

Keywords: environmental circadian disruption, phase shift, shift work, SHR-SP, stroke-prone spontaneously hypertensive rats, urine volume
Abstract
Nontraditional work schedules, such as shift work, have been associated with numerous health issues, including cardiovascular and metabolic disease. These work schedules can chronically misalign environmental timing cues with internal circadian clock systems in the brain and in peripheral organs, leading to dysfunction of those systems and their associated biological processes. Environmental circadian disruption in the kidney may be an important factor in the increased incidence of hypertension and adverse health outcomes in human shift workers. The relationship between renal rhythmicity and injury resilience is not well understood, especially in the context of environmental, rather than genetic, manipulations of the circadian system. We conducted a longitudinal study to determine whether chronic shifting of the light cycle that mimics shift work schedules would disrupt output rhythms of the kidney and accelerate kidney injury in salt-loaded male spontaneously hypertensive, stroke-prone rats. We observed that chronic shifting of the light-dark (LD) cycle misaligned and decreased the amplitude of urinary volume rhythms as the kidney phase-shifted to match each new lighting cycle. This schedule also accelerated glomerular and tubular injury marker excretion, as quantified by nephrin and KIM-1 compared with rats kept in a static LD cycle. These data suggest that disrupted rhythms in the kidney may decrease resilience and contribute to disease development in systems dependent on renal and cardiovascular functions.
INTRODUCTION
Late-night eating, sleeping during the day, and light exposure at night are common among people who engage in shift work. With nearly 29% of the United States population engaging in some version of shift work (1), this labor practice is broadly defined as a work schedule outside of the 9 AM to 5 PM constraints. Obesity (2), stroke (3), kidney disease (4), cancer (5), and sleep loss (1) are more common among shift workers than in the general population. Although the mechanisms linking shift work exposure to disease are unknown, a potential explanation for this association may be the misalignment of diurnal and circadian rhythms in biological function among multiple central and peripheral organs (6–9).
The suprachiasmatic nucleus (SCN) is a central clock that synchronizes the timing of peripheral clocks throughout the body to coordinate biological processes. Located in the hypothalamus, the SCN receives external light cues from the retina to correctly time the endogenously rhythmic transcription of clock genes (10–13). These oscillating genes are organized into several interacting positive and negative feedback loops, whose output regulates the timing of sleep-wake behavior and clocks in peripheral organs. Present in nearly every cell type, peripheral clocks coordinate local biological functions. In addition to receiving instructions from the central clock, peripheral clocks have the potential to be influenced by a variety of cues independent of light, including activity or food intake (13). Desynchronization among the peripheral and central clocks drive circadian misalignment of biological processes (14, 15). Repeated long-term disruption and misalignment of these myriad rhythms is the likely consequence of a typical shift work lifestyle and is believed to be the underlying cause of the associated morbidities (6, 16).
Blood pressure, like many biological processes, is highly influenced by the circadian system. In humans, a nighttime dip in blood pressure is associated with a decrease in cardiac output, arterial resistance, and heart rate (17) and is considered healthy. Nondipping, on the other hand, occurs when nocturnal blood pressure values fail to fall at least 10% from day-time blood pressure values (18, 19). Several cross-sectional studies in both general and hypertensive populations have revealed that cardiac hypertrophy, microalbuminuria, and cerebral infarction are prevalent among individuals with a nondipping blood pressure profile compared with individuals with robust blood pressure rhythms (20–22). Furthermore, the absence of a distinct trough in blood pressure rhythm at night has been demonstrated longitudinally to be associated with organ damage, including renal disease development and progression (23–25).
The maintenance of blood pressure depends on the kidney’s capacity to regulate Na+ content in the extracellular space (26). Decreased renal excretion of Na+ during the daytime has been shown to correlate with nocturnal hypertension (27). Clock genes modulate renal excretory function through the expression of Na+ transporters and channels (28–30). Dysfunction and inappropriate regulation of these channels in the kidney have been shown to consequently result in improper renal Na+ handling and blood pressure disorders (31–35). Period 1 (Per 1), a repressor within the circadian clock mechanism, has been demonstrated to facilitate aldosterone function, increase Na+ reabsorption, and maintain rhythmicity in renal excretion (29). Constitutive activation of the positive leg of the circadian clock via deletion of the suppressors cryptochrome 1 (Cry 1) and cryptochrome 2 (Cry 2) in mice resulted in salt-sensitive hypertension (36, 37). Although these studies illustrate the complexities involved in circadian regulation of blood pressure, little is known about the relationship between renal rhythms and injury. Elevated salt intake is associated with the development of hypertension and is an independent risk factor for chronic kidney disease (38). Notably, a high-salt diet appears to induce region-specific suppression and desynchronization of clock genes in the kidney (13). However, how the kidney may handle high salt conditions in the presence of a disrupted circadian clock is still unknown.
To address these questions, we investigated urine excretory rhythms and kidney health in male spontaneously hypertensive stroke-prone rats (SHR-SP) in both a stable and circadian-disrupted environment. Here, we provide evidence showing that chronic circadian disruption targets the renal system and its ability to regulate excretion, likely contributing to the development of diseases associated with hypertension.
METHODS
Circadian Disruption Protocol
Male SHR-SP (Charles River Laboratories, Wilmington, MA) were used in this experiment. Rats arrived at 7 wk of age and were housed in groups of three to four rats until 11 wk of age to acclimate to the housing facilities and handling. Animals were individually housed at 12 wk of age in a standard 12:12-h light-dark (LD) cycle. At 16 wk, rats were randomly assigned into experimental groups and placed in sleep chambers for the remainder of the study. Rats were assigned to remain under standard 12:12-h LD schedule (n = 6; Fig. 1A) or placed in chronic LD shift schedule (n = 6) for a 44-day (6-wk) study (Fig. 1A). This lighting schedule induces environmental circadian disruption (ECD) and has been previously described (6). In this schedule, the 12:12-h LD cycle is advanced by 6 h, once every 7 days, by shortening the dark phase. Rats were given standard rat chow upon arrival into the facility. Rats were switched to high-salt chow at the beginning of the study and remained on high-salt chow until the end of the experiment. Rat chow and water were available to animals ad libitum (in some cases, salt enhanced; see salt load protocol). Animal care procedures and experimental protocols were approved by Morehouse School of Medicine Institutional Animal Care and Use Committee.
Figure 1.

Study protocol. A: control light schedule and environmental circadian disruption (ECD) light schedule. Each block represents 6 h; white blocks represent lights on, whereas dark gray blocks represent lights off. Rows illustrate each week of the study. *Weeks where 28-h urine series were collected. Light gray shading over weeks 5 and 6 represents the replacement of drinking water with 1% saline solution. At the start of the experiment, rats were 16 wk of age. B: expanded view of week 2 as an example of the daily schedule. Open triangles represent one sample time for the time-series urine collections reflecting two series each week: on the third day (early week) and sixth day (late week). Rats were 16 wk of age at the beginning of the study.
Salt Load Protocol
Beginning at 16 wk of age and for the entirety of the study, rats were given high-salt chow at a 4% NaCl-0.75% K+ diet (Zeigler Feed P#528801-12-01, Gardners, PA) to accelerate the development of hypertension and associated renal damage (39). At 20 wk of age (day 31 of the experimental protocol), rats were additionally salt loaded with 1% saline drinking water until the end of the 44-day study. Saline (1%) drinking water replaced regular drinking water to further expedite hypertension (Fig. 1A, gray-shaded region).
