

Abstract
Prostate cancer is primarily fatal after it becomes metastatic and castration-resistant despite novel combined hormonal and chemotherapeutic regimens. Hence, new therapeutic concepts and drug delivery strategies are urgently needed for the eradication of this devastating disease. Here we report the highly specific, in situ click chemistry driven pretargeted delivery of cytotoxic drug carriers to PSMA(+) prostate cancer cells. Anti-PSMA 5D3 mAb and its F(ab′)2 fragments were functionalized with trans-cyclooctene (TCO), labeled with a fluorophore, and used as pretargeting components. Human serum albumin (ALB) was loaded with the DM1 antitubulin agent, functionalized with PEGylated tetrazine (PEG4-Tz), labeled with a fluorophore, and used as the drug delivery component. The internalization kinetics of components and the therapeutic efficacy of the pretargeted click therapy were studied in PSMA(+) PC3-PIP and PSMA(−) PC3-Flu control cells. The F(ab′)2 fragments were internalized faster than 5D3 mAb in PSMA(+) PC3-PIP cells. In the two-component pretargeted imaging study, both components were colocalized in a perinuclear location of the cytoplasm of PC3-PIP cells. Better colocalization was achieved when 5D3 mAb was used as the pretargeting component. Consecutively, the in vitro cell viability study shows a significantly higher therapeutic effect of click therapy in PC3-PIP cells when 5D3 mAb was used for pretargeting, compared to its F(ab′)2 derivative. 5D3 mAb has a longer lifetime on the cell surface, when compared to its F(ab′)2 analogue, enabling efficient cross-linking with the drug delivery component and increased efficacy. Pretargeting and drug delivery components were cross-linked via multiple bioorthogonal click chemistry reactions on the surface of PSMA(+) PC cells forming nanoclusters, which undergo fast cellular internalization and intracellular transport to perinuclear locations.
Keywords: pretargeted therapy, drug delivery, PSMA(+) prostate cancer, nanomedicine, bioorthogonal click chemistry
Graphical Abstract
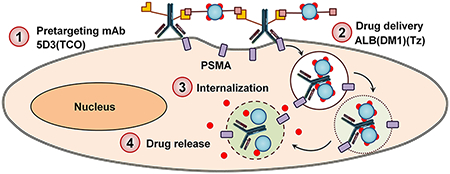
1. INTRODUCTION
Prostate cancer (PC) is the most common noncutaneous malignancy in American men.1–3 Over 170 000 new cases of PC are diagnosed, and approximately 30 thousand patients die from PC each year in the US.4 Conventional treatments such as surgery, radiation, and androgen deprivation are effective; however, PC is essentially incurable once it becomes metastatic and castration-resistant.5–9 That outcome persists despite the approval of several new drugs and combined hormonal and chemotherapeutic regimens.10,11 Novel therapeutic approaches and cancer-specific drug delivery strategies are urgently needed for the improved management and eventual complete eradication of this devastating disease.12–14
The prostate-specific membrane antigen (PSMA) is a type II membrane protein that is highly expressed in aggressive PC.15–18 Due to the cell-surface and cancer-specific nature of PSMA, and its rapid cellular internalization and recycling kinetics, PSMA is a leading target for primary and metastatic PC diagnosis, imaging, and therapy.19–21 PSMA has glutamate carboxypeptidase activity and can be targeted with small-molecule substrate analogues as well as engineered peptides, aptamers, and antibodies.22–31 Nanosize hyperbranched polymers conjugated with PSMA-specific small molecules, peptides, and anti-PSMA antibodies have also been successfully studied as therapeutic delivery platforms targeting PSMA(+) PC in mouse models.32 Anti-PSMA antibodies typically enhance the internalization of PSMA and can be used for the delivery of isotopes and drugs targeting PSMA(+) prostate cancers.33 Only two monoclonal antibodies, J591 and 7E11, have moved to clinical trials, and radiolabeled variants of J591 have progressed to Phase II.34–36 The 5D3 mAb is a novel anti-PSMA mAb, recently developed and used for imaging PC.37 In preclinical binding and imaging studies, the 5D3 mAb has demonstrated higher PSMA binding affinity when compared to J591.37,38
Antibody–drug conjugate (ADC) drug delivery systems have shown promising results in cancer therapy over recent decades.39–42 Target specificity and the use of a minimum amount of highly cytotoxic chemotherapeutics increase ADC efficacy.43 Moving forward, pretargeted drug delivery can be an even more effective strategy by circumventing the use of highly cytotoxic chemotherapeutics with significant off-target effects in cancer treatment.44,45 We have previously developed a bioorthogonal pretargeted two-component drug delivery system driven by trans-cyclooctene (TCO) and PEGylated tetrazine (PEG4-Tz) in situ click chemistry for specific delivery of therapeutics to HER2(+) breast cancer.44 This strategy can be applied to other cancer types such as PC, which overexpress cancer-specific PSMA on the cell surface.
The trans-cyclooctene-tetrazine (TCO-Tz) click chemistry is a strain-promoted reaction that does not require catalysts and can proceed under mild conditions. TCO-Tz click chemistry is ideal for biological applications, as it can be performed under physiological conditions and releases no toxic byproducts.46,47 This inverse-electron demand Diels–Alder (IEDDA) cycloaddition reaction exhibits exceptional kinetics (k > 100 000 M−1 s−1) and chemoselectivity. We have utilized this bioorthogonal click reaction for in situ conjugations of two components in physiological conditions to enhance the cellular internalization of drugs, and this strategy has shown enhanced therapeutic efficacy in preclinical breast cancer models. In this study, we have applied and extended the strategy of bioorthogonal pretargeted drug delivery to PSMA(+) PC using novel functionalized 5D3 mAb or its F(ab′)2 domains as the pretargeting component, and the highly cytotoxic drug, mertansine (DM1)-loaded human serum albumin (ALB), as the drug delivery component. This approach has demonstrated high efficacy in PSMA(+) PC cells.
2. MATERIALS AND METHODS
2.1. Antibody, Chemotherapeutics, and Chemicals.
5D3 mAb in PBS with 0.02% NaN3 was prepared as described previously and used after buffer exchange to pure BupH phosphate buffered saline (PBS).37 Mertansine (DM1) was purchased from Abcam, Inc. The amine-reactive linkers, TCO-NHS and methyltetrazine-PEG4-NHS esters, were purchased from Sigma-Aldrich Corp. and KeraFast, Inc., respectively. Sulfo-SMCC heterobifunctional crosslinker, NHS esters of fluorophores, and Dulbecco’s phosphate-buffered saline (DPBS) were purchased from ThermoFisher, Inc.
2.2. 5D3 mAb Fragmentation.
The F(ab′)2 fragments of 5D3 mAb were prepared using the Pierce Mouse IgG1 Fab and F(ab′)2 Preparation Kit (Thermo Scientific) following the manufacturer’s protocol. Briefly, 5D3 antibody (3 mg in 0.5 mL of PBS) was digested for 24 h at 37 °C in mouse IgG1 digestion buffer with the provided Immobilized Ficin resin in the presence of cysteine·HCl (0.5 mL of 0.7 mg/mL). Digested F(ab′)2 fragments were separated from the resin by centrifugation (5000g, 1 min) followed by 3 separate washes with provided protein A binding buffer to collect a total of 2.0 mL. The F(ab′)2 fragments were purified using a Nab protein A plus spin column and protein A binding buffer.
2.3. Synthesis of Pretargeting Components.
The 5D3 mAb or F(ab′)2 fragments (2 mg in 500 μL of PBS) were treated with TCO-NHS ester (250 mol equiv in 10–20 μL of anhydrous DMSO) and incubated for 2 h. The 2–4% DMSO/PBS reaction mixture was initially cloudy, and TCO-NHS ester was not completely soluble. However, the reaction mixture was clear after 1-2 h, and excess TCO-NHS and byproducts were removed by ultrafiltration (Scheme 1). The product was further purified by size-exclusion column chromatography (SEC). The comparatively higher equivalent of TCO-NHS ester was required for this reaction, possibly due to poor solubility. The number of TCO groups attached per antibody or antibody fragments was determined based on the change of molecular weight measured by Voyager DE-STR MALDI-ToF mass spectrometer (Figure S1). The change of molecular weight corresponds to the total molecular weights of TCO groups attached to 5D3 or F(ab′)2.
Scheme 1. Synthesis of 5D3 mAb and Its F(ab′)2-Based Pretargeting Componentsa.
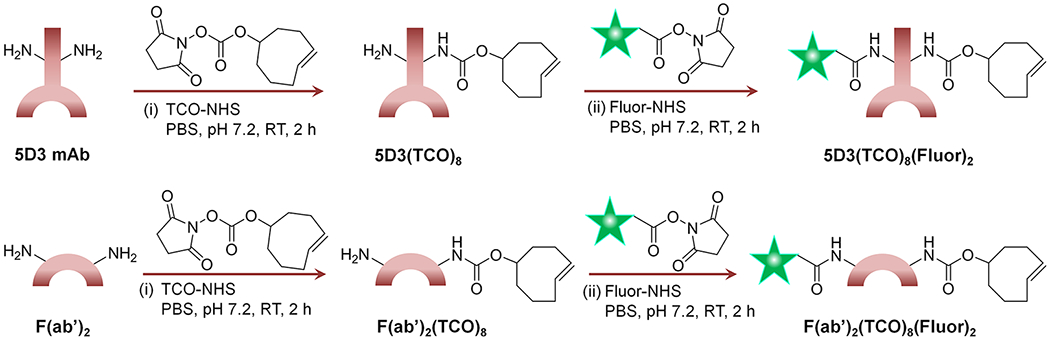
a(i) 5D3 mAb was treated with TCO-NHS to obtain 5D3(TCO)8 and then (ii) labeled with a suitable fluorophore to obtain the final product, 5D3(TCO)8(Fluor)2. The same synthetic route was followed starting with F(ab′)2 of 5D3 to obtain (i) F(ab′)2(TCO)8 and (ii) F(ab′)2(TCO)8(Fluor)2. Subscripted numbers in the formulas indicate the number of TCO or fluorophores attached per biomolecule.
