

Abstract
Colorectal cancer (CRC) is one of the most common malignant tumors, with the second-highest mortality of all 36 cancers worldwide. The roles of fatty acid metabolism in CRC were investigated to explore potential therapeutic strategies. The data files were downloaded from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases. Univariate and least absolute shrinkage and selection operator (LASSO) Cox regression analyses were used to construct a prognostic risk score model with fatty acid metabolism-related genes for predicting prognosis in CRC. Patients with a high-risk score had a poorer prognosis in TCGA cohort than those with a low-risk score and were confirmed in the GEO cohort. Further analysis using the “pRRophetic” R package revealed that low-risk patients were more sensitive to 5-fluorouracil. A comprehensive evaluation of the association between prognostic risk score model and tumor microenvironment (TME) characteristics showed that high-risk patients were suitable for activating a type I/II interferon (IFN) response and inflammation-promoting function. Tumor Immune Dysfunction and Exclusion (TIDE) and SubMap algorithm results also demonstrated that high-risk patients are more suitable for anti-CTLA4 immunotherapy. Therefore, the evaluation of the fatty acid metabolism pattern promotes our comprehension of TME infiltration characteristics, thus guiding effective immunotherapy regimens.
Keywords: colorectal cancer, fatty acid metabolism, immune, prognosis
Graphical Abstract
A fatty acid prognostic risk score model is developed to enhance our comprehension of the role of fatty acid metabolism in CRC and guide more effective drug therapy strategies.
Introduction
Colorectal cancer (CRC) is one of the most common malignant tumors in humans, accounting for about 10% of diagnosed cancers and cancer-related deaths yearly. CRC mortality ranks second of all 36 cancers worldwide, with about 900,000 people dying yearly.1 Moreover, it has the third-highest incidence in men and the second in women.2 However, there are regional differences in CRC incidences. Its incidence in developed countries is higher than in developing countries.3 Arnold et al.4 have predicted that CRC cases could increase to 2.5 million in 2035 because of the rapid growth in developing countries. Nationwide screening, including colonoscopy, can stabilize or reduce CRC incidence.5 However, tumor recurrence occurs in 25%–40% of postoperative patients, and chemotherapy does not improve prognosis.6
The tumor microenvironments (TMEs) are characterized by hypoxia, high oxidation, acidity, and malnutrition because of the rapid proliferation of tumor cells and inadequate angiogenesis. Therefore, cancer cells exhibit unique metabolic characteristics different from normal cells to deal with various adverse microenvironments through a metabolic reprogramming process that maintains the proliferation and survival of cancer cells when the carcinogenic signal is blocked.7, 8, 9 Reprogramming energy metabolism is a hallmark in cancers and is essential in cell proliferation and division.10 The metabolism of carbohydrate, lipid, and amino acid in cancer cells is significantly different from normal cells.11 For instance, normal cells catabolize most glucose to pyruvate via glycolysis, and glucose catabolism coupled with oxidative phosphorylation in mitochondria produces high energy. However, cancer cells catabolize glucose into lactate with insufficient energy production, making cancer cells need a higher glucose consumption for growth.12 In CRC, cellular pyruvate metabolism changes, including mitochondrial pyruvate carrier (MPC) reduction, promote cancer initiation.13 Besides glucose metabolism, lipid metabolism is a potential hallmark in malignant tumors. Upregulated uptake, storage, and synthesis of the lipid promote the rapid growth of the tumor.14 In recent years, fatty acid metabolism, essential for many biological activities, such as cell membrane formation, energy storage, and signaling molecule generation in oncogenesis, has attracted much attention.15,16 For instance, Qi et al.17 have demonstrated that a specific expression pattern of fatty acid catabolic metabolism-related genes is associated with malignancy, prognosis, and immune phenotype in glioma. Enhanced lipolysis and fatty acid synthesis induce lymph node metastasis in cervical cancer patients via activated nuclear factor κB (NF-κB) signaling.18 Activated fatty acid oxidation can promote acute myeloid leukemia cell survival by remodeling and lipolysis of bone marrow adipocytes.19 Moreover, a study has illustrated that metabolic pathway analysis of CRC could help us to understand the molecular mechanism of CRC better and further develop new treatment methods.20 However, the fatty acid metabolism-related gene set in CRC has not been systematically studied.
In this study, the genomic information of 548 CRC samples was analyzed to comprehensively assess the fatty acid metabolism pattern and construct a fatty acid prognostic risk score model. The prognostic risk score model independently predicted the survival outcome of CRC patients and effectively distinguished CRC patients resistant to 5-fluorouracil (5-FU). Moreover, the relationship between the prognostic risk score model and TME cell-infiltrating characteristics was investigated. The prognostic risk score model could effectively define CRC patients suitable for anti-CTLA4 antibody immunotherapy, suggesting that fatty acid metabolism is essential in shaping individual TME characterizations. These findings can provide a novel perspective for exploring the metabolic mechanism and treatment of CRC.
