

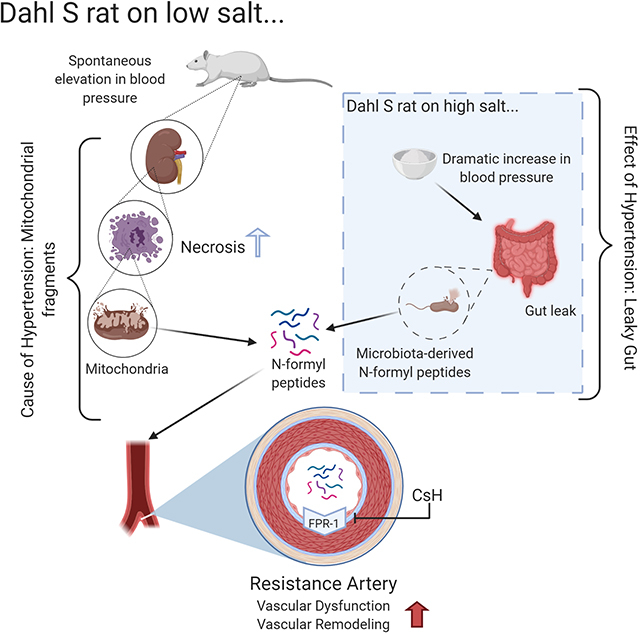
Abstract
Cell death has long been a characteristic phenotype of organ damage in hypertension and recently, leaky gut has been revealed as a novel hypertensive phenotype. However, despite the increase in bacterial and damaged mitochondrial products in the circulation of hypertensive patients and animals, the mechanistic contribution of these two phenomena to hypertension pathophysiology is unknown. Mitochondria and bacteria both start protein translation with an N-formyl methionine residue and thus are the only sources of N-formyl peptides (NFPs) which activate the formyl peptide receptor-1 (FPR-1). We hypothesized that the synergistic action of bacterial and mitochondrial N-formyl peptides would cause the spontaneous elevation of blood pressure and vascular remodeling in male Dahl salt-sensitive (S) rats via FPR-1. We observed that mitochondria-derived peptides originating from cell death in the kidneys are responsible for FPR-1-induced vascular hypercontractility and remodeling and premature elevation of BP in Dahl S rats fed a low salt diet. However, a high-salt diet leads to gut barrier disruption and, subsequently, a synergistic action of mitochondria and bacterial-derived leaky gut NFPs leads to a severe and established hypertension. Administration of an FPR-1 antagonist lowered blood pressure in Dahl S rats on a low salt diet, but amoxicillin administration did not. These results reveal for the first time that cell death can be a cause of hypertensive pathophysiology, whereas leaky gut is a consequence.
Keywords: Hypertension, Dahl Salt Sensitive Rats, Vascular Function, Mitochondria, Microbiota
Graphical Abstract
1. Introduction
Hypertension is a global health concern (1). Although newly updated guidelines for hypertension diagnosis promote early intervention (2), there are a number of patients with resistant hypertension, whose elevated blood pressure remains uncontrolled despite therapy (3). A critical barrier in advancing the development of novel therapeutic approaches for hypertension is the gap in understanding of the precise cause-effect mechanism. Recent evidence implicates immune mechanisms in the pathophysiology of hypertension (4–6). In fact, immune system activation and inflammation have been proposed as a unifying mechanism linking the major organ systems involved in the development of hypertension – the cardiovascular system, the kidneys, the autonomic nervous system (7,8), and more recently, the gut (9). However, it is still unclear if the activation of the innate immune system precedes hypertension.
The formyl peptide receptor (FPR-1) is a pattern recognition receptor that plays a crucial role in the function of the innate immune system. FPR-1 is a G-protein-coupled receptor that can bind N-formyl peptides such as N-formylmethionine-leucyl-phenylalanine (fMLP), produced by bacterial degradation (10–15). Interestingly, mitochondria carry hallmarks of their bacterial ancestry including the usage of N-formyl-methionyl-tRNA as an initiator of protein synthesis (10–15). Any injury that causes plasma membrane lysis in vivo can lead to the release of mitochondrial N-formyl peptides (NFPs). Consequently, both mitochondrial and bacterial-produced peptides have a formyl group at their N-terminus. Therefore, NFPs, regardless of origin, are recognized by FPR-1 and thus play a role in the initiation of inflammation.
One of the most potent inducers of actin polymerization is FPR-1 (10, 16). Interestingly, FPR-1 is also expressed in endothelial and vascular smooth muscle cells (15, 17), and it is important for vascular remodeling and motility via actin polymerization, independent of bacteria or mitochondrial fragments (15). This role is like the one observed in sentinel cells of the innate immune system, such as neutrophils. In physiological conditions, FPR-1 responds to tension and contributes to arterial myogenic tone (18). Nonetheless, the precise mechanism linking FPR-1 activation in the vascular-immune network in hypertension remains unknown.
Hypertensive patients are known to have “leaky gut” (19–22), whereby circulating levels of bacterial products are increased in hypertension. Therefore, we postulated that both leaky gut-derived bacterial NFPs (from microbiota) and cell damage-derived mitochondrial NFPs (from the host) could cause vascular dysfunction and remodeling in hypertension. Specifically, we hypothesized that mitochondrial-derived NFPs and FPR-1 activation would lead to vascular remodeling and the genesis of high BP. A high-salt diet would further exacerbate this response by causing gut barrier disruption (23). Subsequently, a synergistic action of mitochondria and bacteria-derived leaky gut NFPs would be the major driving force in the maintenance of hypertension.
2. Materials and Methods
The data that support the findings of this study are available from the corresponding author upon request.
2.1. Animals
All animals were from a colony maintained at the University of Toledo College of Medicine and Life Sciences. All animal procedures and protocols used were approved by the University of Toledo Institutional Animal Care and Use Committee (IACUC protocol approval numbers 108854, 104573, 108390). Experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and Animal Research Reporting of In Vivo Experiments (ARRIVE) guidelines. Male inbred Dahl salt-resistant (R) and salt-sensitive (S) rat strains were from the University of Toledo College of Medicine and Life Sciences. All rats were weaned at 4 weeks of age and maintained on a 12-hour light cycle with water ad libitum and a low-salt diet (0.3% NaCl, Harlan Teklad diet TD 7034; Madison, WI) until 5 weeks of age. At this time, the R and S were divided into two groups high-salt diet (2% NaCl, Harlan Teklad diet; TD 94217; Madison, WI) or maintained on a low-salt diet for a further 5–6 weeks.
