

Abstract
Implanted porous precision templated scaffolds (PTS) with 40-μm spherical pores reduce inflammation and foreign body reaction (FBR) while increasing vascular density upon implantation. Larger or smaller pores, however, promote chronic inflammation and FBR. While macrophage (MØ) recruitment and polarization participates in perpetuating this pore-size-mediated phenomenon, the driving mechanism of this unique pro-healing response is poorly characterized. We hypothesized that the primarily myeloid PTS resident cells release small extracellular vesicles (sEVs) that induce pore-size-dependent pro-healing effects in surrounding T cells. Upon profiling resident immune cells and their sEVs from explanted 40-μm-(pro-healing) and 100-μm-pore diameter (inflammatory) PTS, we found that PTS pore size did not affect PTS resident immune cell population ratios or the proportion of myeloid sEVs generated from explanted PTS. However, quantitative transcriptomic assessment indicated cell and sEV phenotype were pore size dependent. In vitro experiments demonstrated the ability of PTS cell-derived sEVs to stimulate T cells transcriptionally and proliferatively. Specifically, sEVs isolated from cells inhabiting explanted 100 μm PTS significantly upregulated Th1 inflammatory gene expression in immortalized T cells. sEVs isolated from cell inhabiting both 40- and 100-μm PTS upregulated essential Treg transcriptional markers in both primary and immortalized T cells. Finally, we investigated the effects of Treg depletion on explanted PTS resident cells. FoxP3+ cell depletion suggests Tregs play a unique role in balancing T cell subset ratios, thus driving host response in 40-μm PTS. These results indicate that predominantly 40-μm PTS myeloid cell-derived sEVs affect T cells through a distinct, pore-size-mediated modality.
Keywords: biomaterial implant, extracellular vesicles, immune polarization, macrophages, sEV, T cells
1 |. INTRODUCTION
We have developed porous precision templated scaffolds (PTS) for tissue regeneration that are polymer constructs with scalable, ordered, and uniformly sized interconnecting pores (Fukano et al., 2006, 2010; Galperin, Long, Garty, & Ratner, 2013; Isenhath et al., 2007; Madden et al., 2010; Teng et al., 2014). PTS made with 40-μm-diameter pores of various polymers all exhibit reduced chronic inflammation and foreign body reaction (FBR) while inducing a prohealing response in skin (Fukano et al, 2006, 2010; Isenhath et al., 2007; Knowles et al., 2005), cornea (Teng et al., 2014), heart muscle (Madden et al., 2010), and bone (Galperin, Oldinski, et al., 2013). The pro-healing response consists of an optimal balance of tissue repair (primarily fibrotic wound resolution) and tissue regeneration (primarily returning tissue function). This pro-healing response is unique to PTS with pores between 30 and 40 μm, which causes the recruitment and differentiation of macrophages (MØ) toward an M2-like phenotype (Madden et al., 2010; Marshall et al., 2004). Despite these findings, pore-size-dependent MØ phenotype does not adequately explain the functional mechanisms of the unique pro-healing immune response observed in 40-μm PTS implants.
The limited success achieved in regenerating tissues is due in part to the tendency of therapeutic approaches to target late-stage processes in healing and regeneration. Conversely, the immune system is a highly complex network that orchestrates tissue integrity and adapts to features of the local microenvironment throughout the healing process. The immune system participates in tissue repair by scavenging debris and dead cells, recruiting and supporting the proliferation of tissue progenitor cells, and inducing vascularization. While previous studies have investigated the host immune response to biomaterials within the context of the FBR, MØ and T cells have been identified as important mediators of scaffold-driven tissue remodeling, and thus should be investigated within the context of immune-mediated tissue regeneration (Brown et al., 2012; Sadtler et al., 2016; Sicari et al., 2014).
Differential T cell subtypes significantly affect wound healing outcome (Engelhardt et al., 1998). CD4+ T cell populations are particularly important due to their presence within the PTS implant microenvironment and influential role in deciding implant fate through phenotypically determined key cell signaling modalities (Julier, Park, Briquez, & Martino, 2017). T Helper 1 (Th1, indicated by TNF±, TBX21, and IFN-γ) cells inhibit collagen deposition and traditionally pro-reparative effects by increasing local inflammatory signaling, while regulatory T cells (Tregs indicated by FoxP3 and IL-10) are instrumental in initiating and maintaining pro-regenerative responses (Julier et al., 2017; Liu et al., 2011; Nosbaum et al., 2016). Particularly, Tregs both influence T cell subtype and modulate neutrophil, macrophages, and local progenitor cell activation (Ali et al., 2017; Castiglioni et al., 2015; D’Alessio et al., 2009; Murphy, Ni Choileain, Zang, Mannick, & Lederer, 2005; Sakaguchi, Yamaguchi, Nomura, & Ono, 2008; Weirather et al., 2014). Recent evidence suggests that T Helper 2 cells (Th2 indicated by GATA3), which modulate classical Th1-driven inflammation while enhancing fibrosis, eosinophil activation, and IgE-mediated responses, are required to elicit the pro-healing response intrinsic to certain biomaterial scaffolds (Allen & Wynn, 2011; Sadtler et al., 2016). T Helper 3 and T Helper 17 (Th3 and Th17 indicated by TGF-β1 and RORγ, respectively) cells are traditionally ambiguous in wound healing: Th3 cells can release varying amounts of IL-10 and TGF-β1 depending on external stimuli, and Thl7 cells can release both classically pro-inflammatory and anti-inflammatory cytokines (Beissert, Schwarz, & Schwarz, 2006; Brockmann, Giannou, Gagliani, & Huber, 2017). Despite this ambiguity, recent evidence suggests that IL-17 released by Th17 cells are instrumental in enabling the inflammatory FBR (Chung et al., 2020).
Growing evidence suggests that the contents of membrane vesicles (i.e., exosomes, microvesicles) can modify the phenotype of recipient cells. Small extracellular vesicles (sEVs) have recently been recognized as promising therapeutic vehicles in many applications - particularly the modulation of immune and regenerative responses. The heterogeneous biological contents of sEVs, comprising functional lipids, proteins, and nucleic acids, are reflective of (though not identical to) their parent cells (Quesenberry, Aliotta, Deregibus, & Camussi, 2015). Further, utilization of the endocytic pathway means that sEVs enable unique, important, and understudied methods of cell signaling (Robbins & Morelli, 2014). The discovery of sEV’s cell signaling potential has recently explained previously misinterpreted immunological phenomena, as sEVs have been shown responsible for both inflammatory and pro-healing host immune responses in a variety of conditions (Lamichhane et al., 2014; Nazimek et al., 2015; Okoye et al., 2014).
Current cytokine signaling theories cannot fully describe immune communication within the PTS environment. However, MØ have demonstrated the ability to affect T cell phenotype in wound healing independent of or in combination with antigen presentation using both soluble (cytokine) and particulate signaling mechanisms (Bhatnagar, Shinagawa, Castellino, & Schorey, 2007; Engelhardt et al., 1998; Giri & Schorey, 2008; Qu, Franchi, Nunez, & Dubyak, 2007). Despite this evidence, the understanding of non-MHC II mediated MØ sEV signaling to CD4+ T cell populations remains lacking, especially in wound healing. We believe that sEVs are responsible for communication between MØ and T cells in implanted PTS. Therefore, this manuscript attempts to quantify the role of MØ sEV signaling in pore-size-dependent scaffold-mediated CD4+ T cell signaling in the implant microenvironment.
2 |. MATERIALS AND METHODS
2.1 |. PTS synthesis
PTS were fabricated (Figure S1a) using a sphere-templating method that generates 3-D porous polymer scaffolds with scalable, uniformly sized pores and pore interconnects (Madden et al., 2010; Marshall et al., 2004; Ratner & Marshall, 2008; Patent US208/075752A1). Polymethyl methacrylate (PMMA) beads (40 and 100 μm OD) were obtained from Microbeads AS. A mold was created by placing 1-mm-thick Teflon strips (2 mm wide) to serve as a retaining gasket between two 75 mm × 25 mm × 1 mm glass slides. Beads of the desired pore size were poured into the cavity. Molds filled with beads were placed in a water bath sonicator for 2 h to ensure uniform packing. Beads were then sintered to each other at their contacts points by heating at 175°C (40 μm) or 179°C (100 μm) for 24 h. The sintering procedure was optimized to obtain PMMA templates with neck sizes (interconnects between the beads) that were approximately 30% of the bead diameter. Poly (hydroxyethyl methyacrylate) (pHEMA) was selected over other polymers for its superior performance in histology dissection, biological inertness, and stability. pHEMA precursor consisted of 5 ml 2-hydroxyethyl methacrylate (HEMA; Polysciences, ophthalmic grade), 0.23 ml tetraethyleneglycol dimethacrylate (TEGDMA; Polysciences), 2.0 ml deionized water, 3.6 ml ethylene glycol, and 20 mg 2,2-dimethoxy-2-phenylacetophenone (Irgacure 651; BASF). The reaction mixture was infiltrated into the glass mold surrounding the bead template and degassed under vacuum for 30 min. HEMA monomer was free radical polymerized to pHEMA under a 450-W broad-spectrum UV lamp for 15 min. After polymerization, the PMMA bead template surrounded by the polymerized pHEMA was removed from the mold and placed in acetone to dissolve the PMMA microspheres. The resulting porous scaffolds, composed of pHEMA, were cut into 5-mm discs using a biopsy punch.
