

Abstract
Background
Digitaria exilis, white fonio, is a minor but vital crop of West Africa that is valued for its resilience in hot, dry, and low-fertility environments and for the exceptional quality of its grain for human nutrition. Its success is hindered, however, by a low degree of plant breeding and improvement.
Findings
We sequenced the fonio genome with long-read SMRT-cell technology, yielding a ∼761 Mb assembly in 3,329 contigs (N50, 1.73 Mb; L50, 126). The assembly approaches a high level of completion, with a BUSCO score of >99%. The fonio genome was found to be a tetraploid, with most of the genome retained as homoeologous duplications that differ overall by ∼4.3%, neglecting indels. The 2 genomes within fonio were found to have begun their independent divergence ∼3.1 million years ago. The repeat content (>49%) is fairly standard for a grass genome of this size, but the ratio of Gypsy to Copia long terminal repeat retrotransposons (∼6.7) was found to be exceptionally high. Several genes related to future improvement of the crop were identified including shattering, plant height, and grain size. Analysis of fonio population genetics, primarily in Mali, indicated that the crop has extensive genetic diversity that is largely partitioned across a north-south gradient coinciding with the Sahel and Sudan grassland domains.
Conclusions
We provide a high-quality assembly, annotation, and diversity analysis for a vital African crop. The availability of this information should empower future research into further domestication and improvement of fonio.
Keywords: domestication, gene amplification, gene loss, millet, polyploidy
Data Description
Background
White fonio (Digitaria exilis, NCBI:txid1010633) is a vital cereal crop of West Africa, where it is commonly known as fonio or acha. A related Digitaria species, black fonio (Digitaria iburura), is a very minor crop, mostly of Nigeria, Benin, and Togo. Fonio (D. exilis) has an exceptionally small but very nutritious grain, with both high protein and high dietary fiber content [1–3]. Fonio can mature in as little as 8 weeks after planting and is commonly grown without fertilizer or irrigation on poor-quality soils in dry regions of the Sudan grasslands and Sahel. Although yields are low, the West African crop is harvested in early summer, where it fills a vital dietary gap before the maturation of sorghum or pearl millet crops in the same region. Perhaps no other crop deserves the title “orphan” more, because research attention on fonio has been minimal [4].
Wild D. exilis (sometimes called “hungry rice”) and other West African Digitaria have been harvested by farmers in times of famine throughout recorded history [4], but very little improvement has been made to the domesticated crop, at least partly evidenced by the fact that no controlled cross between fonio varieties has been substantiated. Fonio was probably domesticated in West Africa, presumably before the arrival of pearl millet or sorghum from Central and East Africa [5], as is suggested by the importance of fonio in Dogon and other creation myths [4]. Applying the term “domesticated” to fonio cultivars is, however, something of a stretch. Fonio cultivars do not exhibit the full set of domestication traits, in that they exhibit the shattering (grain release at maturity) and day-length dependence traits that have been selected against by early farmers across virtually all cereal crops [6, 7]. The selected mutations to non-shattering and day-length independence are routinely recessive, so the absence of these agricultural improvements may be an outcome of the polyploid nature of the fonio genome [8]. As an orphan crop, fonio has received very little research attention. Over the past 20 years, for instance, only 9 refereed publications report any new investigation of any aspect of fonio biology, although an additional ≥30 publications in that period investigated fonio agronomy, cultural significance, or nutritional properties [9, 10]. In 2007, Adoukonou-Sagbadja and colleagues [11] published a DNA marker-based analysis of fonio genetic diversity, and there are some transcript sequence data at NCBI [12]. Beyond this, most fonio investigations have been conducted in West Africa to determine appropriate conditions for subsistence farmers to grow and/or process the grain from local landraces. In contrast, several other orphan cereal crops of Africa and Asia have begun to receive extensive attention, including comprehensive analyses of germplasm resources, even to the extent of full genome sequence analysis. Three of these cereals with relatively deep recent analyses are, like fonio, panicoid grasses: foxtail millet (Setaria italica), pearl millet (Cenchrus americanus), and proso millet (Panicum miliaceum) [13–15]. With these panicoid grass resources, and a comparative genomics strategy [16], it should be possible to rapidly elevate fonio research to benefit fonio consumers and producers. This article describes our genomic sequence analysis of the fonio landrace Niatia, and a genetic comparison of fonio germplasms from across West Africa.
Plant material and nucleic acid preparation
Fonio millet (cv. Niatia) seed was obtained from Dr. Sara Patterson (University of Wisconsin, Madison, WI, USA), which was collected in Mali at GPS coordinates 3.9861 W, 17.5739 N. Niatia is a popular local variety in Mali [17] (see Genetic Diversity for Nagoya protocol and germplasm access). The seeds were multiplied in a University of Georgia greenhouse. Seeds collected from a single plant were used for all DNA isolation. The seeds were surface sterilized with 8% sodium hypochlorite (Bioworld, Visalia, CA, USA) for 10 min, followed by 3 rinses with sterile distilled water. The plants were grown in standard potting soil (Fafard® 4M Sungro Professional Growing Mix, Sungro Horticulture, McClellan Park, CA, USA) in a greenhouse (with 14 h daylight and day/night temperatures of 26/20°C). They were watered daily to ∼70% soil water-holding capacity. The leaves of 4-week-old plants were used for DNA isolation, using a previously described protocol [18]. Briefly, leaf tissue (2.5 g) was ground in liquid nitrogen. After lysing in 15 mL of 2X extraction buffer (100 mM Tris–HCl pH 8.0, 1.4 M NaCl, 20 mM EDTA, 2% w/v CTAB with 10 μL/mL β-mercaptoethanol) and extraction with chloroform/isoamyl alcohol twice, the aqueous phase was then transferred to 3–3.5 volumes of precipitation buffer (50 mM Tris–HCl pH 8.0, 10 mM EDTA, 1% w/v CTAB). The sample was incubated overnight at room temperature to precipitate the DNA. After centrifugation at 3,500 rpm for 15 min, the DNA pellet was washed with ddH2O and centrifuged for 10 min. Then, 5 mL of 1.5 M NaCl and 6 μL of 10 mg/mL RNaseA was added to the pellet and incubated at 37°C until completely resuspended. A chloroform extraction was performed as above to remove RNaseA and any additional contaminants. The aqueous phase was collected and DNA was precipitated and washed with ethanol. The pellet was then resuspended in 100 μL ddH2O.
PacBio SMRT sequencing, sequence polishing, and genome assembly
DNA samples were used to construct a PacBio (Pacific Biosciences, Menlo Park, CA, USA) SMRT sequencing library according to manufacturer recommendations at the University of California at Davis Genome Center. Fragments >10 kb were selected for sequencing via BluePippen (Sage Science, LLC, Beverly, MA, USA). A total of 88 Gb of raw PacBio reads from 76 SMRT cells were passed through the secondary analysis pipeline in SMRT Link (v6.0 [19]) and filtered for read quality >0.75 and length >1 kb. The resultant 75 Gb of filtered reads were assembled in Canu v1.8 (Canu, RRID:SCR_015880) [20] with the default settings for raw PacBio reads.
Racon (Racon, RRID:SCR_017642) was used to polish the original assembly for 2 rounds with the Canu-corrected PacBio reads. Sequentially, Arrow (VariantCaller v2.3.3) and Pilon v1.23 (Pilon, RRID:SCR_014731) were used to further polish the assembly with 36 Gb of Illumina paired-end reads obtained on the HiSeq 4000 (Illumina HiSeq 3000/HiSeq 4000 System, RRID:SCR_016386) at the Georgia Genomics and Bioinformatics Core at the University of Georgia.
