

Abstract
The brain requires a continuous supply of energy in the form of ATP, most of which is produced from glucose by oxidative phosphorylation in mitochondria, complemented by aerobic glycolysis in the cytoplasm. When glucose levels are limited, ketone bodies generated in the liver and lactate derived from exercising skeletal muscle can also become important energy substrates for the brain. In neurodegenerative disorders of ageing, brain glucose metabolism deteriorates in a progressive, region-specific and disease-specific manner — a problem that is best characterized in Alzheimer disease, where it begins presymptomatically. This Review discusses the status and prospects of therapeutic strategies for countering neurodegenerative disorders of ageing by improving, preserving or rescuing brain energetics. The approaches described include restoring oxidative phosphorylation and glycolysis, increasing insulin sensitivity, correcting mitochondrial dysfunction, ketone-based interventions, acting via hormones that modulate cerebral energetics, RNA therapeutics and complementary multimodal lifestyle changes.
Increased longevity over the past 50 years has contributed to a rising prevalence of neurodegenerative disorders of ageing (NDAs), particularly Alzheimer disease (AD) and Parkinson disease (PD). These disorders are a major socio-economic and medical challenge with little prospect of a solution so far. Some drugs provide a degree of symptomatic relief, but disease-modifying treatments for NDAs remain elusive despite concerted attempts to counter the pathological processes of neurotoxic protein accumulation, neuroinflammation, axonal or synaptic dysfunction and neuronal death1,2.
Neuroinflammation.
An inflammatory response or state in the brain that involves functional, morphological and energetic shifts in microglia and ‘reactive’ astrocytes, as well as macrophages that migrate into the brain from the periphery. It is a characteristic of neurodegenerative disorders and brain response to infectious agents or injury.
Since the concept was first reported 40 years ago, evidence has been accumulating that impaired brain energetics is involved in the cause and progression of NDAs, especially AD3–7. Brain energy metabolism declines subtly during ageing and this decline is frequently present before diagnosis of NDAs; it both drives and is driven by functional impairment and neurodegeneration in a destructive cycle3,6.
Accordingly, a broad range of ‘brain energy rescue’ strategies have recently been explored and are the focus of this Review. These strategies aim to impede the onset and progress of NDAs by improving, preserving and/or restoring brain energy status. We first summarize how fuel is supplied and used across various cell types in the brain, including central, peripheral and endocrine mechanisms that modulate brain energy homeostasis as well as cognitive and neuronal function. The core features of disrupted brain energetics in the five main NDAs — AD, PD, Huntington disease (HD), frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) — are then outlined. This forms the basis for the brain energy rescue strategies reported in preclinical and clinical studies, including promoting mitochondrial function, alternative fuel sources such as ketone bodies (ketones), hormonal interventions to increase insulin sensitivity and brain glucose metabolism and complementary lifestyle approaches. Finally, we highlight genetic and other emerging approaches to enhance and restore brain energetics and we consider the challenges of translating promising preclinical results towards the dual goals of symptom relief and disease modification in NDAs.
Brain energetics
Despite representing slightly more than 2% of adult body weight, the human brain accounts for 20% of the body’s total energy requirement8. The brain’s main competitors for energy are the liver, kidneys and heart, which have as high or higher rates of energy consumption per gram, but their overall energy consumption is lower that of the brain. The immune system also consumes considerable energy, especially when activated in NDAs9. Brain energy metabolism is influenced by the endocrine modulation of appetite and whole-body energetics — processes that are compromised during healthy ageing and in NDAs10 (Supplementary Table 1). Food intake, glucose-sensing mechanisms and energy homeostasis are themselves regulated by a complex set of neural networks that in turn modulate autonomic function, appetite, reward and executive functioning. These aspects are beyond the scope of this Review, and have been reviewed elsewhere11,12.
Ketone bodies.
(Ketones). β-Hydroxybutyrate and acetoacetate. Produced endogenously by fatty acid β-oxidation during caloric or severe carbohydrate restriction, and from medium-chain fatty acids. Exogenous ketones are mostly salts or esters of β-hydroxybutyrate. Acetone is a breakdown product of acetoacetate that is measurable in plasma and on breath.
ATP
ATP is the main currency of brain energy metabolism. Most ATP is used by Na+/K+-ATPase and Ca2+-ATPase, the cell membrane pumps that reset ion gradients during neuronal signalling8,13,14. Excitatory (glutamatergic) neurons consume 80–85% of the brain’s ATP, with inhibitory neurons and other cells accounting for the remainder15–17. Although non-signalling pathways and cellular processes, including axonal transport, maintenance of cytoskeletal architecture, proton leakage, microglial motility, DNA repair, RNA translation and phospholipid remodelling, consume less energy than neurotransmission, the energetic requirements of these processes remain hard to define13,16. ATP is also a neurotransmitter released from neurons, astrocytes and microglia18.
Glucose
Glucose metabolism fuels 95% or more of ATP production in the brain8. Within the brain, glucose uptake is orchestrated across several cell types collectively known as the neurovascular unit: brain capillary endothelial cells, pericytes, astrocytes, oligodendrocytes, microglia and neurons — the final beneficiary of glucose uptake19,20 (FIG. 1). The normally tight spatial and temporal association of local blood flow, oxygen consumption and glucose consumption in the brain is termed ‘neurovascular coupling’, and is the basis for functional magnetic resonance imaging20.
Fig. 1 |. Energy supply and use by neurons and other brain cells.

a | Organization of the neurovascular unit, which functions to supply glucose (Glc) to neurons. b | Astrocytes produce ATP mainly via aerobic glycolysis (glucose to pyruvate (Pyr)), yet also exploit oxidative phosphorylation in mitochondria (ovals) to generate ATP using the tricarboxylic acid (TCA) cycle. Astrocytes supply glucose to neurons and oligodendrocytes from capillaries and endogenous stores of glycogen, a reversible transformation. Astrocytes take up glutamate (Glu) released from synapses and convert it into glutamine (Gln), which is sent to neurons: this metabolically costly operation relieves neurons of an energetic burden. Some glutamate and glutamine contributes carbon to the TCA cycle via the intermediate α-ketoglutarate (α-KG). Astrocytic generation of ketones from acetyl coenzyme A (acetyl-CoA) and uptake of lactate (Lac) and medium-chain fatty acids is not shown for clarity. c | Oligodendrocytes insulate axons with myelin and deliver lactate to axons, which is transformed into pyruvate and then ATP by mitochondria. Axons promote their own energy supply by releasing glutamate to stimulate N-methyl-d-aspartate receptors (NMDARs) on oligodendrocytes; this promotes membrane insertion of GLUT1 and increased oligodendrocyte uptake of glucose delivered as lactate to axons. d | Microglia support neurons by clearing pathogens, waste and toxic proteins but do not provide them with energy. They generate ATP mainly from glucose but also from free fatty acids and glutamine. Fatty acid and fructose (prominent in ‘modern diets’) uptake by microglia is linked to neuroinflammation in neurodegenerative disorders of ageing. e | Neurons use transporters to acquire glucose and lactate from astrocytes or the vasculature. Food, adipose tissue and gut microbiota yield short-chain fatty acids (SCFAs) and triglycerides (TGs), which are transformed by the liver into the ketones d-β-hydroxybutyrate (BHB) and acetoacetate (AcAc), which are taken up by neurons. Glucose enters the TCA cycle via pyruvate and acetyl coenzyme A to yield ATP or the pentose phosphate pathway to provide (via glucose 6-phosphate (G6P)) the antioxidant glutathione (GSH) and nucleosides. Neurons also generate some ATP by aerobic glycolysis. Microtubule transport of mitochondria and other cargo is driven by ATP, partially generated by ‘onboard’ glycolytic enzymes. See the main text and Supplementary Fig. 1 for details. AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; EAAT, excitatory amino acid transporter; FABP, fatty acid-binding protein; FFA, free fatty acid; GLUT, glucose transporter; HK, hexokinase; LDH, lactate dehydrogenase; LPL, lipoprotein lipase; MCT, monocarboxylate transporter; mGluR, metabotropic glutamate receptor; Rib5P, ribonucleoside 5-phosphate; SLC1A5, solute carrier family 1 member 5; SLC38A1, solute carrier family 38 member 1; SNAT, sodium-coupled neutral amino acid transporter.
Brain uptake of glucose from the circulation is driven by the energy demand of activated neurons not by the level of circulating glucose. Indeed, under normal conditions, the capacity to transport glucose into the brain exceeds the brain’s energy requirement by twofold to threefold15. Simply put, glucose is actively ‘pulled’ into an area of the brain in response to increased local neuronal activity. Glucose transport is achieved by the coordinated activity of glucose transporters on the capillary endothelium (GLUT1) and plasma membrane of astrocytes (GLUT1, GLUT2, and GLUT7), oligodendrocytes (GLUT1) and neurons (GLUT3 and GLUT4) in the cortex, hippocampus and cerebellum10,17,21. Only GLUT4 is mobilized as a direct response to sustained synaptic activity; its membrane insertion is stimulated by the metabolic sensor, AMP-activated protein kinase (AMPK)17. Membrane translocation of GLUT4 is insulin dependent in muscle and adipose tissue and probably also in neurons, so insulin resistance, as occurs in NDAs, is characterized by reduced neuronal glucose uptake17,22.
To reach neurons from capillaries, blood glucose either diffuses directly through the extracellular space or is channelled through astrocytes via their end-feet, which surround the capillary walls. It is taken up by astrocytic GLUT1 and exits through GLUT1 on perisynaptic processes adjacent to neurons and oligodendrocytes (FIG. 1). Some glucose that enters astrocytes is metabolized to ATP and some is converted to lactate, which can act both as a neurotransmitter (discussed later) and as an alternative energy source18.
Energy use by brain cells
The ATP required by neurons is predominantly generated within mitochondria by oxidative phosphorylation of glucose via the tricarboxylic acid cycle14 (TCA cycle; also known as the citric acid or Krebs cycle; BOX 1). Additional ATP is generated by aerobic glycolysis in the cytoplasm, which is required to support the high energy demands of synaptic transmission17. Glutamate is the neurotransmitter in most excitatory neurons and is recycled by astrocytes and delivered back to neurons as glutamine for reconversion into glutamate or (to a lesser degree) for use in energy generation by the TCA cycle13,23. Compared with astrocytes, neurons have more unphosphorylated (active) pyruvate dehydrogenase, more rapid TCA cycle activity and favour oxidative phosphorylation over aerobic glycolysis8. In contrast, the energy requirements of astrocytes are predominantly met by aerobic glycolysis, so some microdomains in astrocytes contain relatively few mitochondria23.
Box 1 |. Generating ATP in the brain.
Glucose entering brain cells is phosphorylated to glucose 6-phosphate and then enters one of two pathways14: generation of ATP via aerobic glycolysis and the mitochondrial tricarboxylic acid (TCA) cycle, or the pentose phosphate pathway, which generates riboses, nucleic acids and NADPH for antioxidant defence and anabolic reactions (FIGS 1,3). The final step in aerobic glycolysis is generation of pyruvate, which is converted either into acetyl coenzyme A (acetyl-CoA) via pyruvate dehydrogenase for entry into the TCA cycle or into lactate via lactate dehydrogenase (FIGS 1,3). The TCA cycle pathway predominates in neurons and the lactate pathway predominates in astrocytes, but this is not an absolute distinction. The TCA cycle occurs in the mitochondrial matrix, with oxidative phosphorylation occurring in the inner mitochondrial membrane. Several TCA cycle steps generate NADH and flavin adenine dinucleotide hydride (FADH2), which are oxidized and donate high-energy electrons to the mitochondrial electron transport chain, which in turn drives conversion of ADP into ATP by oxidative phosphorylation. More than 90% of brain ATP is generated in mitochondria by oxidative phosphorylation. Each molecule of glucose consumed during oxidative phosphorylation generates about 33 ATP molecules272, compared with 2 molecules of ATP produced by aerobic glycolysis. However, oxidative phosphorylation is slower than aerobic glycolysis and occurs at the price of generating reactive oxygen species60.
The TCA cycle not only feeds the mitochondrial electron transport chain but also provides carbon for synthesis of glutamate and acetylcholine, a process called ‘cataplerosis’. Carbon exiting the TCA cycle needs to be replaced to maintain TCA cycle activity, a process called ‘anaplerosis’. Via pyruvate, glucose is an important contributor to anaplerosis, such that when brain glucose uptake is impaired, not only ATP production but also anaplerosis is adversely affected. Pyruvate carboxylase generates oxaloacetate, mostly in astrocytes but also in oligodendrocytes27. Certain branched-chain amino acids and odd-chain fatty acids (such as heptanoate) are anaplerotic273. Ketones can replace glucose as a source of acetyl-CoA, but they are not anaplerotic.
The astrocyte–neuron lactate shuttle hypothesis postulates that activated glutamatergic neurons stimulate astrocytes to increase their supply of lactate to neurons8. Astrocytes release lactate through the low-affinity perisynaptic monocarboxylate transporter 1 (MCT1) and MCT4 (REF.18). Lactate is taken up by high-affinity neuronal MCT2 and transformed into ATP in neurons by oxidative phosphorylation (FIG. 1). ATP produced by metabolism of glucose or glycogen to lactate does not require oxygen, so this route reduces net brain oxygen consumption in cells or organelles with a high level of aerobic glycolysis, such as hippocampal synapses15. Lactate derived from glycogen in astrocytes can stimulate neuronal plasticity and learning, but this effect may occur only at relatively high levels of lactate (2–5 mM)8. Moreover, some types of neurons, including inhibitory GABAergic interneurons, do not necessarily depend on lactate8. Hence, the functional importance of the astrocyte–neuron lactate shuttle in vivo requires additional study15,274,275.
Lactate may act as a paracrine regulator through the lactate receptor, hydroxycarboxylic acid receptor 1 (HCAR1; also known as GPR81). During intense exercise, plasma lactate levels increase and lactate enters the brain through MCTs at the blood–brain barrier (FIG. 3), activating HCAR1 and promoting angiogenesis in the hippocampus and neocortex253,254. Exogenously administered lactate enhances hippocampal synaptic plasticity, neurogenesis and memory formation in rodents8.
Microglia.
Resident brain macrophages of mesodermal origin that clear neurotoxic proteins and protect neurons from damaging exogenous molecules, toxins, infectious agents or pathogens. Excess and persistent microglial activation is associated with neuroinflammation, neuronal energetic deterioration and progression of neurodegenerative diseases of ageing.
Neuronal activation transiently triggers aerobic glycolysis in astrocytes, thereby generating lactate. The astrocyte–neuron lactate shuttle hypothesis proposes that neuronal release of glutamate during neural transmission stimulates glucose uptake, glycogen catabolism, aerobic glycolysis and lactate production in neighbouring astrocytes8,24. The lactate produced by astrocytes is posited to support neuroplasticity, although its precise contribution and the conditions of lactate exploitation remain unclear8,10,15,23 (BOX 2).
Box 2 |. Measuring brain energy metabolism.
In vitro models
To measure high-energy phosphates in the brain, fresh mitochondria are fixed and visualized by electron microscopy, and adenine nucleotides are measured by high-performance liquid chromatography. Frozen mitochondria can be used to evaluate antioxidants, oxidative damage and the activity of respiratory chain complexes. Frozen brain homogenates are used to determine the activity of glycolytic, tricarboxylic acid cycle and electron transport chain complexes, and markers of mitochondrial biogenesis, dynamics and autophagy.
Regulatory mechanisms of energy homeostasis can be studied in single neurons and glia using nanosensors based on fluorescence resonance energy transfer, which detect changes in intracellular concentrations of cyclic AMP, protein kinase A, glucose and lactate in response to activation of G-protein-coupled receptors276. Intact brain cell respiration can be studied using an extracellular flux analyser to establish rates of oxygen consumption, glycolysis, proton leakage, mitochondrial reserve and other bioenergetic parameters129. The use of reprogrammed human cells and brain organoids for advancing bioenergetic drug discovery is considered in Supplementary Box 1.
Fluorescence lifetime imaging microscopy of cells or brain slices measures mitochondrial NADH production in real time and reveals a role for astrocytes in the glycolytic deficits of a mouse model of Huntington disease expressing mutant huntingtin65. This technique identifies defective glycolysis as leading to mitochondrial dysfunction in Alzheimer disease (AD) neurons and supports the potential of pyruvate to bypass impaired glycolysis and maintain mitochondrial respiration147. The energetics of neuronal network activity can also be studied in cortical slices34. Gamma oscillations have high energy expenditure34, in which pyruvate and d-β-hydroxybutyrate can partially replace glucose277.
Brain imaging in vivo
In living organisms, brain energy metabolism is usually evaluated by positron emission tomography (PET) in which the glucose analogue deoxyglucose is labelled with 18F to make [18F]fluorodeoxyglucose (FDG). To quantify brain uptake of FDG (or ketones or oxygen) by PET — that is, cerebral metabolic rate (micromoles per 100 g per minute) — blood samples to measure the tracer must be obtained from as near to the brain as possible. Cerebral metabolic rates obtained by PET reflect values obtained by arteriovenous difference across the brain3, but PET provides a visual image of both global and regional brain energy metabolism. A new PET tracer of mitochondrial complex I function, 18F-labelled 2-tert-butyl-4-chloro-5–2H-pyridazin-3-one278, has been proposed as a marker of mitochondrion-specific energy failure arising before the onset of impaired glycolysis in AD, and could be used to validate new therapeutics aiming to correct mitochondrial function.
In healthy ageing individuals, the brain glucose deficit (energy gap; FIG. 2) is about 8% and occurs mostly in the frontal cortex, whereas in AD the parietal and temporal lobes are more adversely affected270. A glucose-specific brain energy deficit is also present in young adults with insulin resistance279. Indeed, brain FDG uptake might be a better marker of declining cognitive function in mild cognitive impairment and AD than the amyloid-β PET marker [18F]florbetapir280.
Metabolism of multiple energy substrates has been assessed in humans and animals by in vivo 13C magnetic resonance spectroscopy, including brain uptake of 13C-labelled glucose and ketones281 and medium-chain fatty acids153 and non-invasive assessment of mitochondrial redox status in the rat282.
Oligodendrocytes.
Cells that produce myelin to insulate the axon and increase the speed of action potential propagation. They energetically support and communicate with neurons and astrocytes.
Neurovascular coupling.
Coordinated response to brain activation involving local capillary dilation and a transitory surge in the flow of oxygenated, glucose-containing blood across the neurovascular unit, thereby replenishing ATP used in neurotransmission.
Oligodendrocytes obtain ATP primarily by aerobic glycolysis. They use lactate for their own energy needs and also supply neighbouring axons with lactate, a process modulated locally by glutamate release from neurons (FIG. 1). This metabolic support of neuronal function by oligodendrocytes is important for effective spatial and temporal information processing in neuronal networks25. Oligodendrocytes are responsible for myelination of axons, which speeds up action potential conduction. However, the insulating myelin sheath restricts access of axons to glucose and other metabolites that would otherwise diffuse across the extracellular space25. The intermittent pattern of axonal myelination in cortical grey matter helps maintain access to extracellular nutrients26. Therefore, the supply of glucose and lactate to myelinated axons by oligodendrocytes requires a highly specialized architecture of the myelin sheath, including a continuum of nanometre-wide cytosolic channels that flank compacted, mature myelin25 (FIG. 1). These channels connect the oligodendrocyte soma with the periaxonal space. Neurons reciprocally deliver N-acetylaspartate to the oligodendrocyte soma via these channels; this N-acetylaspartate (as part of the aspartate–oxaloaspartate–malate shuttle) stimulates the TCA cycle and mitochondrial ATP production and is used to generate lipids and myelin27.
Insulin resistance.
A state in which insulin is ineffective in stimulating glucose use by peripheral tissues and certain populations of neurons in the brain, due mainly to receptor-signalling desensitization. It is associated with glucose intolerance and type 2 diabetes, and increases the risk of neurodegenerative disorders, particularly Alzheimer disease.
Oxidative phosphorylation.
Process by which mitochondria generate ATP by conveying electrons through enzyme complexes (I to IV), thereby creating a proton gradient that powers phosphorylation of ADP to ATP by ATP synthase.
Tricarboxylic acid cycle.
(TCA cycle). Process by which acetyl coenzyme A is oxidized to form GTP, FADH2 and NADH. NADH and FADH2 feed electrons to the electron transport chain to produce ATP by oxidative phosphorylation. Several neurotransmitters (acetylcholine, glutamate and GABA) are produced by carbon leaving the TCA cycle.
Axons also transport mitochondria, RNA, proteins, vesicles and other cargo to presynaptic terminals. This transport is an ATP-dependent process regulated by calcium and involves motor proteins and microtubules. Retrograde transport of vesicles to the cell body for lysosomal degradation is ATP driven2. Fast-conducting axons release trace amounts of glutamate, which stimulates local N-methyl-d-aspartate (NMDA) receptors in oligodendrocytes, promoting surface expression of GLUT1 on axonal myelin sheaths and thereby increasing glucose uptake and the rate of aerobic glycolysis. This in turn increases local provision of lactate to axons for ATP generation21,28 (FIG. 1). In addition, the molecular motors driving fast axonal transport are equipped with glycolytic enzymes, allowing them to generate their own energy ‘onboard’29 (FIG. 1).
