

Abstract
Cardiovascular disease (CVD) is the leading cause of death worldwide, causing over 17 million deaths per year, which outpaces global cancer mortality rates. Despite these sobering statistics, most bioinformatics and computational biology research and funding to date has been concentrated predominantly on cancer research, with a relatively modest footprint in CVD. In this paper, we review the existing literary landscape and critically assess the unmet need to further develop an emerging field at the multidisciplinary interface of bioinformatics and precision cardiovascular medicine, which we refer to as ‘cardioinformatics’.
Keywords: cardiovascular disease, bioinformatics, cardiology, computational biology
Introduction
According to the World Health Organization, ischemic heart disease and stroke have remained the top two global killers in the past 15 years. The Global Burden of Diseases, Injuries, and Risk Factors Study shows that heart disease is still the dominant cause of death globally for both genders [1], with a projection that by 2030, almost half of the adult population will have a CVD diagnosis [2, 3]. In the United States, cardiovascular diseases (CVDs) have been the leading cause of death by non-communicable diseases, consistently surpassing cancer for the past many decades (Figure 1A). However, federal National Institutes of Health (NIH) funding on CVD research has consistently been less than that on cancer research, at least half a billion dollars annually since 2008 (Figure 1B). Meanwhile, there has been a 12.5% increase in the global number of deaths from CVD in the past decade [4].
Figure 1.
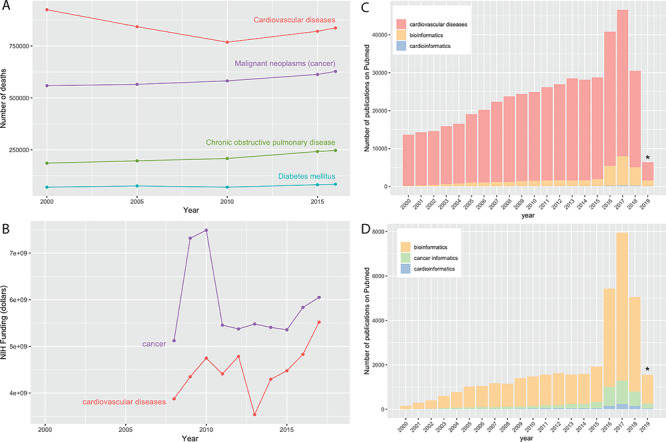
The status of CVD research. (A) Number of deaths by non-communicable diseases in the United States and (B) funding by the NIH for research on cancer and CVDs. (C) PubMed queries reveal a large body of CVD research, out of which only a small percentage involve bioinformatics. (D) Relative to the total pool of bioinformatics papers (in any field), there are far more cancer papers that utilize bioinformatics methods than CVD papers that utilize such methods. 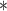 Since all the queries are based on the manual MeSH catalog, more recent tallies will lag behind the true volume of publication.
Since all the queries are based on the manual MeSH catalog, more recent tallies will lag behind the true volume of publication.
Research in CVD has steadily increased since the year 2000, as measured by the body of publications indexed in PubMed over this time (Figure 1C). In 2017 alone, there were more than 40 000 primary research (non-review) articles classified with the subject heading ‘cardiovascular disease’, defined according to the Medical Subject Heading (MeSH) terms (Figure 1C). However, the share of bioinformatics research has remained modest among these CVD outputs at least relative to comparable work done in cancer biology (Figure 1D). For example, multi-omics data integration reveals novel disease pathways and therapeutic targets, but its implementation in CVD research areas like cardiovascular (CV) calcification is failing to keep pace with other research fields, such as oncology [5]. While bioinformatics is at the center of precision medicine [6] and CV research is involved in several existing precision medicine initiatives [7, 8], the field of cardioinformatics is still in its early days with ample opportunities to benefit from cutting-edge data science techniques and machine learning (ML) methodologies, as has been the case in precision oncology. Even now, the application of ML is already being recognized as an indispensable component of the practice of cardiology in the future [9, 10] and, therefore, given the availability of increasingly performant ML implementations [11], cardioinformatics is better positioned to tackle domain-specific research questions by developing clinical applications to enhance compute-intensive tasks such as those found in medical imaging, CVD risk prediction modeling, among other active research areas. For instance, current methods for CV calcification imaging are mostly limited to advanced calcification and miss clinically relevant early microcalcifications, creating an unmet need for implementation of advanced imaging tools and artificial intelligence to improve diagnostics and risk assessment [5].
In general, efficient implementations of advanced computational algorithms that optimize for time, cost and accuracy measures across broad domains of biological data science, such as single-cell sequencing [12] (e.g. to investigate cellular heterogeneity in transcription [13]) or long-read mapping [14] (e.g. to reconstruct full-length isoform transcripts in high resolution [13]), will find increasingly more adoption in CVD research throughout the next few years as journals begin gearing up for the release of special issues dedicated exclusively to performance benchmarking of new and existing software tools. In addition, the availability of open-access benchmarking data and guidelines to evaluate ML methods across a broad range of application areas including biomedical studies, signal processing and image classification will catalyze the precipitation of the most appropriate bioinformatics software tools for any given research task [15, 16]. Taken together, programmatic need for bioinformatics benchmarking and awareness of state-of-the-art tools for performing CVD research will bridge across multiple areas of expertise (e.g. single-cell sequencing technologies, long-read mapping, 3D genome visualization, etc.), making cardioinformatics research a truly multidisciplinary initiative for dissecting the molecular mechanisms behind complex CVD traits.
The American Heart Association (AHA) Institute for Precision Cardiovascular Medicine recently partnered with Amazon Web Services to provide a variety of grant funding opportunities for testing and refining artificial intelligence (AI) and ML algorithms using healthcare system data and multiple longitudinal data sources to fund research that improves our understanding of all CVD data related to precision medicine. Therefore, we expect that grant funding initiatives such as these will gradually begin narrowing the gap between cardioinformatics and cancer research in terms of the availability of improved computational tools, infrastructure and analysis resources. Some recent positive trends in this direction include large-scale infrastructure and knowledge portal development [17–20] for working with CVD data, as well as population-wide multi-omics initiatives such as the NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium [21] for integrating whole-genome sequencing (WGS) and other -omics data (e.g. metabolic profiles, protein and RNA expression patterns) with molecular, behavioral, imaging, environmental and clinical data. In this review, we highlight these contemporary opportunities and perspectives for CVD genomic and precision medicine research, introduce the bountiful resources available and propose ways to advance this field further by promoting a culture steeped in computation vis-à-vis modern bioinformatics and computational biology methodologies.
The review is structured as follows: an overview of the current informatics landscape is provided in The democratization of data and the rise of knowledge bases. To explore what has been learned about CVDs, we reviewed an extensive body of CVD research, pivoting around genetics. Emerging from this survey is a central theme of the diseases’ enormous complexity, which is elaborated in the section called Complexity of CVDs. Despite decades of applying and extending statistical methods to study CVD, our knowledge of the diseases barely extends beyond genetic associations into causal, mechanistic insights. Given the exciting expansion of biological datasets, the advances of knowledge bases and the current status of CVD research, we then propose three areas where bioinformaticians and CVD researchers may want to prioritize in pushing this field forward, in The challenges of cardioinformatics.
The democratization of data and the rise of knowledge bases
The past few years have seen a substantial rise in the availability of computational resources and infrastructure that provide access to aggregate genetic data and genomic summary results to facilitate rapid and open sharing of individual level data and summary statistics pertinent to various biological diseases and data types. One of the early pioneers of web-based knowledge portals has been a Memorial Sloan Kettering Cancer Center resource called cBioPortal [22, 23], which provides intuitive visualization and analysis of large-scale cancer genomics datasets from large consortium efforts such as TCGA [24] and TARGET [25] as well as publications from individual labs. Other major players in the cancer knowledge base arena include the National Cancer Institute’s Genomic Data Commons (GDC) Portal [26, 27], which provides full download and access to all raw data (e.g. mRNA expression files, full segmented copy number variant [CNV] files, etc.) generated by TCGA and TARGET. In addition, resources such as the Broad Institute’s Single Cell Portal [28] provide an unprecedented view into the biology of different diseases, including cancers like glioblastoma, at the single-cell sequencing level.
More recently, the Knowledge Portal Framework, an infrastructure sponsored by the Accelerating Medicines Partnership and developed at the Broad Institute, has empowered a variety of disease-focused portals, including those for type II diabetes [29], amyotrophic lateral sclerosis [30], sleep disorders [31], CV [32] and cerebrovascular diseases [33]. The purpose of these resources is to aggregate and store statistical data for hundreds of millions of genetic variants and organize them to be rapidly queried and visualized by biologists, statistical geneticists, pharmaceutical researchers and clinicians. Other such knowledge bases focused on exploring large-scale genetic association data in the context of, for instance, drug/treatment targets include the OpenTargets initiative [25], which is a public–private venture that generates evidence on the validity of therapeutic targets based on genome-scale experiments and analysis. Another public–private partnership—the Accelerating Medicines Partnership-Alzheimer’s Disease (AMP-AD) Target Discovery and Preclinical Validation Project—has developed an AMP-AD Knowledge Portal to help researchers identify potential drug targets to accelerate pre-competitive Alzheimer’s disease treatment and prevention [34]. Interactive, web-based tools such as the Agora platform [35] bring together both AMP-AD analyses and OpenTargets knowledge under one umbrella to help explore and ultimately assist the validation of early AD candidate drug targets.
In addition to these various portals anchored on the results of population genetic association studies, CVD knowledge bases such as HeartBioPortal [17] have begun organizing and integrating the large volume of publicly available gene expression data with genetic association content, motivated by the stimulus that transcriptomic data provide powerful insights into the effects of genetic variation on gene expression and alternative splicing in both health and disease. Other knowledge portals such as COPaKB [36] and large-scale initiatives such as HeartBD2K [37] have taken a parallel focus on CVD proteomics datasets [38]. Such integrative multilevel efforts to dissect the molecular mechanisms behind complex disease traits now also extend beyond academia into biotech startup companies, non-profits and other initiatives such as SVAI, Quiltomics, Omicsoft, Sage Bionetworks, Omics Data Automation, Occamzrazor, NextBio, BenevolentAI, Insitro, Researchably and others—several of which have recently been effectively integrated into the workflow of larger biotech companies such as Illumina and Qiagen. Parallel to these academic and industry initiatives, several government-led genomic sequencing programs to collect a nation’s data have appeared over the years, setting the stage for centralized databases serving disease prevention, health management and discovery. Among those programs are the recently completed 100K Genomes Project in the UK [39], the ongoing 100K Wellness Pioneer Project in China [40], NIH’s All of Us Research Program [41] and the Department of Veterans Affairs Million Veteran Program [42] in the United States, many of which contain vast quantities of population-wide race/ethnic group-specific CVD data. Most recently, the All of Us program released a public data commons browser [43] to explore the prevalence of specific conditions, drug exposures and other clinically relevant factors on a demographically diverse cohort of participants, including populations historically underrepresented in biomedical research. The data in the All of Us Data browser include many CVD phenotypes and come from participant electronic health records (EHRs) and from survey responses (e.g. on basic demographics, overall health and lifestyle) as well as physical measurements (e.g. blood pressure, heart rate, height, weight, waist circumference and hip circumference) taken at the time the participants enroll in the All of Us program.
Complexity of CVDs
The number of genetic actors
There is a certain genetic component in all major categories of heart disease (Figure 2). The increase in genome-wide association studies (GWASs) has led to the associations of more and more genetic variants to human traits and diseases [44], fueling the identification of hundreds of novel drug targets and the development of polygenic risk scores that may help improve the ability to predict a person’s pre-disposition to various CV ailments [45–49] and facilitate early and preventative care [50]. Although CVD is significantly broad and encompasses diseases related to blood vessels, the myocardium, heart valves, the conduction system and developmental abnormalities, there are only a few CV disorders that can be attributed to a single pathogenic gene (as covered in detail within a recent review [2]). Although GWAS is, by definition, designed to implicate a single pathogenic gene, or a limited number of pathogenic genes, Leopold and Loscalzo [2] present a cogent argument against the theory of a single causal pathogenic gene/gene product as a mediator of CVD phenotypes, even in cases of certain classic Mendelian disorders. Nevertheless, discovering new rare and common variants that may control individual drug responses in different race/ethnic populations may elucidate not only disease mechanisms but also improve clinical trial design whereby drug candidates can be tested in more targeted subpopulations, in which drug efficacy is not masked by the inclusion of predicted nonresponders [51]. To this end, enriching clinical trial selection and enrollment is one of the target outcomes of precision medicine [2]. This is motivated by, for example, case studies of CV pathologies that are prominently characterized by biomarkers that do not reveal the underlying complexity of the disease or its etiology. For instance, although atherosclerosis is strongly clinically associated with elevated low-density lipoprotein levels, the underlying biology is more complex, as suggested by the clinical failure of evacetrapib despite significant effects on low-density lipoprotein [52]. Likewise, since clinical trials tend to focus on the mean response to an intervention instead of examining variability in response, current therapies for clinical indications like essential hypertension are still unsatisfactory because most clinical trials generally examine outcome effects as the sample mean blood pressure is decreased, not personalized differential treatment approaches tailored to patients’ individual hypertension profiles [52]. As a result, it is estimated that 44% of patients with essential hypertension were unable to achieve blood pressure control despite pharmacological therapy [52, 53].
Figure 2.
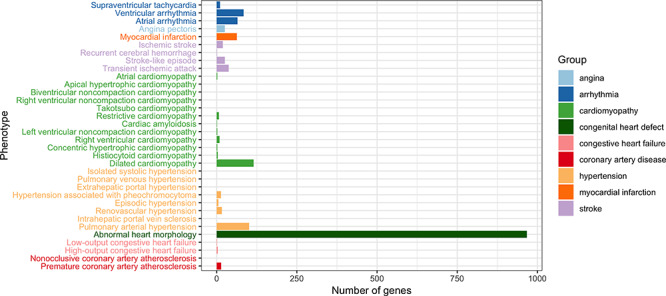
The number of genes associated with CVD. CVD is defined to include all phenotypes under the term ‘Abnormality of the cardiovascular system’ (HP:0001626) in the Human Phenotype Ontology [248]. Annotations of each phenotype was pooled from OMIM [249], Orphanet [250] and DECIPHER [251].
