

Abstract
Gasdermins were recently identified as the mediators of pyroptosis — inflammatory cell death triggered by cytosolic sensing of invasive infection and danger signals. Upon activation, gasdermins form cell membrane pores, which release pro-inflammatory cytokines and alarmins and damage the integrity of the cell membrane. Roles for gasdermins in autoimmune and inflammatory diseases, infectious diseases, deafness and cancer are emerging, revealing potential novel therapeutic avenues. Here, we review current knowledge of the family of gasdermins, focusing on their mechanisms of action and roles in normal physiology and disease. Efforts to develop drugs to modulate gasdermin activity to reduce inflammation or activate more potent immune responses are highlighted.
Subject terms: Inflammasome, Drug discovery
Gasdermins (GSDMs) are a recently characterized protein family that mediate a programmed inflammatory cell death termed pyroptosis. Here, Lieberman and colleagues review current understanding of the expression, activation and regulation of GSDMs, highlighting their roles in cell death, cytokine secretion and inflammation. Emerging opportunities to develop GSDM-targeted drugs and the associated challenges are highlighted.
Introduction
Gasdermins (GSDMs) are a recently characterized protein family encoded by six paralogous genes: GSDMA, GSDMB, GSDMC, GSDMD, GSDME (also known as DFNA5) and DFNB59 (also known as pejvakin)1. Proteolytic cleavage of the GSDMs liberates an N-terminal (NT) fragment, which can assemble in membranes to form pores (verified for most, but not all, of the family). GSDM pores can disrupt cell membrane integrity to trigger an inflammatory cell death in which cellular contents, including inflammatory cytokines, are released into the extracellular space2–4 (Box 1; Figs 1,2). GSDM-mediated cell death is termed pyroptosis (fiery death).
Fig. 1. Key events in the history of gasdermins.

The purple boxes indicate milestone discoveries of gasdermin (GSDM) research.
Fig. 2. Gasdermins function as gatekeepers of pyroptosis.

In response to invasive pathogens, sterile danger signals or cytotoxic T cell attack, gasdermins (GSDMs) are activated by proteolytic cleavage, which releases the N-terminal (NT) fragment, which forms large cell membrane pores. The GSDM pore behaves as a gatekeeper for initiating downstream inflammatory cascades and pyroptotic cell death. Pyroptotic cells form large balloon-like membrane structures. Small intracellular molecules, including cytokines and cellular alarmins, are released through GSDM pores, causing inflammation. Some cells, termed ‘hyperactivated’, repair GSDM pores by shedding the damaged membrane and survive, but still induce inflammation by releasing IL-1 family cytokines.
The name ‘gasdermin’ comes from GSDM expression in the gastroinstestinal tract and skin. Individual GSDMs are selectively expressed with varying abundance wherever the body encounters, detects and responds to infection3,5, being predominantly expressed in specific mucosal sites (Table 1). GSDMA is found in skin and the gastrointestinal tract, GSDMB is located in the lung, oesophagus, gastrointestinal tract and immune cells, GSDMC is found in keratinocytes and the gastrointestinal tract, and GSDMD is found in the gastrointestinal epithelia and in the sentinel cells of the immune system, macrophages and dendritic cells3. GSDME has a different pattern of expression in mesenchymal cells — muscle (both skeletal and cardiac), central nervous system (CNS) and placenta6. The physiological role of GSDME in these tissues, where inflammation might be harmful, is not entirely clear, although it may be involved in development7.
Table 1.
Human and mouse gasdermin genes
| Human gene (chromosomal location) | Mouse gene (chromosomal location) | Expression in normal tissues | Expression in tumours | Activation pathway | Biological function | Disease links | Refs |
|---|---|---|---|---|---|---|---|
| GSDMA (17q21.1) | Gsdma1–3 (11D) | Gastric and skin epithelia | Silenced in gastric cancer tissues and cell lines | Not known | Not known | Systemic sclerosis in humans, alopecia in mice | 1,27,28,30,31,61 |
| GSDMB (17q21.1) | None | Airway, oesophagus, gastrointestinal tract, liver and colon epithelium, neuroendocrine cells, immune cells | Expressed in colon, rectal, pancreatic and cervical cancers, and barely expressed in breast, lung and liver cancers | Granzyme A | Not known | Inflammatory bowel disease, asthma, type I diabetes | 19,32,72,74–80 |
| GSDMC (8q24.21) | Gsdmc1–4 (15D1) | Keratinocytes, trachea, spleen, oesophagus, small intestine, caecum, and colon | Upregulated in colorectal cancer and melanoma | TNFR–caspase 8 | Not known | Not known | 20,82–85 |
| GSDMD (8q24.3) | Gsdmd (15D3-E1) | Immune cells, placenta, oesophagus and gastrointestinal tract epithelium | Expressed in oesophageal and gastric, pancreatic, prostate cancers, melanoma, salivary gland tumours, Jurkat T cells, Ramos B cells | Inflammasome inflammatory caspases; neutrophil elastase; cathepsin G; RIPK1–caspase 8 | Pyroptosis; NETosis | Sepsis, experimental autoimmune encephalomyelitis, macular degeneration, neonatal onset multisystem inflammatory disease | 13,14,25,43,105,106,131,144 |
| GSDME (7p15.3) (a.k.a. DFNA5) | Gsdme (6B2.3) (a.k.a. Dfna5) | Cochlea, placenta, heart, brain, kidney | Epigenetically inactivated by DNA methylation in gastric, colorectal and breast cancer, and most human cancer cell lines | Granzyme B, caspase 3 | Pyroptosis | Autosomal dominant nonsyndromic hearing loss | 16–18,33–38 |
| DFNB59 (2q31.2) (a.k.a. PJVK) | Dfnb59 (2C3) (a.k.a. Pjvk) | Brain, eye, inner ear, heart, lung, kidney, liver, intestine, testis | Not known | Not known | Hair cell maintenance | Recessive nonsyndromic hearing impairment | 39 |
a.k.a., also known as.
GSDMs A–E share highly conserved NT and C-terminal (CT) domains separated by a variable linker (Fig. 3a). The NT domain of GSDMs binds to acidic phospholipids in the inner leaflet of cell membranes and forms pores8–11. In the full-length protein, the CT domain folds back on the NT domain to auto-inhibit pore formation9,12. GSDM cleavage in the linker region liberates the active, pore-forming NT domain. GSDMD is the most studied GSDM as it is the key executioner of inflammasome-induced pyroptosis and is probably the most important GSDM mediating disease-associated inflammation. GSDMD cleavage and pyroptosis are activated when cytosolic danger and infection sensors in macrophages and dendritic cells assemble into large supramolecular complexes called inflammasomes that recruit and activate inflammatory caspases13–15. The inflammatory caspases activate GSDMD by cleaving after Asp275 (mouse Asp276) in the linker13,14. By contrast, GSDME is activated during classical apoptosis (Box 2), by caspase 3 cleavage after Asp270, which converts noninflammatory apoptotic death into inflammatory pyroptotic death in cells that express GSDME16,17. Granzymes (Gzms) A and B, death-inducing proteases in natural killer (NK) cells and cytotoxic T lymphocytes (CTLs), also cleave GSDMB and GSDME, respectively, to activate pyroptosis during killer cell attack, which was previously thought of as noninflammatory18,19. A recent study showed that GSDMC can be cleaved by caspase 8 after TNF-mediated death receptor signalling to trigger pyroptosis20. How GSDMA is activated is currently unknown.
Fig. 3. Molecular mechanisms that activate gasdermins.

In response to apoptotic signalling, gasdermin C (GSDMC) and GSDME are processed by caspase 8 and caspase 3, respectively, converting apoptosis into pyroptosis. Granzymes (Gzms) secreted by killer cells are delivered by perforin into target cells, where GzmA and GzmB can directly cleave and activate GSDMB and GSDME, respectively, to trigger pyroptotic cell death. In immune sentinel cells, both cytosolic canonical inflammasomes that respond to microbial infection or danger signals and the noncanonical inflammasome that responds directly to lipopolysaccharide (LPS) or endogenous oxidized phospholipids activate the inflammatory caspases (caspases 1, 4, 5 and 11), which cleave GSDMD and generate pore-forming N-terminal GSDMD (GSDMD-NT). TAK1 inhibition by the Yersinia effector protein YopJ or the small molecule 5z7 triggers caspase 8-dependent GSDMD cleavage and activation. GSDMD can also be directly processed and activated by neutrophil elastase (ELANE) and cathepsin G. In addition to protease-mediated release of active GSDM-NT, mutations in Gsdma3 lead to abolition of C-terminal GSDM inhibition and trigger GSMDA3 pore-forming activity. Diagram at bottom right indicates the proteases known to cleave and activate each of the gassdermins (yellow, caspases; purple, lymphocyte granzymes; blue, myeloid cell granule proteases). ALR, AIM-2 like receptor; CLR, C type lectin receptor; DAMP, damage-associated molecular pattern; NLR, NOD-like receptor; MOMP, mitochondrial outer membrane permeabilization; PAMP, pathogen-associated molecular pattern; TLR, Toll-like receptor.
GSDM pores in the plasma membrane act as channels through which low-molecular-weight cellular contents are released into the extracellular space to initiate inflammation (Fig. 2). Importantly, the pores mediate the ‘unconventional protein secretion’ of the pro-inflammatory cytokines (IL-1β and IL-18) that lack a signal peptide for secretion via the endoplasmic reticulum to Golgi secretory pathway21. Cellular alarmins, including ATP and HMGB1, cleaved GSDMs and other cytokines and chemokines, are released at the same time to amplify inflammation in the tissue and recruit immune cells to respond to the infection or damage. Many small molecules and other proteins, which may differ between cell types, are probably released from pyroptotic cells. Pore formation triggers rapid pyroptotic cell death in which the damaged membrane forms large ballooning bubbles and dying cells appear to flatten as their cytoplasmic contents are released22. Both the rapidity of cell death and the morphological changes are hallmarks that distinguish pyroptotic from apoptotic cell death3. In some circumstances, GSDM-mediated cell membrane damage can be repaired by the ubiquitous damaged cell membrane repair response — the cell survives but inflammatory mediators are released through GSDM pores before the pores are removed from the membrane23. Macrophages that survive despite GSDMD cleavage have been termed ‘hyperactivated’, because they both stimulate inflammation and preserve their other antigen-presenting functions24.
The diverse physiological and pathological roles of GSDMs as executioners of pyroptosis are still being uncovered. However, associations of GSDMs with several diseases have emerged. GSDMD has been implicated in the pathogenesis of many inflammasome-associated diseases and tissue damage14,25,26. In addition, spontaneous mutations of GSDMs lead to disorders such as alopecia (GSDMA)27–31, asthma (GSDMB)32 and hearing impairment (GSDME/DFNB59)6,33–39 in mice and/or humans. Moreover, accumulating evidence suggests that GSDMs can inhibit or promote infection and cancers, implying complicated links between inflammation and pyroptosis and the development and progression of those diseases18–20,40–47.
The key roles of GSDMs in infection, inflammation and cancer have stimulated investigation of GSDM-targeting therapeutics. Strategies to therapeutically modulate GSDM activity are beginning to emerge, and several inhibitors of GSDMD have now been reported45,48,49.
This Review encapsulates our current understanding of the expression, activation and regulation of GSDMs and highlights their important physiological and pathological roles as perpetrators of cell death, cytokine secretion and inflammation. Emerging opportunities to develop GSDM-targeted drugs, particularly GSDMD inhibitors, and the associated challenges, are discussed.
Box 1 Discovery of pyroptosis.
Pyroptosis, a programmed, inflammatory cell death, is mediated by N-terminal (NT) gasdermin pores (Fig. 2). Pyroptotic cells lose cell membrane integrity, which leads to release of inflammatory cellular contents and formation of large ballooning bubbles, which are much larger than apoptotic blebs22,186. The dying cell appears to flatten as its cytoplasmic contents are released.
The first descriptions of pyroptosis were in the 1990s in macrophages that died after infection with invasive Shigella flexneri or Salmonella enterica subsp. Typhimurium. At that time, invasive bacteria-induced cell destruction was regarded as apoptosis, as markers of apoptosis (DNA fragmentation and membrane blebbing) were observed187–190. Further studies showed that macrophage death in response to bacterial infection was mediated by inflammatory caspase 1 rather than apoptotic caspases and was associated with pro-inflammatory cytokine secretion190–195. In 2001, Cookson and Brennan coined the term pyroptosis (fiery death) to distinguish inflammatory caspase-mediated cell death from apoptosis and unprogrammed necrosis caused by lack of membrane integrity196. Gasdermin D (GSDMD) was identified in 2015 as the inflammatory caspase substrate responsible for pyroptotic cell death in immune cells, and the following year the mechanism responsible for GSDMD-mediated death was identified as pore formation by the inflammatory caspase-generated GSDMD-NT8–11,13,14,22. Identifying the molecular basis of pyroptosis has prompted an explosion of research to understand the role of pyroptosis in disease.
Box 2 Overview of apoptosis.
Classical apoptosis, a physiological noninflammatory programme of cell suicide, is triggered by a variety of stimuli including death receptor signalling, cytotoxic drugs, radiation and a variety of cellular stresses (including growth factor deprivation and hypoxia) that lead to mitochondrial outer membrane permeabilization (MOMP) and the activation of apoptotic effector caspases (caspases 3, 6 and 7)197,198. During apoptosis, mitochondria generate rective oxygen species (ROS), lose their transmembrane potential and release cytochrome c into the cytosol, cell membrane lipids become scrambled and externalize phosphatidyl serine, chromatin condenses, genomic DNA is fragmented, mRNA is globally degraded and new protein synthesis is blocked197,199–201. Phagocytic cells, which recognize cell surface phosphatidylserine on apoptotic cells, rapidly engulf and remove apoptotic cells in vivo before their plasma membrane loses its integrity202,203. Thus, apoptotic cells do not release their cellular contents into the extracellular milieu, and apoptotic death is usually noninflammatory and immunologically silent. Although pyroptosis and apoptosis have historically been considered separate pathways, recent studies discussed in this Review show that apoptotic stimuli can trigger pyroptosis under certain circumstances. For example, activated caspase 3 can cleave gasdermin E (GSDME) to cause pyroptosis instead of apoptosis16,17. Similarly, in cells infected with pathogenic Yersinia, TNF receptor activation activates caspase 8 to trigger GSDMD-dependent pyroptosis105,106.
The gasdermin family
Humans have six GSDM genes: GSDMA, GSDMB, GSDMC, GSDMD, GSDME and DFNB59 (Table 1; Fig. 1). GSDME on human chromosome 7p15.3 is the most ancient gene with orthologues found in early metazoa and invertebrates17,50,51. The other GSDM genes likely arose as gene duplications — GSDMA and GSDMB on human chromosome 17q21 and GSDMC and GSDMD on human chromosome 8q2. Although in metazoa DFNB59 is not truncated and is closely homologous to GSDME, mammalian DFNB59 is a more distant and truncated gene, whose N terminus is homologous to the NT, pore-forming region of the other family members, but whether it possesses pore-forming activity is unknown. Mice lack a GSDMB orthologue, but have one Gsdmd on chromosome 15, one Gsdme on chromosome 6 and three Gsdmas (Gsdma1–3) and four Gsdmcs (Gsdmc1–4) clustered on chromosomes 11D and 15D1, respectively1.
Although published studies and web-based gene expression atlases are available for GSDM expression, these resources are inadequate for accurately predicting GSDM expression, which can be induced by cytokines and immune signalling in cells that are not ordinarily thought to express GSDMs, including lymphocytes. For example, GSDMA is upregulated by TGFβ52, GSDMB by interferon-α (IFNα), IFNβ, IFNγ and TNF19, GSDMD by IRF2 (ref.53) and GSDME by corticosteroids and forskolin54. Furthermore, expression of some of the GSDMs (especially of GSDMA and GSDME in cancer cell lines) is inferred to be epigenetically suppressed by DNA methylation, as DNA methyltransferase inhibitors can induce their expression55–60. Perhaps because of their expression in barrier cells, mutations in GSDM genes have been associated with skin defects, asthma and deafness in mice and humans (Table 2).
Table 2.
