

Abstract
Soluble epoxide hydrolase (sEH) is an α/β hydrolase fold protein and widely distributed in numerous organs including the liver, kidney, and brain. The inhibition of sEH can effectively maintain endogenous epoxyeicosatrienoic acids (EETs) levels and reduce dihydroxyeicosatrienoic acids (DHETs) levels, resulting in therapeutic potentials for cardiovascular, central nervous system, and metabolic diseases. Therefore, since the beginning of this century, the development of sEH inhibitors is a hot research topic. A variety of potent sEH inhibitors have been developed by chemical synthesis or isolated from natural sources. In this review, we mainly summarized the interconnected aspects of sEH with cardiovascular, central nervous system, and metabolic diseases, and then focus on representative inhibitors, which would provide some useful guidance for the future development of potential sEH inhibitors.
Keywords: Soluble epoxide hydrolase (sEH), Natural products, Pharmacology, Structure-activity relationship, Biological activity
Graphical Abstract
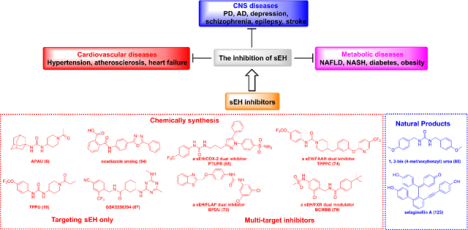
1. Introduction
Epoxide hydrolases are widely distributed enzymes responsible for the rapid hydrolysis of epoxides to the corresponding vicinal diols.1,2 In mammals, soluble epoxide hydrolase (sEH), an α/β hydrolase fold protein, is expressed in the cytosol, and sometimes also in the peroxisome, of numerous tissues, such as liver, kidney, lungs, heart, brain, spleen, adrenals, and intestine.3 Beside mammals, the sEH is present in all vertebrates; however, only the mammalian sEH has phosphatase activity.4–14 Expression levels varies among species; for example, the sEH is much lower in rats than in mice.15 Furthermore, the sEH expression can be induced by angiotensin II and peroxisome proliferator-activated receptor α (PPARα) and PPARγ agonists, such as fibrates and glitazones;3, 16 but also by sexual hormones.17 Notably, the expression of sEH is upregulated by PPARγ agonists in adipose tissues, while the oppositive result is observed in cardiomyocytes.18 Additionally, inflammation also increases its expression, therefore, sEH can be considered a marker of inflammation.19
The mammalian sEH comprises of two 62.5 kDa monomers, containing 555 amino acids each, and it is a bifunctional enzyme that possesses a 25 kDa N-terminal phosphatase region and a 35 kDa C-terminal hydrolase domain (Figure 1A).20 The C-terminal hydrolase catalyzes the hydrolysis of epoxy-fatty acids (EpFAs), such as epoxyeicosatrienoic acids (EETs), metabolized from arachidonic acid (AA), to the corresponding diols, such as dihydroxyeicosatrienoic acids (DHETs) (Figure 2).20 The hydrolase catalytic pocket of the C-terminal hydrolase consists of two tyrosine residues (Tyr381 and Tyr465) that interact with the oxygen atom of the epoxide via two hydrogen bonds. The nucleophilic carboxylic acid Asp333 of the catalytic triad Asp333-Asp495-His523, located on the opposite side of Tyr381 and Tyr465, is oriented and activated by His523 and Asp495, and attacks the epoxide carbon backbone to form an ester bond with the opened epoxide (Figure 1C). The following hydrolysis of this ester results in the production of the corresponding diols.21, 22 Compared with the well-understood function of the C-terminal hydrolase domain, the role of the N-terminal phosphatase domain is still unclear.21, 22 Cronin et al. and Newman et al. independently discovered that the N-terminal domain of sEH exhibits phosphatase activity to hydrolyze diverse lipid phosphates in vitro, including farnesyl pyrophosphate, sphingosine-1-phosphate, and lysophosphatidic acid, whereas its physiological and possibly pathophysiological role are still not well understood.23, 24
Figure 1.
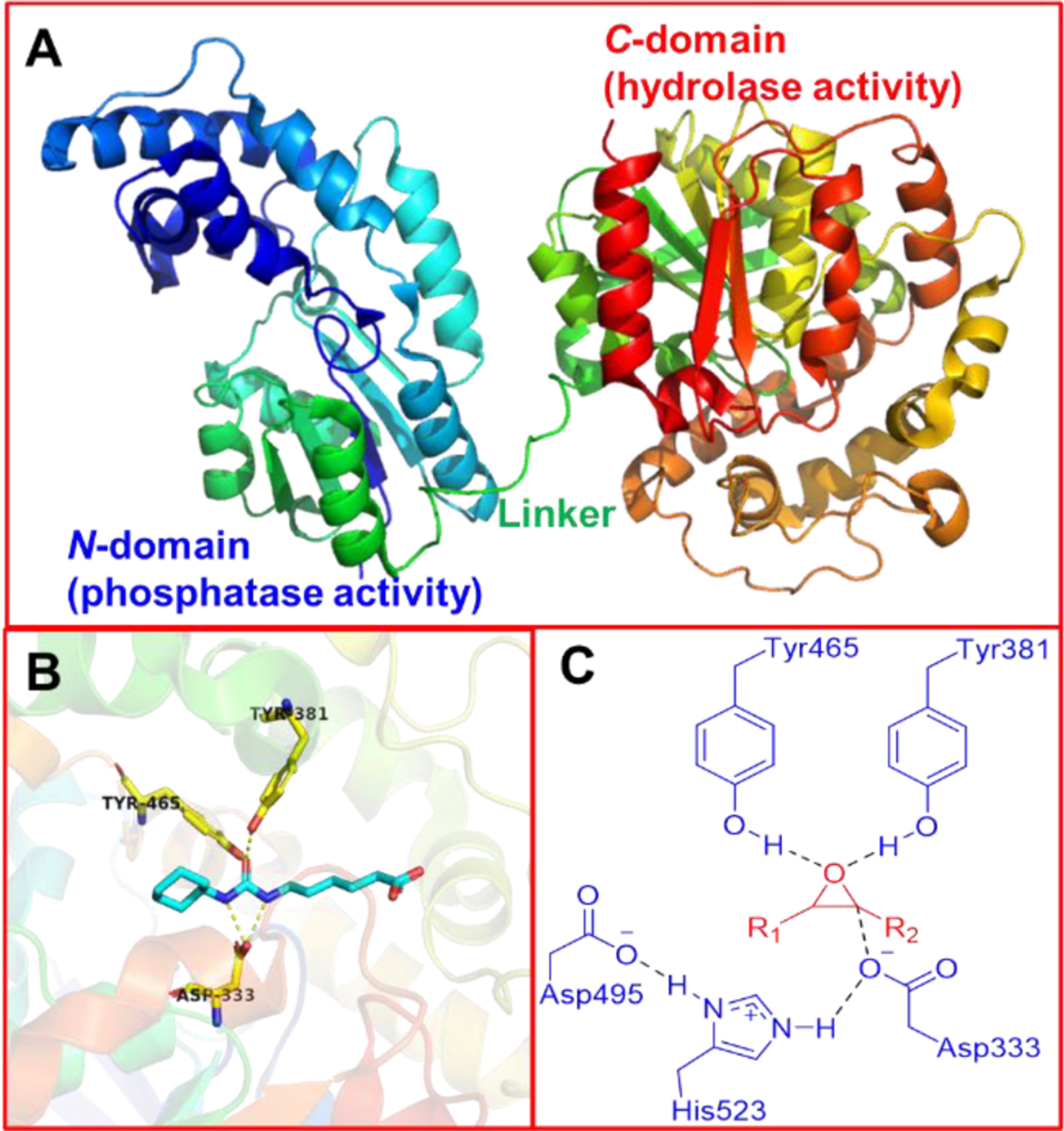
(A) X-ray cocrystal structure of a single subunit of the human sEH (PDB code 1ZD4). (B) X-ray cocrystal structure of human sEH C-domain showing the binding site (PDB code 1ZD4). (C) The potential interactions between epoxides and human sEH C-domain.
Figure 2.
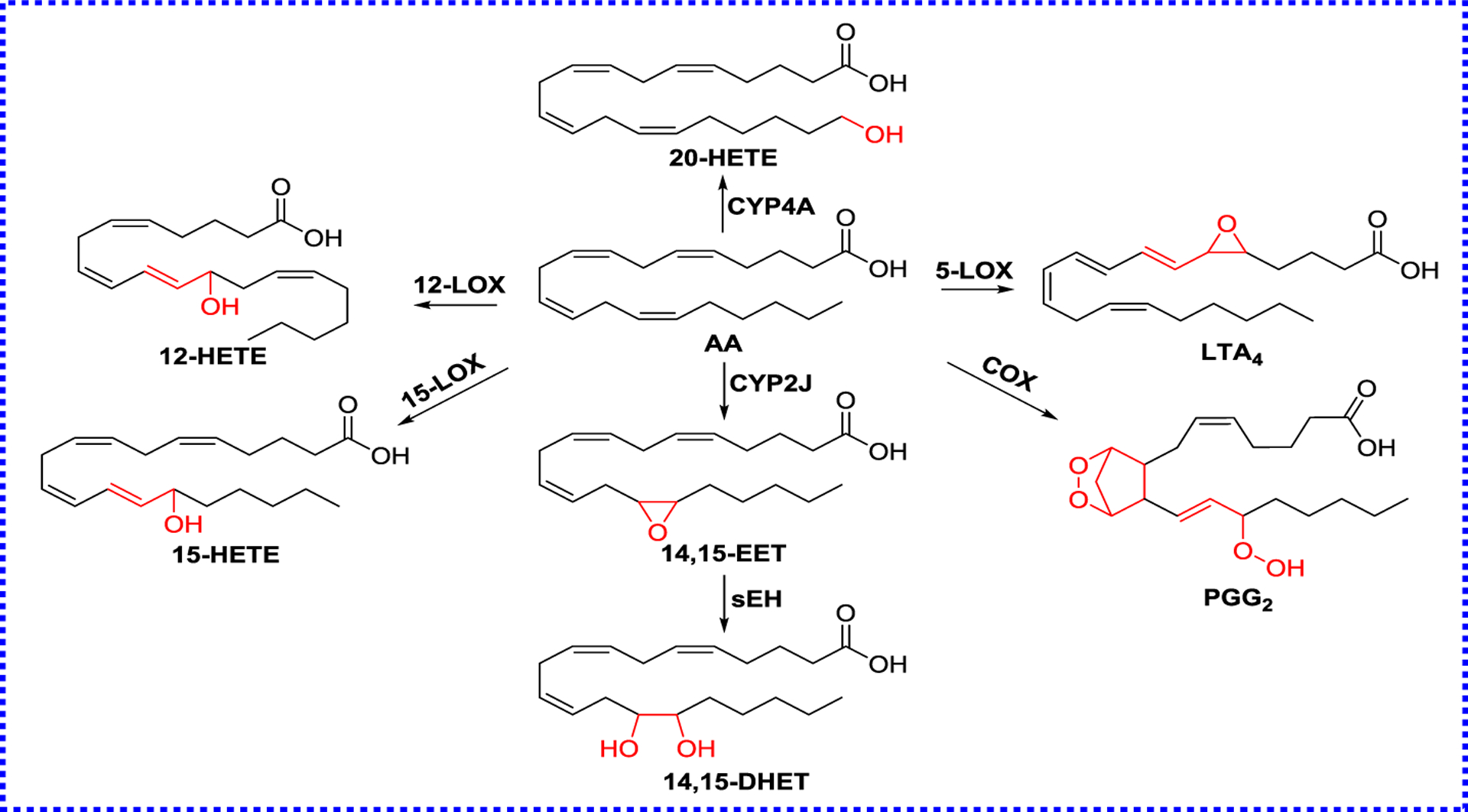
The metabolic pathways of AA
EPHX2, the human gene encoding sEH, is a 19-exon gene (45 kb) localized in the chromosomal region 8p21–12.25 So far, seven single-nucleotide polymorphisms (SNPs) of sEH have been discovered, including K55R, R103C, C154Y, R287E, R287E/R103C, V422A, and E470G.26 Compared with the wild-type (WT) protein, the mutants the strong reduction of its phosphatase activity in R103C and R287E significantly decreased the phosphatase activity of sEH, and its hydrolase activity is increased in R287E/R103C, while the increasing of its N-terminal activity in K55R and C154Y.12, 27 Among them, K55R, R103C, and R287Q have been verified in connection with several diseases,28, 29 including heart failure, ischemic stroke, anorexia nervosa, and nephropathy.
In response to infection or damage, the body generates an inflammatory response. The central to this response is the production of bioactive lipids from AA (Figure 2).30 Eicosanoids possess a series of protective effects and are defined to be vital metabolites from AA via three metabolic pathways: 1) the cytochrome P450 (CYP) pathway forms EETs and hydroxy eicosatetraenoic acids (HETEs); 2) Lipoxygenases (LOXs) catalyze the formation of lipoxins (LXs) and leukotrienes;31 3) Cyclooxygenases (COXs) pathway produces prostanoids.32 The above-mentioned pathways can also metabolize eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and other unsaturated fatty acids.30, 32–34
All these bioactive lipids, called oxylipins, have strong roles in the inflammatory response.35 For example, EETs, synthesized by CYP450s, such as CYP2C8, CYP2C9, and CYP2J2, activate guanine nucleotide-binding protein (G-protein) Gαs via ADP-ribosylation, resulting in the activation of the smooth muscle large conductance Ca2+-activated K+ channels (BKCa), to produce vasodilation and lowering blood pressure.36–39 Furthermore, EETs increase Ca2+ influx, enhance fibrinolysis, and stimulate tube formation in the endothelium.40,41 In addition, EETs prevent IκB degradation via suppressing IκB kinase, inhibit nuclear transcription factor-κB (NF-κB) responsible for cytokine-mediated inflammation, and further inhibits NF-κB-mediated gene transcription, allowing in the anti-inflammatory effect.42, 43 Compared with bioactive substrate EETs, DHETs are inactivation products of EETs,37, 44 because their activities are reduced in many systems, such as relaxation of the preglomerular vasculature and the bovine coronary artery,45, 46 inhibition of Na+ channel and cyclooxygenase,47, 48 and Ca2+ uptake.49 In addition, DHETs produce functional effects in several other systems as well, including the inhibition of hydroosmotic action of arginine vasopressin,50 relaxation of canine and porcine coronary arterioles51, 52 and activation of the BKCa channel.48–51
Because sEH can rapidly metabolize or inactivate EETs to produce DHETs, nowadays, sEH inhibition or knockout has become an experimental approach to investigate the biological roles of EpFAs, especially EETs. For example, inflammatory pain can be ameliorated by increasing levels of EETs or inhibiting sEH to stabilize EETs.53 Furthermore, the sEH expression level is upregulated in angiotensin II (Ang II)-induced lung injury mice, levels of tumor necrosis factor-α (TNF-α) and interleukin-1 beta (IL-1β) are significantly increased, and the Ephx2 (the gene name of mouse sEH) knockout can remarkably alleviate the lung injury.54 In addition, sEH is found to be expressed in the cerebral cortex and hippocampus as well as striatum (STR). Therefore, it is speculated that sEH is closely related to central nervous systems (CNS) diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), depression, and stroke.30 In this review, we summarized recent findings of sEH inhibitors from chemical synthesis and natural products, which provided useful guidance for the future development of potential sEH inhibitors, and the relationship between sEH and cardiovascular, CNS, and metabolic diseases.
2. sEH inhibitors
EETs exhibit potential biological functions in dilating blood vessels, and protecting neurons. Numerous studies indicated that inhibiting sEH to stabilize EETs can significantly reduce inflammation and pain.54, 55 Therefore, sEH inhibitors are considered as promising and effective treatment for various diseases and its development has become a hot topic in the research field since the beginning of this century. So far, sEH inhibitors have been discovered from a variety of pathways, such as synthesis and natural products,56 therefore, we summarized the recent progress in the design and development of sEH inhibitors.
2.1. Targeting sEH only
2.1.1. Urea sEH inhibitors
The earliest selective sEH inhibitors reported are substituted chalcone oxides (such as compound 1) and phenylglycidols, rather than ureas. 4-Phenyl-chalcone oxide (1, Figure 3) is a sEH substrate, and forms a covalent intermediate, allowing in low turnover and transient inhibition. However, they are unstable in the presence of glutathione, and slowly hydrolyzed by sEH, therefore, their use in vivo is limited.57 The first potent and stable inhibitors of sEH were reported in 1999 based on the urea skeleton (I) (Figure 3), such as N,N′-dicyclohexyl-urea (2, DCU) and N-cyclohexyl-N′-(3-phenylpropyl)-urea (3, CPU).58 They display significant inhibitory potency against the mouse and human sEH with IC50 values in the tens of nanomolar. Their kinetic constants (Ki) are in the low nanomolar and are 20- to 150-fold better than 1 (Kd = 430 nM), revealing that compounds like DCU (2) or CPU59 act as competitive inhibitors at stoichiometric concentrations,56, 60 and have a tight association with the target enzyme.59 DCU (2) was first to be used in vivo, and could significantly inhibit the conversion from 14,15-EET to 14,15-DHET in spontaneously hypertensive rat, then leading to an antihypertensive effect.61 In porcine coronary endothelial cells, DCU (2) led to the decrease of the EETs conversion rate by 3-fold.62 While it demonstrated the usefulness of the urea inhibitors to affect the metabolism of EpFAs in vitro and in vivo, the early sEH inhibitors, such as DCU (2), had poor water solubility and high melting point (mp) due to its rigid structure and non-polar groups, which affected its oral availability and bioavailability. To date, the urea derivatives are the most abundant and effective sEH inhibitors, because the central urea group can bind strongly to the catalytic pocket of sEH (Figure 1B).63 The oxygen atom on the urea group formed two hydrogen bonds with amino acid residues Tyr381 and Tyr465 in the catalytic pocket of sEH.63 Moreover, the N-H of the urea group acts as the hydrogen bond donor of Asp333.63 Because of their high potency, urea derivatives have been continuously developed, especially to improve solubility and bioavailability to apply them for the treatment of EpFA-related diseases.56, 64
Figure 3.
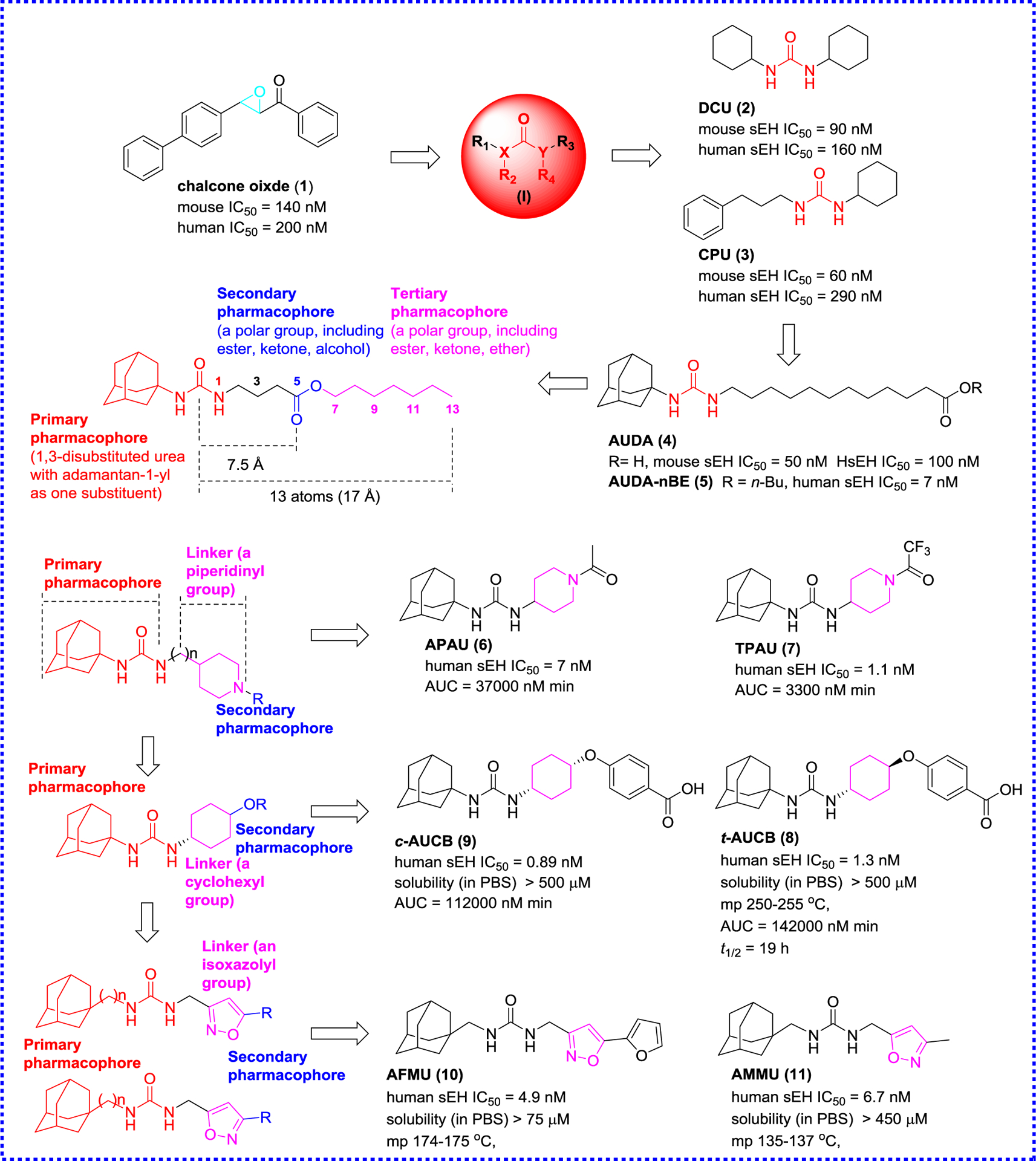
Representative sEH inhibitors and development of early sEH inhibitors
From DCU (2), the first improvement in the design of sEH inhibitors was the introduction on one side of the urea function of a flexible chain, such as 12-(3-adamantan-1-yl-ureido)dodecanoic acid (4, AUDA). The introduction of a flexible side chain significantly improved the water solubility of AUDA (4) and reduced the mp while maintaining the inhibitory effect.65 AUDA (4) continuously administered to mice via drinking water (25 mg/L) for 14 days, and finally achieved the purpose of reducing blood pressure. Although AUDA (4) was easier to formulate and to dose animals with (it could be given in the drinking water at 25 mg/L),66 it was extremely sensitive to β-oxidation and therefore it was easily metabolized and excreted,32 thus limiting its application in clinic. Nevertheless, AUDA n-butyl ester prodrug (5, AUDA-nBE) has a relatively good pharmacokinetic67 allowing in vivo application. AUDA-nBE (5) and its in vivo metabolite AUDA (4) reduced ischemic cerebral infarct size in stroke-prone spontaneously hypertensive rats, and prevented death and restore systolic blood pressure in lipopolysaccharide (LPS)-stimulated mice as well as the inhibition on the tumor growth.68, 69
Based on the analysis of the structure activity of 348 urea-based inhibitors,70 and in order to improve physical properties while reducing metabolism, polar functional groups were incorporated into one of the alkyl chains of 1,3-disubstituted ureas.64 This study revealed that a representative sEH inhibitor could possess a secondary pharmacophore (a polar functional group) that is incorporated at least five atoms (about 7.5 Å) from the primary pharmacophore (the central urea carbonyl) which effectively improved aqueous solubility and PK properties (Figure 3) while maintaining potency. Solubility could even be enhanced with a polar tertiary pharmacophore that is 13 atoms or ~17 Å away from the primary pharmacophore, without affecting much the inhibitor potency.64
To reduce metabolism, in addition to the introduction of polar groups, Jones et al. proposed in 2006 conformationally constrained human sEH inhibitors,71 which have the central pharmacophore and secondary pharmacophore connected by saturated rings, such as piperidine or other unsaturated rings. APAU (6) and TPAU (7) (Figure 3) are examples of the first series of piperidine-based urea inhibitors. These compounds are not only very potent inhibitors (IC50s in the low nanomolar) but have better PK characteristics, such as 10-fold higher area under the curve (AUC)72 compared to AUDA (4) (Figure 3), underlying the benefits of conformationally constrained inhibitors.71
In order to further investigate the influence of conformationally constrained groups towards sEH inhibition, Hwang et al. used a cyclohexyl group as a linker instead of a piperidinyl group because of their structural similarity, and designed a series of cis- or trans-1,4-cyclohexyloxy-N,N′-disubstituted urea derivatives based on the skeleton of APAU (6, Figure 3).73 These inhibitors not only have a good water solubility, but also resolve the problem that flexible alkyl chains are easily oxidized and metabolized rapidly, increase bioavailability in vivo and significantly improve metabolic stability.73 Among them, a pair of epimers trans-4-[4-(3-adamantan-1- ylureido)cyclohexyloxy]benzoic acid (8, t-AUCB) and cis-4-[4-(3-adamantan-1- ylureido)cyclohexyloxy]benzoic acid (9, c-AUCB) shows low nanomolar inhibitory effects (8, IC50 = 1.3 nM; 9, IC50 = 0.89 nM) and desired PK properties, especially t-AUCB (8). It is notable that the 1,4-trans cyclohexane urea t-AUCB (8) is more stable than its isomer c-AUCB (9) in human liver microsome (HLM), and the AUC of the former in dogs increases about by 40-fold compared with that of AUDA (4). t-AUCB (8) at a dose of 1 mg/kg also shows the same therapeutic effect of returning blood pressure to normal as AUDA-nBE (5) at a dose of 10 mg/kg in LPS-induced hypotension model. An analysis of their structures indicates that the introduced free carboxyl group forms an additional hydrogen bond with Met418 in addition of the classical hydrogen bonds of the urea function with Asp333 and Tyr381.73 In addition, t-AUCB (8) displays remarkable biological effects, including anti-fibrosis and anti-inflammatory activities, as well as protective effects against liver and cardiovascular diseases.74–78 t-AUCB (8) can down-regulate muscle-specific miR-133 expression level in the myocardial infarction mouse established by ligating the coronary artery, allowing in the reducing infarct size and preventing the development of cardiac arrhythmias.74 Moreover, t-AUCB (8) reduces adipose tissue inflammation, prevents hepatic steatosis and the increased fasting glycemia, improved glucose tolerance, and decreases gluconeogenesis in high-fat diet (HFD) mice.77
In addition to the cyclohexyl group, an aromatic ring and an isoxazolyl group were also used as the replacement of the piperidinyl group at the right side of N-adamantyl-N′-piperidinyl-ureas such as APAU (6) in view of their limited solubility and rapid metabolism (Figure 3), which reduced mp of the inhibitors and should enhance the stability.79 Despite the potent inhibitory activity observed in this series of compounds, such as 1-(adamant-1-ylmethyl)-3-{[5-(furan-2-yl)isoxazol-3-yl]methyl}urea (10, AFMU), and 1-(adamant-1-ylmethyl)-3-[(3-methylisoxazol-5-yl)methyl]urea (11, AMMU), their stability was not improved in HLM with nicotinamide adenine dinucleotide phosphate (NADPH).79
The next step in sEH inhibitor development involved the optimization of the adamantyl- ureas via adding the substitution to the bridgehead of the adamantane to afford a library of this type compounds (Figure 4).80 Interestingly, the substitution influenced greatly the potency, physical properties, and metabolic stability of ureas and diureas. Interestingly, a methyl or chloride substitution on the bridgehead of the adamantane is to the benefit of the potency, and the adamantane and urea functions are linked via a methylene, which is in favor of potency and physical properties. These findings provide a new path to further design sEH inhibitors.80
Figure 4.
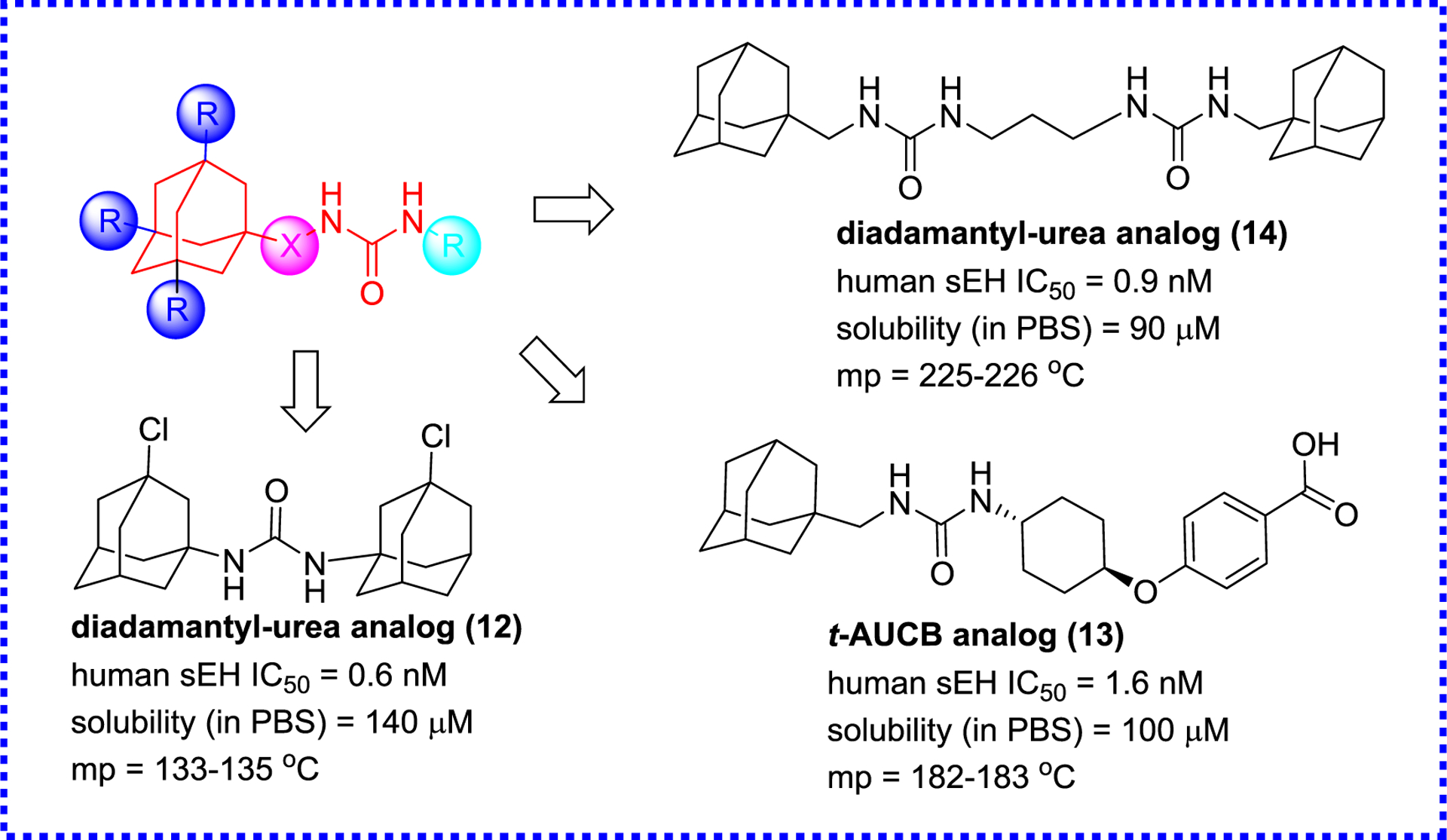
Optimization of adamantyl-ureas
Although the above-mentioned adamantyl-urea sEH inhibitors have excellent potency, this type of inhibitors is easily oxidized and metabolized, resulting in low drug concentrations and short in vivo half-life (t1/2). Therefore, in the next iteration of compounds design, aromatic or other aliphatic moieties were used instead of the adamantyl group based on the structure of t-AUCB (8).81 A library of aromatic- and other aliphatic-ureas were synthesized (Figure 5; compounds 14–20). While the replacement of the adamantyl group by aromatic groups did not significantly improve the inhibitory potencies, the PK properties were dramatically improved, especially for TPPU (19) and TPCU (20). The AUC/IC50 of TPCU (20), a metric of the inhibitory effect, is 5,500-fold higher than that of APAU (6), and a similar result is obtained in the PK of TPPU (19) as well. These compounds have become commercial inhibitors of sEH and been used in a wide variety of studies to test for sEH biological activities.81
Figure 5.