Urine Collection and Analysis
On specific days across the study protocol, rats were removed from housing cages and placed in metabolic cages in the exact same position in the sleep chamber to maintain consistent light exposure. Urine was collected for 28 h at 4-h intervals. In between these 28-h collection periods, rats were replaced in their home cages. To prevent stress during metabolic cage exposure, animals were acclimated to cages using several previous exposures once a week for 3 wk in increasing duration (up to 18 h) before the start of the study at 16 wk of age. Urine collection occurred biweekly on the third day (referred to as “early”) and sixth day (referred to as “late”) of weeks 2, 4, 5, and 6, starting at the end of lights on (Fig. 1B). Collection times were assigned/labeled according to the end of each 4-h collection window. Urine excretion volumes were determined by weighing collection tubes. Samples were immediately centrifuged at 4°C at 5,300 rpm for 10 min to separate particulate debris. Urine was aliquoted into 2.0-mL centrifuge tubes and stored at −80°C until analysis. Urinary endothelin-1 (ET-1) concentration was quantified by ELISA (Quantikine ELISA Endothelin-1, kit no. DET100, R&D Systems, Minneapolis, MN). ET-1 concentration was multiplied by urine volume for each time point to determine the excretion rate per 4 h.
Renal Injury Measurements
To examine the potential impact of ECD on renal damage, urinary kidney injury molecule-1 (KIM-1) excretion was measured to evaluate tubular injury, and nephrin excretion was quantified to detect glomerular injury. Urinary KIM-1 and nephrin concentrations were quantified by ELISA (Quantikine ELISA KIM-1, kit no. RKM100, R&D Systems, and Nephrin NPHN Competitive ELISA, kit no. MBS723583, My BioSource, San Diego, CA). Peak and trough urine volume time points for each group were chosen to investigate KIM-1 and nephrin excretion to ensure that injury was examined at the same circadian phase across groups. Reported levels reflect the mean of two time points reflecting maximum and minimum excretion of each day in the urine volume sampling sequence.
At the end of the 44-day study, we examined renal structure to validate urinary evidence of tubular and glomerular injury. Control and ECD rats were euthanized by 100% CO2 followed by secondary cervical dislocation. The left kidney was removed and cut into two transverse sections through the papilla and incubated in 4% paraformaldehyde-PBS for 48 h. Tissue was then transferred to a 70% ethanol solution. Kidney sections were processed and embedded in paraffin blocks. Slices (4 µm) were cut and mounted on a glass slide. Kidney slices were stained with Masson’s trichrome according to the manufacturer’s instructions (Thermo Scientific Richard-Allen Scientific Chromaview). Sections were scored by a researcher blinded from the treatment groups and hypothesis. An entire stained kidney slice was observed for each subject by light microscopy (×40). The percentage of injured glomeruli that displayed sclerosis or necrosis and the percentage of injured tubules that displayed proteinaceous casts or atrophy were estimated for each animal.
Statistics and Data Analysis
MetaCycle (https://cran.r-project.org/web/packages/MetaCycle/index.html) (40) was used to screen for diurnal/circadian rhythmicity of urine volume and ET-1 excretion with these limiters: ARSER(ARS) default period = 24, maximum period = 28, and minimum period = 20. Rhythmicity was defined by P < 0.05 for the combined statistic, which includes ARSER, JTK_CYCLE, and Lomb-Scargle algorithms. Relative amplitude of urine volume and ET-1 excretion rhythms were calculated incorporating the curve fit amplitude of ARSER, JTK_CYCLE, and Lomb-Scargle divided by the mean excretion of the urinary analyte (41). Relative amplitude data were included in the analysis whether or not an individual 24-h rhythm was present, but peak phase was only included for series with a significant rhythm. Except where stated, phase was reported as clock time to prevent confusion that might result from using Zeitgeber time (ZT) in cases where the light cycle is changing over weeks. The degree of misalignment in urine excretion phase was determined in ZT time. Pairwise phase comparisons were made using t tests. Longitudinal urinary injury markers are compared by two-way ANOVA followed by multiple comparison analysis. The significance threshold was set at α = 0.05. Post hoc power continuous calculation was used to determine effect size. Main and interaction effects were assessed using GraphPad software, single statistical model two-way ANOVA.
RESULTS
ECD Shifts Circadian Phase and Suppresses Rhythms in Urine Volume and ET-1
In this 44-day study, we found that urinary excretion rates of control animals were consistently highest during late night, at the end of the active phase in these nocturnal rats, and reached minimum levels before lights off (Fig. 2A). The phase of urinary excretion in this study was determined by the timing of peak urine volume using MetaCycle and was plotted for all collection days across the study in Fig. 2C. There was evidence of a phase change in the control group from the beginning of the study to the end [week 2 early vs. week 6 late, P = 0.0103, difference in mean 0.91 h ± 0.30, t = 3.2, degrees of freedom (df) =10, effect size = 1.6, power = 1.0]. As rats became more salt loaded, an increase in urine excretion volume was seen (week 2 early vs. week 6 late, P < 0.0001, difference in mean 111.3 g ± 6.5, t = 17.1, df = 10, effect size = 12.0, power = 1.0). Urine excretion phase within each week remained consistent in the control group with no significant differences (week 2: P = 0.5663, mean difference: 0.24 h ± 0.4, t = .5, df = 10, effect size = 0.40, power = 0.09; week 4: P = 0.8288, mean difference: 0.08 h ± 0.35, t = .2, df = 10, effect size = 0.12, power = 0.04; week 5: P = 0.6867, mean difference: 0.23 h ± 0.54, t = .4, df = 10, effect size = 0.27, power = 0.06; and week 6: P = 0.1076, mean difference: 0.81 h ± 0.45, t = 1.8, df = 10, effect size = 0.76, power = 0.07). Rats subjected to a weekly 6-h phase advance (ECD) exhibited a leftward shift in urine excretion rate indicative of phase advancing transients to follow the LD cycle both within and across weeks (Fig. 2, B and C). ECD rats showed significant early-to-late week phase differences in peak urine excretion in weeks 2 and 5 (week 2: P < 0.0001 difference in mean: 3.50 h ± 0.30, t = 11.54, df = 6, effect size = 6.22, power = 1.0; week 4: P = 0.3313, mean difference: 5.50 h ± 5.11, t = 1.1, df = 5, effect size = 0.82, power = 0.25; week 5: P < 0.0001, mean difference: 7.72 h ± 0.35, t = 21.78, df = 6, effect size = 18.43, power = 1.0; and week 6: P = 0.6340, mean difference: 0.64 h ± 1.3, t = .5, df = 8, effect size = 0.23, power = 0.07). These dynamics reflect persistent misalignment between the urinary excretion rhythms and the current LD cycle of intermittent degree (Table 1), using the phase measured in unshifted controls on the same day as the expected, aligned phase.
Figure 2.

