

Summary
Immune checkpoint therapy to reverse natural killer (NK) and T cell exhaustion has emerged as a promising treatment in various cancers. While anti‐programmed cell death 1 (PD‐1) pembrolizumab has recently gained Food and Drug Administration (FDA) approval for use in recurrent or metastatic cervical cancer, other checkpoint molecules, such as T cell immunoreceptor with immunoglobulin (Ig) and immunoreceptor tyrosine‐based inhibition motif (ITIM) domains (TIGIT) and T cell immunoglobulin and mucin‐domain containing‐3 (Tim‐3), have yet to be fully explored in this disease. We report expression of TIGIT, Tim‐3 and PD‐1 on subsets of peripheral blood NK (CD56dim/negCD16bright/dim/neg and CD56brightCD16dim/neg) and T cells. The percentages of these cells were increased in women with cervical cancer and pre‐malignant lesions. PD‐1+ NK and T cells were likely to co‐express TIGIT and/or Tim‐3. These cells, with an apparently ‘exhausted’ phenotype, were augmented in patients. A subset of cells were also natural killer group 2 member D (NKG2D)‐ and DNAX accessory molecule 1 (DNAM‐1)‐positive. PD‐1int and PD‐1high T cells were notably increased in cervical cancer. Soluble programmed cell death ligand 1 (PD‐L1) was higher in cancer patient blood versus healthy donors and we observed a positive correlation between sPD‐L1 and PD‐1+ T cells in women with low‐grade lesions. Within the cancer group, there were no significant correlations between sPD‐L1 levels and cervical cancer stage. However, when comparing cancer versus healthy donors, we observed an inverse association between sPD‐L1 and total T cells and a correlation between sPD‐L1 and CD56dim NK cells. Our results may show an overview of the immune response towards pre‐cancerous lesions and cervical cancer, perhaps giving an early clue as to whom to administer blocking therapies. The increase of multiple checkpoint markers may aid in identifying patients uniquely responsive to combined antibody therapies.
Keywords: cervical carcinoma, checkpoint, CIN, DNAM, immune evasion, NKG2D, PBMC, PD1, soluble ligands, TIGIT, TIM3
Overview of immune cells in the blood of patients with cervical cancer and precursor lesions. The co‐expression of checkpoint molecules PD‐1, TIGIT and Tim‐3 describe distinct NK and T cell populations that increase in accordance with disease progression. Tumor expression of checkpoint ligands, or soluble PD‐L1, may influence the development of these cells.
Introduction
In recent years the role of immune checkpoint molecules has emerged as one of the most promising avenues of biomolecular and clinical discovery [1]. The ability to regulate these molecular regulators represents a new paradigm in directed treatment. Fundamental to these advances are the ideas of exhausted effector cells; that is, putatively cytotoxic cells that, for one reason or another, have lost their capacity to effectively participate in anti‐tumor responses [2]. Increases in regulatory T cells, chronic inflammation and myeloid and other lineage suppressor cells have all been implicated in the suppression of immune response and evasion of tumor cells [3]. More recently, the so‐called checkpoint molecules have risen to the fore. These include stimulatory molecules (not the subject of this paper) and inhibitory molecules such as cytotoxic T lymphocyte antigen (CTLA)‐4, programmed cell death 1 (PD‐1), T cell immunoreceptor with immunoglobulin (Ig) and immunoreceptor tyrosine‐based inhibition motif (ITIM) domains (TIGIT), lymphocyte‐activation gene 3 (LAG3), T cell Ig and mucin‐domain containing‐3 (Tim‐3), V domain‐containing immunoglobulin suppressor of T cell activation (VISTA), Siglec‐7, human endogenous retrovirus‐H long terminal repeat‐associating protein 2 (HHLA2), B and T lymphocyte attenuator (BTLA) and adenosine A2aR [4, 5]. Together, or in various combinations, these molecules have been postulated to form patterns that describe dysfunctional or exhausted T and NK cells [6, 7]. Additionally, the anti‐tumor activity of these immune cells that express different inhibitory receptors is also regulated by the expression of their cognate ligands in antigen‐presenting cells [CD80/86, PD‐L2, programmed cell death ligand 1 (PD‐L1] or in the tumor (PD‐L1, CD112, CD155, galectin 9) [8]. For this reason, some protocols for cancer treatment with immune checkpoint inhibitors include monoclonal antibodies that target both the receptor and its ligand; for example, blocking the PD‐1/PD‐L1 inhibitory pathway. Despite the success of anti‐PD‐1/PD‐L1 therapies, the existence of immune‐related adverse events must be considered, together with the fact that only a fraction of patients benefit from this treatment [9]. Therefore, the use of predictive biomarkers may help to select the patients who are the most promising candidates for this therapy and to predict clinical outcomes [10]. The over‐expression of PD‐L1 in tumor tissues has been considered as a predictive biomarker; however, some authors have concluded that this molecule may not be a robust indicator of treatment response due to the fact that PD‐L1‐negative tumors can also respond to PD‐1/PD‐L1 targeting [11, 12]. The soluble form of PD‐L1 (sPD‐L1) has been reported to be an improved predictor of metastasis versus membrane‐bound PD‐L1 and a prognostic factor for the survival of first‐line chemotherapy, and may also mediate resistance to PD‐L1 blockade [13, 14, 15, 16].
The analysis of checkpoint molecules on peripheral blood lymphocytes may also be used as a non‐invasive method to predict treatment outcomes [10]. Additionally, the expression of immune checkpoint receptors on cytotoxic lymphocytes from the blood of cancer patients may indicate, or hint at, the profile of these cells in the tumor [17]. The roles and distributions of immune checkpoints may vary between diseases and disease models, and previous studies in one disease may not be applicable to other, similar diseases; for this reason, investigation on these molecules continues even when utility and clinical efficacy have already been proven for these molecules as therapeutic targets in some diseases.
The primary targets for many immune checkpoint therapy studies have been T cells; however, not all patients respond to treatment. There are multiple reasons for this non‐responsiveness, and one possibility is that in certain tumors natural killer (NK) cells, rather than cytotoxic clonally specific T cells, may play a more important role. In some tumors the transformed cells develop the ability to down‐regulate major histocompatibility complex (MHC)‐I, and thus escape T cell‐mediated immunity. The absence of MHC‐I molecules then, in turn, makes these tumor cells ideal targets for NK cells. Furthermore, some checkpoint molecules that are well characterized in T cells, such as PD‐1, LAG‐3, TIGIT and Tim‐3, are also present in NK cells, and their blockade has been shown to restore NK cytotoxicity; thus it is possible that anti‐checkpoint therapies targeting NK cells may be a promising strategy to improve clinical outcomes in patients who do not respond to T cell‐based immunotherapy, or to augment the reversal of exhausted T cells with the revival of exhausted NK cells as well [18].
In cervical cancer, different immune cells work together to avoid tumor growth. Both NK cells and T cells play an important role in the destruction of human papillomavirus (HPV)‐infected cells and tumor cells [19]. NK cells are one of the first lines of defense against HPV infection; they are present at early stages of the infection and can respond against HPV‐induced lesions [20]. However, similar to other cancers, cervical tumors and advanced dysplasias have also been characterized by various ‘dysfunctions’ of the immune system; that is, failure of immunosurveillance [21]. Interestingly, while down‐regulated MHC‐I has been found in cervical cancer and increased NK cell percentages have been found in patient blood, this increase was not reflected in the tumor, suggesting that this inefficient migration may be due to inhibition of the NK cells [22]. NK cell activity in the HPV‐infected cervix and cervical cancer may be affected in different ways [23, 24]. For example, HPV16 E6 and E7 oncoproteins inhibit NK cell interleukin (IL)‐18‐induced interferon (IFN)‐γ production [25]. Reduced cytotoxic activity of NK cells has also been observed in cervical cancer and precursor lesions, together with a reduction in the expression of important activating receptors [e.g. natural killer group 2 member D (NKG2D), NKp30, NKp46] [26].
NK cells can be subdivided into various subsets based on the relative expression of CD16 and CD56. The two classical NK cell subsets in peripheral blood are CD56dimCD16bright and CD56brightCD16−/dim NK cells [27]. Traditionally, the first population is predominant in peripheral blood and is considered to be the most cytotoxic subset, whereas the second are cytokine producers, and are primarily found in secondary lymphoid tissue and other tissues [28]. CD56brightCD16− NK cells are the precursors of CD56brightCD16dim (or CD16+) NK cells. The CD56brightCD16+ subset has an intermediate profile between CD56brightCD16− and CD56dimCD16+ NK cells. From a functional viewpoint, compared with CD56brightCD16−, CD56dimCD16+ NK cells can more effectively kill target cells by antibody‐dependent cell cytotoxicity and direct lysis [29].
In peripheral blood it is possible to subdivide NK cells further and define other cell subsets. CD56dimCD16dim NK cells may represent an intermediate stage between CD56dimCD16bright and CD56dimCD16− NK cell subsets. CD56dimCD16dim NK cells have a more immature phenotype than CD56dimCD16bright NK cells. However, CD56dimCD16dim cells degranulate in the presence of K562 target cells to a higher level than CD56dimCD16bright cells, but much less than CD56dimCD16− cells [30]. CD56dimCD16− NK cells are more cytotoxic cells that may be previously CD56dimCD16bright cells that lost the expression of CD16 through metalloprotease‐mediated shedding [30, 31, 32]. CD56negCD16bright NK cells are considered an NK cell subset with a phenotype functionally impaired with respect to cytokine production and cytolytic function. These cells are reported to be expanded in patients with chronic hepatitis C virus infection and HIV‐1 infection [33, 34, 35].
Perhaps some of the most promising checkpoint markers of immune system dysfunction are PD‐1, TIGIT and Tim‐3. While these markers have been established on T cells, their role in NK cell activity is less well known. To date, PD‐1 and Tim‐3 have been found in cervical cancer tissues [36, 37, 38]. TIGIT has yet to be described in NK cells in cervical cancer or precursor lesions. The role of these three markers together on the same cells, or on different cell populations, has only begun to be described in certain models [39, 40, 41, 42]. Recent works have begun to explore the role of different subpopulations within effector cells; that is, populations that express sharply higher levels of PD‐1 as a way of distinguishing the expected presence of ‘normal’ checkpoint molecules (which may be physiologically expected in healthy populations) and pathogenic levels that may be encountered in select patients [43]. The accurate distinction between these two groups may define the difference between tumors responsive to, or refractory to, current and future immunotherapy checkpoint blockades.
The goal of this current paper is to identify different NK and T cell populations with respect to checkpoint molecules in cervical cancer patients and to correlate these populations with levels of sPD‐L1 in patient serum.