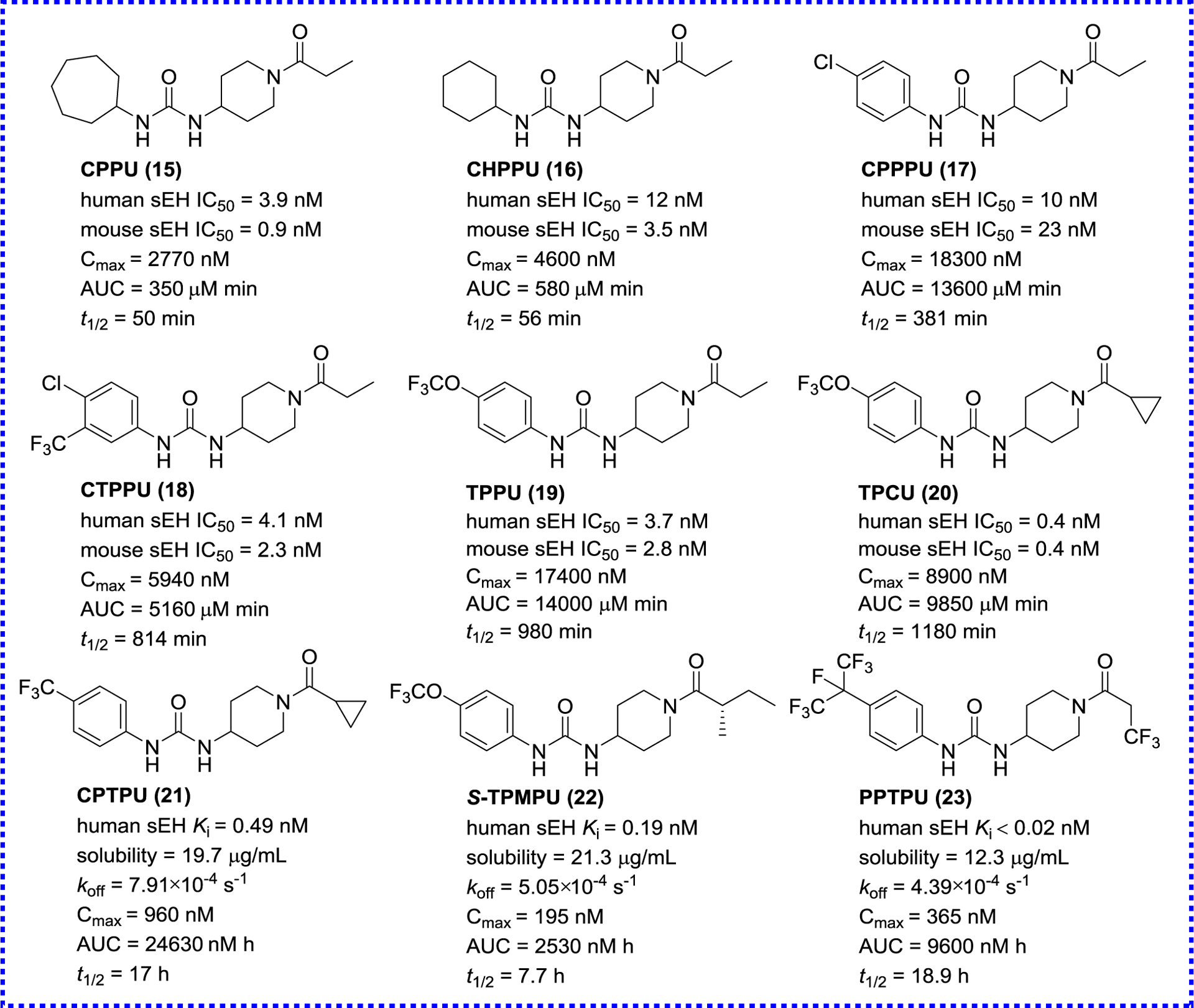
Representative urea sEH inhibitors
Although acyl piperidine ureas have good inhibitory effects, they show unsatisfactory PK properties in dog model.82 Hammock’s team designed a series of piperidyl-urea derivatives for the treatment of diabetic neuropathic pain based on the skeleton of N-phenyl-N′-piperidyl-urea in 2014.83 The potency of sEH inhibitors does not only consider IC50 or Ki, but also depends on how long the target is occupied by the inhibitor, because the catalysis is blocked only when the enzyme is occupied. In these 1,3-disubstituted ureas (Figure 5), the dissociation rate constant (koff) of compounds 21–23 increase 2.4–4.4 fold compared with that of a previous inhibitor TPAU (7, koff = 19.2 × 10−4 s−1). An analysis of the interaction between their analog TPPU (19, koff = 10.2 × 10−4 s−1) and human sEH reveals a small secondary binding site next to the α-carbon of its amide, which provides the possibility of additional binding. The hydrophobic 4-trimethoxyphenyl group binds closely to the right side of the binding pocket with a little extra binding space, which may be the reason why it stays on the target for a long time and shows excellent inhibitory effect.83
CYP enzymes can metabolize AA to produce four regioisomers and each isomer has different proportions of S/R- or R/S-enantiomers (Figure 6). Because the production rate and ratio of stereoisomers of these diol products are closely related to the stereochemistry of substrates and epoxy ring substitution, stereoselectivity is also an important factor in the design of sEH inhibitors.84 Manickam et al. designed a series of pairs of enantiomers with a chiral center close to urea nitrogen atoms.84 There are significant differences in the inhibitory activity of enantiomers, for example, the inhibitory activity of (S)-1-(1-phenylethyl)-3-(3-phenylpropyl) urea (24, S-PPU) is 40-fold higher than that of its enantiomer (25). This similar result is also observed by Lukin et al, which indicates that the chirality of the α-carbon of urea nitrogen atoms can affect the inhibition of inhibitors on sEH.60 Among the synthesized compounds, ITBU (26) possesses a potent sEH inhibitory activity and an excellent bioavailability in mice. However, the further analysis reveals that ITBU (26) is a racemic inhibitor, and can be isolated to afford (+)-ITBU [(+)-26] and (−)-ITBU [(−)-26]. The inhibitory potency of the former is 11-fold higher than that of the latter. All the above-mentioned suggests that the selective sEH inhibition by different enantiomers provides more possibilities for the binding sites of sEH and a new direction for the development of sEH inhibitors.60
Figure 6.
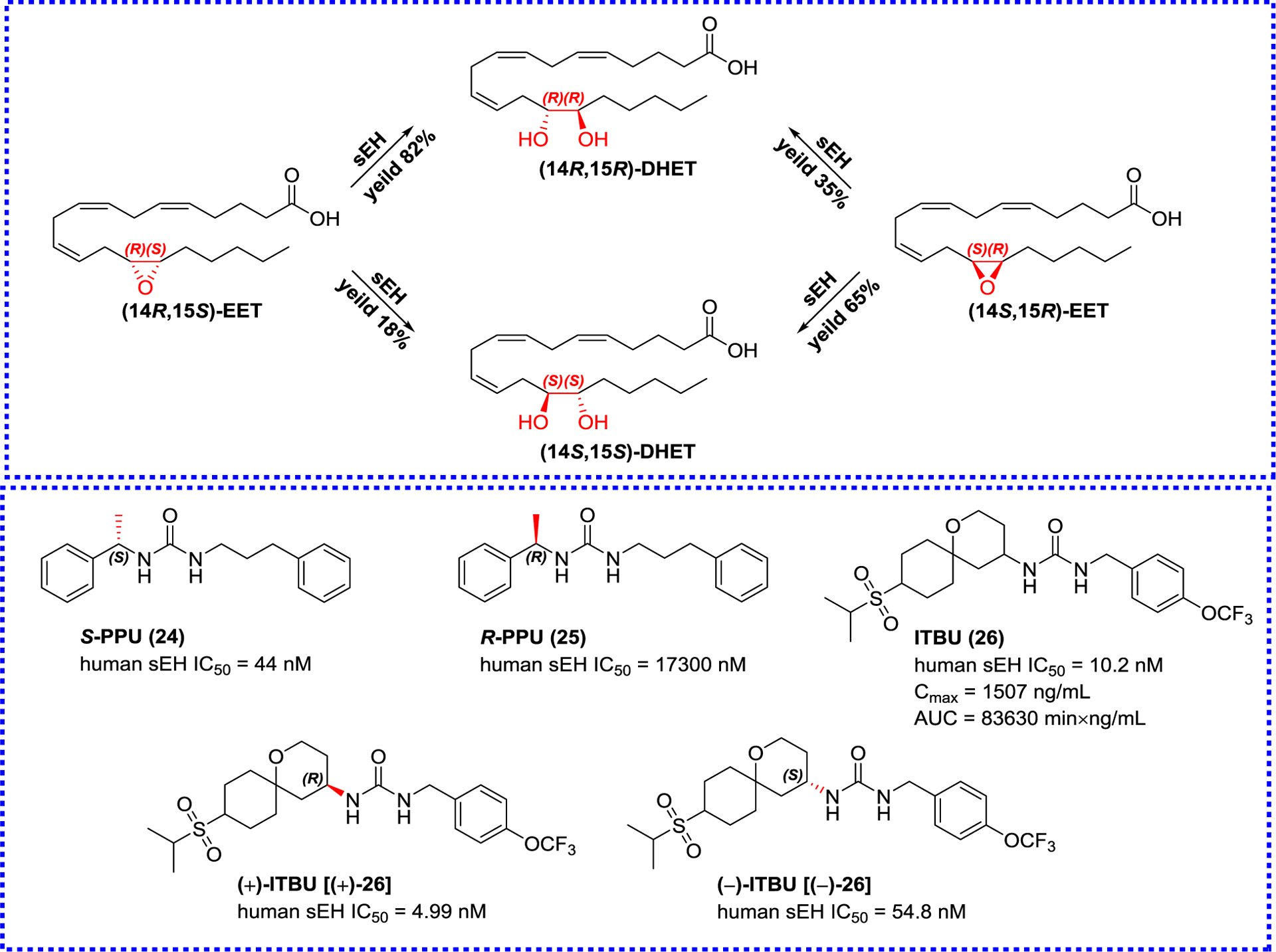
The influence of stereo configurations on inhibitory potentials and representative sEH inhibitors
Except for the above-mentioned modification, the urea moiety was also altered by replacing the oxygen atom of urea with the sulfur atom to afford thiourea-based sEH inhibitors (Figure 7).85 Among the thioureas, 1-(adamantan-2-yl)-3-phenethylthiourea (27, APU) and 1,1’-(heptane-1,7-diyl)bis(3-(adamantan-1-yl)thiourea) (28, HBAU) are found to be potent sEH inhibitors. Although their potencies less than the previous described as TPPU (19), they have better water solubility and are easier to be prepared, which is one of the ideal properties of an ideal inhibitor.85
Figure 7.
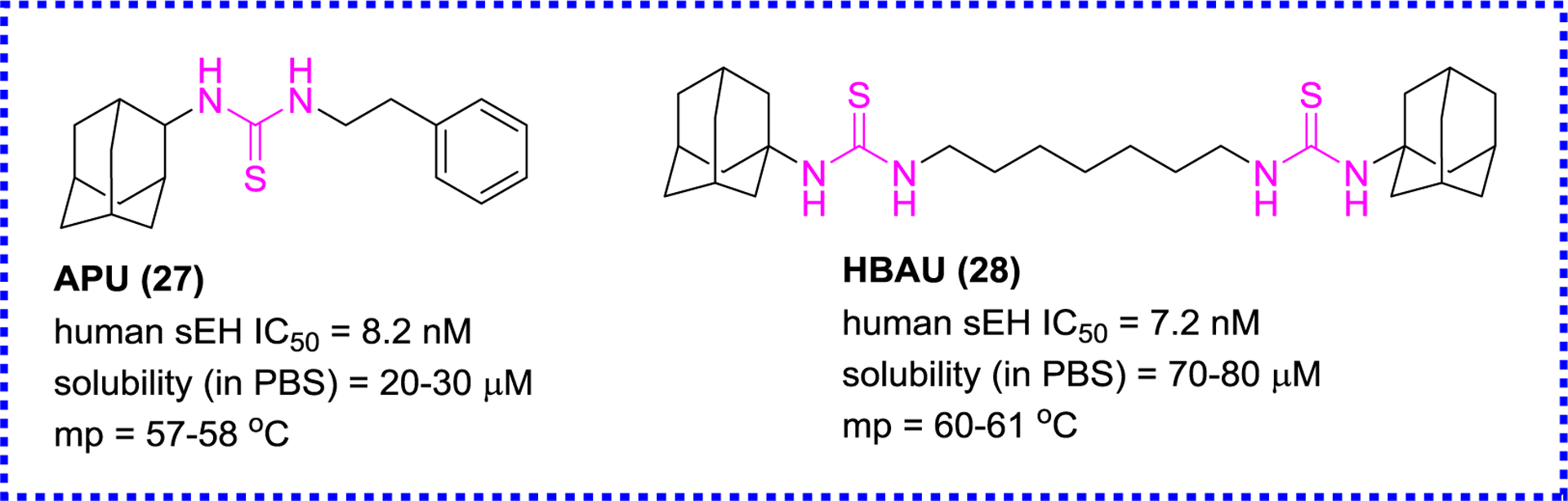
Representative thiourea-based sEH inhibitors
2.1.2. Amide sEH inhibitors
In addition to the urea-based sEH inhibitors, amides have been also widely developed as sEH inhibitors by removing one of the NH group of the ureas (Figure 8), yielding compounds 29–32.86 The primary structure-activity relationship (SAR) indicates that their inhibition potencies almost does not affect for mouse sEH compared to the corresponding ureas, whereas this modification leads to the 2.5-fold decrease inhibition potency for human sEH. On the other hand, the amide function can significantly improve the physical property compared to the corresponding ureas, such as solubility and mp, for example AABA-PE (31; mp, oil; solubility, 17.4 μg/mL).86
Figure 8.
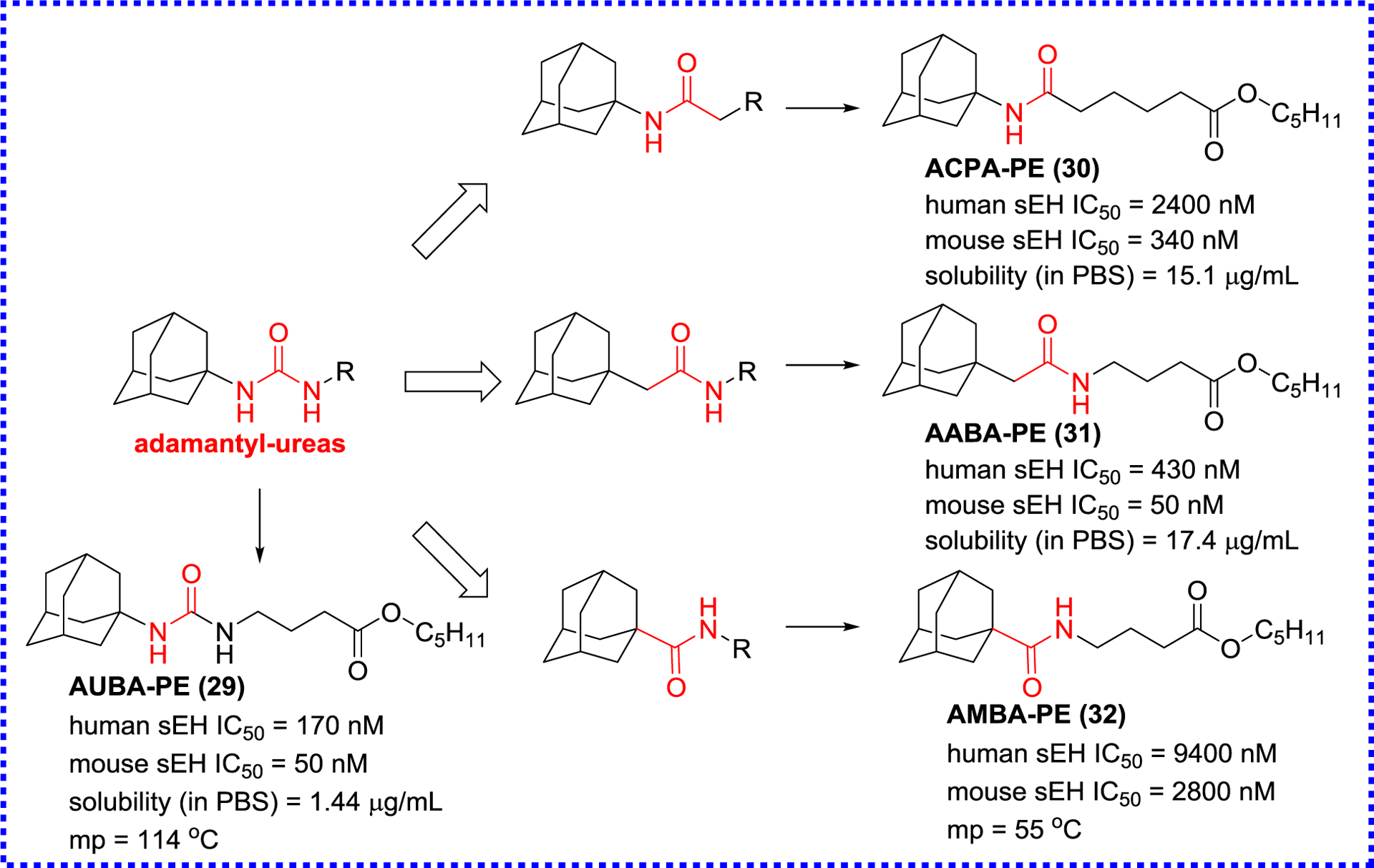
The modification of adamantyl-ureas to afford amide inhibitors
Eldrup and colleagues screened in-house compounds with drug-like characteristics for sEH inhibition, and found that N-(3,3-diphenyl-propyl)-nicotinamide (33, DPN) could be regarded as a new starting point for an optimization process.87 According to the X-ray crystal result of DPN (33) and sEH as shown in Figure 9, a library of amide inhibitors were designed and synthesized via the introduction of methanesulfonyl and fluorine at two benzene rings based on the structural core of DPN (33), yielding compounds 34–37 (Figure 9). They showed similar inhibitory potencies as DPN (33), whereas their metabolic stability was significantly improved in HLM and rat liver microsome.88 CFMB (36) as a potential sEH inhibitor was found to have a low drug-drug interaction potential with selectivity for CYP enzymes.87 In addition, DPN (33) was further optimized based on the structure of urea analog (38) with good metabolic stabilities (t1/2, 185 min in HLM; 105 min in RLM), resulting in the production of a series of nicotinamide analogs.89 Among them, nicotinamide analog (39) showed a low nanomolar inhibitory effect and a great metabolic stability, and had also acceptable (t1/2, 185 min in HLM and 105 min in RLM) plasma exposure.89
Figure 9.
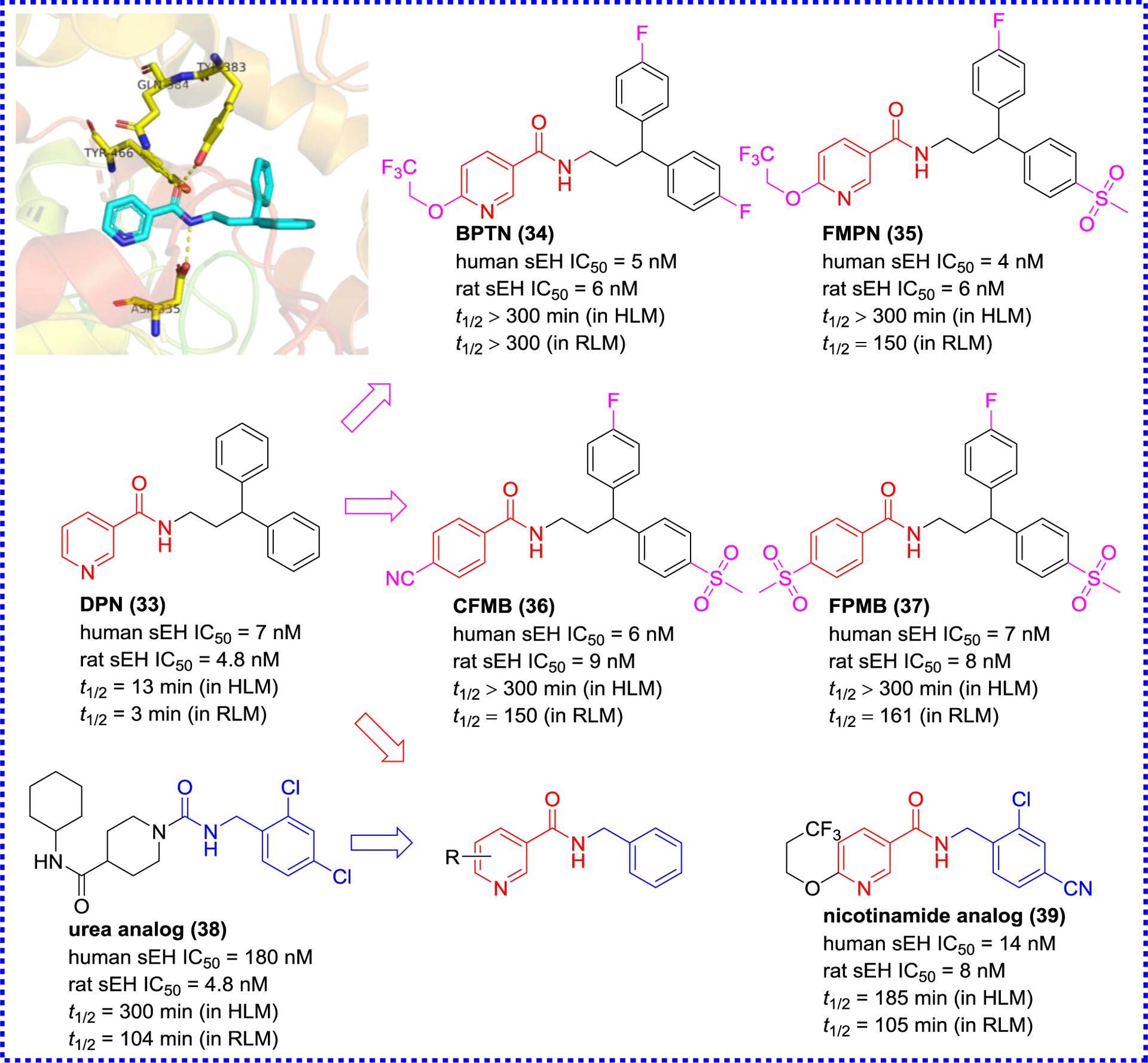
X-ray co-crystal structure of DPN (33) with sEH (PDB code 3I1Y) and its optimization.
Anandan et al. analyzed previous urea inhibitors, such as CPU,59 AUDA (4), and TPPU (19), and summarized the pharmacophore model comprising of a primary, secondary, and tertiary pharmacophores, and two linkers, therefore, they developed a series of urea, amide, sulfonamide, thiourea, sulfonyl urea, hydroxyamide, and ketoamide inhibitors based on the skeleton (II, Figure 10).90 A detailed analysis of SAR reveals that no less of inhibitory potency is observed when the N atom on the left side of the carbonyl group of urea is replaced by a C atom, whereas the decrease of inhibitory potency is present when the urea group is substituted with other groups, such as sulfonamide, thiourea, sulfonyl urea, hydroxyamide, and ketoamide. A methylene group occurs between adamantyl and hydroxyamide or ketoamide group, which will be to the benefit of inhibitory potency, except for amides (40 and 45). However, the inhibition potency is decreased when the adamantyl group is replaced by other groups, including cyclohexyl, 4-trifluoromethylphenyl, 4-chlorophenyl, and 4-trifluoromethoxyphenyl group.91
Figure 10.
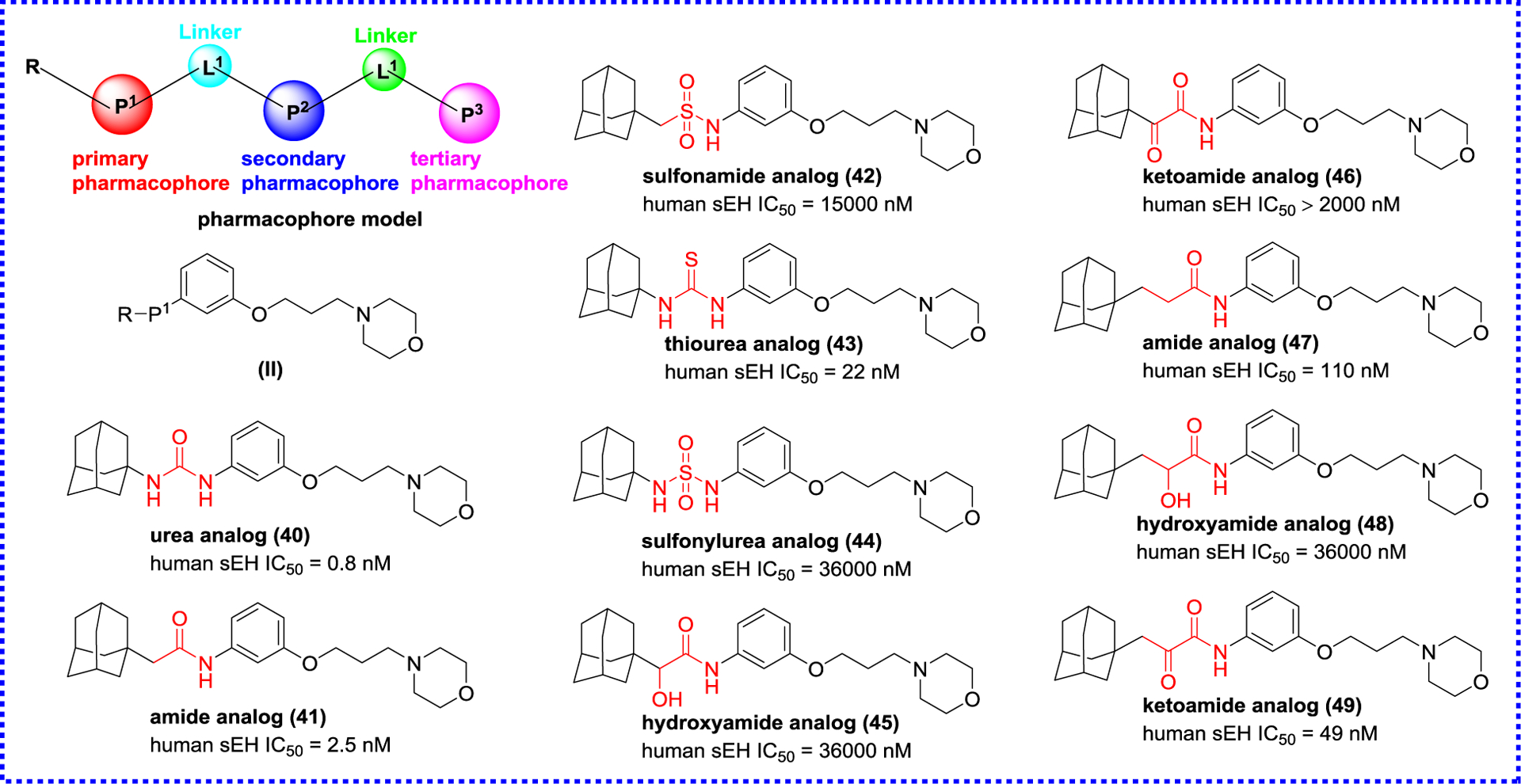
Modification based on the skeleton.
Based on these findings, the subsequent researches focused on the secondary pharmacophore to optimize the inhibition potency of amides.92–96 Firstly, an oxyoxalamide functional group was used to design and develop sEH inhibitors as the secondary pharmacophore according to the skeleton (III) by Kim and co-workers (Figure 11).94 Among the synthesized compounds, 50 (MMMO) and 51 (PMMO) show low nanomolar inhibitory activities as similar as that of AUDA (4), and their physical property has been greatly improved compared to AUDA (4), especially their solubility. The new skeleton (IV) by modifying the secondary pharmacophore was constructed using the phosphonate function in place of the oxyoxalamide group,95 to afford potent sEH inhibitors, such as 52 (DPDP) and 53 (DAPDP). In addition, Zavareh et al. also tried to develop the novel skeleton (V) via using an oxadiazole ring as a novel secondary pharmacophore,93, 96 yielding a series of potent oxadiazole-base amide inhibitors, such as oxadiazole analogs 54 (IC50 = 0.91 nM) and 55 (IC50 = 0.83 nM). Molecular docking indicates that amino acid residues Asp335, Tyr383, and Tyr466 play a crucial role in the interaction with sEH, and Val498, His524, and Trp525 also form additional hydrogen bond interaction with an oxadiazole group. These findings have great importance in the design of sEH inhibitors.96
Figure 11.
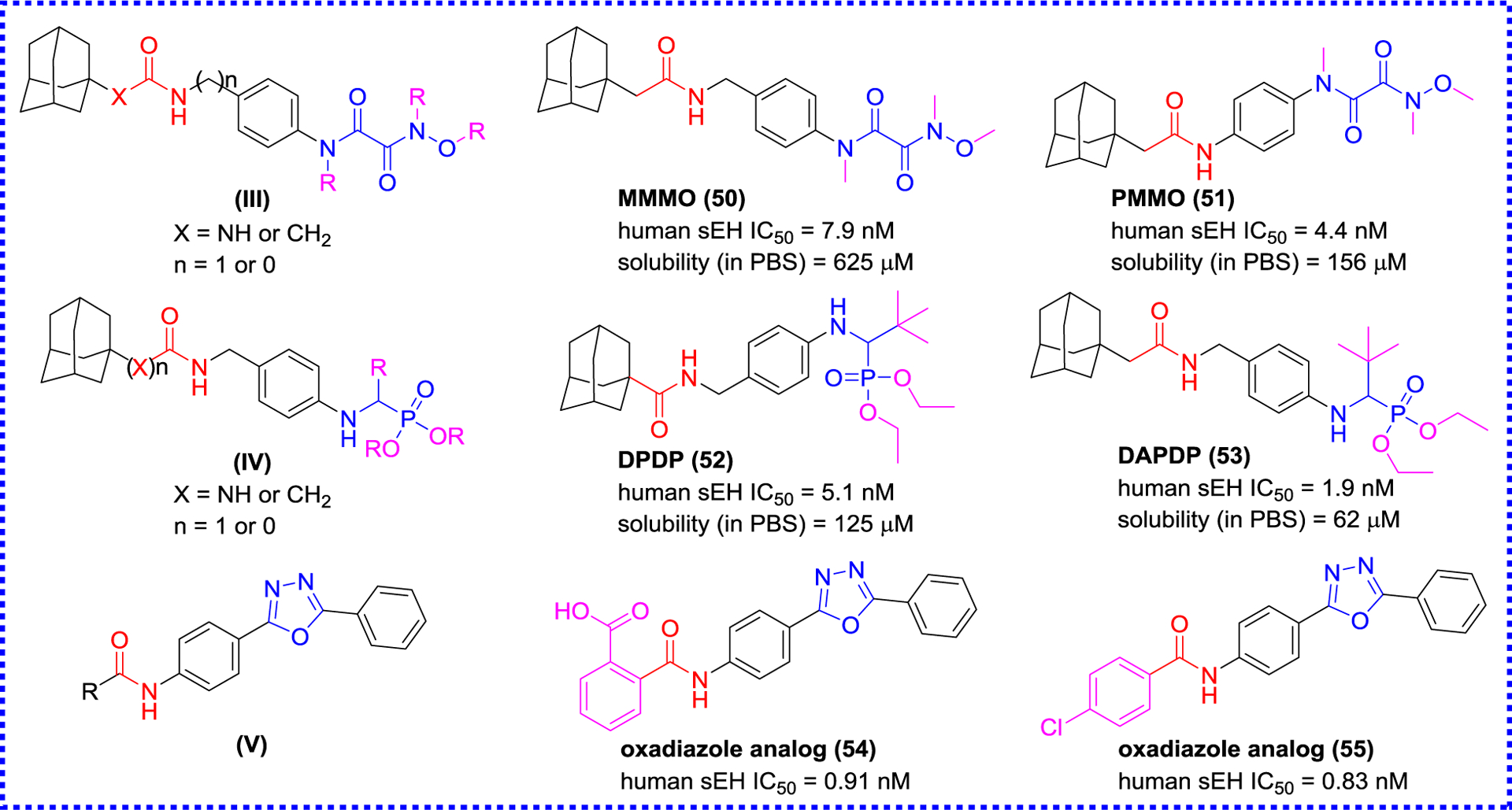
Modification of the secondary pharmacophore.
Kim and co-workers carried out the structural modification on both sides of the amide pharmacophore of 1,3-bicycloalkyl substituted amides to improve the inhibitory effect on sEH.97 According to the SAR result (Figure 12), potent sEH inhibitors 56 (TPNC) and 57 (MTCMB) are obtained with reasonable physical properties. For example, mp of MTCMB (57) is reduced (115 °C) and the water solubility (125 μM) is 2-fold higher than that of AUDA (4). This shows that amide inhibitors can achieve similar inhibitory potency as urea inhibitors while having better physical properties. In addition, this study opened the door to numerous possibilities for the design of potent amide inhibitors, especially through combinatorial chemistry.97
Figure 12.
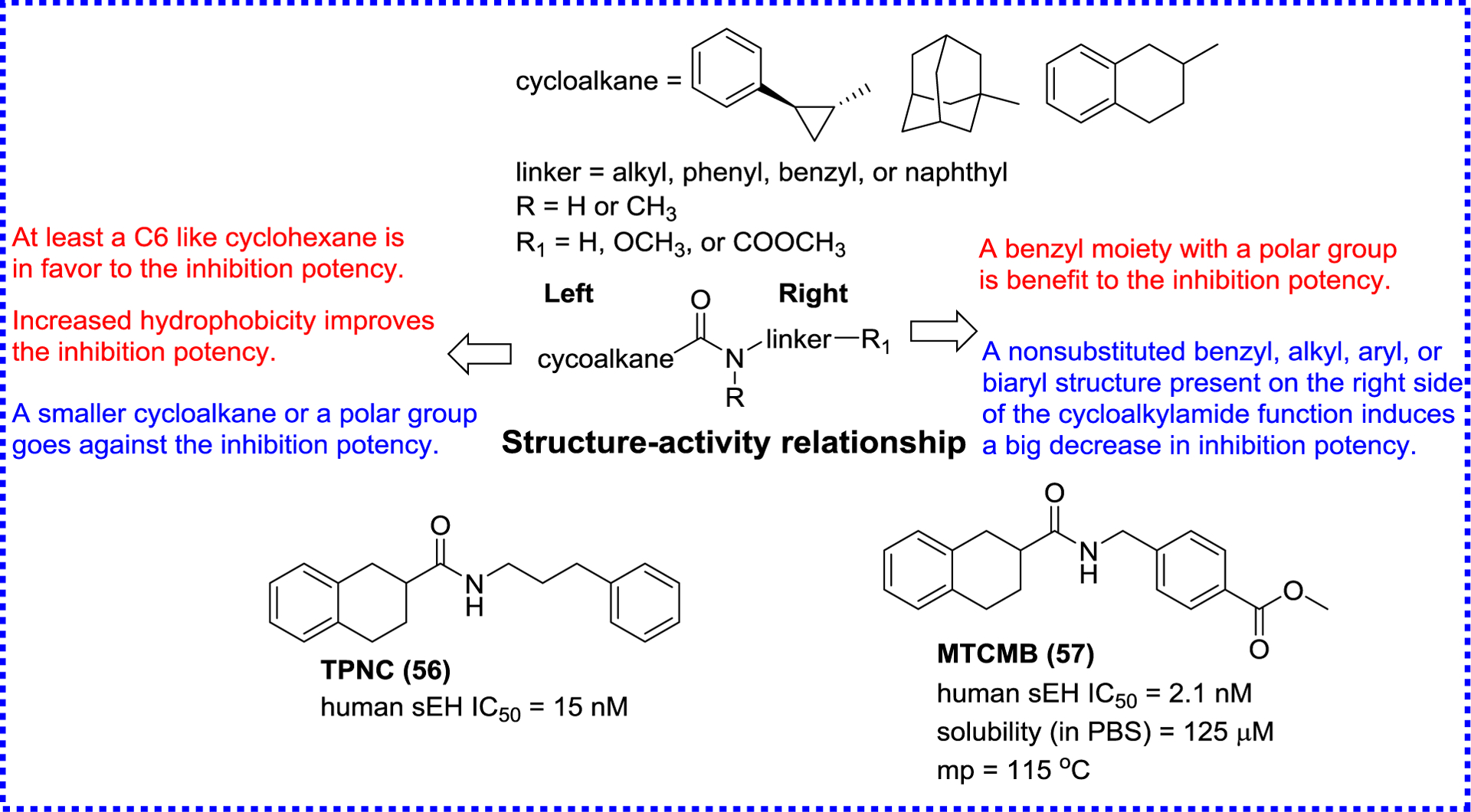
SAR of amide inhibitors summarized by Kim et al.