2.2. Telemetry
Male rats (~ 10–11 weeks old) were surgically implanted with radiotelemetry transmitters (HDS10) (Data Science International, St Paul, MN) as previously described (24–26). For this, all rats were anesthetized with isoflurane (2% in 100% O2 administered via nose cone) and implanted with S10 radiotelemetry transmitters via the femoral artery. Rats under anesthesia were given analgesics (buprenorphine, 0.05mg/kg) as approved by the IACUC. Post-surgery, rats were individually housed and allowed to recover from surgery for four days before baseline BP was recorded (24–26). Post-operative care was provided as per the approved protocol and euthanasia was via carbon dioxide inhalation.
2.3. In vivo treatments
2.3.1. Antibiotic treatment
After the baseline BP was recorded, rats received either normal drinking water (control) or water supplemented with amoxicillin (50 mg/kg/day, Sigma-Aldrich, St. Louis, MO) for 2 weeks. Water bottles were replenished once a week.
2.3.2. Cyclosporin H (CsH) treatment
In another experiment, a group of rats (Dahl R and S on low salt diet) were administered FPR-1 antagonist, cyclosporin H (CsH, 0.3 mg/kg/day), or vehicle by osmotic minipump for 2 weeks. For this, CsH was prepared at the desired concentration (based on animals’ body weight) in the required volume of ethanol (15%) and water. The volume of 100 μl of CsH solution was loaded into the osmotic minipumps (Azlet, model 1002, Cupertino, CA) as directed by the manufacturer using the blunt needle provided. The flow moderator was inserted into the pump, and the osmotic minipumps were stored aseptically in saline in an incubator overnight until implantation into rats. Rats were randomized to receive either CsH or vehicle minipumps.
For mini-pump implantation, rats were anesthetized (2% isoflurane in 100% O2 administered via nose cone). A patch of skin was exposed by shaving fur from the flank using a clipper. The surgical site was disinfected with isopropyl alcohol and iodine swabs. Under sterile conditions, 2–3 cm skin incision was made between the shoulder blades. Blunt artery forceps were inserted into the incision to make a subcutaneous pocket. The osmotic mini pump was inserted into the pocket with the flow moderator away from the incision and the incision was closed using wound clips. Antibiotic ointment was applied to the outside of the incision. While rats were anesthetized, they were given analgesics (meloxicam, 2 mg/kg) as approved by the IACUC policy. Rats were watched closely until they recovered from anesthesia.
2.4. Survival Curve
Starting from birth, mortality was reported in R and S rats on low and high salt diet. Age at time of death (in days) was entered into analysis software and mortality percentage was calculated.
2.5. Necrosis and Apoptosis Assay
Bone marrow-derived macrophage (BMDM), whole kidney cells, and whole mesenteric resistance artery cells were isolated from 7-week old male Dahl R and S rats and were used to measure the basal level of necrosis and apoptosis. For this, we used the Dead Cell Apoptosis Kit with Annexin V Alexa Fluor™ 488 and Propidium Iodide (PI) (Invitrogen™, Eugene, OR) according to the manufacturer’s instruction. Briefly, cells (2.0×106) were resuspended in Annexin V binding buffer (1X) in 100 μl per assay. Alexa Fluor® 488 Annexin V (5 μl) and PI (1 μl of 100 μg/ml) working solution was added to each 100 μl of cell suspension and incubated the cells at room temperature for 15 min without light. After the incubation period, 400 μl of 1X annexin-binding buffer was added to each tube and mixed gently and kept the samples on ice. The % necrosis and apoptosis cells were measured via flow cytometry (Accuri C6, BD Biosciences, San Jose, CA) and analyzed using the BD Accuri C6 Software (Becton Dickinson, Canaan, CT). Results were presented as the percentage of (i) early apoptotic cells: Annexin-V single-positive cells; (ii) late apoptotic cells: Annexin V+ propidium iodide (PI) double-positive cells and (iii) necrotic cells: PI single-positive cells.
2.6. Tissue Collection
After treatment, rats were weighed and euthanized by thoracotomy and exsanguination via cardiac puncture under isoflurane anesthesia (5% in 100% O2 administered via nose cone). Whole blood was first collected from the abdominal aorta and centrifuged. For serum, whole blood was centrifuged for 15 minutes at 2000 rcf in a 4°C centrifuge and the supernatant was collected. For plasma, whole blood was centrifuged for 15 minutes at 1500 rcf in a 4°C centrifuge and the supernatant was collected. Subsequently, mesenteric resistance arteries whole hearts, kidneys, and tibias were all harvested. The left and right ventricles were dissected from the whole hearts and weighed. Lungs were measured wet immediately after removal and allowed to dry in a 37°C oven for a minimum of 4 days and re-weighed.
2.6.1. Left ventricle mass measurements
Body weight is correlated nonlinearly (cubically) with tibia length (TL), but linearly with TL3 (27). The linear relation with the left ventricle (LV) and TL3 showed that LV weight crossed the x-axis at TL3 −26.73 (95% CI −29.83; −23.43). Therefore, TL and LV weight were indexed by dividing the weights by 26.73 + TL3 (27).
2.6.2. Vascular Function
Third or fourth order MRA, 2 mm in length, were mounted on DMT wire myographs (Danish MyoTech, Aarhus, Denmark) and kept in a Krebs solution (Table S1). The MRA were normalized to their optimal lumen diameter for active tension development, as described previously by our group (28, 29). To test vascular smooth muscle cell integrity, the arteries were initially contracted with 120 mmol/L potassium chloride (KCl). Concentration-response curves to acetylcholine (ACh, 1 nmol/L to 10 μmol/L) after initial contraction to phenylephrine (PE, 30 μmol/L) was performed to evaluate relaxation. Relaxation responses to ACh are shown as a percent of the initial PE contraction (30 μmol/L). Concentration-response curves to PE (10 nmol/L to 10 μmol/L) were also performed (force, mN).
