

SUMMARY:
Malignant stem cells have long been considered a key therapeutic target in leukemia. Therapeutic strategies designed to target the fundamental biology of leukemia stem cells while sparing normal hematopoietic cells may provide better outcomes for leukemia patients. One process in leukemia stem cell biology that has intriguing therapeutic potential is energy metabolism. In this article we discuss the metabolic properties of leukemia stem cells and how targeting energy metabolism may provide more effective therapeutic regimens for leukemia patients. In addition, we highlight the similarities and differences in energy metabolism between leukemia stem cells and malignant stem cells from solid tumors.
Introduction
The inability to fully eradicate tumor-initiating cancer stem cell (CSC) populations remains a significant hurdle in cancer therapy. CSCs generally manifest as a subpopulation of cells within heterogeneous tumors that have the ability to initiate disease, are often resistant to chemotherapy and radiation, and result in disease recurrence in cancer patients(Jordan et al., 2006, O’Brien et al., 2009, Batlle and Clevers, 2017, Kreso and Dick, 2014, Prager et al., 2020). Therefore, eradication of CSCs is likely to be an important component of improved cancer therapies. It has been postulated that effective targeting of CSCs could result in deeper clinical remission, more durable responses, and potentially curative therapies(Pollyea and Jordan, 2017). CSCs have been reported in many tumor types including leukemia(Lapidot et al., 1994, Bonnet and Dick, 1997, Uckun et al., 1995, Cox et al., 2007, Chiu et al., 2010), breast cancer(Al-Hajj et al., 2003), bone cancer(Brown et al., 2017), brain cancers(Beier et al., 2007, Singh et al., 2004), colon cancer(O’Brien et al., 2007, Ricci-Vitiani et al., 2007), prostate cancer(Hurt et al., 2008), lung cancer(Kim et al., 2005), liver cancer(Ma et al., 2007, Sun et al., 2016), and pancreatic cancer(Li et al., 2007); therefore, effectively targeting CSCs has the potential to improve patient outcomes across a broad range of cancer types.
Based on the potential benefits of CSC targeting, multiple strategies have been employed including therapies directed towards specific cell surface antigens, self-renewal pathways, signaling pathways, and the epigenome(Desai et al., 2019, Pollyea and Jordan, 2017, Gimple et al., 2019, Batlle and Clevers, 2017, Saygin et al., 2019). However, as yet, relatively little attention has been directed towards the role of energy metabolism as an Achilles Heel of CSC populations. In this article we will discuss the metabolic properties of CSCs with a particular emphasis on acute myeloid leukemia (AML), where recent studies have converged to suggest several approaches to selective targeting of the leukemia stem cell (LSC) population. Specifically, we discuss the unique biology that regulates energy metabolism of LSCs as well as strategies to therapeutically target energy metabolism. In addition, we describe how our knowledge of LSC metabolism compares to what is known about malignant stem cells from solid tumors. Because metabolic properties are highly specific to the cell/tissue context in which they occur, the studies summarized herein will be limited to reports using primary human or murine cancer stem cell experimental systems.
Leukemia and Leukemia Stem Cells
Leukemia arises from mutations in hematopoetic stem or progenitor cells and manifests as a heterogeneous tumor population. AML is the most common acute leukemia in adults, and is an aggressive and devastating disease characterized by the accumulation of myeloid progenitors arrested in their differentiation and failure of normal blood cell production(Löwenberg et al., 1999) Most AML patients who receive intensive chemotherapy achieve a significant clinical response; however, the majority of patients will relapse and succumb to their disease. Relapse is thought to occur due to failure to eradicate the disease-initiating leukemia stem cell (LSC) population(Bonnet and Dick, 1997, Hope et al., 2003) This distinct subpopulation sustains the disease, has proliferative capacity, and is functionally defined by its ability to transplant the malignancy into immunodeficient mouse models(Bonnet and Dick, 1997, Lapidot et al., 1994, Jordan, 2002). Unlike bulk AML, LSCs are mostly quiescent in the G0 phase of the cell cycle(Guzman et al., 2001, Guan et al., 2003), and can in some instances migrate to microenvironments in which they are protected from therapeutic insult (Ishikawa et al., 2007, Ye et al., 2016). LSCs have been functionally demonstrated to drive disease recurrence(Shlush et al., 2017), a major cause of death for AML patients. Therefore, understanding the biology of and developing therapies to more effectively eradicate LSCs is essential to improve AML patient outcomes.
Enrichment of stem cells using reactive oxygen species
For any analysis of stem cell biology, investigators must have means to identify stem cells that reside within a given population. By far the most common approach has been to employ cell surface proteins as a means to identify and in many cases isolate stem cells using flow cytometry or related methods. The most prevalent markers utilized to identfy and enrich for LSCs is CD34+CD38−(Lapidot et al., 1994). Many additional markers have been shown to be upregulated on LSCs compared to normal HSCs including CD123, CD99, CD96, CD93, CD47, CD32, CD25, TIM3, CLL-1, IL1RAP, and GPR65(Majeti, 2011, Jordan, 2010, Kikushige et al., 2010, van Rhenen et al., 2007). However, in the context of cancer, the consistency of surface marker expression can vary widely from patient to patient, or even within the same patient during pathogenesis(Nagare et al., 2017, Murar and Vaidya, 2015, Kreso and Dick, 2014, Clarke et al., 2006, Duan et al., 2013). Consequently, we and others have reported alternative approaches that rely on physiological properties such as oxidative state. Indeed, relatively low levels of reactive oxygen species (termed “ROS-low”) is a phenotype that has been widely associated with both normal and malignant stem cell populations(Murar and Vaidya, 2015, Jang and Sharkis, 2007, Smith et al., 2000, Diehn et al., 2009).
ROS are a byproduct of normal oxygen metabolism and play an important role in cell signaling and homeostasis. The concept that low ROS levels closely correlate with increased self-renewal capacity and reduced differentiation was first established in the hematopoietic system using normal hematopoietic stem cells (HSCs)(Ito et al., 2004, Ito et al., 2006, Jang and Sharkis, 2007, Miyamoto et al., 2007, Tothova et al., 2007). Foundational studies showed that HSCs with relatively low levels of ROS (termed ROS-low) had increased primary and secondary engraftment potential compared to cells with higher ROS levels (termed ROS-high)(Jang and Sharkis, 2007), demonstrating that low levels of ROS enriched for functional stem cells in the hematopoietic system. In AML, relative levels of ROS enrich for leukemia stem cells (LSCs) from both newly diagnosed and relapsed AML patients(Lagadinou et al., 2013, Pei et al., 2018); a characteristic that makes ROS-low a more consistent phenotype for LSC studies. From a physiological perspective, the regulation of redox balance is essential for leukemia stem cell maintenance(Adane et al., 2019, Ito et al., 2006, Juntilla et al., 2010, Pei et al., 2013, Tothova et al., 2007) which may explain why the ROS-low phenotype is so commonly observed in the stem cell biology field. Notably, the ROS-low phenotype has also been described in studies of some solid tumor stem cells(Diehn et al., 2009).
Irrespective of the phenotype used to describe LSCs, use of transplantation models to functionally define stem cell populations circumvents any challenges associated with heterogeneous phenotypes, and is an important tool in providing an unambiguous means to assess the role of metabolic (or any other perturbation) on LSC function.
Mitochondrial Biology and OXPHOS
Mitochondria control many essential cellular functions including production of energy, biomass, modulation of redox balance, ROS production, regulation of calcium signaling, and apoptosis initiation(Wallace, 2012). The main role of the mitochondria in energy production is accomplished through OXPHOS (Figure 1a). OXPHOS is the process of producing energy in the form of ATP within the mitochondrial by transferring electrons from NADH or FADH2 to O2 through a chain of electron carriers called the electron transport chain which is schematically diagramed in figure 1a. In primary human AML, LSCs have increased levels of mitochondrial mass and reduced spare respiratory capacity for OXPHOS compared to normal HSCs(Sriskanthadevan et al., 2015). Spare respiratory capacity is the amount of ATP that can be produced by the mitochondria upon energy demand. In addition, low OXPHOS spare capacity likely contributes to LSC susceptibility to OXPHOS perturbation(Sriskanthadevan et al., 2015). In chronic myeloid leukemia (CML) stem cells have increased oxidative phosphorylation levels and increased catabolism of metabolites in the TCA cycle(Kuntz et al., 2017) potentially contributing to the reduced spare capacity observed in LSCs. Multiple studies that have demonstrated that primary human LSCs are sensitive to perturbation of OXPHOS(Skrtic et al., 2011, Lagadinou et al., 2013, Jones et al., 2018, Pollyea et al., 2018, Jones et al., 2019, Cole et al., 2015). Equally important, in vitro normal HSCs are able to compensate for reduced OXPHOS by a compensatory increase in glycolysis, whereas LSCs have little to no glycolytic reserve capacity(Lagadinou et al., 2013). Consequently, OXPHOS inhibition acts to selectively eradicate LSCs while sparing HSCs(Lagadinou et al., 2013).
Figure 1: LSCs utilize OXPHOS for energy production.
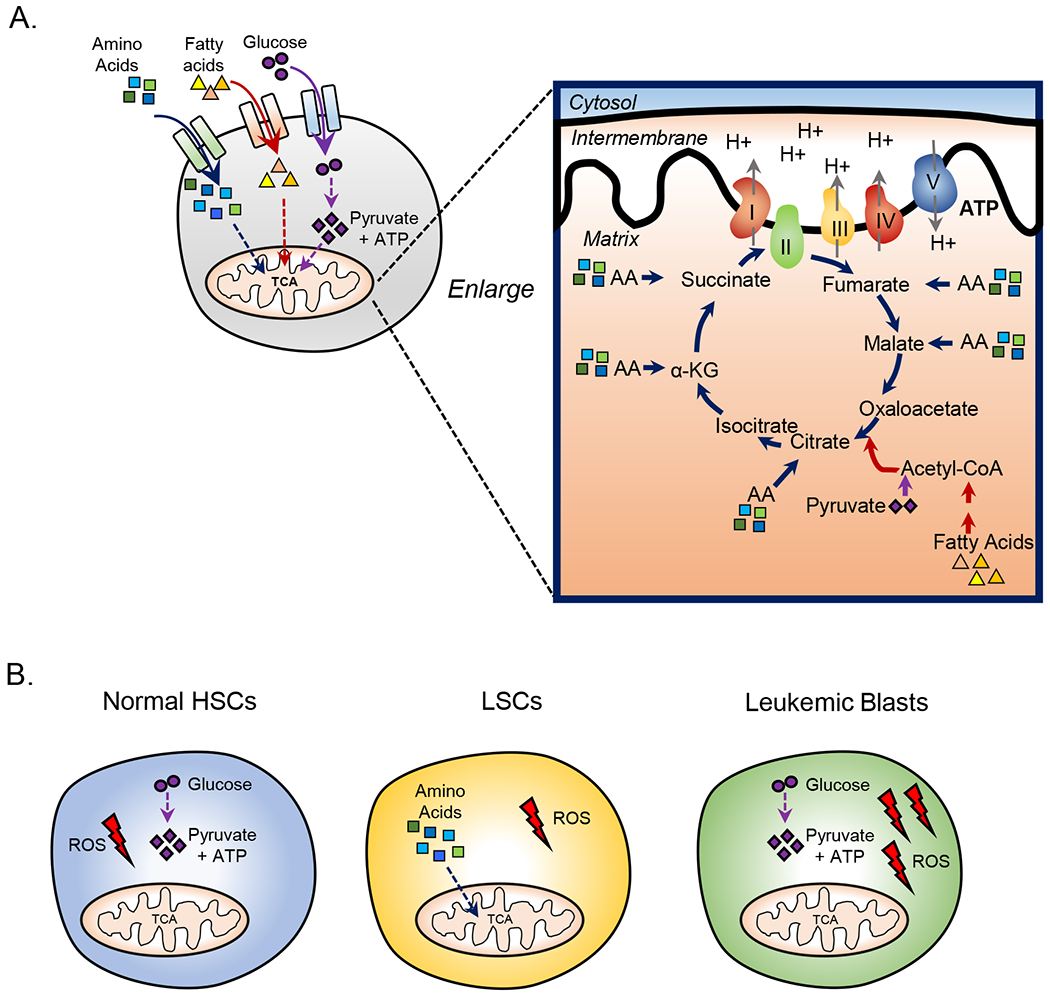
A. Cells generate ATP through glycolysis or oxidative phosphorylation. Each of these pathways rely on the breakdown of metabolites including glucose, fatty acids, and amino acids. B. Figure shows the essential energy pathways used to produce energy (ATP) in the indicated cell types. Normal HSCs and mature leukemic blasts are highly glycolytic and rely on glycolysis for ATP production. LSCs use oxidative phosphorylation to produce ATP and therefore rely on oxidative phosphorylation for survival. Cancer stem cells from several cancer types including pancreatic cancer, glioma, and multiple carcinomas are also highly dependent on OXPHOS. Despite relying on different energy pathways HSCs and LSCs have relatively low levels of reactive oxygen species (ROS) likely due to low levels of OXPHOS and spare capacity in these populations compared to mature leukemic blasts which have increased levels of ROS.
Intriguingly, many of the mitochondrial functions perturbed in cancer cells are important in regulation of OXPHOS in LSCs. For example, mitochondrial proteases have been shown to regulate energy pathways in mitochondria(Glynn, 2017). Genetic and pharmacologic inhibition of mitochondrial protease ClpP results in decreased OXPHOS in human LSCs(Cole et al., 2015). Intriguingly, activation of ClpP has also been shown to decrease OXPHOS in AML through reduced electron transport chain complex I, II, and IV activity(Ishizawa et al., 2019) indicating that a balance of protease activity is needed to support OXPHOS in LSCs. Other mitochondrial functions such as mitochondrial translation have also been shown to regulate OXPHOS in LSCs(Skrtic et al., 2011). Further, glutathione (GSH) levels regulate complex II activity through the post-translational modification, glutathionylation(Pollyea et al., 2018, Jones et al., 2019). Finally, the zinc metalloprotease neurolysin (NLN) has been shown to mediate OXPHOS levels by regulating the formation of respiratory chain supercomplexes(Mirali et al., 2020), which promote efficient oxidative metabolism. Together, several of the LSC-specific mitochondrial properties that regulate OXPHOS are potential therapeutic targets of interest, as further discussed below.
Energy Metabolisms in LSCs
Cells produce energy in the form of ATP predominantly through two pathways, glycolysis and mitochondrial oxidative phosphorylation (OXPHOS). In the 1950’s Otto Warburg reported that upon transformation cancer cells become highly glycolytic and less dependent on OXPHOS for ATP production, despite OXPHOS being a more efficient form of energy production(Warburg, 1956). This is commonly referred to as the Warburg effect(Warburg, 1956). However, cancer is a heterogeneous disease and as our understanding of cancer biology has increased so has our knowledge of the nuances of cancer cell metabolism. Over the past few years, it has become increasingly apparent that metabolism in stem cells is distinct from more mature cells. As described above, the majority of studies have reported that human LSCs are dependent on OXPHOS for survival(Sriskanthadevan et al., 2015, Lagadinou et al., 2013). In the widely used AML MLL-AF9 mouse model, AMPK has been reported to reduce LSC function through reducing glycolysis and increasing oxidative stress(Saito et al., 2015). In the same report, AMPK loss was also shown to reduce OXPHOS(Saito et al., 2015). An important finding from this study was that upon dietary restriction AMPK was activated to maintain leukemic potential(Saito et al., 2015), suggesting that the metabolic pathways LSCs rely on may differ depending on the conditions in which the LSCs exist. This concept has implications for how to design metabolism targeting therapies, as well as strategies to address therapy resistance.