Results
Enrichment analysis of normal and cancer tissue samples
Expression level comparison of fatty acid metabolism-related genes in normal and cancer tissue samples was conducted, and 70 genes with false discovery rate (FDR) < 0.05 were selected in The Cancer Genome Atlas (TCGA) cohort. A total of 23 genes were upregulated and 47 downregulated in the cancer tissue samples. Figure S1A shows the differentially expressed genes (DEGs). Gene Ontology (GO) enrichment analysis was then conducted for the DEGs. Fatty acid metabolic, fatty acid catabolic, and nucleoside bisphosphate metabolic processes were highly enriched GO terms in the biological processes (Figure S1B). Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis results showed that fatty acid degradation, metabolism, biosynthesis, and elongation were highly enriched KEGG terms (Figure S1C). These results illustrate that fatty acid metabolism plays a significant role in CRC development.
Prognostic risk score model development in the training set
Samples from TCGA cohort were classified as the training set. Univariate Cox regression analysis was performed on 70 differentially expressed fatty acid metabolism-related genes. A total of 13 genes related to prognosis were identified with a p value <0.05 (Figure 1A). The somatic mutation profile of the 13 fatty acid metabolism-related genes associated with prognosis was first summarized. A total of 67 of 399 CRC samples experienced mutations of fatty acid metabolism-related genes, with a frequency of 16.79%, as shown in Figure 1B. ACACB had the highest mutation frequency than ACOX1. However, ACAA2 did not exhibit any mutations in CRC samples. Further analyses demonstrated a mutation co-occurrence relationship between ACADM and ADH6, ACACB and ACADM, and OXSM and ACADM (Figure 1C). Least absolute shrinkage and selection operator (LASSO) Cox regression analysis was then used to narrow the number of genes. Finally, eight genes (ELOVL3, ACADL, ACOX1, ACACB, ADH5, CPT2, ACSL6, and ELOVL6) were used for prognostic risk score model construction (Figures 1D and 1E). The risk score of each sample was calculated using the following: risk score = (0.496855878561209) × ELOVL3 + (0.564161973443707) × ACADL +(−0.21155749803107) × ACOX1 + (0.46605598297121) × ACACB + (−0.0411122918767907) × ADH5 + (−0.475528417085679) × CPT2 + (−0.137748231278715) × ACSL6 + (−0.178101766953256) × ELOVL6, which was demonstrated in Table S1.
Figure 1.

Development of prognostic risk score model
(A) Forrest plot of 13 fatty acid metabolism-related genes related with prognosis. (B) The mutation frequency of 13 fatty acid metabolism-related genes in 399 patients with CRC from TCGA cohort. (C) The mutation co-occurrence and exclusion analyses for 13 fatty acid metabolism-related genes. Co-occurrence, green; exclusion, purple. (D) LASSO coefficients of the 13 fatty acid metabolism-related genes. (E) Identification of genes for development of prognostic risk score model. (F) Principal component analysis based on all fatty acid metabolism-related genes in CRC. (G) Principal component analysis based on fatty acid metabolism risk score to distinguish tumors from normal samples in TCGA cohort. The group marked with green represented high-risk patients, and the group marked with red represented low-risk patients.
The risk score model was used to completely distinguish CRC samples (low or high risk) (Figures 1F and 1G)
The relationship between risk score and clinical features
The cutoff value was the median value of risk scores in the training set. The sample risk scores were ranked and classified into low (n = 253)- and high (n = 253)-risk score groups, according to the cutoff value above. The distribution of risk scores in age, gender, pathological stage, and American Joint Committee on Cancer (AJCC) TNM Classification of Malignant Tumors (TNM) stage of corresponding samples was analyzed. Although there were no significant differences in risk score associations with age, gender, and AJCC-T (tumor invasion) stages (Figures 2A−2C), higher risk scores were correlated with higher AJCC-N (lymphoid metastasis) (p < 0.001; Figure 2D) and AJCC-M (distal metastasis) (p = 0.05; Figure 2E) and advanced pathological stages (p = 0.012; Figure 2F). High-risk score group samples had a worse prognosis in TCGA compared with the low-risk score group (p = 2.995e−05; Figure 2G). Time-dependent receiver operating characteristic (ROC) was plotted at 5 years to validate the accuracy of the prognostic risk score model (Figure 2H). For prognostic risk score model validation, test group samples from Gene Expression Omnibus (GEO): GSE39582 were classified into low (n = 270)- and high (n = 285)-risk score groups, according to the cutoff value determined in the training set. High-risk group samples had a poorer prognosis than the low-risk group (Figure 2I), indicating that the prognostic risk score model can predict overall survival (OS) in CRC (Figure 2J). Among factors associated with OS in the univariate analysis, including clinical stage, T stage, lymph nodes status, distal metastasis, CMS (consensus molecular subtype), and risk score, only risk score and clinical T stage were independent predictors of OS in multivariate analysis (Figures 2K and 2L).
Figure 2.

The predictive value of fatty acid metabolism score model in survival status of CRC patients
(A−F) The relationship of risk score and clinicopathological features, including age (A), gender (B), tumor invasion (C), lymphoid metastasis (D), distal metastasis (E), and TNM stage (F). (G and I) The comparison of overall survival (OS) between low- and high-risk score groups in the training set and the test set. (H and J) The predictive value of the risk score measured by ROC curves in the training set and the test set. The area under the curve (AUC) is 0.718 and 0.559. (K and L) The forest plot of the univariate and multivariate Cox regression analysis in TCGA cohort.
