

Abstract
Intramyocardial injection of hydrogels offers great potential for treating myocardial infarction (MI) in a minimally invasive manner. However, traditional bulk hydrogels generally lack microporous structures to support rapid tissue ingrowth and biochemical signals to prevent fibrotic remodeling toward heart failure. To address such challenges, a novel drug-releasing microporous annealed particle (drugMAP) system is developed by encapsulating hydrophobic drug-loaded nanoparticles into microgel building blocks via microfluidic manufacturing. By modulating nanoparticle hydrophilicity and pregel solution viscosity, drugMAP building blocks are generated with consistent and homogeneous encapsulation of nanoparticles. In addition, the complementary effects of forskolin (F) and Repsox (R) on the functional modulations of cardiomyocytes, fibroblasts, and endothelial cells in vitro are demonstrated. After that, both hydrophobic drugs (F and R) are loaded into drugMAP to generate FR/drugMAP for MI therapy in a rat model. The intramyocardial injection of MAP gel improves left ventricular functions, which are further enhanced by FR/drugMAP treatment with increased angiogenesis and reduced fibrosis and inflammatory response. This drugMAP platform represents a new generation of microgel particles for MI therapy and will have broad applications in regenerative medicine and disease therapy.
Keywords: drug delivery, granular hydrogels, microgels, myocardial infarction, tissue engineering
1. Introduction
Ischemic heart disease (IHD) is a leading cause of global mortality, accounting for over nine million deaths per year, according to the World Health Organization (WHO).[1] Acute MI is the most common manifestation of IHD, usually caused by the complete occlusion of a coronary artery with atherosclerotic plaque rupture and thrombosis.[2] Following MI, the damaged myocardium eventually undergoes a remodeling process with cardiomyocyte depletion, tissue fibrosis, cardiac dilatation, and dysfunction, culminating in heart failure.[3] Currently, several therapeutic strategies have been exploited to repair and regenerate the damaged cardiac tissues caused by MI, including pharmaceutic approaches,[4] injectable hydrogels,[5] cardiac patches,[6] cell transplantation,[7] and cell reprogramming.[8] Among them, injectable hydrogels have shown great potential to treat MI by providing mechanical support and tissue integration to increase myocardial thickness and prevent ventricular remodeling through a minimally invasive and cost-effective manner.[5,9] Nevertheless, traditional hydrogels usually have a trade-off between mechanical strength to support cell attachment and porous structure to enable rapid tissue ingrowth before hydrogel degradation. Thus, biomaterials with independently tunable biophysical properties are needed to improve therapeutic outcomes.[5a]
Mechanical properties, porosity and microarchitecture of porous hydrogels can significantly impact in vivo cell behavior and tissue regeneration effects.[10] Currently, a variety of manufacturing techniques have been developed to fabricate structured porous hydrogel scaffolds, including solvent/porogen leaching,[11] gas foaming,[12] freeze-drying,[13] and 3D printing,[14] but these methods are challenging to be delivered via minimally invasive techniques. To decouple porous structure and mechanical support, we have recently developed an injectable microporous annealed particle (MAP) scaffold by crosslinking uniform microgel (μGel) building blocks produced in a microfluidic device.[15] By combining injectability, microporosity and mechanical strength, the porous MAP scaffolds have demonstrated rapid cellular infiltration without bulk material degradation to facilitate wound and stroke healing in vivo.[15a,16] However, the therapeutic efficacy of MAP gel for treating MI and its capabilities as a drug delivery platform to promote functional regeneration remain to be investigated.
Pharmacological treatments are commonly used in clinic to slow down or reverse detrimental cardiac remodeling in MI patients,[17] with specific effects such as proangiogenesis,[18] anti-fibrosis,[18,19] anti-inflammatory,[20] anti-cardiomyocyte death,[4b] antiarrhythmic,[21] and anti-thrombosis.[22] The cyclic adenosine monophosphate (cAMP) is an essential second messenger and mediates many critical intracellular signaling under physiological and pathophysiological conditions.[23] Activation and increased generation of cAMP can markedly increase cardiac LV function and survival, and attenuate cardiac fibrosis and its sequelae after acute MI.[24] Additionally, transforming growth factor-β (TGF-β) signaling plays a pleiotropic role in driving disease progression.[25] TGF-β expression is upregulated in acute MI and cardiac hypertrophy, which leads to fibrosis and diastolic dysfunction with induced myodifferentiation, extracellular matrix (ECM) synthesis, and cardiomyocyte hypertrophy.[26] Here we explored the approach to promote cardiac regeneration by activating cAMP pathway while inhibiting TGF-β signaling. Forskolin (F) is a cAMP agonist, and RepSox (R) is a selective TGF-β inhibitor. Both small molecules have shown the beneficial effects to rescue cardiac dysfunction and ameliorate post-MI remodeling.[27,38] However, it is unclear whether there is a synergistic effect by modulating both signaling pathways for heart repair. Furthermore, the majority of drugs are administrated to patients by simple systemic delivery, which generally leads to adverse off-target effects, drug toxicity, and low treatment efficacy.[28] In addition, a holistic approach is still required to regenerate damaged human heart by targeting multiple tissue pathologies, including remuscularization, electromechanical stability, angiogenesis, resolution of fibrosis, and immunological balance.[29] Therefore, biomaterial-based scaffolds with localized multidrug delivery may be necessary to promote cardiac regeneration by providing pleiotropic pharmaceutic effects.
In this study, we developed a novel injectable, multimodal drugMAP hydrogel for MI therapy. The generation of a porous drugMAP scaffold is shown schematically in Figure 1. Hydrophobic drugs were loaded into nanoparticles (NPs), which were further encapsulated into matrix metalloprotease (MMP) sensitive polyethylene glycol (PEG)-based μGel beads to generate drugMAP building blocks, i.e., drug/NPs-μGel beads, using a flow-focusing microfluidic device. When the drugMAP building blocks were injected into the infarcted heart, endogenous factor XIIIa (FXIIIa) could activate peptide K (Pep-K) and peptide Q (Pep-Q) in μGel to induce surface binding between μGel beads and form contiguous porous drugMAP scaffolds in situ. By coloading hydrophobic drugs of F and R, the injectable drugMAP could endow pleiotropic benefits for heart repair by providing mechanical support, promoting cell migration, and neovascularization, while suppressing fibrosis and immune responses.
Figure 1.

Scheme illustrating the microfluidic generation of drug-releasing MAP (drugMAP) scaffolds for MI therapy. A) Microfluidic generation of drugMAP building blocks by encapsulating drug/NPs into μGel beads to generate drug/NPs-μGel beads in a microfluidic device. The hydrogels are formed by crosslinking pregel solutions via thiol-ene reactions to encapsulate NPs in the gel mesh. B) Injection of cardiac drugMAP scaffolds for MI therapy. Via delivery of specific drugs, the cardiac drugMAP scaffolds endow pleiotropic benefits for heart repair.
2. Results and Discussion
2.1. Development of drugMAP Building Blocks
The microfluidic device for drugMAP gel generation was designed and fabricated with soft lithography (Figure 2A,B). To achieve sustained drug release from drugMAP gel, biodegradable poly(lactic-co-glycolic acid) (PLGA) based polymers were used to make drug/NPs by an emulsification solvent evaporation technique, and mixed with the pregel solutions prior to μGels formation. PLGA is a biodegradable polymer being used in many FDA-approved products, and PLGA-based particles have been widely employed for drug delivery because of their biocompatibility and controllable biodegradation.[30] However, the hydrophobic PLGA NPs aggregated and precipitated quickly in the aqueous pregel solution, leading to failure in the production of NPs-μGels in a microfluidic device, because of blockage or leakage of the microfluidic channels, and unstable processing which caused the generation of heterogenous low-quality μGels (Figure S1, Supporting Information). Thus, we employed two strategies to suspend the PLGA NPs and delay particle aggregation in the pregel solution through improving NP surface hydrophilicity and increasing the viscosity of the pregel solution (Figure S1, Supporting Information). The mean hydrodynamic diameter of NPs was ≈400 nm with a polydispersity index of 0.23 as measured by dynamic light scattering (DLS). We first adjusted the NP surface hydrophilicity in the aqueous pregel solutions by using different PLGA–PEG copolymers, including PLGA 35k, PLGA55k-b-PEG5k, and PLGA25k-b-PEG5k. We found that particle suspension was enhanced with the increase of PEG length, and PLGA55k-b-PEG5k NPs resulted in a stable preparation of NPs-μGel. However, a further increase of hydrophilicity might lead to a lower encapsulation capacity of hydrophobic drugs and cause a burst drug release.[31] Thus, another strategy was implemented in addition to the adjustment of the particle hydrophilicity. Here, through the addition of hyaluronic acid (HA) to the pregel solution, the solution viscosity was increased, resulting in a reduction of NPs aggregation. After optimization, we found that the NPs made by PLGA35k/PLGA55k-PEG5k (1:1 weight ratio) and the addition of 0.25% v/v HA in pregel solution achieved a stable preparation of NPs-μGel beads with uniform size, controlled NPs loading, and uniform NPs distribution (Figure 2C–E). To monitor and visualize the mixing process of the two aqueous phases and the particle distribution in the μGel beads, NPs were labeled with coumarin-6 (green) and the pregel solution was conjugated with AF546-maleimide (red) (Figure 2F). The fluorescent images showed that NPs were uniformly encapsulated in the μGel beads (Figure 2G). Thus, PLGA35k/PLGA55k-PEG5k NPs were used as the drug loading material for all the subsequent studies.