In contrast to LSCs, bulk AML blast cells are dependent on glycolysis to produce energy, and upon glucose deprivation have decreased viability in culture(Jones et al., 2018). The observation that the energy pathways used by LSCs differs from mature blasts is critically important because it demonstrates that heterogeneity of metabolic dependencies exists within cancer (Figure 1b). This may also have important clinical implications in the context of targeting metabolism as an anti-cancer therapy. It is also important to note that many normal stem cells including HSCs preferentially rely on glycolysis (Suda et al., 2011, Simsek et al., 2010, Takubo et al., 2013) (Figure 1b). Interestingly though, relative reliance on glycolysis in HSCs may be dependent on stem cell state (quiescent vs. primed)(Liang et al., 2020). Specifically primed HSCs are dependent on glycolysis whereas quiescent HSCs have increased mitochondrial membrane potential and lysosomal content(Liang et al., 2020), suggesting that metabolism may vary as cells transition in and out of quiescence. In aged murine HSCs, increased mitochondrial membrane potential enhances engraftment of old HSCs(Mansell et al., 2020) indicating that mitochondrial membrane potential is linked to HSC function. Further, it should be noted that disruption of mitochondrial function in mouse HSCs through deletion of essential mitochondrial genes such as fumarate hydratase(Guitart et al., 2017) or succinate dehydrogenase D(Bejarano-Garcia et al., 2016) results in decreased HSC function and HSC (Lin”Sca+Kit+) numbers, respectively. These data suggest that while LSC-specific metabolic properties are readily evident and can be selectively targeted for therapy, the nuances of LSC and HSC energy metabolism remain relatively opaque. Additional studies are required to fully understand the consequences of metabolic intervention.
Unlike glycolysis, which is fueled by glucose metabolism, OXPHOS generates energy through catabolism of several metabolites including amino acids, fatty acids, and glucose. Each of these metabolites can be catabolized into the intermediates of the tricarboxylic acid cycle (TCA) (Figure 1a) an essential component of OXPHOS. In newly diagnosed AML, studies have shown that amino acids, fatty acids, and glucose are differentially utilized to synthesize the TCA cycle intermediates(Jones et al., 2018). Specifically, LSCs are dependent on amino acids to maintain OXPHOS levels and cannot effectively catabolize fatty acids or glucose. In contrast, AML blasts have the ability to upregulate glycolysis and fatty acid oxidation upon amino acid deprivation(Jones et al., 2018). Consistent with these observations, inhibition of OXPHOS in LSCs results in decreased ATP levels, whereas AML blasts simply increase glycolysis which permits sufficient production of ATP(Lagadinou et al., 2013). Similarly, as noted above, normal HSCs can also compensate for loss of OXPHOS via increased glycolysis. These findings suggest that an intrinsic property of LSCs is metabolic inflexibility, a characteristic that provides opportunities for therapeutic intervention. In addition, the concept of metabolic inflexibility has important implications for studying metabolism in other forms of cancer as it demonstrates the need to dissect the metabolic properties of heterogeneous populations that exist within a tumor.
Metabolic Regulation of Gene Expression
In addition to energy production, mitochondrial metabolism also influences LSC biology by mediating the epigenome(Momparler RL, 2020, Dhall et al., 2019). For example, in LSCs from AML specimens, branch chain amino acids transaminase 1 (BCAT1) transfers α-amino groups from branch chain amino acids (BCAA) to α-ketoglutarate, a cofactor for the functions of various α-ketoglutarate-dependent enzymes including the ten-eleven translocation (TET) family of DNA demethylases(Sivanand and Vander Heiden, 2020). High BCAT1 expression correlates with a poor outcome in myeloid leukemias(Hattori et al., 2017). Overexpression of BCAT1 decreases levels of α-ketoglutarate altering TET activity resulting in DNA hypermethylation and changes in gene expression related to stemness(Raffel et al., 2017). TET2 loss of function mutations are also recurrent in AML and associated with DNA hypermethylation(Cimmino et al., 2017). Interestingly, TET2 function can be mediated by the metabolite ascorbate in both leukemia and normal HSCs. (Cimmino et al., 2017, Agathocleous et al., 2017). Another example is neomorphic mutations in isocitrate dehydrogenase genes (IDH1 and IDH2) which occur early in leukemogenesis in approximately in 15–20% of AML cases resulting in increased production of the oncometabolite 2-hydroxyglutarate (2-HG), a competitive inhibitor of α-ketoglutarate-dependent enzymes including TET2(Castro et al., 2019). Consequently, IDH mutations result in DNA and histone hypermethylation, altering gene expression and causing a block in cellular differentiation, tumor initiation and progression(Dang et al., 2009, Figueroa et al., 2010).
The Influence of Disease Pathogenesis on LSC Metabolism
As described above, in considering metabolic properties in AML patients, it is critical to understand that LSCs have unique metabolic properties in comparison to bulk tumor. Another important consideration is the stage of disease pathogenesis. For example, LSCs isolated from AML patients who have relapsed after conventional cytotoxic chemotherapy show distinct properties in comparison to LSCs founds in newly diagnosed patients. Such differences include being more phenotypically diverse, varying gene expression patterns, and increased frequency(Shlush et al., 2017, Ho et al., 2016). In addition, there is evidence that metabolic properties change as a consequence of disease pathogenesis. LSCs that arise in relapsed disease are no longer dependent on amino acids to drive OXPHOS(Jones et al., 2018), but rather acquire the ability to synthesize TCA cycle intermediates from glucose or fatty acids(Jones et al., 2018), thus displaying an increased degree of metabolic flexibility. These findings are consistent with other leukemia studies demonstrating that disease progression in mouse models correlates with increased utilization of glucose(Ye et al., 2018). Importantly, while the dependence of relapsed LSCs on amino acid metabolism is decreased, these cells remain highly reliant on OXPHOS(Jones et al., 2018). Indeed, amino acid deprivation in combination with inhibition of fatty acid oxidation results in decreased OXPHOS in relapsed LSCs(Jones et al., 2018, Stevens et al., 2020) Further, relapsed LSCs have a distinct metabolic profile consisting of increased energy metabolism pathways that include amino acid catabolism, glycolysis, and fatty acid oxidation, all of which is supported through increased synthesis of NAD+(Jones et al., 2020). Notably, relapsed pediatric acute lymphoblastic leukemia (ALL) cells are also metabolically divergent from ALL cells isolated from newly diagnosed patients exhibiting an increased OXPHOS and mitochondrial translation transcriptional profile(Dobson et al., 2020). Indeed, the relapse-iniating subclone present in newly diagnosed disease exhibits a partial relapse metabolic signature, suggesting that the metabolic differences observed between diagnosis and relapse may contribute to disease pathogensis(Dobson et al., 2020). Interestingly, AML cells resistant to the conventional chemotherapy agent cytarabine have increased levels of OXPHOS which is supported by increased fatty acid oxidation through increased CD36 expression(Farge et al., 2017). Further, the microenviornment that LSCs reside in can alter metabolic properties of LSCs resulting in therapy resistance likely contributing to disease pathogenesis. Mouse LSCs located in adipose tissue elevate fatty acid metabolism resulting in chemotherapy resistance(Ye et al., 2016). The hepatic microenvironement supports increased poly-unsaturated fatty acid levels promoting cell survival pathways(Ye et al., 2020) in mouse models of AML. In ALL, a source of disease relapse is the central nervious system (CNS)(Pui and Howard, 2008). ALL cells residing the the CNS have elevated levels of fatty acid synthsis and stearoyl-CoA desaturase(Savino et al., 2020), the rate limiting enzyme for the synthesis of mono-unsaturated fatty acids. Further increasing stearoyl-COA desaturase levels results in increased CNS disease(Savino et al., 2020). Finally, residual mouse AML cells residing the bone marrow post maximal chemotherapy response display increased glutamine metabolism which contributes to chemoresistance through increase GSH and nucleotide synthesis(van Gastel et al., 2020). In addition, increased glutamine levels are utilized by bone marrow stromal cells to synthesize aspartate, which is also required for nucleotide synthesis, through the TCA cycle(van Gastel et al., 2020). Interestingly, the metabolic profile of residual mouse AML cells residing the bone marrow after chemotherapy treatment(van Gastel et al., 2020) and human relapsed LSCs(Jones et al., 2020) share several similarities including enrichment of amino acid metabolism pathways and elevated nicotinamide metabolism.
As hematologic malignancies are increasingly treated with regimens that include targeted and other non-chemotherapeutic agents, determining if disease pathogenesis is correlated or caused by metabolic changes will be an important consideration. For example, AML patients who relapse on the combination of venetoclax with azacitidine exhibit an increased OXPHOS transcriptional profile(Pei et al., 2020). Higher OXPHOS levels correlated with a worse response to venetoclax with azacitidine in laboratory based studies(Pei et al., 2020), suggesting that relative OXPHOS activity in LSCs is a potential predictor of therapy response to both conventional chemotherapy and venetoclax based therapies.
Targeting LSCs Metabolism
To date, many strategies related to metabolism have been reported to impair LSCs and other CSCs include inducing ROS, targeting the CSC microenvironment, inhibiting glycolysis, inducing differentiation, and modulating genetic events that mediate metabolism, all of which have been reviewed elsewhere(Luo and Wicha, 2019, Shi et al., 2012, De Francesco et al., 2018, Vander Heiden and DeBerardinis, 2017). Due the central role of OXPHOS in LSCs(Lagadinou et al., 2013, Sriskanthadevan et al., 2015), this article is focused on the potential therapeutic value of strategies the either directly or indirectly modulate OXPHOS in LSCs (Figure 2a).
Figure 2: Targeting OXPHOS and Therapy Resistance in LSCs.
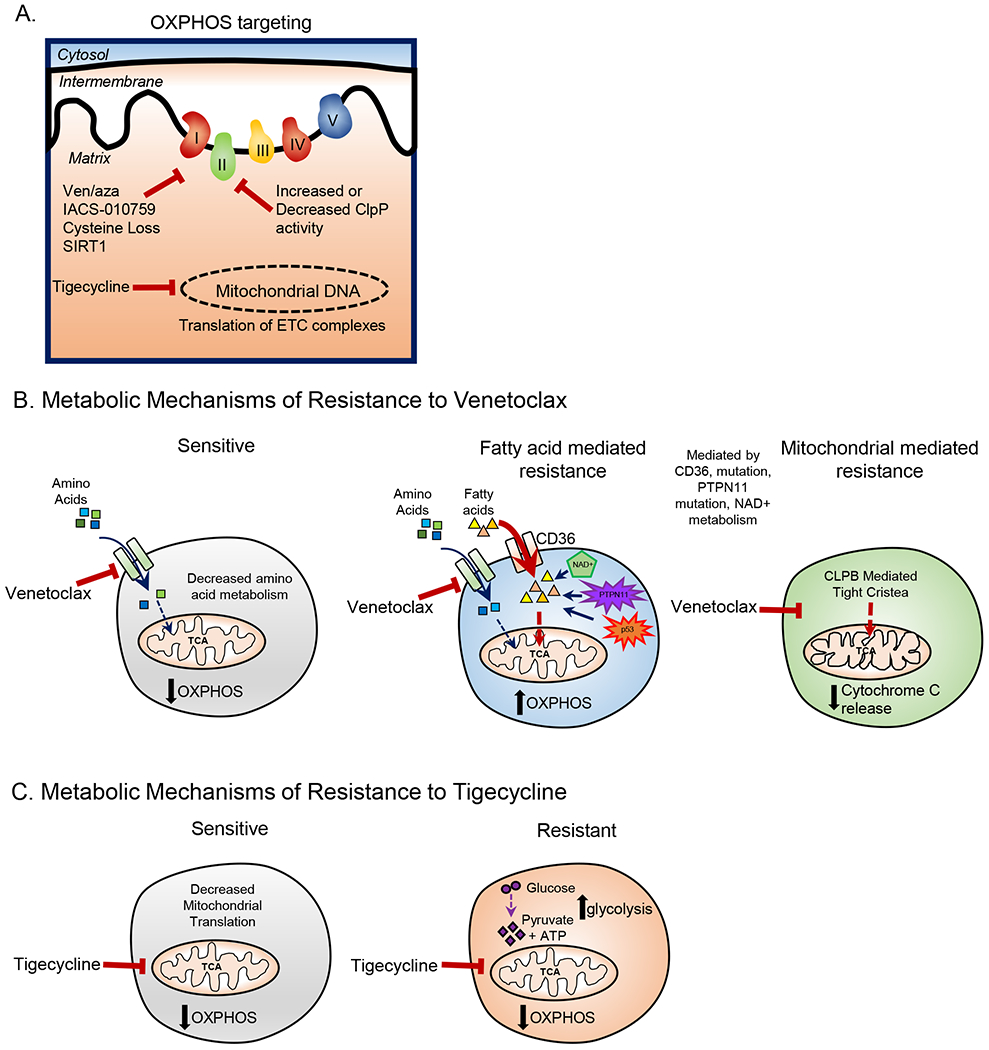
A. OXPHOS in LSCs can be targeted by venetoclax with azacitidine treatment, IACS-010759 treatment, cysteine depletion, ClpP activation or inhibition, inhibition of mitochondrial translation, and SIRT1 deletion. B. In sensitive LSCs venetoclax decreases amino acid metabolism resulting in decreased OXPHOS. Venetoclax resistance in LSCs has been shown to be mediated by increases in fatty acid metabolism which can be caused by increased expression in fatty acid transporter CD36, mutations in TP53, and potentially other mechanisms. Further, venetoclax resistance can be caused by changes in mitochondrial cristae structure which is mediated by CLPB expression. C. In sensitive CSCs tigecycline decreases mitochondrial translation resulting in decreased OXPHOS. Tigecycline resistant CSCs upregulate glycolysis to compensate for the loss of OXPHOS.
Direct Targeting of the Electron Transport Chain in LSCs
The mitochondrial ETC is a series of complexes located in the inner mitochondrial membrane that transfer electrons from donors to acceptors using redox reactions. Electron transfer is coupled with the transfer of protons from the mitochondrial matrix to the inner membrane space which creates a proton gradient that is utilized by ATP synthase to flow protons back into the mitochondrial matrix and generate ATP. Inhibition of ETC Complex I using IACS-010759 or mubritinib targets LSCs by decreasing OXPHOS(Molina et al., 2018, Baccelli et al., 2019). Strategies designed to directly target ETC complex activity have resulted in significant toxicities and therefore have had limited clinical utility(Nayak et al., 2018). Importantly, neither IACS-010759 nor mubritinib treatment targeted normal stem and progenitor cells suggesting that these therapies may be more selective compared to previous ETC complex inhibitors(Molina et al., 2018). IACS-010759 is currently under clinical evaluation in AML and solid tumors(Molina et al., 2018).