For finding new structures, Landry et al. tested a library of 130,000 chemicals, provided by the National Institutes of Health Roadmap project, for sEH inhibition. They obtained a lead compound 58 (Figure 13) with nanomolar potency that contains a sulfonyl isonipecotamide that is resembling APAU (6). Further optimization of 58 resulted in the production of three subnanomolar inhibitors (59–61).98, 99 Xing et al. designed a combinatorial library of benzoxazole derivatives, and screened the potential sEH inhibitors via a virtual screening (Figure 14).100 The X-ray crystal structure (PDB code 3PDC) of the human sEH with compound 62 revealed unique interactions between the enzyme and the benzoxazole group. In addition to the classic hydrogen bonds between the amide group and active site residues (Asp333, Tyr381, and Tyr465), the oxygen of the benzoxazole ring forms a hydrogen bond interaction with His523, one of the catalytic triad Asp333-Asp495-His523 (Figure 14). These additional hydrogen bonds may account for the enhanced potency of the amide-based inhibitors containing a benzoxazolyl group.100
Figure 13.
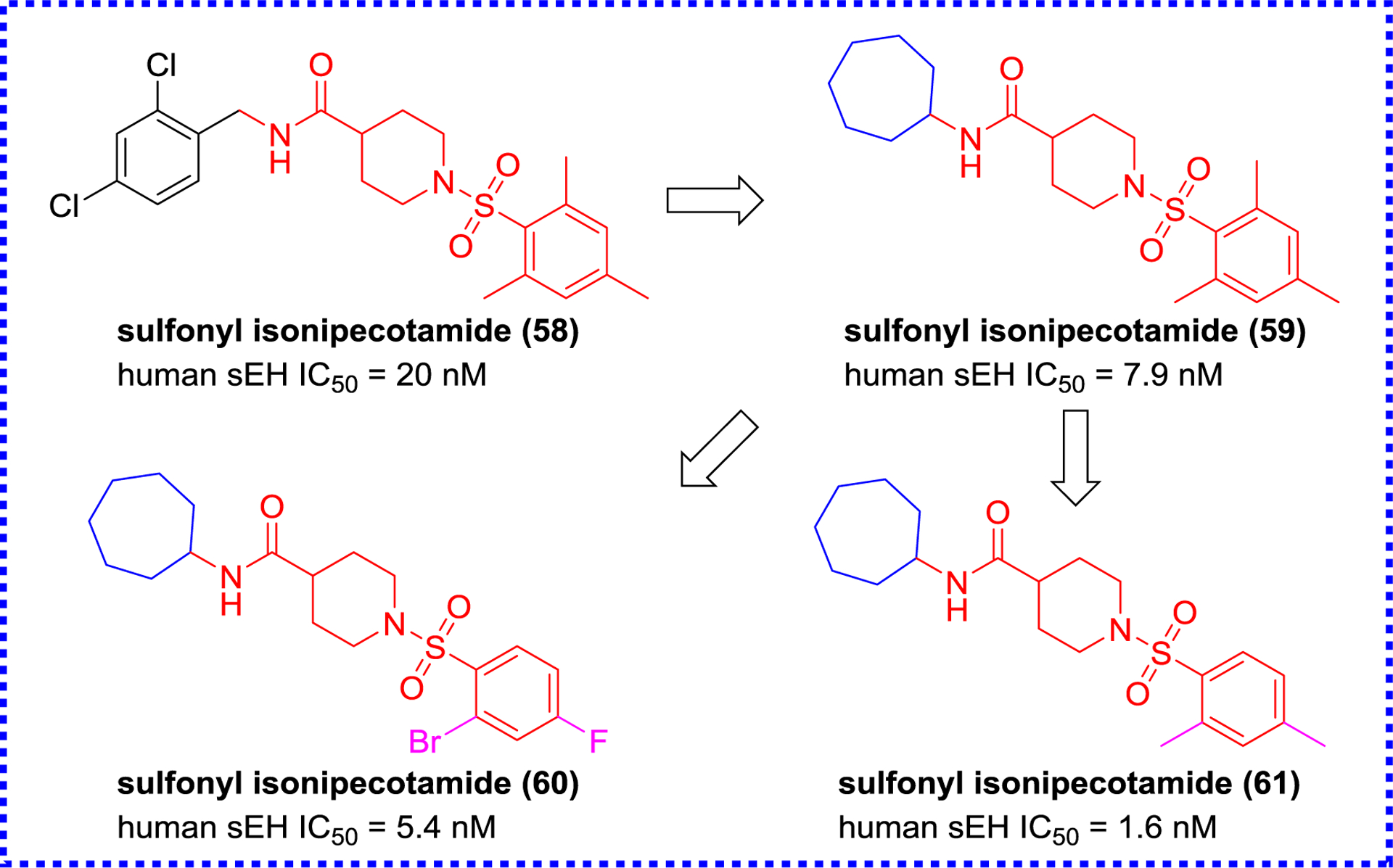
Chemical structures of sulfonyl isonipecotamides.
Figure 14.
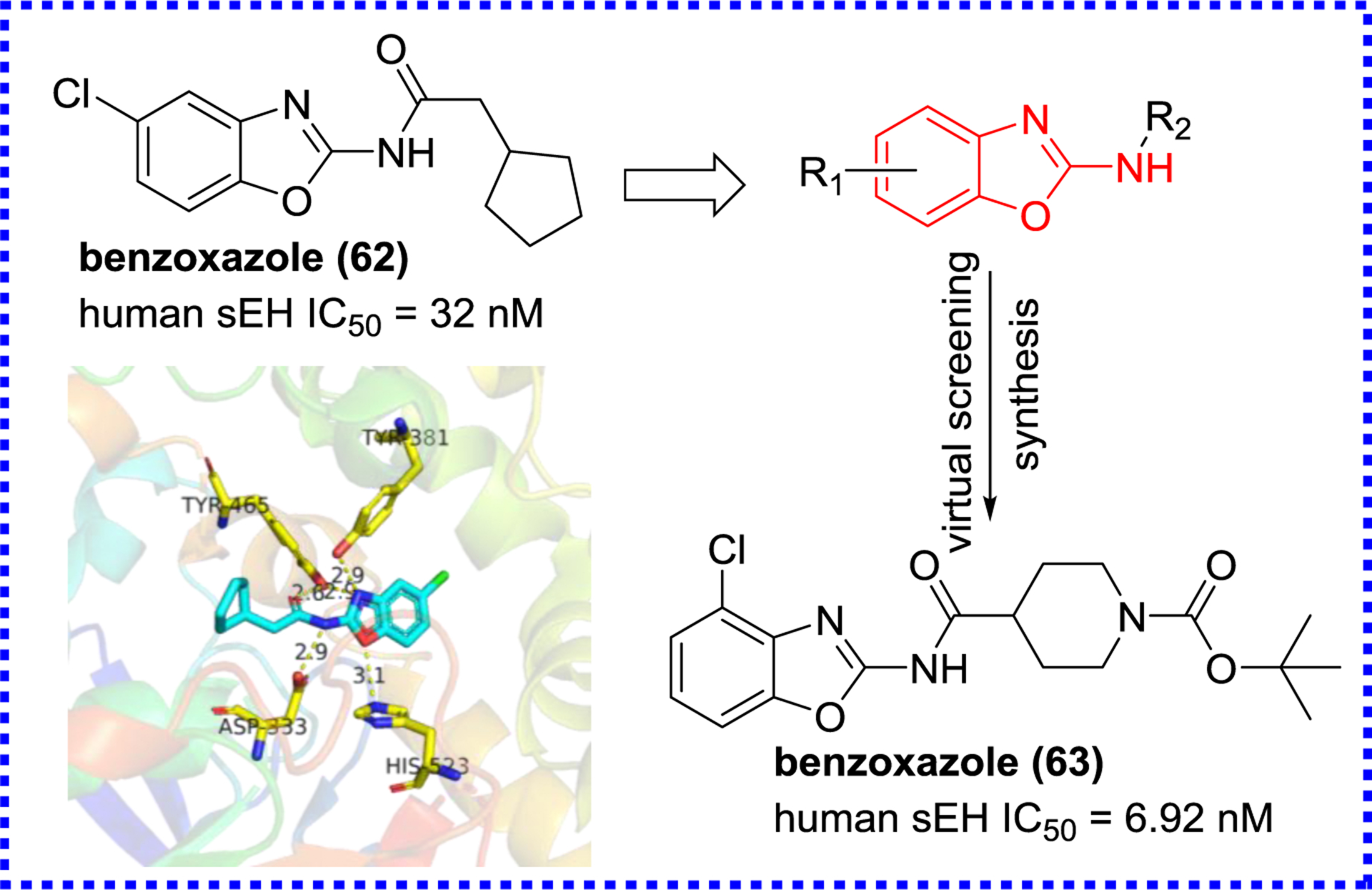
X-ray crystal structure of sEH complexed with the benzoxazole (62) (PDB code 3PDC) and its optimization.
Marino and co-workers used encoded library technology to discover a sEH inhibitor (64) featuring a piperidinecarboxamide skeleton via high throughput screening (HTS, Figure 15),101, 102 and optimized this skeleton to yield compounds 65 and 66 displaying low nanomolar inhibitory potencies.103 The X-ray crystal structure of MTTP (65) with sEH (PDB code 4JNC) showed that 65 formed H-bonds with Asp335, Tyr383, and Tyr466 at the active site (Figure 15). The further optimization of this skeleton led to the production of a low picomolar sEH inhibitor GSK2256294 (67). The result of in vivo study indicated that GSK2256294 (67) could dose-dependent reduce pulmonary leukocyte and keratinocyte chemoattractant levels in cigarette smoke-induced mice, suggesting that GSK2256294 (67) would be an appropriate agent for the treatment of chronic obstructive pulmonary and cardiovascular diseases.59 This compound has been tested in clinical trials.
Figure 15.
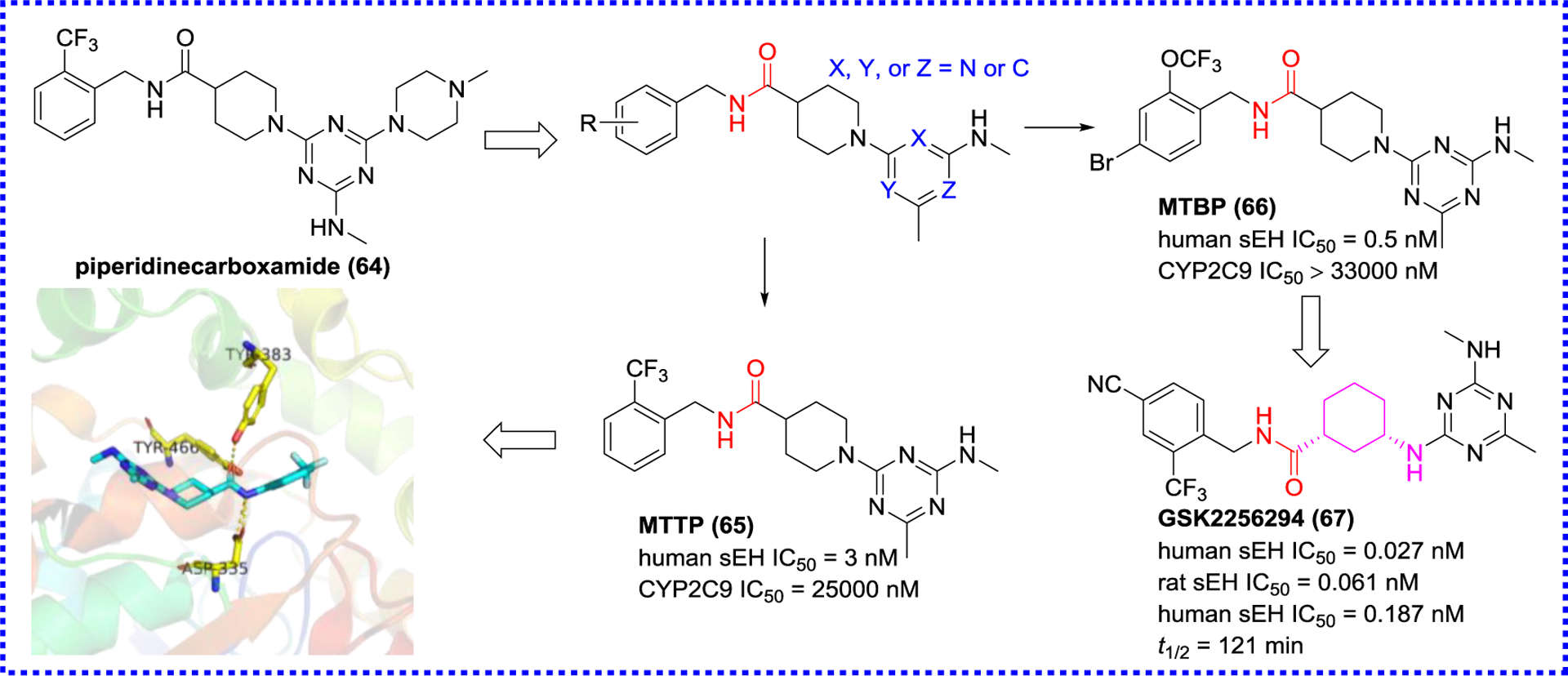
Modification based on the structure of the amide (64) and co-crystal structure of MTTP (65) and sEH (PDB code 4JNC)
2.2. Multi-target inhibitors
Highly effective drugs with a single biological target are the mainstream of drug research and development.104 However, they also have some limitations. By targeting a single enzyme or receptor only, a drug could lack efficacy on complex diseases, and may cause additional adverse reactions when used in combination with other drugs.104, 105 In order to solve these problems, research is focusing now on multi-target drugs with higher safety and effectiveness.105, 106 In recent years, several multi-target drugs have shown excellent therapeutic ability in meddling with the AA cascade. The sEH plays a crucial role in the AA cascade and participates in the pathological process of numerous diseases. In addition, the effectiveness of sEH inhibitors has been verified in vivo.104 By combining the inhibition of sEH with the inhibition of another actor of the AA cascade, such as COX-2, 5-LOX, and FLAP, one could expect a greater effect for the treatment of complex diseases, such as neuro-inflammation or cancer.107, 108
2.2.1. sEH/COX-2 dual inhibitors
COX-2 is an enzyme in which expression is induced by the stimulation of inflammation, hormones, or growth factors.109 COX-2 is highly expressed in inflammation and proliferative diseases, such as various cancers, therefore, it is a target to develop anti-inflammatory and anti-tumor agents.104 A recent study has demonstrated that co-administration of EETs and COX inhibitors exerts a better effect than the single administration of COX inhibitors to reduce LPS-induced pain and hypotension.79, 84 Accordingly, a series of sEH/COX-2 dual inhibitors based on the structural skeletons of celecoxib and t-AUCB (8, Figure 16), were designed and found that 68 (PTUPB) displayed potent inhibitory activities against both sEH and COX-2. Molecular stimulation shows that PTUPB (68) forms hydrogen bond interactions with amino acid residues Asp333, Tyr381, and Tyr465 of human sEH, and His90 and Tyr335 of mouse COX-2. In a nociceptive assay, PTUPB (68) enhances in vivo anti-allodynia effects compared to both the individual and combination therapies of celecoxib and t-AUCB (8).107, 110
Figure 16.
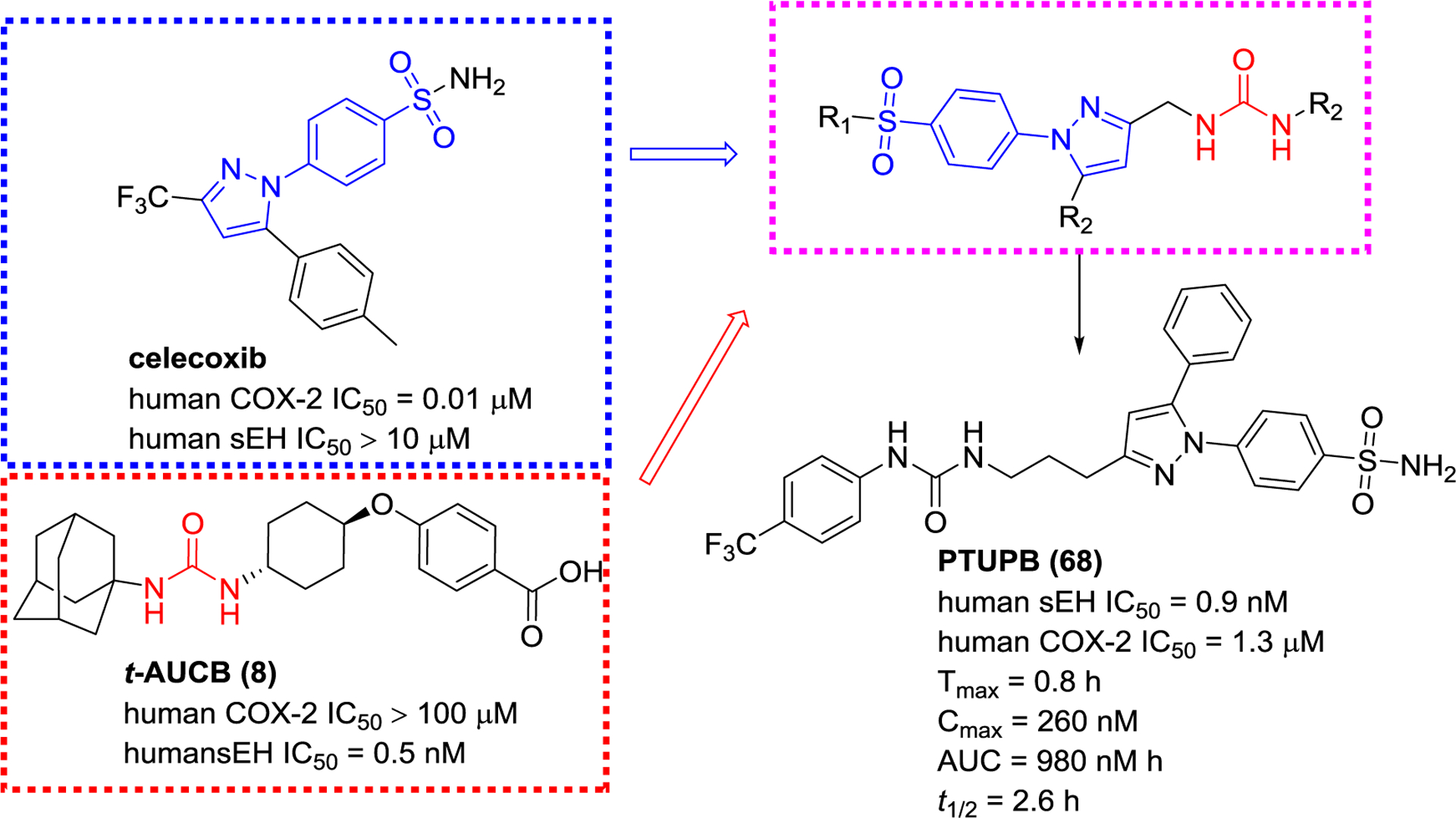
The design principle of an sEH/COX-2 dual inhibitor PTUPB (68)
PTUPB (68) was found to be beneficial to sepsis, non-alcoholic fatty liver disease, pulmonary fibrosis, and cancer.33, 107, 111–116 PTUPB (68) inhibits tumor growth and metastasis by suppressing endothelial cell proliferation, and it also improves survival rate, delays the onset of debris-stimulated ovarian tumor growth, and reduces ascites.107, 115, 116 In addition to the treatment of cancers, PTUPB (68) suppresses the activation of NLRP3 inflammasome,111 allowing in improving the survival rate, reducing the clinical scores and systemic inflammatory response, and alleviating the multiple-organ injury in sepsis mice.112 Therefore, sEH and COX-2 dual inhibition may be an approach to treat inflammation and cancer diseases.
2.2.2. sEH/5-LOX dual inhibitors
5-LOX as a dioxygenase, play a catalytic role in two steps of the biosynthesis of LTs which progress regulate the innate immune response and play a pathophysiological role in chronic inflammatory diseases, such as asthma and atherosclerosis.104 To date, a 5-LOX inhibitor zileuton is used in the clinic for the treatment of asthma with restrictions.117 Co-administration of an sEH inhibitor t-AUCB (8) with a 5-LOX inhibitor showed a significant enhancement of the anti-inflammatory response.109 Therefore, the sEH/5-LOX dual inhibitor has caught the attention of medicinal chemists. Moser and co-workers applied the pharmacophores of sEH and 5-LOX for screening 37,429 compounds in silico to afford a poor sEH/5-LOX dual inhibitor (sEH IC50 = 3.5 μM; 5-LOX IC50 = 36 μM), which, nevertheless, led to the new era of discovery dual inhibitors.118 Based on the key pharmacophores, an imidazo[1,2-a] pyridine motif of a 5-LOX inhibitor EP6 (5-LOX IC50 = 59 nM) and a urea group of AUDA (4), a library of compounds were designed and synthesized by Meirer et al.109 The SAR indicated that two pharmacophores required an n-propyl linker to maintain both sEH and 5-LOX inhibition. Among them, compound 69 showed high inhibitory potency against both 5-LOX and sEH (Figure 17). Although CMMPPU (69) exhibited high potency in vitro, it was not stability in plasma.109 Subsequently, Meirer et al. designed KM55 (70), which showed a strong anti-inflammatory effect in vitro.117
Figure 17.
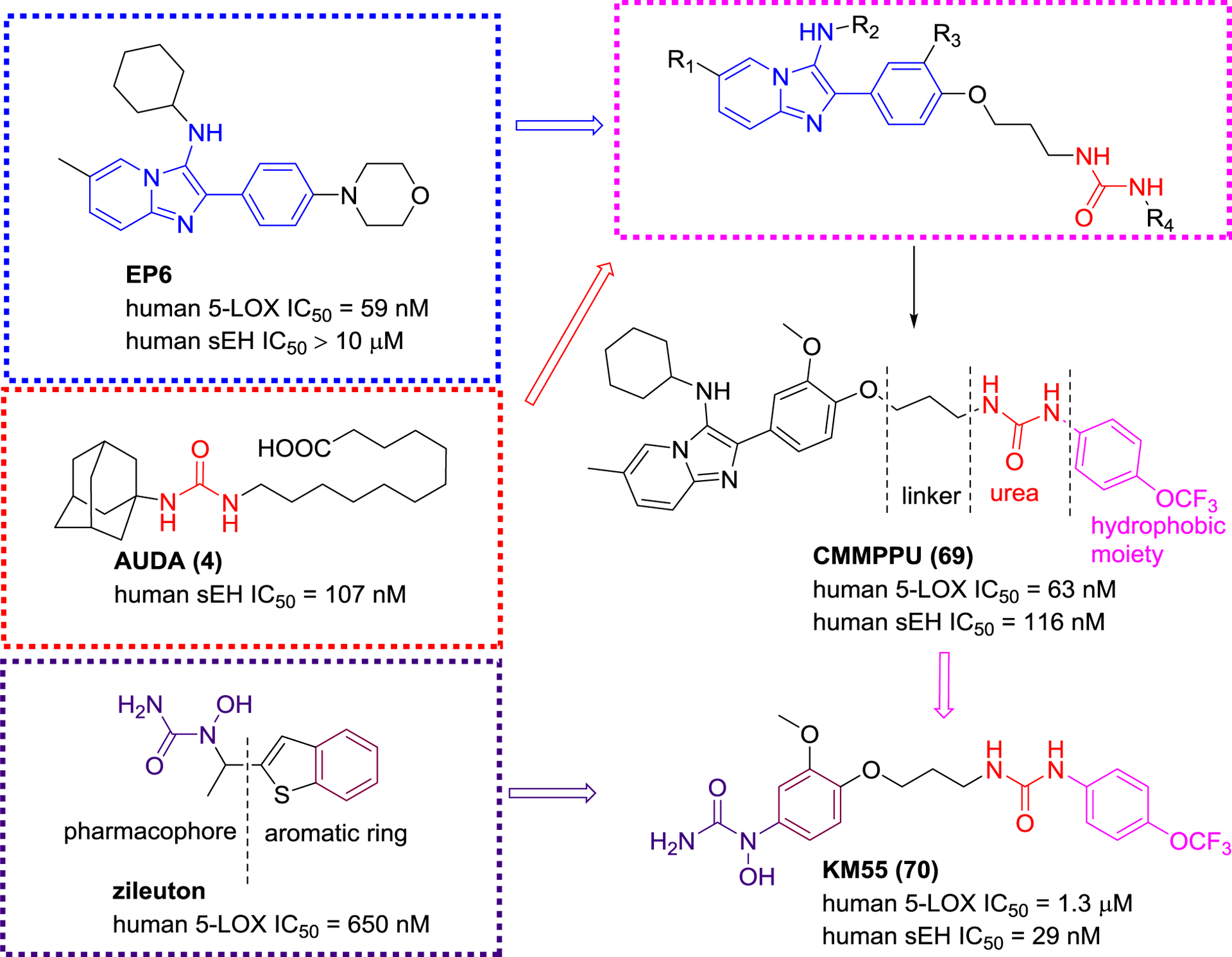
sEH/5-LOX dual inhibitors that linked two pharmacophores for sEH and 5-LOX.
Achenbach et al. used saturation transfer difference (STD)-NMR and activity-based assays (Figure 18), and found that a compound featuring an aminothiazole skeleton had better inhibitory activities towards sEH (IC50 = 170 nM) and 5-LOX (IC50 = 30 nM).119 Nandha et al. designed and developed a potential sEH/5-LOX dual inhibitor (FFBMB, 71) based on the structure of the 5-LOX inhibitor RWJ-63556 (Figure 19), a benzimidazole analog, and the sEH inhibitor t-TUCB. The in vivo study confirmed that FFBMB (71) possessed significant inhibition of carrageenan-induced rat paw edema.120
Figure 18.
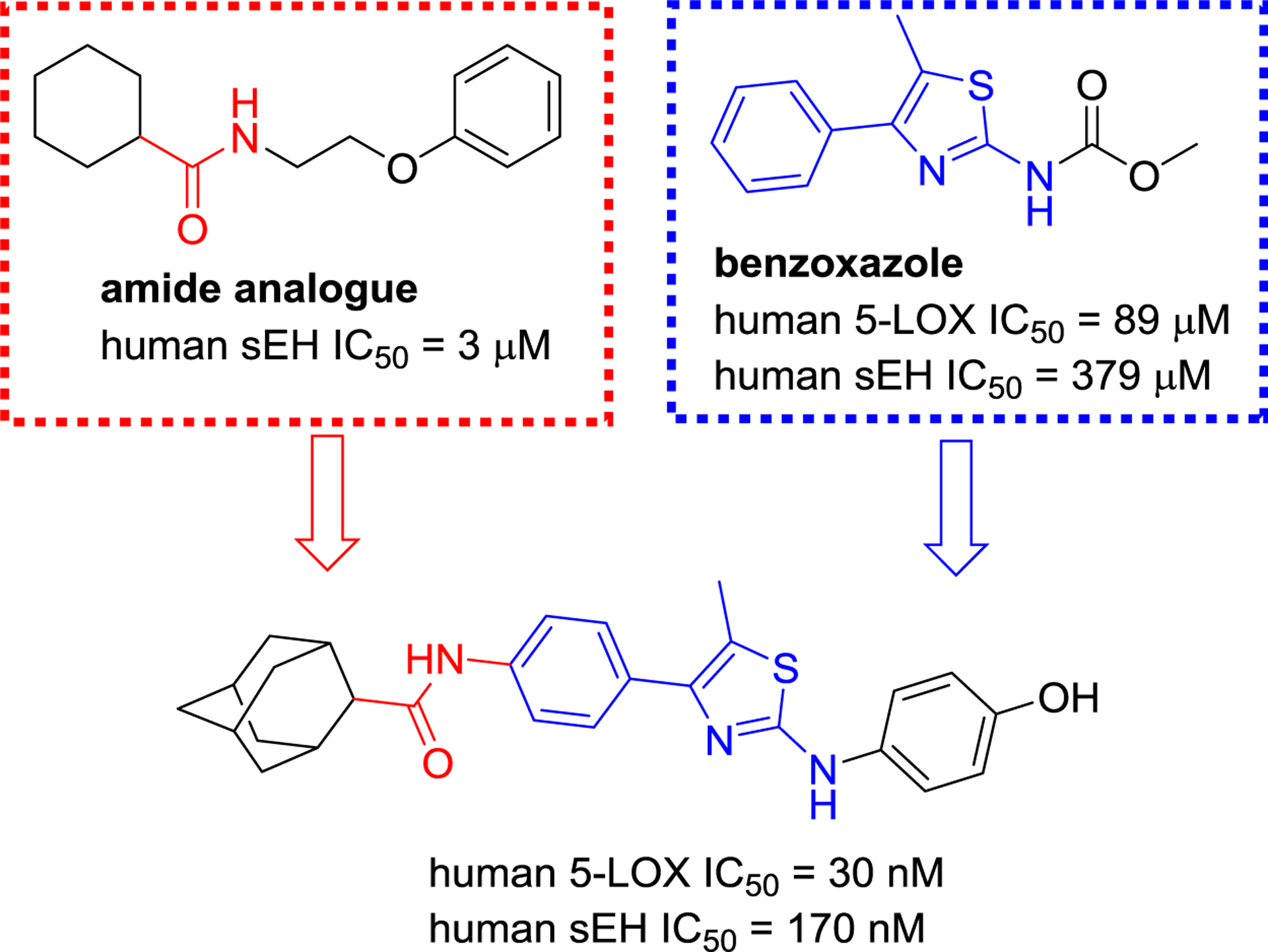
A sEH/5-LOX dual inhibitor with an amide and an aminothiazole moiety.
Figure 19.
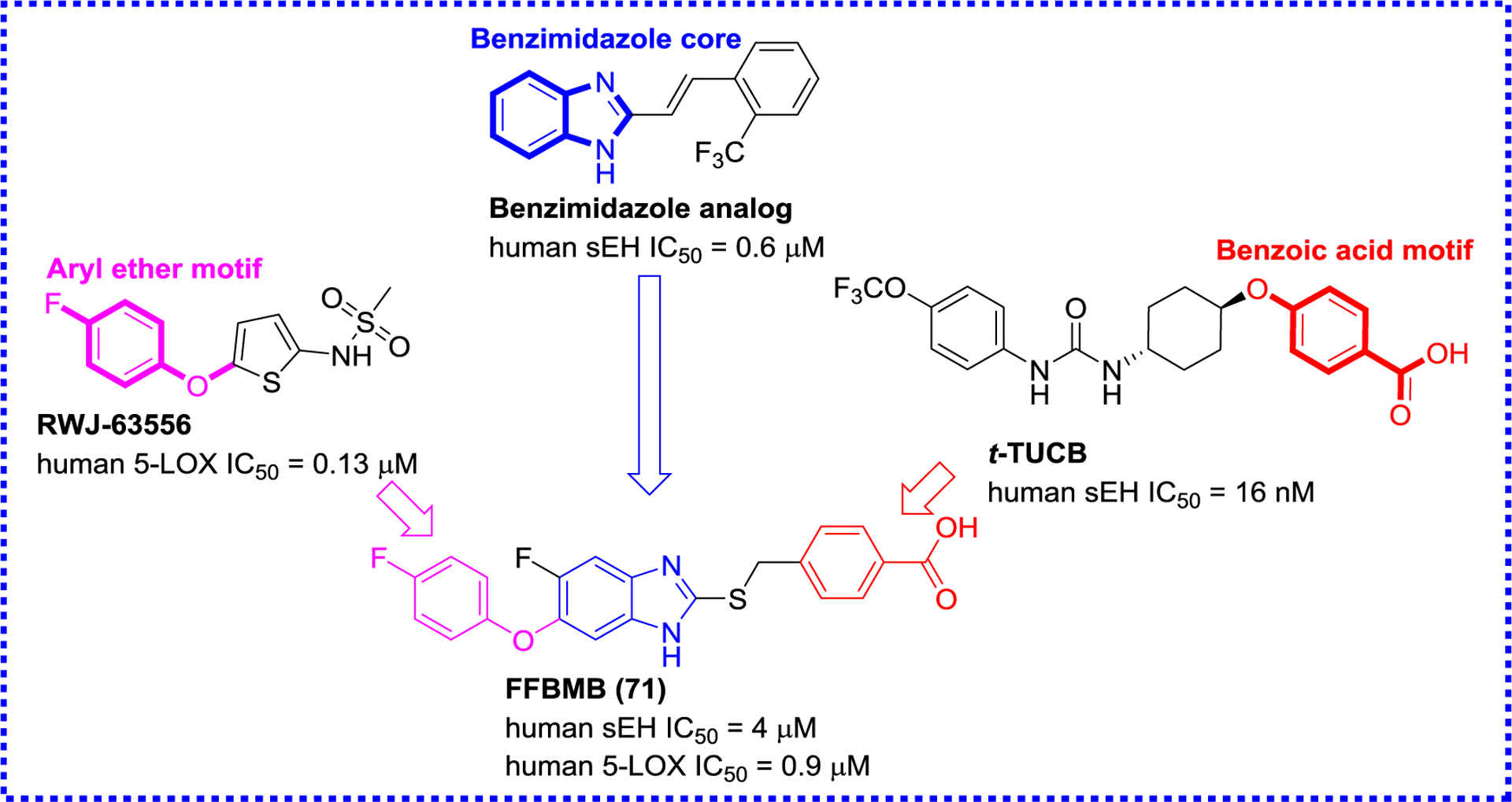
A sEH/5-LOX dual inhibitor FFBMB (71) based on an aryl ether and a benzoic acid motif and a benzimidazole core.
