

Abstract
INTRODUCTION:
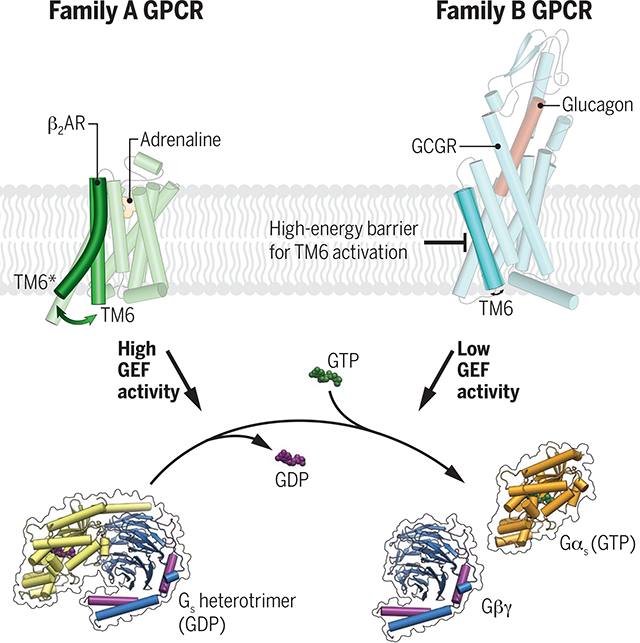
Transmembrane signaling of heterotrimeric guanine nucleotide–binding protein (G protein)–coupled receptors (GPCRs) is mediated by ligand-dependent conformational changes that are transmitted from the extracellular ligand binding site to the intracellular side of the receptor to allow coupling with transducers. One hallmark of GPCR activation is the outward movement of the cytoplasmic end of transmembrane domain 6 (TM6) that opens up an intracellular cavity to accommodate the Gα subunit, leading to nucleotide exchange and activation of the G protein. Comparison of family A and family B receptor-Gs protein complex structures has revealed substantial differences in the conformational changes of TM6 upon activation. In family B GPCRs, TM6 shows a disruption of the helical fold and the formation of a sharp kink. This differs from the gradual bending in TM6 observed in family A GPCRs.
RATIONALE:
Despite the recent surge of determined GPCR–G protein complex structures, the activation mechanism of family B receptors remains poorly understood. The missing conserved structural motifs found in family A GPCRs together with the marked differences in the conformation of TM6 in the active state suggest distinct activation mechanisms between family B and family A GPCRs. In particular, the disruption of the helical fold and the unraveling of the extracellular end of TM6 suggest that the energy required to produce the fully active-state of family B GPCRs is higher than for family A GPCRs. We investigated the functional impact of these structural differences by comparing the structure and function of a prototypical family B receptor, the glucagon receptor (GCGR), with the β2 adrenergic receptor (β2AR), a family A GPCR.
RESULTS:
We present the cryo–electron microscopy structure of the GCGR-Gs complex bound to an engineered soluble glucagon derivative. The structure shows that full activation of GCGR leads to a disruption in the α-helix of TM6 typical for family B GPCRs. Analysis of the functional consequence of this helix break on receptor-mediated G protein dissociation and guanosine triphosphate (GTP) turnover reveals that GCGR exhibits a substantially lower guanine nucleotide exchange factor (GEF) activity in comparison with the family A receptor β2AR. Characterization of G protein association, guanosine diphosphate (GDP) release, and GTP binding kinetics shows that the receptor-mediated GDP dissociation and GTP binding of Gs are slower for GCGR than for β2AR. Measurements of ligand-dependent conformational alterations of GCGR by means of fluorescence and double electron-electron resonance spectroscopy show that agonist binding alone is insufficient to promote TM6 opening, in contrast to previously studied family A GPCRs, including β2AR. The outward movement of TM6 of GCGR is only observed upon interaction with Gs, suggesting that TM6 activation is only triggered by the engagement of the α5 helix of Gαs. Furthermore, TM6 of GCGR remains in the active state for a prolonged time after disengagement of Gs, which might contribute to the persistent and sustained cyclic adenosine monophosphate (cAMP) signaling previously observed for this receptor. A comprehensive comparison of the G protein activation kinetics for a number of other family A and family B GPCRs shows that family B receptors are in general less efficient than family A GPCRs in triggering G protein signaling.
CONCLUSION:
Our findings provide evidence for distinct activation mechanisms between family A and family B GPCRs. We propose that formation of the helix break and the sharp kink in TM6 of GCGR requires overcoming a higher energy barrier than the bending and outward movement of TM6 in family A receptors. Because of this kinetic barrier, ligand binding alone is not sufficient to stabilize the outward movement of TM6 but promotes the initial formation of the receptor–G protein complex and subsequent full engagement of the G protein at a slower time scale. Once activated by the insertion of the α5 helix of Gαs into the receptor core, as seen in the nucleotide-free complex structure, TM6 stays in the active conformation long after full disengagement of the G protein. This may be responsible for the previously described sustained and prolonged signaling of GCGR. Our comprehensive comparison of the G protein activation kinetics of family A and family B receptors suggests that the activation mechanism described for GCGR is generalizable to other family B GPCRs.
Family B heterotrimeric guanine nucleotide–binding protein (G protein)–coupled receptors (GPCRs) play important roles in carbohydrate metabolism. Recent structures of family B GPCR-Gs protein complexes reveal a disruption in the α-helix of transmembrane segment 6 (TM6) not observed in family A GPCRs. To investigate the functional impact of this structural difference, we compared the structure and function of the glucagon receptor (GCGR; family B) with the β2 adrenergic receptor (β2AR; family A). We determined the structure of the GCGR-Gs complex by means of cryo–electron microscopy at 3.1-angstrom resolution. This structure shows the distinct break in TM6. Guanosine triphosphate (GTP) turnover, guanosine diphosphate release, GTP binding, and G protein dissociation studies revealed much slower rates for G protein activation by the GCGR compared with the β2AR. Fluorescence and double electron-electron resonance studies suggest that this difference is due to the inability of agonist alone to induce a detectable outward movement of the cytoplasmic end of TM6.
Graphical Abstract
Difference in the activation mechanism of family A and family B GPCRs affects G protein signaling. In contrast to family A GPCRs such as β2AR, agonist binding alone is not sufficient to induce TM6 outward movement of GCGR, most likely because of the high energy barrier for the formation of the kinked and partially unwound TM6 found in the G protein–coupled state of family B GPCRs. In comparison with family A GPCRs, this leads to a reduced G protein activation rate.
The cytoplasmic end of transmembrane helix 6 (TM6) of heterotrimeric guanine nucleotide–binding protein (G protein)–coupled receptors (GPCRs) opens to allow the activated receptor to accommodate the α5 helix of Gα, leading to G protein coupling and activation. Recent structures of family B receptor–Gs protein complexes have revealed a disruption of the helical fold at the conserved PxxG motif in the middle of TM6 and an unraveling of the extracellular end of the helix (1–5). This contrasts the changes observed in family A GPCR–Gs protein complexes, where there is a gradual bending in TM6 to provide space for the insertion of the α5 helix. The marked differences in the structural changes of TM6 for family A and family B GPCRs suggest that the energy required to produce the active state of TM6 found in family B GPCRs is higher than for family A GPCRs. To explore the functional impact of the structural differences between these two GPCR families, we compared the β2 adrenergic receptor (β2AR), a prototypical family A GPCR, with the glucagon receptor (GCGR), a family B GPCR. The GCGR is an ideal candidate for this comparison because, unlike most other family B GPCRs, it can be purified in a functional state without being complexed with Gs and without thermostabilizing mutations, enabling structure determination as well as biochemical and biophysical studies to characterize Gs activation.
The peptide hormone glucagon, released from the α cells of the pancreas in response to low blood glucose level (6), binds and activates the GCGR (7, 8). Upon activation, GCGR predominantly interacts with the adenylyl cyclase stimulatory G protein, Gs, leading to increased cyclic adenosine monophosphate (cAMP) production (9). This signaling in turn drives glycogenolysis and gluconeogenesis and an increased hepatic glucose output (10). Because of its fundamental role in glucose homeostasis, GCGR represents an important therapeutic target for the treatment of severe hypoglycemia in diabetic patients (11). Furthermore, modulation of GCGR signaling has therapeutic potential for obesity and type 2 diabetes therapies (12). The sympathetic nervous system is also activated by hypoglycemia, but this leads to activation of hepatocyte β2ARs and subsequent release of glucose. Previous studies in hepatocytes suggest differences in the kinetics of cAMP accumulation after activation of the GCGR when compared with the kinetics of cAMP accumulation after activation of the β2AR (13). To explore the structural basis of these functional differences, we determined the structure of the GCGR in complex with Gs by means of cryo–electron microscopy (cryo-EM). Comparison of the active-state GCGR structure with previously determined inactive structures, unliganded and bound to a partial agonist, provides insights into ligand efficacy for family B GPCRs. In addition, comparative functional and biophysical studies of the GCGR and the β2AR revealed marked differences in agonist-induced conformational changes and receptor-mediated G protein activation. Analysis of receptor-mediated G protein dissociation rates of different family A and family B GPCRs suggest that the observed differences in the activation mechanism of GCGR are generalizable to other family B GPCRs.
Results
Development of a soluble glucagon analog, ZP3780
To determine the structure of the GCGR signaling complex, we designed a glucagon analog with improved solubility and stability in aqueous solutions compared with those of the native glucagon peptide that is prone to rapidly form fibrillar aggregates at neutral pH (fig. S1A and tables S1 and S2) (14). Building on our previous efforts in designing glucagon analogs (15), we retained the native sequence in the N terminus of the peptide, which is important for ligand efficacy (16), while improving solubility and stability by iteratively substituting amino acids in the C terminus to change the peptide isoelectric point (pI) (fig. S1B). The resulting glucagon analog, ZP3780, acts as a full agonist and includes four C-terminal mutations that only slightly reduce the affinity while showing similar maximum attainable response (Emax) in comparison with that of wild-type (WT) glucagon (fig. S1, C and D, and tables S3 and S4). Receptor activation with ZP3780 enabled the formation of a GCGR-Gs complex that was stable enough for cryo-EM imaging, yielding a final density map at a global nominal resolution of 3.1 Å (Fig. 1A, fig. S2, and table S5).
Fig. 1. Cryo-EM structure of the ZP3780-bound GCGR-Gs complex.

(A) Cryo-EM density map of the ZP3780-bound GCGR-Gs heterotrimeric complex colored by subunit. Cyan, GCGR; red, ZP3780; yellow, Gαs; dark blue, Gβ; purple, Gγ; gray, Nb35. (B to D) Comparison of the inactive, peptide ligand–free state of GCGR (Fab-bound GCGR, blue, PDB 5XEZ) (19), partial agonist NNC1702-bound GCGR (orange, PDB 5YQZ) (17), and active, full-agonist ZP3780-bound state of GCGR (cyan). Substantial structural change is observed in TM6, which moves outward by 18 Å and partially unwinds to form a kink with an angle of 105°. Additional changes are also observed in TM1, TM3, TM5, and TM7.
Cryo-EM structure of ZP3780-bound GCGR–Gs complex
The cryo-EM map shows well-defined density that allowed unambiguous modeling of the secondary structure and side chain orientations of all components of the GCGR-Gs complex, including the full agonist peptide ZP3780 (figs. S2F and S3A). The glucagon analog forms a continuous α-helix bound between the extracellular domain (ECD) and the transmembrane bundle of the receptor (Fig. 1, A and B, and fig. S3). ZP3780 engages GCGR through extensive van der Waals and hydrophobic interactions with residues in the ECD and mostly polar contacts with residues in the transmembrane domain (Fig. 2A and fig. S3, C and D). The overall peptide binding mode and the relative orientation of the ECD with respect to the transmembrane domain is very similar to the recently reported crystal structure of GCGR bound to the partial agonist peptide NNC1702 (fig. S3B) (17). Out of the four mutated residues (Q20E, Q24K, M27E, and N28S) that were inserted into the WT glucagon peptide to generate the more soluble derivative ZP3780, only two residues (E20 and E27) interact with the receptor (figs. S1B and S3C). (Single-letter abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; and Y, Tyr. In the mutants, other amino acids were substituted at certain locations; for example, Q20E indicates that glutamine at position 20 was replaced by glutamic acid.) The glutamate side chain of the Q20E substitution in ZP3780 forms a hydrogen bond (H-bond) with Q1311.29 in TM1 similarly to the WT glutamine residue in glucagon, as seen in the NNC1702-bound crystal structure of GCGR (17). In contrast to the apolar WT methionine residue in glucagon (M27), which most likely forms a hydrophobic interaction with the nearby side chain of Y65ECD (18), the mutated residue M27E in ZP3780 interacts with Q122ECD of GCGR through a H-bond (fig. S3C). Thus, the nonconservative substitution at position 27 might explain the slightly reduced potency of ZP3780 compared with WT glucagon (fig. S1C).
Fig. 2. ZP3780 binding and activation at GCGR.

(A) The N terminus of ZP3780 (red) is required for full ligand efficacy and penetrates deep in the receptor core to make H-bonds (dotted lines) with residues in TM1, TM3, TM7, and ECL3 (cyan). (B) The difference in receptor recognition by full-agonist ZP3780 (red) and partial-agonist NNC1702 (dark blue) that lacks H1 and has a D9E mutation is shown. NNC1702-bound GCGR is shown in orange (PDB 5YQZ) (17), and the ZP3780-bound GCGR-Gs complex structure is shown in cyan. The polar interactions are shown as dotted lines colored according to the GCGR structures bound to the two ligands, respectively (ZP3780, cyan; NNC1702, orange). (C) Comparison of the structures of the partial agonist NNC1702 (blue)–bound GCGR (orange, PDB 5YQZ) (17) and the full-agonist ZP3780 (red)–bound GCGR (cyan) reveal conformational differences in ECL3 and TM5, TM6, and TM7. The presence of H1 in ZP3780 ensures interaction with Q2323.37 and may induce rearrangement of residues in TM5. The interaction of D9 seems to stabilize TM7 and ECL3 displacement, which might trigger GCGR activation. (D) Mutation of Q2323.37, D370ECL3, R3787.35, and D3857.42 to alanine has a large effect on GCGR-mediated cAMP signaling. All mutants were expressed to similar levels as that of the WT receptor. For (D), data represent mean ± SEM from at least three independent experiments, performed in triplicates. Superscripts are Wootten numbering.