Unlike astrocytes and oligodendrocytes, microglia do not directly provide energy to neurons, but the high amounts of lactate released by activated microglia may well be retrieved by local neurons23. Microglia are predominantly fuelled by oxidative phosphorylation but are metabolically reprogrammed by neuroinflammation in NDAs to an aerobic glycolysis-predominant phenotype associated with upregulation of GLUT1 and GLUT4 (REFS1,2). In parallel with this energetic shift, microglia transition from a protective to a disease-driving role in NDAs. When brain glucose supply is suboptimal for a long time, the high energy demands of activated microglia further limit energy availability to neurons1,30.
Neuronal networks and energy use
The provision of energy substrates from astrocytes to synapses and from oligodendrocytes to axons is critical for communication both within and between brain networks25. Brain regions are connected by tracts of myelinated axons, which are adversely affected by NDAs. For example, corticocortical loops are disrupted in AD and FTD, corticostriatal pathways are disrupted in HD, corticospinal tracts are disrupted in ALS and nigrostriatal projections are disrupted in PD. These tracts consist mainly of long-range excitatory neurons. Inhibitory interneurons, such as fast-spiking interneurons, are mostly present within local networks, such as the CA3 region of the hippocampus and frontal cortex. Local interneuron dysfunction disrupts synchronization between remote neuronal networks and across brain regions31,32.
Gamma oscillations (30–100 Hz) are fast brain rhythms synchronizing the activity of excitatory principal neurons and neuronal networks31. Fast-spiking GABAergic interneurons generating gamma oscillations have a high density of mitochondria in their axons and a specialized kind of myelination that facilitates provision of energy by oligodendrocytes33. The high metabolic needs of fast-spiking interneurons are supported primarily by oxidative phosphorylation. Parvalbumin-positive, GABAergic interneurons are particularly sensitive to deficits in energy and oxygen supply13,34. The decreased ability of oligodendrocytes to provide lactate to axons probably aggravates the decline in fast-spiking interneuron activity observed in NDAs.
Rhythmically firing, highly branched, nigrostriatal dopaminergic neurons in the substantia nigra pars compacta are especially vulnerable to mitochondrial failure and oxidative stress, features characteristic of PD13,35 (Supplementary Table 1).
Neuroendocrine mechanisms
As demonstrated in diverse cellular and animal models, insulin has a globally positive influence on cerebral energy balance and function. It reinforces neuronal energy supply by increasing neuronal glucose uptake by GLUT4 in the hippocampus and cortex17,22 (Supplementary Table 2). Insulin and activation of insulin-like growth factor 1 (IGF1) receptors also promote synaptic plasticity and cognitive processes36. Nevertheless, normal insulin sensitivity is paramount; insulin resistance is a major risk factor for AD because it disrupts both the modulation of energy availability by insulin and insulin signalling pathways in the brain37,38.
Aerobic glycolysis.
Conversion of glucose into pyruvate by the Emden–Meyerhoff pathway. Pyruvate is either converted into acetyl coenzyme A and enters the TCA cycle or reduced to lactate by NADH, a pathway prominent in glia to produce ATP without oxygen. Aerobic glycolysis may also occur in neurons.
Other hormones, including ghrelin, incretins, leptin, amylin and adiponectin, modulate both appetite and energy homeostasis and influence numerous aspects of brain function that are compromised in NDAs. The neurobiology of these hormones and their synthetic agonists in relation to food intake and energy homeostasis, brain energy balance, mitochondrial function, cognition, motor function, neurogenesis, synaptic integrity and neuronal integrity in animal models of NDAs are documented in Supplementary Table 2, with their clinical effects reported in TABLES 1–3 and in the section entitled ‘Therapies based on brain energy rescue’.
Table 1 |.
Treatments that improve brain energetics and/or function in preclinical models of NDAs
| Study characteristics | Primary end points: results | Ref. |
|---|---|---|
| Mitochondrial function | ||
| AD mice (3xTg) receiving MitoQ (100 μM in drinking water) for 5 months | Mitochondrial function: ↓ cognitive decline, ↓oxidative stress, ↓ Aβ accumulation, ↓ astrogliosis, ↓ synaptic loss, ↓ caspase activation, ↓ neuropathology, ↑ mitochondrial function | 257 |
| AD mice (APP/PS1) receiving CP2 at 25 mg kg−1 per day for 14 months | Mitochondrial function:↓ complex I activity, ↑ AMPK level, ↑ mitochondrial bioenergetics | 128 |
| AD mice (APP/PS1) receiving 25–250 μM AP39 in neurons in culture; 100 nmol AP39 per kilogram body weight for 6 weeks | Brain energy status and mitochondrial function: ↑ brain ATP level, protected against mitochondrial DNA damage, ↓ ROS concentration, ↓ brain atrophy | 258 |
| AD mice (APP/PS1) receiving mdivi-1 at 10 or 40 mg kg−1 by gavage for 1 month | Mitochondrial dynamics: ↓ mitochondrial fragmentation, ↓ loss of mitochondrial membrane potential, ↓ ROS concentration, ↓ synaptic dysfunction,↑ ATP concentration, ↑ learning and memory, ↑ mitochondrial function | 136 |
| AD mice (APP/PS1) receiving nicotinamide riboside at 400 mg kg−1 per day for 10 weeks | Mitochondrial function and proteostasis: ↓ Aβ accumulation, ↑ cognitive function, ↑ oxidative phosphorylation activity | 259 |
| PD rats (hA53T-α-syn) receiving mdivi-1 at 20 mg kg−1 by intraperitoneal injection for 8 weeks | Mitochondrial dynamics: ↓ mitochondrial fragmentation, ↓ mitochondrial dysfunction, ↓ oxidative stress, ↓ neurodegeneration and α-syn aggregates, ↑ motor function | 135 |
| HD mice (HD R6/2 and YAC128) receiving DA1 peptides (1 mg kg−1 per day) via osmotic pump for 2–3 months | Mitochondrial dynamics and function: ↑ mitochondrial biogenesis and bioenergetics, ↓ inflammation and neuropathology | 260 |
| ALS mice (Sod1G93A) receiving MitoQ (500 μM in drinking water) for 30–40 days | Mitochondrial function: ↓ nitroxidative stress, ↓ neuropathology, ↑ mitochondrial function and lifespan | 261 |
| SCA1 mice (Sca1154Q/2Q) receiving MitoQ (500 μM in drinking water) for 16 weeks | Mitochondrial function: ↓ neuropathology, ↓ oxidative stress, ↓ DNA damage, ↓ neuronal loss, ↑ mitochondrial function | 262 |
| Seizure model (risk of AD). In vivo study: CD1 mice; 35% of energy from C10 in regular diet. In vitro study: astrocytes exposed to 200 mM C8 or C10 for 10 days | Seizures — in vivo study: ↓ seizures after C10 but not C8; no change in levels of glycolytic enzymes. In vitro study: C8 and C10: ↑ basal respiration and mitochondrial leak; ↑ ATP synthesis, ↑ antioxidant capacity caused by C10 but not C8 | 150 |
| Insulin sensitizers | ||
| AD mice (APP/PS1) receiving metformin at 200 mg kg−1 intraperitoneally for 14 days | Cognitive performance, neuropathology: ↑ cognitive performance (Morris water maze), ↓ hippocampal neuron loss, ↓ Aβ accumulation, ↓ neuroinflammation | 176 |
| HD mice (Hdh150 knock-in) receiving metformin in drinking water at 5 mg ml−1 for 3 weeks | Early network hyperactivation in visual cortex, behaviour: ↓ hyperactive neurons, ↑ normal network patterns, ↓ green fluorescent protein–huntingtin synthesis, ↓ anxiolytic behaviour | 177 |
| Ketogenic molecules | ||
| AD mice (APP/PS1) receiving BHB and pyruvate at 26 mg kg−1 per day for 5 weeks | Brain redox status: ↑ brain nicotinamide adenine dinucleotide phosphate (reduced) level; ↓ network hyperactivity (epileptiform discharges) | 146 |
| AD mice (3xTgAD) receiving 125 g ketone ester per kg in diet for 8 months | Brain TCA cycle activity, mitochondrial function: BHB level ↑ fivefold; 30–40% ↑ in level of brain TCA cycle and glycolytic intermediates; ↑ mitochondrial redox potential; ↓ level of oxidized lipids/proteins in hippocampus | 263 |
| AD mice (Sirt3+/−/AppPs1) receiving ketone ester added at 22% of dietary energy for 20 weeks | Neurodegeneration, neuronal network hyperexcitability: ↑ cortical SIRT3 expression, ↓ loss of GABAergic neurons, ↓ seizures and prevented death of Sirt3+/−/AppPs1 mice | 264 |
| PD mice receiving MPTP at 18 mg kg−1 4 times over 2 h and BHB at 40, 80 or 160 mg kg−1 per day for 7 days | Mitochondrial function: ↑ mitochondrial respiration and ATP at complex II, ↓ dopamine neurodegeneration and motor deficit | 265 |
| ALS mice (SOD1-G93A) receiving 10% of calories as C8 for 10 weeks (7–17 weeks old) | Physical symptoms: ↑ mitochondrial function, ↓ spinal cord motor neuron loss, ↑ mitochondrial O2 consumption, no change in survival | 266 |
| Nutrients and metabolites | ||
| AD mice (Tg2576) receiving nicotinamide riboside at 250 mg kg−1 per day for 3 months | Brain redox status, cognitive performance: ↑ cortical redox status; attenuated cognitive decline | 267 |
| AD mice (3xTgAD/Polb+/−) receiving nicotinamide riboside at 3 g l−1 (12 mM) in drinking water for 6 months | Brain redox status: normalized cortical NAD+/NADH; nicotinamide riboside ↑ cognitive function and restored hippocampal synaptic plasticity | 142 |
| AD mice (treated with streptozotocin) receiving N-acetylcysteine at 50 mg kg−1 for 9 days | Brain glucose uptake, cognitive performance: normalized glucose uptake in hippocampus after streptozotocin treatment; prevented spatial/non-spatial learning and memory impairment | 268 |
| ALS mice (SOD1G93A) receiving 35% of calories as triheptanoin for 5 weeks (35–70 days old) | Physical performance, TCA cycle activity, brain glucose uptake, cognitive performance: ↑ hindlimb grip strength by 2.8 weeks, ↑ time to loss of balance on rotarod, ↑ time before weight loss, ↑ TCA cycle | 269 |
Reports shown here exclude those involving the ketogenic diet and lifestyle interventions (see BOX 3); studies involving antioxidants are documented in other reviews60. Triheptanoin269 is a seven-carbon triglyceride. The APP/PS1 Alzheimer disease (AD) mice bear the APP Swedish mutation plus the PS1-L166P mutation. 3xTgAD mice express three mutant proteins (APP Swedish, PS1-M146L and tau-P301L). ↑, increase; ↓, decrease; 3xTg, triple transgenic (amyloid precursor protein, tau and presenilin-1); Aβ, amyloid-β; AMPK, AMP-activated protein kinase; ALS, amyotrophic lateral sclerosis; AP39, proprietary mitochondrial-targeted H2S donor; BHB, d-β-hydroxybutyrate; C8, octanoic acid; C10, decanonic acid; DA1, dynamin-related protein antagonist 1; hA53T, adenosine to threonine missense mutation at position 53 of human synuclein; HD, Huntington disease; mdivi-1, mitochondrial division inhibitor 1; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; NDAs, neurodegenerative disorders of ageing; PD, Parkinson disease; Pol, DNA polymerase; ROS, reactive oxygen species; SCA1, spinocerebellar ataxia type 1; α-Syn, α-synuclein; TCA, tricarboxylic acid; YAC, yeast artificial chromosome.
Table 3 |.
Trials reporting improved brain function and/or energetics in major risk conditions for NDAs
| Disorder | Study details | Results and comments | Ref. |
|---|---|---|---|
| T2D ± cognitive impairment | Open-label study (n = 205) of DPP4 inhibitor (sitagliptin) at 100 mg per day ± metformin ± insulin (n = 101) vs metformin ± insulin only (n = 104) for 6 months | ↑ MMSE score in DPP4 inhibitor arm; similar glycaemic control in both groups; ↓ insulin level in DPP4 inhibitor arm; RCT needed | 197 |
| MCI or mild AD (non-diabetic) | Placebo-controlled, parallel-group crossover study (n = 10 per group) of metformin at 2 g per day for 8 weeks (NCT01965756) | Metformin measurable in CSF; no change in CSF Aβ42 or phosphorylated tau levels; ↑ CBF in two brain regions at 8 weeks; trend to ↑ executive function, memory and attention. Metformin penetrates brain; underpowered for cognitive outcomes | 186 |
| MCI | RCT of high-carbohydrate (n = 11) or high-fat (n = 12) KD for 6 weeks (NCT00777010) | Feasibility study; cognitive outcomes (executive function, long-term memory), mood: on KD, ↑ memory (paired associate learning); no change in executive function or depression score. Metabolic improvement for KD: ↓ weight, waist circumference, fasting glucose level and fasting insulin level | 155 |
| MCI | RCT of C8C10 at 30 g per day (n = 19) or energy-matched non-ketogenic placebo (n = 20) for 6 months (NCT02551419) | Brain glucose and ketone status: ↑ brain ketone uptake twofold. ↑ executive function, episodic memory, language and processing speed. Several cognitive outcomes improved in direct proportion to ketone concentration and/or brain ketone uptake | 154 |
| SMC and MCI | Crossover RCT with 6-week washout in between 6 weeks of AHAD and 6 weeks of MMKD interventions, in patients with SMC (n = 11) or MCI (n = 9) (NCT2984540) | CSF AD markers, neuroimaging markers, peripheral metabolic status, cognition. For MMKD only: ↑ CSF Aβ42 level, ↓ CSF tau level, ↑ brain ketone levels and ↑ brain perfusion. In both groups: ↑ levels of metabolic markers and ↑ memory. Adherence ≥90% in both groups; MMKD feasible, acceptable and has prevention effect on AD CSF biomarkers | 156 |
↑, increase; ↓, decrease; Aβ42, 42 amino acid isoform of amyloid-β; AD, Alzheimer disease; ADAS-Cog, Alzheimer Disease Assessment Scale - Cognitive Subscale; AHAD, American Heart Association diet; C8C10, octanoic acid plus decanoic acid; CSF, cerebrospinal fluid; DPP4, dipeptidyl peptidase 4; KD, ketogenic diet; MCI, mild cognitive impartment; MMKD, modified Mediterranean ketogenic diet; MMSE, Mini Mental State Examination; NDAs, neurodegenerative disorders of ageing; RCT, randomized controlled trial; SMC, subjective memory impairment; T2D, type 2 diabetes.
Astrocyte–neuron lactate shuttle.
The hypothesis that lactate produced in astrocytes is delivered to neurons to support the energy requirements of neurotransmission.
Brain use of ketones and lactate
Glucose (and glycogen) reserves within the brain can supply its ATP needs for only a few minutes24,39. Much of the brain’s resilience in the face of energetic challenge therefore depends on opportunistic use of alternative fuels sourced from outside the brain. Ketones and lactate are the main alternative fuels to glucose and are delivered to the brain by monocarboxylate transporters on astrocytes and on the capillary endothelium (FIG. 1). The two principal ketones, acetoacetate and d-β-hydroxybutyrate (BHB), are the main alternative brain fuels to glucose in adults under conditions of dietary carbohydrate or energy restriction. In infants, however, ketones are not only an essential brain fuel but are also the main substrate for brain lipid synthesis40.
Fast axonal transport.
Rapid transport of vesicles, mitochondria and other cargo along axonal microtubules. Vesicles are equipped with molecular motors (kinesin and dynein) and glycolytic enzymes, permitting rapid, local ATP production by aerobic glycolysis.
Acetoacetate and BHB are both in equilibrium in the blood, but only acetoacetate is metabolized to acetyl coenzyme A (acetyl-CoA), which then enters the TCA cycle to generate ATP. After an overnight fast, plasma ketone concentrations are usually 0.1–0.2 mM and they supply 3–5% of brain energy requirements. However, during a 3–4-day fast, plasma ketone concentrations can reach 5–6 mM and they provide 50% or more of brain energy requirements3. Unlike the entry rate of glucose, which enters the brain in response to brain cell activity, the rate of entry of ketones to the brain is directly related to their plasma concentration3, which explains the glucose-sparing effect of increased ketone levels41. Unlike glucose, ketones do not undergo aerobic glycolysis and cannot be metabolized to lactate, so they contribute to ATP production only via oxidative phosphorylation.
The gut–brain axis
Gut microbiota are involved in bidirectional communication between the gastrointestinal tract and the brain, generating nutrients and modulating overall energy homeostasis42. Disruption of the gut microbiota (dysbiosis) is implicated in the pathogenesis of NDAs43. Dietary fibre is an important substrate for generation of short-chain fatty acids by the gut microbiota; dietary fibre also slows down glucose absorption, which increases insulin sensitivity. These effects of dietary fibre are partly mediated by short-chain fatty acids, which are ligands for G-protein-coupled hydroxycarboxylic acid receptors (such as HCAR2) in enterocytes42,44. Short-chain fatty acids produced by gut bacteria, particularly butyrate, are key fuels for intestinal cells. Propionate, butyrate and succinate (a precursor of propionate) generated by the microbiota also improve control of peripheral glucose metabolism, adiposity and body weight44. In preclinical studies, exogenous butyrate promoted the development of dendritic spines, long-term potentiation, myelination and memory formation45. However, butyrate levels produced endogenously are usually low, so butyrate would at best be expected to be a minor energy substrate for the brain.
The beneficial effect of intestinal gluconeogenesis on insulin sensitivity and systemic energy metabolism is mediated in part by glucose sensing in the portal vein. This information is relayed via portal sensory nerves to the brain, which suppresses appetite and reduces hepatic gluconeogenesis44,46. Short-chain fatty acids also modulate the immune system, stimulate the release of hormones such as glucagon-like peptide 1 (GLP1) from the gut (Supplementary Table 2) and inhibit histone deacetylases.
Impaired brain energetics
Impaired brain glucose metabolism compromises transmembrane ion transport, vesicle recycling and synaptic signalling17,31,34. Less effective maintenance of transmembrane ion gradients and transmitter release, especially in fast-spiking interneurons, leads to hyperexcitability, excitatory–inhibitory imbalance and functional impairment of cortical networks, which further compromises the brain’s energy efficiency. These changes are exacerbated by disrupted glutamatergic transmission and abnormal astrocyte and oligodendrocyte function13,34,47,48, as well as impaired autophagy, which, in turn, decreases nutrient recycling2. Furthermore, the neuroinflammation that is common to diverse classes of NDAs is energetically costly2,49.
Incretins.
Peptide hormones produced by the small intestine that stimulate pancreatic insulin secretion, regulate glucose metabolism and influence cognition. These include glucagon-like peptide 1 and glucose-dependent insulinotropic polypeptide.
The regional pattern of brain energetic disruption depends on the NDA and its pathophysiological phenotype (Supplementary Table 1). Indeed, brain glucose hypometabolism in NDAs has no single cause; reduction in neuronal glucose uptake, impairment of aerobic glycolysis and the TCA cycle, failure of axonal transport and the loss of glial energetic support to neurons are all implicated. Supplementary Box 1 outlines how induced pluripotent stem cells and organoids are illuminating the cellular substrates of energy dysregulation, and Supplementary Box 2 summarizes how disruption of the cerebral microvasculature exacerbates the disruption of brain energy supply in NDAs.
Alzheimer disease
AD is the most common NDA. It is associated with weight loss and poor appetite but also type 2 diabetes (T2D), all of which contribute to lower brain energy availability. AD is characterized by lower uptake of glucose, TCA activity, mitochondrial function and astrocyte and oligodendrocyte energetic support of neurons. In addition, microglial consumption of glucose due to neuroinflammation is elevated, siphoning energy away from neurons1,4,50–52 (see Supplementary Table 1).
Even before diagnosis of AD, a characteristic regional disruption of glucose metabolism is linked to neuropathology and reduced cerebral blood flow in the brain. Nevertheless, the brain in AD still has normal or near-normal oxygen, lactate and ketone metabolism3,52–54. Many positron emission tomography (PET) studies confirm that the entorhinal cortex and parietal lobes, including the precuneus, have a 10–12% deficit in glucose uptake in mild cognitive impairment (MCI), a deficit that becomes anatomically more widespread with the onset of AD and worsens during its progression (Supplementary Table 1). The regional pattern of the brain glucose deficit distinguishes AD from FTD, PD, Lewy body disease and other disorders associated with dementia3,4,6.
White matter atrophy in AD impairs neuronal network operation and axonal mitochondrial transport. Especially in women, white matter loss reflects reduced maintenance and synthesis of myelin (energy-intensive processes) and catabolism of myelin to provide energy in the face of glucose scarcity37,55,56. However, as with impaired glucose uptake in grey matter, white matter ketone uptake remains normal in AD57.
Parkinson disease
In idiopathic PD, weight loss and low body mass index are common despite increased visceral fat. A decline in glucose metabolism is seen in the striatum (caudate), the frontal cortex and several other cortical regions but not in the cerebellum: this pattern of hypometabolism correlates with specific patterns of motor and cognitive dysfunction and is predictive of disease progression4,58,59. Although PET cannot resolve the substantia nigra pars compacta (where dopaminergic cell bodies degenerate in PD), mitochondrial fragmentation and dysfunction, including decreased glycolysis and reduced complex I activity, is pronounced in this brain region50,60. Energetic deficits have been reproduced in PD-derived induced pluripotent stem cells overexpressing the gene encoding α-synuclein61. There is evidence that neuroinflammation further compromises neuronal fuel supply in PD1.