The number of groups attached is denoted as the subscripted number in the formulas. The hydrodynamic diameter of components was measured by dynamic light scattering (DLS) using a Nano-ZS90 (Malvern Instruments, U.K.) Zetasizer (Figure S2). For imaging experiments and flow cytometry, the native 5D3 antibody, F(ab′)2, and functionalized 5D3(TCO)8, and F(ab′)2(TCO)8 (~500 μL of 1 mg/mL in PBS) were labeled with AlexaFluor 488 or Cy5 by adding AlexaFluor 488 NHS ester or Cy5-NHS ester (10–20 mol equiv in 10 μL of anhydrous DMSO) and stirred for 2 h, and excess dye was removed by ultrafiltration. A fraction of fluorescent 5D3(AF-488)2 (250 μg in 250 μL in PBS) was further labeled with a low-pH fluorescence indicator, pHrodo, by adding pHrodo-NHS (20–50 mol equiv in 10 μL of anhydrous DMSO). After stirring for 2 h, the excess dye was removed by ultrafiltration. After fluorescence labeling, all products were further purified by SEC chromatography. The degree of fluorescence labeling was determined following the manufacturer’s protocol.
2.4. Synthesis of Delivery Components.
Human serum albumin (ALB, 20 mg in 4 mL of PBS) was treated with a 10-fold molar excess of sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (Sulfo-SMCC) in dry DMSO and stirred for 2 h (Scheme 2). The excess SMCC and byproducts were removed by ultrafiltration, and the product was further purified by size-exclusion chromatography (SEC). The ALB functionalized by SMCC (10 mg in 2 mL of PBS with 2 mM EDTA, pH 6.5) was treated with mertansine (DM1, 10 mol equiv in 10 μL of anhydrous DMSO) and stirred for 20 h at room temperature. The excess DM1 was removed by ultrafiltration, and the product, ALB(DM1)3.3, was further purified by SEC chromatography. ALB(DM1)3.3 (5 mg in 1 mL of PBS) was functionalized with PEG4-Tz by adding methyltetrazine-PEG4-NHS ester (100 mol equiv in 10 μL of anhydrous DMSO) and stirring for 2 h. The resulting product ALB(DM1)3.3(PEG4-Tz)10 was purified by ultrafiltration, followed by SEC chromatography. The product, ALB(DM1)3.3(PEG4-Tz)10 (3 mg in 1 mL of PBS), was treated with rhodamine-NHS ester or AlexaFluor 488 NHS ester (20 mol equiv in 10–20 μL of anhydrous DMSO) and stirred for 2 h to obtain ALB(DM1)3.3(PEG4-Tz)10(Rhod)2 and ALB(DM1)3.3(PEG4-Tz)10(AF-488)2. The product was finally purified by ultrafiltration followed by SEC chromatography. After each step, the molecular weights of intermediates and final products were determined using the Voyager DE-STR MALDI-TOF mass spectrometer. The degree of MCC linker conjugation, DM1 drug load, and PEG4-Tz functionalization were determined based on the change of molecular weights (Figure S1) and is denoted as subscripts in formulas. The degree of fluorophore labeling was determined following the manufacturer’s protocol.
Scheme 2. Synthesis of the Albumin (ALB)-Based Drug delivery Componenta.
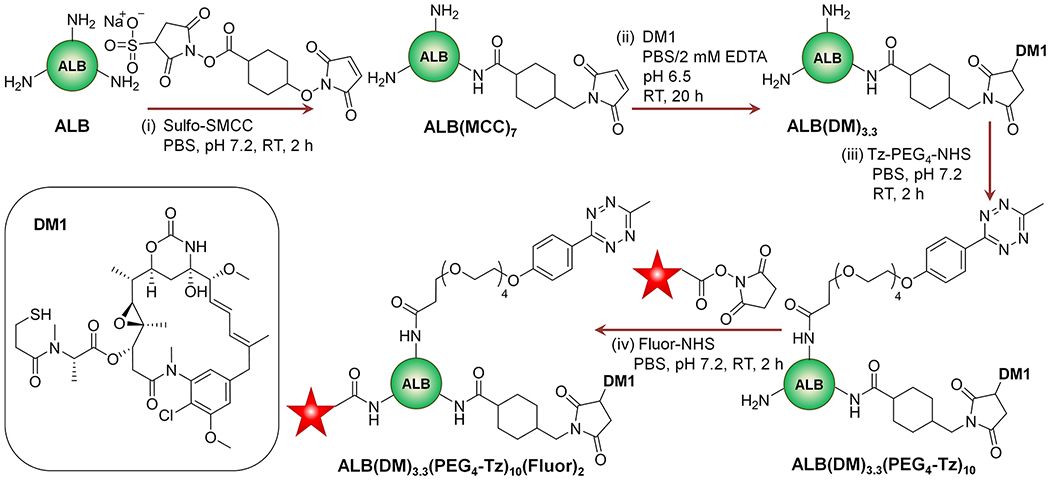
a(i) ALB was treated with the sulfo-SMCC bifunctional linker to obtain ALB(MCC)7. (ii) It was conjugated with DM1 drug molecules, and the resultant ALB(DM1)3.3 was treated with (iii) NHS-PEG4-TZ ester to obtain ALB(DM1)3.3(PEG4-Tz)10. (iv) It was labeled with a suitable fluorophore to obtain the final product, ALB(DM1)3.3(PEG4-Tz)10(Fluor)2. The subscripted numbers in the formulas indicate the number of MCC, DM1, PEG4-Tz groups, or fluorophores attached per protein.
2.5. Purification of the Products.
After each step of the synthesis, the excess small molecular reagents and byproducts were removed by Amicon ultrafiltration using 15 mL, 30 kDa MWCO units (for ≥2 mg samples) or 0.5 mL, 30 kDa MWCO units (for ≤2 mg samples). The products were further purified by SEC chromatography by the Waters binary pump/dual absorbance HPLC system equipped with a YMC-Pack Diol-300 (300 × 8.0 mm I.D.; particle size, 5 μm; pore size, 30 nm) size-exclusion column, using 0.1 M PBS with 0.2 M NaCl (pH 7.2) as the mobile phase.
2.6. Cells.
PSMA(+) PC3-PIP and PSMA(−) PC3-Flu cells were used to prove the strategy in PSMA(+) PC. Cells were grown in RPMI 1640 medium supplemented with 10% FBS and 1% penicillin–streptomycin and maintained in a humidified incubator at 37 °C with 5% CO2. Cells were confirmed to be free of mycoplasma contamination.
2.7. Time-Dependent Internalization of Pretargeting Components.
PC3-PIP or PC3-Flu cells were seeded in 4-well chamber slides (0.2 million cells per chamber) and grown for 24–48 h to 80–90% confluency. Each chamber was treated with 150 μL of 20 μg/mL of 5D3(AF-488)2 or F(ab′)2(AF-488)2 and incubated at 37 °C under CO2 and humidity-controlled conditions. Chambers at each time point (15 min, 1, 6, 24 h) were washed with Dulbecco’s phosphate buffered saline (DPBS) for 5 min and fixed using 4% paraformaldehyde (PFA) for 10 min on ice. The nuclei were counterstained using Hoechst 33342 and wet mounted. Images were taken using an inverted confocal microscope and analyzed using dedicated software developed in the IDL and MatLab environment. Briefly, the program searches for the cell membrane-extracellular medium interface using a user-defined coarse outline of the cell membrane. The algorithm provides the precise position of the plasma membrane based on the intensity gradient analysis of the images. Membrane areas with low fluorescence signals are interpolated using neighboring regions with a high fluorescence intensity. Integral fluorescence localized to the membrane (within the search radius from the interface) and the integral internalized signal within the space enclosed within the membrane was evaluated for each cell within the field of view. The measured values are exported in a tabular format.
2.8. mAb Internalization Inhibition Study.
PC3-PIP cells (0.2 million/chamber) were seeded in 4-well chamber slides and grown for 24–48 h to 80–90% confluency. Each chamber was treated with 150 μL of 2 μg/mL 5D3(AF-488)2 in DPBS in the presence of endocytosis inhibitors, chlorpromazine hydrochloride (10 μg/mL), methyl-β-cyclodextrin (5 mM), or Nocodazole (20 μM). Two control experiments were performed treating PC3-PIP cells with 2 μg/mL 5D3(AF-488) alone and incubated at 4 and 37 °C.48 After 1 h, cells were washed with DPBS and fixed with 4% PFA in PBS. After the nuclear counterstaining by Hoechst 33342, the slide was wet mounted, and cells were imaged using a Zeiss AxioObserver confocal fluorescence microscope with an LSM700 confocal module.
2.9. Flow Cytometry Analysis.
For the determination of PSMA expression levels, PC3-PIP and PC3-Flu cells were seeded in a 6-well plate (0.4 million/well) and grown for 24 h to ~90% confluency. Then cells were treated with 5D3(Cy5)2 or F(ab′)2(Cy5)2 (20 μg/mL) and incubated for 30 min at 4 °C. Cells were washed once using DPBS and harvested by trypsinization. Cell pellets were resuspended in buffer and fixed by 4% PFA for flow cytometry. To prove the click delivery, PSMA(+) PC3-PIP and PSMA(−) PC3-Flu cells were seeded in a 6-well plate (0.4 million/well) and grown for 24 h to ~90% confluency. Then, cells in each well were treated with one of the reactive pretargeting components, 5D3-(TCO)8(Cy5)2, F(ab′)2(TCO)8(Cy5)2, or unreactive 5D3-(Cy5)2, and F(ab′)2(Cy5)2 (20 μg/mL), and incubated for 20 min at 37 °C. Cells were washed once with DPBS, treated with a drug delivery component, ALB(DM1)3.3(PEG4-Tz)10(AF-488)2 (50 μg/mL in DPBS), and incubated for 30 min at 37 °C. Then, the treating solution was replaced by fresh media, and cells were incubated for an additional 1.5–2 h. Cells were washed once using DPBS and harvested by trypsinization. Cell pellets were resuspended in buffer and fixed by 4% PFA. Cells were analyzed on a BD LSR II Flow Cytometer using red (633 nm) and blue (488 nm) lasers for Cy5 and AlexaFluor 488, respectively. Histograms for PSMA expression levels and fluorescence density plots for each click delivery tests are presented in Figure 2.