2.2.3. sEH/FLAP dual inhibitors
5-LOX-activating protein (FLAP) is a nuclear membrane-anchored protein responsible for transferring AA to 5-LOX that produces pro-inflammatory LTs. The inhibition of FLAP by FLAP inhibitors or genetic knockout can abolish the production of pro-inflammatory LTs. Meanwhile, a FLAP inhibitor, GSK2190915, has completed phase II trials for the treatment of asthma.121, 122 Recent studies have indicated that co-administration of a sEH inhibitor t-AUCB (8) and MK886, a FLAP inhibitor, enhances anti-inflammatory activities in a murine model, yielding interest in sEH/FLAP dual inhibitors.123 Temml et al. used a pharmacophore-based virtual screening to afford a first sEH/FLAP dual inhibitor diflapolin (72) (Figure 20), which did not affect other enzymes of the AA cascade, suggesting that (72) is very selective for sEH and FLAP.124 Investigation of its in vivo biological effects indicates that diflapolin (72) ameliorates vascular permeability, inhibits the formation of pro-inflammatory factors cysteinyl-LTs and leukotriene B4 (LTB4), suppresses neutrophil infiltration in the zymosan-induced peritonitis mouse model.124 Subsequently, Vieider and colleagues designed a library of diflapolin derivatives, leading to BPDU (73) (Figure 20), a more potent sEH/FLAP dual inhibitor.125
Figure 20.
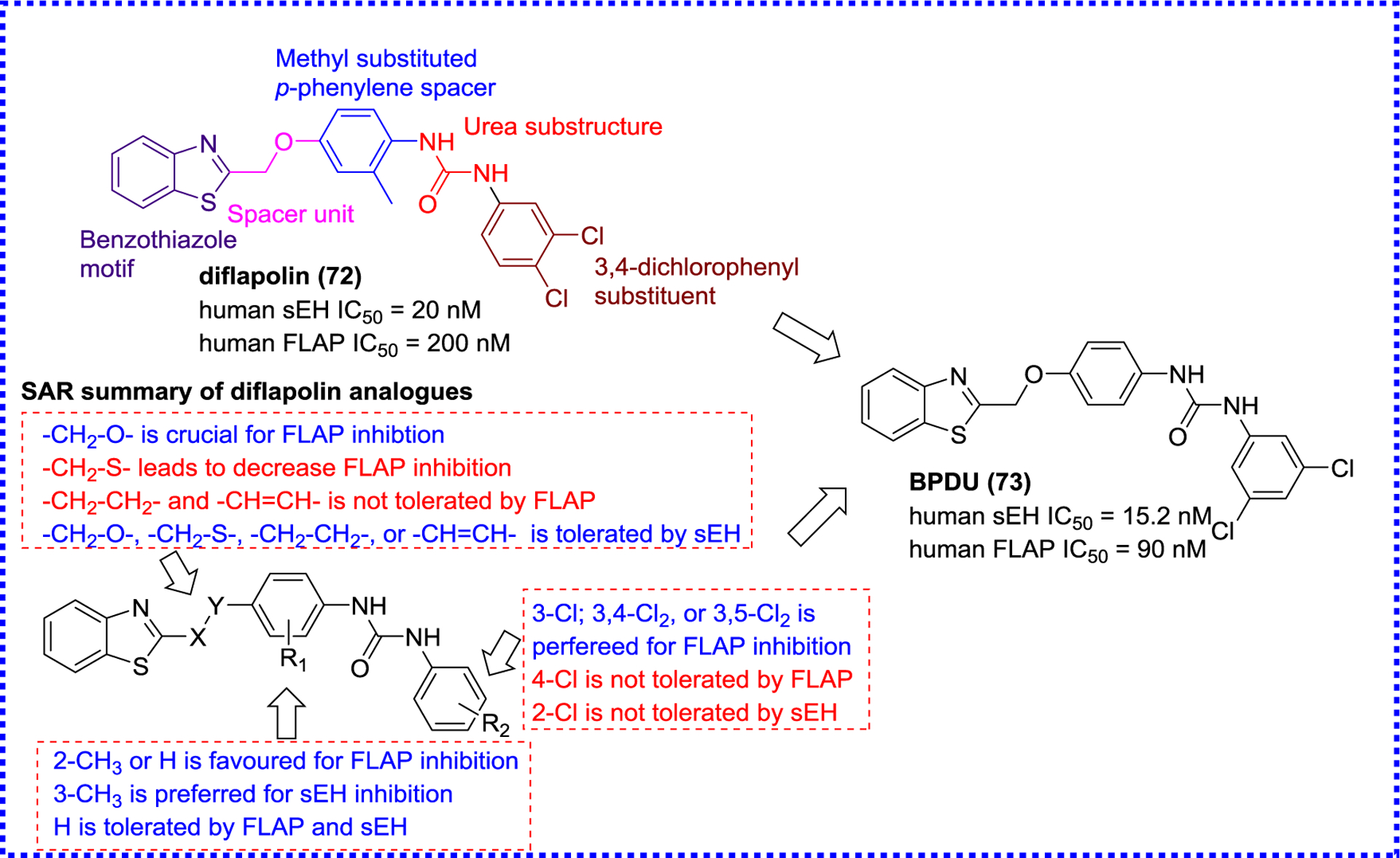
sEH/FLAP dual inhibitors diflapolin analogues.
2.2.4. sEH/FAAH dual inhibitors
Fatty acid amide hydrolase (FAAH) is a membrane-bound serine hydrolase responsible for the deactivating metabolism of endogenous cannabinoids, such as oleamide, anandamide, and myristic amide.104 Both EpFAs and endocannabinoids exert anti-inflammatory effects, therefore, elevating their concentrations by inhibiting both sEH and FAAH should be a good method to treat inflammation and neuropathic pain.104 Indeed, co-administration of a FAAH inhibitor and a sEH inhibitor elicited a synergistic analgesic response in acute inflammatory and chronic pain mice,126 and thus, sEH/FAAH dual inhibitors had attracted more attention. Interestingly, because t-TUCB, a potent sEH inhibitor (IC50 = 0.4 nM), has a N-(3-trifluoromethoxyphenyl) moiety, which is a good leaving group for FAAH, it has also reasonable inhibitory potency against FAAH (IC50 = 260 nM).105 Further chemical modifications resulted in a 6-fold increase potency against FAAH while retaining t-TUCB intrinsic sEH inhibitory effect (Figure 21).105
Figure 21.
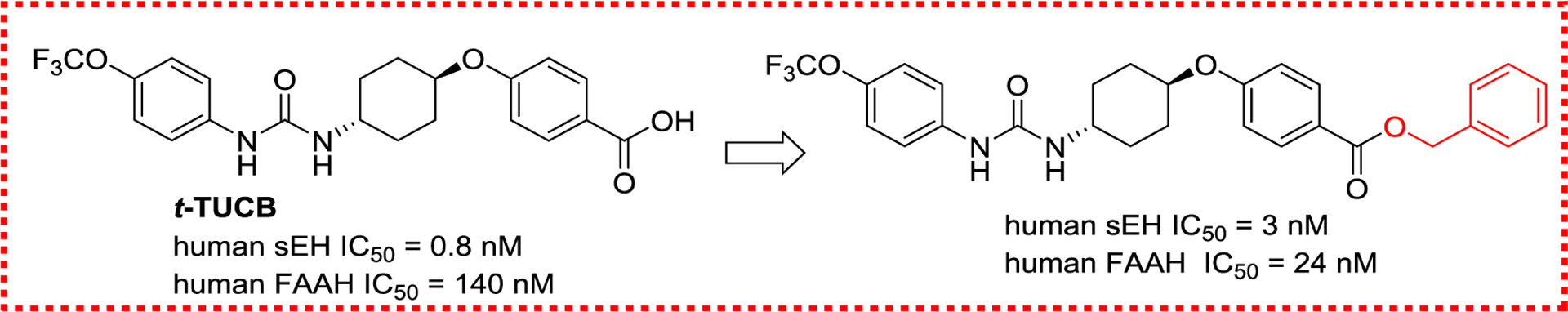
A sEH/FAAH dual inhibitor derived from t-TUCB.
Separately, a series of sEH/FAAH dual inhibitors were developed based on the cores of a FAAH inhibitor PF-3845 and sEH inhibitor TPPU (19, Figure 22). Kodani et al. found that TPPPC (74) is a low nanomolar inhibitor for both FAAH and sEH inhibitions (Figure 22). Further investigation suggested that TPPPC (74) possesses a good target selectivity, PK property (AUC = 1200 nM*h; Cmax = 98 nM; t1/2 = 4.9 h in mice), and in vivo target engagement.127
Figure 22.
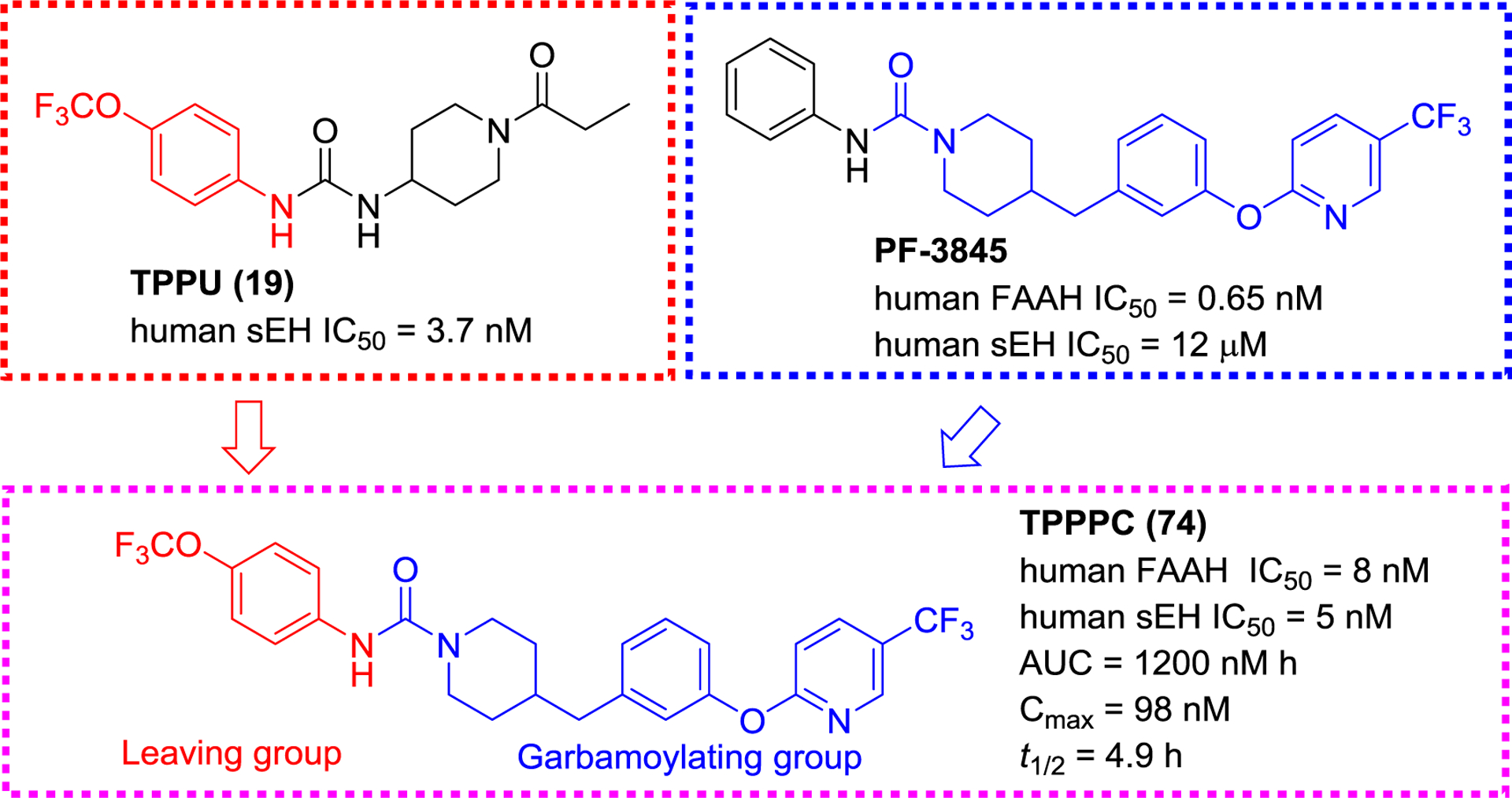
A selective sEH/FAAH dual inhibitor TPPPC (74) derived from TPPU (19) and PF-3845.
2.2.5. sEH/PPARγ dual modulators
PPARγ is a nuclear receptor that can be activated by EETs allowing the inhibition of NF-κB pathway and anti-inflammatory effects.128 Some studies have indicated that the sEH expression level is increased and the PPARγ transcriptional activity is decreased in obese patients, suggesting that simultaneous regulation of sEH and PPARγ has a therapeutic potential for metabolic diseases.128 Recently, Imig and co-workers co-administrated thiazolidinedione (TZD), a PPARγ agonist, and t-AUCB(8), a sEH inhibitor, to spontaneously hypertensive obese rats, resulting in protective effects towards vascular function and kidney,129 supporting the idea of sEH/PPAR dual modulators.104 Proschak et al. designed a library of sEH/PPAR dual modulators based on the sEH pharmacophore 4-(trifluoromethoxy)phenyl ureas and PPAR modulator structures (Figure 23), and obtained compounds, such as TPUBPC (75) and AUBPC (76), that display inhibition on sEH and activation on PPARα/γ (Figure 24).108
Figure 23.
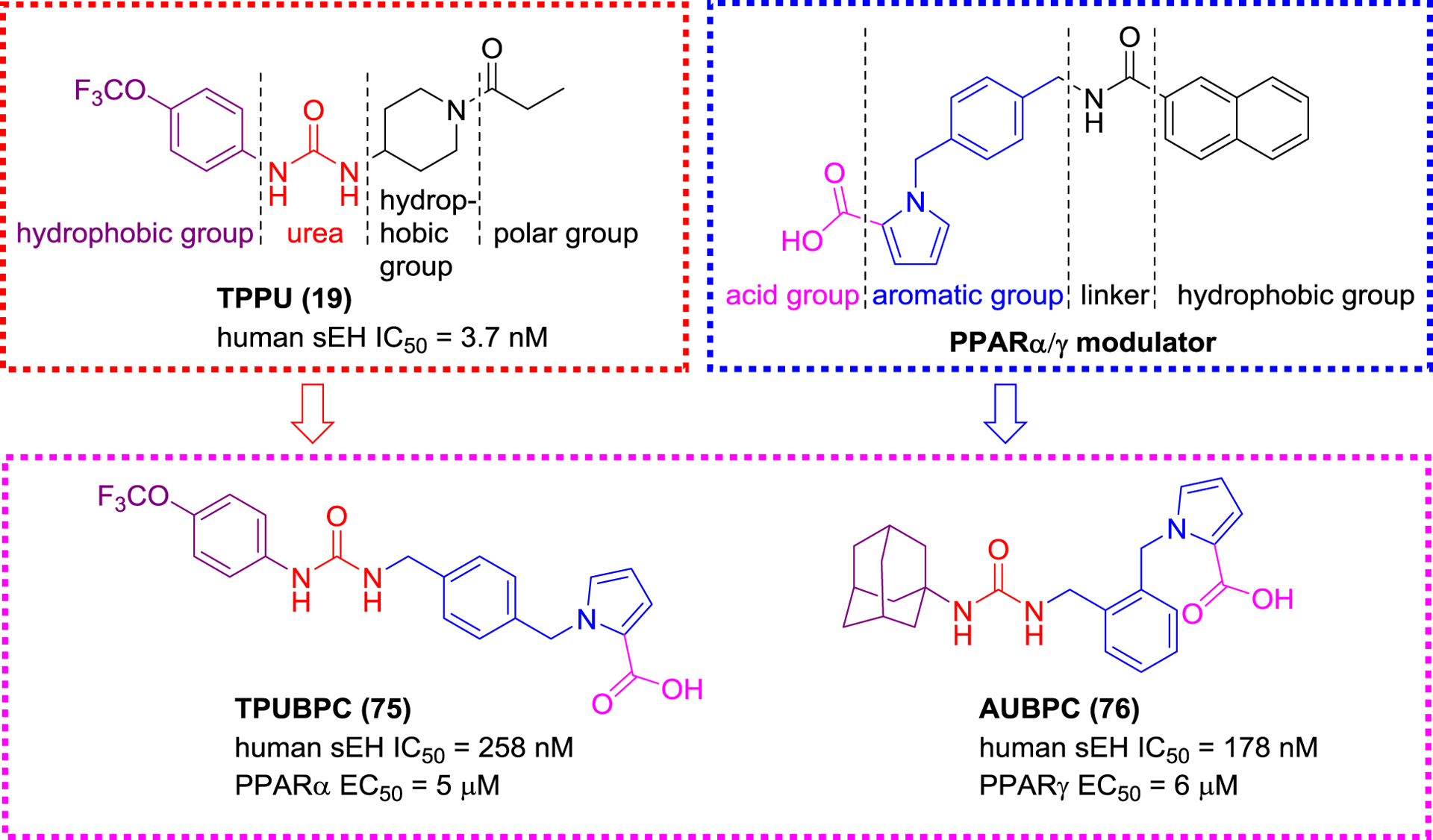
Selective sEH/PPAR dual modulators TPUBPC (75) and AUBPC (76).
Figure 24.
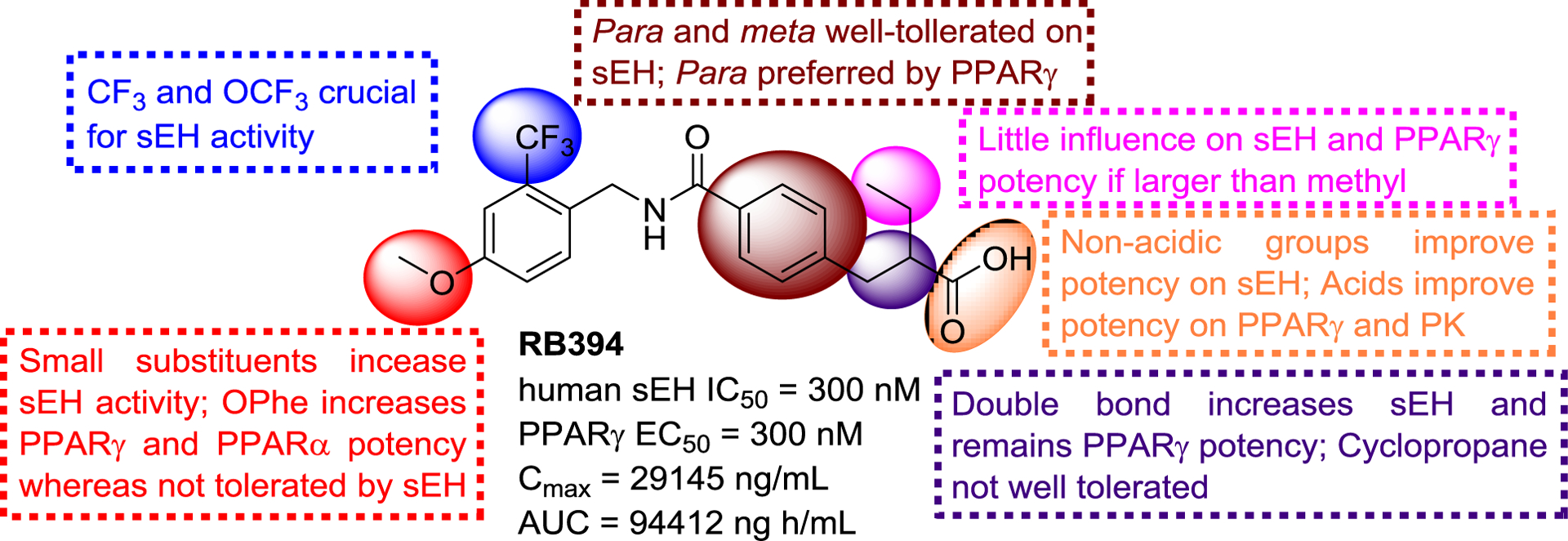
SAR of N-benzylbenzamides with sEH and PPARγ.
Subsequently, Blöcher et al. also designed a series of sEH/PPAR dual modulators with a benzylamide structure, a merged pharmacophore for sEH and PPARγ, on the basis of the structure of the PPARγ agonist GSK1997132B, such as RB394 (Figure 24) with good potencies against sEH and PPARγ. In addition to its good PK and pharmacodynamic profile,130 RB394 could also ameliorate the development of hypertension, insulin resistance, and hyperlipidaemia in obese spontaneously hypertensive and obese diabetic Zucker fatty/spontaneously hypertensive heart failure F1 hybrid rats, which suggested that it could be regarded as a potential agent to treat metabolic syndromes.131
Afterwards Schierle et al. found that zafirlukast, an antagonist of cysteinyl leukotriene receptor 1, also possesses an agonistic effect towards PPARγ and inhibitory effect towards sEH (Figure 25). Further structural optimization led to the production of compound 77. Its potencies on sEH and PPARγ was increased by 46.5- and 8.1- fold compared to zafirlukast, respectively, and it also exerted an anti-inflammatory potency in the zymosan-induced paw edema.132
Figure 25.
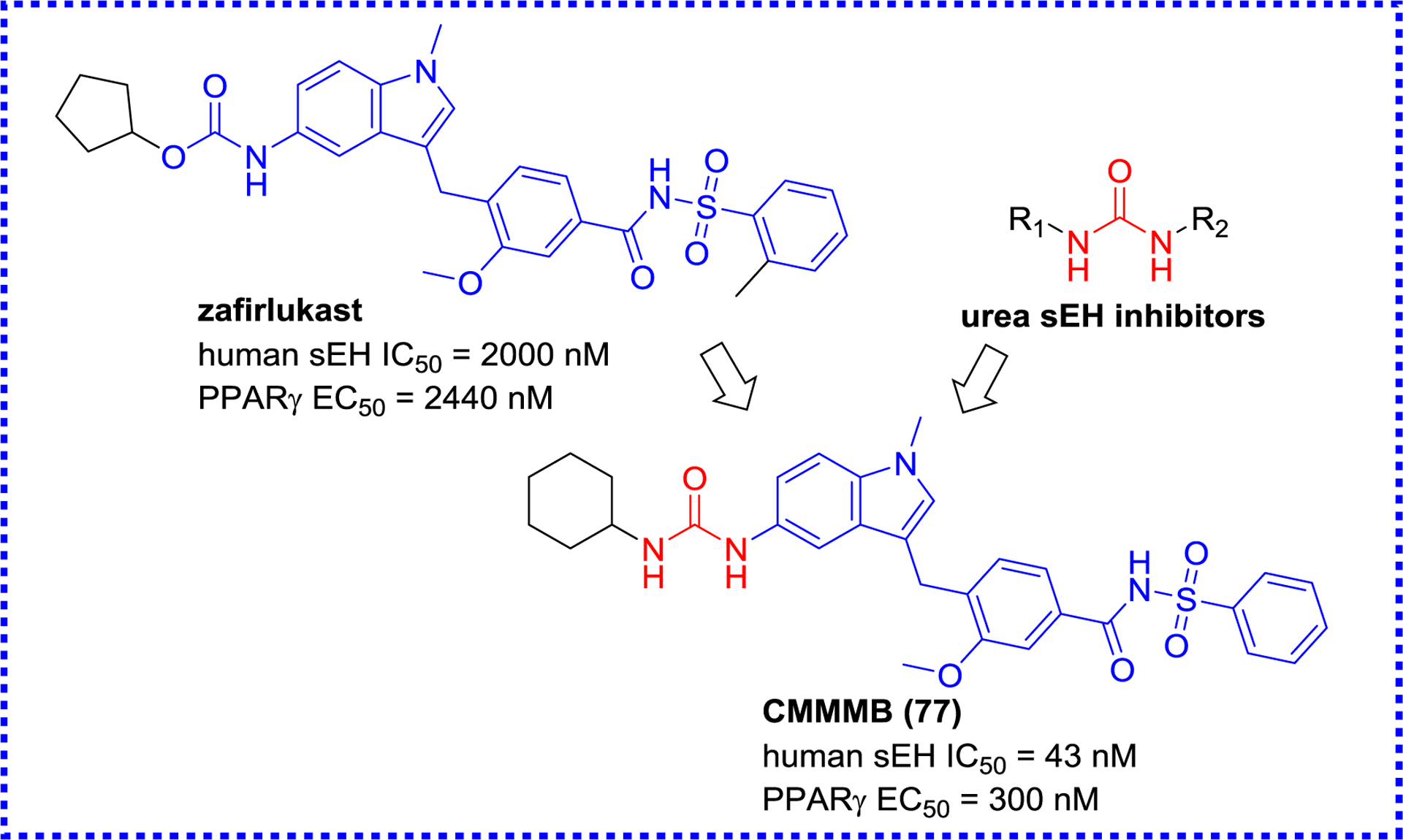
A selective sEH/PPAR dual modulator CMMMB (77) derived from zafirlukast.
2.2.6. sEH/FXR dual modulators
Farnesoid X receptor (FXR), known as the bile acid receptor, is a ligand-activated nuclear receptor.133 Clinical trials have reported that administration of FXR agonists could improve histological features and clinical markers of non-alcoholic fatty liver disease (NAFLD) as well as metabolic parameters, revealing that FXR can be served as a potential target for fatty liver disorders and metabolic diseases.
Interestingly, an antagonist of cysteinyl leukotriene receptor 1, zafirlukast, also has an agonistic effect against FXR (EC50 = 3.9 μM) and inhibitory effect against sEH (IC50 = 2.0 μM). The optimization of zafirlukast structure lead to a 15-fold increase of its agonistic effect against FXR with a similar potency toward sEH (Figure 26).134
Figure 26.
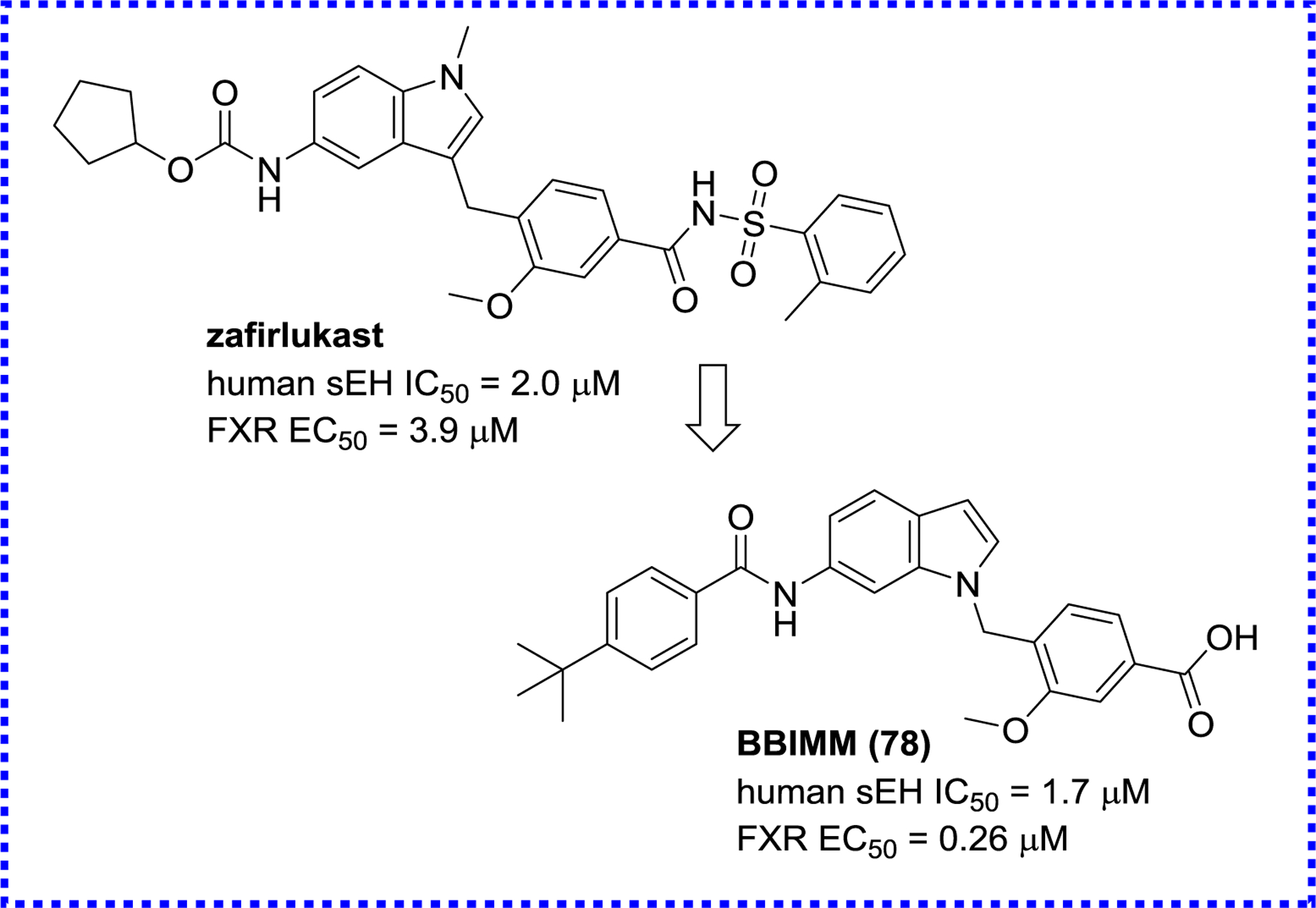
A sEH/FXR dual modulator BBIMM (78) derived from zafirlukast.
Separately, based on the structure of a sEH inhibitor (GSK2188931B, Figure 27) and a FXR agonist, Schmidt et al. extracted their similar structural characteristics and combined them in the dual pharmacophore N-benzylamide. This finding led to the production of a sEH/FXR dual modulator BCMBB (79).135 Extensive in vitro characterization confirmed its potency and favorable PK (Figure 27).134
Figure 27.
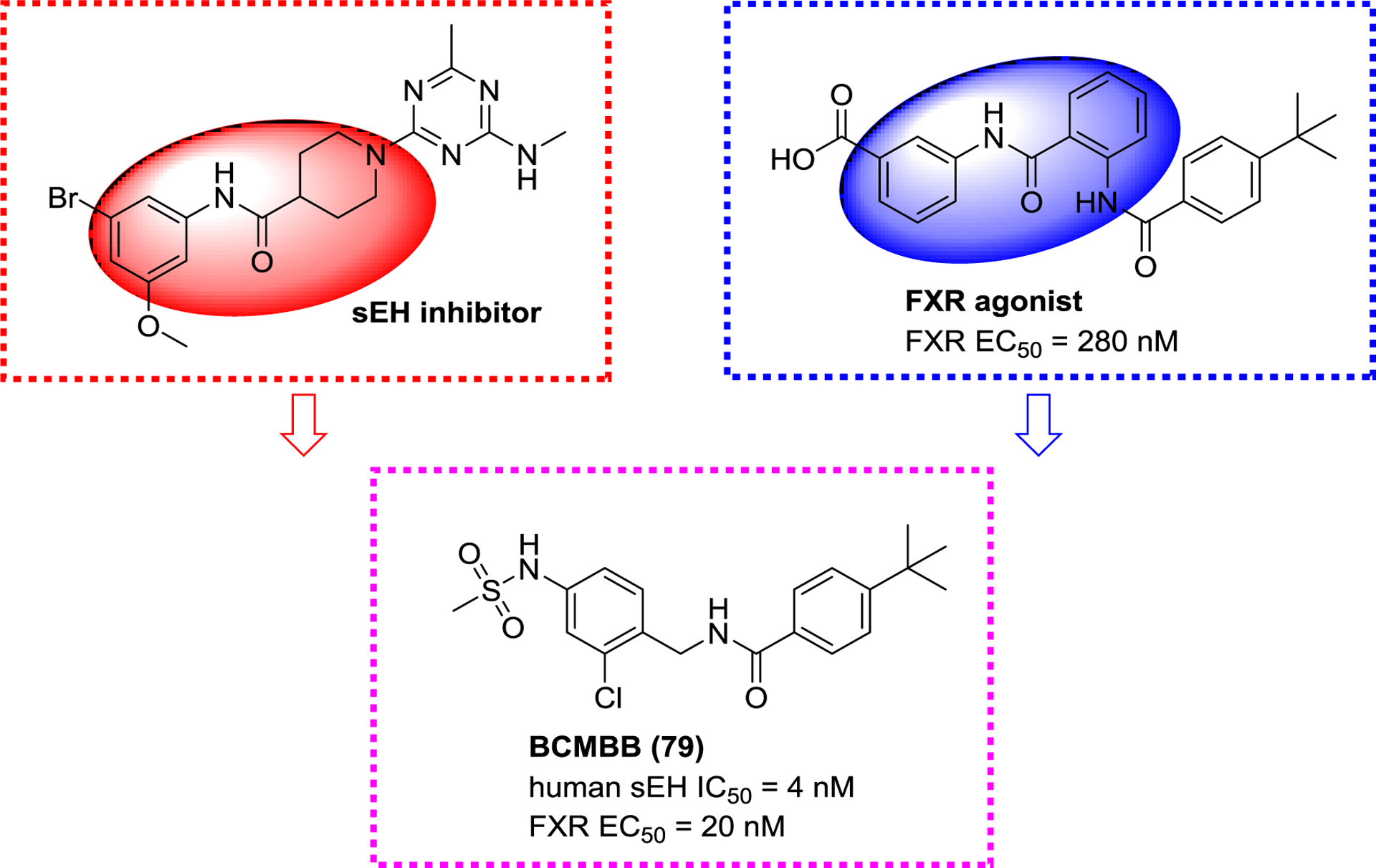
A sEH/FXR dual modulator BCMBB (79).