2.6.3. Vascular Structure
Fifth to seventh order MRA were mounted on DMT pressure-culture myographs (Danish MyoTech, Aarhus, Denmark) to test the mechanical properties of the arteries as previously described by our group (29). Briefly, arteries were mounted on glass cannulas and tied down with nylon suture. Intraluminal pressure was slowly raised to 160 mmHg and the artery was stretched to obtain the optimal parallel view of the walls. The MRA was equilibrated at 60 mmHg in filtered Krebs for 30 min. After testing vascular smooth muscle cell integrity with 120 mmol/L KCl, the extraluminal Krebs was exchanged with high Ca2+ Krebs (2.5 mM). Intraluminal pressure was dropped to 3 mmHg and a pressure curve was obtained by increasing the intraluminal pressure in 20 mmHg steps to 160 mmHg with a two-minute equilibration time at every step. After this curve, the same procedure was followed using an extraluminal buffer with zero Ca2+ Krebs buffer. DMT data capture software was used to capture videos and/or images and subsequently analyzed with the VasoTracker Offline Diameter Analyzer for accurate inner and outer diameter measurements (30). From the internal and external diameter measurements in the passive conditions, structural parameters were calculated as previously described (29, 31). See all Krebs solutions in Table S1.
2.6.4. Leaky gut assay
Zonulin concentration was determined using a sandwich ELISA (MyBioSource, San Diego, CA, Catalog # MBS2606662) following the manufacturer’s protocol.
Samples and the detecting antibody were added into the ELISA plate wells and placed in a 37°C incubator for 90 min. The plate was washed with 1X phosphate buffer saline (PBS) 3 times. The enzyme-conjugate was added to each well and placed in a 37°C incubator for 30 min. The plate was washed with 1X PBS 5 times. The color reagent was added to the wells and the plate was placed in a dark 37°C incubator. When the highest concentration for the standard curve darkened and the color gradient appeared, the plate was removed from the incubator. Color reagent C was then added to each well and mixed well. The plate was read immediately. The intensity of the color was inversely proportional to the Zonulin concentration in the samples, which were read at 450 nm.
2.6.5. Real-time PCR
Total RNA was extracted from aortas using the Qiagen RNeasy Mini Kit following the manufacturer’s protocol (Qiagen, Hilden, Germany). Nucleic acid concentration was quantified with the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). After isolation, cDNA was generated by reverse transcription using the Applied Biosystems cDNA synthesis kit (Cat No. #: 4368814, Darmstadt, Germany). The thermal-cycling settings used were as follows: Step 1: 25°C for 10 minutes, Step 2: 37°C for 120 minutes, Step 3: 85°C for 5 minutes, and Step 4: 4°C for no more than 1 hour. After thermal-cycling, the sample was diluted with 80 μL of RNAse-Free dH2O and stored in −20°C freezer until further analysis with qRT-PCR (23). PCR amplification of cDNA was performed by quantitative RT-PCR with the TrueAmp SYBR Green qPCR SuperMix (Smart Bioscience, Maumee, OH) (25). The annealing temperature for PCR was 56°C and 26 cycles were carried out. Relative gene expression was calculated using the ΔΔCt method with GAPDH as an internal control. Primers were designed using the National Center for Biotechnology Information primer blast software (Bethesda, MD) or previously published (32). Primer sequences are listed in Table S2.
2.6.6. Immunoblotting
Mitochondrial N-formylated protein (NADH dehydrogenase 6) was measured in plasma from all groups. Plasma was diluted (1:20) and 25 uL of the solution was loaded and proteins were separated by SDS-polyacrylamide electrophoresis as previously described (14). PVDF membranes were incubated overnight at 4°C with a primary antibody raised against rat NADH dehydrogenase 6 (ND6, 1:1000, Sigma). Ponceau was used as a loading control.
2.7. Statistical Analysis
All statistical analysis was performed using GraphPad Prism 8.4.2 (La Jolla, CA, USA). Data are presented as mean ± standard error of the mean (SEM) and statistical significance was set at p<0.05 unless noted otherwise. Procedures used include Student’s unpaired t-test, one-way, and two-way analysis of variance (ANOVA), non-linear regression analysis (LogEC50 and Emax). Tukey’s post-hoc testing and the Bonferroni post-hoc testing were used in one-way ANOVA and two-way ANOVA respectively. For the survival curve, we used the Mantel-Cox test. The number (n) of animals per group is described within the graphs.
3. Results
3.1. Survival curve
In Figure 1A, the probability of survival in animals in low and high salt diets was observed. There was no mortality among Dahl R rats (LS and HS diets) and Dahl S-LS. On the other hand, Dahl S-HS rats began to die around 86 days of age.
Figure 1.

Survival curve (A), serum zonulin levels (B), mitochondrial ND6 expression in plasma analysis and representative images (C and D), and FPR-1 gene expression in arteries (E) from male Dahl salt resistant (R) and Dahl salt sensitive (S) rats on low salt (LS, 0.3%) and high (HS, 2%) diet. Data presented in mean ± SEM. Statistics: t-test or One- ANOVA: Number of animals and p value are indicated on graphs.
3.2. Increases in circulating levels of NFPs and, subsequently FPR-1 activation, contribute to the genesis and maintenance of hypertension
Dr. John Rapp (33) observed that the premature spontaneous elevation of BP occurs in inbred Dahl S rats fed a low salt diet (younger 12 weeks old), but not in Dahl R rats. This phenomenon occurred due to unknown genetic influences, and independent of environmental factors. High-salt diet accelerates this process leading to a malignant hypertension (33). Here, we observed that mitochondrial NFPs were increased in the plasma of Dahl S-LS rats at 7–9 weeks of age and in established models of hypertension (Dahl S-HS) (Figure 1B and C).
Due to the dual source of NFPs (from microbiota and host mitochondria), we postulated that leaky gut-derived bacterial NFPs (from microbiota) could also play a role in the genesis of hypertension. However, this hypothesis was refuted given that zonulin (biomarker for leaky gut) was not increased in the circulation of Dahl R-LS, Dahl R-HS, and Dahl S-LS (Figure 1D). On the other hand, zonulin was significantly increased in Dahl S-HS (Figure 1D). These data suggest that high-salt diet leads to leaky gut and the synergistic action of mitochondria and bacterial-derived leaky gut NFPs only maintains hypertension.
In the Figure 1E, we observed that arteries from male Dahl S-LS present increased FPR-1 mRNA expression. These data suggest that FPR-1 expression in Dahl S is independent of high salt. However, high salt diet also induced FPR-1 expression (Figure 1E) in normotensive animals.