Figure 2.
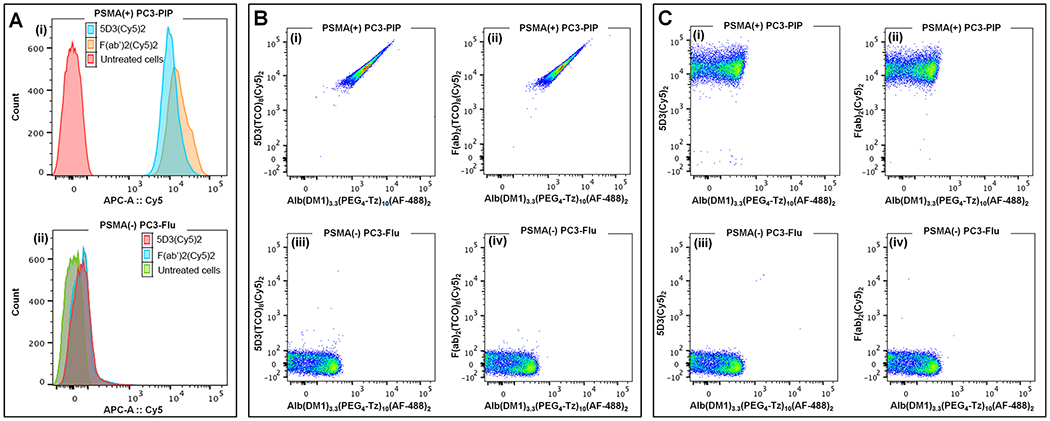
Flow cytometry analysis of the pretargeted click-chemistry delivery. (A) PSMA expression levels in (i) PC3-PIP and (ii) PC3-Flu cells. Combined histograms of untreated cells and cells treated with 5D3(Cy5)2 and F(ab′)2(Cy5)2. (B) Pretargeted delivery in the presence of the click reaction between Alb(DM1)3.3(PEG4-Tz)10(AF-488)2 and reactive (i) 5D3(TCO)8(Cy5)2 in PC3-PIP cells, (ii) F(ab′)2(TCO)8(Cy5)2 in PC3-PIP cells, (iii) 5D3(TCO)8(Cy5)2 in PC3-Flu cells, and (iv) F(ab′)2(TCO)8(Cy5)2 in PC3-Flu cells. (C) Pretargeted delivery in the absence of a click reaction between ALB(DM1)3.3(PEG4-Tz)10(AF-488)2 and unreactive (i) 5D3(Cy5)2 in PC3-PIP cells, (ii) F(ab′)2(Cy5)2 in PC3-PIP cells, (iii) 5D3(Cy5)2 in PC3-Flu cells, and (iv) F(ab′)2(Cy5)2 in PC3-Flu cells.
2.10. In Vitro Two-Component Delivery Imaging Study.
PC3-PIP and PC3-Flu cells grown in 4-well chamber slides were treated with 5D3(TCO)8(AF-488)2 or F-(ab′)2(TCO)8(AF-488)2 (150 μL of 20 μg/mL in each well) and incubated at 37 °C for 30 min, and unbound pretargeting components were washed by DPBS. Cells were then treated with ALB(DM1)3.3(PEG4-Tz)10(Rhod)2 (150 μL of 50 μg/mL in DPBS) at 37 °C for 30 min. After the washing step, DPBS was replaced by the growth medium, and the incubation was continued for 30 min, 1, 2, and 4 h. After each time point, selected chambers of cells were fixed by 4% PFA for 20 min at 4 °C and washed with deionized H2O. Fixed slides were imaged using the Zeiss AxioObserver confocal fluorescence microscope with an LSM700 confocal module and analyzed using NIH ImageJ.
2.11. In Vitro Study of Pretargeted Therapy.
PC3-PIP or PC3-Flu cells (2000 cells/well in 200 μL of a medium) were seeded in a 96-well plate and incubated at 37 °C for 24 h. Cultured cells were treated with or without modified or unmodified 5D3 or its F(ab′)2 fragments (20 μg/mL in 100 μL of DPBS) and incubated at 37 °C for 20 min. Cells were washed with DPBS and incubated for 2 h with or without the drug delivery component ALB(DM1)3.3(PEG4-Tz)10(Rhod)2 or with DM1 (1.5 μg/mL in 100 μL of DPBs). After treatment, cells were washed with DPBS and resupplied with fresh media. After the incubation for 48 h, the cell viability was determined using the WST-8 assay, following the manufacturer’s protocol. Briefly, cells in 100 μL of media were treated with 10 μL of the WST-8 reagent per well and incubated at 37 °C. After 3 h, the absorbance was measured at 450 nm. The WST-8 is tetrazolium salt, which is reduced by dehydrogenase in living cells forming a yellow formazan dye. This dye turns media into a yellow color (450 nm absorption peak). The concentration of the formazan dye in the media produced by the activities of dehydrogenases in living cells is directly proportional to the number of viable cells. The cell viability of treated cells was normalized to readings in untreated control cells, which were considered to have 100% viability.
2.12. Statistical Analysis.
The in vitro therapeutic study was performed in triplicate per plate, and duplicate independent experiments were performed for statistical analysis. The WST-8 assay test was quadruplicated per plate, and triplicate independent experiments were carried out for the statistical analysis. The one-way analysis of variance (ANOVA) was used for the omnibus F-test, and the Scheffé’s test was used for post hoc analysis (StatPlus:mac, AnalystSoft Inc., Alexandria, VA, USA). Changes in the cell viability were considered significant (p-value < 0.05) when the F value is greater than the critical value of the F-distribution.
3. RESULTS
3.1. Design and Synthesis of Components.
Anti-PSMA 5D3 mAb and F(ab′)2 fragments of 5D3 mAb were labeled with AlexaFluor-488 for the in vitro internalization experiments. For pretargeting experiments, 5D3 mAb and F(ab′)2 were first functionalized with TCO bioorthogonal reactive groups. The number of TCO groups was maintained at ~8 for both biomolecules. The second delivery component that was based on human serum ALB was functionalized by the SMCC linker and conjugated with DM1 by maleimide–thiol conjugation. It was further functionalized with tetrazine, the corresponding bioorthogonal click reactive group. The number of drug molecules and tetrazine groups was maintained at ~3 and ~10, respectively. Molecular weights of all intermediate and final components were measured by MALDI-TOF (Figure S1). The number of conjugated groups and drugs was determined based on the change of molecular weight, as shown in Figure S1. The sizes of components were measured by dynamic light scattering using the Zetasizer (Figure S2). Changes in size after modifications were not statistically significant. For optical imaging, both mAb and ALB conjugates were labeled with rhodamine and AlexaFluor 488 fluorophores, respectively. For flow cytometry analysis, pretargeting and drug delivery components were labeled with Cy5 and AlexaFluor 488, respectively, to match with the configuration available in the FACS instrument.
3.2. Analysis of the Internalization of Pretargeting Components.
The internalization of fluorescence-labeled 5D3 and F(ab′)2 fragments was studied in vitro with PSMA(+) PC3-PIP cells (Figure S3). The fluorescence intensity of internalized and cell surface-bound components was measured by a dedicated software developed in IDL and MatLab. The ratio of the internalized and membrane-bound fluorescence was reported for each cell in the field of view. Both components exhibit similar internalization rates; 75% of cell surface-bound 5D3 was internalized within 2.5 h, whereas 75% of the internalization of F(ab′)2 took approximately 1 h. The fluorescence intensity faded with time; however, a significant amount of the pretargeting component was retained in the cytoplasm for up to 24 h. No significant specific binding and/or internalization was detected in PSMA(−) PC3-Flu cells (data not shown).
3.3. Localization of the Pretargeting Component.
5D3(AF-488)2(pHrodo)2 was used to determine the cellular microenvironment in which 5D3 mAb was localized after internalization. pHrodo red is an intracellular fluorescent pH indicator that only becomes fluorescent, at low pH (<6.5). The dye is typically used for the identification of late endosomes. After 4 h of incubation with PSMA(+) PC3-PIP cells, the intracellular compartments were bright red. This signal colocalized with the green 5D3 signal, indicating localization within the acidic late-stage endosomes (Figure 1A). Internalization was further investigated by imaging PC3-PIP cells treated with 5D3(AF-488)2 at 4 °C and at 37 °C in the presence of internalization inhibitors such as chloropromazine hydrochloride, which inhibits clathrin-mediated endocytosis; methyl-β-cyclodextrin, which inhibits caveolae-mediated endocytosis; and nocodazole, which is an antineoplastic agent that interferes with the polymerization of microtubules (Figure 1B). The fluorescence images of cells treated at 4 °C showed no internalization of 5D3. Despite the presence of inhibitors, 5D3 was predominantly internalized at 37 °C and localized in the cytoplasm, proving that internalization is not entirely controlled by clathrin-mediated or caveolae-mediated endocytosis, which agrees well with previously reported results.33,49,50
Figure 1.