Figure 2.
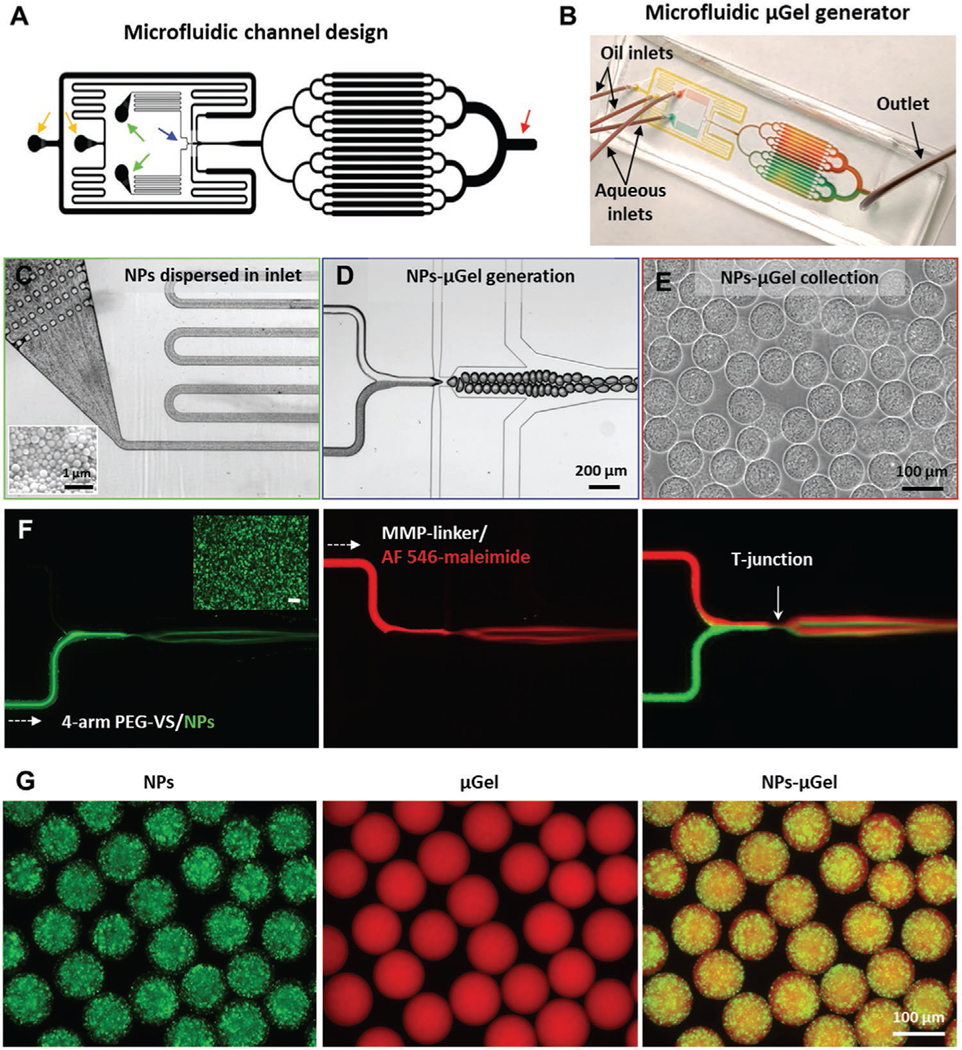
Preparation of drugMAP building blocks. A) Microfluidic channel design for μGel generator. The yellow arrows denote oil inlets. The green arrows denote aqueous inlets. The blue arrow denotes droplet generation region, and the red arrow denotes droplet collection region. B) Photograph of the microfluidic device of μGel generator, channels are highlighted with colored dye solutions. C) PEG-VS pregel solution with dispersed nanoparticles (NPs) flows stably through the inlet filters. Insert image in the lower-left corner is a representative SEM image of PLGA-based NPs. D) Homogeneous droplets containing pregel solution and crosslinker formed at a flow focusing junction of the microfluidic channel. E) NPs-μGel beads with a uniform NP distribution collected at the outlet region. F) Fluorescence images of droplets generated with fluorescent-labeled aqueous solutions, one aqueous channel with coumarin-6 (green) labeled NPs with 4-arm PEG-VS pregel solution and another aqueous channel with AF 546-maleimide (red) with MMP-sensitive crosslinker solution. G) Representative fluorescent images of NPs-μGel beads made under optimized processing conditions, with NPs distributed uniformly in μGel.
2.2. Characterization of drugMAP Building Blocks and Scaffolds
A major advantage of the microfluidic-emulsion technique is the production of highly monodisperse hydrogel microparticles of well-defined size. We were able to produce NPs-μGel beads with diameters ranging from 45 to 120 μm by tuning the flow rate of aqueous solutions (Figure 3A). Some minor differences in μGel bead formation were observed for beads containing NPs. In particular, at an aqueous flow rate of 8 μL min−1, NPs-μGel beads in the oil phase were larger than μGel beads, potentially because the addition of HA and NPs increased the viscosity of aqueous solution and affected the droplet breakup. However, NPs encapsulation slightly decreased the gel swelling ratio in buffer solution, resulting in the final diameter of NPs-μGel beads still being similar to μGel beads (≈100 μm) (Figure 3B).
Figure 3.

Characterization of drugMAP building blocks and annealed scaffolds. A) Generation of NPs-μGel beads with highly defined sizes by altering the aqueous flow rate. B) NPs-μGel beads, made with an aqueous flow rate of 8 μL min−1, and swollen in buffer after aqueous extraction from the oil phase. Qv represents the volumetric swelling ratio of a bead. C) Representative images of NPs-μGel beads loaded with increasing amounts of NPs. The numbers in brackets represent the weight percentages of the NPs to dry pregel components. D) Nanoparticle loading efficiency in different NP-μGel beads as a function of wt%. E) Nanoparticle loading concentration in NPs-μGel beads as a function of initial concentration. F) Microporous drugMAP scaffolds generated by annealing NPs-μGel beads using FXIIIa. G) Pore size and void fraction of MAP and drugMAP scaffolds. H) Storage moduli of bulk hydrogels mixed with different amounts of NPs. Data are shown as mean ± SD. *p < 0.05, NS represents no significant difference.
To adjust the drug delivery capacity of drugMAP, we prepared gel droplets loaded with different amounts of NPs from 0% to 100% (weight of NPs/weight of dry pregel components) (Figure 3C). After gelling and purification, the particle loading efficiency was higher than 90% for all tested NPs-μGel beads (Figure 3D), and the final NP concentration in the gels was highly correlated with the initial loading amount (Figure 3E). The NPs that were not encapsulated in the μGels might be lost in the device during gel fabrication or released during gel purification. Similar to the MAP scaffolds, the drugMAP scaffolds generated from 100 μm NPs-μGel beads maintained an interconnected porous structure after annealing (Figure 3F) with a median pore diameter ≈20 μm and ≈15% average void fraction (Figure 3G). With pores of these dimensions, cells can easily infiltrate and traverse the microporous scaffold even before MAPgel degradation. In addition, the pore diameters could be adjusted by tuning the building-block sizes.[15a] The loading of NPs did not affect the ability of NPs-μGel beads to anneal to form contiguous microporous drugMAP scaffolds. In vitro, the building blocks were annealed via activated FXIIIa, in which a noncanonical amide covalent bond formed between the ε-amine of lysine in peptide-K and the γ-carboxamide of glutamine in peptide-Q on the microbeads.[15a,32] When the beads were injected in vivo, the endogenous thrombin and FXIIIa could induce the crosslinking of μGels to form MAP scaffold in the infarcted heart.[33] Mechanical properties are critical biophysical cues in MI therapy.[34] Therefore, the influence of nanoparticle loading on the mechanical stiffness of MAP gel was investigated. The results demonstrated that the addition of NPs in gels had a negligible effect on the hydrogel stiffness, yielding a storage modulus of ≈600 Pa (Figure 3H). The stiffness is in the same order as the stiffness of other soft hydrogels, which have shown improved therapeutic outcomes in post-MI therapy.[35] In particular, the porous MAP gel could provide a microporous structure for fast cell infiltration and mechanical support immediately after injection, and the mechanical properties of drugMAP could be easily adjusted to achieve stiffness matching between the scaffold and native tissue via modulating the stiffness of individual μGel beads, annealing chemistry, crosslinking degree, and bead-packing density.
