

SUMMARY
The myeloid tumor suppressor KMT2C is recurrently deleted in myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML), particularly therapy-related MDS/AML (t-MDS/t-AML), as part of larger chromosome 7 deletions. Here, we show that KMT2C deletions convey a selective advantage to hematopoietic stem cells (HSCs) after chemotherapy treatment that may precipitate t-MDS/t-AML. Kmt2c deletions markedly enhance murine HSC self-renewal capacity without altering proliferation rates. Haploid Kmt2c deletions convey a selective advantage only when HSCs are driven into cycle by a strong proliferative stimulus, such as chemotherapy. Cycling Kmt2c-deficient HSCs fail to differentiate appropriately, particularly in response to interleukin-1. Kmt2c deletions mitigate histone methylation/acetylation changes that accrue as HSCs cycle after chemotherapy, and they impair enhancer recruitment during HSC differentiation. These findings help explain why Kmt2c deletions are more common in t-MDS/t-AML than in de novo AML or clonal hematopoiesis: they selectively protect cycling HSCs from differentiation without inducing HSC proliferation themselves.
Graphical Abstract
In Brief
Chen et al. use loss-of-function mice to study the role Kmt2c/MLL3 in hematopoietic stem cell (HSC) self-renewal. They show that haploid Kmt2c deletions impede differentiation when HSCs are driven into cycle by chemotherapy, especially in conjunction with interleukin-1 exposure. This conveys a selective advantage that may promote therapy-related leukemias.
INTRODUCTION
Therapy-related myelodysplastic syndrome (t-MDS) and acute myeloid leukemia (t-AML) can arise after patients receive high cumulative doses of chemotherapy for prior, unrelated malignancies. Prognosis for t-MDS/t-AML remains poor even with aggressive modern therapies (Oliai and Schiller, 2020). Thus, it poses one of the most severe long-term complications of chemotherapy. The mutation profiles of t-MDS/t-AML distinguish them from de novo AML. Approximately half of all t-MDSs/t-AMLs carry deletions in all or part of chromosome 7, but only 4%–14% of de novo AMLs carry chromosome 7 deletions (McNerney et al., 2017; Smith et al., 2003). Chromosome 5 deletions are similarly enriched in t-MDS/t-AML (42% t-MDS/t-AML versus 5%–16% de novo AML), as are TP53 mutations (23%–37% t-MDS/t-AML versus 2%–12% de novo AML) (McNerney et al., 2017; Wong et al., 2015). The distinct mutation profiles, coupled with poor survival rates, suggest that t-MDS/t-AML and de novo AML arise via distinct mechanisms.
To initiate t-MDS/t-AML, pre-leukemic hematopoietic stem cells (HSCs) and other myeloid progenitors must acquire driver mutations, either prior to chemotherapy or as a consequence of chemotherapy, that convey a selective advantage through the course of additional treatment cycles. A good example of this phenomenon involves TP53 mutations, which can arise in HSCs prior to therapy and then drive t-MDS/t-AML during and after chemotherapy (Wong et al., 2015). Monosomy 7 (−7) and 7q deletions (del7q) also arise within HSCs and immature myeloid progenitor populations (Will et al., 2012), but less is known about how −7/del7q potentiates t-MDS/t-AML. One possibility is that −7/del7q conveys resistance to DNA damage responses that otherwise limit HSC self-renewal after chemotherapy. A second possibility is that mutations convey resistance to HSC exhaustion, a phenomenon in which self-renewal capacity declines as HSCs undergo multiple self-renewing divisions (Bernitz et al., 2016; Foudi et al., 2009; Qiu et al., 2014; Wilson et al., 2008). DNA damage can contribute to HSC exhaustion (Walter et al., 2015), but other physiological changes, such as mitochondrial dysfunction and epigenetic reprogramming, also restrict self-renewal capacity in multiply-divided HSCs (Beerman et al., 2013; Hinge et al., 2020; Liang et al., 2020). Cycling HSCs must bypass these restrictions to give rise to t-MDS/t-AML.
KMT2C is one of several putative tumor suppressor genes that are recurrently deleted in −7/del7q leukemias, including t-MDS/t-AML (Chen et al., 2014). Mouse models have confirmed that Kmt2c is a bona fide myeloid tumor suppressor, because haploid Kmt2c deletions can greatly accelerate AML pathogenesis (Chen et al., 2014). Despite this function, KMT2C is not recurrently mutated in age-related clonal hematopoiesis, and aged Kmt2c knockout mice do not develop MDS or AML (Arcipowski et al., 2016). These observations suggest that haploid KMT2C deletions do not, by themselves, convey a strong advantage to affected HSCs. This raises the question of whether KMT2C deletions convey a selective advantage only under specific sets of circumstances, such as cumulative chemotherapy cycles that precipitate t-MDS/t-AML.
KMT2C encodes MLL3, a histone methyltransferase that binds enhancer elements and promotes transcription (Hu et al., 2013). MLL3 nucleates COMPASS (complex of proteins associated with Set1) complexes that bind enhancers via interactions with DNA-bound transcription factors and co-factors (Jozwik et al., 2016; Sun et al., 2018; Wang et al., 2018). Once bound, MLL3 monomethylates histone H3, lysine 4 (H3K4me1) to prime enhancers for activation (Herz et al., 2012; Hu et al., 2013; Wang et al., 2016), recruits KDM6A to remove repressive histone H3, lysine 27 tri-methyl marks (H3K27me3) (Wang et al., 2018), recruits CBP/p300 to place activating histone H3, lysine 27 acetyl marks (H3K27ac) (Lai et al., 2017; Wang et al., 2017, 2018), and facilitates long-range interactions between enhancers and their target promoters (Dorighi et al., 2017; Lai et al., 2017; Shilatifard, 2012; Wang et al., 2017; Yan et al., 2018). These observations suggest that MLL3 may help activate differentiation programs in HSCs, similar to what has been described for adipose differentiation (Lee et al., 2013). Alternatively, MLL3 may sensitize HSCs to chemotherapy-induced DNA damage, similar to what has been described in urothelial cells (Lee et al., 2009). In either case, KMT2C deletions could convey a selective advantage to HSCs after chemotherapy but before leukemic transformation.
To understand how Kmt2c/MLL3 regulates HSC self-renewal, we have generated germline and conditional loss-of-function mice. Kmt2c deletions impaired HSC differentiation and conveyed a selective advantage after chemotherapy. This contrasts with a previously described function of Kmt2d/MLL4, which is necessary to preserve HSC self-renewal (Santos et al., 2014), and it highlights how highly homologous COMPASS family proteins can have divergent functions in somatic stem cells. At a mechanistic level, we found that Kmt2c-deficient HSCs resisted differentiation in response to inflammatory cytokines, such as interleukin-1 (IL-1), but only when they were driven into cycle by other stimuli. Furthermore, Kmt2c-deficient HSCs failed to prime and activate enhancers appropriately during HSC differentiation, and they failed to remodel their epigenomes during cumulative division cycles. Thus, the consequences of Kmt2c deletion, in HSCs, are largely restricted to contexts in which strong mitogenic stimuli compel HSCs to divide. This context specificity may explain why KMT2C deletions arise more frequently in t-MDS/t-AML than in de novo AML or age-related clonal hematopoiesis.
RESULTS
Haploid Kmt2c deletion enhances HSC self-renewal capacity
To understand how Kmt2c regulates hematopoiesis, we used CRISPR/Cas9 to create a germline premature stop codon in mouse Kmt2c exon 14 (Figure 1A). This mutation caused complete loss of protein expression (Figure 1B). Kmt2c−/− mice died at birth yet appeared morphologically normal, consistent with prior studies in which an exon 49 deletion or an intron 33 gene trap insertion led to pulmonary failure and perinatal lethality (Ashokkumar et al., 2020; Lee et al., 2013). We measured HSC numbers (CD150+CD48−Lineage−c-kit+Sca1+; Figure S1A) in wild-type, Kmt2c+/−, and Kmt2c−/−embryonic day (E) 18.5 fetal mice and in wild-type and Kmt2c+/− 8-week-old adult mice. HSC numbers were similar in wild-type, Kmt2c+/−, and Kmt2c−/− fetal mice (Figure 1C). In contrast, HSC numbers doubled in Kmt2c+/− adult mice relative to controls (Figure 1D). Cell-cycle distributions and bromodeoxyuridine (BrdU) incorporation were similar in wild-type and Kmt2c+/− adult HSCs (Figures 1E and 1F). Thus, haploid Kmt2c deletion expanded the adult HSC pool without increasing HSC proliferation rates.
Figure 1. Haploid Kmt2c deletion increases HSC numbers and enhances HSC self-renewal capacity.
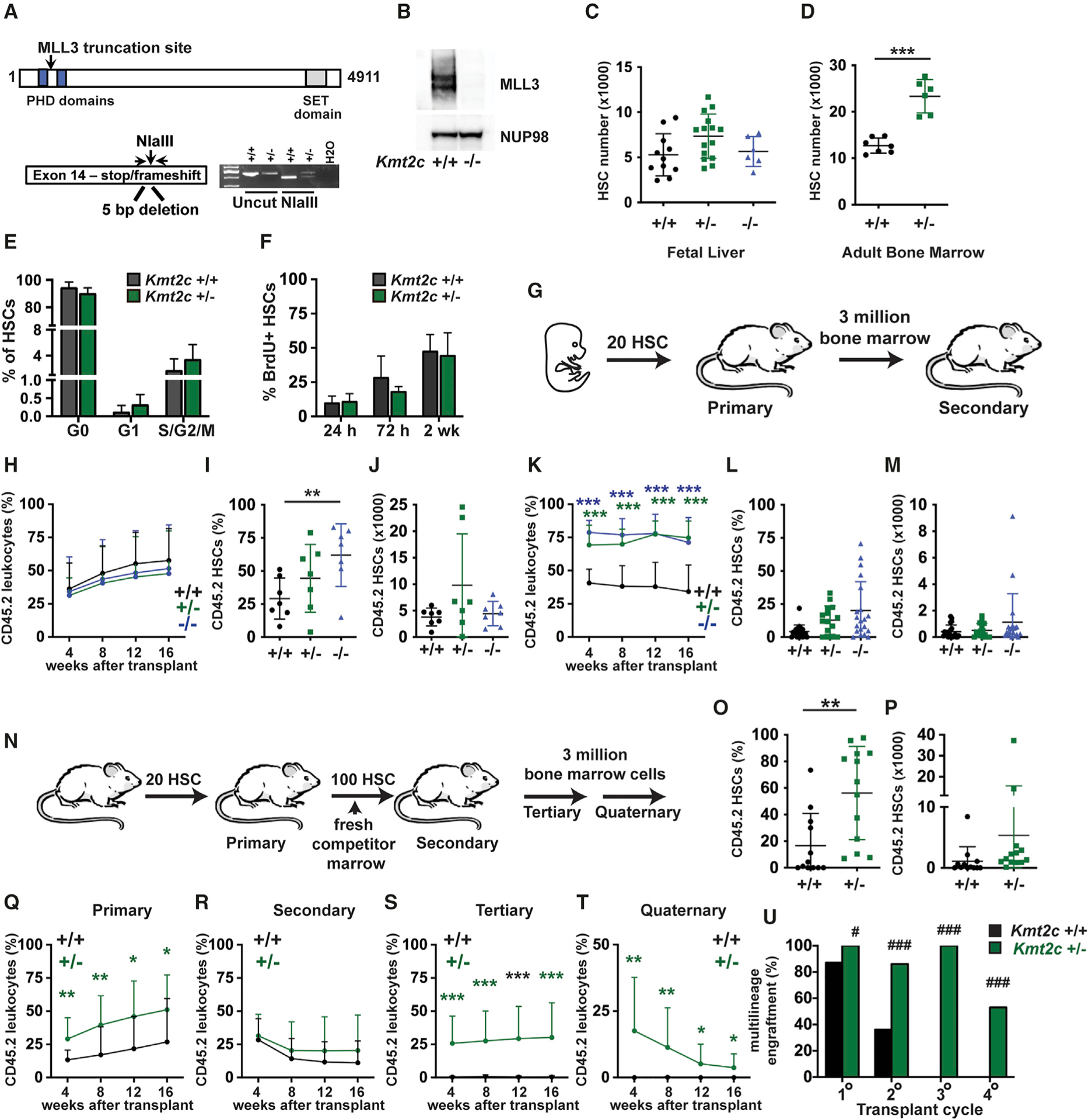
(A) Overview of the germline Kmt2c null allele.
(B) MLL3 expression in lysates from wild-type and Kmt2c−/− MEFs, as assessed by western blot.
(C and D) HSC numbers in E18.5 fetal livers or 8-week-old adult bone marrow (two hindlimbs) from mice of the indicated Kmt2c genotypes. n = 6–15 (fetal) or 6–7 (adult).
(E) Cell-cycle phase distributions of adult Kmt2c+/+ and Kmt2c+/− HSCs as determined by Ki67/DAPI staining. n = 5–6.
(F) HSC BrdU incorporation after 24 h, 72 h, or 2 weeks of BrdU exposure. n = 3–7 per time point and genotype.
(G) Competitive transplantation of fetal HSCs overview.
(H) CD45.2+ donor leukocyte chimerism in peripheral blood from primary recipients at the indicated weeks after transplant. n = 14–15 recipients per genotype from at least three independent donors.
(I and J) Donor HSC chimerism and numbers in primary recipient bone marrow 16 weeks after transplantation. n = 7.
(K) Donor leukocyte chimerism in secondary recipients after transplantation of 3 million primary recipient bone marrow cells. n = 15–20 recipients per genotype from at least 5 independent donors.
(L and M) Donor HSC chimerism and numbers in secondary recipient bone marrow 16 weeks after transplantation. n = 15–20.
(N) Serial transplantation of adult HSCs overview.
(O and P) Donor HSC chimerism and numbers in primary recipient bone marrow. n = 12–13.
(Q–T) CD45.2+ donor leukocyte chimerism in peripheral blood from primary, secondary, tertiary, and quaternary recipient mice at the indicated weeks after transplant. n = 14–15 recipients per genotype and transplant cycle from at least three independent donors.
(U) Percentage of mice with multi-lineage CD45.2 chimerism (myeloid, B, and T cell) at 16 weeks after each of the indicated transplant cycles.