Material and methods
Patients and samples
We collected blood samples from 78 women: patients with cervical carcinoma (n = 21; median age = 44, age range = 47·2 ± 11·5 years), high‐grade lesions (n = 9; median age =37, age range = 42·9 ± 16·6 years), low‐grade lesions (n = 24; median age = 30, age range = 30·2 ± 9·6 years) and healthy donors (n = 24; median age = 38·5, age range = 39·25 ± 7·2 years). The women with pre‐malignant lesions and cancer had their diagnosis confirmed with colposcopy and histopathological analysis. The cancer group included cervical adenocarcinoma and squamous cell carcinoma in situ (stage 0) and FIGO stages I, II, III and IV. These patients were treated at the Instituto Jalisciense de Cancerología and the Hospital Civil Nuevo, Guadalajara, Jalisco, México. Not all samples from all participants were used in every experiment in the study. For this reason, sample numbers change slightly between cytometry and enzyme‐linked immunosorbent assay (ELISA) experiments. All study participants gave their written informed consent. This study was performed according to the guidelines of the local institutional ethics committees and in accordance with the 2013 Declaration of Helsinki.
Venous blood was collected from each donor in silicone‐coated BD (Becton Dickinson, Franklin Lakes, NJ, USA) Vacutainer® tubes (5 ml) for the isolation of the serum and in spray‐coated K2ethylenediamine tetraacetic acid (EDTA) BD Vacutainer® tubes (10 ml) for the separation of peripheral blood mononuclear cells (PBMCs).
PBMCs were separated from the blood samples using Lymphoprep™ (Stemcell Technologies, Vancouver, Canada) density gradient centrifugation. The PBMCs were stored frozen in liquid nitrogen at a temperature below −130°C until the cytometry analysis. The viability of the thawed cells was determined with trypan blue and cells were used when viability was greater than or equal to 90%. Serum from the blood of the same women was isolated and stored at −80°C.
Flow cytometry
A multi‐color flow cytometry protocol was used to analyze the expression of immune checkpoint molecules (PD‐1, TIGIT and Tim‐3) and activating receptors (DNAM‐1, NKG2D, CD16) on study group PBMCs separated into the following populations: CD3−CD56dim, CD3−CD56bright NK and CD56−CD3+ T cells. We used the following antibodies to stain 5 × 105 PBMCs: anti‐CD3‐ε [fluorescein isothiocyanate (FITC), clone UCHT1, anti‐CD56 [phycoerythrin cyanin 7 (PE‐Cy7), clone 5.1 H11], anti‐CD16 [peridinin chlorophyll cyanin 5·5 (PerCP‐Cy5·5), clone 3G8], anti‐TIGIT [allophycocyanin (APC), clone A15153G], anti‐Tim‐3 (BV510, clone F38‐2E2) and anti‐PD‐1 (BV421, clone EH12.2H7) (all antibodies purchased from BioLegend, San Diego, CA, USA). Data acquisition was performed using the BD FACSCanto II flow cytometer (Becton Dickinson). First, for acquisition of each sample we derived an initial dot‐plot (FSC‐A versus FSC‐H) for singlets. This selection generated a dot‐plot with the combination of FSC‐A versus SSC‐A, after which we acquired 250 000 events from the lymphocyte gate.
For the t‐distributed stochastic neighbor‐embedding analysis (t‐SNE), individual donor fcs files containing measured intensities for each marker at single‐cell level were imported into FCS Express 7 Plus (De Novo Software, Glendale, CA, USA). A data merge was conducted to visualize the general expression of the receptors in all the samples from each group. Prior to starting the dimensional analysis, singlets and subpopulation gating were performed manually. NK cell subsets were defined based on CD56 and CD16 expression, while T cells were defined as CD56negCD3+ cells. A total of 50 000 cells were randomly sampled per donor to create a final file containing 500 000 cells. t‐SNE density plots of the peripheral NK cell subpopulations and T cells were generated. Normalized protein expression levels for CD3, CD56, CD16, PD‐1, TIGIT, Tim‐3, DNAM‐1 and NKG2D in t‐SNE fields were represented in red for high expression and in blue for low expression (hot‐to‐cold heat map).
Conventional gating of the different populations was performed in Kaluza software version 2.1 (Beckman Coulter, Brea, CA, USA) to analyze the single expression or co‐expression of the different receptors. Isotype controls were used to adjust background fluorescence. To establish the parameters for gating we employed fluorescence minus one (FMO) staining controls.
ELISA
Soluble PD‐L1 concentrations in the serum samples (female patient and healthy donor serum) were measured by an ELISA kit (ThermoFisher, Waltham, MA, USA), according to the manufacturer’s protocol. Briefly, 50 µl of sample in duplicate was added to the sample wells. Wells were then incubated with the biotin‐conjugated antibody for 2 h at room temperature. Streptavidin‐horseradish peroxidase (HRP) was next added to all the samples and incubated 1 h at room temperature. 3,3′,5,5′‐Tetramethylbenzidine (TMB) substrate solution was added and the wells were incubated for approximately 30 min at room temperature. Finally, the reaction was stopped with stop solution and the color intensity was measured at 450 nm.
Statistical analysis
Data were assessed for normality using the D’Agostino–Pearson normality test and comparison between the groups were performed using analysis of variance (anova) with Dunnett’s multiple comparisons test. Pearson’s correlation analysis was performed to verify the linear associations between the groups. P‐values < 0·05 were considered statistically significant. A convergence table (Fisher’s exact test) was prepared to analyze increased sPD‐L1 with respect to variables identified using a linear regression analysis. For this analysis, the sPD‐L1 levels were divided based on the median of the healthy donor samples (to avoid undue influence of out‐of‐range donor samples); other variables were defined based on the mean of the healthy donor population or when the cancer samples were out of that range, the mean ± 1 standard deviation of the healthy population. Prism version 7, GraphPad software, was used for this analysis. Only significant P‐values are displayed.
Results
Percentages of different NK cell subpopulations and T cells in healthy donors, patients with pre‐malignant lesions and cervical cancer patients
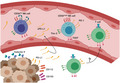
We first analyzed the percentages of T cells and different NK cell subpopulations in healthy donors (HD group), low‐grade lesions (LG group), high‐grade lesions (HG group) and cervical cancer patients (CC group) (Fig. 1). To this end, we gated on CD56−CD3+ cells to select the region of the T cells (Fig. 1a). In the case of the NK cells, we subdivided the two classical NK cell subpopulations, CD56dim and CD56bright NK cells into six different subsets based on the expression of CD56 and CD16.
Fig. 1.
Percentages of peripheral blood natural killer (NK) and T cells in healthy donors (HD) group, low‐grade lesions (LG) group, high‐grade lesions (HG) group and cervical cancer patients (CC) group. (a) Gating strategy for the flow cytometric analysis of NK and T cells in peripheral blood mononuclear cells (PBMCs) from HD (n = 17), LG (n = 19), HG (n = 8) and CC (n = 19) groups. CD56−CD3+ cells were gated to select the region of the T cells. NK cells were subdivided into six different subsets based on the expression of CD56 and CD16. Frequency of (b) T cells, (c) CD56brightCD16neg NK cells, (d) CD56brightCD16dim NK cells, (e) CD56dimCD16neg NK cells, (f) CD56dimCD16dim NK cells, (g) CD56dimCD16bright NK cells and (h) CD56negCD16bright NK cells. (i) Correlation analysis to compare the percentage of CD56dimCD16bright NK cells veraus CD3 T cells in CC patients. Frequency data are shown as individual percentages of expression (taken from the lymphocyte gate) and their mean. Inset example of the six different groups shows percentages based on the total NK cell region. Comparisons between the groups were performed using analysis of variance (anova) with Dunnett’s multiple comparisons test. Pearson’s correlation analysis was performed to verify the linear associations between CD56dimCD16bright NK cells and the rest of the NK cell subpopulations or CD3+ T cells and the NK subpopulations. The correlation coefficient, r and P‐values are shown in the figures. *P ≤ 0·05, **P ≤ 0·01, ***P ≤ 0·001.
We found that the percentages of CD3+T cells were significantly decreased (P < 0·05) in the three patient groups when compared to HD (Fig. 1b). A decrease in the percentages of CD56brightCD16neg NK cells was also seen, but only in the CC group in comparison with HD (Fig. 1c). No significant changes were found when we analyzed the percentages of CD56brightCD16dim or CD56dimCD16neg NK cells in the different groups (Fig. 1d,e).
In contrast, the percentages of CD56dimCD16dim and CD56dimCD16bright NK cells were increased in the CC group in comparison with HD and pre‐malignant lesions (Fig. 1f,g). Regarding the CD56negCD16bright NK cell subpopulation, we did not observe any change in the percentages of these cells in the different groups (Fig. 1h).
A correlation analysis was performed between different cell populations. The CD56dimCD16bright NK cells (the most abundant NK population) were found to be significantly negatively correlated with CD3 cells in CC (Fig. 1i). The correlation analysis was continued to compare the percentage of CD56dimCD16bright NK cells with the rest of the NK cell subpopulations in each patient group (LG, HG, CC). No significant results were found. We found non‐significant positive correlations between CD56dimCD16bright NK cells and CD56dimCD16dim NK cells in LG and CC groups and CD56dimCD16bright NK cells and CD56dimCD16neg NK cells in LG; there was also a weak, but non‐significant positive correlation between CD56brightCD16dim and CD3 cell percentages in CC (Supporting information, Fig. S1).
In summary, we observed that there is a significant decrease in the percentages of T cells and CD56brightCD16neg NK cells in cervical cancer patients and an increase in the percentages of CD56dimCD16dim and CD56dimCD16bright NK cells. Given this observation, we decided to focus our subsequent checkpoint analysis on both NK and T cells, with the idea that some patients might be polarized towards either the NK or T cell responses.
Immune checkpoint profiling of CD56bright and CD56dim NK cell subpopulations
We then aimed to evaluate the expression of inhibitory (PD‐1, TIGIT, Tim‐3) and activating (NKG2D, DNAM‐1) immune checkpoint receptors on the two classical NK cells subsets, CD56dim NK cells and a CD56bright NK cells. We show the most significant of these data in Figs 2 and 3.
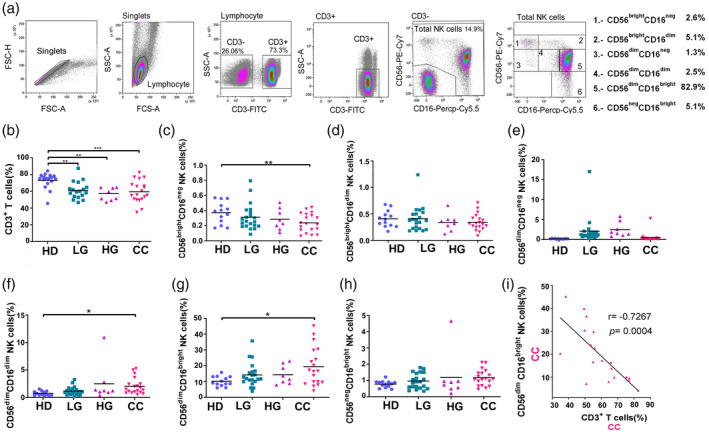
We utilized t‐SNE analysis to assess the general expression patterns in a merged analysis of all the samples from each group (Figs 2a, 3a). t‐SNE analysis helped us to visualize the general expression of the receptors and to define the manual gating strategy to identify populations defined by patterns of multiple receptor expression that might be of potential interest. Starting with PD‐1 expression, we used the t‐SNE blots to suggest the receptor patterns that might overlap with PD‐1 expression or might be over‐expressed in the cancer group. We then evaluated the single and co‐expression using traditional histograms and dot‐plots (Figs 2b,c, 3b,c).