The degradation of biomaterials enables in situ tissue regeneration with cell infiltration and ECM formation. The MAP gel mesh was crosslinked with MMP-sensitive peptide, making it degradable by MMP enzyme.[36] MMPs are highly relevant to cardiac remodeling after MI as the MMP9 level is elevated in plasma and left ventricle after MI in animals and humans.[37] To check the MAP gel degradation in enzyme solution and address whether MAP gel degradation affected the drug release profile, we loaded coumarin-6 into drugMAP beads as a hydrophobic fluorescent model drug and characterized the degradation of pelleted NPs-μGel beads in the presence of MMP enzyme (collagenase II) in vitro (Figure S2A, Supporting Information). We found that the drugMAP beads degraded faster with the increase of collagenase concentration. However, the release of coumarin-6 in NPs was not affected by changing the concentrations of collagenase. Fluorescence imaging of NPs-μGel beads showed a direct correlation between the collagenase concentration and the extent of degradation (represented by diminishing AF546 signal intensity) as well as particle deformability (evidenced by elongation and swelling of the particles) (Figure S2B, Supporting Information). Furthermore, we found that NPs also increased in size during degradation and remained trapped inside μGels. There might be two possible reasons for the particle trapping in drugMAP mesh during degradation. First, the ester bonds of polyester could be hydrolyzed to form hydrophilic carboxyl and hydroxyl groups, so the hydrophilicity of the particles would increase gradually to promote water absorption, thus forming larger swollen particles or clusters. Second, the carboxyl groups of polyester fragments could interact with the amine groups of gel components electrostatically. Overall, these in vitro data suggested that the drug release profile from the drugMAP remained relatively independent of gel degradation.
2.3. In Vitro Evaluation of Drugs and drugMAP
Previous studies have reported that F and R have specific effects on preventing cardiac dysfunction, respectively.[27,38] However, their effects on various cell types in cardiac tissues have not been systematically evaluated, and it is not clear whether the combination of F and R has additive or synergistic effects. In the initial drug evaluation, we found that both F and R or FR combination could maintain cardiomyocyte viability at 80% after 5 days in vitro culture, which was significantly higher than 25% for control cells (Figure S3, Supporting Information). In addition, both F and R significantly enhanced the proliferation of neonatal cardiomyocytes, yielding three and six times as many cells as the control, respectively (Figure S4, Supporting Information). For cardiac fibroblasts (Figure S5A, Supporting Information), F showed dose effects to enhance fibroblast proliferation, in contrast, R showed the opposite inhibitory effects. Nevertheless, the inhibition of fibroblast proliferation can be maintained when both drugs used together. We also found that each F or R, or their combination could prevent myodifferentiation of cardiac fibroblasts (Figure S5B, Supporting Information). Furthermore, for endothelial cell (EC) proliferation and tubule network formation (Figure S6, Supporting Information), both F and R showed dose-dependent effects to enhance EC proliferation, and their optimal concentration was the same (20 μM). Notably, EC proliferation was significantly enhanced with the combination of the two drugs. EC network formation was increased with F or FR treatment, while not with R alone. Altogether, the collected effects of both drugs on cardiac cells were summarized in Figure 4A. Since F and R had additive and complementary benefits in promoting cardiomyocyte survival, inhibiting fibroblast myodifferentiation and enhancing EC proliferation and tubule formation, both hydrophobic agents were loaded into PLGA-based NPs (FR/NPs), which were further encapsulated into μGel beads to generate FR/drugMAP building blocks.
Figure 4.

In vitro cellular evaluations of drugs and drugMAP gels. A) The summarized drug effects of forskolin (F), Repsox (R), and FR on various cardiac remodeling-associated cells. Sign + represents a positive effect, and sign – represents a negative effect. B) Cumulative drug release profiles from FR/NPs (FR loaded NPs) and FR/drugMAP (F and R loaded drugMAP gel). C) Live and dead staining of neonatal cardiomyocytes cultured in the indicated conditions on day 3, and D) cell viability of neonatal cardiomyocytes. E) Myo-differentiation of neonatal cardiac fibroblasts cultured in the indicated conditions on day 5, and F) mean fluorescent intensity of α-SMA in (E). G) Representative fluorescent images of vascular network formation. Human umbilical vein endothelial cells (HUVECs) are cultured at the indicated conditions for 16 h and stained with Calcein-AM. H) Quantification of junction numbers, tube numbers and mesh numbers. Data are shown as mean ± SD. *p < 0.05 and **p < 0.01 indicate comparisons to blank. ##p < 0.01 indicates comparisons to R condition. NS represents no significant difference.
The drug release profiles from FR/drugMAP demonstrated that both hydrophobic chemicals were gradually released throughout 2 weeks (Figure 4B). The in vitro biological evaluations were further performed for the drug-releasing platforms (Figure 4C–F). Similar to FR added directly to the medium, FR/NPs and FR/drugMAP yielded the combined beneficial effects and significantly enhanced cardiomyocyte survival compared to control (FR/NPs: 65%, FR/drugMAP: 75% versus blank: 25% at day 5) (Figure 4C,D). In addition, both alpha-smooth muscle actin (α-SMA) and F-actin were strongly expressed in the blank control (Figure 4E,F), and there were no differences between the blank control and the supernatants from the unloaded NPs or blank μGels (Figure S5, Supporting Information). However, same as adding drugs (F and R) in the medium, both FR/NPs and FR/drugMAP diminished fibroblast myodifferentiation with significantly lower α-SMA expression. In parallel, there was a decrease of F-actin in response to released F and R, suggesting that the formation of actin stress fibers was blunted in parallel with the decrease in α-SMA expression, consistent with a previous finding.[38] Moreover, both FR/NPs and FR/ drugMAP obviously enhanced EC vascular network formation (Figure 4G), and exhibited significant higher number of junctions, tubes, and meshes versus the blank control (Figure 4H). Taken together, these in vitro results demonstrated the beneficial effects of FR/drugMAP on regulating cardiac remodeling cells, including enhancing cardiomyocyte survival, inhibiting fibroblast myodifferentiation and promoting EC proliferation and tubule formation. Beyond the controlled drug release, we found that the cellular uptake of NPs embedded in μGel beads were significantly reduced, compared to free NPs (Figure S7, Supporting Information), which could decrease the cytotoxicity and inflammatory response.[39]
2.4. Cardiac Function Improvement with drugMAP Injection
To investigate the potency for cardiac repair with drugMAP, rat MI models were created by ischemia-reperfusion injury through the ligation of the left anterior descending artery. As previous studies suggested that the best therapeutic outcomes of hydrogel-based approaches were found 2–3 days after MI,[40] we performed injections 2 days after infarction of four randomized groups with the treatments of PBS (n = 9), FR/NPs (n = 6), MAP gel (n = 9), and FR/drugMAP gel (n = 9), respectively.
Solutions were successfully injected into the infarcted zone by ultrasound-guided transthoracic injection (total 100 μL with two sites of 50 μL injections). The commonly used natural and synthetic hydrogels usually undergo a solution-to-gel transition upon stimulus exposure, while it is relatively difficult to control and balance the ideal solution-to-gel transition time.[41] Inappropriate gelation speed may lead to many adverse effects. Slow gelation (from minutes to hours) could increase tissue necrosis or the loss of materials and therapeutic molecules. On the other hand, rapid gelation (from seconds to minutes) leads to quick needle blockage, handling inconveniences and limited tissue integration. Unlike commonly applied bulk hydrogels, the MAP gel building blocks are flowable and can be easily injected into highly motile cardiac tissue and stay at the injection site without gel dislodgment, which might avoid the handling issues and risks of rapid or slow gelation.
The MI therapeutic outcomes of all groups were evaluated at 5 weeks post-treatment by histology and echocardiography analysis. Masson’s trichrome staining showed the gross heart morphology and revealed less MI region, fibrosis, LV dilation, and wall thinning in hearts treated with FR/NPs- or MAP gel-only groups compared with PBS group, with further improvement for hearts treated with integrated FR/drugMAP (Figure 5A; Figure S8, Supporting Information), resulting in the smallest infarct size (FR/drugMAP: 15.4 ± 3.9% vs PBS: 35 ± 6.8%; FR/NPs: 23.1 ± 5.1%; MAP: 24.2 ± 4.7%; Figure 5B) and the thickest minimum LV wall (FR/drugMAP: 1.85±0.14 mm vs PBS: 1.11 ± 0.21 mm; FR/NPs: 1.45± 0.17 mm; MAP: 1.6 ± 0.13 mm; Figure 5C). In addition, the reduced cardiac remodeling of FR/NPs, MAP gel, and FR/drugMAP-treated groups was further demonstrated by the reduction in left ventricle end-diastolic volume (LVEDV) and end-systolic volume (LVESV), respectively, compared with PBS controls (Figure 5D,E). The ventricular ejection fractions (LVEF) at day 2 baseline were similar between all groups, indicating a similar degree of initial MI injury (Figure 5F). However, after 5 weeks, the LVEF of PBS-treated group distinctly declined, while LVEF was well preserved in the FR/NPs, MAP gel, and FR/drugMAP-treated groups. Notably, FR/drugMAP-treated rats displayed the best LV contractility of infarcted hearts with the highest LVEF (FR/drugMAP: 53.6 ± 5.2% vs PBS: 33.7 ± 4.9%; FR/NPs: 44.9 ± 3.1%; MAP: 47.7 ± 5.3%; Figure 5F) and the highest therapeutic efficiencies (change of LVEFs from baseline, Figure 5G). Overall, the cardiac remodeling was significantly attenuated by the treatment with FR/NPs or MAP gel alone, indicating the respective benefits of the drugs (F and R)[27,38] and hydrogel-based mechanical support[5,9] in ameliorating post-MI remodeling and rescuing cardiac dys-function. Compared to treatment alone, the integrated FR/drugMAP showed the best therapeutic outcomes.