2.2 |. PTS characterization
Scanning electron microscopy (SEM) was performed on lyophilized PTS to confirm their pore dimensions (Figure S1b). Lyophilized samples were sputter coated for 120 s with gold/palladium (~20 nm layer). Scaffolds were then imaged on a scanning electron microscope (FEI SEM XL Siron) at 5 kV with a 5 mm working distance. Both cross section and top view were observed. ImageJ™ software measured the size of pores and the interconnecting throats based on the images collected from SEM. For each scaffold and template, three top-view images from different batches were analyzed and 10 sample points were measured from each image. Scaffolds were evaluated for endotoxin using a standard limulis amebocyte lysate gel clot protocol (Lonza) and analyzed by Time-of-Flight secondary ion mass spectroscopy and X-ray photoelectron spectroscopy to assess surface chemistry consistency and quality control between different pore size PTS (data available upon request). All scaffold batches used for biological experiments contained endotoxin concentration ≤0.06 EU/ml. Scaffolds were rehydrated before use in either PBS (in vivo studies) or tissue culture medium (in vitro studies). Figure S1b provides dimensional characteristics and SEM images of polyHEMA scaffolds used in this work.
2.3 |. Double transgenic mouse model for myeloid characterization
NIH guidelines for the care and use of laboratory animals (NIH Publication #85-23 Rev. 1985) have been observed. All breeding, genotyping, and PTS implantation experiments were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Washington.
A double transgenic mouse strain was developed to quantify in vivo myeloid phenotypes within implanted PTS. Cells of myeloid origin express the fluorescent protein, enhanced green fluorescent protein (eGFP), while cells of non-myeloid lineage express mTomato (mT). This recombination is irreversible and extracellular vesicles (EVs) also retain their parent cell’s fluorescence, allowing quantitative lineage tracking of cells and sEVs. To create this strain, LysM-Cre on C57Bl/6 background mice (Jackson labs, stock number 004,781) were bred with mT (mTomato)/mG (eGFP) mice on C57Bl/6 background (Jackson labs, stock number 007,676). Briefly, breeding trios utilizing female C57Bl/6 and homozygous male transgenic mice to generate heterozygous (either LysM-Cre +/0 or mT/mG +/0) were established, and then heterozygous LysM-Cre and heterozygous mT/mG mice were bred to produce offspring of all genotypes for further study. All mice were on a C57Bl/6 background to minimize strain difference. Cell-type-specific expression of mG was confirmed in resident tissue MØs in multiple organs. To verify model efficacy, lungs were retrieved from LysM-Cre+/0:mT/mG+/0 mice and processed for fluorescence microscopy; no synthetic fluorochromes were applied. mT/mG reporter mice contain a single copy of the transgene integrated into the ROSA26 locus (R26R; Figure S2). The transgene cassette consists of a chicken β-actin promoter with a cytomegalovirus enhancer driving the expression of a floxed membrane localized Tomato tandem dimer (mT). Cre-mediated recombination excises the mT transgene, driving expression of membrane-localized eGFP (mG). Genotyping was carried out via fluorescence imaging and polymerase chain reaction (PCR) genotyping.
DNA for genotyping was prepared from ear biopsies. Litters were ear-tagged and clipped at 7-12 days post-birth. Presence of different alleles was assessed by PGR using the primer pairs available in Figure S3 (all purchased from Integrated DNA Technologies). PGR products were electrophoresed on a 1.5% agarose gel containing ethidium bromide, and amplified fragments were visualized under UV transillumination (Figure S4).
2.4 |. In vivo PTS implantation and explant sample collection
PTS were rehydrated and implanted subcutaneously in mice. Using aseptic technique, mice were anesthetized with isoflurane and a mid-sagittal dorsal incision 1.5 cm long was made. PTS were inserted on both sides of the incision under the skin, ensuring that the scaffold laid flat and did not fold. The wounds were sealed with wound clips. The scaffolds were removed at 1 week and cultured for 2 days in Roswell Park Memorial Institute (RPMI) 1640 containing 10% fetal bovine serum (FBS) and 1% penicillin streptomycin in 12-well plates. Cells recovered from the scaffold were separated by centrifugation. The cell-free conditioned medium was processed for sEV isolation and ELISA analysis.
2.5 |. PTS resident cell population profiling
White blood cell differentials were performed on cells recovered from PTS after hemolysis and debris removal with a 40-μm cell strainer. Briefly, 200 μl of cells were put into a Simport plastic funnel (ThermoFischer) with an attached slide and spun for 5 min at 750 rpm on a Cytospin3. The cells were then fixed with methanol and stained using Hema 3 Stat Pack (Fisherbrand). The number of MØ, lymphocytes, basophils, eosinophils, and neutrophils was quantified until 200 total cells were counted.
2.6 |. Tissue histology
Explanted PTS were embedded in optimal cutting temperature compound (OCT)_(Tissue-Tek Sakura, VWR), and snap frozen using liquid nitrogen. OCT blocks were cut with a cryotome into 5-μm sections. The tissue sections were then stained for collagen (blue), nuclei (black), and cytoplasmic spaces (red) using a Masson’s Trichrome stain (Abcam) as per manufacturer’s protocol. Histo-Clear II (National Diagnostics) was used as a clearing agent, the slides were mounted using Omnimount (National Diagnostics) and covered with a 1.5-mm cover slips.
2.7 |. EV isolation, purification, and characterization
PTS were explanted after 1 week and placed in EV-free medium for 48 h. sEVs were isolated using Invitrogen’s Total Exosome Isolation Kit (Catalog number 4484450) after centrifugation of the conditioned medium at 2000 × g for 30 min to eliminate cell debris as per manufacturer instructions. Upon final PBS resuspension, EV quality and concentration were assessed via nanoparticle tracking analysis (NTA) (NanoSight™, Malvern Panalytical Inc.) and ImageStream® imaging flow cytometric analysis. NTA was performed using three 60-s captures per sample after vigorous vortexing, and a camera level of 12 and a detection threshold of 3. Particles were resuspended in PBS and diluted further to reach the acceptable concentration range of NTA quantification. SEV presence and sample integrity were based on measured particle size (under 150 nm without contaminating medium EVs [mEVs] or large EVs [IEVs]). Imaging flow cytometric analysis of sEVs from double transgenic PTS explants was performed on an Amnis Image-Stream® (Luminex, Inc.). sEVs were diluted 1:100 in sterile-filtered PBS. All events were recorded in each sample. Fluidics were set to slow, sensitivity to high, and observations were taken at 60×. Compensation matrices were generated based on the results from control fluorescent beads. Explanted PTS-derived sEVs were gated on size and fluorescence signal obtained from calibration beads and analyzed for the presence of EGFP and mTomato in order to assess myeloid or non-myeloid lineage. RNAseq was also performed on total RNA isolated from PTS cell-derived sEVs (Maverix Biomics) to quantify EV small RNA contents (i.e., miRNA and tRNA).
