

Abstract
Background
Anti–programed cell death 1 checkpoint inhibitors have recently demonstrated effectiveness against metastatic cancers including urothelial carcinoma.
Aims
To identify biomarkers/factors that correlate with the clinical response in advanced bladder cancer patients who received immune checkpoint inhibitor treatment.
Methods and results
We investigated tumors from 18 bladder cancer patients who had received anti–programed cell death 1 (pembrolizumab) or anti–programmed death‐ligand 1 therapy (atezolizumab or durvalumab) and performed exome analysis, T‐cell receptor sequencing of the tumor‐infiltrating lymphocytes (TILs), and immunohistochemical analysis of CD8 and programmed death‐ligand 1 in cancer tissues. Immunohistochemical analysis of bladder cancer tissues demonstrated that a higher number of CD8 T‐cell infiltration into cancer tissues was significantly associated with longer cancer‐specific survival of the patients (P = .0012). T‐cell receptor beta sequencing of TILs using genomic DNAs extracted from the tissues of 15 cases revealed that patients with higher clonal expansion of TILs had some tendency of longer cancer‐specific survival (P = .055), than those with lower clonal expansion. We performed whole exome sequencing of 14 cases and found that patients carrying higher numbers of somatic mutations received greater benefit from immunotherapy (P = .034) and one patient who had high microsatellite instability has survived for 1034 days.
Conclusion
CD8 infiltration in tumors and nonsynonymous mutation load might be useful predictive markers for immune checkpoint inhibitors for bladder cancer patients.
Keywords: bladder cancer, DNA damage repair gene, microsatellite instability, nonsynonymous mutation, T‐cell receptor, whole exome sequencing
1. INTRODUCTION
Bladder cancer is the fourth most common cancer in males and the 11th most common in females, with projected 81 190 new cases and 17 240 deaths in the United States1 in 2018. Approximately 40% of patients who undergo radical cystectomy experience recurrence, and the prognosis of patients with metastatic bladder cancer is very poor with a 5‐year survival2, 3 of 5%. Platinum‐based systemic therapies have been applied widely for relapsed/metastatic bladder cancer, but their efficacy is limited.4 In recent years, immunotherapy has been developed for treatment of bladder cancer and has shown good clinical responses in a subset of patients.
Immune checkpoint inhibitors used in the clinic are monoclonal antibodies targeting the immune inhibitory molecules expressed in tumor cells or T cells.5 These inhibitors are now approved for treatment of patients with various types of cancer including melanoma, non‐small cell lung cancer, Hodgkin lymphoma, head and neck cancer, gastric cancer, kidney cancer, and bladder cancer.6, 7, 8, 9, 10, 11, 12, 13 Pembrolizumab, an antibody targeting programed cell death 1 (PD1), was proven to be clinically beneficial in pivotal trials for locally advanced or metastatic urothelial carcinoma patients.14, 15 Atezolizumab and durvalumab inhibit the PD1/programmed death‐ligand 1 (PD‐L1) pathway through targeting PD‐L1 and were also approved for advanced urothelial carcinoma.10, 16 However, response rates of immune checkpoint inhibitor treatment are only 22% for recurrent metastatic urothelial carcinoma,14, 17 underscoring the need for development of predictive markers.
In this study, to better understand the molecular mechanisms in response to immune checkpoint inhibitor therapy, we characterized bladder tumors through whole exome sequencing, immunohistochemical (IHC) analysis of PD‐L1 and CD8 in cancer tissues, and T‐cell receptor (TCR) analysis of tumor‐infiltrating lymphocytes cells (TILs) for 18 patients who had received anti‐PD1/anti–PD‐L1 treatment.
2. RESULTS
2.1. Clinical characteristics of bladder cancer patients
The clinical information of 18 bladder cancer patients is shown in Table 1. The median age of patients was 66 years old (range, 44‐80 y), and 12 patients had been treated with chemotherapy before receiving immune checkpoint inhibitor therapy. Three of 18 patients had received a platinum‐based treatment for metastatic. The median follow‐up period of these patients was 416 days (range, 44‐1133 d). Six patients received pembrolizumab, 11 patients received atezolizumab (3 of them treated together with the MOXRO916), and 1 patient received durvalumab. Best clinical responses evaluated by the Response Evaluation Criteria in Solid Tumors version 1.1 were partial response (PR) in 8 patients, stable disease in 5 patients, and progressive disease in the remaining 5 patients. As of the time of data cutoff, 10 patients died of tumor progression, and 1 patient died of a non–cancer‐related reason. The median duration of response and cancer‐specific survival period were 195 days (range, 21‐1022 d) and 420 days (range, 44‐1133 d), respectively. No treatment attributed death was observed.
Table 1.
