

Abstract
Major sickle cell disease syndrome (SCD) is a set of potentially serious and disabling constitutional haemoglobin pathologies characterised by chronic haemolysis and vaso‐occlusion phenomena. If expression takes the form of acute vaso‐occlusive crisis, SCD is currently considered to be a chronic systemic pathology, primarily associated with vasculopathy and ischaemia‐reperfusion phenomena. The haemolytic aspect of the disease may be associated with endothelial dysfunctional complications, including leg ulcers, which are a classic spontaneous complication of major SCD. Their frequency, all aetiologies combined, varies considerably according to the series under consideration. Hydroxycarbamide has become the standard treatment for some SCD phenotypes, but has classically been described as one of the causes of leg ulcer. This causality is widely debated and is still difficult to establish because it is a specific complication of the disease. Comorbidity factors (eg, iron deficiency) are also often implicated as causal or aggravating factors so research into all the potential aetiologies of leg ulcers in a sickle cell patient must be exhaustive. We discuss the aetiologies of a leg ulcer in a patient treated by hydrocarbamide for major SCD. The imputation of the drug was established, followed by a marrow allograft in this patient.
Keywords: hydroxycarbamide, leg ulcer, sickle cell disease syndrome
1. INTRODUCTION
The major sickle cell disorders (SCDs) represent the most common monogenic pathology in the world and in France, where there is an average of 1 per 3000 births, 400 new‐borns are affected each year.1 SCD prevalence is constantly increasing and comprises a set of constitutional pathologies of haemoglobin, generated by certain point mutations of the recessive haemoglobin gene. Their presence in the homozygous state (SS phenotype) or heterozygous composite (phenotype SC, Sβ+, Sβ°, SO, SDPunjab, SLepore…) results in the synthesis of abnormal haemoglobin with modified physicochemical properties. The abnormal haemoglobin polymerises in the deoxygenated form, and under the effect of particular physical conditions (including hypoxia, extreme temperatures, dehydration, stress, infection, and acidosis) induces a reversible morphological modification of the red blood cell (a sickle cell). Sickle cell red blood cells have a tendency to spontaneous, maximal sickling in postcapillary venules and are characterised by a shortened lifespan (chronic intravascular constitutional haemolysis). Sickling, associated with hyperviscosity and lack of erythrocyte deformability,2 leads to reversible vaso‐occlusion phenomena of multifactorial origin involving many cellular and humoral actors (endothelial activation and recruitment of inflammatory cells, in particular).3 Although vaso‐occlusion and minimal haemolysis may be asymptomatic, vaso‐occlusive crises cause acute pain in the bones and spleen, and, sometimes, extreme chest pain that can be life threatening. Apart from these acute complications (major SCD is currently regarded as a chronic vascular disease,4 ischaemic, oxidative, and physical lesions eventually lead to degenerative organ dysfunction, and are a source of complex morbidity or even mortality observed in adults at a relatively young age. The haemolytic aspect of the disease is classically described as being associated with complications involving endothelial dysfunction; cerebral vasculopathy, pulmonary arterial hypertension, priapism, and leg ulcers. In practice, however, all clinical profiles are potentially long‐term sources of any type of complication.2, 5
Hydroxycarbamide (HC) is a molecule historically used as a cytostatic agent in the treatment of myeloproliferative disorders.6 Its use in the prevention of vaso‐occlusive incidents in patients with SCD is more recent,7, 8 resulting in a reduction in the frequency and severity of crises.9 It is now established that its impact is not restricted to the elements of the erythrocyte lineage and its efficacy relies on multiple mechanisms: induction of foetal haemoglobin synthesis and increase of the deformability of the red blood cells under treatment.10 but also reduction of leukocyte adhesion to the endothelium, reduction of associated leukocytosis and thrombocytosis, and, finally, affecting the metabolism of nitric oxide.11 HC has thus become the standard long‐term treatment for certain phenotypes (SS and Sβ° in particular).