The survival status and corresponding risk score of each sample were plotted (Figure S2A). Figure S2A demonstrates the different expressions of eight genes in the low- and high-risk score groups. High-risk genes (ELOVL3, ACADL, and ACACB) had higher expressions in the high-risk score group, whereas the low-risk score group had a high level of protective genes (ACOX1, ADH5, CPT2, ACSL6, and ELOVL6), consistent with previous results. The risk score distribution, the survival status of the CRC samples, and the gene expression in the test set are represented in Figure S2B.
Construction of a nomogram for predicting survival
A nomogram with integrated age, gender, pathological stage, carcinoembryonic antigen (CEA) level, and prognostic risk score model was constructed for OS prediction in CRC samples (Figure 3A). The calibration curves at 1 year, 3 years, and 5 years proved that the nomogram could accurately predict the OS of CRC patients, as shown in Figures 3B−3D. Multivariate Cox regression analysis results illustrated that prognostic risk score model, age, and clinical-pathological stage are independent prognostic indicators (Figure 3E). The area under the ROC curves (AUC) demonstrated that the nomogram (AUC = 0.840) had better prognostic value than a single indicator, such as age (AUC = 0.631), pathological stage (AUC = 0.744), and prognostic risk score model (AUC = 0.673; Figure 3F).
Figure 3.

The predictive value of fatty acid metabolism score in combination with clinical pathological characteristics in OS of patients from TCGA cohort
(A) Nomogram predicting OS of patients from TCGA cohort. (B−D) The calibration plots of the nomogram. The x axis is nomogram-predicted survival, and the y axis is actual survival. (E) Multivariate Cox regression analysis of the nomogram. (F) ROC curves for fatty acid metabolism score and clinical pathological characteristics.
Response to chemotherapy response
Since risk score is associated with poor prognosis, the relationship between risk score and chemoresistance was explored. The half-maximal inhibitory concentration (IC50), calculated using the “pRRophetic” R package, was used to predict the treatment response to 5-FU in TCGA cohort. Low-risk score samples were more sensitive to 5-FU, widely used in treating advanced CRC (Figures 4A and 4B). There was a strong correlation between chemotherapy sensitivity and risk score in the GEO: GSE39582 cohort, according to RFS (recurrence-free survival) at 3 years (Figure 4C). The analyses for the activity of stroma-related pathways responsible for chemotherapy resistance revealed that high-risk scores were significantly related to intensive activation of stromal pathways, including epithelial–mesenchymal transition (EMT), transforming growth factor (TGF)-β pathways, angiogenesis, and Wnt target (Figure 4D).
Figure 4.

Fatty acid metabolism model in the role of chemotherapy
(A) The correlation between risk scores of patients and estimated IC50 value of 5-FU. (B) The differences of response to 5-FU between low- and high-risk score groups. (C) The comparison of disease-free survival (DFS) between low- and high-risk score groups in the GEO cohort. (D) Differences in stroma-activated pathways between low- and high-risk score groups (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001). (E) The heatmap of GSVA enrichment between low- and high-risk score groups. (F−K) Differences in fatty metabolism score among different of molecule subtypes, including KRAS mutation (F), BRAF mutation (G), MMR status (H), CIN status (I), TP53 status (J), and CIMP status (K).
Gene set variation analysis (GSVA)
GSVA enrichment was conducted using the gene sets of “c2.cp.kegg.v7.2” downloaded from the Molecular Signatures Database (MSigDB) to explore the biological behaviors in the two groups. Interestingly, most metabolism pathways, including fatty acid metabolism, were enriched in the low-risk score (Figure 4E). The risk score was negatively associated with fatty acid metabolism score, calculated with single-sample gene-set enrichment analysis (ssGSEA) using the expression of fatty metabolism-associated genes in CRC patients, consistent with GSVA (Figures S3A and S3B). Although the time-dependent ROC at 5 years suggested that the fatty metabolism score could also accurately predict the patients’ survival rate, the prognostic value (AUC = 0.652) was inferior to the risk score model (AUC = 0.718) (Figures S3C and S3D). The enrichment pathways of the high-risk group were associated with the immune biological processes, such as the T cell receptor and mammalian target of rapamycin (mTOR) signaling pathways (Figure 4E). Furthermore, patients with mismatch repair proficient (pMMR), chromosome instability (CIN), and CpG island methylator phenotype (CIMP) had higher risk scores (Figures 4F−4K).
Immune-related characteristic in the low- and high-risk score groups
The group was remarkably rich in immune-suppressive cell infiltration, including MDSC (myeloid-derived suppressor cells), T cell regulatory cells (Tregs), and T helper (Th)17 cells, consistent with the survival disadvantage in the high-risk group. Central memory CD8 T cells, effector memory CD8 T cells, and natural killer T cells were also enriched in the high-risk group (Figure 5A). Moreover, type I interferon (IFN) response, type II IFN response, and inflammation-promoting function were also activated in the high-risk group, indicating that patients in the high-risk group with immunity suppression could respond to immunotherapy (Figure 5B). The immunotherapy represented by the CTLA4 and programmed cell death 1 (PD-1) blockade has made a breakthrough in cancer therapy. Immune checkpoint-associated genes were activated in the high-risk group (Figure 5D). The ability of the prognostic risk score model to distinguish patients with different responses to immune checkpoint blockade therapy was investigated. Although there was no difference in clinical response to anti-PD-1 immunotherapy, therapeutic advantages and clinical responses to anti-CTLA4 immunotherapy in high-risk score patients were better than the low-risk score group (Figure 5C; Figures S4B−S4D). Further research revealed that besides immunity checkpoint genes (PD-1, programmed death ligand 1 [PD-L1], and CTLA4), immunity activation- and antigen presentation-associated genes were all significantly upregulated in the high-risk group (Figure S4F). Moreover, CMS1 phenotype patients suitable for immunotherapy had higher risk scores (Figure S4E), indicating that the quantification of fatty acid metabolism risk score is a novel and robust biomarker for evaluating the prognosis and clinical response to immunotherapy.