The clinical characteristics of the bladder cancer patients
| Patient ID | Best Response Modified RECIST | Age at Diagnosis, y | Gender | Histology | Immune Checkpoint Inhibitor | Duration of Response, d | Overall Survival, d |
|---|---|---|---|---|---|---|---|
| A1 | PR | 63 | F | UC with focal sarcomatoid and glandular differentiation | Pembrolizumab | 237 | 601 |
| A2 | PR | 72 | M | UC with micropapillary variant | Durvalumab | 296 | 391 |
| A3 | PR | 57 | M | UC | Atezolizumab | 278 | 681 |
| A4 | PR | 51 | M | UC with plasmacytoid features | Pembrolizumab/ACP196 | 40 | 169 |
| A5 | PR | 69 | M | UC | Atezolizumab | 63 | 99 |
| A6 | PD | 72 | M | UC | Pembrolizumab | 21 | 44 |
| A7 | PD | 72 | M | UC with focal squamous differentiation | Atezolizumab | 63 | 87 |
| A8 | SD | 44 | M | UC with glandular and squamous differentiation | Atezolizumab | 1022 | 1022 |
| A9 | PD | 66 | M | UC | Atezolizumab | 40 | 307 |
| A10 | SD | 61 | M | UC | Atezolizumab/MOXRO916 | 61 | 134 |
| A11 | SD | 66 | F | UC | Atezolizumab/MOXRO916 | 125 | 420 |
| A12 | SD | 69 | F | UC | Pembrolizumab | 931 | 1133 |
| A13 | PD | 59 | M | UC | Pembrolizumab | 21 | 663 |
| A14 | PR | 80 | F | UC | Atezolizumab | 376 | 491 |
| A15 | PR | 69 | M | UC | Atezolizumab/MOXRO916 | 316 | 412 |
| A16 | PD | 54 | M | UC with sarcomatoid differentiation and focal squamous features | Atezolizumab | 230 | 1034 |
| A17 | SD | 69 | M | UC with squamous differentiation | Atezolizumab | 62 | 147 |
| A18 | PR | 59 | M | UC | Pembrolizumab | 195 | 497 |
Abbreviations: PD, progressive disease; PR, partial response; RECIST, Response Evaluation Criteria in Solid Tumors; SD, stable disease; UC, urothelial carcinoma.
2.2. Correlation of immune‐related parameters with prognosis
To investigate a potential correlation between immune‐related parameters and prognosis of immunotherapy‐treated bladder cancer patients, we performed IHC staining to examine the extent of CD8+ cell infiltration and PD‐L1‐positive cells in cancer tissues using formalin‐fixed and paraffin‐embedded (FFPE) analysis. The number of CD8+ cells was quantified by ImageJ software and divided by median value into high and low groups (Figure 1A and 1B). Immunohistochemical results demonstrated that PD‐L1 was primarily expressed on the cytoplasmic membrane of the tumor cells (Figure 1C and 1D). The PD‐L1 positivity was defined as PD‐L1 staining of greater than 1% of the tumor cells. CD8 positivity in the tumor tissues was significantly associated with longer cancer‐specific survival (Figures 2A and S1). By using univariate Cox proportional hazard analysis, the CD8 T‐cell infiltration level was statistically significantly correlated with the cancer‐specific survival (Table 2, P = .013). On the other hand, the PD‐L1 positivity showed no significant association with the cancer‐specific survival (Figure 2B).
Figure 1.
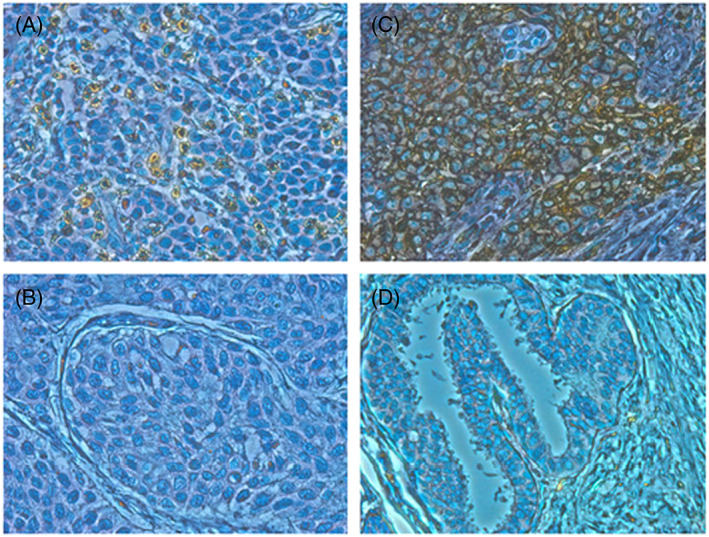
Immunohistochemical staining of CD8 and programmed death‐ligand 1 (PD‐L1) in bladder cancer tissues. Immunohistochemical staining of bladder cancer tissues showed representatives of A, high CD8 staining and B, low CD8 staining. Immunohistochemical staining of bladder cancer tissues showed representatives of C, positive PD‐L1 staining and D, negative PD‐L1 staining (×400)
Figure 2.