Predictably, over time the number of variants found associated with (any given) disease has increased and, in most cases, gone beyond a few implicated genes that could be described in a single-page table or diagram. Dilated cardiomyopathy (DCM), a common cause of heart transplantation [54], is a vivid example of how causal variants and their corresponding genes were discovered over the years. In a recent review [54], 16 disease-causing genes were compiled, along with an additional 41 putative genes. Meanwhile, the NHGRI-EBI GWAS Catalog [55] and annotations on Human Phenotype Ontology [56] suggest a larger number of genes associated with this condition, 69 genes and 115 genes, respectively. Clinical application has been keeping up, with a typical commercial gene panel for DCM genetic testing covering 50 genes on average, and 111 in total [57]. Although these genes were discovered via different approaches, the catalog of DCM-associated loci kept expanding. Similarly for coronary artery disease (CAD), additional loci have been associated with the disease almost every year since 2007, bringing the total number of loci associated with CAD to over 150 [58]. Since it is reasonably expected that when more genes are involved in a disease, the individual effect exerted by each gene will be small; these constantly expanding gene panels suggest that the common mutations in a single gene are not likely to capture substantial disease risk for most cases that are polygenic. From a research perspective, these findings imply that the quest of pinpointing causal variants is getting progressively more challenging, because testing the variant–phenotype association on small-effect variations requires a much larger number of samples for sufficient power [44], or critically different methods of statistical testing and inference. Using atherosclerosis as an example, Cranley and MacRae [59] argue that the slow progress on disease mechanisms comes not from incomplete genotyping to identify associated variants, but rather from the inability to draw causal relationships between identified variants (e.g. 9p21) and disease pathways [51]. Specifically, although SNPs in this region were identified by several independent GWAS, and each risk allele was associated with a 29% increased risk of CVD, these SNPs are in noncoding regions where the nearest genes (CDKN2B, CDKN2A) are >100 Kb away, and the causality between these genes and susceptibility to atherosclerosis has not yet been ascertained [60–62]. In general, many genomic variants implicated in GWAS occur in intervening regions with no immediate connections to known coding genes or biochemical pathways and, therefore, studies using ATAC-seq and other NGS techniques (e.g. RNA-seq, Hi-C, etc.) are linking loci identified by GWAS to epigenetic changes such as enhancer–promoter interactions [51]. In addition, large numbers of GWAS variants are now known to function as expression quantitative trait loci (eQTL), meaning that they regulate the expression level of transcripts (as measured, e.g. by RNA-seq), whereas splice quantitative trait loci regulate the splice ratio of transcript isoforms [51], highlighting how the transcriptome can offer a dynamic view of the functions of genetic variants in response to various acute and cumulative exposures including genetic, metabolic and environmental mediators. Finally, as large-scale data become more readily available for population-level estimation of many genetic variants with low allele frequencies, the high penetrance of many previously labeled ‘pathogenic’ rare variants (minor allele frequency < 0.1%) has been questioned [63]. In other words, genomic sequencing data from large population-level cohorts is uncovering many of the same variants previously annotated as pathogenic mutations [63, 64], prompting the need for variant reclassification and the conclusion that some genes reported to cause inherited heart disease are likely spurious [65]. Alternatively, it can also happen that ostensibly causal genetic variants found in family studies have no related phenotype in the population-level setting [66], highlighting some of the general challenges in attributing causality and understanding disease mechanism at the level of the individual patient [65]. In general, although GWAS studies have been successful in identifying genetic variation implicated in CVDs, they provide little or no molecular evidence of gene causality [67]. These observations open up a small window into the complicated and dynamic landscape of human disease genetics.
Besides the increasing difficulty of discovering these variations, modeling their effects poses another set of challenges. With a potential interaction between every pair of genomic features, be they genes or regulatory sequences, the number of such interactions increases quadratically with the number of actors, leading to the combinatorial explosion of states that a biological system can assume (theoretical calculation: 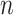 elements leads to
elements leads to 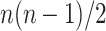 , i.e.
, i.e. 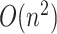 interactions). The plethora of variants associated with a disease or risk of disease do not promise a quick understanding of pathobiological mechanisms, as the functional consequences of a majority of these variants remain unknown. Among the most understood are PCSK9 [68], ANGPTL4 [69, 70] and APOC3 [71] on which association tests and sequencing have been combined to ascertain the linkage to CAD risk, translating to potential vascular protective drugs. The methodology of these studies are still under the influence of mainstream CVD research, i.e. revolving around genotype–phenotype association testing. This top-down approach, i.e. phenotype to gene to variant, has certain limits in its power, requiring more and more samples for less frequent and less penetrant alleles while leaving gaps in mechanistic understanding. Bottom-up approaches in which variations are systematically introduced into a DNA sequence (and their functional consequences are characterized in vitro) will complement the current understanding of these diseases. As exemplified in the novel assays enabled by state-of-the-art experimental techniques and computational processing, this approach has demonstrated utility in cancer variant classification [72], foreshadowing similar progress in CVD research.
interactions). The plethora of variants associated with a disease or risk of disease do not promise a quick understanding of pathobiological mechanisms, as the functional consequences of a majority of these variants remain unknown. Among the most understood are PCSK9 [68], ANGPTL4 [69, 70] and APOC3 [71] on which association tests and sequencing have been combined to ascertain the linkage to CAD risk, translating to potential vascular protective drugs. The methodology of these studies are still under the influence of mainstream CVD research, i.e. revolving around genotype–phenotype association testing. This top-down approach, i.e. phenotype to gene to variant, has certain limits in its power, requiring more and more samples for less frequent and less penetrant alleles while leaving gaps in mechanistic understanding. Bottom-up approaches in which variations are systematically introduced into a DNA sequence (and their functional consequences are characterized in vitro) will complement the current understanding of these diseases. As exemplified in the novel assays enabled by state-of-the-art experimental techniques and computational processing, this approach has demonstrated utility in cancer variant classification [72], foreshadowing similar progress in CVD research.
In addition to the loci that have been directly associated with CVD, a large number of genes or regulatory elements may contribute significantly to CVD risks in an indirect manner, due to highly interconnected biological pathways. For instance, independent research in aging has unraveled the intertwined relationship between heart disease and longevity pathways [73]. With age being the most important factor in conferring CVD risk [74], it is likely that these longevity genes will be involved in future analyses of CVD genetics. The genetic scope of CVD may be enlarged even further to include most of the genome, under the recently proposed omnigenic model for complex traits, in which most heritability is explained by peripheral genes outside of the core pathways [75]. Such expansion calls for a paradigm shift from additive effects of multiple genes to the interactions between them, from the physical genes to the ‘eigen-genes’ that represent biologically functional modules [76].
Such a paradigm shift is, in fact, only part of the potential answer to the long-standing puzzle of missing heritability in CVD as well as other complex diseases [77]. Heritability 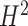 , in the ‘broad sense’, is the proportion of phenotypic variance that can be explained by genetic factors, while the ‘narrow sense’ heritability
, in the ‘broad sense’, is the proportion of phenotypic variance that can be explained by genetic factors, while the ‘narrow sense’ heritability 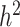 is the proportion attributable to ‘additive’ genetic factors [77]. If all the heritability has been accounted for, the squared correlation between the observed and the predicted phenotype should be equal to
is the proportion attributable to ‘additive’ genetic factors [77]. If all the heritability has been accounted for, the squared correlation between the observed and the predicted phenotype should be equal to 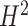 (or
(or 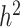 ). The increasing number of loci associated with complex traits still leave a large gap between predicted and observed phenotypes, prompting different strategies to account for the missing heritability. One of the more obvious causes of missing heritability relates to the limited ascertainment of the total pool of rare variants in humans. Even among rare causal variants identified to date, associations with disease has likely been under-appreciated due to insufficient study power to detect modest effects on risk. One addresses this issue by collecting more samples among diverse study populations [44, 63] and improving statistical tests [78, 79]. Even so, conventional models of phenotype prediction have relied almost exclusively on the additive effects of genetic factors, hence can only explain narrow-sense heritability
). The increasing number of loci associated with complex traits still leave a large gap between predicted and observed phenotypes, prompting different strategies to account for the missing heritability. One of the more obvious causes of missing heritability relates to the limited ascertainment of the total pool of rare variants in humans. Even among rare causal variants identified to date, associations with disease has likely been under-appreciated due to insufficient study power to detect modest effects on risk. One addresses this issue by collecting more samples among diverse study populations [44, 63] and improving statistical tests [78, 79]. Even so, conventional models of phenotype prediction have relied almost exclusively on the additive effects of genetic factors, hence can only explain narrow-sense heritability 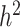 at best. In addition, alterations outside of DNA sequences have been found to be heritable, suggesting another part of the puzzle relies on epigenetics. Thus, to advance our understanding of complex diseases such as CVD requires moving beyond the exome and genome, as discussed in the next section.
at best. In addition, alterations outside of DNA sequences have been found to be heritable, suggesting another part of the puzzle relies on epigenetics. Thus, to advance our understanding of complex diseases such as CVD requires moving beyond the exome and genome, as discussed in the next section.
The diversity of actors
From SNPs to structural variations
Genome-wide association studies have been predominantly conducted on single-nucleotide polymorphisms (SNPs) thanks to the availability of easy-to-produce SNP microarrays. Such technologies have clearly enriched our knowledge base of the effect of single nucleotide variants, while leaving the effect of structural variations (SV) poorly understood. There are now 660 million SNPs documented in the dbSNP database [80], compared to 4.6 million SVs in the DGVa (Database of Genomic Variants archive [81], which also includes studies annotated by the NCBI-hosted database of structural variants, dbVar [82]). SV databases such as dbVar and DGVa are in fact storing each study–publication individually instead of cataloging SVs into data entries. Although the current knowledge base of SVs is not sufficient to create reference entries of SV, the map of SV from 1000 Genomes Project [83] has enabled further studies of the role of SV in cardiac diseases, suggesting the potential impact of SV on the transcriptional regulation of cardiac genes expressed in the heart [84]. As envisioned, SVs might be one of the promising areas to look for the missing heritability in CVD [85]. In fact, the relative lack of SV investigation in CVD has been recognized as one of the key issues that confound the attribution of causality in linking genetic variants to CVD phenotypes [65]. To this end, ongoing projects like TOPMed contain a Structural Variant Working Group to call CNVs within TOPMed, and they have begun incorporating large-scale multi-ancestry studies spanning diverse types of sequencing data from both European and non-European race/ethnic groups. Most recently, the gnomAD browser [63] added 500 000 structural variants from 15 000 genomes, with full VCF and BED files available for download [86].
From coding to noncoding regions
As array-based genotyping was gradually replaced by next-generation sequencing, the cost of sequencing an exome, i.e. the protein-coding part of a genome, became much more affordable and enabled the collection of more than 60 000 exomes [63]. Using this dataset, Walsh and colleagues [64] found that many ‘pathogenic’ genetic variants associated with various cardiomyopathies are equally common in clinical cases as in the control population. Genes that were consistently included on genetic testing panels for DCM such as MYBPC3, MYH6, SCN5A, etc. turned out to be less penetrant than previously thought, in consideration of their frequency in the control population. The rationale for prioritizing the sequencing of exome over that of the entire genome, besides the lower cost, was a regularly cited statement that the exome harbors 85% of disease-causing variants [87] which turned out to be an outdated estimate from 1995. In our own survey of the NHGRI-EBI GWAS Catalog [55], a large fraction of variants tend to occur in non-protein coding regions such as intronic, intergenic and splice junctions (Figure 3). The distribution of CVD-associated variants is similar to that of variants associated with all traits. Previous studies also asserted the prevalence of regulatory regions among variants associated with cardiometabolic risk [88], as well as many other complex traits [89]. As an unprecedented amount of WGS data become available from large-scale genomic projects such as The 1000 Genomes Project [90], UK10K [91], The 100,000 Genomes Project [39], The 100K Wellness Pioneer Project in China [40], All of Us Research Program [41], TOPMed [21] and CCDG [92], we are poised to learn more about this ‘dark matter’ in the human genome and how it works in complex diseases.
Figure 3.
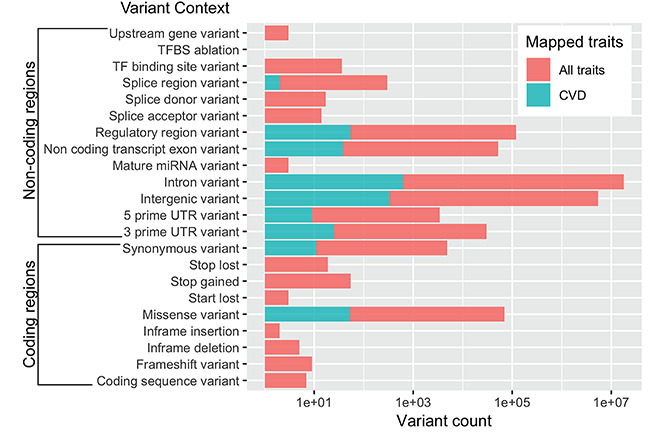
Distribution of SNPs that have been associated with a phenotypic trait. The associations are downloaded from NHGRI-EBI GWAS Catalog in which only those with P-value 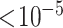 were retained.
were retained.
Beyond genetics: epigenetics and gene–environment interplay
CV risks can be conferred through heritable changes in gene expression without alterations in the underlying DNA sequence. These epigenetic processes traditionally involve DNA methylation, a wide range of histone modifications including acetylation, methylation, phosphorylation, ubiquitylation, sumoylation and biotinylation, and are now encompassing a looselydefined group of processes mediated by long noncoding RNAs (lncRNAs) and microRNAs (miRNAs). Dysregulation in epigenetic processes has been associated with the pathogenesis of cancer and many other diseases. To date, epigenetic mechanisms have been demonstrated to be involved in a variety of CVDs and conditions [93–97]. For instance, early differential epigenomic analysis, albeit on a limited number of samples, established differentiating features in DNA methylation and histone H3 methylation between control and failing hearts [98]. Likewise, abnormal expression and activity of histone deacetylases (HDACs) have been linked to cardiac defects, heart disease and cardiac development [99–102]. For example, HDAC9 is highly expressed in cardiac muscle, and one of the targets of HDAC9 is the transcription factor MEF2, which has been implicated in cardiac hypertrophy [103]. Following these early findings, epigenome-wide association studies have proposed a number of DNA methylation sites associated with blood lipid [104], body mass index [105, 106], heart failure [107] and heart attack history [108]. In addition, alterations in chromatin structure have been shown to induce heart failure [109].
As more lncRNAs were discovered and characterized, the prevalence of these molecules in CV biology also emerged. At least 22 lncRNAs were reportedly dysregulated in CVDs including CAD, myocardial infarction, cardiac hypertrophy and atherosclerosis, affecting a wide range of molecular, cellular and physiological processes [110, 111]. Due to low relative abundance levels and highly tissue-specific expression patterns, lncRNAs remain challenging to study. Some of the functions of lncRNA that have been recognized include imprinting, scaffolding, enhancer activity and molecular sponges. These actions mark the presence of lncRNAs in many CV processes such as cardiac differentiation, macrophage activation and sarcomere development [112]. With 107 039 lncRNAs detected in the human genome so far (reported by LNCipedia [113], as of November 2018), more lncRNAs are likely to be implicated in CV biology in the future, hence promising potential therapeutic targets. In this regard, there is increasing evidence that circulating miRNAs can serve as potential prognostic and diagnostic biomarkers for the prevention and treatment of CVDs [114], since they are critical regulators of CV function and play important roles in almost all aspects of CV biology [115–118] (for historical perspective, Azuaje and colleagues [119] reviewed some of the first CVD biomarkers discovered through integrative omics approaches). For example, miRNAs associated with the diagnosis and prognosis of heart failure, acute myocardial infarction, pulmonary hypertension and arrhythmia are reviewed by Zhou and colleagues [114]. Nevertheless, challenges remain: for example, for a miRNA to be considered a potential therapeutic target or diagnostic marker of CVD, it should be predominantly expressed in cardiac tissue and/or be essential for heart development, function or repair of heart-specific damage (e.g. miR-1, miR133a, miR-208a/b and miR-499) [120, 121], while also normalizing for the fact that miRNA expression levels are often affected by non-cardiac conditions (e.g. cancer, infection, drug use, etc.) and other co-morbidities. However, given the utility of miRNAs in both animal models and human clinic trials for cancer treatment [122–125], miRNA-based therapeutics for the treatment of CVD remain a promising area of research.