2.8 |. In vitro assessment of sEV signaling
Immortalized T lymphocytes (EL-4 Cells, ATCC) were cultured according to manufacturer guidelines. Briefly, EL-4 cells were cultured in T-75 culture flasks with Dulbecco’s Modified Eagle’s medium (containing 4.5 mg/ml, 4 mM L-glutamine, 1 mM sodium pyruvate, and 1.5 mg/ml sodium bicarbonate, Gibco) with 10% horse serum and 1% penicillin streptomycin. The cells were kept at a concentration between 1 × 105 and 1 × 106 cells/ml. sEV-depleted FBS generated via overnight ultracentrifugation at 100,000 g was used when tissue culture for sEV generation or sEV treatment was required. EL-4 cells were treated with explanted PTS-derived sEVs from double transgenic mice. sEVs were added to affect a final concentration of 7.13 × 105 sEVs/ml. EL-4 cells in a 12-well plate were exposed to sEVs for 1 hour in medium as described above. Cells were then centrifuged at 300 g for 15 min, and the pellet was resuspended in PBS. The cell suspensions were analyzed with the ImageStream. While signal from non-myeloid (mTomato red) sEVs was too weak for meaningful analysis due to ImageStream laser limitations, the myeloid (EGFP) signal was strong. The SPOT tool was then used to determine co-localization of fluorescent sEVs with the EL-4 cells.
2.9 |. Primary splenic T cell harvest and viability assessment
Primary splenic T cells were isolated using CD3+ immunomagnetic separation. Briefly, spleens were removed from euthanized C57Bl/6 mice and homogenized in PBS. The homogenized spleen cells were placed in 15 ml of hemolytic buffer and incubated at room temperature for 15 min and strained through a 40-μm cell strainer. The cells were exposed to biotinylated CD3+ antibody and streptavidin coated magnetic beads and run through an LS™ column as per Miltenyi’s instructions. The positive fraction was resuspended in RPMI 1640 (10% FBS, 1% penicillin streptomycin, 100 IU/ml IL-2 [Pro-Leukin from Novartis]), and then cultured in a plate coated with CD3 and CD28 antibodies for costimulation. After 48 h, the cells were removed for experimentation.
Primary splenic T cells were loaded into a 96-well plate at a concentration of 1 × 105 cells/well and stimulated for 48 h in either stimulatory (CD3 and CD28 antibody coated plate) or non-stimulatory conditions (PBS without antibodies). After 48 h, the cells were treated with PTS cell-derived sEVs at an average concentration of 7.13 × 105 sEVs/ml or PBS, and 48 h later, they were stained with trypan blue (Gibco). The live and dead cells were counted.
2.10 |. Transcriptional analysis using RT-qPCR
Cell and sEV samples recovered from the explanted PTS were processed with RNeasy Mini kit as per manufacturer’s protocol (Qiagen) and used to generate cDNA as per manufacturer’s protocol (Applied Biosciences). sEVs were lysed in 2.5% Triton X-100 and 0.05% Tween-20 before transfer into RLT™ buffer (Qiagen) (Álvarez et al., 2018). qPCR was performed with SYBR green (Macrophage panel) and FAM (T Cell Panel) probes on an ABI 7900HT (thermal cycling and primer/probe sequences used included in supplementary information). The T Cell panel included GAPDH, FOXP3, IL-10, IFN-γ, TGFβ1, TNFα, RORC, GATA3, and TBX21 (Table 1a). The MØ panel included PPIA, ARG-1, CD86, CD206, FIZZ-1, IL-10, iNOS, TGFβ, TNFα, and YM1 (Table 1b). Rationale for these panels can be seen in Table S1. Ct was considered undetermined if one or both replicate failed to amplify. Results were expressed in relative fold expression by normalizing against housekeeping genes (GAPDH or PPIA) and expressed as 2(−ΔCt) where −ΔCt is Cttarget — Cthousekeeping*
TABLE 1.
Genes and associated cell types of T cell (a) and macrophage (b) qPCR panels
| T Cell qPCR Panel (a) | |
|---|---|
| Gene | Indicated cell type |
| FoxP3 | Treg |
| IL-10 | Tr1 |
| IFN-γ | Th1 |
| TNFα | Th1 |
| TBX21 | Th1 |
| GATA3 | Th2 |
| TGF-β1 | Th3 |
| RORC | Th17 |
| Macrophage qPCR panel (b) | |
| Gene | Indicated cell type |
| ARG-1 | M2-like |
| CD86 | M1-like |
| CD206 | M2-like |
| FIZZ-1 | M2-like |
| IL-10 | M2-like |
| iNOS | M1-like |
| TGF-β1 | M2-like |
| TNFα | M1-like |
| YM1 | M2-like |
2.11 |. FoxP3+ cell depletion
Foxp3DTR mutant mice express knocked-in human diphtheria toxin receptor and eGFP genes from the Foxp3 locus, without disrupting expression of the endogenous FoxP3 gene. FoxP3DTR mice (Jackson Labs strain B6.129[Cg]-Foxp3tm3 [DTR/GFP]Ayr/J) were treated with 1 μg of diphtheria toxin (Sigma Aldrich) in 100 μL of sterile saline via intraperitoneal injection once every 3 days. Scaffolds were implanted 1-day post-injection. Mice were weighed daily to ensure no more than 15% of body weight was lost during treatment.
2.12 |. Statistical analyses
Shapiro-Wilk tests were performed to assess the normality with α = 0.10. Parametric tests included Studen’s t-test performed at α = 0.05 for comparison between two sample populations. If there were multiple comparisons (three or more sample groups), an ANOVA with Tukey-Kramer post-hoc was performed at α = 0.05.
For data that were non-parametric tests, comparisons between two sample populations were assessed via Mann-Whitney U test performed at α = 0.05. If there were multiple comparisons (three or more sample groups), a Kruskal-Wallis test with a Conover-Iman post-hoc test corrected for false discovery rate using a two-stage Benjamini-Hochberg correction. Figures were created in Microsoft Excel and Python version 3.7.0. In box and whisker plots, boxes show the median bound by the first and third quartiles while whiskers denote the minimum and maximum data points within 1.5 times the interquartile range (IQR). Values not within 1.5 times the IQR were denoted with a diamond. Error bars in bar graphs reflect the standard error of the mean. Asterisks denote significance (i.e., p-values under 0.05).
3 |. RESULTS
3.1 |. Immune cell populations are independent of PTS pore size, but 40-μm PTS induce unique cellular transcriptomic effects consistent with replicated pro-healing responses upon implantation
Immune cell population profiles in PTS explanted after 1 week were not a function of pore size when assessed with a white blood cell differential. Both 40- and 100-μm PTS displayed a predominant (~85%) MØ population and similar lymphocyte, eosinophil, and neutrophil relative populations (Figure 1). 40-μm PTS demonstrated reduced fibrotic encapsulation and increased implant cellularization as visualized with Masson’s Trichrome Stain (Figure S5), which is consistent with previous results (Madden et al., 2010). Further studies examining transcriptional differences between PTS-derived cells and sEVs were carried out, revealing (Figure 2) cells inhabiting 40-μm PTS after 1 week of implantation significantly upregulated ARG-1, FIZZ-1, and CD86 expression (ARG-1 p = 0.041, FIZZ-1 p = 0.03, CD86 p = 0.023, 40 μm n = 6, and 100 μm PTS n = 5).
FIGURE 1.