Huntington disease
Patients with HD characteristically lose weight, even when increasing their calorie intake. In presymptomatic HD, brain glucose hypometabolism is seen in the striatum, frontal cortex and temporal cortex, and is linked to impaired neurotransmission in corticostriatal tracts62. Glucose uptake, ATP generation by aerobic glycolysis, mitochondrial function and oxidative phosphorylation are all decreased in HD35,63,64. Astrocytes in the striatum may oxidize fatty acids as an alternative source of energy, but reactive oxygen species contribute to further tissue damage65. The cerebellum is less severely affected by impaired glucose metabolism than the basal ganglia, perhaps because it uses amino acids for gluconeogenesis65. HD neurons show disrupted glycolysis66 and impaired axonal transport of vesicles owing to the interference of mutant protein huntingtin with molecular motors35,67.
Monocarboxylate transporters.
Transporters in the cell membrane that facilitate unidirectional, proton-linked transport (uptake) of small monocarboxylic acids such as lactate and ketones.
ALS and FTD
FTD and ALS have overlapping genetic risk factors and clinical and pathophysiological features6,68–70. Both are characterized by increased energy expenditure, yet only in FTD is there a distinctive carbohydrate/sweetness preference and weight gain. In contrast, patients with ALS eventually lose weight due to insufficient nutrient and energy intake68,71 (Supplementary Table 1). Brain energetics also deteriorate differently in ALS and FTD. FTD is associated with declining glucose metabolism and cerebral blood flow, especially in the frontal lobes, striatum and thalamus, where mitochondrial function is disrupted with reduced signalling to the endoplasmic reticulum and aberrant mitophagy6,68,69,72. Conversely, ALS is associated with a regionally complex pattern of lower and higher brain glucose metabolism: of particular note are reductions in mitochondrial function and glycolysis in the cortex, spinal cord and motor neurons, and at neuromuscular junctions in muscle68,70,73. In the superoxidase dismutase 1 (SOD1) mouse model of ALS, the pentose phosphate pathway is also impaired71. Furthermore, a loss of mitochondrial energetics and impaired glycolysis in astrocytes is linked to disruption of C9orf72, a genetic risk factor for ALS associated with failure of energetic support of neurons by astrocytes and oligodendrocytes74,75.
Energy deficits and neurotoxic proteins
Brain glucose hypometabolism contributes to synapse loss and neuronal death in AD, with energetic deficits and neurotoxic protein accumulation mutually aggravating one another in a vicious cycle3,52,76,77 (FIG. 2a). Insufficient neuronal glucose and mitochondrial energy generation compromise the clearance of the 42 amino acid isoform of amyloid-β (Aβ42) and tau proteins from the brain. Conversely, accumulation of Aβ42 and tau triggers mitochondrial damage, impairs energy production and increases oxidative stress77,78. These neurotoxic proteins also inhibit GLUT4 (REF.51) and phosphofructokinase, thereby blocking glucose uptake, aerobic glycolysis and ATP synthesis79. Mitochondria accumulate in axonal swellings and are no longer replaced in presynaptic terminals80. Failure to clear dysfunctional mitochondria by mitophagy further compromises the bioenergetics of vulnerable neuronal circuits in AD, PD and other NDAs2. Excitation–inhibition balance is crucial for network operation at optimal energetic efficiency48 and, at the circuit level, an early, neurotoxic protein-driven feature of AD is the energetically expensive hyperexcitability of glutamatergic neurons34,81, which is associated with an imbalance between excitation and inhibition in local cortical and hippocampal networks32,47.
Fig. 2 |. Causes and consequences of the brain energy gap in neurodegenerative disorders.

a | Brain glucose hypometabolism occurs in conditions that increase the risk of Alzheimer disease (AD). The persistent brain energy gap and the neuropathological processes both contribute to a vicious cycle leading to brain energy exhaustion and dysfunction. Brain energy rescue strategies (FIG. 3; TABLES 1–3) attempt to inhibit the positive feedback between the brain energy gap and neuropathology involving amyloid-β and phosphorylated tau (dashed black arrow). Hormones (principally insulin, adipokines and incretins), as well as synthetic agonists and insulin sensitizers, can influence brain energy rescue and inhibit the onset of neuropathology. b | Glucose contributes to about 95% of total brain fuel supply in cognitively healthy young adults, and ketones supply the remaining 5%. In cognitively healthy older adults, brain glucose uptake is decreased by about 9%, in people with mild cognitive impairment (MCI) it is decreased by about 12% and in people with mild-to-moderate AD it is decreased by about 18%. The magnitude of the brain energy gap is the difference in total brain fuel uptake (glucose and ketones combined) between healthy young adults and people with mild-to-moderate AD; that is, the therapeutic target for brain energy rescue in MCI and AD. The brain energy gap has not been rigorously quantified in neurodegenerative disorders of ageing other than AD.
Aβ is involved in a healthy neuronal response to damage and/or infection82, but this protective function is lost when Aβ aggregates into plaques. Aβ exacerbates brain glucose hypometabolism, both in foci of Aβ accumulation and in remote regions, possibly due to a pericyte-mediated constriction of capillary blood flow. In turn, this hypometabolism triggers cellular damage and neuroinflammation77,83. Perturbed astrocytic and oligodendrocyte function, together with accumulation of phosphorylated tau, exacerbates ageing32 and Aβ/phosphorylated tau-induced network hyperexcitability, thereby perpetuating a cycle of neurodegeneration and declining brain glucose metabolism13,47 (FIG. 2a). This vicious cycle driven by energy failure in AD has similarities with the neural circuit disruption seen in schizophrenia84 and in epilepsy4, and contributes not only to deterioration of memory and cognition but also to abnormal behaviour in affected patients.
Short-chain fatty acids.
Acetate (two carbons), propionate (three carbons) and butyrate (four carbons). End products of microbial fermentation of dietary polysaccharides (soluble fibre). Butyrate is ketogenic and propionate is anaplerotic.
Energetics and endocrine dysregulation
Insulin resistance is a common feature of AD, PD, FTD, ALS and probably also HD, with reduced signalling at central insulin and IGF1 receptors contributing to deficits in neural function, synaptic plasticity and cellular integrity36,37,85 (TABLE 1; Supplementary Table 1). Even though insulin itself does not globally promote brain glucose uptake, insulin resistance reduces glucose uptake by corticohippocampal neurons that express insulin-sensitive GLUT4 (REFS17,22).
NDAs are associated with numerous changes in hormones that modulate brain energetics and neuroplasticity (Supplementary Table 2). The following observations may be highlighted. First, plasma leptin and hippocampal leptin signalling are reduced in AD, resulting in a state of leptin resistance mirroring insulin resistance86. This decline in leptin signalling is superimposed on a background of declining plasma leptin concentration with ageing and is linked to impaired learning, memory and long-term potentiation87. Second, an age-related reduction in ghrelin signalling in the temporal cortex may be related to neuronal damage and cognitive deficits in AD88. Circulating ghrelin concentration is reduced in PD, and the loss of its neuroprotective properties is linked to dopaminergic neuron degeneration and motor dysfunction88,89. In addition to blunted neuroprotective properties, antineuro-inflammatory effects of ghrelin involving astrocytes and microglia may be diminished in AD and PD88,90. Third, amylin oligomers and aggregates are suspected to damage neurons and the microvasculature in AD, although amylin has a Janus-faced role as further discussed later91–93. Fourth, an increase in circulating adiponectin levels has been reported in AD and ALS: if centrally expressed, this increase might counter cognitive deficits and exert neuroprotective properties, but this awaits confirmation86,94–96. In contrast to the above-mentioned hormones, there are very few data on the relationship between GLP1 and glucose-dependent insulinotropic polypeptide (GIP) and NDAs97 (Supplementary Table 1). The levels of brain hormones are challenging to measure and cause–effect relationships are hard to disentangle, but changes in the secretion and central actions of these hormones are implicated in the energy imbalance, pathophysiology and functional deficits in NDAs (Supplementary Table 2).
Cataplerosis.
Process by which intermediates (carbon) leave the tricarboxylic acid cycle to support biochemical reactions; that is, acetylcholine and lipid synthesis from citrate, or amino acid synthesis from α-ketoglutarate and oxaloacetate; opposite of anaplerosis.
Mild cognitive impairment.
(MCI). A condition prodromal to Alzheimer disease that is characterized by a subjective memory impairment and modest deficits in at least one of five main cognitive domains (executive function, memory, language, processing speed or attention). About 50% of cases progress to Alzheimer disease within 5 years.
Cerebral metabolic rate.
Quantity of energy substrate consumed by the brain (micromoles per 100 g per minute). Typically refers to glucose, but also used for brain consumption of oxygen, lactate and ketones.
Energetics and disease risk factors
Age
Ageing is the main risk factor for NDAs, but there is an important distinction between the cognitive, structural and neurometabolic changes associated with healthy ageing and those occurring in NDAs. During healthy ageing, some cognitive domains such as episodic and working memory and processing speed show a modest decline, whereas others (such as semantic memory) change relatively little98. Although the decline in brain volume and cortical thickness forms a continuum between cognitively healthy ageing, MCI and AD, regional changes in brain glucose metabolism seen during healthy ageing are quantitatively and qualitatively different from those in MCI and AD99,100. In healthy ageing, brain glucose metabolism decreases mainly in the frontal cortex, whereas in MCI and AD, the parietal lobe and precuneus are the most markedly affected. Decreased aerobic glycolysis101, loss of myelination, network perturbation and attenuation of neurovascular coupling are integral features of the ageing brain that might provide a template for the onset of the more severe brain energetic deficits in NDAs32,56,102,103. Mitochondrial proteins are expressed at lower levels in brains of older people experiencing accelerated cognitive decline104.
Metabolic dysregulation
The risk of NDAs is substantially higher in conditions of systemic metabolic dysregulation, including insulin resistance, obesity and T2D105 (TABLE 3). Most strikingly, poorly controlled type 1 diabetes (T1D) or T2D is associated with increased risk of cognitive impairment and AD106. Intriguingly, similarities exist between AD and T2D with respect to the deleterious effects of Aβ in the AD brain and the disruptive actions of amylin in the pancreas and brain in T2D: both disorders are associated with neurotoxic protein-induced peripheral metabolic and vascular abnormalities107. In young women with polycystic ovary syndrome, mild insulin resistance is associated with a pattern of glucose-specific brain hypometabolism similar to that seen in older people108, suggesting that the adverse effect of insulin resistance on brain energy metabolism is independent of age.
T2D doubles the risk of developing PD, possibly owing to increased expression of α-synuclein85. The risk of ALS is increased in T1D, but obesity and T2D are associated with decreased risk of ALS2,74. The metabolic syndrome associated with insulin resistance and weight gain is also present in ‘atypical’ major depression, itself often co-morbid with NDAs, especially AD and PD109. Effective treatment of T2D, metabolic syndrome and depression would be expected to reduce the risk of developing AD and other NDAs110.
Despite the persistent deficit in brain glucose uptake and utilization in NDAs, the normal ketogenic response to low plasma glucose levels is not stimulated because the main drivers of endogenous ketone production — low blood glucose and low insulin levels — are absent. Mild hyperglycaemia and mild insulin resistance commonly develop as people age, so plasma insulin levels rarely drop for long enough to release the insulin-mediated inhibition of lipolysis in adipose tissue, the source of the endogenous free fatty acids needed for ketogenesis. This metabolic deterioration continues as AD develops, so the brain experiences a persistent, progressive glucose-specific brain energy gap3 (FIG. 2a) that is not corrected by endogenous ketone production as it would be if insulin sensitivity were normal and plasma glucose concentration were decreased by a period of carbohydrate or caloric restriction.
Oestrogen
Menopause is associated with deteriorating systemic and brain glucose metabolism, weight gain, insulin resistance and loss of mitochondrial efficiency111. Ovariectomized rodent models of menopause show metabolic responses similar to fasting, including increased oxidation of long-chain fatty acids and elevated plasma levels of ketones, as well as white matter and myelin degeneration, changes that in part reflect the use of brain lipids as a source of fatty acids for ATP generation56,112. Oestrogen modulates many facets of brain glucose metabolism, including uptake, aerobic glycolysis and oxidative phosphorylation37. Oestrogen also stimulates the catabolism and clearance of Aβ, in part by upregulating insulin-degrading enzyme, so the loss of oestrogen after menopause could directly favour pathological processes leading to AD56,112. Accordingly, low oestrogen concentration in plasma is associated with an increased incidence of AD in women, although this relationship remains controversial37,112.
Genetic risk factors
Possession of two APOE*E4 alleles (encoding the E4 isoform of apolipoprotein E (ApoE4)) confers the highest genetic risk of sporadic AD. In APOE*E4 carriers the brain is hyperexcitable113, has reduced glucose utilization in regions affected by glucose hypometabolism in AD114 and accumulates more aggregated Aβ. Regardless of age, the following parameters all decline in APOE*E4 carriers in response to a high-fat diet: brain insulin signalling115, expression of glucose-regulating enzymes and glucose transporters114, mitochondrial function in the cortex and cognitive function104,116. These effects of ApoE4 on brain energetics are additive to the adverse effects of Aβ77.
Brain energy gap.
Deficit in brain energy metabolism of about 10% in mild cognitive impairment and of about 20% in early Alzheimer disease. Also present in other neurodegenerative disorders of ageing. It appears to be specific to glucose inasmuch as no studies to date have shown that brain ketone metabolism is affected.
Some of the adverse effects of ApoE4 may result from production of a carboxy-terminal fragment of ApoE4, which inhibits the electron transport chain117,118, increases generation of reactive oxygen species and forces neurons to increase their reliance on aerobic glycolysis or alternative energy substrates118. Whether or not ApoE4 affects ketone metabolism in individuals with MCI or AD is unclear. In one AD study, a ketogenic supplement did not raise plasma levels of ketones or improve cognitive outcomes as much in APOE*E4 carriers as it did in non-carriers119. A clinical trial of medium-chain triglycerides in individuals with AD who were specifically selected non-carriers of APOE*E4 showed beneficial cognitive outcomes after 1 month120. However, in transgenic mice expressing human APOE*E4, the presence of ApoE4 did not significantly affect brain ketone uptake in comparison with wild-type controls114.
Caloric restriction.
Limiting food intake to a level that does not permit full satiety. Can be self-determined (usually the case in human studies) or imposed relative to the food consumed by a matched group fed ad libitum (usually only in animal studies).
Electron transport chain.
A series of enzymatic protein complexes in the inner mitochondrial membrane that transfer electrons donated from NADH (complex I) or fatty acid dehydrogenase (complex II) to oxygen (complex IV).
Polymorphisms in major risk genes for PD, including PINK1 (encoding PTEN-induced putative kinase protein 1) and PRKN (encoding E3 ubiquitin-protein ligase parkin) are closely linked to impaired brain ATP production50,121. Phosphorylation of the endocytic sorting protein Rab10 by leucine-rich repeat serine/threonine-protein kinase 2 (LRRK2) is essential for GLUT4 translocation to the neuronal plasma membrane and is defective in PD patients possessing the LRRK2G2019S mutation122. In HD, axonal transport of mitochondria and glycolytic proteins to the synapse is hindered by mutant huntingtin29. In ALS and FTD, the proteins encoded by risk genes such as TARDBP (encoding TAR DNA-binding protein 43) interfere with mitochondrial function and quality control, thereby compromising ATP production123. Furthermore, the most prominent risk gene for ALS and FTD, C9orf72, encodes part of a complex with guanine nucleotide exchange factor activity that is linked to decreased autophagic lysosome-driven nutrient recycling, leading to frontal and thalamic glucose hypometabolism and aberrant lipogenesis2,124. Indeed, many products of risk genes associated with NDAs interfere with autophagic lysosomal clearance, which has a doubly disabling effect because the metabolic end products of carbohydrates, fats and proteins are then lost to energy generation pathways2.
Medium-chain triglycerides.
Edible oils comprising saturated fatty acids of 6–14 carbons in length. These have long been used in clinical nutrition to support energy needs in diseases or conditions involving malabsorption. Eight-carbon medium-chain triglycerides are more ketogenic than those of 10 or 12 carbons.
Mitochondrial biogenesis.
Renewal of mitochondria. In neurons, mitochondrial biogenesis occurs in the cell body with newly formed mitochondria being transported along the axon to dendritic synapses.
Therapies based on brain energy rescue
As outlined already, the prevailing notion that impaired brain glucose metabolism in NDAs is simply a consequence of neuronal dysfunction is now being revised. Most notably in AD, the progressive decline in brain glucose uptake and metabolism creates a persistent brain energy gap that contributes to brain cell dysfunction and accumulation of neurotoxic proteins even before the onset of cognitive and neuropsychiatric deficits3 (FIG. 2b). Once glycolysis is impaired and neuronal function starts to decline, the brain energy deficit cannot be corrected by simply increasing blood glucose concentration; indeed, additional dietary glucose aggravates the insulin resistance already commonly present in older people10. Furthermore, brain glucose uptake is driven by neuronal activity, not by circulating glucose levels3. Conversely, ketones and lactate are an alternative brain energy source41, brain uptake of which is driven by their availability in the circulation.
Because no single common pathway causes brain energy deficits in NDAs, brain energy rescue strategies may need to target different metabolic pathways and processes depending on the disease in question3,4,125 (FIG. 3). Some of these strategies focus on a single enzyme, transporter or metabolite, but others are broader (TABLES 1–3). The following discussion first addresses the energetic dimension of mitochondrial dysfunction in NDAs. Strategies that have broader effects such as modulating redox status and ketone-based approaches are described next, then hormone-based approaches to brain energy rescue, followed by a suite of novel strategies currently under exploration. These strategies should all act synergistically with preventive lifestyle changes, such as increased exercise and dietary improvements that help counter insulin resistance126,127 (BOX 3; TABLE 3). For links between mitochondrial dysfunction, oxidative stress and neurodegeneration, see two recent reviews60,78.
Fig. 3 |. Brain energy disruption and rescue strategies.

a | Several pathways of brain energy metabolism in neurons are disrupted (shown with dashed black arrows) in neurodegenerative disorders of ageing (specific disorders with declines are shown in the red boxes). Increased production of reactive oxygen species (ROS) and neuroinflammation that negatively affect brain energy levels are shown with a thick black arrow. The combination of impaired ATP production and increased levels of ROS contributes to declining brain function. b | Molecules or potential therapies implicated in brain energy rescue strategies target six broad pathways: ATP and redox state (light blue); brain glucose transport and/or aerobic glycolysis (dark blue; interventions indicated with an asterisk: adiponectin, ghrelin, insulin (GLUT4 only), nicotinamide riboside, dichloroacetate, N-acetylcysteine, oxaloacetate, glucagon-like peptide 1 (GLP1), glucose-dependent insulinotropic polypeptide (GIP), leptin, amylin, metformin, liraglutide and sitagliptin); anaplerosis and the tricarboxylic acid (TCA) cycle (purple; propionic acid (C3), heptanoic acid (C7), octanoic acid (C8), d-β-hydroxybutyrate (BHB), and ketone esters (KE)); mitochondrial transport and biogenesis (olive-grey); ketogenesis (orange; interventions indicated with two asterisks: BHB, C8, decanoic acid (C10) and KE); or protection against ROS and inflammation (light green; interventions indicated with three asterisks: ghrelin, GLP1, GIP, leptin, adiponectin, metformin, AP39, mitochondrial division inhibitor 1 (mdivi-1), MitoQ, BHB, ketogenic diet and KE). Details of the molecules or potential therapies are shown in TABLE 1 (preclinical studies) and TABLES 2,3 (clinical studies). Complementary interventions such as caloric restriction, ketogenic diet and exercise are not shown. Neurons take up lactate generated by astrocytes and oligodendrocytes (not shown). Medium-chain fatty acids such as decanoic acid and octanoic acid in the circulation can enter astrocytes and produce ketones and acetyl coenzyme A (acetyl-CoA). AAV, adeno-associated virus; AcAc, acetoacetate; acetyl-CoA, acetyl coenzyme A; Ach, acetylcholine; AD, Alzheimer disease; AG, aerobic glycolysis; ALS, amyotrophic lateral sclerosis; ApoE4, E4 isoform of apolipoprotein E; ASO, antisense oligonucleotide; ATP Syn, ATP synthase; DA1, dynamin-related protein antagonist 1; ETC, electron transport chain; FTD, frontotemporal dementia; HD, Huntington disease; MCT, monocarboxylate transporter; mHTT, mutant huntington protein; NR, nicotinamide riboside; PD, Parkinson disease; PGC1α, peroxisome proliferator-activated receptor-γ coactivator 1α; PDH, pyruvate dehydrogenase; PPP, pentose phosphate pathway.
Box 3 |. Complementary multimodal lifestyle strategies.