Indirect Targeting of the Electron Transport Chain in LSCs
Indirect perturbation of OXPHOS can be accomplished by inhibition of translation of mitochondrial-encoded ETC proteins, dysregulated proteasome activity resulting in decreased ETC Complex II activity, alterations in post-translational modifications that regulate ETC complex function, decreasing energy metabolism pathways that fuel the TCA cycle, and by SIRT1 inhibition. The mitochondrial genome encodes 13 of the 90 proteins within the ETC(Skrtic et al., 2011). Inhibition of mitochondrial translation has been shown to effectively target OXPHOS and decrease LSC function in primary human AML and CML(Skrtic et al., 2011, Kuntz et al., 2017). Inhibition of mitochondrial translation, through treatment with the antibiotic tigecycline, was first shown to effectively target primary human AML LSCs by decreasing synthesis of mitochondrial translated proteins (including enzymes in the electron transport chain), thereby inhibiting OXPHOS(Skrtic et al., 2011). Reduced levels of OXPHOS upon tigecycline treatment correlates with decreased protein levels of mtCOXI and mtCOXII both of which are components of the electron transport chain(Kuntz et al., 2017, Skrtic et al., 2011). Importantly, in these models tigecycline preferentially targets LSCs compared to normal HSCs(Skrtic et al., 2011).
Mitochdondrial protease ClpP has been shown to mediate OXPHOS by regulating the activity of ETC enzymes. Interestingly, both inhibition and activation of ClpP decrease the activity of ETC enzymes and LSC function. Inhibitors of ClpP have been created which target LSCs(Cole et al., 2015, Tan et al., 2019). The first inhibitors of ClpP were developed against the bacterial form of the protein, and were subsequently shown to inhibit human ClpP and reduce primary human AML LSC function(Cole et al., 2015). Decreased LSC function upon ClpP inhibition is caused by reduced activity of ETC Complex II(Cole et al., 2015). The specificity of the inhibitors used in these initial studies was not determined(Cole et al., 2015). In addition, they exhibited poor stability in aqueous solutions(Cole et al., 2015). To address these concerns, investigators have designed more potent ClpP inhibitors(Tan et al., 2019) which may enable translation of these preclinical studies to clinical trials. Activation of ClpP has also been shown to decrease OXPHOS in AML through reduced electron transport chain complex I, II, and IV activity(Ishizawa et al., 2019). Structural changes in ClpP result in its activation which can be accomplished by ClpP binding to Imipridones(Ishizawa et al., 2019). Imipridones selectively kill AML cells in preclinical mouse models(Ishizawa et al., 2019).
GSH has also been shown to be essential in Complex II function in LSCs(Pollyea et al., 2018, Jones et al., 2019). GSH levels are evavated in LSCs compared to leukemic blasts(Lagadinou et al., 2013) and can be decreased in primary human LSCs through treatment with the BCL2 inhibitor, venetoclax, in combination with the hypomethylating agent, azacitidine(Pollyea et al., 2018), or by treatment with a cysteine degrading enzyme (cysteinase)(Jones et al., 2019, Cramer et al., 2017). Decreased GSH levels results in reduced levels of glutathionylation of ETC Complex II causing decreased activity of complex II(Pollyea et al., 2018, Jones et al., 2019). It still remains to be determined if GSH levels could be used as a biomarker for response for these OXPHOS targeting therapies. Venetoclax with azacitidine has also been shown to decrease OXPHOS through a second mechanism, involving decrease of global amino acids which are necessary for the synthesis of TCA cycle intermediates(Jones et al., 2018). Furthermore, venetoclax with azacitidine decreased OXPHOS in LSCs isolated from patients with high-risk myelodysplastic (MDS), and reduced in vivo tumor burden in patient-derived xenograft models(Stevens et al., 2018). Altogether, these data demonstrate that targeting the metabolic pathways required for synthesis of the TCA cycle intermediates or regulation of ETC enzymes are potential therapuetic strategies to target LSCs. Importantly, both direct and indirect inhibition of OXPHOS targets LSCs with diverse genetic backgrounds(Cole et al., 2015, Skrtic et al., 2011, Jones et al., 2018, Pollyea et al., 2018). These data suggest that reliance on OXPHOS is at least partially independent of cancer genotype, and therefore may represent a broadly conserved LSC vulnerability.
SIRT1 regulates OXPHOS and LSC function
Sirtuins are a highly conserved family of NAD+ dependent enzymes that regulate various cellular functions that are essential for stem cell biology(Fang et al., 2019). Sirtuins play a key role in the regulation of metabolic processes(Correia et al., 2017); however, less is known about the role of sirtuins in regulating metabolism in HSCs and LSCs. In CML LSCs, SIRT1 regulates OXPHOS gene expression signatures(Abraham et al., 2019). Previous studies have shown that SIRT1 inhibition in combination with BCR-ABL tyrosine kinase inhibitors (TKIs) results in increased survival and elimination of LSCs in a BCL-ABL driven CML mouse model(Li et al., 2012). Deletion of SIRT1 in hematopoietic cells confirms a functional role for this enzyme, demonstrating a delay of leukemia development and increased survival(Abraham et al., 2019). Further, SIRT1 deletion results in decreased OXPHOS in LSCs but not normal hematopoietic cells, suggesting a selective reliance on SIRT1 for mitochondrial metabolism upon transformation(Abraham et al., 2019). This observation is consistent with the findings that SIRT1 deletion did not influence adult HSC function(Abraham et al., 2019, Leko et al., 2012).
It is important to note that in many of these preclinical studies toxicities to normal stem cells have not been observed, despite the fact that many normal cells utilize OXPHOS for energy production. In the blood system normal stem cells can upregulate other energy metabolism pathways upon OXPHOS inhibition whereas LSCs cannot(Lagadinou et al., 2013), a characteristic which may explain the therapeutic window for OXPHOS inhibitors in the blood. Still, for the OXPHOS targeting therapies that are now being used clinically, it may be important to assess whether prolonged inhibition of OXPHOS impairs normal tissue function.
Clinical Translation of LSC metabolism targeting
Multiple strategies to inhibit metabolism have been tested in clinical trials, but very few have been designed to target CSC specific biology (Table 1). The majority of these studies have been designed to target LSCs. A subset of the preclinical strategies described above have been translated into clinical trials including therapies such as mitochondrial translation inhibitor tigecycline, BCL2 inhibitor venetoclax with hypomethylating agent azacitidine, protein translation inhibitor omacetaxine with azacitidine and electron transport complex I inhibitor ICAS-010759. A phase I study of intravenous infusion of tigecycline in refractory AML patients established the maximum tolerated dose in a cohort of 27 adult AML patients(Reed et al., 2016). No significant clinical responses were observed; however, the half-life of tigecycline was significantly shorter in AML patients than previously reported in non-cancer patients(Reed et al., 2016). New formulations of tigecycline with improved stability may enhance the antileukemic effect of the drug in vivo(Jitkova et al., 2014). Inhibition of BCL2 through venetoclax in combination hypomethylating agents has resulted in superior outcomes for treatment naive AML patients compared to standard therapy(DiNardo et al., 2018a, Pollyea et al., 2018) (NCT02993523). Venetoclax for AML treatment was FDA approved for use in combination with hypomethylating agents in 2018. The preclinical data demonstrates that venetoclax and venetoclax combinations likely target LSCs through various mechanisms including by perturbing energy metabolism. Interestingly, many of the metabolic consequences described upon venetoclax with azacitidine treatment in preclinical models have also been observed in AML patients including decreased levels of OXPHOS, glutathione and amino acids(Pollyea et al., 2018, Jones et al., 2018). Evaluation of omacetaxine with azacitidine in treatment naïve high grade MDS patients is currently ongoing (NCT03564873). This trial was prompted by preclinical data that demonstrated this combination targets malignant stem cells in MDS patient specimens(Stevens et al., 2018). ICAS-010759 is currently being evaluated in clinical trials for relapsed/refractory AML (NCT02882321) based on promising preclinical findings(Molina et al., 2018).
Table 1:
Clinical approaches to targeting OXPHOS in Leukemias
| Therapy | Drug Target | Cancer Type | Trial Reference |
|---|---|---|---|
| Tigecycline | Mitochondrial translation | AML | NCT01332786 |
| Venetoclax with hypomethylating agents | BCL2 and DNA methylation | AML CLL (approved) | NCT02203773 |
| Omacetaxine | Protein Translation | MDS | NCT03564873 |
| ICAS-010759 | Electron transport chain Complex I |
AML | NCT02882321 |
While many trials have been designed to target CSCs or metabolism, few have by design specifically target OXPHOS in LSCs. The studies listed in the table represent initial efforts to modulate CSC metabolism.
Targeting metabolic enzymes IDH1/2 has also been shown to be clinically active in AML. Ivosidenib, an IDH1 inhibitor has been approved for the treatment of AML patients with IDH1 mutation after clinical trials revealed Ivosidenib treatment resulted in durable remissions in relapsed or refractory AML patients with IDH1 mutation(DiNardo et al., 2018c). Similarly, Enasidenib, an IDH2 inhibitor has been approved for the treatment of relapsed AML after durable remissions were achieved in clinical trials(Yen et al., 2017, Pollyea et al., 2019, Stein et al., 2017). Several additional inhibitors are currently being clinically evaluated including mutant IDH1 or IDH2 specific inhibitors and a pan IDH1 and 2 inhibitors(Golub et al., 2019).
Resistance to Metabolism Targeting Therapies in LSCs
Resistance to cancer therapies resulting in disease progression and recurrence has been well documented(Nikolaou et al., 2018), and is also evident for therapies that influence metabolism. AML patients who have relapsed on conventional therapy and then receive venetoclax have inferior outcomes when compared to newly diagnosed AML patients(DiNardo et al., 2018b). These clinical findings correlate with an inability of venetoclax with azacitidine to decrease OXPHOS in relapsed LSCs in laboratory-based studies(Jones et al., 2018). Further, once an AML patient has relapsed on conventional chemotherapy the reliance of LSCs on amino acid metabolism decreases due to an increased utilization of fatty acid oxidation as discussed in the previous section. Therefore, in relapsed LSCs to inhibit the synthesis of TCA cycle intermediates and target LSCs it appears necessary to inhibit additional mechanisms that drive oxidative energy metabolism. This can be accomplished through the combination of venetoclax with azacitidine with inhibition of fatty acid uptake using a CD36 inhibitor(Jones et al., 2018) or through inhibition of fatty acid metabolism in the mitochondria(Stevens et al., 2020). Both of these strategies to inhibit fatty acid catabolism act to restore the sensitivity of relapsed LSCs to treatment with venetoclax and azacitidine. Furthermore, since primed HSCs primarily rely on glycolysis(Suda et al., 2011, Takubo et al., 2013, Liang et al., 2020) to drive energy production, approaches that inhibit amino acid and fatty acid metabolism may be relatively well tolerated in the normal hematopoietic system. Indeed, preclinical modeling suggests that combining venetoclax with inhibitors of fatty acid metabolism does not harm human HSCs in vitroJones et al., 2018, Stevens et al., 2020) and are well tolerated in mice(Stevens et al., 2020), supporting the concept that this strategy should be evaluated in human clinical trials.
Notably, fatty acid metabolism can be increased through the mutation of RAS pathway genes(Stevens et al., 2020) and TP53(Nechiporuk et al., 2019) both of which have been shown to contribute to venetoclax resistance in AML patients and ex vivo screening of primary AML respectively. Furthermore, outgrowth of tumor subclones that bear RAS mutations was recently reported in AML patients who relapse following venetoclax and azacitidine treatment(Pei et al., 2020). Thus, underlying mutations that influence energy metabolism clearly provide a competitive advantage for tumor cells in AML patients, and represent an important consideration for future therapies.
Additional mechanism of resistance related to mitochondria were recently identified using an in vitro CRISPR screen. These studies revealed that genes involved in changes in mitochondrial organization and function structure(Chen et al., 2019) as well as mitochondrial translation and the integrated stress response(Sharon et al., 2019) may contribute to venetoclax resistance. Further, we reported that in relapsed patients LSCs support both amino acid metabolism and fatty acid metabolism by increasing the synthesis of NAD+ from nicotinamide through the salvage pathway(Jones et al., 2020). Notably, inhibition of NAMPT, the rate-limiting enzyme in conversion of nicotinamide to NAD+, decreased both amino acid and fatty acid catabolism into TCA cycle intermediates, and reduced tumor burden and LSCs in patient derived xenograft models(Jones et al., 2020). These findings suggest that inhibition of NAMPT may be an effective strategy to target LSCs in relapsed AML patients. A summary of mechanisms of resistance to venetoclax is provided in Figure 2b.
Tigecycline is another OXPHOS targeting therapy that has been explored clinically, as described above. In an AML in vitro experimental model, prolonged treatment with tigecycline results in a metabolic shift with decreased OXPHOS and increased glycolysis levels which is reversible upon removal of tigecycline treatment(Jhas et al., 2013) (Figure 2c). This preclinical study may provide important insights on mechanisms that could lead to clinical resistance.
Altogether, these data suggest that targeting LSC metabolism is a viable clinical approach that has the potential to result in superior clinical outcomes for many leukemia patients. However, like many other forms of therapy, resistance can emerge, which highlights the need to further understand the basic mechanisms that underlie metabolic targeting of malignant stem cell populations, both in newly diagnosed patients and after relapse following various lines of therapy.
Energy Metabolism in solid tumor CSCs
While leukemia provides a relatively tractable model for analysis of malignant stem cells and metabolic properties, similar questions and studies are equally relevant in solid tumors, where CSCs have been described for many types of cancer(O’Brien et al., 2009, Batlle and Clevers, 2017, Prager et al., 2020). As metabolic targeting of CSCs has been successful in leukemia, it is also an appealing strategy to target solid tumor CSCs. Not surprisingly, the biology and properties of various CSCs can vary substantially, likely a reflection of differing tissue origins. Intriguingly though, dependence on OXPHOS has been reported for multiple independent tumor types. Thus, despite the distinct properties of cancers arising from disparate tissue sources, a common metabolic property may be evident in the pathways utilized by CSCs to regulate energy production (Table 2 summarizes similarities and differences between LSCs and CSCs). This observation may provide insights on fundamental mechanisms of pathogenesis and opportunities for therapeutic intervention.