For all panels, error bars reflect standard deviation. *p < 0.05, **p < 0.01, ***p < 0.001; comparisons were made by two-tailed Student’s t test or one-way ANOVA with Holm-Sidak post hoc test (multiple comparisons). (U) #p < 0.05, ###p < 0.001, by Fisher’s exact test. See also Figures S1 and S2.
To test whether Kmt2c is required for long-term HSC self-renewal, we performed competitive transplantation assays with purified HSCs. Because Kmt2c−/− mice died at birth, we isolated 20 HSCs from wild-type, Kmt2c+/−, and Kmt2c−/− E18.5 fetal mice, all on a CD45.2 background, and transplanted them into wild-type CD45.1 recipient mice along with 300,000 wild-type CD45.1 competitor bone marrow cells (Figure 1G). In primary transplants, Kmt2c deletion had no effect on peripheral blood reconstitution (Figure 1H; Figure S2A). Donor HSC chimerism was marginally higher in recipients of Kmt2c−/− HSCs than in controls (Figure 1I), but the Kmt2c−/− HSC population did not expand appreciably in numbers (Figure 1J). We next performed secondary transplants with 3 million primary recipient bone marrow cells (Figure 1G). Deletion of one or both Kmt2c alleles enhanced reconstitution of all lineages in secondary recipients (Figure 1K; Figure S2B), although Kmt2c−/− HSCs did not expand in numbers relative to controls (Figures 1L and 1M). A similar result was observed when we sorted and transplanted equal numbers of wild-type, Kmt2c+/−, and Kmt2c−/− HSCs from the primary recipients (Figures S2C and S2D). The data show the Kmt2c deletions enhance HSC repopulating activity without markedly increasing HSC numbers.
We next tested whether haploid Kmt2c deletions enhance serial repopulating activity in adult HSCs. We isolated and transplanted 20 adult HSCs from 8-week-old wild-type and Kmt2c+/− mice, along with 300,000 CD45.1 competitor bone marrow cells (Figure 1N). Kmt2c+/− HSCs repopulated primary transplant recipients more effectively than wild-type HSCs, but again with only modest increases in HSC chimerism (Figures 1O–1Q). For secondary transplants, we isolated 100 HSCs from primary recipients and transplanted them with fresh competitor marrow so that secondary recipients received equal numbers of HSCs (Figure 1N). This approach put the donor HSCs at a severe competitive disadvantage relative to the competitor marrow as they had already undergone one round of transplantation, and reconstitution levels were similar for both Kmt2c genotypes (Figure 1R). However, Kmt2c+/− donor HSCs retained multilineage repopulating activity in tertiary and quaternary transplants, whereas wild-type donor HSCs did not (Figures 1S–1U). Donor Kmt2c+/− HSCs were too infrequent to measure after serial rounds of transplantation (data not shown). Collectively, these data show that haploid Kmt2c deletions allow HSCs to retain self-renewal capacity through several cycles of transplantation, without triggering metabolic or differentiation programs that lead to HSC exhaustion.
Homozygous Kmt2c deletion modestly expands the adult HSC pool at the expense of more committed progenitor populations
To better study how MLL3 regulates adult hematopoiesis, we generated a conditional Kmt2c loss-of-function allele. LoxP sites were placed around exon 3 so that Cre-mediated excision could create a frameshift, premature truncation and complete loss of MLL3 protein expression (Figures 2A and 2B). The conditional allele targeted exon 3, whereas the germline allele targeted exon 14. To confirm that the alleles are functionally equivalent, we deleted a floxed exon 3 allele in the germline with CMV-Cre. We then tested whether compound exon 3 and 14 mutations could complement one another and rescue homozygous lethality in late gestation. Crosses of Kmt2c+/− (exon 3) and Kmt2c+/− (exon 14) parents failed to yield any viable compound heterozygotes at post-natal day (P) 0 (Figure 2C). Thus, the exon 3 and 14 mutations are functionally equivalent, at least with regard to perinatal lethality.
Figure 2. Conditional Kmt2c deletion expands the HSC pool at the expense of HPCs.
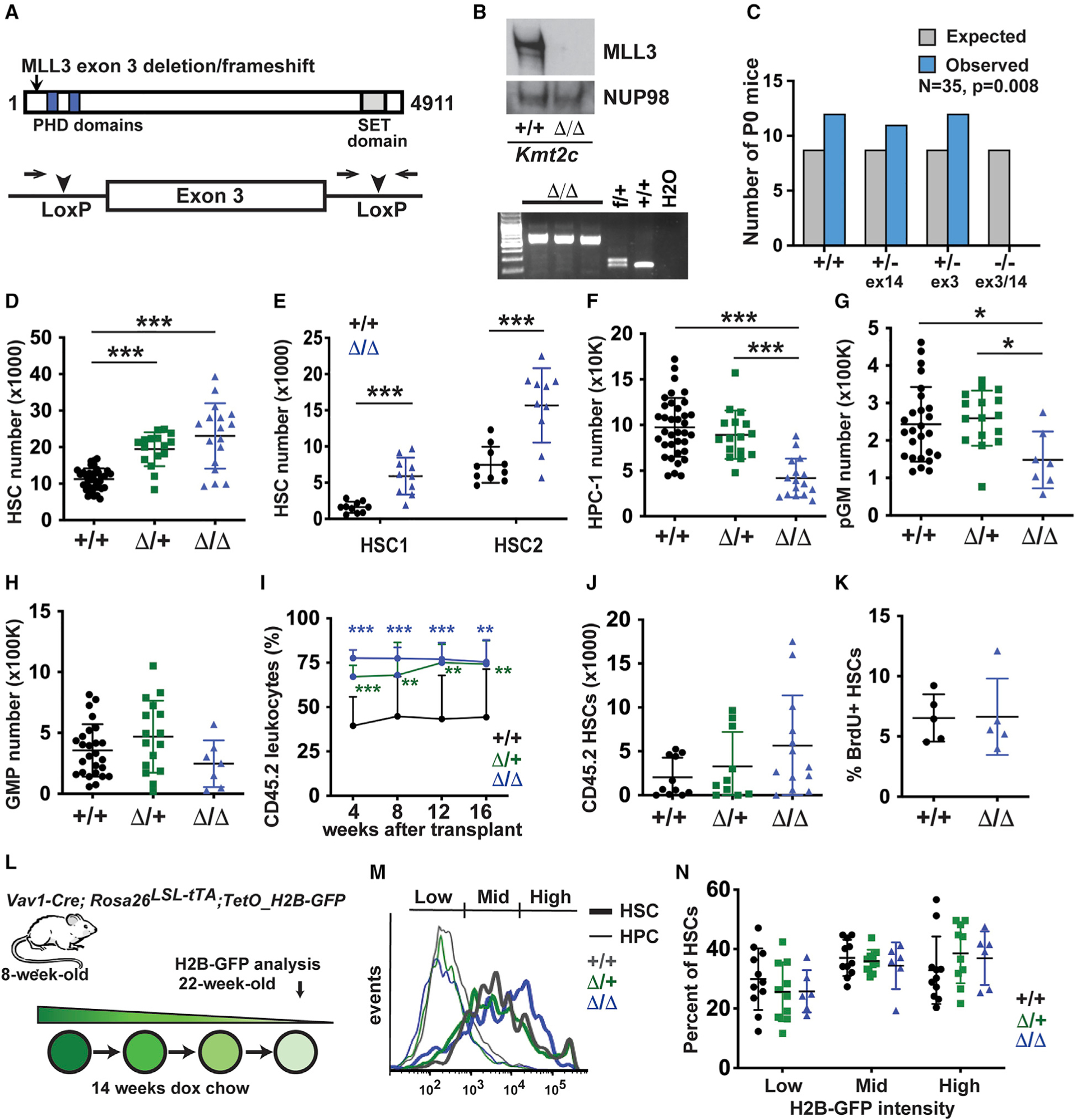
(A) Overview of the conditional Kmt2c-flox allele.
(B) Western blot and genotyping PCR showing loss of MLL3 protein expression in Kmt2cΔ/Δsplenocytes.
(C) Complementation testing of the Kmt2c null (exon 14) and Kmt2cflox (exon 3) alleles, as measured by viable offspring after germline Kmt2cflox deletion. Observed and expected allele frequencies were compared by the chi-square test. n = 35.
(D–H) HSC, HPC-1, pGM, and GMP numbers in 8-week-old mice of the indicated Kmt2c genotypes (two hindlimbs). n = 7–20.
(I) Peripheral blood CD45.2+ leukocyte chimerism after competitive whole bone marrow transplants. n = 10–13 per genotype from three independent donors.
(J) CD45.2+ HSC numbers in primary recipient bone marrow 16 weeks after transplantation.
(K) Twenty-four-hour BrdU incorporation in wild-type and Kmt2cΔ/Δ HSCs. n = 5.
(L) Overview of H2B-GFP pulse-chase experiment.
(M) Representative histograms showing H2B-GFP signal in HSCs and HPCs after 14 weeks of doxycycline exposure.
(N) Percentages of H2B-GFPHigh, H2B-GFPMid, and H2B-GFPLow HSCs after a 14-week doxycycline chase. n = 6–11.
In all panels, error bars reflect standard deviation. *p < 0.05, **p < 0.01, ***p < 0.001; comparisons were made by two-tailed Student’s t test or one-way ANOVA with Holm-Sidak post hoc test (multiple comparisons). See also Figures S1 and S3.
We generated Kmt2cf/f; Vav1-Cre mice (Kmt2cΔ/Δ) to inactivate MLL3 in hematopoietic cells. Spleen weights were marginally enlarged in Kmt2cΔ/+ and Kmt2cΔ/Δ mice (Figure S3A), consistent with a prior observation with MLL3 SET (Su(var)3–9, Enhancer-of-zeste, Trithorax) domain-deficient mice (Arcipowski et al., 2016). Peripheral blood counts were normal in Kmt2cΔ/+ and Kmt2cΔ/Δ mice (Figures S3B–S3D). HSCs expanded to a modest extent in Kmt2cΔ/+ and Kmt2cΔ/Δ mice, consistent with our findings from germline-deleted mice (Figure 2D). Oguro et al. (2013) previously identified two HSC populations based on expression of SLAM surface markers, as well as several distinct populations of multipotent progenitors (MPPs) and committed hematopoietic progenitors (HPCs), and we evaluated these populations after Kmt2c deletion (Figures S1A and S3E). Both the deeply quiescent HSC-1 population and a more frequently cycling HSC-2 population expanded in Kmt2cΔ/Δ mice, as did MPPs (Figure 2E; Figure S3G–S3I). HPC-1 s were depleted in Kmt2cΔ/Δ mice, but HPC-2 s were not (Figure 2F; Figures S3J and S3K). Pre-granulocyte-monocyte progenitors (pGMs), as defined by Pronk et al. (2007) (Figures S1B and S3F), were depleted in Kmt2cΔ/Δ mice, but granulocyte-monocyte progenitors (GMPs) and common lymphoid progenitors (CLPs) were not (Figures 2G and 2H; Figure S3L). Immature megakaryocyte and erythroid progenitors (MkP, pre-MegE, and pre-CFU-E) were depleted in Kmt2cΔ/Δ mice (Figures S3M–S3O), even though more mature CFU-E, red blood cells, and platelets were normal (Figures S3C, S3D, and S3P). Competitive transplantation assays confirmed that Kmt2cΔ/+ and Kmt2cΔ/Δ HSCs repopulate more effectively than wild-type HSCs, consistent with the germline knockout findings (Figures 2I and 2J). Altogether, the data show that modest HSC expansion in Kmt2cΔ/Δ mice comes at the expense of HPC differentiation. However, by later stages of maturation, the lineages recover their numbers.
Because self-renewal capacity declines when HSCs divide multiple times (Bernitz et al., 2016; Foudi et al., 2009; Qiu et al., 2014; Wilson et al., 2008), we considered that Kmt2cΔ/Δ HSCs may cycle less frequently than wild-type adult HSCs. BrdU incorporation was equivalent in wild-type and Kmt2cΔ/Δ mice (Figure 2K; Figure S1C), but the resolution of this assay was limited by the short-term BrdU exposure. To better assess HSC division histories, we used a doxycycline-repressible histone H2B-green fluorescent protein (GFP) allele to monitor HSC divisions over a 14-week period of time, beginning at 8 weeks old (Figure 2L). In this system, GFP-tagged histone H2B was incorporated into the chromatin of all blood cells during development and into adulthood. Doxycycline suppressed H2B-GFP transgene expression, and GFP intensity declined with each cell division. Prior to doxycycline treatment, HSCs expressed high levels of H2B-GFP irrespective of Kmt2c genotype (data not shown). After 14 weeks of doxycycline treatment, rapidly dividing HPCs lost H2B-GFP signal, as expected (Figure 2M). In contrast, HSCs retained H2B-GFP signal to varying degrees. Distributions of H2B-GFP-low, -medium and -high HSCs were equivalent for wild-type, Kmt2cΔ/+, and Kmt2cΔ/Δ mice (Figures 2M and 2N; Figure S1D). Thus, Kmt2c deletion enhances HSC function and impedes differentiation without altering HSC proliferation rates or dormancy. This suggests that Kmt2c deletions might convey a selective advantage primarily in contexts that force quiescent HSCs into cycle.