Fig. 2.
Expression and co‐expression of programmed cell death 1 (PD‐1), T cell immunoreceptor with immunoglobulin (Ig) and immunoreceptor tyrosine‐based inhibition motif (ITIM) domains (TIGIT), T cell immunoglobulin and mucin‐domain containing‐3 (Tim‐3), natural killer group 2 member D (NKG2D) and DNAX accessory molecule 1 (DNAM‐1) on peripheral blood CD56dim NK cells from healthy donors (HD) group, low‐grade lesions (LG) group, high‐grade lesions (HG) group and cervical cancer patients (CC) group. (a) t‐Distributed stochastic neighbor‐embedding analysis (t‐SNE) density plots showing expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3. The PD‐1+ region is highlighted and cross‐over with similar regions within CC is outlined. (b) Representative histogram staining for the antibodies used for each population group. (c) Representative dot‐plot analysis for all single antibody combinations and the multiple antibody combinations that showed significant differences between HD and CC. t‐SNE analysis was used to visualize expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3 in HD (n = 13), LG (n = 19), HG (n = 8) and CC (n = 19) groups. Flow cytometry gating of the data was performed to analyze the single expression of the immune checkpoints: (d) PD‐1, (e) TIGIT, (f) Tim‐3, (g) NKG2D, (h) DNAM‐1. Co‐expression of (i) PD‐1+TIGIT, (j) PD‐1+Tim‐3, (k) PD‐1+NKG2D, (l) TIGIT+DNAM and (m) PD‐1+TIGT+Tim‐3 are shown. Data are the result of analyses from HD (n = 13), LG (n = 19), HG (n = 8) and CC (n = 19) groups. Data are shown as individual percentages of expression of total CD56dim NK cells and their mean. Comparisons between the groups were performed using analysis of variance (anova) with Dunnett’s multiple comparisons test. *P ≤ 0·05, **P ≤ 0·01, ***P ≤ 0·001.
Regarding the classical cytotoxic NK cell subset (CD56dim, the vast majority of which are CD16bright, Fig. 2), we observed a significant increase of PD‐1+CD56dim NK cells in cervical cancer and precursor lesions compared to HD (Fig. 2d). While TIGIT and Tim‐3 alone were not significantly changed between cancer patients and controls, we found that the patient PD‐1+ NK cells were more likely to also co‐express TIGIT or/and Tim‐3, and these double checkpoint‐positive NK cells were significantly increased in CC and precursor lesions. The percentages of NKG2D+PD‐1+, and NKG2D+DNAM‐1+TIGIT+PD‐1+Tim‐3+ CD56dim NK cells were also significantly increased compared to HD. There was also a significant increase of Tim‐3 in only LG, but not HG or CC, versus HD controls. We did not find significant differences between HD and patient populations for other receptor combinations, and for this reason other results are not shown here.
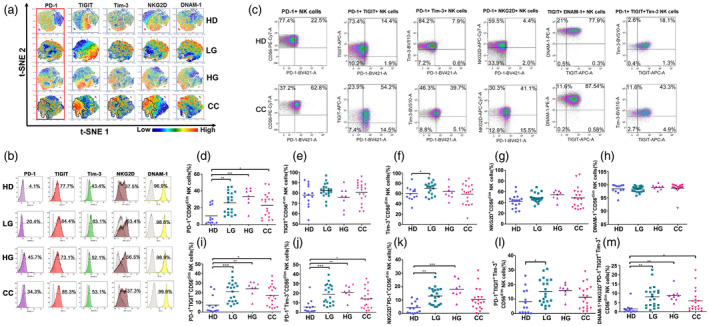
We next analyzed the CD56bright NK cell subset (Fig. 3), and we observed a significant increase of PD‐1+ CD56bright NK cells in cervical cancer and precursor lesions compared to HD. We also observed a significant increase in TIGIT+, PD‐1+Tim3+, PD‐1+TIGIT+ and PD‐1+TIGIT+Tim‐3+ cells in CC versus HD samples.
Fig. 3.
Expression and co‐expression of programmed cell death 1 (PD‐1), T cell immunoreceptor with immunoglobulin (Ig) and immunoreceptor tyrosine‐based inhibition motif (ITIM) domains (TIGIT), T cell immunoglobulin and mucin‐domain containing‐3 (Tim‐3), natural killer group 2 member D (NKG2D) and DNAX accessory molecule 1 (DNAM‐1) on peripheral blood CD56bright NK cells from healthy donors (HD group), low‐grade lesions (LG) group, high‐grade lesions (HG) group and cervical cancer patients (CC) group. (a) t‐Distributed stochastic neighbor‐embedding analysis (t‐SNE) density plots showing expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3. The PD‐1+ region is highlighted and cross‐over with similar regions within CC is outlined. (b) Representative histogram staining for the antibodies used for each population group. (c) Representative dot‐plot analysis for all single antibody combinations and the multiple antibody combinations that showed significant differences between HD and CC. Flow cytometry gating of the data was performed to analyze the single expression of the immune checkpoints: (d) PD‐1, (e) TIGIT, (f) Tim‐3, (g) NKG2D and (h) DNAM‐1. Co‐expression of (i) PD‐1+TIGIT, (j) PD‐1+Tim‐3, (k) PD‐1+NKG2D, (l) TIGIT+DNAM and (m) PD‐1+TIGT+Tim‐3 are shown. Data are the result of analysis from HD (n = 13), LG (n = 19), HG (n = 8) and CC (n = 19) groups. Data are shown as individual percentages of expression of total CD56bright NK cells and their mean. Comparisons between the groups were performed using analysis of variance (anova) with Dunnett’s multiple comparisons test. *P ≤ 0·05, **P ≤ 0·01, ***P ≤ 0·001.
Finally, we continued our checkpoint analysis on the six NK cell subpopulations subdivided based on the expression of CD56 and CD16: CD56brightCD16neg, CD56brightCD16dim, CD56dimCD16neg, CD56dimCD16dim, CD56dimCD16bright and CD56negCD16bright NK cells. We observed very similar expression trends in all the NK subsets, with significantly increased percentages of PD‐1 positive cells found in LG, HG and CC in each subpopulation. These results are shown in Supporting information, Figs S2–S7, as these results are very similar to the results shown for the CD56dim and CD56bright subsets above.
Although we did not find significant changes in the percentage of NK cell subpopulations that expressed TIGIT or Tim‐3 alone we observed that some NK cell subsets may co‐express the three immune checkpoint receptors, and that cells double‐ and triple‐positive for some of these receptors were significantly increased in the patient groups (Supporting information, Figs S2–S7). These cells may also co‐express DNAM‐1 and/or NKG2D and were also increased in the patients. We did not find significant changes in the density of the receptors (MFI) in any of the NK cell subpopulations.
Immune checkpoint profiling of CD56−CD3+ T cells
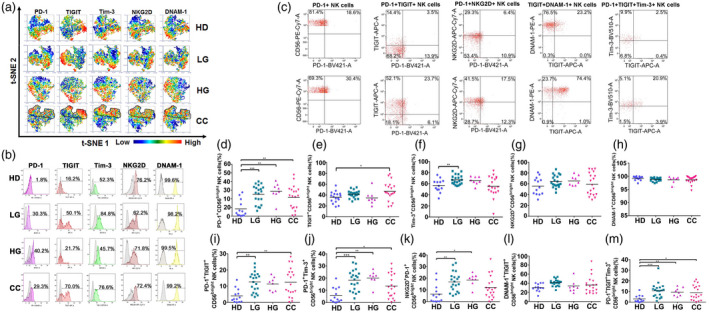
In order to visualize the expression patterns of PD‐1, TIGIT, Tim‐3, NKG2D and DNAM‐1 in T cells at the single‐cell level within the CD56−CD3+ T cells of the study groups, a high‐dimensional t‐SNE analysis was performed. Our results showed a different pattern of immune checkpoint expression between CC, HG, LG and HD groups (Fig. 4a).
Fig. 4.
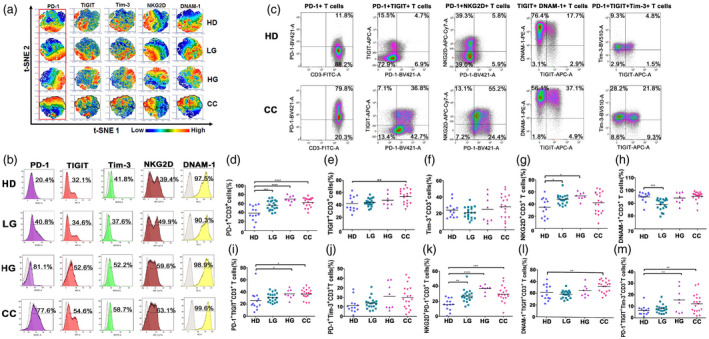
Expression and co‐expression of programmed cell death 1 (PD‐1), T cell immunoreceptor with immunoglobulin (Ig) and immunoreceptor tyrosine‐based inhibition motif (ITIM) domains (TIGIT), T cell immunoglobulin and mucin‐domain containing‐3 (Tim‐3), natural killer group 2 member D (NKG2D) and DNAX accessory molecule 1 (DNAM‐1), on peripheral blood T cells from healthy donors (HD group), low‐grade lesions (LG) group, high‐grade lesions (HG) group and cervical cancer patients (CC) group. (a) t‐Distributed stochastic neighbor‐embedding analysis (t‐SNE) density plots showing expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3. The PD‐1+ region is highlighted and cross‐over with similar regions within CC is outlined. (b) Representative histogram staining for the antibodies used for each population group. (c) Representative dot‐plot analysis for all single antibody combinations and the multiple antibody combinations that showed significant differences between HD and CC. Flow cytometry gating of the data was performed to analyze the single expression of the immune checkpoints: (d) PD‐1, (e) TIGIT, (f) Tim‐3, (g) NKG2D and (h) DNAM‐1. Co‐expression of (i) PD‐1+TIGIT, (j) PD‐1+Tim‐3, (k) PD‐1+NKG2D, (l) TIGIT+DNAM and (m) PD‐1+TIGT+Tim‐3 are shown. Data are shown as individual percentages of expression of total T cells and their mean. Comparisons between the groups were performed using analysis of variance (anova) with Dunnett’s multiple comparisons test. *P ≤ 0·05, **P ≤ 0·01, ***P ≤ 0·001.
Next, we evaluated single expression and co‐expression of the receptors using traditional histograms and dot plots (Fig. 4b,c). When we analyzed PD‐1 expression in these cells, we observed that PD‐1+ T cells were increased in all the groups compared to HD. The percentages of TIGIT+ T cells were also increased in the CC group. Tim‐3+ T cells were not significantly different between the groups; however, PD‐1+TIGIT+Tim‐3+ T cells were found to be significantly increased in patient groups. This may reflect that PD‐1+ T cells significantly increased in the group of patients can also co‐express TIGIT and/or Tim‐3.
