

Abstract
Messenger RNAs (mRNAs) convey genetic information from the DNA genome to proteins and thus lie at the heart of gene expression and regulation of all cellular activities. Live cell single molecule tracking tools enable the investigation of mRNA trafficking, translation and degradation within the complex environment of the cell and in real-time. Over the last 5 years, nearly all tools within the mRNA tracking toolbox have been improved to achieve high-quality multi-color tracking in live cells. For example, the bacteriophage-derived MS2-MCP system has been improved to facilitate cloning and achieve better signal-to-noise ratio, while the newer PP7-PCP system now allows for orthogonal tracking of a second mRNA or mRNA region. The coming of age of epitope-tagging technologies, such as the SunTag, MoonTag and Frankenbody, enables monitoring the translation of single mRNA molecules. Furthermore, the portfolio of fluorogenic RNA aptamers has been expanded to improve cellular stability and achieve a higher fluorescence “turn-on” signal upon fluorogen binding. Finally, microinjection-based tools have been shown to be able to track multiple RNAs with only small fluorescent appendages and to track mRNAs together with their interacting partners. We systematically review and compare the advantages, disadvantages and demonstrated applications in discovering new RNA biology of this refined, expanding toolbox. Finally, we discuss developments expected in the near future based on the limitations of the current methods.
Graphical Abstract
Tools for the intracellular visualization of mRNA metabolism and function.
Introduction
The fundamental role of messenger RNAs (mRNAs) is to act as the transient molecular intermediaries between genetic information stored in DNA and the amino-acid sequence of proteins, making them the key mediators of gene expression. In eukaryotes, the process of gene expression involves transcription, pre-mRNA processing, nucleocytoplasmic trafficking of the matured mRNA, translation, transport to subcellular compartments, and mRNA degradation. Significant effort has been directed towards understanding each of these steps, as each is crucial for regulating cellular processes by fine-tuning cellular protein levels (Figure 1). The steps of gene expression, however, exhibit complex spatiotemporal dynamics so that dissecting their mechanisms in physiological and disease contexts is contingent on the development of techniques capable of addressing these complexities, ideally within their cellular environment (Ben-Yishay & Shav-Tal, 2019; Tourrière, Chebli, & Tazi, 2002; Xie & Ren) (Hinnebusch & Lorsch, 2012). This review focuses on recent developments in the field of intracellular single molecule fluorescence imaging and broadly aims to summarize the various applications of these methods to mRNA biology in live cells (Chao & Lionnet, 2018; Ingolia et al., 2014).
Figure 1. Key steps of the mRNA life cycle can be visualized at the single molecule level.

Transcription (1), mRNA processing (2), small RNA processing (3), export (4), RNP import (5), translation (6), regulation by small RNAs (7), mRNA storage (8) and transport (9) all have been examined using the intracellular single molecule visualization techniques reviewed here.
Classically, mRNA metabolism has been studied using ensemble assays, which average over heterogeneous behaviors, limiting the information they can provide on the spatiotemporal dynamics of biomolecules, especially in the context of live cells or intact tissues. One approach to expanding the information generated by biochemical or genetic assays is to employ transcriptome-wide analyses. For example, high-throughput deep RNA-sequencing of ribosome footprints has revealed a variety of non-canonical mRNA translations events, such as alternative start codon selection, 5’-cap independent translation initiation, frameshifting, internal ribosome entry site selection, and stop codon read-through (Andreev et al., 2016; Ingolia, 2014; Ingolia et al., 2014; Kearse & Wilusz, 2017; Lacerda, Menezes, & Romão, 2017). Such transcriptome-wide approaches are able to reveal biologically interesting subpopulations of transcripts. However, they are incapable of providing refined information on the sub-second timescale events in single cells that drive much of biology.
Single molecule fluorescence microscopy is able to go beyond these general limitations of ensemble-averaging approaches by allowing the study of molecular interactions at the sub-second time and ~20 nm length scales, in live cells and even intact tissues (Pitchiaya, Heinicke, Custer, & Walter, 2014).
A pioneering breakthrough established over 20 years ago in the single molecule RNA imaging field was single molecule fluorescence in situ hybridization (smFISH). smFISH provides a snapshot of the sub-cellular, spatial distribution of RNAs at single molecule resolution in fixed cells (Femino, Fay, Fogarty, & Singer, 1998; George, Indig, Abdelmohsen, & Gorospe, 2018; Pitchiaya et al., 2014; Tutucci, Livingston, Singer, & Wu, 2018). With advances in fluorescence detection hardware and the development of protein-based and chemical probes that allow powerful labeling of mRNAs, several single-color single molecule techniques have been developed to track diverse classes of RNAs in living cells and tissues (Bertrand et al., 1998; Jalihal, Lund, & Walter, 2019; Lim & Peabody, 2002; Lyon & Stasevich, 2017; Pitchiaya et al., 2014; E. Tutucci et al., 2018). These methods have provided significant insights into the dynamic and stochastic nature of gene expression at the single molecule level that leads to a high degree of heterogeneity among individual cells. Additionally, combining these techniques with improved image processing workflows and theoretical integrative models has advanced our understanding of gene expression dynamics (Pitchiaya et al., 2014; Sokabe & Fraser, 2018).
However, such single-color imaging is limited to the study of only one RNA species. Many fundamental questions of the types and origins of heterogeneity among mRNA populations cannot be addressed by imaging or tracking without simultaneous visualization of other segments of the mRNA or of its interaction partners. Thus, there is a compelling need for tools that enable multiplexed detection of RNAs or RNAs and proteins.
In the following, we describe recent technological developments tackling these limitations. We focus on single molecule methods that provide simultaneous readouts of the behavior of multiple components that are involved in the mRNA biology of living eukaryotic cells. Their advantages and limitations are discussed and compared systematically. Finally, based on the limitations of current methods we discuss future directions for the single molecule mRNA tracking field.
RECENT DEVELOPMENTS IN SINGLE MRNA IMAGING IN VIVO
Bacteriophage derived RNA labeling tags
RNA labeling tags derived from bacteriophages utilize the high RNA binding affinity and specificity of bacteriophage-derived coat proteins (CPs) to their corresponding RNA recognition sequences (Buxbaum, Haimovich, & Singer, 2015; Park, Buxbaum, & Singer, 2010). When conjugated to a fluorescent protein (FP), such as GFP, the CP binds to 6 to 96 repeats of the hairpin-structured phage RNA recognition sequence that are incorporated into a specific segment of the target mRNA (Figure 2) and, by accumulation of many dim GPF signals, allow a single molecule to be visualized as a bright fluorescent spot (Katz et al., 2018). Such genetically encoded RNA-protein appendages report on the RNA molecule’s localization and allow for its tracking in live cells due to an excess pool of CP-GFP conjugates that can exchange with photobleached molecules, limiting dimming of the tag but also leading to high background. First implemented by Rob Singer’s group, this strategy has remained a workhorse of single molecule mRNA visualization in live cells (Femino et al., 1998; Evelina Tutucci et al., 2018).