We observed substantial structural rearrangements in the transmembrane region of the activated receptor (Fig. 1, B to D). Comparison of our structure with the previously determined full-length GCGR structures in the inactive, peptide-free (apo), state (19) and in complex with the partial agonist NNC1702 (17) shows that ZP3780 binding induces conformational changes not only of the ECD and the stalk region but also in the extracellular loop 1 (ECL1) and the extracellular ends of TM1, TM2, TM6, and TM7 (Fig. 1, B and C). Some of these rearrangements are also found in the NNC1702-bound inactive structure of GCGR, suggesting that they are important for peptide binding by expanding the ligand-binding site to allow the penetration of the peptide N terminus deep into the receptor core. However, the GCGR-Gs complex structure also reveals structural changes that are different from the NNC1702-bound GCGR structure and that are related to full activation of the receptor. These include movement of the extracellular half of TM7 that bends further toward TM6 (6.5 Å Cα-Cα distance between L3777.34) (receptor residues in superscript are defined by using the family B numbering system) (20), facilitated by the conserved G3937.50 in the center of the transmembrane domain (Fig. 1, B and C). Furthermore, the entire TM3 shifts up by half a helix turn, and its extracellular end moves away from the receptor core. On the intracellular side, TM5 moves toward TM6 by 6.5 Å, followed by the C-terminal end of TM3 (Fig. 1D). The largest structural rearrangement between the NNC1702-bound structure of GCGR and the fully active receptor conformation in the G protein complex involves the formation of a sharp kink in the middle of TM6 (105.5°, measured between V368-G359-K344) (Fig. 1B). As reported for the previously determined active structures of other family B GPCRs [glucagon-like peptide-1 (GLP-1), calcitonin receptor (CTR), calcitonin gene-related peptide (CGRP), and parathyroid hormone receptor (PTH)], the kink pivots the intracellular half of TM6 to move outward by ~18 Å, while the α-helical structure of the extracellular half partially unwinds (1–5).
The conformational differences between the NNC1702- and the ZP3780-bound structures are reflected in the distinct efficacy profiles of the two ligands, which likely result from sequence variations in the N-terminal part of the peptides, and the stabilization of the active state by the receptor-engaged heterotrimeric G protein Gs. In the native glucagon peptide, the N-terminal residues histidine 1 (H1) and aspartate 9 (D9) are critical positions for glucagon activity (21, 22). In contrast to ZP3780, which retains these residues, NNC1702 has a deletion of H1 and substitution of D9 by glutamate (D9E). Substitution of H1 or D9 to alanine in glucagon results in a marked reduction in ligand efficacy (23). Therefore, a comparison between the interaction patterns of the N termini of the peptides ZP3780 and NNC1702 and the transmembrane bundle (TMs 3, 5, 6, 7, and ECL3) of the receptor can provide a structural framework to explain the molecular basis of ligand efficacy for the glucagon receptor.
Structural insights into ligand efficacy in GCGR
In the full agonist ZP3780, the carboxyl group of D9 forms a salt bridge with residue R3787.35 on the N terminus of TM7 that seems to draw TM7 toward TM6 (Fig. 2, A to C). This polar interaction is also present in previously determined complex structures of the family B GPCR GLP-1 with Gs (2, 4). Mutation of R3787.35 to alanine completely inhibits GCGR-mediated cAMP signaling (Fig. 2D and table S6), high-lighting the importance of this interaction for ligand-dependent activation within receptors of the glucagon family. The altered position of TM7 is accompanied by movement of ECL3 toward the receptor core with a H-bond formed between D370ECL3 and T5 of ZP3780 (Fig. 2, B and C). This interaction is part of a polar network that includes R3787.35 and D9, which may stabilize the active TM7 conformation together with the H-bond between S2 and D3857.42. Mutation of D370ECL3 and D3857.42 to alanine reduces glucagon potency from 0.14 nM to more than 100 nM (Fig. 2D and table S6). These results suggest that TM7 and ECL3 play an important role in the process of receptor activation, which is in agreement with previous studies on other family B GPCRs (24). At the peptide N terminus, H1 forms a H-bond with residue Q2323.37 in TM3 on the other side of the ligand binding pocket (Fig. 2A). Formation of this interaction seems to anchor the peptide deep into the ligand-binding pocket and constrains its movement. Alanine substitution of Q2323.37 reduces the glucagon potency from 0.14 to 51 nM (Fig. 2D and table S6), indicating that this interaction is crucial for ligand engagement and receptor activation. Furthermore, W3045.36 in TM5 flips toward the receptor core, which may lead to a stabilization of the H1-Q2323.37 interaction by forming a “cap” that presumably provides a further steric hindrance to peptide movement within the binding pocket (Fig. 2C). The functional importance of W3045.36 is shown by a mutation to glutamine that completely abolishes peptide binding (25). In addition, the inward movement of W3045.36 appears to transmit the rotation through the helix to facilitate a large-scale translation of the cytoplasmic side of TM5 toward TM6 upon GCGR activation (Fig. 1D).
Comparison of the N-terminal positions of the full agonist ZP3780 and the partial agonist NNC1702 suggests that the absence of H1 results in an upward shift of NNC1702 toward TM7 (Fig. 2B). In this position, the partial agonist would clash with TM1 and TM7 in the fully activated receptor conformation (fig. S4, A and B). In addition to the steric restriction of TM1 and TM7 movement, the D9E mutation allows the partial agonist to form additional polar contacts with the extracellular end of TM7 (residues R3787.35 and Q3747.31) in comparison with ZP3780, which may stabilize it in an inactive state and prevent the inward movement of ECL3 (Fig. 2, B and C). In addition, the missing N-terminal H1 and the lack of rearrangement in ECL3 most likely restrict the inward rotation of W3045.36 and, as a consequence, the translational rotation of TM5 that is important for full activation of GCGR (Fig. 2C).
Agonist-induced structural changes in TM6
As noted above, a hallmark of GPCR activation is the outward movement of TM6 that is accompanied in family B GPCRs by the formation of a sharp kink in the middle of the helix at the highly conserved PxxG motif (26). Compared with the NNC1702-bound receptor structure showing an outward movement of the extracellular tip of TM6 induced by peptide binding, the ZP3780-bound GCGR-Gs complex reveals partial unwinding of helix 6 above the PxxG (P3566.47-L3576.48-L3586.49-G3596.50 in GCGR) motif to avoid spatial clashes with both the peptide and the repositioned TM7 (Fig. 3, A and B, and fig. S4, C to E). Although this most likely initiates the destabilization of TM6 helicity, other conformational rearrangements around the PxxG motif may be required for kink formation. Ligand-induced movement of TM5 seems to cause F3225.54 to relocate toward TM6 and probably creates a clash between this bulky side chain and P3566.47 of the PxxG motif in the helical conformation of TM6, as seen in the NNC1702 bound structure (Fig. 3B) (17). As a result, P3566.47 rotates away by approximately 90°, which probably causes the PxxG motif to shift inward toward TM3 and TM7 (Fig. 3, A and B). Substitution of F3225.54 to alanine results in decreased potency of glucagon from 0.14 nM to 110 nM, indicating that the rearrangement of TM5 and packing of F3225.54 against the PxxG motif is important for receptor activation (Fig. 3C and table S6). Furthermore, a slight rotation of TM7 away from TM6, presumably caused by the ligand-induced changes, may help to accommodate the displaced PxxG motif in the active state of the receptor by releasing the steric hindrance between the newly positioned P3566.47 and L3957.52 in TM7 (Fig. 3B). Another obstacle for kink formation is found in TM3, where the side chain of L2423.47 would prevent inward movement of the PxxG motif. The observed upward shift of TM3 may lead to the removal of this restriction by shifting the leucine side chain away from the PxxG motif (Fig. 3B). Individual replacement of the two leucine residues (L2423.47 and L3957.52) in TM3 and TM7 to the smaller alanine side chain causes a decrease in glucagon potency (from 0.14 to 3 nM and 0.82 nM, respectively) as well as a significant decrease in the maximal glucagon response (Fig. 3C and table S6). This highlights the importance of the residues at these positions to produce conformational rearrangements that seem to be necessary for the inward rotation of the PxxG motif upon receptor activation. The repositioning of the PxxG motif is probably further stabilized by a H-bond with TM7 (P3566.47-Q3927.49) (Fig. 3B). The functional relevance of these residues for activation of the glucagon receptor family have been previously shown by mutagenesis studies (20). As a result of the 90° rotation of TM6 at P3566.47, the N terminus of TM6 moves away from TM5, maintaining the 90° rotation (compare the positions of F3456.36 in Fig. 3A and of the α carbon of T3566.42 Fig. 3B), and dislocates from the receptor core by 18 Å to enable the coupling of the G protein (Figs. 1B and 3A).
Fig. 3. Structural changes in the PxxG motif of GCGR induced by full-agonist ZP3780 binding and Gs coupling.

(A) The rearrangement of TM3, TM5, and TM7 allows TM6 outward movement and kink formation at the conserved PxxG motif (P3566.47-L3576.48-L3586.49-G3596.50). The ZP3780-bound GCGR-Gs complex (cyan) is superimposed on the NNC1702-bound GCGR structure (orange) to highlight structural changes that result in the TM6 outward movement in the fully active GCGR-Gs complex structure. (B) Reorientation of residues in TM3, TM5, and TM7 around the PxxG motif that seems to facilitate kink formation in TM6. The polar interactions are shown as dotted lines colored according to the GCGR structure. The Cα atom of T3516.42 is presented as a sphere to highlight the extent of TM6 outward movement. (C) Alanine substitution of residues around the PxxG motif significantly reduces GCGR-mediated cAMP signaling. All mutants were expressed to similar levels as that of the WT receptor. For (C), data represent mean ± SEM from at least three independent experiments, performed in triplicates. Superscripts are Wootten numbering.
Coupling of GCGR to Gs
The conformational rearrangements in the transmembrane bundle of the receptor open up an intracellular cavity that allows engagement of the heterotrimeric G protein, Gs. In the complex structure, the C-terminal α5 helix of Gαs inserts into the receptor core to form extensive hydrophilic interactions with residues in TM5 and TM6 and helix 8 (H8) and hydrophobic contacts with TM3 (Fig. 4, A and B). In general, the GCGR-Gs complex displays high similarity in the receptor–G protein interactions compared with other reported structures of family B GPCRs-Gs complexes (such as GLP-1, CTR, CGRP, and PTH), suggesting a common mechanism for G protein engagement and activation (fig. S5) (1–5, 27).
Fig. 4. Structural changes in Gs upon coupling to GCGR.

(A) Comparison of GDP-bound Gαs (green, PDB 6EG8) (73) and nucleotide-free Gαs (yellow) in complex with GCGR (cyan). Major differences between these two structures are the opening and displacement of the α-helical domain, the rotational translation of the α5 helix to engage the receptor core, and conformational rearrangements in the α1 helix and α5-β6 loop. (B) The α5 helix (yellow) of Gαs in the nucleotide-free GCGR-Gs complex engages the cytoplasmic core of GCGR (cyan) to form extensive polar interactions (dotted lines) with TM5, TM6, and helix 8 (H8). (C) Comparison between the interaction of ICL2 of GCGR (cyan) and β2AR (pink) (PDB 3SN6) (50) and α5 of Gs (yellow and dark yellow, respectively). Close-up view shows that residue F139ICL2 of β2AR engages the hydrophobic pocket lined by residues from the αN-β1 loop, the β2-β3 loop, and the α5 helix. In GCGR, the corresponding residue is A256ICL2, which engages the hydrophobic pocket less efficiently.
A notable difference between GCGR and β2AR, a family A GPCR that couples predominantly to Gs, is the extent of interaction between the intracellular loop 2 (ICL2) and the hydrophobic pocket in Gs formed by the αN-β1 loop, the β2-β3 loop, and the α5 helix of Gαs (Fig. 4C). In the β2AR-Gs complex, ICL2 forms a helix that positions F139 to penetrate deeply into this pocket. This interaction has been shown to be functionally important because the F139A mutation dramatically reduces GDP release even though it still forms a complex with the G protein (28). By contrast, ICL2 in GCGR is a loop where A256 occupies the position of F139 in β2AR, having weaker interactions with the hydrophobic pocket (Fig. 4C, close-up view).
Functional impact of structural differences between family B and family A GPCRs
Despite the lack of sequence homology between GPCR families, the activation of family B receptors involves structural rearrangements that are similar to family A receptors, including the outward movements of TM5 and TM6. However, conserved structural motifs and conformational switches that play important roles for the activation of family A GPCRs (such as the DRY/ERY, NPxxY, and PIF motifs) are absent in family B receptors (26, 29). This implies that the mechanisms of activation between family B and family A receptors are substantially different from each other. In family B GPCRs, peptide binding results in conformational changes on the extracellular side that leads to an expansion rather than a contraction of the ligand binding cavity, as seen in family A receptors. Furthermore, in the structures available to date, the active conformation of family B receptors in complex with Gs shows a distinct sharply kinked TM6 when compared with Gs-coupled family A GPCRs such as the β2AR, where TM6 moves outward without the formation of a helix break (Fig. 5A). We speculated that the energy required to produce the kinked conformation of the active state of TM6 found in family B GPCRs is much higher than the energy required to produce the much smaller change in TM6 observed in family A GPCRs.