Figure 5.

Fatty acid metabolism model in the role of immunotherapy
(A) The immunity infiltration difference between high-risk score and low-risk score. (B) The known function associated with immunity regulation difference between patients with high-risk score and low-risk score. (C) Response prediction to immunotherapy (anti-PD-1 and anti-CTLA4) between the high-risk group and low-risk group according to TIDE and SubMap algorithms. (D) The differences of known gene signatures between the low-risk score group and high-risk score group.
Protein-protein interaction (PPI) network of DEGs in the low- and high-risk score groups
STRING online database was used to analyze the expression profiles of DEGs in the low- and high-risk score groups. The PPI network was constructed using the DEGs is shown in Figure S5A. Cytoscape software was used to process and display the PPI network data. Figure 6A presents DEG interaction, where upregulated genes in the high-risk score group are marked in red and in the low-risk score group are presented in blue. cytoHubba, a plug-in of Cytoscape, was used to determine the hub genes from the DEGs. A total of 10 genes in the network were selected, as shown in Figure 6B. FN1, EGF, CDKN2A, SYP, KRT5, SERPINE1, CDH2, KRT14, ACTC1, and GNG8 were ranked using the degree method. The gene expression differences between normal and tumor were then compared. CRC patients had a higher proportion of SERPINE1, CDKN2A, EGF, and KRT14 and a lower proportion of GNG8, SYP, and CDH2 compared with normal colorectal tissue. GO and KEGG analyses of eight difference hub genes were analyzed using the “GOplot” R package to better understand their function. GO results indicated that the genes participate in replicative senescence, negative regulation of cell-matrix adhesion, positive regulation of receptor-mediated endocytosis, neuroepithelial cell differentiation, and aging (Figure 6C). KEGG results indicated that these genes were enriched in the P53 signaling pathway, (HIF-1) signaling pathway, apelin signaling pathway, cellular senescence, and human cytomegalovirus infection (Figure 6D). Survival analysis indicated that only CDKN2A mRNA expression of the seven hub genes was significantly associated with CRC patients’ prognoses (Figure 6E; Figures S5B−S5J). Besides, CDKN2A expression increases as the tumor stage advances (Figure 6F). CDKN2A expression was associated with poor prognosis. The specific difference of TME immune cell infiltration between high and low CDKN2A expression patients was explored using the CDKN2A median expression value as the cutoff value. Tumors with high CDKN2A expressions had significantly increased infiltration in Tregs than patients with low expressions (Figure 6G).
Figure 6.

Protein-protein interaction (PPI) network
(A) PPI network processed by Cytoscape (red): DEGs that expressed highly in the high-risk score group; blue: DEGs that expressed highly in the low-risk score group. (B) Top 10 hub genes selected by cytoHubba. (C and D) The results of GO and KEGG enrichment analysis on top 10 hub genes. (E) Survival analysis for subgroup patients stratified by CDKN2A mRNA expression. (F) The CDKN2A mRNA expression difference among different clinical stages. (G) The abundance of each TME-infiltrating cell in patients with high and low CDKN2A mRNA expression.
Discussion
The reprogramming of the cellular metabolism plays an essential role in tumor development.21 Cell metabolic activity changes are cancer hallmarks.22 For instance, the upregulated glycolytic metabolism is one of the physiological characteristics of human malignant tumor.23 Some studies have illustrated that metabolic signatures containing the cysteine metabolism, nucleotide metabolism, and 2-hydroxyglutarate can be used to classify and treat gliomas.24, 25, 26 In CRC, increased anaerobic metabolic pathways are essential in cancer stem-like cells (CSCs) related to tumor formation, development, and therapy resistance.27,28 Although most studies have focused on the role of a single regulator of fatty acid metabolism in CRC, the integrated roles of multiple fatty acid metabolism-related genes are unknown. The exploration of the role of distinct fatty acid metabolism patterns in CRC could help understand fatty acid metabolism in CRC progression, thus guiding to an effective therapeutic strategy.
To date, this is the first study to explore the relationship between fatty acid metabolism-related genes and CRC. Herein, a prognostic risk score model with 70 differentially expressed fatty acid metabolism-related genes in tumor and normal CRC tissue samples was established in TCGA cohort and GEO cohort using univariate Cox regression analysis and LASSO Cox regression analysis. A prognostic risk score model was used to predict the OS of CRC patients in the training set to better understand the role of these genes in CRC. There were differences in survival of CRC patients between low- and high-risk score groups. The same result was reported in the test set, indicating that the prognostic risk score model can screen patients with poor survival. The prognostic risk score model was the independent prognostic factor on multivariable analysis. Moreover, the predictive potential of this prognostic risk score model was further improved by combining with a few selected clinicopathological features and CEA levels in a risk-assessment nomogram.