Lifestyle interventions may delay the onset of neurodegenerative diseases of ageing (NDAs), as exemplified by the FINGER trial, which reduced the risk of Alzheimer disease (AD) in a typical older population126,251. Two lifestyle approaches that improve brain energetics and increase insulin sensitivity are attracting considerable attention: caloric restriction (and the variant, intermittent fasting)5 and physical exercise127. Both approaches are neuroprotective and improve cognitive and motor function in preclinical models of AD and Parkinson disease by increasing synaptic spine density98, mitochondrial biogenesis283, neurogenesis in the hippocampus, autophagy of neurotoxic proteins, mitophagy of dysfunctional mitochondria98,284 and activation of ghrelin signalling206. Nutritional ketosis is a feature common to caloric restriction, intermittent fasting and other ketogenic interventions3,98,285.
Exercise helps regulate glucose metabolism and reduces two important risk factors for NDAs: obesity and type 2 diabetes. Exercise also improves executive function and attention and increases processing speed in NDAs — effects related to enhanced cerebral blood perfusion, notably in the hippocampal dentate gyrus. Exercise increases angiogenesis in several brain regions254 and mitigates the age- and NDA-related decline in cerebral blood flow98, which, in turn, may improve synaptic function by providing ketones and lactate5,98,286,287. A 3-month exercise regimen increased brain ketone transport by 30% in AD108, so the improvement in brain energetics caused by ketones is one possible link between exercise, brain-derived neurotrophic factor (BDNF), neurogenesis and cognitive gains in NDAs287.
The angiogenic effect of exercise is partly mediated by vascular endothelial growth factor253,254. Lactate liberated from skeletal muscle during exercise can also be used by the brain5,287 (BOX 1). Exogenous lactate mimicked exercise in inducing brain vascular endothelial growth factor and increasing capillary density, actions dependent on hydroxycarboxylic acid receptor 1 (HCAR1)254. The hippocampal myokine irisin may also be involved; both irisin and its precursor, fibronectin domain 5, contribute to metabolic homeostasis and neuroprotection287. Lactate recruitment of BDNF is dependent on fibronectin domain 5, thereby interlinking the actions of lactate and irisin in the beneficial effect of exercise on the brain222,287,288.
The goal of mimicking the gains of exercise and fasting in a broadly accessible manner by an appropriate pharmacological intervention (‘exercise in a pill’) is analogous to using lactate253 or a ketogenic supplement to mimic and/or augment endogenous ketone production without severely limiting dietary carbohydrate or food intake156,157. Exercise mimetics could include agents acting via myokines, cathepsin B, AMP-activated protein kinase (AMPK) or adiponectin98,132,287,289.
Cognitive reserve is the capacity or resilience of the ageing brain to resist functional decline and is directly correlated with higher education and intellectual occupation both early and later in life. Whether improved cognitive reserve can stall AD is currently under exploration290. [18F]Fluorodeoxyglucose positron emission tomography suggests that cognitive reserve reflects in part the capacity of the brain to maintain normal function in the face of bioenergetic or other deficits291,292. Maintaining and improving cognitive reserve in individuals with NDAs could potentially be enhanced by the brain energy rescue strategies discussed in this article.
Support of mitochondrial function
Despite continued uncertainty about the extent to which mitochondrial damage is a consequence versus cause of the onset or progression of NDAs65, considerable research focuses on improving mitochondrial function by protecting the electron transport chain, promoting mitochondrial biogenesis and/or reducing oxidative damage to mitochondria125. Assessment of mitochondrial integrity is mostly indirect, but histochemical evidence of decreased cytochrome c activity in post-mortem brain samples from young adult APOE*E4 carriers118 demonstrates that impaired mitochondrial function can be present in presymptomatic individuals at risk of AD.
CP2, a proprietary tricyclic pyrone, improves cognitive and behavioural phenotypes in transgenic AD mice, in part by binding to and partially inhibiting the flavin mononucleotide subunit of complex I. This improves mitochondrial bioenergetics and overall brain energy status, possibly because of increased mitochondrial biogenesis128. CP2 also stimulates AMPK, promotes neuronal resistance to oxidative stress, reduces brain levels of phosphorylated tau and Aβ, improves axonal trafficking and increases levels of brain-derived neurotrophic factor (BDNF) and synaptic proteins in vivo128,129. Controlling the activity of complex I specifically seems to underpin this beneficial effect130 because mutations that inhibit both complex I and complex III or both complex I and complex V are detrimental to brain energetics131.
The mitochondrion-targeted antioxidant MitoQ reduces oxidative stress in mitochondria and is neuroprotective in several NDA models (TABLE 1). Resveratrol stimulates mitochondrial biogenesis through the sirtuin 1 (SIRT1)–AMPK–peroxisome proliferator-activated receptor-γ (PPARγ) coactivator 1α (PGC1α) pathway. Resveratrol also recruits AMPK to enhance autophagy, which removes damaged organelles (including mitochondria) and misfolded proteins and recycles their components, thereby promoting ATP generation2,132. Replacement of old and/or damaged mitochondria starts in the neuronal cell body with new mitochondria being transported along axons to presynaptic terminals15. Both ageing and NDAs increase mitochondrial division in a manner decoupled from the normal fission–fusion cycle, suggesting that inhibiting mitochondrial fragmentation could be beneficial in NDAs133. Quinazolinone and its derivatives such as mitochondrial division inhibitor 1 (mdivi-1) were originally described as selective inhibitors of mitochondrial fission, but their neuroprotective effects in both in vitro and in vivo models of AD, PD and traumatic brain injury are now thought to reflect improved mitochondrial fusion and biogenesis134–136, and possibly better function of complex I.
Redox state.
Capacity of a molecule to be reduced or acquire electrons; opposite of oxidation. Many biological reactions involve the reduction of one molecular species while another is being simultaneously oxidized. Energy metabolism is highly dependent on the redox state of the cell.
A pilot clinical study showed that (S)-equol, a selective oestrogen receptor-β agonist, increases cytochrome c oxidase activity in AD137, so treatments that improve mitochondrial function by selective partial inhibition of complex I, target mitochondrial uncoupling proteins or increase mitochondrial biogenesis may result in clinical improvement in NDAs (TABLE 2). Mitochondrial uncoupling proteins could help cells to resist oxidative and metabolic stress98. Low doses of the uncoupling agent dinitrophenol had a neuroprotective effect in preclinical models of AD, PD and HD138. In a mouse model of HD, mitochondrial respiration was improved by bexarotene, a retinoid X receptor agonist and PPARδ activator139.
Table 2 |.
Clinical trials reporting improved brain function and/or energetics in NDAs
| Disorder | Study details | Results and comments | Ref. |
|---|---|---|---|
| AD | Single-blind RCT of (S)-equol (n = 15) or placebo (n = 15) for 2 weeks (NCT02142777) | Well tolerated. More participants showed ↑ cytochrome oxidase activity with (S)-equol than with placebo; no cognitive change. First study of a mitochondrial intervention as a direct biomarker of mitochondrial engagement in AD | 137 |
| Mild-to-moderate AD and MCI | RCT in individuals with AD (n = 21) or MCI (n = 39) receiving long-acting intranasally administered insulin (20 or 40 IU) or placebo for 4 months (NCT01595646) | Dose-dependent ↑ memory composite score on regular but not long-acting intranasal insulin in ApoE4+ individuals. No change in functional autonomy or executive function | 38 |
| AD | RCT of liraglutide (n = 14) or placebo (n = 20) for 26 weeks (NCT01469351) | No change in Aβ load or cognitive scores. ↓ brain glucose uptake over 26 weeks only with placebo. Underpowered for cognitive outcomes. Liraglutide may delay metabolic decline in brain | 194 |
| AD or MCI | RCT (n = 20) of metformin (500 mg) or placebo for 8 weeks (NCT01965756) | ↑ Executive function. No change in cerebral blood flow | 182 |
| AD | Double-blind RCT of C8 at 20 g per day (n = 77) or placebo (n = 63) for 90 days (NCT00142805) | ADAS-Cog score ↑ by 3.4 points in ApoE4− individuals. Cognitive score varied directly as ketones. ↑ cognition in mild-to-moderate AD | 119 |
| Mild-to-moderate AD | Open label study (n = 10) of KD ± C8C10 for 12 weeks (NCT03690193) | ↑ ADAS-Cog score; no cardiovascular safety or other metabolic concerns. First reported clinical use of a KD in AD including medium-chain triglyceride supplementation | 157 |
| AD and MCI | RCT of KD (n = 9) or NIA low-fat diet (n = 5) for 12 weeks (NCT02521818) | ↑ composite cognitive score, particularly memory domain, only in adherent participants and only with KD. First reported clinical use of a KD without medium-chain triglyceride supplementation. Feasibility is very challenging but beneficial effects of ketones were clearly present | 162 |
| Mild-moderate AD | Open-label study of C8C10 (n = 11) or C8 (n = 6) at 30 g per day for 4 weeks (NCT02709356) | ↑ ketones twofold. Brain ketone uptake ↑ in direct proportion to ↑ ketone and brain glucose utilization. In AD, the brain can utilize additional ketones provided as C8C10 | 270 |
| Mild-to-moderate ApoE4− AD | Crossover RCT of medium-chain triglycerides (17.3 g) (n = 24) or placebo (canola oil) (n = 25) for 30 days (ChiCTRIOR6009737) | 2.62-point increase on ADAS-Cog (Chinese version) for medium-chain triglycerides, 2.57-point reduction for placebo. Study restricted to ApoE4− patients. Inverse correlation between cognitive changes and plasma lysophosphatidylcholine species | 120 |
| PD | RCT of exenatide at a dosage of 2 mg once per week (n = 31) or placebo (n = 29) for 48 weeks plus a 12-week washout (NCT 01971242) | UPDRS motor subscale at 60 weeks:↑ by 1.0 point for drug and ↓ by 2.1 points for placebo. ↓ motor symptoms | 97 |
| PD | RCT of KD (n = 20) or low-fat diet (n = 20) for 8 weeks (ACTRN 12617000027314) | ↑ UPDRS score in both groups, but 41% more for the KD. 86% adherence; tremor ± rigidity intermittently ↑ for KD. First RCT of KD in PD. A KD and low-fat diets are safe in PD | 160 |
| HD | Open-label study in individuals with HD (n = 10) and controls (n = 13) receiving triheptanoin at 1 g kg−1 for 1 month (NCT01696708) | MRS: ↑ levels of brain high-energy phosphates including ↑ inorganic phosphate/phosphocreatine during visual stimulation | 271 |
These studies reported statistically significant improvements in primary or secondary end points with novel treatments or drugs approved for other indications and repurposed for treatment of neurodegenerative disorders of ageing (NDAs). The low-fat diet162 was a modified Atkins diet. The three ketogenic diet (KD) trials were all principally feasibility studies not powered for cognitive or metabolic outcomes157,161,163. ↑, increase; ↓, decrease; Aβ, amyloid-β; AD, Alzheimer disease; ADAS-Cog, Alzheimer Disease Assessment Scale — Cognitive Subscale; ApoE4, E4 isoform of apolipoprotein E; C8, octanoic acid; C8C10, octanoic acid plus decanoic acid; HD, Huntington disease; MCI, mild cognitive impairment; MRS, magnetic resonance spectroscopy; NIA, National Institute on Aging; PD, Parkinson disease; RCT, randomized controlled trial; UPDRS; Unified Parkinson Disease Rating Scale.
Redox state, glycolysis and the TCA cycle
The redox state of a cell is typically measured by the ratio of oxidized to reduced nicotinamide adenine dinucleotide (the NAD+ to NADH ratio), which is a non-invasive marker of global brain energy status5,78,140. In general, nutrients and metabolites that raise either blood NAD+ levels or the blood NAD+ to NADH ratio improve the energetic status of the brain141. The NAD+ precursor nicotinamide riboside mitigates cognitive impairment, synaptic degeneration and neuronal death in transgenic mouse models of AD78,98,142. Nicotinamide riboside also improves mitochondrial function in PD neurons and reduces age-related loss of dopaminergic neurons and associated motor deficits in an animal model of PD143. Another potential approach to raising the NAD+ to NADH ratio is dietary supplementation of oxaloacetate144. In several in vitro and animal models of PD, terazosin (a drug approved for treatment of benign prostatic hypertrophy) stimulated phosphoglycerate kinase 1 activity, aerobic glycolysis and ATP production145. Patients taking terazosin to treat other conditions had a decreased risk of developing PD and slower PD progression, so its repurposing to treat PD seems promising.
Supplementation with pyruvate could potentially improve brain energetics by stimulating pyruvate dehydrogenase146,147, a possibility supported by the rescue of defective aerobic glycolysis by pyruvate in HD-derived human induced pluripotent stem cells66. Treatments that improve mitochondrial function have had mixed success in preclinical models of ALS, and these approaches remain largely untested in humans70,74.
Interventions that raise levels of circulating ketones also increase levels of acetyl-CoA, which fuels the TCA cycle independently of aerobic glycolysis. Preclinical studies show that supplementation with BHB, octanoic acid (also known as caprylic acid, an eight-carbon saturated fatty acid), oxaloacetic acid and decanoic acid (also known as capric acid, a ten-carbon saturated fatty acid) as well as a ketogenic diet or caloric restriction (BOX 3) all contribute to increased TCA cycle activity within the brain144,148 (FIG. 3b). In humans, plasma medium-chain fatty acids can be transported into and metabolized by the brain149. TCA cycle intermediates also give rise to bioactive molecules such as the neurotransmitter acetylcholine, whose levels are decreased in AD. These responses are generally reversible and therefore transformation of glutamate into α-ketoglutarate (which enters the TCA cycle) generates ATP in neurons and glia98.
Triheptanoin, a triglyceride of heptanoic acid, delays motor symptoms and is neuroprotective in animal models of ALS74, epilepsy and ischaemic stroke150,151. Triheptanoin also reduces the effort needed to undertake exercise in HD, a beneficial effect associated with increased creatine phosphate concentration in the brain152. Triheptanoin appears to substitute for the branched-chain amino acids that are an endogenous substrate of anaplerosis and levels of which are decreased in HD152,153.
Ketogenic diet.
A very-low-carbohydrate, very-high-fat diet inciting the liver to produce ketones from free fatty acids released from adipose tissue because there is minimal insulin production. The stricter, medical form of the ketogenic diet developed to treat intractable epilepsy usually also limits dietary protein.
Ketone-based strategies
Several clinical trials show that ketogenic interventions result in cognitive and/or functional improvements in MCI154–156, AD119,120,157–159 and PD160,161. These interventions fall into two categories: ketogenic dietary supplements containing medium-chain triglycerides (either octanoic acid (C8) alone or octanoic acid plus decanoic acid (C8C10)) and the very-low-carbohydrate ketogenic diet (BOX 4; Table 2). In the phase I (ref. 119) and phase II (REF.154) placebo-controlled studies of octanoic acid supplementation and octanoic acid plus decanoic acid supplementation, the interventions lasted from 12 weeks119 to 6 months154, respectively. Two subsequent feasibility studies of ketogenic diets in AD showed increased global cognitive scores in the most adherent patients but did not have control groups157,162.
Box 4 |. Deteriorating brain glucose but not brain ketone uptake: an opportunity for brain energy rescue.
The decline in brain glucose metabolism associated with neurodegenerative diseases of ageing has traditionally been assumed to be a consequence of the disease process. The development of positron emission tomography (PET) tracers for assessing ketone uptake (BOX 2) provided an opportunity to assess whether brain ketone metabolism was also disrupted. Dual-tracer PET studies of brain glucose ([18F]fluorodeoxyglucose tracer) and ketone ([11C]acetoacetate tracer) uptake show that whereas brain glucose utilization is impaired, brain ketone metabolism is still normal in Alzheimer disease (AD) and mild cognitive impairment (MCI) (see the figure, part a). These PET images show the rate constant (min−1) for brain glucose uptake (KGlc; left) and brain acetoacetate uptake (KAcAc; right) in cognitively healthy, elderly controls (CTL; n = 24), individuals with MCI (n = 20) and individuals with mild-to-moderate AD (n = 19). The images are paired; that is, one for [18F]fluorodeoxyglucose and one for [11C] acetoacetate obtained from each participant on the same afternoon. Unlike the cerebral metabolic rate (CMR), which is partly dependent on plasma concentrations of the substrate in question, KAcAc is largely independent of plasma levels of glucose or ketones, so it is a better measure of the brain’s capacity to take up these energy substrates. KGlc is significantly lower in the parietal and temporal cortex as MCI develops and progresses to AD, but KAcAc does not decrease in MCI or AD compared with cognitively unimpaired age-matched controls293. The CMR of acetoacetate increases in direct proportion to plasma ketone levels in individuals with AD after 1 month of receiving a daily supplement of 30 g of a ketogenic medium-chain triglyceride (octanoic acid plus decanoic acid (C8C10) or octanoic acid (C8) alone; see figure, part b). However, there was no change in the CMR of glucose270.
Mitochondrial oxidative phosphorylation is the only way of generating ATP from ketones; that is, there is no extramitochondrial pathway for ketones as there is for conversion of glucose into lactate. Accordingly, the fact that brain ketone metabolism is normal in MCI and AD indirectly implies that mitochondrial respiration is relatively normal in a significant proportion of brain mitochondria in order for them to be able to generate ATP. Hence, comparisons of brain glucose and brain ketone metabolism offer an opportunity to determine whether mitochondrial function is markedly impaired (in which case both glucose and ketone metabolism would be decreased, regionally or globally) or whether the defect is more at the level of glycolysis or glucose transport (in which case glucose but not ketone metabolism would be impaired). This PET comparison of brain energy substrate uptake could help to clarify whether the onset of mitochondrial dysfunction is an early event in neurodegenerative diseases of ageing, whether such dysfunction occurs in the brain regions most affected65 and whether a therapeutic agent being tested corrects the dysfunction or promotes mitochondrial biogenesis or other aspects of mitochondrial health.
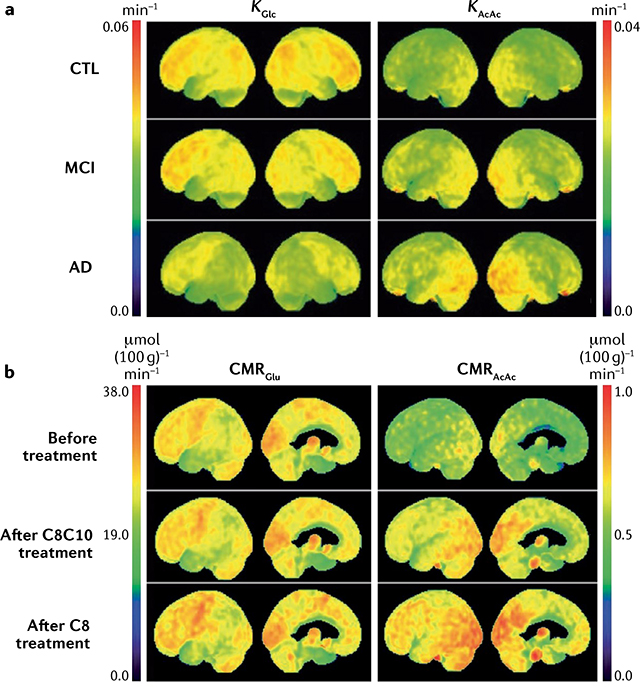
Figure part a is reprinted with permission from REF.293, Elsevier. Part b is republished with permission of IOS Press, from Ketogenic medium chain triglycerides increase brain energy metabolism in Alzheimer’s disease, Croteau, E. et al., J. Alzheimer’s Dis. 64, 551–561 (2018)270; permission conveyed through Copyright Clearance Center, Inc.
A recent 6-month study of octanoic acid plus decanoic acid supplementation in MCI showed a direct and statistically significant dose–response relationship between brain uptake of ketones and/or plasma ketone levels and executive function, verbal fluency and language, strongly implying that ketones were directly and mechanistically linked to cognitive improvement via brain energy rescue154. Because of the short half-life of ketones in the body, however, the main challenge with ketogenic interventions is to achieve a sustained therapeutic level of ketosis. In two studies of ketogenic supplements that had a sample size large enough to provide adequate statistical power to assess cognitive outcomes, the 24-h average plasma ketone level was 0.2 mM or less for octanoic acid (REF.119) and 0.4 mM or less for octanoic acid plus decanoic acid (REF.154); these ketone levels would have only partially corrected the brain energy gap in MCI and less so in AD (FIG. 2b; BOX 4). In other clinical trials with a ketogenic diet in AD157,162, MCI155,156 or PD160,161, higher plasma ketone levels were directly related to improved clinical outcomes, but sample size and patient adherence were inadequate to produce definitive evidence of a cognitive benefit.
Anaplerosis.
Process by which four-carbon or five-carbon units enter the tricarboxylic acid cycle independently of acetyl coenzyme A to replenish intermediates used in the synthesis of acetylcholine or lipids (from citrate) or amino acids (from α-ketoglutarate and oxaloacetate); opposite of cataplerosis.
In individuals with PD, consumption of a ketogenic diet for 8 weeks led to a substantial reduction in urinary problems, pain and fatigue scores versus individuals in a control group consuming a low-fat diet160,161. The ketogenic diet group also showed a trend towards increased motor scores versus the control group. Ketogenic interventions are being explored with some success in animal models of PD163, ALS164 and HD165 (TABLE 1), but randomized, controlled clinical trials of this approach are yet to be reported in these NDAs. A ketogenic diet promotes neurovascular function and metabolic status in mice, along with a healthier intestinal microbiota profile166.