Disease-linked gasdermin mutations
| Genea | Nucleotide change | Amino acid change | Consequence | Disease link | Refs |
|---|---|---|---|---|---|
| GSDMA | 53G>A | R18Q | Unknown | Asthma/systemic sclerosis | 67 |
| 382G>A | V128M | Elevated IgE and bronchial hyperresponsiveness | Asthma | 69 | |
| Gsdma3 | – | Lacks auto-inhibition interfaces I and II: | Disrupts auto-inhibition, and Gsdma3 becomes constitutively active to induce pyroptosis in hair follicle cells | Alopecia | 9,27–31 |
| B2 insertion | 1–259 + RDW | ||||
| – | Disrupts auto-inhibition interface I: | ||||
| 832A>C | T278P | ||||
| 1028T>C | L343P | ||||
| 1030T>C | Y344H | ||||
| 1031A>G | Y344C | ||||
| 1042G>A | A348T | ||||
| – | Lacks auto-inhibition interface II: | ||||
| 1096A>T | K366* (stop codon) | ||||
| – | Disrupts auto-inhibition interface II: | ||||
| 1231_1236dup | Duplication of E411A412 | ||||
| GSDMB | 871G>A; 892C>T |
G291R; P298S Reduce conformational flexibility and increase positive surface charge |
Unknown | Asthma | 133 |
| 236–1199G>A | Intron variant | Elevated IgE and bronchial hyperresponsiveness | 69 | ||
| 661+742T>C | Exon 6 (13 aa) deletion | Unknown | 77 | ||
| GSDME |
990+503_ 990+1691delins132 991-15_991-13del 991-6C>G 1183+4A>G 991-15_991-13del |
Exon 8 skipping and truncated GSDME containing aa 1–330 and an aberrant 41-residue C-terminal sequence due to frameshift (lacks auto-inhibition interfaces I and II) | Truncated GSDME is constitutively active to induce pyroptosis in cochlear hair cells | Autosomal dominant hearing loss | 6,17,33–38,92 |
| 410T>C | I137T | Most GSDME mutants have reduced pore-forming activity and may have reduced tumour inhibition | Cancer | 18 | |
| 650T>A | I217N | ||||
| – | Disrupts oligomerization interface: | ||||
| 53A>T | D18V | ||||
| 70A>G | N24D | ||||
| 635C>T | P212L | ||||
| – | Lacks extension domains I and II: | ||||
| 138G>A | W46* | ||||
| 628G>T | E210* | ||||
| DFNB59 | 113dupT | K41Efs*8 (48 aa truncation) | T54I and R183W mutants result in auditory neuropathy while other truncations lead to defect in cochlear hair cell function | Autosomal recessive hearing loss | 39,97–100 |
| 122delA | K41Sfs*18 | ||||
| 161C>T | T54I | ||||
| 406C>T | R136* | ||||
| 499C>T | T167* | ||||
| 547C>T | R183W | ||||
| 726delT | F242Lfs*7 | ||||
| 988delG | V330Lfs*7 | ||||
| Dfnb59 | 868A>T | K290* | Defect in cochlear hair cell function | Autosomal recessive hearing loss | 98 |
aa, amino acid; GSDM, gasdermin. aUpper case gene names refer to human genes, and lower case refers to mouse genes.
GSDMA
The first GSDM family member was identified by positional cloning of a gene causing abnormal skin and hair development in the mouse Rim3 mutant5,61. Subsequent studies of mice with similar skin phenotypes27–31 and bioinformatics62 identified a Gsdma1, Gsdma2 and Gsdma3 gene cluster on mouse chromosome 11D and found that Gsdma3 mutations caused alopecia (Table 2; Fig. 1). All these mutations localize to the CT domain of Gsdma3 and behave as gain-of-function mutations that unmask the functional NT domain9. Gsdma2 and Gsdma3 are primarily expressed in stomach and skin, respectively27,52. The human GSDMA orthologue is also expressed in stomach and skin but is silenced in gastric cancer tissues and cell lines5 (Table 1). The DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine (5-aza-dC) upregulates GSDMA in these cancer lines, suggesting that GSDMA expression is suppressed by DNA methylation55. In pit cells of the gastric epithelium, TGFβ induces the LIM domain only 1 (LMO1) transcription factor to upregulate GSDMA and trigger pit cell death52. TNF similarly upregulates Gsdma3 and causes death of mouse skin keratinocytes in vitro and in vivo63. It is worth noting that no obvious phenotype was observed in Gsdma–/– or Gsdma3–/– mice64,65, indicating that the Gsdma genes may be redundant in development and in the maintenance of gastric and skin barriers, or that their function is only apparent upon challenge by pathogens or other danger signals, which would be consistent with cleavage-dependent activation. GSDMA mutations have not been reported in human alopecia; however, GSDMA polymorphisms have been linked to systemic sclerosis (an autoimmune fibrotic disease of the skin and visceral organs), inflammatory bowel disease (IBD) and childhood asthma66–69 (Table 2).
GSDMB
The human GSDMB gene clusters with GSDMA62, but no Gsdmb has been identified in rodents. GSDMB is expressed more broadly than GSMDA, especially in airway and gastrointestinal epithelia, liver, and neuroendocrine and immune cells32,70–72 (Table 1). There are four GSDMB isoforms (1–4; Ensembl: ENSG00000073605), but whether these isoforms are expressed in a tissue or cell-specific manner is unknown. Genome-wide association studies (GWAS) have uncovered a strong association between GSDMB and susceptibility to chronic inflammatory diseases including IBD, asthma and type I diabetes32,73–76, and a single nucleotide polymorphism linked to increased GSDMB expression is associated with asthma32 (Table 2). The link to childhood asthma and IBD is shared between GSDMA and GSDMB66,69,77, likely reflecting the close chromosomal locations of these two genes and their potentially coupled expression.
GSDMB isoform 1 is the dominant isoform in human bronchial epithelial cells, and elevated isoform 1 expression is found in patients with asthma compared with healthy controls32. GSDMB isoform 1 overexpression increases the levels of genes involved in airway responsiveness and remodelling including the genes encoding TGFβ1, 5-lipoxygenase and MMP9, which could explain its role in asthma32. Surprisingly, unlike other GSDMs, GSDMB isoform 1 was found exclusively in the nucleus, consistent with a postulated role in amplification of transcription32, but making it uncertain whether GSMDB-mediated pyroptosis is involved in the pathogenesis of asthma or IBD. More in vitro and in vivo evidence is required to understand whether the effects of GSDMB depend on pore formation.
Unlike GSDMA and GSDME, GSDMB expression is markedly increased in various cancers, including cervical, breast, gastrointestinal and hepatic cancers, and its high expression is associated with poor prognosis71,78–80 (Table 1). However, whether different isoforms of GSDMB are expressed in distinct cancers or how GSDMB affects tumour biology is not known. The novel nuclear localization and transcriptional effect of GSDMB32 suggest that its function may be largely independent of pore-forming activity and raise the possibility that other GSDMs might also have unsuspected functions that are independent of their cleavage or ability to form pores.
GSDMC
GSDMC was first cloned from melanoma cells as a marker for melanoma progression81,82, and the mouse cluster (Gsdmc1–4) was identified by phylogenetic analysis1. GSDMC is expressed in trachea, spleen, oesophagus, small intestine, caecum, colon1,70,82 and in skin keratinocytes, where expression is markedly increased by UV radiation via calcium-triggered NFATc1 signalling83,84. GSDMC expression can also be induced in tumour cells by hypoxia-triggered STAT3 phosphorylation and its complexation with nuclear-translocated PDL1 (ref.20) (Table 1). GSDMC contributes to UV-induced MMP1 expression by activating ERK and JNK pathways84. GSDMC is cleaved and activated by caspase 8 in death receptor signalling, converting apoptosis into pyroptosis in cells that express GSDMC20. GSDMC is also upregulated in colorectal cancer and promotes cancer cell proliferation85, suggesting that GSDMC may function as an oncogene.
GSDMD
GSDMD was originally identified by homology to other GSDM genes on chromosome 8 near GSDMC81. A 2010 proteomics study86 identified GSDMD as the most efficient (and also selective) substrate of inflammatory caspases. However, the functional role of GSDMD and its cleavage were not explored further until the discovery of GSDMD as the key executor of inflammasome-induced pyroptosis reported by two groups in 2015, using a chemical mutagenesis screen in mice and a cell-based CRISPR screen13,14.
GSDMD is expressed in immune cells (especially macrophages and dendritic cells)3, placenta, gastrointestinal epithelium, various cancers including oesophageal and gastric cancers, pancreatic cancer, prostate cancer, melanoma and salivary gland tumour, and Jurkat T and Ramos B cancer cell lines70,81,87 (Table 1). Transcription of GSDMD is upregulated by IRF2 (ref.53), which is a transcriptional suppressor of interferons. Although human GSDMD genetic polymorphisms have been identified, including a few that disrupt cleavage or pore formation88, most single nucleotide polymorphisms did not alter pore formation, and so far none has been linked to disease. The lack of known disease links does not mean that none exists, especially as the identification of GSDMD as the mediator of pyroptosis is so recent. Although GSDMD is widely expressed, no overt phenotype has so far been described in Gsdmd–/– mice, suggesting that GSDMD is largely dispensable for development and physiological homeostasis87. However, a recent study showed that the AIM2 cytosolic DNA-sensing inflammasome purges genetically compromised neuronal cells during neurodevelopment through GSDMD activation, preventing the accumulation of genetically aberrant cells and contributing to CNS homeostasis7. Gsdmd–/– mice, like Aim2–/–, Asc–/– or Casp1–/– mice, exhibited increased anxiety in behavioural tests7. In models of infection, Gsdmd–/– mice can be more or less susceptible to infection than wild-type mice, depending on the pathogen40,44,89,90. However, Gsdmd–/– mice are protected from death and morbidity in many models of genetic or induced inflammation, including sepsis, experimental autoimmune encephalomyelitis (EAE, a mouse model for multiple sclerosis (MS)), macular degeneration and neonatal onset multisystem inflammatory disease (NOMID, caused by gain-of-function mutation of the inflammasome gene NLRP3)14,25,26, highlighting the central role of GSDMD in inflammatory disease.
GSDME
GSDME was first described as a mutated gene (DFNA5) that results in an autosomal dominant form of hearing impairment, caused by cochlear hair cell loss6,33–38,91. These mutations in DFNA5 lead to skipping of exon 8, premature termination and activation of GSDME to cause cell death. GSDME mRNA is detected in cochlea, placenta, heart, brain and kidney6,92 (Table 1). GSDME is cleaved and activated by caspase 3 (and possibly other apoptotic effector caspases, caspases 6 and 7) when cells expressing GSDME are triggered to undergo apoptosis. GSDME cleavage converts noninflammatory apoptosis into inflammatory pyroptosis. Why GSDME-activating mutations specifically cause auditory damage, with no apparent effect on other organs in which it is highly expressed, is unclear. One possibility is that because the autoactive GSDME protein is unstable, pyroptosis could be limited to especially vulnerable cochlear hair cells17. The poor stability of autoactive GSDMs could also explain why there are no reports of disease-associated autoactive mutant GSDMs except for human GSDME and mouse GSDMA.
Relative to normal tissue, GSDME expression is epigenetically suppressed by methylation in gastric, colorectal and breast cancers, and most human cancer cell lines. By contrast, p53, which is inactivated in many cancers, reportedly activates GSDME transcription in DNA-damaged cells93, which could promote the removal of cancer cells by inflammatory recruitment and activation of phagocytic macrophages. GSDME is also mutated in a variety of other cancers, consistent with its postulated role as a tumour suppressor gene56–60,94,95. When expression of GSDME is induced by DNA methyltransferase inhibitors in vitro, colony formation in gastric and colorectal cancers and invasiveness in breast cancer are reduced56–59. Notably, cancer-associated GSDME mutations in the NT domain are primarily loss-of-function (LOF) mutations, which lead to impaired pore-forming ability18 (Table 2).
DFNB59
DFNB59 is a more distantly related GSDM family member with a truncated non-homologous CT domain. DFNB59 transcripts are detected in brain, eye, inner ear, heart, lung, kidney, liver, intestine and testis39,96 (Table 1). Like GSDME, DFNB59 is linked by familial genetic mutations to hearing loss in humans, but these mutations are recessive, rather than dominant39. Patients with DFNB59 mutations show either dysfunctional cochlear outer hair cells97 or auditory neuropathy39, which impairs neural transmission of the auditory signal (Table 2). Mutations in mouse Dfnb59 were also found to be associated with deafness by forward genetic screening97. However, Dfnb59–/– mice, but not Gsdme–/– mice, show hearing impairment39,97–99, suggesting that they cause deafness through different mechanisms.
DFNB59 is a peroxisome-associated protein required for oxidative stress-induced peroxisome proliferation in both hair cells and auditory neurons96, which protects against sound-induced oxidative damage. DFNB59 senses sound-induced reactive oxygen species (ROS) and recruits the autophagy machinery to degrade damaged peroxisomes100. It is unclear whether DFNB59 is a pore-forming protein or whether it is constitutively active, as its extremely short CT domain might not inhibit pore formation. It is worth exploring whether DFNB59 forms pores in peroxisome membranes and whether other interacting proteins inhibit or activate it. In support of the latter possibility, DFNB59-CT interacts with the ROCK2 Rho-associated protein kinase and the IQGAP1 scaffold protein99.
Role of gasdermins in pyroptosis
The best-characterized pyroptosis pathway is that induced by GSDMD downstream of inflammasome activation in immune sentinel cells and some epithelial cells at mucosal and skin barriers (Fig. 3). Depending on the upstream inflammasome signals, the nature of the danger molecules released from dying cells may be different. Most canonical inflammasomes first require a transcriptional priming step (for example by lipopolysaccharide (LPS)-mediated Toll-like receptor signalling) to upregulate the expression of inflammasome sensors and pro-forms of IL-1 family cytokines101. When these canonical inflammasomes are activated, such as through the AIM2 sensor by cytosolic double-stranded DNA (dsDNA), the NLRP3 sensor by a large array of stimuli that disturb cell membrane integrity (for example bacterial toxin nigericin, extracellular ATP, uric acid crystals, cholesterol crystals and amyloid aggregates) or the pyrin sensor by the Clostridioides difficile toxin TcdB, IL-1 family cytokines IL-1β and IL-18 are processed to maturity102,103 and GSDMD is cleaved at Asp275 (mouse Asp276) in the interdomain linker13,14. By contrast, noncanonical inflammasomes that activate caspases 4, 5 and 11 proteolyse only GSDMD, but not IL-1 family cytokines even with priming; however, secondary GSDMD pore-induced membrane damage and NLRP3 activation result in cytokine maturation in addition to GSDMD processing14,104.
GSDMD pores in the plasma membrane trigger rapid pyroptotic cell death in which the damaged membrane forms large ballooning bubbles and dying cells appear to flatten as their cytoplasmic contents are released22 (Fig. 2). How or whether GSDMD pore-induced cell death is regulated is not clear, but cells with activated GSDMD pores do not always die. Both the rapidity of cell death and the morphological changes are hallmarks that easily distinguish pyroptotic from apoptotic cell death3. The cellular contents that are released from pyroptosis can include IL-1 family cytokines IL-1β and IL-18, as well as IL-1α that does not require processing for its biological activity. Of note, neither full-length nor processed IL-1 family cytokines have a signal peptide for secretion via the endoplasmic reticulum to Golgi secretory pathway, but pyroptosis provides a mechanism for their release by ‘unconventional protein secretion’. Other released contents include cellular alarmins, such as ATP and HMGB1, cleaved GSDMD, inflammatory caspases, chemokines and other cytokines. These signals amplify inflammation in the tissue and recruit immune cells to respond to infection or danger. Many small molecules and other proteins, which may differ from cell type to cell type and from stimulus to stimulus, are probably released from pyroptotic cells. Detailed unbiased studies are required to elucidate what molecules are released under what condition and which released molecules are particularly important for amplifying inflammation.