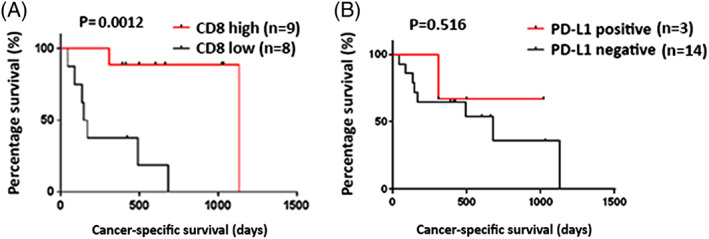
The Kaplan‐Meier analysis of cancer‐specific survival of patients by the immunohistochemical analysis of CD8 and programmed death‐ligand 1 (PD‐L1). A, High levels of CD8+ cell infiltration were significantly associated with longer cancer‐specific survival (P = .0012). B, PD‐L1 positivity on the tumor cells showed no correlation with clinical outcomes (P = .516)
Table 2.
Univariate analysis of the correlations between the cancer‐specific survival, the duration of response, and the clinicopathological factors
| Cancer‐specific Survival | Duration of Response | |||||
|---|---|---|---|---|---|---|
| HR | 95%CI | P value | HR | 95%CI | P value | |
| CD8 cell numbers (high vs low) | 0.068 | 0.008‐0.565 | .013* | 0.299 | 0.060‐1.487 | .140 |
| PD‐L1 expression (negative vs positive) | 0.506 | 0.062‐4.122 | .524 | 0.711 | 0.087‐5.784 | .750 |
| TCRβ DI (<median value vs ≥median value) | 0.585 | 0.107‐3.205 | .537 | 0.645 | 0.143‐2.904 | .568 |
| Nonsynonymous mutation load (<median value vs ≥median value) | 4.204 | 0.412‐42.921 | .226 | 1.615 | 0.269‐9.680 | .600 |
Abbreviations: CI, confidence interval; DI, diversity of index; HR, hazard ratio; PD‐L1, programmed death‐ligand 1; TCRβ, T‐cell receptor beta.
P value < .05.
2.3. T‐cell receptor beta clonality in TILs with the immune clinical parameters
Tumor‐infiltrating lymphocytes (TILs) are one of the key factors representing the tumor microenvironment. Since we previously reported a significant correlation of T‐cell clonality of TILs with the relapse‐free survivals of surgically resected bladder cancer,18 we also analyzed T‐cell receptor beta (TCRβ) sequences to characterize the repertoires of TILs using genomic DNAs from these cases. We successfully obtained the TCRβ information from 15 cases; 3 were unable to be obtained probably because of low DNA quality. We had a total of 609 735 to 1 948 787 TCRβ reads (mean ± standard deviation of 1 151 129 ± 101 908) and identified 1557 to 41 435 unique TCRβ complementary‐determining 3 clonotypes (mean ± standard deviation of 14 954 ± 3428) (Table S1). Patients with lower TCRβ diversity index (DI) showed the tendency to have longer cancer‐specific survival (1 patient who died of non–cancer‐related cause was excluded) (Figure 3A, R = −0.522, P = .055) although we did not find significant correlation of the TCR DI (DI‐high and DI‐low groups) with cancer‐specific survival when we applied Kaplan‐Meier analysis (Figure S2, P = .421).
Figure 3.
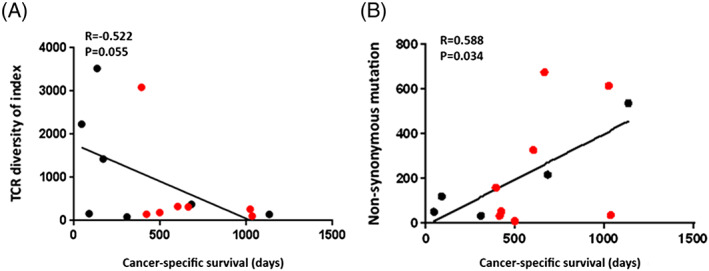
The relationship of the T‐cell receptor (TCR) diversity index and the numbers of nonsynonymous mutations with cancer‐specific survival. A, The extent of T‐cell clonal expansion (TCR diversity index) measured by TCRβ sequences showed tendency of correlation with the prognosis of bladder cancer patients treated with immune checkpoint inhibitors (R = −0.522, P = .055). B, The number of nonsynonymous mutations and cancer‐specific survival showed significant statistical correlation (R = 0.588, P = .034). The red dots stand for the cases that were still alive at the cutoff date
2.4. Nonsynonymous mutation load and DNA damage repair
In the whole exome analysis, we successfully obtained the information from 14 cases. We obtained an average sequencing depth of 73.8× per base and identified a total of 2856 nonsynonymous mutations (8 to 674 mutations per tumor). The numbers of nonsynonymous mutations showed significant correlation with cancer‐specific survival (Figure 3B, R = 0.588, P = .034). The comparison of cancer‐specific survival between a high nonsynonymous mutation group and a low mutation group was also calculated by Kaplan‐Meier analysis (Figure S3, P = .193). We also analyzed the neoantigen burdens due to binding affinity to human leukocyte antigen class I alleles and totally identified 5966 neoantigen burdens (23 to 1432 neoantigens per tumor). The numbers of the neoantigen burdens showed correlationship with cancer‐specific survival (Figure S4, P = .047). In addition, we focused on alterations in genes involved in DNA damage repair and DNA replication and found the mutations listed in Table 3. Eight patients carried mutations in DNA damage repair genes, FANCA, RECQL4, RECQL5, ATM, TP53, ERCC6, or XPA, and 1 showed a mutation in the DNA polymerase epsilon (POLE) gene. Five of these 8 patients had a higher number of somatic mutations (216 to 674 mutations) and showed the tendency of a longer cancer‐specific survival (601 to 1133 d). The patient who carried the POLE mutation had a total of 187 nonsynonymous mutations, showed PR, but was excluded from the survival analysis because of death from a non–cancer‐related disease. Using the Bethesda consensus panel, we evaluated 16 cases for microsatellite instability (MSI) analysis and found MSI‐H positive status in one case A16 (Figure S5). This patient has survived for >1034 days.