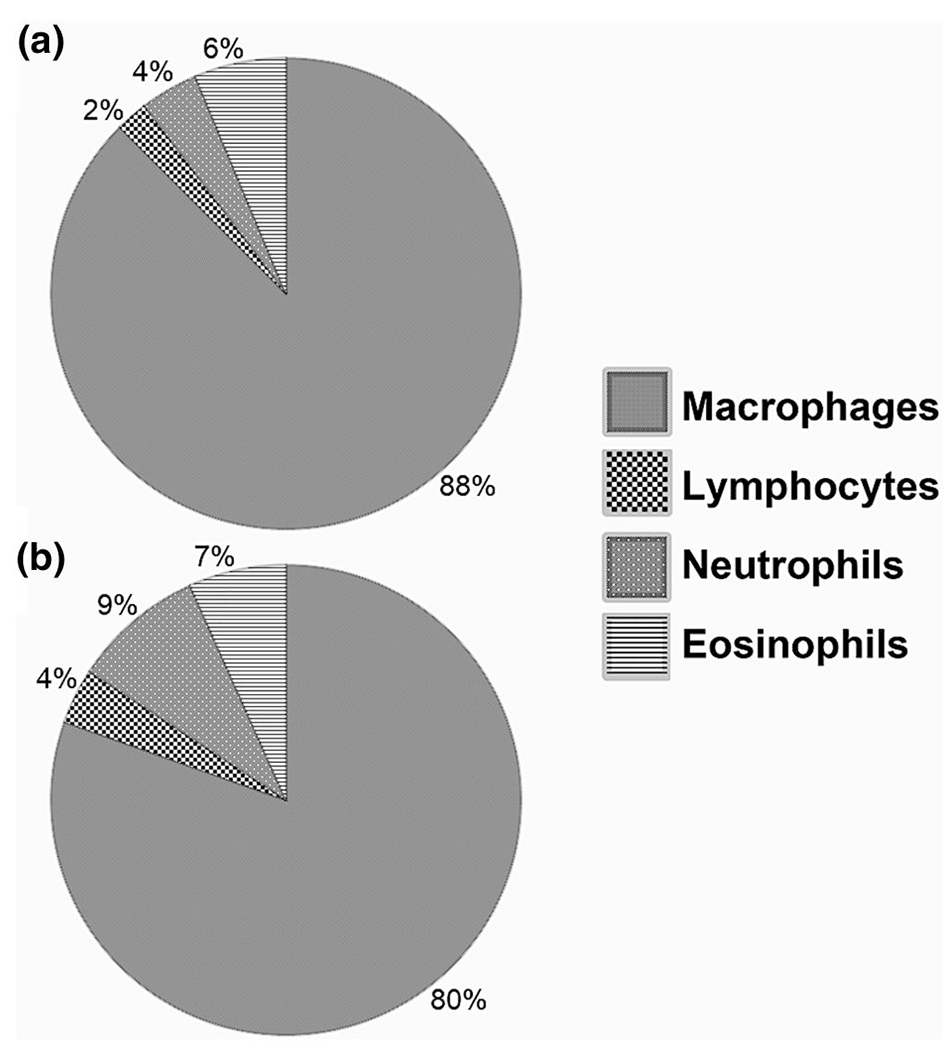
Proportional populations of precision templated scaffolds (PTS) resident leukocytes of different pore sizes as measured by a white blood cell differential after 1 week of subcutaneous murine implantation in 40-μm (a) and 100-μm (b) PTS. No significant difference was found in leukocyte population proportions as a result of changing PTS pore size
FIGURE 2.
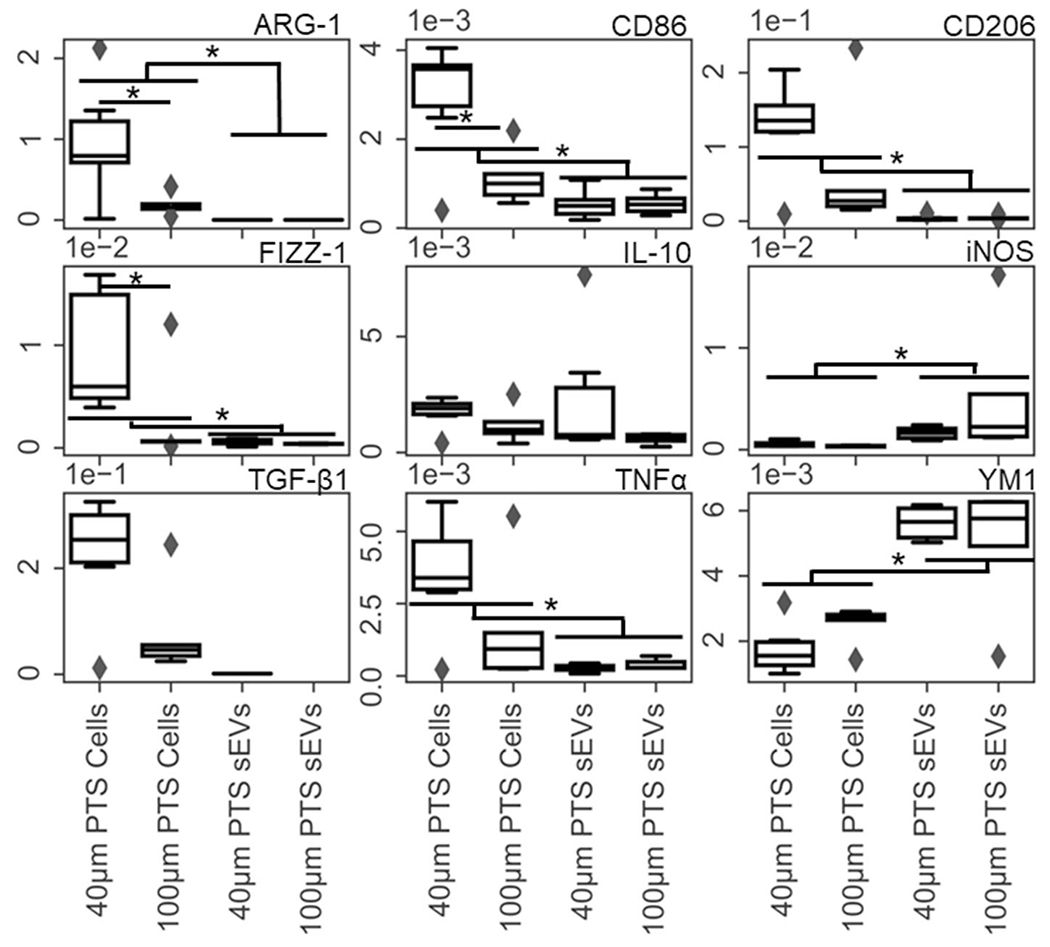
RT-qPCR was performed on precision templated scaffolds (PTS) resident cells and their small extracellular vesicles (sEVs) isolated from 1 week subcutaneously implanted 40- and 100-μm PTS. Gene targets were the macrophage panel denoted in Table 1. Transcriptional differences between cells and sEVs isolated from 40- and 100-μm pore PTS explanted from LysM-Cre+/0:mT/mG+/0 mice after 1 week detailed differential gene expression in both cells and their sEVs as a function of pore size (*p < 0.05, n = 6 for 40-μm PTS and n = 5 for 100-μm PTS)
3.2 |. PTS cell-derived sEV cargo is unique compared to parent cell mRNA contents
sEVs isolated from cells in explanted PTS had average modal sizes of 124.2 nm [109.1-139.3 nm] and 149.7 nm [132.0-164.7 nm] for 40- and 100-μm scaffolds, respectively, when examined via NTA (Figure S6). These results indicate that the sEV populations isolated from cellularized PTS were correctly sized and free of mEVs, IEVs, or cellular debris contamination. ImageStream analysis of sEVs generated from both 40- and 100-μm PTS explanted from double transgenic (LysM-Cre+/0:mT/mG+/0) mice indicates that about ~58% of the sEVs produced were of myeloid lineage (Figure S7). Figure 2 shows sEV iNOS (indicative of M1-like macrophages) and YM1 (indicative of M2-like macrophages) were significantly upregulated in PTS cell-derived sEVs compared to their parent cells (YM1 p = 0.00025, iNOS p = 0.000069, 40-μm PTS n = 6 and 100-μm PTS n = 5). Conversely, FIZZ1, CD206, ARG-1, CD86, and TNFα were upregulated in PTS resident cells compared to their sEVs (FIZZ-1 p = 0.022, CD206 p = 0.000,053, ARG-1 p = 0.00,004, CD86 p = 0.0012, and TNFα p = 0.0092). These findings indicate both the impact of PTS pore size on the cellular transcriptome and preferential packaging of specific nucleic acid cargo to released sEVs.
3.3 |. Myeloid sEVs from explanted PTS co-localized with immortalized EL-4 T cells
PTS cell-derived sEVs explanted from double transgenic mice were applied to EL-4 cells in vitro. We visualized these sEVs co-localized upon the membrane of EL-4 cells using ImageStream imaging, suggesting a potential interaction between T lymphocytes and PTS-derived myeloid sEVs. The data showed that 10%-12% of EL-4 T cells, sometimes in dividing T lymphocytes, were co-localizing with one or more myeloid sEVs (representative image in Figure 3). These findings supported further investigating the capability of PTS cell-derived sEVs to induce immunomodulation by influencing T cell phenotype via non-traditional cellular mechanisms (i.e., free cytokine and direct cell-cell signaling) in a pore-size-dependent manner.
FIGURE 3.