Urine volume rhythms. A and B: traces show means ± SE urine volume every 4 h for male rats exposed to a fixed light schedule (A; control, n = 6) and environmental circadian disruption (ECD) light schedule (B; ECD, n = 6). Shading indicates dark periods. C: rhythmic phase (peak) of urine volume excretion rhythms for each collection series throughout the study. Symbols (squares and circles) indicate individual rats, with whiskers indicating means and SEs. Secondary y-axis labels indicate the sample sizes where the numerator denotes how many rat urine series were rhythmic and are plotted here, and the denominator indicates how many total series were collected including those found to be arrhythmic according to MetaCycle and thus omitted from phase analysis. D: means ± SE relative amplitude of urine volume (UV) for control and ECD animals determined by MetaCycle analysis. The black dotted lines in C and D denote the replacement of regular drinking water with 1% Na+-supplemented drinking water. Two-way ANOVA, multiple comparisons for control vs. ECD: *P < 0.05 and **P < 0.01.
Table 1.
Peak phase of urine excretion rhythms for control and environmental circadian disruption-exposed male rats from Fig. 2, A and B
| Control Rats |
Environmental Circadian Disruption-Exposed Rats |
Effect Size | Power | P Value | Difference | t Ratio | Degrees of Freedom | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Means ± SE | n | Means ± SE | n | |||||||
| Week 2 early | 21.19 ± 0.25 | 6 | 3.40 ± 0.28 | 4 | 10.05 | 1 | <0.000001 | 6.21 ± 0.38 | 16.33 | 8 |
| Week 2 late | 21.43 ± 0.31 | 6 | 23.90 ± 0.23 | 4 | 3.96 | 1 | 0.00028 | 2.47 ± 0.40 | 6.14 | 8 |
| Week 4 early | 21.02 ± 0.26 | 6 | 0.45 ± 3.65 | 4 | 1.5 | 0.46 | 0.04931 | 3.43 ± 1.48 | 2.32 | 8 |
| Week 4 late | 20.94 ± 0.24 | 6 | 0.95 ± 0.32 | 3 | 7.5 | 1 | 0.00001 | 4.01 ± 0.38 | 10.68 | 7 |
| Week 5 early | 21.21 ± 0.34 | 6 | 6.96 ± 0.49 | 2 | 12.48 | 1 | 0.00001 | 9.75 ± 0.64 | 15.28 | 6 |
| Week 5 late | 21.44 ± 0.42 | 6 | 23.23 ± 0.38 | 4 | 2.1 | 0.97 | 0.01154 | 1.80 ± 0.55 | 3.26 | 8 |
| Week 6 early | 21.09 ± 0.43 | 6 | 21.67 ± 2.75 | 5 | 0.3 | 0.07 | 0.64291 | 0.58 ± 1.21 | 0.48 | 9 |
| Week 6 late | 20.28 ± 0.15 | 6 | 22.31 ± 0.39 | 5 | 3.17 | 1 | 0.00053 | 2.03 ± 0.39 | 5.24 | 9 |
Phase is reported in Zeitgeber time to facilitate determination of misalignment between peak urine excretion and the current light-dark cycle, using phase of the control group each week as the expected aligned phase. Shown are mean phase values ± SE, effect size, power, P value, t ratio, and degrees of freedom.
The rhythms of urine excretion from all six animals exposed to a fixed lighting schedule were determined by MetaCycle to be significant for all eight time-series (Fig. 2C, secondary y-axis indicates the number of rats in the group that exhibited rhythmic urine volume excretion/rats in the group). On the other hand, significant urinary excretion rate rhythms in ECD rats were found in fewer animals than controls, most evident in the middle and end of the study after multiple shifts had been accomplished. For example, for the week 5 early time series, control rats had six of six time series found rhythmic, whereas ECD rats’ rhythms were only significant in two of six time series.
In addition to these binary observations of fewer significant rhythms of urinary output in ECD rats than control rats, ECD rats’ time series exhibited lower relative circadian amplitude, as quantified by MetaCycle (Fig. 2D). Two-way ANOVA revealed a main effect of circadian disruption versus control (P < 0.0001, F = 45.2, DFn = 1, DFd = 80), an effect of time (P = 0.0004, F = 4.3, DFn = 7, DFd = 80), and an interaction (P = 0.0017, F = 3.7, DFn = 7, DFd = 80). Specifically, during weeks 4 and 5, urine volume rhythms were significantly suppressed in ECD rats according to post hoc analysis (Fig. 2D, week 4 early: P = 0.03, mean difference: 0.21 ± 0.07, t = 3.0, df = 10, effect size = 1.48, power = 0.79; week 4 late: P < 0.0001, mean difference: 0.44 ± 0.07, t = 6.4, df = 10, effect size = 5.44, power = 1.0, and week 5 early: P = 0.02, mean difference: 0.21 ± 0.07, t = 3.1, df = 10, effect size = 7.45, power = 0.98).
Like urine volume, the urinary ET-1 excretion rate for control animals was the highest late in the active phase and lowest late in the rest phase (Fig. 3A) each week. Phase slowly shifted later throughout the study for the controls (Fig. 3C, week 2 early vs. week 6 late: P = 0.001, 3.13 h ± 0.82, t = 5.3, df = 7, effect size = 3.73, power= 1.0). The number of time series with a significant diurnal rhythm dropped off considerably as salt loading progressed, making conclusions regarding longitudinal phase changes more difficult to draw (Fig. 3C, secondary axis).
Figure 3.

Endothelin-1 (ET-1) excretion rhythms. A and B: traces show means ± SE urine volume every 4 h for male rats exposed to a fixed light schedule (A; control, n = 6) and environmental circadian disruption (ECD) light schedule (B; ECD, n = 6). Shading indicates dark periods. Shown are average ET-1 excretion rhythms. C: peak phase of ET-1 excretion for rhythmic traces. D: mean ± SE relative amplitude of ET-1 excretion of control (n = 6) and ECD (n = 6) male animals determined by MetaCycle. Two-way ANOVA, multiple comparisons for control vs. ECD: not significant.
In ECD rats, ET-1 excretion rate rhythms steadily advanced within the sampling weeks (Fig. 3, B and C). As with both controls, phase was more difficult to assess later in the experiment as the observation of significant diurnal rhythms dropped precipitously across the study (Fig. 3C, secondary axis), with the loss of rhythmicity more common in the ECD group than the control group. Consistent with that observation, relative circadian amplitude trended lower in ECD rats than in control rats near the end of the study, but this difference did not reach significance (Fig. 3D, two-way ANOVA: main effect for group: P = 0.07, F = 3.3, DFn = 1, DFd = 80, main effect for time P < 0.0001, F = 6.3, DFn = 7, DFd = 80, interaction P = 0.33, F = 1.16, DFn = 7, DFd = 80).