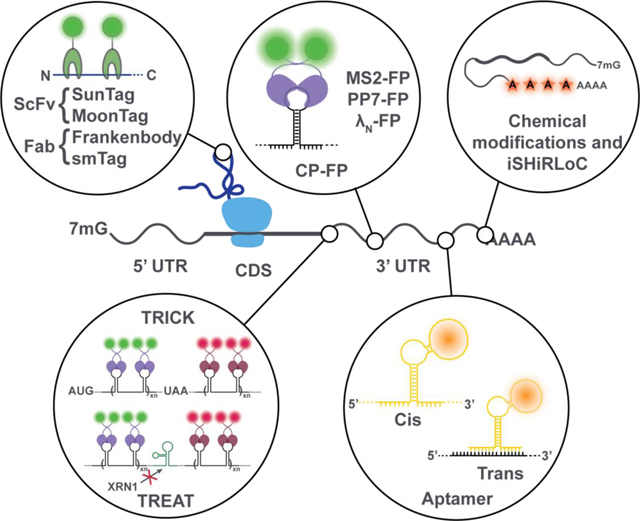
Figure 2. State-of-the-art mRNA and nascent-peptide visualization strategies.

Fluorescently-tagged RNA-binding proteins such as MS2, PP7 and λN bind to specific RNA secondary structures, typically incorporated into the 3’UTR of an mRNA to prevent interference with translation. When placed in the coding sequence (CDS), displacement of these proteins by the translocation of the ribosome can be used to infer protein translation. During translation, the elongating nascent peptide itself can be visualized using nanobodies tagged with fluorescent proteins. Fluorescent RNA aptamers can be used in cis or trans to visualize mRNAs.
Several CP-derived RNA labeling tags, combined with the vast number of spectrally distinct FPs, offer ways to study mRNA functions. First to be deployed, the hairpin of a single-stranded RNA derived from a bacteriophage that infects Enterobacteriaceae, MS2, binds its MS2 coat protein (MCP) (Buxbaum et al., 2015; Katz et al., 2018). A second popular system, PP7, originates from an RNA bacteriophage of Pseudomonas aeruginosa whose RNA hairpins bind its own specific coat protein (PCP) (Buxbaum et al., 2015; Katz et al., 2018). Additional systems have been derived from the coat proteins of λ and Qβ phages (Daigle & Ellenberg, 2007; X. Zhang, Lee, Zhao, Xia, & Qin, 2010).
MS2 and PP7 both share the structural feature of a bulged adenosine within the RNA hairpin that is necessary for binding of their corresponding CP, yet each system varies in other structural features such as loop size and stem length, imparting binding specificity of each RNA-protein pair. RNA appendages have been engineered that contain most commonly 24 repeats of the same RNA hairpin to reach the necessary signal intensity over background noise (Buxbaum et al., 2015; Daigle & Ellenberg, 2007; Katz et al., 2018; E. Tutucci et al., 2018). In newer versions, the hairpin’s stem sequence is slightly varied to facilitate cloning while maintaining the same CP binding specificity (Evelina Tutucci et al., 2018; B. Wu et al., 2015). The MS2-MCP system in particular has been used to determine localization of single mRNAs, track their active transport, and visualize aspects of its cellular life cycle (see below) (Buxbaum et al., 2015; Katz et al., 2018).
Compared to other labeling methods, bacteriophage derived RNA tags have the main advantage of being genetically encoded as part the RNA of interest (Katz et al., 2018). Consequently, both the labeling protein and the modified RNA bearing the recognition hairpins can be transiently or stably expressed in a mammalian cell, which may cause perturbations due to the typically resulting over-expression. Translation of the mRNA is maintained by inserting the recognition hairpin repeats into an area of the RNA that does not perturb normal function and trafficking, typically the 3’ untranslated region (3’UTR) (E. Tutucci et al., 2018). Functionality can be verified by comparing the localization with that of the unmodified RNA, obtained from immunostaining or smFISH of fixed cells (Buxbaum et al., 2015; George et al., 2018; Katz et al., 2018; E. Tutucci et al., 2018; B. Wu, Eliscovich, Yoon, & Singer, 2016). With these considerations in mind, the MS2 system and others like it offer the ability to monitor a range of cellular processes involving mRNAs.
Improvements to the bacteriophage derived RNA labeling tags include the development of similar systems with orthogonal labeling specificity and modifications that improve the functionality of the mRNA. One of the more recent developments is the boxB-λN system that consists of a peptide, just 22 amino acids long, from the N protein of the bacteriophage λ (λN) that binds to the RNA hairpin boxB, only 15 nucleotides in length (Daigle & Ellenberg, 2007; X. Zhang et al., 2010). In addition, an MS2 system with reduced affinity for the MCP facilitates the intrinsic mRNA decay in yeast cells, where exonucleases were observed to degrade only the coding region but not the more structurally stable MS2 RNA hairpin repeats (Evelina Tutucci et al., 2018). The development of multiple, highly sequence-specific bacteriophage-derived RNA labeling tags allows for multiplexing to probe the integrity of an mRNA and its interactions with partner RNAs or RNA binding proteins (RBPs) (Buxbaum et al., 2015; George et al., 2018; Katz et al., 2018).
The primary limitation of the CP-GFP fusion strategy is the low signal-to-noise ratio arising from freely diffusing and unbound fluorescent proteins. This necessitates a large number of hairpin loops to be inserted into the RNA, typically 24, but up to 96, in order to increase the fluorescence intensity over background. Another strategy that helps is subcellular compartmentalization, e.g., using nuclear localization sequences on the CP-FP to sequester it away from the compartment of interest, in this case the cytosol to monitor mRNA function (J. Wu et al., 2019; Yan, Hoek, Vale, & Tanenbaum, 2016).
Fluorogenic RNA aptamers
RNA aptamers are structured RNA modules that bind with high specificity and affinity typically to a small organic compound or an inorganic ion. Fluorogenic RNA aptamers represent a class of RNA aptamers that has been selected or evolved in vitro to bind specific fluorogenic dyes (fluorogens). The binding of fluorogen to a specific aptamer ideally leads to a strong increase in fluorescence, allowing RNA bound fluorogens to be distinguished from unbound ones (Grate & Wilson, 1999; Paige, Wu, & Jaffrey, 2011). This background suppression feature in particular makes fluorogenic aptamers promising tools for single molecule RNA visualization in live cells, with advantages over most CP-GFP-based approaches.
Fluorescent aptamers have been implemented using two orthogonal strategies that differ in whether or not the aptamer is contiguous with the sequence of the RNA to be detected. Thus, the aptamer, relative to the target RNA can be in cis, that is, appended to the target RNA and therefore functioning as an RNA “tag”, or in trans so that the target RNA and fluorogenic RNA aptamer are expressed independently and interact via engineered sequence complementarity (Figure 2) (Autour et al., 2018; Bouhedda, Autour, & Ryckelynck, 2017; Grate & Wilson, 1999; Paige et al., 2011).
The first class of fluorogenic RNA aptamer tags were evolved to bind to triphenylmethane dyes (e.g., Malachite green) (Babendure, Adams, & Tsien, 2003; Grate & Wilson, 1999). Malachite green-based dyes show remarkable fluorescence enhancement (>2,000-fold) in vitro upon binding to the corresponding malachite green RNA aptamer (Babendure et al., 2003). Nevertheless, they suffer from significant background fluorescence and phototoxicity in the cellular context, making them unsuitable for live cell imaging (Ilgu et al., 2016; Kraus et al., 2008; Saurabh, Perez, Comerci, Shapiro, & Moerner, 2016).