Table 2:
Comparison of metabolic properties of LSCs vs. solid tumor CSCs
| CSC Type | Metabolism relative to LSCs | Metabolic Flexibility Compared to LSCs |
|---|---|---|
| Glioblastoma |
Similarities Reliance on OXPHOS for CSC activity80 Differences Increased oxidative capacity and ATP levels, some GSCs are reliant on glycolysis for CSC activity79,96 |
Similarities Metabolically unique CSC populations within same tumor sample, certain GSCs reliant on fatty acid metabolism97,98,99 Differences *Switch from OXPHOS to glycolysis occurs during metabolic stress96,97,98 |
| Breast Cancer |
Similarities Decreased CSC activity upon mitochondrial metabolism inhibition84 Differences Increased mitochondrial membrane potential and ATP levels83 |
Similarities Increased OXPHOS activity and reliance on fatty acid metabolism in CSCs from chemo-resistant samples85,86,101 Differences Some BCSCs reliant on glycolysis100 |
| Ovarian Cancer |
Similarities Not reliant on glycolysis for CSC activity, decreased CSC activity upon mitochondrial metabolism inhibition87 Differences Increased mitochondrial membrane potential87 |
Similarities Increased metabolic demands upon chemotherapy resistance102 Differences *OXPHOS inhibition induces compensatory glycolysis, increased pentose phosphate pathway activity103 |
| Pancreatic Cancer |
Similarities Decreased CSC activity upon OXPHOS inhibition90 Differences *Increased OCR and mitochondrial membrane potential89 |
Similarities Ability to metabolically reprogram upon chemotherapy treatment104 Differences Decreased CSC activity upon glycolysis inhibition, glycolysis inhibition re-sensitizes to chemotherapeutic agents104 |
| Colorectal Cancer |
Similarities Decreased ROS levels, reliance on AMPK for CSC activity91 Differences Increased levels of S-Adenosyl Methionine106 |
Similarities Increased mitochondrial metabolism in chemotherapy resistant CSCs, metabolic reprogramming upon chemotherapy exposure92,93,106 Differences *Increased lysine metabolism upon chemotherapy treatment108 |
Regulation and Targeting of OXPHOS in Solid Tumor CSCs
Similar to LSCs, CSCs from cancers including glioblastoma, lung, breast, ovarian, pancreas, colon, and melanoma have been reported to primarily rely on OXPHOS for energetic demands(Janiszewska et al., 2012, Vlashi et al., 2011, Gao et al., 2016, Ye et al., 2011, Vlashi et al., 2014, Lee et al., 2017, De Luca et al., 2015, Farnie et al., 2015, Pasto et al., 2014, Sato et al., 2016, Viale et al., 2014, Sancho et al., 2015). While this represents a potentially important common feature of many CSCs, it is important to recognize that underlying mechanisms clearly differ. For example, unlike leukemia stem cells, glioblastoma stem cells (GSCs) have higher oxidative capacity and ATP levels compared to more differentiated glioma cells(Vlashi et al., 2011). In GSCs, OXPHOS is mediated by insulin-like growth factor 2 mRNA-binding protein 2 (IMP2)(Janiszewska et al., 2012). GSCs are dependent on this increased OXPHOS levels as inhibition of OXPHOS through inhibition of IMP2 led to decreased stem cell function as measured by gliomasphere formation from primary tumor cultures and decreased engraftment in NOD/SCID mice(Janiszewska et al., 2012). Importantly like LSCs, inhibition of glycolysis did not significantly affect malignant stem cell function(Janiszewska et al., 2012).
Breast cancer stem cells have also been shown to rely on oxidative phosphorylation in multiple studies(Lee et al., 2017, De Luca et al., 2015, Farnie et al., 2015). Breast cancer stem cells (BCSCs) derived from breast cancer cell lines were propagated into mammospheres and compared to differentiated tumor cells(Vlashi et al., 2014). BCSCs had decreased lactate production, increased mitochondrial membrane potential and increased ATP levels, all suggestive of increased dependence of OXPHOS(Vlashi et al., 2014). Further, in a model of chemotherapy resistant BCSCs, compared to non-stem tumor cells, BCSCs had increased OXPHOS activity(Lee et al., 2017). BCSCs also had increased basal respiratory capacity, maximal respiratory capacity and mitochondrial membrane potential compared to non-stem tumor cells(Lee et al., 2017). Inhibition of MYC led to reduced mitochondrial respiration and decreased mammosphere formation establishing a novel role for MYC in mediating BCSC activity through mitochondrial metabolism(Lee et al., 2017). Interestingly, increased mitochondrial mass has been associated with BCSCs isolated from patients with chemo-resistant metastatic breast cancer, PDX (patient-derived xenograft) models of TNBC and BCSCs derived from cell lines(De Luca et al., 2015, Farnie et al., 2015). Further, similar to LSCs(Pei et al., 2018) inhibition of mitochondrial biogenesis leads to decreased oxidative metabolism and decreased mammosphere formation(De Luca et al., 2015, Farnie et al., 2015). Taken together, these studies suggest BCSCs regardless of chemotherapy exposure, rely on OXPHOS and mitochondrial metabolism. Importantly these metabolic properties seem to be analogous to those of LSCs, indicating similarities between liquid and solid tumor CSCs.
Ovarian cancer stem cells (OCSCs) derived from patients with epithelial ovarian cancer revealed overexpression in genes associated with OXPHOS(Pasto et al., 2014). Additionally, OCSCs had increased mitochondrial membrane potential and were not sensitive to glucose deprivation(Pasto et al., 2014). Further, treatment of OCSCs with oligomycin, rotenone, antimycin and metformin all led to decreased viability(Pasto et al., 2014). These data suggest that OCSCs derived from patient samples have unique mitochondrial biology properties which include increased OXPHOS activity.
Pancreatic adenocarcinoma (PDAC) CSCs derived from primary mouse models revealed an increase in expression of genes involved in mitochondrial metabolism compared to non-stem tumor cells(Viale et al., 2014). This finding is similar to Ros-low LSCs which were shown to have increased expression of genes involved in mitochondrial metabolism compared to non-stem cell counterparts (Lagadinou et al. 2013). Further, PDAC CSCs had significantly increased OCR, ETC activity, and increased mitochondrial membrane potential(Viale et al., 2014). Metabolic flux analyses revealed decreased glycolytic activity and increased TCA cycle activity in CSCs. Inhibition of OXPHOS using oligomycin led to decreased sphere formation indicating decreased CSC activity(Viale et al., 2014). In addition, PDAC CSCs derived from PDX models had similar mitochondrial metabolism properties as observed in mouse models(Sancho et al., 2015). When these CSCs were treated with metformin, OXPHOS activity and sphere-forming ability were significantly decreased(Sancho et al., 2015). However, in vivo, metformin resistance occurred suggesting metabolic flexibility in PDAC CSCs. As previously discussed, LSCs have also been shown to have metabolic flexibility indicating both liquid and solid tumor CSCs are able to adapt to changes in metabolic pathways.
Colorectal cancer stem cells isolated from PDX models had decreased ROS levels and increased mitochondrial activity(Guo et al., 2018). Further, inhibition of AMPK led to decreased CSC activity similar to LSCs(Guo et al., 2018). In vivo xenograft studies of stage III human colon adenocarcinoma cells showed increased mitochondrial content and activity in cells resistant to 5-FU(Denise et al., 2015). Further corroborating this work, liver metastases from colon cancer were studied to determine changes in biology in chemotherapy resistant colon cancer cells(Vellinga et al., 2015). RNA-seq revealed increased expression of genes in the mitochondrial biogenesis and OXPHOS pathways in chemo resistant cells(Vellinga et al., 2015). Colonosphere cultures derived from primary colorectal tumors and liver metastases showed increased mitochondrial content and OXPHOS activity in chemotherapy treated cultures(Vellinga et al., 2015). Taken together, these data suggest a selection for cells with stem-cell activity and increased OXPHOS activity in colon cancer cells resistant to chemotherapy.
A subset of melanoma cells have been shown to have cancer stem cell properties such as tumor initiation, tumor maintenance and sphere forming abilities(Petrachi et al., 2017). Further, melanoma cancer stem cells (MCSC) have been shown to have unique metabolic properties(Petrachi et al., 2017). Upon treatment with phenformin, a complex I inhibitor, MCSCs derived from primary cultures and cell lines decreased viability, sphere forming and invasion ability(Petrachi et al., 2017). Further, treatment with phenformin led to decreased stem cell properties in MCSCs including decreased expression SOX2(Roesch et al., 2013).
Comparing data from a variety of solid tumor CSCs versus LSCs has shown many similarities including increased gene expression of mitochondrial metabolism genes, increased OXPHOS activity and decreased activity upon inhibition of mitochondrial metabolism.
Metabolic Flexibility of Solid Tumor CSCs
While there is significant evidence that OXPHOS is the primary fuel source for multiple CSC types and LSCs, it is important to note that not all tumor stem cells consistently utilize OXPHOS. Indeed, a significant degree of metabolic flexibility has been documented, where CSCs can alternate between energy production pathways. For instance, some studies have shown GSCs rely on glycolysis through increased glucose consumption by upregulating GLUT3 expression(Flavahan et al., 2013). Intriguingly, GSCs dependent on either glycolysis or OXPHOS were both found in the same tumor population highlighting the heterogeneity of GSCs(Saga et al., 2014, Shibao et al., 2018). Further, primary mouse GSCs have been shown to switch from OXPHOS dependence to glycolysis dependence upon metabolic stress(Saga et al., 2014, Shibao et al., 2018). Additionally, certain GSC populations have been shown to rely on glutamine metabolism and fatty acid metabolism for energy(Oizel et al., 2017). Analysis of gliomaspheres formed from primary patient samples revealed two distinct GSC populations that were differentially reliant on glutamine metabolism for survival(Oizel et al., 2017).
Similar to the glioblastoma data there is evidence some BCSCs primarily rely on fermentative glycolysis for survival(Ciavardelli et al., 2014). Sphere-forming BCSCs had increased expression of proteins involved in glycolysis compared to non-sphere forming cells(Ciavardelli et al., 2014). Metabolomic analysis revealed increased levels of glycolytic intermediates in BCSC mammospheres and enzymatic activity assays showed increased activity of glycolytic enzymes in BCSCs(Ciavardelli et al., 2014). Further, inhibiting glycolysis using 2-DG decreased BCSC growth and survival indicating reliance on glycolysis(Ciavardelli et al., 2014). While certain BCSC populations have been shown to rely on either OXPHOS or glycolysis for metabolic fuel, BCSCs have also been shown to rely on fatty acid metabolism for survival and chemoresistance(Wang et al., 2018). Using a fatty acid oxidation (FAO) assay, mammospheres and CSCs derived from primary patient samples showed increased rates of FAO compared to non-stem cell tumor cells(Wang et al., 2018). Further, etomoxir treatment led to decreased viability and mammosphere formation indicating a reliance on FAO for BCSC activity(Wang et al., 2018). Additionally, chemo-resistant primary TNBCs had increased tumorsphere formation, expression of FAO enzymes, and FAO activity, suggesting changes in tumor metabolism upon acquiring therapy resistance(Wang et al., 2018). These studies suggest different BCSC sub-populations rely on varying metabolic pathways and these metabolic dependencies can change upon chemotherapy treatment.
In contrast to de novo LSCs, primary mouse OCSCs were found to have increased glycolytic activity, be metabolically flexible and rely on glycolysis upon OXPHOS inhibition(Anderson et al., 2014). Additionally, ovarian spheroid cells have increased glycolytic flux and increased flux of glucose into the pentose phosphate pathway compared to the TCA cycle(Liao et al., 2014). While differing from studies mentioned above showing OCSCs rely on OXPHOS, taken together these studies suggest the microenvironment plays a critical role in determining the metabolic phenotype of OCSCs(Liao et al., 2014, Anderson et al., 2014, Pasto et al., 2014, Sato et al., 2016).
Similar to other solid tumor stem cells, some PDAC CSCs have also been shown to preferentially rely on glycolysis for survival(Isayev et al., 2014). Treatment of PDAC cells with 3-bromopyruvate, a glycolysis inhibitor, led to decreased tumorsphere formation in vitro, decreased viability of primary human PDAC CSCs and decreased engraftment into nude mice(Isayev et al., 2014). While other studies have shown, PDAC CSCs are reliant on OXPHOS for survival and tumor formation, discrepancies in metabolic reliance of PDAC CSCs may be due to metabolic flexibility(Isayev et al., 2014, Viale et al., 2014, Sancho et al., 2015, Perusina Lanfranca et al., 2019). Evidence for this includes gemcitabine resistant PDAC CSCs becoming sensitized to gemcitabine after pre-treatment with 3-bromopyruvate indicating metabolic reprogramming as a mechanism of chemotherapy resistance in PDAC CSCs(Isayev et al., 2014). CSC metabolic flexibility is not exclusive to PDAC CSCs as other gastrointestinal tumor CSCs including colon cancer stem cells (CCSCs) display metabolic flexibility(Chen et al., 2014). Metabolomic analysis of patient-derived CCSCs compared to non-stem tumor cells revealed increased TCA-cycle metabolites and S-Adenosyl Methionine(Chen et al., 2014). Additionally, as mentioned above CCSCs have been shown to reprogram metabolism upon chemotherapy treatment further corroborating the metabolic flexibility of CCSCs(Kahlert et al., 2017, Denise et al., 2015, Vellinga et al., 2015). Further, in primary mouse samples, CCSCs from liver metastases versus primary site CCSCs had increased lysine metabolism activity indicating metabolic reprogramming associated with metastatic CCSCs(Wu et al., 2015). Inhibiting lysine metabolism abolished metastatic potential of CCSCs indicating a reliance on lysine metabolism for survival and migration of primary CCSCs(Wu et al., 2015).
The discrepancy between which metabolic pathway CSCs are primarily dependent on has been partially attributed to the microenvironment in which the CSCs are found and isolated(Snyder et al., 2018, Kreuzaler et al., 2020). The tumor microenvironment is able to influence CSC metabolism by competing with the controlling nutrients available for CSCs(Snyder et al., 2018, Kreuzaler et al., 2020). Furthermore, as noted for LSCs, the stage of pathogenesis can be very important, where drug treatments and/or disease progression is generally associated with increased metabolic flexibility. Thus, while targeting CSC metabolism in as an appealing strategy to eradicate CSCs in patients, it is important to consider factors such as microenvironment and pathogenic stage in devising therapeutic approaches.
Clinical Targeting of Solid Tumor CSC Metabolism
Clinical trials targeting CSC metabolic pathways have been promising. Metformin has been investigated in clinical trials as an OXPHOS inhibitor of ovarian and pancreatic cancer stem cells. In ovarian cancer, a phase II clinical trial showed tumors treated with metformin had a 2.4-fold decrease in CSCs and decreased sphere-forming abilities (NCT01579812) (Brown et al., 2020). Additionally, the median progression free survival (PFS) was 18 months and the overall survival was 57.9 months which was higher than historical controls. These data suggest inhibiting ovarian cancer CSC metabolism has therapeutic potential in solid tumors. In a phase II clinical trial, metformin did not improve outcomes of patients with advanced pancreatic cancer, suggesting potential metabolic flexibility of PDAC CSCs leading to metformin insensitivity (NCT01210911)(Kordes et al., 2015).
In addition to targeting CSC OXPHOS activity in solid tumors, glycolysis inhibitors have also been pursued in clinical trials with limited efficacy(Jagust et al., 2019). In particular, silibinin, a GLUT transporter inhibitor, was investigated in a phase I clinical trial for prostate cancer patients (NCT00487721). There was no response in PSA levels and significant toxicities were present(Flaig et al., 2007) as GLUT transporters are ubiquitously expressed. The above-mentioned studies highlight both the promising and challenging aspects of clinically targeting CSC metabolism.
A comprehensive review of all current clinical trials targeting various pathways important to CSCs was recently published by Yang et al(Yang et al., 2020). LAT1 (SLC7A5, amino acid transporter light chain L system) inhibition is currently being investigated as a strategy to target CSCs in glioblastoma multiforme (NCT03849105). Primary human GBM cells that were orothotopically injected into mice were found to have increased LAT1 expression, indicating LAT1 expression was higher in tumor-initiating cells(Cai et al., 2020). Pegzilarginase, a recombinant pegylated arginase, is being evaluated for use in small-cell lung cancer (NCT03371979). Through metabolomic analysis, MYC-driven small-cell lung cancer cells were found to be uniquely dependent on arginine metabolism, therefore pegzilarginase is being explored as a treatment option(Chalishazar et al., 2019). Further, there is evidence the stem cell compartment is uniquely reliant on arginine as engraftment potential significantly decreased upon arginine depletion(Chalishazar et al., 2019). FASN (fatty acid synthase) inhibition is being evaluated for breast cancer, high grade astrocytoma, colon cancer and non-small cell lung cancer (NCT03179904, NCT03032484, NCT02980029, NCT03808558). In breast cancer, FASN inhibitors were able to inhibit mammosphere forming ability and BCSC survival indicating the importance of FASN for BCSC potential(Giró-Perafita et al., 2019). In patients with advanced solid tumors, AMPK inhibition through IM156 is being evaluated (NCT03272256). IM156 in pre-clinical models was able to reduce OXPHOS activity and be effective in glioblastoma, gastric cancer and other solid tumor models(Rha et al., 2018). In solid tumors, telaglenastat, a glutaminase inhibitor is being evaluated for clinical efficacy due to efficacy against multiple CSCs including glioma stem cells, hepatocellular carcinoma stem cells and sarcoma initiating cells (NCT03965845)(Koch et al., 2020, Li et al., 2019, Lee et al., 2020).
