated on Novex 4%–12% Tris-glycine gel (Invitrogen), and analyzed by western blot using rabbit anti-GFAP monoclonal antibody (CST). GAPDH was stained with rabbit anti-GAPDH monoclonal antibody (CST) and used as loading control.
T7E1 assay
N2A-C9 cells were seeded in a 24-well plate at 1 × 105 cells per well and, on the following day, were transfected with a mixture containing 0.5 μg of plasmid and 1 μL of Lipofectamine 2000. Medium was changed on the next day, and cells were collected 48 h after transfection. Genomic DNA was extracted using QuickExtract DNA extraction solution (Lucigen, Middleton, WI, USA). PCR reactions were performed using PrimeSTAR HS DNA polymerase (Takara, Mountain View, CA, USA), using the respective PCR primers. The PCR products were subjected to denaturation and reannealing using a thermocycler and purified using a Monarch PCR & DNA cleanup kit (NEB, Ipswich, MA, USA). The cycle conditions used for denaturation and reannealing were 95°C for 2 min, ramp at 2°C/s until 85°C was reached followed by 85°C for 2 min, ramp at 0.1°C/s until 25°C, 25°C for 2 min, and then kept at 16°C until used for T7E1 digestion. Purified PCR products (300 ng) were digested by 0.5 μL of T7E1 at 37°C for 20 min. The resulting products were separated on 2% agarose gel and imaged by Axygen gel documentation systems.
EAE induction
LSL-Cas9 mice, 8–10 weeks of age, were used for EAE induction. Mice were immunized subcutaneously at two sites on the back with 200 μg of myelin oligodendrocyte glycoprotein (MOG)35–55 peptide (GenScript, Piscataway, NJ, USA) emulsified in complete Freund’s adjuvant (BD Biosciences, San Jose, CA, USA) supplemented with 10 mg/mL Mycobacterium tuberculosis H37Ra (BD Biosciences). Mice were also injected intraperitoneally with 200 ng of pertussis toxin (Sigma, St. Louis, MO, USA) on days 0 and 2 p.i. Mice were monitored for weight changes and clinical signs until 30 days p.i. Clinical signs were scored by two separate researchers in a blinded manner using a 0–5 scale: 0, no clinical sign; 1, limp tail; 2, limp tail with weak/partially paralyzed hind legs; 3, limp tail with complete paralyzed hind legs; 4, tetraplegia; 5, moribund.
Statistical analysis
Statistical analyses were performed with Prism software (GraphPad). An unpaired two-tailed t test was used for comparison of two groups. Two-way ANOVA was used for comparison of more than two groups. p values of <0.05 were considered significant. All error bars represent SD, as noted in the individual figure legends.
Acknowledgments
This work was supported by the NIH grants NS099594 and AI135601. W.Z. and D.X. were partly supported by Chinese National Natural Science Foundation grant 31601086. We thank Katherine Regan for editorial assistance.
Author contributions
W.Z., D.X., and G.-X.Z. conceived and designed the experiments, analyzed data, and wrote the manuscript. W.Z. and D.X. carried out the experiments. Q.W. performed the immunohistology experiments. X.L. and Y.Z. helped with the experimental design and statistical analysis. J.R. performed flow cytometry experiments. G.C. helped in the preparation of vectors and revised the manuscript. M.C. helped evaluate immunohistological results and revised the manuscript. B.C. and A.R. co-supervised the study and wrote the paper. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.02.012.
Supplemental information
References
- 1.Kim H., Kim M., Im S.K., Fang S. Mouse Cre-LoxP system: general principles to determine tissue-specific roles of target genes. Lab. Anim. Res. 2018;34:147–159. doi: 10.5625/lar.2018.34.4.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horii T., Morita S., Kimura M., Terawaki N., Shibutani M., Hatada I. Efficient generation of conditional knockout mice via sequential introduction of lox sites. Sci. Rep. 2017;7:7891. doi: 10.1038/s41598-017-08496-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yin H., Xue W., Chen S., Bogorad R.L., Benedetti E., Grompe M., Koteliansky V., Sharp P.A., Jacks T., Anderson D.G. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 2014;32:551–553. doi: 10.1038/nbt.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sánchez-Rivera F.J., Papagiannakopoulos T., Romero R., Tammela T., Bauer M.R., Bhutkar A., Joshi N.S., Subbaraj L., Bronson R.T., Xue W., Jacks T. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature. 2014;516:428–431. doi: 10.1038/nature13906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng R., Peng J., Yan Y., Cao P., Wang J., Qiu C., Tang L., Liu D., Tang L., Jin J. Efficient gene editing in adult mouse livers via adenoviral delivery of CRISPR/Cas9. FEBS Lett. 2014;588:3954–3958. doi: 10.1016/j.febslet.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 9.Ding Q., Strong A., Patel K.M., Ng S.L., Gosis B.S., Regan S.N., Cowan C.A., Rader D.J., Musunuru K. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ. Res. 2014;115:488–492. doi: 10.1161/CIRCRESAHA.115.304351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xue W., Chen S., Yin H., Tammela T., Papagiannakopoulos T., Joshi N.S., Cai W., Yang G., Bronson R., Crowley D.G. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature. 