Given that leaky gut is not present in animals fed a low salt diet, suggesting that gut microbiota-derived fragments may not play a role in the genesis of hypertension in Dahl S, we decided to investigate whether increased FPR-1 expression and mitochondrial NFPs play a role in the genesis of hypertension. Therefore, the results demonstrated from this point were collected only from Dahl-R and S on a low salt diet to avoid leaky gut and a possible increase in levels of microbiota-derived NFPs in the circulation. However, for scientific rigor, we still treated one group of Dahl S and R rats with antibiotics to investigate (1) the possible effect, if any, of gut microbiota, but independent of leaky gut, in the genesis of the premature elevation of blood pressure in Dahl S fed a low salt diet; (2) the possible role of microbiota from another source outside of the gut; and (3) possible pleiotropic effect of antibiotics on the cardiovascular system.
3.3. Cell death-derived mitochondrial NFPs are present prior to the onset of hypertension
We observed that Dahl S-LS present with cell necrosis in the kidney when compared to Dahl R-LS (Figure 2A). No differences were observed in cell necrosis from bone marrow derived macrophages (BMDM) and mesenteric resistance arteries (Figure 2 B and C). No differences were observed in early and late cell apoptosis from all tissues evaluated (Figure 2A–C). These data suggest that low-grade trauma is inherited and independent of high salt diet. Further, cell necrosis is present during the development of hypertension and kidneys are the possible initial source of mitochondrial NFPs in the circulation. See Figure S1 and Table S4.
Figure 2.

Cell necrosis, early apotosis, and late apoptosis measured by flow cytometry in kidney cells (A), bone marrow derived macrophages (B), and mesenteric resistant arteries cells (C) from male Dahl salt resistant (R) and Dahl salt sensitive (S) rats on low salt (LS, 0.3%) diet. Data presented in mean ± SEM. Statistics: t-test. Each dot respresents an individual rat and p values are indicated on graphs.
3.4. Formyl peptide receptor-1 antagonist prevents BP increase in Dahl-S fed a low salt diet
FPR-1 antagonist, CsH, prevents the increase of diastolic BP in Dahl S-LS after 4 days of treatment with CsH (Figure 3A). Although, there was a tendency to prevent an increase in systolic BP after treatment with CsH, no statistical differences were observed (Figure 3B). On the other hand, antibiotic amoxicillin (AMO) did not change diastolic and systolic BP in Dahl S-LS diet (Figure 3C and D). These data confirm that leaky gut is not associated with the genesis of premature spontaneous elevation of BP in Dahl S-LS rats. Both CsH and AMO did not change BP in normotensive animals (Dahl R-LS) (Figures 3A–D).
Figure 3.

Diastolic and systolic blood pressure, measured by telemetry, from male Dahl salt resistant (R) and Dahl salt sensitive (S) rats on low salt (LS, 0.3%) diet treated with cyclosporin H (CsH) (A and B) or amoxicillin (AMO) (C and D) for 14 days. The first point (–1) on the graph is the baseline average of BP for 14 days. Arrows indicate the start of the treatment. Data presented in mean ± SEM. Number of animals are indicated on graphs. P value <0.05. Statistics: Two-way ANOVA, * vs. R-LS and R-LS + CsH; # vs. S-LS.
3.5. FPR-1 antagonist treatment decreases heart left ventricle (LV) mass and vascular cross-sectional area (CSA) of resistance arteries
Similar to hypertensive animals, we observed that Dahl S-LS presented with a significant increase in LV mass and CSA of the mesenteric resistance arteries (Figure 4A–D), which suggests vascular hypertrophy, when compared to Dahl R-LS. Treatment with CsH reduced both parameters (Figure 4A, B and D). Interestingly, although AMO treatment did not affect BP in normotensive and hypertensive animals, this antibiotic decreased LV mass in Dahl S-LS animals (Figure 4A). On the other hand, AMO did not change vascular CSA (Figure 4C and D). Both treatments did not affect LV mass and CSA from normotensive animals (Figure 4A–D). There were no changes in lung wet to dry mass ratio, showing no lung edema (Table S2).
Figure 4.

Left ventricle (LV) mass normalized by tibia lenght (TL)3 (A). For this, the linear relation with LV and TL3 showed that LV weight crossed the x-axis at TL3 −26.73 (95% CI −29.83; −23.43). Therefore, TL and LV weight were indexed by dividing the weights by 26.73 + TL3. Cross-sectional area (CSA) measured with increasing intraluminal pressure in mesenteric resistance arteries from male Dahl salt resistant (R) and Dahl salt sensitive (S) rats on low salt (LS, 0.3%) treated with cyclosporin H (CsH) (B) or amoxicillin (AMO) (C) for 14 days. Mesenteric resistance arteries at 80 mmHg from all groups are plotted CSA vs. lumen diameter (D), suggesting vascular hyperthrophy. Data presented in mean ± SEM. Number of animals and p value are indicated on graphs, otherwise p value <0.05. Statistics: One or two- way ANOVA; * vs. R-LS; # vs. S-LS.
3.6. Vascular Function
After FPR-1 antagonist, antibiotic and vehicle treatment, we evaluated vascular function in mesenteric resistance arteries from all groups. We observed that KCl-induced contraction was increased in arteries from Dahl S-LS compared to Dahl R-LS (Figure 5A). Cyclosporine H, but not AMO, decreased this response (Figure 5A). A similar pattern was observed in phenylephrine induced contraction (Figures 5B and C). There were no significant changes in contraction to phenylephrine in arteries from Dahl R-LS treated with CsH or AMO (Figures 5A–C).
Figure 5.

KCl (120 mM) (A) and phenylephrine (B and C)-induced contraction in mesenteric resistance arteries from male Dahl salt resistant (R) and Dahl salt sensitive (S) rats on low salt (LS, 0.3%) treated with cyclosporin H (CsH) or amoxicillin (AMO) for 14 days. Data presented in mean ± SEM. Number of animals and p value are indicated on graphs, otherwise p value <0.05. Statistics: One or two- way ANOVA; * vs. R-LS; # vs. S-LS.