The final assembly (Niatia v1.0) has a total length of 760.66 Mb and 3,329 contigs, with N50 of 1.73 Mb (L50 of 126) and N90 of 75.85 kb (L90 of 889). The longest contig is 10.17 Mb and the shortest contig is 1,013 bp, with a mean of 228.5 kb. We compare the quality of our genome with that of CM05836 [21], which was assembled using short reads, linked reads, and Hi-C. Although scaffold size is larger for the aforementioned genome, our genome has much better contiguity than CM05836 [21] as seen by N50 (1,734 vs 78 kb) and L50 (8 vs 2,624) (Supplementary Table 1). Scaffolding is expected to be higher in the latter genome because Hi-C technology was used that associates contigs on the same histone protein regardless of their size, but the Niatia genome shows much greater contiguity. To see the high contiguity in our genome assembly in detail, we took 2 of our medium-sized contigs (tig00001331 and tig00010942) as examples showing a dramatic improvement in contiguity in our genome, emphasizing the importance of long reads in assembly and annotation. This is further exemplified by comparing 2 random medium-sized contigs, tig00001331 corresponding to 100 consecutive segments anchored on the same chromosome 3B and tig00010942 corresponding to 65 consecutive segments on the chromosome 5A of the CM05836 [21] genome (Supplementary Fig. 1).
Table 1:
Summary of repeat sequence properties in the genome assembly
| Class | Subclass | Type | No. of families | No. of repeats | Length (Mb) | Percent of genome |
|---|---|---|---|---|---|---|
| Class I TEs, retroelements | LTR-RT | Copia | 353 | 45,194 | 22.8 | 3.0 |
| Gypsy | 1,223 | 125,773 | 153.9 | 20.2 | ||
| Other | 824 | 90,110 | 57.8 | 7.6 | ||
| LINE | I | 17 | 3,040 | 1.5 | 0.2 | |
| SINE | 3,790 | 181,505 | 30.6 | 4.0 | ||
| Class II TEs, DNA transposons | TIR | CACTA | 348 | 42,737 | 7.4 | 1.0 |
| Mutator | 34 | 8,493 | 1.8 | 0.2 | ||
| PIF | 120 | 13,973 | 2.4 | 0.3 | ||
| Tc1 | 896 | 124,252 | 21.5 | 2.8 | ||
| hAT | 93 | 13,097 | 2.5 | 0.3 | ||
| Helitron | Helitron | 313 | 104,271 | 21.6 | 2.8 | |
| Tandem repeats | SSRs | 133,570 | 5.9 | 0.8 | ||
| Unclassified repeats | (Repbase) | 48.0 | 6.3 | |||
| Total | 329.8 | 49.7 |
Figure 1:

Whole-genome duplication and polyploidy analysis. (A) Ks estimation of age distribution for paralogs and orthologs of white fonio (Digitaria) and some close relatives. The distribution in light pink represents the entire white fonio paranome, while the distribution in darker pink represents the anchor points (duplicated genes lying in syntenic or collinear regions; see C). Distributions in black, dark green, and light green represent the 1-vs-1 ortholog comparisons between Digitaria-Setaria, Digitaria-Sorghum, and Sorghum-Setaria, respectively. (B) Ks distributions for paralogs of white fonio, sorghum, and Setaria (zoom in), showing an older, likely Poaceae-shared, WGD. (C) Syntenic relationships between putative homoeologous contigs, with colored lines connecting homoeologous gene pairs in the white fonio genome assembly. (D) Time-calibrated phylogenetic tree of several major Poaceae lineages, including white fonio, based on 1,242 gene families consisting of a single gene copy in each lineage and an anchor pair (A and B) in Digitaria. The time scale is shown in million years (My). See text for details.
Estimation of genome size and heterozygosity
Kmer Analysis Toolkits [22] was used to count k-mers in Illumina raw reads and to compare the results with the k-mers counted from the genome assembly at several different k-mer sizes, from 17 to 30. These all yielded similar results but with a somewhat larger fonio genome predicted at smaller k-mer lengths. The distribution of k-mer counts was modeled and the heterozygosity level was estimated using GenomeScope2.0 [23].
Two distinct peaks were observed in the raw read k-mer distribution. We interpret the peaks at ∼50 and ∼100 counts/coverage as the 2 subgenomes in fonio (Supplementary Fig. S2). Genome size estimated from the peaks was 668–707 Mb, depending on the k-mer size used. This range of values is low compared to previous results from flow cytometry that indicated a genome size range of 830–1,000 Mb for a broad selection of D. exilis germplasm [4]. The underestimate is likely due to polyploidy confounding duplicated genes both within and among subgenomes. Single-nucleotide variation was estimated to be 4.3% when comparing the A and B genomes in this tetraploid, but slightly less than 0.01% heterozygosity was observed within either the A or B genomes, as assayed by k-mer allelic ratios. The k-mer counts in the assembled genome suggest that the peak at 100 counts represents common sequences between the 2 subgenomes, and the k-mers under the peak at 50 counts represent the divergent regions between the 2 subgenomes.
Repeat annotation
Repeated sequences were mined and annotated with a combination of de novo and homology-based methods. First, simple sequence repeats (SSRs) were identified and masked with GMATA [24]. Long terminal repeat–retrotransposons (LTR-RTs) were identified de novo using the bioinformatic tools LTR_FINDER (LTR_Finder, RRID:SCR_015247) [25] and LTRharvest (LTRharvest, RRID:SCR_018970) [26], which use structural criteria to find intact LTR-RTs, followed by LTR_retriever analysis [27] to minimize false-positive results. SINE scan (version 1.1.1) [28] was used to find small interspersed nuclear elements (SINEs), a class of retroelements, and these were confirmed by manual investigation. Long interspersed nuclear elements (LINEs), another class of retroelements, were found with MGEscan-nonLTR (version 2) [29]. Small DNA transposable elements (TEs) were found with MITE Tracker [30], and HelitronScanner [31] was used to identify the DNA transposons called "Helitrons." All of the TEs from the genome assembly were used to generate a fonio-specific TE library, with individual TE families named according to the prevalent current nomenclature system [32]. The fonio TE library was compared to the Repbase [33] multispecies repeat repository to validate annotations and to discover any additional candidate repeats represented in Repbase. Then, the fonio TE library was used to identify both full-length and truncated TE elements by homologous search with RepeatMasker version 4.0.7 (RepeatMasker, RRID:SCR_012954) [34] in the genome assembly. Parameter settings were adopted from the analysis described in a previous publication [35]. The predicted insertion dates of intact LTR-RTs were calculated with LTR_retriever (LTR_retriever, RRID:SCR_017623) [27]. The SSRs and TEs were masked by Ns and a TE annotation file in GFF3 format was generated for subsequent gene annotation. Types and abundances of TEs and other repeats discovered in the fonio genome are presented in Table 1.
Transcriptome assembly, candidate gene annotation, and BUSCO quality assessment
Illumina RNA sequencing data (paired-end 100 bp) of D. exilis were downloaded from the NCBI SRA (accession No. SRX1967865 [12]) from RNA consisting of ∼80% inflorescence and ∼20% leaf tissue. FastQC (FastQC, RRID:SCR_014583) [36] was used to evaluate data quality, and low-quality reads and adapter sequences were removed using Trimmomatic (Trimmomatic, RRID:SCR_011848) [37]. The remaining reads were aligned to the genome assembly using HISAT2 (HISAT2, RRID:SCR_015530) [38]. The spliced alignments were used as input for StringTie [39] and assembled into transcripts. TransDecoder, a companion software package of the Trinity platform [40], was used to predict open reading frames (ORFs).