Focusing on the activating receptors NKG2D and DNAM‐1, we found that the percentages of NKG2D+ T cells were very significantly increased in the LG (P < 0·01) and HG and CC (P < 0·001) groups. Similar to what was observed with the NK cells, DNAM‐1 was found to be expressed in approximately 95% of the T cells, thus expression of DNAM‐1 showed no significant change between HD and HG or CC; there was a small but significant decrease in LG patients. However, we found a significant increase of cells that were positive for both DNAM‐1 and TIGIT markers in CC.
These results indicated that, in cervical cancer patients and pre‐malignant lesions, PD‐1+ T cells were significantly increased. The percentages of PD‐1+TIGIT+ and PD‐1+TIGIT+Tim‐3+ T cells were also higher in the groups of patients when compared to HD. These inhibitory receptors were often accompanied by the co‐expression of activating receptors; NKG2D+PD‐1+ and DNAM‐1+TIGIT+ T cells were also significantly increased in the CC patients.
Differing PD‐1 expression on T cells
Additionally, we assessed whether PD‐1‐positive T cells expressed PD‐1 at different levels according to the PD‐1 MFI (Fig. 5a). To this end, we subdivided the region positive for PD‐1 into three subregions labeled as low (PD‐1low), intermediate (PD‐1int) or high (PD‐1high). We found that the percentages of PD‐1low T cells were not significantly increased between controls and patients (data not shown), while PD‐1int and PD‐1high T cells significantly increased in patients with cancer and precursor lesions (Fig. 5b,c).
Fig. 5.
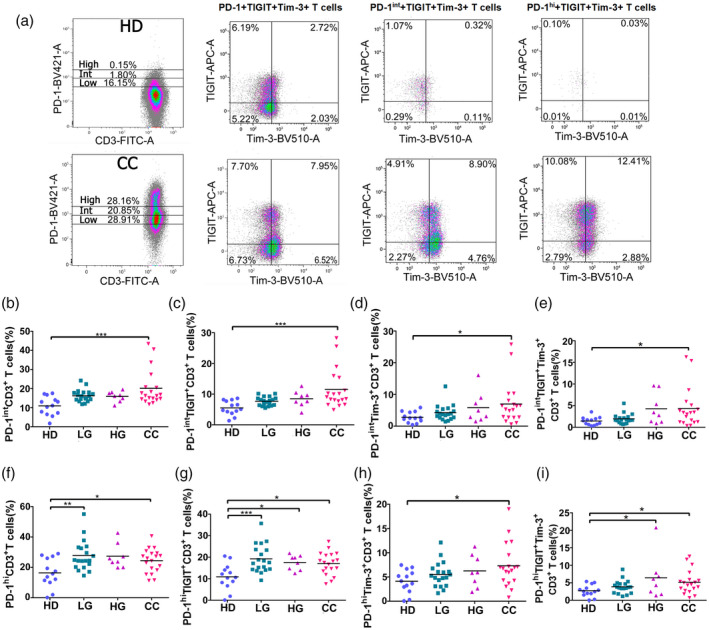
Programmed cell death 1 (PD‐1) intermediate (PD‐1int) and PD‐1 high (PD‐1high) expression on peripheral blood T cells from healthy donors (HD) group, low‐grade lesions (LG) group, high‐grade lesions (HG) group and cervical cancer patients (CC) group. PD‐1 expression was measured by flow cytometry and staining was characterized as high or intermediate depending on mean fluorescence intensity (MFI) expression. (a) Examples of high and intermediate PD‐1 expression in HD and CC; the first plot in each row shows PD‐1 divisions and the subsequent plots show T cell immunoreceptor with immunoglobulin (Ig) and immunoreceptor tyrosine‐based inhibition motif (ITIM) domains (TIGIT) and T cell immunoglobulin and mucin‐domain containing‐3 (Tim‐3) staining in PD‐1low, PD‐1int and PD‐1high subpopulations. Data are shown as individual percentages of total T cells. (b–e) In PD‐1int and (f–i) PD‐1high CD3+ T cells, the expression of TIGIT and/or Tim‐3 was evaluated for HD (n = 13), LG (n = 19), HG (n = 8) and CC (n = 19) groups. Frequency data are shown as individual percentages of expression and their mean. Comparisons between the groups were performed using analysis of variance (anova) with Dunnett’s multiple comparisons test. *P ≤ 0·05, **P ≤ 0·01, ***P ≤ 0·001.
We also investigated whether PD‐1int and PD‐1high T cells co‐expressed TIGIT and Tim‐3. We saw a significant increase in the percentages of the PD‐1intTIGIT+ and PD‐1highTIGIT+ T cells in the CC group in comparison with the HD group. Interestingly, we also found a significantly higher percentage of PD‐1int and PD‐1high CD3+ cells co‐expressing Tim‐3, which was not seen when evaluating total PD‐1 expression (Fig. 4). The percentages of triple‐positive (PD‐1‐TIGIT‐Tim‐3) PD‐1int and PD‐1high CD3+ cells were also increased in cancer patients, which suggest an exhausted T cell phenotype. PD‐1high TIGIT+ percentages were increased in LG and CC and PD‐1highTIGIT+Tim‐3+ cell populations were also significantly increased in both HG and CC.
Checkpoint expression in CD4 and CD8 T cells
We further subdivided the T cells into CD4 and CD8 positive populations and examined PD‐1 expression. We found that CD4+PD‐1+ and CD8+PD1+ cell percentages were significantly increased (up to ×2) in HG and CC with respect to HD (Fig. 6a,b). This increase in patients versus healthy donors was similar in both CD4 and CD8 T cells. The mean fluorescence intensities of the PD‐1 signals in CD4 and CD8 cells were similar (Fig. 6c,d). No significant differences were found in PD‐1 expression between CD4 and CD8 cells; in contrast TIGIT, and to a lesser extent Tim3, and PD1+TIGIT+Tim3 expression were found at significantly higher percentages in CD8 cells (Fig. 6e,f).
Fig. 6.
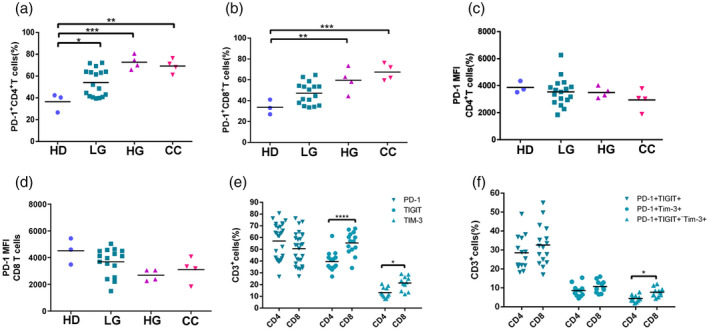
Checkpoint expression in CD4 and CD8 T cells. (a) Programmed cell death 1 (PD‐1) expression in CD4+ T cells and (b) in CD8+ T cells in healthy donors (HD) (n = 3), low‐grade (LG, n = 17), high‐grade (HG, n = 4) and cervical cancer (CC, n = 4) patients. (c) Mean fluorescence intensity (MFI) for PD‐1 expression in CD4+ T cells. (d) MFI for PD‐1 expression in CD8+ T cells. (e) Percentages of CD4 and CD8 T cells positive for PD‐1, T cell immunoreceptor with immunoglobulin (Ig) and immunoreceptor tyrosine‐based inhibition motif (ITIM) domains (TIGIT) and T cell immunoglobulin and mucin‐domain containing‐3 (Tim‐3). (f) Percentages of CD4 and CD8 T cells positive for PD‐1+TIGIT+, PD‐1+Tim3+ and PD‐1+TIGIT+Tim3+. P ≤ 0·05, **P ≤ 0·01, ***P ≤ 0·001.
Soluble PD‐L1 is increased in cervical cancer
We performed an ELISA on serum samples from CC (n = 21), HG (n = 9), LG (n = 24) and HD (n = 24) donors (Fig. 7) to evaluate the soluble levels of PD‐L1 (sPD‐L1). We observed a significant increase between HD and CC and CC and LG in the concentration of sPD‐L1 (Fig. 7a). Of note was that the majority (19 of 21) of the cervical cancer patients had higher sPD‐L1 levels than the mean HD levels (11·02 pg/ml). We then wanted to determine whether sPD‐L1 levels were different between cervical cancer stages; however. we did not find significant changes in the levels of this molecule when we compared stages I–IB versus stages II–III (Fig. 7b). We next performed a correlation test to assess different associations between sPD‐L1 and the percentages of different cell populations. A table of the correlations between sPD‐L1 and all the different cell and patient populations analyzed is shown in Supporting information, Table S1. When we correlated sPD‐L1 with cell populations in different disease stages we observed a significant positive correlation between sPD‐L1 levels and PD‐1+ T cells in LG (Fig. 7c). A significant negative correlation was observed between sPD‐L1 in CC stage IB and the percentage of CD56negCD16bright NK cells (Fig. 7d).
Fig. 7.
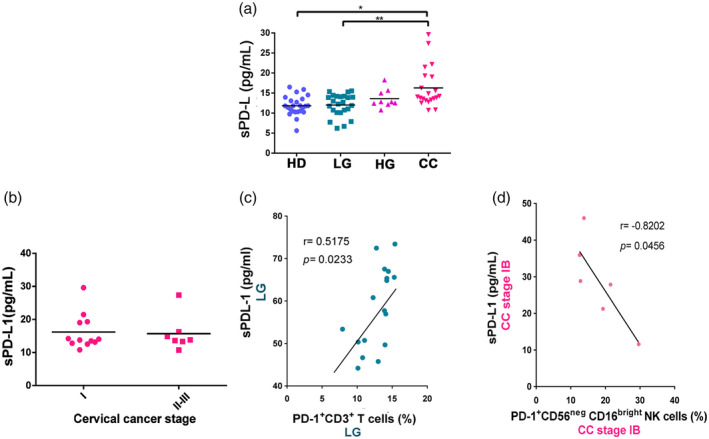
Soluble programmed cell death ligand 1 (sPD‐L1) in serum from healthy donors (HD) group, low‐grade lesions (LG) group, high‐grade lesions (HG) group and cervical cancer patients (CC) group. (a) The concentration of soluble PD‐L1 in the serum of HD (n = 24), LG (n = 24), HG (n = 9) and CC (n = 21) was measured by enzyme‐linked immunosorbent assay (ELISA). Data are shown as pg/ml of sPD‐L1 and their mean. (b) sPD‐L1 in the serum of cervical cancer patients classified according to clinical stage. (c) A correlation analysis was performed between sPD‐L1 and the percentages of PD‐1+CD3+T cells in the LG group. (d) A correlation analysis was performed between sPD‐L1 in cervical cancer patients stage IB and their respective percentages of PD‐1+CD56negCD16bright NK cells. Comparison between the groups were performed using analysis of variance (anova) with Dunnett’s multiple comparisons test. Pearson’s correlation analysis was performed to verify the linear associations between sPD‐L1 and the percentages of PD‐1+NK cells and PD‐1+CD3+ T cells. The correlation coefficient, r and P‐values are shown in the figures. *P ≤ 0·05, **P ≤ 0·01, ***P ≤ 0·001.