Figure 5.

Cardiac functional assessment in the rat acute MI model. A) Representative Masson’s trichrome-stained sections of infarcted rat hearts after 5 weeks treatment with PBS, FR/NPs, MAP gel and FR/drugMAP gel. (Bottom) High-magnification views of the infarcted zones. B) Quantitative analyses of infarcted size (as % of the total LV area). C) Quantitative analyses of infarcted minimum LV wall thickness. D) LVEDV and E) LVESV of infarcted hearts measured by echocardiography at 5 weeks. E) LV ejection fraction (EF) of infarcted hearts at day 2 (baseline) and week 5 after treatment. G) Change in LVEF in comparison to baseline (ΔLVEF). Data are shown as mean ± SD. PBS (n = 9), FR/NPs (n = 6), MAP (n = 9) and FR/drugMAP gel (n = 9). *p < 0.05 and **p < 0.01 indicate significant difference in comparison to the PBS control group. #p < 0.05 and ##p < 0.01 in (F) indicate comparisons of 5 week treated group to the corresponding baseline. NS represents no significant difference.
To reveal the underlying mechanisms for the functional effects of drugMAP, we further performed immunostaining analysis and assessed angiogenesis and immune response in the infarcted hearts (Figure 6). Infarcted hearts were stained with von Willebrand factor (vWF, for ECs) and α-SMA (for smooth muscle cells) (Figure 6A; Figure S9, Supporting Information), and the results showed that the numbers of both capillaries (vWF+) and arterioles (α-SMA+) were significantly increased in FR/NP-treated and FR/drugMAP-treated groups in comparison to PBS and MAP-treated groups (Figure 6C,D). Notably, the FR/drugMAP-treated hearts exhibited prominent angiogenesis, while there was less angiogenesis treated with MAP gel alone, suggesting that the drugs further promote neovascularization. Additionally, in contrast to PBS-treated hearts, other three treatments showed less CD68+ macrophage infiltration in the infarcted hearts, especially for the FR/drugMAP-treated group (Figure 6B,E; Figure S10, Supporting Information), demonstrating that both drug and MAP gel could reduce the inflammatory responses in MI hearts, and their combination and integration could further enhance the efficiency. Together, these in vivo results suggest that the integrated drugMAP could enhance the MI therapeutic effects through the promotion of neovascularization and the inhibition of inflammatory response.
Figure 6.

DrugMAP promotes angiogenesis and reduces inflammatory response in MI therapy. A) Representative images of angiogenesis staining with α-SMA (green) and vWF (magenta) in the central infarct LV zone of hearts treated with PBS, FR/NPs, MAP, and FR/drugMAP gel at 5 weeks. Microgel beads were labeled by AF546 dye (red) for material tracking. B) Representative images of macrophage staining with CD68 (green). Quantification of C) capillary density (vWF+ vessels), D) arteriolar density (α-SMA+ vessels) and E) macrophage density in the central infarct LV zone of hearts treated with PBS (n = 9), FR/NPs (n = 6), MAP (n = 9), and FR/drugMAP (n = 9) at 5 weeks. Data are shown as mean ± SD. *p < 0.05 and **p < 0.01 indicate significant difference in comparison to PBS control group.
To date, numerous injectable hydrogels have been investigated for cardiac repair and regeneration. However, rapid host tissue integration and spatiotemporal control of biologics presentation are challenges for most natural and synthetic bulk hydrogels, which can compromise the efficacy of the hydrogel-based therapy for cardiac repair. In recent years, very few granular hydrogels have been exploited in tissue repair.[15a,16,42] By annealing the μGel building blocks to form porous scaffolds, the granular hydrogel permits several noteworthy features. First, the small size of μGel enables minimally invasive injection. Second, the modular building makes it flexible to engineer multiscale physical properties by varying polymer composition, μGel shape, size and stiffness, and interparticle friction. Third, granular hydrogels possess porosity and diffusivity and can be tuned to support cell proliferation and migration. For example, the injection of granular porous hyaluronic acid hydrogels into myocardial tissues demonstrated the degradation behavior and cell invasion after 3 weeks.[43] However, this study did not evaluate the MI therapeutic outcomes by histology and echocardiography analysis.
Injectable hydrogels are promising for localized drug and cell delivery in many biomedical applications. Current granular hydrogel systems have been used for the sustained delivery of hydrophilic biologics (cells and drugs). For example, heparin has been incorporated into microparticles to sustain the delivery of growth factors through electrostatic associations.[44] Similarly, protein activators or inhibitors such as antibodies can also be delivered, while they are more expensive and may lose activity through proteolytic enzymatic digestion and degradation over time. In contrast, small molecules are generally more stable, cheaper, and easier to be loaded into a drug delivery system. However, it is still challenging to pack hydrophobic drugs into microfluidic-generated granular hydrogel systems. Delivery of hydrophobic drugs or cargos can be controlled by loading the drugs into hydrophobic carriers (such as NPs). However, these hydrophobic particles can aggregate into clusters and precipitate quickly in the hydrophilic pregel solution, resulting in the blockage of microfluidic channels and unstable drug loading, as shown in this study. Here we achieved the uniform encapsulation hydrophobic drug-loaded NPs within microfluidic-generated hydrophilic μGel beads by modulating NP surface hydrophilicity and the viscosity of the pregel solution for controlled hydrophobic drug delivery.
In this study, F and R were evaluated and loaded into the drugMAP for MI therapy. Both hydrophobic drugs can be sustained release in two weeks in vitro. There was a partial release of both drugs during the production phase of drugMAP, due to the burst release occurring when the NPs were suspended in an aqueous pregel solution or embedded in MAP gel. Depending on the therapeutic purpose, the drug release period from NPs can be tailored from hours to months, by tuning the polymer composition, molecular weight, and the content of the hydrophilic block.[31,45] Besides the intrinsic release profile from drug-loaded NPs, the amount of NPs encapsulated in each μGel and the volume of μGels are also critical parameters to determine the overall drug release profile and pharmacologic effects. Furthermore, we have systematically analyzed the MI therapeutic outcomes of drugMAP systems by histology, echocardiography and immunostaining. We found that the integrated FR/drugMAP could significantly ameliorate cardiac remodeling and dysfunction, in comparison to FR/NPs only and MAP-only groups, by inhibiting fibrosis and inflammatory response, and promoting cell migration and neovascularization. It is worth noting that the drugMAP gel has shown partial degradation in vivo after 5 weeks. A longer study is needed to determine the potential long-term benefits on cardiac repair, and large animal studies need to be performed before advancing into clinical studies.
3. Conclusions
In summary, we first developed an annealing drug-releasing drugMAP hydrogel, through overcoming challenges in integrating hydrophobic NPs within microfluidic-generated hydrophilic μGel beads. DrugMAP was loaded with hydrophobic drugs (F and R), and injected into ischemic heart, which promoted cardiac repair by offering multifunctional benefits, including fast cell infiltration, mechanical support, and synergistic pharmacological effects. Our findings suggest that drugMAP has a great potential for MI therapy and broad biomedical applications in soft tissue repairs and disease therapies.
4. Experimental Section
Microfluidic Device Fabrication:
Droplet generating microfluidic devices were fabricated by soft lithography as previously described.[15a] Briefly, master molds were fabricated on silicon wafers (University wafer) using two-layer photolithography with KMPR 1050 photoresist (Microchem Corp). The height for the droplet formation channel was 50 μm, and the height for the collection channel was 150 μm. Devices were molded from the masters using poly(dimethyl)siloxane (PDMS) (Sylgard 184 kit, Dow Corning). The base and crosslinker were mixed at a 10:1 mass ratio, poured over the mold and degassed before curing overnight at 65 °C. Channels were sealed by treating the PDMS mold and a glass microscope slide (VWR) with oxygen plasma (Plasma Cleaner, Harrick Plasma) at 500 mTorr and 80 W for 30 s. Thereafter, the channels were functionalized by injecting 100 μL of Aquapel (88625–47100, Aquapel) and reacting for 30 s until washed by Novec 7500 (9802122937, 3M). The channels were dried by air suction and kept in the oven at 65 °C until used.