Increased Urinary Evidence of Renal Injury in Rats with Disrupted Renal Rhythms
Both urinary nephrin and KIM-1 excretion levels were low in both groups until the introduction of 1% saltwater on day 31 (dotted lines in Fig. 4, A and B). With urinary nephrin levels increasing around weeks 4 and 5, before KIM-1 levels increasing ∼1 wk later, all rats exhibited detectable signs of early kidney damage. ECD rats had a more rapid onset and higher rise in nephrin excretion rate than control rats (Fig. 4A, two-way ANOVA: main effect of group: P = 0.0002, F = 16.2, DFn = 1, DFd = 52, main effect of time: P < 0.0001, F = 15.3, DFn = 7, DFd = 52, interaction: P = 0.03, F = 2.4, DFn = 7, DFd = 52), with a significant difference in multiple comparisons for both time points in week 5 (Fig. 4A, week 5 early: P = 0.01, effect size = 1.80, power = 0.89; and week 5 late: P = 0.0007, effect size = 4.87, power = 0.84). Urinary KIM-1 excretion was overall higher in ECD rats as well (main effect of group: P = 0.02, F = 6.0, DFn = 1, DFd = 67, main effect of time: P < 0.0001, F = 122.2, DFn = 7, DFd = 67, interaction: P < 0.0001, F = 13.15, DFn = 7, DFd = 67), with the bulk of those differences seen during week 6 (Fig. 4B, week 6 early: P = −0.02, effect size = 1.71, power = 1.0; and week 6 late: P < 0.0001, effect size = 8.29, power = 1.0). The week 5 late time point was unique in that control rats had slightly higher excretion of KIM-1 in multiple comparisons (Fig. 4B, p = 0.02, effect size = 11.85, power = 0.41). Unlike the in vivo longitudinal measures of nephrin and KIM-1, end-point measures using Masson’s trichrome staining of kidneys did not reveal differences among the groups (Fig. 4, C–E). There was glomerular and tubular injury apparent in both groups at day 44. However, there was no further increase of renal injury due to circadian disruption.
Figure 4.
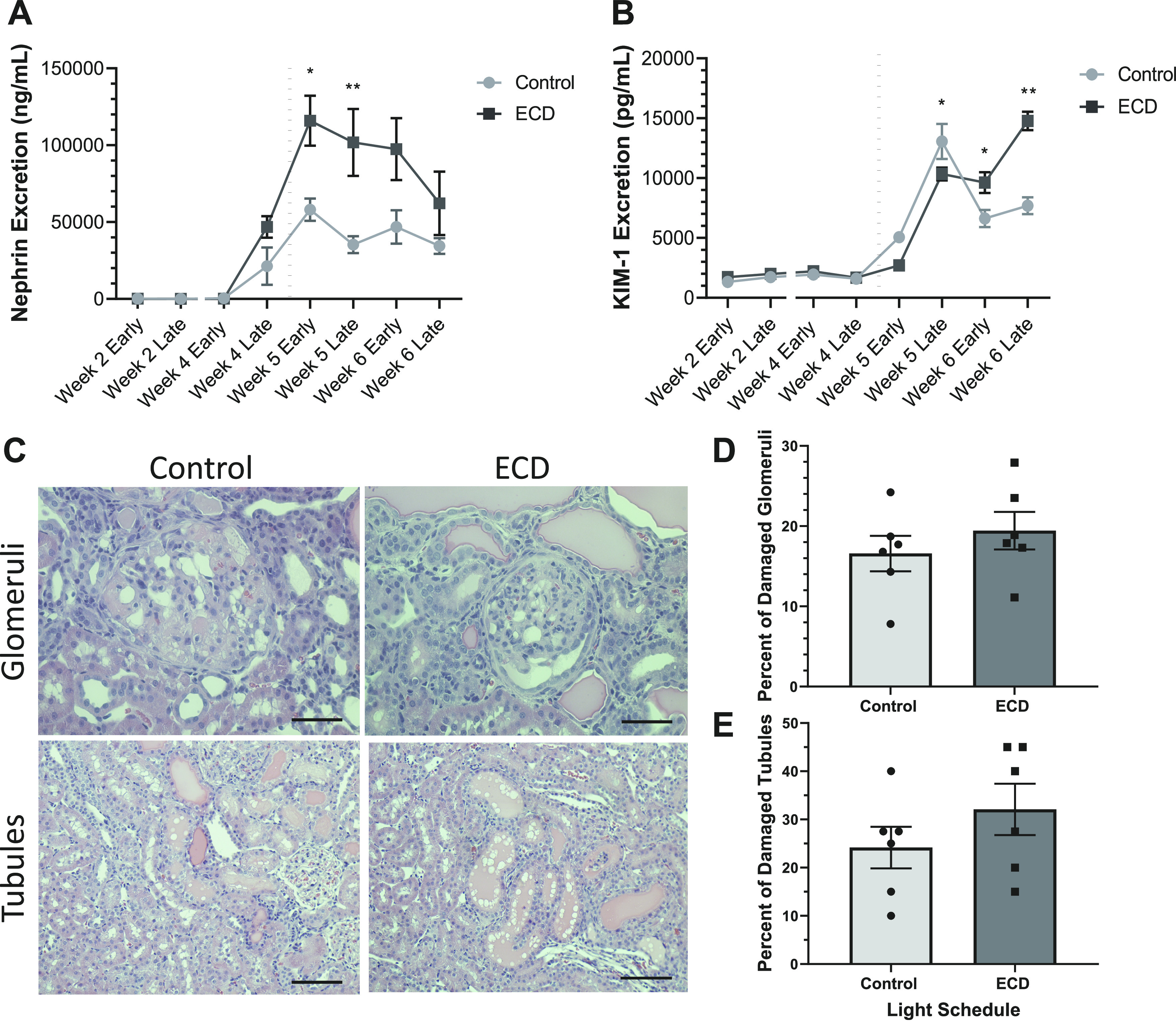
Renal injury markers. A: mean ± SE urinary nephrin excretion rate of male rats exposed to a control (n = 6) or ECD (n = 6) light schedule. B: mean ±SE urinary kidney injury molecule-1 (KIM-1) excretion rate. C: representative images showing the glomerular and tubular injury that was present in both control and ECD rats. Scale bars = 0.05 mm for top images and 0.1 mm for bottom images. D: individual and mean ± SE percentage of damaged glomeruli at the study completion. E: individual and mean ± SE percentage of damaged tubules. The black dotted line represents the replacement of regular drinking water with 1% Na+-supplemented drinking water. Two-way ANOVA, multiple comparisons for control vs. ECD: *P < 0.05 and **P < 0.01.
DISCUSSION
In this study, we assessed how repeated shifting of the environmental LD cycle impacts renal health and rhythmicity in renal function. We collected 28-h urine from rats housed intermittently in metabolic cages in a 44-day longitudinal study. Urine volume and ET-1 were used as indices of rhythmic output of the kidney. Such rhythms are thought to reflect the output of the circadian clock in the kidney since periodic variations in food and water intake have been shown not to drive blood pressure or renal excretory rhythms (42). Consistent with previous findings showing that urine excretion peaks primarily during the active period (43, 44), our study determined that urine and ET-1 excretion is maximal late in the active period in control rats. Renal tissue from animals expressing a circadian reporter-gene oscillate in culture (45, 46) and in vivo (47), so such rhythms in kidney function are likely driven by the renal molecular clock.