Major progress in this technology has been made, inspired by the photochemistry of GFP, by selecting RNA aptamers that mimic fluorescent proteins (Paige et al., 2011). The amino acids Ser65-Tyr66-Gly67 within the photo-active center of GFP undergo autocatalytic intramolecular cyclization, leading to the formation of fluorescent 4-hydroxybenzylidene imidazolinone (HBI) (Conyard et al., 2011). This chemical scaffold has served as the basis for the in vitro selection of new aptamers.
In Paige et al. 2011, column-based systematic evolution of ligands by exponential enrichment (SELEX) against several synthetic HBI derivatives was used to identify a plethora of HBI derivative binding RNA aptamers, covering a large color spectrum (Paige et al., 2011). Among this aptamer pool, an RNA called Spinach showed the best performance (Paige et al., 2011). The cell-permeable, non-toxic fluorogen 3,5-difluoro-4-hydroxylidene imidazolinone (DFHBI) binds to the G-quadruplex of the Spinach aptamer (Paige et al., 2011; Katherine Deigan Warner et al., 2014). This binding enhances DFHBI ‘s fluorescence by allowing the excited fluorophore to adopt a planar conformation, which decreases the dissipation of its excitation energy through non-radiative decay pathways (Han, Leslie, Fei, Zhang, & Ha, 2013; Katherine Deigan Warner et al., 2014).
The Spinach aptamer has been applied in vivo as a genetically encoded RNA imaging probe to tag a 5S rRNA whose trafficking was detected in living mammalian cells by fluorescence microscopy (Paige et al., 2011).
Efforts have been made to improve this initial system in multiple ways. To increase the signal-to-noise ratio and to allow the detection of less abundant cellular RNAs, a genetically encoded tandem Spinach as well as the improved Spinach2 aptamer (Strack et al., 2013) have been implemented in arrays to visualize mRNAs (Ilgu et al., 2016; J. Zhang et al., 2015). Furthermore, the fluorogen itself has been improved for in vivo detection through structure engineering. Addition of a trifluoroethyl substituent to DFHBI displays lower background fluorescence, higher brightness, and improved excitation/emission properties (with a bathochromic spectral shift), making it more adaptive for standard fluorescence microscopes (Filonov el al., 2014).
Besides the improvement of the fluorogen-aptamer system itself, developments in imaging strategies have aided the performance of the Spinach system. Insights into the photophysics of DFHBI deactivation have allowed the development of a pulsed excitation-based imaging strategy, allowing a significant increase in observation time (Han et al., 2013). Using spinning disc confocal microscopy and sophisticated imaging-processing workflows, the tracking of nuclear export of yeast mRNA fused with a single copy of Spinach aptamer has been possible to track (Guet et al., 2015). The primary drawback of the Spinach system lies in the RNA structure itself, which suffers from thermal instability due to low melting temperature (Tm ≈ 34°C) and poor folding capacity (Strack et al., 2013). To overcome these limitations, a portfolio of additional, robust, fluorogenic aptamers have been developed with improved folding, in vivo resistance and fluorogen spectral properties (Autour et al., 2018; Chen et al., 2019; Dolgosheina et al., 2014; Filonov et al., 2014; Song et al., 2017; Strack et al., 2013; Katherine Deigan Warner et al., 2014). Among them, the most refined fluorogenic RNA aptamers are those of the Mango family (Autour et al., 2018; Dolgosheina et al., 2014).
The Mango family consists of so far four different ~40 nt long aptamers, with a high folding capacity in vivo and the ability to bind two different fluorogens with subnanomolar affinities, thiazole orange and thiazole red (TO1-B and TO3-B) biotin conjugates (Autour et al., 2018; Dolgosheina et al., 2014). Their superior photophysical and structural properties have been used in vivo to detect small non-coding RNAs in mammalian cells (Autour et al., 2018; Dolgosheina et al., 2014). Because the TO fluorogens are biotinylated, this system has also been adapted to purify RNAs and RBPs (Panchapakesan et al., 2017).
Yet another tool in the fluorogenic RNA portfolio is the so-called Corn aptamer, which binds its fluorogen (DFHO) as a quasisymmetric homodimer and has the potential to be used in a Split GFP manner, which allows to study RNA binding partners (Kamiyama et al., 2016; K. D. Warner et al., 2017). Staying with the vegetable theme, a recent publication describes a Pepper aptamer that binds a whole series of bright and stable fluorogens with a broad range of fluorescence emission maxima ranging from blue to red, suggesting that the fluorogenic RNA toolbox will continue to expand (Chen et al., 2019).
Most fluorogenic RNA aptamers, perhaps with the exception of Pepper (Chen et al., 2019), contain G-quadruplex structures in the fluorogen binding pocket, which require sufficient concentrations of potassium and can unfold in mammalian systems, leading to a significant decrease in the binding capacity of the fluorogen, consequently resulting in a poor signal-to-noise (Guo & Bartel, 2016). One potential way to overcome this limitation is to conjugate the organic dye with a quencher for a further decrease in background (Sunbul & Jaschke, 2013). In this strategy, when the organic dye is unbound its fluorescence is quenched (Arora, Sunbul, & Jaschke, 2015). Upon RNA binding, the quenching effect is (partially) reversed, leading to an increase in fluorescence (Arora et al., 2015; Braselmann et al., 2018). Another advantage of this strategy is that it allows for greater flexibility in the choice of detection color (Arora et al., 2015; Braselmann et al., 2018). Similarly, Breselmann et al. reported a new genetically encoded labeling strategy, Riboglow, which exploits the broad fluorescence quenching properties of cobalamin together when conjugated with a fluorophore, which is suppressed when the quencher binds to a bacterial cobalamin aptamer. A disadvantage of this system is that the complex cobalamin-fluorophore conjugate is no longer cell permeable and has to be bead loaded to transiently disrupt the cellular membrane (Braselmann et al., 2018).
A final RNA visualization system that was recently developed uses a protein binding RNA. A mutant of the trans-activation response element (TAR) of the human immunodeficiency virus (HIV) is used as a stabilizer of a fluorescence protein-degron tag fusion (FP-tDeg). This RNA appendage, confusingly also termed “Pepper aptamer” (although it was not in vitro selected or evolved as the basic definition of an aptamer), was shown to confer the ability to image mRNA in living cells (J. Wu et al., 2019). In the absence of the stabilizing RNA motif, the FP-tDeg is degraded quickly. Only in the presence of its RNA stabilizer, the half-life of the FP-tDeg is sufficiently long to be detected as bound to the target RNA. Incorporation of multiple copies of this Pepper RNA into the target mRNA and co-expression of the FP-tDeg fluorogen allow the target mRNA to be robustly visualized (J. Wu et al., 2019).
This survey may convey the fact that fluorogen-aptamer approaches are rapidly catching up with the well-developed protein-based fluorescence toolkit (Duwe & Dedecker, 2019).
Visualization of mRNA translation
In addition to enabling direct mRNA tracking, there has been an increased interest in fluorescent labeling strategies that allow the visualization of various functional aspects of mRNA translation. Fully genetically encoded, single molecule protein labeling methodologies have been reported recently that exploit a highly specific interaction between fluorescently labeled single-chain variable fragments (scFVs), or camelid antibodies (“nanobodies”) to nascent peptide epitopes produced during translation elongation (Figure 2). These techniques have been used to observe and quantify, in a cellular context the ribosome loading, translation initiation, elongation kinetics, mobility and even polysome shape associated with single mRNAs (Custer & Walter, 2017; Pichon et al., 2016; Tanenbaum, Gilbert, Qi, Weissman, & Vale, 2014; Wang, Han, Zhou, & Zhuang, 2016).