The differences in patients’ response to drug therapies between the low- and high-risk score groups were compared to further understand the role of the prognostic risk score model in CRC. The risk score was positively associated with 5-FU chemoresistance, consistent with previous research. The CRC patients with higher risk scores had lower RFS, indicating that the fatty acid prognostic risk score model can be used for the personalized treatment of CRC patients. High-risk score patients exhibited a significant stroma activation status, indicating chemoresistance, consistent with the above definitions. Since patients with high-risk scores experience chemoresistance, it was hypothesized that they are not suitable for immunotherapy. Whereas some patients are suitable for the immunotherapy (immune checkpoint blockade [PD-1/L1 and CTLA4]), most CRC patients are not suitable. Therefore, it is important to distinguish patients suitable for immunotherapy in clinical practice. High-risk score patients were enriched with inhibitory immunity cells, including Tregs and MDSCs, and immune-inflamed cells. Moreover, high-risk score patients had the activation of type I IFN response, type II IFN response, and inflammation-promoting function, indicating that high-risk score patients are suitable for the immunotherapy, consistent with the prediction using Tumor Immune Dysfunction and Exclusion (TIDE) and SubMap algorithms. CTLA4, limited to activated T cells and Tregs, is the inhibitory counterpart to CD28, because it shares ligands with the same conserved MYPPPY motif.29 Therefore, anti-CTLA4 antibodies could be suitable for high-risk score patients, especially chemoresistance patients.
Since there are significant differences between low-risk and high-risk score groups, the different genes in the two groups were further explored. CDKN2A, p16, was found to be essential. CDKN2A mRNA expression was not only positively related to the clinical stage but also associated with poorer prognosis. Previous studies have also demonstrated a highly significant correlation between higher p16INK4A expression, especially in the front of invasion, and lower survival. However, mechanical research has shown that CDKN2A is a β-catenin target gene that could inhibit CRC cell proliferation. This paradoxical result could have attributed to the over-estimation that proliferation promotes CRC malignant progression. Interestingly, some tumor cells defined by p16INK4A expressions are components of the budding zone and good prognostic markers for low survival. However, these results should be further explored.30,31
In summary, the fatty acid prognostic risk score model can be used to evaluate the fatty acid metabolism patterns comprehensively. The risk score can characterize patients’ clinicopathological features, including clinical stages, molecular subtypes, microsatellite instability (MSI) status, CIN status, and CIMP status. Besides, the risk score is associated with patients’ prognosis and can also predict chemotherapy sensitivity. Therefore, risk score and clinical stage can effectively guide clinical practice to achieve a more personalized clinical follow-up strategy. Patients suitable for anti-CTLA4 treatment can be distinguished using risk scores. These findings provide a novel, efficient, and accurate predicting model in prognosis and in response to chemotherapy and immunotherapy, thus promoting personalized cancer chemotherapy and immunotherapy in the future.
Materials and methods
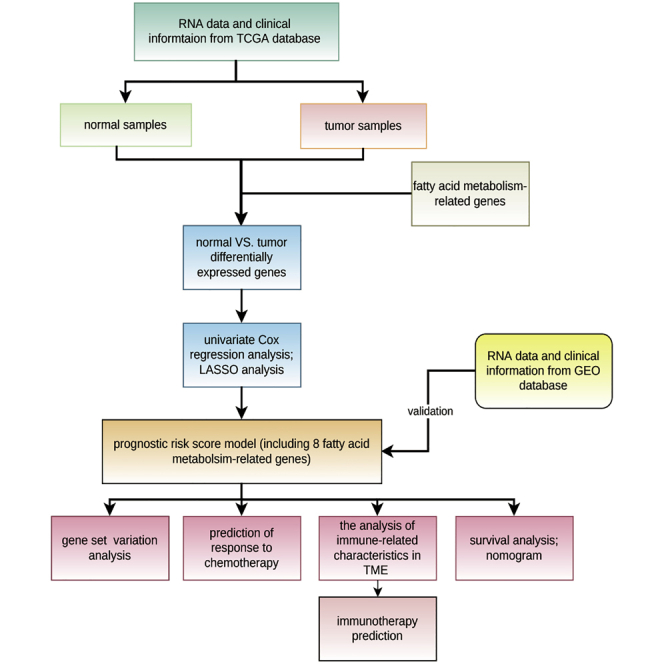
Data processing
The raw RNA sequencing (RNA-seq) data profiles, including 568 CRC and 44 normal colorectum tissue samples, were downloaded in the High Throughput Sequencing (HTSeq)-fragments per kilobase of transcript per million mapped reads (FPKM) workflow type from TCGA database (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga). The clinical information of 548 CRC samples, including gender, age, pathological stage, AJCC TNM stage, and prognostic information, was also obtained from TCGA database. The microarray data profiles of GEO: GSE39582 based on platform GPL570 were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The Entrez Gene IDs of each sample were converted into corresponding gene symbols using the annotation platform. Mean value was adopted if the identical Entrez Gene ID was targeted by more than one probe. Besides, clinical information of each sample in GEO: GSE39582 was downloaded from the GEO database.