Leg ulcer is a classic spontaneous complication of major sickle cell syndromes; the frequency of these ulcers in SCD, all aetiologies combined, varies between regions and ethnic origins from 10% to 75%.4 Leg ulcers are also a rare side effect of HC, with most published series consisting of patients with myeloproliferative disorders.12, 13
The imputation of HD in the occurrence of ulcers in the patient with major SCD is a crucial issue. This hypothesis can lead to the arrest of treatment without a simple therapeutic alternative. The possibility of the drug origin should not exclude the conduct of an exhaustive review of other causes: the occurrence of a leg ulcer is often multifactorial, and the interpretation of causes is always difficult.
2. CASE REPORT
A 45‐year‐old French‐born patient with two parents from the Republic of Congo (Brazzaville) had been followed since childhood for SS major sickle cell syndrome. His base haemoglobin was 90 to 95 g/L (normal range between 134 and 167 g/L), with 92% HbS and his history included cured hepatitis B, cerebral spinal tuberculosis, bilateral pneumonia, and two spontaneous left pneumothoraxes unrelated to constitutional haemoglobinopathy. The patient did not smoke.
In terms of specific degenerative complications, the patient presented with retinopathy requiring multiple laser sessions, and bilateral osteonecrosis of the hips and shoulders that justified conservative treatment by puncture reinjection of haematopoietic stem cells at the humeral level. Since adolescence he has had a history of multiple bone vaso‐occlusive crises (on average six episodes per year requiring hospitalisation), impacting the quality of life because of their frequency, without any threat to life, and notably without acute chest syndrome.
There are also episodes of priapism among his family. He had been treated in an acute episode with simple transfusions and erythrocyte exchanges and had been treated with HC at doses varying between 1000 mg and 1500 mg/d (15‐22 mg/kg/d). A precise chronology could not be reconstructed; however, over a period of 9 years, between 1993 and 2002, the patient had recurrent hyperalgesic leg ulcers at two sites (lower quarter of the leg and anterior ankle) while undergoing HC treatment, which justified dose reduction, suspension of treatment, and local care, and skin grafting. A decision had been taken to discontinue HC treatment in this context. Between 2002 and 2017, there was no recurrence of ulcer, with a preventive program of erythrocyte exchanges set up in 2006, progressively becoming monthly, with six red blood cell concentrates per session. The control of vaso‐occlusive manifestations was imperfect, however, with at least a recurrence of crisis a few days before the expected date of each erythrocyte exchange.
In 2013, this led to the induction of iatrogenic iron deficiency, designed to slow autologous erythropoiesis as much as possible. Thus, the initial iron status of the patient in 2008 was in favour of minimal tissue iron overload: ferritinaemia was around 500 μg/L (normal range 12 to 300 μg/L) (overestimated in a chronic inflammatory context), coefficient of saturation of transferrin (CST) at 42%, reticulocytes 301 G/L on average, normal platelet count, hepatic tissue iron loading estimated in magnetic resonance imaging (MRI) at 95 μMol/g (normal lower than 36 ± 20 μMol/g), and the absence of myocardial overload in MRI. Iron deficiency was measured in 2014 with ferritinaemia around 40 μg/L (normal range 12‐300 μg/L), CST around 10%, and reticulocytes 210 G/L (normal range between 20 and 120 G/L) but this status provided only moderate and transient attenuation of the vaso‐occlusive crises. Without an identifiable aetiology, there was a gradual increase in signs of crisis, which also appeared increasingly early in the cycle, after 3 weeks of the last erythrocyte exchange, despite a maximum HbS of 35% before exchange.
This situation of poorly controlled vaso‐occlusion, despite transfusion management judged to be at a maximum, led to an increase in the rate of erythrocyte exchange and suggested the reintroduction of HC from 05 May 2017, initially at 500 mg/d for 7 days (6.6 mg/kg/d) then at 1000 mg/d or 13.3 mg/kg/d.