The SUperNova Tag (SunTag) utilizes a genetically encoded scFV fused to a superfolder green fluorescent protein GFP (sfGFP) and allows a single molecule readout of translation processes in single cells (Tanenbaum et al., 2014; Wilson et al., 1984). The system relies on four modules: I) up to 56 repeats of the 19-amino acid SunTag peptide epitope, which needs to be integrated into ORF sequence, II) a ScFV-sfGFP fusion targeting the epitope, III) an mRNA modification suitable for labeling (such as the MS2 or PP7 RNA hairpins), and IV) a combined system for mRNA labeling with a distinct color and for membrane tethering (e.g. PCP-mCherry-CAAX expression module) (Boersma et al., 2019; Pichon et al., 2016; Tanenbaum et al., 2014).
As the mRNA is translated, the epitopes emerge from the ribosome and begin to bind the scFV-sfGFP fusion protein, leading to an increase in green GFP fluorescence intensity at the mRNA spot. In addition, this spot is marked by the red mCherry via MS2- or PP7-binding and is localized to a cellular membrane through prenylation of the CAAX protein component, allowing for the mRNA and nascent polypeptide chain to become colocalized as an indication of active translation. In addition, the tethering to a relatively immobile structure such as the endoplasmic reticulum or the outer cell membrane improves the signal-to-noise during imaging and, combined with the equilibrium exchange with over-expressed, unbleached fluorescent proteins, allows individual mRNA molecules to be tracked on long timescales (tens of minutes). The drawback of tethering is that it bears the risk of interfering with the translation process (Boersma et al., 2019; Tanenbaum et al., 2014).
Yan et al. used this two-color translation reporter system to deconvolute initiation, elongation, and ribosome stalling on individual mRNA molecules within single cells (Yan et al., 2016). Improving upon this system, Hoek et al. applied it to nonsense mediated decay (NMD), and reported multiple mRNA subpopulations that rationalize the variability in NMD efficiency (Hoek et al., 2019).
As a step towards increasing the number of translated peptides that can simultaneously be studied, a second ScFV-epitope pair called the MoonTag system was reported by Boersma et al.. Using this system, heterogeneity of initiation frequency during mRNA translation at different start sites could be tracked (Boersma et al., 2019). Additionally, frameshifting and non-canonical out-of-frame translation were investigated by placing SunTag and MoonTag repeats in alternate frames (MashTag) (Boersma et al., 2019). Finally, Lyon et al. combined a semi-genetically encodable multi-frame tag strategy, using both repeat FLAG and SunTag epitopes as readout, to study out-of-frame translation events (Lyon et al., 2019).
An orthogonal strategy for nascent peptide detection was reported by Zhao et al. who developed the hemagglutinin (HA) frankenbody. HA frankenbodies have somewhat higher affinity for their epitopes than scFV antibodies as well as higher specificity (Zhao et al., 2019) and enable swapping of protein-based fluorescence probes, leading to greater multiplexing potential (Zhao et al., 2019).
In contrast to the SunTag system, the 10x FLAG tag of the “spaghetti monster” is nonuniformly distributed and consists of a stable, nonfluorescent GFP-derived core structure with, in total, 10 repeats of the FLAG epitope (Morisaki et al., 2016; Tanenbaum et al., 2014). Finally, degron tags can be incorporated into the peptide sequence, allowing it to be quickly degraded and facilitating the clearance of full-length translation product, which in turn enables prolonged imaging especially of cells with high protein expression levels (Ilgu et al., 2016; B. Wu et al., 2016).
The pioneer round of ribosome scanning is biologically important as it ties cytoplasmic translation to nuclear splicing as well as cytoplasmic mRNA decay by linking it to the NMD pathway. Utilizing the orthogonal PP7 and MS2 systems in a dual-color single mRNA imaging assay, Halstead et al. investigated the dynamics of the pioneer round of translation in living cells (Figure 2). The TRICK (Translating RNA Imaging by Coat protein Knock-off) system takes advantage of the fact that a translating ribosome displaces most RNA binding proteins within the coding region of the mRNA during the first round of translation (James M. Halstead et al., 2015). Appending translatable PP7 hairpins into the coding sequence together with MS2 hairpins into the 3’-UTR of a target mRNA and combining these tags with spectrally distinct fluorescent proteins fused to the corresponding coat proteins allowed untranslated and translated mRNA molecules to be distinguished on the basis of whether they showed color colocalization or not, respectively. Overall, the TRICK reporter system thus reports spatiotemporal information on an mRNA’s first translation round in living cells (James M. Halstead et al., 2015; J. M. Halstead et al., 2016).
TREAT as mRNA turnover sensor
Beyond visualizing mRNA translation based on the TRICK sensor, utilizing similar principles the same group reported a technique that tracks mRNA decay in eukaryotic living cells (Horvathova et al., 2017). This system uses a flavivirus-derived RNA pseudoknot structure that stalls the translocation of the processive eukaryotic 5’→3’ exoribonuclease XRN1, leading to stable mRNA decay intermediates (Kieft, Rabe, & Chapman, 2015). The resulting mRNA decay sensing module, called TREAT (three prime RNA End Accumulation during Turnover), contains a tandem of XRN-1 stalling West Nile virus (WNV) pseudoknot RNA structures between the PP7 and MS2 stem-loops of the target mRNA’s 3’-UTR (Figure 2). This design allows to discriminate between full-length mRNA and mRNA molecules undergoing degradation from the 5’-end, where colocalized colors indicate the more intact mRNA (Horvathova et al., 2017; Russo & Wilusz, 2017). TREAT thus offers the remarkable opportunity to dissect important aspects of a single mRNA’s lifecycle from transcription to decay (Russo & Wilusz, 2017).
Intracellular single-molecule high-resolution localization and counting
The mRNA visualization strategies discussed thus far all involve genetic manipulations to alter the sequence of the target RNA to insert aptamer cassettes or rather large repeats of viral coat protein recognition motifs to which then large numbers of fluorescent protein conjugates bind. Major drawbacks of altering the RNA sequence for implementing these strategies are that the target RNA has a considerably increased size after the inclusion of these appendages, which may alter its intracellular localization or stability, and with most promoters it is difficult to achieve homogeneous and near-endogenous mRNA copy number distributions across cells, which may be a relevant parameter either as a biological variable or for ease of single molecule imaging. Many of these limitations can be addressed with extracellular labeling and physical delivery into cells (Pitchiaya et al., 2012).