As epigenetic processes include various molecular and cellular events, the experimental assays for mapping of the epigenome are accordingly diverse. DNA methylation profiling can be done with methylation-sensitive restriction enzymes, bisulfite sequencing or immunoprecipitation with antibodies against methylated-cytosine [126]. Histone modifications can be profiled by immunoprecipitation with antibodies specific to the modified histone of interest, essentially requiring a ChIP experiment for each of the histone modifications one wants to interrogate [127]. Meanwhile, the noncoding RNA transcripts can be profiled with variations of RNA-seq experiments that are optimized for the target fraction of RNA. Such diversity entails significant difficulty in comprehensive profiling of the epigenome in a single experimental assay, stressing the need for re-collection and re-analysis of dispersed datasets for a more complete multi-omics picture. As epigenetic alterations have been found to be responsive to environmental cues throughout life, the epigenome lays an important bridge between the genetic makeup of an organism and its phenotype by helping to explain the gene–environment interplay. For example, environmental factors have been known for decades to play critical roles in conferring CV risk. Framingham-based risk scores [128], which include variables that can be intervened upon by lifestyle habits (smoking, blood cholesterol, blood pressure, diabetes), have guided clinical practices [129] and shown to perform well in predicting CV risk in many populations [130, 131]. The importance of a healthy lifestyle (absence of obesity, no current tobacco use, a healthy diet, regular physical activity) cannot be understated for CVD risk reduction, and it has been shown to serve as an environmental resilience factor and modify genetic risk of CVD [2]. In fact, over the past 50 years, historical progress towards the eradication of CVD has been achieved primarily through the adoption of lifestyle modifications, including dietary, tobacco and exercise interventions [2], including changes to public health policy (e.g. secondhand smoke legislation) and other health measures. In a recent study of 55 685 people stratified according to a polygenic risk score, it was found that individuals with a high genetic risk of CAD had a 46% reduction in the relative risk of coronary events if they had a healthy lifestyle, compared to individuals who did not [132]. However, the relative performance of phenotype-based risk scores and the genotype-based counterpart is highly variable depending on specific populations and practices in designing score components. There exist lines of evidence favoring both phenotypic variables [133] and genotypic ones in predicting disease risk [131, 134–136]. Clearly, there remains a gap in understanding gene–environment interaction that can now be studied at the molecular level, thanks to advances in experimental techniques to measure the exposome, i.e. all environmental factors/exposures throughout life that influence disease, including an individual’s diet, pollutants and infections [137]. With recent development of wearable devices to collect real-time data in a non-intrusive manner, it is now possible to monitor the exposome for its dynamic compositions of chemical compounds and micro-organisms [138, 139] as well as monitor early identifiers of CVD progression [140, 141] and disease diagnosis [142]. In general, the emergence of mobile health devices and sensors is now ushering in a new era of streaming data collection relevant to CVD metrics at the individual-based level, e.g. an individual’s blood pressure, heart rhythm, oxygen saturation, brain waves, air quality, radiation, among others [143]. Such devices will enable the collection of longitudinal personal omics profiles across different demographics, ultimately not only helping to detect health–disease transitions based on molecular and physiological metrics but also measuring interactions of environment and health outcomes that inform individualized health data [143]. Being among complex traits that are heavily influenced by environmental factors, CVD research is especially well positioned to benefit from these advances. For instance, Cranley and MacRae mention in a recent review [59] how studying the nutritional exposome (e.g. quantified images, purchase data, modern supply chain tracking of food) is likely to identify new triggers of coronary heart disease (CHD) and other disorders across populations. One interesting population–scale application we envision is monitoring the nutritional exposome (e.g. with respect to the temporal trajectory of atherosclerosis) through social media timeline photos (e.g. many Facebook/Instagram users consistently post food photos of their meals over a timespan of several years), which could potentially be tagged via AI/ML image classification algorithms that ultimately track dietary or lifestyle habits in a long-term longitudinal fashion. Therefore, one area of potentially transformational investigative impact could be the integration of social media activity with data streaming from wearable devices as a way to monitor the exposome. Clearly, such large-scale initiatives directly benefit from public–private partnerships between academia and industry, as has been the case for OpenTargets [25], which unites the academic efforts of EMBL-EBI and the Wellcome Sanger Institute with the commercial efforts of Sanofi, GlaxoSmithKline, Takeda and other biopharmaceutical companies. Likewise, the One Brave Idea initiative [144] has brought together the AHA, Verily, Astrazeneca and Quest Diagnostics to collectively work towards detecting the earliest stages of CHD, how it develops and how it can be stopped from leading to heart attacks and strokes. The objective of One Brave Idea centers on measuring biology early enough to define health and its maintenance rather than just disease and to do so longitudinally in a way that enables passive capture of disease trajectories [59].
The challenges of cardioinformatics
Research in CVD faces unique challenges due to many peculiarities of these diseases. One such idiosyncrasy is time-scale, e.g. atherosclerotic plaques and other CV risk factors build up over an extended period of time (often many decades), which puts CV phenotypes on a complex and continuous spectrum of transition from health to disease, from disease onset to progression. In contrast to diseases like cancer, which are characterized by rapid progression often with a clearly delineated before and after-disease state (e.g. stark mutational profile differences due to somatic hypermutation), CV pathologies such as CAD develop over an extended period of decades, beginning with atherosclerosis and manifesting variably along a spectrum from asymptomatic to stable ischemic heart disease, acute coronary syndrome and sudden cardiac death [52]. In general, the transition from one CVD (e.g. hypertension) to another (e.g. atherosclerosis), which may gradually morph into another CVD (e.g. CAD), which may or may not ultimately lead to a clinical episode like myocardial infarction or stroke, all over the time-scale of several (or more) decades poses its own set of unique informatics challenges. Monitoring the complex disease etiology of such a temporal progression influenced by a combination of genetics (omics profiles), environment (socioeconomics—e.g. zip code can often be as or more important than genetic code at predicting CVD risk [145, 146]) and lifestyle [smoking, diet (e.g. lipids, alcohol), etc.] is a very computationally challenging task and calls for new innovative data integration approaches for risk stratification and surveillance at both individual and population levels across different race/ethnic groups. To complicate matters further, CV pathologies frequently present as co-morbid or multi-morbid with other disease phenotypes such as diabetes, cancer, obesity and metabolic syndrome and rheumatologic disease [52]. Innovative systems biology/medicine approaches to increase the understanding of the multifactorial, complex underpinnings of CVD promise to enhance CVD risk assessment and pave the way to tailored therapies [147]. One active area of research to address these issues involves deeper phenotyping to enable better clinical phenomapping—the stratification of different CVDs into etiologically distinct subtypes, such that it becomes possible to define disease throughout its temporal trajectory, thereby allowing the measurement of fundamental underlying traits such as subclinical vascular abnormalities before they evolve into the classical syndrome (i.e. the full-blown clinical indication/manifestation of disease) [59]. In general, parsing phenotypes in this way allows for a finer, more granular approach to CVD management and prevention that can facilitate precision subtyping of pre-symptomatic and at-risk individuals from symptomatic ones in order to stratify patients for optimized care delivery [52]. Other challenges of cardioinformatics, particularly at the level of tailoring individualized CVD treatments and predicting patient outcomes, are highlighted in some recent reviews [148–152].
As illustrated in the previous section, the complexity of CVDs calls for pushing research beyond traditional boundaries. Such expansion implies the inclusion of various data modalities described above, such as genome sequences, DNA-methylation profiles, RNA expression profiles, protein expression profiles, metabolic profiles, etc. (Figure 5) within computational analysis workflows. For instance, the presence of even a single metabolite circulating in the blood can strongly predict myocardial infarction risk on top of clinical models [153]. Leon-Mimila and colleagues discuss in a recent review [67] the unmet need for more metabolomics and metagenomics approaches to identify biomarkers with potential clinical applicability in CVD studies. For instance, some bacterial species are associated with risk of CAD and plasma metabolites, e.g. the bacteria Veillonella is associated with chronic heart failure and is also inversely correlated with known CV protective metabolites such as niacin, cinnamic acid and orotic acid [154]. Such correlation between changes in metabolites and gut microbiome associated with chronic heart failure may also potentially be observed in other CVD phenotypes in the future, inviting exploration of new research avenues in this currently underexplored area. In general, circulating small molecules comprise not only endogenous species encoded by the genome but also various xenobiotics from the ‘envirome’, including ingested nutrients, pollutants and other particulate matter such as volatile organic compounds, heavy metals and air pollutants [51, 155, 156]. This complexity extends further to proteomics, where mass spectrometry methods have identified post-translational modifications such as citrullination and S-nitrosylation as direct modulators of CV biology [157], highlighting the direct role of organic chemistry [158] in conferring CVD risk. In general, these data modalities often represent different classes of biological molecules as well as their interactions (Figure 5A). Computational workflows relevant to CV medicine have been proposed [159], clearly illustrating how CVD research can benefit from existing computing resources, from cloud-computing infrastructures to analytic methods for metadata, search and indexing. Likewise, modular data science architectures for supporting CV investigations have been illustrated [17, 160]. Due to the sheer amount of data obtained from CVD research, ranging from medical records to medical images and high-throughput omics profiles, challenges related to data management and analysis that are generic to many fields become even more pressing for cardioinformatics. While benefiting from two decades of research in bioinformatics, there remain significant challenges that can be addressed to accelerate CVD research. From our own perspective, we suggest three particularly pertinent areas to prioritize cardioinformatics research: data sharing/security, multi-omics analysis and augmented intelligence.
Figure 4.
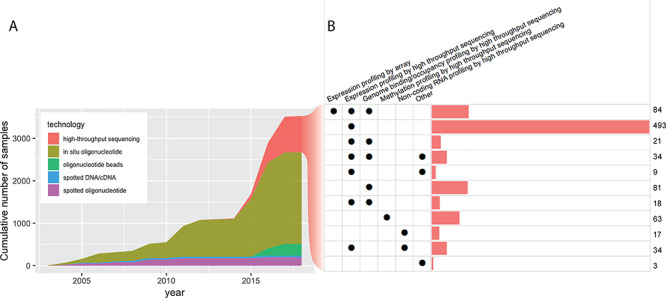
Molecular assays on GEO. (A) The cumulative number of molecular assays (i.e. unique combinations of biosample, study and platform) deposited in GEO by CV research. (B) Breakdown of high-throughput sequencing assays by the type of study. Expression profiling by high-throughput sequencing, i.e. mRNA-seq assays, are often coupled with another profiling technique, for example, to provide functional read-out of transcription factor binding profiled by ChIP-seq. Note that to avoid excessive over-counting of irrelevant samples such as those from plants or unrelated model organisms, we only counted samples deposited with a PubMed ID pointing to a CV study. Surveys were done on the GEOmetadb database [176] updated on 17 November 2018.
To share or not to share
Data sharing is believed to help scientific advances, thus benefiting everyone [161]. The sharing of personal health and medical data, however, comes with the risk of compromising a person’s privacy and subjecting them to discrimination [161–163]. The current data governance practices employ several administrative measures in the hope of minimizing the risk of exposing the data to adversity, or bad intentions. Taking the process of dbGaP data requests as an example: to access 658 305 records of genotype–phenotype data (Table 1) potentially relevant to future biomedical studies, a researcher first needs to browse these datasets, determine whether each dataset is consented for its purpose, obtain IRB approval if necessary, then file a request, prepare the facility, implement the security measures and transfer the data upon approval. Although the data users are advised, for example, to ‘avoid placing data on mobile devices’ and ‘destroy [the data] if they are no longer used or needed’, the only guarantee to such compliance is the vigilant mindset of every researcher involved in the data behind the project(s). In addition, different datasets are associated with different types of consents, dictating what purposes are permitted (e.g. general research, disease-specific research or biomedical research). Therefore, data users are responsible for obtaining the IRB approval compatible with these consents. These regulatory requirements have heightened the barrier to data access without robust mechanisms to enforce data protection nor to revoke the access when necessary. To add to these challenges, before filing a request, one needs to dive into the metadata of individual studies and decide which datasets are useful for the target research. Important information about a dataset such as the list of phenotypic variables are often vastly different from study to study and cannot be filtered against. In addition to those parameters of a study design, researchers need to be aware of the various types of consent forms applied to different datasets, many times within a single study. This procedure to obtain data access is currently applied for all controlled-access data in dbGaP, adding a significant administrative burden to biomedical researchers.
Table 1.
The subject count aggregated from studies deposited in dbGaP, consented for General Research Use (GRU)
| CVD | All | |
|---|---|---|
| 16s rRNA (NGS) | 0 | 92 |
| CNV Genotypes | 0 | 48 972 |
| Chromatin (NGS) | 0 | 139 |
| Genomic Sequence Amplicon (NGS) | 0 | 8 |
| Methylation (CpG) | 0 | 657 |
| Methylome sequencing | 0 | 152 |
| QTL Results | 0 | 281 |
| RNA Seq (NGS) | 333 | 1498 |
| SNP Genotypes (Array) | 6658 | 113 597 |
| SNP Genotypes (NGS) | 4277 | 11 786 |
| SNP Genotypes (PCR) | 0 | 10 |
| SNP Genotypes (imputed) | 0 | 29 693 |
| SNP/CNV Genotypes (NGS) | 0 | 936 |
| SNP/CNV Genotypes (imputed) | 0 | 9291 |
| SNV (.MAF) | 0 | 2 |
| SNV Aggregate (.MAF) | 0 | 570 |
| Targeted Genome (NGS) | 0 | 9918 |
| Whole Exome (NGS) | 5518 | 12 771 |
| Whole Genome (NGS) | 0 | 1245 |
| mRNA Expression (Array) | 0 | 798 |
| miRNA (NGS) | 0 | 228 |
| Total subject count in data consented for GRU | 16 786 | 242 644 |
| All consent groups | 584 884 | Unknown |
NGS: Next-generation sequencing, MAF: Minor Allele Frequency, QTL: Quantitative Trait Loci, SNV: Single-nucleotide variant
As a pioneering effort towards more accessible biomedical data, the AHA’s Precision Medicine Platform [18] has greatly simplified this process by streamlining the search, request and transfer of data. Datasets deposited on this platform were harmonized such that users can query for data across multiple studies by some common parameters, selectively request access to the relevant data and perform analyses on the cloud-based workspace. The platform has lifted significant burden off of data users by having them file one request for multiple datasets, and the data owners, being aware and responsible for complying with the consents on their data, will decide whether access can be granted or not. The cloud-based workspace also allows data to be transferred and analyzed in a controlled environment that can be ensured to comply with regulatory standards. The risk of intellectual property being compromised remains, for the data, once transferred, cannot be withdrawn nor prevented from being copied. As recognized by the authors, the platform is ‘only as good as the researchers make it’ [18]. More secure modes of data sharing have been explored, forming a spectrum of varying balance between analytic power and data protection. ViPAR [164] supports on-memory analysis of pooled data that is transferred to a central system, avoiding the permanent storage of sensitive data outside of the original sites. In all of the platforms above, a strong system for registering users as well as applying sanction measures are critical to enforce data usage agreements and deter malicious intent. Nevertheless, there still remains significant risk associated with data transfer and data protection at the user end. A couple of solutions have been proposed to further reduce the risks and responsibilities associated with direct access to sensitive data. For example, PRINCESS [165] is designed to perform statistical tests within an enclave hosted on a trusted server, in a stream of small data segments (8000 SNPs at a time). On the other end of the spectrum, DataSHIELD [166, 167] and COINSTAC [168] aim to allow data to be analyzed without moving out of the owners’ facility. Current implementations have shown that a variety of analytic tasks such as summary statistics, histograms, generalized linear models (DataSHIELD) and gradient descent (COINSTAC) can be performed in a distributed manner to achieve equally accurate results compared to the physically pooled counterpart and, more importantly, without disclosure of sensitive or personally identifiable information. The increasing prevalence of cloud computing platforms in scientific research [169] implies that forward-looking solutions should be able to work on these cloud environments.
