

Abstract
IMPORTANCE
The patients evaluated in this study, to our knowledge, represent the first complete clinical description of a family with an autosomal dominant inheritance pattern of retinal dystrophy associated with a novel mutation in RAX2.
OBJECTIVES
To clinically evaluate 4 patients and 5 unaffected family members, characterize the disease phenotype over time, and identify the associated genetic mutation.
DESIGN, SETTING, AND PARTICIPANTS
A prospective, longitudinal, observational, case-series analysis of 9 members of an affected family at the Casey Eye Institute, Oregon Health and Science University, Portland. The dates of the study were from July 31, 1992, to August 11, 2014.
INTERVENTIONS
Clinical evaluations included eye examination, color fundus photography, autofluorescence imaging, spectral-domain optical coherence tomography, kinetic visual field testing, and electroretinography. Genetic mutation screening was performed with next-generation sequencing, and identified mutations were confirmed with Sanger sequencing.
MAIN OUTCOMES AND MEASURES
Clinical diagnosis and longitudinal characterization of retinal dystrophy and identification of genetic mutation.
RESULTS
Six members of the family were identified as having retinal dystrophy (4 were examined, and 3 were genetically tested). Five unaffected family members were clinically evaluated (2 were genetically tested). The age at onset of retinal dystrophy was variable. All affected individuals presented with declining visual acuity, central scotomas, waxy disc pallor, attenuated vasculature, small yellow macular deposits and/or macular pigment mottling, and abnormal electroretinograms demonstrating mixed cone and rod dysfunction and a scotopic electronegative response to bright flashes. There were no other causes of an electronegative electroretinogram identified in any of the affected patients. Genetic testing revealed, to our knowledge, a novel frameshift heterozygous mutation in RAX2 in the patients with retinal dystrophy.
CONCLUSIONS AND RELEVANCE
A frameshift heterozygous mutation in RAX2 inherited in an autosomal dominant fashion was associated with mixed cone and rod dysfunction. Among the patients, there was variability in the age at onset and in the specific pattern of photoreceptor dysfunction, but the clinical course was nevertheless slowly progressive. Screening for RAX2 mutation could provide prognostic value for patients and families with scotopic electronegative responses to bright flashes.
Progressive cone and cone-rod dystrophies are a group of disorders with heterogeneous clinical presentations and genotypes that may be inherited in an autosomal dominant, autosomal recessive, or X-linked pattern.1 These disorders generally manifest in childhood or early adulthood with the initial loss of visual acuity and the central visual field, photophobia, difficulty with color discrimination, and cone or cone-rod dysfunction detected by electroretinography. Subsequently, in advanced disease, rod dysfunction becomes more apparent when an individual loses his or her peripheral visual field and experiences difficulty with night vision. There are more than 20 genes associated with nonsyndromic cone or cone-rod dystrophy, including more common genes such as PRPH2, ABCA4, and RPGR. However, much less is known regarding other rare genes, and there is a need for ongoing genotype-phenotype correlations to better understand these disorders and to help guide molecular testing, as well as future therapies. One such poorly-understood gene, RAX2, has previously been associated with cone-rod dystrophy.2 In the present study, we characterize the clinical features of 4 affected members of a family with retinal dystrophy due to an autosomal dominant novel mutation in RAX2.
Methods
Participants
For each participant, a complete medical history was recorded, and a complete ocular examination was performed. All participants underwent full-field electroretinography (ffERG). Our study followed the tenets of the Declaration of Helsinki and was approved by the institutional review board at Oregon Health and Science University in Portland. Written informed consent was obtained from all participants. The institutional review board at Oregon Health and Science University approved the study protocol. The dates of the study were from July 31, 1992, to August 11, 2014.
Full-Field and Multifocal ERG
Full-field ERGs were obtained using Burian-Allen electrodes (Hansen Ophthalmic Development Laboratory) according to the International Society for Clinical Electrophysiology of Vision guidelines3 and as previously described.4 All 30-Hz flicker responses were filtered with a discrete Fourier transform for a fundamental frequency.5 Multifocal ERGs were also obtained using Burian-Allen bipolar contact lens electrodes, and multifocal ERG was performed according to the International Society for Clinical Electrophysiology of Vision guidelines6 using the Veris system (Electro-Diagnostic Imaging) with a protocol that used 103 hexagons, a bandpass of 10 to 300 Hz, and a testing time of 8 minutes divided into 60-second segments.
Additional Testing
Those participants with an abnormal ERG underwent additional testing that included color fundus photography, fundus autofluorescence, spectral-domain optical coherence tomography (SD-OCT; Heidelberg Engineering), and kinetic visual field testing (Goldmann perimetry or Octopus 101/900; Haag-Streit, Inc). To normalize the visual field data from different perimeters for comparison, computerized quantitative digitization of kinetic fields was performed using a computer program as previously described.7
Molecular Analysis
Genetic mutation screening was performed by the Casey Eye Institute Molecular Diagnostics Laboratory, Oregon Health and Science University. The patient’s DNA was isolated from whole blood, amplified by polymerase chain reaction, and used for next-generation sequencing to screen for 26 cone/cone-rod dystrophy mutations as follows: ABCA4, ADAM9, AIPL1, BEST1, C8orf37, CACNA1F, CACNA2D4, CDHR1, CERKL, CNGB3, CNNM4, CRX, GUCA1A, GUCY2D, KCNV2, PDE6C, PDE6H, PITPNM3, PROM1, PRPH2/RDS, RAX2, RDH5, RIMS1, RPGRIP1, SEMA4A, and UNC119. Thereafter, Sanger sequencing was used to confirm the identified mutations.