In addition to inflammatory caspases, caspase 8, which is activated by cell surface death receptor ligation and oligomerization, can trigger GSDMD-dependent pyroptotic cell death (Fig. 3). During pathogenic Yersinia infection, TAK1 or IκB kinase inhibition by the Yersinia effector protein YopJ unleashes RIPK1–caspase 8-dependent GSDMD cleavage, which contributes to host defence against Yersinia infection105–107. Meanwhile, GSDME also gets cleaved and activated by an unknown mechanism and functions in concert with active GSDMD to trigger pyroptosis of infected cells. Similarly, in intestinal epithelial cells that lack FADD, RIPK1 and RIPK3-mediated caspase 8 activation leads to GSDMD-dependent pyroptosis and causes ileitis108. Like FADD, cellular FLICE-like inhibitory protein (cFLIP) negatively regulates pyroptotic cell death and inflammation. Deficiency of cFLIP relieves the inhibition of LPS-induced formation of a death receptor complex, which contains RIPK1 and caspase 8, leading to caspase 8-dependent cleavage of GSDMD109.
Pyroptosis can be induced by other GSDMs that are not activated by inflammasomes and without involving the IL-1 family cytokines (Fig. 3). During classical apoptosis, caspase 3 activates GSDME by cleaving after Asp270 (mouse Asp271), which converts relatively slow noninflammatory apoptotic death into a more rapid inflammatory pyroptotic death in cells that express GSDME16,17. By contrast, activated caspase 3 cleaves GSDMD within the N terminus and destroys its ability to form pores110. GzmB, which is a serine protease that, like caspases, cleaves after Asp residues, is released from killer lymphocytes into target cells and can also directly cleave cytosolic GSDME to trigger pyroptosis18. GzmB also cleaves and activates caspase 3, which amplifies its effectiveness at killing target cells111–113. GzmA, another death-inducing protease in killer lymphocytes, cleaves GSDMB that is expressed in digestive tract epithelial tumours and causes pyroptotic cell death in target tumour cells19. Collectively, although immune-mediated elimination of infected or cancerous target cells has always been thought of as noninflammatory, killer cells cause inflammatory pyroptosis in target cells expressing GSDME or GSDMB. Furthermore, a recent study showed that GSDMC can also be cleaved by caspase 8 after TNF-mediated death receptor signalling to trigger pyroptosis20.
Regulation of gasdermin activity
Structural studies over the past few years have helped to reveal the elegant mechanisms that control and activate GSDMs, from auto-inhibition to processing, and finally to formation of large β-barrel pores (Fig. 4a). The molecular basis of GSDM auto-inhibition has been illustrated by crystal structures of full-length, inactive mouse GSDMA3 and human and mouse GSDMD9,12. Whereas GSDMA3 was readily crystallized9, extensive loop truncations were required to crystallize either human or mouse GSDMD12. In both GSDMA3 and GSDMD full-length structures, the pore-forming NT segments of the proteins comprise a twisted β-sheet surrounded by α-helices, and a large part of the polypeptide chain that inserts as extended β-strands into the membrane is disordered. The corresponding GSDM-CTs, the repressive segment of the proteins, are compact and almost exclusively α-helical and interact extensively with the GSDM-NTs to inhibit pore formation.
Fig. 4. Mechanisms of gasdermin auto-inhibition, processing and pore formation.

a | Cartoon representation of gasdermin (GSDM) pore formation. Interaction between the N-terminal and C-terminal fragments of a GSDM (GSDM-NT, in purple and GSDM-CT, in yellow) auto-inhibits protein activation. Active caspase 1, a dimer of the p10 (in pale blue)–p20 (in silver) complex, recognizes two copies of GSDM through the GSDM-CT. This exosite recognition mechanism places the long interdomain linker (in grey) between GSDM-NT and GSDM-CT near the active site of caspase 1 (red balls). Active caspase 4 and caspase 11 use a similar mechanism to process GSDMD. Linker cleavage liberates GSDM-NT for pore formation at the membrane. The fully formed GSDM pore features a large cytosolic rim in addition to a transmembrane β-barrel. b | Structural illustration of GSDM at each stage in pore formation depicted in part a. In auto-inhibited GSDM (PDB: 5B5R for GSDMA3), GSDM-NT contacts GSDM-CT at two interfaces. Interface I is formed primarily by charged interactions of the α1 helix and the β1–β2 loop of GSDM-NT with the α5–α8–α12 helix cluster of GSDM-CT. Interface II is formed mainly by hydrophobic interactions between α4 of GSDM-NT and α9 and α11 of GSDM-CT. GSDMD recognition (PDB: 6KN0 and 6VIE) by caspase 1 is mediated by a substrate-binding exosite, comprised of two anti-parallel β-strands — βIII of caspase 1-p20 and βIII′ of caspase 1-p10. The exosite binds to α6 and α8 of GSDMD-CT through primarily hydrophobic interactions. The GSDMD NT–CT linker is positioned near the active site Cys285 of caspase 1 for cleavage. The GSDM pore contains roughly 27 subunits according to the structure of the GSDMA3 pore (PDB: 6CB8). With each subunit contributing four transmembrane β-strands, the membrane-inserted β-barrel contains 108 β-strands. The inner and outer diameters of the pore are approximately 180 Å and 280 Å, respectively. A soluble rim, rich in α-helices and loops, decorates the β-barrel on the cytosolic side. c | GSDM-NT conformations before (left) and after (right) pore formation. The positively charged lipid-binding helix α1 (in orange, like a thumb) is masked by GSDM-CT in the auto-inhibited conformation, and in the membrane-inserted conformation it interacts with lipids with negatively charged head groups found on the inner leaflet of the plasma membrane. Drastic conformational changes of GSDM-NT occur at extension domains 1 and 2 (ED1, in magenta and ED2, in red), which are located at the β3–β4–β5 and β7–α4–β8 regions in auto-inhibited GSDM-NT, respectively. Through pore formation, ED1 and ED2 become the two transmembrane β-hairpins (HP1, in magenta and HP2, in red, like four fingers), respectively.
Some mutations, especially in the C terminus of GSDMs, that disrupt the inhibitory interaction between the CT and NT fragments activate GSDMs spontaneously (Table 2). Both spontaneous and mutagenesis-induced Gsdma3 mutants acquire pore-forming activity and induce inflammatory cell death, which leads to mouse skin abnormalities27–31,61. GSDME mutations that cause familial hearing loss result in a truncated GSDME with spontaneous pore-forming activity6,33–38. The pores formed by these mutants are believed to be constructed with the dissociated C terminus still attached, although this has not been formally verified.
Based on the GSDMA3 structure, there are two auto-inhibitory GSDM NT–CT interfaces. Interface I is located near the centre of the protein and interface II at the periphery (Fig. 4b). The biophysical determinants of these two interfaces differ remarkably. Interface I, formed between the positively charged α1 helix and β1–β2 loop of GSDM-NT and an acidic groove of GSDM-CT, is primarily mediated by charge–charge interactions. By contrast, interface II is established through mainly hydrophobic interactions between the α4 helix of GSDM-NT and GSDM-CT. Gain-of-function mutations in GSDM-associated diseases disrupt either of the auto-inhibitory contacts (Table 2), bypassing the requirement of proteolysis in GSDM activation and leading to membrane penetration by full-length GSDMs9,12.
Enzymatic processing of GSDMD for its activation (Fig. 4a) is more complex than classical substrate engagement by inflammatory caspases. The crystal structure of active caspase 1 in complex with a GSDMD interdomain linker fragment suggested that the catalytic pocket of inflammatory caspases recognizes the FLTD tetrapeptide motif (residues 272–275 in human GSDMD)114. However, the crystal structures of active caspases 1/4/11 in complex with GSDMD-CT and further functional characterization revealed a conserved mechanism whereby these inflammatory caspases interact with GSDMD-CT, instead of the tetrapeptide motif in the linker region, to drive the recognition and processing of GSDMD115 (Fig. 4b). The GSDMD mutant with FLTD mutated to AAAD was equally processed in vitro and in cells, and rescued GSDMD deficiency in LPS-induced pyroptosis115. In the structures of the complex between caspase 1/4/11 and GSDMD-CT, the autoprocessed inflammatory caspase forms a two-β-strand protrusion that acts as a substrate-binding exosite to recruit GSDMD by hydrophobic and hydrogen bonding interactions with the CT. This recruitment likely places the interdomain linker near the caspase catalytic site for cleavage, a hypothesis now supported by the crystal structure of full-length GSDMD in complex with caspase 1 (ref.116) (Fig. 4b). Thus, GSDMD-CT not only auto-inhibits GSDMD-NT, it also provides the platform for inflammatory caspase recruitment and GSDMD activation. It remains to be seen whether similar mechanisms regulate activation of other GSDMs.
Mechanism of gasdermin pore formation
The mechanism of GSDM pore formation was revealed from the structure of the GSDMA3-NT pore117. Because GSDM-NT pores are cytotoxic, preparing the GSDMA3-NT protein for structural studies involved expressing an engineered recombinant full-length GSDMA3 in which a human rhinovirus 3C protease site was inserted in the interdomain linker. The recombinant purified protein was then cleaved by the 3C protease in the presence of liposomes containing the acidic lipid cardiolipin. GSDMA3-NT pores formed on the liposomes were then solubilized in detergent and imaged by cryo-electron microscopy (cryo-EM). The high-resolution structure is a strikingly large, predominantly 27-subunit transmembrane β-barrel with 108 extended β-strands and an inner diameter of 18 nm (Fig. 4b). This observed dimension for GSDMA3-NT pores is on a par with the 20 nm ring dimension for GSDMD-NT pores measured by atomic force microscopy10,118. GSDM pores are sufficiently large to allow the passage of small or medium-sized proteins, including the IL-1 family cytokines (Fig. 4b).
The membrane-inserted GSDMA3-NT subunit resembles a left hand with its soluble globular domain as the palm and two membrane-inserted β-hairpins as fingers (Fig. 4c). Comparison of auto-inhibited with membrane-inserted GSDMA3-NT structures indicated drastic conformational changes that result in the extension of the fingers as if a closed fist unfurls to an open hand, including melting of the α4 helix at the tip of a β-hairpin9,117 (Fig. 4c). In close juxtaposition to the inserted β-strands is the positively charged α1 helix, which interacts with cardiolipin in the cryo-EM structure and serves as a major acidic lipid-binding element. This α1 helix looks like a thumb folded over the palm. The α1 and α4 (Fig. 4c) helices of GSDM-NT, which are important in lipid binding and membrane insertion, respectively, are masked in the full-length protein by interactions with GSDM-CT (Fig. 4b), which explains how GSDM-CT represses GSDM activity. Oligomerization in the GSDMA3-NT pore is mediated by the sides of the hand-shaped subunit, from the membrane-inserted fingers, folded thumb to the palm117. In particular, the α1 thumb helices in the oligomerized pore align head to tail on the membrane to form a stabilizing helical belt. Interestingly, phosphorylation of Thr6 of α1 in GSDME (equivalent to Thr8 in GSDMA3) inhibits oligomer formation and pyroptosis119, presumably by altering the α1 helix conformation to interfere with oligomerization. The kinases responsible for phosphorylating GSDME or other GSDMs and how phosphorylation is regulated are unknown.
Gasdermin D-mediated cell hyperactivation
In certain situations, macrophages, dendritic cells and neutrophils survive inflammasome-activated GSDMD cleavage without membrane rupture and pyroptosis, and are called hyperactivated cells because they both stimulate inflammation and preserve their other functions24,120 (Fig. 2). Based on the cryo-EM structure of the GSDMA3-NT pore with an ~18 nm inner diameter117, GSDMD-NT pores are also big enough for chemicals (such as ATP) and small proteins to pass through9,117. Hyperactivation and pyroptosis can be distinguished by measuring cell culture supernatants for lactate dehydrogenase (LDH), which is too large to exit through GSDMD-NT pores and relies on cell lysis for its release, and for mature IL-1 family cytokines and possibly HMGB1, which are small enough to be readily released through these pores. While pyroptotic cells release both LDH and cytokines, hyperactivated cells do not release LDH, but do release cytokines21. Because hyperactivated cells survive, they may even process and pump out more inflammatory mediators than cells that die. An unbiased proteomics analysis of molecules released from hyperactivated cells and an analysis of their roles in infection and inflammation will be informative.
A number of processes that activate live cell cytokine secretion or hyperactivation have been described. In dendritic cells, a mixture of oxidized phosphorylcholine derivatives (oxPAPC) or an isolated component 1-palmitoyl-2-glutaryl-sn-glycero-3-phosphocholine (PGPC) produced in dying mammalian cells induces noncanonical inflammasome-dependent release of IL-1β without causing cell death120. Multiple stimuli, including an N-acetylglucosamine fragment of bacterial peptidoglycan and oxPAPC, can also hyperactivate macrophages and induce inflammasome-mediated IL-1β release from living macrophages in a GSDMD pore-dependent process21,24,121,122. Human, but not murine, circulating monocytes can respond to extracellular LPS to activate an alternative NLRP3–ASC–caspase 1 inflammasome independently of K+ efflux and without ASC speck formation, leading to IL-1β secretion without cell death123. A TLR4–TRIF–RIPK1–FADD–caspase 8 signalling axis was dissected genetically as upstream of alternative inflammasome activation123, but the exact mechanism remains unknown. A hyperactive state can also be brought about by deleting the sterile- and armadillo-motif-containing protein 1 (SARM1), which also contains a Toll–IL-1R (TIR) domain124. Sarm1–/– macrophages essentially recapitulate NLRP3-dependent hyperactivation after priming and nigericin treatment, with release of IL-1β but without cell death124. Potentiation of cell death by SARM1 involves SARM1 clustering at the mitochondria and SARM1-dependent mitochondrial depolarization124.
Interestingly, exposure to Salmonella enterica subsp. Typhimurium, ATP or nigericin, which cause inflammasome activation and pyroptosis in macrophages, induces GSDMD-dependent IL-1β release in neutrophils, without killing them122,125,126. With some stimuli, GSDMD-NT in neutrophils predominantly targets primary granules and LC3+ autophagosomes, instead of the plasma membrane, and IL-1β appears to be secreted via an autophagy-dependent mechanism127. The resistance of neutrophils to pyroptotic death is unique among inflammasome-signalling cells so far described. It allows neutrophils to secrete IL-1β at sites of infection without compromising the crucial inflammasome-independent neutrophil antimicrobial functions that would be impaired if neutrophils died. The inflammasome pathway must somehow be modified in neutrophils to optimize host pro-inflammatory and antimicrobial responses during pathogen challenge.
What determines whether GSDMD cleavage triggers pyroptosis or hyperactivation is not well understood. However, a likely mechanism responsible for cell survival in the face of membrane damage by GSDM pores is activation of the ubiquitous plasma membrane damage repair response in eukaryotic cells, which rapidly restores membrane integrity, allowing the cell to survive. All cells sense damage to the plasma membrane by an immediate increase in intracellular Ca2+, which triggers membrane repair by recruiting the endosomal sorting complexes required for transport (ESCRT) machinery to damaged membrane areas and removes them in ectosomes23. The plasma membrane repair response to other types of membrane damage also involves increased endocytosis of damaged membrane and patching of damaged membrane by donated vesicular membranes (mostly from lysosomes)128. Whether these other membrane repair phenomena occur in cells with GSDMD-damaged plasma membranes has not been examined. The ability of cells to survive and repair the damage likely reflects a contest between how much and how rapidly membrane damage is induced and whether it is spatially localized, versus the robustness of the repair processes, which likely depends on how much GSDMD is in the cell and how quickly and efficiently it is cleaved. The latter will in turn depend on the intensity of inflammatory stimuli and degree of inflammasome and inflammatory caspase expression and activation, as well as the effectiveness of mechanisms for regulating and clearing inflammasomes, activated caspases and GSDMs. Given the importance of IL-1β secretion and pyroptosis in human inflammatory diseases, a better understanding of unconventional cytokine secretion and the regulation of pyroptosis is important.
Processing of IL-1 family cytokines removes an NT acidic region that converts pro-IL-1β from an acidic precursor into a more basic mature cytokine that binds to PIP2-rich acidic regions on the plasma membrane. Plasma membrane association was proposed to be a necessary step for efficient release129. It is worth noting that Gsdmd–/– macrophages can still release caspase 1-processed mature IL-1β but the GSDMD-independent release, which may also rely on PIP2-mediated plasma membrane binding, is much slower than GSDMD-dependent release129.