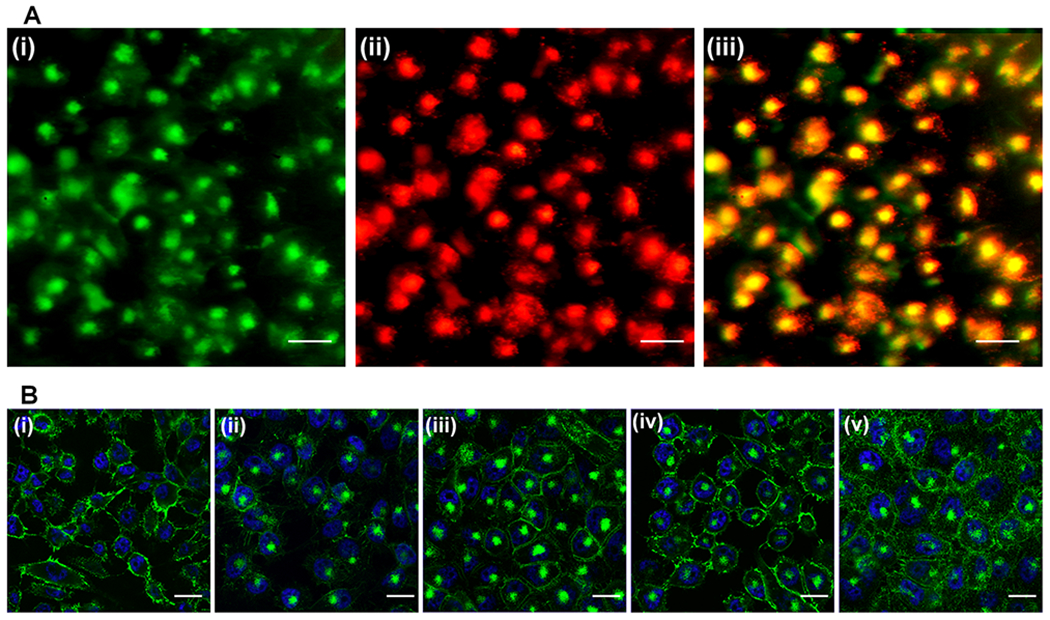
Internalization study of 5D3 mAb. (A) Internalization of 5D3(AF-488)2(pHrodo)2, green channel (i), low-pH-activated red channel (ii), and merged image (iii) (scale bar: 100 μm). (B) Internalization of 5D3 at 4 °C (i) and 37 °C (ii) and internalization of 5D3 in the presence of internalization inhibitors, chlorpromazine hydrochloride (iii), methyl-β-cyclodextrin (iv), and nocodazole (v) (scale bar: 100 μm).
3.4. Flow Cytometric Analysis of Click Delivery.
As shown in FACS histograms in Figure 2A, both 5D3(Cy5)2 and F(ab′)2(Cy5)2 have high binding affinity in PSMA(+) PC3-PIP cells, and no accumulation was observed in PSMA(−) PC3-Flu cells. Figure 2B shows density dot plots for both Cy5 and AF-488 channels. Both pretargeting and drug delivery components accumulated in PC3-PIP cells but not in PC3-Flu cells. Reactive 5D3(TCO)8(Cy5)2 and F(ab′)2(TCO)8(Cy5)2 pretargeting components and the ALB(DM1)3.3(PEG4-Tz)10(AF-488)2 drug carrier were used in these experiments. When the experiment was repeated using click unreactive pretargeting components (without the TCO group), 5D3-(Cy5)2 and F(ab′)2(Cy5)2, accumulation of Cy5-labeled pretargeting components was high in PC3-PIP cells and low in PC3-Flu cells, respectively (Figure 2C). The fluorescence signal corresponding to ALB(DM1)3.3(PEG4-Tz)10(AF-488)2 was low in both cell lines as there was no specific click reaction between the components.
3.5. Imaging Assessment of Click Delivery.
Figure 3A shows images of PC3-PIP cells treated with the two-component pretargeting system. First, the pretargeting component 5D3(TCO)8(AF-488)2 labeled the PSMA receptors on the cell surface. After washing the excess and unbound pretargeting components, cells were treated with the second delivery component, ALB(PEG4-Tz)10(Rhod)2. Both components underwent multiple strain-promoted TCO-Tz click reactions in physiological conditions, self-assembled, and formed nanoscale clusters on the cell surface. The nanoscale clusters can efficiently internalize into the cytoplasm.51,52
Figure 3.
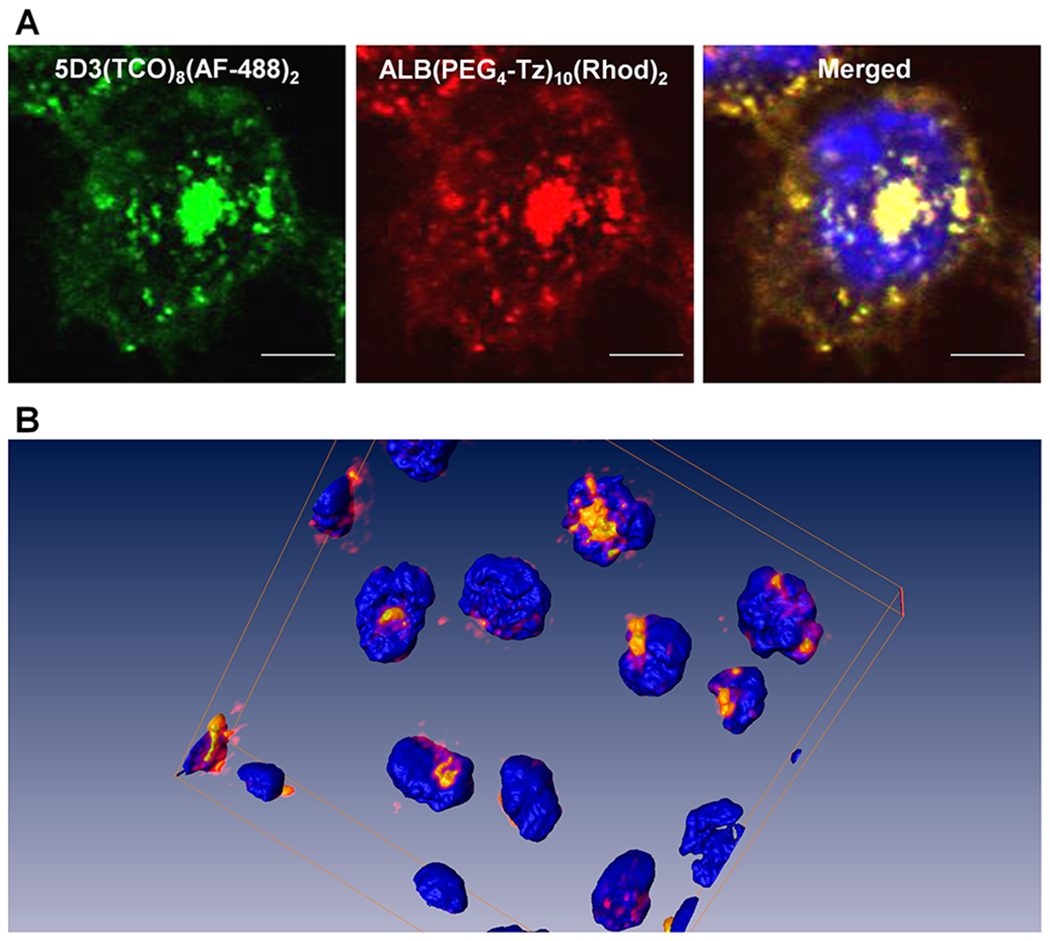
Fluorescence images of the pretargeted two-component delivery in PSMA(+) PC3-PIP cells, (A) using 5D3(TCO)8(AF-488)2 (green), ALB(PEG4-Tz)10(Rhod)2 (red), and nuclear staining by Hoechst 33342 (blue) (magnification 100×, scale bar: 30 μm). (B) 3D rendering of the click therapy components colocalized in the cytoplasm of PSMA(+) cells.
In this experiment, colocalization of two components was observed in the cytoplasm, depicting an efficient bioorthogonal two-component drug delivery in PSMA(+) PC cells using a 5D3 mAb-based pretargeted strategy. Multichannel images were rendered in the Amira 3D software platform (Thermo Fisher) to visualize the colocalization of two components in the cells (Figure 3B). We conducted an in vitro control experiment using the second delivery component, ALB(PEG4-Tz)10(Rhod)2, without the pretargeted step. PSMA(+) PC3-PIP cells treated with ALB(PEG4-Tz)10(Rhod)2 in the absence of pretargeting 5D3(TCO)8(AF-488)2, showed low accumulation of the delivery component on the cell surface or in the cytoplasm (Figure S4).
The pretargeting components, 5D3(TCO)8(AF-488)2 or F(ab′)2(TCO)8(AF-488)2, and the drug-carrier component, ALB(DM1)3.3(PEG4-Tz)10(Rhod)2, were first used to analyze time-dependent delivery by fluorescent imaging. In this strategy, the pretargeting component-labeled cell surface PSMA receptors react with the second component via in situ bioorthogonal TCO-Tz click reactions. Multiple click reactions among the components form nanoscale clusters that internalize faster and more efficiently than individual components.53 Based on the fluorescence image analysis, the time for internalization of 75% of 5D3-based pretargeting and delivery component complexes was 4 h (Figures 4 and 5). On the other hand, F(ab′)2-based pretargeting was significantly faster and resulted in the internalization of 75% of complexes within 2 h (Figures 6 and 7). Further analysis of fluorescence images of F(ab′)2-based delivery shows that a significant amount of F(ab′)2(TCO)8 is dispersed in the cytoplasm without colocalization with the red channel. These free F(ab′)2(TCO)8 were internalized before in situ bioorthogonal click reactions with the drug delivery component.
Figure 4.