In a previous study,32 92 fatty acid metabolism-related genes were identified. A total of 81 common genes were then selected from these genes in TCGA and GEO cohorts.
Enrichment analysis of the DEGs in the normal and cancer tissue samples
“limma” R package was used to analyze differentially expressed fatty acid metabolism-related genes in the normal and cancer tissue samples. Genes with FDR < 0.05 were considered statistically significant. “org.Hs.eg.db” R package was then used to convert the symbol gene of each DEG into a corresponding Entrez Gene ID. The GO and KEGG pathway enrichment analyses were performed on DEGs using “clusterProfiler” R package to identify the major biological features and cell functional pathways. p value (q value) <0.05 was considered a statistically significant difference. Finally, “enrichplot” and “ggplot2” R packages were used to visualize the enrichment analysis results.
Development and verification of a prognostic risk score model
TCGA cohort samples were classified as the training set and GEO: GSE39582 samples as the test set. The expression level of differentially expressed fatty acid metabolism-related genes of each sample was first merged with corresponding prognostic outcomes using the samples’ ID. The genes related to prognosis were screened from differentially expressed fatty acid metabolism-related genes through univariate Cox regression analysis in the training set. The genes with p value <0.05 were selected. The “maftools” R package was used to analyze the mutation and correlation of the genes in CRC samples of the training set. “glmnet” R package was used to further process the prognosis-related genes using LASSO Cox regression analysis to develop a prognostic risk score model for predicting OS of CRC samples. The ten-fold cross-validation was conducted to determine the penalty parameter (λ) of the model. The formula below was used to calculate each sample risk score.
The “Coef” represents non-zero regression coefficients calculated using the LASSO Cox regression analysis, and “ExpGene” is the expression values of genes from the prognostic risk score model. All samples were classified into low- and high-risk score groups based on the median value of risk scores. Kaplan-Meier analysis with the log-rank test was used to compare the OS difference between low-risk and high-risk score groups. The time-dependent ROC curve was plotted using the “survivalROC” package in R to evaluate the predictive accuracy of prognostic risk score mode. Finally, the reliability and applicability of the prognostic risk score model were further validated in the test set.
Principal-component analysis (PCA) comparison before and after prognostic risk score model
limma package in R was successfully used to perform PCA on expression profiles of genes before and after the prognostic risk score model in the training set to understand the prominent distinction in the low- and high-risk score groups. PCA was first performed on the expression profiles of all differentially expressed fatty acid metabolism-related genes. PCA was then used to analyze the expression profiles of genes from the prognostic risk score model. Finally, ggplot2 package was used to present the PCA results on two-dimensional diagrams according to the first two principal components.
Relationship between risk scores and clinical features
In TCGA cohort, “CMScaller” R package was used to classify all samples into CMSs based on their features. The risk score of each sample was merged with corresponding clinical characteristics based on the sample ID. The relationship between risk scores and clinical information, including gender, age, pathological stages, AJCC TNM stages, and CMS, was explored using the limma R package. Besides, the expression level of the immune checkpoint (represented by PD-1, PD-L1, and CTLA4) was obtained from TCGA database. The expression levels of the immune checkpoints in the low- and high-risk score groups were then compared. The clinical information related to CRC in the GEO cohort, including CIMP status, CIN status, MMR status, and KRAS/BRAF/TP53 mutation, was collected to determine the association between risk scores and clinical features. Samples were classified into two groups, according to the clinical features, to compare the risk score differences. Wilcoxon rank-sum and Kruskal-Wallis (K-W) tests were used to compare two groups and more than two groups, respectively. p value <0.05 was considered statistically significant.
GSVA
GSVA was conducted on the gene profile through the “GSVA” R package to compare the distinctions of the biological processes between low- and high-risk score groups. GSVA, a non-parametric and unsupervised method, can evaluate the pathway variations or biological processes through an expression matrix sample.33 The “c2.cp.kegg.v7.1.symbols” gene sets, from the molecular signatures database (https://www.gsea-msigdb.org/gsea/msigdb), was used as the reference gene sets. FDR <0.05 indicated a statistically significant enrichment pathway.
Characteristic comparison between the low- and high-risk score groups
pRRophetic R package was used to predict IC50 of 5-FU in each sample. IC50 indicates the effectiveness of a substance in inhibiting specific biological or biochemical functions. ssGSEA was performed using “GSEABase” and GSVA R packages to quantify the extent of the immune-related infiltration of each sample in TCGA cohort. The gene sets were collected for evaluation of immune-related characteristics in TME from the previous study, including many different types of human immune cell subtypes and immune-related activities, such as CD8+ T cell, B cell, T cell co-stimulation, and so on (Table S1).34,35 The enrichment scores calculated using the ssGSEA algorithm indicated a relative degree of each immune-related characteristic expression in each sample. The difference of enrichment scores in the low- and high-risk score groups was compared. The correlation between genes related to prognosis and immune cells was also evaluated. Finally, TIDE (http://tide.dfci.harvard.edu/) and SubMap (https://cloud.genepattern.org/gp) algorithms were used to predict immune checkpoint response inhibitors of PD-1 and CTLA4 in the low- and high-risk score groups. p value < 0.05 was considered statistically significant.