2014;514:380–384. doi: 10.1038/nature13589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh K., Evens H., Nair N., Rincón M.Y., Sarcar S., Samara-Kuko E., Chuah M.K., VandenDriessche T. Efficient in vivo liver-directed gene editing using CRISPR/Cas9. Mol. Ther. 2018;26:1241–1254. doi: 10.1016/j.ymthe.2018.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swiech L., Heidenreich M., Banerjee A., Habib N., Li Y., Trombetta J., Sur M., Zhang F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 2015;33:102–106. doi: 10.1038/nbt.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Staahl B.T., Benekareddy M., Coulon-Bainier C., Banfal A.A., Floor S.N., Sabo J.K., Urnes C., Munares G.A., Ghosh A., Doudna J.A. Efficient genome editing in the mouse brain by local delivery of engineered Cas9 ribonucleoprotein complexes. Nat. Biotechnol. 2017;35:431–434. doi: 10.1038/nbt.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park H., Oh J., Shim G., Cho B., Chang Y., Kim S., Baek S., Kim H., Shin J., Choi H. In vivo neuronal gene editing via CRISPR-Cas9 amphiphilic nanocomplexes alleviates deficits in mouse models of Alzheimer’s disease. Nat. Neurosci. 2019;22:524–528. doi: 10.1038/s41593-019-0352-0. [DOI] [PubMed] [Google Scholar]
- 15.Gaj T., Ojala D.S., Ekman F.K., Byrne L.C., Limsirichai P., Schaffer D.V. In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci. Adv. 2017;3:eaar3952. doi: 10.1126/sciadv.aar3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan K.Y., Jang M.J., Yoo B.B., Greenbaum A., Ravi N., Wu W.L., Sánchez-Guardado L., Lois C., Mazmanian S.K., Deverman B.E., Gradinaru V. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017;20:1172–1179. doi: 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu Z., Yang H., Colosi P. Effect of genome size on AAV vector packaging. Mol. Ther. 2010;18:80–86. doi: 10.1038/mt.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Platt R.J., Chen S., Zhou Y., Yim M.J., Swiech L., Kempton H.R., Dahlman J.E., Parnas O., Eisenhaure T.M., Jovanovic M. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dayton R.D., Grames M.S., Klein R.L. More expansive gene transfer to the rat CNS: AAV PHP.EB vector dose-response and comparison to AAV PHP.B. Gene Ther. 2018;25:392–400. doi: 10.1038/s41434-018-0028-5. [DOI] [PubMed] [Google Scholar]
- 20.Matias I., Morgado J., Gomes F.C.A. Astrocyte heterogeneity: impact to brain aging and disease. Front. Aging Neurosci. 2019;11:59. doi: 10.3389/fnagi.2019.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.García-Cáceres C., Quarta C., Varela L., Gao Y., Gruber T., Legutko B., Jastroch M., Johansson P., Ninkovic J., Yi C.X. astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell. 2016;166:867–880. doi: 10.1016/j.cell.2016.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cai W., Xue C., Sakaguchi M., Konishi M., Shirazian A., Ferris H.A., Li M.E., Yu R., Kleinridders A., Pothos E.N., Kahn C.R. Insulin regulates astrocyte gliotransmission and modulates behavior. J. Clin. Invest. 2018;128:2914–2926. doi: 10.1172/JCI99366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li X., Commane M., Nie H., Hua X., Chatterjee-Kishore M., Wald D., Haag M., Stark G.R. Act1, an NF-κB-activating protein. Proc. Natl. Acad. Sci. USA. 2000;97:10489–10493. doi: 10.1073/pnas.160265197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qian Y., Liu C., Hartupee J., Altuntas C.Z., Gulen M.F., Jane-Wit D., Xiao J., Lu Y., Giltiay N., Liu J. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat. Immunol. 2007;8:247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- 25.Kang Z., Altuntas C.Z., Gulen M.F., Liu C., Giltiay N., Qin H., Liu L., Qian W., Ransohoff R.M., Bergmann C. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity. 2010;32:414–425. doi: 10.1016/j.immuni.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harpaz I., Abutbul S., Nemirovsky A., Gal R., Cohen H., Monsonego A. Chronic exposure to stress predisposes to higher autoimmune susceptibility in C57BL/6 mice: glucocorticoids as a double-edged sword. Eur. J. Immunol. 2013;43:758–769. doi: 10.1002/eji.201242613. [DOI] [PubMed] [Google Scholar]
- 27.Miller S.D., Karpus W.J., Davidson T.S. Experimental autoimmune encephalomyelitis in the mouse. Curr. Protoc. Immunol. 2010;Chapter 15 doi: 10.1002/0471142735.im1501s88. Unit 15.1. [DOI] [PubMed] [Google Scholar]
- 28.Yan Y., Ding X., Li K., Ciric B., Wu S., Xu H., Gran B., Rostami A., Zhang G.X. CNS-specific therapy for ongoing EAE by silencing IL-17 pathway in astrocytes. Mol. Ther. 2012;20:1338–1348. doi: 10.1038/mt.2012.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zolotukhin S., Byrne B.J., Mason E., Zolotukhin I., Potter M., Chesnut K., Summerford C., Samulski R.J., Muzyczka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- 30.Gray S.J., Choi V.W., Asokan A., Haberman R.A., McCown T.J., Samulski R.J. Production of recombinant adeno-associated viral vectors and use in in vitro and in vivo administration. Curr. Protoc. Neurosci. 2011;Chapter 4 doi: 10.1002/0471142301.ns0417s57. Unit 4.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.