Chemotherapy conveys a selective advantage to Kmt2cΔ/+ HSCs
To test whether haploid Kmt2c deletions convey a selective advantage to non-transplanted HSCs, and whether chemotherapy or inflammatory stress can exacerbate the advantage, we generated Fgd5-CreER; Kmt2cf/+ mice. Fgd5-CreER is expressed exclusively in HSCs (Gazit et al., 2014). We titrated the tamoxifen dose to delete a single Kmt2c allele in only ~20% of the HSC pool, as assessed by sorting single HSCs into methylcellulose and genotyping colonies. One week after tamoxifen treatments concluded, we administered one or three cycles of vehicle (phosphate-buffered saline [PBS]), 5-fluorouracil (5-FU), or cyclophosphamide (CY) to drive HSCs into cycle (Figure 3A) (Morrison et al., 1997; Randall and Weissman, 1997). We also treated a cohort of mice with poly-inosine:poly-cytosine (pIpC) to induce type I interferon expression and create inflammatory stress (Figure 3A) (Walter et al., 2015). None of the treatments significantly altered total HSC numbers (Figure 3B), and single cycles of 5-FU, CY, or pIpC did not increase the percentage of Kmt2cΔ/+ HSCs relative to PBS controls (Figure 3C). In contrast, three cycles of 5-FU and CY did increase Kmt2cΔ/+ HSC frequencies relative to controls, but pIpC did not (Figure 3D). This was not simply due to selection for Fgd5-expressing HSCs: the Fgd5-CreER allele is linked to an IRES-ZsGreen reporter (Gazit et al., 2014), and we did not observe any changes in the percentage of ZsGreen-positive HSCs with 5-FU or CY treatments (Figure 3E). The difference between chemotherapy treatment and pIpC treatment was surprising, and it may reflect a recent observation that pIpC primarily activates a non-repopulating subpopulation of CD150+CD48−LSK cells that does not express Fgd5 (Bujanover et al., 2018), or pIpC may simply be a weaker mitogenic stimulus than 5-FU or CY. Regardless, the data again show that Kmt2c-deleted HSCs can cycle extensively without activating differentiation or exhaustion mechanisms. This conveys a selective advantage, but only in the context of sustained HSC proliferation, as occurs after several cumulative cycles of chemotherapy.
Figure 3. Haploid Kmt2c deletion conveys a selective advantage to HSCs that divide after chemotherapy.
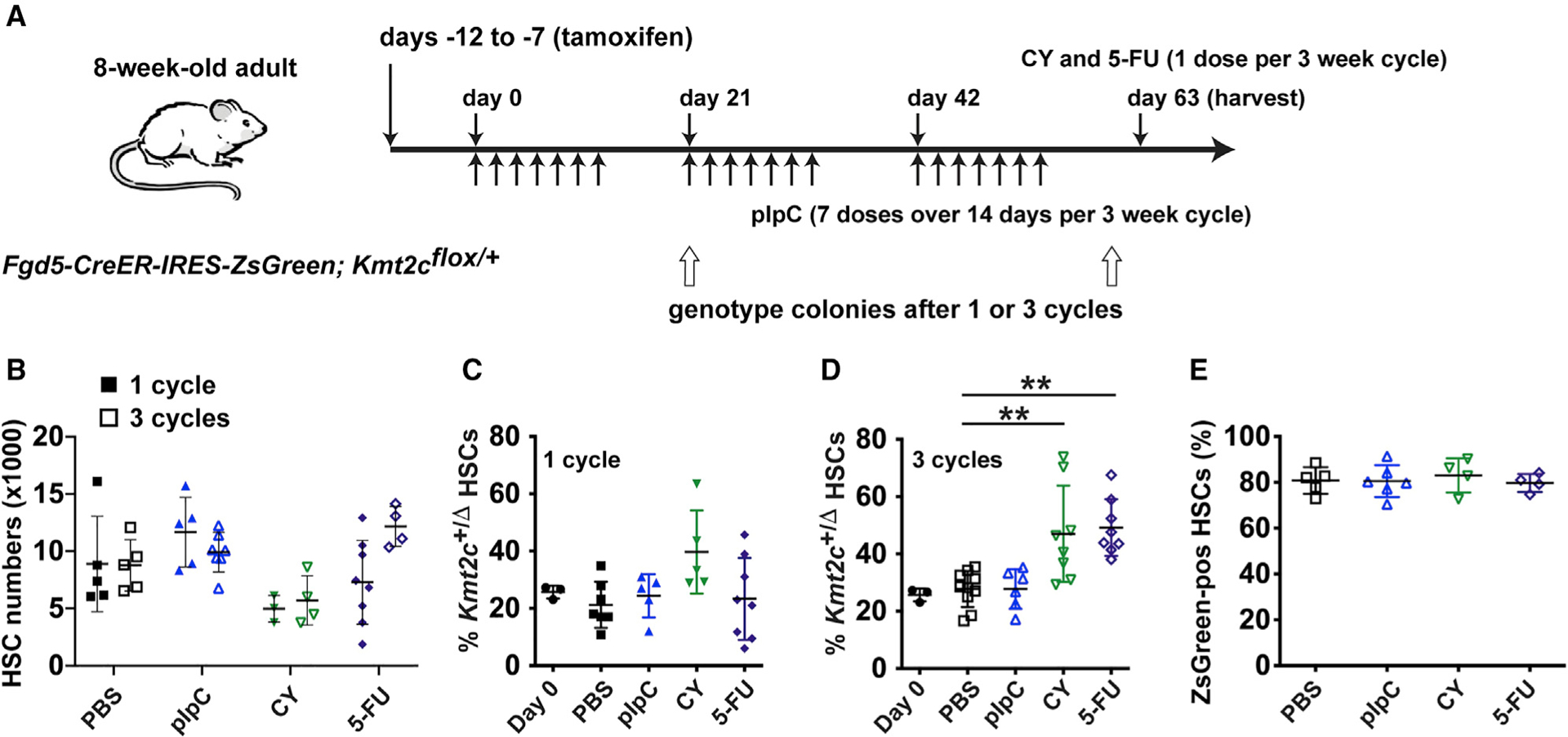
(A) Overview of the experiment. Arrows indicated treatment days for pIpC, CY, and 5-FU.
(B) HSC numbers in Fgd5-CreER; Kmt2cflox/+ mice after 1 or 3 cycles of the indicated treatments. n = 4–8.
(C and D) Percentage of HSCs with Kmt2c deletions after 1 or 3 cycles of the indicated treatments. n = 5–9.
(E) Percentage of ZsGreen-positive HSCs, among all CD150+CD48−LSK cells, after the indicated treatments. n = 4–6. Error bars reflect standard deviation. **p <0.01, ***p < 0.001; comparisons were made by one-way ANOVA with Holm-Sidak post hoc test.
Kmt2c deletion has negligible effects on reactive oxygen species generation and DNA damage
The consequences of Kmt2c/MLL3 inactivation in HSCs differ considerably from previously described consequences of Kmt2d/MLL4 inactivation. HSC self-renewal capacity increases following Kmt2c deletion (Figures 1 and 2), whereas it decreases following Kmt2d deletion (Santos et al., 2014). Loss of self-renewal capacity in Kmt2dΔ/Δ HSCs has been attributed to reduced anti-oxidant gene expression, increased reactive oxygen species (ROS) levels, and elevated DNA damage that promotes differentiation (Santos et al., 2014). We therefore tested whether Kmt2c deletion can enhance self-renewal capacity by suppressing ROS and DNA damage after chemotherapy.
We used 2′,7′-dichlorofluorescein diacetate (DCFDA) and γH2Ax staining to measure ROS levels and DNA damage, respectively. We analyzed wild-type and Kmt2cΔ/Δ HSCs at baseline and 2 days after CY treatment, when the HSCs had entered cycle. We chose CY rather than 5-FU for these experiments because HSC surface marker phenotypes are stable 2 days after CY treatment, in contrast with 5-FU (Morrison et al., 1997; Umemoto et al., 2018). At baseline, we did not observe ROS level changes in Kmt2cΔ/Δ HSCs (Figures S4A and S4B), After CY treatment, ROS levels were slightly, but significantly, attenuated in Kmt2cΔ/Δ HSCs (Figures S4A and S4B). DNA damage was similar in wild-type and Kmt2cΔ/Δ HSCs, irrespective of CY treatment (Figures S4C and S4D). Thus, Kmt2c deletion causes only a modest reduction on ROS levels, and it does not reduce DNA damage after CY.
Kmt2c deletion blunts inflammatory programs and induces glutathione-related gene expression in HSCs
To better understand how Kmt2c/MLL3 regulates HSC self-renewal and hematopoiesis, we performed RNA sequencing (RNA-seq) with wild-type, Kmt2cΔ/+, and Kmt2cΔ/Δ HSCs, HPC-1 s, pGMs, and GMPs. We identified 328 genes that were expressed lower in Kmt2cΔ/Δ HSCs as compared with wild-type (false discovery rate [FDR] < 0.05) and 213 genes that were expressed more highly (Figure 4A). Of these, only five met the same criteria for differential expression in Kmt2cΔ/+ HSCs, although 192 met a less stringent criterion of p < 0.05 (Figure 4B; Table S1). There was surprisingly little overlap between genes that were differentially expressed in Kmt2cΔ/Δ HSCs and those that were differentially expressed in HPC-1 s, pGMs, and GMPs (Figures S5A–S5D). GAGE (generally applicable gene set enrichment) pathway analysis showed reduced expression of genes associated with inflammation, IL-1, and inflammasome signaling in HSCs (Figures 4A and 4C). IL-1 stimulates HSCs to differentiate into myeloid progenitors (Pietras et al., 2016; Rabe et al., 2020). Thus, MLL3 may mediate IL-1-stimulated differentiation.
Figure 4. Kmt2c deletion reduces sensitivity to IL-1 and enhances antioxidant gene expression in HSCs.
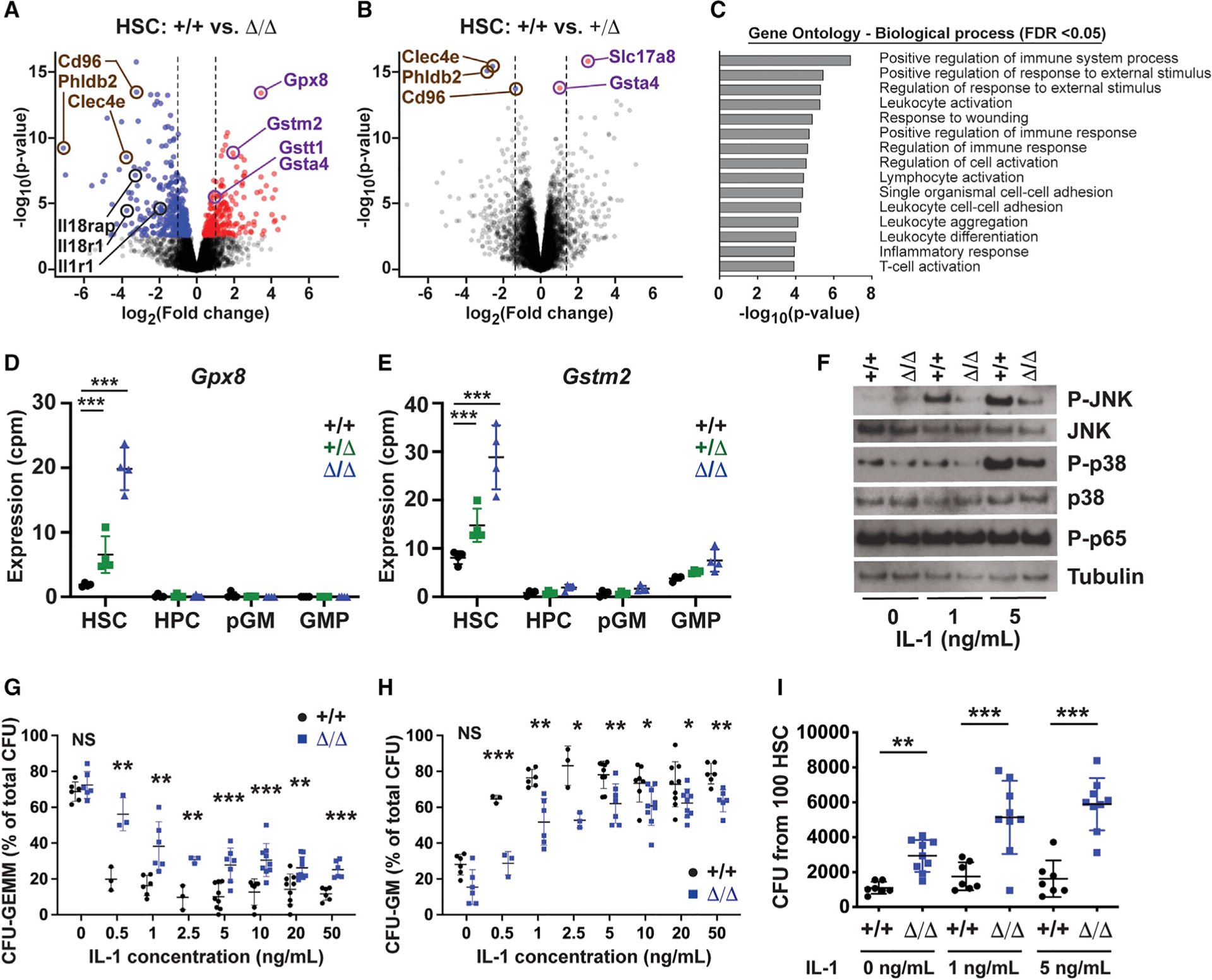
(A and B) Volcano plots showing gene expression changes in Kmt2cΔ/Δ and Kmt2cΔ/+ HSCs. Red and blue denote significantly increased or decreased expression after Kmt2c deletion (FDR < 0.05). n = 4.
(C) GAGE pathway analysis of genes reduced in Kmt2cΔ/Δ HSCs.
(D and E) Gpx8 and Gstm2 expression in HSCs, HPC-1 s, pGMs, and GMPs. n = 4 per genotype.
(F) Western blots showing JNK, p38, and p65 phosphorylation in HSCs after ex vivo IL-1β exposure.
(G and H) CFU-GEMM and CFU-GM percentages for wild-type and Kmt2cΔ/Δ HSCs plated in the indicated IL-1β concentrations. n = 3–8.
(I) Total CFU activity after 100 wild-type and Kmt2cΔ/Δ HSCs were cultured for 7 days with IL-1β at the indicated concentration. n = 7–9. Error bars reflect standard deviation.
*p < 0.05, **p < 0.01, ***p < 0.001; comparisons were made by two-tailed Student’s t test or one-way ANOVA with Holm-Sidak post hoc test (multiple comparisons). See also Figures S4 and S5 and Table S1.