For gene prediction and genome annotation, we used the Maker-P pipeline [41], in combination with Augustus (Augustus, RRID:SCR_008417) [42], SNAP [43], and GeneMark (GeneMark, RRID:SCR_011930) [44]. Augustus gene models came from the BUSCO (BUSCO, RRID:SCR_015008) [45] dataset identified during the assembly (see below). GeneMark_ES was used to produce ab initio gene predictions. Detailed settings for each round of Maker can be found in the Supplemental Methods. The first round of gene prediction with Maker used the following inputs: the RNAseq assembly described in the previous section, protein fasta sequences from S. bicolor and S. italica [46] as well as the repeat models for Digitaria (described above), and the soft-masked genome assembly. A second round of Maker used as input the genome file, the annotation produced by the previous round, and a SNAP species parameter/hmm file based on the prior annotation. Finally, the third round of Maker was run using the following input: the genome assembly, the annotation produced by round 2, and the GeneMark models. Functional annotation was done using the accessory scripts of Maker as described by Campbell and coworkers [47]. Briefly, a BLAST [48] search against the Swissprot database was used to assign putative functions to the newly annotated gene models, while InterProScan 5 (InterProScan, RRID:SCR_005829) [49] was used to obtain domain information.
Following mapping of RNAseq data with HISAT2, 88% of the RNAseq reads could be well aligned to the genome. Transcripts were assembled with Stringtie and ORFs were predicted with TransDecoder (TransDecoder, RRID:SCR_017647). A total of 58,305 candidate transcripts were obtained, of which 50,389 had predicted ORFs.
Our first round of Maker predicted 60,300 protein-coding genes (based only on RNA evidence and protein evidence from sorghum and Setaria). After the second and third round, where Augustus, SNAP, and Genemark-ES models were included, the number of predicted protein-coding genes increased to 67,921 and finally to 68,302. We removed 447 candidate genes that were judged to be spurious because they were fragments of otherwise fully assembled genes in the annotation, so the final number of genes annotated as protein-coding genes is 67,855. The statistics for the gene annotation can be found in Supplementary Table S2. In total, 88.3% of the gene models were supported by RNAseq data. The annotation edit distance (AED) measurements indicate how well an annotation agrees with overlapping evidence (protein, messenger RNA, or expressed sequence tag data). In the fonio assembly, >90% of the gene models have an AED score <0.4%, indicating that gene models are well supported by evidence. The number of genes and gene model lengths are greater than that reported by Abrouk et al. [21] for CM05836 (59,844), indicating the importance of long-read assemblies and contiguity in genome assembly and annotation.
BUSCO v 4.0.2 [45, 50] analysis of the filtered predicted protein sequences against the reference set for plants, on the gVolante platform [51], showed that 98.1% of the BUSCO genes were found as complete genes, while this representation number increased to 99.3% if partially covered BUSCO genes were added compared to the 97.2 reported by Abrouk et al. [21]. A total of 11.6% of the BUSCO genes were single copy, while 86.5% of the BUSCO genes were found in duplicate. Approximately 1.2% of the BUSCO genes were fragmented and ∼0.7% were missing.
A total of 4,741 non-coding RNAs (see Supplementary Table S3) were predicted with Infernal [52] by comparing the genome fasta file with the RFAM CM database, version 14.2 [53], using the protocol described by Kalvari et al. [54]. Most of these non-coding RNAs were found to be transfer RNAs (31.2%), 5S ribosomal RNAs (12.2%), and small nucleolar RNAs (23.4%), as seen in other plant genomes.
Phylogenetic divergence and dating the most recent whole-genome duplication
The coding DNA sequences and annotations for S. bicolor and S. italica were downloaded from the PLAZA database [46]. Ks distribution analyses were performed using the wgd package (v1.1) [47]. For each species, the paranome (entire collection of duplicated genes) was obtained with “wgd mcl” using all-against-all BlastP [47] and MCL clustering [55]. Ks distributions were then constructed using “wgd ksd” with default settings (using MAFFT for multiple sequence alignment [56], codeml for maximum likelihood estimation of pairwise synonymous distances [57], and FastTree [FastTree, RRID:SCR_015501] [58] for inferring phylogenetic trees used in the node-weighting procedure). Anchors or anchor pairs (duplicates lying in collinear or syntenic regions of the genome) were obtained using i-ADHoRe [59] using the default settings in “wgd syn.”
We obtained gene families for a set of 9 species in the Poaceae family using OrthoFinder (OrthoFinder, RRID:SCR_017118) with default settings [60]. All sequence data were obtained from PLAZA [46]. From this set of gene families, we identified all gene families that were single-copy in all species but duplicated in D. exilis, and where the D. exilis duplicates were anchor pairs (1,967 gene families). For these gene families, we performed pre-alignment homology filtering using PREQUAL [61] and multiple sequence alignment of the masked amino acid sequences using MAFFT (MAFFT, RRID:SCR_011811) [56]. For each multiple sequence alignment, we obtained the corresponding codon-level nucleotide alignment. For each obtained nucleotide alignment, we sampled tree topologies from the posterior using MrBayes v3.2 (MrBayes, RRID:SCR_012067) [62] under the GTR model with a discrete Gamma mixture for relative substitution rates across sites (using 4 classes), sampling every 10 iterations, for a total of 250,000 iterations. We then identified all gene families for which the expected species tree topology had posterior probability >0.9, resulting in a set of 1,242 gene families. A concatenated codon alignment was obtained for these families, which was in 3 partitions corresponding to each codon position. We then performed posterior inference of substitution rates and divergence times for the partitioned alignment using MCMCTree [55, 63] using the multivariate normal (MVN) approximation of the likelihood (where the MVN approximation was based on the maximum likelihood estimates under the GTR model with Gamma distributed relative rates across sites [5 categories]). We used a Gamma (2, 11) prior for the mean substitution rate per site per 100 My (million years), based on a rough estimate of the substitution rate under the molecular clock with a root age of 50 My obtained using baseml from the PAML package [53]. We use an independent log-normal rates relaxed molecular clock prior on branch-specific substitution rates, using a Gamma (2, 10) prior for the variance parameter of the clock. We set the birth-death-sampling prior such that a uniform prior over node ages is obtained. We include 2 fossil calibrations. First, we used a minimum age for the Oryza—Hordeum divergence of 34 My based on the review of Iles et al. [64]. Next, a secondary calibration for the root based on previous dating studies included in the TimeTree [65] database was used, where we excluded all time estimates younger than the 34 My constraint and older than 80 My. We then fitted a log-normal distribution to the age estimates in the time tree data, which we approximated by a Gamma (47, 100) distribution. We used MCMCTree to obtain 5,000 from the posterior sampling every 200 iterations after a burn-in of 50,000 iterations. We compared 2 independent runs with each other to verify convergence and with a run of the MCMC algorithm under the prior alone to compare the posterior distribution for the node ages to the effective prior implied by the fossil calibrations (Supplementary Fig. S3). The results of this analysis provide the phylogenetic tree shown in Fig. 1D.
Transposable element properties
The ∼42.6% TE content of the fonio genome is a minimal estimate, given that degraded TE fragments are often missed by the de novo discovery analysis that was used. This underestimation is routine in other plant genome annotations as well [66], so it is reasonable to compare TE descriptions across plant genomes. In fonio, the very high level of Gypsy LTR-RTs compared to Copia LTR-RTs is exceptional. Although most grass genomes have more Gypsy TEs than Copia (e.g., ∼50% Gypsy and ∼25% Copia in the ∼2.4-Gb maize genome [67] or ∼36% Gypsy and ∼33% Copia in the ∼2.8-Gb pearl millet genome [14]), the ∼6.7:1 Gypsy to Copia ratio in the ∼900-Mb fonio genome is unprecedented. One should remember, however, that the diploid constituent genomes of fonio are ∼450 Mb, so somewhat similar results are observed in other small panicoid genomes like sorghum (∼750 Mb) and rice (∼430 Mb), with Gypsy/Copia of ∼3.7 and ∼4.9, respectively [68]. This fonio observation is surprising because the quantity of Gypsy LTR-RTs is the major determinant of genome size in grasses [69], so one would expect higher Gypsy to Copia ratios as genome size increases, rather than the opposite that we observe. These results suggest that either different factors initiate Gypsy amplification bursts than Copia amplifications, or that Copia elements are particularly sensitive to shared activation factors. It would be useful to investigate additional Digitaria species to see whether this Gypsy/Copia ratio trait is shared by other close relatives and thus a possible outcome of common ancestral properties.