Arteries from hypertensive animals displayed an endothelium-dependent acetylcholine-induced contraction when compared to normotensive (Figures 6A and B). Interestingly, AMO treatment significantly decreased the late-phase contraction to acetylcholine (Figure 6B), while CsH treatment was not able to improve endothelial function (Figure 6A). No treatment differences were observed in the concentration response curves to acetylcholine in arteries from Dahl R-LS (Figure 6A and B).
Figure 6.

Concentrations response curves to acetylcholine in mesenteric resistance arteries from male Dahl salt resistant (R) and Dahl salt sensitive (S) rats on low salt (LS, 0.3%) treated with cyclosporin H (CsH) (A) or amoxicillin (AMO) (B) for 14 days. Arteries were pre-contracted with phenylephrine (PE). Data presented in mean ± SEM. Number of the animals are indicated on graphs. P value <0.05. Statistics: Two- way ANOVA; * vs. R-LS; # vs. S-LS.
4. Discussion
Hypertension has been a topic of concern since its discovery over a century ago. Diagnostic guidelines have evolved over time which has led to over half of the American population being considered hypertensive (2). Many scientists debated whether mild hypertension should be treated until the 1960s when mild hypertension was shown to lead to the same negative outcomes as severe hypertension (34).
Inbred Dahl salt-sensitive rats, developed by Dr. John Rapp at the University of Toledo (33), mimic human salt-sensitive hypertension (35). Salt-sensitive hypertension is considered a hallmark of hypertension in the Black population, including African Americans, given that it is found in 73% of male and female hypertensive Blacks (36). Dr. Rapp (33) observed that the premature, spontaneous elevation of BP occurs in Dahl S rats fed a low salt (LS) diet, but not in Dahl R rats, due to unknown genetic influences and independent of environmental factors such as dietary salt. Specifically, premature, spontaneous elevations in BP begin in animals younger than 12 weeks of age (33). A high-salt diet can accelerate this process leading to a malignant hypertension, similar to humans. Here, we also observed that the Dahl S-LS presents premature, spontaneous elevation of BP, vascular dysfunction and increased LV mass. High salt diet worsened these parameters (Figure S2 and 3A–B). These inbred animals are a unique model, because although they present similarities with spontaneous hypertensive rats (SHR), such as cardiovascular injury prior the establishment of hypertension, they are also salt sensitive. Therefore, unknown genetic influences, rather than high salt diet, are the trigger of the vascular injury and genesis of elevated BP and a high salt diet accelerates and exacerbates these phenotypes.
Santisteban et al. (21) have provided provocative data demonstrating that in established hypertension, there is decreased expression of several tight junction proteins in the gut and a concomitant increase in intestinal permeability. In addition, alterations in human gut microbiota have been reported in the prehypertensive state (19, 20), suggesting that changes in the gut microbiota precede the onset of hypertension. This led us to test for intestinal gut leak in normotensive and hypertensive rats fed a low and high salt diet. We measured levels of serum zonulin, which is an intestinal permeability indicator (37, 38). There are multiple stimuli that can cause zonulin (39). One of them is gut dysbiosis which can cause increased amounts of zonulin production and secretion into circulation (40). Surprisingly, we observed that zonulin was not increased in the circulation of Dahl R-LS and HS, and Dahl S-LS. These data suggest that leaky gut-derived fragments are not the cause of the premature spontaneous elevation of BP in Dahl S-LS, despite the elevated BP presented in these rats. However, zonulin is significantly increased in Dahl S-HS, suggesting that high-salt diet, in an animal that is sensitive to salt, leads to gut barrier disruption (23). Therefore, gut disruption, and subsequently, leaky gut-derived fragments, are associated with accelerated and exacerbated cardiovascular injury, and they are important for the maintenance of established hypertension. Given that we did not observe leaky gut in Dahl S-LS animals, we questioned what factors were leading to the development of hypertension in the absence of high salt diet.Because we observed that FPR-1 mRNA expression was increased in arteries from Dahl S-LS, we suggested that mitochondria NFPs could be the factor that initiates immune activation and vascular injury via FPR-1. It has been previously shown that hypertensive animals have increased cell death and subsequently, increased levels of mitochondria fragments in the circulation (28). Therefore, we hypothesized that mitochondrial-derived NFPs and FPR-1 activation lead to vascular dysfunction and remodeling and the genesis of elevated BP. Indeed, here we observed that the mitochondrial protein ND6 is increased in the circulation in hypertensive rats, confirming that gut-independent NFPs would be the activator of FPR-1. In general, cell necrosis can be due to both external and internal forces which target the structure of the cell and its energy provider, the mitochondrion, and causes them to swell and explode releasing their contents, leading to an inflammatory response (41, 42). In opposition, apoptosis is programmed cell death that shrinks cells and leaves mitochondria in normal condition. The different cellular contents are phagocytized, and minimal inflammatory response is initiated (43). We observed an increase in cell necrosis in the kidneys of Dahl S rats. There were no changes in the apoptosis measurements in both normotensive and hypertensive subjects. In opposition to our findings, it has been shown that Dahl S rats on a high salt diet have severe kidney injury including tubular atrophy, tubular cell loss, intraluminal cast formation, and expansion of the interstitium (44) along with increased apoptosis in the kidney cortex, glomerular, and tubular components of the kidney (45,46). This suggests that the kidney, as one of the contributors of cell death in salt-sensitive hypertension, serves as a possible source of mitochondrial NFPs due to necrosis and warrants further investigation in rats on low and high salt.
Both mitochondrial and bacterial NFPs bind the FPR-1 to induce vascular stiffness (14, 47). Here, an FPR-1 antagonist, CsH, decreased the spontaneous elevation of BP seen in Dahl S-LS. We decided to use cyclosporine H (CsH), because this FPR-1 inhibitor, unlike cyclosporin A, does not induce an immunosuppressant response nor bind cyclophilin (48, 49). Along with FPR-1 blockade, we also used antibiotic treatment to indiscriminately reduce bacteria in hypertensive rats. Although we observed that these rats do not have a leaky gut, we investigated the effects that other microbiota sources or gut dysbiosis (in the absence of leak gut), would have on BP. However, no changes in BP were observed with AMO treatment in Dahl-S LS.