On 23 May 2017, 18 days after the introduction of the drug, spontaneous ulceration occurred on a scarred area on the dorsal surface of the right foot. The patient was hospitalised for a vaso‐occlusive attack and treatment with HC was suspended from 26 May 2018, when tests showed a haemoglobin level of 89 g/L (normal range between 134 and 167 g/L), reticulocytes at 61 G/L, a platelet count of 21 G/L (normal range between 161 and 393 G/L), and a polynuclear neutrophil count of 4.6 G/L (normal range between 4.1 and 9.9 G/L). Haemoglobin S was 15% (70% haemoglobin A). Other results included ferritin at 167 μg/L in inflammatory context C‐reactive protein (CRP) 9 mg/mL (previously, before the development of the cutaneous lesion, ferritin was at 74 μg/L), TGO 48 IU/L (normal range below 37 IU/L), TGP 50 IU/L (normal range lower than 61 IU/L), alkaline phosphatase 107 (normal range lower than 117 IU/L), gamma glutamyl transferase 155 IU/L (normal range less than 85 IU/L), lactate dehydrogenase 406 (normal range lower than 241 IU/L), and a 25‐OH vitamin D level at 19 nmoL/L (normal range greater than 75 nmol/L). Renal function was perfectly preserved with creatinine 49μMol (chronic kidney disease ‐ epidemiology collaboration (CKD‐EPI) 119 mL/min/1. 73 m2).
The ulcer became painful on 08 June. It measured 1.5 cm by 1.5 cm, not deep, without bone contact or signs of osteomyelitis, with 20% fibrin and peri‐ulcer induration (Figure 1). The site of the ulcer was one of the scar sites from the earlier ulcers seen under HC treatment (Figure 2). The remainder of the examination showed significant xerosis without other cutaneo‐mucous lesions or pigmentary disorder, absence of mucocutaneous haemorrhage. Venous and arterial macrocirculatory examinations disclosed no abnormalities. Microcirculatory examination of toe pressure was also normal.
Figure 1.
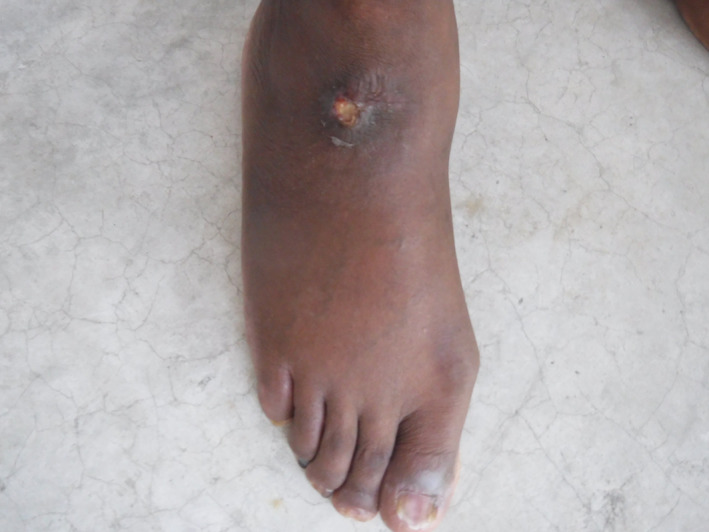
Ulceration of the leg at the dorsal surface of the right foot of a sickle cell patient
Figure 2.
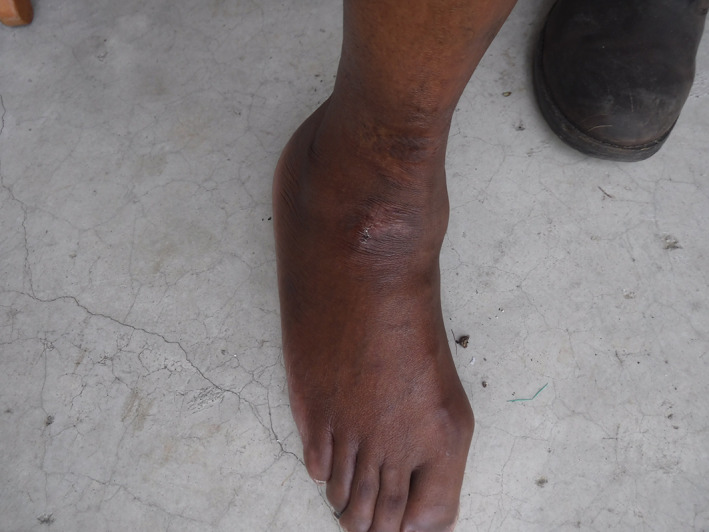
Scarred areas of old ulcers on the same sites as recent ulceration
An MRI was performed to eliminate the hypothesis of osteomyelitis. The patient was treated with morphine (morphine sulphate, Actiskenan, Laboratory Ethypharm, France) 1 hour prior to cleansing, lidocaine/prilocaine under an occlusive dressing 20 minutes before care, and, in addition, a dressing with poly‐absorbent polyacrylate fibres, (Urgoclean, Laboratory Urgo, France), was applied for detergent purposes. His other treatments included oxygen therapy at a rate of 2 L/min for 12 h/d and iron and vitamin D supplements. Transfusion management was temporarily intensified, with haemoglobin S kept below 30% between 02 May 2018 and 29 June 2019, lower than 40% thereafter by means of monthly erythrocyte exchange. The treatment was combined with a compression stocking grade II prophylaxis to prevent oedema because of altered ambulation.