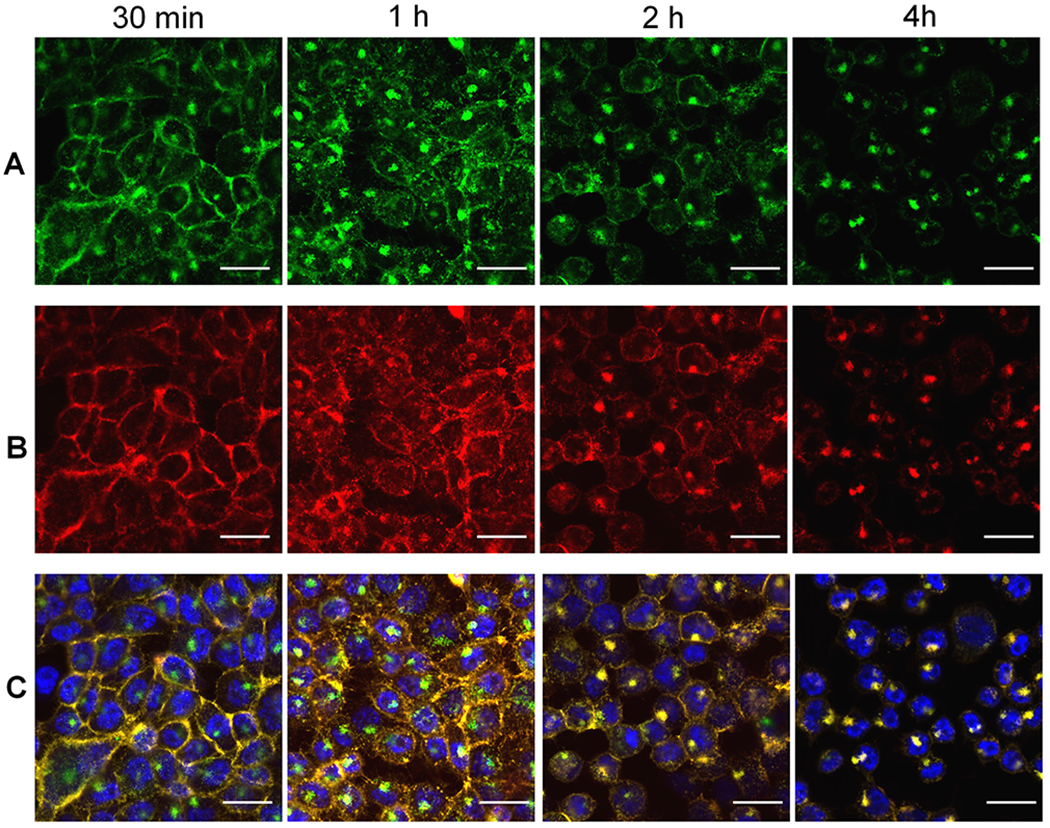
Time-dependent fluorescence images of PSMA(+) PC3-PIP cells in pretargeted drug delivery using 5D3(TCO)8(AF-488)2 and ALB(DM1)3.3(PEG4-Tz)10(Rhod)2. Panel A: Localization of the first component, 5D3(TCO)8(AF-488)2, over time after treatment with the drug delivery component (postdrug treatment time 30 min, 1, 2, 4 h). Panel B: Localization of the second drug delivery component, ALB(DM1)3.3(PEG4-Tz)10(Rhod)2, over time after the treatment (30 min, 1, 2, 4 h). Panel C: Merged images of corresponding green and red channels. (Scale bar: 100 μM.)
Figure 5.
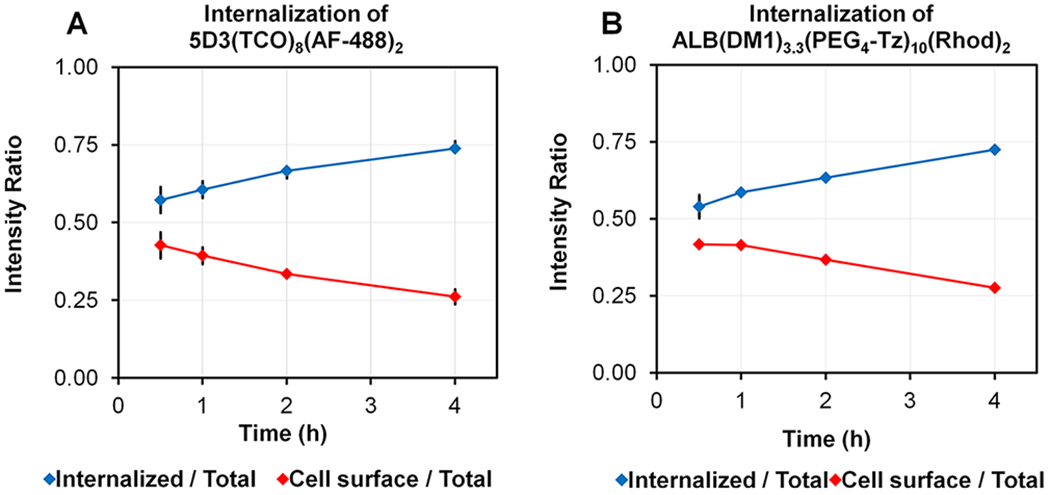
Change of the internalized and cell surface fluorescence intensity ratio of (A) 5D3(TCO)8(AF-488)2 and (B) ALB-(DM1)3.3(PEG4-Tz)10(Rhod)2 with time.
Figure 6.
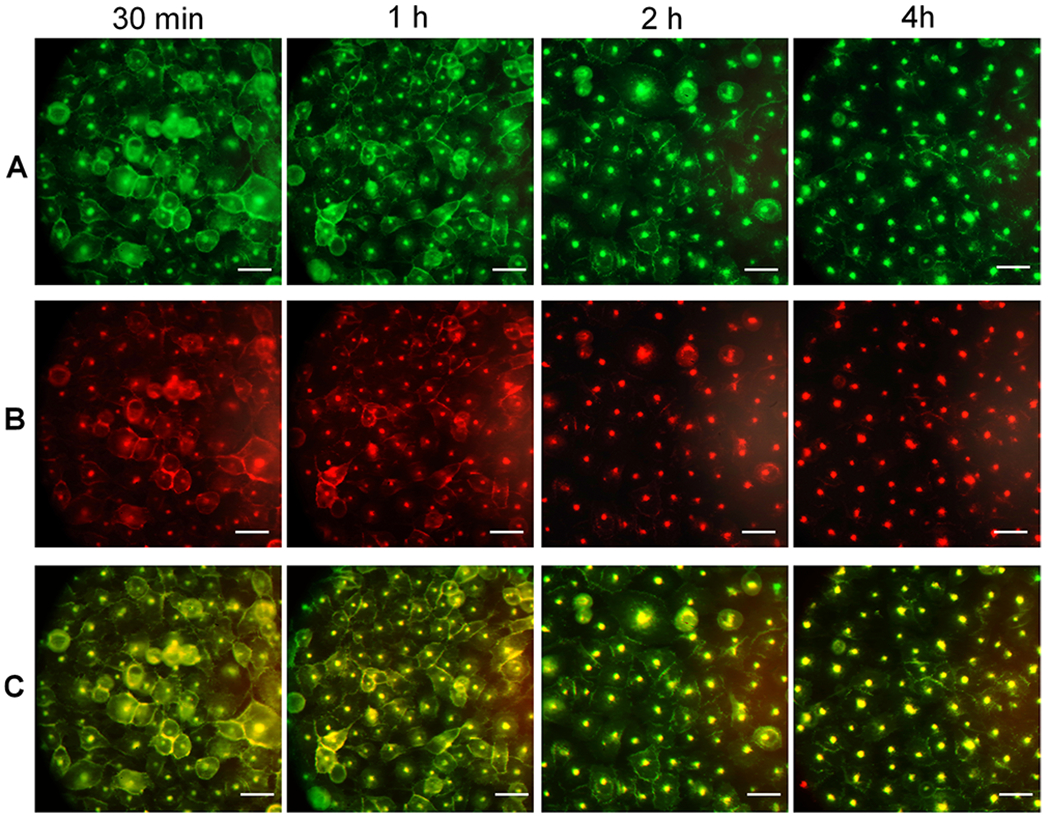
Time-dependent images of PSMA(+) PC3-PIP cells in pretargeted drug delivery using F(ab′)2(TCO)8(AF-488)2 and ALB(DM1)3.3(PEG4-Tz)10(Rhod)2. Panel A: Localization of the first component, F(ab′)2(TCO)8(AF-488)2, over the time after treatment with the drug delivery component (postdrug treatment time 30 min, 1, 2, 4 h). Panel B: Localization of the second drug delivery component, ALB(DM1)3.3(PEG4-Tz)10(Rhod)2, over the time after the treatment. Panel C: Merged images of corresponding green and red channels. (Scale bar: 100 μM.)
Figure 7.
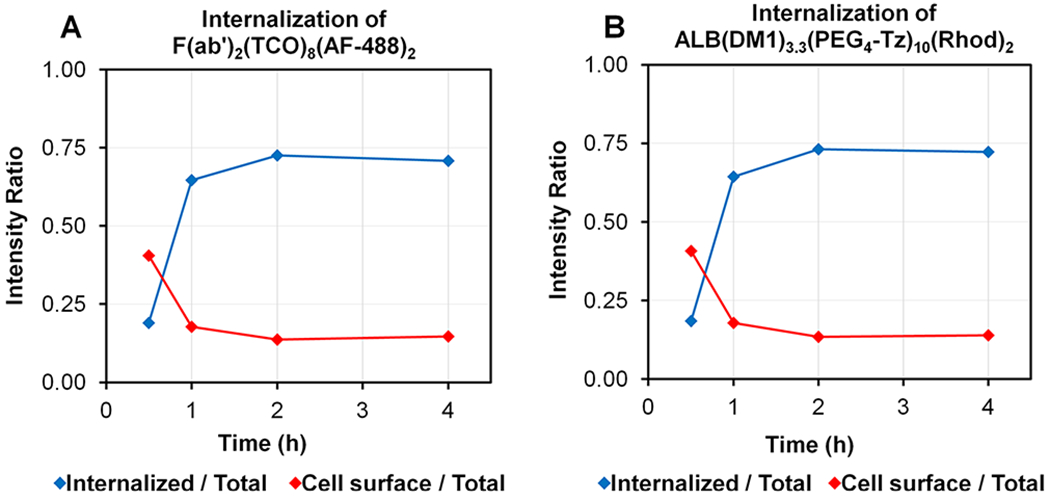
Change of the internalized and cell surface fluorescence intensity ratio of (A) 5D3-F(ab′)2(TCO)8(AF-488)2 and (B) ALB(DM1)3.3(PEG4-Tz)10(Rhod)2 with time.
3.6. Click Therapy on PSMA(±) Cells and Therapeutic Efficacy.
PSMA(+) PC3-PIP and PSMA(−) PC3-Flu cells were treated following the therapeutic schedule shown in Table 1. First, four series of cell chambers were treated with 5D3(TCO)8, F(ab′)2(TCO)8, unconjugated 5D3, or unconjugated F(ab′)2, washed to remove excess pretargeting components, and subsequently treated with the drug delivery component, ALB(DM1)3.3(PEG4-Tz)10. Next, four-cell chambers were treated with 5D3(TCO)8, F(ab′)2(TCO)8, 5D3 mAb, or F(ab′)2 fragments alone to evaluate the effect of pretargeting components on cell viability. Finally, two cell chambers were treated with ALB(DM1)3.3(PEG4-Tz)10 or pure DM1 equiv of the DM1 concentration in ALB(DM1)3.3(PEG4-Tz)10 to measure the effect of the drug or the drug carrier without the pretargeting paradigm.