Arteries from animals with mild hypertension exhibit vascular hypercontractility and vascular remodeling (50, 51). Previously, we observed that the absence of FPR-1 decreases vascular contraction in intrarenal resistance arteries and aorta (15). Direct induction of actin polymerization ameliorates this response (15). In the present study, arteries from Dahl S-LS presented with greater KCl- and phenylephrine-induced contraction and an increase in vascular cross-sectional area when compared to Dahl R-LS. Treatment with FPR-1 antagonist decreased these parameters. These results suggest that FPR-1 activation, via mitochondria-derived NFPs, initiates vascular dysfunction and remodeling in Dahl S-LS. On the other hand, FPR-1 antagonist did not improve endothelium-dependent relaxation, suggesting that FPR-1 activation plays a role mainly in vascular smooth muscle cells. Surprisingly, antibiotic treatment improved endothelium-dependent relaxation in arteries and decreased LV mass from Dahl S-LS. These data suggest that microbiota and/or fragments from microbiota, which are not originating from and thereby are independent of a leaky gut, may act on the endothelium to induce vascular dysfunction in hypertension. This may lead to an increase in vascular resistance and subsequently, an increase in LV mass. Another possible explanation for this phenomenon may be that antibiotics, independently, act on the cardiovascular system impact hypertension.
Limitations of the study
A limitation of the present study was that pharmacological treatments could have unexpected side effects. While knock-out and knock-in mice are powerful tools for probing the functions of specific genes, we decided not use mice in the present study to avoid variations between different species. The Dahl salt-sensitive rat is an inbred rat model that has been widely used to study salt-sensitive hypertension and these rats present with premature and spontaneous hypertension, even in low salt diet. We did not include females in the present study. However, it is well established that there are sex differences in the prevalence of hypertension. Therefore, as a follow up to this study, we will consider understanding whether FPR-1 activation play a role in sex differences, vascular remodeling, and hypertension.
Conclusions/Perspectives
FPR-1 has long been associated with innate immunity, but now has emerged to be vital in the cardiovascular system. In this study, we identified a role for FPR-1 in the genesis of hypertension in Dahl S-LS rats. Specifically, mitochondria-derived NFPs, originating from the kidneys, are responsible for vascular injury and premature elevation of BP in Dahl S rats, independent of high salt diet. The next logical step is to study the mechanism for FPR-1’s critical role in the pathophysiology of salt-sensitive hypertension. Whether pharmacological methods are used to block FPR-1 or used to decrease NFPs’ activating ability, additonal studies need to be conducted to target this receptor in hypertension treatment.
Supplementary Material
Novelty and Significance.
1. What is new?
Mitochondrial N-formyl peptides and FPR-1 activation promote vascular injury and premature, spontaneous blood pressure elevation in Dahl salt-sensitive rats independent of high salt diet.
Activation of FPR-1 induces vascular hypercontractility and hypertrophy in Dahl salt-sensitive rats independent of high salt.
Blocakade of FPR-1 ameliorates vascular injury and prevents the increase of BP.
2. What is relevant?
Innate immune system activation is crucial for the cause of spontaneous elevation of blood pressure in Dahl salt-sensitive rats.
Leaky gut is not associated with the genesis of hypertension in Dahl salt-sensitive rats.
High salt diet induces leaky gut and increases mortality in Dahl salt-sensitive rats.
3. Summary
In conclusion, our study showed that cell injury, and subsequently, mitochondrial-derived NFPs and FPR-1 activation leads to vascular remodeling and the genesis of high BP in Dahl salt-sensitive rats, independent of high salt diet. High-salt diet further exacerbates this response by causing gut barrier disruption. Subsequently, a synergistic action of mitochondria and microbiota-derived NFPs are the major driving force in the maintenance of hypertension.
Acknowledgments
The graphical abstract was created with Biorender.com.
Source(s) of Funding
This work was supported by National Science Foundation (NOA-AGEP: 1432878 – J.M.E.), National Institutes of Health (NIGMS: R00GM11888 – C.F.W.), (NHLBI: HL1430820 - B.J.), (NCI: R01CA219144 – M.V.) and (NHLBI: K99HL151889 – C.G.M.), and American Heart Association (18POST34060003 – C.G.M.)
Footnotes
Conflict(s) of Interest/Disclosure(s)
None declared.
References
- 1.Mills KT, Stefanescu A, He J. The global epidemiology of hypertension. Nat Rev Nephrol. 2020;16(4):223–237. doi: 10.1038/s41581-019-0244-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C, DePalma SM, Gidding S, Jamerson KA, Jones DW, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines [published correction appears in J Am Coll Cardiol. 2018 May 15;71(19):2275–2279]. J Am Coll Cardiol. 2018;71(19):e127–e248. doi: 10.1016/j.jacc.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 3.Patel KV, Li X, Kondamudi N, Vaduganathan M, Adams-Huet B, Fonarow GC, Vongpatanasin W, Pandey A. Prevalence of Apparent Treatment-Resistant Hypertension in the United States According to the 2017 High Blood Pressure Guideline. Mayo Clin Proc. 2019;94(5):776–782. doi: 10.1016/j.mayocp.2018.12.033. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez-Iturbe B, Pons H, Johnson RJ. Role of the Immune System in Hypertension. Physiol Rev. 2017;97(3):1127–1164. doi: 10.1152/physrev.00031.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Higaki A, Caillon A, Paradis P, Schiffrin EL. Innate and Innate-Like Immune System in Hypertension and Vascular Injury. Curr Hypertens Rep. 2019;21(1):4. Published 2019 Jan 18. doi: 10.1007/s11906-019-0907-1. [DOI] [PubMed] [Google Scholar]