In addition to the downregulated genes, several glutathione-related genes were upregulated in Kmt2cΔ/+ and Kmt2cΔ/Δ HSCs, relative to wild-type controls, including Gpx8, Gsta4, Gstt1, and Gstm2 (Figures 4D and 4E; Table S1). These increases were not observed in HPC-1 s, pGMs, or GMPs (Figures 4D and 4E). GPX8 reduces hydrogen peroxide by oxidizing glutathione, and it regulates calcium flux between the mitochondria and endoplasmic reticulum (Brigelius-Flohé and Maiorino, 2013; Yoboue et al., 2017). The glutathione S-transferases (GSTA4, GSTT1, and GSTM2) conjugate glutathione to xenobiotic compounds and mitigate oxidative stress (Ye et al., 2015). These changes contrast with decreases in antioxidant gene expression that occur in Kmt2dΔ/Δ HSCs (Santos et al., 2014).
MLL3 mediates IL-1-induced myeloid differentiation in vitro
Because the RNA-seq data suggest that MLL3 may mediate IL-1-stimulated differentiation, we tested whether Kmt2c deletion could impair IL-1 signal transduction or myeloid commitment in vitro. To assess signal transduction, we isolated 30,000 HSCs and HPC-1 s from wild-type and Kmt2cΔ/Δ mice. We incubated the cells for 30 min with 0, 1, or 5 ng/mL recombinant IL-1β. We then performed western blots to assess signal transduction. IL-1β has been shown to activate NF-κB (p65), p38, and JNK pathways. We did not observe changes in p65 phosphorylation, but p38 and JNK phosphorylation were reduced in IL-1β-treated Kmt2cΔ/Δ HSCs (Figure 4F). These pathways were not suppressed in Kmt2cΔ/Δ HPCs (Figure S5E). To test whether MLL3 primes HSCs to differentiate in response to IL-1, we sorted single HSCs into methylcellulose supplemented with IL-1β ranging from0.5 to 50 ng/mL. Consistent with prior work (Pietras et al., 2016), IL-1β exposure significantly reduced multilineage colony formation (CFU-GEMM) and increased granulocyte-monocyte colony formation (CFU-GM), even at low concentrations (Figures 4G and 4H). Kmt2c deletion blunted this effect, particularly at low IL-1β concentrations (Figures 4G and 4H).
We next tested whether Kmt2c deletion could allow cultured HSCs to retain colony-forming activity, even upon exposure to IL-1β. We sorted 100 HSCs into liquid media supplemented with stem cell factor (SCF), thrombopoietin (TPO), and 0, 1, or 5 ng/mL of IL-1β. We cultured the cells for 7 days and then plated 1% of the culture volume in methylcellulose. Wild-type HSCs yielded similar numbers of colony-forming units (CFUs) after 7 days of exposure to all IL-1β concentrations (Figure 4I). Kmt2cΔ/Δ HSCs gave rise to more CFUs than wild-type HSCs, and CFUs increased with IL-1β supplementation (Figure 4I). Kmt2cΔ/Δ HSCs did not fully retain their function as they failed to repopulate irradiated mice after 7 days in culture (data not shown). Nevertheless, the data show that Kmt2c deletion impedes differentiation in response to IL-1β in vitro.
Quiescent and cycling Kmt2c-deficient HSCs have distinct responses to IL-1 in vivo
We next tested whether Kmt2c deletion conveys a selective advantage to IL-1β-treated quiescent and dividing HSCs in vivo. We first treated adult mice with IL-1β (0.5 μg/day for 3 days). IL-1β treatment unexpectedly reduced Kmt2cΔ/Δ HSC numbers relative to PBS-treated mice (Figure 5A). A similar reduction was seen after 21 days of treatment (data not shown). MPP numbers were dramatically reduced in IL-1β-treated mice, irrespective of Kmt2c genotype (Figure 5B). HPC numbers were not affected by IL-1; they were lower in Kmt2cΔ/Δ mice in both the PBS and IL-1β treatment groups (Figure 5C). We transplanted 20 HSCs from wild-type and Kmt2cΔ/Δ mice after 21 days of PBS or IL-1β treatment and found that IL-1β selectively impaired Kmt2cΔ/Δ HSC function (Figure 5D). Prior work has shown that self-renewing HSCs become depleted within the CD150+CD48−LSK population after IL-1 treatment, as evidenced by a reduction in the EPCR+CD34− HSC sub-fraction (Rabe et al., 2020), and we therefore tested whether Kmt2c deletion exacerbates this effect. Indeed, after IL-1β treatment, non-self-renewing EPCR−CD34+ cells dominated the HSC fraction to a far greater extent in Kmt2cΔ/Δ mice than in wild-type mice (Figure 5E). Thus, Kmt2c deletion mitigates HSC differentiation in vitro (Figure 4), but not in vivo (Figure 5), at least not at steady state.
Figure 5. Kmt2c deletion impairs IL-1β-driven myeloid differentiation and conveys a selective advantage to cycling, IL-1β-stimulated HSCs.
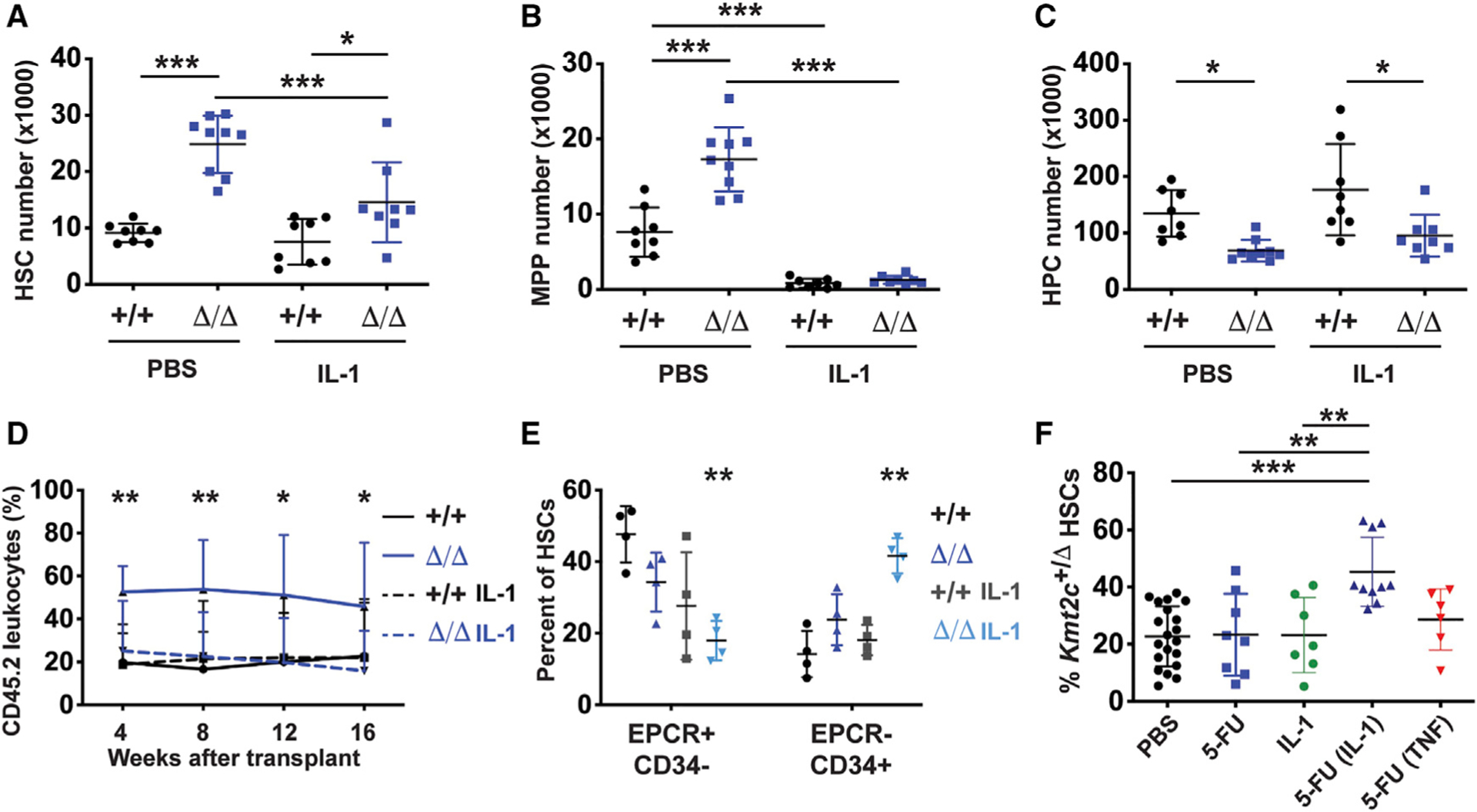
(A–C) HSC, MPP, and HPC numbers in wild-type and Kmt2cΔ/Δ mice after treatment with PBS or IL-1β for 3 days. n = 8–9.
(D) CD45.2+ donor leukocyte chimerism in peripheral blood from primary recipients transplanted with 20 wild-type and Kmt2cΔ/Δ HSCs, after a 21-day treatment with vehicle (PBS) or IL-1β. n = 10–13.
(E) Phenotypic HSC subpopulations after IL-1β treatment based on EPCR and CD34 expression.
(F) Percent of HSCs with Kmt2c deletions after one cycle of PBS or 5-FU, with or without concomitant IL-1β or TNF. n = 6–20. Error bars reflect standard deviation.
*p < 0.05, **p < 0.01, ***p < 0.001; comparisons were made by two-tailed Student’s t test or one-way ANOVA with Holm-Sidak post hoc test (multiple comparisons).
The discrepancy between in vitro and in vivo responses to IL-1β raised the question of whether this reflects differences in proliferation status. HSCs cycle in culture, but they are usually quiescent in the adult bone marrow. To test this, we treated Fgd5-CreER; Kmt2cf/+ mice with tamoxifen as in Figure 3. We subsequently treated the mice with a single cycle of 5-FU, IL-1β (0.5 μg/day for 21 days), or both. As before, a single cycle of 5-FU did not increase Kmt2cΔ/+ HSC frequencies relative to undeleted HSCs (Figure 5F). IL-1β treatment also failed to expand the Kmt2cΔ/+ HSC population by itself. However, 5-FU and IL-1β together conveyed a strong selective advantage to Kmt2cΔ/+ HSCs (Figure 5F). This synergy was not observed with another inflammatory cytokine, TNF. The data show that MLL3 promotes HSC differentiation in response to IL-1β, but only when HSCs are driven into cycle by another stimulus, such as culturing or chemotherapy. Inflammatory signaling does not, by itself, convey a selective advantage to Kmt2cΔ/+ HSCs in vivo.
MLL3 promotes enhancer recruitment during HSC to HPC differentiation, particularly in response to IL-1
The MLL3 COMPASS complex places H3K4me1 marks at enhancer elements, and it facilitates enhancer activation through CBP/p300-mediated H3K27ac (Herz et al., 2012; Lai et al., 2017). This raised the question of whether MLL3 primes enhancers that mediate differentiation, particularly in response to IL-1. To identify MLL3-regulated enhancer elements, we performed ATAC-seq (Assay for Transposase-Accessible Chromatin using Sequencing) and ChIPmentation (a modified chromatin immuno-precipitation with massively parallel DNA sequencing technique). We identified enhancers as regions of open chromatin with overlapping H3K4me1 peaks (Buenrostro et al., 2015; Schmidl et al., 2015). Altogether, we identified 57,590 enhancers that met these criteria (ATAC-seq peak with overlapping H3K4me1 peak) in either wild-type HSCs or HPC-1 s (Figure 6A). Global H3K4me1 levels were largely unaffected by Kmt2c deletions in both the HSC and HPC populations (Figures 6B and 6C; Figure S6A), suggesting that other SET domain proteins, such as MLL4, can monomethylate H3K4 in the absence of MLL3. This raises the question of whether MLL3 regulates a subset of HSC/HPC enhancer elements rather than the global enhancer landscape.
Figure 6. MLL3 promotes enhancer priming and IL-1 target enhancer activation during HSC to HPC differentiation.
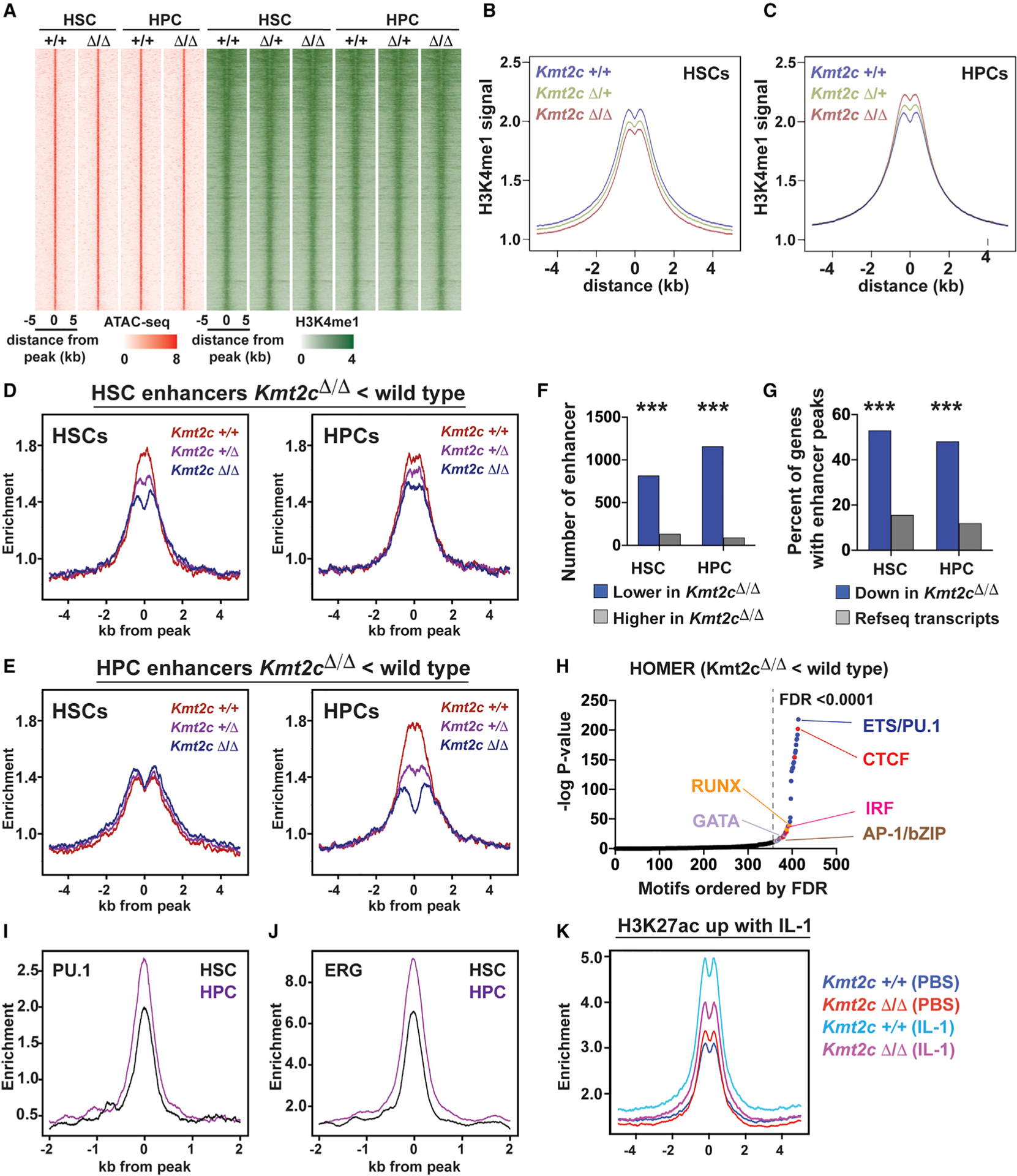
(A) ATAC-seq (red) and H3K4me1 (green) peaks for all identified HSC and HPC enhancers.