Studies in which a single dose of a ketogenic supplement transiently improved cognition in AD167 and in cognitively normal older people (66 years old)158 suggest that ketones reduce the brain energy gap by bypassing glycolysis and providing acetyl-CoA to enter the TCA cycle directly (FIG. 3b). This interpretation is supported by reports that mild-to-moderate ketosis lasting less than 4 h prevents the autonomic, cognitive and behavioural symptoms of acute insulin-induced hypoglycemia in T1D168. In turn, mild ketosis probably spares some glucose to be used by pathways other than glycolysis and oxidative phosphorylation41, including the pentose phosphate pathway, which generates NADPH, but also anaplerosis for the TCA cycle (FIG. 3). Whether glucose sparing is central to the therapeutic effect of ketone supplementation in NDAs remains to be determined.
Metabolism of ketones in the brain not only generates fuel but also provides an important substrate for the synthesis of brain lipids, including myelin40. Ketones are also substrates for post-translational protein modification and activate cell signalling125. The density and activation of HCAR2 are increased in the substantia nigra in PD169 and lactate is neuroprotective in an animal model of PD170. Reducing neuronal hyperexcitability by raising GABAergic tone may contribute to the efficacy of ketone supplementation in individuals with NDAs as it does in epilepsy13,148,171.
Disease modification — that is, retardation or reversal of neuropathology — is a crucial goal in the treatment of NDAs. Studies in transgenic AD mice show that ketones decrease Aβ deposition in the brain172 and reduce the excitatory effect of Aβ42 on neurons146. These findings from preclinical studies were confirmed in a pilot clinical study in MCI156, suggesting that in addition to providing an alternative brain energy substrate that bypasses the brain glucose deficit, ketogenic interventions could potentially improve cognitive outcomes in MCI and AD by slowing pathological processes that result in Aβ accumulation and damage.
High-fat diets are commonly perceived to increase the risk of cardiovascular disease, so it is important to consider their potential risks. Very-high-fat ketogenic diets have been assessed in five clinical trials of durations ranging from 6 to 12 weeks. Three of these trials were conducted in participants with MCI or AD155,157,162 and the other two were conducted in participants with PD160,161. In none of these trials were the levels of common biomarkers of cardiovascular risk increased, including body weight or plasma LDL cholesterol or triglycerides. However, no standard definition of a ketogenic diet exists and some ‘high-fat’ diets used experimentally (and possibly also clinically) might adversely affect cardiovascular health outcomes (in particular during very long-term use) because they contain an excess of refined carbohydrate. In clinical trials, ketogenic medium-chain triglycerides were not associated with increased cardiovascular or metabolic risk (TABLE 2); indeed, like the very-high-fat ketogenic diet, medium-chain triglycerides are commonly used to treat obesity and T2D, which are themselves risk factors for NDAs.
Increasing insulin sensitivity
Brain energy homeostasis is closely linked to peripheral insulin sensitivity, both of which depend on the balance between global energy intake and energy use. The two main determinants of peripheral insulin sensitivity are exercise and intake of refined carbohydrate173. When lifestyle changes are ineffective or difficult to implement, peripheral injections of insulin are commonly used to treat T2D, but the challenge is to avoid episodes of hypoglycaemia and exacerbation of insulin resistance, both of which increase morbidity168.
Intranasally administered insulin and insulin sensitizers
Intranasally administered insulin and intranasal insulin sensitizers could potentially mitigate the deleterious effects of insulin resistance and a glucose deficit (FIG. 3; TABLES 1–3). Intranasally administered insulin enters the brain directly via olfactory neurons, which enables treatment of central nervous system insulin resistance while minimizing systemic hypoglycaemia. Short-term studies show that intranasally administered insulin enhances cognitive function in healthy young adults, individuals with MCI and individuals with mild-to-moderate AD, in part by stimulating brain glucose metabolism38,174. Little or no intranasally administered insulin enters the peripheral circulation, but intranasal insulin delivery still needs to be optimized to achieve a more consistent increase in brain insulin levels before its efficacy for cognitive improvement can fully be assessed38.
Metformin decreases hepatic glucose production, which increases insulin sensitivity in T2D, so it is being investigated for therapeutic use in NDAs175. Metformin reduces neuropathology and corrects memory deficit in AD mice176 and normalizes cortical network disruption and anxious behaviour in HD mice177. Within the brain, metformin also stimulates autophagy, improves synaptic function and reduces neuroinflammation — effects that mimic those of caloric restriction and exercise2,178. Metformin suppresses coupling of the redox and proton transfer domains of complex I, but its overall mechanism of action remains unclear179,180. Recent data suggest that growth/differentiation factor 15 (GDF15) acting via GDNF family receptor-α-like (GFRAL) may mediate the influence of metformin on energy metabolism, suggesting that they could become novel therapeutic targets for safely combating brain energetic deficits associated with NDAs181. Indeed, metformin is potentially protective against cognitive decline in MCI or AD182,183 and cognitive impairment due to stroke184. However, metformin may exacerbate Aβ accumulation185 and has adverse effects linked to overproduction of AMPK and vitamin B12 deficiency132. In addition, studies are needed to further clarify the putative utility of metformin for restoring energetic status in NDAs175,186.
Impaired brain glucose uptake and insulin receptor desensitization could potentially be corrected by targeting nuclear hormone receptors that activate insulin-regulated and IGF-regulated pathways. For example, thiazolidinediones are PPARγ agonists that potentially reduce brain insulin resistance associated with AD and other NDAs36. However, PPARγ agonists, including pioglitazone and rosiglitazone, have produced no cognitive benefit in clinical trials in AD. Inhibitors of sodium–glucose co-transporter 2 such as dapagliflozin increase glucose excretion, improve cardiovascular outcomes and reduce mortality in T2D187. In addition, these agents induce mild ketonaemia188, suggesting that they should be tested in NDAs, perhaps in combination with ketogenic interventions.
Incretin hormones
GLP1 receptor agonists such as liraglutide are approved to treat insulin resistance, obesity and T2D189,190. On the basis of encouraging findings in animal models of AD, PD, HD and ALS (TABLE 1; Supplementary Table 2), they are also being assessed for treatment of NDAs97,191,192. For example, liraglutide and exenatide reduced neuropathology, neuroinflammation and microvascular pathology and improved cognitive outcomes in a transgenic mouse model of AD192 (TABLE 2). A GLP1 receptor agonist reduced Aβ accumulation and reduced mitochondrial proapoptotic signalling, while increasing antiapoptotic signalling and BDNF levels192. Semaglutide, a long-acting GLP1 analogue, was more neuroprotective than liraglutide in an animal model of PD, a beneficial effect related to improved mitochondrial function and lower oxidative stress, apoptosis and neuroinflammation97. Inhibitors of GLP1 breakdown are now in clinical trials to treat NDAs193.
In mild-to-moderate AD, liraglutide administration for 6 months attenuated the decline in brain glucose uptake but had no effect on brain Aβ load or cognitive outcomes194. Exenatide administration reduced symptoms of PD in a phase II trial195. Interestingly, metformin might exert its actions partly via GLP1 (REF.36). Some dipeptidyl peptidase 4 (DPP4) inhibitors (gliptins) approved to treat T2D are known to prolong the activation of GLP1 (REF.196); one such agent, sitagliptin, improved cognition in older individuals with diabetes with or without AD197.
GIP agonists have been shown to have benefits similar to those of GLP1 agonists in mouse models of PD198,199. GIP agonists closely mimic GLP1 agonists in animal models of AD200,201. Dual agonists of both GLP1 and GIP were more effective in rodent models of PD than GLP1 agonists alone200,201. A triple agonist of GLP1, GIP and glucagon receptors had broad neuroprotective activity in a mouse model of AD202. Clinical data are eagerly awaited for these multitargeted agents.
Ghrelin
Ghrelin is neuroprotective and improves cognition in animal models of AD, PD and HD203–205 (Supplementary Table 2). The beneficial effects of ghrelin involve promotion of neuronal glucose uptake, increased expression of uncoupling protein 2, improved mitochondrial function and enhanced mitophagy88,205. AMPK activation in dopaminergic neurons of the substantia nigra may also contribute to the beneficial effects of ghrelin in PD: AMPK activates PGC1α to induce mitochondrial biogenesis and increase ATP production, as well as stimulating autophagy to eliminate α-synuclein2,205,206. Since ghrelin counters gastrointestinal dysfunction in PD, relamorelin, a centrally penetrant and selective agonist of ghrelin receptor (also known as growth hormone secretagogue receptor type 1), is being assessed to treat constipation in PD and T2D. The effects of relamorelin on motor function, neuronal survival and bioenergetics are also being evaluated in PD207.
Leptin and adiponectin
Leptin promotes mitochondrial function, has neuroprotective properties and mitigates the neurotoxic effects of Aβ accumulation in animal models of AD and PD86,87,208. Synergistic beneficial effects on mitochondrial function have been reported for leptin in combination with PPARα agonists209. The risk of ALS is reduced in individuals with T2D, so it is interesting that knocking out the gene encoding leptin (Lep), which suppresses appetite in a mouse model of ALS, slowed the progression of ALS symptoms while decreasing energy expenditure and increasing body weight210. This suggests that leptin antagonists should be evaluated as a potential treatment in ALS. Data on adiponectin are currently limited to mouse models of AD in which adiponectin agonists had neuroprotective properties associated with reduced loads of Aβ and phosphorylated tau and improved cognitive performance — benefits that were related to increased glucose uptake in the hippocampus and, possibly, to increased insulin sensitivity86,211.
Amylin
The status of amylin as a potential target for treating NDAs is controversial92 because native amylin itself is amyloidogenic and aggregated amylin has proapoptotic and neuroinflammatory effects and may seed Aβ aggregation91,93. Moreover, Aβ binds to amylin receptors, and its neurotoxic actions and interference with cognition were blunted by an amylin antagonist91,212. Nevertheless, amylin itself could have potential beneficial properties as a leptin sensitizer, including leptin resistance in AD213. The satiety-stimulating effects of amylin may help to control weight gain as well as increase insulin sensitivity and brain glucose metabolism. Treatment with human amylin or a non-aggregating amylin analogue, pramlintide, has shown both cognitive benefits and reductions in Aβ pathology in animal models of AD214. Amylin and pramlintide also promote Aβ efflux from the brain, regulate synaptic proteins, reduce oxidative stress and inflammation and improve mitochondrial function214,215. Pramlintide protects against the neurotoxic and memory-disrupting actions of Aβ91. Whether amylin-related mechanisms can be harnessed in the treatment of AD and other NDAs remains to be seen.
Antagomirs.
Also known as anti-microRNAs or blockmirs. Synthetic oligonucleotides engineered to silence endogenous microRNAs or prevent other molecules from binding to a specific mRNA.
Locked nucleic acids.
RNAs in which the flexibility of the ribose ring has been restrained by adding a methylene bridge connecting the 2′ oxygen and 4′ carbon. Oligonucleotides containing locked nucleic acids have increased specificity, sensitivity and hybridization stability.
Restoration of downstream signalling
An important fate of glucose distinct from its use as an energy substrate is its utilization to generate O-linked β-N-acetylglucosamine (O-GlcNAc), which is post-translationally and reversibly added to serine and threonine residues of numerous proteins. O-GlcNAcylation occurs via O-GlcNAc transferase, whereas O-GlcNAcase removes O-GlcNAc residues: both of these enzymes are therapeutically targetable in NDAs216. O-GlcNAcylation is important for axonal stability and synaptic plasticity and for the local and dynamic coupling of glucose utilization to glycolysis and mitochondrial function at both presynaptic and postsynaptic sites216. In primary cell culture models of PD, O-GlcNAcylation of α-synuclein reduced its aggregation and toxicity217. In addition, in transgenic mouse models of AD, downregulation of O-GlcNAcylation is implicated in the production of Aβ and phosphorylated tau218. Novel therapeutic agents that aim to restore O-GlcNAcylation are currently under investigation in experimental models of NDAs216–218.
Epigenetic interventions
In addition to driving the TCA cycle, acetyl-CoA is a precursor of brain lipids and a substrate for generation of acetylcholine. Acetyl-CoA is also the source of the acetyl moiety used to acetylate several enzymes that modulate glycolysis, gluconeogenesis and the TCA cycle, tau (acetylation of which promotes its aggregation) and histones219. Acetylation is a core component of histone modification, which regulates gene expression, so cellular energetics are affected by the availability of acetyl-CoA for histone acetyltransferases220,221. Accordingly, acetyl-CoA provides a direct link between brain energy balance and the epigenetic control of gene expression — a link reinforced by other components of the TCA cycle, including the intermediates succinate and citrate219. Furthermore, increased activity of the deacetylating enzyme sirtuin 1 promotes mitochondrial function98 and is implicated in the positive influence of the exercise-induced increase in brain lactate levels on cognition222. Specific histone deacetylases in the hippocampus may contribute to the increased resilience to stress mediated by lactate in mice223.
Short-chain fatty acids produced by the intestinal microbiota also influence the activity of histone deacetylases44. For example, BHB modulates the β-hydroxybutyrylation of histones at lysine residues, which couples metabolic status to the control of gene expression224. Post-translational histone modifications and DNA methylation influence bioenergetic processes that are disrupted in NDAs, so pharmacological modulation of these epigenetic mechanisms could potentially improve brain energy status in NDAs220,221.
RNA-based and DNA-based therapies
Diverse techniques are being developed to alter the level of mRNA encoding proteins that are anomalously expressed in NDAs. These strategies could suppress neurotoxic effects or compensate for a loss of physiological function, thereby improving the energetic status of the brain. Targeting specific classes of microRNAs and long non-coding RNAs that control the translation of dysregulated glycolytic and ATP-generating mitochondrial proteins should also be feasible.
Recent clinical success with oligonucleotide-based therapies in central nervous system disorders such as spinal muscular atrophy225,226 and advances in the manipulation of oligonucleotides, such as antagomirs and locked nucleic acids, make this approach increasingly relevant to NDAs, even for hitherto ‘undruggable’ targets227–229. In HD, clinical trials of antisense oligomers directed against mutant HTT mRNA are under way to prevent its interference with mitochondrial transport and function230. In addition, allele-specific strategies that specifically decrease mutant HTT mRNA while preserving normal HTT mRNA are under investigation. Some of these interventions use zinc-finger nucleases (which act as transcription factors), whereas others rely on small molecules that promote clearance specifically of the mutant proteins231. Antisense oligomers and similar approaches could also be used to target genes containing mutations that disrupt mitochondrial energetics in PD and ALS with FTD232,233 (Supplementary Table 1).
Oligonucleotides and small interfering RNAs that modulate pre-mRNA splicing or neutralize mRNA-directed microRNAs preferentially increase the expression of the intact allele of energy-generating genes downregulated in NDAs226,234. A more direct mode of gene therapy that aims to restore abnormally low or absent gene expression is to transfer copies of the intact gene into the brain using an adeno-associated virus vector. For example, the approved gene therapy onasemnogene abeparvovec uses an adeno-associated virus vector to deliver intact SMN1 gene copies to motor neurons to treat spinal muscular atrophy235. An adeno-associated virus vector loaded with the human GLUT1 promoter injected directly into the brains of GLUT1-deficient mice led to robust GLUT1 expression in corticolimbic regions, together with increased cerebrospinal fluid glucose levels and improved motor function236. A similar strategy that targeted dysfunctional PGC1α helped to restore mitochondrial function in dopaminergic pathways in mouse models of PD237.
DNA and RNA editing might also become options for the treatment of NDAs, for example, using zinc-finger nucleases or CRISPR–Cas9 technologies. One specific approach to improve brain energetics in AD is the conversion of the ApoE4 isoform into ApoE3 as shown in a neuronal cell line234,238,239. Gene editing of APOE has not yet been achieved in vivo but progress is rapid in this field and a broad range of options for improving glucose metabolism and other abnormalities associated with AD based on neutralization of ApoE4 are being investigated240.
Mitochondrial dysfunction and other disturbances associated with HD may also be linked to excessive translation of mRNAs and overproduction of proteins resulting from inactivation of the eukaryotic translation initiation factor 4E (EIF4E) translational repressor complex. Rapamycin and other repressors of this complex or its components should therefore be assessed for restoration of mitochondrial energetics and integrity in NDAs241. Another approach could be so-called autophagy-targeting chimaeras to specifically target and eliminate fragmented mitochondria. In a proof-of-concept study, this approach restored the generation of functional mitochondria and ATP levels in fibroblasts from patients with Down syndrome, who invariably develop AD242.
Photobiomodulation therapy
Low wavelengths of light penetrate brain tissue to a considerable depth, and transcranial (intracranial, intra-aural or intranasal) application of near-infrared light is being studied as a treatment for various brain disorders243,244. The mechanisms underlying the positive effects of photobiomodulation therapy await further elucidation, but increased brain perfusion, energy availability and oxygen supply have been proposed, in addition to neuronal actions implicating light-absorbing cytochrome c and increased ATP production243–245. The use of photobiomodulation to improve brain energetics has received initial support from small-scale clinical trials243,244,246, but rigorous controlled studies with larger sample sizes are needed. An alternative strategy to improve the energy status of the brain could be the use of non-invasive light and/or auditory stimulation regimes. In a mouse model of AD, this approach reduced pathology and reduced the levels of neurotoxic proteins, in part by favouring neurovascular coupling and, by inference, brain energetic status247.
Conclusions and outlook
Impaired brain energy metabolism is now recognized in NDAs and, at least in AD, clearly precedes the onset of clinical symptoms. The metabolic defects occur at multiple levels, including reduced neuronal glucose uptake, impaired glycolysis and suboptimal function of the TCA cycle, all of which adversely impact axonal transport, mitochondrial function and ATP production (FIG. 3a). The multiple faces of brain glucose hypometabolism present challenges for drug development in NDAs. Indeed, in view of the multiple brain energetic pathways affected, a fundamental question is whether pharmacological approaches that target a single enzyme, receptor or protein could ever be truly clinically effective. By analogy to other multifactorial disorders associated with an increased risk of NDAs, such as depression and T2D248, where multimodal interventions are the most effective, the same could turn out to be true for brain energetic rescue in NDAs. An example would be use of agents that simultaneously clear aggregated toxic proteins and/or suppress reactive oxygen species and neuroinflammation2,249,250.
The efficacy of a multimodal approach depends on a better understanding of the cause-and-effect relationships between the brain glucose deficit and pathophysiological processes1,2,103. In any event, attempting to promote energetically expensive processes such as microglial clearance, synaptic remodelling, myelin regeneration or axonal transport seems questionable unless the brain has adequate energy resources to fuel the additional work. Therefore, optimization of brain energetics should become a core component of future clinical trials of potential therapies for NDAs, irrespective of their mechanisms of action, because unless the brain energy gap can be reduced (FIG. 3), potential benefits of new medications may well be missed. Furthermore, a multipronged approach embracing both targeted pharmaceutical treatments and broad improvement in diet and lifestyle is emerging as a viable way to improve prognosis and relieve clinical symptoms of NDAs2,5,249,251. Insights could also be obtained by considering brain energetics in other neurological diseases (BOX 5).
Box 5 |. Brain energy rescue in other central nervous system disorders.
Owing to the success of combinations of antiretroviral drugs, patients infected with HIV are living longer. However, reflecting persistent neurological sequelae of the virus, about half of them develop cognitive deficits involving impaired glucose metabolism, mitochondrial dysfunction and reduced glial support of neurons. Brain energy rescue therefore might be useful to treat cognitive decline in these patients, and in people with long-term neurological repercussions of other classes of viral infection294.
Prions are infectious particles that lack DNA or RNA, and in the degenerative prion disorder Creutzfeldt–Jakob disease, brain glucose hypometabolism is seen in frontal and parietal regions and is related to sensory and motor dysfunction295. Lower brain perfusion and mitochondrial dysfunction are also observed102. Mouse models of prion diseases mimic clinical cases in displaying altered metabolism of glucose and fatty acids296. By binding to oligomeric amyloid-β, prion antagonists suppress synaptic pathology and cognitive deficits in mouse models of AD, underpinning the pertinence of prion disorders to neurodegenerative disorders of ageing297.
Both ischaemic stroke (caused by blood vessel occlusion) and haemorrhagic stroke (caused by blood vessel rupture) involve an abrupt interruption of the supply of energy and oxygen to neurons, triggering neurodegeneration, acute brain energetic failure and functional deficits. Stroke management is driven by the twin goals of restoring blood flow and protecting mitochondrial function60,298, in both of which brain energy rescue with ketones is being assessed299.
Like stroke, traumatic brain injury is associated not only with tissue damage but also with focal interruption of brain nutrient and energy supply. Strategies to restore mitochondrial function are under investigation in traumatic brain injury300. Clinical and preclinical studies suggest that ketogenic interventions might be therapeutically beneficial140,301. Furthermore, hypertonic sodium lactate reduces intracranial pressure and compensates for the acute neuronal energetic crisis in traumatic brain injury302.
Inherited GLUT1 deficiencies (De Vivo disease) are associated with neurodevelopmental delay, motor symptoms and seizures during infancy303. Seizures consume considerable energy, and adult-onset epilepsy is characterized by focal brain hypometabolism, decreased glucose uptake, defective tricarboxylic acid cycling and mitochondrial dysfunction34,299. Seizures also occur with greater frequency in Alzheimer disease (AD), so drugs that reinforce the GABAergic inhibition of hyperactive (energetically costly) networks47 warrant assessment in AD. In mild cognitive impairment, task-induced hyperexcitability in the temporal lobe responded favourably to levetiracetam304, an antiepileptic drug that promotes vesicular release of GABA. In a mouse model of AD that overexpresses amyloid precursor protein, the addition of pyruvate and d-β-hydroxybutyrate to the diet reduced neuronal hyperexcitation and the incidence of epileptiform activity146. Ketone-based interventions are also being studied as a therapy for seizure-related disorders in adults because ketogenic diets are a well-established therapy for De Vivo disease and intractable epilepsy in children4,252,303.