Table 3.
Whole exome sequencing results of 14 tumors and corresponding blood‐derived DNA
| A13 | A8 | A12 | A1 | A3 | A5 | A2 | A7 | A11 | A6 | A16 | A15 | A9 | A18 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FANCA | C269G S90C | R714W | ||||||||||||
| BRCA2 | R2424K | |||||||||||||
| POLE | G1581W | |||||||||||||
| XPA | L162V | |||||||||||||
| ATM | R248G | |||||||||||||
| RECQL4 | K88N | W379X | ||||||||||||
| RECQL5 | S911F | |||||||||||||
| ERCC6 | S418C | |||||||||||||
| Mutation load | 674 | 614 | 536 | 327 | 216 | 187 | 159 | 119 | 53 | 50 | 35 | 33 | 32 | 8 |
Mutations in genes involved in the DNA damage repair process and the DNA replication process (DNA polymerase epsilon—POLE) are listed as above.
3. DISCUSSION
Several recent efforts have been undertaken to identify biomarkers to predict clinical responses to immune checkpoint inhibitors.19, 20 The expression level of PD‐L1 on tumor cells has been reported to be correlated with outcomes of immune checkpoint inhibitor‐treated patients in some advanced solid tumors.7, 21, 22 In addition, PD‐L1 expression on T cells has been suggested to have association with clinical response with atezolizumab treatment.23 However, conflicting data have been also reported from other trials including those in bladder cancer. For example, PD‐L1 expression levels had no significant correlation with the clinical response of patients with non‐small cell lung cancer, melanoma, or gastric cancer.24, 25, 26 In the bladder cancer studies, one open‐label, phase 3 trial indicated that the pembrolizumab appeared to be independent of PD‐L1 expression on tumor and infiltrating immune cells,14 while the other clinical trial (KEYNOTE‐052) showed that PD‐L1 expression cutoff of 10% was associated with a higher frequency of response to pembrolizumab.15 Hence, further investigation for the role of other biomarkers with clinical outcomes is certainly required. In this study, we importantly found significant association of the number of CD8+ cells in tumor tissues and the number of nonsynonymous mutations with the cancer‐specific survival of immunotherapy‐treated bladder cancer patients. We found no significant correlation between clinical outcomes and PD‐L1 expression by IHC.
For PD‐L1, there are several potential hypothesized reasons for the divergent data across different studies and different tumor types. For example, the use of different antibodies and the use of different cutoff values of PD‐L1 positivity are 2 obvious potential reasons.16, 27 In addition, since PD‐L1 expression is known to dynamically change, the time when the biopsy is performed and the anatomical region where the sample is obtained (including intertumor or intratumor heterogeneity) could significantly influence results. We in this study applied a cutoff value for positivity of PD‐L1 expression at 1% of the tumor cells. However, in the future, PD‐L1 might be more useful when it is used together with the other parameters or could be applied by semiquantitative algorithms.28
Nonsynonymous mutational burdens as well as the number of the possible neoantigens have also been reported as possible useful predictors in melanoma, non‐small cell lung cancer, and bladder cancer. Such tumors possessing higher numbers of somatic mutations are expected to have more immunogenic neoantigens attracting T cells, facilitating clinical response by immune checkpoint blockades.29, 30, 31, 32 A phase 2 trial of atezolizumab in locally advanced and metastatic urothelial cancer revealed the importance of mutational load as a biomarker of response.17 In our study, we identified a total of 2856 nonsynonymous mutations in 14 tumors and also found a statistically significant correlation between nonsynonymous mutation load and cancer‐specific survival. We also found the correlationship between the neoantigen burden and cancer‐specific survival. Higher neoantigen might predict the better prognosis of the patients receiving immunotherapy. In addition to somatic mutational load and neoantigen burden, we analyzed MSI. We found MSI‐H positivity in only one of the 16 tested cases. This frequency is similar to a previous report (MSI‐H, 1‐3%).33, 34 It is notable that this MSI‐H patient has lived for 1034 days. Microsatellite instability is now of course an approved qualifying biomarker for identification of pembrolizumab‐eligible patients.35
In our study, when we subdivided tumors based on levels of the CD8 cell infiltration, the CD8 high expression group revealed better cancer‐specific survival of an average of 673 days than the CD8 low group of 271 days (P = .0012), implying that the extent of CD8 infiltration into tumors might be one of the promising predictors for the clinical efficacy by the PD1/PD‐L1 blockade therapy. As similar to the results shown in this article, a study reported by Tumeh et al36 revealed that the CD8+ T‐cell densities in the parenchyma and invasive margin of pretreatment metastatic melanoma tissues were higher in responders than poor responders in the KEYNOTE‐001 trial, in which patients received pembrolizumab.