Results
Clinical Features
At the onset of our study, 6 affected individuals across 4 generations in 1 family were identified, and only 4 were living. The pedigree was consistent with an autosomal dominant pattern of inheritance (Figure 1). A total of 9 family members were seen in the ophthalmic genetics clinic at the Casey Eye Institute, where it was determined that 5 family members were normal and that 4 family members were found to have a retinal dystrophy with an electronegative scotopic waveform response to bright flashes (Figure 2; eFigure 1 in the Supplement). The characteristics of the affected patients are summarized.
Figure 1. Pedigree Showing 4 Generations of a Family With an Autosomal Dominant Inheritance Pattern of Retinal Dystrophy Associated With a Novel RAX2 Mutation.
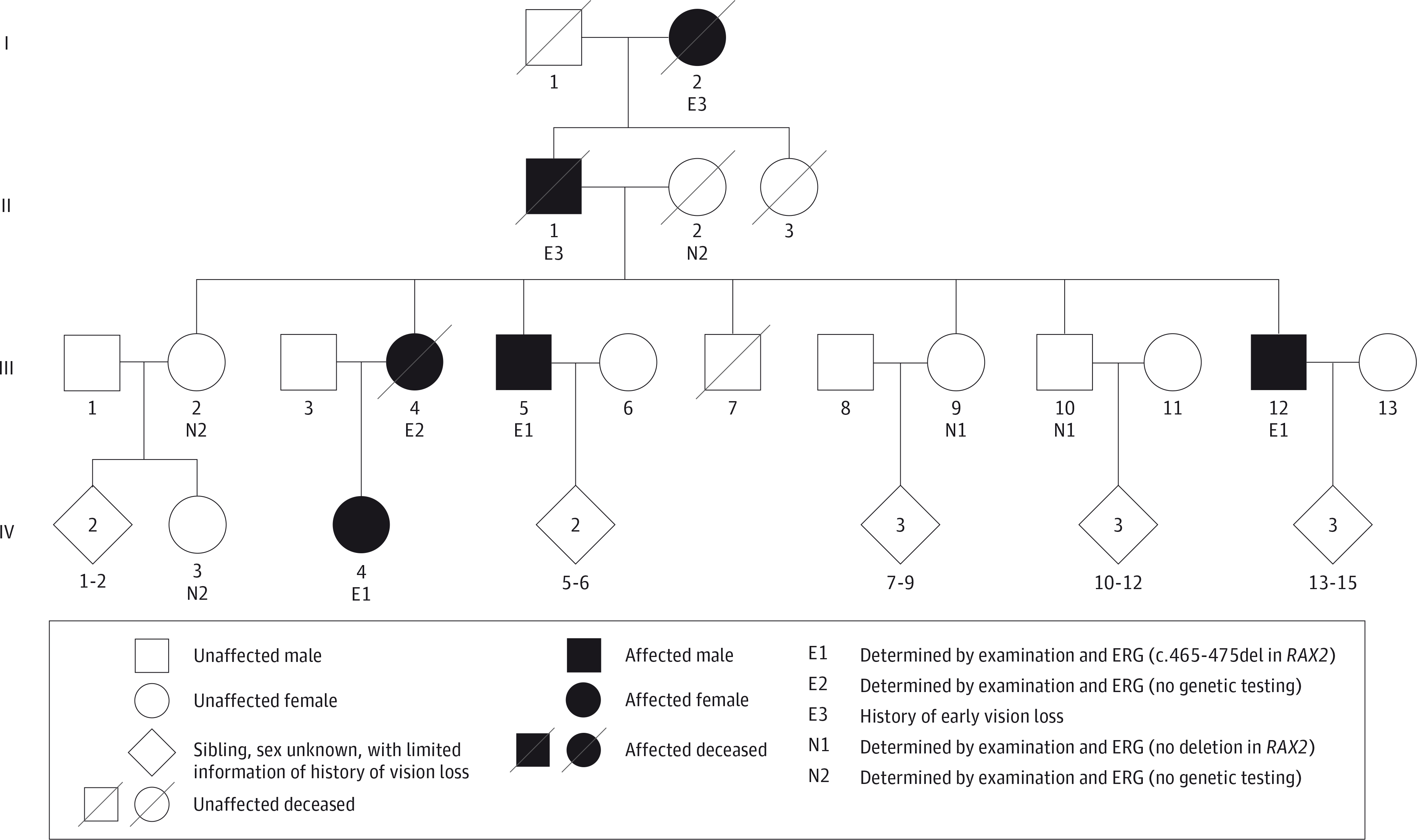
ERG indicates electroretinography.
Figure 2. Full-Field Electroretinograms of the Patients and an Unaffected Family Member.