Besides controlled-access data, a large amount of publicly available human data such as RNA-seq, ChIP-seq, Hi-C, etc. results are freely accessible with no restriction. Without genomic sequences, genotype or phenotype data, the processed output of these assays are deemed anonymous and void of sensitive information. However, recent studies have shown that information leakage is still possible, subjecting individuals to linking attacks that may reveal their identity [170, 171]. With millions of human genomes and thousands of other omics profiles on the not-so-distant horizon, a large fraction of which comes from CVD research programs (Figure 5), it is critical that cardioinformatics researchers pioneer the applications of these security measures, to ensure scientific advances do not compromise human rights to privacy and non-discrimination.
Multi-omics data ocean
The explosion of biological data is manifested in the growth of databases, consortium efforts, repositories, as well as the amount of raw and summary-level data hosted in these warehouses. High-throughput technologies are now available for characterizing and quantifying all major classes of biological molecules including DNA, RNA and protein (Figure 5A), leading to the creation of centralized repositories such as the Gene Expression Omnibus (GEO) [172] for gene expression data, dbGaP [173] for genotypes and phenotypes, ProteomeXchange [174, 175] for proteomics, MetabolomeXchange for metabolomics, among others. GEO, one of the most popular repositories for functional genomics data, has accumulated more than 100 000 datasets [176]. Among these, CVD publications have contributed more than 2000 microarray-based experiments, and about 900 high-throughput sequencing experiments for various purposes (Figure 4). The amount of data potentially reusable for CVD research may be even larger, when taking into account studies that did not focus on CVD but generated a decent number of assays on relevant biospecimens (e.g. heart, blood or blood vessels) such as ENCODE [177] and the Roadmap Epigenomics initiatives [178]. Likewise in dbGaP, where human genotype–phenotype data are deposited, CVD-related research has contributed data on 658 305 subjects, only 16 786 (2.9%) of whom had consented for the data to be employed for general research, leaving a large amount of data locked in field-specific or disease-specific studies (Table 1) (see Supplementary Data for a comprehensive list of CVD studies deposited in dbGaP). An extensive compilation of human genotype–phenotype databases is given in a recent review [179]. In addition to the central repositories for established and popular experimental methods, smaller databases with narrower focus are also budding. For instance, chromatin structure data from 3C, 4C, 5C and Hi-C experiments have been collected in dedicated databases such as 3CDB [180] and 4DGenome [181]. Also, noncoding RNAs are being added into databases such as lncRNAdb [182], NONCODE [183] and LNCipedia [113].
Figure 5.
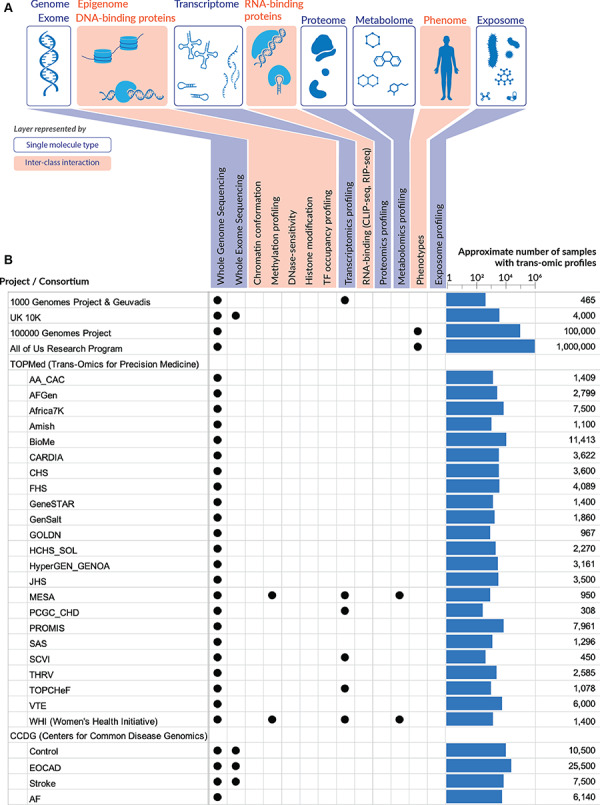
Multi-omics data. (A) The multiple layers of omics data that are now accessible to researchers. Genome/exome, transcriptome, proteome, metabolome, as well as the microbiome and chemical compounds in the exposome can be profiled by assays on a single class of molecules (DNA, RNA, proteins or small molecules), while the other layers depend on the ability to capture DNA–protein or RNA–protein interactions. The phenome is less well defined as phenotypic measures vary greatly from physical measurements to laboratory tests, from descriptive to quantitative traits. Sources of comprehensive phenotypic data comparable to the other omics can be obtained, for example, from EHRs. Beyond the genome, omics datasets become highly complicated, due to the variation across tissues and cell types. (B) Large omics datasets that are (or will be) available for CVD research. For each dataset, the number of samples being assayed across multiple omics are indicated on the right. This number is often smaller than the total number of samples/participants in a given project, because not every sample is run on multiple assays. Sources are provided in Supplementary Data.
Such abundance and diversity of data promises valuable insights once the data are aggregated across studies within a given omics domain (e.g. RNA-seq), or across multiple omics domains (e.g. RNA-seq, ChIP-seq, ATAC-seq, etc.). Efforts to aggregate genomic data (both individual and summary-level statistics) have resulted in valuable collections such as The Cancer Genome Atlas [24] for genomics and functional genomics data in cancer, or ExAC and gnomAD for exome and genome sequencing data [63]. For instance, aggregated exomes/genomes such as ExAC/gnomAD have been a valuable resource for estimating the allele frequencies of the general population as well as within various race/ethnic groups. Although the need for data integration has been identified within the field of CVD research, resulting in some CVD-focused databases dating back to 2015 (Table 2), many of these resources are either discontinued, not well maintained or remain in a primitive stage where database queries are delivered in plain texts and hyperlinks that require substantial efforts to synthesize new integrative insights. With the upcoming wave of trans-omics data spanning diverse populations and sequencing types (Figure 5B), data harmonization will become a more pressing problem. Generic solutions have started to be proposed, e.g. Biochat for GEO metadata [184] or OmicsDI for diverse datasets spanning genomics, transcriptomics, proteomics and metabolomics [185], and are promising for facilitating data integration in specialty fields like CVD.
Table 2.
List of available resources for cardioinformatics research
| Type | Name | URL | Ref |
|---|---|---|---|
| Knowledge portal | Cardiovascular Disease Knowledge Portal | http://www.broadcvdi.org/ | [10] |
| Knowledge portal | Cerebrovascular Disease Knowledge Portal | http://www.cerebrovascularportal.org/ | [2] |
| Knowledge portal | HeartBioPortal | https://heartbioportal.com/ | [5] |
| Analytics platform | AHA Precision Medicine Platform | https://precision.heart.org/ | [4] |
| Analytics platform | DataSTAGE | https://datastage.io/ | In planning |
| Database | HGDB (Heart Gene Database) | http://www.hgdb.ir/ | [7] |
| Database | In-Cardiome (Integrated Cardiome Database) | http://www.tri-incardiome.org/ | [9] |
| Database | Cardio/Vascular Disease Database | http://www.padb.org/cvd/ | [3] |
| Database | CardioGenBase | Discontinued | [11] |
| Review paper | Cloud computing for genomic data analysis and collaboration | [6] | |
| Review paper | Human genotype–phenotype databases: aims, challenges and opportunities | [1] | |
| Review paper | Methods of integrating data to uncover genotype–phenotype interactions | [8] | |
Resource types are categorized as follows: Database: integrated datasets, harmonized and built into a single, searchable database. Query results are usually presented as table of text and hyperlinks. Knowledge portal: integrated datasets, harmonized and built into a single, searchable database. Query results are usually visualized with charts tailored for the biological data and insights. Analytics platform: computing system, usually comes with access to diverse datasets, allowing users to perform various analyses on the hosted data.
While the issues above are generic for all types of research aiming to reuse public data, we believe CVD research benefits even more by expanding beyond traditional methods. Most use of high-throughput data in CVD research has been largely limited to the very first layer of omics data (Figure 5A), i.e. genome/exome. Whole-exome and whole-genome sequencing data have been slowly incorporated into conventional GWAS, bringing more ascertainment to earlier findings [68–71]. When deeper phenotype data such as blood lipid tests and diagnosis (ICD) codes became available, phenome-wide association studies emerged [186], triggering a new line of biomedical research that coupled EHRs with omics data, enabling powerful analyses, as discussed by [187, 188] and exemplified by [189, 190].
Besides existing data, new recent research programs have started to put more focus on high-throughput assays that result in a comprehensive cross-section of biological molecules (DNA, RNA and protein) and their interactions. For instance, the Multi-Ethnic Study of Atherosclerosis (MESA) medical research study [191] within TOPMed includes WGS, RNA-seq, metabolomics, proteomics and methylomics data across a variety of multi-ethnic communities (white, Hispanic, African-American and Asian). Specifically, MESA investigates the characteristics of subclinical CVD (disease detected non-invasively before it has produced clinical signs and symptoms) and the risk factors that predict progression to clinically overt CVD or progression of the subclinical disease. Some traditional CVD risk factors include hyperlipidemia, hypertension, diabetes mellitus, metabolic syndrome and chronic kidney disease [2]. Likewise, TOPMed is generating a second modest size multi-omic resource involving RNA-seq, metabolomics and methylomics in a subset of participants of the Women’s Health Initiative (WHI) study [192] who have undergone WGS already. Figure 5 highlights the large datasets that are (or will be) available for CV research. It is clear that assays for DNA sequences, including WGS and whole-exome sequencing, are still dominant among these studies. However, a modest number of multi-omics experiments are planned to be assayed for transcriptome, methylome and metabolome, as in the MESA and WHI studies. The availability of these datasets, especially at the individual-level, is critical to correlate the variations across multiple omics and bridge the gap from genotype to phenotype. In general, the analysis of these trans-omics datasets is a fascinating problem—although the computational approaches envisioned from the early days of gene expression profiling, i.e. differential gene expression analysis, co-expression analysis and gene clustering with subsequent identification of enriched biological pathways [193] can still bring fruitful analysis [194], cardioinformatics is clearly steering towards the integration of multiple omics layers (Figure 5). Although the potential of data integration had been recognized as early as a decade ago [195], leading to the development of many data integration strategies [196] such as gene expression and summary-level associations of SNPs and phenotypes from GWAS studies, only recently have these successful data integration strategies begun to emerge in CVD research [197]. Such approaches, usually referred to as transcriptome-wide association studies, are now adopted more widely [198]. In contrast to the relatively recent surge of interest in data integration methodologies, systems biology approaches in CVD have existed since at least the mid-2000s [199–201] and continue to pose challenging questions and present interesting results. For instance, modern systems-level approaches that leverage network analysis methods suggest that covariation between molecules (modeled as the reorganization of network nodes and edges) can be more instructive than the differential expression of individual markers, e.g. for conceptualizing molecular changes that occur in the emergence of high glucose levels in the prediabetic state [51, 202]. Since diabetes is a major risk factor for atherosclerosis, re-casting such physiological phenomena in a new light as tipping points and bifurcations of a network with multiple alternative stable states addresses a blind spot of the disease-oriented paradigm of clinical research and practice, which by definition precludes detailed knowledge about early presentations in subclinical populations [51]. One well-known success story of early detection of a subclinical prediabetic state was the result of a longitudinal multi-omics study that monitored the transcriptome, proteome and metabolome of a single individual over 14 months, ultimately helping the individual avoid the clinical indication (diabetes) by early adjustment in diet [203]. Other successful examples of multi-omics studies conducted on longitudinal personal omics profiles in single individuals to provide constant monitoring and preventative intervention include the MyConnectome study [204], P100 Wellness study [205] and the Personalized Nutrition study [206], which were covered in a recent editorial [51] in the context of CVD-related traits such as blood pressure, QT interval, postprandial glycemic response, etc. In general, computationally integrating diverse stores of data such as physiological and environmental information with other omics layers such as genomes, metabolomes and microbiomes can help identify subclinical imbalances or elevated disease risk in otherwise healthy individuals [51], heralding in an era of preventative healthcare/medicine. Historically, although the first draft of the human genome project had brought a lot of hope and excitement about potential advancements in the diagnosis and treatment of cardiac diseases—such as the ability to identify disease genes within the associated loci, to improve risk estimation based on more precise genotypes, or to personalize the prediction of drug effects on a patient [207]—it seems that a collection of the first population-scale ‘drafts’ of the whole ‘multi-ome’ will ultimately be required for gaining a deeper understanding of the phenotypic manifestations of CVD across different race/ethnic groups. A recent cardiac hypertrophy study in mice [208] highlighted the utility of conducting multi-omics investigations for discovering additional disease gene candidates not apparent from studying each omics data type separately in the context of CVD pathogenesis. In humans, large-scale studies such as the MyHeartCounts study [140] and a global physical activity study of nearly 800 000 individuals across 111 countries [209] demonstrated the feasibility of consenting and engaging large populations using smartphone technology, suggesting the potential to create a similar population-wide intercontinental network for multi-omics participation in the near future. For a recent comprehensive review detailing the success stories of multi-omics studies in CVD, see Leon-Mimila et al. [67]. All in all, the ability to combine data from every omics layer depicted in Figure 5, either at the summary level or the individual level, opens up ample opportunities for cardioinformatics to expand and augment the understanding of CVD etiology.