Analysis of LTR-RT insertion dates demonstrated that most of the elements had been inserted within the past 2 My. This high level of recent activity is a standard observation in the grasses, at least partly caused by the fact that the rapid DNA removal by accumulated small deletions quickly excises and otherwise obscures any DNA that is not under positive selection [70, 71].
Whole-genome duplication and subsequent stability
We inferred whole-paranome and 1-vs-1 ortholog Ks distributions and performed syntenic analyses to further assess the clear signature of a relatively recent whole-genome duplication (WGD) in Digitaria exilis.Ks distributions present a clear signature of WGD in the recent evolutionary past of D. exilis, with this event not shared with the closest relative in our analyses (S. italica) (Fig. 1A). We note that a trace of an older, likely Poaceae-shared WGD [72] event was also clearly observed in both the whole-paranome and anchor pair Ks distributions of D. exilis, coinciding with similar signatures in sorghum and Setaria (Fig. 1B). Analysis of collinearity and synteny show that the genome of D. exilis is still largely conserved in duplicate (Fig. 1C). Phylogenetic divergence time estimation (Fig. 1D) estimated the timing of the WGD event (or divergence of parental genomes in the case of an allopolyploidy event) at ∼3.1 million years ago (mya) with a 95% posterior uncertainty interval of (2.2, 4.2 My) and the divergence of Digitaria from Setaria at 17.8 (12.5, 23.1) mya, with these estimates associated with a posterior mean substitution rate across the 3 codon positions of 2.5 × 10−9 (1.1 × 10−9, 5.0 × 10−9) substitutions per year per site. This is consistent with CM05836 [21]. The closest relatives to fonio with whole-genome sequences would be P. miliaceum, S. italica, and C. americanus. The diploid ancestor to D. exilis is not clear [73].
It is interesting that Fig. 1C shows extreme conservation of gene content and order across long scaffolds but also the presence of large rearrangements that differentiate chromosome-size blocks. This suggests a possible selection for major rearrangements after the polyploids were formed, perhaps to minimize tetrasomic inheritance [74, 75].
In the ∼3.1 My since the latest WGD, most of the duplicated genes have had both copies retained. For instance, the BUSCO gene set yielded 86.5% of the genes still in a duplicated state. Our genome assemblies did not yield complete chromosomes, so we could not investigate the details of major chromosomal rearrangements, preferential gene loss (also known as fractionation), or parent-specific gene expression differences that might differentiate the 2 ancestral genomes in this tetraploid [76]. The large stretches of gene content and gene collinearity retention observed between our largest contiguous assemblies (Fig. 1C) do demonstrate, however, that there has been no large number of small rearrangements of these genomes over the past 3.1 My.
Expansions and contraction of gene families
To see the expansions and contractions of gene families, broomcorn millet (Panicum miliaceum L.) was added in the phylogenetic analysis because it experienced a recent tetraploidization estimated at ∼5.8 mya that is similar to fonio.
Based on sequence homology, we assigned 58,459 genes to 20,003 families, 14,549 of which have expanded in the fonio genome. Expansion in a similar number of gene families (11,819) was also observed in the broomcorn millet genome, also an allotetraploid crop. Of the fonio gene families, 57.4% contain 2 copies (the most abundant category in these 10 species) and 30.4% contain >2 copies (Fig. 2). Most (∼90%) of the 2-copy gene families of fonio are located in syntenic blocks, indicating that the expansion was mainly due to the recent WGD event (Fig. 2 and Supplementary Fig. S4).
Figure 2:

The number of gene families that expanded or contracted during evolution mapped to the species phylogenetic tree in related Poaceae species.
In addition to the majority of multi-copy genes, there are many (∼12.1% of the total) that are single-copy genes and thus a likely outcome of at least some deletion after polyploidy. Gene Ontology enrichment analyses of contracted genes (1 copy; Supplementary Fig. S5) and expanded genes (>2 copies; Supplementary Fig. S6) relative to O. sativa were performed. The analysis identifies negative regulators and recognition factors for biotic and abiotic stresses, as well as pollen/fertility recognition, as single-copy genes. In contrast, there is general expansion of gene families encoding positive regulators of multiple-copy genes. These results suggest that further analysis of these genes may reveal their roles in heat and drought stress tolerance, and in understanding of crossing barriers in fonio.
Candidate domestication genes
Improvement of fonio will require further domestication, particularly to solve the issues of shattering and lodging. This process should be greatly assisted by the provision of a comprehensive genome sequence.
In rice, sorghum, and maize, mutations in the gene SSH1 (SUPPRESSION of SEED SHATTERING-1) are associated with panicle retention of the grain after seed maturation (the “non-shattering” trait) in domesticated accessions [77]. Nine sequenced grass genomes were scanned with OrthoFinder (as described in the section “Phylogenetic divergence and dating the most recent whole-genome duplication”) to find the orthologues of this gene. The gene family fasta files were used to construct trees using MAFFT and Iqtree, and trees were visualized in FigTree. Interproscan was used to annotate the proteins with their pFam domains, and alignments were visualized in Geneious Prime [78].
Fonio has 4 genes related to SSH1, but the phylogenetic tree indicated that 2 are more closely related to the rice SSH1 gene associated with shattering than to the other SSH1-like gene in rice (Supplementary Fig. S7). Other species included in our dataset have between 1 and 3 SSH1-like genes (Supplementary Table S4). The extra copies in D. exilis are expected because of its polyploid nature and thus can explain why no ancient or modern farmers have detected recessive single-gene mutations at each of these loci in a single fonio plant. By modern forward or reverse genetic and breeding techniques, inactivation and selection of both of these genes should be targeted to solve the shattering problem in fonio.
Inactivation of the dw3 (Dwarfing-3) genes of sorghum is responsible for the semi-dwarf trait that diminishes lodging and thereby greatly improves yield and input response in this important crop of arid and semi-arid agriculture [79]. Inactivation-mutant orthologues of the same gene are also responsible for the pearl millet cultivars with highest lodging resistance and the highest grain yield [80]. Hence, orthologues of dw3 also should be targets for inactivation-mutation and molecular breeding in fonio. Once again, fonio has more copies of this gene than do any of the other grasses screened, all of which are diploids (Supplementary Fig. S8 and Table S5).
The GW2 (GRAIN WEIGHT-2) gene controls seed weight in wheat and rice, with inactivation of the gene leading to larger grain [81, 82]. Orthofinder results indicated that members of this gene family are present in single copy in all of the examined grass species, except fonio and maize (Supplementary Fig. S9 and Table S6). The 2 copies in D. exilis only differ from each other by 3 amino acid residue substitutions. The fonio genes were found to be nearly identical to the unmutated GW2 version that yields smaller grain in rice and wheat (data not shown). Although increased seed weight does not always increase yield (due to correlated traits, such as seed number), it is a particularly important trait in fonio to enable sowing for uniform stands and mechanical threshing.