A convergence table analyzing sPD‐L1 levels above or below the median of the HD group and select variables is shown in Table 1. In this table we observed an inverse association between high sPD‐L1 and total T cell percentages, and an association between high sPD‐L1 and presence of CC. Additionally, an association between increased CD56dim NK cells and increased sPD‐L1 was also observed; a notable, but not significant (P = 0·064) association between increased PD1+TIGIT+ T cells and increased sPD‐L1 was similarly observed.
Table 1.
Patient variables stratified according to the sPD‐L1 level
| Variable | sPD‐L1 ≤ 11·8 pg/ml | sPD‐L1 > 11·8 pg/ml | P‐value | |
|---|---|---|---|---|
| Disease extent | HD | 9 | 8 | 0·0049 |
| CC | 2 | 19 | ||
| % T cells | ≤ 64·8 | 1 | 12 | 0·0238 |
| > 64·8 | 10 | 11 | ||
| % Total CD56dim NK cells | ≤ 15·5 | 9 | 10 | 0·0045 |
| > 15·5 | 2 | 23 | ||
| % Total CD56bright NK cells | ≤ 0·77 | 6 | 17 | 0·4344 |
| > 0·77 | 5 | 6 | ||
| % PD‐1+CD3+ T cells | ≤ 63 | 9 | 13 | 0·2525 |
| > 63 | 2 | 10 | ||
| % PD‐1+TIGIT+CD3+ T cells | ≤ 21·9 | 9 | 10 | 0·064 |
| >21·9 | 2 | 13 | ||
| % PD‐1hi+CD3+ T cells | ≤ 20·3 | 4 | 7 | > 0·9999 |
| > 20·3 | 7 | 16 | ||
| % Total PD‐1+ CD56dim CD16bright NK cells | ≤ 13·4 | 7 | 10 | 0·4646 |
| > 13·4 | 4 | 13 | ||
| % Total PD‐1+CD56neg CD16bright NK cells | ≤ 16 | 7 | 9 | 0·2743 |
| > 16 | 4 | 14 |
TIGIT = T cell immunoreceptor with immunoglobulin (Ig) and immunoreceptor tyrosine‐based inhibition motif (ITIM) domains; Tim3 = T cell immunoglobulin and mucin‐domain containing‐3; PD‐1 = programmed cell death 1; sPL‐L1 = soluble programmed cell death ligand 1; NK = natural killer; HD = healthy donor; CC = cervical cancer.
Bold indicates significant resluts with P ≤ 0·05.
Furthermore, we performed a linear regression analysis to define which other variables (in addition to sPD‐L1) would best predict the difference between cancer and HD. Using the strongest two variables, we performed a logistic regression test and found that in our model sPD‐L1 levels had an odds ratio of 3·09 (P = 0·015) and PD‐1+ TIGIT+ T cell percentages had an odds ratio of 1·29 (P = 0·049) to together predict cancer versus healthy donor assignments.
Discussion
Immunological checkpoint‐based anti‐tumor therapy has attracted extensive attention. Reversing exhaustion by blocking PD‐1 is a promising target for cervical cancer treatment. Although the blockade of PD‐1 and its ligand may be of use in the treatment of cervical cancer there also exist other checkpoint molecules, such as TIGIT and Tim‐3, that play an important role in T and NK cell exhaustion [44, 45]. However, only a few studies have explored the expression of these immune checkpoints on T cells from cervical cancer patients [37, 38], and the expression of these three receptors on different NK cell subpopulations from cancer patients is still unknown.
The majority of the published studies evaluated the expression of immune checkpoints in the classical cytotoxic CD56dim NK cell population. In our study, we found that PD‐1 was expressed on both CD56dim and CD56bright NK cells. The expression of peripheral blood PD‐1+ NK cells has already been reported in healthy donors and patients with ovarian carcinoma [46]. In that study, PD‐1 expression was found to be restricted to the mature NK cell population (NKG2AnegKIR+CD57+CD56dim NK cells), which is characterized by high CD16, perforin and granzyme expression [47]. However, and similar to our results, in a more recent study, Liu et al. observed PD‐1 to be expressed in both CD56dim and CD56bright NK cells from the blood or tumor infiltrates in patients with digestive cancers, including esophageal, liver, colorectal, gastric and biliary cancer [48].
The role of the expression of different immune checkpoint molecules on the same NK cell is still not well understood, and the majority of the studies evaluate the expression of PD‐1, TIGIT or Tim‐3, but not all together. We found triple‐positive CD56dim NK cells to be significantly increased in cervical cancer patients.
In the case of TIGIT, this receptor is constitutively expressed by NK cells and is up‐regulated in tumor‐infiltrated NK cells [45]. We found significant differences between HD cells and percentages of TIGIT+ cells in the NK CD56bright CC group and NK CD56dim LG group. In experiments performed by Zhang et al. on NK and T cells from tumor‐bearing mice and patients with colon cancer [45], TIGIT expression was observed to be higher on NK cells and was associated with NK cell exhaustion. In line with that report, we found that TIGIT was found in a higher percentage of NK CD56dim cells versus T cells and NK CD56bright cells. In the CC group, an average of 81% of the NKdim cells were positive for TIGIT versus 54% of T cells and 43% of NK CD56bright cells.
Tim‐3 is another immune checkpoint constitutively expressed by NK cells. It is also up‐regulated on these cells in cancer and chronic infections [39, 49, 50]. Here, we did not find changes in the expression of Tim‐3 on peripheral blood NK cells in patients with cancer (although it was increased in both CD56dim and CD56bright NK cells in the group with low‐grade pre‐malignant lesions). The co‐expression of Tim‐3 and TIGIT is also reported and is associated with NK cell dysfunction. Exhausted NK cells from patients with bladder cancer showed up‐regulation of Tim‐3 and TIGIT in both the periphery and tumor [51]. In our work, we did not observe differences between controls and patients in the percentages of peripheral Tim‐3+TIGIT+ NK cells; however, we observed that some PD‐1+TIGIT+Tim‐3+ NK cell subsets were increased in pre‐malignant lesions and cancer.
Regarding the functional activity of the NK cells, the role of PD‐1 expression on cytotoxicity and IFN‐γ production has been investigated in peripheral blood PD1+CD56bright NK cells and PD1+CD56dim NK cells from asymptomatic pediatric thoracic transplant patients with lymphoproliferative disorders [52]. These PD‐1+ cells showed an impaired functionality. This group also observed that those cells down‐modulate the activating receptors NKp46 and NKG2D while up‐regulating PD‐1. In our work, we did not find significant changes on the expression of activating receptors (NKG2D or DNAM‐1) in NK cells. We only saw higher percentages of PD‐1+NKG2D+CD56brightCD16dim/neg in pre‐malignant lesions.
It is also important to note that great variety was observed in the HD controls regarding the PD‐1 population in NK cells; controls recovering from transient viral infections may explain this variation. Indeed, it has been noted that chronic asymptomatic HCMV infections may be associated with persistently high PD‐1 levels in normal controls [46].
T cells are the principal target of immunotherapy. Here we found that in pre‐malignant lesions and cervical cancer patients the percentages of CD3+ T cells were decreased. T cell percentages in individual cervical cancer patients were negatively correlated with CD56dimCD16bright NK cells in the same sample.
When comparing the percentage of immune checkpoints on T cells from cervical cancer patients and healthy donors, we found a significant increase in the percentage of PD‐1+ and TIGIT+ T cells in the cancer group. We also observed a greater proportion of T cells in cervical cancer patients with a double (PD‐1+TIGIT+ and PD‐1+Tim‐3+) or triple‐positive (PD‐1+TIGIT+Tim‐3+) co‐expression of immune checkpoint molecules. It must be noted that analysis of these cells in the tumor environment, not only in peripheral blood, is absolutely essential. A recent study on multiple checkpoint molecules in T cells from blood and tumors from different samples found PD‐1, TIGIT and Tim‐3, among other receptors, to be markedly increased in tumor‐infiltrating cells compared to peripheral blood [11]. This may explain why in peripheral blood T cells from cervical cancer patients we did not see changes in Tim‐3+ T cells.
In our T cell samples, we observed a wide range of PD‐1 expression levels on individual cells. Other groups have also noted the difference between low or basal PD‐1 expression and higher levels, indicating more complete exhaustion [53, 54]. Based on those works, we began by subdividing our PD‐1‐positive T cells into six different groups, which we eventually condensed into three broad gates: PD‐1low, PD‐1int and PD‐1high. Our results show that the difference between patients and controls is most clear when comparing PD‐1 intermediate or PD‐1 high‐expressing cells. No significant changes were observed on the expression of PD‐1low cells when we compared between the groups.
Our data demonstrate, for the first time to the best of our knowledge, that in cervical cancer patients the percentage of PD‐1int and PD‐1high CD3+ cells is significantly higher in patients compared with healthy donors. Interestingly, we also saw a greater percentage of CD3+ cells with intermediate and high expression of PD‐1 co‐expressing TIGIT and Tim‐3. Together, these results suggest that in peripheral blood from cervical cancer patients it is possible to find T cell populations expressing PD‐1 at different levels and co‐expressing other immune checkpoint molecules (TIGIT and Tim‐3). These types of cells show a pattern of immune checkpoint receptor expression previously observed in tumor‐infiltrating CD8 T cells from patients with non‐small‐cell lung cancer, where distinct expression levels of PD‐1 show a different spectrum of exhaustion [43]. Here it is important to note that when we gated on PD‐1+ cells, then analyzed TIGIT versus Tim‐3 expression, the PD‐1high cells were much more likely to be also positive for TIGIT or Tim‐3 and much less likely to have cells in the double‐negative quadrant (PD‐1 cells that were negative for both TIGIT and Tim‐3).
This type of co‐expression has been reported in other models. For example, different levels of PD‐1+ have been reported on CD8 T cell subsets expressing LAG‐3 and PD‐1 after chronic stimulation within tolerizing environments [55]. In that work, the authors found that CD8 T cells could differentiate into either LAG‐3+PD‐1int, LAG‐3negPD‐1int or PD‐1high. These subsets were found to show distinct phenotypes and functional properties as well; CD8 T cells expressing the highest levels of PD‐1 (PD‐1high) were not capable of producing IFN‐γ while, in contrast, cells expressing an intermediate level of PD‐1 could produce IFN‐γ.