High‐throughput sequencing of TCRβ repertoires may be a good approach to capture the immune microenvironment. Monitoring the changes in TCR repertoire during treatment with the immune checkpoint inhibitors in various types of cancer could provide useful information for better understanding of molecular mechanisms in responders and nonresponders.37 T‐cell receptor beta DI that is used to measure the levels of clonal expansion of certain T‐cell population could objectively reflect the microenvironment of tumors. Our previous work in muscle‐invasive bladder cancer showed that the low TCRβ diversity (oligoclonal TIL expansion) was a good biomarker for longer relapse‐free survival in combination with the neoantigen load.18 In this current study, we also show a tendency that oligoclonal expanded TILs in tumor microenvironments might lead to better prognosis among immune checkpoint inhibitor‐treated bladder cancer patients (P = .055), though the number of the patients studied was small.
It has been suggested that the higher mutational load in tumors may be one of the key predictors for good clinical responses in the cancer immunotherapy.31, 32 A phase 2 trial of atezolizumab in locally advanced and metastatic urothelial cancer revealed the importance of mutational load as a biomarker of response.16 In our study, we identified a total of 2856 nonsynonymous mutations in 14 tumors and found the statistically significant correlation between the nonsynonymous mutation load and the cancer‐specific survival. In addition to the somatic mutational load, we analyzed MSI that is caused by the deleterious mutations in the DNA mismatch repair genes. The US FDA already approved the MSI test for selection of patients for anti‐PD1 drug (pembrolizumab) both pediatric and adult patients, regardless to tissue origins of cancer. In this study, we found MSI‐H in an only one of the 16 tested cases; this frequency is similar to a previous report (MSI‐H, 1‐3%).33, 34 It is notable that this MSI‐H patient has lived for 1034 days.
Taken together, we have investigated various immune‐related factors in tumors from patients treated with immune checkpoint inhibitors and found that the extent of CD8+ infiltration into tumor tissues and the number of nonsynonymous mutations, but not PD‐L1 status, were good predictors of clinical outcomes. Because of the limitation of the patients enrolled, further studies using larger cohorts are essential.
4. MATERIALS AND METHODS
4.1. Study design
The study was conducted under a protocol approved by the University of Chicago institutional review board (IRB number: 15550B). Whole blood, tumor tissues, and comparative normal tissues were collected from bladder patients between 2013 and 2016 at the time of surgical resection of the primary tumor. Specimens were FFPE. Whole blood samples were collected and stored at −80°C. To define the effectiveness of immune therapy, cancer‐specific survival was calculated from the treatment start date until disease progression, cancer‐related death, or for patients not progressing, those patients were censored at the date of analysis cutoff of May 16, 2017. To objectively categorize immunotherapy treatment response, complete response, PR, stable disease, and progressive disease were defined according to the Response Evaluation Criteria in Solid Tumors version 1.1.38
4.2. Whole exome sequencing
Genomic DNAs were extracted from FFPE tissues and blood by following the QIAamp DNA FFPE Tissue Kit or blood DNA kit (Qiagen), respectively. Whole exome libraries were built up using SureSelectXT Human All Exon V5 kit (Agilent Technologies, Santa Clara, California) and sequenced by 100‐bp paired‐end reads on HiSeq2500 Sequencer (Illumina, San Diego, California). The obtained sequence data were analyzed using an in‐house pipeline as described previously.39 Briefly, the reads were mapped to the human reference genome GRCh37/hg19 using Burrows‐Wheeler Aligner, and then possible polymerase chain reaction (PCR) duplicates were removed with Picard tool (http://broadinstitute.github.io/picard/). Read pairs with a mapping quality of <30 and with mismatches more than 5% of read length were also removed. Somatic variants (single nucleotide variations and indels) were called using the following parameters: (1) base quality ≥ 15, (2) sequence depth ≥ 10, (3) variant depth ≥ 4, (4) variant frequency in tumor ≥ 10%, (5) variant frequency in normal < 2%, and (6) Fisher P value < .05.40 Single nucleotide variations and indels were annotated based on RefGene using ANNOVAR.41, 42 Neoantigens were predicted for each nonsynonymous variant by defining all novel 8‐ to 11‐mers resulting from the mutation and determining whether the predicted binding affinity to human leukocyte antigen class I was <500nM using NetMHCpanv2.8 software.18
4.3. TCRβ complementary‐determining 3 amplification and sequencing
The TIL samples were assessed from the core punctures obtained from lymphocyte‐enrich regions of FFPE blocks by our pathologist. Genomic DNAs were extracted as previously described.43 Here we applied a novel high‐throughput TCRβ DNA sequencing technique capable of quantifying T cells. Samples were analyzed from 1 μg of genomic DNA. T‐cell receptor beta was amplified by using a primer mix specific to the V and J regions. The PCR products in the 200 to 700 bp range were selected using the Pippin Prep System (Sage Science, Beverly, Massachusetts). Then we added Illumina sequence adapter with barcode sequences using the Nextera XT Index kit (Illumina). Barcoded samples were pooled to form a final library and sequenced by Illumina MiSeq platform using 600v3 reagents (Illumina). Sequencing data analysis was performed by MiXcr. The Simpson DI was used to evaluate the TCRβ clonality as previously described.44
4.4. Microsatellite instability analysis
Genomic DNAs were used for MSI analysis with the Promega panel according to the manufacturer's protocol (5 mononucleotide markers: BAT‐25, BAT‐26, NR‐21, NR‐24, and MONO‐27; and 2 pentanucleotide markers for identifying sample mix‐ups and/or contamination: Penta C and Penta D).45 The PCR products were analyzed by capillary electrophoresis using an ABI 3500 Genetic Analyzer (Applied Biosystems, Forster City, California). Data were analyzed by GeneMapper software from Applied Biosystems to identify predominate allele sizes for each locus. Microsatellite instability positive for a marker was recognized as alteration in the lengths of the microsatellite due to deletion or insertion of a repeating unit, which produces a novel length allele(s) in test DNA samples compared with corresponding normal DNA samples. Samples with greater than 2 of 5 loci altered were classified as MSI‐H, those with 1 or 2 loci altered were MSI‐L, and those with no alterations were microsatellite stable.