(B and C) Aggregate H3K4me1 levels in wild-type, Kmt2cΔ/+, and Kmt2cΔ/Δ HSCs and HPCs, all enhancers.
(D) Histograms showing H3K4me1 at enhancers with reduced H3K4me1 in Kmt2cΔ/Δ HSCs relative to wild-type HSCs (HSC enhancers). Data are shown separately for HSCs and HPCs.
(E) Histograms showing H3K4me1 at enhancers with reduced H3K4me1 in Kmt2cΔ/Δ HPCs relative to wild-type HPCs (HPC enhancers). No Kmt2c-dependent changes are observed for these enhancers in HSCs.
(F) Numbers of enhancers with increased or decreased H3K4me1 levels after Kmt2c deletion.
(G) Percent of Kmt2c-regulated genes (from Figure 4) with enhancers located within 100 kb of the transcriptional start site. All RefSeq genes are shown as a control. ***p < 0.0001 by hypergeometric test.
(H) HOMER motif enrichment showing −log p values for each motif ranked according to decreasing false discovery rate (FDR). Motifs with FDR < 0.0001 are color coded based on unique classes of transcription factor binding domains.
(I and J) PU.1 and ERG binding at HSC and HPC enhancers in HPC-7 cells.
(K) H3K27ac at IL-1 target enhancers in wild-type and Kmt2cΔ/Δ HPCs.
See also Figure S6.
To address this question, we identified enhancers with significantly altered H3K4me1 levels in Kmt2cΔ/Δ HSCs and HPCs as compared with wild-type HSCs and HPCs (Figures 6D and 6E). An overwhelming majority of differentially methylated enhancers showed a reduction in H3K4me1 in Kmt2cΔ/Δ HSCs/HPCs rather than an increase (Figure 6F). Differentially methylated enhancers were significantly enriched near genes that were also downregulated in Kmt2cΔ/Δ HSCs and HPCs (Figure 6G). There was little overlap between enhancers that had significantly lower H3K4me1 in Kmt2cΔ/Δ HSCs, relative to wild-type HSCs, and enhancers that had lower H3K4me1 in Kmt2cΔ/Δ HPCs, relative to wild-type HPCs (HSC and HPC enhancers, respectively). H3K4me1 priming increased at HPC enhancers during HSC to HPC differentiation (Figure 6E, left versus right panel). This priming failed in the absence of Kmt2c/MLL3 (Figure 6E, right). Motif enrichment analysis showed that MLL3-dependent enhancers harbor binding sites for transcription factors that mediate HSC to HPC differentiation, particularly ETS/PU.1, IRF, RUNX, AP-1, and GATA sites. Analysis of previously described ChIP-seq data confirmed that these enhancers bind the ETS domain proteins PU.1 and ERG (Figures 6I and 6J) (Wilson et al., 2016).
We next tested whether IL-1 activates enhancers in an MLL3-dependent manner. We treated wild-type and Kmt2cΔ/Δ mice with PBS and IL-1β (0.5 μg/day for 3 days). We isolated HPCs and performed ChIPmentation to measure H3K27ac levels (a mark of active enhancers). IL-1β activated 2,030 enhancers in HPCs, as evidenced by a significant rise in H3K27ac. Kmt2c deletion reduced H3K27ac at these enhancers (Figure 6K). Altogether, these data show that MLL3 helps prime HPC enhancers, and it promotes IL-1 target enhancer activation during HSC to HPC differentiation.
MLL3 activates a subset of enhancers when HSCs cycle in response to chemotherapy
Given that Kmt2c deletion conveys a selective advantage to cycling HSCs, we tested whether enhancer priming (H3K4me1) and activation (H3K27ac) change in a Kmt2c/MLL3-dependent manner as HSCs cycle in response to chemotherapy. We treated wild-type and Kmt2cΔ/Δ mice with three cycles of PBS or 5-FU, as in Figure 3A. HSCs were isolated from the bone marrow after the mice had recovered from the third 5-FU cycle (cycle 3, day21). HSC numbers increased significantly in 5-FU-treated Kmt2cΔ/Δ mice, relative to vehicle-treated Kmt2cΔ/Δ mice, but they did not expand in wild-type mice (Figure 7A). HPC-1 numbers declined in 5-FU-treated wild-type mice, but they were already reduced in Kmt2cΔ/Δ mice and declined no further with 5-FU treatment (Figure 7B). GMP numbers were similar across all genotypes and treatment groups (Figure 7C). H2B-GFP analysis confirmed that HSCs had cycled extensively in the 5-FU treatment group (Figure 7D). These observations are all consistent with our earlier analyses (Figures 2 and 3).
Figure 7. Kmt2c deletion mitigates enhancer activation in multiply-divided HSCs.
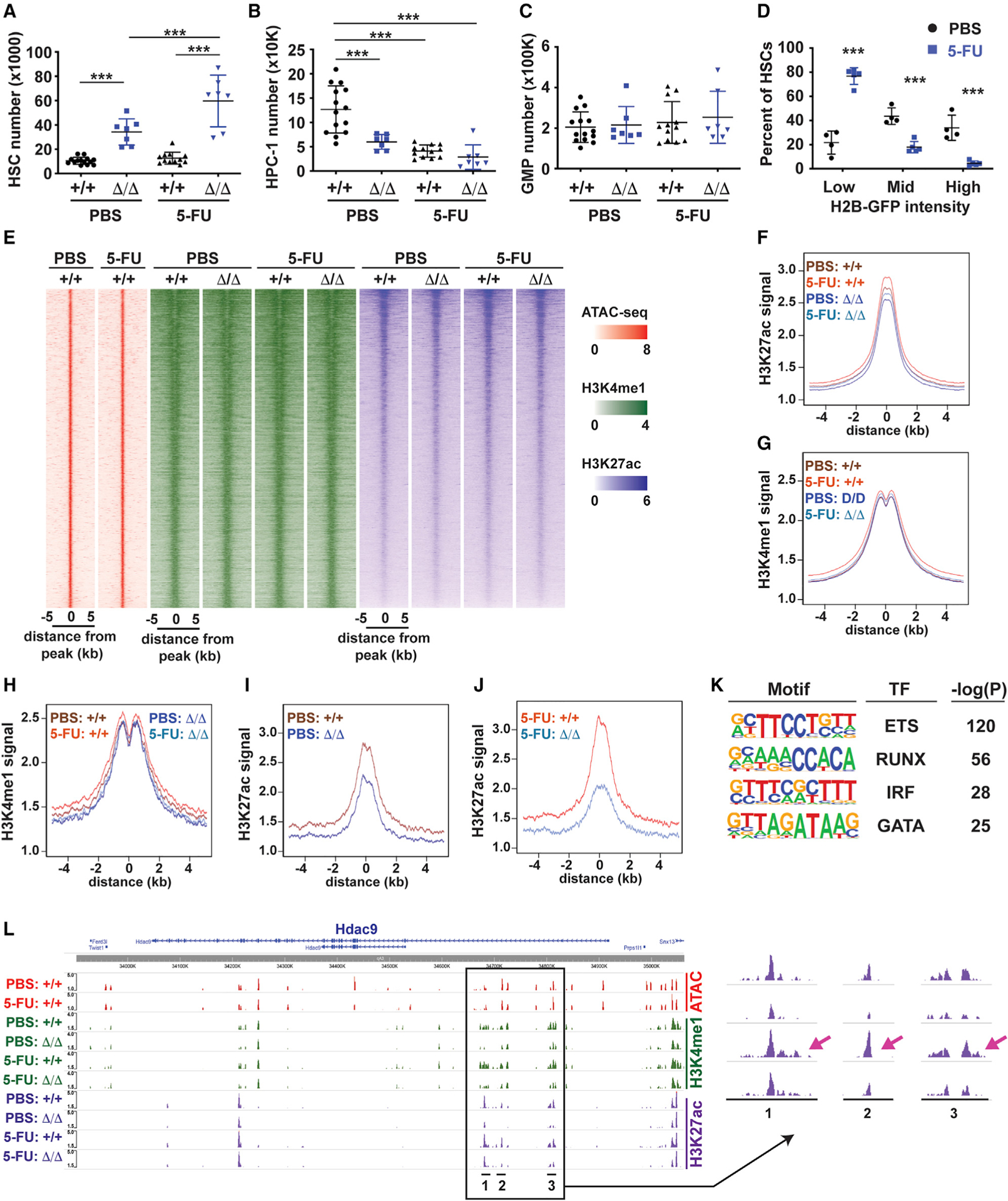
(A–C) HSC, HPC-1, and GMP numbers in wild-type and Kmt2cΔ/Δ mice treated with three cycles of PBS or 5-FU. n = 7–14. ***p < 0.001 one-way ANOVA with Holm-Sidak post hoc test.
(D) Percentage of H2B-GFP-retaining HSCs after three cycles of PBS or 5-FU treatment. N = 4. ***p < 0.001 by two-tailed Student’s t test.
(E) Heatmap showing ATAC-seq, H3K4me1, and H3K27ac at all enhancers in wild-type and Kmt2cΔ/Δ HSCs after three cycles of PBS or 5-FU treatment.
(F and G) Aggregate H3K27ac and H3K4me1 levels in wild-type (+/+) and Kmt2c-deleted (Δ/Δ) HSCs from mice treated with three cycles of PBS and 5-FU.
(H–J) Aggregate H3K4me1 and H3K27ac levels at MLL3-regulated enhancers (based on H3K27ac) after three cycles of 5-FU treatment.
(K) Top four enriched motifs within MLL3 target enhancers by HOMER.
(L) ATAC-seq, H3K4me1, and H3K27ac tracks at the Hdac9 locus. The arrow denotes an increase H3K27ac in wild-type HSCs after 5-FU treatment. n = 2–4 (ATAC) and n = 3–6 (H3K4me1, H3K27ac) biological replicates per genotype/cell type/treatment group.
Error bars in (A)–(D) reflect standard deviations. See also Figures S6 and S7 and Table S2.
We performed ChIPmentation to identify sites of H3K4me1 and H3K27ac in PBS- and 5-FU-treated wild-type and Kmt2cΔ/Δ HSCs (Figure 7E). Global H3K4me1 and H3K27ac levels did not change much with 5-FU treatment or Kmt2c deletion (Figures 7F and 7G; Figures S6B and S6C). However, we identified 838 enhancers (~2% of all enhancers) that exhibited MLL3-dependent changes in H3K27ac levels. Whereas H3K4me1 levels were similar at these enhancers irrespective of genotype or treatment group (Figure 7H), H3K27ac levels increased after 5-FU treatment, and Kmt2c deletion exacerbated the change (Figures 7I and 7J). MLL3-dependent enhancers were enriched for ETS/PU.1, RUNX, IRF, and GATA binding motifs (Figure 7K), and many localized near genes that are downregulated in the absence of Kmt2c (e.g., Hdac9, Bach2, Dab2, Fyb, Phldb2, and Runx1t1) (Figure 7L; Figures S7A–S7D). Further studies are needed to determine which MLL3 target genes contribute to HSC exhaustion and why these enhancers are exquisitely dependent on MLL3. Nevertheless, the data show that as HSCs undergo self-renewing divisions after chemotherapy, their epigenomes change, and previously inactive enhancers become active in an MLL3-dependent manner.
DISCUSSION
Our data provide a framework for understanding how KMT2C deletions convey a selective advantage in the context of larger chromosome 7q deletions. Prior studies have shown that haploid KMT2C deletions accelerate myeloid transformation (Chen et al., 2014). Now we show that it enhances HSC self-renewal capacity, as well. MLL3 primes enhancers for activation during HSC differentiation into HPCs, and it promotes enhancer activation by inflammatory cytokines, most notably IL-1. Furthermore, MLL3 helps reprogram and activate enhancers as HSCs cycle after chemotherapy treatment. Kmt2c-mutated HSCs resist these changes and retain self-renewal capacity through cumulative division cycles even as wild-type HSCs become exhausted. This conveys a selective advantage.
Our findings draw an important distinction between mutations that convey a selective advantage after chemotherapy, such as KMT2C deletions, and those that convey a selective advantage during aging or inflammation, such as DNMT3A or TET2 mutations. DNMT3A mutations suppress HSC cycling and thereby preserve a quiescent HSC pool over decades of life, while simultaneously suppressing differentiation (Challen et al., 2011; Young et al., 2016). TET2 mutations sensitize HSCs to mitogenic cytokines while simultaneously enhancing self-renewal capacity (Moran-Crusio et al., 2011; Muto et al., 2020). KMT2C deletions do neither of these things: HSCs cycle normally, inflammatory target gene expression is suppressed, and the selective advantage becomes most evident when HSCs are driven into cycle by chemotherapy. These observations may help explain why DNMT3A and TET2 mutations are common in age-related clonal hematopoiesis, but KMT2C mutations are not (Steensma and Ebert, 2020; Young et al., 2016).