- 6.Drummond GR, Vinh A, Guzik TJ, Sobey CG. Immune mechanisms of hypertension. Nat Rev Immunol. 2019;19(8):517–532. doi: 10.1038/s41577-019-0160-5. [DOI] [PubMed] [Google Scholar]
- 7.Singh MV, Chapleau MW, Harwani SC, Abboud FM. The immune system and hypertension. Immunol Res. 2014;59(1–3):243–253. doi: 10.1007/s12026-014-8548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCarthy CG, Wenceslau CF, Goulopoulou S, Ogbi S, Matsumoto T, Webb RC. Autoimmune therapeutic chloroquine lowers blood pressure and improves endothelial function in spontaneously hypertensive rats. Pharmacol Res. 2016;113(Pt A):384–394. doi: 10.1016/j.phrs.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mell B, Jala VR, Mathew AV, et al. Evidence for a link between gut microbiota and hypertension in the Dahl rat. Physiol Genomics. 2015;47(6):187–197. doi: 10.1152/physiolgenomics.00136.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol. 2002;23(11):541–548. doi: 10.1016/s1471-4906(02)02316-5. [DOI] [PubMed] [Google Scholar]
- 11.Wenceslau CF, McCarthy CG, Szasz T, Spitler K, Goulopoulou S, Webb RC. Mitochondrial damage-associated molecular patterns and vascular function. Eur Heart J. 2014;35(18):1172–1177. doi: 10.1093/eurheartj/ehu047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wenceslau CF, Szasz T, McCarthy CG, Baban B, NeSmith E, Webb RC. Mitochondrial N-formyl peptides cause airway contraction and lung neutrophil infiltration via formyl peptide receptor activation. Pulm Pharmacol Ther. 2016;37:49–56. doi: 10.1016/j.pupt.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wenceslau CF, McCarthy CG, Webb RC. Formyl Peptide Receptor Activation Elicits Endothelial Cell Contraction and Vascular Leakage. Front Immunol. 2016;7:297. Published 2016 Aug 2. doi: 10.3389/fimmu.2016.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wenceslau CF, McCarthy CG, Szasz T, Goulopoulou S, Webb RC. Mitochondrial N-formyl peptides induce cardiovascular collapse and sepsis-like syndrome. Am J Physiol Heart Circ Physiol. 2015;308(7):H768–H777. doi: 10.1152/ajpheart.00779.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wenceslau CF, McCarthy CG, Szasz T, Calmasini FB, Mamenko M, Webb RC. Formyl peptide receptor-1 activation exerts a critical role for the dynamic plasticity of arteries via actin polymerization. Pharmacol Res. 2019;141:276–290. doi: 10.1016/j.phrs.2019.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Norgauer J, Krutmann J, Dobos GJ, Traynor-Kaplan AE, Oades ZG, Schraufstätter IU. Actin polymerization, calcium-transients, and phospholipid metabolism in human neutrophils after stimulation with interleukin-8 and N-formyl peptide. J Invest Dermatol. 1994;102(3):310–314. doi: 10.1111/1523-1747.ep12371788. [DOI] [PubMed] [Google Scholar]
- 17.Marshall SA, Qin CX, Jelinic M, et al. The Novel Small-molecule Annexin-A1 Mimetic, Compound 17b, Elicits Vasoprotective Actions in Streptozotocin-induced Diabetic Mice. Int J Mol Sci. 2020;21(4):1384. Published 2020 Feb 18. doi: 10.3390/ijms21041384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cipolla MJ, Gokina NI, Osol G. Pressure-induced actin polymerization in vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J. 2002;16(1):72–76. doi: 10.1096/cj.01-0104hyp. [DOI] [PubMed] [Google Scholar]
- 19.Richards EM, Pepine CJ, Raizada MK, Kim S. The Gut, Its Microbiome, and Hypertension. Curr Hypertens Rep. 2017;19(4):36. doi: 10.1007/s11906-017-0734-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Zhao F, Wang Y, Chen J, Tao J, Tian G, Wu S, Liu W, Cui Q, Geng B, Zhang W, Weldon R, Auguste K, Yang L, Liu X, Chen L, Yang X, Zhu B, Cai J. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 2017;5(1):14. Published 2017 Feb 1. doi: 10.1186/s40168-016-0222-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santisteban MM, Qi Y, Zubcevic J, Kim S, Yang T, Shenoy V, Cole-Jeffrey CT, Lobaton GO, Stewart DC, Rubiano A, Simmons CS, Garcia-Pereira F, Johnson RD, Pepine CJ, Raizada MK. Hypertension-Linked Pathophysiological Alterations in the Gut. Circ Res. 2017;120(2):312–323. doi: 10.1161/CIRCRESAHA.116.309006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor WR, Takemiya K. Hypertension Opens the Flood Gates to the Gut Microbiota. Circ Res. 2017;120(2):249–251. doi: 10.1161/CIRCRESAHA.116.310339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu J, Luo H, Wang J, Tang W, Lu J, Wu S, Xiong Z, Yang G, Chen Z, Lan T, Zhou H, Nie J, Jiang Y, & Chen P Enteric dysbiosis-linked gut barrier disruption triggers early renal injury induced by chronic high salt feeding in mice. Exp Mol Med. 2017;49(8):e370. Published 2017 Aug 25. doi: 10.1038/emm.2017.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joe B, Garrett MR, Dene H, Rapp JP. Substitution mapping of a blood pressure quantitative trait locus to a 2.73 Mb region on rat chromosome 1. J Hypertens. 2003;21(11):2077–2084. doi: 10.1097/00004872-200311000-00017. [DOI] [PubMed] [Google Scholar]
- 25.Sherman SB, Sarsour N, Salehi M, Schroering A, Mell B, Joe B, Hill JW. Prenatal androgen exposure causes hypertension and gut microbiota dysbiosis. Gut Microbes. 2018;9(5):400–421. doi: 10.1080/19490976.2018.1441664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumarasamy S, Gopalakrishnan K, Toland EJ, Yerga-Woolwine S, Farms P, Morgan EE, Joe B. Refined mapping of blood pressure quantitative trait loci using congenic strains developed from two genetically hypertensive rat models. Hypertens Res. 2011;34(12):1263–1270. doi: 10.1038/hr.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hagdorn QAJ, Bossers GPL, Koop AC, Piek A, Eijgenraam TR, van der Feen DE, Silljé HHW, de Boer RA, Berger RMF. A novel method optimizing the normalization of cardiac parameters in small animal models: the importance of dimensional indexing. Am J Physiol Heart Circ Physiol. 2019;316(6):H1552–H1557. doi: 10.1152/ajpheart.00182.2019. [DOI] [PubMed] [Google Scholar]