A 6-h phase advance in the light schedule promotes a rapid phase change in the SCN, followed by a shift in peripheral organs, which appear to adjust at different rates (45). The liver, skeletal muscle (48), and lung (45) re-entrain before the sixth day of the advanced light schedule. In line with previous reports of reentrainment to light schedule modifications (6, 47, 49), we observed a steady advance in the timing of the peak, referred to as a phase shift, of daily urine volume of the rats exposed to a weekly shift in light schedule. Our in vivo, longitudinal assessment of a peripheral clock output confirms that, like behavior and core body temperature, the kidney peripheral clock re-entrains to an advanced light cycle over several days with transients. Thus, under these conditions, the renal system rhythms are repeatedly misaligned with the external lighting cycle, attempting to realign after several days in the new light schedule. Similar in vivo data were recently published in mice (47) using a reporter gene, showing that kidney re-entrainment to an 8-h advance takes between 4 and 7 days to complete. Interestingly, the van der Vinne et al. study also reported a reduction in amplitude in renal clock gene rhythms during the phase advance. Urine output rhythms were similarly suppressed in ECD rats in our study. Taken together, these data suggest that peripheral clock re-entrainment may result in the transient dampening of rhythms during misalignment and that repeated shifting results in a persistent disruption in local clock function, which could underlie organ and system dysfunction and decreased resilience to pathology in shift workers and animal models of shift work.
To further assess the influence of environmental circadian disruption on the circadian regulation of renal output, we examined the excretion rate of ET-1. Independent from food intake, ET-1 has been found to facilitate diurnal renal excretion by modulating clock genes responsible for the daily handling of Na+ (50). The phase remained steady for control rats, aside from a slow 3-h delay in peak ET-1 levels as salt loading progressed. This observation is consistent with previous findings showing that high salt promotes a shift in the phase of the kidney circadian clock (13). The loss of rhythms in both groups made further phase analysis difficult. Although our study revealed a reduction of ET-1 diurnal rhythms for both treatment groups toward the middle and end of the experiment, there was a trend toward an ECD-specific loss of relative amplitude. Loss of ET-1 rhythms on an individual rat level was more common in ECD rats than control rats, further validating the disruption of circadian timing by ECD observed for urine volume and the Per2 clock gene (47). Given that disruption of the molecular clock leads to conditions associated with elevated cardiovascular risks (36, 51), we speculate that intermittent disruption of diurnal renal function may contribute to renal injury.
In our longitudinal study, we observed a steady increase of both urine and ET-1 excretion throughout the 44-day study in both groups, suggestive of an increase in blood volume and natriuresis to maintain normal blood pressure, as expected in this salt-loaded hypertensive-stroke-prone rat model. The work of Mills and colleagues (52) in humans demonstrated that renal excretion of Na+ not only follows a rhythmic pattern but that the pattern is independent of Na+ intake. In our study, total urine production was consistent among both exposure groups, allowing us to discount the possibility that the amplitude reduction in the ECD group was caused by salt consumption.
Biweekly urine samples were examined to assess renal injury in response to environmental circadian disruption. As expected in this model, renal damage progressed in both groups. Furthermore, a more rapid onset or higher degree of glomerular and kidney tubule injury markers was observed in the urine of animals exposed to weekly light schedule advances. Urinary nephrin excretion was used to determine early glomerular injury. Serving as a selective filter, the kidney’s glomerular filtration barrier restricts the passage of plasma macromolecules based on their size, shape, and charge (53–55). Nephrin, a scaffolding protein, is a key component of the slit diaphragm, playing an essential role in the preservation and selectivity of the filtration barrier (56–59). The presence of nephrin in urine precedes albuminuria and serves as a marker for renal damage (56). Our study revealed an increase of nephrin excretion in both treatment groups throughout the study, suggestive of glomerular injury (60). Furthermore, we found an accelerated and more profound increase in urinary nephrin in animals exposed to ECD versus controls housed in a consistent light schedule. The subsequent decrease in nephrin seen at the end of our study is thought to be a consequence of filtration slit integrity, promoting the development of further kidney injury and other associated diseases.
In addition to urinary evidence of glomerular injury, we found urinary evidence of increased renal tubule injury in ECD rats. Urinary KIM-1 can reflect severity of renal damage (59). High levels of urinary KIM-1 have been used as an early indicator for proximal tubule injury in both humans and rodents (61–64). In this study, we observed a significant increase of urinary KIM-1 excretion in animals exposed to an advanced LD schedule, late in the study. We speculate that the transient increase in KIM-1 seen in controls is an acute response to salt loading and/or hypertension to which control rats can adapt but ECD rats cannot. To the best of our knowledge, this is the first study to evaluate renal injury markers in response to ECD.
Contrary to the urinary indication of renal injury, we were unable to discern a statistical difference in structural damage to kidney tubules and glomeruli; therefore, we were unable to conclude that ECD directly influences kidney injury. Considering that SHR-SP are genetically prone to malignant hypertension and extensive renal damage (65), the failure to discern a difference between the groups in structural damage at the termination point likely reflects a ceiling effect. The early onset difference between the ECD and control groups, most notably for nephrin excretion, suggests that kidney injury was accelerated in ECD rats, but the injury in the controls eventually caught up. We speculate that an earlier termination of the study would have captured a difference in structural damage as reflected by urinary analytes.
Several laboratories have demonstrated the influence of the circadian clock on sodium handling and the development of hypertension. Global genetic deletion of core circadian clock genes in rodents is known to lead to loss of diurnal control of both Na+ and water handling causing distinct blood pressure phenotypes (29, 66, 67). However, constitutive activation of the positive leg of the circadian clock via deletion of the suppressors Cry 1 and Cry 2 in mice was shown to result in salt-sensitive hypertension (36, 37). It is worth noting that most studies examining the influence of circadian rhythms on renal function have used a genetic knockdown or knockout approach to disturb the circadian clock. The recent development of a rat Bmal 1 knockout demonstrated that blood pressure and urine excretion rhythms, peaking during the active period, persisted despite the loss of a key genetic regulator of rhythms (42). Our study examined renal complications in a progressive salt-loaded rat model with an environmentally disrupted but genetically intact clock. This is presumably reflective of the state of the kidney clock in shift workers.
This study had some limitations. Although many of our measurements had adequate power, some key measures were underpowered due to a sample size limitation. The use of only male animals further limits our findings. As estrogen metabolites have been found to have a protective effect on renal damage (68, 69), another study is needed with female animals to determine the influence of chronic circadian disruption on the protective mechanisms of estrogen. Although urinary evidence suggests that ECD accelerates renal injury, failure to capture a histological difference in glomerular and tubular damage limits our findings. Earlier termination of the study would have allowed for the potential capture of accelerated renal damage and a more decisive conclusion. Furthermore, renal rhythms and injury analysis were limited to the samples collected using a discrete time series during selected weeks across the study. More frequent captures throughout the study would have permitted a greater insight into the effects of ECD on the renal system. Furthermore, an analysis of kidney clock gene rhythms in these contexts would have been difficult but could have provided more insight into the status of the molecular clock in the kidney. Also, key aspects of the rats’ physiology and health are still of interest: what is happening with blood pressure rhythms and hypertension development as kidney rhythms are shifting and suppressed? Do these conditions accelerate stroke onset?
Perspectives
The prevalence of cardiovascular complications such as stroke, cardiovascular disease, and heart failure is greater among hypertensive individuals with a nondipping blood pressure phenotype than individuals with robust blood pressure rhythms. Our study demonstrates the multiple levels at which shifting of the renal clock can influence rhythms and result in the acceleration of organ damage markers and the exacerbation of disease. Our results further confirm accounts in the literature that illustrate the kidney’s role in the development of hypertension and associated cardiovascular complications. Repeated long-term disruption of environmental cues and the subsequent resynchronization of internal processes have been shown in our study to be consistent with events of a shift work lifestyle and are believed to contribute to the associated morbidities. We conclude from this study that the kidney may be a target organ for environmental circadian disruption, subsequently influencing the development of hypertension and associated complications, including renal damage.