2.2.7. sEH/c-RAF dual inhibitors
RAF is a proto-oncogene serine/threonine-protein kinase (c-RAF or RAF1),136 and is the target for antitumor drugs because its inhibition or knockout regulates the RAS-RAF-MEK-ERK pathway to suppress lung and ovarian tumors growth.137 Sorafenib (80), the first FDA-approved inhibitor of c-RAF,138 possesses a similar urea pharmacophore as a selective sEH inhibitor t-TUCB, and it was founded to exhibit potent inhibitory activity towards sEH as well (Figure 28).139 Structural optimization leads to the design and synthesis of a sEH/c-RAF dual inhibitor t-CUPM (81, Figure 28).140 Compared to sorafenib (80), t-CUPM (81) is 25-folds more potent toward sEH while displaying similar potency toward c-RAF, while having satisfying PK properties. The subsequent in vivo study in LSL-KrasG¹²D/Pdx-1-Cre mice indicated that t-CUPM (81) significantly alleviated chronic pancreatitis by suppressing mutant Kras-transmitted phosphorylations of cRAF/MEK/ERK.140
Figure 28.
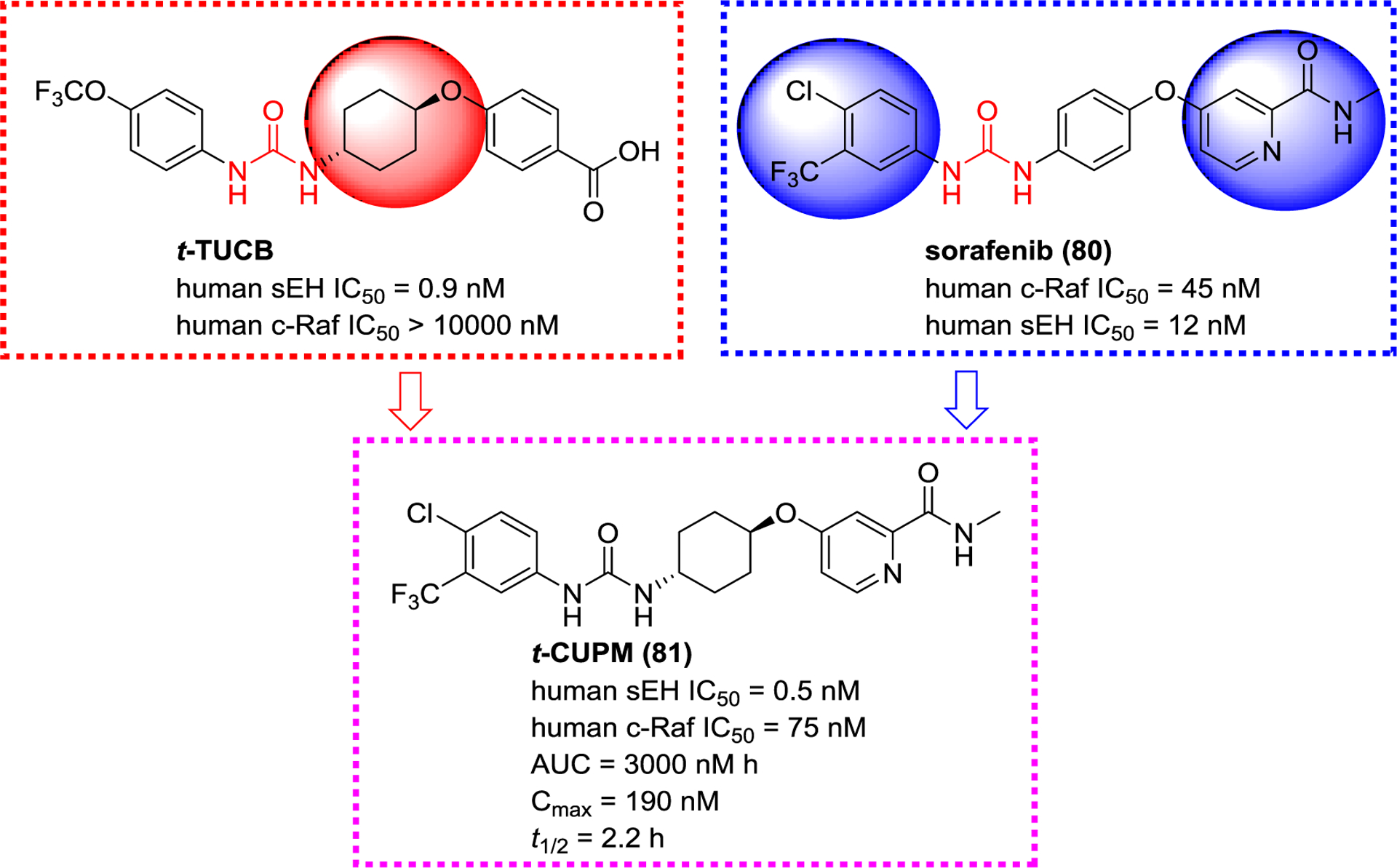
A sEH/c-RAF dual modulator t-CUPM (81) derived from t-TUCB and sorafenib (80)
2.3. Natural (or naturally occurring) products as sEH inhibitors
Natural products play an important role in drug discovery, and about 50% of drugs in clinic are natural products, such as artemisinin, taxol, andrographolide, and penicillin.141, 142 Accordingly, numerous compounds from natural resources also display inhibition of sEH, including natural ureas, triterpenoids, flavones, and phenyl-propionic acids.143–146 Interestingly, numerous compounds reported as sEH inhibitor have a weak potency with IC50 or Ki in the micromolar range, thus limiting their usefulness for eventually treating patients.
2.3.1. Natural ureas and amides
So far, as described above, ureas and amides have been most widely studied as sEH inhibitors. To date, natural urea-containing compounds also are discovered from the genera Pentadiplandra, Salvadora, Lepidium, and Moringa (Figure 29),145, 147–151 whereas only five ureas (82–86), isolated from P. brazzeana and L. meyenii, were screened for their sEH inhibitory activities and possess potent human and rat sEH inhibitory activities (Figure 29).152 These are the only reported natural compounds that inhibit sEH with IC50 in the nanomolar range.
Figure 29.
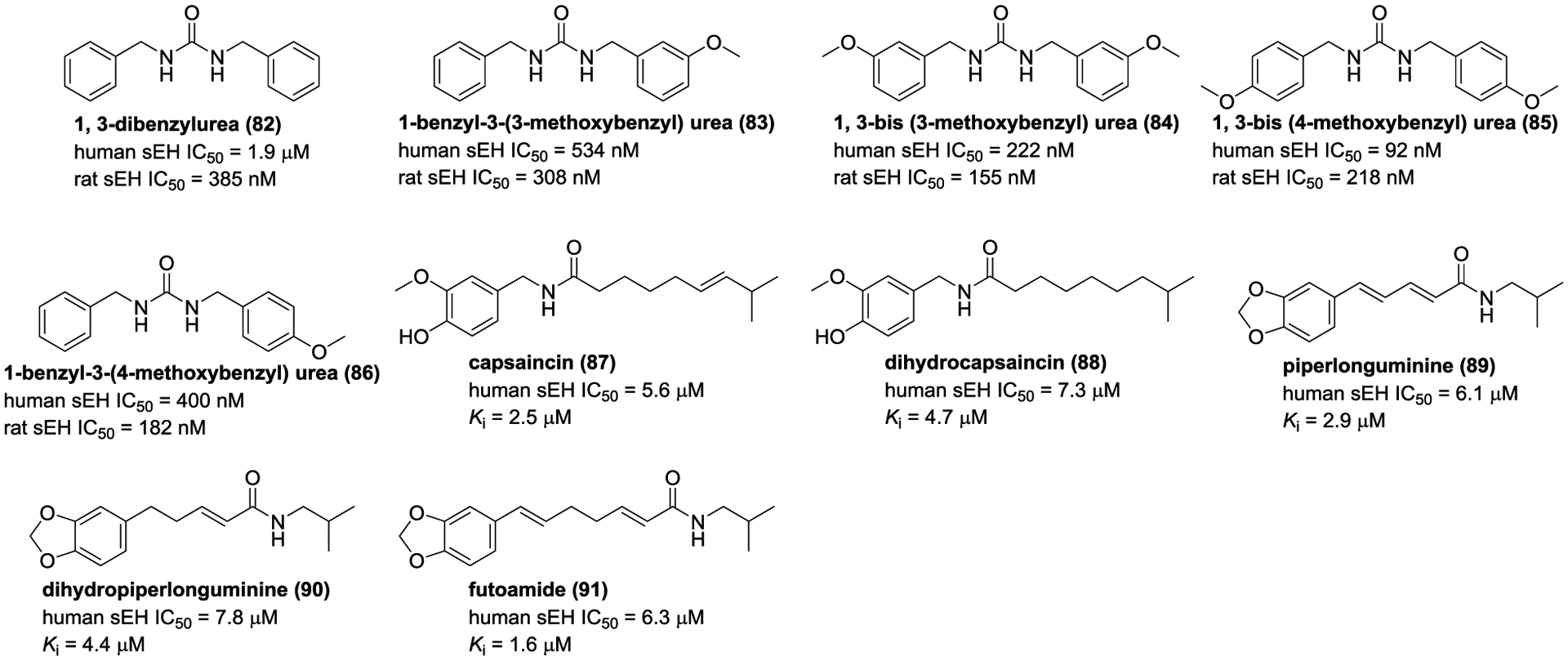
Chemical structures of naturally occurring sEH inhibitors: ureas (82–86) and amides (87–91).
Capsaincin (87) and dihyrocapsainin (88), isolated from Capsicum chinense, are competitive-type inhibitors with Ki values of 7.3 and 4.7 μM, respectively.153 In addition, three amide derivatives piperlonguminine (89), piperlonguminine (90), and futoamide (91) from the traditional Chinese medicine Scutellaria baicalensis, are weak sEH inhibitors.144
2.3.2. Flavonoids
Some of flavonoids, isolated and identified from the genera Scutellaria, Tetrastigma, Epimedium, Apios, Kaempferia, Boscia, and so on, exhibited sEH inhibitory potencies (Figure 30).144, 145, 154–158
Figure 30.
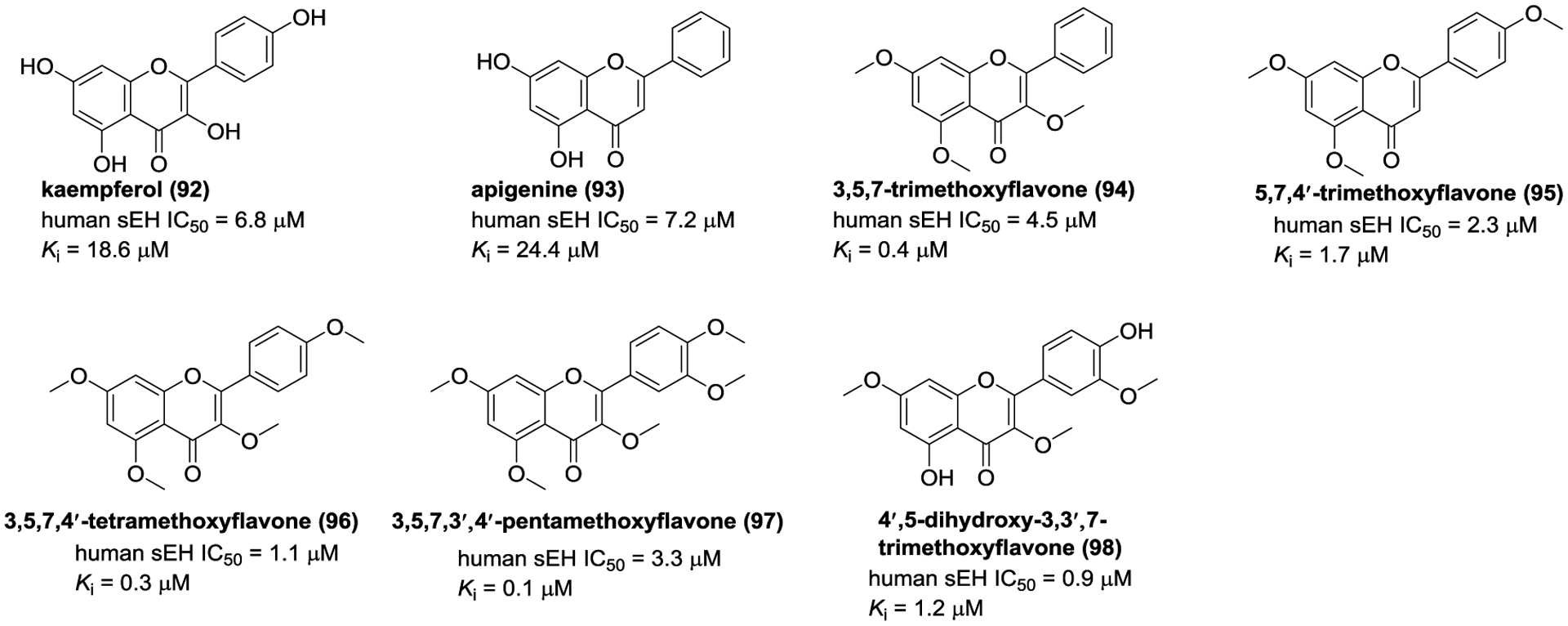
Chemical structures of naturally occurring sEH inhibitors: flavonoids (92–98).
The investigation on T. hemsleyanum led to the isolation of kaempferol (92) and apigenine (93), and they were defined as noncompetitive-type inhibitors with Ki values of 18.6 and 24.4 μM, respectively.145 In addition, 3,5,7-trimethoxyflavone (94), 5,7,4′-trimethoxyflavone (95), 3,5,7,4′-tetramethoxyflavone (96), 3,5,7,3′,4′-pentamethoxyflavone (97), and 4′,5-dihydroxy-3,3′,7-trimethoxyflavone (98), isolated from K. parviflora, displayed potent inhibitory potentials with Ki values ranging from 0.1 μM to 1.7 μM.155
2.3.3. Triterpenoids
Thao and co-workers studied the constituents of Cimicifuga dahurica to afford cycloartuane-type triterpenoids (99–107, Figure 31).159, 160 It was found that compounds 99–107 were mixed-type inhibitors and their IC50 values were 0.4–4.8 μM. Protostane-type triterpenoids are characteristic constituents of the genus Alimsa, and twenty-five of them were assayed for their inhibitory effects against sEH.161 Among them, 11-deoxy-25-anhydro alisol E (108, IC50 = 3.4 μM) and 11-deoxy alisol B (109, IC50 = 5.9 μM) significantly suppressed sEH activity. As mixed-type inhibitors, 11-deoxy-25-anhydro alisol E (108, Ki = 12.6 μM) and 11-deoxy alisol B (109, Ki = 3.5 μM) are hypothesized to form hydrogen bonds with amino acid residues Asp335, Trp336, Tyr383, and Tyr466, respectively.
Figure 31.
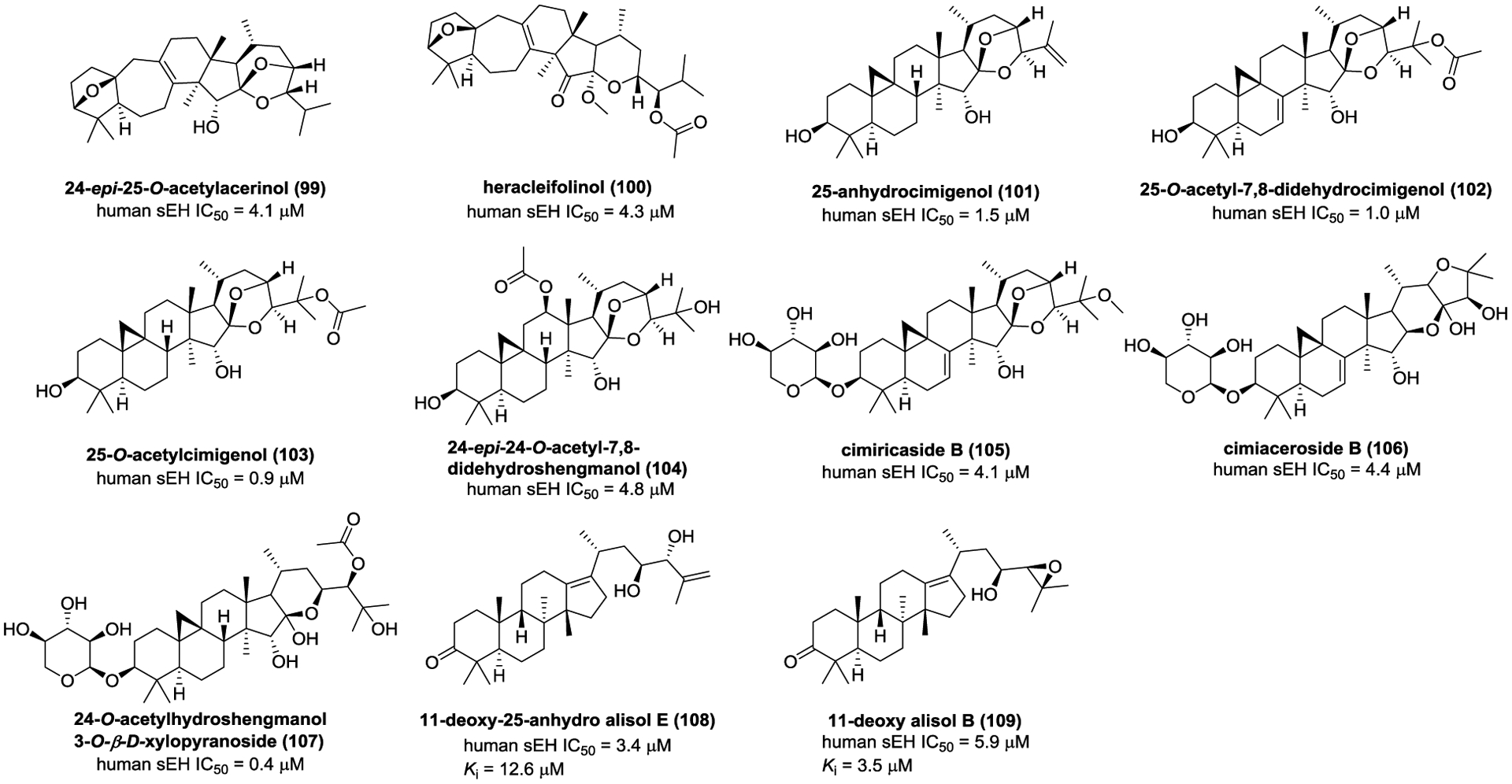
Chemical structures of naturally occurring sEH inhibitors: triterpenoids (99–109).
2.3.4. Phenylpropionic acids
Six phenylpropionic acids from T. hemsleyanum, such as (1α,3R,4α,5R)-4-O-(E)-caffeoylquinic acid methyl ester (110) and caffeic acid (111, Figure 32), showed weak inhibitory activities towards sEH.145 In order to discover more potent sEH inhibitors from natural resources, the subsequent investigation on Polygala tenuifolia led to the isolation of nine analogues, such as 6-O-(O-methyl-pbenzoyl)-3′-O-(O-methylsinapoyl)sucrose (112), and 6,3′-di-O-sinapoylsucrose (113).146 Kim and co-workers investigated the constituents of C. dahurica and Gentiana scabra to obtain cimiciphenone (114), cimiracemate A (115), 3,5-di-O-caffeoylquinic acid (116), and ferulic acid methyl ester (117).162, 163 Compounds 114–117 could significantly inhibit sEH activity (Figure 32).
Figure 32.
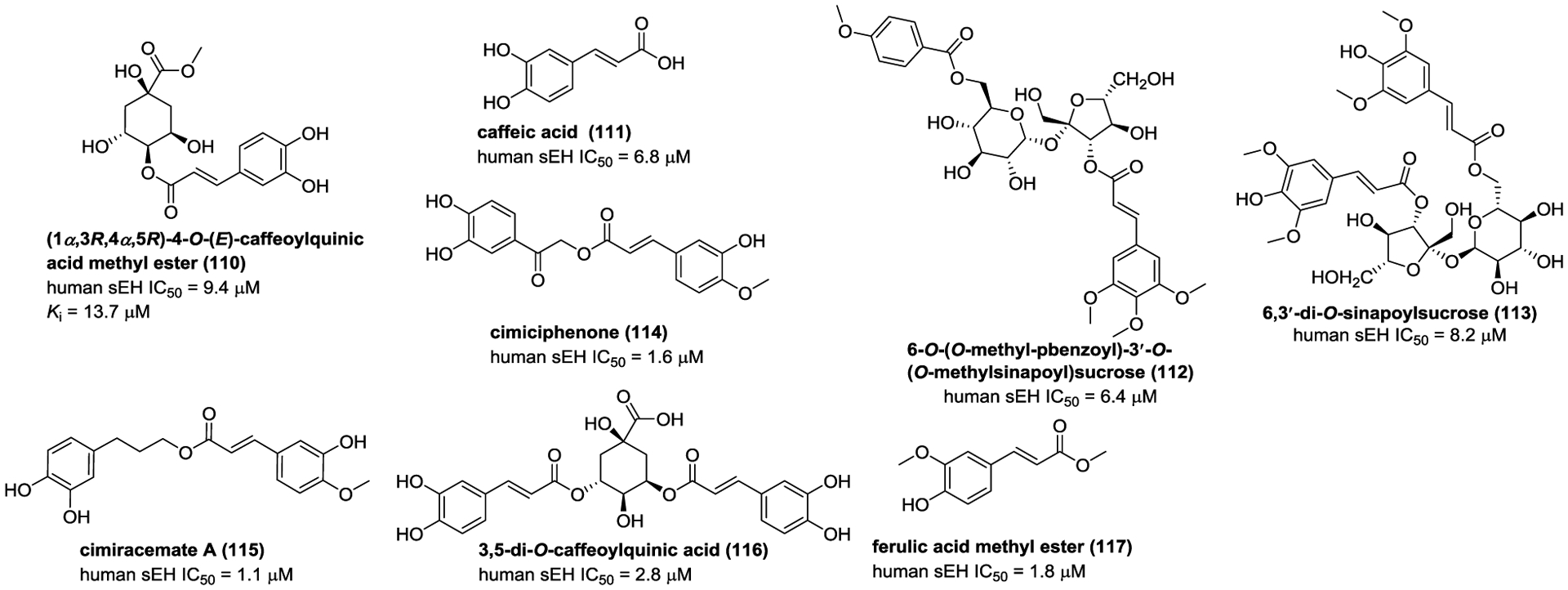
Chemical structures of naturally occurring sEH inhibitors: phenylpropionic acids (110–117).
2.3.5. Anthraquinones
Aloe is a short-stemmed succulent herb mainly used in traditional medicine to treat various diseases. Anthraquinones are considered as the major active constituents of the genus Aloe (Figure 33). Sun et al. obtained three anthraquinones, desoxyaloin (118), aloinosides C (119), and elgonica dimer A (120), from Aloe.164 Among them, desoxyaloin (118) showed the most potent inhibitory activity against sEH with a mixed-type inhibitor behavior. Molecular docking suggested that desoxyaloin (118) could bond to sEH with hydrogen bond interactions with Tyr343 and Pro361. Besides, emodin (121), an anthraquinone derivative isolated from P. multiflorum, also displayed inhibitory potency against sEH.165
Figure 33.
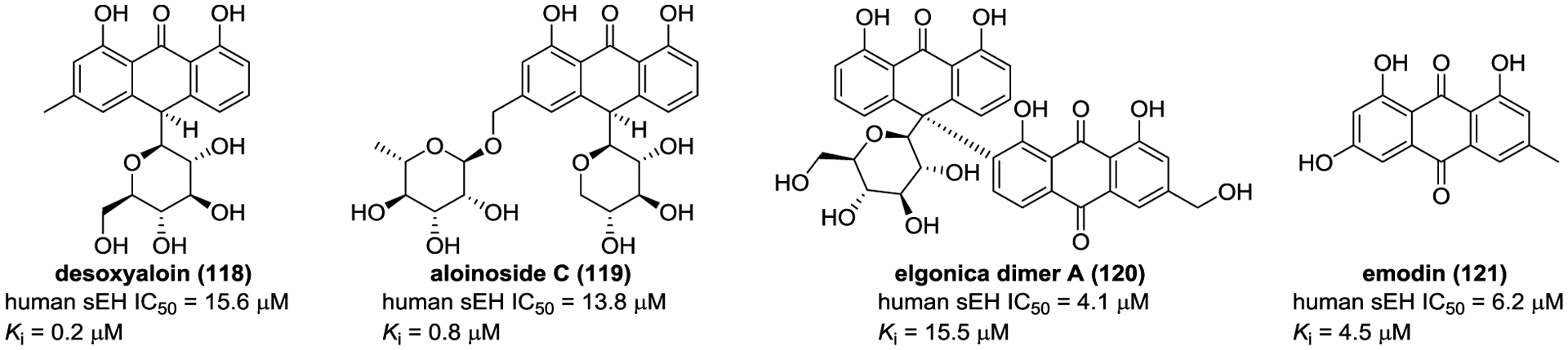
Chemical structures of naturally occurring sEH inhibitors: anthraquinones (118–121).
2.3.6. Stilbenes
The investigation on Rheum undulatum resulted in the isolation of astringin (122, Figure 34), which was found to inhibit sEH.166 Molecular docking suggests seven hydrogen bonds between astringin (122) and Ser415, Leu417, Met419, Tyr466, Lys495, and His524 residues of sEH. In addition, three noncompetitive inhibitors of sEH, selaginellin A (123), selaginellin B (124), and selaginellin (125) were isolated from Selaginella tamariscina.164 In silico investigation suggests that they bonded to a complex of sEH and substrate via hydrogen bond interactions.
Figure 34.
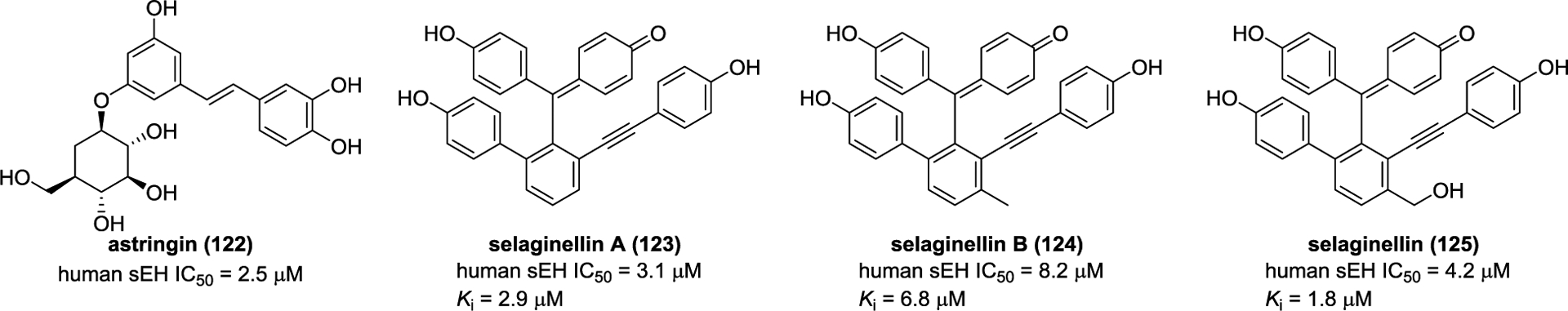
Chemical structures of naturally occurring sEH inhibitors: stilbenes (122–125).
2.3.7. alkaloids
Investigation of the phytochemical constituents of traditional Chinese medicines C. dahurica and Glycosmis stenocarpa, resulted in the isolation and identification of four alkaloids (126–129) (Figure 35) that inhibits sEH.160, 167
Figure 35.
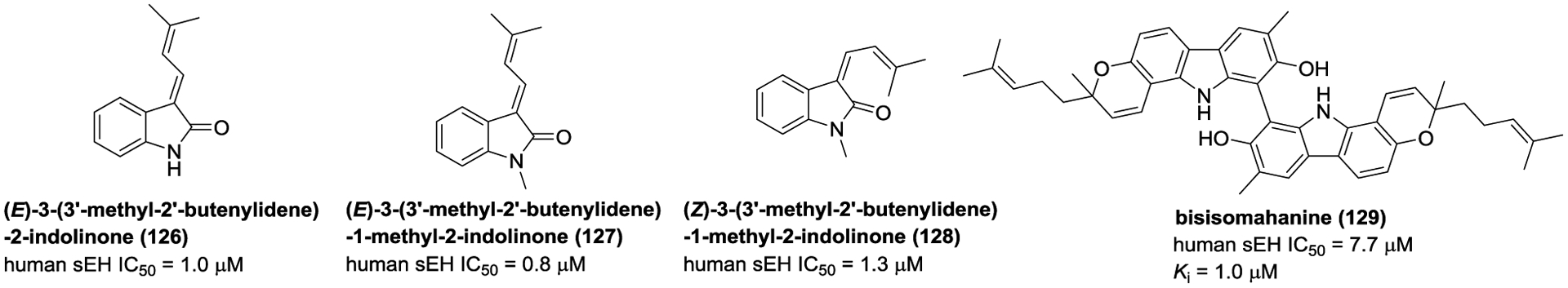
Chemical structures of naturally occurring sEH inhibitors: alkaloids (126–129).
2.3.8. Others
Besides all the above-mentioned compounds, some other type of compounds isolated from G. scabra, Magnolia officinalis, Callistemon citrinus and Cassia tora,163, 168, 169 also displayed some inhibition on sEH, such as berchemol (130), honokiol (131), callistenone B (132), and cassitoroside (133) (Figure 36).
Figure 36.
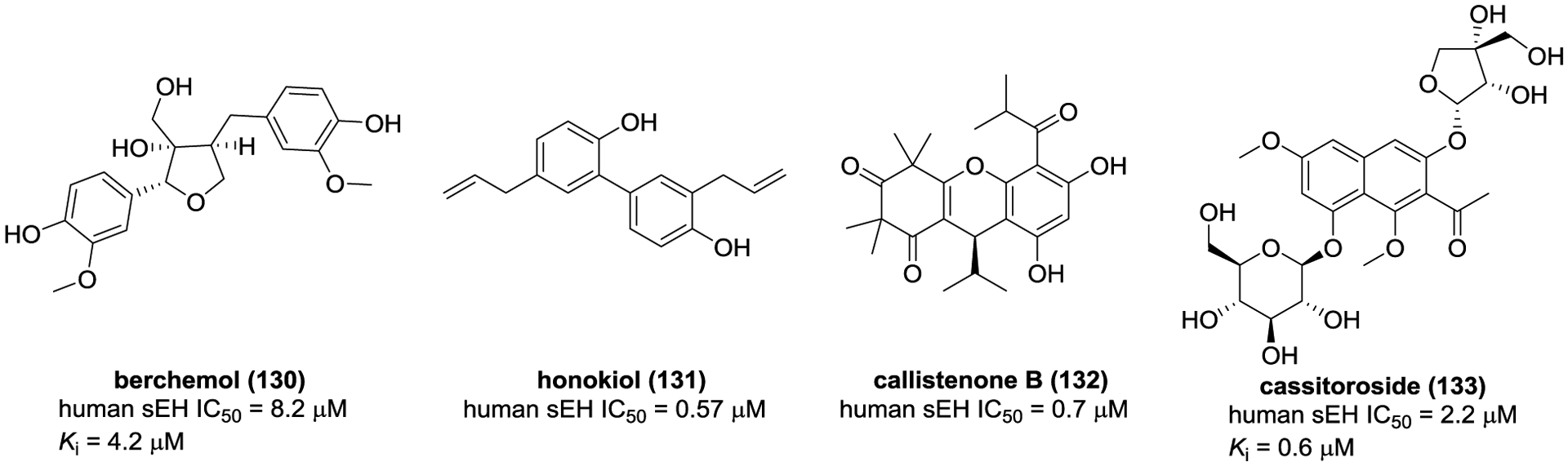
Chemical structures of naturally occurring sEH inhibitors: others (130–133).