Augmented intelligence to advance cardiology
As alluded to in the Introduction, AI and ML will play an increasingly important role in cardioinformatics research. Recent trends in this direction include studies for cardiac arrhythmia detection [210, 211], heart failure prediction and classification [141, 212, 213], CV risk stratification [214], individualized treatment effect estimates from clinical trial data [215], among various other active CVD-related research areas [206, 216–218] that utilize ML techniques ranging from deep learning [210, 213, 219–224], class imbalance learning [225, 226], active learning [227], probabilistic graphical models [228, 229] and other areas. An emerging area of AI/ML applications ripe for early adoption is the field of augmented and virtual reality (AR/VR), specifically its applications to cardiology. For instance, surgeons at Children’s Mercy Kansas City, a hospital in Missouri, have been exploring the use of augmented reality to view CT scans of patients’ hearts before an operation to understand patient-specific blood vessel anatomy in different chambers of the heart (e.g. right atrium or left ventricle), ultimately facilitating a safer, more informed surgical intervention/procedure [230]. Other emerging applications of AR/VR in CV medicine include advances for education, pre-procedural planning, intraprocedural visualization and patient rehabilitation [231].
Nevertheless, in an era of big data analytics to improve CV care [232] and an accelerated adoption of ML methodologies to facilitate these objectives, the role of human experts may turn out to be even more indispensable. Among tasks that still require considerable human judgment include understanding and processing of free text data as well as recognizing visual patterns, especially corner/edge cases (e.g. in CVD medical imaging [233, 234]) that may be missed by algorithms trained on conventional datasets. As with other areas, CV research requires expert-level domain knowledge to make the best use of ML applications, for instance in properly labeling CVD data or harmonizing it across epidemiological cohorts. Without a doubt, domain expertise in cardiology is by no means a tractable problem at scale, exemplified by the insurmountable pile of CV publications accumulating over the years (Figure 1C) and the intrinsic difficulty in keeping up with this momentum. With active research in text-mining and natural language processing (NLP), the goal is to liberate human researchers from the time-consuming tasks associated with reading new CVD literature and making sense of free texts in metadata and publications at scale, including tables, figures and charts. Recently, a novel text-mining NLP-based approach was used to analyze over 1 million literature abstracts to uncover novel extracellular matrix functions, pathways and molecular relationships implicated across six CVDs [235]. Since different subdomains in biomedical literature vary along many linguistic dimensions, making text-mining systems that perform well on one subdomain is not guaranteed to perform well on another [184, 236, 237]; we believe that development of cardioNLP algorithms and dedicated large-scale comprehensively labeled CVD training datasets will be essential for progress in tasks such as harmonized patient-data meta analyses in CV precision medicine. To this end, we envision a framework like the Kipoi model zoo for genomics [238] but with a focus on CVD knowledge (both text and data) that can be used to train ML models in cardioinformatics research.
Recognition of visual patterns, on the other hand, remains a powerful human faculty that needs to be fully exploited rather than entirely replaced with automation. With the rise of various visualization techniques across diverse biological data types [239, 240], it will be an exciting challenge for cardioinformatics researchers to leverage them for an integrative representation of heterogeneous data layers towards extracting deeper CVD insights. In addition, the visualization of many experimental assays and biological processes remains a significant challenge, e.g. visualizing alternative splicing events [241, 242], fast implementations for biological heatmaps [243] or interactive matrices for chromatin conformation data from Hi-C experiments [244, 245]. Moving into the clinical setting, gearing up towards large-scale precision medicine will entail the requirement of providing more comprehensive and integrated data, in a more comprehensible manner to assist clinicians. To this end, visualization technology and software design will be critical in improving CVD biomedical software, e.g. for designing robust clinical decision support systems or validating prediction models for critical care outcomes. For instance, by integrating multiple measures of clinical trajectories together with NLP of clinical free text notes from EHR data, more accurate prediction of critical care outcomes were observed among patients in intensive care units across three major hospital systems [246]. Such studies suggest that automated algorithms, particularly those using unstructured data from notes and other sources, can augment clinical research and quality improvement initiatives.
Closing remarks
As we further our quest to understand the genetics and molecular biology of heart disease, many complex clinical CVD indications and pathophenotypes have become too nuanced for traditional computational approaches. Reflecting on the current body of knowledge, we recognize that many aspects of this complexity can be addressed with more (and improved) computational methods, as has been the case for bioinformatics tools and their impact on cancer genomics research. But bioinformatics techniques for conducting CVD studies will require progressively more sophisticated strategies for identifying and monitoring elevated disease risk and intermediary molecular endophenotypes (including CVD-related risk factors and quantitative traits) as CVD poses an unprecedented and unique set of computational challenges with respect to clinical phenomapping and the large-scale integration of multiple diverse sources of population-level biomedical data for understanding the progression of a subclinical imbalance into the clinical manifestation of the disease itself. In this review, we discussed some of the important works evolving at the multidisciplinary interface of bioinformatics and cardiology and advocate for shining a brighter spotlight on cardioinformatics as an emerging field, in its own right. We suggest some future insights based on our understanding of historical perspectives and ongoing work in current CVD research and welcome feedback and ideas from the broader scientific community.
Methods
PubMed queries
Queries are all based on MeSH terms. For reproducibility purposes, the exact queries for each category in Figure 1 are listed below:
Cardiovascular disease: ”cardiovascular diseases”[MeSH Terms]
Cancer: (*cancer[MeSH Terms])
Bioinformatics: bioinformatics[MeSH Terms] OR genomics[MeSH Terms]
Cardioinformatics: (bioinformatics[MeSH Terms] OR genomics[MeSH Terms]) AND (”cardiovascular diseases”[MeSH Terms])
Cancer informatics: (bioinformatics[MeSH Terms] OR genomics[MeSH Terms]) AND (*cancer[MeSH Terms])
All queries were appended with a filtering term to reduce the count of non-primary research items. The filtering term is
AND (hasabstract[text] AND English[lang]))) NOT ((‘autobiography’[Publication Type] OR ‘biography’[Publication Type] OR ‘corrected and republished article’[Publication Type] OR ‘duplicate publication’[Publication Type] OR ‘electronic supplementary materials’[Publication Type] OR ‘interactive tutorial’[Publication Type] OR ‘interview’[Publication Type] OR ‘lectures’[Publication Type] OR ‘legal cases’[Publication Type] OR ‘legislation’[Publication Type] OR ‘meta analysis’[Publication Type] OR ‘news’[Publication Type] OR ‘newspaper article’[Publication Type] OR ‘patient education handout’[Publication Type] OR ‘published erratum’[Publication Type] OR ‘retracted publication’[Publication Type] OR ‘retraction of publication’[Publication Type] OR ‘review’[Publication Type] OR ‘scientific integrity review’[Publication Type] OR ‘support of research’[Publication Type] OR ‘video audio media’[Publication Type] OR ‘webcasts’[Publication Type])).
The MeSH Database entry for ‘cardiovascular disease’ includes many types of CV abnormalities that may occur in organs outside the immediate circulatory system.
Research funding statistics
Data on research funding were provided by the NIH, via the NIH Research Portfolio Online Reporting Tool [247], as a table of awards for each fiscal year and research category. Categorization was done by NIH starting in 2008, through the Research, Condition, and Disease Categorization system. To calculate the research funding for cancer and CVDs, we retrieved tables of awards for all related categories, removed duplicate entries and summed up all the amounts of awards greater than $100.
For ‘Cancer’ funding, relevant categories are Brain Cancer, Breast Cancer, Cancer, Cancer Genomics, Cervical Cancer, Colo-Rectal Cancer, HPV and/or Cervical Cancer Vaccines, Liver Cancer, Lung Cancer, Lymphoma, Neuroblastoma, Ovarian Cancer, Pancreatic Cancer, Pediatric Cancer, Prostate Cancer, Uterine Cancer, Vaginal Cancer.
For ‘Cardiovascular disease’ funding, relevant categories are Aging, Cardiovascular, Cerebrovascular, Congenital Heart Disease, Heart Disease, Heart Disease - Coronary Heart Disease, Hypertension, Pediatric Cardiomyopathy, Stroke.
Reproducibility
All data and source code powering the data-driven visualizations and quantitative analyses presented in this review are provided at http://doi.org/10.5281/zenodo.2622064
Acknowledgments
B.B.K. acknowledges and thanks the AHA for financial support through the AHA Postdoctoral Fellowship program.
Bohdan Khomtchouk is currently an Instructor at University of Chicago, previously an American Heart Association Postdoctoral Fellow at Stanford University. His principal research focus has been on creating artificial intelligence and machine learning software to organize and better understand the world's cardiovascular disease knowledge (text and data) at a massive scale—working at the multidisciplinary interface of healthcare, algorithm design, software engineering, integrative bioinformatics, multi-omics, natural language processing and statistical learning.
Diem-Trang Tran is a doctoral student in Data Management and Analysis at the School of Computing, University of Utah, where she works on a variety of bioinformatics analyses, ranging from processing, visualizing to quantitative analyses of high-throughput sequencing data.
Kasra Vand is a Research Scientist at Quiltomics and one of the lead developers of the HeartBioPortal project. He holds a Bachelor's degree in Software Engineering and his research interests span broadly across artificial intelligence, machine learning and natural language processing.
Matthew Might has been the Director of the Hugh Kaul Precision Medicine Institute at the University of Alabama at Birmingham (UAB) since 2017. At UAB, Dr. Might is the Hugh Kaul Endowed Chair of Personalized Medicine, a Professor of Internal Medicine and a Professor of Computer Science.
Or Gozani is the Morris Herzstein Professor of Biology at Stanford University. The main focus of his laboratory is understanding the molecular mechanisms by which chromatin signaling networks affect nuclear and epigenetic programs and how disruption in these mechanisms contribute to cancer and other pathologies including heart disease.
Themistocles (Tim) Assimes is an Associate Professor at Stanford University. He is a board–certified clinical cardiologist whose laboratory investigates the genomic determinants of coronary heart disease (CHD) and risk factors of CHD. Dr. Assimes is also interested in clinical prediction modeling using genetic and non-genetic biomarkers.
Funding
This work has been supported by the AHA Postdoctoral Fellowship grant #18POST34030375 (B.B.K.) and partially by the National Science Foundation (CAREER grant 1350344 to M.M.). The US Government is authorized to reproduce and distribute reprints for Governmental purposes notwithstanding any copyright notation thereon.
References
- 1. Roth GA, Abate D, Abate KH, et al. Global, regional, and national age–sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018;392(10159):1736–88. doi: 10.1016/S0140-6736(18)32203-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Leopold JA, Loscalzo J. Emerging role of precision medicine in cardiovascular disease. Circ Res 2018;122(9):1302–15. doi: 10.1161/CIRCRESAHA.117.310782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Heidenreich PA, Trogdon JG, Khavjou OA, et al. Forecasting the future of cardiovascular disease in the United States. Circulation 2011;123(8):933–44. doi: 10.1161/CIR.0b013e31820a55f5. [DOI] [PubMed] [Google Scholar]
- 4. Joseph P, Leong D, McKee M, et al. Reducing the global burden of cardiovascular disease, part 1: the epidemiology and risk factors. Circ Res 2017;121(6):677–94. [DOI] [PubMed] [Google Scholar]
- 5. Rogers MA, Aikawa E. Cardiovascular calcification: artificial intelligence and big data accelerate mechanistic discovery. Nat Rev Cardiol 2019;16(5):261–74. [DOI] [PubMed] [Google Scholar]
- 6. Gómez-López G, Dopazo J, Cigudosa JC, et al. Precision medicine needs pioneering clinical bioinformaticians. Brief Bioinform 2017;20: 752–66. [DOI] [PubMed] [Google Scholar]
- 7. Houser SR. The American Heart Association’s new institute for precision cardiovascular medicine. Circulation 2016;134(24):1913–4. doi: 10.1161/CIRCULATIONAHA.116.022138. [DOI] [PubMed] [Google Scholar]
- 8. czbiohub . Chan Zuckerberg Biohub Awards $13.7 Million to Fund New Intercampus Collaborative Research Programs to Advance Human health. Chan Zuckerberg Biohub. https://www.czbiohub.org/intercampus-research-programs/, 2018.
- 9. Shameer K, Badgeley MA, Miotto R, et al. Translational bioinformatics in the era of real-time biomedical, health care and wellness data streams. Brief Bioinform 2017;18(1):105–24. doi: 10.1093/bib/bbv118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shameer K, Johnson KW, Glicksberg BS, et al. Machine learning in cardiovascular medicine: are we there yet? Heart 2018;104(14):1156–64. doi: 10.1136/heartjnl-2017-311198. [DOI] [PubMed] [Google Scholar]
- 11. MLPerf . MLPerf. https://mlperf.org/, 2018.
- 12. Becht E, McInnes L, Healy J, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol 2019;37:38–44. [DOI] [PubMed] [Google Scholar]
- 13. Wirka RC, Pjanic M, Quertermous T. Advances in transcriptomics: investigating cardiovascular disease at unprecedented resolution. Circ Res 2018;122(9):1200–20. doi: 10.1161/CIRCRESAHA.117.310910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 2018;34(18):3094–100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Olson RS, La Cava W, Orzechowski P, et al. PMLB: a large benchmark suite for machine learning evaluation and comparison. BioData Min 2017;10(1):36. doi: 10.1186/s13040-017-0154-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weber LM, Saelens W, Cannoodt R, et al. Essential guidelines for computational method benchmarking. Genome Biol 2019;20:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Khomtchouk B, Vand KA, Koehler WC, et al. HeartBioPortal: an internet-of-omics for human cardiovascular disease data. Circ Genom Precis Med 2019;12(4):e002426. doi: 10.1161/CIRCGEN.118.002426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kass-Hout TA, Stevens LM, Hall JL. American Heart Association precision medicine platform. Circulation 2018;137(7):647–9. doi: 10.1161/CIRCULATIONAHA.117.032041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crawford KM, Gallego-Fabrega C, Kourkoulis C, et al. Cerebrovascular Disease Knowledge Portal: an open-access data resource to accelerate genomic discoveries in stroke. Stroke 2018;49(2):470–5. doi: 10.1161/STROKEAHA.117.018922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fernandes M, Patel A, Husi H. C/VDdb: a multi-omics expression profiling database for a knowledge-driven approach in cardiovascular disease (CVD). PLoS One 2018;13(11):e0207371. doi: 10.1371/journal.pone.0207371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. National Heart, Lung, and Blood Institute . Trans-Omics for Precision Medicine (TOPMed) Program. https://www.nhlbi.nih.gov/science/trans-omics-precision-medicine-topmed-program, 2014. [DOI] [PMC free article] [PubMed]
- 22. Cerami E, Gao J, Dogrusoz U, et al. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6(269):l1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. The Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, et al. The Cancer Genome Atlas pan-cancer analysis project. Nat Genet 2013;45:1113–20. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koscielny G, An P, Carvalho-Silva D, et al. Open Targets: a platform for therapeutic target identification and validation. Nucleic Acids Res 2017;45(D1):D985–94. doi: 10.1093/nar/gkw1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grossman RL, Heath AP, Ferretti V, et al. Toward a shared vision for cancer genomic data. N Engl J Med 2016;375(12):1109–12. doi: 10.1056/NEJMp1607591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jensen MA, Ferretti V, Grossman RL, et al. The NCI Genomic Data Commons as an engine for precision medicine. Blood 2017;130(4):453–9. doi: 10.1182/blood-2017-03-735654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. The Broad Institute of MIT & Harvard . Single Cell Portal. https://portals.broadinstitute.org/single∖_cell, 2018a.