GRANTS
This work was supported by National Institutes of Health Grants R25GM058268 (to Morehouse School of Medicine), Grants SC1GM112567, R21NS108197, and R35GM136661 (to A. J. Davidson), Grant SC2GM125493 (to O. Castanon-Cervantes), R01DK109570 (to M. L. Gumz), and F32DK121424 (to G. R. Crislip). Further support was provided by American Heart Association Grants P0133392 (to M. L. Gumz) and 19POST34450134 (to G. R. Crislip) and the Gatorade Trust through the University of Florida Department of Medicine.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.M.H., A.R., O.C-C., M.L.G., and A.J.D. conceived and designed research; A.M.H., G.R.C., A.S., I.E., and A.R. performed experiments; A.M.H. and G.R.C. analyzed data; A.M.H., G.R.C., A.S., A.R., O.C-C., M.L.G., and A.J.D. interpreted results of experiments; A.M.H. and G.R.C. prepared figures; A.M.H. drafted manuscript; A.M.H., G.R.C., A.S., I.E., A.R., O.C-C., M.L.G., and A.J.D. edited and revised manuscript; A.M.H., G.R.C., A.S., I.E., A.R., O.C-C., M.L.G., and A.J.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the Morehouse School of Medicine’s CLAR staff for their technical assistance and Xueying Zhao Lab for the gracious donation of metabolic cages.
REFERENCES
- 1.Kecklund G, Axelsson J. Health consequences of shift work and insufficient sleep. BMJ 355: i5210, 2016. doi: 10.1136/bmj.i5210. [DOI] [PubMed] [Google Scholar]
- 2.Karlsson B, Knutsson A, Lindahl B. Is there an association between shift work and having a metabolic syndrome? Results from a population based study of 27,485 people. Occup Environ Med 58: 747–752, 2001. doi: 10.1136/oem.58.11.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morikawa Y, Nakagawa H, Miura K, Soyama Y, Ishizaki M, Kido T, Naruse Y, Suwazono Y, Nogawa K. Shift work and the risk of diabetes mellitus among Japanese male factory workers. Scand J Work Environ Health 31: 179–183, 2005. doi: 10.5271/sjweh.867. [DOI] [PubMed] [Google Scholar]
- 4.Uhm JY, Kim H-R, Kang GH, Choi YG, Park TH, Kim SY, Chang SS, Choo WO. The association between shift work and chronic kidney disease in manual labor workers using data from the Korea National Health and Nutrition Examination Survey (KNHANES 2011-2014). Ann Occup Environ Med 30: 69–69, 2018. doi: 10.1186/s40557-018-0279-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drake CL, Roehrs T, Richardson G, Walsh JK, Roth T. Shift work sleep disorder: prevalence and consequences beyond that of symptomatic day workers. Sleep 27: 1453–1462, 2004. doi: 10.1093/sleep/27.8.1453. [DOI] [PubMed] [Google Scholar]
- 6.Castanon-Cervantes O, Wu M, Ehlen JC, Paul K, Gamble KL, Johnson RL, Besing RC, Menaker M, Gewirtz AT, Davidson AJ. Dysregulation of inflammatory responses by chronic circadian disruption. J Immunol 185: 5796–5805, 2010. doi: 10.4049/jimmunol.1001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costa G, Haus E, Stevens R. Shift work and cancer – considerations on rationale, mechanisms, and epidemiology. Scand J Work Environ Health 36: 163–179, 2010. doi: 10.5271/sjweh.2899. [DOI] [PubMed] [Google Scholar]
- 8.Golombek DA, Casiraghi LP, Agostino PV, Paladino N, Duhart JM, Plano SA, Chiesa JJ. The times they’re a-changing: Effects of circadian desynchronization on physiology and disease. J Physiol 107: 310–322, 2013. doi: 10.1016/j.jphysparis.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Karatsoreos IN, Bhagat S, Bloss EB, Morrison JH, McEwen BS. Disruption of circadian clocks has ramifications for metabolism, brain, and behavior. Proc Natl Acad Sci USA 108: 1657–1662, 2011. doi: 10.1073/pnas.1018375108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Albrecht U. The mammalian circadian clock: a network of gene expression. Front Biosci 9: 48–55, 2004. doi: 10.2741/1196. [DOI] [PubMed] [Google Scholar]
- 11.Albrecht U, Eichele G. The mammalian circadian clock. Curr Opin Genet Dev 13: 271–277, 2003. doi: 10.1016/S0959-437X(03)00055-8. [DOI] [PubMed] [Google Scholar]
- 12.Richards J, Gumz ML. Mechanism of the circadian clock in physiology. Am J Physiol Regul Integr Comp Physiol 304: R1053–R1064, 2013. doi: 10.1152/ajpregu.00066.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Speed JS, Hyndman KA, Roth K, Heimlich JB, Kasztan M, Fox BM, Johnston JG, Becker BK, Jin C, Gamble KL, Young ME, Pollock JS, Pollock DM. High dietary sodium causes dyssynchrony of the renal molecular clock in rats. Am J Physiol Renal Physiol 314: F89–F98, 2018. doi: 10.1152/ajprenal.00028.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morris CJ, Purvis TE, Mistretta J, Scheer FAJL. Effects of the internal circadian system and circadian misalignment on glucose tolerance in chronic shift workers. J Clin Endocrinol Metab 101: 1066–1074, 2016. doi: 10.1210/jc.2015-3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morris CJ, Yang JN, Scheer FAJL. The impact of the circadian timing system on cardiovascular and metabolic function. Prog Brain Res 199: 337–358, 2012. doi: 10.1016/B978-0-444-59427-3.00019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Potter GDM, Skene DJ, Arendt J, Cade JE, Grant PJ, Hardie LJ. Circadian rhythm and sleep disruption: Causes, metabolic consequences, and countermeasures. Endocr Rev 37: 584–608, 2016. doi: 10.1210/er.2016-1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sherwood A, Steffen PR, Blumenthal JA, Kuhn C, Hinderliter AL. Nighttime blood pressure dipping: the role of the sympathetic nervous system. Am J Hypertens 15: 111–118, 2002. doi: 10.1016/S0895-7061(01)02251-8. [DOI] [PubMed] [Google Scholar]
- 18.Cuspidi C, Tadic M, Sala C, Gherbesi E, Grassi G, Mancia G. Blood pressure non-dipping and obstructive sleep apnea syndrome: a meta-analysis. J Clin Med 8: 1367, 2019. doi: 10.3390/jcm8091367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Brien E, Sheridan J, O'Malley K. Dippers and non-dippers. Lancet 332: 397, 1988. doi: 10.1016/S0140-6736(88)92867-X. [DOI] [PubMed] [Google Scholar]
- 20.Kario K, Matsuo T, Kobayashi H, Imiya M, Matsuo M, Shimada K. Nocturnal fall of blood pressure and silent cerebrovascular damage in elderly hypertensive patients. Hypertension 27: 130–135, 1996. doi: 10.1161/01.HYP.27.1.130. [DOI] [PubMed] [Google Scholar]