Genetic diversity
Fonio genetic diversity was assessed using 184 samples from ∼130 accessions collected from Mali and Niger, signatories to the Cartagena Protocol on Biosafety (Supplementary Table S7). Consistent with the Nagoya Protocol and the third objective of the Convention on Biological Diversity of access and benefit sharing, fonio materials from Mali were collected in Mali by Institut d'Economie Rurale (IER) while those from Niger were collected in Niger by Institut National la de Recherche Agronomique du Niger (INRAN) and conserved at the ICRISAT Niamey genebank. M.D.S., F.H., and P.G. were involved in the germplasm collection, seed conservation at the genebank, and/or DNA extraction from young seedlings. All DNA samples or seed were sent to the USA for analysis for research purposes only. This research has no direct commercial application.
Seedlings of each sample were grown at the respective institutions in West Africa, and DNA was extracted from young leaves with a QIAGEN DNeasy Plant Mini Kit (Germantown, MD, USA). Lyophilized DNA was then sent to Data2Bio (Ames, IA, USA) for tunable genotyping-by-sequencing using 2-bp selection and 5 runs on an Ion Torrent Ion Proton Instrument (Thermo Fisher Scientific, Waltham, MA, USA). The resulting raw sequences were quality-trimmed by Data2Bio, which removed bases with PHRED quality scores <15. These trimmed sequences were then aligned to the genome assembly with GSNAP v2020–04-08 [83] using default parameters. Single-nucleotide polymorphisms were called using the bcftools mpileup command v1.9 [84] with max-depth set to 1,000 and minimum base quality set to 20. Raw single-nucleotide polymorphisms were then filtered using TASSEL v5.2.40 [85], custom R scripts with R v3.5.1 [86], and bcftools to include only sites with ≤25% heterozygosity, ≤500 total read depth, ≤60% missing data, and ≥2.5% minor allele frequency (Supplementary Table S8). Population substructure was determined with fastStructure v1.0 [87], testing from 1 to 10 population clusters and identifying the optimal number with the included chooseK.py program. This identified 3 clear clusters of material, with genetic separation strongly correlated with geography (Fig. 3A). The genetic distinctions among these clusters are clear when plotting the genetic principal coordinates and relationship dendrogram (Fig. 3B). A small number of accessions (<5) appear “misplaced” on the geographic map, which could be due to recent transfer of germplasm or human error during collection, storage, or processing. Geographic clustering can reflect both human trafficking of seed stocks and the genetic basis of local adaptation. Further (both broader and deeper) germplasm analyses will be useful for resolving these issues.
Figure 3:
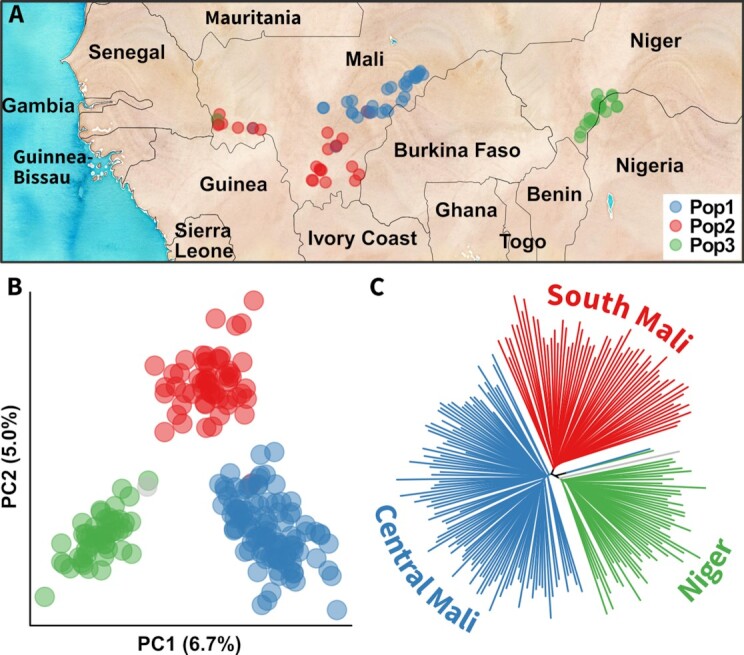
Fonio genetic diversity. The genetic diversity of fonio samples was surveyed by genotyping-by-sequencing. (A) Fonio samples originated from Mali and Niger. They separate into 3 primary subpopulations based on population structure analysis. Both principal coordinate analysis of the genetic diversity (B) and a neighbor-joining tree of the population (C) confirm these groupings. A few discrepancies between population assignment and geography may be due to recent long-distance germplasm exchanges or labeling errors during collection and storage.
Conclusions
Genome analysis of any polyploid is challenging, especially when no diploid ancestors are known. Our sequence of the white fonio (D. exilis) genome indicates its recent tetraploid origin and the retention of most of the genes duplicated in this process. This retention of duplicated genes likely explains why recessive mutations for important agronomic traits like shattering, seed size, semi-dwarfism, and others like day-length dependence have not yet been detected in fonio. However, it is now possible to identify such mutations by using modern mutation detection schemes, like those used for the tetraploid cereal Eragrostis tef [88]. One purpose for generating a fonio genome sequence was to attract molecular genetics researchers to the study of this crop and thereby enable hypothesis-driven breeding through genomics-assisted selection. If future researchers develop a transformation technology for fonio [89] or develop other genome-editing strategies [90], then directed mutagenesis could be used to knock out pairs of these domestication genes in a single step [91].
The importance of correcting such problems as shattering, seed size, and lodging in fonio cannot be overestimated. Until shattering is solved, farmers will continue to be required to harvest before grains fully mature, thus dramatically decreasing overall yield. Without semi-dwarf varieties, already serious lodging problems in fonio will continue to prohibit the use of more inputs (because fertilizer increases plant height and thus lodging) or even the selection of larger grain yield from the panicles because greater weight on the top of the plant can cause more lodging. The same will almost certainly be true for fonio, hence providing a partial explanation for its tiny seed size in cultivated landraces. With domestication traits fully penetrant into fonio cultivars, one can expect dramatic increases in fonio performance, with expectations of a 2-fold or greater yield enhancement easily within the short-term range of possibilities.
The absence of an outcrossing protocol for fonio is another technical deficiency that severely limits this crop's potential for improvement. Our diversity analysis on cultivar Niatia indicates <0.01% heterozygosity, showing that crosses occur very rarely by natural processes. Hence, generating controlled crosses will probably require a serious dedication to this pursuit. Our results indicate a great deal of genetic variability within fonio landraces, so we have no doubt that hybridization could be used in breeding projects to optimize fonio germplasm quality for future West African and other farmers.
Data Availability
The genome and annotation underlying this article are available in the African Orphan Crops Consortium–specific branch of the ORCAE platform [92, 93] athttps://bioinformatics.psb.ugent.be/orcae/aocc/overview/Digex. The GenBank project number for the assembly is PRJNA640067. All scripts for diversity analysis and data tables are available at [94] including full genotyping table. Genotyping table is also available at GenBank Project No. PRJNA644458. All supporting data and materials are available at the GigaScience GigaDB database [95].
Additional Files
Supplementary Methods.
Supplementary Figure S1. A. Comparison of the contiguity of the Niatia Genome and CM05836 [21] genome. B. Comparison of contig tig00001331 corresponding to 100 consecutive segments anchored on the same chromosome 3B and tig00010942 corresponding to 65 consecutive segments on the chromosome 5A on the Abrouk et al. [21] genome.
Supplementary Figure S2. The k-mer distribution of raw Illumina reads at k-mer value 33 bp.