Active walking and exercises with compression were used to compensate for the lack of feasibility of tissue mobilisation in the wound area. After eliminating the possibility of macrovascular and infectious causes, the first diagnostic hypothesis was a microcirculatory ulcer, despite the normal toe pressures and atypical topography for a microcirculatory ulcer. The very painful character of the condition supported this hypothesis and implied that a skin biopsy was not performed. The second hypothesis was an iatrogenic cause of the ulcer from (Siklos, Laboratory Addmedica, france). Siklos had already been interrupted (less than a month after its introduction) because of skin disorders and haematological toxicity. This measure was marked by the complete scarring of the ulcer after 5 months of follow‐up (Figure 3). The Siklos‐induced leg ulcer hypothesis was accepted, particularly as a result of the recurrent history of ulcers under (Hydrea, Laboratory Bristol‐Myers Squibb, France), the scarring of the last event after stopping Siklos, and the weight of probability regarding vascular and infection causation. The patient did not experience a recurrence after 8 months of follow‐up. The participation of a cofactorial element causing the ulcer was not excluded, especially considering the martial status of the patient. He is currently being treated with a monthly prophylactic erythrocyte exchange program and iron substitution, and was treated several months later by a haematopoietic stem cell allograft.
Figure 3.
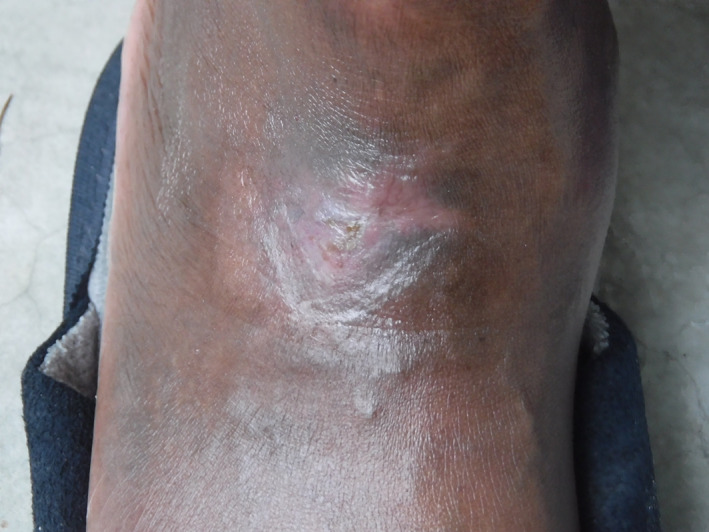
Ulcer healing 5 months after stopping hydroxycarbamide
3. DISCUSSION
We have presented a rare case of HC‐induced leg ulcer in a sickle patient with therapeutic consequences leading to an allograft. Leg ulcer is a classic, frequent, and spontaneous complication of SCD syndrome. The frequency of all ulcers, from whatever cause, in SCD varies by series and is estimated to be about 75% of cases in Jamaica and between 8% and 10% in North America.4 The majority of patients will never encounter this complication. The risk factors for developing leg ulcers are age of over 20 years, male gender, an HbF haemoglobin level less than 5%, a haemoglobin level lower than 6 g/L,14 certain HLA types, and antithrombin deficiency.15 Leg ulcers in this context often develop on a site already affected. The exact pathogenesis in SCD is uncertain but it appears to be a multifactorial mechanism including mechanical obstruction by sickling, venous insufficiency, potential additional infection, poor feedback of autonomic nervous system with excessive vasoconstriction, possible thrombosis, decreased oxygen transport capacity because of anaemia, and impairment of endothelial function. Sickle cells increase blood viscosity, are less deformable,2 and induce a slowing of blood flow, especially postcapillary, resulting in occlusion and consequent tissue ischaemia.