Fig. 5. Engagement and activation of Gs by GCGR and β2AR.

(A) Conformation of TM6 in the GCGR-Gs (cyan) and β2AR-Gs (pink, PDB 3SN6) (50) complex structures. (B) The GTP-turnover assay shows that β2AR (blue) activates Gs much faster than GCGR (red), with GCGR inducing a maximal GTP-turnover rate of 0.11 GTP min−1 Gs−1 compared with (inset) 8 GTP min−1Gs−1 for β2AR. (C) Monitoring of GPCR-Gs association by means of FRET between Cy3B-labeled GCGR (red) or Cy3B-labeled β2AR (blue) with Sulfo-Cy5-labeled Gs. The decrease in donor fluorescence shows comparable rates of association between the receptors and Gs. (D) The rate of receptor-induced [3H]-GDP dissociation from Gs shows faster release for β2AR (blue; koff = 0.042 s−1) compared with GCGR (red; koff = 0.0022 s−1). (E) Bodipy-GTPγS binding to nucleotide-free complex is slower for the GCGR-Gs complex (kon = 0.001 s−1) compared with the β2AR-Gs complex (kon = 0.003 s−1). (F) Concentration-response curves for Gs dissociation rates in HEK293 cells show slower Gs activation by GCGR compared with β2AR. In (B) and (D), data represent mean ± SEM from at least three independent experiments performed in triplicates. In (C) and (E), data represent mean ± SEM of triplicate measurements. In (F), data represent mean ± SEM of three to seven independent experiments.
To better understand the impact of the TM6 helix kink on receptor activation and signaling of family B GPCRs, we compared the activity of GCGR to β2AR in in vitro GTP-turnover assays using purified receptors reconstituted into high-density lipoprotein (HDL) particles (also known as nanodiscs) (3:2 molar ratio of POPC: POPG) and purified Gs heterotrimer. The functionality of both receptors in HDL particles was tested by means of radioligand filtration binding to ensure that the specific activity was comparable between the two receptors (fig. S6, A to D, and table S8). GCGR shows significantly lower guanine nucleotide exchange factor (GEF) activity than β2AR, with a maximal GTP turnover rate of 0.11 GTP min−1 Gs−1 versus ~8 GTP min−1 Gs−1 seen for β2AR (Fig. 5B and table S7). The reduced GCGR-dependent G protein activation might be related to any or all of the following: less efficient G protein association, reduced receptor-induced GDP release, reduced GTP binding, and slower complex dissociation. To distinguish between these possibilities, we performed several biophysical and functional assays. First, we tested the rate of G protein association to the receptors. For this purpose, we measured Förster resonance energy transfer (FRET) between Cy3B-labeled receptors and Sulfo-Cy5-labeled heterotrimeric G protein, Gs, in bulk detergent solutions to avoid nonspecific adsorption of lipidated G proteins to HDL particles. The result shows that the G protein association rate for GCGR with an association rate constant (kon) of 0.0044 s−1 is only slightly slower than for β2AR (kon = 0.0077 s−1) (Fig. 5C and table S7) and hence cannot be the only cause for the substantially slower GTP-turnover rate of GCGR described above. We next tested whether the receptor-induced GDP release is significantly different between the two receptors by using a [3H]-GDP dissociation assay in HDL particles. GCGR catalyzed nucleotide release from [3H]-GDP loaded Gs with a substantially slower rate [dissociation rate constant (koff ) = 0.002 s−1] compared with β2AR (koff = 0.042 s−1) (Fig. 5D and table S7). Together, these results suggest that GCGR forms a more stable intermediate state with GDP-bound Gs and shows lower activity toward the formation of the nucleotide-free complex than that of β2AR. This might be explained by the weaker interaction between ICL2 of the GCGR and the hydrophobic pocket formed by the αN-β1 loop, the β2-β3 loop, and the α5 helix of Gαs (Fig. 4C).
Single-molecule FRET studies on β2AR reveal that one or more nucleotide-bound transient intermediate states play an important role in the process of receptor-mediated G protein activation (30). Supported by the inhibitory effect of GDP on the GTP turnover of GCGR (fig. S8A) and the previously reported higher potency of GDP in destabilizing high-affinity glucagon binding of the GCGR in comparison with GTP (31), we propose that the longer-lived GDP-bound complex of GCGR is at least in part responsible for the overall lower GTP turnover rate of GCGR in comparison with β2AR. Another rate-limiting step for G protein activation was found to be the GTP loading of the receptor–G protein complex, as evidenced by Bodipy-GTPγS binding studies on nucleotide-free receptor complexes of GCGR and β2AR (Fig. 5E and table S7). GCGR shows a slower Bodipy-GTPγS (kon = 0.001 s−1) binding rate compared with the β2AR (kon = 0.003s−1), which demonstrates that the efficiency of GDP release and GTP binding are significantly reduced compared with those of β2AR and thus represents the major underlying rate limiting steps for GCGR-mediated G protein activation. In addition, these results also suggest that differences in the conformations of the nucleotide-free receptor–G protein complex of GCGR and β2AR are responsible for differences in nucleotide binding. This is further supported by the previously observed higher affinity of the GCGR-Gs complex for GDP over GTP (31), which is in contrast to the higher GTP affinity found for Gs coupled to the full-agonist-bound β2AR (30, 32).
To provide evidence for the less efficient G protein activation mediated by GCGR in comparison with β2AR in intact cells, we performed a NanoBiT complementation-based G protein dissociation assay in human embryonic kidney (HEK) 293 cells. In this in vivo assay, an agonist-dependent decrease in signal was measured because of the receptor-mediated dissociation of the Gαs and Gβγ subunits (33). With similar expression levels of both receptors (table S9), concentration-response curves allowed us to determine the maximum rate of Gs activation, which was found to be slower for GCGR (2.0 % relative light unit (RLU) change s−1) compared with the β2AR (3.24 RLU change s−1) (Fig. 5F and table S9). The observed difference in the Gs dissociation rate in our cell-based assay between GCGR and β2AR is not as large as in the in vitro assays described above (Fig. 5B). This could be due to several reasons, including differences in the lipid composition of cellular membranes in comparison with that of HDL particles, the presence of accessory proteins such as receptor activity–modifying proteins (RAMPs) (34, 35), or variations in the internalization rates. Despite these differences, the results from whole cells confirm the lower GEF activity of GCGR in comparison with the β2AR observed in our in vitro studies.
Agonist binding alone does not stabilize an active conformation of the GCGR
Dissimilarities in the GEF activity between GCGR and β2AR might suggest some differences in the process involved in the formation of the fully active conformation of the receptor. For GCGR, this process includes the kink formation and outward movement of the cytoplasmatic end of TM6, a conformation that is further stabilized by movement of TM5 toward TM6 (Figs. 1D and 3B). To explore the effect of agonists on conformational changes associated with receptor activation of GCGR, we used double electron-electron resonance (DEER) spectroscopy (36). We established a minimal cysteine GCGR construct (mC-GCGR) to enable site-specific labeling through thiol-based chemistry. Four cysteine residues in the TM domains, not involved in disulfide bonding, were mutated (C171T, C240A, C287A, and C401V). These modifications did not substantially affect ligand binding or G protein signaling (fig. S7A). Guided by the GCGR-Gs complex structure and the available inactive structure of GCGR, we engineered cysteine residues, pairwise, at the cytoplasmic ends of TM4 and TM5 (265C to 335C) and TM4 and TM6 (265C to 345C) to probe distance changes upon agonist binding (fig. S7B). Introducing double cysteines into the mC-GCGR background did not substantially alter the pharmacological properties of the receptor (fig. S7A). For the DEER experiments, we labeled the receptor with bis-(2,2,5,5-tetramethyl-3-imidazoline-1-oxyl-4-yl)disulfide (IDSL), which provided reduced background labeling of the mC-GCGR than did other commonly used spin labels. DEER distance distributions on IDSL-labeled TM4-TM5 and TM4-TM6 constructs were collected for receptor both in detergents and in HDL particles. In the apo (unliganded) state, the distance distributions between TM4-TM6 and TM4-TM5 show single conformations in detergent (Fig. 6, A and B, gray shading) as well as HDL particles (fig. S7C, dark gray shading), which are in good agreement with the inactive state of the receptor. Surprisingly, addition of the full agonist ZP3780 at saturating concentration (5x molar excess, 0.5 mM) did not change the distance distribution, indicating that TM5 and TM6 do not undergo ligand-induced conformational changes upon agonist binding. This is in contrast to DEER, single-molecule FRET, and nuclear magnetic resonance (NMR) studies on family A GPCRs (30, 37–42). In the case of β2AR, for example, DEER distance measurements between TM4 and TM6 have shown that agonist binding increases the population of the active state by stabilizing the outward conformation of TM6, as seen in the β2AR-Gs crystal structure (38). That no such DEER distance population for an active conformation was observed for agonist-bound GCGR implies a substantially higher energy level of the fully active conformational state of GCGR in the presence of agonist alone compared with the β2AR and other characterized family A GPCRs.
Fig. 6. Conformational changes in GCGR and β2AR upon ligand binding, G protein coupling, and G protein dissociation.

(A and B) DEER data showing no change in the distance distribution, between Apo (gray shading) and ZP3780 binding (red), of the TM4-TM6 and TM4-TM5 pairs. The upper limit of reliable distance (r) and width (σ) determination are shown as gray and black bars, respectively. (C) The apo (gray) spectrum of bimane-labeled GCGR (TM6) does not change upon ZP3780 addition (red). Further addition of Gs (purple) results in a decrease in fluorescence intensity and a 4-nm λmax shift owing to outward movement of TM6. (D) β2AR labeled with bimane in TM6 shows a decrease in fluorescence and a redshift in λmax when agonist, epinephrine (Epi) (blue), is added to apo (black), which changes further upon Gs (green) addition. (E) The addition of GDP (orange) or GTPγS (cyan) to NBD-labeled GCGR (TM6) bound to ZP3780 and coupled to Gs (purple) does not result in an increase of the fluorescence intensity to the level of ZP3780-bound GCGR in the absence of G protein (red). (F) The addition of GDP (brown) or GTPγS (light purple) to bimane-labeled β2AR bound to Epi and coupled to Gs (green) leads to an increase in fluorescence intensity and a blueshift in λmax to the same values as that for for Epi-bound β2AR in the absence of G protein (blue). In (C) to (F), data represent mean ± SEM of triplicate measurements.
For the β2AR, as well as other family A GPCRs, full stabilization of the active state requires binding of the G protein or a G protein–mimetic nanobody, in addition to the agonist (29). Because of the nature of the IDSL spin label used for labeling of mC-GCGR—which gets stripped from the receptor upon engagement of the G protein, as evidenced by a decrease in modulation depth (fig. S7D)—we were unable to use DEER to study conformational changes of the receptor in the presence of the heterotrimer Gs. As an alternative, we used the environmentally sensitive fluorophore monobromobimane (bimane) that has been previously utilized as a conformational reporter of TM6 activation of β2AR and other GPCRs (43–45). We labeled the mC-GCGR construct at position 349C in TM6 with bimane and compared it with β2AR modified at the endogenous cysteine residue 265 in TM6. Using this reporter system, we observed a small but statistically significant difference in the wavelength of maximum emission (λmax) of the fluorescence spectrum of bimane upon addition of ZP3780 to GCGR (Fig. 6C, red, and fig. S8B). This small redshift in λmax suggests that ligand binding induces some conformational alterations on the intracellular side of the receptor that slightly change the local environment at around the fluorophore attached to TM6. Because the DEER data show no ligand-dependent distance change in TM5 and TM6, we hypothesize that ligand binding induces conformational changes in other regions of the receptor in close vicinity of TM6 that might trigger initial engagement of the GDP-bound Gs to the receptor. By contrast, addition of the agonist epinephrine (Epi) to β2AR results in a decrease in intensity and a more pronounced redshift in λmax, demonstrating TM6 outward movement (Fig. 6D, blue). These results are consistent with the DEER data that shows that TM6 displacement occurs on agonist binding in β2AR but not in GCGR (Fig. 6A) (38). Adding Gs to the ZP3780-bound GCGR results in a decrease in intensity and a moderate 5-nm redshift in λmax (Fig. 6C, pink). This suggests that GCGR interaction with both agonist and Gs is required to induce conformational changes in TM6 that are necessary for the formation of the nucleotide-free receptor–G protein complex. For β2AR, addition of Gs to Epi-bound β2AR leads to an additional reduction of the fluorescence intensity and λmax, indicating that Gs interaction with the receptor further stabilizes the active state of the receptor, as shown previously (45). Taken together, the DEER and fluorescence experiments point to a mechanism for GCGR in which agonist binding to the receptor must trigger conformational changes in regions other than TM5 and TM6 that are important for initial G protein coupling. Subsequent full engagement of the G protein is required to induce the kink formation and outward movement of TM6 that are necessary for establishing the nucleotide-free receptor–G protein complex. Although no DEER or fluorescence data are available for other family B GPCRs, the kinked TM6 that pivots away from the core of the receptor is a common feature of all family B GPCR complexes. Hence, the requirement of Gs binding to initiate structural changes in TM6 might be a general mechanism in family B GPCRs.