Gasdermin-NTs and mitochondrial damage
The activated NT domain has also been shown to bind to internal cellular membranes119,127,130,131. GSDMD-NT binds with apparently higher affinity to the mitochondrial and bacterial lipid, cardiolipin, than to plasma membrane lipids8,9. GSDMD-NT and GSDME-NT can translocate to mitochondria, permeabilize the outer membrane and disrupt mitochondrial function, leading to ROS generation, loss of transmembrane potential and cytochrome c release, which also activates caspase 3 and enhances overall cell death119. As cardiolipin resides in the mitochondrial inner membrane, which is not accessible to the cytosol132, further study of mitochondrial targeting by GSDMs and its consequences is needed. NT fragments of GSDMA3, GSDMA and GSDME bind to the same phospholipids as GSDMD-NT9,17. Intriguingly, an in vitro lipid-binding assay showed that both full-length GSDMB and its NT domain bind to sulfatide and phosphoinositides, but not to cardiolipin133. The somewhat distinct phospholipid-binding properties of GSDMB suggest that it might bind to different membranes from the other GSDMs, but this has not been shown in cells.
Role of gasdermin D in NETosis
GSDMD plays a key role in NETosis, a regulated form of neutrophil cell death triggered by microbial infection, activation and danger signals, during which the nuclear envelope and plasma membrane are perforated to release large web-like structures, called neutrophil extracellular traps (NETs), composed of genomic DNA, histone and antimicrobial proteins43,131,134 (Fig. 3). In phorbol 12-myristate 13-acetate (PMA)-induced NET formation, serine proteases such as neutrophil elastase (ELANE) are released from granules, which cleave and activate GSDMD. GSDMD in turn permeabilizes primary azurophilic granules to further release ELANE in a feed-forward loop, allowing its nuclear translocation, histone processing, chromatin expansion and neutrophil lysis43. Similarly, GSDMD is required for Gram-negative bacterial infection-triggered NETosis whereby LPS activates the noncanonical inflammasome, which activates GSDMD, which permeabilizes primary granules and the neutrophil plasma membrane to facilitate protease release, nuclear decondensation and NET extrusion131. Although chromatin is extruded through the nuclear envelope during NETosis, there is no evidence that activated GSDMD localizes to the nuclear envelope or is directly involved in its permeabilization.
In another study, the granule-associated protease cathepsin G (CatG) present in both macrophages and neutrophils cleaves GSDMD directly at Leu274, only two residues upstream of the caspase cleavage site at Asp276, to activate its pore-forming activity135 (Fig. 3). The catalytic activity of CatG on GSDMD appears to be stronger than that of other myeloid granule enzymes, including ELANE and proteinase 3, and even exceeds that of caspase 1 and caspase 11, based on measured kcat/KM values. Sublethal LPS-induced systemic inflammation in mice depends on CatG and GSDMD and is suppressed by CatG-inhibiting serpins. Canonical NLRP3 inflammasome activation-dependent IL-1β release in bone marrow-derived macrophages (BMDMs) by nigericin or ATP also depends on CatG and GSDMD. Thus, GSDMD activation in both neutrophils and macrophages permeabilizes their granules to release CatG, which in turn further processes GSDMD in a feed-forward loop to enhance secretion of processed pro-inflammatory cytokines135.
Gasdermins and disease
Gasdermin D in infection
The affinity of GSDMD for bacterial cell membrane lipid cardiolipin allows it to bind, permeabilize and kill bacteria in the cytosol or phagosomes, as well as cell-free bacteria when it is released from pyroptotic cells8,9,89. Indeed, GSDMD was shown to mediate caspase 11-dependent defence against S. Typhimurium ΔsifA41 and Brucella abortus42. Similarly, Gsdmd deficiency compromises AIM2-mediated host defence and makes mice susceptible to Francisella novicida infection40. GSDMD protects against Burkholderia pseudomallei using both canonical and noncanonical pathways89. For Legionella pneumophila infection, Gsdmd–/– mice showed only mildly increased susceptibility compared with wild-type mice and failed to recapitulate the greater susceptibility in Nlrc4–/– mice46. The NAIP5/NLRC4 inflammasome also activates caspase 8, which cleaves and activates caspase 7, and GSDMD and caspase 7 together are responsible for restricting L. pneumophila infection46.
However, even though GSDMD-NT can permeabilize and kill bacteria, GSDMD is not always protective in infection and even exacerbates some bacterial infections by diverse mechanisms. In one study, Gsdmd–/– mice were better able to survive pathogenic Escherichia coli infection because neutrophils, which are the main cellular defenders against bacteria43–45, survived longer without Gsdmd44. In another study, Mycobacterium tuberculosis infection in a human macrophage cell line induced plasma membrane damage and secondary activation of NLRP3 and GSDMD-dependent pyroptosis and inflammatory cytokine release, which increased bacterial cell-to-cell spread in vitro136. However, the overall effect of GSDMD and pyroptosis on mycobacterial infection would need to be studied in vivo.
In the case of parasites, Toxoplasma gondii has been reported in one study to induce AIM2, ASC and caspase 8-dependent, but GSDMD-independent, death in human macrophages137. However, another study suggests that T. gondii infection in human monocytes activates a Syk–CARD9–NF-κB signalling pathway and the NRLP3 inflammasome to release IL-1β in a GSDMD-independent manner138. IL-1α, but not IL-1β, has been shown to be crucial for parasite control. IL-1α release from microglia depends on GSDMD, and Gsdmd deficiency in mice reduces IL-1α secretion and compromises immune protection in response to T. gondii infection in the brain139. GSDMD is activated in macrophages in vitro when another parasite, Entamoeba histolytica, binds and activates the noncanonical caspase 4 inflammasome by sensing an unknown ligand140. In vitro, E. histolytica causes caspase 4 and GSDMD-dependent macrophage hyperactivation with IL-1β secretion but without pyroptosis. Determining whether the net physiological impact of GSDMD is to improve immune control or exacerbate tissue damage in E. histolytica infection will require in vivo experiments.
The role of inflammasome activation and GSDMD in viral infection is not well studied. Viruses activate Toll-like receptors that sense viral nucleic acids on the cell surface or in endosomes to prime expression of innate immune genes. Cytoplasmic DNA and RNA sensors can trigger an interferon response (that is, cGAS, RIGI and MDA5) and/or an inflammatory response (IFI16, AIM2, NLRP6 and NLRP9) to viruses. Many viral proteins inhibit the antiviral interferon response, but so far viral mechanisms to inhibit inflammasome activation and pyroptosis have not been identified. In one study, the NLRP9 inflammasome, which recognizes stretches of viral dsRNA, protected intestinal epithelial cells from rotavirus infection in a GSDMD-dependent manner141. By contrast, inflammasome and GSDMD activation might contribute to viral pathogenesis for viruses whose disease course has a strong inflammatory component (such as HIV, influenza A or SARS coronaviruses). In rhinovirus and norovirus infections, NLRP3- and GSDMD-dependent pyroptosis increases pathological inflammation, contributing to viral pathogenesis90,142.
Gasdermin D in autoinflammatory genetic diseases
Autoactivation of inflammasome pathways causes or contributes to multiple sterile inflammatory diseases. Familial Mediterranean fever (FMF), the most prevalent monogenic autoinflammatory disease worldwide, is caused by missense mutations in MEFV. MEFV encodes the pyrin inflammasome, which senses bacterial toxin inactivation of RhoA GTPases143. MEFV mutation reduces the threshold for activating the pyrin inflammasome, causing recurrent bouts of fever and pain. GSDMD is crucial for FMF autoinflammatory pathology. In a mouse FMF model that expresses a hypersensitive pyrin, Gsdmd deficiency abolishes auto-inflammatory symptoms144. Similarly, autoactive NLRP3 mutants result in autoinflammatory disorders, including NOMID, which features severe systemic inflammation that is not adequately treated with current cytokine inhibitor drugs. Ablation of Gsdmd in NOMID mice prevents disease symptoms26 (Fig. 5).
Fig. 5. Role and function of gasdermins in human disease.

Autoactive gasdermin A (GSDMA) triggers pyroptosis in hair follicle cells and causes alopecia in mice but so far no human link to alopecia has been reported. GSDMB is implicated in autoimmune diseases such as childhood asthma and inflammatory bowel disease (IBD), but the underlying mechanism remains unclear. Granzyme A (GzmA) in killer lymphocytes activates GSDMB in target cells to cause pyroptosis. TNF can activate caspase 8 to trigger GSDMC-mediated pyroptosis in tumours. Upon sensing infection or danger, assembly of inflammasomes activates inflammatory caspases to cleave GSDMD and trigger pyroptosis, initiating immune responses. However, excessive pyroptosis leads to lethal sepsis, cytokine release syndrome (CRS) and severe inflammation and tissue damage. Autoactive pyrin or NLRP3 constitutively activate GSDMD and cause pyroptosis leading to familial Mediterranean fever (FMF) and neonatal-onset multisystem inflammatory disease (NOMID), respectively. Natural killer (NK) and CD8+ killer lymphocytes and chimeric antigen receptor (CAR) T cells activate pyroptosis in GSDME-expressing tumours when GzmB cleaves and activates GSDME and caspase 3. Alarmins released from pyroptotic tumour cells can also activate GSDMD-mediated pyroptosis in tumour-infiltrating macrophages and dendritic cells to enhance antigen presentation and functional activation of tumour-infiltrating lymphocytes (TILs). Secondary activation of macrophages can also mediate CRS during CAR T cell therapy. Chemotherapy drugs and radiotherapy activate caspase 3 in GSDME-expressing cells to convert apoptosis into pyroptosis, increasing their effectiveness at suppressing tumour growth by activating antitumour immunity; at the same time normal tissue cell damage and cytotoxicity is increased by activating GSDME-dependent pyroptosis especially in gut epithelial cells and haematopoietic precursor cells. Autoactive GSDME induces cochlear hair cell pyroptosis, leading to nonsyndromic hearing impairment. Loss-of-function mutation of DFNB59 also causes recessive nonsyndromic hearing loss. CAPS, cryopyrin-associated periodic syndromes; MS, multiple sclerosis; SS, systemic sclerosis.
Gasdermin D in complex diseases exacerbated by inflammation
GSDMD is probably the most important GSDM in mediating the inflammation that is associated with many diseases. It is the convergent target of canonical and noncanonical inflammasome signalling in immune sentinel cells, and excessive activation of GSDMD-mediated pyroptosis and IL-1 family cytokine secretion can lead to inflammatory diseases and pathological consequences (Figs 4,5). An unrestrained inflammatory response to infection is responsible for cytokine release syndrome (CRS), sepsis, disseminated intravascular coagulation (DIC) and acute organ damage, including acute respiratory distress syndrome (ARDS) and acute kidney injury (AKI). Release of IL-1 family cytokines is crucially dependent on GSDMD activation21,122. Binding of these cytokines to the IL-1 receptor triggers the activation and release of other pro-inflammatory cytokines, including IL-6 and TNF145. GSDMD-mediated pyroptosis in macrophages also triggers release of tissue factor, which activates coagulation, leading to DIC in sepsis146,147. Similarly, GSDMD-dependent neutrophil NETs capture platelets and serve as a nidus for thrombus formation, which can also contribute to DIC148. The noncanonical inflammasome plays a key role in sepsis in mouse models14,104,149. In LPS and caecal ligation and puncture (CLP)-induced sepsis, Gsdmd deficiency protects mice from lung, intestine, liver and kidney injury and DIC14,150. CRS and ARDS, triggered in patients with clinical deterioration during pathogenic influenza and SARS respiratory infections, could also be mediated or exacerbated by pyroptosis in tissue-resident macrophages, but this has not been shown.
GSDMD has been clearly linked to noninfectious diseases that are not caused by a single genetic mutation, in which inflammation plays an important pathogenic role. The NLRP3 inflammasome is implicated in EAE in mice. Gsdmd deficiency suppresses EAE neuroinflammation, and GSDMD inhibitors disulfiram (DSF) and dimethyl fumarate (DMF) attenuate EAE25,48. In EAE, GSDMD-mediated pyroptosis is activated in peripheral myeloid cells as well as in microglia and oligodendrocytes in the CNS25,151, but which cell types are most crucial for neuroinflammation is unclear. In a mouse bone marrow transplantation model, a recent study indicated that radiation causes severe damage to bones and spleen through NLRP3- and AIM2-mediated GSDMD-dependent pyroptosis, which is prevented only when GSDMD is deficient in both donor and host cells152. Activation of the noncanonical inflammasome pathway in mice worsens graft-versus-host disease, and Gsdmd deficiency reduces intestinal inflammation, tissue damage, donor T cell expansion and mortality in allogeneic haematopoietic stem cell transplantation153. Genetic deficiency or knockdown studies have also implicated GSDMD in the pathogenesis of alcoholic hepatitis, nonalcoholic steatohepatitis, noninfectious liver injury and ischaemia–reperfusion injury in humans and mice154–158. Both caspase 8 and GSDMD have also been implicated in mixed-lineage kinase domain-like protein (MLKL)-independent ileitis in mice with intestinal epithelial FADD deficiency108. The serum of children with acute vasculitis in Kawasaki disease contains elevated levels of GSDMD and IL-1β and induces endothelial cell pyroptosis in vitro, suggesting that GSDMD-mediated pyroptosis plays an important role in endothelial damage in Kawasaki disease159. GSDMD-mediated pyroptosis has also been implicated in diabetic kidney disease160, diabetic cardiomyopathy161, contrast-induced kidney injury162, early brain injury after subarachnoid haemorrhage163 and traumatic brain injury164. Although activation of canonical and noncanonical inflammasomes has been implicated in the inflammation and pathogenesis of many noninfectious human diseases, the role of GSDMD in all these conditions has not yet been verified by studies in Gsdmd-deficient mice.
Gasdermins in cancer
The role of GSDMs in cancer is complicated (reviewed elsewhere47). The 2000 paper5 that named GSDMA based on its restricted expression in the gastrointestinal tract and skin hinted at a possible role of GSDMs in cancer by noting that GSDMA expression is suppressed in human gastric cancers and that it might be a tumour suppressor gene5. In fact, GSDMA, GSDMC and GSDMD all reportedly suppress gastric cancer cell proliferation70,165. However, reduced GSDMD expression attenuates tumour proliferation and predicts a favourable prognosis in non-small-cell lung cancer166. GSDMB is amplified and highly expressed in cervical, hepatic, colon and HER2+ breast cancer and may function as an oncogene in those cancers71,79. GSDMB expression in HER2+ breast cancer correlates with increased metastasis and poor prognosis78,80. Overexpression of GSMDB in HER2+ MCF7 cells promotes cell mobility and invasion, probably by activating Rac1 and Cdc42 (ref.78). Moreover, introducing anti-GSDMB into HER2+ tumour cells using a hyaluronan-coated nanoparticle reduces xenograft growth and metastasis in vivo167. By contrast, GSDMB expression is suppressed in primary oesophageal and gastric tumours19, and its expression correlates with better outcome in patients with bladder carcinoma or melanoma. GSDMC is upregulated in melanoma, colorectal cancer and lung adenocarcinoma, promotes tumour growth and metastasis, and might function as an oncogene in those cancers82,85,168. These studies suggest that the roles of GSDMs in cancer are context, GSDM and cancer type specific, reflecting the complex role of inflammation in tumorigenesis, cancer proliferation and antitumour immunity169. Because of this complexity, these studies need to be interpreted with caution. In vitro experiments and studies in immunodeficient mice do not take into account the interaction between the tumour and the immune system, where GSDMs may play their most important in vivo role.
GSDME, which is activated by caspase 3 during apoptosis, is more strongly implicated in tumour suppression. GSDME is epigenetically suppressed by promoter DNA methylation in gastric, colorectal and breast cancers and most human cancer cell lines; and most tumour-associated GSDME mutations studied compromise its ability to induce pyroptosis17,18,56–60 (Table 2). Moreover, GSDME expression, induced by 5-aza-dC in vitro, suppresses colony formation and tumour cell proliferation in gastric cancer, melanoma and colorectal cancer, and reduces invasivity of breast cancer56,57,60,119. Worse 5-year survival and an increase in lymph node metastases are associated with reduced GSDME in breast cancer57. Moreover, lack of GSDME promotes melanoma cell line resistance to etoposide, which is rescued by GSDME overexpression170. In addition, treatment of lung cancer cells with KRAS, EGFR or ALK inhibitors leads to caspase 3-mediated GSDME activation, which increases the antitumour effectiveness of these drugs171.