Conclusions and Future Directions
Based on the evidence presented in this article and elsewhere, targeting metabolic vulnerabilities appears to represent a useful strategy for cancer therapy. This approach addresses three important issues: 1) Targeting metabolic properties may facilitate eradication of disease-initiating cells and thereby inhibit disease recurrence and metastasis. 2) Targeting metabolic dependencies of CSCs may allow for eradication of cells independent of genotype or tissue of origin. 3) Metabolic properties of CSCs can also be quite different than normal stem cells, thereby providing a potential therapeutic index for therapies designed to selectively target CSC metabolism. Altogether, these considerations suggest that targeting CSC metabolism may improve cancer patient outcomes. A particularly noteworthy vulnerability reported for multiple types of CSCs is reliance on OXPHOS. Thus far, OXPHOS dependency has been targeted clinically through four unique approaches including inhibition of mitochondrial translation using tigecycline(Reed et al., 2016), inhibition of amino acid and glutathione metabolism through venetoclax with azacitidine treatment(Pollyea et al., 2018, Jones et al., 2018, DiNardo et al., 2018b), inhibition of protein translation through omacetaxine (NCT03564873), and direct ETC Complex I inhibition through ICAS-010759 treatment (NCT02882321 and NCT03291938). Notably, the results of venetoclax with azacitidine treatment have demonstrated high response rates in newly diagnosed AML patients suggesting that targeting OXPHOS may have significant therapeutic potential in leukemia(DiNardo et al., 2018b).
Current and Potential Roadblocks
Going forward, additional preclinical and clinical studies evaluating novel metabolic vulnerabilities of CSCs as well as mechanisms of resistance to metabolism targeting therapies will serve to validate the overall approach. As metabolism is a ubiquitous process, toxicities from metabolism-based therapies will be important to evaluate. Indeed, unacceptable toxicities have previously been reported for clinical trials evaluating inhibition of glycolysis in solid tumors(Jagust et al., 2019). Further, in some clinical trials targeting CSC metabolism has not resulted in significant response. This could be attributed to metabolic rewiring as multiple models of CSCs have been shown to be metabolically flexible. Therefore finding specific and consistent metabolic vulnerabilities of CSCs will be crucial in developing more robust therapeutic strategies.
Additional studies are needed for the field to better understand commonalities and differences among CSC from varying tumor types. The conditions in which these studies are done should be carefully considered. Whenever possible, defining the metabolism of CSCs isolated from primary human specimens is preferable in creating CSC-targeted therapies. Further, pharmacodynamic and correlative endpoints designed to evaluate the clinical effect of metabolic targeting therapies on CSC metabolism and CSC function will be important as a means to accurately assesss the consequences of CSC therapy. More in-depth and frequent patient monitoring and specimen collection should also help to identify the mechanisms of resistance to metabolic targeting therapies. Therapy resistance is currently a major limitation in achieving long-term clinical responses for the majority of cancer patients. However, together with the advent of increasingly effective targeted agents, and the ability to identify and clinically modulate metabolic properties, addressing CSC-specific vulnerabilities appears to be a promising direction in the treatment of cancer.
Cancer stem cells have unique metabolic biology that can in some instances be exploited for therapeutic intervention. This review summarizes the progress and challenges inherent to this field, including metabolic flexibility, inter and intra-patient heterogeneity and commonalities and differences in energy metabolism between leukemia and solid tumors.
Acknowledgements:
We would like to thank Drs. James DeGregori and Daniel Pollyea for their critical review of this article. This work was supported by the Leukemia and Lymphoma Society Special Fellow award and The Princess Margaret Cancer Centre, The Princess Margaret Cancer Foundation, and the Ontario Ministry of Health (C.L.J.), the National Institutes of Health under Ruth L. Kirschstein National Research Service Award T32CA190216 (A.I.), as well as National Institutes of Health grants R01CA200707, R01CA243452, R35CA242376, and P30CA046934 (to C.T.J.); and a Leukemia and Lymphoma Society Specialized Center of Research grant (principal investigator, C.T.J.). C.T.J. is supported by the Nancy Carroll Allen Endowed Chair.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest:
The authors declare no conflict of interest.
References:
- ABRAHAM A, QIU S, CHACKO BK, LI H, PATERSON A, HE J, AGARWAL P, SHAH M, WELNER R, DARLEY-USMAR VM & BHATIA R 2019. SIRT1 regulates metabolism and leukemogenic potential in CML stem cells. The Journal of clinical investigation, 129, 2685–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ADANE B, YE H, KHAN N, PEI S, MINHAJUDDIN M, STEVENS BM, JONES CL, D’ALESSANDRO A, REISZ JA, ZABEREZHNYY V, GASPARETTO M, HO TC, KELLY KK, MYERS JR, ASHTON JM, SIEGENTHALER J, KUME T, CAMPBELL EL, POLLYEA DA, BECKER MW & JORDAN CT 2019. The Hematopoietic Oxidase NOX2 Regulates Self-Renewal of Leukemic Stem Cells. Cell Rep, 27, 238–254.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AGATHOCLEOUS M, MEACHAM CE, BURGESS RJ, PISKOUNOVA E, ZHAO Z, CRANE GM, COWIN BL, BRUNER E, MURPHY MM, CHEN W, SPANGRUDE GJ, HU Z, DEBERARDINIS RJ & MORRISON SJ 2017. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature, 549, 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AL-HAJJ M, WICHA MS, BENITO-HERNANDEZ A, MORRISON SJ & CLARKE MF 2003. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A, 100, 3983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ANDERSON AS, ROBERTS PC, FRISARD MI, HULVER MW & SCHMELZ EM 2014. Ovarian tumor-initiating cells display a flexible metabolism. Exp Cell Res, 328, 44–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BACCELLI I, GAREAU Y, LEHNERTZ B, GINGRAS S, SPINELLA JF, CORNEAU S, MAYOTTE N, GIRARD S, FRECHETTE M, BLOUIN-CHAGNON V, LEVEILLE K, BOIVIN I, MACRAE T, KROSL J, THIOLLIER C, LAVALLEE VP, KANSHIN E, BERTOMEU T, COULOMBE-HUNTINGTON J, ST-DENIS C, BORDELEAU ME, BOUCHER G, ROUX PP, LEMIEUX S, TYERS M, THIBAULT P, HEBERT J, MARINIER A & SAUVAGEAU G 2019. Mubritinib Targets the Electron Transport Chain Complex I and Reveals the Landscape of OXPHOS Dependency in Acute Myeloid Leukemia. Cancer Cell, 36, 84–99.e8. [DOI] [PubMed] [Google Scholar]
- BATLLE E & CLEVERS H 2017. Cancer stem cells revisited. Nat Med, 23, 1124–1134. [DOI] [PubMed] [Google Scholar]
- BEIER D, HAU P, PROESCHOLDT M, LOHMEIER A, WISCHHUSEN J, OEFNER PJ, AIGNER L, BRAWANSKI A, BOGDAHN U & BEIER CP 2007. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res, 67, 4010–5. [DOI] [PubMed] [Google Scholar]
- BEJARANO-GARCÍA JA, MILLÁN-UCLÉS Á, ROSADO IV, SÁNCHEZ-ABARCA LI, CABALLERO-VELÁZQUEZ T, DURÁN-GALVÁN MJ, PÉREZ-SIMÓN JA & PIRUAT JI 2016. Sensitivity of hematopoietic stem cells to mitochondrial dysfunction by SdhD gene deletion. Cell Death & Disease, 7, e2516–e2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BONNET D & DICK JE 1997. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature Medicine, 3, 730–737. [DOI] [PubMed] [Google Scholar]
- BROWN HK, TELLEZ-GABRIEL M & HEYMANN D 2017. Cancer stem cells in osteosarcoma. Cancer Letters, 386, 189–195. [DOI] [PubMed] [Google Scholar]
- BROWN JR, CHAN DK, SHANK JJ, GRIFFITH KA, FAN H, SZULAWSKI R, YANG K, REYNOLDS RK, JOHNSTON C, MCLEAN K, UPPAL S, LIU JR, CABRERA L, TAYLOR SE, ORR BC, MODUGNO F, MEHTA P, BREGENZER M, MEHTA G, SHEN H, COFFMAN LG & BUCKANOVICH RJ 2020. Phase II clinical trial of metformin as a cancer stem cell-targeting agent in ovarian cancer. JCI Insight, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAI L, KIRCHLEITNER SV, ZHAO D, LI M, TONN JC, GLASS R & KÄLIN RE 2020. Glioblastoma Exhibits Inter-Individual Heterogeneity of TSPO and LAT1 Expression in Neoplastic and Parenchymal Cells. Int J Mol Sci, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CASTRO I, SAMPAIO-MARQUES B & LUDOVICO P 2019. Targeting Metabolic Reprogramming in Acute Myeloid Leukemia. Cells, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHALISHAZAR MD, WAIT SJ, HUANG F, IRELAND AS, MUKHOPADHYAY A, LEE Y, SCHUMAN SS, GUTHRIE MR, BERRETT KC, VAHRENKAMP JM, HU Z, KUDLA M, MODZELEWSKA K, WANG G, INGOLIA NT, GERTZ J, LUM DH, COSULICH SC, BOMALASKI JS, DEBERARDINIS RJ & OLIVER TG 2019. MYC-Driven Small-Cell Lung Cancer is Metabolically Distinct and Vulnerable to Arginine Depletion. Clin Cancer Res, 25, 5107–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN KY, LIU X, BU P, LIN CS, RAKHILIN N, LOCASALE JW & SHEN X 2014. A metabolic signature of colon cancer initiating cells. Conf Proc IEEE Eng Med Biol Soc, 2014, 4759–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN X, GLYTSOU C, ZHOU H, NARANG S, REYNA DE, LOPEZ A, SAKELLAROPOULOS T, GONG Y, KLOETGEN A, YAP YS, WANG E, GAVATHIOTIS E, TSIRIGOS A, TIBES R & AIFANTIS I 2019. Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment. Cancer Discov, 9, 890–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHIU PP, JIANG H & DICK JE 2010. Leukemia-initiating cells in human T-lymphoblastic leukemia exhibit glucocorticoid resistance. Blood, 116, 5268–79. [DOI] [PubMed] [Google Scholar]
- CIAVARDELLI D, ROSSI C, BARCAROLI D, VOLPE S, CONSALVO A, ZUCCHELLI M, DE COLA A, SCAVO E, CAROLLO R, D’AGOSTINO D, FORLÌ F, D’AGUANNO S, TODARO M, STASSI G, DI ILIO C, DE LAURENZI V & URBANI A 2014. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis, 5, e1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CIMMINO L, DOLGALEV I, WANG Y, YOSHIMI A, MARTIN GH, WANG J, NG V, XIA B, WITKOWSKI MT, MITCHELL-FLACK M, GRILLO I, BAKOGIANNI S, NDIAYE-LOBRY D, MARTIN MT, GUILLAMOT M, BANH RS, XU M, FIGUEROA ME, DICKINS RA, ABDEL-WAHAB O, PARK CY, TSIRIGOS A, NEEL BG & AIFANTIS I 2017. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell, 170, 1079–1095.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLARKE MF, DICK JE, DIRKS PB, EAVES CJ, JAMIESON CH, JONES DL, VISVADER J, WEISSMAN IL & WAHL GM 2006. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res, 66, 9339–44. [DOI] [PubMed] [Google Scholar]
- COLE A, WANG Z, COYAUD E, VOISIN V, GRONDA M, JITKOVA Y, MATTSON R, HURREN R, BABOVIC S, MACLEAN N, RESTALL I, WANG X, JEYARAJU DV, SUKHAI MA, PRABHA S, BASHIR S, RAMAKRISHNAN A, LEUNG E, QIA YH, ZHANG N, COMBES KR, KETELA T, LIN F, HOURY WA, AMAN A, AL- AWAR R, ZHENG W, WIENHOLDS E, XU CJ, DICK J, WANG JC, MOFFAT J, MINDEN MD, EAVES CJ, BADER GD, HAO Z, KORNBLAU SM, RAUGHT B & SCHIMMER AD 2015. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell, 27, 864–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORREIA M, PERESTRELO T, RODRIGUES AS, RIBEIRO MF, PEREIRA SL, SOUSA MI & RAMALHO-SANTOS J 2017. Sirtuins in metabolism, sternness and differentiation. Biochimica et Biophysica Acta (BBA) - General Subjects, 1861, 3444–3455. [DOI] [PubMed] [Google Scholar]
- COX CV, MARTIN HM, KEARNS PR, VIRGO P, EVELY RS & BLAIR A 2007. Characterization of a progenitor cell population in childhood T-cell acute lymphoblastic leukemia. Blood, 109, 674–82. [DOI] [PubMed] [Google Scholar]
