

Abstract
The intestinal microbiota comprises diverse fungal and viral components, in addition to bacteria. These microbes interact with the immune system and affect human physiology. Advances in metagenomics have associated inflammatory and autoimmune diseases with alterations in fungal and viral species in the gut. Studies of animal models have found that commensal fungi and viruses can activate host-protective immune pathways related to epithelial barrier integrity, but can also induce reactions that contribute to events associated with inflammatory bowel disease. Changes in our environment associated with modernization and the COVID-19 pandemic have exposed humans to new fungi and viruses, with unknown consequences. We review the lessons learned from studies of animal viruses and fungi commonly detected in the human gut and how these might affect health and intestinal disease.
Keywords: mycobiome, virome, Microbiome, virus, fungi, mucosal immunity, inflammatory bowel disease
The intestinal microbiota contains bacteria, archaea, fungi, protozoans, and viruses. Among these, bacteria have received the most attention. The human intestine contains hundreds of bacterial species, reaching a density of 1012 cells per gram of intestinal content. Disruptions to microbial communities (dysbiosis) have been associated with diseases, and strategies to restore a composition associated with health are being developed as therapies. As demonstrated in mice, intestinal colonization by individual and groups of bacteria affect physiology via production of metabolites and/or direct effects on the immune system. However, bacteria are not the only microbes in the gut, and are not sufficient to mediate all the functions attributed to the intestinal microbiota.
We review viruses and fungi of the gut microbiota and their effects on the immune system (Figure 1). Increasing evidence suggest that viruses and fungi regulate physiology via multiple interactions with host cells during symbiosis with the human gastrointestinal tract, despite their relative low abundance compared with bacteria. Studies of the total genetic content of stool specimens have found that viral and fungal genomes each account for 1% or less of the total. Nevertheless, the absolute number of fungi and viruses are much more impressive (Figure 1). Each gram of stool contains 108–109 virus-like particles (VLPs), most of which are from phages that infect bacteria1. Phages shape the functions and composition of bacteria by lysing them or integrating into their genomes. Only few examples in the current literature demonstrate phages directly interacting with immune cells2,3, thus this review will focus on viruses that infect animal cells.
Figure 1. Fungi and viruses in the intestinal microbiota.
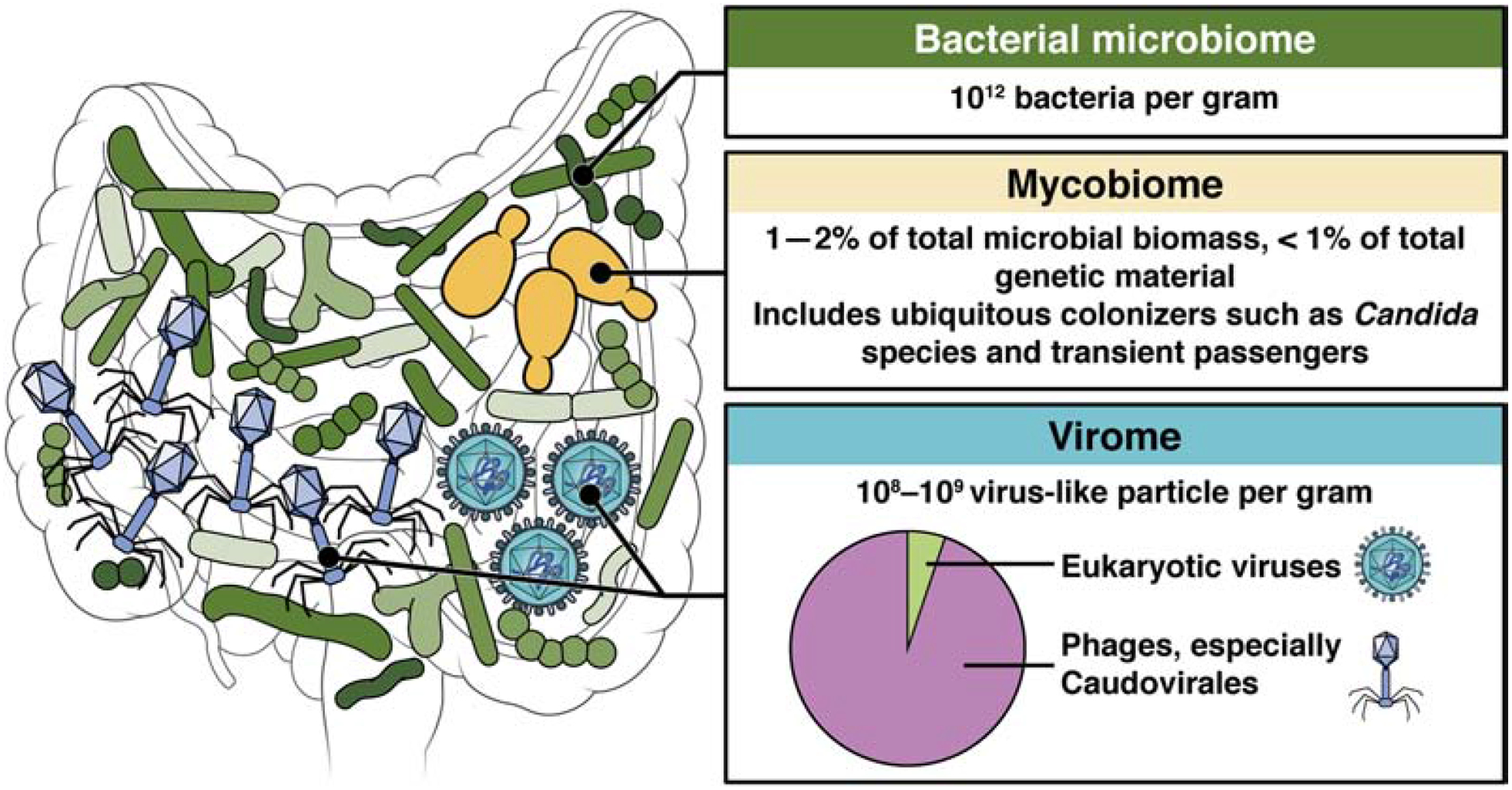