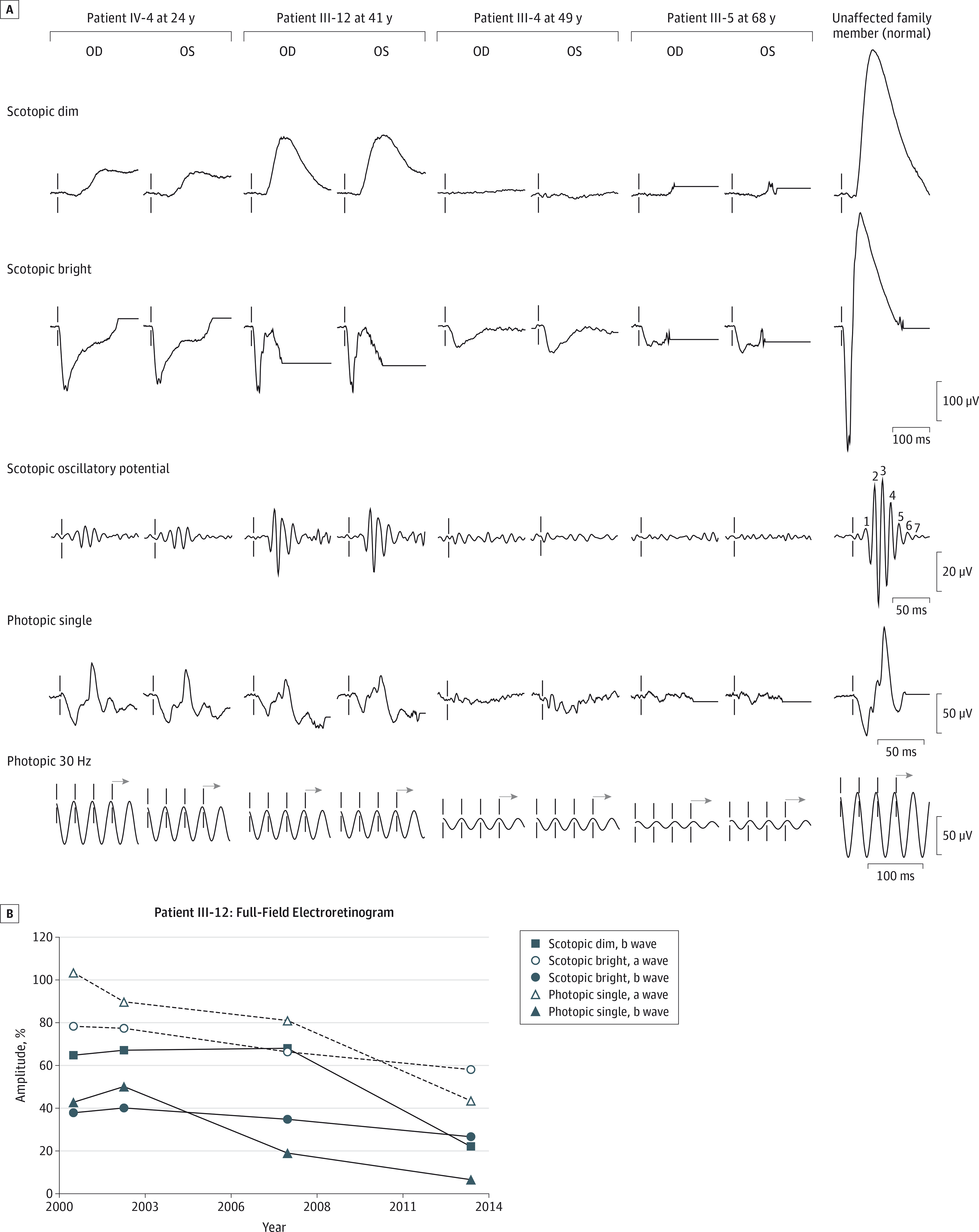
Amplitudes were the average of the right- and left-eye responses for all measurements and are presented as a percentage of the lower limit of age-matched normal response.
Patient IV-4
Patient IV-4 is the affected daughter of affected patient III-4. She began to experience a slow progressive decline of her central vision at 15 years of age, and by 16 years of age, her visual acuity was 20/60 OD and 20/70 OS. Fundus photography revealed attenuated vessels; a few small, yellow, deep macular deposits; and fine granularity of the periphery (Figure 3). Intact peripheral isopters to all test targets with relative central scotoma to I4e, I3e, and I2e test targets were seen on kinetic visual fields (Figure 3). The ffERG showed rod-cone dysfunction with an electronegative scotopic waveform response to bright flashes and a normal photopic 30-Hz flicker. At 20 to 21 years of age, her visual acuity and fundus appearance remained unchanged, and there were no significant changes in her kinetic visual field. The ffERG showed a worsening delay of the rod-dependent response, and the oscillatory potentials were more attenuated. At 24 years of age, her visual acuity was stable, but there was development of a waxy disc pallor and a bull’s eye maculopathy with worsening of vascular attenuation and progressive constriction of the I3e isopter on her kinetic visual field (Figure 3). The ffERG still showed a similar rod-cone dysfunction with an electronegative waveform, but with attenuated photopic 30-Hz flicker responses (Figure 2A; Table).
Figure 3. Longitudinal Color Fundus Photographs and Digitized Kinetic Visual Fields of the Patients.