Supplementary Figure S3. A. Marginal posterior distributions for 2 independent chains (green and orange) and induced marginal prior distributions (blue) for internal node ages (t_n11 to t_n19, see panel C), overall mean substitution rate (mu), mean substitution rate for different codon positions (mu1, mu2, and mu3), and variance parameter of the uncorrelated relaxed clock (sigma2_1, sigma2_2, and sigma2_3) for the 3 codon positions. B. Trace plots for the MCMC chains associated with panel (A).
Supplementary Figure S4. There are 10,075 families that have 2 copies in fonio and 1 copy in Setaria italica, and 90% of 2-copy families are located in synteny blocks. The above 4 examples indicate the high degree of collinearity and synteny between S. italica and fonio.
Supplementary Figure S5. GO of single-copy, contracted genes in fonio.
Supplementary Figure S6. GO enrichment for expanded genes in D. exilisand relative to O. sativa.
Supplementary Figure S7. Phylogenetic tree of the SSH-like genes from fonio and related species. The genes shaded in light blue are the family members most closely related to SSH-1 in O. sativa and D. exilis. Genes are named according to their PLAZA identifiers. Abbreviations for species names are as follows: Bradi (Brachypodium distachyon), pgl_GLEAN (Cenchrus amercianus), Digex (Digitaria exilis), Oropetium (Oropetium thomaeum), OsR (Oryza sativa), Seita (Setaria italica), Sobic (Sorghum bicolor), and Zm (Zea mays).
Supplementary Figure S8. Phylogenetic tree of thedw3 gene family of fonio and related species.
Supplementary Figure S9. Gene family tree for GW2-A-like genes in fonio and related species. This figure also includes the genes from 2 additional Pooid species, barley (Hordeum vulgare) (HORV) and wheat (Triticum turgidum) (TRITD).
Supplementary Table S1. Comparison of genome assembly statistics of fonio.
Supplementary Table S2. Statistics for the gene annotation.
Supplementary Table S3. Annotated non-coding RNA genes.
Supplementary Table S4. Orthologs for suppression of Shattering1 genes.
Supplementary Table S5. Orthologs of Dwarf Gene-3.
Supplementary Table S6. Orthologs of Grain Weight-2 genes.
Supplementary Table S7. Passport data for accessions and samples used for diversity study (see Supplementary Tables Excel file).
Supplementary Table S8. Single-nucleotide polymorphism database used for diversity study (see Supplementary Tables Excel file).
Abbreviations
AED: annotation edit distance; BLAST: Basic Local Alignment Search Tool; BUSCO: Benchmarking Universal Single-Copy Orthologs; CTAB: cetyl trimethylammonium bromide; Dw3: dwarf3; EDTA: ethylenediaminetetraacetic acid; Gb: gigabase pairs; GO: Gene Ontogeny; GW2: grain weight2; kb: kilobase pairs; LINE: long interspersed nuclear element; LTR: long terminal repeat; LTR-RT: long terminal repeat retrotransposon; MAFFT: Multiple Alignment with Fast Fourier Transform; Mb: megabase pairs; MITE: miniature inverted repeat transposable element; My: million years; mya: million years ago; NCBI: National Center for Biotechnology Information; ORF: open reading frame; PacBio: Pacific Biosciences; PAML: Phylogenetic Analysis by Maximum Likelihood; SINE: small interspersed nuclear element; SMRT: single-molecule, real-time sequencing; SRA: Sequence Read Archive; SSH1: suppression of shattering1; SSR: simple sequence repeat; TE: transposable element; TIR: terminal inverted repeat transposable element; WGD: whole-genome duplication.
Competing Interests
The authors declare that they have no competing interests.
Funding
J.L.B. acknowledges the Giles Fellowship from the University of Georgia as a source of funding for this project. Y.V.d.P. acknowledges funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement No. 833,522). A.V. acknowledges funding from the Seed Biotechnology Center, University of California. J.G.W. acknowledges funding from the International Crops Research Institute for the Semi-Arid Tropics (ICRISAT) and the University of Georgia. M.D.S. acknowledges funding from the McKnight foundation.
Authors' Contributions
J.L.B., J.W., Y.V.d.P., and A.V.D. conceived, designed, and interpreted the study; S.C., X.M., X.W., A.E.J.Y., S.R.C., M.S.J., P.G., F.H., M.D.S., and A.Z. prepared the materials, conducted the experiments, and analyzed all data; J.L.B. and A.V. led on manuscript preparation, while all other authors revised the manuscript and approved the final version.
Supplementary Material
Xupo Ding -- 8/24/2020 Reviewed
Xupo Ding -- 12/28/2020 Reviewed
Fanna Maina -- 9/3/2020 Reviewed
Fanna Maina -- 12/31/2020 Reviewed
Gincy P. Thottathil -- 9/13/2020 Reviewed
ACKNOWLEDGEMENTS
We thank Shu-Min Kao for providing scripts and Sara Patterson for providing Niatia seed and for helpful discussions, Oanh Nguyen from the UC Davis Genome and Biomedical Sciences Facility for technical expertise for sequencing with Pacific Biosciences, and Armando Garcia-Llanos for DNA and library quality control for sequencing.
Contributor Information
Xuewen Wang, Department of Genetics, University of Georgia, Athens, GA 30602, USA.
Shiyu Chen, Department of Plant Sciences, Seed Biotechnology Center, University of California, 1 Shields Ave. Davis, CA 95616, USA.
Xiao Ma, Bioinformatics & Systems Biology, VIB / Ghent University, Technologiepark 71, 9052 Zwijnaarde, Belgium.
Anna E J Yssel, Centre for Microbial Ecology and Genomics, Department of Biochemistry, Genetics and Microbiology, University of Pretoria, Pretoria 0028, South Africa; Centre for Bioinformatics and Computational Biology, Department of Biochemistry, Genetics and Microbiology, University of Pretoria, Pretoria 0028, South Africa.
Srinivasa R Chaluvadi, Department of Genetics, University of Georgia, Athens, GA 30602, USA.
Matthew S Johnson, Institute of Plant Breeding, Genetics, and Genomics, University of Georgia, 111 Riverbend Rd, Athens, GA 30602, USA.
Prakash Gangashetty, International Crops Research Institute for the Semi-Arid Tropics (ICRISAT), BP 12404, Niamey, Niger.
Falalou Hamidou, International Crops Research Institute for the Semi-Arid Tropics (ICRISAT), BP 12404, Niamey, Niger.
Moussa D Sanogo, Institut d'Economie Rurale, Ministere de l'Agriculture, Cinzana, BP 214, Ségou, Mali.
Arthur Zwaenepoel, Bioinformatics & Systems Biology, VIB / Ghent University, Technologiepark 71, 9052 Zwijnaarde, Belgium.
Jason Wallace, Department of Crop and Soil Sciences, University of Georgia, 3111 Carlton St Bldg, Athens, GA 30602, USA.
Van Yves de Peer, Bioinformatics & Systems Biology, VIB / Ghent University, Technologiepark 71, 9052 Zwijnaarde, Belgium; Centre for Microbial Ecology and Genomics, Department of Biochemistry, Genetics and Microbiology, University of Pretoria, Pretoria 0028, South Africa; College of Horticulture, Nanjing Agricultural University, Nanjing, China.
Jeffrey L Bennetzen, Department of Genetics, University of Georgia, Athens, GA 30602, USA.
Allen Van Deynze, Department of Plant Sciences, Seed Biotechnology Center, University of California, 1 Shields Ave. Davis, CA 95616, USA.