2.4. Selectivity of sEH inhibitors
High selectivity of pharmacological inhibitors as an important factor limit off-target non-desirable side effects in clinic. Here, we are mostly concentrating on the urea-based sEH inhibitors. Because urea function is also the central pharmacophore for compounds that inhibit proteases and kinases,170 and bind to canabinoid receptors 1,171 it is possible that inhibitors for sEH could also alter the function of these other proteins. It is interesting that the Raf-1 kinase inhibitor sorafenib (80), used to treat some cancer, was found to inhibit sEH also.139 A commonly use sEH inhibitor, TPPU (19), was recently reported to also inhibit significantly the P38 kinase, influencing its action in Alzheimer disease model.172 The selectivity of a particular compound is not only dependent of the central pharmacopohore but also of the chemical groups present on both side of the urea. As these groups are becoming more complex and establishing additional bond with sEH, the likelihood of a compound to bind to another protein decrease. However, any compound developed as a pharmacological sEH inhibitor should be tested for it selectivity especially for its ability to inhibit or not proteases and kinases.173
3. Biological potentials for sEH inhibition
EpFAs play a crucial role in biological functions, such as reducing inflammation and pain, dilating blood vessels, and protecting neurons. A growing body of studies indicated that many cardiovascular, CNS, and metabolic diseases, such as hypertension, arteriosclerosis, AD, PD, NAFLD, and diabetes, are related to the level of EpFAs in the human body and the expression of sEH.32, 174–176 Moreover, chemical inhibition or genetic knockout sEH, which yield a steady state of EpFAs level in the body, have beneficial effects on the physiological environment and greatly influence the above-mentioned diseases.177
3.1. Cardiovascular diseases
The abnormal function of vascular endothelial cells plays a crucial role in the occurrence and development of cardiovascular diseases. The prevention of endothelial cell dysfunction is one of the key problems related to cardiovascular diseases to solve. EETs synthesized in endothelial cells act as an autocrine and paracrine mediator of cardiovascular system, which has vasodilation and anti-inflammatory effects.174 Therefore, sEH is considered in the treatment of cardiovascular diseases via improving the bioavailability of endothelium-derived hyperpolarization and relaxing factor EETs.32
Hypertension is caused by a complex interaction of genes and several environmental factors, and is also one of the important causes of heart remodeling. Hypertension not only reduces the quality of life, but also increases the risk of other cardiovascular diseases.178 Extensive researches have verified that endogenic EETs contribute to vasodilation in various organs, such as brain, kidney, intestine, and heart,45, 179, 180 especially 14,15-EET. They activate BKCa in vascular smooth muscle cells to promote K+ efflux and membrane hyperpolarization via modulating cAMP-mediated protein kinase A and ADP ribosylation of Gαs pathway, leading to the relaxation of blood vessels.40, 181–185 However, DHETs, sEH-mediated hydrolysates of EETs, exhibit weak relaxant effect,184 which suggests that inhibition of sEH to stabilize levels of EETs is a therapeutic strategy for hypertension.179 Present series of studies provide evidence that the expression level of sEH is increased in hypertensive rats.61, 186 For example, the increasing of sEH expression was observed in the kidney of spontaneously hypertensive rats and Ang II-induced hypertension rodent model.187, 188 The similar result was also found in aortic specimens from saline-fed and Ang II-infused rats,189, 190 demonstrating the presence of the relationship between sEH and hypertension. The Ephx2 abolishment significantly decreases systolic blood pressure, and enhances the ratio of EETs/DHETs in a saline-fed-induced hypertensive model.191 Moreover, a great increase of evidence demonstrated that administration of sEH inhibitors, such as AUDA (4), TPPU (19), and c-AUCB (9), lowered blood pressure in the hypertensive mice model.67,192, 193 An orally administered sEH inhibitor AR9281 (APAU, 6) developed by Arête Therapeutics Inc finished the Phase I and II clinical trials. AR9281 is safe and well tolerated for healthy volunteers, even administration with a high dose (single oral dose, 1000 mg; multiple dose, 400 mg every 8 hours for 7 days) in the Phase I clinical trial, whereas does not display thrilling efficacy in early-stage hypertension during the Phase II clinical trial.194
Atherosclerotic diseases are the major causes of mortality worldwide. Atherosclerosis is a progressive disease characterized by the formation of foam cells, dyslipidemia, and accumulation of lipid plaques in the arterial wall.195 Extensive epidemiological studies clearly revealed the correlation of the plasma cholesterol profile with atherosclerosis. In the course of atherosclerosis, low-density lipoprotein (LDL), especially oxidized-LDL, activates inflammation and foam cell formation, but high-density lipoprotein (HDL) has a protective effect on atherosclerosis. sEH inhibitors reduced atherosclerotic lesion formation in the deficient of apolipoprotein E (apoE−/−) or LDL receptor (Ldlr−/−) mice; these mice exhibited advanced lesions and increased in the LDL level.196 In apoE−/− mice, administration of a sEH inhibitor significantly decreased serum LDL concentrations and secretion of inflammatory factors.196 In order to reveal the protective mechanism for inhibition of sEH, a study by Xu et al. reported that administration of t-AUCB (8) could enhance CD36-mediated recognition and interpretation of oxidized-LDL through the activation of PPARγ, and further improve cholesterol outflow by increasing the ATP cassette A1 expression level, which reduced the formation of atherosclerotic lesions in Ldlr−/− mice.197 Similarly, Shen et al. found that t-AUCB (8) treatment alleviated atherosclerosis symptoms in Ldlr−/− mice as well via modulating the expression of ATP binding cassette transporter A1, cholesterol efflux, and plasma HDL levels.198 In addition, inhibition of sEH with t-TUCB delayed the formation of hyperlipidemia and atherosclerosis in Ldlr−/− mice, and Ephx2 genetic abolishment significantly alleviated the symptoms of atherosclerosis as well.195
Heart failure is caused by blocking blood flow to the heart, results in coronary heart disease, myocardial infarction, arrhythmia, viral myocarditis, and genetic cardiomyopathies.199 A study by Seubert et al. has demonstrated the increasing of sEH activity in animal models of myocardial infarction, and the increasing of sEH expression was observed in the human hearts of patients with ischemic cardiomyopathy. Moreover, increasing EET levels protected against myocardial infarction and left ventricular dysfunction after ischemic injury in Ephx2−/− mice.200 Inhibition of sEH with GSK2188931B and TUPS both protected heart failure.201, 202 For example, TUPS treatment reduced the thickness of heart wall, down-regulated the expression of hypertrophy markers atrial and brain natriuretic peptides, and regulated negative autophagy via activating mammalian target of rapamycin (mTOR) signaling, allowing in preventing myocardial hypertrophy induced by isoproterenol.202 These data all suggested the therapeutic potential of sEH in heart failure.
Preclinical results have demonstrated that inhibition of sEH by inhibitors or Ephx2 genetic knockout possesses beneficial effects, such as vasodilation, antihypertension, and anti-atherosclerotic effects. Although inhibition of sEH to treat human diseases still remains to be demonstrated in clinical trials, the increasing of positive effects of sEH inhibitors in cardiovascular diseases strongly supports the clinical role of sEH in cardiovascular diseases.
3.2. CNS diseases
EETs play an important role in CNS diseases, it can regulate angiogenesis and cerebral blood flow (CBF), and affect signal transduction process. Because of the abundant expression of sEH in various parts of the brain, the neuroprotective effect of sEH inhibitors has become an important research direction in the treatment of CNS diseases. In recent years, sEH inhibitors have been used in the treatment of CNS diseases, such as AD, PD, and depression with satisfactory performance.175, 203–205
PD, one of the most common neurodegenerative diseases, leads to progressive motor deficits and non-motor symptoms, including speech, motor, and mental disorders.203, 204 In the course of onset, dopaminergic neurons in substantia nigra (SN) prematurely die and Lewy bodies accumulate in the brain.71 The current treatment works by increasing dopamine concentration and/or stimulating its receptors, but this treatment only targets basal ganglia and dopamine, but does not involve more neurons associated with PD. Therefore, this therapy is generally not very effective. In human and animal PD models, the expression level of sEH in brain regions is associated with dopaminergic death, endoplasmic reticulum (ER) stress, and tyrosine hydroxylase positive cell death, which can be alleviated by administration of AUDA (4), TPPU (19) or 14,15-EET.206, 207 EETs stimulate astrocytes to secrete nerve growth factors (NGFs), including vascular endothelial growth factor and brain-derived neurotrophic factor, which promotes the growth and differentiation of nerve cells and protects neurons.177 Administration of AUDA (4) reduced MPTP-mediated loss of TH positive cells in SN.207 Furthermore, the Ephx2 abolishment protected against MPTP-induced neurotoxicity in STR, while overexpression of sEH in STR significantly enhanced MPTP-induced neurotoxicity.206 Moreover, the expression of the sEH protein in STR from MPTP-treated mice was significantly higher than control group. Interestingly, there was a positive correlation between sEH expression and p-α-synuclein in STR, suggesting that sEH may play a role in the phosphorylation of α-synuclein in STR. In model animals, administration of TPPU (19) increased dopamine, 3,4-dihydroxyphenylacetic acid, and homovanillic acid levels, and inhibited the apoptosis of parkinson disease protein 2 (PARK2) neurons. Accordingly, it is likely that sEH can act as a potential target for the treatment of PD.
AD is a neurodegenerative disease characterized by impaired learning, memory, and cognitive abilities. As our acknowledge, neuroinflammation is a critical factor causing AD and plays a crucial role in the development course of AD,208–210 therefore, 16% of currently on-going clinical trials for the treatment of AD is correlate with inflammation.211 Neuroinflammation is intimately linked to the oxidative stress associated with AD,212, 213 and controls the interactions between the immune system and the nervous system.214 The senescence-accelerated mouse prone 8 (SAMP8) is a paradigm of late-onset AD and cognitive impairment in age, causing oxidative stress, neuroinflammation, tau hyperphosphorylation, and proamilodogenic APP processing.215–219 Griñán-Ferré et al. found the increasing of sEH expression in the hippocampus of SAMP8 mice, and inhibition of sEH with TPPU (19), AS2586114, or UB-EV52 could reduce biomarkers of inflammation, oxidative stress, and ER stress.220 Similarly, these positive results also were observed in a 5×FAD mouse model of early-onset AD after administration of sEH inhibitors.220 A recent study has revealed that sEH level was increased in the brain and predominantly appeared in hippocampal astrocytes of APP/PS1 Tg mice.221 Genetic deletion of sEH alleviates behavior outcomes and amyloid β (Aβ) accumulation, increases astrogliosis and the production of anti-inflammatory cytokines IL-4 and IL-10, and inhibits NF-κB signaling pathway, resulting in a protective effect against AD.175 Additionally, compared with healthy human, the analysis of clinical specimens demonstrated that sEH were overexpressed in Braak III and V phases of AD patients’ brains,220 which confirmed the correlation of sEH with AD.
Depression and schizophrenia both have high mortality rates and involve a wide range of severe chronic mental illness.205 The pro-inflammatory cytokines in serum and cerebrospinal fluid are increased, and the sEH level is significantly increased in the parietal cortex of the brain of deceased-patients suffering of these diseases.222 It strongly suggests that sEH is likely involved in the pathophysiological process of depression and schizophrenia. In LPS-treated depressed mice, TPPU (19), a sEH inhibitor, can significantly improve depressive symptoms by reducing the level of TNF-α, up-regulating the expression of BDNF in mouse hippocampus and PC12 cells, and enhancing NGF-induced neuronal growth in PC12 cells. Moreover, sEH knockout ameliorates symptoms of depression as well.223 Chemical inhibition on sEH by AS2586114 decreases levels of 11,12-DHET and 14,15-DHET, and improves hyperactivity and pre-pulse inhibition defects in phencyclidin-induced schizophrenia.224 Thus, the inhibition of sEH appears to exert anti-depressant and anti-schizophrenic effects by increasing the concentration of EETs, regulating BDNF-tropomyosin receptor kinase B (BDNF-TrkB) signaling pathway, and preventing oxidative stress.222
Additionally, sEH is also involved in epilepsy and stroke. Administration of sEH inhibitors could prevent neuroinflammation and reduce the number and duration of seizures of epilepsy.55, 225 After treatment with AUDA (4), the expression of inflammatory factors decreased and the EETs/DHETs ratio increased, while at the same time, the epileptic induction threshold was increased and the epileptic sensitivity was decreased. There is a correlation between the mutation of Arg287Gln of sEH, which led to a decrease of sEH activity and a reduced risk of stroke.30 Chemical inhibition can reduce astrocyte infiltration, glial scar formation, microglial activation, neuronal death, insufficient blood flow, and behavioral disorder after cerebral ischemia.226 Put together, sEH has a clear role in CNS health and diseases, and represent a potential target for treatment.
3.3. Metabolic diseases
Besides cardiovascular and CNS diseases, sEH is closely related to metabolic diseases, including NAFLD, diabetes, and obesity. Many studies have shown overexpression of sEH in many liver diseases, such as NAFLD, non-alcoholic steatohepatitis227, and hepatic fibrosis.227–231 NAFLD is caused by excessive fat accumulation in hepatocytes, affecting approximately 25% of the global adult population. Some of preclinical results verified that sEH inhibition was to the benefit of NAFLD and NASH. Iyer et al. investigated the role of sEH inhibition in high-carbohydrate and high-fat diet (HCHFD)-induced rat model.33 The inhibition of sEH with t-AUCB (8) alleviated HCHF -induced liver hypertrophy and steatosis, had significant improvements in plasma lipid levels and insulin sensitivity, and suppressed the increasing of aspartate aminotransferase (AST) and lactate dehydrogenase (LDH) levels.33 Similarly, chemical inhibition of sEH or Ephx2 genetic abolishment has a protective role in high-fat diet (HFD)-induced NAFLD.232 t-AUCB (8) could reduce HFD-induced macrophage accumulation, downregulate mRNA expression levels of pro-inflammatory cytokines TNF-α, IL-6, MCP-1, and IFN, leading to the improvement of HFD-induced inflammation via the activation of MAPK pathway (JNK and p38).232 However, overexpression of sEH by the injection of adenoviruses encoding human sEH resulted in the aggravation of metabolic syndromes, such as the increasing of triglyceride and pro-inflammatory cytokine and the upregulation of JNK and p38.232 Conversely, the Ephx2 deletion alleviated hepatic steatosis and tissue inflammatory described as administration of t-AUCB (8), which suggested the role of sEH-mediated hepatic steatosis. The role of sEH in NAFLD also was supported in a high methionine diet-induced hyperhomocysteinemia and hepatic steatosis.233 Accumulating evidence revealed that hyperhomocysteinemia disturbs lipid metabolism in the liver and is served as a significant risk factor. A study by Yao et al. demonstrated that sEH inhibition by TPPU (19) ameliorated hyperhomocysteinemia-induced lipid accumulation, further mechanism investigation suggested that the increase of EETs through the inhibition of sEH by TPPU (19), especially 11,12-EET, elevated the expression levels of PPARα target genes and activated PPARα.233 These findings suggested that the increase of sEH activity was a critical threaten in hyperhomocysteinemia-induced hepatic steatosis, and sEH inhibition might be regarded as an effective means to treat hyperhomocysteinemia-induced hepatic steatosis.
Inflammasome is responsible for the modulation of the adaptive response of the liver to pathogenic challenge, and a library of evidences demonstrated that sEH inhibition could suppress inflammasome activation to reduce hepatic inflammation.230 Administration of PTUPB (68), a sEH/COX-2 dual inhibitor, could block fibrotic progression by suppressing collagen deposition and down-modulating Col1a1, Col1a11a3, and α-SMA expression levels. The dual inhibition of sEH and COX-2 also suppressed the NLRP3 inflammasome activation through the decrease of Nlrp3/NLRP3 and Asc expression, and reduced the downstream target protein of NLRP3, pro-caspase 1, pro-IL-1β, and pro-IL-18. Besides, PTUPB (68) treatment decreased the release of pro-inflammatory cytokines IL-6 and MCP-1.230 Accordingly, it was worth noting that the role of sEH in inflammasome activation still need further be studied.
Diabetes is mainly due to the lack of insulin produced by pancreatic β-cells or the insensitivity of the target tissue to insulin, leading to the severe injuries to organs.234 A growing body of evidences have demonstrated that the stabilization of EETs, inhibition of sEH by sEH inhibitors or Ephx2 genetic deletion plays a crucial role in diabetes. A study by Schaefer et al. verified that EETs enhanced insulin signaling through the regulation of Akt expression level rather than affecting levels of insulin receptor.72 Furthermore, the insulin release was also promoted by 5,6-EET, while 8,9-EET, 11,12-EET, and 14,15-EET might promote the release of glucagon and increase the level of blood glucose, which suggested that increasing the concentration of EETs through inhibiting sEH might be a potential pathway to treat diabetes.32 In streptozotocin (STZ)-induced diabetic mice, administration of t-AUCB (8) attenuated hyperglycemia and had protective effects against β-cell damage.235 An investigation by Pardeshi et al. has been reported that TPPU (19) reduced the level of fasting blood glucose, improved the cognitive impairment and memory impairment by stabilizing EETs to dilate arterioles, and increased cerebral blood perfusion in STZ-induced diabetes.236 A similar result was also observed after administration of t-AUCB (8).237 In addition, inhibition of sEH or Ephx2 knockout has been found to alleviate symptoms of hyperglycemia and insulin resistance in HFD-induced type 2 diabetes,238 which might activate insulin signaling in the liver and adipose tissue.
Diabetes also causes many complications, such as retinopathy, nephropathy, and cardiovascular diseases, and seriously affect a patient’s quality of life. A great number of studies have verified that inhibition of sEH by inhibitors or genetic knockout sEH is beneficial to diabetic complications.228, 239 Diabetic retinopathy is one of diabetic complications caused by progressive loss of vascular cells and slow dissolution of vascular junctions.240 The expression of sEH is increased in human retina and vitreous of diabetic retinopathy. Furthermore, overexpression of sEH in retinal Müller glial cells can result in vascular abnormalities in diabetic retinopathy.239 Therefore, the overexpression of sEH is a key factor in the pathogenesis of diabetic retinopathy. Jouihan et al. found that STZ treatment promoted sEH mRNA expression and reduced EET level in cerebral vessels.228 Furthermore, brain infarction was exacerbated in STZ-treated mice after middle cerebral artery occlusion, which was reversed through the inhibition of sEH by t-AUCB (8). Moreover, sEH is related to diabetic cardiomyopathy since sEH is highly expressed in the heart tissue of type 1 diabetes mice.231 In addition, sEH genetic disruption reduced levels of blood urea nitrogen and creatinine as well via suppressing renal tubular apoptosis and enhancing renal endothelial function, allowing in the alleviation of diabetic nephropathy.229, 230
So far, a selective sEH inhibitor GSK2256294 (67) has finished its Phase I clinical trial, which clinical experimental data demonstrated its safety and sustained inhibitory effect against sEH.173 These results supported further clinical trials in patients with endothelial dysfunction, such as diabetes. In 2018, Vanderbilt University Medical Center sponsored the Phase II clinical trial of GSK2256294 (67) to treat diabetes, endocrine system diseases, glucose metabolism disorders, and obesity, and started volunteer recruitment.
Obesity is a global public health problem and a major risk factor for diseases as same as cancer and diabetes. In metabolic disorders, chemical inhibition of sEH or Ephx2 deletion is capable of lessening body weight gain, lowering blood pressure, and suppressing the production of inflammatory cytokines.78, 241, 242 Moreover, the increasing of sEH expression has been found in adipose tissue of obese mice,78, 241, 242 revealing the potential correlation of sEH with obese. A growing body of evidence indicates that ER is a known cellular consequence of metabolic diseases, such as obesity and NAFLD.243 Importantly, sEH deficiency or inhibition in mice attenuated HFD-induced ER stress in liver and adipose tissue. Similarly, the inhibition of sEH in 3T3-L1 preadipocytes mitigated chemical-induced ER stress and activation of JNK, p38, and cell death.54 A study by Lopez-Vicario et al. has reported that t-TUCB could stabilize the levels of EpFAs, 17,18-epoxyeicosatetraenoic acid (17,18-EEQ) and 19,20-epoxydocosapentaenoic (19,20-EDP) derived from omega-3.244 Moreover, t-TUCB promoted the polarization of macrophage to anti-inflammatory M2-type cells, induced hepatic autophagy via increasing Atg12-Atg5 and LC3-II expression levels, reduced p62 expression and ER stress via suppressing p-IRE-1α and p-eIF2α expression levels.244 The inhibition of sEH to exert a protective effect was also confirmed in palmitate-primed hepatocytes and adipocytes through the incubation of 19,20-EDP or 17,18-EEQ,244 which suggested that sEH was a physiological modulator of ER stress and a potential target for the treatment of obesity. In short, the inhibition on sEH blocks the progression of obesity, and is of great significance for obesity and related diseases.
4. Conclusion and perspective
In this review, we described a complete picture of chemically synthesized or naturally obtained sEH inhibitors. Among these inhibitors, urea-based compounds are the most prominent because they can accommodate well the active pocket of sEH hydrolase and form stable hydrogen bonds, and have in general a good selectivity for sEH. Besides potency, the main drivers for improvement have been to increase water solubility and other physical properties as well as better bioavailability and PK parameters. Toward solving these problems, amides are good alternatives to ureas for sEH inhibition. In addition, the introduction of secondary polar groups and tertiary pharmacophore into the structure of the inhibitors would greatly help with the design of “Better” inhibitor. Nowadays, Nature is providing more and more structural skeletons on which to build potent sEH inhibitors besides ureas and amides, such as flavones, and triterpenoids. These compounds have their advantages and disadvantages, which provide more space for the development and optimization of future sEH inhibitors.
Currently, sEH is a potential therapeutic target in multiple diseases related to inflammation, such as cardiovascular (hypertension and arteriosclerosis), CNS (AD, PD, and depression), and metabolic (diabetes, NAFLD, NASH, and obesity) diseases. Moreover, some sEH inhibitors, such as AR9281, EC5026, and GSK2256294, are underway clinical trials to treat heart failure, insulin resistance, glucose intolerance, hypertension, endothelial dysfunction, and pain, while future researches in some key areas are needed to further elucidate its applications and action mechanisms. Compared with the function of C-terminal hydrolase, the role of the N-terminal phosphatase domain is still undetermined. Although Kramer et al. has firstly discovered in vivo active inhibitors of sEH phosphatase with favorable PK, its N-terminal physiological function needs to be further in-depth investigated. Future studies should consider that inhibition of sEH C-terminal activity remains its phosphatase activity, discovering any differences in phenotype between sEH hydrolase inhibitor-treated and Ephx2 deficiency animals.
ACKNOWLEDGMENTS
This work was supported by National Natural Science Foundation of China (No. 81930112 and 81703769), National Key Research and Development Program of China (2018YFC1705900), Liaoning Provincial Key Research and Development Program (2019JH2/10300022), Distinguished professor of Liaoning Province, Revolutionizing Innovative, Visionary Environmental Health Research Program of the National Institute of Environmental Health Sciences (R35 ES030443), Superfund Basic Research Program of the National Institutes of Environmental Health Sciences (P42 ES04699), Liaoning Revitalization Talents Program, Natural Science Foundation of Liaoning Province (No. 2020-MS-256), and Dalian Young Star of Science and Technology.
ABBREVIATIONS USED
- AA
arachidonic acid
- Aβ
amyloid β
- AD
Alzheimer’s disease
- AFIMU
1-(adamant-1-ylmethyl)-3-{[5-(furan-2-yl)isoxazol-3-yl]methyl}urea
- AMMU
1-(adamant-1-ylmethyl)-3-[(3-methylisoxazol-5-yl)methyl]urea
- Ang II
angiotensin Ⅱ
- APU
1-(adamantan-2-yl)-3-phenethylthiourea
- AUC
area under the curve
- AUDA
12-(3-adamantan-1-yl-ureido) dodecanoic acid
- AUDA-nBE
AUDA n-butyl ester
- BDNF-TrkB
BDNF-tropomyosin receptor kinase B
- c-AUCB
cis-4-[4-(3-adamantan-1- ylureido)cyclohexyloxy]benzoic acid
- CBF
cerebral blood flow
- CNS
central nervous system
- CPU
N-cyclohexyl-N′-(3-phenylpropyl)-urea
- COXs
cyclooxygenases
- CYP
cytochrome P450
- DCU
N,N′-dicyclohexyl-urea
- DHA
docosahexaenoic acid
- DHETs
dihydroxyeicosatrienoic acids
- DPN
N-(3,3-diphenyl-propyl)-nicotinamide
- EETs
epoxyeicosatrienoic acids
- EpFAs
epoxy-fatty acids
- FAAH
fatty acid amide hydrolase
- FLAP
5-LOX-activating protein
- HBAU
1,1’-(heptane-1,7-diyl)bis(3-(adamantan-1-yl)thiourea)
- HCHFD
high-carbohydrate and high-fat diet
- HDL
high-density lipoprotein
- HETEs
hydroxy eicosatetraenoic acids
- HFD
high-fat diet
- HLM
human liver microsome
- ITBU
N-[9-(isopropylsulfonyl)-1-oxa-9-azaspiro[5.5]undec-4-yl]-N′-[4-(trifluoromethoxy)benzyl]urea
- Ki
kinetic constant
- koff
dissociation rate constant
- LDL
low-density lipoprotein
- IL-1β
interleukin-1 beta
- LOXs
lipoxygenases
- LXs
lipoxins
- LPS
lipopolysaccharide
- LTB4
leukotriene B4
- LTs
leukotrienes
- mp
melting point
- mTOR
mammalian target of rapamycin
- NADPH
nicotinamide adenine dinucleotide phosphate
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- NF-κB
nuclear factor-kappa B
- PD
Parkinson’s disease
- PK
pharmacokinetic
- PARK2
parkinson disease protein 2
- PPARγ
peroxisome proliferator-activated receptor gamma
- RLM
rat live microsome
- sEH
soluble epoxide hydrolase
- S-PPU
(S)-1-(1-phenylethyl)-3-(3-phenylpropyl) urea
- STZ
streptozotocin
- t-AUCB
trans-4-[4-(3-adamantan-1-ylureido)cyclohexyloxy]benzoic acid
- TNF-α
tumor necrosis factor-α
- TZD
thiazolidinedione
Biographies
Dr. Cheng-Peng Sun graduated from Shenyang Pharmaceutical University in 2016 and received Ph.D. in Natural Medicinal Chemistry under the guidance of Prof. Feng Qiu. Afterward, he joined the Department of Medicinal Chemistry in Dalian Medical University. In January 2019, he was promoted to Associate Professor of Natural Medicinal Chemistry at Dalian Medical University. So far, he has published more than 40 papers on journals indexed by the Science Citation Index. His major research interests include extraction and isolation of chemical constituents from natural medicines, structural modification of active compositions, and natural modulators of sEH, FXR, and PXR. In addition, he is focusing on the prevention and treatment of neurodegenerative diseases.
Dr. Xin-Yue Zhang graduated from Dalian Medical University and received her Bachelor’s degree in 2019. She is pursuing a Master’s degree at the Dalian Medical University, in the research group of Professor Xiao-Chi Ma. Her research focuses on the active substances of traditional Chinese medicine and the discovery of sEH inhibitors.
Dr. Christophe Morisseau obtained his Ph.D. in organic chemistry in 1995 from the Universitée de la Mditerranée in Marseille, and since he has been working with Prof. Hammock at UC Davis. He is now a Professional Researcher faculty. He is working at understanding the mechanism and biological role of EHs and developing selective tight binding inhibitors for EHs. He developed the first stable highly potent inhibitor of human sEH, obtained the first crystal structures for sEH, the first high-throughput assays for the human sEH and mEH, and developed more biochemical tools to study EHs. Some of the inhibitors he obtained are in development for the treatment of inflammation, cardiovascular disease, pain, and cancer. His work has resulted in more than 250 papers and 20 patents.
Dr. Sung Hee Hwang obtained his Ph.D. in organic chemistry in 2004 from the University of California, Davis. He is an accomplished medicinal chemist. He leads the development of highly potent inhibitors of epoxide hydrolase and cyclooxygenase enzymes. In addition, he has synthesized various fattyacids and their cytochrome P450-metabolite analogs to study their biological activities. His work has resulted in more than 100 publications.
Prof. Zhan-Jun Zhang received his Ph.D. in medicine from Beijing University of Chinese Medicine in 2005, and is the director of the Center for Beijing Aging Brain Rejuvenation Institute and the vice president of State Key Laboratory of Cognitive Neuroscience and Learning Beijing Normal University. He has been engaged in a long-term research of digging the pattern of varied human advanced cognitive functions decline, the degradation mode of brain’s structure and function network, the mechanism of genetic variation towards brain and cognitive aging, and the contribution of Chinese medicine in protecting the brain from aging. He is the recipient of the National Science Fund for Distinguished Young Scholars and the New Century Excellent Talents Program of China’s Ministry of Education.
Prof. Bruce D. Hammock received his Ph.D. in entomology and toxicology in 1974 from the University of California Berkeley. He is now a Distinguished Professor at the University of California, Davis. Dr. Hammock is an elected member of the National Academy of Sciences, US since 1999. He discovered the sEH and has devoted his carreer studying the biological roles of this enzyme. He has over 30 years of extensive experience in the field of eicosanoids that has yielded over 1,200 publications and 100 issued patents on synthesis, pharmacology, and biological action of eicosanoids and sEH inhibitors.
Prof. Xiao-Chi Ma received his Ph.D. in phytochemistry from School of Traditional Chinese Medicine, Shenyang Pharmaceutical University in July 2005, under the guidance of Prof. De-An Guo and Li-Jun Wu. He worked as a postdoctor in School of Pharmaceutical Sciences, Peking Unversity, and mainly focused on Biological fermentation, tissue culture of endangered plant medicine, biotransformation of natural products by microorganism and plant suspension cells and phytochemistry. In 2008, he joined in the Dalian medical university, and was promoted to Professor of Natural Medicinal Chemistry at Dalian Medical University in 2012. He is the recipient of the National Science Fund for Excellent Young Scholars. His major research interests include effective substances from traditional Chinese medicine, drug metabolism, development and application of small molecular probe.
Footnotes
The authors declare no competing financial interest.
Contributor Information
Zhan-Jun Zhang, State Key Laboratory of Cognitive Neuroscience and Learning, Beijing Normal University, Beijing 100875, People’s Republic of China..
Bruce D. Hammock, Department of Entomology and Nematology, UC Davis Comprehensive Cancer Center, University of California, Davis 95616, CA, USA
Xiao-Chi Ma, Dalian Key Laboratory of Metabolic Target Characterization and Traditional Chinese Medicine Intervention, College (Institute) of Integrative Medicine, College of Pharmacy, Dalian Medical University, Dalian 116044, People’s Republic of China.