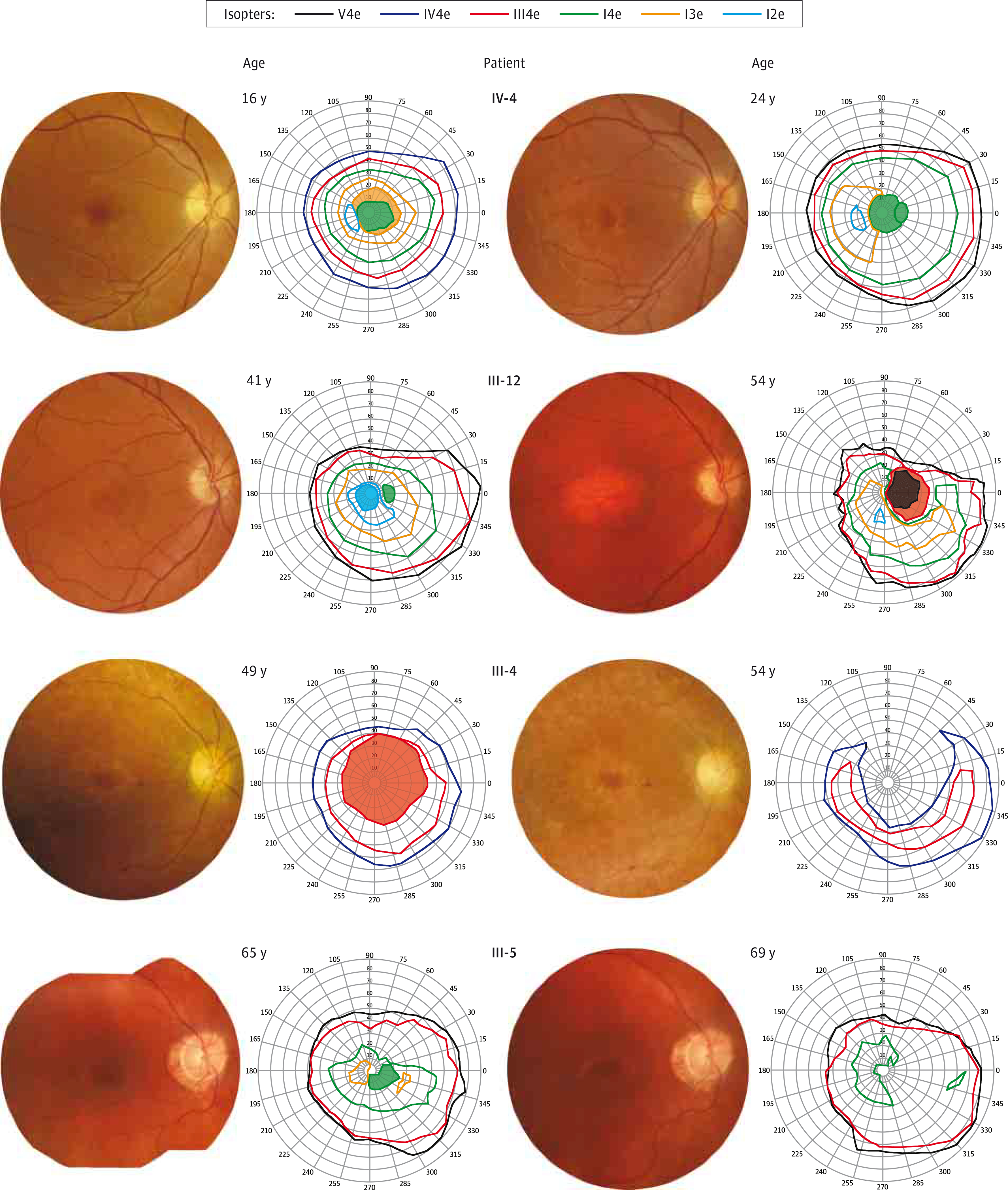
The unit in which visual fields are presented is degrees.
Table.
Amplitudes and Implicit Times for Full-Field ERGs of 4 Affected Patients
| ERG Stimuli | Patient IV-4 at 24 y | Patient III-12 at 41 y | Patient III-4 at 49 y | Patient III-5 at 68 y | ||||
|---|---|---|---|---|---|---|---|---|
| Amplitude,a μV | Implicit Time, ms | Amplitude,a μV | Implicit Time, ms | Amplitude,a μV | Implicit Time, ms | Amplitude,a μV | Implicit Time, ms | |
| Scotopic dim | ||||||||
| b Wave | 65 (24%)b | 124c | 145 (64%)b | 88 | 9 (4%)b | 129c | 9 (6%)b | Blinkd |
| Scotopic bright | ||||||||
| a Wave | 153b | 25c | 166b | 22c | 62b | 40c | 59b | 30c |
| b Wave | 97 (20%)b | ENW | 160 (38%)b | ENW | 49 (12%)b | ENW | 17 (5%)b | ENW |
| b- to a-Wave amplitude ratio | 0.6 | 1.0 | 0.8 | 0.3 | ||||
| Scotopic OPe | 25b | 69b | 23b | 6b | ||||
| Photopic single flash | ||||||||
| a Wave | 36 | 16c | 30 | 15 | 14b | 16c | 10b | 19c |
| b Wave | 72 (54%)b | 34.4c | 38 (42%)b | 34.2c | 15 (19%)b | 38.1c | 12 (17%)b | 35.4c |
| b- to a-Wave amplitude ratio | 2.0 | 1.3 | 1.1 | 1.3 | ||||
| Photopic 30-Hz flicker | 58b | 29.0 | 45b | 27.6 | 26b | 32.7c | 11b | 37.8c |
| Pattern of dysfunction | Rod-cone dysfunction | Cone-rod dysfunction | Rod-cone dysfunction | Rod and cone dysfunction | ||||
Abbreviations: ENW, electronegative waveform; ERG, electroretinogram; OP, oscillatory potential.
Amplitudes were the average of the right- and left-eye responses for all measurements and are also presented in parentheses as a percentage of the lower limit of age-matched normal response.
Amplitude attenuated.
Timing prolonged.
Waveform truncated by blink artifact.