- 21.Shimada K, Kawamoto A, Matsubayashi K, Nishinaga M, Kimura S, Ozawa T. Diurnal blood pressure variations and silent cerebrovascular damage in elderly patients with hypertension. J Hypertens 10: 875–878, 1992. [PubMed] [Google Scholar]
- 22.Verdecchia P, Schillaci G, Guerrieri M, Gatteschi C, Benemio G, Boldrini F, Porcellati C. Circadian blood pressure changes and left ventricular hypertrophy in essential hypertension. Circulation 81: 528–536, 1990. doi: 10.1161/01.CIR.81.2.528. [DOI] [PubMed] [Google Scholar]
- 23.Davidson MB, Hix JK, Vidt DG, Brotman DJ. Association of impaired diurnal blood pressure variation with a subsequent decline in glomerular filtration rate. Arch Intern Med 166: 846–852, 2006. doi: 10.1001/archinte.166.8.846. [DOI] [PubMed] [Google Scholar]
- 24.Lurbe E, Redon J, Kesani A, Pascual JM, Tacons J, Alvarez V, Batlle D. Increase in nocturnal blood pressure and progression to microalbuminuria in type 1 diabetes. N Engl J Med 347: 797–805, 2002. doi: 10.1056/NEJMoa013410. [DOI] [PubMed] [Google Scholar]
- 25.Timio M, Venanzi S, Lolli S, Lippi G, Verdura C, Monarca C, Guerrini E. Non-dipper” hypertensive patients and progressive renal insufficiency: a 3-year longitudinal study. Clin Nephrol 43: 382–387, 1995. [PubMed] [Google Scholar]
- 26.Nikolaeva S, Pradervand S, Centeno G, Zavadova V, Tokonami N, Maillard M, Bonny O, Firsov D. The circadian clock modulates renal sodium handling. J Am Soc Nephrol 23: 1019–1026, 2012. doi: 10.1681/ASN.2011080842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burnier M, Coltamai L, Maillard M, Bochud M. Renal sodium handling and nighttime blood pressure. Semin Nephrol 27: 565–571, 2007. doi: 10.1016/j.semnephrol.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 28.Gumz ML, Cheng K-Y, Lynch IJ, Stow LR, Greenlee MM, Cain BD, Wingo CS. Regulation of αENaC expression by the circadian clock protein Period 1 in mpkCCD(c14) cells. Biochim Biophys Acta 1799: 622–629, 2010. doi: 10.1016/j.bbagrm.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gumz ML, Stow LR, Lynch IJ, Greenlee MM, Rudin A, Cain BD, Weaver DR, Wingo CS. The circadian clock protein Period 1 regulates expression of the renal epithelial sodium channel in mice. J Clin Invest 119: 2423–2434, 2009. doi: 10.1172/JCI36908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richards J, Greenlee MM, Jeffers LA, Cheng K-Y, Guo L, Eaton DC, Gumz ML. Inhibition of αENaC expression and ENaC activity following blockade of the circadian clock-regulatory kinases CK1δ/ε. Am J PhysiolRenal Physiol 303: F918–F927, 2012. doi: 10.1152/ajprenal.00678.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abriel H, Loffing J, Rebhun JF, Pratt JH, Schild L, Horisberger JD, Rotin D, Staub O. Defective regulation of the epithelial Na+ channel by Nedd4 in Liddle's syndrome. J Clin Invest 103: 667–673, 1999. doi: 10.1172/JCI5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hummler E. Implication of ENaC in salt-sensitive hypertension. J Steroid Biochem Mol Biol 69: 385–390, 1999. doi: 10.1016/S0960-0760(99)00073-4. [DOI] [PubMed] [Google Scholar]
- 33.Hummler E, Horisberger JD. Genetic disorders of membrane transport. V. The epithelial sodium channel and its implication in human diseases. Am J Physiol 276: G567–G571, 1999. doi: 10.1152/ajpgi.1999.276.3.G567. [DOI] [PubMed] [Google Scholar]
- 34.Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, Gill JR, Ulick S, Milora RV, Findling JW. Liddle's syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell 79: 407–414, 1994. doi: 10.1016/0092-8674(94)90250-X. [DOI] [PubMed] [Google Scholar]
- 35.Snyder PM, Price MP, McDonald FJ, Adams CM, Volk KA, Zeiher BG, Stokes JB, Welsh MJ. Mechanism by which Liddle's syndrome mutations increase activity of a human epithelial Na+ channel. Cell 83: 969–978, 1995. doi: 10.1016/0092-8674(95)90212-0. [DOI] [PubMed] [Google Scholar]
- 36.Doi M, Takahashi Y, Komatsu R, Yamazaki F, Yamada H, Haraguchi S, Emoto N, Okuno Y, Tsujimoto G, Kanematsu A, Ogawa O, Todo T, Tsutsui K, van der Horst GTJ, Okamura H. Salt-sensitive hypertension in circadian clock-deficient Cry-null mice involves dysregulated adrenal Hsd3b6. Nat Med 16: 67–74, 2010. doi: 10.1038/nm.2061. [DOI] [PubMed] [Google Scholar]
- 37.Rudic RD, Fulton DJ. Pressed for time: the circadian clock and hypertension. J Appl Physiol 107: 1328–1338, 2009. doi: 10.1152/japplphysiol.00661.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bakris GL, Williams M, Dworkin L, Elliott WJ, Epstein M, Toto R, Tuttle K, Douglas J, Hsueh W, Sowers J. Preserving renal function in adults with hypertension and diabetes: a consensus approach. National Kidney Foundation Hypertension and Diabetes Executive Committees Working Group. Am J Kidney Dis 36: 646–661, 2000. doi: 10.1053/ajkd.2000.16225. [DOI] [PubMed] [Google Scholar]
- 39.Henning EC, Warach S, Spatz M. Hypertension-induced vascular remodeling contributes to reduced cerebral perfusion and the development of spontaneous stroke in aged SHRSP rats. J Cereb Blood Flow Metab 30: 827–836, 2010. doi: 10.1038/jcbfm.2009.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu G, Anafi RC, Hughes ME, Kornacker K, Hogenesch JB. MetaCycle: an integrated R package to evaluate periodicity in large scale data. Bioinformatics 32: 3351–3353, 2016. doi: 10.1093/bioinformatics/btw405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stubblefield JJ, Gao P, Kilaru G, Mukadam B, Terrien J, Green CB. Temporal control of metabolic amplitude by nocturnin. Cell Rep 22: 1225–1235, 2018. doi: 10.1016/j.celrep.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnston JG, Speed JS, Becker BK, Kasztan M, Soliman RH, Rhoads MK, Tao B, Jin C, Geurts AM, Hyndman KA, Pollock JS, Pollock DM. Diurnal control of blood pressure is uncoupled from sodium excretion. Hypertension 75: 1624–1634, 2020. doi: 10.1161/HYPERTENSIONAHA.119.13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Firsov D, Bonny O. Circadian regulation of renal function. Kidney Int 78: 640–645, 2010. doi: 10.1038/ki.2010.227. [DOI] [PubMed] [Google Scholar]
- 44.Herrera GM, Meredith AL. Diurnal variation in urodynamics of rat. PLoS One 5: e12298-e12298, 2010. doi: 10.1371/journal.pone.0012298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamazaki S, Numano R, Abe M, Hida A, Takahashi R-I, Ueda M, Block GD, Sakaki Y, Menaker M, Tei H. Resetting central and peripheral circadian oscillators in transgenic rats. Science 288: 682–685, 2000. doi: 10.1126/science.288.5466.682. [DOI] [PubMed] [Google Scholar]