- CRAMER SL, SAHA A, LIU J, TADI S, TIZIANI S, YAN W, TRIPLETT K, LAMB C, ALTERS SE, ROWLINSON S, ZHANG YJ, KEATING MJ, HUANG P, DIGIOVANNI J, GEORGIOU G & STONE E 2017. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nature medicine, 23, 120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DANG L, WHITE DW, GROSS S, BENNETT BD, BITTINGER MA, DRIGGERS EM, FANTIN VR, JANG HG, JIN S, KEENAN MC, MARKS KM, PRINS RM, WARD PS, YEN KE, LIAU LM, RABINOWITZ JD, CANTLEY LC, THOMPSON CB, VANDER HEIDEN MG & SU SM 2009. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature, 462, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE FRANCESCO EM, SOTGIA F & LISANTI MP 2018. Cancer stem cells (CSCs): metabolic strategies for their identification and eradication. The Biochemical journal, 475, 1611–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE LUCA A, FIORILLO M, PEIRIS-PAGES M, OZSVARI B, SMITH DL, SANCHEZ-ALVAREZ R, MARTINEZ-OUTSCHOORN UE, CAPPELLO AR, PEZZI V, LISANTI MP & SOTGIA F 2015. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget, 6, 14777–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DENISE C, PAOLI P, CALVANI M, TADDEI ML, GIANNONI E, KOPETZ S, KAZMI SM, PIA MM, PETTAZZONI P, SACCO E, CASELLI A, VANONI M, LANDRISCINA M, CIRRI P & CHIARUGI P 2015. 5-fluorouracil resistant colon cancer cells are addicted to OXPHOS to survive and enhance stem-like traits. Oncotarget, 6, 41706–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DESAI A, YAN Y & GERSON SL 2019. Concise Reviews: Cancer Stem Cell Targeted Therapies: Toward Clinical Success. Stem Cells Transl Med, 8, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DHALL A, ZEE BM, YAN F & BLANCO MA 2019. Intersection of Epigenetic and Metabolic Regulation of Histone Modifications in Acute Myeloid Leukemia. Frontiers in Oncology, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIEHN M, CHO RW, LOBO NA, KALISKY T, DORIE MJ, KULP AN, QIAN D, LAM JS, AILLES LE, WONG M, JOSHUA B, KAPLAN MJ, WAPNIR I, DIRBAS FM, SOMLO G, GARBEROGLIO C, PAZ B, SHEN J, LAU SK, QUAKE SR, BROWN JM, WEISSMAN IL & CLARKE MF 2009. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature, 458, 780–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DINARDO CD, PRATZ KW, LETAI A, JONAS BA, WEI AH, THIRMAN M, ARELLANO M, FRATTINI MG, KANTARJIAN H, POPOVIC R, CHYLA B, XU T, DUNBAR M, AGARWAL SK, HUMERICKHOUSE R, MABRY M, POTLURI J, KONOPLEVA M & POLLYEA DA 2018a. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol, 19, 216–228. [DOI] [PubMed] [Google Scholar]
- DINARDO CD, RAUSCH CR, BENTON C, KADIA T, JAIN N, PEMMARAJU N, DAVER N, COVERT W, MARX KR, MACE M, JABBOUR E, CORTES J, GARCIA-MANERO G, RAVANDI F, BHALLA KN, KANTARJIAN H & KONOPLEVA M 2018b. Clinical experience with the BCL2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies. Am J Hematol, 93, 401–407. [DOI] [PubMed] [Google Scholar]
- DINARDO CD, STEIN EM, DE BOTTON S, ROBOZ GJ, ALTMAN JK, MIMS AS, SWORDS R, COLLINS RH, MANNIS GN, POLLYEA DA, DONNELLAN W, FATHI AT, PIGNEUX A, ERBA HP, PRINCE GT, STEIN AS, UY GL, FORAN JM, TRAER E, STUART RK, ARELLANO ML, SLACK JL, SEKERES MA, WILLEKENS C, CHOE S, WANG H, ZHANG V, YEN KE, KAPSALIS SM, YANG H, DAI D, FAN B, GOLDWASSER M, LIU H, AGRESTA S, WU B, ATTAR EC, TALLMAN MS, STONE RM & KANTARJIAN HM 2018c. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N Engl J Med, 378, 2386–2398. [DOI] [PubMed] [Google Scholar]
- DOBSON SM, GARCIA-PRAT L, VANNER RJ, WINTERSINGER J, WAANDERS E, GU Z, MCLEOD J, GAN OI, GRANDAL I, PAYNE-TURNER D, EDMONSON MN, MA X, FAN Y, VOISIN V, CHAN-SENG-YUE M, XIE SZ, HOSSEINI M, ABELSON S, GUPTA P, RUSCH M, SHAO Y, OLSEN SR, NEALE G, CHAN SM, BADER G, EASTON J, GUIDOS CJ, DANSKA JS, ZHANG J, MINDEN MD, MORRIS Q, MULLIGHAN CG & DICK JE 2020. Relapse fated latent diagnosis subclones in acute B lineage leukaemia are drug tolerant and possess distinct metabolic programs. Cancer Discovery, CD-19-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUAN JJ, QIU W, XU SL, WANG B, YE XZ, PING YF, ZHANG X, BIAN XW & YU SC 2013. Strategies for isolating and enriching cancer stem cells: well begun is half done. Stem Cells Dev, 22, 2221–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FANG Y, TANG S & LI X 2019. Sirtuins in Metabolic and Epigenetic Regulation of Stem Cells. Trends Endocrinol Metab, 30, 177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FARGE T, SALAND E, DE TONI F, AROUA N, HOSSEINI M, PERRY R, BOSC C, SUGITA M, STUANI L, FRAISSE M, SCOTLAND S, LARRUE C, BOUTZEN H, FELIU V, NICOLAU-TRAVERS ML, CASSANT-SOURDY S, BROIN N, DAVID M, SERHAN N, SARRY A, TAVITIAN S, KAOMA T, VALLAR L, IACOVONI J, LINARES LK, MONTERSINO C, CASTELLANO R, GRIESSINGER E, COLLETTE Y, DUCHAMP O, BARREIRA Y, HIRSCH P, PALAMA T, GALES L, DELHOMMEAU F, GARMY-SUSINI BH, PORTAIS JC, VERGEZ F, SELAK M, DANET-DESNOYERS G, CARROLL M, RECHER C & SARRY JE 2017. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. CancerDiscov, 7, 716–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FARNIE G, SOTGIA F & LISANTI MP 2015. High mitochondrial mass identifies a subpopulation of stem-like cancer cells that are chemo-resistant. Oncotarget, 6, 30472–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FIGUEROA ME, ABDEL-WAHAB O, LU C, WARD PS, PATEL J, SHIH A, LI Y, BHAGWAT N, VASANTHAKUMAR A, FERNANDEZ HF, TALLMAN MS, SUN Z, WOLNIAK K, PEETERS JK, LIU W, CHOE SE, FANTIN VR, PAIETTA E, LOWENBERG B, LICHT JD, GODLEY LA, DELWEL R, VALK PJ, THOMPSON CB, LEVINE RL & MELNICK A 2010. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell, 18, 553–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLAIG TW, GUSTAFSON DL, SU LJ, ZIRROLLI JA, CRIGHTON F, HARRISON GS, PIERSON AS, AGARWAL R & GLODé LM 2007. A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Invest New Drugs, 25, 139–46. [DOI] [PubMed] [Google Scholar]
- FLAVAHAN WA, WU Q, HITOMI M, RAHIM N, KIM Y, SLOAN AE, WEIL RJ, NAKANO I, SARKARIA JN, STRINGER BW, DAY BW, LI M, LATHIA JD, RICH JN & HJELMELAND AB 2013. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci, 16, 1373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAO C, SHEN Y, JIN F, MIAO Y & QIU X 2016. Cancer Stem Cells in Small Cell Lung Cancer Cell Line H446: Higher Dependency on Oxidative Phosphorylation and Mitochondrial Substrate-Level Phosphorylation than Non-Stem Cancer Cells. PLoS One, 11, e0154576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIMPLE RC, BHARGAVA S, DIXIT D & RICH JN 2019. Glioblastoma stem cells: lessons from the tumor hierarchy in a lethal cancer. Genes Dev, 33, 591–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIRÉ-PERAFITA A, RABIONET M, PLANAS M, FELIU L, CIURANA J, RUIZ-MARTÍNEZ S & PUIG T 2019. EGCG-Derivative G28 Shows High Efficacy Inhibiting the Mammosphere-Forming Capacity of Sensitive and Resistant TNBC Models. Molecules, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GLYNN SE 2017. Multifunctional Mitochondrial AAA Proteases. Frontiers in Molecular Biosciences, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOLUB D, IYENGAR N, DOGRA S, WONG T, BREADY D, TANG K, MODREK AS & PLACANTONAKIS DG 2019. Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics. Frontiers in oncology, 9, 417–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUAN Y, GERHARD B & HOGGE DE 2003. Detection, isolation, and stimulation of quiescent primitive leukemic progenitor cells from patients with acute myeloid leukemia (AML). Blood, 101, 3142–9. [DOI] [PubMed] [Google Scholar]
- GUITART AV, PANAGOPOULOU TI, VILLACRECES A, VUKOVIC M, SEPULVEDA C, ALLEN L, CARTER RN, VAN DE LAGEMAAT LN, MORGAN M, GILES P, SAS Z, GONZALEZ MV, LAWSON H, PARIS J, EDWARDS-HICKS J, SCHAAK K, SUBRAMANI C, GEZER D, ARMESILLA-DIAZ A, WILLS J, EASTERBROOK A, COMAN D, SO CWE, O’CARROLL D, VERNIMMEN D, RODRIGUES NP, POLLARD PJ, MORTON NM, FINCH A & KRANC KR 2017. Fumarate hydratase is a critical metabolic regulator of hematopoietic stem cell functions. Journal of Experimental Medicine, 214, 719–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUO B, HAN X, TKACH D, HUANG SG & ZHANG D 2018. AMPK promotes the survival of colorectal cancer stem cells. Animal Model Exp Med, 1, 134–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUZMAN ML, NEERING SJ, UPCHURCH D, GRIMES B, HOWARD DS, RIZZIERI DA, LUGER SM & JORDAN CT 2001. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood, 98, 2301–7. [DOI] [PubMed] [Google Scholar]
- HATTORI A, TSUNODA M, KONUMA T, KOBAYASHI M, NAGY T, GLUSHKA J, TAYYARI F, MCSKIMMING D, KANNAN N, TOJO A, EDISON AS & ITO T 2017. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature, 545, 500–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HO TC, LAMERE M, STEVENS BM, ASHTON JM, MYERS JR, O’DWYER KM, LIESVELD JL, MENDLER JH, GUZMAN M, MORRISSETTE JD, ZHAO J, WANG ES, WETZLER M, JORDAN CT & BECKER MW 2016. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood, 128, 1671–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOPE KJ, JIN L & DICK JE 2003. Human acute myeloid leukemia stem cells. Arch Med Res, 34, 507–14. [DOI] [PubMed] [Google Scholar]
- HURT EM, KAWASAKI BT, KLARMANN GJ, THOMAS SB & FARRAR WL 2008. CD44+ CD24(−) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br J Cancer, 98, 756–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISAYEV O, RAUSCH V, BAUER N, LIU L, FAN P, ZHANG Y, GLADKICH J, NWAEBURU CC, MATTERN J, MOLLENHAUER M, ROCKERT F, ZACH S, HABERKORN U, GROSS W, SCHÖNSIEGEL F, BAZHIN AV & HERR I 2014. Inhibition of glucose turnover by 3-bromopyruvate counteracts pancreatic cancer stem cell features and sensitizes cells to gemcitabine. Oncotarget, 5, 5177–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISHIKAWA F, YOSHIDA S, SAITO Y, HIJIKATA A, KITAMURA H, TANAKA S, NAKAMURA R, TANAKA T, TOMIYAMA H, SAITO N, FUKATA M, MIYAMOTO T, LYONS B, OHSHIMA K, UCHIDA N, TANIGUCHI S, OHARA O, AKASHI K, HARADA M & SHULTZ LD 2007. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol, 25, 1315–21. [DOI] [PubMed] [Google Scholar]
- ISHIZAWA J, ZARABI SF, DAVIS RE, HALGAS O, NII T, JITKOVA Y, ZHAO R, ST- GERMAIN J, HEESE LE, EGAN G, RUVOLO VR, BARGHOUT SH, NISHIDA Y, HURREN R, MA W, GRONDA M, LINK T, WONG K, MABANGLO M, KOJIMA K, BORTHAKUR G, MACLEAN N, MA MCJ, LEBER AB, MINDEN MD, HOURY W, KANTARJIAN H, STOGNIEW M, RAUGHT B, PAI EF, SCHIMMER AD & ANDREEFF M 2019. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell, 35, 721–737.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ITO K, HIRAO A, ARAI F, MATSUOKA S, TAKUBO K, HAMAGUCHI I, NOMIYAMA K, HOSOKAWA K, SAKURADA K, NAKAGATA N, IKEDA Y, MAK TW & SUDA T 2004. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature, 431, 997–1002. [DOI] [PubMed] [Google Scholar]
- ITO K, HIRAO A, ARAI F, TAKUBO K, MATSUOKA S, MIYAMOTO K, OHMURA M, NAKA K, HOSOKAWA K, IKEDA Y & SUDA T 2006. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med, 12, 446–51. [DOI] [PubMed] [Google Scholar]
- JAGUST P, DE LUXÁN-DELGADO B, PAREJO-ALONSO B & SANCHO P 2019. Metabolism-Based Therapeutic Strategies Targeting Cancer Stem Cells. Frontiers in pharmacology, 10, 203–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JANG YY & SHARKIS SJ 2007. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood, 110, 3056–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JANISZEWSKA M, SUVA ML, RIGGI N, HOUTKOOPER RH, AUWERX J, CLEMENT-SCHATLO V, RADOVANOVIC I, RHEINBAY E, PROVERO P & STAMENKOVIC I 2012. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev, 26, 1926–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JHAS B, SRISKANTHADEVAN S, SKRTIC M, SUKHAI MA, VOISIN V, JITKOVA Y, GRONDA M, HURREN R, LAISTER RC, BADER GD, MINDEN MD & SCHIMMER AD 2013. Metabolic adaptation to chronic inhibition of mitochondrial protein synthesis in acute myeloid leukemia cells. PLoS One, 8, e58367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JITKOVA Y, GRONDA M, HURREN R, WANG X, GOARD CA, JHAS B & SCHIMMER AD 2014. A novel formulation of tigecycline has enhanced stability and sustained antibacterial and antileukemic activity. PLoS One, 9, e95281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JONES CL, STEVENS BM, D’ALESSANDRO A, CULP-HILL R, REISZ JA, PEI S, GUSTAFSON A, KHAN N, DEGREGORI J, POLLYEA DA & JORDAN CT 2019. Cysteine depletion targets leukemia stem cells through inhibition of electron transport complex II. Blood. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JONES CL, STEVENS BM, D’ALESSANDRO A, REISZ JA, CULP-HILL R, NEMKOV T, PEI S, KHAN N, ADANE B, YE H, KRUG A, REINHOLD D, SMITH C, DEGREGORI J, POLLYEA DA & JORDAN CT 2018. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell, 34, 724–740.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JONES CL, STEVENS BM, POLLYEA DA, CULP-HILL R, REISZ JA, NEMKOV T, GEHRKE S, GAMBONI F, KRUG A, WINTERS A, PEI S, GUSTAFSON A, YE H, INGUVA A, AMAYA M, MINHAJUDDIN M, ABBOTT D, BECKER MW, DEGREGORI J, SMITH CA, D’ALESSANDRO A & JORDAN CT 2020. Nicotinamide Metabolism Mediates Resistance to Venetoclax in Relapsed Acute Myeloid Leukemia Stem Cells. Cell Stem Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JORDAN CT 2002. Unique molecular and cellular features of acute myelogenous leukemia stem cells. Leukemia, 16, 559–62. [DOI] [PubMed] [Google Scholar]
- JORDAN CT 2010. Targeting myeloid leukemia stem cells. Sci Transl Med, 2, 31ps21. [DOI] [PubMed] [Google Scholar]
- JORDAN CT, GUZMAN ML & NOBLE M 2006. Cancer stem cells. N Engl J Med, 355, 1253–61. [DOI] [PubMed] [Google Scholar]
- JUNTILLA MM, PATIL VD, CALAMITO M, JOSHI RP, BIRNBAUM MJ & KORETZKY GA 2010. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood, 115, 4030–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAHLERT UD, MOONEY SM, NATSUMEDA M, STEIGER HJ & MACIACZYK J 2017. Targeting cancer stem-like cells in glioblastoma and colorectal cancer through metabolic pathways. Int J Cancer, 140, 10–22. [DOI] [PubMed] [Google Scholar]