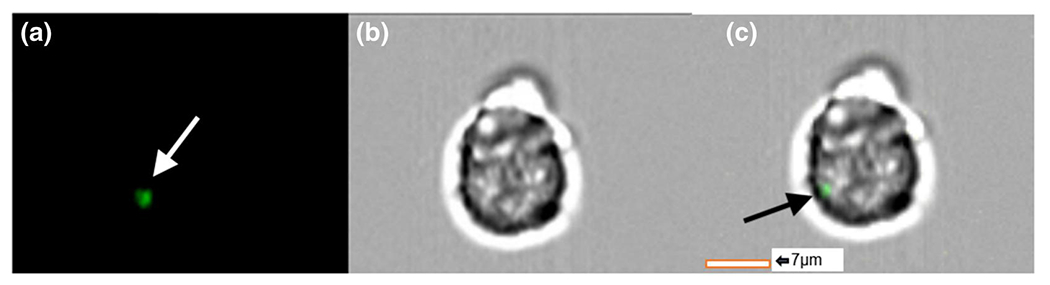
Imaging flow cytometric images of EL-4 immortalized T cells co-cultured with precision templated scaffolds cell-derived small extracellular vesicles (sEVs) explanted after 1 week from double transgenic LysM-Cre+/0:mTmG+/0 mice. Green myeloid sEVs can be seen co-localized with EL-4 cells; (a) green fluorescent protein channel, (b) brightfield, (c) overlay [Colour figure can be viewed at wileyonlinelibrary.com]
3.4 |. SEVs from PTS resident cells affect the transcriptome and viability of T cells in pore-size-dependent and-independent manners
When compared to an untreated control, EL-4 cells treated with either 40-μm or 100-μm PTS cell-derived sEVs significantly upregulated IL-10 (Figure 4) (40-μm PTS cell-derived sEVs p = 0.00012 and 100-μm PTS cell-derived sEVs p = 0.000043) and IFN-γ (40- and 100-μm PTS cell-derived sEVs; p = 0.001) (sEV treated cells n = 4 and untreated cells n = 10). TBX21 expression was significantly upregulated in EL-4 cells treated with explanted 100-μm PTS cell-derived sEVs when compared with untreated control (Figure 4) (p = 0.0086, sEV treated cells n = 4 and untreated cells n = 10). These results demonstrate that sEVs generated from PTS resident cells can broadly stimulate T cell transcriptomes independent of pore size. Furthermore, these results indicate an increased inflammatory response shown by TBX21 upregulation as a result of 100-μm PTS cell-derived sEV treatment. Regrettably, FoxP3 (Treg indicator) was not measured in this experiment as EL-4 cells do not express FoxP3 unless stimulated with both CD3 and TGF-β1 agonists (Tone et al., 2008).
FIGURE 4.
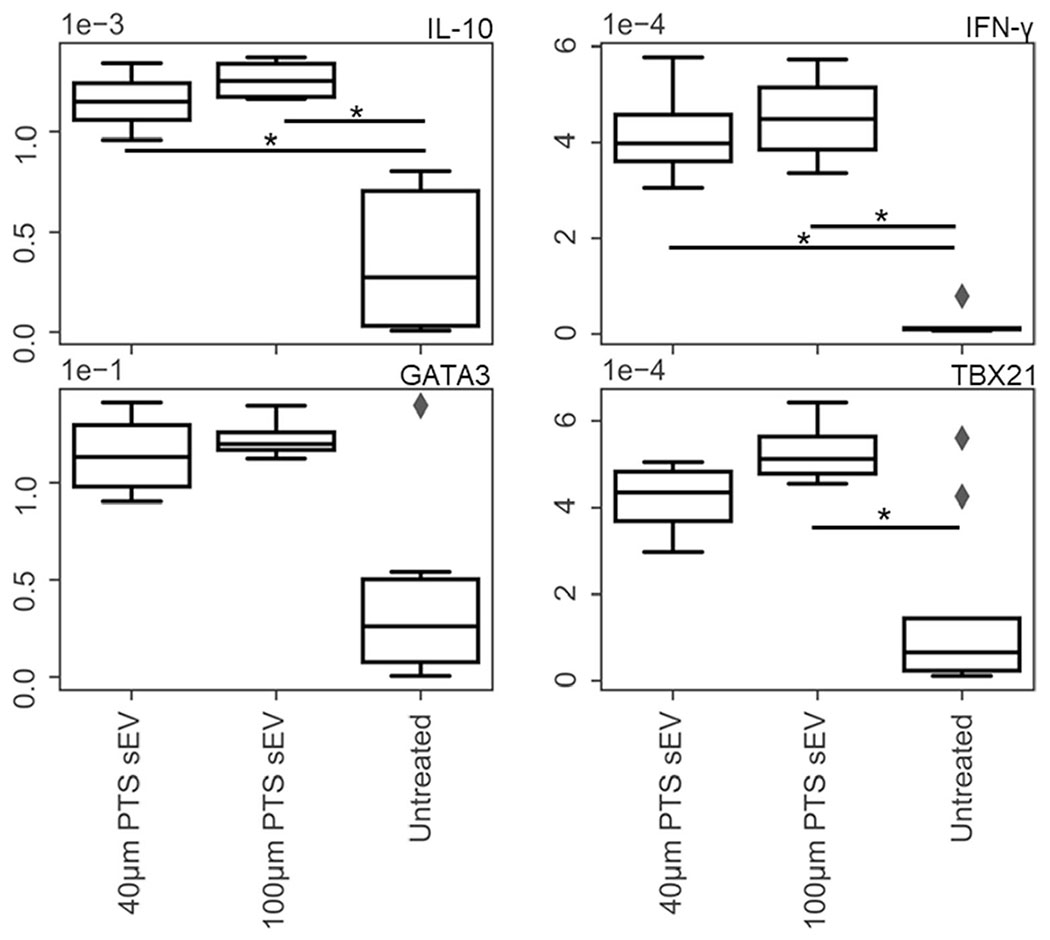
RT-qPCR performed on EL-4 (immortalized T cell line) cells treated with precision templated scaffolds (PTS) cell-derived small extracellular vesicles (sEVs) from PTS implanted for 1 week. Gene targets were chosen using the T cell panel denoted in Table 1. The transcriptional response of the immortalized T cells exposed to 40- and 100-μm PTS cell-derived sEVs is upregulated compared to the untreated cells, and the 40-μm PTS cell-derived sEV treatment caused differential gene regulation compared to the 100 μm PTS cell-derived sEV treatment (*p < 0.05, n = 4 for sEV treated and 10 for untreated)
Transcriptomic differences in primary T cells were characterized via RT-qPCR. Naïve splenic T cells cultured with sEVs generated from cells inhabiting explanted 40- and 100-μm PTS both upregulated FoxP3 in comparison to untreated cells (N = 5, p = 0.035, and p = 0.027, respectively). SEVs from both cells inhabiting explanted 40- and 100-μm PTS upregulated TBX21 compared to untreated samples (N = 5, p = 0.014, and p = 0.001, respectively). These data (Figure 5) demonstrate PTS cell-derived sEVs mediate transcriptomic stimulation on primary T cells. Afterwards, we quantified the stimulatory effects of PTS cell-derived sEVs on primary T cell proliferation. We collected/harvested sEVs derived from 40- and 100-μm PTS cell-derived sEVs from LysM-Cre+/0: mTmG+/0 mice were applied to CD3/CD28 stimulated and unstimulated (PBS only) primary splenic T cells. Samples treated with both the stimulatory condition and PTS cell-derived sEVs demonstrated statistically significant increased cellular viability compared to untreated primary T cells (Figure 6) (p = 0.007 and p = 0.0051 for 40- and 100-μm PTS cell-derived sEV treated, co-stimulated n = 12 and non-co-stimulated n = 4). These data further support our assertion that PTS cell-derived sEVs stimulate primary T cells regardless of PTS pore size.
FIGURE 5.