Glucose metabolism and the function of mitochondria and GABAergic interneurons are all impaired in the cortex, basal ganglia and other brain structures in schizophrenia34,227,305. As in epilepsy, ketone-based interventions have been proposed as a therapy for schizophrenia306,307 and have shown encouraging results in two clinical case reports308.
Migraine is a highly debilitating and widespread form of headache. Energy deficits and/or excessive oxidative stress within the brain are attracting attention as possible triggers and, hence, as targets for metabolically focused therapeutic interventions309.
Finally, the retina is an outpost of the brain, and considerable progress has been made in understanding and potentially treating energetic disorders of the eye, such as age-related macular degeneration310 (see also Supplementary Box 3).
Preclinical studies have demonstrated that brain energy rescue can delay the onset and/or progression of NDAs at two levels: first, by improving neuronal integrity, synaptic plasticity and neuronal–glial interactions linked to cognitive and functional deficits; and second, by disease modification, at least for metformin176 and certain ketone-based interventions146,172. The benefit of ketone-based interventions in NDAs resembles that seen in other brain disorders, including schizophrenia84 and epilepsy4,252. It is interesting that ketones and the other traditional ‘black sheep’ of energy metabolism — lactate — which have complementary roles both as signalling molecules and as neuroprotective molecules in brain energetics, have much to offer in developing brain energetic rescue strategies in NDAs3,8,253,254. Hormone-based interventions that modulate appetite and energy expenditure should also have both an early preventive influence by delaying the onset of neuropathology and symptoms and a later benefit by delaying further decline in cognitive function or other functional outcomes. Genetic, epigenetic and other novel strategies are also showing promise for improving brain energetics in NDAs.
Being able to link clinical symptomatic relief to a clinical readout or a measurable biomarker (that is, imaging, metabolite or hormone76,255) would accelerate validation of clinical effectiveness and product development. In addition, an ideal biomarker would be able to predict disease modification by an intervention. Ketone PET links brain energy status to cognitive outcomes in MCI (BOX 4), but it does not demonstrate whether disease modification occurred. Given the probable need for a long-term multimodal strategy including lifestyle intervention, successful adherence and retention in future clinical trials are ultimately likely to depend on the intervention being personalized; for example, exercise or insulin sensitizers only for those who are insulin resistant256.
In conclusion, just as normal neurocognitive development during infancy depends on adequate brain energy supply, the maintenance of cognitive performance and cerebral function during ageing is contingent on the brain continuing to successfully meet its energy needs. Guaranteeing the energy status of the brain should become a cornerstone for trials attempting to delay the onset and progression of NDAs. The observations discussed herein should help us move towards this important goal.
Supplementary Material
Acknowledgements
This article is based on the proceedings of a small, focused symposium organized by M.J.M. and supported by an unrestricted grant from Advances in Neuroscience for Medical Innovation, which is affiliated with the Institut de Recherche Servier. S.C.C. is supported by the Alzheimer’s Association (USA), the Canadian Institutes of Health Research, the Fonds de Recherche du Québec – Santé, the Natural Sciences and Engineering Research Council of Canada and Nestlé, and thanks V. St-Pierre, M. Fortier, A. Castellano, É. Myette-Côté, E. Croteau, M. Roy, M.-C. Morin and C. Vandenberghe in particular for outstanding help. M.J.M. thanks J.-M. Rivet for help with preparation of the original graphic artwork and M. Gaillot and her team for the ordering and provision of PDF documents consulted in the preparation of the manuscript. R.H.S. is supported by grants P30 AG035982, R01 AG060733 and R01 AG061194 from the US National Institutes of Health (NIH). E.T. is supported by the NIH National Institute on Aging (grant RF1AG55549) and National Institute of Neurological Disorders and Stroke (grants R01NS107265 and RO1AG062135). Z.B.A. is supported by a Senior Research Fellowship from the National Health and Medical Research Council of Australia (APP1154974). P.I.M. is supported by funding from the Alzheimer’s Association (NIRG-13-282387), European Regional Development Fund funds through the operational programme ‘Thematic Factors of Competitiveness’ and by the Portuguese Foundation for Science and Technology (grants PEst-C/SAU/LA0001/2013-2014 and UIDB/04539/2020). G.C. is supported by the NIH (grants 1R15AG050292 and 1R21AG064479). R.D.B. is supported by the National Institute on Aging (grants R37AG053589, R01AG057931 and P01-AG026572). J.H.J. is supported by grants from the Canadian Institutes of Health Research, Alberta Prion Research Institute, the Alzheimer’s Society of Alberta and Northwest Territories and the University Hospital Foundation (Edmonton, AB, Canada). L.H.B. holds grants from Nasjonalforeningen-Demensforbundet, Norway. A.E. is supported by the Swiss National Science Foundation (grant 31003A-179294). O.K. is supported by the Deutsche Forschungsgemeinschaft (grant CRC 1134, B02). M.P.M. is supported by the UK Medical Research Council (MC U105663142) and by a Wellcome Trust Investigator Award (110159/Z/15/Z). F.S. is funded by the European Research Council under the European Union’s Horizon 2020 research and innovation programme (ADG grant agreement no. 834317). W.F.M. is supported by the European Research Council (ADG 666053 and VW 93046). A.P. is supported by the Deutsche Forschungsgemeinschaft (PR1527/5-1) and the German Federal Ministry of Education and Research (AZ.031A318 and 031L0211). K.A.N. is supported by the European Research Council (ADG 671048) and the Adelson Medical Research Foundation. C.M. was supported by the Research Council of Norway: 262647/F20.
Competing interests
S.C.C. declares that he has consulted for and has received honoraria, test products and/or research funding from Abitec, Accera, Bulletproof, Nestlé and Servier, is the founder of Senotec and is co-inventor on a patent for a medium chain triglyceride formulation. M.J.M. declares that he is a full-time employee of Servier and has no other interests to declare. M.P.M. declares that he holds patents related to therapies targeted at decreasing oxidative damage to mitochondria. G.C. declares that she holds a patent related to compositions and methods for treating cognitive deficits using amylin and other hormones. A.E. declares that she has received honoraria, test products and/or research funding from Schwabe and Vifor. F.S. declares that he has consulted for Servier and TEVA. R.D.B. declares that she holds patents for therapeutics targeting Alzheimer disease and neurodegenerative disorders of ageing and is the founder of NeuTherapeutics. E.T. declares that she holds a patent related to compositions and methods for treating cognitive deficit using complex I inhibitors. All other authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/s41573-020-0072-x.
References
- 1.Aldana BI Microglia-specific metabolic changes in neurodegeneration. J. Mol. Biol. 431, 1830–1842 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Boland B et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug. Discov. 17, 660–688 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cunnane SC et al. Can ketones help rescue brain fuel supply in later life? Implications for cognitive health during aging and the treatment of Alzheimer’s disease. Front. Mol. Neurosci 10.3389/fnmol.2016.00053 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zilberter Y & Zilberter M The vicious circle of hypometabolism in neurodegenerative diseases: ways and mechanisms of metabolic correction. J. Neurosci. Res. 95, 2217–2235 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Camandola S & Mattson MP Brain metabolism in health, aging, and neurodegeneration. EMBO J. 36, 1474–1492 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilson H, Pagano G & Politis M Dementia spectrum disorders: lessons learnt from decades with pet research. J. Neural Transm. 126, 233–251 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson ECB et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med 10.1038/s41591-020-0815-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Magistretti PJ & Allaman I Lactate in the brain: from metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 19, 235–249 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Wang A, Luan HH & Medzhitov R An evolutionary perspective on immunometabolism. Science 363, eaar3932 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tups A, Benzler J, Sergi D, Ladyman SR & Williams LM Central regulation of glucose homeostasis. Compr. Physiol. 7, 741–764 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Caron A & Richard D Neuronal systems and circuits involved in the control of food intake and adaptive thermogenesis. Ann. N. Y. Acad. Sci. 1391, 35–53 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Dodd GT et al. Insulin regulates POMC neuronal plasticity to control glucose metabolism. Elife 10.7554/eLife.38704 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oyarzabal A & Marin-Valencia I Synaptic energy metabolism and neuronal excitability, in sickness and health. J. Inherit. Metab. Dis. 42, 220–236 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Bordone MP et al. The energetic brain - a review from students to students. J. Neurochem. 151, 139–165 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Dienel GA Brain glucose metabolism: integration of energetics with function. Physiol. Rev. 99, 949–1045 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Engl E & Attwell D Non-signalling energy use in the brain. J. Physiol. 593, 3417–3429 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ashrafi G, Wu Z, Farrell RJ & Ryan TA GLUT4 mobilization supports energetic demands of active synapses. Neuron 93, 606–615.e603 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gundersen V, Storm-Mathisen J & Bergersen LH Neuroglial transmission. Physiol. Rev. 95, 695–726 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Cheng J et al. Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol. 136, 507–523 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lecrux C, Bourourou M & Hamel E How reliable is cerebral blood flow to map changes in neuronal activity? Autonomic Neurosci 217, 71–79 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Saab Aiman S. et al. Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron 91, 119–132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pearson-Leary J, Jahagirdar V, Sage J & McNay EC Insulin modulates hippocampally-mediated spatial working memory via glucose transporter-4. Behav. Brain Res. 338, 32–39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barros LF, Brown A & Swanson RA Glia in brain energy metabolism: a perspective. Glia 66, 1134–1137 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waitt AE, Reed L, Ransom BR & Brown AM Emerging roles for glycogen in the CNS. Front. Mol. Neurosci 10.3389/fnmol.2017.00073 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nave K-A & Werner HB Myelination of the nervous system: mechanisms and functions. Annu. Rev. Cell Dev. Biol. 30, 503–533 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Tomassy GS et al. Distinct profiles of myelin distribution along single axons of pyramidal neurons in the neocortex. Science 344, 319–324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amaral AI, Hadera MG, Kotter M & Sonnewald U Oligodendrocytes do not export NAA-derived aspartate in vitro. Neurochem. Res. 42, 827–837 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trevisiol A et al. Monitoring ATP dynamics in electrically active white matter tracts. eLife 10.7554/elife.24241 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hinckelmann M-V et al. Self-propelling vesicles define glycolysis as the minimal energy machinery for neuronal transport. Nat. Commun 10.1038/ncomms13233 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deczkowska A et al. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell 173, 1073–1081 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Frere S & Slutsky I Alzheimer’s disease: from firing instability to homeostasis network collapse. Neuron 97, 32–58 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Jessen SB, Mathiesen C, Lind BL & Lauritzen M Interneuron deficit associates attenuated network synchronization to mismatch of energy supply and demand in aging mouse brains. Cereb. Cortex 27, 646–659 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Micheva KD et al. Distinctive structural and molecular features of myelinated inhibitory axons in human neocortex. eNeuro 10.1523/eneuro.0297-18.2018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kann O The interneuron energy hypothesis: implications for brain disease. Neurobiol. Dis. 90, 75–85 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Illarioshkin SN, Klyushnikov SA, Vigont VA, Seliverstov YA & Kaznacheyeva EV Molecular pathogenesis in Huntington’s disease. Biochemistry 83, 1030–1039 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Griffith CM, Eid T, Rose GM & Patrylo PR Evidence for altered insulin receptor signaling in Alzheimer’s disease. Neuropharmacology 136, 202–215 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Duarte AI, Santos MS, Oliveira CR & Moreira PI Brain insulin signalling, glucose metabolism and females’ reproductive aging: a dangerous triad in Alzheimer’s disease. Neuropharmacology 136, 223–242 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Craft S et al. Effects of regular and long-acting insulin on cognition and Alzheimer’s disease biomarkers: a pilot clinical trial. J. Alzheimers Dis. 57, 1325–1334 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bak LK, Walls AB, Schousboe A & Waagepetersen HS Astrocytic glycogen metabolism in the healthy and diseased brain. J. Biol. Chem. 293, 7108–7116 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cunnane SC & Crawford MA Energetic and nutritional constraints on infant brain development: implications for brain expansion during human evolution. J. Hum. Evol. 77, 88–98 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Courchesne-Loyer A et al. Inverse relationship between brain glucose and ketone metabolism in adults during short-term moderate dietary ketosis: a dual tracer quantitative positron emission tomography study. J. Cereb. Blood Flow. Metab. 37, 2485–2493 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cani PD Is colonic propionate delivery a novel solution to improve metabolism and inflammation in overweight or obese subjects? Gut 68, 1352–1353 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spielman LJ, Gibson DL & Klegeris A Unhealthy gut, unhealthy brain: the role of the intestinal microbiota in neurodegenerative diseases. Neurochem. Int. 120, 149–163 (2018). [DOI] [PubMed] [Google Scholar]
- 44.de Vadder F & Mithieux G Gut-brain signaling in energy homeostasis: the unexpected role of microbiota-derived succinate. J. Endocrinol. 236, R105–R108 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Olson CA et al. The gut microbiota mediates the anti-seizure effects of the ketogenic diet. Cell 174, 497 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soty M, Gautier-Stein A, Rajas F & Mithieux G Gut-brain glucose signaling in energy homeostasis. Cell Metab. 25, 1231–1242 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Zott B, Busche MA, Sperling RA & Konnerth A What happens with the circuit in Alzheimer’s disease in mice and humans? Annu. Rev. Neurosci. 41, 277–297 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu L, Shen Z, Wang C & Yu Y Efficient coding and energy efficiency are promoted by balanced excitatory and inhibitory synaptic currents in neuronal network. Front. Cell Neurosci. 12, 123 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ta T-T et al. Priming of microglia with IFN-γ slows neuronal gamma oscillations in situ. Proc. Natl Acad. Sci. USA 116, 4637–4642 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Briston T & Hicks AR Mitochondrial dysfunction and neurodegenerative proteinopathies: mechanisms and prospects for therapeutic intervention. Biochem. Soc. Trans. 46, 829–842 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oliveira LT et al. Exogenous β-amyloid peptide interferes with GLUT4 localization in neurons. Brain Res. 1615, 42–50 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Ryu JC, Zimmer ER, Rosa-Neto P & Yoon SO Consequences of metabolic disruption in Alzheimer’s disease pathology. Neurotherapeutics 16, 600–610 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.An Y et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimers Dement. 14, 318–329 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Toppala S et al. Midlife insulin resistance as a predictor for late-life cognitive function and cerebrovascular lesions. J. Alzheimers Dis. 72, 215–228 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bartzokis G Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiol. Aging 32, 1341–1371 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klosinski LP et al. White matter lipids as a ketogenic fuel supply in aging female brain: Implications for Alzheimer’s disease. EBioMedicine 2, 1888–1904 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roy M et al. Fascicle- and glucose-specific deterioration in white matter energy supply in Alzheimer’s disease. J. Alzheimer’s Dis, in the press. [DOI] [PubMed] [Google Scholar]
- 58.Matthews DC et al. FDG PET Parkinson’s disease-related pattern as a biomarker for clinical trials in early stage disease. Neuroimage Clin 20, 572–579 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chu JS et al. The metabolic activity of caudate and prefrontal cortex negatively correlates with the severity of idiopathic Parkinson’s disease. Aging Dis. 10, 847–853 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murphy MP & Hartley RC Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug. Discov. 17, 865–886 (2018). [DOI] [PubMed] [Google Scholar]
- 61.Zambon F et al. Cellular α-synuclein pathology is associated with bioenergetic dysfunction in Parkinson’s iPSC-derived dopamine neurons. Hum. Mol. Genet. 28, 2001–2013 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McColgan P et al. Brain regions showing white matter loss in Huntington’s disease are enriched for synaptic and metabolic genes. Biol. Psychiatry 83, 456–465 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morea V et al. Glucose transportation in the brain and its impairment in Huntington disease: one more shade of the energetic metabolism failure? Amino Acids 49, 1147–1157 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Liot G, Valette J, Pepin J, Flament J & Brouillet E Energy defects in Huntington’s disease: why “in vivo” evidence matters. Biochem. Biophys. Res. Commun. 483, 1084–1095 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Polyzos AA et al. Metabolic reprogramming in astrocytes distinguishes region-specific neuronal susceptibility in Huntington mice. Cell Metab. 29, 1258–1273.e1211 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kedaigle AJ et al. Bioenergetic deficits in Huntington’s disease iPSC-derived neural cells and rescue with glycolytic metabolites. Hum. Mol. Genet. 10.1093/hmg/ddy430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saudou F & Humbert S The biology of huntingtin. Neuron 89, 910–926 (2016). [DOI] [PubMed] [Google Scholar]
- 68.Ahmed RM et al. Amyotrophic lateral sclerosis and frontotemporal dementia: distinct and overlapping changes in eating behaviour and metabolism. Lancet Neurol. 15, 332–342 (2016). [DOI] [PubMed] [Google Scholar]
- 69.Jawaid A, Khan R, Polymenidou M & Schulz PE Disease-modifying effects of metabolic perturbations in ALS/FTLD. Mol. Neurodegener 10.1186/s13024-018-0294-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vandoorne T, De Bock K & Van Den Bosch L Energy metabolism in ALS: an underappreciated opportunity? Acta Neuropathol. 135, 489–509 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tefera TW, Bartlett K, Tran SS, Hodson MP & Borges K Impaired pentose phosphate pathway in the spinal cord of the hSOD1G93A mouse model of amyotrophic lateral sclerosis. Mol. Neurobiol. 56, 5844–5855 (2019). [DOI] [PubMed] [Google Scholar]
- 72.Lau DHW et al. Disruption of ER−mitochondria signalling in fronto-temporal dementia and related amyotrophic lateral sclerosis. Cell Death Dis 10.1038/s41419-017-0022-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Delic V et al. Discrete mitochondrial aberrations in the spinal cord of sporadic ALS patients. J. Neurosci. Res. 96, 1353–1366 (2018). [DOI] [PubMed] [Google Scholar]
- 74.Tefera TW & Borges K Metabolic dysfunctions in amyotrophic lateral sclerosis pathogenesis and potential metabolic treatments. Front. Neurosci 10.3389/fnins.2016.00611 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Allen SP et al. C9orf72 expansion within astrocytes reduces metabolic flexibility in amyotrophic lateral sclerosis. Brain 142, 3771–3790 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sintini I et al. Regional multimodal relationships between tau, hypometabolism, atrophy, and fractional anisotropy in atypical Alzheimer’s disease. Hum. Brain Mapp 10.1002/hbm.24473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carbonell F, Zijdenbos AP & Bedell BJ Spatially distributed amyloid-β reduces glucose metabolism in mild cognitive impairment. J. Alzheimers Dis. 73, 543–557 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Butterfield DA & Halliwell B Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 20, 148–160 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Velliquette RA, O’Connor T & Vassar R Energy inhibition elevates β-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer’s disease pathogenesis. J. Neurosci. 25, 10874–10883 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Correia SC, Perry G & Moreira PI Mitochondrial traffic jams in Alzheimer’s disease - pinpointing the roadblocks. Biochim. Biophys. Acta - Mol. Basis Dis 1862, 1909–1917 (2016). [DOI] [PubMed] [Google Scholar]
- 81.Ashraf A, Fan Z, Brooks DJ & Edison P Cortical hypermetabolism in mci subjects: a compensatory mechanism? Eur. J. Nucl. Med. Mol. Imaging 42, 447–458 (2014). [DOI] [PubMed] [Google Scholar]
- 82.Li H, Liu C-C, Zheng H & Huang TY Amyloid, tau, pathogen infection and antimicrobial protection in Alzheimer’s disease –conformist, nonconformist, and realistic prospects for ad pathogenesis. Transl. Neurodegener 10.1186/s40035-018-0139-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fülöp T, Larbi A & Witkowski JM Human inflammaging. Gerontology 65, 495–504 (2019). [DOI] [PubMed] [Google Scholar]