- 46.Yoo S-H, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong H-K, Oh WJ, Yoo OJ, Menaker M, Takahashi JS. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci USA 101: 5339–5346, 2004. doi: 10.1073/pnas.0308709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van der Vinne V, Martin Burgos B, Harrington ME, Weaver DR. Deconstructing circadian disruption: assessing the contribution of reduced peripheral oscillator amplitude on obesity and glucose intolerance in mice. J Pineal Res 69: e12654, 2020. [32243642] doi: 10.1111/jpi.12654. [DOI] [PubMed] [Google Scholar]
- 48.Davidson AJ, Castanon-Cervantes O, Leise TL, Molyneux PC, Harrington ME. Visualizing jet lag in the mouse suprachiasmatic nucleus and peripheral circadian timing system. Eur J Neurosci 29: 171–180, 2009. doi: 10.1111/j.1460-9568.2008.06534.x. [DOI] [PubMed] [Google Scholar]
- 49.Johnston JG, Pollock DM. Circadian regulation of renal function. Free Radic Biol Med 119: 93–107, 2018. doi: 10.1016/j.freeradbiomed.2018.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bugaj V, Mironova E, Kohan DE, Stockand JD. Collecting duct-specific endothelin B receptor knockout increases ENaC activity. Am J Physiol Cell Physiol 302: C188–C194, 2012. doi: 10.1152/ajpcell.00301.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Solocinski K, Holzworth M, Wen X, Cheng KY, Lynch IJ, Cain BD, Wingo CS, Gumz ML. Desoxycorticosterone pivalate-salt treatment leads to non-dipping hypertension in Per1 knockout mice. Acta Physiol (Oxf) 220: 72–82, 2017. doi: 10.1111/apha.12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mills JN, Stanbury SW. Persistent 24-hour renal excretory rhythm on a 12-hour cycle of activity. J Physiol 117: 22–37, 1952. [PMC free article] [PubMed] [Google Scholar]
- 53.Brenner BM, Hostetter TH, Humes HD. Molecular basis of proteinuria of glomerular origin. N Engl J Med 298: 826–833, 1978. doi: 10.1056/NEJM197804132981507. [DOI] [PubMed] [Google Scholar]
- 54.Kanwar YS, Liu ZZ, Kashihara N, Wallner EI. Current status of the structural and functional basis of glomerular filtration and proteinuria. Semin Nephrol 11: 390–413, 1991. [PubMed] [Google Scholar]
- 55.Venkatachalam MA, Rennke HG. The structural and molecular basis of glomerular filtration. Circ Res 43: 337–347, 1978. doi: 10.1161/01.RES.43.3.337. [DOI] [PubMed] [Google Scholar]
- 56.Aaltonen P, Holthöfer H. The nephrin-based slit diaphragm: new insight into the signalling platform identifies targets for therapy. Nephrol Dial Transplant 22: 3408–3410, 2007. doi: 10.1093/ndt/gfm403. [DOI] [PubMed] [Google Scholar]
- 57.Kestilä M, Lenkkeri U, Männikkö M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan CE, Peltonen L, Holmberg C, Olsen A, Tryggvason K. Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol Cell 1: 575–582, 1998. doi: 10.1016/S1097-2765(00)80057-X. [DOI] [PubMed] [Google Scholar]
- 58.Ruotsalainen V, Ljungberg P, Wartiovaara J, Lenkkeri U, Kestilä M, Jalanko H, Holmberg C, Tryggvason K. Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc Natl Acad Sci USA 96: 7962–7967, 1999. doi: 10.1073/pnas.96.14.7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tryggvason K. Unraveling the mechanisms of glomerular ultrafiltration: nephrin, a key component of the slit diaphragm. J Am Soc Nephrol 10: 2440–2445, 1999. [DOI] [PubMed] [Google Scholar]
- 60.Chang JH, Paik SY, Mao L, Eisner W, Flannery PJ, Wang L, Tang Y, Mattocks N, Hadjadj S, Goujon JM, Ruiz P, Gurley SB, Spurney RF. Diabetic kidney disease in FVB/NJ Akita mice: temporal pattern of kidney injury and urinary nephrin excretion. PLoS One 7: e33942, 2012. doi: 10.1371/journal.pone.0033942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. Kidney injury molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int 62: 237–244, 2002. doi: 10.1046/j.1523-1755.2002.00433.x. [DOI] [PubMed] [Google Scholar]
- 62.Ichimura T, Asseldonk EJPV, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest 118: 1657–1668, 2008. doi: 10.1172/JCI34487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ren H, Zhou X, Dai D, Liu X, Wang L, Zhou Y, Luo X, Cai Q. Assessment of urinary kidney injury molecule-1 and interleukin-18 in the early post-burn period to predict acute kidney injury for various degrees of burn injury. BMC Nephrol 16: 142–142, 2015. doi: 10.1186/s12882-015-0140-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van Timmeren MM, van den Heuvel MC, Bailly V, Bakker SJ, van Goor H, Stegeman CA. Tubular kidney injury molecule-1 (KIM-1) in human renal disease. J Pathol 212: 209–217, 2007. doi: 10.1002/path.2175. [DOI] [PubMed] [Google Scholar]
- 65.Churchill PC, Churchill MC, Griffin KA, Picken M, Webb RC, Kurtz TW, Bidani AK. Increased genetic susceptibility to renal damage in the stroke-prone spontaneously hypertensive rat. Kidney Int 61: 1794–1800, 2002. doi: 10.1046/j.1523-1755.2002.00321.x. [DOI] [PubMed] [Google Scholar]
- 66.Nikolaeva S, Ansermet C, Centeno G, Pradervand S, Bize V, Mordasini D, Henry H, Koesters R, Maillard M, Bonny O, Tokonami N, Firsov D. Nephron-specific deletion of circadian clock gene Bmal1 alters the plasma and renal metabolome and impairs drug disposition. J Am Soc Nephrol 27: 2997–3004, 2016. doi: 10.1681/ASN.2015091055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tokonami N, Mordasini D, Pradervand S, Centeno G, Jouffe C, Maillard M, Bonny O, Gachon F, Gomez RA, Sequeira-Lopez MLS, Firsov D. Local renal circadian clocks control fluid-electrolyte homeostasis and BP. J Am Soc Nephrol 25: 1430–1439, 2014. doi: 10.1681/ASN.2013060641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Antus B, Hamar P, Kokeny G, Szollosi Z, Mucsi I, Nemes Z, Rosivall L. Estradiol is nephroprotective in the rat remnant kidney. Nephrol Dial Transplant 18: 54–61, 2003. doi: 10.1093/ndt/18.1.54. [DOI] [PubMed] [Google Scholar]
- 69.Gross M-L, Adamczak M, Rabe T, Harbi NA, Krtil J, Koch A, Hamar P, Amann K, Ritz E. Beneficial effects of estrogens on indices of renal damage in uninephrectomized SHRsp rats. J Am Soc Nephrol 15: 348–358, 2004. doi: 10.1097/01.ASN.0000105993.63023.D8. [DOI] [PubMed] [Google Scholar]