The intestine contains microbes that include bacteria, archaea, fungi, protozoans, and viruses. Most of these microbes are bacteria, which interact with the mucosal immune system. Fungi account for less than 1% of the total genetic material in fecal samples, but they are larger organisms that might occupy different niches along the gastrointestinal tract. Certain genera, such as Candida, are ubiquitous and establish long-term colonization. Viruses include phages, which infect bacteria, and viruses that infect eukaryotic cells. Phages can affect immune and other cell types via effects in bacteria they infect. Although viruses account for a small proportion of the mammalian gut microbiota, and the degree to which they establish long-term colonization is unclear, they could have large effects in that they directly invade and replicate in host cells. There is evidence fungal and viral members of the intestinal microbiota affect differentiation and function of the immune system. In this way, intestinal fungi and viruses might increase or decrease risks of diseases ranging from nosocomial infections to autoimmune diseases or IBD.
The gastrointestinal tract within the first few months after birth is exposed to animal RNA and DNA viruses from at least 16 families, including anelloviruses, picornaviruses (a large family that includes enteroviruses and coxsackieviruses), caliciviruses (noroviruses and sapoviruses), parvoviruses, adenoviruses, astroviruses, circoviruses, polyomaviruses, and papillomaviruses4,5. Although the degree to which these viruses establish long-term infections and affect human health is an active area of investigation, experiments in animal models indicate these viruses are not silent passengers. Shotgun sequencing studies of VLPs (to enrich for viruses) in feces have shown that individuals with inflammatory bowel diseases (IBD), celiac disease, colorectal cancer, or enteric graft vs host disease have alterations in viromes associated with increased phage diversity and the presence of certain animal viruses6–11. One study identified a new family of eukaryotic small circular DNA viruses of oro-respiratory viromes linked with periodontal and respiratory disease severity12. However, methodology hurdles have made it difficult to define a normal virome in a way that is quantitatively rigorous and inclusive of all RNA and DNA animal viruses. HIV-positive individuals with low CD4+ T-cell counts have increases in adenoviruses and anelloviruses in fecal samples13; it is likely that the immune system normally suppresses these viruses to levels below the threshold of detection by metagenomics.
Like bacteria and viruses, fungi are members of the normal microbiota. Metagenomic studies have found fungi to constitute 0.01%–0.1% of the human gut microbiome14,15. There has been no accurate estimate of the fungal biomass in the gastrointestinal tract—such an endeavor would be confounded by differences in genome sizes, the lack of many annotated genomes, and different sizes of fungal cells. Exposure to fungal commensals, pathobionts, or environmental and food-associated species has effects on the immune system that should be studied, regardless of the absolute numbers of fungi in the gut or their degree of stable colonization16,17.