It is important to note that the DNAM‐1 receptor plays a role in T cell and NK cell‐mediated cytotoxicity against tumors via its interaction with CD155 and CD112 [56]. For CD8 T cells, the TIGIT/DNAM‐1 axis has emerged as an inhibitory/stimulatory receptor pair of importance for CD8 T cell function, similar to CTLA‐4 and CD28 [57]. When comparing healthy donors and cervical cancer patients, we did not see significant changes in the expression of DNAM‐1 on NK cells and T cells (DNAM‐1 expression was uniformly high, with a mean above 95% of T cells and 98% of NK cells); however, a decreased percentage (down to a mean of 88%) of T cells expressing this receptor was observed in the LG group. It has been reported that the expression of DNAM‐1 is down‐regulated during chronic HIV infection [58]. Minnie et al. found a notable increase in DNAM‐1‐negative CD8+ T cells that were also PD‐1‐ and TIGIT‐positive in mice with relapsed myeloma [59]. However, we did not observe significant quantities of DNAM‐1‐negative T cells in our different groups. Thus, we conclude that in PBMCs from cervical cancer and precursor lesions cells with checkpoint markers cannot be readily defined by the lack of DNAM‐1 expression. In contrast, we found a significant increase in the percentage of DNAM‐1+TIGIT+CD3+ cells (and a decrease in the DNAM‐1+TIGITnegCD3+ population, data not shown) in our cervical cancer group. This may indicate that emerging TIGIT is playing a dominant role in some of these DNAM‐1+ T cells. Perhaps in the tumor environment DNAM‐1 loss will be more marked.
The increase in soluble PD‐L1 in cervical cancer was notable. Our data showed that 19 of 21 cervical cancer patients had sPD‐L1 levels above the mean of the controls. sPD‐L1 levels have been increased in different types of cancer and are associated with disease severity and prognosis [13, 60, 61]. It is also possible that the presence of sPD‐L1 may indicate patients who would not respond well to some forms of PD‐L1 blockade antibody therapy, as it has been reported that secreted PD‐L1 variants can mediate resistance to PD‐L1 blockade by acting as a decoy [10]. Continuing with the idea that sPD‐L1 may serve as a marker for advancement of the tumor development, we classified our samples with respect to tumor subtype and grade. We observed that the frequency of PD‐1+CD56negCD16bright NK cells was negatively correlated with sPD‐L1 levels in cervical cancer stage IB. The PD1+ T cell percentage was positively correlated with sPD‐L1 only in LG samples. As this result was not observed in HD, this may indicate the beginning of a perturbed immune response in patients with low‐grade infections. It is possible that, with advancing disease growth, PD‐1‐positive T cells become less common in the blood. Indeed, lower T cell percentages overall were observed in the blood of CC patients, and a negative correlation was also observed between the total soluble PD‐L1 levels and the frequency of T cells in cervical cancer. We speculate that T cells may be trafficking to the tumor in these cases.
In conclusion, this work showed a pattern of expression of important immune checkpoints on different NK cell subpopulations and T cells. In general, our data suggest that the pattern of immune checkpoint expression was very similar between NK cell subsets. The frequencies of PD‐1+ NK cells (different subsets) and PD‐1+ T cells were increased in pre‐malignant lesions and cervical cancer. These PD‐1+ cells often co‐express TIGIT and/or Tim‐3. Soluble PD‐L1 levels were also increased in cervical cancer patients and were not correlated with cervical cancer stage.
We propose as a model that cytotoxic cells, either CD8 T cells, CD56dim NK cells or CD56bright NK cells, begin to express different checkpoint molecules during the response to persistent HPV infection and cervical cancer. In the course of the progression of the disease, the expression of these molecules will accumulate and identify the most afflicted or exhausted cells. Additionally, soluble PD‐L1, which was strongly increased in a subset of cervical cancer patients and correlated with NK and T cell populations, may also identify patients with tumors producing checkpoint ligands. Therefore, identification of the patients with such characteristics may highlight those uniquely able to respond to combined antibody therapies.
Disclosures
The authors state no commercial or financial conflicts of interest.
Supporting information
Fig. S1. Gating strategy for the flow cytometric analysis of NK cells in PBMCs from patients and controls. (a) representative workflow showing identification of NK subpopulations and representative analysis for six NK subpopulations in HD, LG, HG and CC samples. Inset examples of the six different subpopulations in different groups shows percentages based on the Total NK Cells region shown on the plot. (b‐g) Correlation analysis between the percentages of CD56dim NK cell and other NK cell subpopulations or NK cell subpopulations and T cells.
Fig. S2. Expression and co‐expression of PD‐1, TIGIT, Tim‐3, NKG2D and DNAM‐1 on peripheral blood CD56brightCD16neg NK cells from healthy donors (HD group), low grade lesions (LG group), high grade lesions (HG group) and cervical cancer patients (CC group). (a) Gating strategy for the t‐SNE analysis within CD56brightCD16neg cells. t‐SNE analysis was used to visualize expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3 in HD (n = 13), LG (n = 19), HG (n = 8) and CC (n = 19) groups. (b) t‐SNE density plots showing expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3. Flow cytometry gating of the data was performed to analyze the single expression of the immune checkpoints: (c) PD‐1. Co‐expression of (d) PD‐1+Tim‐3, (e) NKG2D+PD‐1, (f) DNAM‐1+TIGIT+PD‐1+, (g) PD‐1+TIGIT+Tim‐3 and (h) DNAM‐1+TIGIT+PD‐1+Tim‐3 are shown. (i) Heat map of single receptor expression and (j) Heat map of the co‐expression of the receptors are show for all groups. Data are shown as individual percentages of expression and their mean. Comparisons between the groups were performed using ANOVA with Dunnett’s multiple comparisons test. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Fig. S3. Expression and co‐expression of PD‐1, TIGIT, Tim‐3, NKG2D and DNAM‐1 on peripheral blood CD56brightCD16dim NK cells from healthy donors (HD group), low grade lesions (LG group), high grade lesions (HG group) and cervical cancer patients (CC group). (A) Gating strategy for the t‐SNE analysis within CD56brightCD16dim cells. t‐SNE analysis was used to visualize expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3 in HD (n = 13), LG (n = 19), HG (n = 8) and CC (n = 19) groups. (B) t‐SNE density plots showing expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3. Flow cytometry gating of the data was performed to analyze the single expression of the immune checkpoints: (C) PD‐1. Co‐expression of (D) PD‐1+TIGIT, (E) PD‐1+Tim‐3, (F) NKG2D+PD‐1, (G) DNAM‐1+TIGIT+PD‐1+, and (H) PD‐1+TIGIT+Tim‐3 are shown. (I) Heat map of single receptor expression and (J) Heat map of the co‐expression of the receptors are show for all groups. Data are shown as individual percentages of expression and their mean. Comparisons between the groups were performed using ANOVA with Dunnett’s multiple comparisons test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Fig. S4. Expression and co‐expression of PD‐1, TIGIT, Tim‐3, NKG2D and DNAM‐1 on peripheral blood CD56dimCD16neg NK cells from healthy donors (HD group), low grade lesions (LG group), high grade lesions (HG group) and cervical cancer patients (CC group). (a) Gating strategy for the t‐SNE analysis within CD56dimCD16neg cells. t‐SNE analysis was used to visualize expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3 in HD (n = 13), LG (n = 19), HG (n = 8) and CC (n = 19) groups. (b) t‐SNE density plots showing expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3. Flow cytometry gating of the data was performed to analyze the single expression of the immune checkpoints: (c) PD‐1. Co‐expression of (d) PD‐1+TIGIT, (e) PD‐1+Tim‐3, (f) NKG2D+PD‐1, (g) DNAM‐1+TIGIT+PD‐1+, (h) PD‐1+TIGIT+Tim‐3, and (i) DNAM‐1+TIGIT+PD‐1+Tim‐3+ are shown. (j) Heat map of single receptor expression and (K) Heat map of the co‐expression of the receptors are show for all groups. Data are shown as individual percentages of expression and their mean. Comparisons between the groups were performed using ANOVA with Dunnett’s multiple comparisons test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Fig. S5. Expression and co‐expression of PD‐1, TIGIT, Tim‐3, NKG2D and DNAM‐1 on peripheral blood CD56dimCD16dim NK cells from healthy donors (HD group), low grade lesions (LG group), high grade lesions (HG group) and cervical cancer patients (CC group). (a) Gating strategy for the t‐SNE analysis within CD56dimCD16dim cells. t‐SNE analysis was used to visualize expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3 in HD (n = 13), LG (n = 19), HG (n = 8) and CC (n = 19) groups. (b) t‐SNE density plots showing expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3. Flow cytometry gating of the data was performed to analyze the single expression of the immune checkpoints: (c) PD‐1. Co‐expression of (d) PD‐1+TIGIT, (e) PD‐1+Tim‐3, (f) NKG2D+PD‐1, (g) DNAM‐1+TIGIT+PD‐1+, and (h) PD‐1+TIGIT+Tim‐3, are shown. (i) Heat map of single receptor expression and (j) Heat map of the co‐expression of the receptors are show for all groups. Data are shown as individual percentages of expression and their mean. Comparisons between the groups were performed using ANOVA with Dunnett’s multiple comparisons test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Fig. S6. Expression and co‐expression of PD‐1, TIGIT, Tim‐3, NKG2D and DNAM‐1 on peripheral blood CD56dimCD16bright NK cells from healthy donors (HD group), low grade lesions (LG group), high grade lesions (HG group) and cervical cancer patients (CC group). (a) Gating strategy for the t‐SNE analysis within CD56dimCD16bright cells. t‐SNE analysis was used to visualize expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3 in HD (n = 13), LG (n = 19), HG (n = 8) and CC (n = 19) groups. (b) t‐SNE density plots showing expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3. Flow cytometry gating of the data was performed to analyze the single expression of the immune checkpoints: (c) PD‐1. Co‐expression of (d) PD‐1+TIGIT, (e) PD‐1+Tim‐3, (f) PD‐1+TIGIT+Tim‐3, (g) NKG2D+PD‐1, (h) DNAM‐1+TIGIT+PD‐1+, (i) DNAM‐1+NKG2D+TIGIT+PD‐1+Tim‐3+, and (j) DNAM‐1+TIGIT+PD‐1+Tim‐3+ are shown. (K) Heat map of single receptor expression and (l)Heat map of the co‐expression of the receptors are show for all groups. Data are shown as individual percentages of expression and their mean. Comparisons between the groups were performed using ANOVA with Dunnett’s multiple comparisons test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Fig. S7. Expression and co‐expression of PD‐1, TIGIT, Tim‐3, NKG2D and DNAM‐1 on peripheral blood CD56negCD16bright NK cells from healthy donors (HD group), low grade lesions (LG group), high grade lesions (HG group) and cervical cancer patients (CC group). (a) Gating strategy for the t‐SNE analysis within CD56negCD16bright cells. t‐SNE analysis was used to visualize expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3 in HD (n = 13), LG (n = 19), HG (n = 8) and CC (n = 19) groups. (b) t‐SNE density plots showing expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3. Flow cytometry gating of the data was performed to analyze the single expression of the immune checkpoints: (c) PD‐1. Co‐expression of (d) PD‐1+TIGIT, (e) PD‐1+Tim‐3, (f) NKG2D+PD‐1, (g) DNAM‐1+TIGIT+PD‐1+, and (h) DNAM‐1+TIGIT+PD‐1+Tim‐3+ are shown. (i) Heat map of single receptor expression and (j) Heat map of the co‐expression of the receptors are show for all groups. Data are shown as individual percentages of expression and their mean. Comparisons between the groups were performed using ANOVA with Dunnett’s multiple comparisons test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Table S1. Correlations Between sPD‐L1 and Cell Populations.