Table 1.
Click Therapy Treatment Schedule
| Treat group | Pretargeting comp. (20 μg/mL, 130/180 nM) | Drug delivery comp. (50 μg/mL, 600 nM) |
|---|---|---|
| 1 | 5D3(TCO)8 | ALB(DM1)3.3(PEG4-Tz)10 |
| 2 | F(ab′)2(TCO)8 | ALB(DM1)3.3(PEG4-Tz)10 |
| 3 | 5D3 | ALB(DM1)3.3(PEG4-Tz)10 |
| 4 | F(ab′)2 | ALB(DM1)3.3(PEG4-Tz)10 |
| 5 | 5D3(TCO)8 | |
| 6 | F(ab′)2(TCO)8 | |
| 7 | 5D3 | |
| 8 | F(ab′)2 | |
| 9 | DM1 (2 μM)a | |
| 10 | ALB(DM1)3.3(PEG4-Tz)10 |
The concentration of pure DM1 was equivalent for DM1 content in a drug delivery component.
As shown in Figure 8, the combination of 5D3(TCO)8 and ALB(DM1)3.3(PEG4-Tz)10 showed selective and enhanced toxicity in PSMA(+) PC3-PIP cells when compared to the combination of nonfunctionalized 5D3 and ALB-(DM1)3.3(PEG4-Tz)10, or any other signal treatment component alone. This enhanced toxicity was not observed in PSMA(−) PC3-Flu cells, suggesting PSMA specificity. Surprisingly, the smaller F(ab′)2 fragments could not replicate this PSMA-specific toxicity. Click therapy using 5D3(TCO)8 and F(ab′)2(TCO)8 with ALB(DM1)3.3(PEG4-Tz)10 as a drug delivery component exhibits a reduction of cell viability of 30% and 55% of PSMA(+) PC3-PIP cells respectively, whereas the corresponding cell viabilities of PSMA(−) PC3-Flu cells were 49% and 40% when using 5D3(TCO)8 and F(ab′)2(TCO)8 as pretargeting components, respectively. The therapeutic effect of the combination of 5D3(TCO)8 and ALB(DM1)3.3(PEG4-Tz)10 was significantly higher compared to the treatment using ALB(DM1)3.3(PEG4-Tz)10 alone and the nonclick combination of 5D3 mAb and ALB(DM1)3.3(PEG4-Tz)10 in PC3-PIP cells. The treatment schedule of PSMA(−) PC3-Flu cells showed a significantly lower decrease in cell viability. Control experiments with ALB(DM1)3.3(PEG4-Tz)10 and its equivalent DM1 treatment result in comparable 57% and 55% cell viabilities in PC3-PIP cells and 63% and 55% cell viabilities in PC3-Flu cells, respectively. The controlled, targeted two-component delivery without click conjugation using pure 5D3 and F(ab′)2 with ALB(DM1)3.3(PEG4-Tz)10 reduces the cell viability to 48% and 52% in PC3-PIP cells and 45% to 42% in PC3-Flu cells; however, this is not significantly different compared to ALB(DM1)3.3(PEG4-Tz)10 alone.
Figure 8.
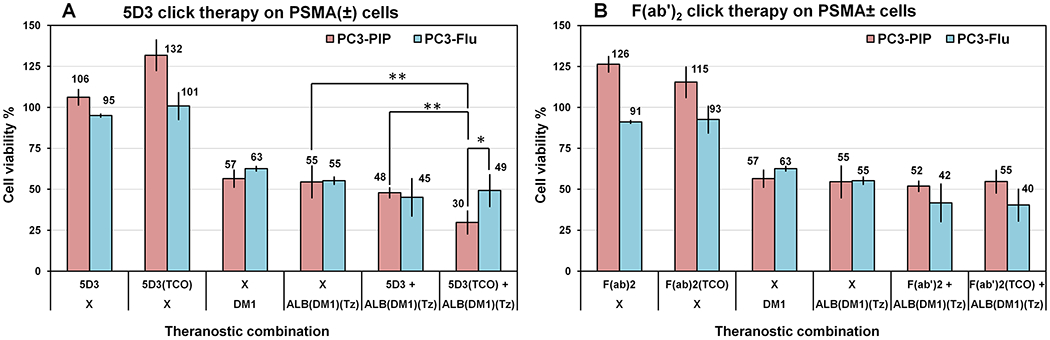
Cell viability-based in vitro therapeutic study of pretargeted two-component drug delivery. 5D3(TCO)8 (A) and F(ab′)2(TCO)8 (B) were used as a pretargeting component. Control experiments were performed with the pretargeting component alone; pure DM1 or with ALB(DM1)3.3(PEG4-Tz)10 without pretargeting components. The combination of 5D3(TCO)8 and ALB(DM1)3.3(PEG4-Tz)10 showed selective and enhanced toxicity in PSMA(+) PC3-PIP cells when compared to the combination of nonfunctionalized 5D3 and ALB(DM1)3.3(PEG4-Tz)10 or treatment with a free drug or drug-carrier component alone. This enhanced toxicity was not observed in PSMA(−) PC3-Flu cells, suggesting PSMA specificity. PSMA-specific toxicity of the smaller F(ab′)2 fragments, used as the pretargeting component, could not replicate this of complete 5D3 mAb (*p < 0.05, **p < 0.005).
4. DISCUSSION
We selected anti-PSMA 5D3 as a pretargeting mAb due to its high binding affinity compared to other existing/commercially available anti-PSMA mAbs. Antibodies that have been used as imaging agents and drug carriers are biocompatible, and their intrinsic amine groups can be easily used for modification and conjugation purposes. Approximately eight TCO groups per mAb provide the optimal substitution ratio for receptor binding and multiple click reactions with the delivery component to produce cross-linked nanoclusters without interfering with the binding affinity.45 Human serum albumin is used in the clinic, as the drug-carrier molecule (Abraxane),54 and as a platform for the development of drug delivery components.55,56 Albumin is a hydrophilic globular protein, which is highly stable in a broad pH range (pH 4–9), up to 40% ethanol, and temperature up to 60 °C. Albumin is also used as a drug carrier for delivery of hydrophobic low-molecular-weight chemotherapeutics in cancer therapy.57,58
We have used PC3-PIP and PC3-Flu cells in this study to validate the concept in PC. PSMA is highly overexpressed in PC3-PIP cells compared to PC3-Flu cells, as proven in Figure 2A. In our experiments, F(ab′)2 alone was internalized faster than 5D3 mAb (75% of 5D3 in 2.5 h vs 75% of F(ab′)2 in 1 h), presumably due to their small size and efficient cross-linking of PSMA receptors on the cell surface. However, the fast internalization is a disadvantage in pretargeted delivery since the time window spent by the pretargeting component on the cell surface is critical for click reactions with the second drug delivery component. After internalization, the receptors could be recycled, and the pretargeting component was retained in the cytoplasm, rendering it unavailable for reaction with the drug carrier.
Next, we assessed the pretargeted delivery driven by TCO-Tz click chemistry in PSMA(+) PC3-PIP and PSMA(−) PC3-Flu cells. When the click delivery has taken place in PSMA(+) cells, the colocalized Cy5 and AF-488 fluorescence signals corresponding to 5D3(TCO)8 (Cy5)2 or F-(ab′)2(TCO)8(Cy5)2 and ALB(DM1)3.3(PEG4-Tz)10(AF-488)2 are detected in each cell (Figure 2B); both fluorescence signals are higher in the FACS density plot. However, when nonreactive compounds are used, high fluorescence signals of the pretargeting components only are detected in PSMA(+) cells but not in PSMA(−) cells (Figure 2C). The pretargeting component 5D3(TCO)8(AF-488)2 labeled the cell surface PSMA receptors and stayed on the membrane for at least 30 min. The second-component, ALB(PEG4-Tz)10(Rhod)2, undergoes multiple in situ TCO-Tz click reactions with the pretargeting components on the cell surface, making cross-linked nanoclusters, leading to their rapid internalization. Multiple functional groups (TCO or Tz) per component, the close proximity of PSMA receptors in PSMA(+) cell surface, and fast kinetics (~300 000 M−1 s−1) of TCO-Tz click reaction facilitate the formation of nanoclusters of two components on the cell surface.59 If solutions of both components (~10 mg/mL) are mixed at room temperature, the mixture rapidly becomes cloudy because of multiple click reactions between components and the formation of large protein–protein complexes.
The cross-linking is not feasible on cells with a low or absent expression of PSMA, and therefore, this strategy can be used to selectively deliver drugs to malignant cells. Pretargeted click therapy was further investigated by a time-dependent study of PC3-PIP cells using 5D3(TCO)8(AF-488)2 (Figure 4) and F(ab′)2(TCO)8(AF-488)2 (Figure 6) with ALB-(DM1)3.3(PEG4-Tz)10(Rhod)2. In these studies, both components were colocalized on the cell surface. However, a significant amount of unconjugated or unreacted F-(ab′)2(TCO)8(AF-488)2 was observed in endosomes in the cell cytoplasm, due to its fast internalization, and was not available for in situ click conjugation and the formation of nanoclusters.