A systematic review of gut mycobiota studies found only 15 of the 267 identified species were detected in multiple studies18. Only 13 of those species, belonging to Candida, Saccharomyces, Aspergillus, Cryptococcus, Malassezia, Cladosporium, Galactomyces, and Trichosporon, can grow at 37°C and thereby potentially inhabit the gastrointestinal tract18,19. Studies that aimed to broadly define the human intestinal mycobiome analyzed fecal samples from 317 healthy donors from the Human Microbiome Project20 or 98 healthy volunteers21. Both studies found a high prevalence of Candida and Saccharomyces, followed by Malassezia or Cladosporium14,21. In addition to these most prevalent taxa, the studies reported another 177 and 63 fungal genera, respectively, that could be environmental and dietary transients22. These findings indicate that healthy humans might have a core mycobiome, with a highly diverse and variable remainder of less-represented fungi.
Effects of viruses on the immune response
When disease is absent, the presence of eukaryotic viruses in humans is frequently described as an asymptotic infection. However, viruses are intracellular parasites, so active viral replication can affect the immune system. Experiments with murine norovirus (MNV) indicate that enteric viruses can engage the immune system without causing diarrhea, with outcomes that are beneficial or adverse, depending on the circumstances. MNV is a positive-strand RNA virus related to human noroviruses and was discovered in an animal facility in 2003 as a transmissible virus that killed immunocompromised mice23. Related strains of MNV, many of which establish robust persistent infection without causing overt disease, were subsequently identified in a multitude of institutions and vendors worldwide. Observations made in microbiota-deficient mice support the concept that MNV is a viral member of the gut microbiota. Inoculating germ-free mice or mice that have been given antibiotics with a persistent strain of MNV reverses many of the intestinal defects observed in mice without microbiota, including aberrant morphology of the small intestinal villi, reduced numbers of local T cells, and susceptibility to chemical injury by dextran sodium sulfate (DSS)24.
The effects of intestinal viral infections are frequently mediated by interferons. Type I interferons (IFNs), such as IFNAs and IFNB, and type III IFNs, such as IFNLs, are produced in response to viral nucleic acids. Although best known as cytokines that induce expression of antiviral genes, IFNs have functions other than reducing viral burden. As an example, production of type I IFNs in response to chronic infection by the herpesvirus murine cytomegalovirus (CMV) induces production of APOL9 by macrophages, which stimulates epithelial proliferation and wound repair in multiple organs, including the colon25. In vulnerable humans, CMV in the gut is associated with colitis. CMV infection of blood monocytes that are precursors of gut macrophages leads to upregulation of SMAD7 and makes these cells resistant to TGFB signaling, promoting inflammation and differentiation26. It is possible that the IFN response is necessary to counteract such an inflammatory response for viruses that establish symbiotic relationships. Consistent with this possibility, the ability of MNV to compensate for the absence of intestinal bacteria is mediated by interleukin 22 (IL22)-producing innate lymphoid cells (ILCs), which are indirectly activated by type I IFN to protect epithelial cells27.
Much of our knowledge about how viral nucleic acid is sensed through pattern recognition receptors in the intestine comes from studies of noroviruses and rotaviruses as pathogens rather than commensals. Rotaviruses are double-stranded RNA viruses that were a leading cause of viral gastroenteritis and death in children until the introduction of the vaccine; although rotaviruses remain a major health concern, noroviruses have become the main threat in many regions of the world. Virus RNA activates toll-like receptor 3 (TLR3) and TLR7 signaling via MYD88 and TRIF, or RIG-I and MDA5, which interact with MAVS, to induce production of IFNs by infected cells28,29. However, rotaviruses and noroviruses encode virulence factors that block IFN signaling30–32. IFN-independent effector mechanisms potentially compensate, such as the NOD-like receptor (NLR) NLRP9b, which induces death of epithelial cells to prevent rotaviruses from completing replication33. Other cytokines can mediate mutually beneficial outcomes for viruses and their hosts. IL1A recruits monocytes and neutrophils that serve as new targets for propagation of the virus34. IL22 is produced as a consequence of a complex immune response involving multiple cell types coordinated by MAVS and chemotactic monocytes during MNV infection, which protects intestinal epithelial cells from intestinal injury27. Further studies of these innate immune pathways are needed to elucidate the symbiotic effects of intestinal viruses.
The mechanisms that inform vaccination strategy and whether it is possible to launch an effective adaptive immune response against enteric viruses is a subject of intense research (for a review, see35). Norovirus infections can persist and cause serious illness in immunocompromised individuals, but less is known about how prolonged shedding occurs in asymptomatic people with presumably intact adaptive immune responses36. In contrast to a strain that establishes an acute infection in myeloid cells, persistent strains of MNV infect intestinal epithelial tuft cells and evoke weak responses by CD8+ T cells37,38. Studies of the encephalomyocarditis virus have found that NLRP6 and its binding partner DHX15 are intestinal epithelium-specific sensors of RNA that might also detect noroviruses39,40. It will be important to elucidate how cell tropism, such as infection of goblet cells by enteroviruses, astroviruses, or adenoviruses41–43, affects innate and adaptive immune responses in the gut and long-term outcomes of colonization.