The OP wavelet index is the arithmetic sum of the 4 largest OP peaks (2–5).
Patient III-12
Patient III-12 is the youngest affected brother of affected patients III-4 and III-5. He began to experience sensitivity to light, difficulty with night vision, and decreased pericentral and peripheral vision at 40 years of age, and at 41 years of age, his visual acuity was 20/20 OD and 20/400 OS (his amblyopic eye). Fundus photography revealed waxy disc pallor, attenuated vessels, macular pigment mottling, and granular pigment changes inferiorly below the arcades (Figure 3). Mild superior constriction of peripheral isopters to all test targets with relative central scotoma to I4e and I2e test targets was seen on kinetic visual fields (Figure 3). The ffERG showed cone-rod dysfunction with an electronegative scotopic waveform response to bright flashes (Figure 2A; Table). He continued to experience photophobia and decline in central vision at 49 years of age, and by 54 years of age, his visual acuity was 20/150 OD and count fingers in the left eye. Fundus photography revealed progressive macular atrophy (Figure 3), as well as peripheral atrophy and reticular pigment changes of the inferior and nasal retinal areas that were seen as mottled hyperautofluorescence and hypoautofluorescence on wide-field fundus autofluorescence images (eFigure 2 in the Supplement). In addition, there was macular hypoautofluorescence that correlated with atrophy of the retinal pigment epithelium (RPE) on SD-OCT scans and perimacular hyperautofluorescence (Figure 4A). Spectral-domain OCT of the macula also revealed subfoveal focal hyperreflective deposits and severe diffuse attenuation of the outer retina. These retinal changes were directly correlated with the enlargement and deepening of the central scotomas and with progressive superotemporal constriction of peripheral isopters on kinetic visual fields to all test targets by 54 years of age (Figure 3). Full-field ERG between 41 and 54 years of age revealed that cone function declined continuously, while the rod function was stable for the first 6 years before declining rapidly over the last 6 years (Figure 2B). The last ffERG at 54 years of age still showed an electronegative waveform and severe progression of cone-rod dysfunction, with cone function that was barely recordable.
Figure 4. Multimodal Imaging of Patients III-12 and III-5.
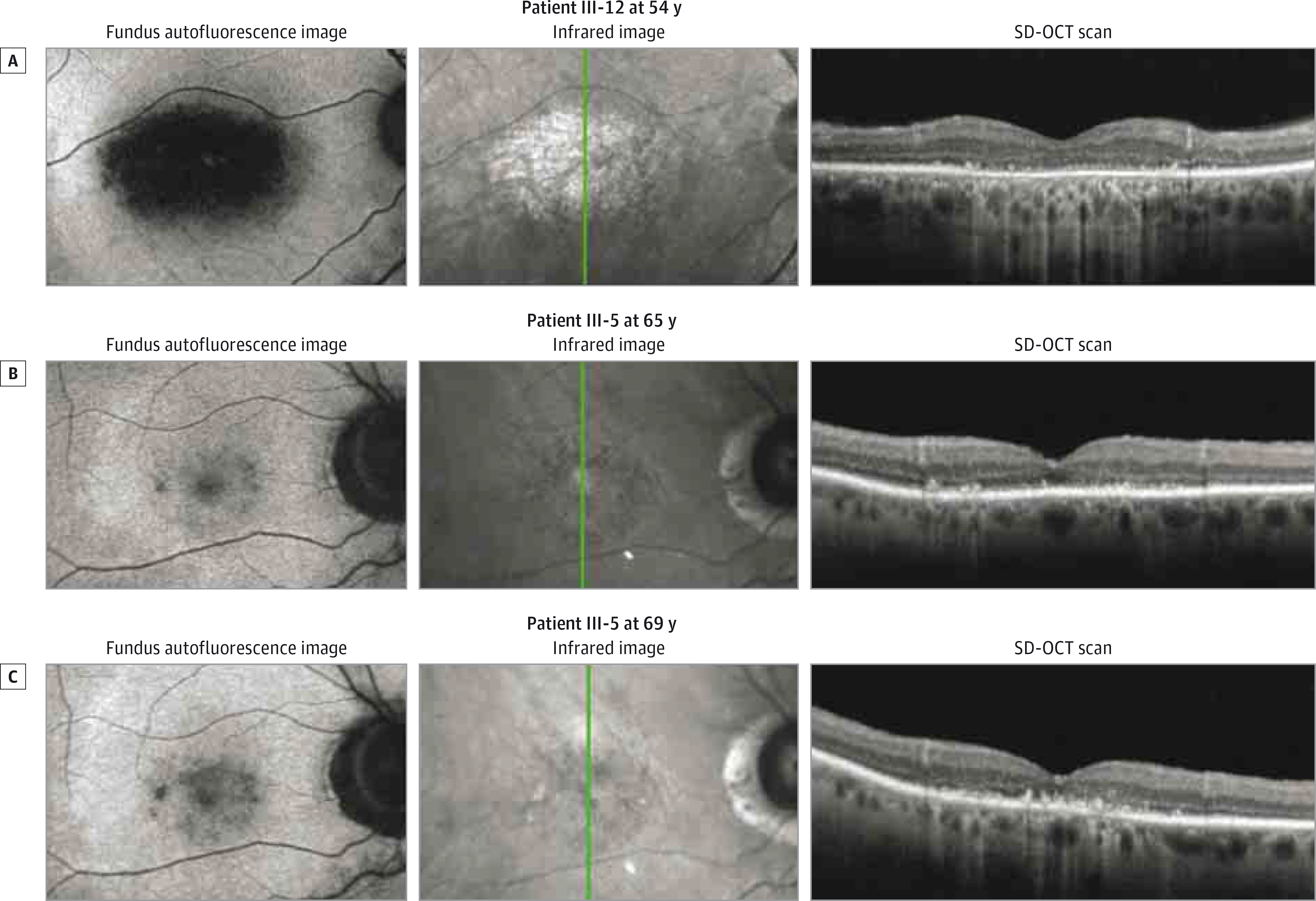
The green lines indicate where the spectral-domain optical coherence tomographic (SD-OCT) scans were taken.