Patients exhibit increases in pain because of microcirculatory ischaemia.14, 15, 16, 17 Recently, vascular anomalies, including tone anomalies and activation of endothelial adhesions molecules, have been identified.4 Optimisation of local treatments, such as debridement, the control of possible infections, and the maintenance of a humid environment, is essential for healing. Some systemic treatments specific to this disease are sometimes discussed.15 Transfusion management can be considered (reduction in haemoglobin levels, partial correction of anaemia), but to our knowledge neither its objectives nor its actual contribution to scarring have been assessed, and there is no consensus on this point (potentially involving risks of iron overload and risks of alloimmunisation).16 Other treatments have been mentioned such as arginine butyrate or hypomethylating agents but the level of evidence remains low for this indication.15, 17, 18
Leg ulcer is also a classic complication, although rare, of treatments by HC if the treatment continues for a long time. HC is a molecule that has historically been used in the treatment of myeloproliferative syndromes as a cytostatic agent.6 HC is an antimetabolite that acts primarily on the bone marrow: it is a non‐competitive inhibitor of an enzyme, ribonucleotide reductase, which is necessary for the synthesis of deoxyribonucleotides from ribonucleotides.19 Its use in the prevention of vaso‐occlusive incidents in patients with major sickle syndrome is more recent,7, 8 producing a reduction in the frequency and severity of crises.9 Observations suggest it has an impact on mortality.20 Favourable impact on the prevention of degenerative complications is more difficult to pin down, but it seems probable and requires further long‐term studies.21 HC leads to an increase in the synthesis of haemoglobin F in the precursor erythroids, with a protective effect.22 It now seems that its effectiveness is actually based on multiple mechanisms with a systemic effect: reduction of leukocyte adhesion to the endothelium, reduction of leukocytosis and thrombocytosis that are often associated, increase of the deformability of red blood cells under treatment, and it may have an impact on the metabolism of NO.11 HC has thus become the standard background treatment for certain phenotypes (SS and Sβ° in particular). No risk of haematological pathology has been observed in the long term, and the main reservation in practice concerns potential impact on male fertility.6 The tendency over time has been to prescribe this treatment more widely, and even in young children if the severity of the condition justifies it, with the immediate objective of improving the quality of life and long‐term life expectancy.6 Its dermatological side effects are multiple and cases of HC‐induced skin ulcer, although rare and contended, have been described12, 23, 24, 25 and some authors estimate their frequency to be up to 9%.26 The mechanism by which this molecule leads to the genesis of the ulcer remains poorly understood and seems to be multifactorial.15, 19 It seems that HC generates cumulative toxicity on the basal layer of the epidermis and thus causes cutaneous atrophy by inhibition of DNA synthesis and repair.23, 27, 28 HC may alter microcirculation and induce tissue anoxia, explaining the occurrence of cutaneous ulcers after minimal trauma and their often‐painful character.29, 30 HC‐induced macrocytosis has been implicated by some studies,13, 19, 26 but subsequently it has been proven that HC induced a better deformability of erythrocytes and reduced the viscosity of blood flow.10
Hydoxycarbamide‐related lesions are most often malleolar. They have a fibrinous aspect, with inflammatory edges, but without necrosis. They are very painful and sometimes require the use of major analgesics. They appear, on average, after 6 years of treatment with high doses of at least 1 g/d,23, 25 but sometimes with doses of 500 mg. The termination of HC in association with local care most often leads to the healing of ulcers within an average of 5 months, but they reappear if HC is re‐introduced.13