- KIKUSHIGE Y, SHIMA T, TAKAYANAGI S, URATA S, MIYAMOTO T, IWASAKI H, TAKENAKA K, TESHIMA T, TANAKA T, INAGAKI Y & AKASHI K 2010. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell, 7, 708–17. [DOI] [PubMed] [Google Scholar]
- KIM CF, JACKSON EL, WOOLFENDEN AE, LAWRENCE S, BABAR I, VOGEL S, CROWLEY D, BRONSON RT & JACKS T 2005. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell, 121, 823–35. [DOI] [PubMed] [Google Scholar]
- KOCH K, HARTMANN R, TSIAMPALI J, UHLMANN C, NICKEL AC, HE X, KAMP MA, SABEL M, BARKER RA, STEIGER HJ, HÄNGGI D, WILLBOLD D, MACIACZYK J & KAHLERT UD 2020. A comparative pharmaco-metabolomic study of glutaminase inhibitors in glioma stem-like cells confirms biological effectiveness but reveals differences in target-specificity. Cell Death Discov, 6, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KORDES S, POLLAK MN, ZWINDERMAN AH, MATHÔT RA, WETERMAN MJ, BEEKER A, PUNT CJ, RICHEL DJ & WILMINK JW 2015. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol, 16, 839–47. [DOI] [PubMed] [Google Scholar]
- KRESO A & DICK JE 2014. Evolution of the cancer stem cell model. Cell Stem Cell, 14, 275–91. [DOI] [PubMed] [Google Scholar]
- KREUZALER P, PANINA Y, SEGAL J & YUNEVA M 2020. Adapt and conquer: Metabolic flexibility in cancer growth, invasion and evasion. Mol Metab, 33, 83–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUNTZ EM, BAQUERO P, MICHIE AM, DUNN K, TARDITO S, HOLYOAKE TL, HELGASON GV & GOTTLIEB E 2017. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat Med, 23, 1234–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAGADINOU ED, SACH A, CALLAHAN K, ROSSI RM, NEERING SJ, MINHAJUDDIN M, ASHTON JM, PEI S, GROSE V, O’DWYER KM, LIESVELD JL, BROOKES PS, BECKER MW & JORDAN CT 2013. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell, 12, 329–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAPIDOT T, SIRARD C, VORMOOR J, MURDOCH B, HOANG T, CACERES-CORTES J, MINDEN M, PATERSON B, CALIGIURI MA & DICK JE 1994. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature, 367, 645–648. [DOI] [PubMed] [Google Scholar]
- LEE KM, GILTNANE JM, BALKO JM, SCHWARZ LJ, GUERRERO-ZOTANO AL, HUTCHINSON KE, NIXON MJ, ESTRADA MV, SANCHEZ V, SANDERS ME, LEE T, GOMEZ H, LLUCH A, PEREZ-FIDALGO JA, WOLF MM, ANDREJEVA G, RATHMELL JC, FESIK SW & ARTEAGA CL 2017. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab, 26, 633–647.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE P, MALIK D, PERKONS N, HUANGYANG P, KHARE S, RHOADES S, GONG YY, BURROWS M, FINAN JM, NISSIM I, GADE TPF, WELJIE AM & SIMON MC 2020. Targeting glutamine metabolism slows soft tissue sarcoma growth. Nat Commun, 11, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEKO V, VARNUM-FINNEY B, LI H, GU Y, FLOWERS D, NOURIGAT C, BERNSTEIN ID & BEDALOV A 2012. SIRT1 is dispensable for function of hematopoietic stem cells in adult mice. Blood, 119, 1856–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI B, CAO Y, MENG G, QIAN L, XU T, YAN C, LUO O, WANG S, WEI J, DING Y & YU D 2019. Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine, 39, 239–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI C, HEIDT DG, DALERBA P, BURANT CF, ZHANG L, ADSAY V, WICHA M, CLARKE MF & SIMEONE DM 2007. Identification of pancreatic cancer stem cells. Cancer Res, 67, 1030–7. [DOI] [PubMed] [Google Scholar]
- LI L, WANG L, LI L, WANG Z, HO Y, MCDONALD T, HOLYOAKE TL, CHEN W & BHATIA R 2012. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell, 21, 266–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIANG R, ARIF T, KALMYKOVA S, KASIANOV A, LIN M, MENON V, QIU J, BERNITZ JM, MOORE K, LIN F, BENSON DL, TZAVARAS N, MAHAJAN M, PAPATSENKO D & GHAFFARI S 2020. Restraining Lysosomal Activity Preserves Hematopoietic Stem Cell Quiescence and Potency. Cell Stem Cell, 26, 359–376.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIAO J, QIAN F, TCHABO N, MHAWECH-FAUCEGLIA P, BECK A, QIAN Z, WANG X, HUSS WJ, LELE SB, MORRISON CD & ODUNSI K 2014. Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through hypoxia-resistant metabolism. PLoS One, 9, e84941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LÖWENBERG B, DOWNING JR & BURNETT A 1999. Acute myeloid leukemia. N Engl J Med, 341, 1051–62. [DOI] [PubMed] [Google Scholar]
- LUO M & WICHA MS 2019. Targeting Cancer Stem Cell Redox Metabolism to Enhance Therapy Responses. Semin Radiat Oncol, 29, 42–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MA S, CHAN KW, HU L, LEE TK, WO JY, NG IO, ZHENG BJ & GUAN XY 2007. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology, 132, 2542–56. [DOI] [PubMed] [Google Scholar]
- MAJETI R 2011. Monoclonal antibody therapy directed against human acute myeloid leukemia stem cells. Oncogene, 30, 1009–19. [DOI] [PubMed] [Google Scholar]
- MANSELL E, SIGURDSSON V, DELTCHEVA E, BROWN J, JAMES C, MIHARADA K, SONEJI S, LARSSON J & ENVER T 2020. Mitochondrial Potentiation Ameliorates Age-Related Heterogeneity in Hematopoietic Stem Cell Function. Cell Stem Cell. [DOI] [PubMed] [Google Scholar]
- MIRALI S, BOTHAM A, VOISIN V, XU C, ST-GERMAIN J, SHARON D, HOFF FW, QIU Y, HURREN R, GRONDA M, JITKOVA Y, NACHMIAS B, MACLEAN N, WANG X, ARRUDA A, MINDEN MD, HORTON TM, KORNBLAU SM, CHAN SM, BADER GD, RAUGHT B & SCHIMMER AD 2020. The mitochondrial peptidase, neurolysin, regulates respiratory chain supercomplex formation and is necessary for AML viability. Science Translational Medicine, 12, eaaz8264. [DOI] [PubMed] [Google Scholar]
- MIYAMOTO K, ARAKI KY, NAKA K, ARAI F, TAKUBO K, YAMAZAKI S, MATSUOKA S, MIYAMOTO T, ITO K, OHMURA M, CHEN C, HOSOKAWA K, NAKAUCHI H, NAKAYAMA K, NAKAYAMA KI, HARADA M, MOTOYAMA N, SUDA T & HIRAO A 2007. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell, 1, 101–12. [DOI] [PubMed] [Google Scholar]
- MOLINA JR, SUN Y, PROTOPOPOVA M, GERA S, BANDI M, BRISTOW C, MCAFOOS T, MORLACCHI P, ACKROYD J, AGIP AA, AL-ATRASH G, ASARA J, BARDENHAGEN J, CARRILLO CC, CARROLL C, CHANG E, CIUREA S, CROSS JB, CZAKO B, DEEM A, DAVER N, DE GROOT JF, DONG JW, FENG N, GAO G, GAY J, DO MG, GREER J, GIULIANI V, HAN J, HAN L, HENRY VK, HIRST J, HUANG S, JIANG Y, KANG Z, KHOR T, KONOPLEV S, LIN YH, LIU G, LODI A, LOFTON T, MA H, MAHENDRA M, MATRE P, MULLINAX R, PEOPLES M, PETROCCHI A, RODRIGUEZ-CANALE J, SERRELI R, SHI T, SMITH M, TABE Y, THEROFF J, TIZIANI S, XU Q, ZHANG Q, MULLER F, DEPINHO RA, TONIATTI C, DRAETTA GF, HEFFERNAN TP, KONOPLEVA M, JONES P, DI FRANCESCO ME & MARSZALEK JR 2018. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med, 24, 1036–1046. [DOI] [PubMed] [Google Scholar]
- MOMPARLER RL CS, MOMPARLER LF 2020. Epigenetic Modulation of Self-Renewal Capacity of Leukemic Stem Cells and Implications for Chemotherapy. Epigenomes, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURAR M & VAIDYA A 2015. Cancer stem cell markers: premises and prospects. Biomark Med, 9, 1331–42. [DOI] [PubMed] [Google Scholar]
- NAGARE RP, SNEHA S, PRIYA SK & GANESAN TS 2017. Cancer Stem Cells - Are Surface Markers Alone Sufficient? Curr Stem Cell Res Ther, 12, 37–44. [DOI] [PubMed] [Google Scholar]
- NAYAK AP, KAPUR A, BARROILHET L & PATANKAR MS 2018. Oxidative Phosphorylation: A Target for Novel Therapeutic Strategies Against Ovarian Cancer. Cancers, 10, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NECHIPORUK T, KURTZ SE, NIKOLOVA O, LIU T, JONES CL, D’ALESSANDRO A, CULP-HILL R, D’ALMEIDA A, JOSHI SK, ROSENBERG M, TOGNON CE, DANILOV AV, DRUKER BJ, CHANG BH, MCWEENEY SK & TYNER JW 2019. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov, 9, 910–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIKOLAOU M, PAVLOPOULOU A, GEORGAKILAS AG & KYRODIMOS E 2018. The challenge of drug resistance in cancer treatment: a current overview. Clin Exp Metastasis, 35, 309–318. [DOI] [PubMed] [Google Scholar]
- O’BRIEN CA, KRESO A & DICK JE 2009. Cancer stem cells in solid tumors: an overview. Semin Radiat Oncol, 19, 71–7. [DOI] [PubMed] [Google Scholar]
- O’BRIEN CA, POLLETT A, GALLINGER S & DICK JE 2007. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature, 445, 106–10. [DOI] [PubMed] [Google Scholar]
- OIZEL K, CHAUVIN C, OLIVER L, GRATAS C, GERALDO F, JARRY U, SCOTET E, RABE M, ALVES-GUERRA MC, TEUSAN R, GAUTIER F, LOUSSOUARN D, COMPAN V, MARTINOU JC, VALLETTE FM & PECQUEUR C 2017. Efficient Mitochondrial Glutamine Targeting Prevails Over Glioblastoma Metabolic Plasticity. Clin Cancer Res, 23, 6292–6304. [DOI] [PubMed] [Google Scholar]
- PASTO A, BELLIO C, PILOTTO G, CIMINALE V, SILIC-BENUSSI M, GUZZO G, RASOLA A, FRASSON C, NARDO G, ZULATO E, NICOLETTO MO, MANICONE M, INDRACCOLO S & AMADORI A 2014. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget, 5, 4305–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PEI S, MINHAJUDDIN M, ADANE B, KHAN N, STEVENS BM, MACK SC, LAI S, RICH JN, INGUVA A, SHANNON KM, KIM H, TAN A-C, MYERS JR, ASHTON JM, NEFF T, POLLYEA DA, SMITH CA & JORDAN CT 2018. AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells. Cell Stem Cell, 23, 86–100.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PEI S, MINHAJUDDIN M, CALLAHAN KP, BALYS M, ASHTON JM, NEERING SJ, LAGADINOU ED, CORBETT C, YE H, LIESVELD JL, O’DWYER KM, LI Z, SHI L, GRENINGER P, SETTLEMAN J, BENES C, HAGEN FK, MUNGER J, CROOKS PA, BECKER MW & JORDAN CT 2013. Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J Biol Chem, 288, 33542–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PEI S, POLLYEA DA, GUSTAFSON A, STEVENS BM, MINHAJUDDIN M, FU R, RIEMONDY KA, GILLEN AE, SHERIDAN RM, KIM J, COSTELLO JC, AMAYA ML, INGUVA A, WINTERS A, YE H, KRUG A, JONES CL, ADANE B, KHAN N, PONDER J, SCHOWINSKY J, ABBOTT D, HAMMES A, MYERS JR, ASHTON JM, NEMKOV T, D’ALESSANDRO A, GUTMAN JA, RAMSEY HE, SAVONA MR, SMITH CA & JORDAN CT 2020. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Acute Myeloid Leukemia Patients. Cancer Discovery, CD-19-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PERUSINA LANFRANCA M, THOMPSON JK, BEDNAR F, HALBROOK C, LYSSIOTIS C, LEVI B & FRANKEL TL 2019. Metabolism and epigenetics of pancreatic cancer stem cells. Semin Cancer Biol, 57, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PETRACHI T, ROMAGNANI A, ALBINI A, LONGO C, ARGENZIANO G, GRISENDI G, DOMINICI M, CIARROCCHI A & DALLAGLIO K 2017. Therapeutic potential of the metabolic modulator phenformin in targeting the stem cell compartment in melanoma. Oncotarget, 8, 6914–6928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POLLYEA DA & JORDAN CT 2017. Therapeutic targeting of acute myeloid leukemia stem cells. Blood, 129, 1627–1635. [DOI] [PubMed] [Google Scholar]
- POLLYEA DA, STEVENS BM, JONES CL, WINTERS A, PEI S, MINHAJUDDIN M, D’ALESSANDRO A, CULP-HILL R, RIEMONDY KA, GILLEN AE, HESSELBERTH JR, ABBOTT D, SCHATZ D, GUTMAN JA, PUREV E, SMITH C & JORDAN CT 2018. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med, 24, 1859–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POLLYEA DA, TALLMAN MS, DE BOTTON S, KANTARJIAN HM, COLLINS R, STEIN AS, FRATTINI MG, XU Q, TOSOLINI A, SEE WL, MACBETH KJ, AGRESTA SV, ATTAR EC, DINARDO CD & STEIN EM 2019. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia, 33, 2575–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRAGER BC, BHARGAVA S, MAHADEV V, HUBERT CG & RICH JN 2020. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer, 6, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUI CH & HOWARD SC 2008. Current management and challenges of malignant disease in the CNS in paediatric leukaemia. Lancet Oncol, 9, 257–68. [DOI] [PubMed] [Google Scholar]
- RAFFEL S, FALCONE M, KNEISEL N, HANSSON J, WANG W, LUTZ C, BULLINGER L, POSCHET G, NONNENMACHER Y, BARNERT A, BAHR C, ZEISBERGER P, PRZYBYLLA A, SOHN M, TONJES M, EREZ A, ADLER L, JENSEN P, SCHOLL C, FROHLING S, COCCIARDI S, WUCHTER P, THIEDE C, FLORCKEN A, WESTERMANN J, EHNINGER G, LICHTER P, HILLER K, HELL R, HERRMANN C, HO AD, KRIJGSVELD J, RADLWIMMER B & TRUMPP A 2017. BCAT1 restricts alphaKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature, 551, 384–388. [DOI] [PubMed] [Google Scholar]
- REED GA, SCHILLER GJ, KAMBHAMPATI S, TALLMAN MS, DOUER D, MINDEN MD, YEE KW, GUPTA V, BRANDWEIN J, JITKOVA Y, GRONDA M, HURREN R, SHAMAS-DIN A, SCHUH AC & SCHIMMER AD 2016. A Phase 1 study of intravenous infusions of tigecycline in patients with acute myeloid leukemia. Cancer Med, 5, 3031–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RHA SY, BEOM S-H, JUNG M, SHIN YG, CHEONG J-H, KIM HS, KIM GM, CHANG JH, LEE YW, CHONG Y, YOO S, O’NEIL V & JANKU F 2018. Phase 1 study of an oxidative phosphorylation inhibitor IM156 in patients with advanced solid tumors. Journal of Clinical Oncology, 36, TPS2620–TPS2620. [Google Scholar]