Intracellular single-molecule high-resolution localization and counting (iSHiRLoC, Figure 3) is a microinjection-based single molecule RNA tracking method that allows multicolor particle tracking and spot counting in live and fixed cells, respectively (Michelini et al., 2017; Pitchiaya et al., 2012; Pitchiaya et al., 2014; Pitchiaya et al., 2017; Pitchiaya, Krishnan, Custer, & Walter, 2013; Pitchiaya et al., 2019; Shankar et al., 2016). For live cell imaging, RNA synthesized and labeled in vitro is introduced to desired sub-cellular compartments by microinjection, and single molecule RNA tracking is performed using, for example, highly inclined and laminated optical sheet (HILO) microscopy (Tokunaga et al., 2008). For fixed cell imaging, step-wise photobleaching data is collected to determine the number of single molecules within an imaged particle, which supplements the live cell imaging by providing stoichiometric information. Recently, iSHiRLoC has been applied to study the dynamics of non-coding RNAs like microRNAs (miRNAs) and DNA damage response RNAs (DDRNAs), but it can also be applied to mRNA and long non-coding RNAs (lncRNAs) (Michelini et al., 2017; Pitchiaya et al., 2012; Pitchiaya et al., 2017; Pitchiaya et al., 2013; Pitchiaya et al., 2019).
Figure 3. iSHiRLoC presents a set of tools that enables the probing of the intracellular behaviors of mRNPs.

Exogenously labeled mRNAs, containing fluorescent nucleotides, are delivered into the cell by microinjection. Co-injecting labeled mRNAs with a fluorescent injection marker such as high-molecular weight Dextran allows the injected cellular compartment to be marked (left). Single particle tracking and photobleaching analyses provide a range of physiologically relevant parameters to be extracted from live and fixed-cell images (right).
As a microinjection-based single molecule tracking tool, iSHiRLoC has many distinct advantages over tools based on other delivery methods (Figure 3): (1) iSHiRLoC enables the precise control over the number of molecules delivered to each cell in contrast to transient or stable transfection of a plasmid, where expression is much more stochastic and hence produces more cell-to-cell heterogeneity. (2) iSHiRLoC tracks near-native mRNA instead of overly extended mRNA. For example, in the bacteriophage CP-derived tracking systems, the target mRNA is longer than the original transcript due to the addition of 24 or more copies of the recognitions hairpin loops. Furthermore, occupancy of these stem loops by the respective CP-FPs can add significant mass that can affect the diffusion behavior of the transcript. In contrast, chemically labeled mRNA molecules are comparable in length and molecular weight to the original transcript. (3) A clearly defined start point after microinjection enables study of kinetics in a way similar to in vitro experiments, especially in the study of mRNA decay. (4) iSHiRLoC allows control over the cellular compartment to which the RNA is delivered, and thus allows dynamic processes like nucleocytoplasmic transport to be studied (Pitchiaya et al., 2017).
To prepare an mRNA for iSHiRLoC, it is typically first in vitro transcribed, then m7G capped and polyadenylated. The latter modifications allow the exogenously prepared mRNAs to mimic the functions of endogenous mRNAs, by permitting cap- and tail-dependent functions and showing stability comparable to endogenous mRNAs. Custer et al. tested three different mRNA labeling strategies, in which chemically modified nucleotides were incorporated using T7 RNA polymerase or yeast poly(A) polymerase (Custer & Walter, 2017). These strategies are: (1) random incorporation into the body of the mRNA, i.e. 5’ UTR, including coding region and untranslated regions during transcription; (2) random incorporation into the poly(A) tail; and (3) specific incorporation between the body and poly-A tail (BTT). Modified nucleotides can either be directly dye-labeled nucleotides (e.g., Cy5-UTP) or 2’-modified nucleotides (e.g., azido-ATP) that are subsequently coupled to fluorophores via click chemistry (Custer & Walter, 2017). The presence of modified nucleotides in the “body” of the RNA was found to significantly hamper translation activity and thus this labeling strategy was found to not be suitable for preparing functional mRNA for microinjection. Both tail- and BBT-labeling efficiently yield functional mRNA molecules in terms of translation activity and regulation by miRNA and were thus found to be suitable for iSHiRLoC experiments.
The number of labels per RNA molecule has both positive and negative impacts on particle tracking during iShiRLoC experiments. On the positive side, multiple fluorescence labels per mRNA molecule provide a higher signal-to-noise ratio and prolong the imaging time before a molecule is completely photobleached. However, the negative is that the typically imprecise (since broadly distributed) number of labels per RNA complicates the accurate counting of single molecules via stepwise photobleaching. The single molecule counting works most reliably if each dye corresponds to exactly one molecule of interest, as has been demonstrated for miRNAs (Pitchiaya et al., 2012).
While iSHiRLoC has clear advantages over existing systems for mRNA visualization, it has several limitations. While highly advantageous as a delivery method, microinjection is of low throughput by virtue of the time it takes to inject individual cells, and limited by a low success rate of injection. Other techniques such as bead loading and electroporation are possible, but have other disadvantages such as variable efficiency per cell and limited experimental control over the payload, and may stress the cells in ways that change their physiology (Stewart et al., 2016).
In summary, iSHiRLoC (Figure 3) allows for the visualization of near-native single mRNAs along with regulatory both RNA and protein factors in live and fixed cells at controlled abundance, enabling investigations of the kinetics of mRNA functions that may be less accessible to expression-based systems.
Conclusions and Future direction
Here we have presented recent advances in the field of single molecule mRNA tracking in cells. These developments can be summarized as progress on three main fronts: better tracking, multiplexed detection, and minimized perturbation of the cell.
Tracking quality is now being further enhanced by aiming for higher signal-to-noise ratios and longer observation times for single molecule spots. Higher signal-to-noise ratios can be achieved by, for example, the use of self-labeling protein tags (i.e., the SNAP-, Halo-, and CLIP-tags) coupled with the development of particularly bright, photostable, cell-permeable ligands (Banaz, Mäkelä, & Uphoff, 2019; Liss, Barlag, Nietschke, & Hensel, 2015). Fluorogenic RNA aptamers utilizing newly developed higher-affinity fluorogens that undergo a more dramatic increase in fluorescence signal upon binding to aptamer can similarly help improve the signal-to-noise ratio (Autour et al., 2018; Bouhedda et al., 2017). Chemical dyes linked with a strong quencher could further increase the difference in fluorescence between the bound and unbound states in dye-aptamer systems. Longer tracking then is becoming possible by the frequent exchange of fluorogens between the aptamers in the mRNA and the pool of dark fluorogens in the cell. The fluorescence of such a fluorogenic RNA aptamer system can be rendered relatively immune to photobleaching because fluorogens are exchanged often before they bleach. Such development of orthogonal labeling strategies will increasingly enable the simultaneous, multi-species tracking of single particles. Even so, multiplexing beyond two RNA or protein species remains a challenge due to the necessity of collecting a maximum number of photons across the typically very broad fluorophore emission spectra.
Another focus of recent work has been on addressing the risk of tracking artefacts introduced by the massive RNP appendages of the now-classical CP-FP systems. In the MS2-MCP system, approximately 2.5 MDa of protein-RNA complex is expected to be appended to the target mRNA if 24 copies of MS2 stem loops were all occupied, which can lead to slowed diffusion and aberrant cellular stability (Evelina Tutucci et al., 2018). In terms of reducing the size of such modifications, fluorogenic RNA aptamer systems reduce the contribution of proteins, while iSHiRLoC avoids the need for sequence-modification altogether by direct covalent labeling with small molecule dyes.