In our patient, the finding that HC caused the ulcer is based on: consistent clinical characteristics (recurrence on the same site as the first ulcer and hyperalgesic, with early recurrence after reintroduction of HC, and with minimal dose of HC), the absence of renal function disorder, association with haematological toxicity, occurrence in a context of deep martial deficiency, in the absence of other aetiology, particularly vascular or infectious. The recurrence of ulcers soon after the introduction of Siklos in this patient with a history of ulcers associated with Hydrea, and without an ulcer for 4 years without HC, strongly supported the drug as the cause. The site of ulcer recurrence with Siklos was identical to that of Hydrea, which is also considered to be an argument for causation. Studies have associated the resurgence of ulcer despite the cessation of treatment with a higher risk of developing an ulcer on the scars of previous ulcers.31, 32
The diagnosis of HC‐induced ulcer in our patient was made after eliminating other possible causes. However, we did not do a skin biopsy because of invasive risk in the case of microcirculatory impairment. The rapid healing of the ulcer after cessation of treatment has reinforced our hypothesis. Management in cases of the molecule's imputation should be reduction of dosage or even the complete cessation of the drug when possible.33 Imputation of the drug led to the decision to perform a marrow allograft in this patient. Our case shows cutaneous toxicity leading to the type of leg ulcer that is a rare complication of HC and cessation of HC is then indicated. Investigations into the patient history and the causal record must be exhaustive in order to conclude that HC is accountable for the condition because the aetiology of leg ulcers in the patient SCD are most often multifactorial.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
Soya E, Makowski C, Blaise S. Leg ulcer induced by hydroxycarbamide in sickle cell disease: What is the therapeutic impact? Int Wound J. 2019;16:897–902. 10.1111/iwj.13115
REFERENCES
- 1. La Drépanocytose mise à jour 2011 . Encyclopédie Orphanet Grand Public Maladies Rares www.orpha.net/data/patho/Pub/fr/Drepanocytose-FRfrPub125v01, Janvier 2019.
- 2. Chien S, Usami S, Bertles JF. Abnormal rheology of oxygenated blood in sickle cell anemia. J Clin Invest. 1970;49(4):623‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. N Engl J Med. 2008;359(21):2254‐2265. [DOI] [PubMed] [Google Scholar]
- 4. Minniti CP, Eckman J, Sebastiani P, Steinberg MH, Ballas SK. Leg ulcers in sickle cell disease. Am J Hematol. 2010;85(10):831‐833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med. 2017;377(3):305. [DOI] [PubMed] [Google Scholar]
- 6. Meier ER. Treatment options for sickle cell disease. Pediatr Clin North Am. 2018;65(3):427‐443. [DOI] [PubMed] [Google Scholar]
- 7. Lanzkron S, Strouse JJ, Wilson R, et al. Systematic review: hydroxyurea for the treatment of adults with sickle cell disease. Ann Intern Med. 2008;148(12):939‐955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.8.Platt OS, Orkin SH, Dover G, Beardsley GP, Miller B, Nathan DG. Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. J Clin Invest. 1984;74(2):652‐656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the multicenter study of hydroxyurea in sickle cell anemia. N Engl J Med. 1995;332(20):1317‐1322. [DOI] [PubMed] [Google Scholar]
- 10. Lemonne N, Möckesch B, Charlot K, et al. Effects of hydroxyurea on blood rheology in sickle cell anemia: a two‐years follow‐up study. Clin Hemorheol Microcirc. 2017;67(2):141‐148. [DOI] [PubMed] [Google Scholar]
- 11. Maiocchi SL, Morris JC, Rees MD, Thomas SR. Regulation of the nitric oxide oxidase activity of myeloperoxidase by pharmacological agents. Biochem Pharmacol. 2017;135:90‐115. [DOI] [PubMed] [Google Scholar]