- RICCI-VITIANI L, LOMBARDI DG, PILOZZI E, BIFFONI M, TODARO M, PESCHLE C & DE MARIA R 2007. Identification and expansion of human colon-cancer-initiating cells. Nature, 445, 111–5. [DOI] [PubMed] [Google Scholar]
- ROESCH A, VULTUR A, BOGESKI I, WANG H, ZIMMERMANN KM, SPEICHER D, KORBEL C, LASCHKE MW, GIMOTTY PA, PHILIPP SE, KRAUSE E, PATZOLD S, VILLANUEVA J, KREPLER C, FUKUNAGA-KALABIS M, HOTH M, BASTIAN BC, VOGT T & HERLYN M 2013. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell, 23, 811–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAGA I, SHIBAO S, OKUBO J, OSUKA S, KOBAYASHI Y, YAMADA S, FUJITA S, URAKAMI K, KUSUHARA M, YOSHIDA K, SAYA H & SAMPETREAN O 2014. Integrated analysis identifies different metabolic signatures for tumor-initiating cells in a murine glioblastoma model. Neuro Oncol, 16, 1048–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAITO Y, CHAPPLE RH, LIN A, KITANO A & NAKADA D 2015. AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell Stem Cell, 17, 585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANCHO P, BURGOS-RAMOS E, TAVERA A, BOU KHEIR T, JAGUST P, SCHOENHALS M, BARNEDA D, SELLERS K, CAMPOS-OLIVAS R, GRANA O, VIERA CR, YUNEVA M, SAINZ B JR. & HEESCHEN C 2015. MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab, 22, 590–605. [DOI] [PubMed] [Google Scholar]
- SATO M, KAWANA K, ADACHI K, FUJIMOTO A, YOSHIDA M, NAKAMURA H, NISHIDA H, INOUE T, TAGUCHI A, TAKAHASHI J, EGUCHI S, YAMASHITA A, TOMIO K, WADA-HIRAIKE O, ODA K, NAGAMATSU T, OSUGA Y & FUJII T 2016. Spheroid cancer stem cells display reprogrammed metabolism and obtain energy by actively running the tricarboxylic acid (TCA) cycle. Oncotarget, 7, 33297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAVINO AM, FERNANDES SI, OLIVARES O, ZEMLYANSKY A, COUSINS A, MARKERT EK, BAREL S, GERON I, FRISHMAN L, BIRGER Y, ECKERT C, TUMANOV S, MACKAY G, KAMPHORST JJ, HERZYK P, FERNÁNDEZ-GARCÍA J, ABRAMOVICH I, MOR I, BARDINI M, BARIN E, JANAKI-RAMAN S, CROSS JR, KHARAS MG, GOTTLIEB E, IZRAELI S & HALSEY C 2020. Metabolic adaptation of acute lymphoblastic leukemia to the central nervous system microenvironment depends on stearoyl-CoA desaturase. Nature Cancer, 1, 998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAYGIN C, MATEI D, MAJETI R, REIZES O & LATHIA JD 2019. Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem Cell, 24, 25–40. [DOI] [PubMed] [Google Scholar]
- SHARON D, CATHELIN S, MIRALI S, DI TRANI JM, YANOFSKY DJ, KEON KA, RUBINSTEIN JL, SCHIMMER AD, KETELA T & CHAN SM 2019. Inhibition of mitochondrial translation overcomes venetoclax resistance in AML through activation of the integrated stress response. Science Translational Medicine, 11, eaax2863. [DOI] [PubMed] [Google Scholar]
- SHI X, ZHANG Y, ZHENG J & PAN J 2012. Reactive oxygen species in cancer stem cells. Antioxidants & redox signaling, 16, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIBAO S, MINAMI N, KOIKE N, FUKUI N, YOSHIDA K, SAYA H & SAMPETREAN O 2018. Metabolic heterogeneity and plasticity of glioma stem cells in a mouse glioblastoma model. Neuro Oncol, 20, 343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHLUSH LI, MITCHELL A, HEISLER L, ABELSON S, NG SWK, TROTMAN-GRANT A, MEDEIROS JJF, RAO-BHATIA A, JACIW-ZURAKOWSKY I, MARKE R, MCLEOD JL, DOEDENS M, BADER G, VOISIN V, XU C, MCPHERSON JD, HUDSON TJ, WANG JCY, MINDEN MD & DICK JE 2017. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature, 547, 104–108. [DOI] [PubMed] [Google Scholar]
- SIMSEK T, KOCABAS F, ZHENG J, DEBERARDINIS RJ, MAHMOUD AI, OLSON EN, SCHNEIDER JW, ZHANG CC & SADEK HA 2010. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell, 7, 380–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SINGH SK, HAWKINS C, CLARKE ID, SQUIRE JA, BAYANI J, HIDE T, HENKELMAN RM, CUSIMANO MD & DIRKS PB 2004. Identification of human brain tumour initiating cells. Nature, 432, 396–401. [DOI] [PubMed] [Google Scholar]
- SIVANAND S & VANDER HEIDEN MG 2020. Emerging Roles for Branched-Chain Amino Acid Metabolism in Cancer. Cancer Cell, 37, 147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SKRTIC M, SRISKANTHADEVAN S, JHAS B, GEBBIA M, WANG X, WANG Z, HURREN R, JITKOVA Y, GRONDA M, MACLEAN N, LAI CK, EBERHARD Y, BARTOSZKO J, SPAGNUOLO P, RUTLEDGE AC, DATTI A, KETELA T, MOFFAT J, ROBINSON BH, CAMERON JH, WRANA J, EAVES CJ, MINDEN MD, WANG JC, DICK JE, HUMPHRIES K, NISLOW C, GIAEVER G & SCHIMMER AD 2011. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell, 20, 674–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH J, LADI E, MAYER-PROSCHEL M & NOBLE M 2000. Redox state is a central modulator of the balance between self-renewal and differentiation in a dividing glial precursor cell. Proc Natl Acad Sci U S A, 97, 10032–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SNYDER V, REED-NEWMAN TC, ARNOLD L, THOMAS SM & ANANT S 2018. Cancer Stem Cell Metabolism and Potential Therapeutic Targets. Front Oncol, 8, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SRISKANTHADEVAN S, JEYARAJU DV, CHUNG TE, PRABHA S, XU W, SKRTIC M, JHAS B, HURREN R, GRONDA M, WANG X, JITKOVA Y, SUKHAI MA, LIN FH, MACLEAN N, LAISTER R, GOARD CA, MULLEN PJ, XIE S, PENN LZ, ROGERS IM, DICK JE, MINDEN MD & SCHIMMER AD 2015. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood, 125, 2120–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEIN EM, DINARDO CD, POLLYEA DA, FATHI AT, ROBOZ GJ, ALTMAN JK, STONE RM, DEANGELO DJ, LEVINE RL, FLINN IW, KANTARJIAN HM, COLLINS R, PATEL MR, FRANKEL AE, STEIN A, SEKERES MA, SWORDS RT, MEDEIROS BC, WILLEKENS C, VYAS P, TOSOLINI A, XU Q, KNIGHT RD, YEN KE, AGRESTA S, DE BOTTON S & TALLMAN MS 2017. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood, 130, 722–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEVENS BM, JONES CL, POLLYEA DA, CULP-HILL R, D’ALESSANDRO A, WINTERS A, KRUG A, ABBOTT D, GOOSMAN M, PEI S, YE H, GILLEN AE, BECKER MW, SAVONA MR, SMITH C & JORDAN CT 2020. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nature Cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEVENS BM, KHAN N, D’ALESSANDRO A, NEMKOV T, WINTERS A, JONES CL, ZHANG W, POLLYEA DA & JORDAN CT 2018. Characterization and targeting of malignant stem cells in patients with advanced myelodysplastic syndromes. Nat Commun, 9, 3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUDA T, TAKUBO K & SEMENZA GL 2011. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell, 9, 298–310. [DOI] [PubMed] [Google Scholar]
- SUN J-H, LUO Q, LIU L-L & SONG G-B 2016. Liver cancer stem cell markers: Progression and therapeutic implications. World journal of gastroenterology, 22, 3547–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKUBO K, NAGAMATSU G, KOBAYASHI CHIHARUI, NAKAMURA-ISHIZU A, KOBAYASHI H, IKEDA E, GODA N, RAHIMI Y, JOHNSON RANDALLS, SOGA T, HIRAO A, SUEMATSU M & SUDA T 2013. Regulation of Glycolysis by Pdk Functions as a Metabolic Checkpoint for Cell Cycle Quiescence in Hematopoietic Stem Cells. Cell Stem Cell, 12, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAN J, GROULEFF JJ, JITKOVA Y, DIAZ DB, GRIFFITH EC, SHAO W, BOGDANCHIKOVA AF, PODA G, SCHIMMER AD, LEE RE & YUDIN AK 2019. De Novo Design of Boron-Based Peptidomimetics as Potent Inhibitors of Human ClpP in the Presence of Human ClpX. J Med Chem. [DOI] [PubMed] [Google Scholar]
- TOTHOVA Z, KOLLIPARA R, HUNTLY BJ, LEE BH, CASTRILLON DH, CULLEN DE, MCDOWELL EP, LAZO-KALLANIAN S, WILLIAMS IR, SEARS C, ARMSTRONG SA, PASSEGUE E, DEPINHO RA & GILLILAND DG 2007. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell, 128, 325–39. [DOI] [PubMed] [Google Scholar]
- UCKUN F, SATHER H, REAMAN G, SHUSTER J, LAND V, TRIGG M, GUNTHER R, CHELSTROM L, BLEYER A & GAYNON P 1995. Leukemic cell growth in SCID mice as a predictor of relapse in high- risk B-lineage acute lymphoblastic leukemia. 85, 873–878. [PubMed] [Google Scholar]
- VAN GASTEL N, SPINELLI JB, SHARDA A, SCHAJNOVITZ A, BARYAWNO N, RHEE C, OKI T, GRACE E, SOLED HJ, MILOSEVIC J, SYKES DB, HSU PP, VANDER HEIDEN MG, VIDOUDEZ C, TRAUGER SA, HAIGIS MC & SCADDEN DT 2020. Induction of a Timed Metabolic Collapse to Overcome Cancer Chemoresistance. Cell Metabolism, 32, 391–403.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN RHENEN A, VAN DONGEN GA, KELDER A, ROMBOUTS EJ, FELLER N, MOSHAVER B, STIGTER-VAN WALSUM M, ZWEEGMAN S, OSSENKOPPELE GJ & JAN SCHUURHUIS G 2007. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood, 110, 2659–66. [DOI] [PubMed] [Google Scholar]
- VANDER HEIDEN MG & DEBERARDINIS RJ 2017. Understanding the Intersections between Metabolism and Cancer Biology. Cell, 168, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VELLINGA TT, BOROVSKI T, DE BOER VC, FATRAI S, VAN SCHELVEN S, TRUMPI K, VERHEEM A, SNOEREN N, EMMINK BL, KOSTER J, RINKES IH & KRANENBURG O 2015. SIRT1/PGC1alpha-Dependent Increase in Oxidative Phosphorylation Supports Chemotherapy Resistance of Colon Cancer. Clin Cancer Res, 21 , 2870–9. [DOI] [PubMed] [Google Scholar]
- VIALE A, PETTAZZONI P, LYSSIOTIS CA, YING H, SANCHEZ N, MARCHESINI M, CARUGO A, GREEN T, SETH S, GIULIANI V, KOST-ALIMOVA M, MULLER F, COLLA S, NEZI L, GENOVESE G, DEEM AK, KAPOOR A, YAO W, BRUNETTO E, KANG Y, YUAN M, ASARA JM, WANG YA, HEFFERNAN TP, KIMMELMAN AC, WANG H, FLEMING JB, CANTLEY LC, DEPINHO RA & DRAETTA GF 2014. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature, 514, 628–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VLASHI E, LAGADEC C, VERGNES L, MATSUTANI T, MASUI K, POULOU M, POPESCU R, DELLA DONNA L, EVERS P, DEKMEZIAN C, REUE K, CHRISTOFK H, MISCHEL PS & PAJONK F 2011. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci U S A, 108, 16062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VLASHI E, LAGADEC C, VERGNES L, REUE K, FROHNEN P, CHAN M, ALHIYARI Y, DRATVER MB & PAJONK F 2014. Metabolic differences in breast cancer stem cells and differentiated progeny. Breast Cancer Res Treat, 146, 525–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALLACE DC 2012. Mitochondria and cancer. Nature reviews. Cancer, 12, 685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG T, FAHRMANN JF, LEE H, LI YJ, TRIPATHI SC, YUE C, ZHANG C, LIFSHITZ V, SONG J, YUAN Y, SOMLO G, JANDIAL R, ANN D, HANASH S, JOVE R & YU H 2018. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab, 27, 1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WARBURG O 1956. On the origin of cancer cells. Science, 123, 309–14. [DOI] [PubMed] [Google Scholar]
- WU Z, WEI D, GAO W, XU Y, HU Z, MA Z, GAO C, ZHU X & LI Q 2015. TPO-Induced Metabolic Reprogramming Drives Liver Metastasis of Colorectal Cancer CD110+ Tumor-Initiating Cells. Cell Stem Cell, 17, 47–59. [DOI] [PubMed] [Google Scholar]
- YANG L, SHI P, ZHAO G, XU J, PENG W, ZHANG J, ZHANG G, WANG X, DONG Z, CHEN F & CUI H 2020. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther, 5, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YE H, ADANE B, KHAN N, ALEXEEV E, NUSBACHER N, MINHAJUDDIN M, STEVENS BM, WINTERS AC, LIN X, ASHTON JM, PUREV E, XING L, POLLYEA DA, LOZUPONE CA, SERKOVA NJ, COLGAN SP & JORDAN CT 2018. Subversion of Systemic Glucose Metabolism as a Mechanism to Support the Growth of Leukemia Cells. Cancer Cell, 34, 659–673.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YE H, ADANE B, KHAN N, SULLIVAN T, MINHAJUDDIN M, GASPARETTO M, STEVENS B, PEI S, BALYS M, ASHTON JM, KLEMM DJ, WOOLTHUIS CM, STRANAHAN AW, PARK CY & JORDAN CT 2016. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell, 19, 23–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YE H, MINHAJUDDIN M, KRUG A, PEI S, CHOU CH, CULP-HILL R, PONDER J, DE BLOOIS E, SCHNIEDEWIND B, AMAYA ML, INGUVA A, STEVENS BM, POLLYEA DA, CHRISTIANS U, GRIMES HL, D’ALESSANDRO A & JORDAN CT 2020. The hepatic microenvironment uniquely protects leukemia cells through induction of growth and survival pathways mediated by LIPG. Cancer Discov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YE XQ, LI Q, WANG GH, SUN FF, HUANG GJ, BIAN XW, YU SC & QIAN GS 2011. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int J Cancer, 129, 820–31. [DOI] [PubMed] [Google Scholar]
- YEN K, TRAVINS J, WANG F, DAVID MD, ARTIN E, STRALEY K, PADYANA A, GROSS S, DELABARRE B, TOBIN E, CHEN Y, NAGARAJA R, CHOE S, JIN L, KONTEATIS Z, CIANCHETTA G, SAUNDERS JO, SALITURO FG, QUIVORON C, OPOLON P, BAWA O, SAADA V, PACI A, BROUTIN S, BERNARD OA, DE BOTTON S, MARTEYN BS, PILICHOWSKA M, XU Y, FANG C, JIANG F, WEI W, JIN S, SILVERMAN L, LIU W, YANG H, DANG L, DORSCH M, PENARD-LACRONIQUE V, BILLER SA & SU SM 2017. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov, 7, 478–493. [DOI] [PubMed] [Google Scholar]