4.5. IHC staining
From FFPE tumor resection specimens, adjacent 4‐μm sections were prepared. Immunohistochemical staining was performed on a semiautomated stainer. All antibodies were applied in optimized dilutions using positive and negative controls. Briefly, slides were deparaffinized and rehydrated. Monoclonal antibodies from Cell Signaling Technology (Danvers, Massachusetts) were used (anti–PD‐L1 and anti‐CD8). Immunohistochemical staining was counterstained with hematoxylin. In addition, a section stained for hematoxylin‐eosin of each case was prepared.
4.6. Histology scoring
All cases were reviewed and scored by T.A. (specialized genitourinary pathologist) who was blinded to the clinical outcome data. For each case, the presence of tumor and the extent of the tumor compartment in relation to surrounding normal tissue were evaluated on hematoxylin‐eosin sections. The expression levels of PD‐L1 and CD8 were scored for proportion of IHC reactivity. The PD‐L1 positivity was classified by positivity of ≥1% of tumor cells. In the analysis of CD8 infiltration, histology sections were scanned with a microscope (Carl Zeiss, Jena, Germany) to create digital images. ImageJ software was applied to measure CD8+ cells in each section (https://imagej.nih.gov/). The cutoff value to separate high and low expressions was defined as the median value based on the distribution of the score.
4.7. Statistical analysis
Pearson correlation (R) was used to analyze the association between all parameters examined. Statistical analysis was performed using GraphPad Prism 7.0 and SPSS 16.0. The Kaplan‐Meier method was applied to calculate correlation between various parameters and cancer‐specific survival. P < .05 was considered to be statistically significant.
CONFLICT OF INTEREST
Y.N. is a stockholder and an adviser of OncoTherapy Science Ltd.
AUTHORS' CONTRIBUTION
All authors had full access to the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Conceptualization, Y.N. and P.O'D.; Methodology, B.D. and J.‐H.P.; Investigation, B.D. and J.‐H.P.; Formal Analysis, K.K., P.Y., and B.D.; Resources, T.A., K.O'C., and P.O'D.; Writing ‐ Original Draft, B.D.; Writing ‐ Review & Editing, Y.N.; Supervision, Y.N.; Funding Acquisition, Y.N. and P.O'D.
Supporting information
Supplementary Figure 1. The relationship between the CD8 expression values and the cancer‐specific survival of the patients (R = 0.630, P = 0.0067). The red dots stand for cases that were still alive at the cut‐off date.
Supplementary Figure 2. The Kaplan‐Meier analysis of cancer‐specific survival of patients by the TCR diversity index (P = 0.421).
Supplementary Figure 3. The Kaplan‐Meier analysis of cancer‐specific survival of patients by the non‐synonymous mutation load (P = 0.193).
Supplementary Figure 4. The Kaplan‐Meier analysis of cancer‐specific survival of patients by the neoantigen load. And the relationship between the neoantigen burden and the cancer‐specific survival of the patients.
Supplementary Figure 5: A case showing MSI‐H by MSI analysis. A tumor of a patient A16 showed one‐base shifts for all of the 5 loci tested and was classified as MSI‐H.
Supplementary Table1. TCRβ sequencing of 15 bladder cancer samples. We identified a total of 1,557 to 41,435 (mean ± standard deviation [SD] 14,954 ± 3,428) unique TCRβ clonotypes of complementarity determining region 3 (CDR3) in 609,735 to 1,948,787 (mean ± standard deviation [SD] 1,151,129 ± 101,908) total reads which obtained from sequencing data.
ACKNOWLEDGEMENTS
We thank Dr Rui Yamaguchi, Seiya Imoto, and Satoru Miyano at The University of Tokyo for developing the algorithm of TCR repertoire analysis and helpful support in data management. The super‐computing resource was provided by Human Genome Center, Institute of Medical Science, The University of Tokyo (http://sc.hgc.jp/shirokane.html). This work was supported in part by a Johns Hopkins Greenberg Bladder Cancer Institute award (P.H.O., J.‐H.P., and Y.N.) (https://www.hopkinsmedicine.org/news/media/releases/johns_hopkins_greenberg_bladder_cancer_institute_awards_first_research_grants).