Our findings also highlight divergent functions for Kmt2c and its close paralog, Kmt2d (MLL4), in HSCs. Prior studies have shown that Kmt2d deletion impairs HSC self-renewal by reducing antioxidant gene expression, thereby exacerbating DNA damage (Santos et al., 2014). In contrast, Kmt2c deletion increases antioxidant gene expression, has no effect on DNA damage burden even after chemotherapy, and enhances HSC self-renewal capacity. Prior work has shown that in many cell types, such as ESCs, adipocytes, and urothelial cells, Kmt2c and Kmt2d are at least partially redundant (Dorighi et al., 2017; Lee et al., 2008, 2013). It is therefore interesting that the pheno-types of Kmt2c- and Kmt2d-deficient HSCs are not only different but also almost reciprocal to one another. This raises the question of whether MLL3 and MLL4 act on shared pathways. For example, it is possible that one or more shared components of the COMPASS complex are rate limiting in HSCs. When MLL3 is lost, stoichiometry could favor MLL4 target gene activation. Likewise, when MLL4 is lost, stoichiometry could favor MLL3 target gene activation. Going forward, it will be important to evaluate compound mutant mice for epistasis and to evaluate the role of other MLL3/4 COMPASS components, such as KDM6A, in HSC self-renewal.
Our data raise additional mechanistic questions. Foremost among these, it is not clear which MLL3 target genes and target enhancers are most responsible for promoting differentiation and impairing self-renewal as HSCs cycle. Furthermore, it is not clear why only a small fraction of all enhancers (~2%) selectively depend on MLL3 and whether their regulation requires SET domain activity. Prior work has shown that N-terminal plant homeodomain (PHD) domains have unique chromatin binding functions that are not evident in MLL4 (Wang et al., 2018), and ongoing studies will test whether these domains mediate MLL3-specific functions in HSCs. Finally, we recognize that KMT2C is one of several putative 7q tumor suppressor genes. Thus, the consequences of haploid Kmt2c deletion in mice do not capture the full effects of −7/del7q mutations in pre-leukemic HSCs or t-MDS/t-AML. Future studies must interrogate interactions between Kmt2c and other putative 7q tumor suppressors.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents and resources should be directed to and will be fulfilled by the Lead Contact, Jeffrey Magee (mageej@wustl.edu).
Materials availability
Kmt2c+/− and Kmt2cflox mice are available for unrestricted use, upon request, with a standard institutional material transfer agreement.
Data and code availability
The datasets generated during this study are available at Gene Expression Omnibus under the SuperSeries accession GSE158162. SubSeries accession numbers are GSE158158 (ATAC-Seq), GSE158159 (ChIPmentation) and GSE158160 (RNA-seq). No new code was generated for this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Kmt2c+/− and Kmt2cflox mice were generated using guide RNAs generated by the Washington University Genome Engineering and iPSC Center. The guide RNAs were complexed with recombinant CAS9 and nucleofected into C57BL/6J embryos by the Washington University Department of Pathology Micro-injection Core. Single strand oligonucleotides were used to insert LoxP sites through homologous recombination. Successfully targeted founder mice were identified by next generation sequencing. Cis-incorporation of the LoxP sites was confirmed by long-range PCR in the founders and linkage of the insertions in F1 progeny. Founders were back-crossed to C57BL/Ka-Thy-1.1 (CD45.2) mice for at least 3 generations prior to use. PCR assays were developed for subsequent genotyping. Vav1-Cre – RRID:IMSR_JAX:008610, Fgd5-CreER – RRID:IMSR_JAX:027789, RosaLSL-tTA – RRID:IMSR_JAX:011008, Col1a1TetO_H2B-GFP – RRID:IMSR_JAX:016836 and CMV-Cre – RRID:IMSR_JAX:006054 mice were obtained from the Jackson Laboratory and genotyped with primers described on the Jackson Laboratory website. All experiments were performed with 8–10-week-old adult mice except as indicated in the figure legends (e.g., E18.5 fetal mice). For experiments requiring longitudinal monitoring (e.g., H2B-GFP lineage tracing, chemotherapy treatments), the treatments began 8 weeks after birth. Male and female mice were used equivalently in all experiments. Mice were housed in a standard pathogen free barrier facility. All procedures were performed according to an IACUC approved protocol at Washington University School of Medicine. All mice were healthy and immune competent. All mice were naive to prior procedures, drugs or tests. Cell lines were not used in these studies.
METHOD DETAILS
Kmt2c genotyping
Mice were genotyped by tail biopsies or fetal liver specimen. For the exon 14 germline Kmt2c mutation (Figure 1), primers 5′−GTGACTTAATACGACTCACTATAGGGCCAACAACATACATCTCAGTC-3′ and 5′-TGTATTGCAGGGAAAGTAAATGTT-3′ were used to amplify the cut site. The PCR product was then cut with NlaIII to distinguish wild-type and mutant PCR products by size. The NlaIII site was destroyed by the 5-base pair deletion that produced the Kmt2c null allele, and the product therefore ran larger after cutting (Figure 1A). For the exon 3 conditional allele, primers 5′-TCTGGTTGCTTATGGGTTGAT-3′ and 5′-AGCCATTAAACTTAGTGAAGACCA-3′ were used to follow identify the floxed allele after sequencing had been used to confirm that the loxP sites had been correctly inserted in cis to one another. Primers 5′-GCTCCTCCTCTGGCTTCCAAGGTCA-3′ and 5′-AGGGCATCTAATCTGACAGCTGTAAGC-3′ were used to identify the deleted allele.
Flow cytometry
Bone marrow cells were obtained by flushing the long bones (tibias and femurs) or by crushing long bones, pelvic bones and vertebrae with a mortar and pestle in calcium and magnesium-free Hank’s buffered salt solution (HBSS), supplemented with 2% heat inactivated bovine serum (GIBCO). Fetal and P0 liver cells were obtained by macerating livers with frosted slides. Single cell suspensions were filtered through a 40 μm cell strainer (Fisher). The cells were then stained sequentially, for 20 minutes each, with biotin conjugated anti-CD117 (c-kit; 2B8) and then Streptavidin-conjugated paramagnetic beads (Biolegend). c-kit+ cells were enriched by magnetic selection with an Automacs or LS magnetic columns (Miltenyi Biotec). Cells were stained for flow cytometry using the following antibodies, all of which were from Biolegend except as indicated: CD150 (TC15–12F12.2), CD48 (HM48–1), Sca1 (D7), c-kit (2B8), Ter119 (Ter-119), CD3 (17A2), CD11b (M1/70), Gr1 (RB6–8C5), B220 (RA3–6B2), CD4 (GK1.5), CD8a (53–6.7), CD2 (RM2–5), CD45.1 (A20), CD45.2 (104), surface IgM (RMM-1), CD5 (53–7.3), CD21 (7E9), CD23 (B3B4), CD135 (eBioscience; A2F10), CD105 (MJ7/18), CD41 (MWReg30), CD16/32 (93). Lineage stains for all experiments included CD2, CD3, CD8a, Ter119, B220 and Gr1. Antibodies to CD4 and CD11b were omitted from the lineage stains because they are expressed on fetal HSCs at low levels. The following surface marker phenotypes were used to define cell populations at all ages: HSCs (CD150+, CD48−, Lineage−, Sca1+, c-kit+), HPCs (CD48+, Lineage−, Sca1+, c-kit+), MPP (CD150−, CD48−, Lineage−, Sca1+, c-kit+), HPC-1 (CD150−, CD48+, Lineage−, Sca1+, c-kit+), HPC-2 (CD150+, CD48+, Lineage−, Sca1+, c-kit+), FLK2+ HPC-1 (CD135+, CD150−, CD48+, Lineage−, Sca1+, c-kit+), pGM (Lineage−, Sca1−, c-kit+, CD150−, CD105−, CD16/32−) and GMP (Lineage−, Sca1−, c-kit+, CD150−, CD105−, CD16/32+). Non-viable cells were excluded from analyses by 4’,6-diamidino-2-phenylindone (DAPI) staining (1 μg/ml except as indicated below). Flow cytometry was performed on a BD FACSAria Fusion flow cytometer (BD Biosciences).
Long-term repopulation assays and serial transplantation
Eight- to ten-week old C57BL/6Ka-Thy-1.2 (CD45.1) recipient mice were given two doses of 550 rad delivered at least 3 hours apart from a Cesium-137 source. HSCs or unfractionated bone marrow cells were transplanted at the ratios indicated in the text and figures. Cells were injected via the retroorbital sinus. To assess donor chimerism, peripheral blood was obtained from the submandibular veins of recipient mice at the indicated times after transplantation. Blood was subjected to ammonium-chloride lysis of the red blood cells and leukocytes were stained with antibodies to CD45.2, CD45.1, B220, CD3, CD11b and Gr1 to assess multilineage engraftment.
Western blots
To assess MLL3 protein expression, nuclei were isolated from 1 million mouse embryonic fibroblasts (Kmt2c+/+ and Kmt2c−/−; Figure 1B) or adult splenocytes (Kmt2c+/+ and Kmt2cf/f; Vav1-Cre; Figure 2B) in nuclear lysis buffer (10 mM NaCl, 10 mM Tris pH 8.0, 3 mM MgCl2 + COMPLETE protease inhibitor; Sigma). The nuclei were then resuspended in 1x LDS sample buffer (Thermo) plus 1 mM dithiothreitol (DTT). Specimens were heated at 70°C for 10 minutes. Lysates from an equivalent of 100,000 cells were run on NuPage 3%–8% Tris-acetate gel (Thermo) and transferred overnight to PVDF. Membranes were blocked with 5% BSA and incubated with anti-MLL3 (Millipore, ABE1851) or anti-NUP98 (Cell Signaling, 2288) as a loading control. Membranes were washed with Tris buffered saline (TBS) + 0.05% Tween, incubated with HRP-conjugated secondary antibodies (Cell Signaling, 7076 and 7074) and developed with the SuperSignal Femto kit (Thermo). For signal transduction analyses, 30,000 HSCs or HPCs were isolated by flow cytometry as described above. The cells were pelleted in PBS + 0.05% BSA and resuspended in Iscove’s Modified Dubelco’s Media (IMDM) + 0.05% BSA and the indicated concentrations of IL-1β. The cells were incubated for 30 minutes at 37°C and then pelleted. They were resuspended in 10 μL of cold PBS, transferred to 10% Trichloroacetic acid, and precipitated by centrifugation. The precipitates were washed twice with acetone and resuspended in solubilization buffer (9M urea, 2% Triton X-100, 1% DTT). LDS sample buffer (Thermo) was subsequently added to 1x final concentration and the specimen was heated at 700°C for 10 minutes. Samples were run on 4%–12% NuPage Bis-Tris gels (Thermo) and processed as described above. Primary antibodies to P-JNK (Cell Signaling, 4688), P-p38 (Cell Signaling, 4511), P-p65 (Cell Signaling, 3033), JNK (Cell Signaling, 9252), p38 (Cell Signaling, 9212) and alpha-tubulin (Cell Signaling, 2144) were added in succession. After each analysis, the membrane was stripped (1% sodium dodecyl sulfate, 25 mM glycine pH 2.0).
H2B-GFP pulse-chase assays
Vav1-Cre, RosaLSL-tTA, and Col1a1TetO_H2B-GFP mice were crossed with Kmt2cflox mice to generate the alleles indicated in the text. Mice were fed doxycycline chow (200 ppm, Bioserv) beginning at 8 weeks after birth to suppress H2B-GFP expression. HSCs were then isolated and analyzed as described above.
Assessment of Kmt2c-deleted HSC frequency after stress
Eight-week-old Fgd5-CreER; Kmt2cf/+ mice were treated with tamoxifen (Cayman Chemical) at 100 mg/kg/day for five days. The tamoxifen was administered in corn oil by intraperitoneal injection. This dose was established empirically, based on our goal of deleting the Kmt2cflox allele in approximately 20% of HSCs. After tamoxifen treatment, the mice were given 7 days to recover. They were treated intraperitoneally with PBS, 5-FU (150 mg/kg), CY (200 mg/kg) or pIpC (20 μg every other day; Sigma). Mice were treated on the schedules indicated in Figure 3. When IL-1β was administered (Figure 6F), mice were given 0.5 μg of IL-1β (Peprotech) in 100 μL PBS with 0.1% BSA, intraperitoneally once daily for 21 days beginning immediately after 5-FU treatment.
To measure deletion frequencies, single ZsGreen-positive HSCs were sorted into individual wells of a 96-well plate that contained MethoCult GF M3434 methylcellulose media (Stem Cell Technologies). Colonies were isolated 12 days later by washing the well with PBS, pelleting cells and lysing with Direct PCR lysis reagent (Viagen). Genotyping was performed as described above. In the absence of tamoxifen treatment (i.e., – vehicle only), we did not observe any Kmt2c-deleted HSCs even after three cycles of 5-FU (data not shown).
BrdU, cell cycle and γH2AX assays
Bromo-deoxyuridine (BrdU; Sigma) was administered by intraperitoneal injection (100 mg/kg/dose) once, and mice were then fed water containing 0.8 mg/mL BrdU for the next 24 hours. HSCs were stained and enriched by c-kit selection as described above. BrdU incorporation was measured by flow cytometry using the APC BrdU Flow Kit (BD Biosciences). For cell cycle and γH2AX analyses, HSCs were stained and enriched by c-kit selection as described above. They were then fixed and permeabilized with cytofix/cytoperm buffer (BD Biosciences), washed, and incubated with streptavidin-PE-CY7 along with Ki67- or γH2AX-AF647 antibodies. Cells were then washed, stained with DAPI (20 μg/mL), and analyzed by flow cytometry.
DCFDA assays
DCFDA staining was performed as described previously (Nakada et al., 2010). Bone marrow was isolated, c-kit enriched and stained as described above, leaving the FITC channel available for DCFDA analysis. We then incubated 2 million stained cells with 5 μM DCFDA (Sigma) for 15 minutes at 37°C. The cells were washed once with staining media, re-suspended in DAPI-containing staining media and immediately analyzed by flow cytometry.