- 12. Best PJ, Daoud MS, Pittelkow MR, Petitt RM. Hydroxyurea induced leg ulceration in 14 patients. Ann Intern Med. 1998;128(1):29‐32. [DOI] [PubMed] [Google Scholar]
- 13. Salmon‐Ehr V, Leborgne G, Vilque JP, Potron G, Bernard P. Secondary cutaneous effects of hydroxyurea: prospective study of 26 patients from a dermatologic consultation. Rev Med Interne. 2000;21:30‐34. [DOI] [PubMed] [Google Scholar]
- 14. Koshy M, Entsuah R, Koranda A, et al. Leg ulcers in patients with sickle cell disease. Blood. 1989;74(4):1403‐1408. [PubMed] [Google Scholar]
- 15. Ladizinski B, Bazakas A, Mistry N, Alavi A, Sibbald RG, Salcido R. Sickle cell disease and leg ulcers. Adv Skin Wound Care. 2012;25:420‐428. [DOI] [PubMed] [Google Scholar]
- 16. Wright JA, Richards T, Srai SK. The role of iron in the skin and cutaneous wound healing. Front Pharmacol. 2014;10(5):156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alavi A, Kirsner RS. Hemoglobinopathies and leg ulcers. Int J Low Extrem Wounds. 2015;14(3):213‐216. [DOI] [PubMed] [Google Scholar]
- 18. Telen MJ. Developing new pharmacotherapeutic approaches to treating sickle‐cell disease. ISBT Sci Ser. 2017;12(1):239‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Quattrone F, Dini V, Barbanera S, Zerbinati N, Romanelli M. Cutaneous ulcers associated with hydroxyurea therapy. J Tissue Viability. 2013;22(4):112‐121. [DOI] [PubMed] [Google Scholar]
- 20. Serjeant GR, Chin N, Asnani MR, et al. Causes of death and early life determinants of survival in homozygous sickle cell disease: the Jamaican cohort study from birth. PLoS One. 2018;13(3):e0192710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sethy S, Panda T, Jena RK. Beneficial effect of low fixed dose of hydroxyurea in Vaso‐occlusive crisis and transfusion requirements in adult HbSS patients: a prospective study in a tertiary care center. Indian J Hematol Blood Transfus. 2018;34(2):294‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baliga BS, Pace BS, Chen HH, Shah AK, Yang YM. Mechanism for fetal hemoglobin induction by hydroxyurea in sickle cell erythroid progenitors. Am J Hematol. 2000. Nov;65(3):227‐233. [DOI] [PubMed] [Google Scholar]
- 23. Dissemond J, Hoeft D, Knab J, Franckson T, Kroger K, Goos M. Leg ulcer in a patient associated with hydroxyurea therapy. Int J Dermatol. 2006;45:158‐160. [DOI] [PubMed] [Google Scholar]
- 24. Kersgard C, Osswald MB. Hydroxyurea and sickle cell leg ulcers. Am J Hematol. 2001;68:215‐216. [DOI] [PubMed] [Google Scholar]
- 25. Saravu K, Velappan P, Lakshmi N, Shastry BA, Thomas J. Hydroxyurea induced perimalleolar ulcers. J Korean Med Sci. 2006;21:177‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hwang SW, Hong SK, Kim SH, Seo JK, Lee D, Sung HS. A hydroxyurea‐induced leg ulcer. Ann Dermatol. 2009;21:39‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sastre JL, Bravo A, Tembrás S, Gómez R, Ulibarrena C. Leg ulcers associated with hydroxyurea therapy. Haematologica. 2007;88(1):e13. [PubMed] [Google Scholar]
- 28. Jang SJ, Moon YJ, Choi YH, Kim JH, Won JY. A case of cutaneous manifestations associated with hydroxyurea therapy. Korean J Dermatol. 2003;41:965‐967. [Google Scholar]
- 29. Flores F, Eaglstein WA, Kirsner RS. Hydroxyurea‐induced leg ulcers treated with Apligraf. Ann Intern Med. 2000;132:417‐418. [DOI] [PubMed] [Google Scholar]
- 30. Chaine B, Neonato MG, Girot R, Aractingi S. Cutaneous adverse reactions to hydroxyurea in patients with sickle cell disease. Arch Dermatol. 2001;137:467‐470. [PubMed] [Google Scholar]
- 31. Kikuchi K, Arita K, Tateishi Y, Onozawa M, Akiyama M, Shimizu H. Recurrence of hydroxyurea‐induced leg ulcer after discontinuation of treatment. Acta Derm Venereol. 2011;91:373‐374. [DOI] [PubMed] [Google Scholar]
- 32. Eckman JR. Leg ulcers in sickle cell disease. Hematol Oncol Clin North Am. 1996;10:1333‐1344. [DOI] [PubMed] [Google Scholar]
- 33. Delaney KMH, Axelrod KC, Buscetta A, et al. Leg ulcers in sickle cell disease: current patterns and practices. Hemoglobin. 2013;37(4):325‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]