The tumour-suppressive effect of GSDME is dependent on immune surveillance in vivo by converting NK and CD8+ T killer cell-mediated noninflammatory apoptosis into inflammatory pyroptosis in the tumour, which augments antitumour immunity18 (Figs 3,5). This effect is abrogated in tumours that express noncleavable GSDME and in mice that lack all lymphocytes, only killer lymphocytes or perforin that is needed for cytotoxic granule-mediated killing18. Gsdme expression in mouse tumours suppresses tumour growth and enhances the number and functions of tumour-infiltrating NK and CD8+ T lymphocytes18. When killer cells recognize a tumour target, they deliver the cytotoxic protease GzmB into the tumour, which induces GSDME-mediated pyroptosis, both directly because GzmB cleaves and activates GSDME and indirectly because GzmB also cleaves and activates caspase 3 (ref.18). Similar findings were reported using chimeric antigen receptor (CAR) T cells, which induce pyroptosis in GSDME-expressing target cells, but how much GSDME pyroptosis contributes to the effectiveness of CAR T cell therapy has not been evaluated172. Caspase 3 activation by GzmB generates a positive feedback mechanism as caspase 3 also cleaves and activates GSDME. As GSDME can be cleaved and activated by both caspase 3 and GzmB, GSDME may be a more potent tumour suppressor than the other GSDMs. Caspase 3 activation is common in cancers, triggered by an inadequate supply of oxygen or nutrients, especially in poorly vascularized regions, by treatment with chemotherapy or radiation and by killer cell attack. Because GSDME is suppressed by promoter hypermethylation in many cancers53–57, epigenetic modifying drugs could promote antitumour immunity. Indeed, combining 5-aza-dC with chemotherapy in one study enhanced the immunogenic effect of chemotherapy173. However, it is unclear how much pyroptosis contributes to the 5-aza-dC-induced antitumour effect as 5-aza-dC should derepress many genes, including tumour suppressor genes.
The potential therapeutic role of inducing pyroptosis in tumours has been demonstrated in a number of examples. Like GzmB-induced GSDME activation, the killer lymphocyte cytotoxic protease GzmA cleaves GSDMB and induces pyroptosis in tumour cells targeted for immune elimination19 (Figs 3,5). When combined with anti-PD1, ectopic expression of human GSDMB in murine colorectal cancer CT26 and melanoma B16 promotes tumour clearance in mice. The effectiveness of BRAF and MEK inhibitors against GSDME-expressing BRAFV600E/K mutant melanoma was shown to depend on the induction of GSDME-mediated pyroptosis to activate CD8+ T cell-dependent antitumour immunity174. In another study, the serine dipeptidase DPP8/9 inhibitor Val-boroPro, which activates the CARD8 inflammasome in human myeloid cells, induced GSDMD-mediated pyroptosis in most human acute myeloid leukaemia (AML) cell lines and primary patient samples, but not cell lines from other lineages175.
These data suggest that GSDM-mediated pyroptosis plays a crucial role in antitumour immunity for some tumours, and harnessing this process could be an effective way to treat cancer. However, activating GSDMs in GSDM-expressing normal tissues and immune cells can also cause severe tissue damage, such as in chemotherapy-induced caspase 3-mediated GSDME activation in normal tissues17 (Figs 3,5). Although a DPP8/9 inhibitor induces pyroptosis in AML cells, it may also activate GSDMD-mediated pyroptosis in B cells and other CD34+ cells175 and cause treatment-associated toxicity. Activation of GSDME-mediated pyroptosis in normal mouse tissues also causes extensive tissue damage after chemotherapy17. During CAR T cell anticancer therapy of B cell leukaemia, GzmB activates both GSDME and caspase 3, causing pyroptosis. Moreover, culture supernatants from B-ALL human cell lines or primary leukaemias cocultured with CAR T cells activate caspase 1–GSDMD-mediated pyroptosis in macrophages. Activation of pyroptosis by CAR T cells is responsible for CRS, one of the serious side effects of this therapy172 (Figs 3,5). Thus, selective activation of GSDMs in tumours rather than normal cells could be crucial for developing pyroptosis-inducing cancer therapy.
Therapeutically targeting gasdermins
The key roles of GSDMs in infection, inflammation and tumour suppression have prompted investigation of GSDM-targeting therapeutics (Fig. 6). Inflammasome pathways have already been extensively targeted indirectly for disease modulation by inhibiting IL-1β, the signature cytokine released in a GSDMD activation-dependent manner, or its receptor, using the FDA-approved drugs anakinra, a recombinant natural human IL-1 receptor antagonist, and canakinumab, an anti-IL-1β monoclonal antibody176. These drugs are used to treat severe manifestations of autoinflammatory diseases, including FMF, NOMID, severe systemic autoimmune diseases (rheumatoid arthritis, systemic juvenile idiopathic arthritis, adult-onset Still’s disease) and bronchiolitis obliterans that do not respond to other treatments. Small-molecule inhibitors of caspase 1, such as VX-765, have also been developed151, although they have not proved useful for treating human disease. Inhibitors of inflammasome sensors are under clinical development177. However, GSDMD is an especially attractive drug target because it is the bottleneck for all inflammasome effects, acting upstream of IL-1 and alarmin secretion and downstream of all inflammasome sensors (Figs 2,3,5). Inhibition of GSDMD and possibly other GSDMs may therefore be the most efficacious strategy for treating diseases that are primarily inflammatory and/or autoimmune and may also be useful for treating diseases exacerbated by pyroptosis, such as cardiovascular disease and CRS in severe COVID-19 infection. On the other hand, induction of GSDM-mediated cell death may be useful for amplifying immune responses, such as in vaccination or cancer.
Fig. 6. Strategies to treat gasdermin-associated diseases.

a | Steps in gasdermin (GSDM) activation that can be targeted, and the mechanisms used by existing GSDMD inhibitors45,48. Other yet unused potential mechanisms are shown with a question mark. b | Cellular delivery of GSDM for cancer cell killing and activation of antitumour immunity180. See text for details. CT, C-terminal; NT, N-terminal.
Small-molecule gasdermin inhibitors
A few studies have identified small-molecule GSDMD inhibitors (Table 3). DSF, an FDA-approved drug for treating chronic alcoholism (Antabuse), was identified in a high-throughput liposome leakage-based screen as an inhibitor of GSDMD pore formation on liposomes with a half-maximal inhibitory concentration (IC50) in the nanomolar range48. In human THP-1 cells and mouse immortalized BMDMs (iBMDMs), stimulated by the canonical NLRP3 and AIM2 inflammasomes and the noncanonical inflammasome, respectively, DSF blocked pyroptosis and cytokine release without affecting inflammasome speck formation, caspase activation or GSDMD cleavage, suggesting that it was acting by blocking pore formation. In a mouse model of LPS-induced septic shock, DSF protected wild-type and caspase 1-knockout mice from death and greatly reduced serum inflammatory cytokine levels. The potency of DSF was enhanced when it was combined with copper gluconate, which reportedly stabilizes diethyldithiocarbamate (DTC), the cellular metabolite of DSF178. The efficacy of DSF in protecting against GSDMD-mediated inflammatory disorders was also demonstrated in EAE25. Necrosulfonamide (NSA), a drug previously known to inhibit MLKL in necroptotic cell death, was also shown to inhibit GSDMD pore formation, with low micromolar cellular IC50 in blocking cytokine release and pyroptosis downstream of NLRP3 and NLRC4 inflammasomes45. Like DSF, NSA extended survival of wild-type mice in a model of LPS-induced sepsis.
Table 3.
Gasdermin D inhibitors
| GSDMD inhibitor | Description | In vitro IC50 | Cellular IC50 | Mechanism of action | In vivo effect | Refs |
|---|---|---|---|---|---|---|
|
Disulfiram
|
Commonly known as Antabuse, an FDA-approved alcoholism drug | ~300 nM alone in GSDMD-induced liposome leakage; ~200 nM in combination with Cu(ii) | ~7.7 µM in canonical inflammasome-dependent pyroptosis in THP-1; ~10.3 µM in noncanonical inflammasome-dependent pyroptosis in murine iBMDMs; ~400 nM in combination with Cu(ii) in THP-1 | Binds GSDMD (Kd = ~12.8 µM), covalently modifies Cys191 in GSDMD, and inhibits GSDMD-NT oligomerization/insertion | Increased survival and suppression of inflammatory responses in murine models of sepsis and EAE | 25,48 |
|
Necrosulfonamide
|
An inhibitor of MLKL in necroptosis | ~9.5 µM in GSDMD-induced liposome leakage | ~10–15 µM in canonical inflammasome-dependent pyroptosis in iBMDMs | Binds GSDMD (Kd = ~32 µM), potentially modifies Cys191 in GSDMD, and inhibits GSDMD-NT oligomerization/insertion | Increased survival and suppression of inflammatory responses in a murine model of sepsis | 45,48 |
|
Dimethyl fumarate
|
An FDA-approved drug (Tecfidera) for the treatment of MS | NA | NA | Succinates GSDMD at Cys191 and other reactive cysteines, prevents interaction between GSDMD and caspases, and inhibits GSDMD-NT oligomerization/insertion | Increased survival and suppression of inflammatory responses in murine models of sepsis, EAE and FMF; decreased levels of GSDMD-NT and IL-1β in patients with MS treated with Tecfidera | 48,49 |
|
LDC7559
|
A compound based on the pyrazolo-oxazepine scaffold | NA | ~5.6 µM in PMA-induced NETosis in murine neutrophils; ~300 nM in cholesterol crystal-induced NETosis in murine neutrophils | Binds GSDMD in pulldown experiments using cell lysates; mechanism of action unclear | NA | 43,48 |
|
Punicalagin
|
A complex polyphenol from pomegranate extract | NA | ~3.9, 3.7 and 7.5 µM in IL-1β secretion, LDH release and membrane permeabilization, respectively, in ATP-treated murine BMDMs | Potentially interferes with membrane fluidity and inhibits GSDMD-NT insertion | NA | 179 |
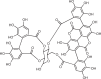
The half-maximal inhibitory concentration (IC50) and equilibrium dissociation constant (Kd) values were taken from the referenced studies. EAE, experimental autoimmune encephalomyelitis; FMF, Familial Mediterranean fever; GSDM, gasdermin; iBMDMs, immortalized murine bone marrow-derived macrophages; LDH, lactate dehydrogenase; MLKL, mixed-lineage kinase domain-like protein; NA, not available; NT, N-terminal; MS, multiple sclerosis; PMA, phorbol myristate acetate; THP-1, a human monocytic cell line.
DMF, FDA-approved Tecfidera for the treatment of MS, was reported to prevent GSDMD from interacting with caspases, limit GSDMD processing and oligomerization, and block pyroptosis; endogenous fumarate similarly inhibited GSDMD49. In murine models of sepsis, EAE and FMF, DMF extended survival and alleviated inflammation. Patients with MS treated with DMF exhibited decreased plasma GSDMD-NT and IL-1β compared with untreated controls, consistent with a model in which GSDMD exacerbates MS, as was shown in mouse EAE. These strong inhibitory effects of DMF contrast with the lack of inhibition of GSDMD-induced liposome leakage by DMF reported in an earlier study48. These two studies suggest that DMF may inhibit GSDMD cleavage, while DSF and NSA work downstream in the pathway to inhibit pore formation. Further characterization of DMF is necessary to evaluate its potency as a GSDMD inhibitor.
DSF, NSA and DMF are all Cys-reactive drugs that inhibit GSDMD by modifying Cys residues (Table 3). DSF and NSA covalently modify Cys191, but not other cysteines, in GSDMD to inhibit pore formation45,48, while DMF and endogenous fumarate modify multiple Cys residues, including Cys191, leading to GSDMD succination49. Consistently, GSDMD with mutated Cys191 and other GSDMs that lack this reactive cysteine are not inhibited by DSF or NSA45,48. GSDMD Cys191 is an exposed cysteine on the α4 helix that plays an important role in membrane penetration (Fig. 4b). Modification of Cys191 may interfere with membrane insertion and therefore abrogate formation of transmembrane β-hairpins (Figs 4c,6a). Although the exposed position of Cys191 may facilitate its modification, a downside of Cys-reactive compounds is that they are less selective and likely also target molecules other than GSDMD. Therefore, the mechanisms of action of DSF, NSA and DMF in vivo should be interpreted with caution.
Two other molecules, LDC7559, a pyrazolo-oxazepine-based compound, and punicalagin, an antioxidant polyphenol from pomegranates, have been reported to inhibit PMA-induced NETosis and NLRP3/AIM2 inflammasome-dependent pyroptosis, respectively, with activity in the low micromolar range43,179. In pulldown experiments using cell lysates, LDC7559 specifically enriched GSDMD detection by mass spectrometry. However, the interaction between LDC7559 and GSDMD was not directly verified, and LDC7559 does not inhibit GSDMD pore formation on liposomes48, hence leaving its mechanism of action unexplained. Likewise, considering that punicalagin also protects macrophages from detergents and may indirectly affect GSDMD pore formation by rigidifying the plasma membrane, further studies are needed to elucidate whether punicalagin directly and specifically binds to GSDMD.
On the basis of a structural understanding of GSDM auto-inhibition, molecules that stabilize the GSDM NT–CT interface may inhibit the full-length protein by locking it in its inactive conformation (Fig. 6a). The recent identification of an exosite for inflammatory caspase recognition of GSDMD suggests that another possible way of inhibiting GSDMD would be to identify a compound that blocks the exosite (Fig. 6a). It is likely that other post-translational modifications and mechanisms to regulate pore formation will be uncovered that might be attractive drug targets.
Manipulating gasdermin expression in cells
In an effort to induce GSDM-mediated cancer cell pyroptosis to prime adaptive immunity, intratumoural delivery of nanoparticle-conjugated, pre-cleaved GSDMA3 (NT + CT) into cells was combined with the cancer-imaging compound phenylalanine trifluoroborate (Phe-BF3), which catalyses the release of GSDMA3 from nanoparticles, causing selective tumour cell pyroptosis180 (Fig. 6b). Pyroptosis of <15% of tumour cells was sufficient to suppress the entire tumour graft in mice, because the treatment activated the adaptive immune response to the tumour. Immunodeficiency or T cell depletion abrogated the tumour-suppressive response. Further, a partially effective intravenous treatment with the nanoparticles synergized with checkpoint blockade and sensitized the tumours to anti-PD1, suggesting that pyroptosis could be exploited to trigger or enhance antitumour immunity. This study provides a proof of concept for targeting GSDMs in cancer; however, care needs to be taken to avoid excessive pyroptosis, which could be toxic to normal cells or cause cytokine release with systemic consequences. Delivery of GSDM-NT or identification of GSDM agonists could also in principle be used as vaccine adjuvants or to enhance the immune response to infection.
Considerations for therapeutic GSDMD inhibition
The wide variety of diseases in which GSDMD likely mediates inflammation and its crucial role as a final bottleneck for pyroptosis suggest that GSDMD inhibition could be an extremely effective therapeutic strategy. However, interfering with the antimicrobial roles of GSDMD could lead to increased susceptibility to infection. In addition, innate immune responses are complex, and each potential therapeutic indication will need to be examined carefully in preclinical models. In a study using the monosodium urate (MSU)-induced gout model, although GSDMD was activated through the NLRP3 inflammasome, it was not needed for cell death or IL-1β release, and the GSDMD inhibitor NSA did not ameliorate disease181. Suppressing pyroptosis can activate other innate immune responses. For instance, GSDMD inhibition in a cell can activate the interferon response182. In a mouse study that modelled systemic lupus erythematosus (SLE) using injected imiquimod, the severity of SLE was unexpectedly exacerbated in Gsdmd–/– mice, leading to more severe kidney and pulmonary pathology, more autoantibodies and reduced mortality183. Type I interferons play an important driving role in many SLE models and in patients with SLE. GSDMD may have suppressed interferon induction in these mice, although a change in interferon signalling in this SLE model in Gsdmd–/– mice was not examined.