In vivo IL-1β responses
To assess HSC numbers and function after IL-1 treatment, adult mice were treated with PBS or recombinant mouse IL-1β (Peprotech) at 0.5 μg/dose (in 100 mL PBS with 0.1% BSA, intraperitoneally) for 3 or 21 days as previously described (Pietras et al., 2016). Bone marrow was then harvested for analysis and transplantation as described above. To identify compare IL-1 target gene expression in wild-type and Kmt2cΔ/ΔHSCs, we treated mice for three days with IL-1β (0.5 μg/dose intraperitoneally) and then isolated HSCs for RNaseq as described below.
RNA-seq
For each replicate, 10,000 HSCs, HPCs, pGMs or GMPs of the indicated genotypes were double sorted into PBS with 0.1% BSA. Cells were pelleted by centrifugation and resuspended in RLT-plus RNA lysis buffer (QIAGEN). RNA was isolated with RNAeasy micro-plus columns (QIAGEN). RNaseq libraries were generated with Clontech SMRTer kits, and sequencing was performed on a HiSeq3000.
ATAC-seq and Chipmentation
For ATAC-seq, 50,000 HSCs or HPCs of the indicated Kmt2c genotypes and treatment groups were double sorted into PBS + 0.1% BSA. At least 3 independent replicates were analyzed per time point and population, with each replicate utilizing cells from multiple mice. Cells were processed, and libraries were generated, using the method described by Corces et al. (2017). Library amplification was performed using barcoded primers as described by Buenrostro et al. (2013). After library amplification, fragments were size selected with SPRI beads (Beckman Coulter) with ratios of 0.9x and 1.8x for right- and left-sided selection. Sequencing was performed on a HiSeq2500.
For ChIPmentation assays, 30,000 HSCs or HPCs of the indicated Kmt2c genotypes and treatment groups were double sorted into PBS + 0.1% BSA. ChIPmentation was performed as described by Schmidl et al. (2015). Sonication was performed with a Covaris E220 using empirically defined setting (Peak incident power 105, duty factor 2%, cycles per burst 200, duration 840 s). ChIP-seq grade H3K4me1 and H3K27ac antibodies were purchased from Diagenode. Library amplification was performed using barcoded primers as described by Buenrostro et al. (2013). After library amplification, fragments were size selected with SPRI beads with ratios of 0.65x and 0.9x for right- and left-sided selection. Sequencing was performed on a HiSeq2500.
Colony formation assays
For CFU-assays in which we assessed colony morphology in the presence or absence of IL-1β, we plated 150 freshly sorted HSCs in1.5 mL of MethoCult GF M3434 media, supplemented with Penicillin-Streptomycin and IL-1β at the indicated concentrations. Colonies were scored 12 days later by an observer blinded to treatment group and genotype.
For assays in which HSCs were cultured ex vivo culture followed by colony formation assays (Figure 6C), 100 HSCs from wild-type or Kmt2cΔ/Δmice were sorted directly into 96-well plates and were grown in StemSpan SFEM (Stem Cell Technologies) supplemented with Penicillin-Streptomycin (GIBCO), recombinant TPO (100 ng/ml; Peprotech), recombinant SCF (100 ng/ml; Peprotech), and IL-1β at the indicated concentrations. The culture cells were counted after 7 days. We then transferred 1% of the culture volume to MethoCult GF M3434 and scored colonies as described above.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical comparisons
Group sizes and statistical tests are indicated in the text, with the exception of the bioinformatic analyses that are described below. In all figures, error bars indicate standard deviations. Sample sizes and numbers of replicates are indicated in the figure legends. In all cases in which multiple comparisons were made, a Holm-Sidak posthoc test was used to correct for multiple comparisons. CFU as-says were performed blinded as described above. All other comparisons were unblinded.
RNA-seq analysis
Sequences were aligned to the mouse genome (Ensembl release 76 top-level assembly) using STAR version 2.0.4b (Dobin et al., 2013). Linear modeling (limma/voom) was used to compare gene expression across samples (Ritchie et al., 2015). False discovery rates were calculated using the Benjamini and Hochberg method. Pathway analysis was performed with Generally Applicable Gene-set Enrichment (Luo et al., 2009) or Gene Set Enrichment Analysis (Subramanian et al., 2005) or Reactome (Joshi-Tope et al., 2005). Heatmaps for visualizing RNA-seq data were generated with the Spotfire Omics package. RNA-seq data can be accessed from Gene Expression Omnibus (GSE158160).
ATAC-seq and ChIPmentation analyses
ATAC-seq reads were demultiplexed and mapped to mm10 using bowtie2. Peaks were identified and ChIP-seq coverage tracks were generated with MACS2 using the ATAC-seq and DNase-seq processing pipeline developed by the Kundaje lab (https://github.com/kundajelab/atac_dnase_pipelines, Version 0.3.3) (Kundaje et al., 2015). Consistency among replicates was assessed based on Irreproducible Discovery Rates (Li et al., 2011). Differential binding peaks were identified with the R package DiffBind (FDR < 0.1). Signal tracks were visualized with the WashU Epigenome browser (Zhou et al., 2013).
ChIPmentation Reads were demultiplexed and mapped to mm10. Signal enrichment was normalized to the corresponding input samples that were sequenced on each independent run. Peaks were identified and ChIP-seq coverage tracks were generated with MACS2 using the AQUAS TF and histone ChIP-seq pipeline with the “histone” option (https://github.com/kundajelab/chipseq_pipeline, version 0.3.3) (Kundaje et al., 2015). Differential binding peaks were identified with the R package DiffBind (FDR < 0.1). Signal tracks were visualized with the WashU Epigenome browser (Zhou et al., 2013). HOMER was used to identify transcription factor binding sites that were enriched within MLL3-regulated enhancers (Heinz et al., 2010).
ATAC-seq and Chipmentation data can be accessed from Gene Expression Omnibus (GSE158158 and GSE158159, respectively).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | DENTIFIER |
|---|---|---|
| Antibodies | ||
| CD150-PE | Biolegend | 115904; RRID: AB_313683 |
| CD48-APC | Biolegend | 103412; RRID: AB_571997 |
| Sca-1-PercpCy5.5 | Biolegend | 108124; RRID: AB_893615 |
| CD201(EPCR)-APC | Biolegend | 141505; RRID: AB_ 2561361 |
| CD48-BV711 | Biolegend | 103439; RRID: AB_2650824 |
| CD2-FITC | Biolegend | 100105; RRID: AB_312652 |
| CD3-FITC | Biolegend | 100204; RRID: AB_312661 |
| CD8a-FITC | Biolegend | 100706; RRID: AB_312745 |
| B220-FITC | Biolegend | 103206; RRID: AB_312991 |
| Gr-1-FITC | Biolegend | 108406; RRID: AB_313371 |
| Ter119-FITC | Biolegend | 116206; RRID: AB_313707 |
| CD2-APC | Biolegend | 100111; RRID: AB_2563089 |
| CD3-APC | Biolegend | 100235; RRID: AB_2561455 |
| CD8a-APC | Biolegend | 100711; RRID: AB_312750 |
| B220-APC | Biolegend | 103211; RRID: AB_312996 |
| Gr-1-APC | Biolegend | 108411; RRID: AB_313376 |
| Ter119-APC | Biolegend | 116211; RRID: AB_313712 |
| CD2-PE-Cy7 | Biolegend | 300221; RRID: AB_2572065 |
| CD3-PE-Cy7 | Biolegend | 100219; RRID: AB_1732068 |
| CD8a-PE-Cy7 | Biolegend | 100721; RRID: AB_312760 |
| B220-PE-Cy7 | Biolegend | 103221; RRID: AB_313004 |
| Gr-1-PE-Cy7 | Biolegend | 108415; RRID: AB_313380 |
| Ter119-PE-Cy7 | Biolegend | 116223; RRID: AB_2137788 |
| CD48-PE-Cy7 | Biolegend | 103424; RRID: AB_2075049 |
| CD16/32-BV711 | Biolegend | 101337; RRID: AB_2565637 |
| CD105-APC | Biolegend | 120414; RRID: AB_2277914 |
| CD45.1-APC-Cy7 | Biolegend | 110716; RRID: AB_313505 |
| CD45.2-FITC | Biolegend | 109806; RRID: AB_313443 |
| CD45.2-AF700 | Biolegend | 109822; RRID: AB_493731 |
| CD11b-APC | Biolegend | 101212; RRID: AB_312795 |
| Gr-1-PE-Cy7 | Biolegend | 108416; RRID: AB_313381 |
| B220-PercpCy5.5 | Biolegend | 103236; RRID: AB_893354 |
| CD41-AF700 | Biolegend | 133925; RRID: AB_2572129 |
| CD3-PE | Biolegend | 100206; RRID: AB_312663 |
| CD135-Biotin | Biolegend | 135307; RRID: AB_1953266 |
| CD117-APC-Cy7 | Biolegend | 105826; RRID: AB_1626278 |
| H2A.X Phospho(Ser139) -AF647 | Biolegend | 613407; RRID: AB_2114994 |
| CD117-Biotin | Biolegend | 135804; RRID: AB_313213 |
| CD117-PE-Cy7 | Biolegend | 105814; RRID: AB_313223 |
| CD244.2-AF647 | Biolegend | 133509; RRID: AB_2072854 |
| CD229-Biotin | Biolegend | 122903; RRID: AB_830724 |
| CD41-AF700 | Biolegend | 133925;RRID: AB_2572129 |
| CD34-FITC | Thermo Fisher | 16-0341-85; RRID: AB_468937 |
| H3K4me1 | Diagenode | C15410194; RRID: AB_2637078 |
| H3K27ac | Diagenode | C15410196; RRID: AB_2637079 |
| Anti-MLL3 | Sigma-Aldrich | ABE1851 |
| Anti-NUP98 | Cell Signaling | 2288;RRID: AB_561204 |
| P-JNK | Cell Signaling | 4688; RRID: AB_823588 |
| P-p38 | Cell Signaling | 4511; RRID: AB_2139682 |
| P-p65 | Cell Signaling | 3033;RRID: AB_331284 |
| JNK | Cell Signaling | 9252; RRID: AB_2250373 |
| α-Tubulin | Cell Signaling | 2144;RRID: AB_2210548 |
| Chemicals, peptides, and recombinant proteins | ||
| Streptavidin-PE-Cy7 | Biolegend | 405206 |
| Streptavidin-APC-Cy7 | Biolegend | 405208 |
| Steptavidin-BV711 | Biolegend | 405241 |
| Mojosort Streptavidin Nanobeads | Biolegend | 76447 |
| Tamoxifen | Cayman Chemical | 13258 |
| Cyclophosphamide for injection (500mg/vial) | Sandoz | |
| 5-Fluorouracil | Sigma-Aldrich | F6627–5G |
| Recombinant Murine IL-1β | Peprotech | 211–11B |
| Recombinant Murine TNF-α | Peprotech | 315–01A |
| Bromo-deoxyuridine | Sigma-Aldrich | B5002 |
| Recombinant murine TPO | Peprotech | 315–14 |
| Recombinant murine SCF | Peprotech | 250–03 |
| Iscove’s Modified Dulbecco’s Medium | Thermo Fisher Scientific | 31980030 |
| MethoCult™ GF M3434 | Stem cell technologies | 03434 |
| DirectPCR lysis reagent (Mouse Tail) | Viagen | 101-T |
| StemSpan™ SFEM | Stem cell technologies | 09650 |
| 2’,7’-Dichlorofiuorescin diacetate | Sigma-Aldrich | D6883–50MG |
| Polyinosinic-polycytidylic acid sodium salt | Sigma-Aldrich | P1530 |
| NuPAGE™ LDS Sample Buffer (4X) | Thermo Fisher Scientific | NP0007 |
| SuperSignal™ West Femto Maximum sensitivity Substrate | Thermo Fisher Scientific | 34095 |
| Critical commercial assays | ||
| APC BrdU Flow Kit | BD | 51–9000019AK |
| RNeasy Plus Micro Kit | QIAGEN | 74034 |
| MinElute Reaction Cleanup Kit | QIAGEN | 28204 |
| Illumina Tagment DNA Enzyme and Buffer (Small Kit) | Illumina | 20034210 |
| Sera-Mag Speedbeads | Fisher | 09-981-123 |
| NEBNext High-Fidelity 2X PCR Master Mix | NEB | M0541S |
| Coomassie Plus (Bradford) Assay Kit | Thermo | 23236 |
| Ampure XP SPRI Beads | Beckman | B23318 |
| NuPage™ 4 to 12% Bis-Tris Gel (10-well) | Thermo Fisher Scientific | NP0335PK2 |
| Deposited data | ||
| ATAC-seq | This paper | GEO: GSE158158 |
| RNA-seq | This paper | GEO: GSE158160 |
| ChIPmentation | This paper | GEO: GSE158159 |
| Series entry (encompassing all datasets) | This paper | GEO: GSE158162 |
| Experimental models: organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:006965 |
| Mouse: B6;129S4-Gt(ROSA)26Sortm1(rtTA*M2)JaeCol1a1tm7(tetO-HIST1H2BJ/GFP)Jae/j | The Jackson Laboratory | RRID:IMSR_JAX:016836 |
| Mouse: C57BL/6N-Fgd5tm3(cre/ERT2)Djr/J | The Jackson Laboratory | RRID:IMSR_JAX:027789 |
| Mouse: B6.Cg-Commd10TgVav1-icre)A2Ko/J | The Jackson Laboratory | RRID:IMSR_JAX:008610 |
| Mouse: B6.C-Tg(CMV-cre)1Cgn/J | The Jackson Laboratory | RRID:IMSR_JAX:006054 |
| Mouse: Kmt2c-null | This paper | N/A |
| Mouse: Kmt2c-fiox | This paper | N/A |
| Oligonucleotides | ||
| Primer for the exon 14 germline Kmt2c mutation genotyping: Forward: GTGACTTAATACGACTC ACTATAGGGCCAACAACAT ACATCTCAGTC | This paper | N/A |
| Primer for the exon 14 germline Kmt2c mutation genotyping: Reverse: TGTATTGCAGGGAAAGT AAATGTT | This paper | N/A |
| Primer for the exon 3 conditional fioxed allele genotyping: Forward: GAACCTGAGGCTGT CGAAGG | This paper | N/A |
| Primer for the exon 3 conditional fioxed allele genotyping: Reverse: AGCCATTAAACTTAGTGAAGACCA | This paper | N/A |
| Primer for the exon 3 conditional to identify deletec allele: Forward: GCTCCTCCTCTGGCTTCCA AGGTCA | This paper | N/A |
| Primer for the exon 3 conditional to identify deleted allele: Reverse: AGGGCATCTAATCTGACAGCTGTAAGC | This paper | N/A |
| ATAC-seq and ChIPmentation primers | Buenrostro et al., 2013 | N/A |
| Software and algorithms | ||
| STAR version 2.0.4b | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| GAGE (Generally Applicable Gene-set Enrichment) | (Luo et al., 2009) | https://bioconductor.org/packages/release/bioc/html/gage.html |
| Reactome | (Joshi-Tope et al., 2005) | https://reactome.org |
| limma/voom | (Ritchie et al., 2015) | https://www.bioconductor.org/packages/release/bioc/html/limma.html |
| ATAC-seq and DNase-seq processing pipeline, v0.3.3 | (Kundaje et al., 2015) | https://github.com/kundajelab/atac_dnase_pipelines |
| AQUAS TF and histone ChIP-seq pipeline, v0.3.3 | (Kundaje et al., 2015) | https://github.com/kundajelab/chipseq_pipeline |
| Bowtie2 | Bowtie, v2.2.26 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| DiffBind | DiffBind | http://www.bioconductor.org/packages/2.13/bioc/html/DiffBind.html |
| Irreproducible Discovery Rate (IDR) | (Li et al., 2011) | https://www.encodeproject.org/software/idr |
| MACS2 | MACS2, V2.1.0 | https://github.com/macs3-project/MACS |
| HOMER | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/motif |
| BD FACSDiva | BD | N/A |
| Flowjo v.10 | Treestar | N/A |
| GraphPad Prism v.8 | GraphPad Software | N/A |
| WashU Epigenome browser | (Zhou et al., 2013) | https://epigenomegateway.wustl.edu |
Highlights.