REFERENCES
- 1.El-Sherbeni AA; El-Kadi AO The role of epoxide hydrolases in health and disease. Arch. Toxicol 2014, 88, 2013–2032. [DOI] [PubMed] [Google Scholar]
- 2.Saini P; Sareen D An overview on the enhancement of enantioselectivity and stability of microbial epoxide hydrolases. Mol. Biotechnol 2017, 59, 98–116. [DOI] [PubMed] [Google Scholar]
- 3.Newman JW; Morisseau C; Hammock BD Epoxide hydrolases: their roles and interactions with lipid metabolism. Prog. Lipid. Res 2005, 44, 1–51. [DOI] [PubMed] [Google Scholar]
- 4.Newman JW; Denton DL; Morisseau C; Koger CS; Wheelock CE; Hinton DE; Hammock BD Evaluation of fish models of soluble epoxide hydrolase inhibition. Environ. Health Perspect 2001, 109, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lauren DJ; Halarnkar PP; Hammcock BD; Hinton DE Microsomal and cytosolic epoxide hydrolase and glutathione S-transferase activities in the gill, liver, and kidney of the rainbow trout, Salmo gairdneri. Baseline levels and optimization of assay conditions. Biochem. Pharmacol 1989, 38, 881–887. [DOI] [PubMed] [Google Scholar]
- 6.Schlezinger JJ; Parker C; Zeldin DC; Stegeman JJ Arachidonic acid metabolism in the marine fish Stenotomus chrysops (Scup) and the effects of cytochrome P450 1A inducers. Arch. Biochem. Biophys 1998, 353, 265–275. [DOI] [PubMed] [Google Scholar]
- 7.Ota K; Hammock BD Cytosolic and microsomal epoxide hydrolases: differential properties in mammalian liver. Science 1980, 207, 1479–1481. [DOI] [PubMed] [Google Scholar]
- 8.Waechter F; Merdes M; Bieri F; Staubli W; Bentley P Purification and characterization of a soluble epoxide hydrolase from rabbit liver. Eur. J. Biochem 1982, 125, 457–461. [DOI] [PubMed] [Google Scholar]
- 9.Meijer J; Lundqvist G; DePierre JW Comparison of the sex and subcellular distributions, catalytic and immunochemical reactivities of hepatic epoxide hydrolases in seven mammalian species. Eur. J. Biochem 1987, 167, 269–279. [DOI] [PubMed] [Google Scholar]
- 10.Miki I; Shimizu T; Seyama Y; Kitamura S; Yamaguchi K; Sano H; Ueno H; Hiratsuka A; Watabe T Enzymic conversion of 11,12-leukotriene A4 to 11,12-dihydroxy-5,14-cis-7,9-trans-eicosatetraenoic acid. Purification of an epoxide hydrolase from the guinea pig liver cytosol. J. Biol. Chem 1989, 264, 5799–5805. [PubMed] [Google Scholar]
- 11.Lakritz J; Winder BS; Noorouz-Zadeh J; Huang TL; Buckpitt AR; Hammock BD; Plopper CG Hepatic and pulmonary enzyme activities in horses. Am. J. Vet. Res 2000, 61, 152–157. [DOI] [PubMed] [Google Scholar]
- 12.Beetham JK; Tian T; Hammock BD cDNA cloning and expression of a soluble epoxide hydrolase from human liver. Arch. Biochem. Biophys 1993, 305, 197–201. [DOI] [PubMed] [Google Scholar]
- 13.Pacifici GM; Lindberg B; Glaumann H; Rane A Styrene oxide metabolism in rhesus monkey liver: enzyme activities in subcellular fractions and in isolated hepatocytes. J. Pharmacol. Exp. Ther 1983, 226, 869–875. [PubMed] [Google Scholar]
- 14.Newman JW; Stok JE; Vidal JD; Corbin CJ; Huang Q; Hammock BD; Conley AJ Cytochrome p450-dependent lipid metabolism in preovulatory follicles. Endocrinology 2004, 145, 5097–5105. [DOI] [PubMed] [Google Scholar]
- 15.Gill SS; Hammock BD Distribution and properties of a mammalian soluble epoxide hydrase. Biochem. Pharmacol 1980, 29, 389–395. [DOI] [PubMed] [Google Scholar]
- 16.Liu Y; Zhang Y; Schmelzer K; Lee TS; Fang X; Zhu Y; Spector AA; Gill S; Morisseau C; Hammock BD; Shyy JY The antiinflammatory effect of laminar flow: the role of PPARgamma, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 16747–16752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pinot F; Grant DF; Spearow JL; Parker AG; Hammock BD Differential regulation of soluble epoxide hydrolase by clofibrate and sexual hormones in the liver and kidneys of mice. Biochem. Pharmacol 1995, 50, 501–508. [DOI] [PubMed] [Google Scholar]
- 18.Pang W; Li N; Ai D; Niu XL; Guan YF; Zhu Y Activation of peroxisome proliferator-activated receptor-gamma downregulates soluble epoxide hydrolase in cardiomyocytes. Clin. Exp. Pharmacol. Physiol 2011, 38, 358–364. [DOI] [PubMed] [Google Scholar]
- 19.Sjodin MOD; Checa A; Yang M; Dahlen SE; Wheelock AM; Eklund A; Grunewald J; Wheelock CE Soluble epoxide hydrolase derived lipid mediators are elevated in bronchoalveolar lavage fluid from patients with sarcoidosis: a cross-sectional study. Respir. Res 2018, 19, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morisseau C; Hammock BD Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu. Rev. Pharmacol 2005, 45, 311–333. [DOI] [PubMed] [Google Scholar]
- 21.Kramer J; Proschak E Phosphatase activity of soluble epoxide hydrolase. Prostaglandins Other Lipid Mediat. 2017, 133, 88–92. [DOI] [PubMed] [Google Scholar]
- 22.Gurung AB; Mayengbam B; Bhattacharjee A Discovery of novel drug candidates for inhibition of soluble epoxide hydrolase of arachidonic acid cascade pathway implicated in atherosclerosis. Comput. Biol. Chem 2018, 74, 1–11. [DOI] [PubMed] [Google Scholar]
- 23.Cronin A; Mowbray S; Durk H; Homburg S; Fleming I; Fisslthaler B; Oesch F; Arand M The N-terminal domain of mammalian soluble epoxide hydrolase is a phosphatase. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 1552–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newman JW; Morisseau C; Harris TR; Hammock BD The soluble epoxide hydrolase encoded by EPXH2 is a bifunctional enzyme with novel lipid phosphate phosphatase activity. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 1558–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sandberg M; Meijer J Structural characterization of the human soluble epoxide hydrolase gene (EPHX2). Biochem. Biophys. Res. Commun 1996, 221, 333–339. [DOI] [PubMed] [Google Scholar]
- 26.Przybyla-Zawislak BD; Srivastava PK; Vazquez-Matias J; Mohrenweiser HW; Maxwell JE; Hammock BD; Bradbury JA; Enayetallah AE; Zeldin DC; Grant DF Polymorphisms in human soluble epoxide hydrolase. Mol. Pharmacol 2003, 64, 482–490. [DOI] [PubMed] [Google Scholar]
- 27.Morisseau C; Hammock BD Gerry Brooks and epoxide hydrolases: four decades to a pharmaceutical. Pest. Manag. Sci 2008, 64, 594–609. [DOI] [PubMed] [Google Scholar]
- 28.Scott-Van Zeeland AA; Bloss CS; Tewhey R; Bansal V; Torkamani A; Libiger O; Duvvuri V; Wineinger N; Galvez L; Darst BF; Smith EN; Carson A; Pham P; Phillips T; Villarasa N; Tisch R; Zhang G; Levy S; Murray S; Chen W; Srinivasan S; Berenson G; Brandt H; Crawford S; Crow S; Fichter MM; Halmi KA; Johnson C; Kaplan AS; La Via M; Mitchell JE; Strober M; Rotondo A; Treasure J; Woodside DB; Bulik CM; Keel P; Klump KL; Lilenfeld L; Plotnicov K; Topol EJ; Shih PB; Magistretti P; Bergen AW; Berrettini W; Kaye W; Schork NJ Evidence for the role of EPHX2 gene variants in anorexia nervosa. Mol. Psychiatry 2014, 19, 724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JP; Yang SH; Kim DK; Lee H; Kim B; Cho JY; Yu KS; Paik JH; Kim M; Lim CS; Kim YS In vivo activity of epoxide hydrolase according to sequence variation affects the progression of human IgA nephropathy. Am. J. Physiol. Renal. Physiol 2011, 300, F1283–F1290. [DOI] [PubMed] [Google Scholar]
- 30.Zarriello S; Tuazon JP; Corey S; Schimmel S; Rajani M; Gorsky A; Incontri D; Hammock BD; Borlongan CV Humble beginnings with big goals: Small molecule soluble epoxide hydrolase inhibitors for treating CNS disorders. Prog. Neurobiol 2019, 172, 23–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Afanador L; Roltsch EA; Holcomb L; Campbell KS; Keeling DA; Zhang Y; Zimmer DB The Ca2+ sensor S100A1 modulates neuroinflammation, histopathology and Akt activity in the PSAPP Alzheimer’s disease mouse model. Cell Calcium. 2014, 56, 68–80. [DOI] [PubMed] [Google Scholar]
- 32.Duflot T; Roche C; Lamoureux F; Guerrot D; Bellien J Design and discovery of soluble epoxide hydrolase inhibitors for the treatment of cardiovascular diseases. Expert. Opin. Drug Discov 2014, 9, 229–243. [DOI] [PubMed] [Google Scholar]
- 33.Dileepan M; Rastle-Simpson S; Greenberg Y; Wijesinghe DS; Kumar NG; Yang J; Hwang SH; Hammock BD; Sriramarao P; Rao SP Effect of dual sEH/COX-2 inhibition on allergen-induced airway inflammation. Front. Pharmacol 2019, 10, 1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hashimoto K Role of soluble epoxide hydrolase in metabolism of PUFAs in psychiatric and neurological disorders. Front. Pharmacol 2019, 10, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nayeem MA Role of oxylipins in cardiovascular diseases. Acta Pharmacol. Sin 2018, 39, 1142–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harder DR; Campbell WB; Roman RJ Role of cytochrome P-450 enzymes and metabolites of arachidonic acid in the control of vascular tone. J. Vasc. Res 1995, 32, 79–92. [DOI] [PubMed] [Google Scholar]
- 37.Roman RJ P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol. Rev 2002, 82, 131–185. [DOI] [PubMed] [Google Scholar]
- 38.Hu S; Kim HS Activation of K+ channel in vascular smooth muscles by cytochrome P450 metabolites of arachidonic acid. Eur. J. Pharmacol 1993, 230, 215–221. [DOI] [PubMed] [Google Scholar]
- 39.Kroetz DL; Zeldin DC Cytochrome P450 pathways of arachidonic acid metabolism. Curr. Opin. Lipidol 2002, 13, 273–283. [DOI] [PubMed] [Google Scholar]
- 40.Node K; Ruan XL; Dai J; Yang SX; Graham L; Zeldin DC; Liao JK Activation of Galpha s mediates induction of tissue-type plasminogen activator gene transcription by epoxyeicosatrienoic acids. J. Biol. Chem 2001, 276, 15983–15989. [DOI] [PubMed] [Google Scholar]
- 41.Munzenmaier DH; Harder DR Cerebral microvascular endothelial cell tube formation: role of astrocytic epoxyeicosatrienoic acid release. Am. J. Physiol. Heart Circ. Physiol 2000, 278, H1163–H1167. [DOI] [PubMed] [Google Scholar]
- 42.Node K; Huo Y; Ruan X; Yang B; Spiecker M; Ley K; Zeldin DC; Liao JK Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 1999, 285, 1276–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campbell WB New role for epoxyeicosatrienoic acids as anti-inflammatory mediators. Trends Pharmacol. Sci 2000, 21, 125–127. [DOI] [PubMed] [Google Scholar]
- 44.Zeldin DC Epoxygenase pathways of arachidonic acid metabolism. J. Biol. Chem 2001, 276, 36059–36062. [DOI] [PubMed] [Google Scholar]
- 45.Imig JD; Navar LG; Roman RJ; Reddy KK; Falck JR Actions of epoxygenase metabolites on the preglomerular vasculature. J. Am. Soc. Nephrol 1996, 7, 2364–2370. [DOI] [PubMed] [Google Scholar]
- 46.Falck JR; Krishna UM; Reddy YK; Kumar PS; Reddy KM; Hittner SB; Deeter C; Sharma KK; Gauthier KM; Campbell WB Comparison of vasodilatory properties of 14,15-EET analogs: structural requirements for dilation. Am. J. Physiol. Heart Circ. Physiol 2003, 284, H337–H349. [DOI] [PubMed] [Google Scholar]
- 47.Lee HC; Lu T; Weintraub NL; VanRollins M; Spector AA; Shibata EF Effects of epoxyeicosatrienoic acids on the cardiac sodium channels in isolated rat ventricular myocytes. J. Physiol 1999, 519, 153–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fang X; Moore SA; Stoll LL; Rich G; Kaduce TL; Weintraub NL; Spector AA 14,15-Epoxyeicosatrienoic acid inhibits prostaglandin E2 production in vascular smooth muscle cells. Am. J. Physiol 1998, 275, H2113–H2121. [DOI] [PubMed] [Google Scholar]
- 49.Fang X; Weintraub NL; Stoll LL; Spector AA Epoxyeicosatrienoic acids increase intracellular calcium concentration in vascular smooth muscle cells. Hypertension 1999, 34, 1242–1246. [DOI] [PubMed] [Google Scholar]
- 50.Hirt DL; Capdevila J; Falck JR; Breyer MD; Jacobson HR Cytochrome P450 metabolites of arachidonic acid are potent inhibitors of vasopressin action on rabbit cortical collecting duct. J. Clin. Invest 1989, 84, 1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weintraub NL; Fang X; Kaduce TL; VanRollins M; Chatterjee P; Spector AA Potentiation of endothelium-dependent relaxation by epoxyeicosatrienoic acids. Circ. Res 1997, 81, 258–267. [DOI] [PubMed] [Google Scholar]
- 52.Oltman CL; Weintraub NL; VanRollins M; Dellsperger KC Epoxyeicosatrienoic acids and dihydroxyeicosatrienoic acids are potent vasodilators in the canine coronary microcirculation. Circ. Res 1998, 83, 932–939. [DOI] [PubMed] [Google Scholar]
- 53.Jones RD; Liao J; Tong X; Xu DD; Sun LY; Li HN; Yang GY Epoxy-oxylipins and soluble epoxide hydrolase metabolic pathway as targets for NSAID-induced gastroenteropathy and inflammation-associated carcinogenesis. Front. Pharmacol 2019, 10, 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harris TR; Hammock BD Soluble epoxide hydrolase: gene structure, expression and deletion. Gene 2013, 526, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wagner KM; McReynolds CB; Schmidt WK; Hammock BD Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol. Ther 2017, 180, 62–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shen HC Soluble epoxide hydrolase inhibitors: a patent review. Expert. Opin. Ther. Pat 2010, 20, 941–956. [DOI] [PubMed] [Google Scholar]
- 57.Mullin CA; Hammock BD Chalcone oxides - potent selective inhibitors of cytosolic epoxide hydrolase. Arch. Biochem. Biophys 1982, 216, 423–439. [DOI] [PubMed] [Google Scholar]
- 58.Morisseau C; Goodrow MH; Dowdy D; Zheng J; Greene JF; Sanborn JR; Hammock BD Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 8849–8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Podolin PL; Bolognese BJ; Foley JF; Long E 3rd; Peck B; Umbrecht S; Zhang X; Zhu P; Schwartz B; Xie W; Quinn C; Qi H; Sweitzer S; Chen S; Galop M; Ding Y; Belyanskaya SL; Israel DI; Morgan BA; Behm DJ; Marino JP Jr.; Kurali E; Barnette MS; Mayer RJ; Booth-Genthe CL; Callahan JF In vitro and in vivo characterization of a novel soluble epoxide hydrolase inhibitor. Prostaglandins Other Lipid Mediat. 2013, 25–31. [DOI] [PubMed] [Google Scholar]
- 60.Lukin A; Kramer J; Hartmann M; Weizel L; Hernandez-Olmos V; Falahati K; Burghardt I; Kalinchenkova N; Bagnyukova D; Zhurilo N; Rautio J; Forsberg M; Ihalainen J; Auriola S; Leppanen J; Konstantinov I; Pogoryelov D; Proschak E; Dar’in D; Krasavin M Discovery of polar spirocyclic orally bioavailable urea inhibitors of soluble epoxide hydrolase. Bioorg. Chem 2018, 80, 655–667. [DOI] [PubMed] [Google Scholar]
- 61.Yu Z; Xu F; Huse LM; Morisseau C; Draper AJ; Newman JW; Parker C; Graham L; Engler MM; Hammock BD; Zeldin DC; Kroetz DL Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ. Res 2000, 87, 992–998. [DOI] [PubMed] [Google Scholar]
- 62.Fang X; Kaduce TL; Weintraub NL; Harmon S; Teesch LM; Morisseau C; Thompson DA; Hammock BD; Spector AA Pathways of epoxyeicosatrienoic acid metabolism in endothelial cells. Implications for the vascular effects of soluble epoxide hydrolase inhibition. J. Biol. Chem 2001, 276, 14867–14874. [DOI] [PubMed] [Google Scholar]
- 63.Gomez GA; Morisseau C; Hammock BD; Christianson DW Human soluble epoxide hydrolase: structural basis of inhibition by 4-(3-cyclohexylureido)-carboxylic acids. Protein Sci. 2006, 15, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim IH; Morisseau C; Watanabe T; Hammock BD Design, synthesis, and biological activity of 1,3-disubstituted ureas as potent inhibitors of the soluble epoxide hydrolase of increased water solubility. J. Med. Chem 2004, 47, 2110–2122. [DOI] [PubMed] [Google Scholar]
- 65.Morisseau CG, M.H.; Dowdy D; Zheng J; Greene JF; Sanborn JR and Hammock BD Structural refinement of inhibitors of urea-based souble epoxide hydrolases. Biochem. Pharmacol 2002, 63, 1599–1608. [DOI] [PubMed] [Google Scholar]
- 66.Imig JD; Zhao X; Capdevila JH; Morisseau C; Hammock BD Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension 2002, 39, 690–694. [DOI] [PubMed] [Google Scholar]
- 67.Honetschlagerova Z; Sporkova A; Kopkan L; Huskova Z; Hwang SH; Hammock BD; Imig JD; Kramer HJ; Kujal P; Vernerova Z; Chabova VC; Tesar V; Cervenka L Inhibition of soluble epoxide hydrolase improves the impaired pressure-natriuresis relationship and attenuates the development of hypertension and hypertension-associated end-organ damage in Cyp1a1-Ren-2 transgenic rats. J. Hypertens 2011, 29, 1590–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gross GJ; Nithipatikom K Soluble epoxide hydrolase: a new target for cardioprotection. Curr. Opin. Investig. Drugs 2009, 10, 253–258. [PMC free article] [PubMed] [Google Scholar]
- 69.Schmelzer KR; Kubala L; Newman JW; Kim IH; Eiserich JP; Hammock BD Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 9772–9777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McElroy NR; Jurs PC QSAR and classification of murine and human soluble epoxide hydrolase inhibition by urea-like compounds. J. Med. Chem 2003, 46, 1066–1080. [DOI] [PubMed] [Google Scholar]
- 71.Jones PD; Tsai HJ; Do ZN; Morisseau C; Hammock BD Synthesis and SAR of conformationally restricted inhibitors of soluble epoxide hydrolase. Bioorg. Med .Chem. Lett 2006, 16, 5212–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schafer A; Neschen S; Kahle M; Sarioglu H; Gaisbauer T; Imhof A; Adamski J; Hauck SM; Ueffing M The epoxyeicosatrienoic acid pathway enhances hepatic insulin signaling and is repressed in insulin-resistant mouse liver. Mol. Cell. Proteomics 2015, 14, 2764–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hwang SH; Tsai HJ; Liu JY; Morisseau C; Hammock BD Orally bioavailable potent soluble epoxide hydrolase inhibitors. J. Med. Chem 2007, 50, 3825–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gui Y; Li D; Chen J; Wang Y; Hu J; Liao C; Deng L; Xiang Q; Yang T; Du X; Zhang S; Xu D Soluble epoxide hydrolase inhibitors, t-AUCB, downregulated miR-133 in a mouse model of myocardial infarction. Lipids Health. Dis 2018, 17, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li J; Tang C; Li L; Li R; Fan Y Quercetin sensitizes glioblastoma to t-AUCB by dual inhibition of Hsp27 and COX-2 in vitro and in vivo. J. Exp. Clin. Cancer Res 2016, 35, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shen L; Peng HC; Zhao SP; Xu DY A potent soluble epoxide hydrolase inhibitor, t-AUCB, modulates cholesterol balance and oxidized low density lipoprotein metabolism in adipocytes in vitro. Biol. Chem 2014, 395, 443–451. [DOI] [PubMed] [Google Scholar]
- 77.Roche C; Guerrot D; Harouki N; Duflot T; Besnier M; Remy-Jouet I; Renet S; Dumesnil A; Lejeune A; Morisseau C; Richard V; Bellien J Impact of soluble epoxide hydrolase inhibition on early kidney damage in hyperglycemic overweight mice. Prostaglandins Other Lipid Mediat. 2015, 120, 148–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Iyer A; Kauter K; Alam MA; Hwang SH; Morisseau C; Hammock BD; Brown L Pharmacological inhibition of soluble epoxide hydrolase ameliorates diet-induced metabolic syndrome in rats. Exp. Diabetes Res 2012, 2012, 758614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Burmistrov V; Morisseau C; Danilov D; Harris TR; Dalinger I; Vatsadze I; Shkineva T; Butov GM; Hammock BD 1,3-Disubstituted and 1,3,3-trisubstituted adamantyl-ureas with isoxazole as soluble epoxide hydrolase inhibitors. Bioorg. Med. Chem. Lett 2015, 25, 5514–5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Burmistrov V; Morisseau C; Harris TR; Butov G; Hammock BD Effects of adamantane alterations on soluble epoxide hydrolase inhibition potency, physical properties and metabolic stability. Bioorg. Chem 2018, 76, 510–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rose TE; Morisseau C; Liu JY; Inceoglu B; Jones PD; Sanborn JR; Hammock BD 1-Aryl-3-(1-acylpiperidin-4-yl)urea inhibitors of human and murine soluble epoxide hydrolase: structure-activity relationships, pharmacokinetics, and reduction of inflammatory pain. J. Med. Chem 2010, 53, 7067–7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tsai HJ; Hwang SH; Morisseau C; Yang J; Jones PD; Kasagami T; Kim IH; Hammock BD Pharmacokinetic screening of soluble epoxide hydrolase inhibitors in dogs. Eur. J. Pharm. Sci 2010, 40, 222–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee KS; Liu JY; Wagner KM; Pakhomova S; Dong H; Morisseau C; Fu SH; Yang J; Wang P; Ulu A; Mate CA; Nguyen LV; Hwang SH; Edin ML; Mara AA; Wulff H; Newcomer ME; Zeldin DC; Hammock BD Optimized inhibitors of soluble epoxide hydrolase improve in vitro target residence time and in vivo efficacy. J. Med. Chem 2014, 57, 7016–7030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Manickam M; Pillaiyar T; Boggu P; Venkateswararao E; Jalani HB; Kim ND; Lee SK; Jeon JS; Kim SK; Jung SH Discovery of enantioselectivity of urea inhibitors of soluble epoxide hydrolase. Eur. J. Med. Chem 2016, 117, 113–124. [DOI] [PubMed] [Google Scholar]
- 85.Burmistrov V; Morisseau C; Pitushkin D; Karlov D; Fayzullin RR; Butov GM; Hammock BD Adamantyl thioureas as soluble epoxide hydrolase inhibitors. Bioorg. Med. Chem. Lett 2018, 28, 2302–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim IH; Heirtzler FR; Morisseau C; Nishi K; Tsai HJ; Hammock BD Optimization of amide-based inhibitors of soluble epoxide hydrolase with improved water solubility. J. Med. Chem 2005, 48, 3621–3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eldrup AB; Soleymanzadeh F; Taylor SJ; Muegge I; Farrow NA; Joseph D; McKellop K; Man CC; Kukulka A; De Lombaert S Structure-based optimization of arylamides as inhibitors of soluble epoxide hydrolase. J. Med. Chem 2009, 52, 5880–5895. [DOI] [PubMed] [Google Scholar]
- 88.Han F; Perrin RJ; Wang Q; Wang Y; Perlmutter JS; Morris JC; Benzinger TLS; Xu JB Neuroinflammation and myelin status in Alzheimer’s disease, Parkinson’s disease, and normal aging brains: a small sample study. Parkinsons Dis. 2019, 2019, 7975407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Taylor SJ; Soleymanzadeh F; Eldrup AB; Farrow NA; Muegge I; Kukulka A; Kabcenell AK; De Lombaert S Design and synthesis of substituted nicotinamides as inhibitors of soluble epoxide hydrolase. Bioorg. Med. Chem. Lett 2009, 19, 5864–5868. [DOI] [PubMed] [Google Scholar]
- 90.Anandan SK; Do ZN; Webb HK; Patel DV; Gless RD Non-urea functionality as the primary pharmacophore in soluble epoxide hydrolase inhibitors. Bioorg. Med. Chem. Lett 2009, 19, 1066–1070. [DOI] [PubMed] [Google Scholar]
- 91.Anandan SK; Webb HK; Do ZN; Gless RD Unsymmetrical non-adamantyl N,N’-diaryl urea and amide inhibitors of soluble expoxide hydrolase. Bioorg. Med. Chem. Lett 2009, 19, 4259–4263. [DOI] [PubMed] [Google Scholar]
- 92.Piirainen S; Youssef A; Song C; Kalueff AV; Landreth GE; Malm T; Tian L Psychosocial stress on neuroinflammation and cognitive dysfunctions in Alzheimer’s disease: the emerging role for microglia? Neurosci. Biobehav. Rev 2017, 77, 148–164. [DOI] [PubMed] [Google Scholar]
- 93.Karami L; Saboury AA; Rezaee E; Tabatabai SA Investigation of the binding mode of 1, 3, 4-oxadiazole derivatives as amide-based inhibitors for soluble epoxide hydrolase (sEH) by molecular docking and MM-GBSA. Eur. Biophys. J 2017, 46, 445–459. [DOI] [PubMed] [Google Scholar]
- 94.Kim IH; Lee IH; Nishiwaki H; Hammock BD; Nishi K Structure–activity relationships of substituted oxyoxalamides as inhibitors of the human soluble epoxide hydrolase. Bioorg. Med Chem 2014, 22, 1163–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim IH; Park YK; Nishiwaki H; Hammock BD; Nishi K Structure-activity relationships of amide-phosphonate derivatives as inhibitors of the human soluble epoxide hydrolase. Bioorg. Med. Chem 2015, 23, 7199–7210. [DOI] [PubMed] [Google Scholar]
- 96.Zavareh ER; Hedayati M; Rad LH; Azin K; Soraya S; Faizi Soraya; Mehrdad F; Sayyed AT Design, synthesis and biological evaluation of some oxadiazole derivatives as novel amide-based inhibitors of soluble epoxide hydrolase. Lett. Drug Des. Discov 2014, 11, 721–730. [Google Scholar]
- 97.Kim IH; Park YK; Hammock BD; Nishi K Structure-activity relationships of cycloalkylamide derivatives as inhibitors of the soluble epoxide hydrolase. J. Med. Chem 2011, 54, 1752–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xie Y; Liu Y; Gong G; Smith DH; Yan F; Rinderspacher A; Feng Y; Zhu Z; Li X; Deng SX; Branden L; Vidovic D; Chung C; Schurer S; Morisseau C; Hammock BD; Landry DW Discovery of potent non-urea inhibitors of soluble epoxide hydrolase. Bioorg. Med. Chem. Lett 2009, 19, 2354–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pecic S; Deng SX; Morisseau C; Hammock BD; Landry DW Design, synthesis and evaluation of non-urea inhibitors of soluble epoxide hydrolase. Bioorg. Med. Chem. Lett 2012, 22, 601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xing L; McDonald JJ; Kolodziej SA; Kurumbail RG; Williams JM; Warren CJ; O’Neal JM; Skepner JE; Roberds SL Discovery of potent inhibitors of soluble epoxide hydrolase by combinatorial library design and structure-based virtual screening. J. Med. Chem 2011, 54, 1211–1222. [DOI] [PubMed] [Google Scholar]
- 101.Deng H; O’Keefe H; Davie CP; Lind KE; Acharya RA; Franklin GJ; Larkin J; Matico R; Neeb M; Thompson MM; Lohr T; Gross JW; Centrella PA; O’Donovan GK; Bedard KL; van Vloten K; Mataruse S; Skinner SR; Belyanskaya SL; Carpenter TY; Shearer TW; Clark MA; Cuozzo JW; Arico-Muendel CC; Morgan BA Discovery of highly potent and selective small molecule ADAMTS-5 inhibitors that inhibit human cartilage degradation via encoded library technology (ELT). J. Med. Chem 2012, 55, 7061–7079. [DOI] [PubMed] [Google Scholar]
- 102.Belyanskaya SL; Ding Y; Callahan JF; Lazaar AL; Israel DI Discovering drugs with DNA-encoded library technology: from concept to clinic with an inhibitor of soluble epoxide hydrolase. Chembiochem. 2017, 18, 837–842. [DOI] [PubMed] [Google Scholar]
- 103.Thalji RK; McAtee JJ; Belyanskaya S; Brandt M; Brown GD; Costell MH; Ding Y; Dodson JW; Eisennagel SH; Fries RE; Gross JW; Harpel MR; Holt DA; Israel DI; Jolivette LJ; Krosky D; Li H; Lu Q; Mandichak T; Roethke T; Schnackenberg CG; Schwartz B; Shewchuk LM; Xie W; Behm DJ; Douglas SA; Shaw AL; Marino JP Discovery of 1-(1,3,5-triazin-2-yl)piperidine-4-carboxamides as inhibitors of soluble epoxide hydrolase. Bioorg. Med. Chem. Lett 2013, 23, 3584–3588. [DOI] [PubMed] [Google Scholar]
- 104.Hiesinger K; Wagner KM; Hammock BD; Proschak E; Hwang SH Development of multitarget agents possessing soluble epoxide hydrolase inhibitory activity. Prostaglandins Other Lipid Mediat. 2019, 140, 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kodani SD; Bhakta S; Hwang SH; Pakhomova S; Newcomer ME; Morisseau C; Hammock BD Identification and optimization of soluble epoxide hydrolase inhibitors with dual potency towards fatty acid amide hydrolase. Bioorg. Med. Chem. Lett 2018, 28, 762–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hefke L; Hiesinger K; Zhu WF; Kramer JS; Proschak E Computer-aided fragment growing strategies to design dual inhibitors of soluble epoxide hydrolase and LTA4 hydrolase. ACS Med. Chem. Lett 2020, 11, 1244–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang G; Panigrahy D; Hwang SH; Yang J; Mahakian LM; Wettersten HI; Liu JY; Wang Y; Ingham ES; Tam S; Kieran MW; Weiss RH; Ferrara KW; Hammock BD Dual inhibition of cyclooxygenase-2 and soluble epoxide hydrolase synergistically suppresses primary tumor growth and metastasis. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 11127–11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.la Buscato E; Blocher R; Lamers C; Klingler FM; Hahn S; Steinhilber D; Schubert-Zsilavecz M; Proschak E Design and synthesis of dual modulators of soluble epoxide hydrolase and peroxisome proliferator-activated receptors. J. Med. Chem 2012, 55, 10771–10775. [DOI] [PubMed] [Google Scholar]
- 109.Meirer K; Rodl CB; Wisniewska JM; George S; Hafner AK; Estel la Buscató; Klingler FM; Hahn S; Berressem D; Wittmann SK; Steinhilber D; Hofmann B; Proschak E Synthesis and structure-activity relationship studies of novel dual inhibitors of soluble epoxide hydrolase and 5-lipoxygenase. J. Med. Chem 2013, 56, 1777–1781. [DOI] [PubMed] [Google Scholar]
- 110.Hwang SH; Wagner KM; Morisseau C; Liu JY; Dong H; Wecksler AT; Hammock BD Synthesis and structure-activity relationship studies of urea-containing pyrazoles as dual inhibitors of cyclooxygenase-2 and soluble epoxide hydrolase. J. Med. Chem 2011, 54, 3037–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sun CC; Zhang CY; Duan JX; Guan XX; Yang HH; Jiang HL; Hammock BD; Hwang SH; Zhou Y; Guan CX; Liu SK; Zhang J PTUPB ameliorates high-fat diet-induced non-alcoholic fatty liver disease via inhibiting NLRP3 inflammasome activation in mice. Biochem. Biophys. Res. Commun 2020, 523, 1020–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang YF; Sun CC; Duan JX; Yang HH; Zhang CY; Xiong JB; Zhong WJ; Zu C; Guan XX; Jiang HL; Hammock BD; Hwang SH; Zhou Y; Guan CX A COX-2/sEH dual inhibitor PTUPB ameliorates cecal ligation and puncture-induced sepsis in mice via anti-inflammation and anti-oxidative stress. Biomed. Pharmacother 2020, 126, 109907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang CY; Duan JX; Yang HH; Sun CC; Zhong WJ; Tao JH; Guan XX; Jiang HL; Hammock BD; Hwang SH; Zhou Y; Guan CX COX-2/sEH dual inhibitor PTUPB alleviates bleomycin-induced pulmonary fibrosis in mice via inhibiting senescence. FEBS. J 2019, 287, 1666–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lakkappa N; Krishnamurthy PT; Yamjala K; Hwang SH; Hammock BD; Babu B Evaluation of antiparkinson activity of PTUPB by measuring dopamine and its metabolites in Drosophila melanogaster: LC-MS/MS method development. J. Pharm. Biomed. Anal 2018, 149, 457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li J; Zhou Y; Wang H; Gao Y; Li L; Hwang SH; Ji X; Hammock BD COX-2/sEH dual inhibitor PTUPB suppresses glioblastoma growth by targeting epidermal growth factor receptor and hyaluronan mediated motility receptor. Oncotarget 2017, 8, 87353–87363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gartung A; Yang J; Sukhatme VP; Bielenberg DR; Fernandes D; Chang J; Schmidt BA; Hwang SH; Zurakowski D; Huang S; Kieran MW; Hammock BD; Panigrahy D Suppression of chemotherapy-induced cytokine/lipid mediator surge and ovarian cancer by a dual COX-2/sEH inhibitor. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 1698–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Meirer K; Glatzel D; Kretschmer S; Wittmann SK; Hartmann M; Blöcher R; Angioni C; Geisslinger G; Steinhilber D; Hofmann B; Fürst R; Proschak E Design, synthesis and cellular characterization of a dual inhibitor of 5-lipoxygenase and soluble epoxide hydrolase. Molecules 2016, 22, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Moser D; Wisniewska JM; Hahn S; Achenbach J; Buscató EL; Klingler FM; Hofmann B; Steinhilber D; Proschak E Dual-target virtual screening by pharmacophore elucidation and molecular shape filtering. ACS Med. Chem. Lett 2012, 3, 155–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Achenbach J; Klingler FM; Blöcher R; Moser D; Häfner AK; Rödl CB; Kretschmer S; Krüger B; Löhr F; Stark H; Hofmann B; Steinhilber D; Proschak E Exploring the chemical space of multitarget ligands using aligned self-organizing maps. ACS Med. Chem. Lett 2013, 4, 1169–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nandha B; Ramareddy SA; Kuntal H Synthesis of substituted fluorobenzimidazoles as inhibitors of 5-lipoxygenase and soluble epoxide hydrolase for anti-inflammatory activity. Arch. Pharm 2018, 351, e1800030. [DOI] [PubMed] [Google Scholar]
- 121.Klegeris A. a. M., P.L. Toxicity of human monocytic THP-1 cells and microglia toward SH-SY5Y neuroblastoma cells is reduced by inhibitors of 5-lipoxygenase and its activating protein FLAP. J. Leukoc. Biol 2003, 73, 369–378. [DOI] [PubMed] [Google Scholar]
- 122.Kent SE; Boyce M; Diamant Z; Singh D; O’Connor BJ; Saggu PS; Norris S The 5-lipoxygenase-activating protein inhibitor, GSK2190915, attenuates the early and late responses to inhaled allergen in mild asthma. Clin. Exp. Allergy 2013, 43, 177–186. [DOI] [PubMed] [Google Scholar]