Mouse models may not always accurately mirror human disease pathogenesis, and mouse models of inflammatory diseases can be affected by small differences in experimental protocol or differences in the microbiome of mouse colonies or other poorly defined environmental or genetic differences. For example, two recent studies of dextran sodium sulfate-induced colitis found opposite results: in one study Gsdmd deficiency exacerbated colitis and enhanced cGAS–STING-mediated interferon induction184, while in another study lack of Gsdmd reduced disease severity185.
The role of GSDMD in various inflammatory diseases and potential interactions of GSDMD activation with other innate immune responses will need to be examined carefully to select disease indications and anticipate possible toxicity of GSDM-modulating compounds as they become available.
Concluding remarks and future perspectives
Since the initial discovery of GSDMD as the substrate of inflammatory caspases, much progress has been made in understanding the activation mechanisms of the GSDM family. Two ways to activate GSDMs have so far been identified: first, protease-mediated cleavage in the linker region to liberate the NT active domain, and second, CT domain mutations including many germline-encoded disease mutations that disrupt the interaction of the NT and CT domains and diminish the ability of the CT domain to inhibit NT domain pore formation (Figs 2–5). Although the physiological stimuli that activate GSDMs are still being uncovered, a theme of GSDMs as sensors of cytosolic protease activation has emerged. Currently these proteases include caspases, granule-associated proteases, such as neutrophil elastase and cathepsin G, and granzymes delivered to target cells by killer T cells. The flexibility and accessibility of the GSDM linker region increases its vulnerability to protease attack. Direct protease activation of a GSDM could induce inflammatory death in diverse types of cells beyond immune and barrier cells. It will not be surprising if GSDMs are found to be cleaved and activated by additional cellular proteases as well as by pathogen-encoded proteases.
As inflammation causes or contributes to many poorly controlled and sometimes lethal diseases and strongly augments antigen-specific immunity, there is a need to further elucidate the regulation of GSDMs at the root of inflammation. For example, inflammasome activation also exacerbates many mouse models of disease, including atherosclerosis, type II diabetes, gout, nonalcoholic steatohepatitis and several neurodegenerative diseases, including Alzheimer disease. GSDMs not only cause cell death, but are also required for unconventional protein secretion of the IL-1 family cytokines and other intracellular danger molecules such as HMGB1 that lack a secretion signal. What regulates whether GSDM activation causes lytic cell death or hyperactivation without cell death remains unclear. A related question is whether pyroptosis or hyperactivation generates a stronger innate immune signal. Factors released by GSDM activation must be quite different in pyroptotic versus hyperactive cells. There is no doubt that future studies will link the GSDM family to more physiological and pathological processes and uncover how these processes are controlled.
As the key perpetrators of inflammation, GSDMs are strategically situated for therapeutical manipulation of inflammation. Developing GSDM agonists or antagonists could transform the treatment of inflammatory disease and potentially lead to better vaccine adjuvants or immune therapy. So far, identifying specific compounds that inhibit GSDMs has been challenging. The currently available GSDMD inhibitors, NSA, DSF and DMF, all covalently modify reactive cysteines and are thus not completely specific. Further mechanistic understanding of GSDM pore formation and the structural intermediates that form in the transition from the inactive to the active state will undoubtedly inform drug identification and optimization. Similarly, understanding the molecular pathways that regulate pore formation could provide additional therapeutic targets. Given the crucial function of GSDMs in inflammation, the search for GSDM-modulating drugs is likely to be very active in the coming years.
Acknowledgements
This work was supported by NIH R01AI139914 (H.W.), R01CA240955 (J.L.), Key Research Program of the Chinese Academy of Sciences (ZDBS-LY-SM008), National Key R&D Program of China (2020YFA0509600), National Natural Science Foundation of China (31972897), Strategic Priority Research Program of Chinese Academy of Sciences (XDB29030300) and Shanghai Municipal Science and Technology Major Project (2019SHZDZX02) (X.L.).
Glossary
- Gasdermins
A family of homologous pore-forming proteins whose N-terminal fragments permeabilize cell membranes when the protein is cleaved in the linker region connecting the active N-terminal and inhibitory C-terminal domains.
- Pyroptosis
A programmed inflammatory cell death, which occurs after the cleavage and activation of a gasdermin, leading to loss of cell membrane integrity and release of pro-inflammatory cytokines and cellular proteins that can act as danger signals.
- Inflammasome
Intracellular complex of cytosolic sensors of invasive infection that recognize pathogen-associated molecular patterns (PAMPs) or of danger that recognize damage-associated molecular patterns (DAMPs) and then assemble into supramolecular complexes that activate inflammatory caspases; the canonical inflammasome activates caspase 1 and the noncanonical inflammasome activates caspase 4 or caspase 5 in humans or caspase 11 in mice.
- Inflammatory caspases
Cysteine proteases that are activated by proximity-induced autocatalytic cleavage when they dimerize after recruitment to canonical (caspase 1) or noncanonical (caspase 4, 5 or 11) inflammasomes and are responsible for processing inflammatory cytokines and gasdermin D to induce pyroptosis or hyperactivation.
- Apoptosis
A noninflammatory programmed cell death mediated by mitochondrial outer membrane permeabilization and activation of the effector caspases (3, 6 and 7).
- Granzymes
Serine proteases stored in the cytotoxic granules of killer lymphocytes that are released during killer cell attack to cause programmed cell death in the target cell.
- Pro-inflammatory cytokines
Small molecular weight proteins secreted by immune cells and some other cells that evoke inflammatory responses, activate immune cells and cause inflammation, fever and tissue damage.
- Alarmins
Host molecules released from damaged or dying cells that act as danger signals to stimulate inflammation, also known as damage-associated molecular patterns (DAMPs).
- AIM2
(Absent in melanoma 2). An inflammasome that senses microbial or mislocalized host DNA in the cytosol to form a canonical inflammasome.
- Experimental autoimmune encephalomyelitis
(EAE). Animal models of autoinflammatory disease in the central nervous system that resemble human multiple sclerosis, which are triggered by injecting immunogenic self peptides that sensitize CD4+ T cells.
- Neonatal onset multisystem inflammatory disease
(NOMID). A rare severe autoinflammatory disease, caused by constitutively active mutations of the NLRP3 inflammasome gene, that occurs in infancy with symptoms of fever, chronic meningitis, skin rash and arthropathy, also known as chronic infantile neurologic cutaneous articular (CINCA) syndrome.
- IL-1 family
A group of 11 pro-inflammatory cytokines and their natural antagonists (IL-1α, IL-1β, IL-1RA, IL-18, IL-33, IL-36α, IL-36β, IL-36γ, IL-36RA, IL-37 and IL-38) that share a conserved β-trefoil structure and have important roles in promoting or regulating inflammatory responses to infection or sterile insults.
- NLRP3
A member of the NOD-like receptor subfamily of pattern recognition receptors, which recruits adaptor protein ASC and caspase 1 to assemble a large protein complex named the inflammasome in response to danger signals. Also known as NALP3 and cryopyrin.
- Pyrin
A cytosolic inflammasome sensor encoded by the MEFV gene, predominantly expressed in phagocytes, that is triggered by bacterial toxins or effectors.
- Exosite
Secondary binding sites of an enzyme that are remote from the active site but modulate substrate binding and enzymatic activity.
- Hyperactivation
A response to gasdermin activation and pore formation in which cells release IL-1 and other inflammatory mediators, but do not die.
- oxPAPC
A mixture of oxidized phospholipids generated by the oxidation of 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine (PAPC), some of which can bind to and activate or antagonize innate immune sensors of lipopolysaccharide.
- NETosis
A unique form of neutrophil cell death that is characterized by release of decondensed chromatin to form neutrophil extracellular traps (NETs) that trap microbes but can also capture and activate platelets to trigger clotting.
- Familial Mediterranean fever
(FMF). A rare spontaneous autosomal recessive genetic disease, characterized by recurrent fever and peritonitis, caused by mutations in the pyrin inflammasome gene (MEFV).
- Autoinflammatory disorders
A diverse group of diseases characterized by recurrent episodes of inflammation, which result from poorly controlled activation of innate immunity.
- Cytokine release syndrome
(CRS). A systemic life-threatening inflammatory response syndrome caused by massive inflammatory cytokine release.
- Cys-reactive drugs
Drugs that covalently bind to the sulfhydryl (SH) group of Cys in cysteine-containing proteins.
Competing interests
H.W. and J.L. are co-founders of Ventus Therapeutics. The other authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Xing Liu, Shiyu Xia, Zhibin Zhang.
Contributor Information
Xing Liu, Email: xingliu@ips.ac.cn.
Hao Wu, Email: wu@crystal.harvard.edu.
Judy Lieberman, Email: judy.lieberman@childrens.harvard.edu.
References
- 1.Tamura M, et al. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics. 2007;89:618–629. doi: 10.1016/j.ygeno.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci. 2017;42:245–254. doi: 10.1016/j.tibs.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 3.Liu X, Lieberman J. Knocking ’em dead: pore-forming proteins in immune defense. Annu. Rev. Immunol. 2020;38:455–485. doi: 10.1146/annurev-immunol-111319-023800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu X, Lieberman J. A mechanistic understanding of pyroptosis: the fiery death triggered by invasive infection. Adv. Immunol. 2017;135:81–117. doi: 10.1016/bs.ai.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saeki N, Kuwahara Y, Sasaki H, Satoh H, Shiroishi T. Gasdermin (Gsdm) localizing to mouse chromosome 11 is predominantly expressed in upper gastrointestinal tract but significantly suppressed in human gastric cancer cells. Mamm. Genome. 2000;11:718–724. doi: 10.1007/s003350010138. [DOI] [PubMed] [Google Scholar]
- 6.Van Laer L, et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat. Genet. 1998;20:194–197. doi: 10.1038/2503. [DOI] [PubMed] [Google Scholar]
- 7.Lammert CR, et al. AIM2 inflammasome surveillance of DNA damage shapes neurodevelopment. Nature. 2020;580:647–652. doi: 10.1038/s41586-020-2174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu X, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 10.Sborgi L, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35:1766–1778. doi: 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aglietti RA, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl Acad. Sci. USA. 2016;113:7858–7863. doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Z, et al. Crystal structures of the full-length murine and human gasdermin D reveal mechanisms of autoinhibition, lipid binding, and oligomerization. Immunity. 2019;51:43–49 e44. doi: 10.1016/j.immuni.2019.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi J, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 14.Kayagaki N, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 15.He WT, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25:1285–1298. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogers C, et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017;8:14128. doi: 10.1038/ncomms14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103. doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Z, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579:415–420. doi: 10.1038/s41586-020-2071-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Z, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. 2020;368:eaaz7548. doi: 10.1126/science.aaz7548. [DOI] [PubMed] [Google Scholar]
- 20.Hou J, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 2020;22:1264–1275. doi: 10.1038/s41556-020-0575-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evavold CL, et al. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity. 2018;48:35–44 e36. doi: 10.1016/j.immuni.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen X, et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 2016;26:1007–1020. doi: 10.1038/cr.2016.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruhl S, et al. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science. 2018;362:956–960. doi: 10.1126/science.aar7607. [DOI] [PubMed] [Google Scholar]
- 24.Zanoni I, Tan Y, Di Gioia M, Springstead JR, Kagan JC. By capturing inflammatory lipids released from dying cells, the receptor CD14 induces inflammasome-dependent phagocyte hyperactivation. Immunity. 2017;47:697–709.e693. doi: 10.1016/j.immuni.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li S, et al. Gasdermin D in peripheral myeloid cells drives neuroinflammation in experimental autoimmune encephalomyelitis. J. Exp. Med. 2019;216:2562–2581. doi: 10.1084/jem.20190377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao J, et al. Gasdermin D mediates the pathogenesis of neonatal-onset multisystem inflammatory disease in mice. PLoS Biol. 2018;16:e3000047. doi: 10.1371/journal.pbio.3000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Runkel F, et al. The dominant alopecia phenotypes Bareskin, Rex-denuded, and Reduced Coat 2 are caused by mutations in gasdermin 3. Genomics. 2004;84:824–835. doi: 10.1016/j.ygeno.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 28.Lunny DP, et al. Mutations in gasdermin 3 cause aberrant differentiation of the hair follicle and sebaceous gland. J. Invest. Dermatol. 2005;124:615–621. doi: 10.1111/j.0022-202X.2005.23623.x. [DOI] [PubMed] [Google Scholar]
- 29.Tanaka S, et al. A new Gsdma3 mutation affecting anagen phase of first hair cycle. Biochem. Biophys. Res. Commun. 2007;359:902–907. doi: 10.1016/j.bbrc.2007.05.209. [DOI] [PubMed] [Google Scholar]
- 30.Li J, et al. Gsdma3 is required for hair follicle differentiation in mice. Biochem. Biophys. Res. Commun. 2010;403:18–23. doi: 10.1016/j.bbrc.2010.10.094. [DOI] [PubMed] [Google Scholar]
- 31.Kumar S, et al. Gsdma3(I359N) is a novel ENU-induced mutant mouse line for studying the function of gasdermin A3 in the hair follicle and epidermis. J. Dermatol. Sci. 2012;67:190–192. doi: 10.1016/j.jdermsci.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 32.Das S, et al. GSDMB induces an asthma phenotype characterized by increased airway responsiveness and remodeling without lung inflammation. Proc. Natl Acad. Sci. USA. 2016;113:13132–13137. doi: 10.1073/pnas.1610433113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu C, et al. A 3-nucleotide deletion in the polypyrimidine tract of intron 7 of the DFNA5 gene causes nonsyndromic hearing impairment in a Chinese family. Genomics. 2003;82:575–579. doi: 10.1016/S0888-7543(03)00175-7. [DOI] [PubMed] [Google Scholar]
- 34.Bischoff AM, et al. A novel mutation identified in the DFNA5 gene in a Dutch family: a clinical and genetic evaluation. Audiol. Neurootol. 2004;9:34–46. doi: 10.1159/000074185. [DOI] [PubMed] [Google Scholar]
- 35.Cheng J, et al. A novel DFNA5 mutation, IVS8+4 A>G, in the splice donor site of intron 8 causes late-onset non-syndromic hearing loss in a Chinese family. Clin. Genet. 2007;72:471–477. doi: 10.1111/j.1399-0004.2007.00889.x. [DOI] [PubMed] [Google Scholar]
- 36.Park HJ, et al. Evidence for a founder mutation causing DFNA5 hearing loss in East Asians. J. Hum. Genet. 2010;55:59–62. doi: 10.1038/jhg.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishio A, et al. A DFNA5 mutation identified in Japanese families with autosomal dominant hereditary hearing loss. Ann. Hum. Genet. 2014;78:83–91. doi: 10.1111/ahg.12053. [DOI] [PubMed] [Google Scholar]
- 38.Li-Yang MN, et al. IVS8+1 DelG, a novel splice site mutation causing DFNA5 deafness in a chinese family. Chin. Med. J. 2015;128:2510–2515. doi: 10.4103/0366-6999.164980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delmaghani S, et al. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nat. Genet. 2006;38:770–778. doi: 10.1038/ng1829. [DOI] [PubMed] [Google Scholar]