In vitro therapeutic study reveals no reduction in cell viability by 5D3 mAb or its TCO conjugated analogues (Figure 8). However, treatment of PC3-PIP cells with ALB-(DM1)3.3(PEG4-Tz)10 or its equivalent concentration of pure DM1 almost equally reduced cell viability. An efficient cross-linking of two components, 5D3(TCO)8 and ALB-(DM1)3.3(PEG4-Tz)10, on the targeted cell surface leads to enhanced cellular internalization and results in the highest therapeutic effects in PSMA(+) PC3-PIP cells compared to PSMA(−) PC3-Flu cells. Substituting the complete mAb with its F(ab′)2 fragment as a pretargeting component does not enhance the therapeutic efficacy of ALB(DM1)3.3(PEG4-Tz)10. Free DM1 is a highly cytotoxic molecule (IC50 in a low nanomolar range for most cancer cells)60 and reduces cell viability to ~60% after 2 h incubation at a concentration of 2 μM. As a naturally abundant carrier and transporting protein in the blood, albumin is one of the most important proteins taken nonspecifically by cells. Hence, we observed the therapeutic effects of untargeted ALB(DM1)3.3(PEG4-Tz)10(Rhod)2 in both PC3-PIP and PC3-Flu cells. Interestingly, in the control study shown in Figure S4, we observe a minimal uptake of ALB(DM1)3.3(PEG4-Tz)10(Rhod)2 in PC3-PIP cells. For comparison, in similar experiments with HER2-positive and HER2-negative breast cancer cells and albumin–paclitaxel conjugates (paclitaxel IC50 = 2.5–7.5 nM),61 we observed moderate cytotoxicity of the carrier, which reduced cell viability to ~70–75%.45 Enhanced cell-kill effects of the free and albumin-bound DM1 even with limited uptake of the free form and albumin-bound drug after 2 h pulse treatment of cells can be attributed to the significantly higher cytotoxicity of the drug (IC50 = 1.1 nM).60 We have also observed an enhancement (not statistically significant) of cell viability after pure 5D3, F(ab′)2, and 5D3(TCO)8, and F(ab′)2(TCO)8 treatment. It appeared to be a slight enhancement of cell growth by anti-PSMA agents, possibly due to interference with signaling pathways in cells. As proven by the mAb and F(ab′)2 internalization study, fast internalization kinetics of the F(ab′)2 domain, and its small size, has diminished the efficient cross-linking with the drug delivery component and therefore resulted in the reduced efficacy.
Our previous study of HER2(+) breast cancer in orthotopic mouse models using anti-HER2 mAb, trastuzumab proved that this strategy can be successfully applied in vivo because HER2 receptors inherently exhibit poor internalization even after ligand binding. Ideally, internalization and pharmacokinetics of the pretargeting agent should have a similar time scale. In the case of PSMA receptors, both 5D3 mAb and its F(ab′)2 fragments have fast internalization rates, which may present a problem for in vivo applications. While the system provided an enhanced specific kill of PSMA(+) PC cells in vitro, the internalization kinetics of the pretargeting agent has been identified as a critical parameter, affecting the treatment efficacy. This will become an issue for in vivo applications, where the timing of administration of the drug-carrier component should be optimized based on pharmacokinetics and tumor accumulation of the pretargeting component. We envision that for successful translation of the technology in vivo, further advances in the design of pretargeting components with a relatively fast clearance time and sufficient persistence on the cellular membrane is required.
5. CONCLUSION
Based on our experimental results, the overall efficacy of the pretargeted approach critically depends on several factors: (1) efficient binding of the pretargeting moiety to the target receptor; (2) sufficient time on the cell surface before internalization to enable click chemistry cross-linking with the drug delivery component; (3) highly cytotoxic therapeutic cargo, such as the DM1 microtubulin inhibitor of microtubule polymerization. On the basis of encouraging results obtained in vitro experiments, we are currently extending our study to investigate strategies for delaying internalization and evaluate the therapeutic effects of the pretargeted strategy in vivo in mouse models of human PC.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the Department of Defense (DoD) grant (W81XWH-16-1-0595), Czech Science Foundation grant (18-04790S), the European Regional Development Fund (Project BIOCEV-CZ.1.05/1.1.00/02.0109), and Czech Academy of Sciences grant (RVO: 86652036). This work also partially supported by R01 grants (CA209884, CA134675, and CA184228) from the National Cancer Institute, grant (EB024495) from the Institute National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, and Emerson Collective grant (128821) from Emerson Collective Cancer Research Fund. The authors acknowledge Dixie Hoyle for her assistance in flow cytometry.
ABBREVIATIONS
- ADC
antibody–drug conjugate
- ALB
human serum albumin
- DM1
mertansine
- DPBS
Dulbecco’s phosphate buffered saline
- HPLC
high-performance liquid chromatography
- IEDDA
inverse-electron demand Diels–Alder
- mAb
monoclonal antibody
- MALDI-TOF
matrix-assisted laser desorption ionization time-of-flight mass spectrometry
- PBS
BupH phosphate buffered saline
- PC
prostate cancer
- PSMA
prostate-specific membrane antigen
- SEC
size-exclusion chromatography
- Sulfo-SMCC
sulfosuccinimidyl 4-(N-maleimiddomethyl)cyclohexane-1-carboxylate
- TCO
trans-cyclooctene
- Tz
tetrazine
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.9b00788.
MALDI-TOF analysis of components, dynamic light scattering (DLS) analysis of components, internalization of anti-PSMA 5D3 mAb and F(ab′)2, and imaging experiment with ALB(PEG4-Tz)10(Rhod)2 (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Reddy A; Roberts R; Shenoy D; Packianathan S; Giri S; Vijayakumar S Prostate Cancer Screening Guidelines for African American Veterans: A New Perspective. J. Natl. Med. Assoc 2018. [DOI] [PubMed] [Google Scholar]
- (2).Ahlborg H; Nightingale AJ Mismatch between Scales of Knowledge in Nepalese Forestry: Epistemology, Power, and Policy Implications. Ecology and Society 2012, 17, 1. [Google Scholar]
- (3).Siegel RL; Miller KD; Jemal A Cancer Statistics, 2019. Ca-Cancer J. Clin 2019, 69, 7–34. [DOI] [PubMed] [Google Scholar]
- (4).Fang JC; Faerber G; Samadder J Digital Rectal Examination for Prostate Cancer Screening Performed with Colonoscopy for Colon Cancer Screening: 2 for the Price of 1. Gastrointestinal Endoscopy 2017, 86, 1147–1150. [DOI] [PubMed] [Google Scholar]
- (5).Zerbib M; Zelefsky MJ; Higano CS; Carroll PR Conventional Treatments of Localized Prostate Cancer. Urology 2008, 72, 25–35. [DOI] [PubMed] [Google Scholar]
- (6).Frieling JS; Basanta D; Lynch CC Current and Emerging Therapies for Bone Metastatic Castration-Resistant Prostate Cancer. Cancer Control 2015, 22, 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gaya JM; Huguet J; Breda A; Palou J [Surgical Treatment of Local Disease in Metastatic Prostate Cancer.]. Arch Esp Urol 2018, 71, 288–297. [PubMed] [Google Scholar]
- (8).Vanneste BG; Van Limbergen EJ; van Lin EN; van Roermund JG; Lambin P Prostate Cancer Radiation Therapy: What Do Clinicians Have to Know? BioMed Res. Int 2016, 2016, 6829875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Das M Androgen Deprivation Therapy for Prostate Cancer. Lancet Oncol. 2017, 18, e567. [DOI] [PubMed] [Google Scholar]
- (10).Sartor O; de Bono JS Metastatic Prostate Cancer. N. Engl. J. Med 2018, 378, 1653–1654. [DOI] [PubMed] [Google Scholar]
- (11).Lee CH; Kantoff P Treatment of Metastatic Prostate Cancer in 2018. JAMA Oncol 2019, 5, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Osanto S; Van Poppel H Emerging Novel Therapies for Advanced Prostate Cancer. Ther. Adv. Urol 2012, 4, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Clarke JM; Armstrong AJ Novel Therapies for the Treatment of Advanced Prostate Cancer. Curr. Treat Options Oncol 2013, 14, 109–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Poon DM; Ng J; Chan K Importance of Cycles of Chemotherapy and Postdocetaxel Novel Therapies in Metastatic Castration-Resistant Prostate Cancer. Prostate Int. 2015, 3, 51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bouchelouche K; Choyke PL; Capala J Prostate Specific Membrane Antigen - a Target for Imaging and Therapy with Radionuclides. Discovery Medicine 2010, 9, 55–61. [PMC free article] [PubMed] [Google Scholar]
- (16).Bravaccini S; Puccetti M; Bocchini M; Ravaioli S; Celli M; Scarpi E; De Giorgi U; Tumedei MM; Raulli G; Cardinale L Psma Expression: A Potential Ally for the Pathologist in Prostate Cancer Diagnosis. Sci. Rep 2018, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Mease RC; Foss CA; Pomper MG Pet Imaging in Prostate Cancer: Focus on Prostate-Specific Membrane Antigen. Curr. Top. Med. Chem 2013, 13, 951–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chatalic KLS; Veldhoven-Zweistra J; Bolkestein M; Hoeben S; Koning GA; Boerman OC; de Jong M; van Weerden WM A Novel in-111-Labeled Anti-Prostate-Specific Membrane Antigen Nanobody for Targeted Spect/Ct Imaging of Prostate Cancer. J. Nucl. Med 2015, 56, 1094–1099. [DOI] [PubMed] [Google Scholar]
- (19).Chang SS Overview of Prostate-Specific Membrane Antigen. Rev. Urol 2004, 6 Suppl 10, S13–18. [PMC free article] [PubMed] [Google Scholar]
- (20).Langut Y; Talhami A; Mamidi S; Shir A; Zigler M; Joubran S; Sagalov A; Flashner-Abramson E; Edinger N; Klein S; et al. Psma-Targeted Polyinosine/Polycytosine Vector Induces Prostate Tumor Regression and Invokes an Antitumor Immune Response in Mice. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 13655–13660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wustemann T; Haberkorn U; Babich J; Mier W Targeting Prostate Cancer: Prostate-Specific Membrane Antigen Based Diagnosis and Therapy. Med. Res. Rev 2019, 39, 40–69. [DOI] [PubMed] [Google Scholar]