- 84.Millan MJ, Rivet J-M & Gobert A The frontal cortex as a network hub controlling mood and cognition: probing its neurochemical substrates for improved therapy of psychiatric and neurological disorders. J. Psychopharmacol. 30, 1099–1128 (2016). [DOI] [PubMed] [Google Scholar]
- 85.Yang L, Wang H, Liu L & Xie A The role of insulin/IGF-1/PI3K/AKT/GSK3β signaling in Parkinson’s disease dementia. Front. Neurosci. 10.3389/fnins.2018.00073 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Forny-Germano L, De Felice FG & Vieira M. N. d. N. The role of leptin and adiponectin in obesity-associated cognitive decline and Alzheimer’s disease. Front. Neurosci 12, 1027 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McGregor G & Harvey J Regulation of hippocampal synaptic function by the metabolic hormone, leptin: implications for health and neurodegenerative disease. Front. Cell. Neurosci 10.3389/fncel.2018.00340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shi L, Du X, Jiang H & Xie J Ghrelin and neurodegenerative disorders—a review. Mol. Neurobiol. 54, 1144–1155 (2016). [DOI] [PubMed] [Google Scholar]
- 89.Suda Y et al. Down-regulation of ghrelin receptors on dopaminergic neurons in the substantia nigra contributes to Parkinson’s disease-like motor dysfunction. Mol. Brain 10.1186/s13041-018-0349-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Frago L & Chowen J Involvement of astrocytes in mediating the central effects of ghrelin. Int. J. Mol. Sci. 18, 536 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fu W, Patel A, Kimura R, Soudy R & Jhamandas JH Amylin receptor: a potential therapeutic target for Alzheimer’s disease. Trends Mol. Med. 23, 709–720 (2017). [DOI] [PubMed] [Google Scholar]
- 92.Grizzanti J, Corrigan R, Servizi S & Casadesus G Amylin signaling in diabetes and Alzheimer’s disease: therapy or pathology? J. Neurol. Neuromedicine 4, 12–16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mietlicki-Baase EG Amylin in Alzheimer’s disease: pathological peptide or potential treatment? Neuropharmacology 136, 287–297 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim MW et al. Suppression of adiponectin receptor 1 promotes memory dysfunction and Alzheimer’s disease-like pathologies. Sci. Rep 10.1038/s41598-017-12632-9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ngo ST et al. Altered expression of metabolic proteins and adipokines in patients with amyotrophic lateral sclerosis. J. Neurological. Sci. 357, 22–27 (2015). [DOI] [PubMed] [Google Scholar]
- 96.Ma J et al. Peripheral blood adipokines and insulin levels in patients with Alzheimer’s disease: a replication study and meta-analysis. Curr. Alzheimer Res. 13, 223–233 (2016). [DOI] [PubMed] [Google Scholar]
- 97.Athauda D & Foltynie T The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson’s disease: mechanisms of action. Drug Discov. Today 21, 802–818 (2016). [DOI] [PubMed] [Google Scholar]
- 98.Mattson MP, Moehl K, Ghena N, Schmaedick M & Cheng A Intermittent metabolic switching, neuroplasticity and brain health. Nat. Rev. Neurosci. 19, 81–94 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nugent S et al. Glucose hypometabolism is highly localized, but lower cortical thickness and brain atrophy are widespread in cognitively normal older adults. Am. J. Physiol. Endocrinol. Metab 306, E1315–E1321 (2014). [DOI] [PubMed] [Google Scholar]
- 100.Castellano C-A et al. Links between metabolic and structural changes in the brain of cognitively normal older adults: a 4-year longitudinal follow-up. Front. Aging Neurosci 10.3389/fnagi.2019.00015 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Goyal MS et al. Loss of brain aerobic glycolysis in normal human aging. Cell Metab. 26, 353–360.e353 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.de la Torre JC Are major dementias triggered by poor blood flow to the brain? Theoretical considerations. J. Alzheimers Dis. 57, 353–371 (2017). [DOI] [PubMed] [Google Scholar]
- 103.Sweeney MD et al. Vascular dysfunction-the disregarded partner of Alzheimer’s disease. Alzheimers Dement. 15, 158–167 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wingo AP et al. Large-scale proteomic analysis of human brain identifies proteins associated with cognitive trajectory in advanced age. Nat. Commun 10.1038/s41467-019-09613-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.de la Monte SM The full spectrum of Alzheimer’s disease is rooted in metabolic derangements that drive type 3 diabetes. Adv. Exp. Med. Biol. 1128. 45–83 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li S & Laher I Exercise pills: at the starting line. Trends Pharmacol. Sci. 36, 906–917 (2015). [DOI] [PubMed] [Google Scholar]
- 107.Vieira MNN, Lima-Filho RAS & De Felice FG Connecting Alzheimer’s disease to diabetes: underlying mechanisms and potential therapeutic targets. Neuropharmacology 136, 160–171 (2018). [DOI] [PubMed] [Google Scholar]
- 108.Castellano C-A et al. A 3-month aerobic training program improves brain energy metabolism in mild Alzheimer’s disease: preliminary results from a neuroimaging study. J. Alzheimers Dis. 56, 1459–1468 (2017). [DOI] [PubMed] [Google Scholar]
- 109.Penninx B & Lange SMM Metabolic syndrome in psychiatric patients: overview, mechanisms, and implications. Dialogues Clin. Neurosci. 20, 63–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Verdile G, Fuller SJ & Martins RN The role of type 2 diabetes in neurodegeneration. Neurobiol. Dis. 84, 22–38 (2015). [DOI] [PubMed] [Google Scholar]
- 111.Mosconi L et al. Increased Alzheimer’s risk during the menopause transition: a 3-year longitudinal brain imaging study. PLoS ONE 13, e0207885 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Brinton RD, Yao J, Yin F, Mack WJ & Cadenas E Perimenopause as a neurological transition state. Nat. Rev. Endocrinol 11, 393–405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nuriel T et al. Neuronal hyperactivity due to loss of inhibitory tone in apoe4 mice lacking Alzheimer’s disease-like pathology. Nat. Commun 10.1038/s41467-017-01444-0 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wu L, Zhang X & Zhao L Human ApoE isoforms differentially modulate brain glucose and ketone body metabolism: implications for Alzheimer’s disease risk reduction and early intervention. J. Neurosci. 38, 6665–6681 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhao N et al. Apolipoprotein e4 impairs neuronal insulin signaling by trapping insulin receptor in the endosomes. Neuron 96, 115–129 e115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Johnson LA et al. Apolipoprotein E4 mediates insulin resistance-associated cerebrovascular dysfunction and the post-prandial response. J. Cereb. Blood Flow. Metab. 39, 770–781 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nakamura T, Watanabe A, Fujino T, Hosono T & Michikawa M Apolipoprotein E4 (1–272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Mol. Neurodegener. 4, 35 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Orr AL et al. Neuronal apolipoprotein E4 expression results in proteome-wide alterations and compromises bioenergetic capacity by disrupting mitochondrial function. J. Alzheimers Dis. 68, 991–1011 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Henderson ST et al. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 6, 31 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xu Q et al. Medium-chain triglycerides improved cognition and lipid metabolomics in mild to moderate Alzheimer’s disease patients with APOE4−/−: a double-blind, randomized, placebo-controlled crossover trial. Clin. Nutr 10.1016/j.clnu.2019.10.017 (2019). [DOI] [PubMed] [Google Scholar]
- 121.Larsen SB, Hanss Z & Krüger R The genetic architecture of mitochondrial dysfunction in Parkinson’s disease. Cell Tissue Res. 373, 21–37 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Funk N et al. The Parkinson’s disease-linked leucine-rich repeat kinase 2 (LRRK2) is required for insulin-stimulated translocation of GLUT4. Sci. Rep 9, 4515 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Joardar A, Manzo E & Zarnescu DC Metabolic dysregulation in amyotrophic lateral sclerosis: challenges and opportunities. Curr. Genet. Med. Rep. 5, 108–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Diehl-Schmid J et al. FDG-PET underscores the key role of the thalamus in frontotemporal lobar degeneration caused by C9ORF72 mutations. Transl. Psychiatry 10.1038/s41398-019-0381-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Koppel SJ & Swerdlow RH Neuroketotherapeutics: a modern review of a century-old therapy. Neurochem. Int. 117, 114–125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ngandu T et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet 385, 2255–2263 (2015). [DOI] [PubMed] [Google Scholar]
- 127.Gaitan JM et al. Two weeks of interval training enhances fat oxidation during exercise in obese adults with prediabetes. J. Sports Sci. Med. 18, 636–644 (2019). [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang L et al. Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer’s disease. EBioMedicine 2, 294–305 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Flannery PJ & Trushina E Mitochondrial dynamics and transport in Alzheimer’s disease. Mol. Cell. Neurosci. 98, 109–120 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Baumgart M et al. Longitudinal RNA-seq analysis of vertebrate aging identifies mitochondrial complex I as a small-molecule-sensitive modifier of lifespan. Cell Syst. 2, 122–132 (2016). [DOI] [PubMed] [Google Scholar]
- 131.Raule N et al. The co-occurrence of mtDNA mutations on different oxidative phosphorylation subunits, not detected by haplogroup analysis, affects human longevity and is population specific. Aging Cell 13, 401–407 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Herzig S & Shaw RJ AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 19, 121–135 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Burte F, Carelli V, Chinnery PF & Yu-Wai-Man P Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 11, 11–24 (2015). [DOI] [PubMed] [Google Scholar]
- 134.Manczak M, Kandimalla R, Yin X & Reddy PH Mitochondrial division inhibitor 1 reduces dynamin-related protein 1 and mitochondrial fission activity. Hum. Mol. Genet. 10.1093/hmg/ddy335 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bido S, Soria FN, Fan RZ, Bezard E & Tieu K Mitochondrial division inhibitor-1 is neuroprotective in the A53T-α-synuclein rat model of Parkinson’s disease. Sci. Rep. 7, 7495 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Baek SH et al. Inhibition of Drp1 ameliorates synaptic depression, Aβ deposition, and cognitive impairment in an Alzheimer’s disease model. J. Neurosci. 37, 5099–5110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wilkins HM et al. A mitochondrial biomarker-based study of s-equol in Alzheimer’s disease subjects: results of a single-arm, pilot trial. J. Alzheimers Dis. 59, 291–300 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wu B et al. 2,4 DNP improves motor function, preserves medium spiny neuronal identity, and reduces oxidative stress in a mouse model of Huntington’s disease. Exp. Neurol. 293, 83–90 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Dickey AS et al. PPARδ activation by bexarotene promotes neuroprotection by restoring bioenergetic and quality control homeostasis. Sci. Transl. Med 10.1126/scitranslmed.aal2332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xin L et al. Nutritional ketosis increases NAD+/NADH ratio in healthy human brain: an in vivo study by 31P-MRS. Front. Nutr 10.3389/fnut.2018.00062 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dellinger RW et al. Repeat dose NRPT (nicotinamide riboside and pterostilbene) increases NAD+ levels in humans safely and sustainably: a randomized, double-blind, placebo-controlled study. NPJ Aging Mech. Dis. 10.1038/s41514-017-0016-9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hou Y et al. NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new ad mouse model with introduced DNA repair deficiency. Proc. Natl Acad. Sci. USA 115, E1876–E1885 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Schöndorf DC et al. The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and fly models of Parkinson’s disease. Cell Rep. 23, 2976–2988 (2018). [DOI] [PubMed] [Google Scholar]
- 144.Wilkins HM et al. Oxaloacetate enhances neuronal cell bioenergetic fluxes and infrastructure. J. Neurochem. 137, 76–87 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cai R et al. Enhancing glycolysis attenuates Parkinson’s disease progression in models and clinical databases. J. Clin. Invest. 129, 4539–4549 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zilberter M et al. Dietary energy substrates reverse early neuronal hyperactivity in a mouse model of Alzheimer’s disease. J. Neurochem. 125, 157–171 (2013). [DOI] [PubMed] [Google Scholar]
- 147.Theurey P et al. Systems biology identifies preserved integrity but impaired metabolism of mitochondria due to a glycolytic defect in Alzheimer’s disease neurons. Aging Cell 18, e12924 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Roy M et al. Rapid adaptation of rat brain and liver metabolism to a ketogenic diet: an integrated study using 1H- and 13C-NMR spectroscopy. J. Cereb. Blood Flow. Metab. 35, 1154–1162 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.St-Pierre V et al. Plasma ketone and medium chain fatty acid response in humans consuming different medium chain triglycerides during a metabolic study day. Front. Nutr. 6, 46 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tan KN, Carrasco-Pozo C, McDonald TS, Puchowicz M & Borges K Tridecanoin is anticonvulsant, antioxidant, and improves mitochondrial function. J. Cereb. Blood Flow. Metab. 37, 2035–2048 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Schwarzkopf TM, Koch K & Klein J Reduced severity of ischemic stroke and improvement of mitochondrial function after dietary treatment with the anaplerotic substance triheptanoin. Neuroscience 300, 201–209 (2015). [DOI] [PubMed] [Google Scholar]
- 152.Mochel F Triheptanoin for the treatment of brain energy deficit: a 14-year experience. J. Neurosci. Res. 95, 2236–2243 (2017). [DOI] [PubMed] [Google Scholar]
- 153.Marin-Valencia I, Good LB, Ma Q, Malloy CR & Pascual JM Heptanoate as a neural fuel: energetic and neurotransmitter precursors in normal and glucose transporter I-deficient (G1D) brain. J. Cereb. Blood Flow. Metab. 33, 175–182 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fortier M et al. A ketogenic drink improves brain energy and some measures of cognition in mild cognitive impairment. Alzheimers Dement. 15, 625–634 (2019). [DOI] [PubMed] [Google Scholar]
- 155.Krikorian R et al. Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 33, 425.e419–425.e427 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Neth BJ et al. Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion, and cerebral ketone body uptake in older adults at risk for Alzheimer’s disease: a pilot study. Neurobiol. Aging 86, 54–63 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Taylor MK, Sullivan DK, Mahnken JD, Burns JM & Swerdlow RH Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimers Dement 4, 28–36 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ota M et al. Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neurosci. Lett. 690, 232–236 (2019). [DOI] [PubMed] [Google Scholar]
- 159.Avgerinos KI, Egan JM, Mattson MP & Kapogiannis D Medium chain triglycerides induce mild ketosis and may improve cognition in Alzheimer’s disease. A systematic review and meta-analysis of human studies. Ageing Res. Rev. 58, 101001 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Phillips MCL, Murtagh DKJ, Gilbertson LJ, Asztely FJS & Lynch CDP Low-fat versus ketogenic diet in Parkinson’s disease: a pilot randomized controlled trial. Mov. Disord. 33, 1306–1314 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Krikorian R et al. Nutritional ketosis for mild cognitive impairment in Parkinson’s disease: a controlled pilot trial. Clin. Parkinsonism Relat. Disord. 1, 41–47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Brandt J et al. Preliminary report on the feasibility and efficacy of the modified atkins diet for treatment of mild cognitive impairment and early Alzheimer’s disease. J. Alzheimers Dis. 68, 969–981 (2019). [DOI] [PubMed] [Google Scholar]
- 163.Shaafi S et al. The efficacy of the ketogenic diet on motor functions in Parkinson’s disease: a rat model. Iran. J. Neurol. 15, 63–69 (2016). [PMC free article] [PubMed] [Google Scholar]
- 164.Ari C et al. Metabolic therapy with Deanna protocol supplementation delays disease progression and extends survival in amyotrophic lateral sclerosis (ALS) mouse model. PLoS ONE 9, e103526 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ruskin DN et al. A ketogenic diet delays weight loss and does not impair working memory or motor function in the R6/2 1J mouse model of Huntington’s disease. Physiol. Behav. 103, 501–507 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ma D et al. Ketogenic diet enhances neurovascular function with altered gut microbiome in young healthy mice. Sci. Rep 10.1038/s41598-018-25190-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Reger MA et al. Effects of β-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 25, 311–314 (2004). [DOI] [PubMed] [Google Scholar]
- 168.Page KA et al. Medium-chain fatty acids improve cognitive function in intensively treated type 1 diabetic patients and support in vitro synaptic transmission during acute hypoglycemia. Diabetes 58, 1237–1244 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wakade C, Chong R, Bradley E, Thomas B & Morgan J Upregulation of GPR109A in Parkinson’s disease. PLoS ONE 9, e109818 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Fu S-P et al. Anti-inflammatory effects of BHBA in both in vivo and in vitro parkinson’s disease models are mediated by GPR109A-dependent mechanisms. J. Neuroinflammation 12, 9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Masino SA & Rho JM Metabolism and epilepsy: ketogenic diets as a homeostatic link. Brain Res. 1703, 26–30 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kashiwaya Y et al. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 34, 1530–1539 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Cucuzzella M, Hite A, Patterson K, Saslow L & Heath R A clinician’s guide to inpatient low carbohydrate diets for remission of type 2 diabetes: toward a standard of care protocol. Diabetes Manag. 9, 7–19 (2019). [Google Scholar]
- 174.Ritze Y et al. Metabolic and cognitive outcomes of subchronic once-daily intranasal insulin administration in healthy men. Front. Endocrinol 10.3389/fendo.2018.00663 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Rotermund C, Machetanz G & Fitzgerald JC The therapeutic potential of metformin in neurodegenerative diseases. Front. Endocrinol 10.3389/fendo.2018.00400 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ou Z et al. Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain Behav. Immun 69, 351–363 (2018). [DOI] [PubMed] [Google Scholar]
- 177.Arnoux I et al. Metformin reverses early cortical network dysfunction and behavior changes in Huntington’s disease. Elife 10.7554/eLife.38744 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Campbell JM et al. Metformin use associated with reduced risk of dementia in patients with diabetes: a systematic review and meta-analysis. J. Alzheimer’s Dis. 65, 1225–1236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Cameron AR et al. Metformin selectively targets redox control of complex I energy transduction. Redox Biol. 14, 187–197 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Rena G, Hardie DG & Pearson ER The mechanisms of action of metformin. Diabetologia 60, 1577–1585 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Coll AP et al. Gdf15 mediates the effects of metformin on body weight and energy balance. Nature 578, 444–448 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Koenig AM et al. Effects of the insulin sensitizer metformin in Alzheimer disease: pilot data from a randomized placebo-controlled crossover study. Alzheimer Dis. Assoc. Disord. 31, 107–113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Luchsinger JA et al. Metformin in amnestic mild cognitive impairment: results of a pilot randomized placebo controlled clinical trial. J. Alzheimers Dis. 51, 501–514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lin Y et al. Corrigendum: evaluation of metformin on cognitive improvement in patients with non-dementia vascular cognitive impairment and abnormal glucose metabolism. Front. Aging Neurosci. 10, 322 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Moore EM et al. Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care 36, 2981–2987 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Arnold SE et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat. Rev. Neurol 14, 168–181 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wiviott SD, Raz I & Sabatine MS Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 380, 1881–1882 (2019). [DOI] [PubMed] [Google Scholar]
- 188.Ferrannini E et al. Shift to fatty substrate utilization in response to sodium–glucose cotransporter 2 inhibition in subjects without diabetes and patients with type 2 diabetes. Diabetes 65, 1190–1195 (2016). [DOI] [PubMed] [Google Scholar]
- 189.Holst JJ & Madsbad S Semaglutide seems to be more effective the other GLP-1RAS. Ann. Transl. Med 10.21037/atm.2017.11.10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Goldenberg RM & Steen O Semaglutide: review and place in therapy for adults with type 2 diabetes. Can. J. Diabetes 43, 136–145 (2019). [DOI] [PubMed] [Google Scholar]
- 191.Batista AF, Bodart-Santos V, De Felice FG & Ferreira ST Neuroprotective actions of glucagon-like peptide-1 (GLP-1) analogues in Alzheimer’s and Parkinson’s diseases. CNS Drugs 33, 209–223 (2018). [DOI] [PubMed] [Google Scholar]
- 192.Yildirim Simsir I, Soyaltin UE & Cetinkalp S Glucagon like peptide-1 (GLP-1) likes Alzheimer’s disease. Diabetes Metab. Syndrome Clin. Res. Rev. 12, 469–475 (2018). [DOI] [PubMed] [Google Scholar]
- 193.Sayed NH et al. Vildagliptin attenuates Huntington’s disease through activation of GLP-1 receptor/PI3K/Akt/BDNF pathway in 3-nitropropionic acid rat model. Neurotherapeutics 17, 252–268 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Gejl M et al. In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: randomized, placebo-controlled, double-blind clinical trial. Front. Aging Neurosci 10.3389/fnagi.2016.00108 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Athauda D et al. Post hoc analysis of the Exenatide-PD trial—factors that predict response. Eur. J. Neurosci. 49, 410–421 (2018). [DOI] [PubMed] [Google Scholar]