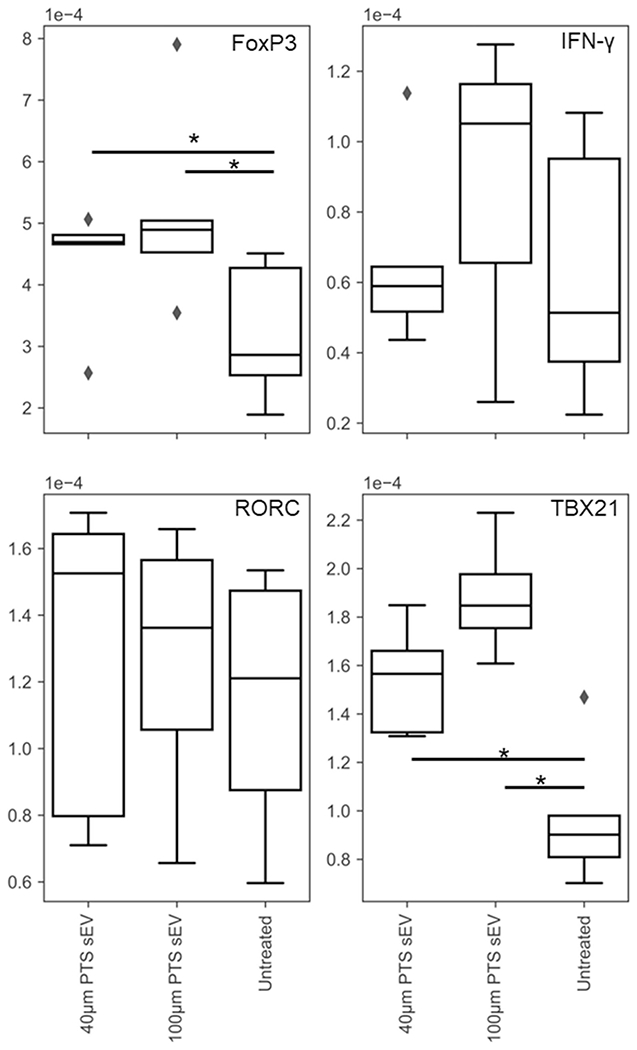
RT-qPCR performed on stimulated murine primary splenic T cells treated with 40- and 100-μm precision templated scaffolds resident cell-derived sEVs for 48 h. Gene regulation of transcripts detailed in Table 1 indicate differential responses as a result of small extracellular vesicles treatment in pore-size-dependent and -independent manners (* < 0.05, n = 5)
FIGURE 6.
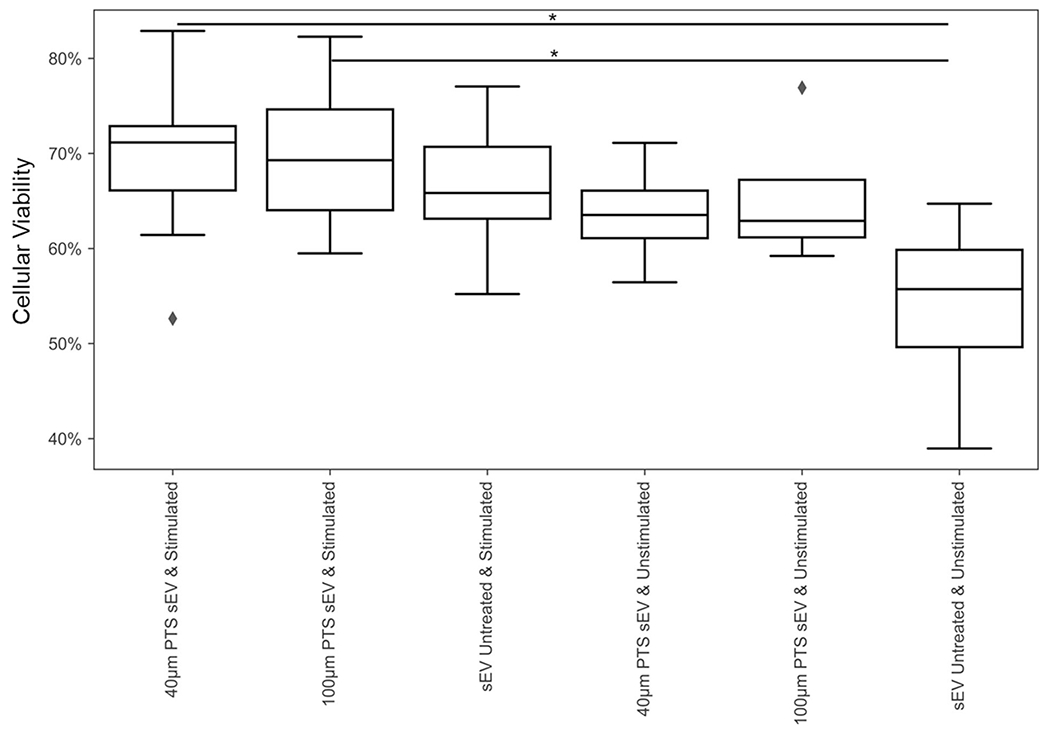
In vitro effects of CD3/CD28 antibody stimulation and precision templated scaffolds derived small extracellular vesicles treatment on CD3+ splenic T cell viability measured via trypan blue cell counts after 48-h culture post-isolation. No significant difference in the total number of cells was noted (data not shown; *p < 0.05. n = 4 for treated and n = 12 for untreated)
3.5 |. FoxP3+ cells play a role in PTS-mediated pro-healing response
Given the importance of regulatory T cells in modulating immune response, we implanted 40- and 100-μm PTS into wild-type and FoxP3DTR mice for 1 week. In cells from wild-type (C57/BL6) mice, 40-μm pores caused the upregulation of TBX21 (p = 0.0055, n = 4), GATA3 (p = 0.0043, n = 6), and FoxP3 (p = 0.041, n = 5; Figure 7). FoxP3+ depletion caused a down-regulation of TBX21 mRNA in cells inhabiting 40-μm PTS (p = 0.00015, n = 4 wild type and n = 3 double transgenic) and abrogated the differences in TBX21 and GATA3 expression between 40- and 100-μm PTS cells. While not statistically significant (p = 0.074), it appears that GATA3 expression may also tend toward upregulation in cells inhabiting 100-μm PTS upon FoxP3 depletion. Finally, FoxP3 depletion also caused a significant down-regulation in IL-10 in cells within both sizes of PTS (40-μm PTS, p = 0.0022; and 100-μm PTS, p = 0.016, n = 4). These results indicate that Tregs are important in maintaining pro-healing immune conditions in PTS implants.
FIGURE 7.
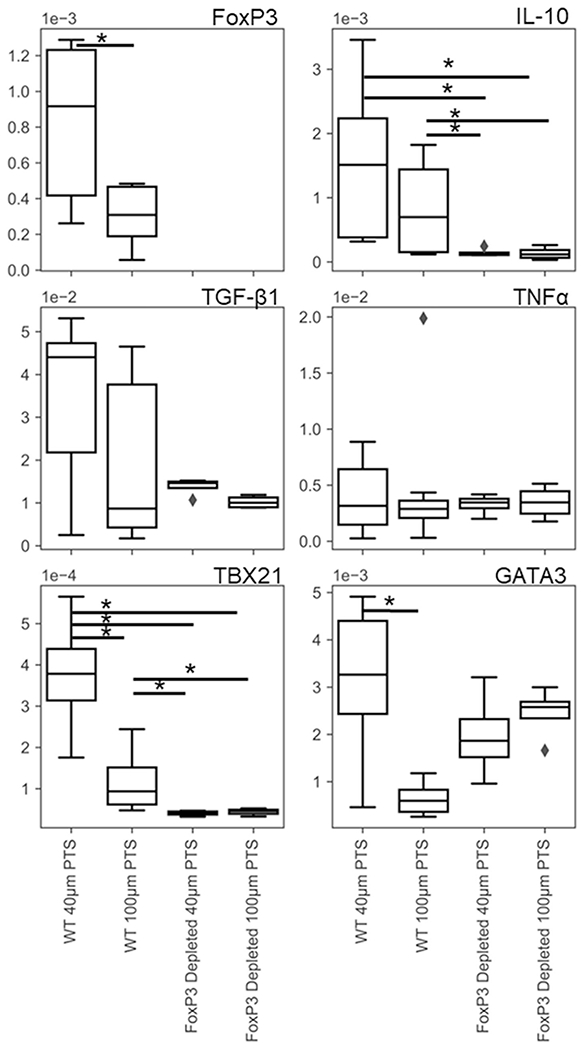
RT-qPCR on 40- and 100-μm precision templated scaffolds resident cells after 1 week of implantation in wild-type and FoxP3DTR mice treated with diphtheria toxin reveals FoxP3 T-cell-dependent transcriptional regulation in primary splenic T cells. Genes of interest were determined using the T cell panel in Table 1 (*p < 0.05, n = 6 wild type and n = 4 FoxP3DTR)
4 | DISCUSSION
Although pore size had no effect on broad immune population recruitment to the PTS after 1 week (Figure 1), our data corroborate previously demonstrated pro-healing responses in 40-μm PTS (Figure S5) (Fukano et al, 2006, 2010; Isenhath et al., 2007; Sussman, Halpin, Muster, Moon, & Ratner, 2014). These observations are supported by transcriptional data (Figure 2) that reveals that PTS resident cell populations and sEV characteristics were pore size dependent. Total RNA-seq expression analysis was carried out on SEVs isolated from explanted 40- and 100-μm-pore-size PTS. Figure S8 shows a dramatic abundance of miRNAs sequences within the 40-μm PTS-derived sEVs versus sEVs generated by cells within 80-μm PTS. Cells in 40-μm PTS upregulated traditionally anti-inflammatory and T cell stimulatory mRNA expression (ARG-1, FIZZ-1, and CD86). Additionally, PTS cell-derived sEVs demonstrated differential nucleic acid contents compared to their parent cells through increased mRNA expression of the traditionally anti-inflammatory and inflammatory markers YM1 and iNOS. However, PTS resident cells themselves demonstrated unique transcriptomic contents through greater expression of other transcripts (ARG-1, CD86, CD206, FIZZ-1, and TNFα). It is important to note that while the proportion of myeloid cells in both 40- and 100-μm PTS totaled ~85%, they produced ~55%-60% of the observed sEVs. Thus, while the majority of PTS cell-derived sEVs were myeloid, the number of sEVs generated per cell is not necessarily a linear constant, as sEVs of the same immune lineage may have markedly unique biological contents and can affect multiple cells via the endocytic pathway.