Patient III-4
Patient III-4 is the affected mother of affected patient IV-4. She began to experience slow progressive loss of her central vision and difficulty with color discrimination at 19 years of age and was legally blind by 33 years of age. At 49 years of age, her visual acuity was 20/400 in both eyes. Fundus photography revealed waxy disc pallor, attenuated vessels with some scattered perivascular pigment accumulation, fine yellow deposits with pigment granularity and atrophy of the macula, and pigment granularity and atrophy of the peripheral retina, which correlated with kinetic visual fields that showed large relative central scotoma to the III4e test target but intact peripheral isopters (Figure 3). The ffERG showed severe rod-cone dysfunction with an electronegative scotopic waveform response to bright flashes and an undetectable rod-system response (Figure 2A; Table). At 54 years of age, her visual acuity was counting fingers eccentrically in both eyes. Fundus photography revealed worsening of disc pallor, vascular attenuation, macular atrophy, and peripheral atrophy with bone spicule pigmentation, while the kinetic visual field showed the enlargement and deepening of the large central scotoma continuous with loss of the peripheral isopters in the superior fields to all test targets (Figure 3).
Patient III-5
Patient III-5 is the brother of affected patients III-4 and III-12. He began to experience slow progressive loss of vision at 60 years of age and difficulty with night vision at 64 years of age. At 65 years of age, his visual acuity was 20/80 OD and 20/40 OS. Fundus photography revealed disc pallor, peripapillary atrophy, attenuated vessels, macular pigment mottling with few small yellow deposits, and pigment granularity of the peripheral retina that correlated with hyperautofluorescence and hypoautofluorescence on fundus autofluorescence images (Figure 3; eFigure 2 in the Supplement). In addition, there was macular hypoautofluorescence and perimacular hyperautofluorescence. Spectral-domain OCT of the macula showed severe attenuation of the outer retina with a residual foveal ellipsoid layer and a few small areas of RPE atrophy (Figure 4B). Kinetic visual field testing revealed relative eccentric scotoma to I4e and I3e test targets, moderate constriction of the I4e isopter, and intact peripheral isopters to V4e and III4e test targets (Figure 3). The ffERG revealed severe dysfunction of both cone and rod-system responses, with an electronegative scotopic waveform response to bright flashes. Multifocal ERG revealed severe attenuation and prolongation of responses that were barely detectable, with relatively worse attenuation centrally in both eyes (eFigure 3 in the Supplement). At 68 years of age, his visual acuity was 20/400 OD and 20/250 OS. There were worsened areas of pigment mottling in the macula. Another ffERG continued to show severe dysfunction of both cone and rod-system responses, with an electronegative scotopic waveform response to bright stimuli (Figure 2A; Table). At 69 years of age, he continued to complain of worsening vision, and his visual acuity was 20/200 eccentrically in both eyes. Fundus photography showed more pigment mottling of the macula, which correlated with worse hypoautofluorescent mottling of the macula and hyperautofluorescence of the perimacula (Figure 3; eFigure 2 in the Supplement). Spectral-domain OCT of the macula revealed worse attenuation of the outer retina and a foveal ellipsoid layer with increased accumulation of subfoveal hyperreflective deposits (Figure 4C). Kinetic visual field testing revealed progressive enlargement of the eccentric scotoma (Figure 3).
Genetic Features
At the time of genetic testing, 2 unaffected and 3 affected family members consented, but the other affected family member, patient III-4, could not be tested because she had received a diagnosis of ovarian cancer at 52 years of age and died of metastatic complications at 54 years of age. The patients IV-4, III-5, and III-12 all had the same novel frameshift heterozygous mutation in RAX2, c.465_475delCGCAGATGGCT. The unaffected family members III-9 and III-10 did not possess the mutation in RAX2.
Discussion
The purpose of our study was to identify the mutation in a family with autosomal dominant retinal dystrophy and to characterize their clinical features. There was some variability in the phenotype, but all patients had obvious macular changes and central scotoma on visual fields, which were consistent with a cone or cone-rod dystrophy. Although only 1 patient actually had a cone-rod pattern of dysfunction on an ffERG, all patients had an electronegative scotopic response to bright stimuli, indicating the possibility of an electronegative cone-rod dystrophy due to mutations in the cone-rod homeobox gene (CRX),8–11 GUCY2D,12 or PRPH2.13 However, there were no mutations found in these genes on molecular testing. Instead, we show here, for the first time to our knowledge, an association of RAX2 with autosomal dominant retinal dystrophy and an electronegative ERG.