Kmt2c deletions enhance HSC self-renewal capacity
Haploid Kmt2c deletions convey a selective advantage to HSCs after chemotherapy
MLL3 promotes HSC differentiation and enhancer priming in response to IL-1
MLL3 activates enhancer elements in multiply-divided HSCs
ACKNOWLEDGMENTS
This work was supported by grants to J.A.M. from the NHLBI (R01 HL152180 and R01 HL136504), Alex’s Lemonade Stand Foundation (‘A’ Award), Gabrielle’s Angel Foundation, The V Foundation, the American Society of Hematology, Hyundai Hope on Wheels, and the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital. We thank the Genome Engineering and iPSC Center and the Department of Pathology Micro-injection Core at Washington University for assistance in generating mouse lines. We thank Todd Druley for helpful discussions.
Footnotes
Supplemental information
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108751.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Arcipowski KM, Bulic M, Gurbuxani S, and Licht JD (2016). Loss of Mll3 Catalytic Function Promotes Aberrant Myelopoiesis. PLoS ONE 11, e0162515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashokkumar D, Zhang Q, Much C, Bledau AS, Naumann R, Alexopoulou D, Dahl A, Goveas N, Fu J, Anastassiadis K, et al. (2020). MLL4 is required after implantation, whereas MLL3 becomes essential during late gestation. Development 147, dev186999. [DOI] [PubMed] [Google Scholar]
- Beerman I, Bock C, Garrison BS, Smith ZD, Gu H, Meissner A, and Rossi DJ (2013). Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 12, 413–425. [DOI] [PubMed] [Google Scholar]
- Bernitz JM, Kim HS, MacArthur B, Sieburg H, and Moore K (2016). Hematopoietic Stem Cells Count and Remember Self-Renewal Divisions. Cell 167, 1296–1309.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigelius-Flohé R, and Maiorino M (2013). Glutathione peroxidases. Biochim. Biophys. Acta 1830, 3289–3303. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol 109, 21.29.1–21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujanover N, Goldstein O, Greenshpan Y, Turgeman H, Klainberger A, Scharff Y, and Gazit R (2018). Identification of immune-activated hematopoietic stem cells. Leukemia 32, 2016–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, et al. (2011). Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet 44, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Liu Y, Rappaport AR, Kitzing T, Schultz N, Zhao Z, Shroff AS, Dickins RA, Vakoc CR, Bradner JE, et al. (2014). MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 25, 652–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorighi KM, Swigut T, Henriques T, Bhanu NV, Scruggs BS, Nady N, Still CD 2nd, Garcia BA, Adelman K, and Wysocka J (2017). Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation. Mol. Cell 66, 568–576.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foudi A, Hochedlinger K, Van Buren D, Schindler JW, Jaenisch R, Carey V, and Hock H (2009). Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat. Biotechnol 27, 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazit R, Mandal PK, Ebina W, Ben-Zvi A, Nombela-Arrieta C, Silberstein LE, and Rossi DJ (2014). Fgd5 identifies hematopoietic stem cells in the murine bone marrow. J. Exp. Med 211, 1315–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz HM, Mohan M, Garruss AS, Liang K, Takahashi YH, Mickey K, Voets O, Verrijzer CP, and Shilatifard A (2012). Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev. 26, 2604–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinge A, He J, Bartram J, Javier J, Xu J, Fjellman E, Sesaki H, Li T, Yu J, Wunderlich M, et al. (2020). Asymmetrically Segregated Mitochondria Provide Cellular Memory of Hematopoietic Stem Cell Replicative History and Drive HSC Attrition. Cell Stem Cell 26, 420–430.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Gao X, Morgan MA, Herz HM, Smith ER, and Shilatifard A (2013). The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol. Cell. Biol 33, 4745–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi-Tope G, Gillespie M, Vastrik I, D’Eustachio P, Schmidt E, de Bono B, Jassal B, Gopinath GR, Wu GR, Matthews L, et al. (2005). Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. 33, D428–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozwik KM, Chernukhin I, Serandour AA, Nagarajan S, and Carroll JS (2016). FOXA1 Directs H3K4 Monomethylation at Enhancers via Recruitment of the Methyltransferase MLL3. Cell Rep. 17, 2715–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. ; Roadmap Epigenomics Consortium (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai B, Lee JE, Jang Y, Wang L, Peng W, and Ge K (2017). MLL3/MLL4 are required for CBP/p300 binding on enhancers and super-enhancer formation in brown adipogenesis. Nucleic Acids Res. 45, 6388–6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Saha PK, Yang QH, Lee S, Park JY, Suh Y, Lee SK, Chan L, Roeder RG, and Lee JW (2008). Targeted inactivation of MLL3 histone H3-Lys-4 methyltransferase activity in the mouse reveals vital roles for MLL3 in adipogenesis. Proc. Natl. Acad. Sci. USA 105, 19229–19234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kim DH, Lee S, Yang QH, Lee DK, Lee SK, Roeder RG, and Lee JW (2009). A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc. Natl. Acad. Sci. USA 106, 8513–8518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Wang C, Xu S, Cho YW, Wang L, Feng X, Baldridge A, Sartorelli V, Zhuang L, Peng W, and Ge K (2013). H3K4 mono- and dimethyltransferase MLL4 is required for enhancer activation during cell differentiation. eLife 2, e01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brown JB, Huang H, and Bickel PJ (2011). Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat 5, 1752–1779. [Google Scholar]
- Liang R, Arif T, Kalmykova S, Kasianov A, Lin M, Menon V, Qiu J, Bernitz JM, Moore K, Lin F, et al. (2020). Restraining Lysosomal Activity Preserves Hematopoietic Stem Cell Quiescence and Potency. Cell Stem Cell 26, 359–376.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Friedman MS, Shedden K, Hankenson KD, and Woolf PJ (2009). GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNerney ME, Godley LA, and Le Beau MM (2017). Therapy-related myeloid neoplasms: when genetics and environment collide. Nat. Rev. Cancer 17, 513–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, et al. (2011). Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20, 11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ, Wright DE, and Weissman IL (1997). Cyclophosphamide/granulocyte colony-stimulating factor induces hematopoietic stem cells to proliferate prior to mobilization. Proc. Natl. Acad. Sci. USA 94, 1908–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto T, Walker CS, Choi K, Hueneman K, Smith MA, Gul Z, Garcia-Manero G, Ma A, Zheng Y, and Starczynowski DT (2020). Adaptive response to inflammation contributes to sustained myelopoiesis and confers a competitive advantage in myelodysplastic syndrome HSCs. Nat. Immunol 21, 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada D, Saunders TL, and Morrison SJ (2010). Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature 468, 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro H, Ding L, and Morrison SJ (2013). SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell 13, 102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliai C, and Schiller G (2020). How to address second and therapy-related acute myelogenous leukaemia. Br. J. Haematol 188, 116–128. [DOI] [PubMed] [Google Scholar]
- Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP, Techner JM, Will B, et al. (2016). Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol 18, 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk CJ, Rossi DJ, Månsson R, Attema JL, Norddahl GL, Chan CK, Sigvardsson M, Weissman IL, and Bryder D (2007). Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell Stem Cell 1, 428–442. [DOI] [PubMed] [Google Scholar]
- Qiu J, Papatsenko D, Niu X, Schaniel C, and Moore K (2014). Divisional history and hematopoietic stem cell function during homeostasis. Stem Cell Reports 2, 473–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabe JL, Hernandez G, Chavez JS, Mills TS, Nerlov C, and Pietras EM (2020). CD34 and EPCR coordinately enrich functional murine hematopoietic stem cells under normal and inflammatory conditions. Exp. Hematol 81, 1–15.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall TD, and Weissman IL (1997). Phenotypic and functional changes induced at the clonal level in hematopoietic stem cells after 5-fluorouracil treatment. Blood 89, 3596–3606. [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos MA, Faryabi RB, Ergen AV, Day AM, Malhowski A, Canela A, Onozawa M, Lee JE, Callen E, Gutierrez-Martinez P, et al. (2014). DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature 514, 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidl C, Rendeiro AF, Sheffield NC, and Bock C (2015). ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nat. Methods 12, 963–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A (2012). The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem 81, 65–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Le Beau MM, Huo D, Karrison T, Sobecks RM, Anastasi J, Vardiman JW, Rowley JD, and Larson RA (2003). Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood 102, 43–52. [DOI] [PubMed] [Google Scholar]
- Steensma DP, and Ebert BL (2020). Clonal hematopoiesis as a model for premalignant changes during aging. Exp. Hematol 83, 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Zhou B, Mao F, Xu J, Miao H, Zou Z, Phuc Khoa LT, Jang Y, Cai S, Witkin M, et al. (2018). HOXA9 Reprograms the Enhancer Landscape to Promote Leukemogenesis. Cancer Cell 34, 643–658.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemoto T, Hashimoto M, Matsumura T, Nakamura-Ishizu A, and Suda T (2018). Ca2+-mitochondria axis drives cell division in hematopoietic stem cells. J. Exp. Med 215, 2097–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, Moehrle B, Brocks D, Bayindir I, Kaschutnig P, et al. (2015). Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 520, 549–552. [DOI] [PubMed] [Google Scholar]
- Wang C, Lee JE, Lai B, Macfarlan TS, Xu S, Zhuang L, Liu C, Peng W, and Ge K (2016). Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition. Proc. Natl. Acad. Sci. USA 113, 11871–11876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SP, Tang Z, Chen CW, Shimada M, Koche RP, Wang LH, Nakadai T, Chramiec A, Krivtsov AV, Armstrong SA, and Roeder RG (2017). A UTX-MLL4-p300 Transcriptional Regulatory Network Coordinately Shapes Active Enhancer Landscapes for Eliciting Transcription. Mol. Cell 67, 308–321.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhao Z, Ozark PA, Fantini D, Marshall SA, Rendleman EJ, Cozzolino KA, Louis N, He X, Morgan MA, et al. (2018). Resetting the epigenetic balance of Polycomb and COMPASS function at enhancers for cancer therapy. Nat. Med 24, 758–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will B, Zhou L, Vogler TO, Ben-Neriah S, Schinke C, Tamari R, Yu Y, Bhagat TD, Bhattacharyya S, Barreyro L, et al. (2012). Stem and progenitor cells in myelodysplastic syndromes show aberrant stage-specific expansion and harbor genetic and epigenetic alterations. Blood 120, 2076–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, et al. (2008). Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135, 1118–1129. [DOI] [PubMed] [Google Scholar]
- Wilson NK, Schoenfelder S, Hannah R, Sánchez Castillo M, Schütte J, Ladopoulos V, Mitchelmore J, Goode DK, Calero-Nieto FJ, Moignard V, et al. (2016). Integrated genome-scale analysis of the transcriptional regulatory landscape in a blood stem/progenitor cell model. Blood 127, e12–e23. [DOI] [PubMed] [Google Scholar]
- Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, Lamprecht TL, Shen D, Hundal J, Fulton RS, et al. (2015). Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 518, 552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Chen SA, Local A, Liu T, Qiu Y, Dorighi KM, Preissl S, Rivera CM, Wang C, Ye Z, et al. (2018). Histone H3 lysine 4 monomethylation modulates long-range chromatin interactions at enhancers. Cell Res. 28, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZW, Zhang J, Townsend DM, and Tew KD (2015). Oxidative stress, redox regulation and diseases of cellular differentiation. Biochim. Biophys. Acta 1850, 1607–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoboue ED, Rimessi A, Anelli T, Pinton P, and Sitia R (2017). Regulation of Calcium Fluxes by GPX8, a Type-II Transmembrane Peroxidase Enriched at the Mitochondria-Associated Endoplasmic Reticulum Membrane. Antioxid. Redox Signal 27, 583–595. [DOI] [PubMed] [Google Scholar]
- Young AL, Challen GA, Birmann BM, and Druley TE (2016). Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun 7, 12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Lowdon RF, Li D, Lawson HA, Madden PA, Costello JF, and Wang T (2013). Exploring long-range genome interactions using the WashU Epigenome Browser. Nat. Methods 10, 375–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available at Gene Expression Omnibus under the SuperSeries accession GSE158162. SubSeries accession numbers are GSE158158 (ATAC-Seq), GSE158159 (ChIPmentation) and GSE158160 (RNA-seq). No new code was generated for this study.