Preparation and Characterization of Drug-Loaded NPs:
An emulsification solvent evaporation technique was applied to prepare NPs.[46] Briefly, different PLGA based polymers, including PLGA (Mw = 35 kDa, acid-terminated, cat# 26270, Polysciences), PLGA55k-b-PEG5k (PLGA average Mn = 55 kDa, PEG average Mn = 5 kDa, cat# 764752, Sigma), PLGA25k-b-PEG5k (PLGA average Mn = 25 kDa, PEG average Mn = 5 kDa, cat# 764 752, Sigma), and mixed PLGA35k/PLGA55k-b-PEG5k (50/50 wt/wt) were dissolved in dichloromethane to make 10% w/v solutions. The resulting solution (1 mL) was added to stirred 3 mL 1% (w/v) poly(vinyl alcohol) (PVA, Mw = 25 kDa, 88% hydrolyzed, cat# 15132, Polysciences) solution using a vortex mixer at 2000 rpm for 2 min, and the emulsified polymer solution was immediately sonicated with a 20% amplitude (Sonic Dismembrator 500, Thermo Fisher Scientific) in six 10 s bursts. The test tube was immersed in ice water during sonication. After sonication, the emulsion was added dropwise into 30 mL 1% (w/v) PVA solution and stirred for 3 h at room temperature to remove the residual organic solvent. NPs were collected and washed three times with distilled water by centrifugation at 10 000 × g for 5 min at 4 °C, and the NPs were stored at −80 °C refrigerator. Particle diameter was measured by dynamic light scattering (DLS), and the surface morphology was observed by SEM with gold electrospray.
To prepare the fluorescence-labeled NPs, 0.02% (w/v) coumarin-6 (green fluorescence, Sigma) was added and dissolved in the polymer solution for NPs fabrication. In addition, forskolin (cat# 11018, Cayman Chemical) and Repsox (cat# 14794, Cayman Chemical) were selected and loaded into NPs to generate FR/NPs. The aforementioned protocol was used, but 5% (wt/wt) of hydrophobic drugs with the same molar ratio of F and R were added and dissolved in the polymer solution of PLGA35k/PLGA55k-b-PEG5k (50/50 wt/wt).
Preparation of drugMAP Building Blocks:
The MMP sensitive PEG-based microgel (μGel) beads were prepared by a customized microfluidic device with two separate pregel aqueous solutions, as previously described.[15a] Aqueous solution 1: 10% (w/v) 4-arm PEG vinyl sulfone (Mw = 20 kDa, JenKem Technology USA Inc.) in 300 × 10−3 M triethylamine (Sigma), pH 8.25, prereacted with 250 × 10−6 M K-peptide (Ac-FKGGERCG-NH2, Genscript), 250 × 10−6 M Q-peptide (Ac-NQEQVSPLGGERCG-NH2, Genscript), and 500 × 10−6 M RGD peptide (Ac-RGDSPGERCG-NH2, Genscript). Aqueous solution 2: 8 × 10−3 M dicysteine modified metalloprotease-sensitive peptide crosslinker (MMP-sensitive crosslinker, Ac-GCRDGPQGIWGQDRCG-NH2, Genscript), prereacted with 10 × 10−6 M Alexa-fluor 568-maleimide (Life Technologies).
Both aqueous solutions were injected at the defined flow rates in a 1:1 volume mixture. Meanwhile, Novec 7500 Engineered Fluid (cat# 7100025016, 3M) with 0.1% Pico-Surf (SF-000149, Sphere Fluidics) acting as a surfactant was used as the continuous oil phase, with the flow rate at 150 μL mL−1. μGel beads were collected into a Corning centrifuge tube and cured at 37 °C for two hours. Thereafter, the cured μGel beads were extracted and purified from the oil phase with a mixed solution of hydroxyethyl piperazineethanesulfonic acid (HEPES) buffer (100 × 10−3 M HEPES, 40 × 10−3 M NaCl, pH 7.4) and hexane in a 1:1 volume, and centrifuged at 3000 rpm for 5 min at 4 °C. The μGel pellets were further washed in HEPES buffer with 0.01% w/v Pluronic F-127 (Sigma) for five times to move the resident oil components. The μGel aqueous solution was further allowed to swell and equilibrate with HEPES buffer at 4 °C.
To make NPs encapsulated microgel (NPs-μGel), different amounts of NPs (0%, 25%, 50%, 100% weight percentage of NPs to the weight of dry pregel components) was dispersed in aqueous solution 1. To enhance the uniform distribution of NPs in the μGel and stable droplet preparation, 0.25% (w/v) hyaluronic acid (HA700K, Lifecore Biomedical, LLC) was added in the dispersed particle solution.
For in vitro cell culture and in vivo evaluation, all μGel beads (pure μGel, NPs-μGel, FR/drugMAP) were prepared with sterilized devices (PDMS device, connecting tubes) and sterile filtered pregel components by a 0.2 μm polyethersulfone membrane. All procedures were performed in a biosafety cabinet.
Size and Swelling Ratio:
To determine the operational regime of droplet generation, at least five images of droplets in the channel were taken using a high-speed camera (Phantom) at each flow rate condition. The size distribution was analyzed by a custom-developed MATLAB code. The size of swollen μGel droplets in buffer solution was also measured in the same manner, and the volume swelling ratio was calculated by the following equation
| (1) |
where Qv is the volume swelling ratio of a single droplet, daq is the diameter of droplets in the aqueous phase (HEPES buffer), and doil is the diameter in the oil phase (Novec 7500).
NP Loading Concentration and Efficiency in μGel:
The NP loading concentration in μGel was quantified by measuring fluorescent intensity of coumarin-labeled NPs. Briefly, the concentrated coumarin-labeled NPs-μGel beads were diluted with HEPES buffer, and 100 μL solution was transferred to a 96-well plate to measure the fluorescent intensity (excitation: 485 nm, emission: 528 nm) by using a plate-reader. Meanwhile, the coumarin-labeled NPs were diluted in HEPES buffer (0 to 8 mg mL−1, 10 serial dilution points) to make the standard curve. The NPs loading efficiency was calculated by the following equation
| (2) |
Degradation of drugMAP Building Blocks:
To study the degradation and model drug release profiles of drugMAP building blocks, NPs were labeled by coumarin-6 dye and μGels were labeled by AF546 dye. 100 μL of NPs-μGel beads were added to the 1 mL PBS or collagenase II solutions (ranging from 1.6 mU mL−1 to 1 U mL−1 in PBS with calcium and magnesium) in centrifuge tubes and incubated at 37 °C with rotation at 20 rpm (n = 4 for each group). Three days later, hydrogel beads were centrifuged at 6000 rpm for 5 min, and 200 μL of the supernatant was transferred to a 96-well plate for measuring the release of coumarin-6 (excitation: 485 nm, emission: 528 nm) and AF546 (excitation: 556, emission: 573 nm) using a plate reader as surrogates for model drug release and hydrogel degradation, respectively. The μGel beads were washed three times with PBS, and pushed through a 110 μm × 110 μm square microfluidic channel and imaged with fluorescence microscopy to measure the remaining model drug and AF546 as well as the deformability and swollen shape of the μGel beads.
Pore Size and Void Fraction of MAP and drugMAP Scaffolds:
Fully swollen and equilibrated MAP or drugMAP building blocks (20 μL) were activated by with 5 U mL−1 FXIIIa (Sigma) and 1 U mL−1 thrombin (Sigma), and the mixture was pipetted into a 3 mm diameter PDMS well on a glass coverslip, and annealed in a humidified incubator at 37 °C for 1.5 h to form porous MAP or drugMAP scaffolds. Thereafter, the scaffolds were placed into HEPES buffer (pH 7.4) overnight to reach equilibrium. Samples were 3D imaged using a Leica TCS SP8 confocal microscope with 10× objective, spanning 1.16 mm × 1.16 mm (in x- and y-axis) × 200 μm (in z-axis). The pore size was analyzed using a custom script written in MATLAB and the void fraction was calculated using ImageJ (stack function).
Rheology Properties:
To determine the effects of particle loading amount on the gelation and gel rheology properties, rheological measurements were performed on bulk gel samples using a DHR-2 rheometer (TA Instruments). Briefly, different amounts of PLGA35k/PLGA55k-b-PEG5k (50/50) NPs (0%, 25%, 50%, 100%, 200% of PEG weight) were quickly vortexed with two pregel aqueous solutions (basic aqueous solution 1 and 2 in 1:1 volume). To make a disk gel sample, a 40 μL mixed particle-containing solution was pipetted onto sterile slide glass siliconized with Sigmacote (SL2–25ML, Sigma-Aldrich), and covered with another glass slides with 1 mm spacer, followed with curing at 37 °C for 2 h. Disc gels were swollen to equilibrium in HEPES buffer overnight before rheological measurements. A frequency sweep of 0.1–10 Hz was performed by using an 8 mm Peltier Plate-Crosshatched surface (TA Instruments), and the storage modulus and loss modulus were calculated from the average of the linear range. At least, four-disc gel samples were measured for each condition.