Persistent active state of GCGR after G protein dissociation
Although molecular dynamics (MD) simulations and fluorescence studies of family A GPCRs have shown fast relaxation of TM6 upon transducer dissociation, less is known about the process of receptor inactivation of family B GPCRs (30, 46). To better understand this process, we used fluorescence spectroscopy to probe conformational changes in TM6 of GCGR upon nucleotide addition to 4-chloro-7-nitrobenz-2-oxa-1,3-diazole (NBD)–labeled mC-GCGR-349C and bimane-labeled β2AR in complex with Gs. We used the NBD fluorophore instead of bimane for GCGR labeling for this experiment because it showed larger responses to small conformational differences. In parallel, we performed FRET studies to directly detect nucleotide-dependent dissociation of the G protein from both receptors (fig. S8C). To first establish the signaling complexes, we added Gs to ZP3780-bound GCGR (Fig. 6E, purple) or Epi-bound β2AR (Fig. 6F, green) and followed the decrease in NBD or bimane fluorescence intensity due to TM6 outward movement in the G protein–coupled fluorescently labeled receptors in comparison with the spectra of ligand-bound receptors in the absence of G protein. Once the steady-state level was reached, addition of GDP (Fig. 6F, brown) or GTPγS (Fig. 6F, violet) to β2AR-Gs, resulted in Gs dissociation (also shown by means of FRET in fig. S8C, brown and violet), and the fluorescence spectrum returned to the wavelength and intensity of the agonist-bound receptor (Fig. 6F, blue). By contrast, although we observed almost complete dissociation of GCGR-Gs complexes by changes in FRET after the addition of GDP or GTPγS, (fig. S8C, orange and teal), no change in NBD fluorescence was observed after 10 min of incubation (Fig. 6E, orange and teal), indicating that TM6 does not return to the inactive state and remains open. But after 1 hour of incubation, the fluorescence intensity increased back to the level of the spectra of receptor bound to ligand only (fig. S8D). This suggests that the active state of GCGR persists for minutes after the dissociation of the G protein before it retracts back to the transmembrane bundle. A minute-scale-long active conformation of TM6 after Gα-α5 dissociation has also been reported previously for the β2AR, although the duration time of 90 s was markedly shorter than the one observed here for GCGR (47). MD simulation suggests that TM6 remains in a more open conformation in the presence of ZP3780 than in the presence of the partial agonist (fig. S8E), suggesting that the efficacy of the ligand might play a role in stabilizing TM6 in the kinked outward conformation.
Receptor-mediated Gs dissociation of family B receptors is slower compared with family A GPCRs
The similarity in all recently solved family B GPCR-Gs complex structures indicate that the mechanism of receptor activation and receptor-mediated G protein signaling is conserved across family B receptors but distinct from family A GPCRs. However, so far no comprehensive studies have been conducted to compare the G protein signaling kinetics of different family B GPCRs among each other and with members of family A. We used the NanoBiT complementation-based G protein dissociation assay to measure the Gs dissociation kinetics of a set of family A and family B GPCRs in order to compare their G protein activation rates. In total, we tested three family B GPCRs, in addition to GCGR—GLP1, PAC1, and PTH—and four family A GPCRs, other than β2AR, that couple primarily to Gs: 5-hydroxytryptamine receptor 4 (5-HT4), histamine H2 receptor (H2R), prostaglandin D2 receptor (DP), and vasopressin receptor 2 (V2R). As described above, we determined the maximum receptor-mediated G protein dissociation rates by measuring the time-dependent Gs dissociation at increasing ligand concentrations and fitting of the plotted concentration-dependent rates (fig. S9). Comparison of the maximum Gs dissociation rates shows that family B receptors in general catalyze a significantly slower Gs dissociation rate relative to all class B receptors tested (P = 0.019) (Fig. 7A and table S9). The expression levels of all GPCRs were determined for each experiment to ensure similar receptor densities in the cytoplasmic membrane (Fig. 7B). The differences in the Gs dissociation rates between the two GPCR families suggest a conserved mechanism of activation among family B GPCRs that is distinct to the previously described signaling mechanism of family A receptors.
Fig. 7. Comparison of Gs activation kinetics between family A and family B GPCRs.

(A) Gs dissociation rates among family A and family B GPCRs. GPCR ligand-induced Gs dissociation was measured by monitoring real-time, luminescent signals from NanoBiT-Gs protein and by fitting the plot to a one-phase decay curve. (B) Surface expression levels of tested GPCRs. Cell-surface GPCRs were fluorescently labeled with anti-FLAG epitope-tagged antibody and analyzed by means of flow cytometry. Each dot represents data from individual independent experiments. P values were obtained by means of two-tailed Student’s t test with Welch’s correction.
Discussion
Here, we report the structure of the GCGR-Gs signaling complex bound to an engineered glucagon derivative with improved aqueous solubility. The structure provides a framework to explain the wealth of existing structure-activity relationship data for glucagon peptides that has accumulated since the discovery of glucagon by Kimball and Murlin almost 100 years ago (48). One hallmark of GPCR activation is the outward movement of TM6. In the GCGR-Gs complex, the conformational change of TM6 is accompanied by a partial unwinding of the secondary structure and formation of a sharp kink in the middle of the helix. This kink represents a characteristic structural feature seen in all recently solved family B GPCR-Gs protein complex structures, but not in family A complexes such as the β2AR-Gs. Characterization of the functional impact of the structural differences observed between GCGR and β2AR revealed that GCGR possesses a lower GEF activity compared with that of β2AR. We show that the two main rate-limiting steps are the GDP release and GTP binding, which are significantly slower for GCGR-Gs than for the β2AR-Gs complex. This suggests that GCGR induces a specific conformation of the receptor-coupled heterotrimeric G protein with different nucleotide sensitivity compared with Gs bound to β2AR. Biophysical characterization of the ligand-induced activation of the GCGR indicates that the low GEF activity could be related to the mechanism of TM6 activation. Our data shows a distinct activation mechanism of GCGR in which Gs coupling is necessary to induce kink formation and outward movement of TM6. This is in contrast to the β2AR, where agonist binding alone promotes some outward movement of TM6 (38). Furthermore, once activated, TM6 of GCGR stays in the active kinked conformation longer than TM6 of β2AR before it eventually relaxes back toward the inactive state.
On the basis of these results, we propose an activation model for GCGR and illustrate the effect of agonist, G protein, and GTP on the equilibrium of three different key functional states: inactive, intermediate, and fully active, with energy landscape diagrams (Fig. 8, A to C). In the unliganded (apo) state, GCGR predominantly exists in the low-energy inactive state, with a large energy barrier toward the intermediate and active states of the receptor, which is in agreement with the low reported basal activity of the receptor (49). By contrast, the β2AR exhibits a moderate level of basal activity, suggesting a higher energy level in the apo state (Fig. 8C). Glucagon binding induces some conformational change on the intracellular side of the receptor without triggering TM6 outward movement. This promotes the formation of an “intermediate state” that we propose plays an important role for the initial engagement with the GDP-bound G protein. This is reflected in the energy landscape where agonist binding results in a decrease in the energy of the intermediate state, with little change in the energy of the fully active receptor conformation with the kinked and unraveled TM6. By contrast, DEER and NMR studies show that in β2AR, agonist binding stabilizes an intermediate state in which TM6 moves outward and also produces a small proportion of the receptor in a fully active state (Fig. 8C) (38). Therefore, agonist binding to β2AR not only lowers the energy of the intermediate state substantially but also decreases the energy of the fully active state.
Fig. 8. Proposed model for GCGR activation and signaling in comparison with β2AR.

(A) Glucagon binding to GCGR induces conformational change on the extracellular side of the receptor (ECD, TM1, TM2, TM6, and TM7) without inducing outward movement of TM6 on the intracellular side. Coupling of GDP-bound Gs enables TM6 outward movement. The putative high energy required to produce the kinked and outward-moved TM6 may result in slower rates for the receptor-catalyzed nucleotide release of GCGR in comparison with β2AR. Another rate-limiting step for GCGR-mediated G protein activation is GTP binding to the nucleotide-free G protein that leads to dissociation of the G protein from the receptor. After disengagement of the G protein, relaxation of TM6 to the inactive state is very slow, which might lead to the previously observed prolonged G protein signaling of GCGR in comparison with β2AR (13). (B) β2AR activation by an agonist increases the active state population of the receptor with an outward-moved TM6. Gs coupling to β2AR fully stabilizes the active state and leads to rapid GDP release. The very transient nucleotide-free complex exhibits a high affinity for GTP that readily binds and dissociates the complex. After disengagement of the G protein, β2AR relaxes back to the more conformational heterogeneous agonist-bound but G protein–free state. (C) Model of the simplified free energy landscapes for GCGR and β2AR. Shown are the effects of agonist, G protein coupling, and GTP binding to the receptor–G protein complex on the equilibrium between the inactive and active states of the receptors.
G protein engagement and establishment of the nucleotide-free state of the GCGR-Gs complex is required to decrease the energy level of the active state. This leads to kink formation and movement of TM6 away from the receptor core in order to accommodate the C-terminal α5 helix of Gαs (Fig. 8, A and C, and fig. S4E). On the basis of the observation that the nucleotide release and subsequent GTP binding are the most rate-limiting steps in the process of GCGR-mediated G protein activation, we speculate that the intermediate and the fully active states are separated by a higher activation barrier for GCGR than for β2AR (Fig. 8C). This most likely leads to a slow transition from the GDP-bound receptor complex to the nucleotide-free complex because of the high energy barrier for the unraveling and formation of the helix break in TM6. However, once outward movement of TM6 occurs, we propose that the G protein can engage the receptor more tightly, establishing the functionally important interactions with ICL2 and the intracellular cavity of GCGR that eventually trigger nucleotide release. Subsequent binding of GTP to the nucleotide-free complex, which results in G protein dissociation from the receptor, converts the energy landscape back to the one of the agonist-bound receptor in the absence of the G protein. However, on the basis of the proposed high energy barrier between the active and intermediate receptor conformation, TM6 of GCGR persists in the fully active conformation for an extended time compared with the β2AR (Fig. 8, A and B). The prolonged active state of TM6 after disengagement of Gs might contribute to the persistent and sustained cAMP accumulation that was previously reported after glucagon addition to perfused rat livers (13).
In β2AR, probably owing to the intermediate state already showing an outward displacement of TM6, full engagement of Gs occurs more rapidly in comparison with GCGR. This proposed rapid tight interaction of Gs with the β2AR most likely leads to a faster decrease in GDP affinity and thus to a more rapid nucleotide release than for GCGR. As shown previously in DEER and NMR studies, G protein coupling further lowers the energy level of the active state of β2AR to fully stabilize the receptor in the conformation observed in the x-ray structure of the nucleotide-free β2AR-Gs complex (38, 50). GTP-induced dissociation of the β2AR signaling complex fully reverses the effect of the G protein, resulting in a similar energy landscape of the receptor as in the presence of agonist alone. In comparison with GCGR, the relaxation is relatively fast because of the lower energy barrier between the fully active and intermediate states (Fig. 8C).
Together, the observed differences in the signaling behavior of GCGR and β2AR and the comprehensive comparison between the G protein activation kinetics of a number of family A and family B receptors suggest that the activation mechanism described for GCGR might be conserved in other members of family B GPCRs. Thus, our results provide insight into the distinct mechanism of receptor and G protein activation of family A and family B GPCRs.
Materials and methods
Solubility assay
Stock solutions of 2.0 mg ml−1 native glucagon and ZP3780 were prepared by dissolving the compounds in milliQ water adjusted to pH 2.5 by HCl. The concentration of compound was determined by absorbance measurement at 280 nm (Nanodrop 2000c, Thermo Scientific) using a calculated extinction coefficient of 8480 M−1 cm−1. Aliquots of 50 μl buffer (table S1) were transferred to a clear bottom, UV compatible 96 wells plate and mixed with 50 μl aliquots of the peptide stock. The final peptide concentrations were 1 and 0.5 mg ml–1 and the final buffer concentration 50 mM. ZP3780 solubility was also determined at 5 mg ml–1 using a stock solution of 10 mg ml–1. The samples were incubated at room temperature for 15 min and the UV absorbance at 325 nm was measured with an absorbance microplate reader (Spectramax 190). The samples were considered fully dissolved if the absorbance at 325 nm was below 0.02 arbitrary units (a.u.), which corresponds to 5 to 6 times the standard deviation of 8 buffer samples.
Aggregation assay
Samples were prepared according to the solubility assay, with the addition of a final concentration of 40 μM ThT. Samples were loaded in a 96-well black fluorescence plate (clear bottom) in triplicates of 150 μl in each well at ambient temperature and subsequently the plate was covered with UV compatible sealing tape to avoid evaporation. The plates were placed in a Spectramax Gemini XS fluorescence plate reader. The plate reader was programmed to read fluorescence from the top of the plate at 355 nm (excitation 295 nm) and at 485 nm (excitation 450 nm) at fixed intervals of 10 min for 96 hours at 40°C preceded by 300 s of automixing.
[125I]-glucagon competition binding assay
Membranes containing the human GCGR (hGCGR) were prepared as described (51) from a HEK293 cell line stably overexpressing the hGCGR. In brief, for generation of the overexpressing cell line, cDNA encoding the hGCGR (P47871) was amplified by means of polymerase chain reaction (PCR) using primers encoding terminal restriction sites for subcloning. The 5′-end primer additionally contained a near Kozak consensus sequence. The fidelity of the DNA encoding the hGCGR was confirmed by DNA sequencing. The PCR product encoding the hGCGR was subcloned into a mammalian expression vector containing a neomycin (G418) resistance marker. The mammalian expression construct was transfected into HEK293 cells by a standard liposome transfection method. 48 hours post-transfection, cells were seeded for limited dilution cloning and selected with 0.5 mg ml–1 G418 in the culture medium. After 2 weeks, surviving colonies of hGCGR-expressing cells were picked, propagated and tested for cAMP accumulation. One hGCGR-expressing clone was selected and used for membrane preparation.