On the one hand, the tracking of single fluorophore-labeled target molecules in cells is challenging due to rapid photobleaching in the oxygenated cell, rapid diffusion in the third dimension and thus out of focus, and various sources of noise (camera noise, motion blur, localization error, missed steps, etc.). As trajectory length increases, the objective of single-particle tracking will increasingly be to understand the complex diffusion patterns within the cellular context. A long trajectory may increasingly encompass different types of diffusion, including active transport that results in near-linear tracks (Monnier et al., 2015). Consequently, the commonly used mean-square displacement (MSD) method can become misleading because it does not consider stochastic switching between diffusive modes. The classical cumulative probability distribution (CPD) analysis of squared displacements can recognize some diffusive behaviors, but does not provide information on transitions between modes within a single track (Schütz, Schindler, & Schmidt, 1997). An HMM-Bayesian model software was therefore developed to annotate stochastic phases of active transport and random motion on the basis of common motion types and parameter values (Monnier et al., 2015). More recently, nonparametric methods have been reported that uncover a range of possible diffusion states within a single particle track, independent of pre-defined parameters. For example, the single molecule analysis by unsupervised Gibbs sampling (SMAUG) algorithm provides the number of mobility states, the fraction, and the average diffusion coefficient of each state, as well as the probability of transitioning between two states (Karslake et al., 2019). Alternatively, background fluctuations arising from out-of-focus fluorescence or diffusion in the Z-direction can be addressed by the newly developed Single-Molecule Accurate LocaLization by LocAl Background Subtraction (SMALL-LABS) algorithm (Isaacoff, Li, Lee, & Biteen, 2019). Finally, a motion-blur filtering approach has been tested for extracting confinement forces and diffusivity from tracks (Calderon, 2016).
Taken together, recent developments in experimental and computational tools have improved our ability to extract information from single mRNA tracks. Further developments promise to help uncover ever more details of individual mRNA behavior in live cells as confined by cellular structures such as membraneless organelles. Only the entire sum of all such single molecule behaviors in a cell will help us reconstitute the full repertoire and complexity of cellular RNA metabolism and thus life.
Acknowledgments
We thank SMART Center Manager Dr. Damon Hoff with facilitating studies related to this review.
Funding Information
N.G.W. acknowledges funding by NIH/National Institute of General Medical Sciences grants GM062357, GM118524, GM122803 and GM131922, National Cancer Institute grants CA204560 and CA229023. AJ was supported by NIH cellular and molecular biology training grant T32-GM007315.
Research Resources
We acknowledge NSF MRI-ID grant DBI-0959823 (to N.G.W.) for seeding the Single Molecule Analysis in Real-Time (SMART) Center, whose Single Particle Tracker TIRFM equipment was used in studies related to this review.
Contributor Information
Andreas Schmidt, Single Molecule Analysis Group, Department of Chemistry, University of Michigan, Ann Arbor, MI 48109-1055, USA.
Guoming Gao, Single Molecule Analysis Group, Department of Chemistry, University of Michigan, Ann Arbor, MI 48109-1055, USA; Biophysics Graduate Program, University of Michigan, Ann Arbor, Michigan 48109, USA.
Saffron R. Little, Single Molecule Analysis Group, Department of Chemistry, University of Michigan, Ann Arbor, MI 48109-1055, USA; Program in Chemical Biology, University of Michigan, Ann Arbor, Michigan 48109, USA.
Ameya P. Jalihal, Single Molecule Analysis Group, Department of Chemistry, University of Michigan, Ann Arbor, MI 48109-1055, USA; Cell and Molecular Biology Program, University of Michigan, Ann Arbor, Michigan 48109, USA.
Nils G. Walter, Single Molecule Analysis Group, Department of Chemistry, University of Michigan, Ann Arbor, MI 48109-1055, USA.
References
- Andreev DE, O’Connor, Patrick BF, Loughran G, Dmitriev SE, Baranov PV, & Shatsky IN (2016). Insights into the mechanisms of eukaryotic translation gained with ribosome profiling. Nucleic Acids Research, 45(2), 513–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora A, Sunbul M, & Jaschke A. (2015). Dual-colour imaging of RNAs using quencher- and fluorophore-binding aptamers. Nucleic Acids Res, 43(21), e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autour A, Jeng CY, S. D. Cawte A, Abdolahzadeh A, Galli A, Panchapakesan SSS, . . . Unrau PJ (2018). Fluorogenic RNA Mango aptamers for imaging small non-coding RNAs in mammalian cells. Nature Communications, 9(1), 656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babendure JR, Adams SR, & Tsien RY (2003). Aptamers Switch on Fluorescence of Triphenylmethane Dyes. Journal of the American Chemical Society, 125(48), 14716–14717. [DOI] [PubMed] [Google Scholar]
- Banaz N, Mäkelä J, & Uphoff S. (2019). Choosing the right label for single-molecule tracking in live bacteria: side-by-side comparison of photoactivatable fluorescent protein and Halo tag dyes. Journal of physics D: Applied physics, 52(6), 064002–064002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Yishay R, & Shav-Tal Y. (2019). The dynamic lifecycle of mRNA in the nucleus. Current Opinion in Cell Biology, 58, 69–75. [DOI] [PubMed] [Google Scholar]
- Bertrand E, Chartrand P, Schaefer M, Shenoy SM, Singer RH, & Long RM (1998). Localization of ASH1 mRNA Particles in Living Yeast. Molecular Cell, 2(4), 437–445. [DOI] [PubMed] [Google Scholar]
- Boersma S, Khuperkar D, Verhagen BMP, Sonneveld S, Grimm JB, Lavis LD, & Tanenbaum ME (2019). Multi-Color Single-Molecule Imaging Uncovers Extensive Heterogeneity in mRNA Decoding. Cell, 178(2), 458–472.e419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhedda F, Autour A, & Ryckelynck M. (2017). Light-Up RNA Aptamers and Their Cognate Fluorogens: From Their Development to Their Applications. International journal of molecular sciences, 19(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braselmann E, Wierzba AJ, Polaski JT, Chromiński M, Holmes ZE, Hung S-T, . . . Palmer AE (2018). A multicolor riboswitch-based platform for imaging of RNA in live mammalian cells. Nature Chemical Biology, 14(10), 964–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum AR, Haimovich G, & Singer RH (2015). In the right place at the right time: visualizing and understanding mRNA localization. Nat Rev Mol Cell Biol, 16(2), 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon CP (2016). Motion blur filtering: A statistical approach for extracting confinement forces and diffusivity from a single blurred trajectory. Physical Review E, 93(5), 053303–053303. [DOI] [PubMed] [Google Scholar]
- Chao JA, & Lionnet T. (2018). Imaging the Life and Death of mRNAs in Single Cells. Cold Spring Harb Perspect Biol, 10(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zhang D, Su N, Bao B, Xie X, Zuo F, . . . Yang Y. (2019). Visualizing RNA dynamics in live cells with bright and stable fluorescent RNAs. Nat Biotechnol. [DOI] [PubMed] [Google Scholar]
- Conyard J, Kondo M, Heisler IA, Jones G, Baldridge A, Tolbert LM, . . . Meech SR (2011). Chemically modulating the photophysics of the GFP chromophore. J Phys Chem B, 115(6), 1571–1577. [DOI] [PubMed] [Google Scholar]