After visualizing physical contact of myeloid PTS cell-derived sEVs with EL-4 cells (Figure 3), transcriptional analysis demonstrates (Figure 4) that sEVs from cells in 40- and 100-μm PTS impact T cell gene expression. Independent of pore size, PTS cell-derived sEVs broadly stimulated the immortalized T cell gene expression (upregulated IL-10 and IFN-γ) while sEVs from cells inhabiting 100-μm PTS cause a significant upregulation of inflammatory gene expression (TBX21). While initially counterintuitive, concomitant IL-10 and IFN-γ expression has been linked to bystander Treg population activation in chronic inflammatory environments (Trinchieri, 2001). Broad sEV stimulation thus occurs in all pore sizes in a manner implicating Treg activation while the transcriptome of T cells treated with 100-μm PTS cell-derived sEVs was more classically inflammatory, quantified by Th1 marker mRNA.
When naïve primary T cells were treated with sEVs from cells in explanted 40- and 100-μm PTS, they demonstrated both increased proliferative stimulation (Figure 6) and broad transcriptional upregulation (higher FoxP3 and TBX21 expression than untreated cells in Figure 5). Consistent with prior results in immortalized T cells, these results indicate that sEVs from PTS were causing broad changes in transcriptional regulation in resident T cells, the balance between Th1 cells, and other T cell subsets affects the healing response.
Due to their potent regulation of CD4+ T cell subsets, we investigated the role of Tregs in the host response to PTS. The change in Th1 markers upon Treg depletion (Figure 7) in cells inhabiting 40-μm PTS concurrent with the abrogation of pore-size-mediated Th1 and Th2 marker expression in FoxP3 depleted PTS cells implicates Tregs as instrumental in the pore-size-mediated pro-healing response to PTS. Th2 cells enhance fibrosis and counteract Th1 inflammation, whereas Th1 cells enhance inflammation and counteract Th2 mediated fibrosis, so a balance of the two is necessary for optimal healing response (Wynn, 2008). Regulatory T cells in the PTS counteract the tendency of 40-μm pores to reduce the inflammatory, anti-fibrotic Th1 response which would otherwise enable excessive Th2 mediated fibrosis. Regulatory T cells may also counteract the tendency of 100-μm PTS cells to enhance Th2 responses, which would otherwise become highly fibrotic - though additional experimentation is necessary to prove this definitively. Tregs are thus crucial to the pore-size-mediated healing responses observed in the PTS, through the regulation of the Th1/Th2 ratio to enable healing without fibrosis or excessive inflammation. These data both elucidate the role of Tregs in pore-size-dependent host response and distinguish the transcriptional changes in T cells observed as a result of primarily myeloid PTS cell-derived sEV treatment.
In this study, we quantified the cellular origin and internal composition of sEVs isolated from cells inhabiting explanted 40- and 100-μm PTS and assessed the signaling effect of those sEVs on T cell response. We demonstrated that despite immune cell populations and sEV lineage of explanted PTS remaining unaffected by pore size, gene expression between cells and sEVs differs between 40- and 100-μm PTS. Further, we showed sEVs, primarily myeloid in lineage isolated from PTS, stimulate both the transcriptome and viability of immortal and primary naïve splenic T cells in a pore-size-dependent manner. Pointedly, while sEVs generated from cells in either pore size PTS stimulate immunologically important genes such as FoxP3, IL-10, GATA3, and IFN-γ in T cells, 100-μm PTS cell-derived sEVs cause increased inflammatory transcriptional effects through elevated TBX21 expression. Finally, we demonstrated the importance of FoxP3+ cells in implanted PTS through the distinct ability of Tregs to balance transcriptomically indicated inflammatory Th1 populations with fibrotic Th2. We have thus shown that the effect of PTS pore size on regeneration capacity may be partially mediated by predominantly myeloid sEV signaling to T cells in the implant environment. On-going work will examine the ability of sEVs generated from cells inhabiting 40-μm PTS to induce functional immunomodulation and explore possible mechanisms of sEV interaction with T cells.
Supplementary Material
ACKNOWLEDGMENTS
Funding for this work was provided by the NIH (Grant #: NIDCR 5R01DE018701-10 and NIGMS 1R01GM128991-01). Thank you to the UW Molecular Analysis Facility and Cell Analysis Facility for SEM and flow cytometry expertise and instrumentation respectively. The authors also express gratitude to the Buddy Ratner lab (University of Washington Engineered Biomaterials, UWEB) for the use of PTS synthesis lab equipment.
Funding information
National Institute of Dental and Craniofacial Research, Grant/Award Number: 5R01DE018701-10; National Institute of General Medical Sciences, Grant/Award Number: 1R01GM128991-01
Footnotes
CONFLICT OF INTEREST
Healionics’ is a clinical stage Seattle-based company developing proprietary STAR® (Sphere Templated Angiogenic Regeneration) silicone scaffolds for various applications in biointegration and tissue regeneration. Buddy Ratner is on the Board of Directors of Healionics. James Bryers is a scientific advisor to Healionics. The authors have no conflicts of interest to report at this time.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
REFERENCES
- Ali N, Zirak B, Rodriguez RS, Pauli ML, Truong H-A, Lai K, … Rosenblum MD (2017). Regulatory T cells in skin facilitate epithelial stem cell differentiation. Cell, 169(6), 1119–1129. 10.1016/j.cell.2017.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JE, & Wynn TA (2011). Evolution of Th2 immunity: A rapid repair response to tissue destructive pathogens. PLoS Pathogens, 7(5), e1002003. 10.1371/journal.ppat.1002003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Álvarez V, Sánchez-Margallo FM, Macías-García B, Gómez-Serrano M, Jorge I, Vázquez J, … Casado JG (2018). The immunomodulatory activity of extracellular vesicles derived from endometrial mesenchymal stem cells on CD4+ T cells is partially mediated by TGFbeta. Journal of Tissue Engineering and Regenerative Medicine, 12(10), 2088–2098. 10.1002/term.2743 [DOI] [PubMed] [Google Scholar]
- Beissert S, Schwarz A, & Schwarz T (2006). Regulatory T cells. Journal of Investigative Dermatology, 126(1), 15–24. 10.1038/sj.jid.5700004 [DOI] [PubMed] [Google Scholar]
- Bhatnagar S, Shinagawa K, Castellino FJ, & Schorey JS (2007). Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo. Blood, 110(9), 3234–3244. 10.1182/blood-2007-03-079152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockmann L, Giannou AD, Gagliani N, & Huber S (2017). Regulation of TH17 cells and associated cytokines in wound healing, tissue regeneration, and carcinogenesis. International Journal of Molecular Sciences, 18(5), 1033. 10.3390/ijms18051033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown BN, Londono R, Tottey S, Zhang L, Kukla KA, Wolf MT, … Badylak SF (2012). Macrophage phenotype as a predictor of constructive remodeling following the implantation of biologically derived surgical mesh materials. Acta Biomaterialia, 8(3), 978–987. 10.1016/j.actbio.2011.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castiglioni A, Corna G, Rigamonti E, Basso V, Vezzoli M, Monno A,, … Rovere-Querini P (2015). FOXP3+ T cells recruited to sites of sterile skeletal muscle injury regulate the fate of satellite cells and Guide effective tissue regeneration. PloS One, 10(6), e0128094. 10.1371/journal.pone.0128094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung L, Maestas D, Lebid A, Mageau A, Rosson GD, Wu X, … Elisseeff JH (2020). Interleukin-17 and senescence regulate the foreign body response. Science Translational Medicine, 15(12), 539. 10.1101/583757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH,Britos MF, … King LS (2009). CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. Journal of Clinical Investigation, 119(10), 2898–2913. 10.1172/JCI36498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt E, Toksoy A, Goebeler M, Debus S, Bröcker E-B, & Gillitzer R (1998). Chemokines IL-8, GROα, MCP-1, IP-10, and MIG are sequentially and differentially expressed during phase-specific infiltration of leukocyte subsets in human wound healing. American Journal of Pathology, 153(6), 1849–1860. 10.1016/S0002-9440(10)65699-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukano Y, Knowles NG, Usui ML, Underwood RA, Hauch KD, Marshall AJ, … Olerud JE (2006). Characterization of an in vitro model for evaluating the interface between skin and percutaneous biomaterials. Wound Repair and Regeneration, 14(4), 484–491. 10.1111/j.1743-6109.2006.00138.x [DOI] [PubMed] [Google Scholar]