- 40.Zhu Q, et al. Promotes AIM2 inflammasome activation and is required for host protection against Francisella novicida. J. Immunol. 2018;201:3662–3668. doi: 10.4049/jimmunol.1800788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thurston TL, et al. Growth inhibition of cytosolic Salmonella by caspase-1 and caspase-11 precedes host cell death. Nat. Commun. 2016;7:13292. doi: 10.1038/ncomms13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cerqueira DM, et al. Guanylate-binding protein 5 licenses caspase-11 for gasdermin-D mediated host resistance to Brucella abortus infection. PLoS Pathog. 2018;14:e1007519. doi: 10.1371/journal.ppat.1007519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sollberger G, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018;3:eaar6689. doi: 10.1126/sciimmunol.aar6689. [DOI] [PubMed] [Google Scholar]
- 44.Kambara H, et al. Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep. 2018;22:2924–2936. doi: 10.1016/j.celrep.2018.02.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rathkey JK, et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol. 2018;3:eaat2738. doi: 10.1126/sciimmunol.aat2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goncalves AV, et al. Gasdermin-D and caspase-7 are the key caspase-1/8 substrates downstream of the NAIP5/NLRC4 inflammasome required for restriction of Legionella pneumophila. PLoS Pathog. 2019;15:e1007886. doi: 10.1371/journal.ppat.1007886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Z, Zhang Y, Lieberman J. Lighting a fire: can we harness pyroptosis to ignite anti-tumor immunity? Cancer Immunol. Res. 2021;9:2–7. doi: 10.1158/2326-6066.CIR-20-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu JJ, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020;21:736–745. doi: 10.1038/s41590-020-0669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Humphries F, et al. Succination inactivates gasdermin D and blocks pyroptosis. Science. 2020;369:1633–1637. doi: 10.1126/science.abb9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang S, Zhou Z, Sun Y, Zhang T, Sun L. Coral gasdermin triggers pyroptosis. Sci. Immunol. 2020;5:eabd2591. doi: 10.1126/sciimmunol.abd2591. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Z, Lieberman J. Lighting a fire on the reef. Sci Immunol. 2020;5:eabf0905. doi: 10.1126/sciimmunol.abf0905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saeki N, et al. GASDERMIN, suppressed frequently in gastric cancer, is a target of LMO1 in TGF-beta-dependent apoptotic signalling. Oncogene. 2007;26:6488–6498. doi: 10.1038/sj.onc.1210475. [DOI] [PubMed] [Google Scholar]
- 53.Kayagaki N, et al. IRF2 transcriptionally induces GSDMD expression for pyroptosis. Sci. Signal. 2019;12:eaax4917. doi: 10.1126/scisignal.aax4917. [DOI] [PubMed] [Google Scholar]
- 54.Webb MS, Miller AL, Thompson EB. In CEM cells the autosomal deafness gene dfna5 is regulated by glucocorticoids and forskolin. J. Steroid Biochem. Mol. Biol. 2007;107:15–21. doi: 10.1016/j.jsbmb.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moussette S, et al. Role of DNA methylation in expression control of the IKZF3-GSDMA region in human epithelial cells. PLoS ONE. 2017;12:e0172707. doi: 10.1371/journal.pone.0172707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Akino K, et al. Identification of DFNA5 as a target of epigenetic inactivation in gastric cancer. Cancer Sci. 2007;98:88–95. doi: 10.1111/j.1349-7006.2006.00351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim MS, et al. Methylation of the DFNA5 increases risk of lymph node metastasis in human breast cancer. Biochem. Biophys. Res. Commun. 2008;370:38–43. doi: 10.1016/j.bbrc.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Croes L, et al. DFNA5 promoter methylation a marker for breast tumorigenesis. Oncotarget. 2017;8:31948–31958. doi: 10.18632/oncotarget.16654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Croes L, et al. Large-scale analysis of DFNA5 methylation reveals its potential as biomarker for breast cancer. Clin. Epigenetics. 2018;10:51. doi: 10.1186/s13148-018-0479-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim MS, et al. Aberrant promoter methylation and tumor suppressive activity of the DFNA5 gene in colorectal carcinoma. Oncogene. 2008;27:3624–3634. doi: 10.1038/sj.onc.1211021. [DOI] [PubMed] [Google Scholar]
- 61.Sato H, et al. A new mutation Rim3 resembling Re(den) is mapped close to retinoic acid receptor alpha (Rara) gene on mouse chromosome 11. Mamm. Genome. 1998;9:20–25. doi: 10.1007/s003359900673. [DOI] [PubMed] [Google Scholar]
- 62.Katoh M, Katoh M. Evolutionary recombination hotspot around GSDML-GSDM locus is closely linked to the oncogenomic recombination hotspot around the PPP1R1B-ERBB2-GRB7 amplicon. Int. J. Oncol. 2004;24:757–763. [PubMed] [Google Scholar]
- 63.Lei M, et al. Gsdma3 is a new factor needed for TNF-alpha-mediated apoptosis signal pathway in mouse skin keratinocytes. Histochem. Cell Biol. 2012;138:385–396. doi: 10.1007/s00418-012-0960-1. [DOI] [PubMed] [Google Scholar]
- 64.Tanaka S, Mizushina Y, Kato Y, Tamura M, Shiroishi T. Functional conservation of Gsdma cluster genes specifically duplicated in the mouse genome. G3. 2013;3:1843–1850. doi: 10.1534/g3.113.007393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shi P, et al. Loss of conserved Gsdma3 self-regulation causes autophagy and cell death. Biochem. J. 2015;468:325–336. doi: 10.1042/BJ20150204. [DOI] [PubMed] [Google Scholar]
- 66.Soderman J, Berglind L, Almer S. Gene expression-genotype analysis implicates GSDMA, GSDMB, and LRRC3C as contributors to inflammatory bowel disease susceptibility. BioMed. Res. Int. 2015;2015:834805. doi: 10.1155/2015/834805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moreno-Moral A, et al. Changes in macrophage transcriptome associate with systemic sclerosis and mediate GSDMA contribution to disease risk. Ann. Rheum. Dis. 2018;77:596–601. doi: 10.1136/annrheumdis-2017-212454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Terao C, et al. Transethnic meta-analysis identifies GSDMA and PRDM1 as susceptibility genes to systemic sclerosis. Ann. Rheum. Dis. 2017;76:1150–1158. doi: 10.1136/annrheumdis-2016-210645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu J, et al. Polymorphisms in GSDMA and GSDMB are associated with asthma susceptibility, atopy and BHR. Pediatr. Pulm. 2011;46:701–708. doi: 10.1002/ppul.21424. [DOI] [PubMed] [Google Scholar]
- 70.Saeki N, et al. Distinctive expression and function of four GSDM family genes (GSDMA-D) in normal and malignant upper gastrointestinal epithelium. Genes Chromosomes Cancer. 2009;48:261–271. doi: 10.1002/gcc.20636. [DOI] [PubMed] [Google Scholar]
- 71.Carl-McGrath S, Schneider-Stock R, Ebert M, Rocken C. Differential expression and localisation of gasdermin-like (GSDML), a novel member of the cancer-associated GSDMDC protein family, in neoplastic and non-neoplastic gastric, hepatic, and colon tissues. Pathology. 2008;40:13–24. doi: 10.1080/00313020701716250. [DOI] [PubMed] [Google Scholar]
- 72.Hu Y, Jin S, Cheng L, Liu G, Jiang Q. Autoimmune disease variants regulate GSDMB gene expression in human immune cells and whole blood. Proc. Natl Acad. Sci. USA. 2017;114:E7860–E7862. doi: 10.1073/pnas.1712127114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen Q, et al. GSDMB promotes non-canonical pyroptosis by enhancing caspase-4 activity. J. Mol. Cell Biol. 2019;11:496–508. doi: 10.1093/jmcb/mjy056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Verlaan DJ, et al. Allele-specific chromatin remodeling in the ZPBP2/GSDMB/ORMDL3 locus associated with the risk of asthma and autoimmune disease. Am. J. Hum. Genet. 2009;85:377–393. doi: 10.1016/j.ajhg.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu H, et al. Genetic variation in ORM1-like 3 (ORMDL3) and gasdermin-like (GSDML) and childhood asthma. Allergy. 2009;64:629–635. doi: 10.1111/j.1398-9995.2008.01912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kang MJ, et al. GSDMB/ORMDL3 variants contribute to asthma susceptibility and eosinophil-mediated bronchial hyperresponsiveness. Hum. Immunol. 2012;73:954–959. doi: 10.1016/j.humimm.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 77.Moffatt MF, et al. A large-scale, consortium-based genomewide association study of asthma. N. Engl. J. Med. 2010;363:1211–1221. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hergueta-Redondo M, et al. Gasdermin-B promotes invasion and metastasis in breast cancer cells. PLoS ONE. 2014;9:e90099. doi: 10.1371/journal.pone.0090099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sun Q, et al. Expression of GSDML associates with tumor progression in uterine cervix cancer. Transl. Oncol. 2008;1:73–83. doi: 10.1593/tlo.08112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hergueta-Redondo M, et al. Gasdermin B expression predicts poor clinical outcome in HER2-positive breast cancer. Oncotarget. 2016;7:56295–56308. doi: 10.18632/oncotarget.10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Katoh M, Katoh M. Identification and characterization of human DFNA5L, mouse Dfna5l, and rat Dfna5l genes in silico. Int. J. Oncol. 2004;25:765–770. [PubMed] [Google Scholar]
- 82.Watabe K, et al. Structure, expression and chromosome mapping of MLZE, a novel gene which is preferentially expressed in metastatic melanoma cells. Jpn. J. Cancer Res. 2001;92:140–151. doi: 10.1111/j.1349-7006.2001.tb01076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kusumaningrum N, Lee DH, Yoon HS, Park CH, Chung JH. Ultraviolet light-induced gasdermin C expression is mediated via TRPV1/calcium/calcineurin/NFATc1 signaling. Int. J. Mol. Med. 2018;42:2859–2866. doi: 10.3892/ijmm.2018.3839. [DOI] [PubMed] [Google Scholar]
- 84.Kusumaningrum N, et al. Gasdermin C is induced by ultraviolet light and contributes to MMP-1 expression via activation of ERK and JNK pathways. J. Dermatol. Sci. 2018;90:180–189. doi: 10.1016/j.jdermsci.2018.01.015. [DOI] [PubMed] [Google Scholar]
- 85.Miguchi M, et al. Gasdermin C is upregulated by inactivation of transforming growth factor β receptor type II in the presence of mutated Apc, promoting colorectal cancer proliferation. PLoS ONE. 2016;11:e0166422. doi: 10.1371/journal.pone.0166422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Agard NJ, Maltby D, Wells JA. Inflammatory stimuli regulate caspase substrate profiles. Mol. Cell Proteom. 2010;9:880–893. doi: 10.1074/mcp.M900528-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fujii T, et al. Gasdermin D (Gsdmd) is dispensable for mouse intestinal epithelium development. Genesis. 2008;46:418–423. doi: 10.1002/dvg.20412. [DOI] [PubMed] [Google Scholar]
- 88.Rathkey JK, Xiao TS, Abbott DW. Human polymorphisms in GSDMD alter the inflammatory response. J. Biol. Chem. 2020;295:3228–3238. doi: 10.1074/jbc.RA119.010604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang J, Deobald K, Re F. Gasdermin D protects from melioidosis through pyroptosis and direct killing of bacteria. J. Immunol. 2019;202:3468–3473. doi: 10.4049/jimmunol.1900045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dubois H, et al. Nlrp3 inflammasome activation and gasdermin D-driven pyroptosis are immunopathogenic upon gastrointestinal norovirus infection. PLoS Pathog. 2019;15:e1007709. doi: 10.1371/journal.ppat.1007709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.de Beeck KO, Van Laer L, Van Camp G. DFNA5, a gene involved in hearing loss and cancer: a review. Ann. Otol. Rhinol. Laryngol. 2012;121:197–207. doi: 10.1177/000348941212100310. [DOI] [PubMed] [Google Scholar]
- 92.Thompson DA, Weigel RJ. Characterization of a gene that is inversely correlated with estrogen receptor expression (ICERE-1) in breast carcinomas. Eur. J. Biochem. 1998;252:169–177. doi: 10.1046/j.1432-1327.1998.2520169.x. [DOI] [PubMed] [Google Scholar]
- 93.Masuda Y, et al. The potential role of DFNA5, a hearing impairment gene, in p53-mediated cellular response to DNA damage. J. Hum. Genet. 2006;51:652–664. doi: 10.1007/s10038-006-0004-6. [DOI] [PubMed] [Google Scholar]
- 94.Yokomizo K, et al. Methylation of the DFNA5 gene is frequently detected in colorectal cancer. Anticancer. Res. 2012;32:1319–1322. [PubMed] [Google Scholar]
- 95.Stoll G, et al. Pro-necrotic molecules impact local immunosurveillance in human breast cancer. Oncoimmunology. 2017;6:e1299302. doi: 10.1080/2162402X.2017.1299302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Delmaghani S, et al. Hypervulnerability to sound exposure through impaired adaptive proliferation of peroxisomes. Cell. 2015;163:894–906. doi: 10.1016/j.cell.2015.10.023. [DOI] [PubMed] [Google Scholar]
- 97.Schwander M, et al. A forward genetics screen in mice identifies recessive deafness traits and reveals that pejvakin is essential for outer hair cell function. J. Neurosci. 2007;27:2163–2175. doi: 10.1523/JNEUROSCI.4975-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Van Laer L, et al. Mice lacking Dfna5 show a diverging number of cochlear fourth row outer hair cells. Neurobiol. Dis. 2005;19:386–399. doi: 10.1016/j.nbd.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 99.Harris SL, et al. Conditional deletion of pejvakin in adult outer hair cells causes progressive hearing loss in mice. Neuroscience. 2017;344:380–393. doi: 10.1016/j.neuroscience.2016.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Defourny J, et al. Pejvakin-mediated pexophagy protects auditory hair cells against noise-induced damage. Proc. Natl Acad. Sci. USA. 2019;116:8010–8017. doi: 10.1073/pnas.1821844116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bauernfeind FG, et al. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009;182:787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Black RA, et al. Generation of biologically active interleukin-1 beta by proteolytic cleavage of the inactive precursor. J. Biol. Chem. 1988;263:9437–9442. doi: 10.1016/S0021-9258(19)76559-4. [DOI] [PubMed] [Google Scholar]
- 103.Thornberry NA, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 104.Kayagaki N, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 105.Orning P, et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science. 2018;362:1064–1069. doi: 10.1126/science.aau2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sarhan J, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl Acad. Sci. USA. 2018;115:E10888–E10897. doi: 10.1073/pnas.1809548115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Demarco B, et al. Caspase-8-dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci. Adv. 2020;6:eabc3465. doi: 10.1126/sciadv.abc3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schwarzer R, Jiao H, Wachsmuth L, Tresch A, Pasparakis M. FADD and Caspase-8 regulate gut homeostasis and inflammation by controlling MLKL- and GSDMD-mediated death of intestinal epithelial cells. Immunity. 2020;52:978–993. doi: 10.1016/j.immuni.2020.04.002. [DOI] [PubMed] [Google Scholar]
- 109.Muendlein HI, et al. cFLIPL protects macrophages from LPS-induced pyroptosis via inhibition of complex II formation. Science. 2020;367:1379–1384. doi: 10.1126/science.aay3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Taabazuing CY, Okondo MC, Bachovchin DA. Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem. Biol. 2017;24:507–514 e504. doi: 10.1016/j.chembiol.2017.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chowdhury D, Lieberman J. Death by a thousand cuts: granzyme pathways of programmed cell death. Annu. Rev. Immunol. 2008;26:389–420. doi: 10.1146/annurev.immunol.26.021607.090404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Heusel JW, Wesselschmidt RL, Shresta S, Russell JH, Ley TJ. Cytotoxic lymphocytes require granzyme B for the rapid induction of DNA fragmentation and apoptosis in allogeneic target cells. Cell. 1994;76:977–987. doi: 10.1016/0092-8674(94)90376-X. [DOI] [PubMed] [Google Scholar]
- 113.Darmon AJ, Nicholson DW, Bleackley RC. Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B. Nature. 1995;377:446–448. doi: 10.1038/377446a0. [DOI] [PubMed] [Google Scholar]
- 114.Yang J, et al. Mechanism of gasdermin D recognition by inflammatory caspases and their inhibition by a gasdermin D-derived peptide inhibitor. Proc. Natl Acad. Sci. USA. 2018;115:6792–6797. doi: 10.1073/pnas.1800562115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang K, et al. Structural mechanism for GSDMD targeting by autoprocessed caspases in pyroptosis. Cell. 2020;180:941–955 e920. doi: 10.1016/j.cell.2020.02.002. [DOI] [PubMed] [Google Scholar]