- Custer TC, & Walter NG (2017). In vitro labeling strategies for in cellulo fluorescence microscopy of single ribonucleoprotein machines. Protein Sci, 26(7), 1363–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle N, & Ellenberg J. (2007). LambdaN-GFP: an RNA reporter system for live-cell imaging. Nat Methods, 4(8), 633–636. [DOI] [PubMed] [Google Scholar]
- Dolgosheina EV, Jeng SCY, Panchapakesan SSS, Cojocaru R, Chen PSK, Wilson PD, . . . Unrau PJ (2014). RNA Mango Aptamer-Fluorophore: A Bright, High-Affinity Complex for RNA Labeling and Tracking. ACS Chemical Biology, 9(10), 2412–2420. [DOI] [PubMed] [Google Scholar]
- Duwe S, & Dedecker P. (2019). Optimizing the fluorescent protein toolbox and its use. Curr Opin Biotechnol, 58, 183–191. [DOI] [PubMed] [Google Scholar]
- Femino AM, Fay FS, Fogarty K, & Singer RH (1998). Visualization of single RNA transcripts in situ. Science, 280(5363), 585–590. [DOI] [PubMed] [Google Scholar]
- Filonov GS, Moon JD, Svensen N, & Jaffrey SR (2014). Broccoli: rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution. Journal of the American Chemical Society, 136(46), 16299–16308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George L, Indig FE, Abdelmohsen K, & Gorospe M. (2018). Intracellular RNA-tracking methods. Open biology, 8(10), 180104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grate D, & Wilson C. (1999). Laser-mediated, site-specific inactivation of RNA transcripts. Proceedings of the National Academy of Sciences of the United States of America, 96(11), 6131–6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guet D, Burns LT, Maji S, Boulanger J, Hersen P, Wente SR, . . . Dargemont C. (2015). Combining Spinach-tagged RNA and gene localization to image gene expression in live yeast. Nat Commun, 6, 8882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, & Bartel DP (2016). RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science, 353(6306). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halstead JM, Lionnet T, Wilbertz JH, Wippich F, Ephrussi A, Singer RH, & Chao JA (2015). Translation. An RNA biosensor for imaging the first round of translation from single cells to living animals. Science, 347(6228), 1367–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halstead JM, Wilbertz JH, Wippich F, Lionnet T, Ephrussi A, & Chao JA (2016). Chapter Six - TRICK: A Single-Molecule Method for Imaging the First Round of Translation in Living Cells and Animals. In Filonov GS & Jaffrey SR (Eds.), Methods Enzymol (Vol. 572, pp. 123–157): Academic Press. [DOI] [PubMed] [Google Scholar]
- Han KY, Leslie BJ, Fei J, Zhang J, & Ha T. (2013). Understanding the Photophysics of the Spinach–DFHBI RNA Aptamer–Fluorogen Complex To Improve Live-Cell RNA Imaging. Journal of the American Chemical Society, 135(50), 19033–19038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG, & Lorsch JR (2012). The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harb Perspect Biol, 4(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek TA, Khuperkar D, Lindeboom RGH, Sonneveld S, Verhagen BMP, Boersma S, . . . Tanenbaum ME (2019). Single-Molecule Imaging Uncovers Rules Governing Nonsense-Mediated mRNA Decay. Mol Cell, 75(2), 324–339.e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvathova I, Voigt F, Kotrys AV, Zhan Y, Artus-Revel CG, Eglinger J, . . . Chao JA (2017). The Dynamics of mRNA Turnover Revealed by Single-Molecule Imaging in Single Cells. Mol Cell, 68(3), 615–625.e619. [DOI] [PubMed] [Google Scholar]
- Ilgu M, Ray J, Bendickson L, Wang T, Geraskin IM, Kraus GA, & Nilsen-Hamilton M. (2016). Light-up and FRET aptamer reporters; evaluating their applications for imaging transcription in eukaryotic cells. Methods (San Diego, Calif.), 98, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT (2014). Ribosome profiling: new views of translation, from single codons to genome scale. Nature Reviews Genetics, 15, 205. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Stern-Ginossar N, Harris MS, Talhouarne GJS, Jackson SE, . . . Weissman JS (2014). Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell reports, 8(5), 1365–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacoff BP, Li Y, Lee SA, & Biteen JS (2019). SMALL-LABS: Measuring Single-Molecule Intensity and Position in Obscuring Backgrounds. Biophysical Journal, 116(6), 975–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalihal AP, Lund PE, & Walter NG (2019). Coming Together: RNAs and Proteins Assemble under the Single-Molecule Fluorescence Microscope. Cold Spring Harb Perspect Biol, 11(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiyama D, Sekine S, Barsi-Rhyne B, Hu J, Chen B, Gilbert LA, . . . Huang B. (2016). Versatile protein tagging in cells with split fluorescent protein. Nat Commun, 7, 11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karslake JD, Donarski ED, Shelby SA, Demey LM, DiRita VJ, Veatch SL, & Biteen JS (2019). SMAUG: Analyzing single-molecule tracks with nonparametric Bayesian statistics. bioRxiv, 578567–578567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz N, Cohen R, Solomon O, Kaufmann B, Atar O, Yakhini Z, . . . Amit R. (2018). An in Vivo Binding Assay for RNA-Binding Proteins Based on Repression of a Reporter Gene. ACS Synth Biol, 7(12), 2765–2774. [DOI] [PubMed] [Google Scholar]
- Kearse MG, & Wilusz JE (2017). Non-AUG translation: a new start for protein synthesis in eukaryotes. Genes Dev, 31(17), 1717–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieft JS, Rabe JL, & Chapman EG (2015). New hypotheses derived from the structure of a flaviviral Xrn1-resistant RNA: Conservation, folding, and host adaptation. RNA Biol, 12(11), 1169–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus GA, Jeon I, Nilsen-Hamilton M, Awad AM, Banerjee J, & Parvin B. (2008). Fluorinated analogs of malachite green: synthesis and toxicity. Molecules (Basel, Switzerland), 13(4), 986–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacerda R, Menezes J, & Romão L. (2017). More than just scanning: the importance of cap-independent mRNA translation initiation for cellular stress response and cancer. Cellular and Molecular Life Sciences, 74(9), 1659–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim F, & Peabody DS (2002). RNA recognition site of PP7 coat protein. Nucleic Acids Research, 30(19), 4138–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liss V, Barlag B, Nietschke M, & Hensel M. (2015). Self-labelling enzymes as universal tags for fluorescence microscopy, super-resolution microscopy and electron microscopy. Scientific reports, 5, 17740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon K, Aguilera LU, Morisaki T, Munsky B, & Stasevich TJ (2019). Live-Cell Single RNA Imaging Reveals Bursts of Translational Frameshifting. Molecular Cell, 75(1), 172–183.e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon K, & Stasevich TJ (2017). Imaging Translational and Post-Translational Gene Regulatory Dynamics in Living Cells with Antibody-Based Probes. Trends in Genetics, 33(5), 322–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelini F, Pitchiaya S, Vitelli V, Sharma S, Gioia U, Pessina F, . . . d’Adda di Fagagna F. (2017). Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nature Cell Biology, 19, 1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnier N, Barry Z, Park HY, Su KC, Katz Z, English BP, . . . Bathe M. (2015). Inferring transient particle transport dynamics in live cells. Nat Methods, 12(9), 838–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisaki T, Lyon K, DeLuca KF, DeLuca JG, English BP, Zhang Z, . . . Stasevich TJ (2016). Real-time quantification of single RNA translation dynamics in living cells. Science, 352(6292), 1425–1429. [DOI] [PubMed] [Google Scholar]