For the competition binding experiments 0.06 nM [125I]-glucagon (PerkinElmer, NEX207) was added together with 10 μg hGCGR containing membranes, 0.5 mg SPA-beads (PerkinElmer, RPNQ0001) and varying concentrations of native glucagon or ZP3780 in a buffer consisting of 50 mM HEPES pH 7.4, 5 mM EGTA, 5 mM magnesium chloride, 0.005% Tween-20 and 0.05% casein (Sigma, C4765). After shaking at 600 rpm for 2 hours at room temperature the plate was transferred to a MicroBeta2 scintillation counter (PerkinElmer) and left for 4 hours to allow the SPA-beads to settle before quantifying 125I radioactivity. To determine the respective Ki-values, data were fitted by nonlinear regression to a one-site competition model using GraphPad Prism.
Radioligand binding to receptor reconstituted HDL-particles
The binding capacity of HDL particles reconstituted with the β2AR or the GCGR were determined by radioligand saturation binding. HDL particles were incubated in a buffer consisting of 20 mM Tris pH 7.5, 100 mM sodium chloride, 1 mM magnesium chloride and 0.013% n-dodecyl-β-d-maltopyranoside (DDM) with increasing concentrations of radioligand in absence or presence of 10 μM alprenolol or 50 μM ZP3780, respectively, to detect nonspecific binding, in a final volume of 100 μl in 5 ml falcon PP tubes (Corning #352063). For the β2AR [3H]-Dihydroalprenolol (DHA) with a specific activity of 105 Cimmol−1 (NET720, Perkin Elmer) was used. For the GCGR, [125I]-glucagon with a specific activity of 2200 Cimmol−1 (NEX207, Perkin Elmer) was diluted with unlabeled native glucagon to a specific activity of around 27.7 Cimmol−1. Gs was added in 38-fold excess of GCGR to detect the maximal specific binding capacity of GCGR. Reactions were incubated for 3 hours at room temperature. Then, receptor-bound radioligand was captured on 25 mm GF/B Whatman filters (Sigma #WHA1821025) by washing with a total of 7.5 ml icecold above mentioned buffer supplemented with 0.1% DDM. The GF/B filters were preincubated with 0.5% PEI at 4°C for 3 hours and washed twice with 2 ml icecold buffer containing 0.1% DDM prior to the capture on a vacuum manifold. Radioactivity was detected on a Hidex Triathler scintillation counter. Specific binding was determined by subtraction of nonspecific binding counts from total counts and Bmax and Kd determined by fitting data to a one-site binding isotherm. Bmax of GCGR and β2AR HDL particles was calculated as the ratio of captured sites (i.e., detectable sites) per reconstituted GCGR or β2AR HDL particle. To determine the amount of Gs protein required to obtain maximal [125I]-glucagon binding, HDL-particles were incubated with fixed amounts of GCGR and [125I]-glucagon (8 nM) and increasing amounts of Gs. Specific binding was determined as described above.
Functional cAMP accumulation assay
The in vitro potencies and Emaxs of glucagon and ZP3780 were assessed in a cAMP accumulation assay using HEK293 cells transiently transfected with the hGCGR. In brief, HEK293 cells were brought into suspension (0.25 × 106 cells ml–1 ) by trypsination. For transfection of 1 ml of cells in suspension, a total DNA amount of 1 μg, consisting of 0.2 μg hGCGR (accession no P47871) expression vector DNA and 0.8 μg pcDNA3.1(zeo), in 25 μl OptiMEM was mixed with 3 μl FuGene6 in 57 μl OptiMEM and incubated for 20 min. Hereafter the 85 μl DNA/FuGene6 mix was added to 1 ml cell suspension, mixed and seeded in a clear poly-D-lysine coated 96-well in a volume of 100 μl/well (final cell density of 2.5 × 105 cells/well). The amounts were scaled up to the number of data points needed to generate concentration response curves for glucagon and ZP3780. Twenty-four hours after transfection the assay was performed by washing wells once in 100 μl HBSS buffer [HBSS Gibco 14025, 20 mM HEPES pH 7.5, 1 mM calcium chloride, 1 mM magnesium chloride and 0.1% bovine serum albumin (BSA)]. After the wash, 100 μl/well of HBSS buffer supplemented with IBMX (100 μM final concentration) were added, quickly followed by addition of 50 μl of HBSS buffer containing compound dilutions of glucagon and ZP3780. After incubation at 37°C for 15 min, the reactions were stopped by addition of 160 μl lysis buffer. The accumulated cAMP levels were quantified using the CisBIO cAMP dymanic HTRF kit (Cisbio, 62AM4PEC) according to the manufacturer’s instructions and measured on an EnVision plate reader (PerkinElmer). Data were fitted by non-linear regression using GraphPad Prism.
Functional testing of the hGCGR mutations was, in general, conducted as described above. The constructs used for the mutational studies all contained a N-terminal FLAG-tag preceded by a HA-signal peptide and were inserted into the pcDNA3.1(neo) expression vector. The following DNA amounts were transfected to ensure similar surface expression to WT hGCGR: WT GCGR was transfected with 0.2 μg ml–1 cell suspension, Q232A and D385A GCGR with 0.05 μg DNA ml–1 cell suspension, and finally L242A, F322A, D370A, R378A, L395A, and Y400F GCGR were transfected with 0.035 μgml−1 cell suspension. Again, in all cases, the total DNA amount was adjusted to 1 μg ml–1 cell suspension by addition of pcDNA3.1(zeo). Twenty-four hours after transfection the assay was performed by washing wells once in 100 μl DPBS + 1 mM calcium chloride (DPBS Gibco 14190). After the wash, 50 μl/well HBSS buffer (HBSS Gibco 14025, 20 mM HEPES pH 7.5, 1 mM calcium chloride, 1 mM magnesium chloride and 0.1% BSA) was added and the plates were incubated for 30 min at 37°C. Hereafter, 50 μl/well glucagon dilutions were added, which were prepared in HBSS buffer supplemented with IBMX to a final concentration of 100 μM. After incubation at 37°C for 15 min, the reactions were stopped and the accumulated cAMP levels were quantified as described above.
Enzyme-linked immunosorbent assay
Surface expression levels of WT and mutated hGCGR variants were tracked using a direct enzyme-linked immunosorbent assay (ELISA) against the N-terminal FLAG tag. In brief, cells were transfected and seeded in a white Poly-D-Lysine-coated 96-well plate as described for the functional cAMP accumulation assay. Twenty-four hours after transfection, cells were washed with 100 μl/well DPBS + 1 mM calcium chloride and fixed with 50 μl/well 4% paraformaldehyde solution for 5 min at room temperature. The wells were washed twice with 100 μl DPBS + 1 mM calcium chloride and blocked with 100 μl/well blocking solution (3% drymilk, 1 mM calcium chloride 50 mM Tris-HCl, pH 7.5) for 30 min at room temperature. Hereafter 75 μl/well HRP-conjugated anti-FLAG antibody (Sigma Aldrich, A8592), diluted 1:2000 in blocking solution, was added and the plate was incubated for 1 hour at room temperature. The plate was washed four times with 100 μl/well blocking solution followed by four washes with 100 μl/well DPBS + 1 mM calcium chloride. The relative amount of present HRP was detected by adding 60 μl/well DPBS + 1 mM calcium chloride and 20 μl/well HRP substrate (Bio-Rad, 170–5060). The plate was incubated for 10 min at room temperature before recording of luminescence on an EnVision plate reader (Perkin Elmer).
Purification of GCGR
Human GCGR (Q27-F477) with N-terminal FLAG and C-terminal octahistidine tag was expressed in Spodoptera frugiperda Sf9 insect cells using the baculovirus method (Expression Systems) in the presence of the L-168,049 (Tocris Bioscience). Cells were collected 48 hours after infection and stored at −80°C until further use. GCGR was extracted with 1% lauryl maltose neopentyl glycol (L-MNG; Anatrace), 0.1% cholesteryl hemisuccinate (CHS; Sigma) in 20 mM HEPES pH 7.5, 150 mM sodium chloride, 20% glycerol, and protease inhibitors and purified by nickel-chelating sepharose chromatography in the presence of the ligand NNC0640. The eluate from the nickel resin was supplemented with 2 mM calcium chloride and applied to the M1 anti-FLAG immunoaffinity resin and washed with progressively decreasing concentration of NNC0640. The receptor was eluted in a buffer consisting of 20 mM HEPES pH 7.5, 150 mM sodium chloride, 0.05% L-MNG, 0.005% CHS, FLAG peptide and 5 mM EDTA. The final step of purification was size exclusion chromatography on Superdex 200 10/300 gel filtration column (GE Healthcare) in 20 mM HEPES pH 7.5, 150 mM sodium chloride, 0.02% MNG and 0.002% CHS. Finally, apo-GCGR was concentrated to ~400 μM and stored at −80°C.
To perform DEER spectroscopy, human GCGR (Q27-F477) with N-terminal FLAG and C-terminal octahistidine tag was cloned into pcDNA-Zeo-tetO (52). Four native cysteine residues were mutated to make the minimal cysteine (mC) construct (C171T, C240A, C287A, C401V). Engineered cysteines were introduced pairwise into TM4/TM5 and TM4/TM6, respectively. Constructs were transfected into Expi293F (Thermo Fisher) cells stably expressing the tetracycline repressor using Expifectamine transfection kit (Thermo Fisher) following the manufacturers recommendations with the following modifications. Two days post-transfection, GCGR expression was induced with doxycycline (4 μg ml–1 and 5 mM sodium butyrate) in the presence of 1 μM L-168,049 (Tocris Bioscience). Cells were harvested 30 hours post-induction and stored at −80°C until use. Pellets were thawed and solubilized with 20 mM HEPES pH 7.5, 150 mM sodium chloride, 20% glycerol, 1% lauryl maltose neopentyl glycol (L-MNG, Anatrace), 0.1% CHS (Sigma), and protease inhibitors and purified by anti-FLAG immunoaffinity chromatography. From the FLAG resin, GCGR was eluted with 20 mM HEPES pH 7.5, 150 mM sodium chloride, 0.05% L-MNG, 0.005% CHS, FLAG peptide and 5 mM EDTA and labeled with 20-fold molar excess of bis-(2,2,5,5-tetramethyl-3-imidazoline-1-oxyl-4-yl)disulfide (IDSL) for 2 hours at room temperature. IDSL-labeled GCGR was purified from free spin label and buffer exchanged into 40 mM HEPES pH 7.5, 150 mM sodium chloride, 0.02% MNG and 0.002% CHS made with D2O (Cambridge Isotopes) using size exclusion chromatography on Superdex 200 10/300 gel filtration column (GE Healthcare).
Expression and purification of heterotrimeric Gs
Heterotrimeric Gs was expressed and purified as previously described (30). Briefly, heterotrimeric Gs was expressed in Trichoplusia ni (T. ni) insect cells using baculoviruses generated by the BestBac method (Expression Systems). Two separate baculoviruses were used, one encoding the human Gαs short splice variant and the other encoding both the Gβ1 and Gγ2 subunits, with an histidine tag and HRV 3C protease site inserted at the amino terminus of the β-subunit. T. ni cells were infected with the baculoviruses followed by an incubation of 48 hours at 27°C. Cells were harvested by centrifugation and lysed in a buffer comprised of 10 mM Tris, pH 7.5, 100 μM magnesium chloride, 5 mM β-mercaptoethanol (βME), 20 μM GDP and protease inhibitors. The membrane fraction was collected by centrifugation and solubilized with a buffer comprised of 20 mM HEPES, pH 7.5, 100 mM sodium chloride, 1% sodium cholate, 0.05% DDM, 5 mM magnesium chloride, 5 mM βME, 5 mM imidazole, 20 μM GDP and protease inhibitors. The solubilization reaction was incubated for 45 min at 4°C after homogenization with a Dounce homogenizer. After centrifugation, the soluble fraction was loaded onto Ni-chelated sepharose followed by a gradual detergent exchange into 0.1% DDM.
The protein was eluted in buffer supplemented with 200 mM imidazole and dialyzed overnight in 20 mM HEPES, pH 7.5, 100 mM sodium chloride, 0.1% DDM, 1 mM magnesium chloride, 5 mM βME and 20 μM GDP together with HRV 3C protease to cleave off the amino- terminal 6xHis tag. The cleaved 6xHis tag, uncleaved fractions and 3C protease were removed by Ni-chelated sepharose and the G protein was dephosphorylated by lambda protein phosphatase (NEB), calf intestinal phosphatase (NEB), and antarctic phosphatase (NEB) in the presence of 1 mM manganese chloride. Lipidated Gs heterotrimer was isolated using a MonoQ 10/100 GL column (GE Healthcare). After binding of the protein to the column in buffer A [20 mM hydroxyethylpiperazine ethane sulfonic acid (HEPES), pH 7.5, 50 mM sodium chloride, 1 mM magnesium chloride, 0.05% DDM, 100 μM tris(2-carboxyethyl)phosphine (TCEP), and 20 μM GDP], the column was washed with buffer A and the G protein heterotrimer was eluted with a linear gradient of 0–50% buffer B (buffer A with 1 M sodium chloride). The main peak containing isoprenylated G protein heterotrimer was collected and the protein was dialyzed into 20 mM HEPES, pH 7.5, 100 mM soidum chloride, 0.02% DDM, 100 μM TCEP and 20 μM GDP. After concentrating the protein to 250 μM, 20% glycerol was added and the protein was flash frozen in liquid nitrogen and stored at −80°C until use.
Purification of Nb35
Nb35 was expressed and purified as described previously (50). Briefly, Nb35 was expressed in Escherichia coli BL21 cells. After lysis, it was purified on a nickel affinity chromatography and finally subjected to size exclusion chromatography on a Superdex 200 10/300 gel filtration column (GE Healthcare) in 20 mM HEPES pH 7.5, 150 mM sodium chloride. Purified Nb35 was concentrated, flash frozen, and stored at −80°C until further use.