PPI network
The RNA-seq data profiles of low- and high-risk score groups were first compared via the limma R package. Genes with an adjusted p value <0.05 were identified as DEGs. The analysis of the DEGs was performed using the STRING online database (version: 11.0; https://string-db.org/) to generate the PPI network data with an interaction score >0.40 (median confidence) (Table S2). The PPI network data were then further processed and displayed using Cytoscape software (version: 3.7.2). The cytoHubba (version: 0.1), a Cytoscape plug-in, was used to search hub genes from all DEGs applying topological algorithms. Genes with differential expression in the normal colorectum tissue and CRC tissue were then collected. GO and KEGG enrichment analyses were performed on the genes using clusterProfiler R package. Finally, all samples were classified into low- and high-expression groups based on the median expression value of the hub genes. Kaplan-Meier analysis was used to determine if there was a difference in survival between the two groups. The comparison of immune cell infiltration was conducted in hub genes related to prognosis.
Development of a nomogram for predicting OS
A nomogram with age, gender, pathological stage, CEA level, and prognostic risk score model was developed using “rms” package in R based on TCGA cohort for OS prediction in CRC. Time-dependent calibration curves were plotted to predict the accuracy of the nomogram. Moreover, multivariate Cox regression analysis was used to validate if the prognostic risk score model could serve as an independent indicator for OS prediction in CRC. The AUC was then calculated to represent the prognostic value of the nomogram through the online ROC curves.
Statistical analysis
Wilcoxon rank-sum test was used to compare the difference between the two groups. K-W test was performed to compare three or more groups. Kaplan-Meier analysis was used to evaluate the survival differences between the low- and high-risk score groups. Multivariate Cox regression analysis was conducted to determine independent indicators for predicting OS in CRC. The ROC curves were plotted to assess the predictive effectiveness of the prognostic risk score mode and nomogram. All of the statistical analyses were conducted using R 4.0.0 (p < 0.05).
Acknowledgments
This work was supported by funding from Science and Technology Commission of Shanghai Municipality (number 16140900302).
Author contributions
C.D. and Z.S. collected and analyzed research data. M.L. was responsible for the interpretation of the results. C.D. drafted the manuscript. H.C., X.L., and Z.J. performed the study design and revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2021.02.010.
Contributor Information
Hongqi Chen, Email: hqchen08@163.com.
Xinxiang Li, Email: 1149lxx@sina.com.
Zhiming Jin, Email: jzmgyp@aliyun.com.
Supplemental information
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Dekker E., Tanis P.J., Vleugels J.L.A., Kasi P.M., Wallace M.B. Colorectal cancer. Lancet. 2019;394:1467–1480. doi: 10.1016/S0140-6736(19)32319-0. [DOI] [PubMed] [Google Scholar]
- 3.Brenner H., Chen C. The colorectal cancer epidemic: challenges and opportunities for primary, secondary and tertiary prevention. Br. J. Cancer. 2018;119:785–792. doi: 10.1038/s41416-018-0264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnold M., Sierra M.S., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66:683–691. doi: 10.1136/gutjnl-2015-310912. [DOI] [PubMed] [Google Scholar]
- 5.Ait Ouakrim D., Pizot C., Boniol M., Malvezzi M., Boniol M., Negri E., Bota M., Jenkins M.A., Bleiberg H., Autier P. Trends in colorectal cancer mortality in Europe: retrospective analysis of the WHO mortality database. BMJ. 2015;351:h4970. doi: 10.1136/bmj.h4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tjandra J.J., Chan M.K.Y. Follow-up after curative resection of colorectal cancer: a meta-analysis. Dis. Colon Rectum. 2007;50:1783–1799. doi: 10.1007/s10350-007-9030-5. [DOI] [PubMed] [Google Scholar]
- 7.Yi M., Li J., Chen S., Cai J., Ban Y., Peng Q., Zhou Y., Zeng Z., Peng S., Li X. Emerging role of lipid metabolism alterations in Cancer stem cells. Journal of experimental & clinical cancer research. J Exp Clin Cancer Res. 2018;37:118. doi: 10.1186/s13046-018-0784-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Viale A., Pettazzoni P., Lyssiotis C.A., Ying H., Sánchez N., Marchesini M., Carugo A., Green T., Seth S., Giuliani V. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–632. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lue H.W., Podolak J., Kolahi K., Cheng L., Rao S., Garg D., Xue C.H., Rantala J.K., Tyner J.W., Thornburg K.L. Metabolic reprogramming ensures cancer cell survival despite oncogenic signaling blockade. Genes Dev. 2017;31:2067–2084. doi: 10.1101/gad.305292.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 11.Yu T., Wang Y., Fan Y., Fang N., Wang T., Xu T., Shu Y. CircRNAs in cancer metabolism: a review. J. Hematol. Oncol. 