The RAX2 gene (previously known as Q50-type retinal homeobox [QRX] and retina and anterior neural fold homeobox-like 1 [RAXL1]) (OMIM 610362) has a cytogenetic location of 19p13.3, is composed of 3 exons, and has a homeobox domain that shares a sequence homologous to one in the CRX gene that is involved in nuclear localization.2 In addition, the RAX2 protein physically interacts with the CRX protein synergistically to recruit other components of the basal transcription machinery to activate transcription in the outer and inner nuclear layers of the retina. The novel frameshift mutation in RAX2 that was identified in our study, c.465_475delCGCAGATGGCT, spares the homeobox domain but affects the majority of the third exon and alters a large portion of the N-terminal coding region. Although binding affinity assays were beyond the scope of our study, there is a high likelihood that such an alteration to the RAX2 protein may affect its affinity for the CRX protein and thus perturb transcriptional regulation. Thus, it is logical that any disturbance in retinal gene transactivation activity due to mutations in RAX2, a synergistic cofactor of CRX, could cause a cone-rod clinical phenotype with both outer retinal attenuation and electronegative waveforms. Only 2 unrelated cases of cone-rod dystrophy (CORD11) (OMIM 610381) and 1 case with macular degeneration (ARMD6) (OMIM 613757) have been previously described to be associated with mutations in the RAX2 gene.2 The 3 cases were minimally characterized clinically; thus, to our knowledge, the present study represents the first complete description of the clinical phenotype associated with mutations in RAX2.
Although there was variability in the age at onset and in the chief complaints, all affected patients in our study presented with small yellow macular deposits and/or macular pigment mottling, but also with signs of diffuse dystrophy such as optic disc pallor and attenuated retinal vessels. The macular yellow deposits correlated with hyperreflective deposits at the level of the outer retina detected on SD-OCT scans. Macular atrophy occurred later in the older patients and correlated with hypoautofluorescence, RPE thinning and outer retinal attenuation detected on SD-OCT scans, central scotoma on the visual field, and diffuse dysfunction of the cone system on ffERGs and multifocal ERGs. Perimacular hyperautofluorescent rings were observed, which is indicative of transition zones that are commonly found in retinal degenerations.14 In addition, autofluorescent abnormalities in the mid- and far-periphery indicate that the rod photoreceptors were also affected.
Indeed, ffERG testing showed variable patterns of both cone and rod-system dysfunction among the patients. The mixed cone and rod scotopic responses to bright flashes were attenuated and prolonged for both the a and b waves, but the b wave was predominantly affected, producing electronegative waveforms and b-wave to a-wave amplitude ratios of less than 1.0 in all patients. Thus, the ERG results showed both inner-retinal and photoreceptor dysfunction, as predicted by the fact that the RAX2 protein is preferentially expressed in both the inner and outer nuclear layers.2 The photopic responses to single flashes were attenuated and prolonged for the b waves in all patients, although the a waves were only attenuated at older ages. Thus, for the later time points in the older patients, there was a correlation between cone-photoreceptor dysfunction on ffERGs and macular outer-retinal attenuation on SD-OCT scans. Longitudinal ffERG data, as illustrated for patient III-12, further illustrates the complex course of this disease, with inner-retinal dysfunction being predominant during the early phase of the disease, while both rod-system and cone-photoreceptor function deteriorate more quickly during the late phase of the disease.
Despite the variability of the presenting symptoms and patterns of dysfunction on ffERGs, all patients presented with a relative central scotoma on kinetic visual fields that correlated with the macular changes seen on multimodal imaging. Over time, the central scotomas worsened as macular atrophy developed. In the later time points of the older patients, mid- and far-peripheral scotomas developed in correlation to RPE changes and atrophy of the mid- and far-peripheral retina, respectively. Interestingly, the spectrum and variability of the clinical phenotype of inner-retinal and photoreceptor dysfunction as assessed by imaging, ERG, and visual fields in our study have also been reported among case series8–11,15–17 of cone-rod dystrophy associated with mutations in CRX, which further supports a synergistic relationship between RAX2 and CRX and a mutual deficit of activity in mutant RAX2 and CRX proteins.
Conclusions
In conclusion, there are multiple reasons that argue in favor of the pathogenicity of the novel RAX2 missense mutation found in our study. The RAX2 mutation segregates with the disease phenotype, but not with the unaffected family members. In addition, the missense mutation affects the majority of the third exon and a large portion of the N-terminal coding region. Thus, the translated mutant RAX2 protein is likely to have reduced transactivation as a synergistic cofactor of the CRX protein, which may explain some of the phenotypic similarities between the cone dystrophies associated with mutations in CRX and those described in our study. Finally, the patients evaluated in our study represent, to our knowledge, the first complete clinical description of a family with an autosomal dominant retinal dystrophy associated with mutations in RAX2.
Supplementary Material
Acknowledgments
Funding/Support: This work was supported by grants CD-NMT-0714–0648-OHSU (Dr Yang) and CD-CL-0808–0469-OHSU (Dr Pennesi) from the Foundation Fighting Blindness, grant 1K08 EY0231186–01 from the National Institutes of Health (Dr Pennesi), Research to Prevent Blindness (Dr Pennesi), core grant P30EY010572 and unrestricted grant C-CL-0711–0534-OHSU01 from the Foundation Fighting Blindness (Casey Eye Institute), and an unrestricted grant from Research to Prevent Blindness (Casey Eye Institute).