Formation and purification of the GCGR-Gs-Nb35 complex
Purified Gαsβ1γ2 in 0.02% DDM was incubated with 1% MNG for one hour on ice and, simultaneously, GCGR was incubated with 5-fold molar excess ZP3780 (500 μM) at room temperature. A 1.5-fold molar excess of detergent-exchanged Gαsβ1γ2 was incubated with ZP3780-bound GCGR at room temperature for two hours, after which a 2-fold molar excess (in terms of Gαsβ1γ2) of Nb35 was added and incubated on ice for 1.5 hours. To stabilize the nucleotide-free complex, Apyrase (1 unit, NEB) was added and incubated overnight at 4°C. A 4-fold volume of 20 mM HEPES pH 7.5, 100 mM sodium chloride, 0.8% L-MNG/0.08% CHS, 0.27% GDN/0.027% CHS, 1 mM magnesium chloride, 5 μM ZP3780, and 2 mM calcium chloride was added to the complexing reaction and complex was purified by M1 anti-FLAG affinity chromatography. The complex was eluted in 20 mM HEPES pH 7.5, 100 mM sodium chloride, 0.01% MNG/0.001% CHS, 0.0033% GDN (Anatrace)/0.00033% CHS, 5 μM ZP3780, 5 mM EDTA, and FLAG peptide. The eluted complex was supplemented with 100 μM TCEP and subjected to size exclusion chromatography on a Superdex 200 10/300 Increase column in 20 mM HEPES pH 7.5, 100 mM sodium chloride, 5 μM ZP3780, 0.00075% MNG, and 0.00025% GDN. Peak fractions were concentrated to ~16 mg ml–1 for electron microscopy studies.
Cryo-EM data acquisition and data processing
An aliquot of 3.5 μl GCGR-Gs-Nb35 complex was applied to glow-discharged 200 mesh grids (Quantifoil R1.2/1.3), at a concentration of 17 mg ml–1 and subsequently vitrified using a Vitrobot Mark IV (Thermo Fischer Scientific) at 100% humidity and 4°C. CryoEM images were collected on a Titan Krios operated at 300 kV at a nominal magnification of 130,000× with a Gatan GIF Quantum LS Imaging energy filter using a Gatan K2 Summit direct electron camera in counted mode, corresponding to a pixel size of 1.06 Å. A total number of 3724 movie stacks were obtained with a dose rate of 7 e−/pix/s and total exposure time of 8 s with 0.2 s per frame, resulting in a total dose of 50 electron per Å2. The defocus range was set to 1.2–2.2 μm.
Dose-fractionated image stacks were subjected to beam-induced motion correction using MotionCor2 (53). Contrast transfer function parameters for each micrograph were determined by Gctf v1.06 (54). Data processing was performed in RELION3.0 (55). A total number of 2,039,910 particles were selected from a template-based auto-picking. A subset of 296,516 particles were selected after one round of two-dimensional (2D) and 3D classification. Particles projections from micrographs with signal better than 3.5 Å were subjected to Bayesian polishing, CTF refinement and 3D reconstruction. The final subset of 266,267 particles was imported to cisTEM for 3D local refinement, resulting in a reconstruction with global resolution of 3.1 Å at FSC 0.143 (56). Local resolution was determined using the Bsoft package at cutoff FSC of 0.5 (57).
Model building and refinement
The initial template of GCGR was derived from the crystal structure of GCGR (PDB 5YQZ) (17). The structural coordinates of PDB 5VAI (4) were used as initial models for the Gs and Nb35. Models were docked into the EM density map using UCSF Chimera (58), followed by iterative manual building in Coot (59). The final model was subjected to global refinement and minimization in real space using phenix.real_space_refine in Phenix (60). Molprobity was used to evaluate model geometry (61). FSC curves were calculated between the resulting model and the half map used for refinement as well as between the resulting model and the other half map for cross-validation.
β2AR purification
β2AR was expressed in Sf9 insect cells using the baculovirus method (Expression Systems) and purified as described previously (50).
Purification of Gαs and Gβγ for GDP release assay
Human Gαs subunit with an amino-terminal 6x histidine tag followed by an HRV 3C protease site were expressed in Rosetta 2 (DE3) cells (EMD Millipore) using pET28a. Cells were grown in Terrific Broth to OD600 of 0.6, and protein expression was induced by addition of 0.5 mM IPTG. After 15 hours of incubation at room temperature, cells were harvested and resuspended in lysis buffer (50 mM HEPES pH 7.5, 100 mM sodium chloride, 1 mM magnesium chloride, 50 μM GDP, 5 mM βME, 5 mM imidazole, and protease inhibitors). Cells were disrupted by sonification using a 50% duty cycle, 70% power for four times 45 s. Intact cells and cell debris were subsequently removed by centrifugation and the supernatant was incubated with Ni-chelated sepharose for 1.5 hours at 4°C. The Ni-chelated sepharose resin was washed multiple times with lysis buffer in batch and then loaded into a wide-bore glass column, and protein was eluted with lysis buffer containing 200 mM imidazole. The eluted protein was dialyzed overnight in dialysis buffer (20 mM HEPES pH 7.5, 100 mM sodium chloride, 1 mM magnesium chloride, 20 μM GDP, 5 mM βME, and 5 mM imidazole). The amino terminal histidine tag was cleaved by adding 1:1000 w/w 3C protease into the dialysis bag. Uncleaved protein, cleaved histidine tag, and 3C protease were subsequently removed by incubation with Ni-chelated sepharose for 45 min at 4°C. The resin was loaded into a wide-bore glass column and the flow-through containing the Gα subunit was collected. The protein was concentrated and run on a Superdex 200 10/300 GL column in SEC buffer (20 mM HEPES pH 7.5, 100 mM sodium chloride, 1 mM magnesium chloride, 20 μM GDP, and 100 μM TCEP).
The Gβγ heterodimer was expressed in Tni cells using a baculovirus generated by the BestBac (Expression systems) method. We used one virus containing the genes for Gβ1 and Gγ2 subunits. The sequence for the Gβ subunit contains an amino terminal 6x histidine tag followed by an HRV 3C protease sequence, allowing us to cleave off the histidine tag after purificiation. T. ni cells were infected at a density of 3.0 × 106 cells ml–1 and incubated at 27°C for 48 hours. After centrifugation, cells were resuspended in lysis buffer (10 mM Tris, pH 7.5, 5 mM βME, and protease inhibitors). The membrane fraction was collected by centrifugation and solubilized by a dounce homogenizer with a buffer comprised of 20 mM HEPES pH 7.5, 100 mM sodium chloride, 1.0% sodium cholate, 0.05% DDM, 5 mM βME, and protease inhibitors. The solubilization reaction was stirred at 4°C for 40 min, and then centrifuged to remove insoluble debris. Ni-chelating sepharose was added to the supernatant and stirred for 1.5 hours at 4°C followed by multiple washes of the Ni-chelating sepharose resin in batch with solubilization buffer. The resin was collected into a wide-bore glass column, and the detergent was gradually exchanged from sodium cholate to 0.1% DDM. The protein was eluted with Ni-chelating sepharose elution buffer (20 mM HEPES pH 7.5, 100 mM sodium chloride, 0.1% DDM, 5 mM βME, 200 mM imidazole and protease inhibitors). To cleave off the amino terminal histidine tag, HRV 3C protease (1:1000 w/w) was added and the sample dialyzed overnight in 20 mM HEPES pH 7.5, 100 mM sodium chloride, 0.1% DDM, 5 mM βME, and 5 mM imidazole. The cleaved histidine tag, uncleaved fractions, and HRV 3C protease were removed by loading of the sample on Ni-chelating sepharose resin and the flow-through containing the Gβγ heterodimer was collected. Lambda protein phosphatase (2000 units, NEB), calf intestinal phosphatase (10 units, NEB), and Antarctic phosphatase (5 units, NEB) were added together with 1 mM manganese chloride, followed by a 1 hour incubation at 4°C. After adjusting the sodium chloride concentration to 50 mM using dilution buffer composed of 20 mM HEPES pH 7.5, 0.02% DDM, and 100 μM TCEP, the sample was passed through a 0.22 μm filter and loaded onto a MonoQ 10/100 GL column (GE Healthcare) equilibrated in buffer A (20 mM HEPES pH 7.5, 50 mM sodium chloride, 0.02% DDM, and 100 μM TCEP). The column was washed with 5 CV of buffer A and bound protein was eluted over 7.5 CV with a linear gradient from 0–25% buffer B (buffer A with 1 M sodium chloride). The fractions containing prenylated heterodimer were pooled and dialyzed in buffer containing 20 mM HEPES pH 7.5, 100 mM sodium chloride, 0.02% DDM, 100 μM TCEP. After concentrating the protein, 20% glycerol was added and protein aliquots were flash frozen in liquid nitrogen.
HDL reconstitution
Reconstitution of GCGR and β2AR into high-density lipoprotein particles was performed using a 3:2 mixture of POPC/POPG and MSP1E3D1 as belt protein. Receptor:lipid:MSP was mixed at 1:10:60 and incubated for 2 hour at 4°C. Biobeads (Biorad) at a ratio of lipid: beads, 1:8 was added and rotated at 4°C for 2 hours. Further addition of biobeads at ratio of 1:8 of lipid:beads was done and incubated overnight, rotating at 4°C. Reconstituted HDL-GCGR and HDL-β2AR were separated from empty discs by FLAG-M1 chromatography and further purified by SEC using a Superdex 200 10/300 Increase column into 20 mM HEPES pH 7.5, 100 mM sodium chloride.
GTP turnover assay
The GTP turnover assay was performed by using a modified protocol of the GTPase-Glo™ assay (Promega) as described previously (30) with the following modification. The final reaction buffer consisted of 20 mM Hepes pH 7.5, 100 mM sodium chloride, 10 mM magnesium chloride, 100 uM TCEP, 0.02% DDM, 0.1 μM GDP, 10 μM GTP. Before the reaction was started, HDL-reconstituted GCGR (300 nM) or β2AR (300 nM) was incubated in the presence of 100 μM full agonist (ZP3780 or epinephrine) in buffer containing 20 mM HEPES pH 7.5, 100 mM sodium chloride, 20 μM GTP. After incubation for 60 min at room temperature, G protein (500 nM) was added in buffer containing 20 mM HEPES pH 7.5, 100 mM sodium chloride, 20 mM magnesium chloride, 200 μM TCEP, 0.04% DDM, and 0.2 μM GDP. After incubation for a given time (Fig. 5C), reconstituted GTPase-Glo reagent supplemented with 10 μM adenosine 5′-diphosphate (ADP) was added to the sample and incubated for 30 min at room temperature. Luminescence was measured after the addition of detection reagent and incubation for 10 min at room temperature using a SpectraMax Paradigm plate reader. The GTP concentration in the samples was calculated by using the relative light units (RLUs) of GTP at different concentrations and the data was analyzed using GrapPad Prism.
To calculate the maximum rate for the GTP turnover, the GTPase-Glo™ assay (Promega) was performed at different concentrations of receptor reconstituted into HDL particles (GCGR: 0–7.1 uM; β2AR: 0–7.5 uM). After addition of Gs at concentrations of 0.1 μM and 0.5 μM to β2AR and GCGR, respectively, reactions proceeded for 10 min (β2AR) or 90 min (GCGR) before addition of GTPase Glo reagent and ADP, as described in the manufacturer’s protocol. Rates were calculated using the RLUs of a GTP standard curve and analyzed using GraphPad Prism.
GDP release assay
The single turnover GDP release assay was performed with GCGR and β2AR reconstituted into HDL particles and purified G protein subunits (Gαs and Gβγ) in detergent. For this purpose, Gαs in 20 mM HEPES pH 7.5, 100 mM sodium chloride, 1 mM magnesium chloride, 100 μM TCEP, and 5 μM GDP was diluted to 0.8 μM in GDP-loading buffer (20 mM HEPES pH 7.5, 150 mM sodium chloride, 1 mM EDTA pH 8.0, 100 μM TCEP) and [3H]-GDP (39.8 Cimmol−1, Perkin Elmer) was added to a final concentration of 2.5 μM. After incubation for 60 min at room temperature, Gβγ in 20 mM HEPES pH 7.5, 100 mM sodium chloride, 0.08% DDM, 100 μM TCEP was added with a 1.2-fold molar access and incubated for 30 min to allow formation of the heterotrimer. GCGR and β2AR in HDL particles were diluted to 10 μM in 20 mM HEPES pH 7.5, 100 mM sodium chloride, and 2 mM GTP and incubated with 100 μM of agonist (ZP3780 or epinephrine) for 1 hour at room temperature. The reaction was started by mixing the ligand-bound receptor with [3H]-GDP loaded heterotrimer to reach final concentrations of 200 nM Gs and 5 μM receptor. At various time points, 500 μl of ice-cold wash buffer (20 mM HEPES pH 7.5, 150 mM sodium chloride, and 20 mM magnesium chloride) was added to 20 μl of the reaction and the mixture was immediately filtered using a microanalysis filter holder (EMD Milipore) and pre-wet mixed cellulose filters (25 mm, 0.22 μm). The filter was washed three times with 500 uL ice-cold wash buffer and dried for at least 1 hour at room temperature. The amount of radioactivity that remained bound to the filters was determined by liquid scintillation spectrometry. GDP release data were analyzed using GraphPad Prism.