References
- 1. Ballogou V, Soumanou M, Toukourou F, et al. Structure and nutritional composition of fonio (Digitaria exilis) grains: a review. Int Res J Biol Sci. 2013;2(1):73–9. [Google Scholar]
- 2. Fanou N, Hulshof P, Koreissi Y, et al. Nutritive values of fonio and fonio products: P110–08. Ann Nutr Metab. 2009;55. [Google Scholar]
- 3. Temple VJ, Bassa JD. Proximate chemical composition of Acha (Digitaria exilis) grain. J Sci Food Agric. 1991;56(4):561–3. [Google Scholar]
- 4. Vietmeyer N, Borlaugh N, Axtell J, et al. Lost Crops of Africa. Washington, DC: The National Academies Press; 1996:59. [Google Scholar]
- 5. De Wet J. The three phases of cereal domestication. In: Chapman GP, ed. Grass Evolution and Domestication. Cambridge: Cambridge University Press; 1992:176–98. [Google Scholar]
- 6. Aliero A, Morakinyo J. Photoperiodism in Digitaria exilis(Kipp) Stapf accessions. Afr J Biotechnol. 2005;4(2):241–3. [Google Scholar]
- 7. Patterson SE, Bolivar-Medina JL, Falbel TG, et al. Are we on the right track: can our understanding of abscission in model systems promote or derail making improvements in less studied crops?. Front Plant Sci. 2016;6:1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Adoukonou-Sagbadja H, Schubert V, Dansi A, et al. Flow cytometric analysis reveals different nuclear DNA contents in cultivated fonio (Digitariaspp.) and some wild relatives from West-Africa. Plant Syst Evol. 2007;267(1):163–76. [Google Scholar]
- 9. Chukwurah PN, Uyoh EA, Usen IN, et al. Assessment of intra and inter species variation in antioxidant composition and activity in marginalized fonio millet (Digitaria spp.). J Cereals Oilseeds. 2016;7(1), doi: 10.5897/JCO2016.0146. [DOI] [Google Scholar]
- 10. Gigou J, Stilmant D, Diallo TA, et al. Fonio millet (Digitaria exilis) response to N, P and K fertilizers under varying climatic conditions in West Africa. Exp Agric. 2009;45(4):401–15. [Google Scholar]
- 11. Adoukonou-Sagbadja H, Wagner C, Dansi A, et al. Genetic diversity and population differentiation of traditional fonio millet (Digitaria spp.) landraces from different agro-ecological zones of West Africa. Theor Appl Genet. 2007;115(7):917–31. [DOI] [PubMed] [Google Scholar]
- 12. Sarah G, Homa F, Pointet S, et al. A large set of 26 new reference transcriptomes dedicated to comparative population genomics in crops and wild relatives. Mol Ecol Resour. 2017;17(3):565–80. [DOI] [PubMed] [Google Scholar]
- 13. Bennetzen JL, Schmutz J, Wang H, et al. Reference genome sequence of the model plant Setaria. Nat Biotechnol. 2012;30(6):555–61. [DOI] [PubMed] [Google Scholar]
- 14. Varshney RK, Shi C, Thudi M, et al. Pearl millet genome sequence provides a resource to improve agronomic traits in arid environments. Nat Biotechnol. 2017;35(10):969–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zou C, Li L, Miki D, et al. The genome of broomcorn millet. Nat Commun. 2019;10(1):436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bennetzen JL, Freeling M. The unified grass genome: synergy in synteny. Genome Res. 1997;7(4):301–6. [DOI] [PubMed] [Google Scholar]
- 17. Cruz J-F. Fonio. Upgrading quality and competitiveness of fonio for improved livelihoods in West Africa: Second activity report. Montpellier: CIRAD, 108 2008. https://agritrop.cirad.fr/545752/ [Google Scholar]
- 18. Murray M, Thompson WF. Rapid isolation of high molecular weight plant DNA. Nucl Acids Res. 1980;8(19):4321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pacific Biosciences: Software Downloads. SMRT Link V 6.0. 2020. https://www.pacb.com/support/software-downloads. Accessed 21 June 2020. [Google Scholar]
- 20. Koren S, Walenz BP, Berlin K, et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Abrouk M, Ahmed HI, Cubry P, et al. Fonio millet genome unlocks African orphan crop diversity for agriculture in a changing climate. Nat Commun. 2020;11(1), doi: 10.1038/s41467-020-18329-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mapleson D, Garcia Accinelli G, Kettleborough G, et al. KAT: a K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics. 2017;33(4):574–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ranallo-Benavidez TR, Jaron KS, Schatz MC. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 2020;11(1):1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang X, Wang L. GMATA: an integrated software package for genome-scale SSR mining, marker development and viewing. Front Plant Sci. 2016;7:1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35(suppl_2):W265–W8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ellinghaus D, Kurtz S, Willhoeft U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics. 2008;9(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ou S, Jiang N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018;176(2):1410–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mao H, Wang H. SINE_scan: an efficient tool to discover short interspersed nuclear elements (SINEs) in large-scale genomic datasets. Bioinformatics. 2017;33(5):743–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rho M, Tang H. MGEScan-non-LTR: computational identification and classification of autonomous non-LTR retrotransposons in eukaryotic genomes. Nucleic Acids Res. 2009;37(21):e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Crescente JM, Zavallo D, Helguera M, et al. MITE Tracker: an accurate approach to identify miniature inverted-repeat transposable elements in large genomes. BMC Bioinformatics. 2018;19(1):348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xiong W, He L, Lai J, et al. HelitronScanner uncovers a large overlooked cache of Helitron transposons in many plant genomes. Proc Natl Acad Sci U S A. 2014;111(28):10263–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wicker T, Sabot F, Hua-Van A, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8(12):973–82. [DOI] [PubMed] [Google Scholar]
- 33. Jurka J, Kapitonov VV, Pavlicek A, et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110(1–4):462–7. [DOI] [PubMed] [Google Scholar]
- 34. RepeatMasker version 4.0.7. http://www.repeatmasker.org/. Accessed 15 March 2020. [Google Scholar]
- 35. Luo M-C, Gu YQ, Puiu D, et al. Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature. 2017;551(7681):498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Andrews S. FastQC a quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 19 June 2020. [Google Scholar]
- 37. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pertea M, Pertea GM, Antonescu CM, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Haas BJ, Papanicolaou A, Yassour M, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8(8):1494–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Campbell MS, Law M, Holt C, et al. MAKER-P: a tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol. 2014;164(2):513–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stanke M, Steinkamp R, Waack S, et al. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 2004;32(suppl_2):W309–W12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Borodovsky M, Mills R, Besemer J, et al. Prokaryotic gene prediction using GeneMark and GeneMark.hmm. Curr Protoc Bioinformatics. 2003, doi: 10.1002/0471250953.bi0405s01. [DOI] [PubMed] [Google Scholar]
- 45. Simao Neto F, Waterhouse R, Ioannidis P, et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–2. [DOI] [PubMed] [Google Scholar]
- 46. Van Bel M, Diels T, Vancaester E, et al. PLAZA 4.0: an integrative resource for functional, evolutionary and comparative plant genomics. Nucleic Acids Res. 2018;46(D1):D1190–D6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Campbell MS, Holt C, Moore B, et al. Genome annotation and curation using MAKER and MAKER-P. Curr Protoc Bioinformatics. 2014;48(1), doi: 10.1002/0471250953.bi0411s48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jones P, Binns D, Chang H-Y, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30(9):1236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Waterhouse RM, Seppey M, Simão FA, et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol. 2018;35(3):543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nishimura O, Hara Y, Kuraku S. gVolante for standardizing completeness assessment of genome and transcriptome assemblies. Bioinformatics. 2017;33(22):3635–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29(22):2933–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kalvari I, Argasinska J, Quinones-Olvera N, et al. Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018;46(D1):D335–D42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kalvari I, Nawrocki EP, Argasinska J, et al. Non-coding RNA analysis using the Rfam database. Curr Protoc Bioinformatics. 2018;62(1):e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dongen S. Graph Clustering by Flow Simulation. Amsterdam, Netherlands: University of Utrecht; 2000. [Google Scholar]