Small DNA viruses, such as adenoviruses, circoviruses, and anelloviruses, are ubiquitous in the gut4,5,44. It will be important to determine how DNA-sensing pathways downstream of TLR9 and cGAS and STING mediate the effects of the gut virome. The finding that α-defensins produced by Paneth cells, which are secretory epithelial cells in the small intestine with bactericidal activity, promote responses of neutralizing antibodies against 1 strain of mouse adenovirus while promoting infectivity of another strain indicates that we have much to learn about the interactions between the intestinal barrier and these ubiquitous viruses45,46.
Effects of fungi on the immune response
Gut fungi, their metabolites and toxins, and their effects on bacterial populations can directly or indirectly influence host immunity16,17. Pattern recognition receptors interact with carbohydrates on the surface of the fungal cell wall (Figure 2). C-type lectin receptors (CLRs) are mainly involved, but TLRs are considered to have secondary or additive effects. CLRs such as dectin 1, dectin 2, dectin 3, mincle, and the mannose receptor, recognize fungal cell wall components including β-glucan, mannans, and mannose-associated complexes. With the exception of the mannose receptor and MelLec, these CLRs signal via spleen associated tyrosine kinase (SYK), a CARD9–BCL10–MALT1 complex, and/or RAF147. Activation of SYK signaling requires an immunoreceptor tyrosine-based activation motif (ITAM) or ITAM-like motif within the receptor tail (dectin 1) or an associated FcRγ adapter molecule (dectin 1, dectin 2, dectin 3, mincle).
Figure 2. Innate and adaptive immune responses to fungi in the gut.
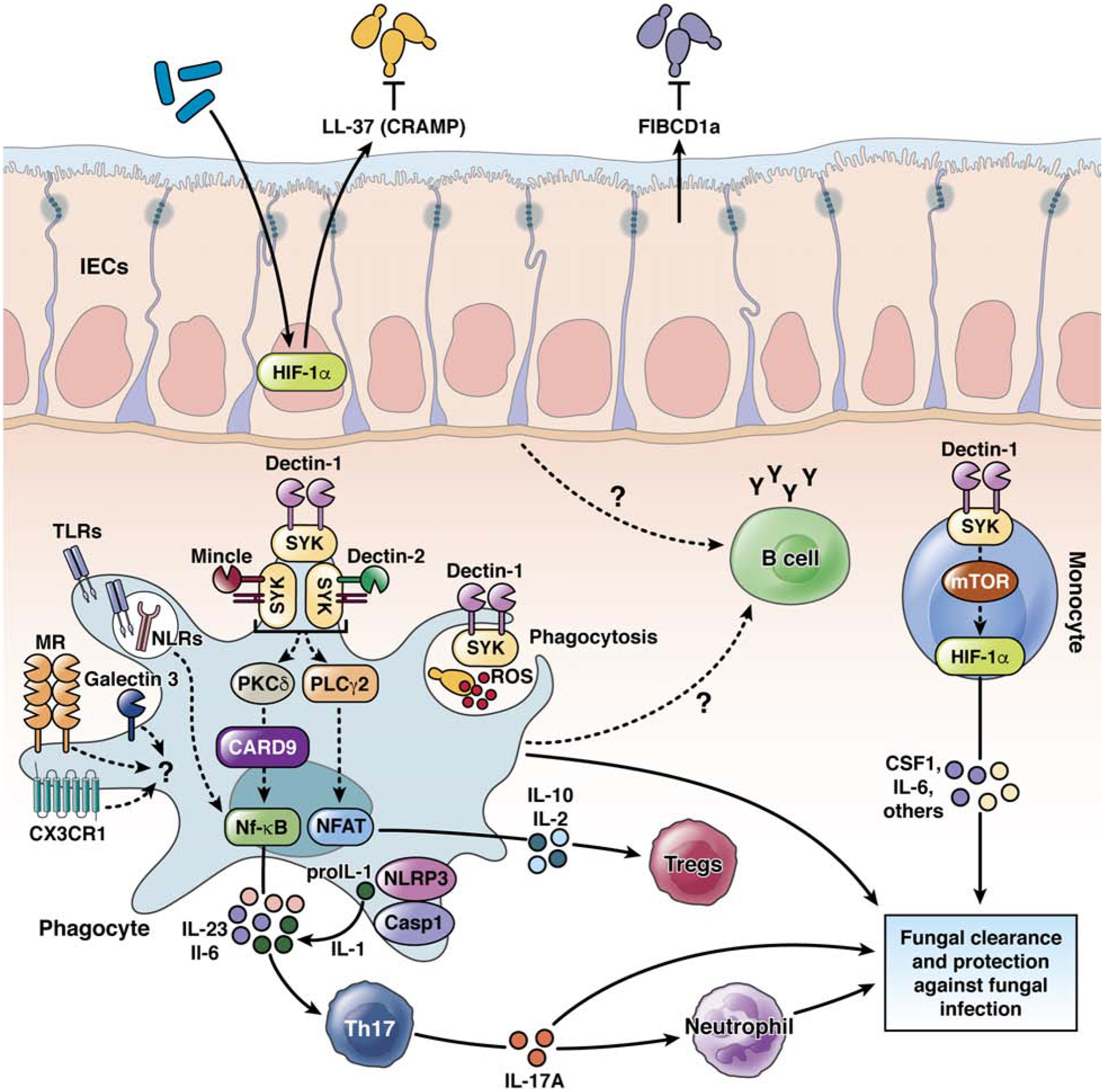
At the mucosal surface of the intestines, CLRs (such as dectin 1, dectin 2, and mincle) interact with fungal cell wall components, resulting in activation of SYK. SYK activates protein kinase C delta (PKCD) to signal via CARD9 and activate nuclear factor (NF)-kB, phospholipase C gamma 2 (PLCG2) to activate NFAT, production of reactive oxygen species (ROS), or MTOR to activate hypoxia inducible factor 1 subunit alpha (HIF1A). These signaling pathways activate production of IL23, IL6, IL10, IL2, IL1 by phagocytes (macrophages, monocytes, DCs, and neutrophils) and CSF1 and IL6 by monocytes (“trained immunity”). The interaction with fungi-charged phagocytes results in the development of Th17 cells and recruitment of neutrophils to the intestinal lamina propria. Receptor-independent functions of phagocytes supports eradication of specific fungal products and cells. TLRs, NLRs, inflammasome components, and galectin 3 also recognize fungal components. These combined effects result in killing of fungi or tolerance to them and a balanced gut fungal community or disease development.