DEER
Sample preparation
IDSL-labeled GCGR in detergent or HDL was diluted to 300 nM and incubated with 500 μM ZP3780 for one hour on ice. Labeled-GCGR was then concentrated to ~100 μM and additional 100 μM ZP3780 was added and frozen with 20% D8-glycerol (Camebridge Isotope). Sample was transferred to 1.4/1.7 mm (i.d./o.d.) borosilicate capillaries (VitroCom, Mountain Lakes, NJ) and flash frozen.
Data Collection
DEER data were acquired on a Bruker ELEXSYS E580 spectrometer equipped with a SuperQ-FT bridge and 10 W AmpQ amplifier operated at Q-band frequency (~33.5 GHz) with an ER 5107D2 resonator (Bruker Biospin). Sample temperature was maintained at 50 K during data collection using a recirculating helium cryo-cooling system (ColdEdge Technologies). A dead-time free four-pulse DEER sequence (62) was used with an 8-step suppression of the deuterium nuclear modulation (63) and 8-step phase cycle. The pump pulse (rectangular; ~36 ns) was applied at the maximum of the low-field line of the absorbance spectrum. The observe pulses (rectangular; ~20 ns π/2 and ~40 ns π) were applied at a −70 MHz offset from the pump pulse. Optimal pulse lengths for each DEER experiment were determined by an echo nutation experiment. Background correction and model-free fitting analysis of dipolar evolution functions were performed with the program LongDistances933 (developed by Christian Altenbach and available at www.biochemistry.ucla.edu/biochem/Faculty/Hubbell/). The regularization (smoothness) parameter for distance distribution determination was selected using the L-curve criterion (64), and the background signal was fitted assuming a three-dimensional distribution of remote spins. The upper limit of reliable distance (r) and width determination (σ) for each mutant in nanometers was calculated using the following equations (36):
where tmax is the maximum time domain recorded for each sample. All distributions were area-normalized and plotted in GraphPad Prism.
Bimane and NBD Fluorescence
Sample preparation
Receptors (minimal cysteine-GCGR (mC-GCGR) 349C and minimal cysteine-β2AR (mC-β2AR) 265C) at 10 μM were incubated with 5-molar excess of bimane or NBD at room temperature for one hour. After concentration the labeled receptors were further purified, to remove excess label, using size exclusion chromatography on a Superdex 200 10/300 Increase column in 20 mM HEPES pH 7.5, 100 mM sodium chloride, 0.01% MNG/0.001% CHS.
Data Collection
Bimane- and NBD-labeled receptor were used at a concentration of 0.1 μM in buffer containing 20 mM HEPES, pH 7.5, 100 mM sodium chloride, 0.01% MNG/0.001% CHS. The concentration of Gs added was 10 μM. Fluorescence data was collected in a 150 μl cuvette with a FluorEssence v3.8 software on a Fluorolog instrument (Horiba) in photon-counting mode. Bimane fluorescence was measured by excitation at 370 nm with excitation and emission bandwidth passes of 4 nm. The emission spectra were recorded from 410 to 510 nm with 1 nm increment and 0.1 s integration time. NBD was measured by excitation at 420 nm with excitation and emission bandwidth passes of 4 nm, and the emission spectra were recorded from 510 nm to 610 nm with 1 nm increment and 0.1 s integration time.
The statistical analysis for the λmax change on ligand and Gs binding, was carried out by independently fitting a single Gaussian between 440 and 485 nm for each of the three measurements of 349C-bimane-labeled GCGR in the Apo state, ZP3780-bound and Gs- bound states. All fluorescence profiles were well-represented by the single Gaussians (R2 > 0.98). Fitted λmax values were taken as the mean of the Gaussian fits and subsequent statistical analyses were performed on the triplicate values for each experimental condition. The λmax values for each condition were compared with unpaired t tests assuming a Gaussian distribution using Welch’s correction so as to not assume equal standard deviations.
Ensemble FRET
Sample preparation
Receptors (minCys-GCGR S265C and minCys-β2AR) at 10 μM were incubated with 5-fold molar excess of Cy3B maleimide (GE Healthcare) at room temperature for 45 min. After quenching with cysteine, size exclusion chromatography on a Superdex 200 10/300 Increase column in 20 mM HEPES pH 7.5, 100 mM sodium chloride, 0.01% MNG/0.001% CHS was performed to remove excess label.
Gs at 10 μM were incubated with 5 molar equivalence of Sulfo-Cy5-NHS ester (Lumiprobe) at room temperature for 45 min before quenching with TRIS-HCl pH 7.5. To remove excess label, size exclusion chromatography was performed on a Superdex 200 10/300 Increase column in 20 mM HEPES pH 7.5, 100 mM sodium chloride, 0.05% DDM/0.001% CHS. 10 μM GDP, 1 mM MgCl2 and 100 μM TCEP.
Data Collection
Cy3B and Sulfo-Cy5 labeled receptors and Gs, respectively, were used to measure the rate of association using FRET. Fluorescence spectra was recorded on a Fluorolog instrument (Horiba) in photon-counting mode. All data were collected with emission (567 nm) and excitation (520 nm) bandpass of 2 nm and integration time of 0.1 s nm−1. The baseline was recorded with Cy3B-labeled receptor (100 nM) till it stabilized (~ 100 s), then 1 μM Sulfo-Cy5-labeled Gs was added and the drop in fluorescence intensity was recorded as a function of time. When the spectra reached steady-state (~800 s), to analyze Gs dissociation GDP or GTPγS was added and the spectra was monitored till 1200 s.
Bodipy-GTPγS binding
Bodipy-GTPγS is a non-hydrolyzable fluorescently labeled GTP analog, which is self-quenched in solution. However, upon binding to G proteins, Bodipy-GTPγS fluorescence increases due to unquenching of the fluorophore. This property was used to investigate the rate of GTP binding to nucleotide-free GPCR–G protein complexes. Nucleotide-free GCGR-Gs and β2AR-Gs complexes were prepared as described in this manuscript and previously (50). For the nucleotide-binding experiment, fluorescence form BODIPY-FL-GTPyS (Thermo Fisher) was recorded in 500 uL quarz cuvettes using the Fluorolog spectrophotometer (HORIBA). The fluorophore was exited at 495 nm and emission was detected at 508 nm at 22°C. All experiments were performed in a buffer comprised of 20 mM HEPES, pH 7.5, 100 mM sodium chloride, 0.01%L-MNG, 0.001% CHS, 10 mM magnesium chloride, and 100 μM TCEP. Typically, kinetics data were collected for 150 nM BODIPY-FL-GTPγS in the absence of receptor-G protein complexes for 100 s to establish the baseline fluorescence intensity. Receptor-G protein complexes were added to 1 μM and rapidly mixed in the fluorescence cuvette without halting data collection. The resulting kinetics spectra were plotted and fit to a one-phase association function using GraphPad Prism.
System setup and protocols for molecular dynamics simulations
We performed all-atom MD simulations for both ZP3780- and NNC1702-bound GCGR. Simulations were initiated from the cryo-EM structure after removing the G protein. The NNC1702-bound structure was obtained by deleting H1 and mutating D9 to E. An initial system containing the peptide bound GCGR and POPC membrane was assembled with Visual Molecular Dynamics 1.9.3 (VMD) (65) Membrane Builder module with 0.15 M sodium chloride in the solution. Bilayer dimensions were chosen to maintain at least a 30Å buffer between protein images in the x–y plane and a 20Å buffer between protein images in the z direction. Final system dimensions were approximately 90 × 90 × 130 Å3. There were 5 replicas for each system.
Simulations were performed with the Compute Unified Device Architecture (CUDA) version of NAMD 2.12 (66), the CHARMM36m force field (67) for proteins, lipids and ions, and TIP3P water. Long-range electrostatic forces were calculated with the Particle Mesh Ewald method (68), and an interaction cutoff of 12 Å was applied within periodic boundary conditions. Van der Waals (vdW) forces were smoothly shifted to zero between 10 and 12 Å. Equations of motion were integrated with a time step of 2 fs. Langevin dynamics with a damping coefficient of 5 ps−1 was used to keep the temperature at 310 K. The pressure was maintained at 1 atm using a Nosé−Hoover−Langevin piston (69, 70). Bond lengths were constrained using the SHAKE algorithm (71).
Simulation systems were equilibrated in three stages. In stage one, a 1000 step minimization followed by 0.5 ns simulation in which everything except lipid tails was fixed was performed to equilibrate lipid layer. In stage two, another 1000 step minimization followed by a 1 ns equilibration with the protein constrained was performed to permit the environment to relax. In stage three, a 5-ns-long simulation was performed to release harmonic constraints and to further equilibrate the whole system.
After equilibration, each replicate was simulated for 300 ns. Atomic coordinates of all atoms were saved every 2 ps. We visualized and aligned trajectories by using VMD to eliminate all rotational and translational motions.
NanoBiT–G protein dissociation assay
GPCR-induced Gs dissociation was measured by a NanoBiT–G protein dissociation assay (33), in which interaction between a Gα subunit and a Gβγ subunit was monitored by a NanoLuc-based enzyme complementation system called NanoBiT (Promega). Specifically, a NanoBiT-Gs protein consisting of Gαs subunit fused with a large fragment (LgBiT) at the alpha helical domain and an N-terminally small fragment (SmBiT)-fused Gγ2 subunit with a C68S mutation was expressed along with untagged Gβ1 subunit, RIC8B and a test GPCR. HEK293A cells (Thermo Fisher Scientific) were seeded in a 6-well culture plate at a concentration of 2 × 105 cells ml−1 [2 ml per well in Dulbecco’s modified Eagle’s medium (DMEM; Nissui) supplemented with 10% fetal bovine serum (Gibco), glutamine, penicillin, and streptomycin] 1 day before transfection. Transfection solution was prepared by combining 5 μl (per well in a 6-well plate hereafter) of polyethylenimine Max solution (Polysciences; 1 mg ml−1), 200 μl of Opti-MEM (Thermo Fisher Scientific) and a plasmid mixture consisting of 100 ng LgBiT-containing Gαs subunit, 500 ng Gβ1, 500 ng SmBiT-fused Gγ2 (C68S) 100 ng, RIC8B and a test GPCR that was optimized to match an expression level. After incubation for 1 day, transfected cells were harvested with 0.5 mM EDTA-containing Dulbecco’s PBS, centrifuged and suspended in 2 ml of HBSS containing 0.01% bovine serum albumin (BSA; fatty acid–free grade; SERVA) and 5 mM HEPES pH 7.4 (assay buffer). The cell suspension was dispensed in a white 96-well plate at a volume of 80 μl per well and loaded with 20 μl of 50 μM coelenterazine (Carbosynth) diluted in the assay buffer. After 2 hours of incubation at room temperature, the plate was measured for baseline luminescence (Spectramax L, Molecular Devices) and a titrated test ligand (20 μl; 6X of final concentrations) were manually added. Ligands and their sources were as follows: (−)-isoproterenol hydrochloride (Sigma-Aldrich) for β2AR; serotonin (5-hydroxytryptamine hydrochloride; FUJIFILM Wako Pure Chemical) for 5-HT4; prostaglandin D2 (Cayman Chemical) for DP; histamine dihydrochloride (FUJIFILM Wako Pure Chemical) for H2R; arginine vasopressin (Peptide Institute) for V2R; glucagon (Zealand Pharma) for GCGR; glucagon-like peptide 1 (7–37) (Peptide Institute) for GLP-1; PACAP27 (Peptide Institute) for PAC1; parathyroid hormone (1–34) (Peptide Institute) for PTH. The plate was immediately read at room temperature for the following 3 min at a measurement interval of 7 s with an accumulation time of 0.17 s per read. The kinetics luminescence counts were normalized to the initial count and fold-change signals over vehicle treatment were used to plot G protein dissociation response. G protein dissociation kinetics were calculated by fitting the normalized luminescent data to a one-phase dissociation model built in Prism 8 software (GraphPad Prism). Initial G protein dissociation speed was calculated by a formula of (Plateau – 1)*K where “Plateau” represents a saturated normalized luminescent counts whereas “K” denotes a rate constant in a unit that is reciprocal of time (sec). The resulting dissociation speed data were fitted to a three-parameter sigmoidal concentration-response curve (“Bottom” is fixed to a value of 0), from which an Emax value (“Top”) was used to represent G protein dissociation for a given experiment. When a sigmoidal curve did not converge due to a lack of saturating data points, a dissociation speed at the highest ligand concentration was used as an Emax value.
Flow cytometry analysis
HEK293A cells (Thermo Fisher Scientific) were seeded in a 6-well culture plate and transfection was performed as following the same procedure as described in the “NanoBiT–G protein dissociation assay” section. Our optimization of GPCR plasmid volume to match surface expression level found following GPCR to be used in the study (volume is denoted as per well in the 6-well plate): β2AR (200 ng), 5-HT4 (40 ng), DP (200 ng), H2R (20 ng), V2R (500 ng), GCGR (40 ng), GLP-1R (40 ng), PAC1R (500 ng), PTH1R (40 ng). Except for PAC1R, all of the eight GPCRs (β2AR, 5-HT4, DP, H2R, V2R, GCGR, GLP-1R, PTH1R) were N-terminally fused to a hemagglutinin signal sequence (KTIIALSYIFCLVFA) and a FLAG epitope (DYKDDDDK) tag with or without a linker sequence between the FLAG epitope tag and a GPCR. For construction of the Class A GPCRs, a full-length coding sequence except for the initial methionine was used. For the N-terminal modification, a signal sequence of the Class B GPCRs was removed as following: GCGR (amino acid positions 1–24), GLP-1R (1–23) and PTH1R (1–23). A PAC1R construct contained the FLAG epitope tag at the amino acid position between 23 and 24 of the native PAC1R sequence. One day after transfection, the cells were collected by adding 200 μl of 0.53 mM EDTA-containing Dulbecco’s PBS (D-PBS), followed by 200 μl of 5 mM HEPES pH 7.4-containing Hank’s Balanced Salt Solution (HBSS). The cell suspension was transferred to a 96-well V-bottom plate in duplicate and fluorescently labeled with an anti-FLAG epitope (DYKDDDDK) tag monoclonal antibody (Clone 1E6, FujiFilm Wako Pure Chemicals; 10 μg ml−1 diluted in 2% goat serum- and 2 mM EDTA-containing D-PBS (blocking buffer)) and a goat anti-mouse IgG secondary antibody conjugated with Alexa Fluor 488 (Thermo Fisher Scientific, 10 μg ml−1 diluted in the blocking buffer). After washing with D-PBS, the cells were resuspended in 200 μl of 2 mM EDTA-containing-D-PBS and filtered through a 40-μm filter. The fluorescent intensity of single cells was quantified by an EC800 flow cytometer equipped with a 488 nm laser (Sony). The fluorescent signal derived from Alexa Fluor 488 was recorded in an FL1 channel, and the flow cytometry data were analyzed with the FlowJo software (FlowJo). Live cells were gated with a forward scatter (FS-Peak-Lin) cutoff at the 390 setting, with a gain value of 1.7. Values of mean fluorescence intensity (MFI) from approximately 20,000 cells per sample were used for analysis.