- 28.McCarthy CG, Wenceslau CF, Goulopoulou S, Ogbi S, Baban B, Sullivan JC, Matsumoto T, Webb RC. Circulating mitochondrial DNA and Toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc Res. 2015;107(1):119–130. doi: 10.1093/cvr/cvv137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edwards JM, Roy S, Tomcho JC, Schreckenberger ZJ, Chakraborty S, Bearss NR, Saha P, McCarthy CG, Vijay-Kumar M, Joe B, Wenceslau CF. Microbiota are critical for vascular physiology: Germ-free status weakens contractility and induces sex-specific vascular remodeling in mice. Vascul Pharmacol. 2020;125–126:106633. doi: 10.1016/j.vph.2019.106633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawton PF, Lee MD, Saunter CD, Girkin JM, McCarron JG, Wilson C. VasoTracker, a Low-Cost and Open Source Pressure Myograph System for Vascular Physiology. Front Physiol. 2019;10:99. Published 2019 Feb 21. doi: 10.3389/fphys.2019.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Briones AM, Salaices M, Vila E. Mechanisms underlying hypertrophic remodeling and increased stiffness of mesenteric resistance arteries from aged rats. J Gerontol A Biol Sci Med Sci. 2007;62(7):696–706. doi: 10.1093/gerona/62.7.696. [DOI] [PubMed] [Google Scholar]
- 32.Wang G, Zhang L, Chen X, Xue X, Guo Q, Liu M, Zhao J. Formylpeptide Receptors Promote the Migration and Differentiation of Rat Neural Stem Cells. Sci Rep. 2016;6:25946. Published 2016 May 13. doi: 10.1038/srep25946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rapp JP, Dene H. Development and characteristics of inbred strains of Dahl salt-sensitive and salt-resistant rats. Hypertension. 1985;7(3 Pt 1):340–349. [PubMed] [Google Scholar]
- 34.Saklayen MG, Deshpande NV. Timeline of History of Hypertension Treatment. Front Cardiovasc Med. 2016;3:3. Published 2016 Feb 23. doi: 10.3389/fcvm.2016.00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanders PW. Salt-sensitive hypertension: lessons from animal models. Am J Kidney Dis. 1996;28(5):775–782. doi: 10.1016/s0272-6386(96)90265-6 [DOI] [PubMed] [Google Scholar]
- 36.Svetkey LP, McKeown SP, Wilson AF. Heritability of salt sensitivity in black Americans. Hypertension. 1996;28(5):854–858. doi: 10.1161/01.hyp.28.5.854 [DOI] [PubMed] [Google Scholar]
- 37.Fasano A, Not T, Wang W, et al. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet. 2000;355(9214):1518–1519. doi: 10.1016/S0140-6736(00)02169-3 [DOI] [PubMed] [Google Scholar]
- 38.Wang W, Uzzau S, Goldblum SE, Fasano A. Human zonulin, a potential modulator of intestinal tight junctions. J Cell Sci. 2000;113 Pt 24:4435–4440. [DOI] [PubMed] [Google Scholar]
- 39.El Asmar R, Panigrahi P, Bamford P, Berti I, Not T, Coppa GV, Catassi C, Fasano A. Host-dependent zonulin secretion causes the impairment of the small intestine barrier function after bacterial exposure [published correction appears in Gastroenterology 2003 Jan;124(1):275. El Asmar Rahzi [corrected to El Asmar Ramzi]]. Gastroenterology. 2002;123(5):1607–1615. doi: 10.1053/gast.2002.36578. [DOI] [PubMed] [Google Scholar]
- 40.Fasano A All disease begins in the (leaky) gut: role of zonulin-mediated gut permeability in the pathogenesis of some chronic inflammatory diseases. F1000Res. 2020;9:F1000 Faculty Rev-69. Published 2020 Jan 31. doi: 10.12688/f1000research.20510.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buemi M, Corica F, Marino D, Medici MA, Aloisi C, Di Pasquale G, Ruello A, Sturiale A, Senatore M, Frisina N. Cardiovascular remodeling, apoptosis, and drugs. Am J Hypertens. 2000;13(4 Pt 1):450–454. doi: 10.1016/s0895-7061(99)00213-7. [DOI] [PubMed] [Google Scholar]
- 42.Priante G, Gianesello L, Ceol M, Del Prete D, Anglani F. Cell Death in the Kidney. Int J Mol Sci. 2019;20(14):3598. Published 2019 Jul 23. doi: 10.3390/ijms20143598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ying WZ, Sanders PW. Cytochrome c mediates apoptosis in hypertensive nephrosclerosis in Dahl/Rapp rats. Kidney Int. 2001;59(2):662–672. doi: 10.1046/j.1523-1755.2001.059002662.x. [DOI] [PubMed] [Google Scholar]
- 45.Ying WZ, Wang PX, Sanders PW. Induction of apoptosis during development of hypertensive nephrosclerosis. Kidney Int. 2000;58(5):2007–2017. doi: 10.1111/j.1523-1755.2000.00373.x [DOI] [PubMed] [Google Scholar]
- 46.Sanders PW, Wang PX. Activation of the Fas/Fas ligand pathway in hypertensive renal disease in Dahl/Rapp rats. BMC Nephrol. 2002;3:1. Published 2002 Jan 7. doi: 10.1186/1471-2369-3-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martinez-Quinones P, Komic A, McCarthy CG, Webb RC, Wenceslau CF. Targeting Endothelial Barrier Dysfunction Caused by Circulating Bacterial and Mitochondrial N-Formyl Peptides With Deformylase. Front Immunol. 2019;10:1270. Published 2019 Jun 6. doi: 10.3389/fimmu.2019.01270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Paulis A, Ciccarelli A, de Crescenzo G, Cirillo R, Patella V, Marone G. Cyclosporin H is a potent and selective competitive antagonist of human basophil activation by N-formyl-methionyl-leucyl-phenylalanine. J Allergy Clin Immunol. 1996;98(1):152–164. doi: 10.1016/s0091-6749(96)70237-3. [DOI] [PubMed] [Google Scholar]
- 49.Handschumacher RE, Harding MW, Rice J, Drugge RJ, Speicher DW. Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science. 1984;226(4674):544–547. doi: 10.1126/science.6238408. [DOI] [PubMed] [Google Scholar]
- 50.Wilson C, Zhang X, Buckley C, Heathcote HR, Lee MD, McCarron JG. Increased Vascular Contractility in Hypertension Results From Impaired Endothelial Calcium Signaling. Hypertension. 2019;74(5):1200–1214. doi: 10.1161/HYPERTENSIONAHA.119.13791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Touyz RM, Alves-Lopes R, Rios FJ, et al. Vascular smooth muscle contraction in hypertension. Cardiovasc Res. 2018;114(4):529–539. doi: 10.1093/cvr/cvy023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.