- 29. Type 2 Diabetes Knowledge Portal . Type 2 Diabetes Knowledge Portal. http://www.type2diabetesgenetics.org/, 2018.
- 30. ALS Knowledge Portal . Als Knowledge Portal. http://alskp.org/, 2019.
- 31. Sleep Disorder Knowledge Portal . Sleep Disorder Knowledge Portal, http://sleepdisordergenetics.org/, 2018.
- 32. The Broad Institute of MIT & Harvard . Cardiovascular Disease Knowledge Portal. http://www.broadcvdi.org/, 2018b.
- 33. The Broad Institute of MIT & Harvard . Cerebrovascular Disease Knowledge Portal. http://www.cerebrovascularportal.org/, 2018c.
- 34. Hodes RJ, Buckholtz N. Accelerating Medicines Partnership: Alzheimer’s Disease (AMP-AD) Knowledge Portal Aids Alzheimer’s Drug Discovery through Open Data Sharing. Expert Opinion on Therapeutic Targets, 2016;20: 389–91. [DOI] [PubMed] [Google Scholar]
- 35. Sage Bionetworks . Agora. http://sagebionetworks.org/research-projects/agora/, 2018.
- 36. Zong NC, Li H, Li H, et al. Integration of cardiac proteome biology and medicine by a specialized knowledgebase. Circ Res 2013;113(9):1043–53. doi: 10.1161/CIRCRESAHA.113.301151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. UCLA . Heartbd2k—A Community Effort to Translate Protein Data to Knowledge: An Integrated Platform. https://www.heartbd2k.org, 2019.
- 38. Lau E, Cao Q, Ng DCM, et al. A large dataset of protein dynamics in the mammalian heart proteome. Sci Data 2016;3:160015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Caulfield M, Davies J, Dennys M, et al. The National Genomics Research and Healthcare Knowledgebase, 2017.
- 40. Kalia P, Charles S. China’s 100K Wellness Pioneer Project uses UniteGen and SapientiaTM integrated platform. https://www.cambridgenetwork.co.uk/news/chinas-100k-wellness-pioneer-project-uses-sapientia/, 2017.
- 41. The All of Us Research Program Investigators. N Engl J Med 2019;381(7):668–76. doi: 10.1056/NEJMsr1809937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gaziano JM, Concato J, Brophy M, et al. Million Veteran Program: a mega-biobank to study genetic influences on health and disease. J Clin Epidemiol 2016;70:214–23. doi: 10.1016/j.jclinepi.2015.09.016. [DOI] [PubMed] [Google Scholar]
- 43. National Institutes of Health . All of Us Data Browser. https://databrowser.researchallofus.org, 2019.
- 44. Visscher PM, Wray NR, Zhang Q, et al. 10 Years of GWAS discovery: biology, function, and translation. Am J Hum Genet 2017;101(1):5–22. doi: 10.1016/j.ajhg.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ganna A, Magnusson PK, Pedersen NL, et al. Multilocus genetic risk scores for coronary heart disease prediction. Arterioscler Thromb Vasc Biol 2013;33(9):2267–72. doi: 10.1161/ATVBAHA.113.301218. [DOI] [PubMed] [Google Scholar]
- 46. Goldstein BA, Knowles JW, Salfati E, et al. Simple, standardized incorporation of genetic risk into non-genetic risk prediction tools for complex traits: coronary heart disease as an example. Front Genet 2014;5:254. doi: 10.3389/fgene.2014.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krarup NT, Borglykke A, Allin KH, et al. A genetic risk score of 45 coronary artery disease risk variants associates with increased risk of myocardial infarction in 6041 Danish individuals. Atherosclerosis 2015;240(2):305–10. doi: 10.1016/j.atherosclerosis.2015.03.022. [DOI] [PubMed] [Google Scholar]
- 48. Tada H, Melander O, Louie JZ, et al. Risk prediction by genetic risk scores for coronary heart disease is independent of self-reported family history. Eur Heart J 2016;37(6):561–7. doi: 10.1093/eurheartj/ehv462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Abraham G, Havulinna AS, Bhalala OG, et al. Genomic prediction of coronary heart disease. Eur Heart J 2016;37(43):3267–78. doi: 10.1093/eurheartj/ehw450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Assimes TL, Goldstein BA. Genetic cardiovascular risk prediction: are we already there? Eur Heart J 2016;37(43):3279–81. doi: 10.1093/eurheartj/ehw498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lau E, Wu JC. Omics, big data, and precision medicine in cardiovascular sciences. Circ Res 2018;122(9):1165–8. doi: 10.1161/CIRCRESAHA.118.313161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Johnson KW, Shameer K, Glicksberg BS, et al. Enabling precision cardiology through multiscale biology and systems medicine. JACC Basic Transl Sci 2017;2(3):311–27. doi: 10.1016/j.jacbts.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moran AE, Odden MC, Thanataveerat A, et al. Cost-effectiveness of hypertension therapy according to 2014 guidelines. N Engl J Med 2015;372(5):447–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Burke MA, Cook SA, Seidman JG, et al. Clinical and mechanistic insights into the genetics of cardiomyopathy. J Am Coll Cardiol 2016;68(25):2871–86. doi: 10.1016/j.jacc.2016.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. MacArthur J, Bowler E, Cerezo M, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res 2017;45(D1):D896–901. doi: 10.1093/nar/gkw1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Köhler S, Vasilevsky NA, Engelstad M, et al. The human phenotype ontology in 2017. Nucleic Acids Res 2017;45(Database issue):D865–76. doi: 10.1093/nar/gkw1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McNally EM, Mestroni L. Dilated cardiomyopathy. Circ Res 2017;121(7):731–48. doi: 10.1161/CIRCRESAHA.116.309396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Clarke SL, Assimes TL. Genome-wide association studies of coronary artery disease: recent progress and challenges ahead. Curr Atheroscler Rep 2018;20(9):47. doi: 10.1007/s11883-018-0748-4. [DOI] [PubMed] [Google Scholar]
- 59. Cranley J, MacRae CA. A new approach to an old problem: one brave idea. Circ Res 2018;122(9):1172–5. doi: 10.1161/CIRCRESAHA.118.310941. [DOI] [PubMed] [Google Scholar]
- 60. Helgadottir A, Thorleifsson G, Manolescu A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 2007;316(5830):1491–3. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 61. McPherson R, Pertsemlidis A, Kavaslar N, et al. A common allele on chromosome 9 associated with coronary heart disease. Science 2007;316(5830):1488–91. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Samani NJ, Erdmann J, Hall AS, et al. Genomewide association analysis of coronary artery disease. N Engl J Med 2007;357(5):443–53 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536(7616):285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Walsh R, Thomson KL, Ware JS, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med 2017;19(2):192–203. doi: 10.1038/gim.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. MacRae CA, Seidman CE. Closing the genotype–phenotype loop for precision medicine. Circulation 2017;136(16):1492–4. doi: 10.1161/CIRCULATIONAHA.117.030831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Manrai AK, Ioannidis JPA, Kohane IS. Clinical genomics: from pathogenicity claims to quantitative risk estimates. JAMA 2016;315(12):1233. doi: 10.1001/jama.2016.1519. [DOI] [PubMed] [Google Scholar]
- 67. Leon-Mimila P, Wang J, Huertas-Vazquez A. Relevance of multi-omics studies in cardiovascular diseases. Front Cardiovasc Med 2019;91(07):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cohen JC, Boerwinkle E, Mosley TH, et al. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006;354(12):1264–72. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 69. Dewey FE, Gusarova V, O’Dushlaine C, et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. N Engl J Med 2016a;374(12):1123–33. doi: 10.1056/NEJMoa1510926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators . Coding variation in ANGPTL4, LPL, and SVEP1 and the risk of coronary disease. N Engl J Med 2016;374(12):1134–44. doi: 10.1056/NEJMoa1507652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. The TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute, Crosby J, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med 2014;371(1):22–31. doi: 10.1056/NEJMoa1307095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Findlay GM, Daza RM, Martin B, et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018;1. doi: 10.1038/s41586-018-0461-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. North BJ, Sinclair DA. The intersection between aging and cardiovascular disease. Circ Res 2012;110(8):1097–108. doi: 10.1161/CIRCRESAHA.111.246876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Steenman M, Lande G. Cardiac aging and heart disease in humans. Biophys Rev 2017;9(2):131–7. doi: 10.1007/s12551-017-0255-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Boyle EA, Li YI, Pritchard JK. An expanded view of complex traits: from polygenic to omnigenic. Cell 2017;169(7):1177–86. doi: 10.1016/j.cell.2017.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Weiss JN, Karma A, MacLellan WR, et al. “Good enough solutions” and the genetics of complex diseases. Circ Res 2012;111(4):493–504. doi: 10.1161/CIRCRESAHA.112.269084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature 2009;461(7265):747–53. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zuk O, Schaffner SF, Samocha K, et al. Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci 2014;111(4):E455–64. doi: 10.1073/pnas.1322563111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kaakinen M, Mägi R, Fischer K, et al. A rare-variant test for high-dimensional data. Eur J Hum Genet 2017;25(8):988–94. doi: 10.1038/ejhg.2017.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. NCBI . dbSNP. https://www.ncbi.nlm.nih.gov/snp, 2018a.
- 81. EMBL-EBI . Database of genomic variants archive. https://www.ebi.ac.uk/dgva, 2018.
- 82. NCBI . dbVar. https://www.ncbi.nlm.nih.gov/dbvar/, 2018b.
- 83. Sudmant PH, Rausch T, Gardner EJ, et al. An integrated map of structural variation in 2,504 human genomes. Nature 2015;526(7571):75–81. doi: 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Haas J, Mester S, Lai A, et al. Genomic structural variations lead to dysregulation of important coding and non-coding RNA species in dilated cardiomyopathy. EMBO Mol Med 2018;10(1):107–20. doi: 10.15252/emmm.201707838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Eichler EE, Flint J, Gibson G, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet 2010;11(6):446–50. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Collins RL, Brand H, Karczewski KJ, et al. An open resource of structural variation for medical and population genetics. bioRxiv 2019;578674. doi: 10.1101/578674. [Google Scholar]
- 87. Antonarakis SE, Krawczak M, Cooper DN. The nature and mechanisms of human gene mutation. In: Scriver CR, Beaudet AL, Sly WS et al. (eds). The Metabolic and Molecular Bases of Inherited Disease, 8th edn. New York: McGraw-Hill, 2001, 343–77. [Google Scholar]
- 88. Franzén O, Ermel R, Cohain A, et al. Cardiometabolic risk loci share downstream cis- and trans-gene regulation across tissues and diseases. Science 2016;353(6301):827–30. doi: 10.1126/science.aad6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Pickrell JK. Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am J Hum Genet 2014;94(4):559–73. doi: 10.1016/j.ajhg.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. 1000 Genomes Project Consortium, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature 2015;526(7571):68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. The UK10K Consortium . The UK10K project identifies rare variants in health and disease. Nature 2015;526(7571):82–90. doi: 10.1038/nature14962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. National Human Genome Research Institute . Centers for Common Disease Genomics (CCDG). https://www.genome.gov/27563570/centers-for-common-disease-genomics-ccdg/, 2016.