Drug Release Assay:
Briefly, 2 mg of FR/NPs or 200 μL FR/drugMAP beads was dispersed in a 0.22 μm filters inserted in a centrifuge tube (Corning Costar Spin-X Centrifuge Tube, Thermo Fisher Scientific) with 1 mL PBS (pH 7.4) at 37 °C, with continuous shaking. At discrete time intervals (16 h, 1, 2, 4, 6 days), 0.5 mL of the sample solution was collected from the tube and frozen for the later analysis. Aliquots of the solutions were analyzed by reversed-phase separation and detection using tandem mass spectrometry with multiple reactions monitoring with previously optimized conditions for parent ion production and fragment ion detection on a triple quadrupole mass spectrometer (Agilent 6460). Quantification was achieved with the external standards for both analytes. All experimental samples were analyzed in triplicate and all results were reported as mean ± standard error of the mean.
Cellular Uptake of NPs:
To check cellular uptake of NPs released from NPs-μGels, primary mice skin fibroblasts were seeded in 24-well plates at the density of 10 000 cells cm−2 and coincubated with 0.1 mg coumarin-labeled NPs or 20 μL NPs-μGel (50) beads (around the same weight of NPs) in the inserted Transwell (8 μm pore size), and cultured in the Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). Cells were incubated in a humidified atmosphere containing 5% CO2 at 37 °C. Adhered cells were washed twice with PBS and fixed with 4% paraformaldehyde (PFA) on day 1 and day 4. The samples were stained with phalloidin F-actin and DAPI. The fluorescent images were taken by Zeiss Axio Observer Z1 inverted microscope and the fluorescent intensity was measured by Image J.
Cell Isolation:
Primary neonatal rat cardiomyocytes and fibroblasts were isolated from the hearts of 1–2 day old Sprague–Dawley rat pups as described previously with minor modifications.[47] Briefly, the cardiac tissue was minced and digested with 80 units mL−1 collagenase II (Worthington) and 0.8 mg mL−1 pancreatin (Sigma) at 37 °C in a water bath. Neonatal calf serum (NCS) was applied to inactivate enzymatic activity in the digested cell mixture. The cell solution was filtered through 100 μm mesh and centrifuged at 2200 rpm for 3 min. The cell pellets were suspended in 1 mL NCS and further separated by Percoll density gradient centrifugation. A two-layer density gradient was formed consisting of 40.5% Percoll (GE17–0891-01, Sigma) solution in the top layer and 58.5% Percoll solution in the bottom layer. The cell suspension was layered on top of the gradient and centrifuged at 3000 rpm at room temperature for 30 min. Fibroblasts equilibrated and collected form the top of the transparent Percoll solution. Cardiomyocytes could subsequently be removed from the newly formed layer between the Percoll solutions and harvested separately. Both cells were washed with warm DMEM medium containing 10% FBS and 1% P/S and used immediately.
Cell Culture and Evaluation of Drug Effects In Vitro:
The drug effects on cardiomyocyte viability and proliferation were evaluated. Isolated cardiomyocytes were calculated and seeded on 0.1% gelatin-coated twenty-four well tissue culture plate with a density of 20 000 cells cm−2, and cultured at 37 °C in a humidified, 5% CO2 incubator overnight in DMEM/medium 199 (4/1) containing 10% FBS, 1% non-essential amino acids (NEAAs), and 1% P/S. The next day, the culture medium was replaced by fresh medium containing 20 × 10−6 M F, R or their combination. The medium was changed every other day. Cell viability assay and proliferation assay of cardiomyocytes were performed at days 1, 3, and 5. A live/dead kit (Invitrogen) was for cell viability assay, and images were taken using inverted microscope fluorescence microscopy (Zeiss Axio Observer Z1) to determine the cell numbers and the percentage of dead cells. To analyze the proliferation of cardiomyocyte, cells were stained by the Click-iT EdU assay (Invitrogen) as the vendor-provided protocol. Briefly, cells treated with EdU concentration of 10 ×10−6 M for 24 h before fixing with 4% PFA in PBS, followed with EdU detection and immunofluorescent staining with cTnT antibody (DSHB).
The drug effects on cardiac fibroblast proliferation and myodifferentiation were evaluated. Fibroblasts were seeded on 24-well tissue culture plate with a density of 5000 cells cm−2 and cultured overnight in DMEM containing 10% FBS and 1% P/S. On the next day, the culture medium was replaced by the fresh medium containing F and R at the determined concentrations and combinations, and the medium was changed every other day. MTS cell proliferation assay (cat# PR-G3582, Thermo Fisher Scientific) was performed on days 1, 3, and 5 by following the protocol from the manufacturer. Meanwhile, some cells were fixed with 4% PFA for myodifferentiation assay by fluorescent staining using α-SMA antibody (Abcam) and phalloidin (for F-actin) (Thermo Fisher Scientific).
The drug effects on EC proliferation and network formation were evaluated. Human umbilical vein endothelial cells (HUVECs) were seeded on 0.1% gelatin-coated 24-well plates with a density of 5000 cells cm−2 and cultured overnight in DMEM containing 10% FBS and 1% P/S. On the next day, the culture medium was replaced by the fresh medium containing F and R at the determined concentrations and combinations, and the medium was changed every other day. MTS cell proliferation assay was performed on day 1, 3, and 5. In addition, the ECs network formation was examined on growth factor reduced Matrigel according to the manufacturer’s instructions (cat# CB-40230C, Thermo Fisher Scientific). Briefly, 24-well plates were coated with Matrigel. ECs were digested and plated onto a layer of Matrigel at a density of 3 × 105 cells per well in M199 medium containing 1% FBS and 1% P/S, with the addition of F and R. Vascular endothelial growth factor (VEGF, 20 ng mL−1) was used as a positive control. After 16 h of culture, cells were stained with calcein acetoxymethyl ester (calcein-AM) and observed with an inverted fluorescent microscope. The number of tubular structure, junctions and meshes were analyzed by Image J with the Angiogenesis Analyzer plugin (n = 4–6 per group).
The effects of drugs released from FR/NPs and FR/NPs-μGel beads were evaluated using the methods mentioned above. During cell culture, 2 mg FR/NPs or 100 μL FR/NPs-μGel (100) (theoretical loading weight of NPs ≈2 mg) was added in the inserted Transwell (0.4 μm pore size) in 24-well plates with cultured cells.
MI Model and Intramyocardial Injection of drugMAP:
All animal work was conducted under protocols approved by the University of California Los Angeles (#2016–101-11) and the University of California San Francisco (#AN176681–02) and was performed in accordance with the recommendations of the American Association for Accreditation of Laboratory Animal Care. The ischemia-reperfusion MI model was established as previously described.[40b] Briefly, the left anterior descending coronary artery of female Sprague–Dawley rats (200–250 g, 8–10 weeks) underwent ligation for 30 min, followed by reperfusion. The intramyocardial injections (50 μL, twice) of sterile PBS, FR/NPs (20 mg mL−1 in PBS), MAP gel and FR/drugMAP gel were performed 2 days post-MI via ultrasound-guided transthoracic injection using a 27-gauge syringe. The successful injection was confirmed by a slight local increase of ultrasound signal in the LV wall.
Echocardiographic Assessment:
Echocardiography was performed at 2-day post-MI and five weeks post-injection using standard methods as previously described.[40b,48] Transthoracic echocardiography was performed with a 15-MHz linear array transducer system (Sequoia c256, Acuson, Erlangen, Germany) on all animals anesthetized with isoflurane. The left ventricular end-diastolic volume (LVEDV), left ventricular end-systolic volume (LVESV), and ejection fraction (LVEF) were measured. All measurements were the averages of three consecutive cardiac cycles. Cases where ejection fraction was above 45% at day 2 were excluded from echocardiographic and histological analyses because they indicated an insufficient infarct model.
Histology and Immunostaining:
At 5 weeks after the injection, all rats were sacrificed for tissue harvesting. The hearts were embedded in optimal cutting temperature (OCT) compound, fresh frozen by dry ice immediately, and stored at −80 °C. All tissue blocks were cryosectioned at a thickness of 10 μm by a cryostat microtome (HM525 NX, Thermo Fisher Scientific) starting from the apex of the left ventricle, and 10 serial sections were collected for every 500 μm intervals. All slides were kept at −20 °C for later staining.
Sections were stained with Masson’s trichrome staining using standard protocols and images were captured with an inverted microscope (Nikon, Eclipse Ti-S fluorescence microscope). Masson’s-trichrome staining images were used to evaluate the infarct size, fibrosis area and LV wall thickness with Image J software. The infarct size or scar area (% LV) was calculated by dividing the collagen deposited area to the entire left ventricle area. LV wall thickness was calculated by averaging the minimum infarcted LV wall thickness of all samples for each group.