Acknowledgements
This work was supported by a grant from the Consejo Nacional de Ciencia y Tecnología (CONACYT) through the Convocatoria del Fondo Sectorial de Investigación en Salud y Seguridad Social SS/IMSS/ISSSTE‐CONACYT (2017‐290195).
Contributor Information
S. del Toro‐Arreola, Email: susana@cucs.udg.mx.
J. Haramati, Email: jharamati@gmail.com.
Data Availability Statement
Data are available upon request to the corresponding author.
References
- 1. Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer 2011; 11:805–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wherry EJ. T cell exhaustion. Nat Immunol 2011; 12:492–9. [DOI] [PubMed] [Google Scholar]
- 3. Teng MW, Galon J, Fridman WH, Smyth MJ. From mice to humans: developments in cancer immunoediting. J Clin Invest 2015; 125:3338–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. De Sousa LA, Leitner J, Grabmeier‐Pfistershammer K, Steinberger P. Not all immune checkpoints are created equal. Front Immunol 2018; 9:1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marin‐Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol 2018; 11:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bi J, Tian Z. NK cell exhaustion. Front Immunol 2017; 8:760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zarour HM. Reversing T‐cell dysfunction and exhaustion in cancer. Clin Cancer Res 2016; 22:1856–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marhelava K, Pilch Z, Bajor M, Graczyk‐Jarzynka A, Zagozdzon R. Targeting negative and positive immune checkpoints with monoclonal antibodies in therapy of cancer. Cancers 2019; 11:1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ramos‐Casals M, Brahmer JR, Callahan MK et al. Immune‐related adverse events of checkpoint inhibitors. Nat Rev Dis Primers 2020; 6:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med 2018; 50:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maleki Vareki S, Garrigós C, Duran I. Biomarkers of response to PD‐1/PD‐L1 inhibition. Crit Rev Oncol Hematol 2017; 116:116–24. [DOI] [PubMed] [Google Scholar]
- 12. Aguiar PN Jr, Santoro IL, Tadokoro H et al. The role of PD‐L1 expression as a predictive biomarker in advanced non‐small‐cell lung cancer: a network meta‐analysis. Immunotherapy 2016; 8:479–88. [DOI] [PubMed] [Google Scholar]
- 13. Asanuma K, Nakamura T, Hayashi A et al. Soluble programmed death‐ligand 1 rather than PD‐L1 on tumor cells effectively predicts metastasis and prognosis in soft tissue sarcomas. Sci Rep 2020; 10:9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takahashi N, Iwasa S, Sasaki Y et al. Serum levels of soluble programmed cell death ligand 1 as a prognostic factor on the first‐line treatment of metastatic or recurrent gastric cancer. J Cancer Res Clin Oncol 2016; 142:1727–38. [DOI] [PubMed] [Google Scholar]
- 15. Park H, Bang J‐H, Nam A‐R et al. Prognostic implications of soluble programmed death‐ligand 1 and its dynamics during chemotherapy in unresectable pancreatic cancer. Sci Rep 2019; 9:11131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gong B, Kiyotani K, Sakata S et al. Secreted PD‐L1 variants mediate resistance to PD‐L1 blockade therapy in non‐small cell lung cancer. J Exp Med 2019; 216:982–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Holl EK, Frazier VN, Landa K, Beasley GM, Hwang ES, Nair SK. Examining peripheral and tumor cellular immunome in patients with cancer. Front Immunol 2019; 10:1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun H, Sun C. The rise of NK cell checkpoints as promising therapeutic targets in cancer immunotherapy. Front Immunol 2019; 10:2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sasagawa T, Takagi H, Makinoda S. Immune responses against human papillomavirus (HPV) infection and evasion of host defense in cervical cancer. J Infect Chemother 2012; 18:807–15. [DOI] [PubMed] [Google Scholar]
- 20. Renoux VM, Bisig B, Langers I et al. Human papillomavirus entry into NK cells requires CD16 expression and triggers cytotoxic activity and cytokine secretion. Eur J Immunol 2011; 41:3240–52. [DOI] [PubMed] [Google Scholar]
- 21. Tindle RW. Immune evasion in human papillomavirus‐associated cervical cancer. Nat Rev Cancer 2002; 2:59–65. [DOI] [PubMed] [Google Scholar]
- 22. Céspedes MA, Rodríguez JA, Medina M, Bravo M, Cómbita AL. Analysis of NK cells in peripheral blood and tumor infiltrating lymphocytes in cervical cancer patients. Rev Colomb Cancerol 2012; 16:16–26. [Google Scholar]
- 23. Zhang J, Jin S, Li X et al. Human papillomavirus type 16 disables the increased natural killer cells in early lesions of the cervix. J Immunol Res 2019; 2019:9182979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Uppendahl LD, Dahl CM, Miller JS, Felices M, Geller MA. Natural killer cell‐based immunotherapy in gynecologic malignancy. A review. Front Immunol 2018; 8:1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee SJ, Cho YS, Cho MC et al. Both E6 and E7 oncoproteins of human papillomavirus 16 inhibit IL‐18‐induced IFN‐gamma production in human peripheral blood mononuclear and NK cells. J Immunol 2001; 167:497–504. [DOI] [PubMed] [Google Scholar]
- 26. Garcia‐Iglesias T, Del Toro‐Arreola A, Albarran‐Somoza B et al. Low NKp30, NKp46 and NKG2D expression and reduced cytotoxic activity on NK cells in cervical cancer and precursor lesions. BMC Cancer 2009; 9:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Del Zotto G, Antonini F, Pesce S, Moretta F, Moretta L, Marcenaro E. Comprehensive phenotyping of human PB NK cells by flow cytometry. Cytometry Part A 2020; 97:891–9. [DOI] [PubMed] [Google Scholar]
- 28. Poli A, Michel T, Thérésine M, Andrès E, Hentges F, Zimmer J. CD56bright natural killer (NK) cells: an important NK cell subset. Immunology 2009; 126:458–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Béziat V, Duffy D, Quoc SN et al. CD56brightCD16+ NK cells: a functional intermediate stage of NK cell differentiation. J Immunol 2011; 186:6753–61. [DOI] [PubMed] [Google Scholar]
- 30. Amand M, Iserentant G, Poli A et al. Human CD56dimCD16dim cells as an individualized natural killer cell subset. Front Immunol 2017; 8:699. 10.3389/fimmu.2017.00699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Romee R, Foley B, Lenvik T et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease‐17 (ADAM17). Blood 2013; 121:3599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Penack O, Gentilini C, Fischer L et al. CD56dimCD16neg cells are responsible for natural cytotoxicity against tumor targets. Leukemia 2005; 19:835–40. [DOI] [PubMed] [Google Scholar]
- 33. Gonzalez VD, Falconer K, Björkström NK et al. Expansion of functionally skewed CD56‐negative NK cells in chronic hepatitis C virus infection: correlation with outcome of pegylated IFN‐α and ribavirin treatment. J Immunol 2009; 183:6612–8. [DOI] [PubMed] [Google Scholar]
- 34. Mavilio D, Lombardo G, Benjamin J et al. Characterization of CD56–/CD16+ natural killer (NK) cells: a highly dysfunctional NK subset expanded in HIV‐infected viremic individuals. Proc Natl Acad Sci USA 2005; 102:2886–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Milush JM, López‐Vergès S, York VA et al. CD56negCD16⁺ NK cells are activated mature NK cells with impaired effector function during HIV‐1 infection. Retrovirology 2013; 10:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cao Y, Zhou X, Huang X et al. Tim‐3 expression in cervical cancer promotes tumor metastasis. PLOS ONE 2013; 8:e53834. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37. Yang W, Song Y, Lu YL, Sun JZ, Wang HW. Increased expression of programmed death (PD)‐1 and its ligand PD‐L1 correlates with impaired cell‐mediated immunity in high‐risk human papillomavirus‐related cervical intraepithelial neoplasia. Immunology 2013; 139:513–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang W, Lu YP, Yang YZ, Kang JR, Jin YD, Wang HW. Expressions of programmed death (PD)‐1 and PD‐1 ligand (PD‐L1) in cervical intraepithelial neoplasia and cervical squamous cell carcinomas are of prognostic value and associated with human papillomavirus status. J Obstetrics Gynaecol Res 2017; 43:1602–12. [DOI] [PubMed] [Google Scholar]
- 39. Audenet F, Farkas AM, Anastos H, Galsky MD, Bhardwaj N, Sfakianos JP. Immune phenotype of peripheral blood mononuclear cells in patients with high‐risk non‐muscle invasive bladder cancer. World J Urol 2018; 36:1741–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brummelman J, Mazza EMC, Alvisi G et al. High‐dimensional single cell analysis identifies stem‐like cytotoxic CD8(+) T cells infiltrating human tumors. J Exp Med 2018; 215:2520–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liao Z, Lv X, Liu S et al. Different aberrant expression pattern of immune checkpoint receptors in patients with PTCL and NK/T‐CL. Asia‐Pacific J Clin Oncol 2018; 14:e252–e258. [DOI] [PubMed] [Google Scholar]
- 42. Lu X, Liu J, Cui P et al. Co‐inhibition of TIGIT, PD1, and Tim3 reverses dysfunction of Wilms tumor protein‐1 (WT1)‐specific CD8+ T lymphocytes after dendritic cell vaccination in gastric cancer. Am J Cancer Res 2018; 8:1564–75. [PMC free article] [PubMed] [Google Scholar]
- 43. Thommen DS, Koelzer VH, Herzig P et al. A transcriptionally and functionally distinct PD‐1(+) CD8(+) T cell pool with predictive potential in non‐small‐cell lung cancer treated with PD‐1 blockade. Nat Med 2018; 24:994–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim‐3 and PD‐1 pathways to reverse T cell exhaustion and restore anti‐tumor immunity. J Exp Med 2010; 207:2187–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Q, Bi J, Zheng X et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti‐tumor immunity. Nat Immunol 2018; 19:723–32. [DOI] [PubMed] [Google Scholar]
- 46. Pesce S, Greppi M, Tabellini G et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: a phenotypic and functional characterization. J Allergy Clin Immunol 2017; 139:335–46.e3. [DOI] [PubMed] [Google Scholar]
- 47. Della Chiesa M, Pesce S, Muccio L et al. Features of memory‐like and PD‐1(+) human NK cell subsets. Front Immunol 2016; 7:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu Y, Cheng Y, Xu Y et al. Increased expression of programmed cell death protein 1 on NK cells inhibits NK‐cell‐mediated anti‐tumor function and indicates poor prognosis in digestive cancers. Oncogene 2017; 36:6143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Komita H, Koido S, Hayashi K et al. Expression of immune checkpoint molecules of T cell immunoglobulin and mucin protein 3/galectin‐9 for NK cell suppression in human gastrointestinal stromal tumors. Oncol Rep 2015; 34:2099–2105. [DOI] [PubMed] [Google Scholar]