- 93. Udali S, Guarini P, Moruzzi S, et al. Cardiovascular epigenetics: from DNA methylation to microRNAs. Mol Aspects Med 2013;34(4):883–901. doi: 10.1016/j.mam.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 94. Abi Khalil C. The emerging role of epigenetics in cardiovascular disease. Ther Adv Chronic Dis 2014;5(4):178–87. doi: 10.1177/2040622314529325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Muka T, Koromani F, Portilla E, et al. The role of epigenetic modifications in cardiovascular disease: a systematic review. Int J Cardiol 2016;212:174–83. doi: 10.1016/j.ijcard.2016.03.062. [DOI] [PubMed] [Google Scholar]
- 96. Gidlöf O, Johnstone AL, Bader K, et al. Ischemic preconditioning confers epigenetic repression of Mtor and induction of autophagy through G9a-dependent H3K9 dimethylation. J Am Heart Assoc Cardiovasc Cerebrovasc Dis 2016;5(12):1–12. doi: 10.1161/JAHA.116.004076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Haitjema S, Meddens CA, van der Laan SW, et al. Additional candidate genes for human atherosclerotic disease identified through annotation based on chromatin organization. Circ Cardiovasc Genet 2017;10(2):e001664. doi: 10.1161/CIRCGENETICS.116.001664. [DOI] [PubMed] [Google Scholar]
- 98. Movassagh M, Choy M-K, Knowles DA, et al. Distinct epigenomic features in end-stage failing human hearts. Circulation 2011;124(22):2411–22. doi: 10.1161/CIRCULATIONAHA.111.040071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 2009;10(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Trivedi CM, Luo Y, Yin Z, et al. Hdac2 regulates the cardiac hypertrophic response by modulating gsk3activity. Nat Med 2007;13:324. [DOI] [PubMed] [Google Scholar]
- 101. Chang S, McKinsey TA, Zhang CL, et al. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol 2004;24(19):8467. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. McBurney MW, Yang X, Jardine K, et al. The mammalian sir2protein has a role in embryogenesis and gametogenesis. Mol Cell Biol 2003;23(1):38. doi: 10.1128/MCB.23.1.38-54.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Allis C, Caparros M, Jenuwein T, et al. Epigenetics, 2nd edn. Cold Spring Harbor Laboratory Press, 2015. [Google Scholar]
- 104. Irvin MR, Zhi D, Joehanes R, et al. Epigenome-wide association study of fasting blood lipids in the genetics of lipid-lowering drugs and diet network study. Circulation 2014;130(7):565–72. doi: 10.1161/CIRCULATIONAHA.114.009158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Dick KJ, Nelson CP, Tsaprouni L, et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet 2014;383(9933):1990–8. doi: 10.1016/S0140-6736(13)62674-4. [DOI] [PubMed] [Google Scholar]
- 106. Wahl S, Drong A, Lehne B, et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 2017;541(7635):81–6. doi: 10.1038/nature20784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Meder B, Haas J, Sedaghat-Hamedani F, et al. Epigenome-wide association study identifies cardiac gene patterning and a novel class of biomarkers for heart failure. Circulation 2017;136(16):1528–44. doi: 10.1161/CIRCULATIONAHA.117.027355. [DOI] [PubMed] [Google Scholar]
- 108. Rask-Andersen M, Martinsson D, Ahsan M, et al. Epigenome-wide association study reveals differential DNA methylation in individuals with a history of myocardial infarction. Hum Mol Genet 2016;25(21):4739–48. doi: 10.1093/hmg/ddw302. [DOI] [PubMed] [Google Scholar]
- 109. Rosa-Garrido M, Chapski DJ, Schmitt AD, et al. High-resolution mapping of chromatin conformation in cardiac myocytes reveals structural remodeling of the epigenome in heart failure. Circulation 2017;136(17):1613–25. doi: 10.1161/CIRCULATIONAHA.117.029430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Das A, Samidurai A, Salloum FN. Deciphering non-coding RNAs in cardiovascular health and disease. Front Cardiovasc Med 2018;5:73. doi: 10.3389/fcvm.2018.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Xu S, Kamato D, Little PJ, et al. Targeting epigenetics and non-coding RNAs in atherosclerosis: from mechanisms to therapeutics. Pharmacol Ther 2019;196:15–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sallam T, Sandhu J, Tontonoz P. Long noncoding RNA discovery in cardiovascular disease. Circ Res 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Volders P-J, Anckaert J, Verheggen K, et al. LNCipedia 5: towards a reference set of human long non-coding RNAs. Nucleic Acids Res 2018;47:D135–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Zhou S-s, Jin J-p, Wang J-q, et al. mirnas in cardiovascular diseases: potential biomarkers, therapeutic targets and challenges. Acta Pharmacol Sin 2018;39(7):1073–84. doi: 10.1038/aps.2018.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Elia L, Contu R, Quintavalle M, et al. Reciprocal regulation of microrna-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation 2009;120(23):2377–85. doi: 10.1161/CIRCULATIONAHA.109.879429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Marques FZ, Campain AE, Tomaszewski M, et al. Gene expression profiling reveals renin mrna overexpression in human hypertensive kidneys and a role for micrornas. Hypertension 2011;58(6):1093–8. doi: 10.1161/HYPERTENSIONAHA.111.180729. [DOI] [PubMed] [Google Scholar]
- 117. Gupta SK, Foinquinos A, Thum S, et al. Preclinical development of a microrna-based therapy for elderly patients with myocardial infarction. J Am Coll Cardiol 2016;68(14): 1557–71. doi: 10.1016/j.jacc.2016.07.739. [DOI] [PubMed] [Google Scholar]
- 118. Barwari T, Joshi A, Mayr M. Micrornas in cardiovascular disease. J Am Coll Cardiol 2016;68(23):2577–84. [DOI] [PubMed] [Google Scholar]
- 119. Azuaje F, Devaux Y, Wagner D. Computational biology for cardiovascular biomarker discovery. Brief Bioinform 2009;10(4):367–77. doi: 10.1093/bib/bbp008. [DOI] [PubMed] [Google Scholar]
- 120. Yu P, Wang H, Xie Y, et al. Deregulated cardiac specific micrornas in postnatal heart growth. Biomed Res Int 2016;6241763:2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Chistiakov DA, Orekhov AN, Bobryshev YV. Cardiac-specific mirna in cardiogenesis, heart function, and cardiac pathology (with focus on myocardial infarction). J Mol Cell Cardiol 2016;94:107–21. [DOI] [PubMed] [Google Scholar]
- 122. Lujambio A, Lowe SW. The microcosmos of cancer. Nature 2012;482(7385):347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Tyagi N, Arora S, Deshmukh SK, et al. Exploiting nanotechnology for the development of microrna-based cancer therapeutics. J Biomed Nanotechnol 2016;12(1):28–42. [DOI] [PubMed] [Google Scholar]
- 124. Drusco A, Croce CM. Micrornas and cancer: a long story for short rnas. Adv Cancer Res 2017;135:1–24. [DOI] [PubMed] [Google Scholar]
- 125. Catela Ivkovic T, Voss G, Cornella H, et al. micrornas as cancer therapeutics: a step closer to clinical application. Cancer Lett 2017;407:113–22. [DOI] [PubMed] [Google Scholar]
- 126. Bibikova M, Fan J-B. Genome-wide DNA methylation profiling. Wiley Interdiscip Rev Syst Biol Med 2010;2(2):210–23. doi: 10.1002/wsbm.35. [DOI] [PubMed] [Google Scholar]
- 127. Kimura H. Histone modifications for human epigenome analysis. J Hum Genet 2013;58(7):439–45. doi: 10.1038/jhg.2013.66. [DOI] [PubMed] [Google Scholar]
- 128. Sheridan S, Pignone M, Mulrow C. Framingham-based tools to calculate the global risk of coronary heart disease. J Gen Intern Med 2003;18(12):1039–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. ALA: Joint British recommendations on prevention of coronary heart disease in clinical practice . Joint British recommendations on prevention of coronary heart disease in clinical practice. British Cardiac Society, British Hyperlipidaemia Association, British Hypertension Society, endorsed by the British Diabetic Association. (1998). Heart (British Cardiac Society), 80 Suppl 2(Suppl 2):S1–29. [PMC free article] [PubMed] [Google Scholar]
- 130. Knuiman MW, Vu HT. Prediction of coronary heart disease mortality in Busselton, Western Australia: an evaluation of the Framingham, national health epidemiologic follow up study, and WHO ERICA risk scores. J Epidemiol Community Health 1997;51(5):515–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Eichler K, Puhan MA, Steurer J, et al. Prediction of first coronary events with the Framingham score: a systematic review. Am Heart J 2007;153(5):722–731.e8. doi: 10.1016/j.ahj.2007.02.027. [DOI] [PubMed] [Google Scholar]
- 132. Khera AV, Emdin CA, Drake I, et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med 2016;375(24):2349–58. doi: 10.1056/NEJMoa1605086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Talmud PJ, Hingorani AD, Cooper JA, et al. Utility of genetic and non-genetic risk factors in prediction of type 2 diabetes: Whitehall II prospective cohort study. BMJ 2010;340:b4838. doi: 10.1136/bmj.b4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. D’Agostino RB, Grundy S, Sullivan LM, et al. Validation of the Framingham coronary heart disease prediction scores: results of a multiple ethnic groups investigation. JAMA 2001;286(2):180–7. doi: 10.1001/jama.286.2.180. [DOI] [PubMed] [Google Scholar]
- 135. Empana JP, Ducimetière P, Arveiler D, et al. Are the Framingham and PROCAM coronary heart disease risk functions applicable to different European populations? The PRIME study. Eur Heart J 2003;24(21):1903–11. [DOI] [PubMed] [Google Scholar]
- 136. Zomer E, Liew D, Owen A, et al. Cardiovascular risk prediction in a population with the metabolic syndrome: Framingham vs. UKPDS algorithms. Eur J Prev Cardiol 2014;21(3):384–90. doi: 10.1177/2047487312449307. [DOI] [PubMed] [Google Scholar]
- 137. Wild CP. Complementing the genome with an “exposome”: the outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol Prev Biomark 2005;14(8):1847–50. doi: 10.1158/1055-9965.EPI-05-0456. [DOI] [PubMed] [Google Scholar]
- 138. Warth B, Spangler S, Fang M, et al. Exposome-scale investigations guided by global metabolomics, pathway analysis, and cognitive computing. Anal Chem 2017;89(21):11505–13. doi: 10.1021/acs.analchem.7b02759. [DOI] [PubMed] [Google Scholar]
- 139. Jiang C, Wang X, Li X, et al. Dynamic human environmental exposome revealed by longitudinal personal monitoring. Cell 2018;175(1):277–291.e31. doi: 10.1016/j.cell.2018.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. McConnell MV, Shcherbina A, Pavlovic A, et al. Feasibility of obtaining measures of lifestyle from a smartphone app: the MyHeart counts cardiovascular health study. JAMA Cardiol 2017;2(1):67–76. doi: 10.1001/jamacardio.2016.4395. [DOI] [PubMed] [Google Scholar]
- 141. Shah SJ, Katz DH, Selvaraj S, et al. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation 2015;131(3):269–79. doi: 10.1161/CIRCULATIONAHA.114.010637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Li X, Dunn J, Salins D, et al. Digital health: tracking physiomes and activity using wearable biosensors reveals useful health-related information. PLoS Biol 2017;15(1):e2001402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Kellogg RA, Dunn J, Snyder MP. Personal omics for precision health. Circ Res 2018;122(9):1169–71. doi: 10.1161/CIRCRESAHA.117.310909. [DOI] [PubMed] [Google Scholar]
- 144. One Brave Idea . One Brave Idea. https://www.onebraveidea.org/, 2019.
- 145. American Heart Association News . Neighborhoods play big role in heart health, study says. https://newsarchive.heart.org/neighborhoods-play-big-role-in-heart-health-study-says/, 2017.
- 146. Ward MP. A Persons Zip Code Is the Number 1 Factor that Predicts Coronary Heart Disease. Here’s How Technology Can Change That. https://www.circulation.com/blog/a-persons-zip-code-is-the-number-1-factor-that-predicts-coronary-heart-disease.-heres-how-technology-can-change-that, 2018.
- 147. Kramer F, Just S, Zeller T. New perspectives: systems medicine in cardiovascular disease. BMC Syst Biol 2018;12(1):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Meder B, Katus HA, Keller A. Computational cardiology—a new discipline of translational research. Genomics Proteomics Bioinformatics 2016;14(4):177–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Krittanawong C, Johnson KW, Hershman SG, et al. Big data, artificial intelligence, and cardiovascular precision medicine. Expert Rev Precis Med Drug Dev 2018;3(5): 305–17. [Google Scholar]
- 150. Niederer SA, Lumens J, Trayanova NA. Computational models in cardiology. Nat Rev Cardiol 2019;16(2):100–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Krittanawong C, Johnson KW, Tang WW. How artificial intelligence could redefine clinical trials in cardiovascular medicine: lessons learned from oncology. Pers Med 2019a;16(2):87–92. [DOI] [PubMed] [Google Scholar]
- 152. Trayanova N. From genetics to smart watches: developments in precision cardiology. Nat Rev Cardiol 2019;16(2):72–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. McGarrah RW, Crown SB, Zhang G-F, et al. Cardiovascular metabolomics. Circ Res 2018;122(9):1238–58. doi: 10.1161/CIRCRESAHA.117.311002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Cui X, Ye L, Li J, et al. Metagenomic and metabolomic analyses unveil dysbiosis of gut microbiota in chronic heart failure patients. Sci Rep 2018;8(1):635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Riggs DW, Yeager RA, Bhatnagar A. Defining the human envirome. Circ Res 2018;122(9):1259–75. doi: 10.1161/CIRCRESAHA.117.311230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Brook RD, Rajagopalan S, Pope CA, et al. Particulate matter air pollution and cardiovascular disease. Circulation 2010;121(21):2331–78. doi: 10.1161/CIR.0b013e3181dbece1. [DOI] [PubMed] [Google Scholar]
- 157. Fert-Bober J, Murray CI, Parker SJ, et al. Precision profiling of the cardiovascular post-translationally modified proteome. Circ Res 2018;122(9):1221–37. doi: 10.1161/CIRCRESAHA.118.310966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. McMahon P, Khomtchouk B, Wahlestedt C. Survival Guide to Organic Chemistry: Bridging the Gap from General Chemistry. Routledge: Taylor & Francis Group, 2017. [Google Scholar]
- 159. Ping P, Hermjakob H, Polson JS, et al. Biomedical informatics on the cloud: a treasure hunt for advancing cardiovascular medicine. Circ Res 2018;122(9):1290–301. doi: 10.1161/CIRCRESAHA.117.310967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Scruggs SB, Watson K, Su AI, et al. Harnessing the heart of big data. Circ Res 2015;116(7):1115–9. doi: 10.1161/CIRCRESAHA.115.306013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Global Alliance for Genomics and Health . Framework for responsible sharing of genomic and health-related data, 2014. https://www.ga4gh.org/genomic-data-toolkit/regulatory-ethics-toolkit/framework-for-responsible-sharing-of-genomic-and-health-related-data/. [DOI] [PMC free article] [PubMed]
- 162. P3g Consortium, Church G, Heeney C, et al. Public access to genome-wide data: five views on balancing research with privacy and protection. PLoS Genet 2009;5(10):e1000665. doi: 10.1371/journal.pgen.1000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Shringarpure SS, Bustamante CD. Privacy risks from genomic data-sharing beacons. Am J Hum Genet 2015;97(5):631–46. doi: 10.1016/j.ajhg.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Carter KW, Francis RW, Carter KW, et al. ViPAR: a software platform for the virtual pooling and analysis of research data. Int J Epidemiol 2016;45(2):408–16. doi: 10.1093/ije/dyv193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Chen F, Wang S, Jiang X, et al. PRINCESS: privacy-protecting rare disease international network collaboration via encryption through software guard extensions. Bioinformatics 2017;33(6):871–8. doi: 10.1093/bioinformatics/btw758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Gaye A, Marcon Y, Isaeva J, et al. DataSHIELD: taking the analysis to the data, not the data to the analysis. Int J Epidemiol 2014;43(6):1929–44. doi: 10.1093/ije/dyu188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Wilson R, Butters O, Avraam D, et al. DataSHIELD—new directions and dimensions. Data Sci J 2017;16(0):21. doi: 10.5334/dsj-2017-021. [Google Scholar]
- 168. Plis SM, Sarwate AD, Wood D, et al. COINSTAC: a privacy enabled model and prototype for leveraging and processing decentralized brain imaging data. Front Neurosci 2016;10. doi: 10.3389/fnins.2016.00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Langmead B, Nellore A. Cloud computing for genomic data analysis and collaboration. Nat Rev Genet 2018;19(4):208–19. doi: 10.1038/nrg.2017.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Harmanci A, Gerstein M. Quantification of private information leakage from phenotype-genotype data: linking attacks. Nat Methods 2016;13(3):251–6. doi: 10.1038/nmeth.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Harmanci A, Gerstein M. Analysis of sensitive information leakage in functional genomics signal profiles through genomic deletions. Nat Commun 2018;9(1):2453. doi: 10.1038/s41467-018-04875-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets-update. Nucleic Acids Res 2013;41(D1):D991–5. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Tryka KA, Hao L, Sturcke A, et al. NCBIs database of genotypes and phenotypes: dbGaP. Nucleic Acids Res 2014;42(Database issue):D975–9. doi: 10.1093/nar/gkt1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Vizcaíno JA, Deutsch EW, Wang R, et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol 2014;32:223–6. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Deutsch EW, Csordas A, Sun Z, et al. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res 2017;45(D1):D1100–6. doi: 10.1093/nar/gkw936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Zhu Y, Davis S, Stephens R, et al. GEOmetadb: powerful alternative search engine for the gene expression omnibus. Bioinformatics 2008;24(23):2798–800. doi: 10.1093/bioinformatics/btn520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Consortium TEP. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, et al. Integrative analysis of 111 reference human epigenomes. Nature 2015;518(7539):317–30. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Brookes AJ, Robinson PN. Human genotype–phenotype databases: aims, challenges and opportunities. Nat Rev Genet 2015;16(12):702–15. doi: 10.1038/nrg3932. [DOI] [PubMed] [Google Scholar]
- 180. Yun X, Xia L, Tang B, et al. 3CDB: a manually curated database of chromosome conformation capture data. Database 2016;2016. doi: 10.1093/database/baw044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Teng L, He B, Wang J, et al. 4DGenome: a comprehensive database of chromatin interactions. Bioinformatics 2015;31(15):2560–4. doi: 10.1093/bioinformatics/btv158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Quek XC, Thomson DW, Maag JLV, et al. lncRNAdb v2.0: expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res 2015;43(D1):D168–73. doi: 10.1093/nar/gku988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Fang S, Zhang L, Guo J, et al. NONCODEV5: a comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res 2018;46(D1):D308–14. doi: 10.1093/nar/gkx1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Khomtchouk BB, Dyomkin V, Vand KA, et al. Biochat: a database for natural language processing of gene expression omnibus data. bioRxiv 2018;480020. doi: 10.1101/480020. [Google Scholar]
- 185. Perez-Riverol Y, Bai M, da Veiga Leprevost F, et al. Discovering and linking public omics data sets using the Omics Discovery Index. Nat Biotechnol 2017;35:406–9. doi: 10.1038/nbt.3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Denny JC, Bastarache L, Ritchie MD, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol 2013;31(12):1102–11. doi: 10.1038/nbt.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Denaxas SC, Morley KI. Big biomedical data and cardiovascular disease research: opportunities and challenges. Eur Heart J Qual Care Clin Outcomes 2015;1(1):9–16. doi: 10.1093/ehjqcco/qcv005. [DOI] [PubMed] [Google Scholar]
- 188. Wu P, Cheng C, Kaddi CD, et al. Omic and electronic health record big data analytics for precision medicine. IEEE Trans Biomed Eng 2017;64(2):263–73. doi: 10.1109/TBME.2016.2573285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Dewey FE, Murray MF, Overton JD, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 2016b;354(6319):aaf6814. doi: 10.1126/science.aaf6814. [DOI] [PubMed] [Google Scholar]
- 190. Li J, Pan C, Zhang S, et al. Decoding the genomics of abdominal aortic aneurysm. Cell 2018;174(6):1361–1372.e10. doi: 10.1016/j.cell.2018.07.021. [DOI] [PubMed] [Google Scholar]
- 191. Bild DE, Bluemke DA, Burke GL, et al. Multi-ethnic study of atherosclerosis: objectives and design. Am J Epidemiol 2002;156(9):871–81. [DOI] [PubMed] [Google Scholar]
- 192. National Heart, Lung, and Blood Institute . Women’s Health Initiative. www.whi.org, 1991.