Deng B, Park J‐H, Ren L, et al. CD8 lymphocytes in tumors and nonsynonymous mutational load correlate with prognosis of bladder cancer patients treated with immune checkpoint inhibitors. Cancer Reports. 2018;1:e1002. 10.1002/cnr2.1002
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7‐30. [DOI] [PubMed] [Google Scholar]
- 2. Babjuk M, Böhle A, Burger M, et al. EAU guidelines on non‐muscle‐invasive urothelial carcinoma of the bladder: update 2016. Eur Urol. 2017;71(3):447‐461. [DOI] [PubMed] [Google Scholar]
- 3. Moschini M, Karnes RJ, Sharma V, et al. Patterns and prognostic significance of clinical recurrences after radical cystectomy for bladder cancer: a 20‐year single center experience. Eur J Surg Oncol. 2016;42(5):735‐743. [DOI] [PubMed] [Google Scholar]
- 4. von der Maase H, Hansen SW, Roberts JT, et al. Gemcitabine and cisplatin versus methotrexate, vinblastine, doxorubicin, and cisplatin in advanced or metastatic bladder cancer: results of a large, randomized, multinational, multicenter, phase III study. J Clin Oncol. 2000;18(17):3068‐3077. [DOI] [PubMed] [Google Scholar]
- 5. Alexander W. The checkpoint immunotherapy revolution: what started as a trickle has become a flood, despite some daunting adverse effects; new drugs, indications, and combinations continue to emerge. P T. 2016;41(3):185‐191. [PMC free article] [PubMed] [Google Scholar]
- 6. Schachter J, Ribas A, Long GV, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open‐label phase 3 study (KEYNOTE‐006). Lancet. 2017;390(10105):1853‐1862. [DOI] [PubMed] [Google Scholar]
- 7. Reck M, Rodríguez‐Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD‐L1‐positive non‐small‐cell lung cancer. N Engl J Med. 2016;375(19):1823‐1833. [DOI] [PubMed] [Google Scholar]
- 8. Brower V. Pembrolizumab in advanced head and neck cancer. Lancet Oncol. 2017;18(5):e248. [DOI] [PubMed] [Google Scholar]
- 9. Burki TK. Pembrolizumab for classical Hodgkin's lymphoma. Lancet Oncol. 2016;17(8):e324. [DOI] [PubMed] [Google Scholar]
- 10. Balar AV, Galsky MD, Rosenberg JE, et al. Atezolizumab as first‐line treatment in cisplatin‐ineligible patients with locally advanced and metastatic urothelial carcinoma: a single‐arm, multicentre, phase 2 trial. Lancet. 2017;389(10064):67‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Powles T, O'Donnell PH, Massard C, et al. Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma: updated results from a phase 1/2 open‐label study. JAMA Oncol. 2017;3(9):e172411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McDermott DF, Sosman JA, Sznol M, et al. Atezolizumab, an anti‐programmed death‐ligand 1 antibody, in metastatic renal cell carcinoma: long‐term safety, clinical activity, and immune correlates from a phase Ia study. J Clin Oncol. 2016;34(8):833‐842. [DOI] [PubMed] [Google Scholar]
- 13. Muro K, Chung HC, Shankaran V, et al. Pembrolizumab for patients with PD‐L1‐positive advanced gastric cancer (KEYNOTE‐012): a multicentre, open‐label, phase 1b trial. Lancet Oncol. 2016;17(6):717‐726. [DOI] [PubMed] [Google Scholar]
- 14. Bellmunt J, de Wit R, Vaughn DJ, et al. Pembrolizumab as second‐line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376(11):1015‐1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Balar AV, Castellano D, O'Donnell PH, et al. First‐line pembrolizumab in cisplatin‐ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE‐052): a multicentre, single‐arm, phase 2 study. Lancet Oncol. 2017;18(11):1483‐1492. [DOI] [PubMed] [Google Scholar]
- 16. Rosenberg JE, Hoffman‐Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum‐based chemotherapy: a single‐arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909‐1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sharma P, Callahan MK, Bono P, et al. Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): a multicentre, open‐label, two‐stage, multi‐arm, phase 1/2 trial. Lancet Oncol. 2016;17(11):1590‐1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Choudhury NJ, Kiyotani K, Yap KL, et al. Low T‐cell receptor diversity, high somatic mutation burden, and high neoantigen load as predictors of clinical outcome in muscle‐invasive bladder cancer. Eur Urol Focus. 2016;2(4):445‐452. [DOI] [PubMed] [Google Scholar]
- 19. Nishino M, Ramaiya NH, Hatabu H, Hodi FS. Monitoring immune‐checkpoint blockade: response evaluation and biomarker development. Nat Rev Clin Oncol. 2017;14(11):655‐668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Teng MW, Khanna R, Smyth MJ. Checkpoint immunotherapy: picking a winner. Cancer Discov. 2016;6(8):818‐820. [DOI] [PubMed] [Google Scholar]
- 21. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med. 2012;366(26):2443‐2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Daud AI, Wolchok JD, Robert C, et al. Programmed death‐ligand 1 expression and response to the anti‐programmed death 1 antibody pembrolizumab in melanoma. J Clin Oncol. 2016;34(34):4102‐4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320‐330. [DOI] [PubMed] [Google Scholar]