- 56. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol Biol Evol. 2007;24(8):1586–91. [DOI] [PubMed] [Google Scholar]
- 58. Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5(3):e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Proost S, Fostier J, De Witte D, et al. i-ADHoRe 3.0—fast and sensitive detection of genomic homology in extremely large data sets. Nucleic Acids Res. 2012;40(2):e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20(1):238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Whelan S, Irisarri I, Burki F. PREQUAL: detecting non-homologous characters in sets of unaligned homologous sequences. Bioinformatics. 2018;34(22):3929–30. [DOI] [PubMed] [Google Scholar]
- 62. Ronquist F, Teslenko M, Van Der Mark P, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rannala B, Yang Z. Inferring speciation times under an episodic molecular clock. Syst Biol. 2007;56(3):453–66. [DOI] [PubMed] [Google Scholar]
- 64. Iles WJ, Smith SY, Gandolfo MA, et al. Monocot fossils suitable for molecular dating analyses. Bot J Linn Soc. 2015;178(3):346–74. [Google Scholar]
- 65. Kumar S, Stecher G, Suleski M, et al. TimeTree: a resource for timelines, timetrees, and divergence times. Mol Biol Evol. 2017;34(7):1812–9. [DOI] [PubMed] [Google Scholar]
- 66. Bennetzen JL, Park M. Distinguishing friends, foes, and freeloaders in giant genomes. Curr Opin Genet Dev. 2018;49:49–55. [DOI] [PubMed] [Google Scholar]
- 67. Schnable PS, Ware D, Fulton RS, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326(5956):1112–5. [DOI] [PubMed] [Google Scholar]
- 68. Paterson AH, Bowers JE, Bruggmann R, et al. The sorghum bicolor genome and the diversification of grasses. Nature. 2009;457(7229):551–6. [DOI] [PubMed] [Google Scholar]
- 69. Bennetzen JL, Wang H. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu Rev Plant Biol. 2014;65:505–30. [DOI] [PubMed] [Google Scholar]
- 70. Devos KM, Brown JK, Bennetzen JL. Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res. 2002;12(7):1075–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ma J, Devos KM, Bennetzen JL. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 2004;14(5):860–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jiao Y, Li J, Tang H, et al. Integrated syntenic and phylogenomic analyses reveal an ancient genome duplication in monocots. Plant Cell. 2014;26(7):2792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Abdul SD, Jideani AIO. Fonio (Digitaria spp.) Breeding. Springer; 2019:47–81. [Google Scholar]
- 74. Soltis D, Soltis P, Rieseberg LH. Molecular data and the dynamic nature of polyploidy. Crit Rev Plant Sci. 1993;12(3):243–73. [Google Scholar]
- 75. Sybenga J. Allopolyploidization of autopolyploids I. Possibilities and limitations. Euphytica. 1969;18(3):355–71. [Google Scholar]
- 76. Bird KA, VanBuren R, Puzey JR, et al. The causes and consequences of subgenome dominance in hybrids and recent polyploids. New Phytol. 2018;220(1):87–93. [DOI] [PubMed] [Google Scholar]
- 77. Lin Z, Li X, Shannon LM, et al. Parallel domestication of the Shattering1 genes in cereals. Nat Genet. 2012;44(6):720–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Geneious Prime. 2020. https://www.geneious.com.Accessed June 21, 2020. [Google Scholar]
- 79. Multani DS, Briggs SP, Chamberlin MA, et al. Loss of an MDR transporter in compact stalks of maize br2 and sorghum dw3 mutants. Science. 2003;302(5642):81–4. [DOI] [PubMed] [Google Scholar]
- 80. Parvathaneni RK, Jakkula V, Padi FK, et al. Fine-mapping and identification of a candidate gene underlying the d2 dwarfing phenotype in pearl millet, Cenchrus americanus (L.) Morrone. G3 (Bethesda). 2013;3(3):563–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Simmonds J, Scott P, Brinton J, et al. A splice acceptor site mutation in TaGW2-A1 increases thousand grain weight in tetraploid and hexaploid wheat through wider and longer grains. Theor Appl Genet. 2016;129(6):1099–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Song X-J, Huang W, Shi M, et al. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet. 2007;39(5):623–30. [DOI] [PubMed] [Google Scholar]
- 83. Wu TD, Reeder J, Lawrence M, et al. GMAP and GSNAP for genomic sequence alignment: enhancements to speed, accuracy, and functionality. Methods Mol Biol. 2016;1418:283–334. [DOI] [PubMed] [Google Scholar]
- 84. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27(21):2987–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bradbury PJ, Zhang Z, Kroon DE, et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23(19):2633–5. [DOI] [PubMed] [Google Scholar]
- 86. Kahle D, Wickham H. ggmap: spatial visualization with ggplot2. R J. 2013;5(1):144–61. [Google Scholar]
- 87. Raj A, Stephens M, Pritchard JK. fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics. 2014;197(2):573–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhu Q, Smith SM, Ayele M, et al. High-throughput discovery of mutations in tef semi-dwarfing genes by next-generation sequencing analysis. Genetics. 2012;192(3):819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ntui VO, Azadi P, Supaporn H, et al. Plant regeneration from stem segment-derived friable callus of “Fonio” (Digitaria exilis (L.) Stapf.). Sci Hortic. 2010;125(3):494–9. [Google Scholar]
- 90. Ji X, Yang B, Wang D. Achieving plant genome editing while bypassing tissue culture. Trends Plant Sci. 2020;25(5):427–9. [DOI] [PubMed] [Google Scholar]
- 91. Hu N, Xian Z, Li N, et al. Rapid and user-friendly open-source CRISPR/Cas9 system for single-or multi-site editing of tomato genome. Hortic Res. 2019;6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sterck L, Billiau K, Abeel T, et al. ORCAE: online resource for community annotation of eukaryotes. Nat Methods. 2012;9(11):1041. [DOI] [PubMed] [Google Scholar]
- 93. Yssel AE, Kao S-M, Van de Peer Y, et al. ORCAE-AOCC: a centralized portal for the annotation of African orphan crop genomes. Genes. 2019;10(12):950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wallace J. Fonio diversity 2020. https://github.com/wallacelab/paper-fonio-diversity-2020. 2020. Accessed 21 June 2020. [Google Scholar]
- 95. Bennetzen JL, Chen S, Ma X WX, et al. Supporting data for “Genome sequence and genetic diversity analysis of an under-domesticated orphan crop, white fonio (Digitaria exilis).”. GigaScience Database. 2021. 10.5524/100857. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Bennetzen JL, Chen S, Ma X WX, et al. Supporting data for “Genome sequence and genetic diversity analysis of an under-domesticated orphan crop, white fonio (Digitaria exilis).”. GigaScience Database. 2021. 10.5524/100857. [DOI] [PMC free article] [PubMed]
Supplementary Materials
Xupo Ding -- 8/24/2020 Reviewed
Xupo Ding -- 12/28/2020 Reviewed
Fanna Maina -- 9/3/2020 Reviewed
Fanna Maina -- 12/31/2020 Reviewed
Gincy P. Thottathil -- 9/13/2020 Reviewed
Data Availability Statement
The genome and annotation underlying this article are available in the African Orphan Crops Consortium–specific branch of the ORCAE platform [92, 93] athttps://bioinformatics.psb.ugent.be/orcae/aocc/overview/Digex. The GenBank project number for the assembly is PRJNA640067. All scripts for diversity analysis and data tables are available at [94] including full genotyping table. Genotyping table is also available at GenBank Project No. PRJNA644458. All supporting data and materials are available at the GigaScience GigaDB database [95].