- 193. Claverie J-M. Computational methods for the identification of differential and coordinated gene expression. Hum Mol Genet 1999;8(10):1821–32. doi: 10.1093/hmg/8.10.1821. [DOI] [PubMed] [Google Scholar]
- 194. Santolini M, Romay MC, Yukhtman CL, et al. A personalized, multiomics approach identifies genes involved in cardiac hypertrophy and heart failure. NPJ Syst Biol Appl 2018;4(1):12. doi: 10.1038/s41540-018-0046-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Hawkins RD, Hon GC, Ren B. Next-generation genomics: an integrative approach. Nat Rev Genet 2010;11(7):476–86. doi: 10.1038/nrg2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Ritchie MD, Holzinger ER, Li R, et al. Methods of integrating data to uncover genotype-phenotype interactions. Nat Rev Genet 2015;16(2):85–97. doi: 10.1038/nrg3868. [DOI] [PubMed] [Google Scholar]
- 197. Gusev A, Ko A, Shi H, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet 2016;48(3):245–52. doi: 10.1038/ng.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Klarin D, Damrauer SM, Cho K, et al. Genetics of blood lipids among 300,000 multi-ethnic participants of the million veteran program. Nat Genet 2018;1. doi: 10.1038/s41588-018-0222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Watson A. Thematic review series: systems biology approaches to metabolic and cardiovascular disorders. Lipidomics: a global approach to lipid analysis in biological systems. J Lipid Res 2006;10(47):2101–11. [DOI] [PubMed] [Google Scholar]
- 200. Wu S, Lusis AJ, Drake TA. A systems-based framework for understanding complex metabolic and cardiovascular disorders. J Lipid Res 2009;04(50 Suppl):S358–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. J. LA, N. WJ. Cardiovascular networks. Circulation 2010;121(1):157–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Trachana K, Bargaje R, Glusman G, et al. Taking systems medicine to heart. Circ Res 2018;122(9):1276–89. doi: 10.1161/CIRCRESAHA.117.310999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Chen R, Mias GI, Li-Pook-Than J, et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 2012;148(6):1293–307. doi: 10.1016/j.cell.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Poldrack RA, Laumann TO, Koyejo O, et al. Long-term neural and physiological phenotyping of a single human. Nat Commun 2015;6:8885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Price ND, Magis AT, Earls JC, et al. A wellness study of 108 individuals using personal, dense, dynamic data clouds. Nat Biotechnol 2017;35:747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Zeevi D, Korem T, Zmora N, et al. Personalized nutrition by prediction of glycemic responses. Cell 2015;163(5):1079–94. doi: 10.1016/j.cell.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 207. Komajda M, Charron P. The heart of genomics. Nat Med 2001;7(3):287–8. doi: 10.1038/85420. [DOI] [PubMed] [Google Scholar]
- 208. Lau E, Cao Q, Lam MPY, et al. Integrated omics dissection of proteome dynamics during cardiac remodeling. Nat Commun 2018;9(1):120. doi: 10.1038/s41467-017-02467-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Althoff T, Sosič R, Hicks JL, et al. Large-scale physical activity data reveal worldwide activity inequality. Nature 2017;547:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Hannun AY, Rajpurkar P, Haghpanahi M, et al. Cardiologist-level arrhythmia detection and classification in ambulatory electrocardiograms using a deep neural network. Nat Med 2019;25(1):65–9. doi: 10.1038/s41591-018-0268-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Attia ZI, Noseworthy PA, Lopez-Jimenez F, et al. An artificial intelligence-enabled ecg algorithm for the identification of patients with atrial fibrillation during sinus rhythm: a retrospective analysis of outcome prediction. Lancet 2019;394:861–7. [DOI] [PubMed] [Google Scholar]
- 212. Awan SE, Sohel F, Sanfilippo FM, et al. Machine learning in heart failure: ready for prime time. Curr Opin Cardiol 2018;33(2):190–5. [DOI] [PubMed] [Google Scholar]
- 213. Choi E, Schuetz A, Stewart WF, et al. Using recurrent neural network models for early detection of heart failure onset. J Am Med Inform Assoc 2017;24(2):361–70. doi: 10.1093/jamia/ocw112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Singh, A. and Guttag, J. V. A comparison of non-symmetric entropy-based classification trees and support vector machine for cardiovascular risk stratification. In: 2011 Annual International Conference of the IEEE Engineering in Medicine and Biology Society. pp 79–82, 2011. IEEE; Boston, MA, USA. [DOI] [PubMed] [Google Scholar]
- 215. Duan T, Rajpurkar P, Laird D, et al. Clinical value of predicting individual treatment effects for intensive blood pressure therapy. Circ Cardiovasc Qual Outcomes 2019;12(3):e005010. doi: 10.1161/CIRCOUTCOMES.118.005010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 216. Johnson KW, Torres Soto J, Glicksberg BS, et al. Artificial intelligence in cardiology. J Am Coll Cardiol 2018;71(23):2668. [DOI] [PubMed] [Google Scholar]
- 217. Al'Aref SJ, Anchouche K, Singh G, et al. Clinical applications of machine learning in cardiovascular disease and its relevance to cardiac imaging. Eur Heart J 2019;40:1975–86. [DOI] [PubMed] [Google Scholar]
- 218. Krittanawong C, Zhang H, Wang Z, et al. Artificial intelligence in precision cardiovascular medicine. J Am Coll Cardiol 2017;69(21):2657–64. doi: 10.1016/j.jacc.2017.03.571. [DOI] [PubMed] [Google Scholar]
- 219. Krittanawong C, Johnson KW, Rosenson RS, et al. Deep learning for cardiovascular medicine: a practical primer. Eur Heart J 2019b;40(25):2058–73. doi: 10.1093/eurheartj/ehz056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Bello GA, Dawes TJW, Duan J, et al. Deep-learning cardiac motion analysis for human survival prediction. Nat Mach Intell 2019;1(2):95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Bizopoulos P, Koutsouris D. Deep learning in cardiology. IEEE Rev Biomed Eng 2019;12:168–93. doi: 10.1109/RBME.2018.2885714. [DOI] [PubMed] [Google Scholar]
- 222. Madani A, Ong JR, Tibrewal A, et al. Deep echocardiography: data-efficient supervised and semi-supervised deep learning towards automated diagnosis of cardiac disease. NPJ Digit Med 2018;1(1):59. doi: 10.1038/s41746-018-0065-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Lee Y, Kwon J-M, Lee Y, et al. Deep learning in the medical domain: predicting cardiac arrest using deep learning. Acute Crit Care 2018;33(3):117–20. doi: 10.4266/acc.2018.00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Kwon J-M, Lee Y, Lee Y, et al. An algorithm based on deep learning for predicting in-hospital cardiac arrest. J Am Heart Assoc 2018;7(13):e008678. doi: 10.1161/JAHA.118.008678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Liu N, Koh ZX, Chua EC, et al. Risk scoring for prediction of acute cardiac complications from imbalanced clinical data. IEEE J Biomed Health Inform 2014;18(6):1894–902. doi: 10.1109/JBHI.2014.2303481. [DOI] [PubMed] [Google Scholar]
- 226. Rahman MM, Davis DN. Addressing the class imbalance problem in medical datasets. Int J Mach Learn Comput 2013;3:224–8. [Google Scholar]
- 227. Wiens J, Guttag JV. Active learning applied to patient-adaptive heartbeat classification. In: Lafferty JD, CKI W, Shawe-Taylor J et al. (eds). Advances in Neural Information Processing Systems 23. 2010, 2442–50. Curran Associates, Inc.: Red Hook, NY. [Google Scholar]
- 228. Orphanou K, Stassopoulou A, Keravnou E. Dbn-extended: a dynamic bayesian network model extended with temporal abstractions for coronary heart disease prognosis. IEEE J Biomed Health Inform 2016;20(3):944–52. doi: 10.1109/JBHI.2015.2420534. [DOI] [PubMed] [Google Scholar]
- 229. Gong W, Koyano-Nakagawa N, Li T, et al. Inferring dynamic gene regulatory networks in cardiac differentiation through the integration of multi-dimensional data. BMC Bioinformatics 2015;16(1):74. doi: 10.1186/s12859-015-0460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Matthews D. Virtual-reality applications give science a new dimension. Nature 2018;557:127–8. [DOI] [PubMed] [Google Scholar]
- 231. Silva JN, Southworth M, Raptis C, et al. Emerging applications of virtual reality in cardiovascular medicine. JACC Basic Transl Sci 2018;3(3):420–30. doi: 10.1016/j.jacbts.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Rumsfeld JS, Joynt KE, Maddox TM. Big data analytics to improve cardiovascular care: promise and challenges. Nat Rev Cardiol 2016;13:350. [DOI] [PubMed] [Google Scholar]
- 233. Slomka PJ, Dey D, Sitek A, et al. Cardiac imaging: working towards fully-automated machine analysis & interpretation. Expert Rev Med Devices 2017;14(3):197–212. doi: 10.1080/17434440.2017.1300057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Fonseca CG, Backhaus M, Bluemke DA, et al. The cardiac atlas project–an imaging database for computational modeling and statistical atlases of the heart. Bioinformatics 2011;27(16):2288–95. doi: 10.1093/bioinformatics/btr360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Liem DA, Murali S, Sigdel D, et al. Phrase mining of textual data to analyze extracellular matrix protein patterns across cardiovascular disease. Am J Physiol Heart Circ Physiol 2018;315(4):H910–24. doi: 10.1152/ajpheart.00175.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Lippincott T, Séaghdha DO, Korhonen A. Exploring subdomain variation in biomedical language. BMC Bioinformatics 2011;12(1):212. doi: 10.1186/1471-2105-12-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Kilicoglu H. Biomedical text mining for research rigor and integrity: tasks, challenges, directions. Brief Bioinform 2018;19(6):1400–14. doi: 10.1093/bib/bbx057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Avsec ž, Kreuzhuber R, Israeli J, et al. The kipoi repository accelerates community exchange and reuse of predictive models for genomics. Nat Biotechnol 2019;37(6): 592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Pavlopoulos GA, Malliarakis D, Papanikolaou N, et al. Visualizing genome and systems biology: technologies, tools, implementation techniques and trends, past, present and future. GigaScience 2015;4(1). doi: 10.1186/s13742-015-0077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. O’Donoghue SI, Baldi BF, Clark SJ, et al. Visualization of biomedical data. Annu Rev Biomed Data Sci 2018; 1(1):275–304. doi: 10.1146/annurev-biodatasci-080917-013424. [Google Scholar]
- 241. Katz Y, Wang ET, Silterra J, et al. Quantitative visualization of alternative exon expression from RNA-seq data. Bioinformatics 2015;31(14):2400–2. doi: 10.1093/bioinformatics/btv034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Strobelt H, Alsallakh B, Botros J, et al. Vials: visualizing alternative splicing of genes. IEEE Trans Vis Comput Graph 2016;22(1):399–408. doi: 10.1109/TVCG.2015.2467911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Khomtchouk BB, Hennessy JR, Wahlestedt C. Shinyheatmap: ultra fast low memory heatmap web interface for big data genomics. PLoS One 2017;12(5):e0176334. doi: 10.1371/journal.pone.0176334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Kerpedjiev P, Abdennur N, Lekschas F, et al. HiGlass: web-based visual exploration and analysis of genome interaction maps. Genome Biol 2018;19(1):125. doi: 10.1186/s13059-018-1486-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Lekschas F, Bach B, Kerpedjiev P, et al. HiPiler: visual exploration of large genome interaction matrices with interactive small multiples. IEEE Trans Vis Comput Graph 2018;24(1):522–31. doi: 10.1109/TVCG.2017.2745978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Marafino BJ, Park M, Davies JM, et al. Validation of prediction models for critical care outcomes using natural language processing of electronic health record data. JAMA Netw Open 2018;1(8):e185097–7. doi: 10.1001/jamanetworkopen.2018.5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. National Institute of Health . Estimates of funding for various research, condition, and disease categories (RCDC). https://report.nih.gov/categorical∖_spending.aspx, 2018.
- 248. Köhler S, Doelken SC, Mungall CJ, et al. The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data. Nucleic Acids Res 2014;42(D1):D966–74. doi: 10.1093/nar/gkt1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD) . OMIM—Online Mendelian Inheritance in Man. https://omim.org/, 2018.
- 250. INSERM . Orphanet: An Online Database of Rare Diseases and Orphan Drugs. http://www.orpha.net/, 1997.
- 251. Firth HV, Richards SM, Bevan AP, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet 2009;84(4): 524–33. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]