Role of the Funder/Sponsor: The funding organizations had no role in the design and conduct of the study; collection, management, analysis, or interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Conflict of Interest Disclosures: All authors have completed and submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest, and Dr Weleber is a consultant to Novartis, Pfizer, and Wellstat; is a member of the scientific advisory board for Applied Genetic Technologies Corp; and serves on the scientific advisory board for the Foundation Fighting Blindness (the relationship has been reviewed and managed by Oregon Health and Science University). Dr Weleber also reports having received grants and personal fees from the Foundation Fighting Blindness and Applied Genetic Technologies Corp, and other support from Sanofi-Fovea, all outside the submitted work. In addition, Dr Weleber has a patent (US patent 8,657,446, Method and apparatus for visual field monitoring, also known as Visual Field Monitoring and Analysis, or VFMA, which has not been issued). Dr Pennesi is on the advisory board for Sucampo Pharmaceuticals and received travel support from Imagine Eyes. Dr Pennesi also reports having received personal fees from Sucampo Pharmaceuticals outside the submitted work. No other disclosures are reported.
REFERENCES
- 1.Michaelides M, Hardcastle AJ, Hunt DM, Moore AT. Progressive cone and cone-rod dystrophies: phenotypes and underlying molecular genetic basis. Surv Ophthalmol. 2006;51(3): 232–258. [DOI] [PubMed] [Google Scholar]
- 2.Wang QL, Chen S, Esumi N, et al. QRX, a novel homeobox gene, modulates photoreceptor gene expression. Hum Mol Genet. 2004;13(10): 1025–1040. [DOI] [PubMed] [Google Scholar]
- 3.Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach M; International Society for Clinical Electrophysiology of Vision. ISCEV Standard for full-field clinical electroretinography (2008 update). Doc Ophthalmol. 2009;118(1):69–77. [DOI] [PubMed] [Google Scholar]
- 4.Oh KT, Weleber RG, Stone EM, Oh DM, Rosenow J, Billingslea AM. Electroretinographic findings in patients with Stargardt disease and fundus flavimaculatus. Retina. 2004;24(6): 920–928. [DOI] [PubMed] [Google Scholar]
- 5.van der Tweel LH, Estevez O. Analytical techniques. In: Heckenlively J, Arden GB, eds. Principles and Practice of Clinical Electrophysiology of Vision. 2nd ed. Cambridge, MA: MIT Press; 2006: 439–459. [Google Scholar]
- 6.Hood DC, Bach M, Brigell M, et al. ; International Society For Clinical Electrophysiology of Vision. ISCEV standard for clinical multifocal electroretinography (mfERG) (2011 edition). Doc Ophthalmol. 2012;124(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weleber RG, Tobler WR. Computerized quantitative analysis of kinetic visual fields. Am J Ophthalmol. 1986;101(4):461–468. [DOI] [PubMed] [Google Scholar]
- 8.Lines MA, Hébert M, McTaggart KE, Flynn SJ, Tennant MT, MacDonald IM. Electrophysiologic and phenotypic features of an autosomal cone-rod dystrophy caused by a novel CRX mutation. Ophthalmology. 2002;109(10):1862–1870. [DOI] [PubMed] [Google Scholar]
- 9.Swain PK, Chen S, Wang QL, et al. Mutations in the cone-rod homeobox gene are associated with the cone-rod dystrophy photoreceptor degeneration. Neuron. 1997;19(6):1329–1336. [DOI] [PubMed] [Google Scholar]
- 10.Itabashi T, Wada Y, Sato H, Kawamura M, Shiono T, Tamai M. Novel 615delC mutation in the CRX gene in a Japanese family with cone-rod dystrophy. Am J Ophthalmol. 2004;138(5): 876–877. [DOI] [PubMed] [Google Scholar]
- 11.Kitiratschky VB, Nagy D, Zabel T, et al. Cone and cone-rod dystrophy segregating in the same pedigree due to the same novel CRX gene mutation. Br J Ophthalmol. 2008;92(8): 1086–1091. [DOI] [PubMed] [Google Scholar]
- 12.Gregory-Evans K, Kelsell RE, Gregory-Evans CY, et al. Autosomal dominant cone-rod retinal dystrophy (CORD6) from heterozygous mutation of GUCY2D, which encodes retinal guanylate cyclase. Ophthalmology. 2000;107(1):55–61. [DOI] [PubMed] [Google Scholar]
- 13.Ba-Abbad R, Robson AG, Yap YC, Moore AT, Webster AR, Holder GE. Prph2 mutations as a cause of electronegative ERG. Retina. 2014;34(6): 1235–1243. [DOI] [PubMed] [Google Scholar]
- 14.Oishi M, Oishi A, Ogino K, et al. Wide-field fundus autofluorescence abnormalities and visual function in patients with cone and cone-rod dystrophies. Invest Ophthalmol Vis Sci. 2014;55(6): 3572–3577. [DOI] [PubMed] [Google Scholar]
- 15.Jacobson SG, Cideciyan AV, Huang Y, et al. Retinal degenerations with truncation mutations in the cone-rod homeobox (CRX) gene. Invest Ophthalmol Vis Sci. 1998;39(12):2417–2426. [PubMed] [Google Scholar]
- 16.Itabashi T, Wada Y, Sato H, Kunikata H, Kawamura M, Tamai M. Ocular findings in a Japanese family with an Arg41Trp mutation of the CRX gene. Graefes Arch Klin Exp Ophthalmol. 2003; 241(7):535–540. [DOI] [PubMed] [Google Scholar]
- 17.Paunescu K, Preising MN, Janke B, Wissinger B, Lorenz B. Genotype-phenotype correlation in a German family with a novel complex CRX mutation extending the open reading frame. Ophthalmology 2007;114(7):1348–1357. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.