2019;12:90. doi: 10.1186/s13045-019-0776-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vazquez A., Kamphorst J.J., Markert E.K., Schug Z.T., Tardito S., Gottlieb E. Cancer metabolism at a glance. J. Cell Sci. 2016;129:3367–3373. doi: 10.1242/jcs.181016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bensard C.L., Wisidagama D.R., Olson K.A., Berg J.A., Krah N.M., Schell J.C., Nowinski S.M., Fogarty S., Bott A.J., Wei P. Regulation of Tumor Initiation by the Mitochondrial Pyruvate Carrier. Cell Metab. 2020;31:284–300.e7. doi: 10.1016/j.cmet.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng C., Geng F., Cheng X., Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. (Lond.) 2018;38:27. doi: 10.1186/s40880-018-0301-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carracedo A., Cantley L.C., Pandolfi P.P. Cancer metabolism: fatty acid oxidation in the limelight. Nat. Rev. Cancer. 2013;13:227–232. doi: 10.1038/nrc3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Currie E., Schulze A., Zechner R., Walther T.C., Farese R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18:153–161. doi: 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qi Y., Chen D., Lu Q., Yao Y., Ji C. Bioinformatic Profiling Identifies a Fatty Acid Metabolism-Related Gene Risk Signature for Malignancy, Prognosis, and Immune Phenotype of Glioma. Dis. Markers. 2019;2019:3917040. doi: 10.1155/2019/3917040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang C., Liao Y., Liu P., Du Q., Liang Y., Ooi S., Qin S., He S., Yao S., Wang W. FABP5 promotes lymph node metastasis in cervical cancer by reprogramming fatty acid metabolism. Theranostics. 2020;10:6561–6580. doi: 10.7150/thno.44868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tabe Y., Konopleva M., Andreeff M. Fatty Acid Metabolism, Bone Marrow Adipocytes, and AML. Front. Oncol. 2020;10:155. doi: 10.3389/fonc.2020.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kahlert U.D., Mooney S.M., Natsumeda M., Steiger H.J., Maciaczyk J. Targeting cancer stem-like cells in glioblastoma and colorectal cancer through metabolic pathways. Int. J. Cancer. 2017;140:10–22. doi: 10.1002/ijc.30259. [DOI] [PubMed] [Google Scholar]
- 21.Pavlova N.N., Thompson C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeBerardinis R.J., Chandel N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016;2:e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei X., Mao T., Li S., He J., Hou X., Li H., Zhan M., Yang X., Li R., Xiao J. DT-13 inhibited the proliferation of colorectal cancer via glycolytic metabolism and AMPK/mTOR signaling pathway. Phytomedicine. 2019;54:120–131. doi: 10.1016/j.phymed.2018.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Pandey R., Caflisch L., Lodi A., Brenner A.J., Tiziani S. Metabolomic signature of brain cancer. Mol. Carcinog. 2017;56:2355–2371. doi: 10.1002/mc.22694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mörén L., Bergenheim A.T., Ghasimi S., Brännström T., Johansson M., Antti H. Metabolomic Screening of Tumor Tissue and Serum in Glioma Patients Reveals Diagnostic and Prognostic Information. Metabolites. 2015;5:502–520. doi: 10.3390/metabo5030502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dang L., White D.W., Gross S., Bennett B.D., Bittinger M.A., Driggers E.M., Fantin V.R., Jang H.G., Jin S., Keenan M.C. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vincent Z., Urakami K., Maruyama K., Yamaguchi K., Kusuhara M. CD133-positive cancer stem cells from Colo205 human colon adenocarcinoma cell line show resistance to chemotherapy and display a specific metabolomic profile. Genes Cancer. 2014;5:250–260. doi: 10.18632/genesandcancer.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeki S.S., Graham T.A., Wright N.A. Stem cells and their implications for colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2011;8:90–100. doi: 10.1038/nrgastro.2010.211. [DOI] [PubMed] [Google Scholar]
- 29.Edner N.M., Carlesso G., Rush J.S., Walker L.S.K. Targeting co-stimulatory molecules in autoimmune disease. Nat. Rev. Drug Discov. 2020;19:860–883. doi: 10.1038/s41573-020-0081-9. [DOI] [PubMed] [Google Scholar]
- 30.Ueno H., Murphy J., Jass J.R., Mochizuki H., Talbot I.C. Tumour ‘budding’ as an index to estimate the potential of aggressiveness in rectal cancer. Histopathology. 2002;40:127–132. doi: 10.1046/j.1365-2559.2002.01324.x. [DOI] [PubMed] [Google Scholar]
- 31.Wassermann S., Scheel S.K., Hiendlmeyer E., Palmqvist R., Horst D., Hlubek F., Haynl A., Kriegl L., Reu S., Merkel S. p16INK4a is a beta-catenin target gene and indicates low survival in human colorectal tumors. Gastroenterology. 2009;136:196–205.e2. doi: 10.1053/j.gastro.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 32.Yang C., Huang X., Liu Z., Qin W., Wang C. Metabolism-associated molecular classification of hepatocellular carcinoma. Mol. Oncol. 2020;14:896–913. doi: 10.1002/1878-0261.12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Charoentong P., Finotello F., Angelova M., Mayer C., Efremova M., Rieder D., Hackl H., Trajanoski Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017;18:248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 35.Ru B., Wong C.N., Tong Y., Zhong J.Y., Zhong S.S.W., Wu W.C., Chu K.C., Wong C.Y., Lau C.Y., Chen I. TISIDB: an integrated repository portal for tumor-immune system interactions. Bioinformatics. 2019;35:4200–4202. doi: 10.1093/bioinformatics/btz210. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.