- (22).Yao V; Berkman CE; Choi JK; O’Keefe DS; Bacich DJ Expression of Prostate-Specific Membrane Antigen (Psma), Increases Cell Folate Uptake and Proliferation and Suggests a Novel Role for Psma in the Uptake of the Non-Polyglutamated Folate, Folic Acid. Prostate 2009, 70, 305–316. [DOI] [PubMed] [Google Scholar]
- (23).Mesters JR; Barinka C; Li W; Tsukamoto T; Majer P; Slusher BS; Konvalinka J; Hilgenfeld R Structure of Glutamate Carboxypeptidase Ii, a Drug Target in Neuronal Damage and Prostate Cancer. EMBO J. 2006, 25, 1375–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Barinka C; Rojas C; Slusher B; Pomper M Glutamate Carboxypeptidase Ii in Diagnosis and Treatment of Neurologic Disorders and Prostate Cancer. Curr. Med. Chem 2012, 19, 856–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Pinto JT; Suffoletto BP; Berzin TM; Qiao CH; Lin S; Tong WP; May F; Mukherjee B; Heston WD Prostate-Specific Membrane Antigen: A Novel Folate Hydrolase in Human Prostatic Carcinoma Cells. Clin. Cancer Res 1996, 2, 1445–1451. [PubMed] [Google Scholar]
- (26).Carter RE; Feldman AR; Coyle JT Prostate-Specific Membrane Antigen Is a Hydrolase with Substrate and Pharmacologic Characteristics of a Neuropeptidase. Proc. Natl. Acad. Sci. U. S. A 1996, 93, 749–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rahbar K; Afshar-Oromieh A; Jadvar H; Ahmadzadehfar H Psma Theranostics: Current Status and Future Directions. Mol. Imaging 2018, 17, 153601211877606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Azad BB; Banerjee SR; Pullambhatla M; Lacerda S; Foss CA; Wang YC; Ivkov R; Pomper MG Evaluation of a Psma-Targeted Bnf Nanoparticle Construct. Nanoscale 2015, 7, 4432–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Chandran SS; Banerjee SR; Mease RC; Pomper MG; Denmeade SR Characterization of a Targeted Nanoparticle Functionalized with a Urea-Based Inhibitor of Prostate-Specific Membrane Antigen (Psma). Cancer Biol. Ther 2008, 7, 974–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Jin W; Qin B; Chen ZJ; Liu H; Barve A; Cheng K Discovery of Psma-Specific Peptide Ligands for Targeted Drug Delivery. Int. J. Pharm 2016, 513, 138–147. [DOI] [PubMed] [Google Scholar]
- (31).Liu TC; Nedrow-Byers JR; Hopkins MR; Wu LSY; Lee J; Reilly PTA; Berkman CE Targeting Prostate Cancer Cells with a Multivalent Psma Inhibitor-Guided Streptavidin Conjugate. Bioorg. Med. Chem. Lett 2012, 22, 3931–3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Fuchs AV; Tse BW; Pearce AK; Yeh MC; Fletcher NL; Huang SS; Heston WD; Whittaker AK; Russell PJ; Thurecht KJ Evaluation of Polymeric Nanomedicines Targeted to Psma: Effect of Ligand on Targeting Efficiency. Biomacromolecules 2015, 16, 3235–3247. [DOI] [PubMed] [Google Scholar]
- (33).Liu H; Rajasekaran AK; Moy P; Xia Y; Kim S; Navarro V; Rahmati R; Bander NH Constitutive and Antibody-Induced Internalization of Prostate-Specific Membrane Antigen. Cancer Res. 1998, 58, 4055–4060. [PubMed] [Google Scholar]
- (34).Tagawa ST; Milowsky MI; Morris M; Vallabhajosula S; Christos P; Akhtar NH; Osborne J; Goldsmith SJ; Larson S; Taskar NP; et al. Phase Ii Study of Lutetium-177-Labeled Anti-Prostate-Specific Membrane Antigen Monoclonal Antibody J591 for Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res 2013, 19, 5182–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Tagawa ST; Milowsky MI; Morris MJ; Vallabhajosula S; Goldsmith S; Matulich D; Kaplan J; Berger F; Scher HI; Bander NH Phase Ii Trial of 177lutetium Radiolabeled Anti-Prostate-Specific Membrane Antigen (Psma) Monoclonal Antibody J591 (177lu-J591) in Patients (Pts) with Metastatic Castrate-Resistant Prostate Cancer (Metcrpc). J. Clin. Oncol 2008, 26, 5140.18838701 [Google Scholar]
- (36).Han DH; Wu JH; Han YH; Wei M; Han S; Lin RH; Sun ZY; Yang F; Jiao D; Xie P; et al. A Novel Anti-Psma Human Scfv Has the Potential to Be Used as a Diagnostic Tool in Prostate Cancer. Oncotarget 2016, 7, 59471–59481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Novakova Z; Foss CA; Copeland BT; Morath V; Baranova P; Havlinova B; Skerra A; Pomper MG; Barinka C Novel Monoclonal Antibodies Recognizing Human Prostate-Specific Membrane Antigen (Psma) as Research and Theranostic Tools. Prostate 2017, 77, 749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ray Banerejee S; Kumar V; Lisok A; Plyku D; Novakova Z; Wharram B; Brummet M; Barinka C; Hobbs RF; Pomper MG Evaluation of (111)in-Dota-5d3, a Surrogate Spect Imaging Agent for Radioimmunotherapy of Prostate-Specific Membrane Antigen. J. Nucl. Med 2019, 60, 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Beck A; Goetsch L; Dumontet C; Corvaia N Strategies and Challenges for the Next Generation of Antibody Drug Conjugates. Nat. Rev. Drug Discovery 2017, 16, 315–337. [DOI] [PubMed] [Google Scholar]
- (40).Lyon R Drawing Lessons from the Clinical Development of Antibody-Drug Conjugates. Drug Discovery Today: Technol 2018, 30, 105–109. [DOI] [PubMed] [Google Scholar]
- (41).Nicolaou KC; Rigol S Total Synthesis in Search of Potent Antibody-Drug Conjugate Payloads. From the Fundamentals to the Translational. Acc. Chem. Res 2019, 52, 127. [DOI] [PubMed] [Google Scholar]
- (42).Diamantis N; Banerji U Antibody-Drug Conjugates-an Emerging Class of Cancer Treatment. Br. J. Cancer 2016, 114, 362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Bae YH; Park K Targeted Drug Delivery to Tumors: Myths, Reality and Possibility. J. Controlled Release 2011, 153, 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Hapuarachchige S; Kato Y; Artemov D Bioorthogonal Two-Component Drug Delivery in Her2(+) Breast Cancer Mouse Models. Sci. Rep 2016, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Hapuarachchige S; Zhu W; Kato Y; Artemov D Bioorthogonal, Two-Component Delivery Systems Based on Antibody and Drug-Loaded Nanocarriers for Enhanced Internalization of Nanotherapeutics. Biomaterials 2014, 35, 2346–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).McKay CS; Finn MG Click Chemistry in Complex Mixtures: Bioorthogonal Bioconjugation. Chem. Biol 2014, 21, 1075–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Karver MR; Weissleder R; Hilderbrand SA Bioorthogonal Reaction Pairs Enable Simultaneous, Selective, Multi-Target Imaging. Angew. Chem., Int. Ed 2012, 51, 920–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Chen Z; Krishnamachary B; Penet MF; Bhujwalla ZM Acid-Degradable Dextran as an Image Guided Sirna Carrier for Cox-2 Downregulation. Theranostics 2018, 8, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Liu J; Kopeckova P; Buhler P; Wolf P; Pan H; Bauer H; Elsasser-Beile U; Kopecek J Biorecognition and Subcellular Trafficking of Hpma Copolymer-Anti-Psma Antibody Conjugates by Prostate Cancer Cells. Mol. Pharmaceutics 2009, 6, 959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Vercauteren D; Vandenbroucke RE; Jones AT; Rejman J; Demeester J; De Smedt SC; Sanders NN; Braeckmans K The Use of Inhibitors to Study Endocytic Pathways of Gene Carriers: Optimization and Pitfalls. Mol. Ther 2010, 18, 561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Shang L; Nienhaus K; Nienhaus GU Engineered Nanoparticles Interacting with Cells: Size Matters. J. Nanobiotechnol 2014, 12, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Yameen B; Choi WI; Vilos C; Swami A; Shi JJ; Farokhzad OC Insight into Nanoparticle Cellular Uptake and Intracellular Targeting. J. Controlled Release 2014, 190, 485–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Zhu W; Okollie B; Artemov D Controlled Internalization of Her-2/ Neu Receptors by Cross-Linking for Targeted Delivery. Cancer Biol. Ther 2007, 6, 1960–1966. [DOI] [PubMed] [Google Scholar]
- (54).Kundranda MN; Niu J Albumin-Bound Paclitaxel in Solid Tumors: Clinical Development and Future Directions. Drug Des., Dev. Ther 2015, 9, 3767–3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Rabbani G; Ahn SN Structure, Enzymatic Activities, Glycation and Therapeutic Potential of Human Serum Albumin: A Natural Cargo. Int. J. Biol. Macromol 2019, 123, 979–990. [DOI] [PubMed] [Google Scholar]
- (56).Karimi M; Bahrami S; Ravari SB; Zangabad PS; Mirshekari H; Bozorgomid M; Shahreza S; Sori M; Hamblin MR Albumin Nanostructures as Advanced Drug Delivery Systems. Expert Opin. Drug Delivery 2016, 13, 1609–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Larsen MT; Kuhlmann M; Hvam ML; Howard KA Albumin-Based Drug Delivery: Harnessing Nature to Cure Disease. Mol. Cell Ther 2016, 4, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Umbricht CA; Benesova M; Schibli R; Muller C Preclinical Development of Novel Psma-Targeting Radioligands: Modulation of Albumin-Binding Properties to Improve Prostate Cancer Therapy. Mol. Pharmaceutics 2018, 15, 2297–2306. [DOI] [PubMed] [Google Scholar]
- (59).Darko A; Wallace S; Dmitrenko O; Machovina MM; Mehl RA; Chin JW; Fox JM Conformationally Strained Trans-Cyclooctene with Improved Stability and Excellent Reactivity in Tetrazine Ligation. Chem. Sci 2014, 5, 3770–3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Widdison WC; Wilhelm SD; Cavanagh EE; Whiteman KR; Leece BA; Kovtun Y; Goldmacher VS; Xie H; Steeves RM; Lutz RJ; et al. Semisynthetic Maytansine Analogues for the Targeted Treatment of Cancer. J. Med. Chem 2006, 49, 4392–4408. [DOI] [PubMed] [Google Scholar]
- (61).Liebmann JE; Cook JA; Lipschultz C; Teague D; Fisher J; Mitchell JB Cytotoxic Studies of Paclitaxel (Taxol) in Human Tumour Cell Lines. Br. J. Cancer 1993, 68, 1104–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.