- Fukano Y, Usui ML, Underwood RA, Isenhath S, Marshall AJ, Hauch KD, … Fleckman P (2010). Epidermal and dermal integration into sphere-templated porous poly (2-hydroxyethyl methacrylate) implants in mice. Journal of Biomedical Materials Research A, 94(4), 1172–1186. 10.1002/jbm.a.32798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin A, Long TJ, Garty S, & Ratner BD (2013). Synthesis and fabrication of a degradable poly (N-isopropyl acrylamide) scaffold for tissue engineering applications. Journal of Biomedical Materials Research Part A, 101(3), 775–786. 10.1002/jbm.a.34380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin A, Oldinski RA, Florczyk SJ, Bryers JD, Zhang M, & Ratner BD (2013). Integrated Bi-layered scaffold for osteochondral tissue engineering. Advanced Healthcare Materials, 2(6), 872–883. 10.1002/adhm.201200345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri PK, & Schorey JS (2008). Exosomes derived from M. Bovis BCG infected macrophages activate antigen-specific CD4+ and CD8+ T cells in vitro and in vivo. PLoS One, 3(6), e2461. 10.1371/journal.pone.0002461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenhath SN, Fukano Y, Usui ML, Underwood RA, Irvin CA, Marshall AJ, … Olerud JE (2007). A mouse model to evaluate the interface between skin and a percutaneous device. Journal of Biomedical Materials Research Part A, 83(4), 915–922. 10.1002/jbm.a.31391 [DOI] [PubMed] [Google Scholar]
- Julier Z, Park AJ, Briquez PS, & Martino MM (2017). Promoting tissue regeneration by modulating the immune system. Acta Biomaterialia, 53, 13–28. 10.1016/j.actbio.2017.01.056 [DOI] [PubMed] [Google Scholar]
- Knowles NG, Miyashita Y, Usui ML, Marshall AJ, Pirrone A, Hauch KD, … Olerud JE (2005). A model for studying epithelial attachment and morphology at the interface between skin and percutaneous devices. Journal of Biomedical Materials Research Part A, 74(3), 482–488. 10.1002/jbm.a.30384 [DOI] [PubMed] [Google Scholar]
- Lamichhane TN, Sokic S, Schardt JS, Raiker RS, Lin JW, & Jay SM (2014). Emerging roles for extracellular vesicles in tissue engineering and regenerative medicine. Tissue Engineering B Reviews, 21(1), 45–54. 10.1089/ten.teb.2014.0300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang L, Kikuiri T, Akiyama K, Chen C, Xu X, … Shi S (2011). Mesenchymal stem cell-based tissue regeneration is governed by recipient T lymphocytes via IFN-γ and TNF-α. Nature Medicine, 17(12), 1594–1601. 10.1038/nm.2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden LR, Mortisen DJ, Sussman EM, Dupras SK, Fugate JA, Cuy JL, … Ratner BD (2010). Proangiogenic scaffolds as functional templates for cardiac tissue engineering. Proceedings of the National Academy of Sciences of the United States of America, 107(34), 15211–15216. 10.1073/pnas.1006442107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall A, Irvin CA, Barker T, Sage EH, Hauch KD, & Ratner B (2004). Biomaterials with tightly controlled pore size that promote vascular in-growth. Polymer Preprints (American Chemical Society, Division of Polymer Chemistry), 228, U386. [Google Scholar]
- Murphy TJ, Ni Choileain N, Zang Y, Mannick JA, & Lederer JA (2005). CD4+CD25+ regulatory T cells control innate immune reactivity after injury. The Journal of Immunology, 174(5), 2957–2963. 10.4049/jimmunol.174.5.2957 [DOI] [PubMed] [Google Scholar]
- Nazimek K, Ptak W, Nowak B, Ptak M, Askenase PW, & Bryniarski K (2015). Macrophages play an essential role in antigen-specific immune suppression mediated by T CD8+ cell-derived exosomes. Immunology, 146(1), 23–32. 10.1111/imm.12466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosbaum A, Prevel N, Truong H-A, Mehta P, Ettinger M, Scharschmidt TC, … Rosenblum MD (2016). Cutting edge: Regulatory T cells facilitate cutaneous wound healing. The Journal of Immunology, 196(5), 2010–2014. 10.4049/jimmunol.1502139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okoye IS, Coomes SM, Pelly VS, Czieso S, Papayannopoulos V, Tolmachova T, … Wilson MS (2014). MicroRNA-containing T-regulatory-cell-derived exosomes suppress pathogenic T helper 1 cells. Immunity, 41(1), 89–103. 10.1016/j.immuni.2014.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Franchi L, Nunez G, & Dubyak GR (2007). Nonclassical IL-1β secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. The Journal of Immunology, 179(3), 1913–1925. 10.4049/jimmunol.179.3.1913 [DOI] [PubMed] [Google Scholar]
- Quesenberry PJ, Aliotta J, Deregibus MC, & Camussi G (2015). Role of extracellular RNA-carrying vesicles in cell differentiation and reprogramming. Stem Cell Research & Therapy, 6. 10.1186/s13287-015-0150-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner BD, & Marshall A (2008). Novel porous biomaterials (United States Patent No. US20080075752A1). Retrieved from https://patents.google.com/patent/US20080075752A1/en
- Robbins PD, & Morelli AE (2014). Regulation of immune responses by extracellular vesicles. Nature Reviews Immunology, 14(3), 195–208. 10.1038/nri3622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadtler K, Estrellas K, Allen BW, Wolf MT, Fan H, Tam AJ, … Elisseeff JH (2016). Developing a pro-regenerative biomaterial scaffold microenvironment requires T helper 2 cells. Science, 352(6283), 366–370 10.1126/science.aad9272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, & Ono M (2008). Regulatory T cells and immune tolerance. Cell, 133(5), 775–787. 10.1016/j.cell.2008.05.009 [DOI] [PubMed] [Google Scholar]
- Sicari BM, Rubin JP, Dearth CL, Wolf MT, Ambrosio F, Boninger M, … Badylak SF (2014). An acellular biologic scaffold promotes skeletal muscle formation in mice and humans with volumetric muscle loss. Science Translational Medicine, 6(234) 234ra58. 10.1126/scitranslmed.3008085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman EM, Halpin MC, Muster J, Moon RT, & Ratner BD (2014). Porous implants modulate healing and induce shifts in local macrophage polarization in the foreign body reaction. Annals of Biomedical Engineering, 42(7), 1508–1516. 10.1007/S10439-013-0933-0 [DOI] [PubMed] [Google Scholar]
- Teng W, Long TJ, Zhang Q, Yao K, Shen TT, & Ratner BD (2014). A tough, precision-porous hydrogel scaffold: Ophthalmologic applications. Biomaterials, 35(32), 8916–8926. 10.1016/j.biomaterials.2014.07.013 [DOI] [PubMed] [Google Scholar]
- Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, & Tone M (2008). Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nature Immunology, 9(2), 194–202. 10.1038/ni1549 [DOI] [PubMed] [Google Scholar]
- Trinchieri G (2001). Regulatory role of T cells producing both interferon γ and interleukin 10 in persistent infection. Journal of Experimental Medicine, 194(10), f53–f57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirather J, Hofmann UDW, Beyersdorf N, Ramos GC, Vogel B, Frey A, … Frantz S (2014). Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circulation Research, 115(1), 55–67. 10.1161/CIRCRESAHA.115.303895 [DOI] [PubMed] [Google Scholar]
- Wynn T (2008). Cellular and molecular mechanisms of fibrosis. The Journal of Pathology, 214(2), 199–210. 10.1002/path.2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.