- Paige JS, Wu KY, & Jaffrey SR (2011). RNA mimics of green fluorescent protein. Science, 333(6042), 642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchapakesan SSS, Ferguson ML, Hayden EJ, Chen X, Hoskins AA, & Unrau PJ (2017). Ribonucleoprotein purification and characterization using RNA Mango. Rna, 23(10), 1592–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HY, Buxbaum AR, & Singer RH (2010). Single mRNA tracking in live cells. Methods Enzymol, 472, 387–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichon X, Bastide A, Safieddine A, Chouaib R, Samacoits A, Basyuk E, . . . Bertrand E. (2016). Visualization of single endogenous polysomes reveals the dynamics of translation in live human cells. 214(6), 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitchiaya S, Androsavich JR, & Walter NG (2012). Intracellular single molecule microscopy reveals two kinetically distinct pathways for microRNA assembly. EMBO reports, 13(8), 709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitchiaya S, Heinicke LA, Custer TC, & Walter NG (2014). Single Molecule Fluorescence Approaches Shed Light on Intracellular RNAs. Chemical Reviews, 114(6), 3224–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitchiaya S, Heinicke LA, Park JI, Cameron EL, & Walter NG (2017). Resolving Subcellular miRNA Trafficking and Turnover at Single-Molecule Resolution. Cell reports, 19(3), 630–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitchiaya S, Krishnan V, Custer TC, & Walter NG (2013). Dissecting non-coding RNA mechanisms in cellulo by Single-molecule High-Resolution Localization and Counting. Methods (San Diego, Calif.), 63(2), 188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitchiaya S, Mourao MDA, Jalihal AP, Xiao L, Jiang X, Chinnaiyan AM, . . . Walter NG (2019). Dynamic Recruitment of Single RNAs to Processing Bodies Depends on RNA Functionality. Molecular Cell, 74(3), 521–533.e526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo J, & Wilusz J. (2017). Trick or TREAT: A Scary-Good New Approach for Single-Molecule mRNA Decay Analysis. Mol Cell, 68(3), 476–477. [DOI] [PubMed] [Google Scholar]
- Saurabh S, Perez AM, Comerci CJ, Shapiro L, & Moerner WE (2016). Super-resolution Imaging of Live Bacteria Cells Using a Genetically Directed, Highly Photostable Fluoromodule. J Am Chem Soc, 138(33), 10398–10401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütz GJ, Schindler H, & Schmidt T. (1997). Single-molecule microscopy on model membranes reveals anomalous diffusion. Biophysical Journal, 73(2), 1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar S, Pitchiaya S, Malik R, Kothari V, Hosono Y, Yocum AK, . . . Chinnaiyan AM (2016). KRAS Engages AGO2 to Enhance Cellular Transformation. Cell reports, 14(6), 1448–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokabe M, & Fraser C. (2018). Toward a Kinetic Understanding of Eukaryotic Translation. Cold Spring Harb Perspect Biol, 11, a032706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Filonov GS, Kim H, Hirsch M, Li X, Moon JD, & Jaffrey SR (2017). Imaging RNA polymerase III transcription using a photostable RNA-fluorophore complex. Nat Chem Biol, 13(11), 1187–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart MP, Sharei A, Ding X, Sahay G, Langer R, & Jensen KF (2016). In vitro and ex vivo strategies for intracellular delivery. Nature, 538(7624), 183–192. [DOI] [PubMed] [Google Scholar]
- Strack RL, Disney MD, & Jaffrey SR (2013). A superfolding Spinach2 reveals the dynamic nature of trinucleotide repeat-containing RNA. Nat Methods, 10(12), 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunbul M, & Jaschke A. (2013). Contact-mediated quenching for RNA imaging in bacteria with a fluorophore-binding aptamer. Angew Chem Int Ed Engl, 52(50), 13401–13404. [DOI] [PubMed] [Google Scholar]
- Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, & Vale RD (2014). A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell, 159(3), 635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga M, Imamoto N, & Sakata-Sogawa K. (2008). Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat Methods, 5(2), 159–161. [DOI] [PubMed] [Google Scholar]
- Tourrière H, Chebli K, & Tazi J. (2002). mRNA degradation machines in eukaryotic cells. Biochimie, 84(8), 821–837. [DOI] [PubMed] [Google Scholar]
- Tutucci E, Livingston NM, Singer RH, & Wu B. (2018). Imaging mRNA In Vivo, from Birth to Death. Annu Rev Biophys, 47, 85–106. [DOI] [PubMed] [Google Scholar]
- Tutucci E, Vera M, Biswas J, Garcia J, Parker R, & Singer RH (2018). An improved MS2 system for accurate reporting of the mRNA life cycle. Nat Methods, 15(1), 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Han B, Zhou R, & Zhuang X. (2016). Real-Time Imaging of Translation on Single mRNA Transcripts in Live Cells. Cell, 165(4), 990–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner KD, Chen MC, Song W, Strack RL, Thorn A, Jaffrey SR, & Ferré-D’Amaré AR (2014). Structural basis for activity of highly efficient RNA mimics of green fluorescent protein. Nature structural & molecular biology, 21(8), 658–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner KD, Sjekloca L, Song W, Filonov GS, Jaffrey SR, & Ferre-D’Amare AR (2017). A homodimer interface without base pairs in an RNA mimic of red fluorescent protein. Nat Chem Biol, 13(11), 1195–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson IA, Niman HL, Houghten RA, Cherenson AR, Connolly ML, & Lerner RA (1984). The structure of an antigenic determinant in a protein. Cell, 37(3), 767–778. [DOI] [PubMed] [Google Scholar]
- Wu B, Eliscovich C, Yoon YJ, & Singer RH (2016). Translation dynamics of single mRNAs in live cells and neurons. 352(6292), 1430–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Miskolci V, Sato H, Tutucci E, Kenworthy CA, Donnelly SK, . . . Hodgson L. (2015). Synonymous modification results in high-fidelity gene expression of repetitive protein and nucleotide sequences. Genes Dev, 29(8), 876–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Zaccara S, Khuperkar D, Kim H, Tanenbaum ME, & Jaffrey SR (2019). Live imaging of mRNA using RNA-stabilized fluorogenic proteins. Nat Methods, 16(9), 862–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, & Ren Y. Mechanisms of nuclear mRNA export: A structural perspective. 0(0). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Hoek TA, Vale RD, & Tanenbaum ME (2016). Dynamics of Translation of Single mRNA Molecules In Vivo. Cell, 165(4), 976–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Fei J, Leslie BJ, Han KY, Kuhlman TE, & Ha T. (2015). Tandem Spinach Array for mRNA Imaging in Living Bacterial Cells. Scientific reports, 5, 17295–17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Lee SW, Zhao L, Xia T, & Qin PZ (2010). Conformational distributions at the N-peptide/boxB RNA interface studied using site-directed spin labeling. Rna, 16(12), 2474–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N, Kamijo K, Fox PD, Oda H, Morisaki T, Sato Y, . . . Stasevich TJ (2019). A genetically encoded probe for imaging nascent and mature HA-tagged proteins in vivo. Nat Commun, 10(1), 2947. [DOI] [PMC free article] [PubMed] [Google Scholar]