For immunofluorescent staining, cell samples or air-dried tissue slides were fixed with 4% PFA for 10 min at room temperature, permeabilized with 0.1% Triton X-100 for 15 min, and blocked with 10% normal goat serum for 1 h at room temperature. The cell samples and slides were incubated with primary antibodies, against α-SMA (rabbit, Abcam, ab5694, 1:300), Von Willebrand Factor (vWF, sheep, Abcam, ab11713), CD68 (mouse, Abcam, ab955, 1:300), cardiac Troponin T (cTnT, mouse, DSHB, 1:200) or Ki67 (Rabbit, Abcam, ab16667, 1:200) overnight at 4 °C. Thereafter, appropriate Alexafluor 488- or Alexafluor 546- or Alexa fluor 637-conjugated secondary antibodies (Thermo Fisher Scientific) was added and incubated for 1 h at room temperature. Thereafter, nuclei were stained with 4′,6-diamindino-2-phenylindole (DAPI, 1:2500 in sterilized deionized water, Sigma) for 10 minutes in the dark. All fluorescent images were taken with Zeiss Axio Observer Z1 inverted microscope and confocal Inverted Leica TCS-SP8-SMD Confocal Microscope.
Statistical Analysis:
Data are presented as means ± standard deviations, calculated from the average of at least three biological replicates unless otherwise specified. Statistical analysis was performed using one-way analysis of variance (ANOVA), followed by post-hoc analysis with Turkey’s test using Origin 8 software. p values < 0.05 were considered statistically significant.
Supplementary Material
Acknowledgements
J.F., J.K., and Q.F. contributed equally to this work. The authors were supported in part by a grant from the National Institutes of Health (HL121450 to S.L.), the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the NIH under the Ruth L. Kirschstein National Research Service Award (T32AR059033 to J.S.), UCLA Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research Innovation Award (to S.L.), UCLA startup funding, by the Presidential Early Career Award for Scientists and Engineers (N00014-16-1-2997 to D.D.C.), Kwanjeong Graduate Scholarship (to J.K.), and UCLA MSTP training grant (NIH NIGMS training grant GM008042). SEM was performed at the California NanoSystems Institute (CNSI) Electron Imaging Center for NanoMachines (EICN) Shared Resource Facility at UCLA. Confocal laser scanning microscopy was performed at the California NanoSystems Institute (CNSI) Advanced Light Microscopy/Spectroscopy Shared Resource Facility at UCLA.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
Jun Fang, Jaekyung Koh, Dino Di Carlo and Song Li have applied for a patent related to this study. Dino Di Carlo have financial interests in Tempo Therapeutics. The remaining authors declare no competing interests.
The ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/adfm.202004307.
Contributor Information
Dr. Jun Fang, Department of Bioengineering, University of California, Los Angeles, CA 90095, USA Department of Medicine, University of California, Los Angeles, CA 90095, USA.
Dr. Jaekyung Koh, Department of Bioengineering, University of California, Los Angeles, CA 90095, USA
Dr. Qizhi Fang, Department of Medicine, Cardiovascular Research Institute and Institute for Regeneration Medicine University of California, San Francisco, CA 94143, USA
Dr. Huiliang Qiu, Department of Medicine, Cardiovascular Research Institute and Institute for Regeneration Medicine University of California, San Francisco, CA 94143, USA
Maani M. Archang, Department of Bioengineering, University of California, Los Angeles, CA 90095, USA Department of Medicine, University of California, Los Angeles, CA 90095, USA.
Dr. Mohammad Mahdi Hasani-Sadrabadi, Department of Bioengineering, University of California, Los Angeles, CA 90095, USA California NanoSystems Institute (CNSI), University of California, Los Angeles, CA 90095, USA.
Hiromi Miwa, Department of Bioengineering, University of California, Los Angeles, CA 90095, USA.
Xintong Zhong, Department of Bioengineering, University of California, Los Angeles, CA 90095, USA.
Dr. Richard Sievers, Department of Medicine, Cardiovascular Research Institute and Institute for Regeneration Medicine University of California, San Francisco, CA 94143, USA
Dr. Dong-Wei Gao, Department of Medicine, Cardiovascular Research Institute and Institute for Regeneration Medicine University of California, San Francisco, CA 94143, USA
Randall Lee, Department of Medicine, Cardiovascular Research Institute and Institute for Regeneration Medicine University of California, San Francisco, CA 94143, USA UC Berkeley-UCSF Graduate Program in Bioengineering, University of California, San Francisco, San Francisco, CA 94158, USA.
Dino Di Carlo, Department of Bioengineering, University of California, Los Angeles, CA 90095, USA California NanoSystems Institute (CNSI), University of California, Los Angeles, CA 90095, USA; Department of Mechanical and Aerospace Engineering, University of California, Los Angeles, CA 90095, USA; Jonsson Comprehensive Cancer Centre University of California, Los Angeles, CA 90024, USA.
Song Li, Department of Bioengineering, University of California, Los Angeles, CA 90095, USA Department of Medicine, University of California, Los Angeles, CA 90095, USA; California NanoSystems Institute (CNSI), University of California, Los Angeles, CA 90095, USA.
References
- [1].Nowbar AN, Gitto M, Howard JP, Francis DP, Al-Lamee R, Circ. Cardiovasc. Qual. Outcomes 2019, 12, e005375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hausenloy DJ, Yellon DM, Nat. Rev. Cardiol 2016, 13, 193. [DOI] [PubMed] [Google Scholar]
- [3].Anderson JL, Morrow DA, Engl N. J. Med 2017, 376, 2053. [DOI] [PubMed] [Google Scholar]
- [4] a).Rodgers K, Papinska A, Mordwinkin N, Adv. Drug Delivery Rev 2016, 96, 245; [DOI] [PubMed] [Google Scholar]; b) Cahill TJ, Choudhury RP, Riley PR, Nat. Rev. Drug Discovery 2017, 16, 699. [DOI] [PubMed] [Google Scholar]
- [5] a).Hasan A, Khattab A, Islam MA, Abou Hweij K, Zeitouny J, Waters R, Sayegh M, Hossain MM, Paul A, Adv. Sci 2015, 2, 2198; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Seif-Naraghi SB, Singelyn JM, Salvatore MA, Osborn KG, Wang JJ, Sampat U, Kwan OL, Strachan GM, Wong J, Schup-Magoffin PJ, Braden RL, Bartels K, DeQuach JA, Preul M, Kinsey AM, DeMaria AN, Dib N, Christman KL, Sci. Transl. Med 2013, 5, 173ra25; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Carlini AS, Gaetani R, Braden RL, Luo C, Christman KL, Gianneschi NC, Nat. Commun 2019, 10, 1735; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Matsumura Y, Zhu Y, Jiang HB, D’Amore A, Luketich SK, Charwat V, Yoshizumi T, Sato H, Yang B, Uchibori T, Healy KE, Wagner WR, Biomaterials 2019, 217, 119289. [DOI] [PubMed] [Google Scholar]
- [6] a).Lin X, Liu Y, Bai A, Cai H, Bai Y, Jiang W, Yang H, Wang X, Yang L, Sun N, Gao H, Nat. Biomed. Eng 2019, 3, 632; [DOI] [PubMed] [Google Scholar]; b) Montgomery M, Ahadian S, Huyer LD, Lo Rito M, Civitarese RA, Vanderlaan RD, Wu J, Reis LA, Momen A, Akbari S, Pahnke A, Li RK, Caldarone CA, Radisic M, Nat. Mater 2017, 16, 1038; [DOI] [PubMed] [Google Scholar]; c) Shadrin IY, Allen BW, Qian Y, Jackman CP, Carlson AL, Juhas ME, Bursac N, Nat. Commun 2017, 8, 1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7] a).Passier R, van Laake LW, Mummery CL, Nature 2008, 453, 322; [DOI] [PubMed] [Google Scholar]; b) Menasche P, Nat. Rev. Cardiol 2018, 15, 659; [DOI] [PubMed] [Google Scholar]; c) Weinberger F, Breckwoldt K, Pecha S, Kelly A, Geertz B, Starbatty J, Yorgan T, Cheng KH, Lessmann K, Stolen T, Scherrer-Crosbie M, Smith G, Reichenspurner H, Hansen A, Eschenhagen T, Sci. Transl. Med 2016, 8, 363ra148. [DOI] [PubMed] [Google Scholar]