- 196.Chalichem NSS, Gonugunta C, Krishnamurthy PT & Duraiswamy B DPP4 inhibitors can be a drug of choice for type 3 diabetes: a mini review. Am. J. Alzheimers Dis. Other Demen 32, 444–451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Isik AT, Soysal P, Yay A & Usarel C The effects of sitagliptin, a DPP-4 inhibitor, on cognitive functions in elderly diabetic patients with or without Alzheimer’s disease. Diabetes Res. Clin. Pract. 123, 192–198 (2017). [DOI] [PubMed] [Google Scholar]
- 198.Jalewa J, Sharma MK & Hölscher C Novel incretin analogues improve autophagy and protect from mitochondrial stress induced by rotenone in SH-SY5Y cells. J. Neurochem. 139, 55–67 (2016). [DOI] [PubMed] [Google Scholar]
- 199.Verma MK, Goel R, Nandakumar K & Nemmani KVS Effect of D-Ala 2 GIP, a stable GIP receptor agonist on MPTP-induced neuronal impairments in mice. Eur. J. Pharmacol. 804, 38–45 (2017). [DOI] [PubMed] [Google Scholar]
- 200.Pathak NM et al. Novel dual incretin agonist peptide with antidiabetic and neuroprotective potential. Biochem. Pharmacol. 155, 264–274 (2018). [DOI] [PubMed] [Google Scholar]
- 201.Hölscher C Novel dual GLP-1/GIP receptor agonists show neuroprotective effects in Alzheimer’s and Parkinson’s disease models. Neuropharmacology 136, 251–259 (2018). [DOI] [PubMed] [Google Scholar]
- 202.Tai J, Liu W, Li Y, Li L & Hölscher C Neuroprotective effects of a triple GLP-1/GIP/glucagon receptor agonist in the APP/PS1 transgenic mouse model of Alzheimer’s disease. Brain Res. 1678, 64–74 (2018). [DOI] [PubMed] [Google Scholar]
- 203.Rudenko O et al. Ghrelin-mediated improvements in the metabolic phenotype in the R6/2 mouse model of Huntington’s disease. J. Neuroendocrinol 10.1111/jne.12699 (2019). [DOI] [PubMed] [Google Scholar]
- 204.Jeong Y.-o et al. MK-0677, a ghrelin agonist, alleviates amyloid beta-related pathology in 5XFAD mice, an animal model of Alzheimer’s disease. Int. J. Mol. Sci 19, 1800 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Morgan AH, Rees DJ, Andrews ZB & Davies JS Ghrelin mediated neuroprotection - a possible therapy for Parkinson’s disease? Neuropharmacology 136, 317–326 (2018). [DOI] [PubMed] [Google Scholar]
- 206.Bayliss JA et al. Ghrelin-AMPK signaling mediates the neuroprotective effects of calorie restriction in Parkinson’s disease. J. Neurosci. 36, 3049–3063 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Parkinson Study Group. A randomized trial of relamorelin for constipation in Parkinson’s disease (MOVE-PD): trial results and lessons learned. Parkinsonism Relat. Disord. 37, 101–105 (2017). [DOI] [PubMed] [Google Scholar]
- 208.Malekizadeh Y et al. A leptin fragment mirrors the cognitive enhancing and neuroprotective actions of leptin. Cereb. Cortex 27, 4769–4782 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Fernandez-Martos CM, Atkinson RAK, Chuah MI, King AE & Vickers JC Combination treatment with leptin and pioglitazone in a mouse model of Alzheimer’s disease. Alzheimers Dement. 3, 92–106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lim MA et al. Genetically altering organismal metabolism by leptin-deficiency benefits a mouse model of amyotrophic lateral sclerosis. Hum. Mol. Genet. 23, 4995–5008 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Yoon G, Shah SA, Ali T & Kim MO The adiponectin homolog osmotin enhances neurite outgrowth and synaptic complexity via ADIPOR1/NGR1 signaling in Alzheimer’s disease. Mol. Neurobiol. 55, 6673–6686 (2018). [DOI] [PubMed] [Google Scholar]
- 212.Soudy R et al. Short amylin receptor antagonist peptides improve memory deficits in Alzheimer’s disease mouse model. Sci. Rep. 9, 10942 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Levin BE & Lutz TA Amylin and leptin: co-regulators of energy homeostasis and neuronal development. Trends Endocrinol. Metab. 28, 153–164 (2017). [DOI] [PubMed] [Google Scholar]
- 214.Patrick S et al. Neuroprotective effects of the amylin analog, pramlintide, on Alzheimer’s disease are associated with oxidative stress regulation mechanisms. J. Alzheimer’s Dis 69, 157–168 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Wang E et al. Amylin treatment reduces neuroinflammation and ameliorates abnormal patterns of gene expression in the cerebral cortex of an Alzheimer’s disease mouse model. J. Alzheimer’s Dis. 56, 47–61 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Pinho TS, Correia SC, Perry G, Ambrósio AF & Moreira PI Diminished O-GlcNAcylation in Alzheimer’s disease is strongly correlated with mitochondrial anomalies. Biochim. Biophys. Acta Mol. Basis Dis 1865, 2048–2059 (2019). [DOI] [PubMed] [Google Scholar]
- 217.Levine PM et al. α-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson’s disease. Proc. Natl Acad. Sci. USA 116, 1511–1519 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Yuzwa SA & Vocadlo DJ O-GlcNAc and neurodegeneration: biochemical mechanisms and potential roles in Alzheimer’s disease and beyond. Chem. Soc. Rev. 43, 6839–6858 (2014). [DOI] [PubMed] [Google Scholar]
- 219.Haas R et al. Intermediates of metabolism: from bystanders to signalling molecules. Trends Biochem. Sci. 41, 460–471 (2016). [DOI] [PubMed] [Google Scholar]
- 220.Cobos SN, Bennett SA & Torrente MP The impact of histone post-translational modifications in neurodegenerative diseases. Biochim. Biophys. Acta Mol. Basis Dis 1865, 1982–1991 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Narayan P & Dragunow M Alzheimer’s disease and histone code alterations. Adv. Med. Biol. 1128. 321–336 (2017). [DOI] [PubMed] [Google Scholar]
- 222.El Hayek L et al. Lactate mediates the effects of exercise on learning and memory through SIRT1-dependent activation of hippocampal brain-derived neurotrophic factor (BDNF). J. Neurosci 10.1523/jneurosci.1661-18.2019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Karnib N et al. Lactate is an antidepressant that mediates resilience to stress by modulating the hippocampal levels and activity of histone deacetylases. Neuropsychopharmacology 44, 1152–1162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Xie Z et al. Metabolic regulation of gene expression by histone lysine β-hydroxybutyrylation. Mol. Cell 62, 194–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Poletti V & Biffi A Gene-based approaches to inherited neurometabolic diseases. Hum. Gene Ther. 30, 1222–1235 (2019). [DOI] [PubMed] [Google Scholar]
- 226.Mercuri E et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 378, 625–635 (2018). [DOI] [PubMed] [Google Scholar]
- 227.Millan MJ Linking deregulation of non-coding RNA to the core pathophysiology of Alzheimer’s disease: an integrative review. Prog. Neurobiol. 156, 1–68 (2017). [DOI] [PubMed] [Google Scholar]
- 228.Fumagalli M, Lombardi M, Gressens P & Verderio C How to reprogram microglia toward beneficial functions. Glia 66, 2531–2549 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Maniati MS, Maniati M, Yousefi T, Ahmadi-Ahangar A & Tehrani SS New insights into the role of microRNAs and long noncoding RNAs in most common neurodegenerative diseases. J. Cell. Biochem. 120, 8908–8918 (2019). [DOI] [PubMed] [Google Scholar]
- 230.Wild EJ & Tabrizi SJ One decade ago, one decade ahead in Huntington’s disease. Mov. Disord. 34, 1434–1439 (2019). [DOI] [PubMed] [Google Scholar]
- 231.Zeitler B et al. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat. Med. 25, 1131–1142 (2019). [DOI] [PubMed] [Google Scholar]
- 232.Cappella M, Ciotti C, Cohen-Tannoudji M & Biferi MG Gene therapy for ALS—a perspective. Int. J. Mol. Sci 10.3390/ijms20184388 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Savitt D & Jankovic J Targeting alpha-synuclein in Parkinson’s disease: progress towards the development of disease-modifying therapeutics. Drugs 79, 797–810 (2019). [DOI] [PubMed] [Google Scholar]
- 234.Deverman BE, Ravina BM, Bankiewicz KS, Paul SM & Sah DWY Gene therapy for neurological disorders: progress and prospects. Nat. Rev. Drug. Discov. 17, 641–659 (2018). [DOI] [PubMed] [Google Scholar]
- 235.Al-Zaidy SA et al. AVXS-101 (onasemnogene abeparvovec) for SMA1: comparative study with a prospective natural history cohort. J. Neuromuscul. Dis. 6, 307–317 (2019). [DOI] [PubMed] [Google Scholar]
- 236.Nakamura S et al. Gene therapy for Glut1-deficient mouse using an adeno-associated virus vector with the human intrinsic GLUT1 promoter. J. Gene Med 20, e3013 (2018). [DOI] [PubMed] [Google Scholar]
- 237.Choong C-J & Mochizuki H Gene therapy targeting mitochondrial pathway in Parkinson’s disease. J. Neural Transm. 124, 193–207 (2016). [DOI] [PubMed] [Google Scholar]
- 238.Rohn TT, Kim N, Isho NF & Mack JM The potential of CRISPR/Cas9 gene editing as a treatment strategy for Alzheimer’s disease. J. Alzheimers Dis. Parkinsonism 10.4172/2161-0460.1000439 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Safieh M, Korczyn AD & Michaelson DM ApoE4: an emerging therapeutic target for Alzheimer’s disease. BMC Med. 17, 64 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Uddin MS et al. APOE and Alzheimer’s disease: evidence mounts that targeting APOE4 may combat Alzheimer’s pathogenesis. Mol. Neurobiol. 56, 2450–2465 (2019). [DOI] [PubMed] [Google Scholar]
- 241.Creus-Muncunill J et al. Increased translation as a novel pathogenic mechanism in Huntington’s disease. Brain 142, 3158–3175 (2019). [DOI] [PubMed] [Google Scholar]
- 242.Takhashi D et al. AUTACS: cargo-specific degraders using selective autophagy. Mol. Cell 76, 797–810 (2019). [DOI] [PubMed] [Google Scholar]
- 243.de la Torre JC Treating cognitive impairment with transcranial low level laser therapy. J. Photochem. Photobiol. B Biol. 168, 149–155 (2017). [DOI] [PubMed] [Google Scholar]
- 244.Hamblin MR Shining light on the head: photobiomodulation for brain disorders. BBA Clin. 6, 113–124 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Salehpour F et al. Transcranial low-level laser therapy improves brain mitochondrial function and cognitive impairment in D-galactose–induced aging mice. Neurobiol. Aging 58, 140–150 (2017). [DOI] [PubMed] [Google Scholar]
- 246.Berman MH et al. Photobiomodulation with near infrared light helmet in a pilot, placebo controlled clinical trial in dementia patients testing memory and cognition. J. Neurol. Neurosci 10.21767/2171-6625.1000176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Martorell AJ et al. Multi-sensory gamma stimulation ameliorates Alzheimer’s-associated pathology and improves cognition. Cell 177, 256–271.e222 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Clemmensen C et al. Emerging hormonal-based combination pharmacotherapies for the treatment of metabolic diseases. Nat. Rev. Endocrinol. 15, 90–104 (2018). [DOI] [PubMed] [Google Scholar]
- 249.Cummings J, Ritter A & Rothenberg K Advances in management of neuropsychiatric syndromes in neurodegenerative diseases. Curr. Psychiatry Rep 10.1007/s11920-019-1058-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.de Freitas Silva M, Dias KST, Gontijo VS, Ortiz CJC & Viegas C Multi-target directed drugs as a modern approach for drug design towards Alzheimer’s disease: an update. Curr. Med. Chem. 25, 3491–3525 (2018). [DOI] [PubMed] [Google Scholar]
- 251.Kivipelto M, Mangialasche F & Ngandu T Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat. Rev. Neurol. 14, 653–666 (2018). [DOI] [PubMed] [Google Scholar]
- 252.McDonald T, Puchowicz M & Borges K Impairments in oxidative glucose metabolism in epilepsy and metabolic treatments thereof. Front. Cell Neurosci. 12, 274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.E L, Lu J, Selfridge JE, Burns JM & Swerdlow RH Lactate administration reproduces specific brain and liver exercise-related changes. J. Neurochem. 127, 91–100 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Morland C et al. Exercise induces cerebral VEGF and angiogenesis via the lactate receptor HCAR1. Nat. Commun 8, 15557 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Insel PS et al. Time to amyloid positivity and preclinical changes in brain metabolism, atrophy, and cognition: evidence for emerging amyloid pathology in Alzheimer’s disease. Front. Neurosci. 10.3389/fnins.2017.00281 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Liyanage SI, Santos C & Weaver DF The hidden variables problem in Alzheimer’s disease clinical trial design. Alzheimers Dement. 4, 628–635 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.McManus MJ, Murphy MP & Franklin JL The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 31, 15703–15715 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Zhao F. l. et al. AP39, a mitochondria-targeted hydrogen sulfide donor, supports cellular bioenergetics and protects against Alzheimer’s disease by preserving mitochondrial function in APP/PS1 mice and neurons. Oxid. Med. Cell. Longev. 2016, 1–19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Sorrentino V et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 552, 187–193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Zhao Y et al. ATAD3A oligomerization causes neurodegeneration by coupling mitochondrial fragmentation and bioenergetics defects. Nat. Commun. 10, 1371 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Miquel E et al. Neuroprotective effects of the mitochondria-targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free. Radic. Biol. Med. 70, 204–213 (2014). [DOI] [PubMed] [Google Scholar]
- 262.Stucki DM et al. Mitochondrial impairments contribute to spinocerebellar ataxia type 1 progression and can be ameliorated by the mitochondria-targeted antioxidant MitoQ. Free. Radic. Biol. Med. 97, 427–440 (2016). [DOI] [PubMed] [Google Scholar]
- 263.Pawlosky RJ et al. Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 141, 195–207 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Cheng A et al. SIRT3 haploinsufficiency aggravates loss of GABAergic interneurons and neuronal network hyperexcitability in an Alzheimer’s disease model. J. Neurosci. 40, 694–709 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Tieu K et al. D-β-Hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Invest. 112, 892–901 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Zhao W et al. Caprylic triglyceride as a novel therapeutic approach to effectively improve the performance and attenuate the symptoms due to the motor neuron loss in ALS disease. PLoS ONE 7, e49191 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Gong B et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 34, 1581–1588 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Costa M et al. N-Acetylcysteine protects memory decline induced by streptozotocin in mice. Chem. Biol. Interact. 253, 10–17 (2016). [DOI] [PubMed] [Google Scholar]
- 269.Tefera TW et al. Triheptanoin protects motor neurons and delays the onset of motor symptoms in a mouse model of amyotrophic lateral sclerosis. PLoS ONE 11, e0161816 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Croteau E et al. Ketogenic medium chain triglycerides increase brain energy metabolism in Alzheimer’s disease. J. Alzheimer’s Dis. 64, 551–561 (2018). [DOI] [PubMed] [Google Scholar]
- 271.Adanyeguh IM et al. Triheptanoin improves brain energy metabolism in patients with Huntington disease. Neurology 84, 490–495 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Mookerjee SA, Gerencser AA, Nicholls DG & Brand MD Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 292, 7189–7207 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Roe CR & Brunengraber H Anaplerotic treatment of long-chain fat oxidation disorders with triheptanoin: review of 15 years experience. Mol. Genet. Metab. 116, 260–268 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Hyder F et al. Uniform distributions of glucose oxidation and oxygen extraction in gray matter of normal human brain: no evidence of regional differences of aerobic glycolysis. J. Cereb. Blood Flow. Metab. 36, 903–916 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Jha MK & Morrison BM Glia-neuron energy metabolism in health and diseases: new insights into the role of nervous system metabolic transporters. Exp. Neurol. 309, 23–31 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Vardjan N et al. Enhancement of astroglial aerobic glycolysis by extracellular lactate-mediated increase in camp. Front. Mol. Neurosci 10.3389/fnmol.2018.00148 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Vodovozov W et al. Metabolic modulation of neuronal gamma-band oscillations. Pflügers Arch. 470, 1377–1389 (2018). [DOI] [PubMed] [Google Scholar]
- 278.Terada T et al. In vivo mitochondrial and glycolytic impairments in patients with Alzheimer disease. Neurology 94, e1592–e1604 (2020). [DOI] [PubMed] [Google Scholar]
- 279.Castellano C-A et al. Regional brain glucose hypometabolism in young women with polycystic ovary syndrome: possible link to mild insulin resistance. PLoS ONE 10, e0144116 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Khosravi M et al. 18F-FDG is a superior indicator of cognitive performance compared to 18F-florbetapir in Alzheimer’s disease and mild cognitive impairment evaluation: a global quantitative analysis. J. Alzheimers Dis. 70, 1197–1207 (2019). [DOI] [PubMed] [Google Scholar]
- 281.Chowdhury GMI, Jiang L, Rothman DL & Behar KL The contribution of ketone bodies to basal and activity-dependent neuronal oxidation in vivo. J. Cereb. Blood Flow. Metab. 34, 1233–1242 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.von Morze C et al. Direct assessment of renal mitochondrial redox state using hyperpolarized 13C-acetoacetate. Magn. Reson. Med. 79, 1862–1869 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Hasan-Olive MM et al. A ketogenic diet improves mitochondrial biogenesis and bioenergetics via the PGC1α-SIRT3-UCP2 axis. Neurochem. Res. 44, 22–37 (2018). [DOI] [PubMed] [Google Scholar]
- 284.McCarty MF, DiNicolantonio JJ & O’Keefe JH Ketosis may promote brain macroautophagy by activating Sirt1 and hypoxia-inducible factor-1. Med. Hypotheses 85, 631–639 (2015). [DOI] [PubMed] [Google Scholar]
- 285.Stekovic S et al. Alternate day fasting improves physiological and molecular markers of aging in healthy, non-obese humans. Cell Metab. 31, 878–881 (2020). [DOI] [PubMed] [Google Scholar]
- 286.Lourenco MV et al. Exercise-linked FNDC5/irisin rescues synaptic plasticity and memory defects in Alzheimer’s models. Nat. Med. 25, 165–175 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Pedersen BK Physical activity and muscle–brain crosstalk. Nat. Rev. Endocrinol. 15, 383–392 (2019). [DOI] [PubMed] [Google Scholar]
- 288.Cao B et al. Comparative efficacy and acceptability of antidiabetic agents for Alzheimer’s disease and mild cognitive impairment: a systematic review and network meta-analysis. Diabetes Obes. Metab. 20, 2467–2471 (2018). [DOI] [PubMed] [Google Scholar]
- 289.Li A, Yau S.-y., Machado S, Yuan T-F & So K-F Adult neurogenic and antidepressant effects of adiponectin: a potential replacement for exercise? CNS Neurol. Disord. Drug Targets 14, 1129–1144 (2015). [DOI] [PubMed] [Google Scholar]
- 290.Kremen WS et al. Influence of young adult cognitive ability and additional education on later-life cognition. Proc. Natl Acad. Sci. USA 116, 2021–2026 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Carapelle E et al. How the cognitive reserve interacts with β-amyloid deposition in mitigating FDG metabolism. Medicine 96, e5876 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Arenaza-Urquijo EM et al. The metabolic brain signature of cognitive resilience in the 80+: beyond Alzheimer pathologies. Brain 142, 1134–1147 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Croteau E et al. A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer’s disease. Exp. Gerontol. 107, 18–26 (2018). [DOI] [PubMed] [Google Scholar]
- 294.Cotto B, Natarajanseenivasan K & Langford D HIV-1 infection alters energy metabolism in the brain: contributions to HIV-associated neurocognitive disorders. Prog. Neurobiol. 181, 101616 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Renard D, Castelnovo G, Collombier L, Thouvenot E & Boudousq V FDG-PET in Creutzfeldt-Jakob disease: analysis of clinical-PET correlation. Prion 11, 440–453 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Bourgognon J-M et al. Alterations in neuronal metabolism contribute to the pathogenesis of prion disease. Cell Death Differ. 25, 1408–1425 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Gunther EC et al. Rescue of transgenic Alzheimer’s pathophysiology by polymeric cellular prion protein antagonists. Cell Rep. 26, 1368 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Norrving B Stroke management — recent advances and residual challenges. Nat. Rev. Neurol. 15, 69–71 (2019). [DOI] [PubMed] [Google Scholar]
- 299.Bazzigaluppi P et al. Imaging the effects of β-hydroxybutyrate on peri-infarct neurovascular function and metabolism. Stroke 49, 2173–2181 (2018). [DOI] [PubMed] [Google Scholar]
- 300.Lamade AM et al. Aiming for the target: mitochondrial drug delivery in traumatic brain injury. Neuropharmacology 145, 209–219 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Deng-Bryant Y, Prins ML, Hovda DA & Harris NG Ketogenic diet prevents alterations in brain metabolism in young but not adult rats after traumatic brain injury. J. Neurotrauma 28, 1813–1825 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Arifianto M, Ma’ruf A, Ibrahim A & Bajamal A Role of hypertonic sodium lactate in traumatic brain injury management. Asian J. Neurosurg. 13, 971 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Koch H & Weber YG The glucose transporter type 1 (glut1) syndromes. Epilepsy Behav. 91, 90–93 (2019). [DOI] [PubMed] [Google Scholar]
- 304.Bakker A, Albert MS, Krauss G, Speck CL & Gallagher M Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. NeuroImage Clin 7, 688–698 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Dean B, Thomas N, Scarr E & Udawela M Evidence for impaired glucose metabolism in the striatum, obtained postmortem, from some subjects with schizophrenia. Transl. Psychiatry 6, e949–e949 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Kraeuter AK, Loxton H, Lima BC, Rudd D & Sarnyai Z Ketogenic diet reverses behavioral abnormalities in an acute NMDA receptor hypofunction model of schizophrenia. Schizophrenia Res. 169, 491–493 (2015). [DOI] [PubMed] [Google Scholar]
- 307.Wlodarczyk A, Wiglusz MS & Cubala WJ Ketogenic diet for schizophrenia: Nutritional approach to antipsychotic treatment. Med. Hypotheses 118, 74–77 (2018). [DOI] [PubMed] [Google Scholar]
- 308.Palmer CM Ketogenic diet in the treatment of schizoaffective disorder: two case studies. Schizophrenia Res 189, 208–209 (2017). [DOI] [PubMed] [Google Scholar]
- 309.Gross EC, Lisicki M, Fischer D, Sandor PS & Schoenen J The metabolic face of migraine - from pathophysiology to treatment. Nat. Rev. Neurol. 15, 627–643 (2019). [DOI] [PubMed] [Google Scholar]
- 310.Country MW Retinal metabolism: a comparative look at energetics in the retina. Brain Res. 1672, 50–57 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.