Figure preparation
Figures were created using the PyMOL Molecular Graphics System, Version 2.20 Schrödinger, (http://pymol.org), and the UCSF Chimera X package (72). In vitro graphs were created using GraphPad Prism.
Supplementary Material
ACKNOWLEDGMENTS
We thank S. Reedtz-Runge (Novo Nordisk A/S) for sharing the ligand NNC0640. We thank K. Sato, S. Nakano, and A. Inoue (Tohoku University) for their assistance of plasmid preparation and cell-based GPCR assays. We thank P. Aschauer for initial characterization of glucagon analogs.
Funding: D.H. was supported by the German Academic Exchange Service (DAAD). K.K.K. was supported by the American Diabetes Association (ADA) Postdoctoral Fellowship. A.I. was funded by the PRIME 18 g5910013 from the Japan Agency for Medical Research and Development (AMED) and the Japan Society for the Promotion of Science (JSPS) KAKENHI 17K08264. The work is supported by NIH grants R01GM083118 and R01NS028471 to B.K.K.; B.K.K. is a Chan Zuckerberg Biohub investigator.
Footnotes
Competing interests: B.K.K. is a cofounder of and consultant for ConfometRx. J.M.M. and L.G. are employees of Zealand Pharma. M.F.P. is a former employee of Zealand Pharma.
Data and materials availability:
The cryo-EM density map has been deposited in the Electron Microscopy Data Bank (EMDB) under accession code EMD-21866, and model coordinates have been deposited in the Protein Data Bank (PDB) under accession no. 6WPW. All other data are available in the manuscript or supplementary materials. Materials described in this study are available upon request sent to the corresponding authors.
REFERENCES AND NOTES
- 1.Liang Y-L et al. , Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nature 561, 492–497 (2018). doi: 10.1038/s41586-018-0535-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liang Y-L et al. , Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor-Gs complex. Nature 555, 121–125 (2018). doi: 10.1038/nature25773 [DOI] [PubMed] [Google Scholar]
- 3.Liang Y-L et al. , Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 546, 118–123 (2017). doi: 10.1038/nature22327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y et al. , Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 546, 248–253 (2017). doi: 10.1038/nature22394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao L-H et al. , Structure and dynamics of the active human parathyroid hormone receptor-1. Science 364, 148–153 (2019). doi: 10.1126/science.aav7942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang G, Zhang BB, Glucagon and regulation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab 284, E671–E678 (2003). doi: 10.1152/ajpendo.00492.2002 [DOI] [PubMed] [Google Scholar]
- 7.Hollenstein K et al. , Insights into the structure of class B GPCRs. Trends Pharmacol. Sci 35, 12–22 (2014). doi: 10.1016/j.tips.2013.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jelinek LJ et al. , Expression cloning and signaling properties of the rat glucagon receptor. Science 259, 1614–1616 (1993). doi: 10.1126/science.8384375 [DOI] [PubMed] [Google Scholar]
- 9.Mayo KE et al. , International Union of Pharmacology. XXXV. The glucagon receptor family. Pharmacol. Rev 55, 167–194 (2003). doi: 10.1124/pr.55.1.6 [DOI] [PubMed] [Google Scholar]
- 10.Müller TD, Finan B, Clemmensen C, DiMarchi RD, Tschöp MH, The New Biology and Pharmacology of Glucagon. Physiol. Rev 97, 721–766 (2017). doi: 10.1152/physrev.00025.2016 [DOI] [PubMed] [Google Scholar]
- 11.Cryer PE, Davis SN, Shamoon H, Hypoglycemia in diabetes. Diabetes Care 26, 1902–1912 (2003). doi: 10.2337/diacare.26.6.1902 [DOI] [PubMed] [Google Scholar]
- 12.Cho YM, Merchant CE, Kieffer TJ, Targeting the glucagon receptor family for diabetes and obesity therapy. Pharmacol. Ther 135, 247–278 (2012). doi: 10.1016/j.pharmthera.2012.05.009 [DOI] [PubMed] [Google Scholar]
- 13.Exton JH, Robison GA, Sutherland EW, Park CR, Studies on the role of adenosine 3′,5′-monophosphate in the hepatic actions of glucagon and catecholamines. J. Biol. Chem 246, 6166–6177 (1971). [PubMed] [Google Scholar]
- 14.Pedersen JS et al. , The changing face of glucagon fibrillation: Structural polymorphism and conformational imprinting. J. Mol. Biol 355, 501–523 (2006). doi: 10.1016/j.jmb.2005.09.100 [DOI] [PubMed] [Google Scholar]
- 15.Hövelmann U et al. , Pharmacokinetic and pharmacodynamic characteristics of dasiglucagon, a novel soluble and stable glucagon analog. Diabetes Care 41, 531–537 (2018). doi: 10.2337/dc17-1402 [DOI] [PubMed] [Google Scholar]
- 16.Unson CG, Molecular determinants of glucagon receptor signaling. Biopolymers 66, 218–235 (2002). doi: 10.1002/bip.10259 [DOI] [PubMed] [Google Scholar]
- 17.Zhang H et al. , Structure of the glucagon receptor in complex with a glucagon analogue. Nature 553, 106–110 (2018). doi: 10.1038/nature25153 [DOI] [PubMed] [Google Scholar]
- 18.Koth CM et al. , Molecular basis for negative regulation of the glucagon receptor. Proc. Natl. Acad. Sci. U.S.A 109, 14393–14398 (2012). doi: 10.1073/pnas.1206734109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H et al. , Structure of the full-length glucagon class B G-protein-coupled receptor. Nature 546, 259–264 (2017). doi: 10.1038/nature22363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wootten D, Simms J, Miller LJ, Christopoulos A, Sexton PM, Polar transmembrane interactions drive formation of ligand-specific and signal pathway-biased family B G protein-coupled receptor conformations. Proc. Natl. Acad. Sci. U.S.A 110, 5211–5216 (2013). doi: 10.1073/pnas.1221585110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Unson CG, Andreu D, Gurzenda EM, Merrifield RB, Synthetic peptide antagonists of glucagon. Proc. Natl. Acad. Sci. U.S.A 84, 4083–4087 (1987). doi: 10.1073/pnas.84.12.4083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Unson CG, Gurzenda EM, Merrifield RB, Biological activities of des-His1[Glu9]glucagon amide, a glucagon antagonist. Peptides 10, 1171–1177 (1989). doi: 10.1016/0196-9781(89)90010-7 [DOI] [PubMed] [Google Scholar]
- 23.Unson CG, Macdonald D, Ray K, Durrah TL, Merrifield RB, Position 9 replacement analogs of glucagon uncouple biological activity and receptor binding. J. Biol. Chem 266, 2763–2766 (1991). [PubMed] [Google Scholar]
- 24.Dal Maso E et al. , Extracellular loops 2 and 3 of the calcitonin receptor selectively modify agonist binding and efficacy. Biochem. Pharmacol 150, 214–244 (2018). doi: 10.1016/j.bcp.2018.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siu FY et al. , Structure of the human glucagon class B G-protein-coupled receptor. Nature 499, 444–449 (2013). doi: 10.1038/nature12393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Graaf C et al. , Extending the structural view of class B GPCRs. Trends Biochem. Sci 42, 946–960 (2017). doi: 10.1016/j.tibs.2017.10.003 [DOI] [PubMed] [Google Scholar]
- 27.Glukhova A et al. , Rules of engagement: GPCRs and G proteins. ACS Pharmacol. Transl. Sci 1, 73–83 (2018). doi: 10.1021/acsptsci.8b00026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du Y et al. , Assembly of a GPCR-G protein complex. Cell 177, 1232–1242.e11 (2019). doi: 10.1016/j.cell.2019.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manglik A, Kruse AC, Structural Basis for G Protein-Coupled Receptor Activation. Biochemistry 56, 5628–5634 (2017). doi: 10.1021/acs.biochem.7b00747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gregorio GG et al. , Single-molecule analysis of ligand efficacy in β2AR-G-protein activation. Nature 547, 68–73 (2017). doi: 10.1038/nature22354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rojas FJ, Birnbaumer L, Regulation of glucagon receptor binding. Lack of effect of Mg and preferential role for GDP. J. Biol. Chem 260, 7829–7835 (1985). [PubMed] [Google Scholar]
- 32.Seifert R, Gether U, Wenzel-Seifert K, Kobilka BK, Effects of guanine, inosine, and xanthine nucleotides on β2-adrenergic receptor/Gs interactions: Evidence for multiple receptor conformations. Mol. Pharmacol 56, 348–358 (1999). doi: 10.1124/mol.56.2.348 [DOI] [PubMed] [Google Scholar]
- 33.Inoue A et al. , Illuminating G-protein-coupling selectivity of GPCRs. Cell 177, 1933–1947.e25 (2019). doi: 10.1016/j.cell.2019.04.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Serafin DS, Harris NR, Nielsen NR, Mackie DI, Caron KM, Dawn of a New RAMPage. Trends Pharmacol. Sci. 41, 249–265 (2020). doi: 10.1016/j.tips.2020.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lorenzen E et al. , Multiplexed analysis of the secretin-like GPCR-RAMP interactome. Sci. Adv 5, eaaw2778 (2019). doi: 10.1126/sciadv.aaw2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jeschke G, DEER distance measurements on proteins. Annu. Rev. Phys. Chem. 63, 419–446 (2012). doi: 10.1146/annurevphyschem-032511-143716 [DOI] [PubMed] [Google Scholar]
- 37.Sušac L, Eddy MT, Didenko T, Stevens RC, Wüthrich K, A2A adenosine receptor functional states characterized by 19F-NMR. Proc. Natl. Acad. Sci. U.S.A 115, 12733–12738 (2018). doi: 10.1073/pnas.1813649115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manglik A et al. , Structural insights into the dynamic process of β2-adrenergic receptor signaling. Cell 161, 1101–1111 (2015). doi: 10.1016/j.cell.2015.04.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okude J et al. , Identification of a conformational equilibrium that determines the efficacy and functional selectivity of the μ-opioid receptor. Angew. Chem. Int. Ed 54, 15771–15776 (2015). doi: 10.1002/anie.201508794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sounier R et al. , Propagation of conformational changes during μ-opioid receptor activation. Nature 524, 375–378 (2015). doi: 10.1038/nature14680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wingler LM et al. , Angiotensin analogs with divergent bias stabilize distinct receptor conformations. Cell 176, 468–478. e11 (2019). doi: 10.1016/j.cell.2018.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ye L, Van Eps N, Zimmer M, Ernst OP, Prosser RS, Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 533, 265–268 (2016). doi: 10.1038/nature17668 [DOI] [PubMed] [Google Scholar]
- 43.Fay JF, Farrens DL, Purification of functional CB1 and analysis by site-directed fluorescence labeling methods. Methods Enzymol. 593, 343–370 (2017). doi: 10.1016/bs.mie.2017.06.026 [DOI] [PubMed] [Google Scholar]
- 44.Schafer CT, Fay JF, Janz JM, Farrens DL, Decay of an active GPCR: Conformational dynamics govern agonist rebinding and persistence of an active, yet empty, receptor state. Proc. Natl. Acad. Sci. U.S.A 113, 11961–11966 (2016). doi: 10.1073/pnas.1606347113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yao XJ et al. , The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex. Proc. Natl. Acad. Sci. U.S.A 106, 9501–9506 (2009). doi: 10.1073/pnas.0811437106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dror RO et al. , Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. U.S.A 108, 18684–18689 (2011). doi: 10.1073/pnas.1110499108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gupte TM, Ritt M, Dysthe M, Malik RU, Sivaramakrishnan S, Minute-scale persistence of a GPCR conformation state triggered by non-cognate G protein interactions primes signaling. Nat. Commun 10, 4836 (2019). doi: 10.1038/s41467-019-12755-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kimball CP, Murlin JR, Aqueous extracts of pancreas III. Some precipitation reactions of insulin. J. Biol. Chem 58, 337–346 (1923). [Google Scholar]
- 49.Yin Y et al. , Rearrangement of a polar core provides a conserved mechanism for constitutive activation of class B G protein–coupled receptors. J. Biol. Chem 292, 9865–9881 (2017). doi: 10.1074/jbc.M117.782987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rasmussen SGF et al. , Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 (2011). doi: 10.1038/nature10361 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM density map has been deposited in the Electron Microscopy Data Bank (EMDB) under accession code EMD-21866, and model coordinates have been deposited in the Protein Data Bank (PDB) under accession no. 6WPW. All other data are available in the manuscript or supplementary materials. Materials described in this study are available upon request sent to the corresponding authors.