- [8] a).Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu JD, Srivastava D, Nature 2012, 485, 593; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mohamed TMA, Ang YS, Radzinsky E, Zhou P, Huang Y, Elfenbein A, Foley A, Magnitsky S, Srivastava D, Cell 2018, 173, 104; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hashimoto H, Olson EN, Bassel-Duby R, Nat. Rev. Cardiol 2018, 15, 585; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Mahmoudi M, Yu M, Serpooshan V, Wu JC, Langer R, Lee RT, Karp JM, Farokhzad OC, Nat. Nanotechnol 2017, 12, 845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9] a).Lee LC, Wall ST, Klepach D, Ge L, Zhang ZH, Lee RJ, Hinson A, Gorman JH, Gorman RC, Guccione JM, Int. J. Cardiol 2013, 168, 2022; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lee RJ, Hinson A, Bauernschmitt R, Matschke K, Fang Q, Mann DL, Dowling R, Schiller N, Sabbah HN, Int. J. Cardiol 2015, 199, 18. [DOI] [PubMed] [Google Scholar]
- [10] a).Annabi N, Nichol JW, Zhong X, Ji CD, Koshy S, Khademhosseini A, Dehghani F, Tissue Eng., Part B 2010, 16, 371; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) De France KJ, Xu F, Hoare T, Adv. Healthcare Mater 2018, 7, 1700927. [DOI] [PubMed] [Google Scholar]
- [11].Huebsch N, Lippens E, Lee K, Mehta M, Koshy ST, Darnell MC, Desai RM, Madl CM, Xu M, Zhao XH, Chaudhuri O, Verbeke C, Kim WS, Alim K, Mammoto A, Ingber DE, Duda GN, Mooney DJ, Nat. Mater 2015, 14, 1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Barbetta A, Rizzitelli G, Bedini R, Pecci R, Dentini M, Soft Matter 2010, 6, 1785. [Google Scholar]
- [13].Brougham CM, Levingstone TJ, Shen NA, Cooney GM, Jockenhoevel S, Flanagan TC, O’Brien FJ, Adv. Healthcare Mater 2017, 6, 1700598. [DOI] [PubMed] [Google Scholar]
- [14] a).Ying GL, Jiang N, Mahar S, Yin YX, Chai RR, Cao X, Yang JZ, Miri AK, Hassan S, Zhang YS, Adv. Mater 2018, 30, e1805460; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Che LB, Lei ZY, Wu PY, Song DW, Adv. Funct. Mater 2019, 9, 1904450. [Google Scholar]
- [15] a).Griffin DR, Weaver WM, Scumpia PO, Di Carlo D, Segura T, Nat. Mater 2015, 14, 737; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Koh J, Griffin DR, Archang MM, Feng AC, Horn T, Margolis M, Zalazar D, Segura T, Scumpia PO, Di Carlo D, Small 2019, 15, e1903147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nih LR, Sideris E, Carmichael ST, Segura T, Adv. Mater 2017, 29, 1606471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dorn GW, Nat. Rev. Cardiol 2009, 6, 283. [DOI] [PubMed] [Google Scholar]
- [18].van der Laan AM, Piek JJ, van Royen N, Nat. Rev. Cardiol 2009, 6, 515. [DOI] [PubMed] [Google Scholar]
- [19].Zhao X, Kwan JYY, Yip K, Liu PP, Liu FF, Nat. Rev. Drug Discovery 2020, 19, 57. [DOI] [PubMed] [Google Scholar]
- [20].Back M, Hansson GK, Nat. Rev. Cardiol 2015, 12, 199. [DOI] [PubMed] [Google Scholar]
- [21].Burashnikov A, Antzelevitch C, Nat. Rev. Cardiol 2010, 7, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Michelson AD, Nat. Rev. Drug Discovery 2010, 9, 154. [DOI] [PubMed] [Google Scholar]
- [23].Pierre S, Eschenhagen T, Geisslinger G, Scholich K, Nat. Rev. Drug Discovery 2009, 8, 321. [DOI] [PubMed] [Google Scholar]
- [24] a).Lai NC, Tang T, Gao MH, Saito M, Takahashi T, Roth DM, Hammond HK, J. Am. Coll. Cardiol 2008, 51, 1490; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Takahashi T, Tang T, Lai NC, Roth DM, Rebolledo B, Saito M, Lew WYW, Clopton P, Hammond HK, Circulation 2006, 114, 388. [DOI] [PubMed] [Google Scholar]
- [25].Akhurst RJ, Hata A, Nat. Rev. Drug Discovery 2012, 11, 790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dobaczewski M, Chen W, Frangogiannis NG, Mol J. Cell Cardiol. 2011, 51, 600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27] a).Namkoong S, Kim CK, Cho YL, Kim JH, Lee H, Ha KS, Choe J, Kim PH, Won MH, Kwon YG, Shim EB, Kim YM, Cell. Signal 2009, 21, 906; [DOI] [PubMed] [Google Scholar]; b) Tan SM, Zhang Y, Connelly KA, Gilbert RE, Kelly DJ, Am. J. Physiol.: Heart Circ. Physiol 2010, 298, H1415. [DOI] [PubMed] [Google Scholar]
- [28].Hastings CL, Roche ET, Ruiz-Hernandez E, Schenke-Layland K, Walsh CJ, Duffy GP, Adv. Drug Delivery Rev 2015, 84, 85. [DOI] [PubMed] [Google Scholar]
- [29].Bertero A, Murry CE, Nat. Rev. Cardiol 2018, 15, 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Swider E, Koshkina O, Tel J, Cruz LJ, de Vries IJM, Srinivas M, Acta Biomater. 2018, 73, 38. [DOI] [PubMed] [Google Scholar]
- [31].Mitragotri S, Burke PA, Langer R, Nat. Rev. Drug Discovery 2014, 13, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hu BH, Messersmith PB, J. Am. Chem. Soc 2003, 125, 14298. [DOI] [PubMed] [Google Scholar]
- [33] a).Dallabrida SM, Falls LA, Farrell DH, Blood 2000, 95, 2586; [PubMed] [Google Scholar]; b) Gemmati D, Vigliano M, Burini F, Mari R, Abd El Mohsein HH, Parmeggiani F, Serino ML, Curr. Pharm. Design 2016, 22, 1449. [DOI] [PubMed] [Google Scholar]
- [34] a).Matsumura Y, Zhu Y, Jiang H, D’Amore A, Luketich SK, Charwat V, Yoshizumi T, Sato H, Yang B, Uchibori T, Healy KE, Wagner WR, Biomaterials 2019, 217, 119289; [DOI] [PubMed] [Google Scholar]; b) Ifkovits JL, Tous E, Minakawa M, Morita M, Robb JD, Koomalsingh KJ, Gorman JH, Gorman RC, Burdick JA, Proc. Natl. Acad. Sci. USA 2010, 107, 11507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35] a).Bao R, Tan B, Liang S, Zhang N, Wang W, Liu W, Biomaterials 2017, 122, 63; [DOI] [PubMed] [Google Scholar]; b) Hernandez MJ, Christman KL, JACC Basic Transl. Sci 2017, 2, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lutolf MP, Hubbell JA, Nat. Biotechnol 2005, 23, 47. [DOI] [PubMed] [Google Scholar]
- [37].Lindsey ML, Nat. Rev. Cardiol 2018, 15, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Swaney JS, Roth DM, Olson ER, Naugle JE, Meszaros JG, Insel PA, Proc. Natl. Acad. Sci. USA 2005, 102, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Behzadi S, Serpooshan V, Tao W, Hamaly MA, Alkawareek MY, Dreaden EC, Brown D, Alkilany AM, Farokhzad OC, Mahmoudi M, Chem. Soc. Rev 2017, 46, 4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40] a).Yoshizumi T, Zhu Y, Jiang H, D’Amore A, Sakaguchi H, Tchao J, Tobita K, Wagner WR, Biomaterials 2016, 83, 182; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Le LV, Mohindra P, Fang Q, Sievers RE, Mkrtschjan MA, Solis C, Safranek CW, Russell B, Lee RJ, Desai TA, Biomaterials 2018, 169, 11; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhu Y, Matsumura Y, Wagner WR, Biomaterials 2017, 129, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pena B, Laughter M, Jett S, Rowland TJ, Taylor MRG, Mestroni L, Park D, Macromol. Biosci 2018, 18, e1800079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Daly AC, Riley L, Segura T, Burdick JA, Nat. Rev. Mater 2020, 5, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mealy JE, Chung JJ, Jeong HH, Issadore D, Lee D, Atluri P, Burdick JA, Adv. Mater 2018, 30, e1705912. [DOI] [PubMed] [Google Scholar]
- [44].Hettiaratchi MH, Miller T, Temenoff JS, Guldberg RE, McDevitt TC, Biomaterials 2014, 35, 7228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Makadia HK, Siegel SJ, Polymers 2011, 3, 1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V, J. Controlled Release 2012, 161, 505. [DOI] [PubMed] [Google Scholar]
- [47].Ehler E, Moore-Morris T, Lange S, J. Vis. Exp 2013, 6, e50154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Mihardja SS, Gonzales JA, Gao DW, Sievers RE, Fang QZ, Stillson CA, Yu JS, Peng M, Lee RJ, Biomaterials 2013, 34, 8869. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.