- 50. Finney CA, Ayi K, Wasmuth JD et al. HIV infection deregulates Tim‐3 expression on innate cells: combination antiretroviral therapy results in partial restoration. J Acquir Immune Defic Syndr 2013; 63:161–7. [DOI] [PubMed] [Google Scholar]
- 51. Farkas AM, Audenet F, Anastos H, Galsky M, Sfakianos J, Bhardwaj N. Tim‐3 and TIGIT mark natural killer cells susceptible to effector dysfunction in human bladder cancer. J Immunol 2018; 200:124.14. [Google Scholar]
- 52. Wiesmayr S, Webber SA, Macedo C et al. Decreased NKp46 and NKG2D and elevated PD‐1 are associated with altered NK‐cell function in pediatric transplant patients with PTLD. Eur J Immunol 2012; 42:541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ma J, Zheng B, Goswami S et al. PD1(Hi) CD8(+) T cells correlate with exhausted signature and poor clinical outcome in hepatocellular carcinoma. J Immunother Cancer 2019; 7:331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kim HD, Song GW, Park S et al. Association between expression level of PD1 by tumor‐infiltrating CD8(+) T cells and features of hepatocellular carcinoma. Gastroenterology 2018; 155:1936–50.e17. [DOI] [PubMed] [Google Scholar]
- 55. Grosso JF, Goldberg MV, Getnet D et al. Functionally distinct LAG‐3 and PD‐1 subsets on activated and chronically stimulated CD8 T cells. J Immunol 2009; 182:6659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sanchez‐Correa B, Valhondo I, Hassouneh F et al. DNAM‐1 and the TIGIT/PVRIG/TACTILE axis: novel immune checkpoints for natural killer cell‐based cancer immunotherapy. Cancers 2019; 11:877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 2015; 27:450–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cella M, Presti R, Vermi W et al. Immunology NCfHAV. Loss of DNAM‐1 contributes to CD8+ T‐cell exhaustion in chronic HIV‐1 infection. Eur J Immunol 2010; 40:949–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Minnie SA, Kuns RD, Gartlan KH et al. Myeloma escape after stem cell transplantation is a consequence of T‐cell exhaustion and is prevented by TIGIT blockade. Blood 2018; 132:1675–88. [DOI] [PubMed] [Google Scholar]
- 60. Buderath P, Schwich E, Jensen C et al. Soluble programmed death receptor ligands sPD‐L1 and sPD‐L2 as liquid biopsy markers for prognosis and platinum response in epithelial ovarian cancer. Front Oncol 2019; 9:1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Finkelmeier F, Canli Ö, Tal A et al. High levels of the soluble programmed death‐ligand (sPD‐L1) identify hepatocellular carcinoma patients with a poor prognosis. Eur J Cancer 1990; 2016:152–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Gating strategy for the flow cytometric analysis of NK cells in PBMCs from patients and controls. (a) representative workflow showing identification of NK subpopulations and representative analysis for six NK subpopulations in HD, LG, HG and CC samples. Inset examples of the six different subpopulations in different groups shows percentages based on the Total NK Cells region shown on the plot. (b‐g) Correlation analysis between the percentages of CD56dim NK cell and other NK cell subpopulations or NK cell subpopulations and T cells.
Fig. S2. Expression and co‐expression of PD‐1, TIGIT, Tim‐3, NKG2D and DNAM‐1 on peripheral blood CD56brightCD16neg NK cells from healthy donors (HD group), low grade lesions (LG group), high grade lesions (HG group) and cervical cancer patients (CC group). (a) Gating strategy for the t‐SNE analysis within CD56brightCD16neg cells. t‐SNE analysis was used to visualize expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3 in HD (n = 13), LG (n = 19), HG (n = 8) and CC (n = 19) groups. (b) t‐SNE density plots showing expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3. Flow cytometry gating of the data was performed to analyze the single expression of the immune checkpoints: (c) PD‐1. Co‐expression of (d) PD‐1+Tim‐3, (e) NKG2D+PD‐1, (f) DNAM‐1+TIGIT+PD‐1+, (g) PD‐1+TIGIT+Tim‐3 and (h) DNAM‐1+TIGIT+PD‐1+Tim‐3 are shown. (i) Heat map of single receptor expression and (j) Heat map of the co‐expression of the receptors are show for all groups. Data are shown as individual percentages of expression and their mean. Comparisons between the groups were performed using ANOVA with Dunnett’s multiple comparisons test. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Fig. S3. Expression and co‐expression of PD‐1, TIGIT, Tim‐3, NKG2D and DNAM‐1 on peripheral blood CD56brightCD16dim NK cells from healthy donors (HD group), low grade lesions (LG group), high grade lesions (HG group) and cervical cancer patients (CC group). (A) Gating strategy for the t‐SNE analysis within CD56brightCD16dim cells. t‐SNE analysis was used to visualize expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3 in HD (n = 13), LG (n = 19), HG (n = 8) and CC (n = 19) groups. (B) t‐SNE density plots showing expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3. Flow cytometry gating of the data was performed to analyze the single expression of the immune checkpoints: (C) PD‐1. Co‐expression of (D) PD‐1+TIGIT, (E) PD‐1+Tim‐3, (F) NKG2D+PD‐1, (G) DNAM‐1+TIGIT+PD‐1+, and (H) PD‐1+TIGIT+Tim‐3 are shown. (I) Heat map of single receptor expression and (J) Heat map of the co‐expression of the receptors are show for all groups. Data are shown as individual percentages of expression and their mean. Comparisons between the groups were performed using ANOVA with Dunnett’s multiple comparisons test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Fig. S4. Expression and co‐expression of PD‐1, TIGIT, Tim‐3, NKG2D and DNAM‐1 on peripheral blood CD56dimCD16neg NK cells from healthy donors (HD group), low grade lesions (LG group), high grade lesions (HG group) and cervical cancer patients (CC group). (a) Gating strategy for the t‐SNE analysis within CD56dimCD16neg cells. t‐SNE analysis was used to visualize expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3 in HD (n = 13), LG (n = 19), HG (n = 8) and CC (n = 19) groups. (b) t‐SNE density plots showing expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3. Flow cytometry gating of the data was performed to analyze the single expression of the immune checkpoints: (c) PD‐1. Co‐expression of (d) PD‐1+TIGIT, (e) PD‐1+Tim‐3, (f) NKG2D+PD‐1, (g) DNAM‐1+TIGIT+PD‐1+, (h) PD‐1+TIGIT+Tim‐3, and (i) DNAM‐1+TIGIT+PD‐1+Tim‐3+ are shown. (j) Heat map of single receptor expression and (K) Heat map of the co‐expression of the receptors are show for all groups. Data are shown as individual percentages of expression and their mean. Comparisons between the groups were performed using ANOVA with Dunnett’s multiple comparisons test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Fig. S5. Expression and co‐expression of PD‐1, TIGIT, Tim‐3, NKG2D and DNAM‐1 on peripheral blood CD56dimCD16dim NK cells from healthy donors (HD group), low grade lesions (LG group), high grade lesions (HG group) and cervical cancer patients (CC group). (a) Gating strategy for the t‐SNE analysis within CD56dimCD16dim cells. t‐SNE analysis was used to visualize expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3 in HD (n = 13), LG (n = 19), HG (n = 8) and CC (n = 19) groups. (b) t‐SNE density plots showing expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3. Flow cytometry gating of the data was performed to analyze the single expression of the immune checkpoints: (c) PD‐1. Co‐expression of (d) PD‐1+TIGIT, (e) PD‐1+Tim‐3, (f) NKG2D+PD‐1, (g) DNAM‐1+TIGIT+PD‐1+, and (h) PD‐1+TIGIT+Tim‐3, are shown. (i) Heat map of single receptor expression and (j) Heat map of the co‐expression of the receptors are show for all groups. Data are shown as individual percentages of expression and their mean. Comparisons between the groups were performed using ANOVA with Dunnett’s multiple comparisons test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Fig. S6. Expression and co‐expression of PD‐1, TIGIT, Tim‐3, NKG2D and DNAM‐1 on peripheral blood CD56dimCD16bright NK cells from healthy donors (HD group), low grade lesions (LG group), high grade lesions (HG group) and cervical cancer patients (CC group). (a) Gating strategy for the t‐SNE analysis within CD56dimCD16bright cells. t‐SNE analysis was used to visualize expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3 in HD (n = 13), LG (n = 19), HG (n = 8) and CC (n = 19) groups. (b) t‐SNE density plots showing expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3. Flow cytometry gating of the data was performed to analyze the single expression of the immune checkpoints: (c) PD‐1. Co‐expression of (d) PD‐1+TIGIT, (e) PD‐1+Tim‐3, (f) PD‐1+TIGIT+Tim‐3, (g) NKG2D+PD‐1, (h) DNAM‐1+TIGIT+PD‐1+, (i) DNAM‐1+NKG2D+TIGIT+PD‐1+Tim‐3+, and (j) DNAM‐1+TIGIT+PD‐1+Tim‐3+ are shown. (K) Heat map of single receptor expression and (l)Heat map of the co‐expression of the receptors are show for all groups. Data are shown as individual percentages of expression and their mean. Comparisons between the groups were performed using ANOVA with Dunnett’s multiple comparisons test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Fig. S7. Expression and co‐expression of PD‐1, TIGIT, Tim‐3, NKG2D and DNAM‐1 on peripheral blood CD56negCD16bright NK cells from healthy donors (HD group), low grade lesions (LG group), high grade lesions (HG group) and cervical cancer patients (CC group). (a) Gating strategy for the t‐SNE analysis within CD56negCD16bright cells. t‐SNE analysis was used to visualize expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3 in HD (n = 13), LG (n = 19), HG (n = 8) and CC (n = 19) groups. (b) t‐SNE density plots showing expression distribution of DNAM‐1, NKG2D, TIGIT, PD‐1 and Tim‐3. Flow cytometry gating of the data was performed to analyze the single expression of the immune checkpoints: (c) PD‐1. Co‐expression of (d) PD‐1+TIGIT, (e) PD‐1+Tim‐3, (f) NKG2D+PD‐1, (g) DNAM‐1+TIGIT+PD‐1+, and (h) DNAM‐1+TIGIT+PD‐1+Tim‐3+ are shown. (i) Heat map of single receptor expression and (j) Heat map of the co‐expression of the receptors are show for all groups. Data are shown as individual percentages of expression and their mean. Comparisons between the groups were performed using ANOVA with Dunnett’s multiple comparisons test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Table S1. Correlations Between sPD‐L1 and Cell Populations.
Data Availability Statement
Data are available upon request to the corresponding author.