- 25. Borghaei H, Paz‐Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non‐small‐cell lung cancer. N Engl J Med. 2015;373(17):1627‐1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kang YK, Boku N, Satoh T, et al. Nivolumab in patients with advanced gastric or gastro‐oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO‐4538‐12, ATTRACTION‐2): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet. 2017;390(10111):2461‐2471. [DOI] [PubMed] [Google Scholar]
- 27. Diggs LP, Hsueh EC. Utility of PD‐L1 immunohistochemistry assays for predicting PD‐1/PD‐L1 inhibitor response. Biomark Res. 2017;5(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Festino L, Botti G, Lorigan P, et al. Cancer treatment with anti‐PD‐1/PD‐L1 agents: is PD‐L1 expression a biomarker for patient selection? Drugs. 2016;76(9):925‐945. [DOI] [PubMed] [Google Scholar]
- 29. Galon J, Fox BA, Bifulco CB, et al. Immunoscore and immunoprofiling in cancer: an update from the melanoma and immunotherapy bridge 2015. J Transl Med. 2016;14(1):273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69‐74. [DOI] [PubMed] [Google Scholar]
- 31. Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA‐4 blockade in melanoma. N Engl J Med. 2014;371(23):2189‐2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carbone DP, Reck M, Paz‐Ares L, et al. First‐line nivolumab in stage IV or recurrent non‐small‐cell lung cancer. N Engl J Med. 2017;376(25):2415‐2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Giedl J, Schneckenpointner R, Filbeck T, et al. Low frequency of HNPCC‐associated microsatellite instability and aberrant MMR protein expression in early‐onset bladder cancer. Am J Clin Pathol. 2014;142(5):634‐639. [DOI] [PubMed] [Google Scholar]
- 34. Catto JW, Xinarianos G, Burton JL, Meuth M, Hamdy FC. Differential expression of hMLH1 and hMSH2 is related to bladder cancer grade, stage and prognosis but not microsatellite instability. Int J Cancer. 2003;105(4):484‐490. [DOI] [PubMed] [Google Scholar]
- 35. Le DT, Uram JN, Wang H, et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med. 2015;372(26):2509‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tumeh PC, Harview CL, Yearley JH, et al. PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Inoue H, Park JH, Kiyotani K, et al. Intratumoral expression levels of PD‐L1, GZMA, and HLA‐A along with oligoclonal T cell expansion associate with response to nivolumab in metastatic melanoma. Oncoimmunology. 2016;5(9):e1204507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228‐247. [DOI] [PubMed] [Google Scholar]
- 39. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25(14):1754‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64‐69. [DOI] [PubMed] [Google Scholar]
- 41. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leisegang M, Engels B, Schreiber K, et al. Eradication of large solid tumors by gene therapy with a T‐cell receptor targeting a single cancer‐specific point mutation. Clin Cancer Res. 2016;22(11):2734‐2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kiyotani K, Park JH, Inoue H, et al. Integrated analysis of somatic mutations and immune microenvironment in malignant pleural mesothelioma. Oncoimmunology. 2017;6(2):e1278330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yew PY, Alachkar H, Yamaguchi R, et al. Quantitative characterization of T‐cell repertoire in allogeneic hematopoietic stem cell transplant recipients. Bone Marrow Transplant. 2015;50(9):1227‐1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bacher JW, Flanagan LA, Smalley RL, et al. Development of a fluorescent multiplex assay for detection of MSI‐high tumors. Dis Markers. 2004;20(4–5):237‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. The relationship between the CD8 expression values and the cancer‐specific survival of the patients (R = 0.630, P = 0.0067). The red dots stand for cases that were still alive at the cut‐off date.
Supplementary Figure 2. The Kaplan‐Meier analysis of cancer‐specific survival of patients by the TCR diversity index (P = 0.421).
Supplementary Figure 3. The Kaplan‐Meier analysis of cancer‐specific survival of patients by the non‐synonymous mutation load (P = 0.193).
Supplementary Figure 4. The Kaplan‐Meier analysis of cancer‐specific survival of patients by the neoantigen load. And the relationship between the neoantigen burden and the cancer‐specific survival of the patients.
Supplementary Figure 5: A case showing MSI‐H by MSI analysis. A tumor of a patient A16 showed one‐base shifts for all of the 5 loci tested and was classified as MSI‐H.
Supplementary Table1. TCRβ sequencing of 15 bladder cancer samples. We identified a total of 1,557 to 41,435 (mean ± standard deviation [SD] 14,954 ± 3,428) unique TCRβ clonotypes of complementarity determining region 3 (CDR3) in 609,735 to 1,948,787 (mean ± standard deviation [SD] 1,151,129 ± 101,908) total reads which obtained from sequencing data.