- 116.Liu Z, et al. Caspase-1 engages full-length gasdermin D through two distinct interfaces that mediate caspase recruitment and substrate cleavage. Immunity. 2020;53:106–114. doi: 10.1016/j.immuni.2020.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ruan J, Xia S, Liu X, Lieberman J, Wu H. Cryo-EM structure of the gasdermin A3 membrane pore. Nature. 2018;557:62–67. doi: 10.1038/s41586-018-0058-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mulvihill E, et al. Mechanism of membrane pore formation by human gasdermin-D. EMBO J. 2018;37:e98321. doi: 10.15252/embj.201798321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rogers C, et al. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 2019;10:1689. doi: 10.1038/s41467-019-09397-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zanoni I, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science. 2016;352:1232–1236. doi: 10.1126/science.aaf3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wolf AJ, et al. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell. 2016;166:624–636. doi: 10.1016/j.cell.2016.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Heilig R, et al. The gasdermin-D pore acts as a conduit for IL-1β secretion in mice. Eur. J. Immunol. 2018;48:584–592. doi: 10.1002/eji.201747404. [DOI] [PubMed] [Google Scholar]
- 123.Gaidt MM, et al. Human monocytes engage an alternative inflammasome pathway. Immunity. 2016;44:833–846. doi: 10.1016/j.immuni.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 124.Carty M, et al. Cell survival and cytokine release after inflammasome activation is regulated by the Toll-IL-1R protein SARM. Immunity. 2019;50:1412–1424.e1416. doi: 10.1016/j.immuni.2019.04.005. [DOI] [PubMed] [Google Scholar]
- 125.Chen KW, et al. The neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation without pyroptosis during acute Salmonella challenge. Cell Rep. 2014;8:570–582. doi: 10.1016/j.celrep.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 126.Karmakar M, Katsnelson MA, Dubyak GR, Pearlman E. Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1β secretion in response to ATP. Nat. Commun. 2016;7:10555. doi: 10.1038/ncomms10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Karmakar M, et al. N-GSDMD trafficking to neutrophil organelles facilitates IL-1beta release independently of plasma membrane pores and pyroptosis. Nat. Commun. 2020;11:2212. doi: 10.1038/s41467-020-16043-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.McNeil PL, Kirchhausen T. An emergency response team for membrane repair. Nat. Rev. Mol. Cell Biol. 2005;6:499–505. doi: 10.1038/nrm1665. [DOI] [PubMed] [Google Scholar]
- 129.Monteleone M, et al. Interleukin-1β maturation triggers its relocation to the plasma membrane for gasdermin-D-dependent and -independent secretion. Cell Rep. 2018;24:1425–1433. doi: 10.1016/j.celrep.2018.07.027. [DOI] [PubMed] [Google Scholar]
- 130.Lin PH, Lin HY, Kuo CC, Yang LT. N-terminal functional domain of gasdermin A3 regulates mitochondrial homeostasis via mitochondrial targeting. J. Biomed. Sci. 2015;22:44. doi: 10.1186/s12929-015-0152-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chen KW, et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol. 2018;3:eaar6676. doi: 10.1126/sciimmunol.aar6676. [DOI] [PubMed] [Google Scholar]
- 132.Schlame M. Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J. Lipid Res. 2008;49:1607–1620. doi: 10.1194/jlr.R700018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chao KL, Kulakova L, Herzberg O. Gene polymorphism linked to increased asthma and IBD risk alters gasdermin-B structure, a sulfatide and phosphoinositide binding protein. Proc. Natl Acad. Sci. USA. 2017;114:E1128–E1137. doi: 10.1073/pnas.1616783114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018;18:134–147. doi: 10.1038/nri.2017.105. [DOI] [PubMed] [Google Scholar]
- 135.Burgener SS, et al. Cathepsin G inhibition by Serpinb1 and Serpinb6 prevents programmed necrosis in neutrophils and monocytes and reduces GSDMD-driven inflammation. Cell Rep. 2019;27:3646–3656 e3645. doi: 10.1016/j.celrep.2019.05.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Beckwith KS, et al. Plasma membrane damage causes NLRP3 activation and pyroptosis during Mycobacterium tuberculosis infection. Nat. Commun. 2020;11:2270. doi: 10.1038/s41467-020-16143-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fisch D, et al. Human GBP1 is a microbe-specific gatekeeper of macrophage apoptosis and pyroptosis. EMBO J. 2019;38:e100926. doi: 10.15252/embj.2018100926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pandori WJ, et al. Toxoplasma gondii activates a Syk-CARD9-NF-kappaB signaling axis and gasdermin D-independent release of IL-1beta during infection of primary human monocytes. PLoS Pathog. 2019;15:e1007923. doi: 10.1371/journal.ppat.1007923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Batista SJ, et al. Gasdermin-D-dependent IL-1alpha release from microglia promotes protective immunity during chronic Toxoplasma gondii infection. Nat. Commun. 2020;11:3687. doi: 10.1038/s41467-020-17491-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Quach J, Moreau F, Sandall C, Chadee K. Entamoeba histolytica-induced IL-1beta secretion is dependent on caspase-4 and gasdermin D. Mucosal Immunol. 2019;12:323–339. doi: 10.1038/s41385-018-0101-9. [DOI] [PubMed] [Google Scholar]
- 141.Zhu S, et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature. 2017;546:667–670. doi: 10.1038/nature22967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liu T, et al. NOD-like receptor family, pyrin domain containing 3 (NLRP3) contributes to inflammation, pyroptosis, and mucin production in human airway epithelium on rhinovirus infection. J. Allergy Clin. Immunol. 2019;144:777–787 e779. doi: 10.1016/j.jaci.2019.05.006. [DOI] [PubMed] [Google Scholar]
- 143.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016;16:407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 144.Kanneganti A, et al. GSDMD is critical for autoinflammatory pathology in a mouse model of familial mediterranean fever. J. Exp. Med. 2018;215:1519–1529. doi: 10.1084/jem.20172060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.O’Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci. STKE. 2000;2000:re1. doi: 10.1126/stke.442000re1. [DOI] [PubMed] [Google Scholar]
- 146.Yang X, et al. Bacterial endotoxin activates the coagulation cascade through gasdermin D-dependent phosphatidylserine exposure. Immunity. 2019;51:983–996 e986. doi: 10.1016/j.immuni.2019.11.005. [DOI] [PubMed] [Google Scholar]
- 147.Wu C, et al. Inflammasome activation triggers blood clotting and host death through pyroptosis. Immunity. 2019;50:1401–1411 e1404. doi: 10.1016/j.immuni.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.McDonald B, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. 2017;129:1357–1367. doi: 10.1182/blood-2016-09-741298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kayagaki N, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 150.Chen H, et al. RIPK3 collaborates with GSDMD to drive tissue injury in lethal polymicrobial sepsis. Cell Death Differ. 2020;27:2568–2585. doi: 10.1038/s41418-020-0524-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.McKenzie BA, et al. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc. Natl Acad. Sci. USA. 2018;115:E6065–E6074. doi: 10.1073/pnas.1722041115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Xiao J, et al. Radiation causes tissue damage by dysregulating inflammasome-gasdermin D signaling in both host and transplanted cells. PLoS Biol. 2020;18:e3000807. doi: 10.1371/journal.pbio.3000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lu Y, et al. Caspase-11 signaling enhances graft-versus-host disease. Nat. Commun. 2019;10:4044. doi: 10.1038/s41467-019-11895-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Xu B, et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 2018;68:773–782. doi: 10.1016/j.jhep.2017.11.040. [DOI] [PubMed] [Google Scholar]
- 155.Khanova E, et al. Pyroptosis by caspase11/4-gasdermin-D pathway in alcoholic hepatitis in mice and patients. Hepatology. 2018;67:1737–1753. doi: 10.1002/hep.29645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yang C, et al. Gasdermin D protects against noninfectious liver injury by regulating apoptosis and necroptosis. Cell Death Dis. 2019;10:481. doi: 10.1038/s41419-019-1719-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Li J, et al. Blocking GSDMD processing in innate immune cells but not in hepatocytes protects hepatic ischemia-reperfusion injury. Cell Death Dis. 2020;11:244. doi: 10.1038/s41419-020-2437-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhang D, et al. Gasdermin D serves as a key executioner of pyroptosis in experimental cerebral ischemia and reperfusion model both in vivo and in vitro. J. Neurosci. Res. 2019;97:645–660. doi: 10.1002/jnr.24385. [DOI] [PubMed] [Google Scholar]
- 159.Jia C, et al. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis. 2019;10:778. doi: 10.1038/s41419-019-2021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wang Y, et al. TLR4/NF-kappaB signaling induces GSDMD-related pyroptosis in tubular cells in diabetic kidney disease. Front. Endocrinol. 2019;10:603. doi: 10.3389/fendo.2019.00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yang F, et al. Silencing long non-coding RNA Kcnq1ot1 alleviates pyroptosis and fibrosis in diabetic cardiomyopathy. Cell Death Dis. 2018;9:1000. doi: 10.1038/s41419-018-1029-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhang Z, et al. Caspase-11-mediated tubular epithelial pyroptosis underlies contrast-induced acute kidney injury. Cell Death Dis. 2018;9:983. doi: 10.1038/s41419-018-1023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yuan B, et al. Inhibition of AIM2 inflammasome activation alleviates GSDMD-induced pyroptosis in early brain injury after subarachnoid haemorrhage. Cell Death Dis. 2020;11:76. doi: 10.1038/s41419-020-2248-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hu X, et al. Role of pyroptosis in traumatic brain and spinal cord injuries. Int. J. Biol. Sci. 2020;16:2042–2050. doi: 10.7150/ijbs.45467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wang WJ, et al. Downregulation of gasdermin D promotes gastric cancer proliferation by regulating cell cycle-related proteins. J. Dig. Dis. 2018;19:74–83. doi: 10.1111/1751-2980.12576. [DOI] [PubMed] [Google Scholar]
- 166.Gao J, et al. Downregulation of GSDMD attenuates tumor proliferation via the intrinsic mitochondrial apoptotic pathway and inhibition of EGFR/Akt signaling and predicts a good prognosis in nonsmall cell lung cancer. Oncol. Rep. 2018;40:1971–1984. doi: 10.3892/or.2018.6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Molina-Crespo A, et al. Intracellular delivery of an antibody targeting gasdermin-B reduces HER2 breast cancer aggressiveness. Clin. Cancer Res. 2019;25:4846–4858. doi: 10.1158/1078-0432.CCR-18-2381. [DOI] [PubMed] [Google Scholar]
- 168.Wei J, et al. Overexpression of GSDMC is a prognostic factor for predicting a poor outcome in lung adenocarcinoma. Mol. Med. Rep. 2020;21:360–370. doi: 10.3892/mmr.2019.10837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lage H, Helmbach H, Grottke C, Dietel M, Schadendorf D. DFNA5 (ICERE-1) contributes to acquired etoposide resistance in melanoma cells. FEBS Lett. 2001;494:54–59. doi: 10.1016/S0014-5793(01)02304-3. [DOI] [PubMed] [Google Scholar]
- 171.Lu H, et al. Molecular targeted therapies elicit concurrent apoptotic and GSDME-dependent pyroptotic tumor cell death. Clin. Cancer Res. 2018;24:6066–6077. doi: 10.1158/1078-0432.CCR-18-1478. [DOI] [PubMed] [Google Scholar]
- 172.Liu Y, et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci. Immunol. 2020;5:eaax7969. doi: 10.1126/sciimmunol.aax7969. [DOI] [PubMed] [Google Scholar]
- 173.Fan JX, et al. Epigenetics-based tumor cells pyroptosis for enhancing the immunological effect of chemotherapeutic nanocarriers. Nano Lett. 2019;19:8049–8058. doi: 10.1021/acs.nanolett.9b03245. [DOI] [PubMed] [Google Scholar]
- 174.Erkes DA, et al. Mutant BRAF and MEK inhibitors regulate the tumor immune microenvironment via pyroptosis. Cancer Discov. 2020;10:254–269. doi: 10.1158/2159-8290.CD-19-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Johnson DC, et al. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat. Med. 2018;24:1151–1156. doi: 10.1038/s41591-018-0082-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012;11:633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Skrott Z, et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature. 2017;552:194–199. doi: 10.1038/nature25016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Martin-Sanchez F, et al. Inflammasome-dependent IL-1beta release depends upon membrane permeabilisation. Cell Death Differ. 2016;23:1219–1231. doi: 10.1038/cdd.2015.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wang Q, et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature. 2020;579:421–426. doi: 10.1038/s41586-020-2079-1. [DOI] [PubMed] [Google Scholar]
- 181.Rashidi M, et al. The pyroptotic cell death effector gasdermin D is activated by gout-associated uric acid crystals but is dispensable for cell death and IL-1beta release. J. Immunol. 2019;203:736–748. doi: 10.4049/jimmunol.1900228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Banerjee I, et al. Gasdermin D restrains type I interferon response to cytosolic DNA by disrupting ionic homeostasis. Immunity. 2018;49:413–426 e415. doi: 10.1016/j.immuni.2018.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wang X, et al. Effects of gasdermin D in modulating murine lupus and its associated organ damage. Arthritis Rheumatol. 2020;72:2118–2129. doi: 10.1002/art.41444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ma C, et al. Gasdermin D in macrophages restrains colitis by controlling cGAS-mediated inflammation. Sci. Adv. 2020;6:eaaz6717. doi: 10.1126/sciadv.aaz6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bulek K, et al. Epithelial-derived gasdermin D mediates nonlytic IL-1beta release during experimental colitis. J. Clin. Invest. 2020;130:4218–4234. doi: 10.1172/JCI138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Fink SL, Cookson BT. Pyroptosis and host cell death responses during Salmonella infection. Cell Microbiol. 2007;9:2562–2570. doi: 10.1111/j.1462-5822.2007.01036.x. [DOI] [PubMed] [Google Scholar]
- 187.Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358:167–169. doi: 10.1038/358167a0. [DOI] [PubMed] [Google Scholar]
- 188.Lindgren SW, Stojiljkovic I, Heffron F. Macrophage killing is an essential virulence mechanism of Salmonella typhimurium. Proc. Natl Acad. Sci. USA. 1996;93:4197–4201. doi: 10.1073/pnas.93.9.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Monack DM, Raupach B, Hromockyj AE, Falkow S. Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc. Natl Acad. Sci. USA. 1996;93:9833–9838. doi: 10.1073/pnas.93.18.9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Chen Y, Smith MR, Thirumalai K, Zychlinsky A. A bacterial invasin induces macrophage apoptosis by binding directly to ICE. EMBO J. 1996;15:3853–3860. doi: 10.1002/j.1460-2075.1996.tb00759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hilbi H, et al. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J. Biol. Chem. 1998;273:32895–32900. doi: 10.1074/jbc.273.49.32895. [DOI] [PubMed] [Google Scholar]
- 192.Hersh D, et al. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc. Natl Acad. Sci. USA. 1999;96:2396–2401. doi: 10.1073/pnas.96.5.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zychlinsky A, Fitting C, Cavaillon JM, Sansonetti PJ. Interleukin 1 is released by murine macrophages during apoptosis induced by Shigella flexneri. J. Clin. Invest. 1994;94:1328–1332. doi: 10.1172/JCI117452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Fernandez-Prada CM, Hoover DL, Tall BD, Venkatesan MM. Human monocyte-derived macrophages infected with virulent Shigella flexneri in vitro undergo a rapid cytolytic event similar to oncosis but not apoptosis. Infect. Immun. 1997;65:1486–1496. doi: 10.1128/iai.65.4.1486-1496.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol. Microbiol. 2000;38:31–40. doi: 10.1046/j.1365-2958.2000.02103.x. [DOI] [PubMed] [Google Scholar]
- 196.Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113–114. doi: 10.1016/S0966-842X(00)01936-3. [DOI] [PubMed] [Google Scholar]
- 197.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 198.Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004;5:897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- 199.Thomas MP, Lieberman J. Live or let die: posttranscriptional gene regulation in cell stress and cell death. Immunol. Rev. 2013;253:237–252. doi: 10.1111/imr.12052. [DOI] [PubMed] [Google Scholar]
- 200.Thomas MP, et al. Apoptosis triggers specific, rapid, and global mRNA decay with 3′ uridylated intermediates degraded by DIS3L2. Cell Rep. 2015;11:1079–1089. doi: 10.1016/j.celrep.2015.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Liu X, et al. PNPT1 release from mitochondria during apoptosis triggers decay of poly(A) RNAs. Cell. 2018;174:187–201 e112. doi: 10.1016/j.cell.2018.04.017. [DOI] [PubMed] [Google Scholar]
- 202.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/S0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 203.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]