- 123.Liu JY; Yang J; Inceoglu B; Qiu H; Ulu A; Hwang SH; Chiamvimonvat N; Hammock BD Inhibition of soluble epoxide hydrolase enhances the anti-inflammatory effects of aspirin and 5-lipoxygenase activation protein inhibitor in a murine model. Biochem. Pharmacol 2010, 79, 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Garscha U; Romp E; Pace S; Rossi A; Temml V; Schuster D; König S; Gerstmeier J; Liening S; Werner M; Atze H; Wittmann S; Weinigel C; Rummler S; Scriba GK; Sautebin L; Werz O Pharmacological profile and efficiency in vivo of diflapolin, the first dual inhibitor of 5-lipoxygenase-activating protein and soluble epoxide hydrolase. Sci. Rep 2017, 7, 9398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vieider L; Romp E; Temml V; Fischer J; Kretzer C; Schoenthaler M; Taha A; Hernández-Olmos V; Sturm S; Schuster D; Werz O; Garscha U; Matuszczak B Synthesis, biological evaluation and structure-activity relationships of diflapolin analogues as dual sEH/FLAP inhibitors. ACS Med. Chem. Lett 2018, 10, 62–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kodani SD; Wan D; Wagner KM; Hwang SH; Morisseau C; Hammock BD Design and potency of dual soluble epoxide hydrolase/fatty acid amide hydrolase inhibitors. ACS Omega 2018, 3, 14076–14086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sasso O; Wagner K; Morisseau C; Inceoglu B; Hammock BD; Piomelli D Peripheral FAAH and soluble epoxide hydrolase inhibitors are synergistically antinociceptive. Pharmacol. Res 2015, 97, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Spector AA; Norris AW Action of epoxyeicosatrienoic acids on cellular function. Am. J. Physiol. Cell. Physiol 2007, 292, C996–C1012. [DOI] [PubMed] [Google Scholar]
- 129.Imig JD; Walsh KA; Hye Khan MA; Nagasawa T; Cherian-Shaw M; Shaw SM; Hammock BD Soluble epoxide hydrolase inhibition and peroxisome proliferator activated receptor γ agonist improve vascular function and decrease renal injury in hypertensive obese rats. Exp. Biol. Med 2012, 237, 1402–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Blocher R; Lamers C; Wittmann SK; Merk D; Hartmann M; Weizel L; Diehl O; Bruggerhoff A; Boss M; Kaiser A; Schader T; Gobel T; Grundmann M; Angioni C; Heering J; Geisslinger G; Wurglics M; Kostenis E; Brune B; Steinhilber D; Schubert-Zsilavecz M; Kahnt AS; Proschak E N-Benzylbenzamides: a novel merged scaffold for orally available dual soluble epoxide hydrolase/peroxisome proliferator-activated receptor gamma modulators. J. Med. Chem 2016, 59, 61–81. [DOI] [PubMed] [Google Scholar]
- 131.Hye Khan MA; Kolb L; Skibba M; Hartmann M; Blocher R; Proschak E; Imig JD A novel dual PPAR-gamma agonist/sEH inhibitor treats diabetic complications in a rat model of type 2 diabetes. Diabetologia 2018, 61, 2235–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Schierle S; Flauaus C; Heitel P; Willems S; Schmidt J; Kaiser A; Weizel L; Goebel T; Kahnt AS; Geisslinger G; Steinhilber D; Wurglics M; Rovati GE; Schmidtko A; Proschak E; Merk D Boosting anti-inflammatory potency of zafirlukast by designed polypharmacology. J. Med. Chem 2018, 61, 5758–5764. [DOI] [PubMed] [Google Scholar]
- 133.Luan ZL; Huo XK; Dong PP; Tian XG; Sun CP; Lv X; Feng L; Ning J; Wang C; Zhang BJ; Ma XC Highly potent non-steroidal FXR agonists protostane-type triterpenoids: structure-activity relationship and mechanism. Eur. J. Med. Chem 2019, 182, 111652. [DOI] [PubMed] [Google Scholar]
- 134.Schierle S; Helmstädter M; Schmidt J; Hartmann M; Horz M; Kaiser A; Weizel L; Heitel P; Proschak A; Hernandez-Olmos V; Proschak E; Merk D Dual farnesoid X receptor/soluble epoxide hydrolase modulators derived from zafirlukast. ChemMedChem. 2020, 15, 50–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Schmidt J; Rotter M; Weiser T; Wittmann S; Weizel L; Kaiser A; Heering J; Goebel T; Angioni C; Wurglics M; Paulke A; Geisslinger G; Kahnt A; Steinhilber D; Proschak E; Merk D A dual modulator of farnesoid X receptor and soluble epoxide hydrolase to counter nonalcoholic steatohepatitis. J. Med. Chem 2017, 60, 7703–7724. [DOI] [PubMed] [Google Scholar]
- 136.Nagao M; Ishikawa F; Tahira T; Ochiai M; Sugimura T Activation of rat and human c-raf(−1) by rearrangement. Princess Takamatsu Symp. 1986, 17, 75–84. [PubMed] [Google Scholar]
- 137.Chang F; Steelman LS; Shelton JG; Lee JT; Navolanic PM; Blalock WL; Franklin R; McCubrey JA Regulation of cell cycle progression and apoptosis by the Ras/Raf/MEK/ERK pathway (review). Int. J. Oncol 2003, 22, 469–480. [PubMed] [Google Scholar]
- 138.Wilhelm S; Chien DS BAY 43–9006: Preclinical data. Curr. Pharm. Des 2002, 8, 2255–2257. [DOI] [PubMed] [Google Scholar]
- 139.Liu JY; Park SH; Morisseau C; Hwang SH; Hammock BD; Weiss RH Sorafenib has soluble epoxide hydrolase inhibitory activity, which contributes to its effect profile in vivo. Mol. Cancer Ther 2009, 8, 2193–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Liao J; Hwang SH; Li H; Yang Y; Yang J; Wecksler AT; Liu JY; Hammock BD; Yang GY Inhibition of mutant KrasG12D-initiated murine pancreatic carcinoma growth by a dual c-Raf and soluble epoxide hydrolase inhibitor t-CUPM. Cancer Lett. 2016, 371, 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Baell JB Feeling nature’s PAINS: natural products, natural product drugs, and pan assay interference compounds (PAINS). J. Nat. Prod 2016, 79, 616–628. [DOI] [PubMed] [Google Scholar]
- 142.Butler MS The role of natural product chemistry in drug discovery. J. Nat. Prod 2004, 67, 2141–2153. [DOI] [PubMed] [Google Scholar]
- 143.Lee GH; Oh SJ; Lee SY; Lee JY; Ma JY; Kim YH; Kim SK Discovery of soluble epoxide hydrolase inhibitors from natural products. Food Chem. Toxicol 2014, 64, 225–230. [DOI] [PubMed] [Google Scholar]
- 144.Liu ZB; Sun CP; Xu JX; Morisseau C; Hammock BD; Qiu F Phytochemical constituents from Scutellaria baicalensis in soluble epoxide hydrolase inhibition: Kinetics and interaction mechanism merged with simulations. Int. J. Biol. Macromol 2019, 133, 1187–1193. [DOI] [PubMed] [Google Scholar]
- 145.Wang CY; Lee S; Jang HJ; Su XD; Wang HS; Kim YH; Yang SY Inhibition potential of phenolic constituents from the aerial parts of Tetrastigma hemsleyanum against soluble epoxide hydrolase and nitric oxide synthase. J. Enzyme Inhib. Med. Chem 2019, 34, 753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Le BV; Kim JH; Lee JS Soluble epoxide hydrolase inhibitory activity of phenolic glycosides from Polygala tenuifolia and in silico approach. Med. Chem. Res 2018, 27, 726–734. [Google Scholar]
- 147.Kitamura S; Morisseau C; Inceoglu B; Kamita SG; De Nicola GR; Nyegue M; Hammock BD Potent natural soluble epoxide hydrolase inhibitors from Pentadiplandra brazzeana Baillon: Synthesis, quantification, and measurement of biological activities in vitro and in vivo. PLoS One 2015, 10, e0117438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tsopmo A; Ngnokam D; Ngamga D; Ayafor JF; Sterner O Urea derivatives from Pentadiplandra brazzeana. J. Nat. Prod 1999, 62, 1435–1436. [DOI] [PubMed] [Google Scholar]
- 149.Ray A; Chand L; Salvadourea DS New urea derivative from Salvadora persica. Chem. Ind 1975, 12, 517–518. [Google Scholar]
- 150.Bahroun A; Damak M Contribution to the study of Lepidium sativum (Cruciferae). Structure of a new compound isolated from the seed: lepidine. J. de la Société Chimique de Tunisie 1985, 2, 15–24. [Google Scholar]
- 151.Sashidhara KV; Rosaiah JN; Tyagi E; Shukla R; Raghubir R; Rajendran SM Rare dipeptide and urea derivatives from roots of Moringa oleifera as potential anti-inflammatory and antinociceptive agents. Eur. J. Med. Chem 2009, 44, 432–436. [DOI] [PubMed] [Google Scholar]
- 152.Kitamura S; Morisseau C; Harris TR; Inceoglu B; Hammock BD Occurrence of urea-based soluble epoxide hydrolase inhibitors from the plants in the order Brassicales. PLoS One. 2017, 12, e0176571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kim JH; Jo YD; Jin CH Isolation of soluble epoxide hydrolase inhibitor of capsaicin analogs from Capsicum chinense Jacq. cv. Habanero. Int. J. Biol. Macromol 2019, 135, 1202–1207. [DOI] [PubMed] [Google Scholar]
- 154.Kim JH; Kim HY; Kang SY; Kim YH; Jin CH Soluble epoxide hydrolase inhibitory activity of components isolated from Apios americana Medik. Molecules 2017, 22, 1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Thao NP; Luyen BTT; Kim JH Soluble epoxide hydrolase inhibitory activity by rhizomes of Kaempferia parviflora Wall. ex Baker. Med. Chem. Res 2016, 25, 704–711. [Google Scholar]
- 156.Morgan AMA; Kim JH; Kim SK A new flavonol glycoside from the leaves of Boscia senegalensis. Bull. Korean Chem. Soc 2014, 35, 3447–3452. [Google Scholar]
- 157.Su XD; Li W; Kim JE Prenyl-flavonoids from Epimedium koreanum Nakai and their soluble epoxide hydrolase and tyrosinase inhibitory activities. Med. Chem. Res 2017, 26, 2761–2767. [Google Scholar]
- 158.Bai MM; Shi W; Tian JM; Lei M; Kim JH; Sun YN; Kim YH; Gao JM Soluble epoxide hydrolase inhibitory and anti-inflammatory components from the leaves of Eucommia ulmoides Oliver (duzhong). J. Agric. Food Chem 2015, 63, 2198–2205. [DOI] [PubMed] [Google Scholar]
- 159.Thao NP; Luyen BTT; Lee JS; Kim JH; Dat NT; Kim YH Inhibition potential of cycloartane-type glycosides from the roots of Cimicifuga dahurica against soluble epoxide hydrolase. J. Nat. Prod 2017, 80, 1867–1875. [DOI] [PubMed] [Google Scholar]
- 160.Thao NP; Kim JH; Thuy Luyen BT; Dat NT; Kim YH In silico investigation of cycloartane triterpene derivatives from Cimicifuga dahurica (Turcz.) Maxim. roots for the development of potent soluble epoxide hydrolase inhibitors. Int. J. Biol. Macromol 2017, 98, 526–534. [DOI] [PubMed] [Google Scholar]
- 161.Sun CP; Zhang J; Zhao WY; Yi J; Yan JK; Wang YL; Morisseau C; Liu ZB; Hammock BD; Ma XC Protostane-type triterpenoids as natural soluble epoxide hydrolase inhibitors: Inhibition potentials and molecular dynamics. Bioorg. Chem 2020, 96, 103637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Thao NP; Luyen BT; Lee JS; Kim JH; Kim YH Soluble epoxide hydrolase inhibitors of indolinone alkaloids and phenolic derivatives from Cimicifuga dahurica (Turcz.) Maxim. Bioorg. Med. Chem. Lett 2017, 27, 1874–1879. [DOI] [PubMed] [Google Scholar]
- 163.Li W; Kim JH; Zhou W; Shim SH; Ma JY; Kim YH Soluble epoxide hydrolase inhibitory activity of phenolic components from the rhizomes and roots of Gentiana scabra. Biosci. Biotechnol. Biochem 2015, 79, 907–911. [DOI] [PubMed] [Google Scholar]
- 164.Sun YN; Kim JH; Li W; Jo AR; Yan XT; Yang SY; Kim YH Soluble epoxide hydrolase inhibitory activity of anthraquinone components from Aloe. Bioorg. Med. Chem 2015, 23, 6659–6665. [DOI] [PubMed] [Google Scholar]
- 165.Sun YN; Li W; Kim JH; Yan XT; Kim JE; Yang SY; Kim YH Chemical constituents from the root of Polygonum multiflorum and their soluble epoxide hydrolase inhibitory activity. Arch. Pharm. Res 2015, 38, 998–1004. [DOI] [PubMed] [Google Scholar]
- 166.Jo AR; Kim JH; Yan XT; Yang SY; Kim YH Soluble epoxide hydrolase inhibitory components from Rheum undulatum and in silico approach. J. Enzyme Inhib. Med. Chem 2016, 31, 70–78. [DOI] [PubMed] [Google Scholar]
- 167.Kim JH; Morgan AM; Tai BH; Van DT; Cuong NM; Kim YH Inhibition of soluble epoxide hydrolase activity by compounds isolated from the aerial parts of Glycosmis stenocarpa. J. Enzyme Inhib. Med. Chem 2016, 31, 640–644. [DOI] [PubMed] [Google Scholar]
- 168.Khanh PN; Duc HV; Huong TT; Son NT; Ha VT; Van DT; Tai BH; Kim JE; Jo AR; Kim YH; Cuong NM Alkylphloroglucinol derivatives and triterpenoids with soluble epoxide hydrolase inhibitory activity from Callistemon citrinus. Fitoterapia 2016, 109, 39–44. [DOI] [PubMed] [Google Scholar]
- 169.Lee GY; Kim JH; Choi SK; Kim YH Constituents of the seeds of Cassia tora with inhibitory activity on soluble expoxide hydrolease. Bioorg. Med. Chem. Lett 2015, 25, 5097–5101. [DOI] [PubMed] [Google Scholar]
- 170.Li HQ; Lv PC; Yan T; Zhu HL Urea derivatives as anticancer agents. Anticancer Agents Med. Chem 2009, 9, 471–480. [DOI] [PubMed] [Google Scholar]
- 171.Jagla CAD; Scott CE; Tang Y; Qiao C; Mateo-Semidey GE; Yudowski GA; Lu D; Kendall DA Pyrimidinyl biphenylureas act as allosteric modulators to activate cannabinoid receptor 1 and initiate beta-arrestin-dependent responses. Mol. Pharmacol 2019, 95, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Liang Z; Zhang B; Xu M; Morisseau C; Hwang SH; Hammock BD; Li QX 1-Trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea, a selective and potent dual inhibitor of soluble epoxide hydrolase and p38 kinase intervenes in Alzheimer’s signaling in human nerve cells. ACS Chem. Neurosci 2019, 10, 4018–4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lazaar AL; Yang L; Boardley RL; Goyal NS; Robertson J; Baldwin SJ; Newby DE; Wilkinson IB; Tal-Singer R; Mayer RJ; Cheriyan J Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br. J. Clin. Pharmacol 2016, 81, 971–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Imig JD Epoxyeicosatrienoic acids and 20-hydroxyeicosatetraenoic acid on endothelial and vascular function. Adv. Pharmacol 2016, 77, 105–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lee HT; Lee KI; Chen CH; Lee TS Genetic deletion of soluble epoxide hydrolase delays the progression of Alzheimer’s disease. J. Neuroinflamm 2019, 16, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kodani SD; Morisseau C Role of epoxy-fatty acids and epoxide hydrolases in the pathology of neuro-inflammation. Biochimie 2019, 159, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Inceoglua B; Bettaiebb A; Hajd FG; Gomesf AV; Hammock BD Modulation of mitochondrial dysfunction and endoplasmic reticulum stress are key mechanisms for the wide-ranging actions of epoxy fatty acids and soluble epoxide hydrolase inhibitors. Prostaglandins Other Lipid Mediat. 2017, 133, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Marshall IJ; Wolfe CD; McKevitt C Lay perspectives on hypertension and drug adherence: systematic review of qualitative research. BMJ. 2012, 345, e3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Imig JD; Hammock BD Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat. Rev. Drug Discov 2009, 8, 794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Gebremedhin D; Ma YH; Falck JR; Roman RJ; VanRollins M; Harder DR Mechanism of action of cerebral epoxyeicosatrienoic acids on cerebral arterial smooth muscle. Am. J. Physiol 1992, 263, H519–H525. [DOI] [PubMed] [Google Scholar]
- 181.Li PL; Zhang DX; Ge ZD; Campbell WB Role of ADP-ribose in 11,12-EET-induced activation of K(Ca) channels in coronary arterial smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol 2002, 282, H1229–H1236. [DOI] [PubMed] [Google Scholar]
- 182.Li PL; Chen CL; Bortell R; Campbell WB 11,12-Epoxyeicosatrienoic acid stimulates endogenous mono-ADP-ribosylation in bovine coronary arterial smooth muscle. Circ. Res 1999, 85, 349–356. [DOI] [PubMed] [Google Scholar]
- 183.Archer SL; Gragasin FS; Wu X; Wang S; McMurtry S; Kim DH; Platonov M; Koshal A; Hashimoto K; Campbell WB; Falck JR; Michelakis ED Endothelium-derived hyperpolarizing factor in human internal mammary artery is 11,12-epoxyeicosatrienoic acid and causes relaxation by activating smooth muscle BK(Ca) channels. Circulation 2003, 107, 769–776. [DOI] [PubMed] [Google Scholar]
- 184.Fisslthaler BPR, Kiss L, Potente M, Harder DR, Fleming I, Busse R,. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature 1999, 6752, 493–497. [DOI] [PubMed] [Google Scholar]
- 185.Imig JD; Inscho EW; Deichmann PC; Reddy KM; Falck JR Afferent arteriolar vasodilation to the sulfonimide analog of 11, 12-epoxyeicosatrienoic acid involves protein kinase A. Hypertension 1999, 33, 408–413. [DOI] [PubMed] [Google Scholar]
- 186.Zhao XY; Yamamoto T; Newman JW; Kim IH; Watanabe T; Hammock BD; Stewart J; Pollock JS; Pollock DM; Imig JD Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J. Am. Soc. Nephrol 2004, 15, 1244–1253. [PubMed] [Google Scholar]
- 187.Ashraf JM; Ansari MA; Fatma S; Abdullah SMS; Iqbal J; Madkhali A; Hamali AH; Ahmad S; Jerah A; Echeverria V; Barreto GE; Ashraf GM Inhibiting effect of zinc oxide nanoparticles on advanced glycation products and oxidative modifications: a potential tool to counteract oxidative stress in neurodegenerative diseases. Mol. Neurobiol 2018, 55, 7438–7452. [DOI] [PubMed] [Google Scholar]
- 188.Imig JD Cardiovascular therapeutic aspects of soluble epoxide hydrolase inhibitors. Cardiovasc. Drug Rev 2006, 24, 169–188. [DOI] [PubMed] [Google Scholar]
- 189.Ai D; Shyy JY; Zhu Y Linking an insect enzyme to hypertension: angiotensin II-epoxide hydrolase interactions. Kidney Int. 2010, 77, 88–92. [DOI] [PubMed] [Google Scholar]
- 190.Ai D; Fu Y; Guo DL; Tanaka H; Wang NP; Tang CS; Hammock BD; Shyy JYJ; Zhu Y Angiotensin II up-regulates soluble epoxide hydrolase in vascular endothelium in vitro and in vivo. Proc. Natl. Acad. Sci. U. S .A 2007, 104, 9018–9023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sinal CJ; Miyata M; Tohkin M; Nagata K; Bend JR; Gonzalez FJ Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J. Biol. Chem 2000, 275, 40504–40510. [DOI] [PubMed] [Google Scholar]
- 192.Bukhari IA; Alorainey BI; Al-Motrefi AA; Mahmoud A; Campbell WB; Hammock BD 1-Trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea (TPPU), a soluble epoxide hydrolase inhibitor, lowers L-NAME-induced hypertension through suppression of angiotensin-converting enzyme in rats. Eur. Rev. Med. Pharmacol. Sci 2020, 15, 8143–8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Imig JD; Zhao X; Zaharis CZ; Olearczyk JJ; Pollock DM; Newman JW; Kim IH; Watanabe T; Hammock BD An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension 2005, 46, 975–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Aga EB; Li HJ; Chen J; Li P Chemical constituents from the aerial parts of Codonopsis nervosa. Chin. J. Nat. Med 2012, 10, 366–369. [Google Scholar]
- 195.Li D; Liu Y; Zhang X; Lv H; Pang W; Sun X; Gan LM; Hammock BD; Ai D; Zhu Y Inhibition of soluble epoxide hydrolase alleviated atherosclerosis by reducing monocyte infiltration in Ldlr(−/−) mice. J. Mol. Cell. Cardiol 2016, 98, 128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zhang LN; Vincelette J; Cheng Y; Mehra U; Chen D; Anandan SK; Gless R; Webb HK; Wang YX Inhibition of soluble epoxide hydrolase attenuated atherosclerosis, abdominal aortic aneurysm formation, and dyslipidemia. Arterioscl. Throm. Vas 2009, 29, 1265–1270. [DOI] [PubMed] [Google Scholar]
- 197.Xu DY; Davis BB; Wang ZH; Zhao SP; Wasti B; Liu ZL; Li N; Morisseau C; Chiamvimonvat N; Hammock BD A potent soluble epoxide hydrolase inhibitor, t-AUCB, acts through PPARgamma to modulate the function of endothelial progenitor cells from patients with acute myocardial infarction. Int. J. Cardiol 2013, 167, 1298–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Shen L; Peng H; Peng R; Fan Q; Zhao S; Xu D; Morisseau C; Chiamvimonvat N; Hammock BD Inhibition of soluble epoxide hydrolase in mice promotes reverse cholesterol transport and regression of atherosclerosis. Atherosclerosis 2015, 239, 557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Qiu H; Li N; Liu JY; Harris TR; Hammock BD; Chiamvimonvat N Soluble epoxide hydrolase inhibitors and heart failure. Cardiovasc. Ther 2011, 29, 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Seubert JM; Sinal CJ; Graves J; DeGraff LM; Bradbury JA; Lee CR; Goralski K; Carey MA; Luria A; Newman JW; Hammock BD; Falck JR; Roberts H; Rockman HA; Murphy E; Zeldin DC Role of soluble epoxide hydrolase in postischemic recovery of heart contractile function. Circ. Res 2006, 99, 442–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kompa AR; Wang BH; Xu G; Zhang Y; Ho PY; Eisennagel S; Thalji RK; Marino JP Jr.; Kelly DJ; Behm DJ; Krum H Soluble epoxide hydrolase inhibition exerts beneficial anti-remodeling actions post-myocardial infarction. Int. J. Cardiol 2013, 167, 210–219. [DOI] [PubMed] [Google Scholar]
- 202.Zhang H; Zhang K; Liang J; Yan W; Wu F; Xu W; Wu Z; Chen Y; Pan R; Wu G Soluble epoxide hydrolase inhibitor, TUPS, attenuates isoproterenol/angiotensin II-induced cardiac hypertrophy through mammalian target of rapamycin-mediated autophagy inhibition. J. Pharm. Pharmacol 2019, 71, 1291–1300. [DOI] [PubMed] [Google Scholar]
- 203.Lan YL; Zhou JJ; Liu J; Huo XK; Wang YL; Liang JH; Zhao JC; Sun CP; Yu ZL; Fang LL; Tian XG; Feng L; Ning J; Zhang BJ; Wang C; Zhao XY; Ma XC Uncaria rhynchophylla ameliorates Parkinson’s disease by inhibiting HSP90 expression: insights from quantitative proteomics. Cell. Physiol. Biochem 2018, 47, 1453–1464. [DOI] [PubMed] [Google Scholar]
- 204.Liang JH; Luan ZL; Tian XG; Zhao WY; Wang YL; Sun CP; Huo XK; Deng S; Zhang BJ; Zhang ZJ; Ma XC Uncarialins A-I, monoterpenoid indole alkaloids from Uncaria rhynchophylla as natural agonists of the 5-HT1A receptor. J. Nat. Prod 2019, 82, 3302–3310. [DOI] [PubMed] [Google Scholar]
- 205.Bosch P; van den Noort M; Staudte H; Lim S Schizophrenia and depression: a systematic review of the effectiveness and the working mechanisms behind acupuncture. Explore 2015, 11, 281–91. [DOI] [PubMed] [Google Scholar]
- 206.Ren Q; Ma M; Yang J; Nonaka R; Yamaguchi A; Ishikawa K.-i.; Kobayashi K; Murayama S; Hwang SH; Saiki S; Akamatsu W; Hattori N; Hammock BD; Hashimoto K Soluble epoxide hydrolase plays a key role in the pathogenesis of Parkinson’s disease. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E5815–E5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Qin X; Wu Q; Lin L; Sun A; Liu S; Li X; Cao X; Gao T; Luo P; Zhu X; Wang X Soluble epoxide hydrolase deficiency or inhibition attenuates MPTP-induced parkinsonism. Mol. Neurobiol 2015, 52, 187–195. [DOI] [PubMed] [Google Scholar]
- 208.Krstic D; Knuesel I Deciphering the mechanism underlying late-onset Alzheimer disease. Nat. Rev. Neurol 2013, 9, 25–34. [DOI] [PubMed] [Google Scholar]
- 209.Calsolaro V; Edison P Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [DOI] [PubMed] [Google Scholar]
- 210.Fu WY; Wang XJ; Ip NY Targeting neuroinflammation as a therapeutic strategy for Alzheimer’s disease: mechanisms, drug candidates, and new opportunities. ACS Chem. Neurosci 2019, 10, 872–879. [DOI] [PubMed] [Google Scholar]
- 211.Kodamullil AT; Zekri F; Sood M; Hengerer B; Canard L; McHale D; Hofmann-Apitius M Trial watch: tracing investment in drug development for Alzheimer disease. Nat. Rev. Drug Discov 2017, 16, 819. [DOI] [PubMed] [Google Scholar]
- 212.Tonnies E; Trushina E Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimers Dis 2017, 57, 1105–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Barnham KJ; Masters CL; Bush AI Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov 2004, 3, 205–214. [DOI] [PubMed] [Google Scholar]
- 214.Macmillan P Review of rethinking health care ethics by Stephen Scher and Kasia Kozlowska. Monash. Bioeth. Rev 2020, 38, 83–86. [DOI] [PubMed] [Google Scholar]
- 215.Morley JE; Farr SA; Flood JF Antibody to amyloid beta protein alleviates impaired acquisition, retention, and memory processing in SAMP8 mice. Neurobiol. Learn Mem 2002, 78, 125–138. [DOI] [PubMed] [Google Scholar]
- 216.Takeda T Senescence-accelerated mouse (SAM) with special references to neurodegeneration models, SAMP8 and SAMP10 mice. Neurochem. Res 2009, 34, 639–659. [DOI] [PubMed] [Google Scholar]
- 217.Akiguchi I; Pallas M; Budka H; Akiyama H; Ueno M; Han J; Yagi H; Nishikawa T; Chiba Y; Sugiyama H; Takahashi R; Unno K; Higuchi K; Hosokawa M SAMP8 mice as a neuropathological model of accelerated brain aging and dementia: Toshio Takeda’s legacy and future directions. Neuropathology 2017, 37, 293–305. [DOI] [PubMed] [Google Scholar]
- 218.Grinan-Ferre C; Palomera-Avalos V; Puigoriol-Illamola D; Camins A; Porquet D; Pla V; Aguado F; Pallas M Behaviour and cognitive changes correlated with hippocampal neuroinflammaging and neuronal markers in female SAMP8, a model of accelerated senescence. Exp. Gerontol 2016, 80, 57–69. [DOI] [PubMed] [Google Scholar]
- 219.Pallàs M Senescence-accelerated mice P8: a tool to study brain aging and Alzheimer’s disease in a mouse model. ISRN Cell Biology 2012, 2012, 917167. [Google Scholar]
- 220.Grinan-Ferre C; Codony S; Pujol E; Yang J; Leiva R; Escolano C; Puigoriol-Illamola D; Companys-Alemany J; Corpas R; Sanfeliu C; Perez B; Loza MI; Brea J; Morisseau C; Hammock BD; Vazquez S; Pallas M; Galdeano C Pharmacological inhibition of soluble epoxide hydrolase as a new therapy for Alzheimer’s disease. Neurotherapeutics 2020, DOI: 10.1007/s13311-020-00854-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Hung CC; Lee YH; Kuo YM; Hsu PC; Tsay HJ; Hsu YT; Lee CC; Liang JJ; Shie FS Soluble epoxide hydrolase modulates immune responses in activated astrocytes involving regulation of STAT3 activity. J. Neuroinflamm 2019, 16, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Ren Q Soluble epoxide hydrolase inhibitor: a novel potential therapeutic or prophylactic drug for psychiatric disorders. Front. Pharmacol 2019, 10, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Wu Q; Cai H; Song J; Chang Q The effects of sEH inhibitor on depression-like behavior and neurogenesis in male mice. J. Neurosci. Res 2017, 95, 2483–2492. [DOI] [PubMed] [Google Scholar]
- 224.Ma M; Ren Q; Fujita Y; Ishima T; Zhang JC; Hashimoto K Effects of AS2586114, a soluble epoxide hydrolase inhibitor, on hyperlocomotion and prepulse inhibition deficits in mice after administration of phencyclidine. Pharmacol. Biochem. Behav 2013, 110, 98–103. [DOI] [PubMed] [Google Scholar]
- 225.Swardfager W; Hennebelle M; Yu D; Hammock BD; Levitt AJ; Hashimoto K; Taha AY Metabolic/inflammatory/vascular comorbidity in psychiatric disorders; soluble epoxide hydrolase (sEH) as a possible new target. Neurosci. Biobehav. Rev 2018, 87, 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Tu R; Armstrong J; Lee KSS; Hammock BD; Sapirstein A; Koehler RC Soluble epoxide hydrolase inhibition decreases reperfusion injury after focal cerebral ischemia. Sci. Rep 2018, 8, 5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Finneran DJ; Nash KR Neuroinflammation and fractalkine signaling in Alzheimer’s disease. J. Neuroinflamm 2019, 16, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Jouihan SA; Zuloaga KL; Zhang W; Shangraw RE; Krasnow SM; Marks DL; Alkayed NJ Role of soluble epoxide hydrolase in exacerbation of stroke by streptozotocin-induced type 1 diabetes mellitus. J. Cereb. Blood Flow Metab 2013, 33, 1650–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Chen GZ; Xu RF; Wang YN; Wang PH; Zhao G; Xu XZ; Gruzdev A; Zeldin DC; Wang DW Genetic disruption of soluble epoxide hydrolase is protective against streptozotocin-induced diabetic nephropathy. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E563–E575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Elmarakby AA; Faulkner J; Al-Shabrawey M; Wang MH; Maddipati KR; Imig JD Deletion of soluble epoxide hydrolase gene improves renal endothelial function and reduces renal inflammation and injury in streptozotocin-induced type 1 diabetes. Am. J. Physiol. Regul. Integr. Comp. Physiol 2011, 301, R1307–R1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Dewey S; Lai XY; Witzmann FA; Sohal M; Gomes AV Proteomic analysis of hearts from akita mice suggests that increases in soluble epoxide hydrolase and antioxidative programming are key changes in early stages of diabetic cardiomyopathy. J. Proteome Res 2013, 12, 3920–3933. [DOI] [PubMed] [Google Scholar]
- 232.Liu Y; Dang H; Li D; Pang W; Hammock BD; Zhu Y Inhibition of soluble epoxide hydrolase attenuates high-fat-diet-induced hepatic steatosis by reduced systemic inflammatory status in mice. PLoS One 2012, 7, e39165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Yao L; Cao B; Cheng Q; Cai W; Ye C; Liang J; Liu W; Tan L; Yan M; Li B; He J; Hwang SH; Zhang X; Wang C; Ai D; Hammock BD; Zhu Y Inhibition of soluble epoxide hydrolase ameliorates hyperhomocysteinemia-induced hepatic steatosis by enhancing beta-oxidation of fatty acid in mice. Am. J. Physiol. Gastrointest. Liver Physiol 2019, 316, G527–G538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Moreira da Silva R; Verjee S; de Gaitani CM; Moraes de Oliveira AR; Pires Bueno PC; Cavalheiro AJ; Peporine Lopes N; Butterweck V Evaluation of the intestinal absorption mechanism of casearin X in Caco-2 cells with modified carboxylesterase activity. J. Nat. Prod 2016, 79, 1084–1090. [DOI] [PubMed] [Google Scholar]
- 235.Chen L; Fan C; Zhang Y; Bakri M; Dong H; Morisseau C; Maddipati KR; Luo P; Wang CY; Hammock BD; Wang MH Beneficial effects of inhibition of soluble epoxide hydrolase on glucose homeostasis and islet damage in a streptozotocin-induced diabetic mouse model. Prostaglandins Other Lipid Mediat. 2013, 104, 42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Pardeshi R; Bolshette N; Gadhave K; Arfeen M; Ahmed S; Jamwal R; Hammock BD; Lahkar M; Goswami SK Docosahexaenoic acid increases the potency of soluble epoxide hydrolase inhibitor in alleviating streptozotocin-induced Alzheimer’s disease-like complications of diabetes. Front. Pharmacol 2019, 10, 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Luo PC; Chang HH; Zhou YQ; Zhang SL; Hwang SH; Morisseau C; Wang CY; Inscho EW; Hammock BD; Wang MH Inhibition or deletion of soluble epoxide hydrolase prevents hyperglycemia, promotes insulin secretion, and reduces islet apoptosis. J. Pharmacol. Exp. Ther 2010, 334, 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Luria A; Bettaieb A; Xi YN; Shieh GJ; Liu HC; Inoue H; Tsai HJ; Imig JD; Haj FG; Hammock BD Soluble epoxide hydrolase deficiency alters pancreatic islet size and improves glucose homeostasis in a model of insulin resistance. Proc. Natl. Acad. Sci .U. S. A 2011, 108, 9038–9043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Hu J; Popp R; Frömel T; Ehling M; Awwad K; Adams RH; Hammes HP; Fleming I Müller glia cells regulate Notch signaling and retinal angiogenesis via the generation of 19,20-dihydroxydocosapentaenoic acid. J. Exp. Med 2014, 211, 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Hu J; Dziumbla S; Lin J; Bibli SI; Zukunft S; de Mos J; Awwad K; Fromel T; Jungmann A; Devraj K; Cheng Z; Wang L; Fauser S; Eberhart CG; Sodhi A; Hammock BD; Liebner S; Muller OJ; Glaubitz C; Hammes HP; Popp R; Fleming I Inhibition of soluble epoxide hydrolase prevents diabetic retinopathy. Nature 2017, 552, 248–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Morisseau C; Hammock BD Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu. Rev. Pharmacol. Toxicol 2013, 53, 37–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Sodhi K; Inoue K; Gotlinger KH; Canestraro M; Vanella L; Kim DH; Manthati VL; Koduru SR; Falck JR; Schwartzman ML; Abraham NG Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of HO-2-null mice. J. Pharmacol. Exp. Ther 2009, 331, 906–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Warner J; Hardesty J; Zirnheld K; McClain C; Warner D; Kirpich I Soluble epoxide hydrolase inhibition in liver diseases: a review of current research and knowledge gaps. Biology 2020, 9, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Lopez-Vicario C; Alcaraz-Quiles J; Garcia-Alonso V; Rius B; Hwang SH; Titos E; Lopategi A; Hammock BD; Arroyo V; Claria J Inhibition of soluble epoxide hydrolase modulates inflammation and autophagy in obese adipose tissue and liver: role for omega-3 epoxides. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]