Dectin1 binds fungi in the intestines48. Mice lacking this receptor or dectin 3 develop more severe colitis following administration of DSS than wild-type mice, accompanied by expansion of opportunistic fungal genera such as Candida and Trichosporon; the antifungal drug fluconazole reduces the severity of colitis48,49. Dectin 1 deficiency in humans is associated with increased colonization of the gut by Candida and graft vs host disease following bone marrow transplantation50,51. Experiments with multi-CLR knockout mice revealed combined functions of dectin 1, dectin 2, and mincle in the immune response to Candida albicans, compared with any single receptor52. Activated dectin 3 induces the expression of mincle or forms heterodimers with dectin 2, which signals through FcRγ53. Mincle, alternatively, can affect dectin 1-mediated production of IL-12 by promoting degradation of interferon regulatory factor 1 (IRF1) via PI3K, AKT, and E3 ubiquitin ligase activation54. The E3 ubiquitin ligase CBLB mediates degradation of dectin 1 and dectin 2 to serve as a negative regulator of antifungal immunity55,56. There are therefore synergistic and antagonistic interactions among CLRs that regulate the antifungal immune response in the gut.
CLR signaling pathways are not redundant, but all include CARD9, based on studies of Card9−/− mice47. Gut fungal and bacterial communities are altered in Card9−/− mice, with notable loss of Lactobacillus spp57. Tryptophan-metabolizing bacteria, including lactobacilli, produce ligands for the aryl hydrocarbon receptor, which stimulate release of IL22 by group 3 ILCs and T-helper (Th) cells57,58. These abnormalities in the intestines of CARD9-deficient mice partially explain the decreased levels of aryl hydrocarbon receptor ligands, reduced expression of IL22, REG3G, and REG3B in colon. These phenotypes are partially rescued by supplementation with 3 strains of Lactobacilli57. CARD9 deficiency also affects immune modulation by gut fungi, which can affect colon cancer development59–61.
Inflammasomes are cytosolic oligomers that mediate processing of IL1B and are part of the mucosal immune response to fungal infections. NLRP3 and NLRC4 inflammasomes protect mice from vaginal Candida infection via activation of IL1B by caspase 162,63. Detection by NLRP3 seems to depend on morphologic features of the fungi (yeast vs hyphae)—NLRP3 is activated by Candida hyphae and hyphae-produced secretory molecules64,65.
CLRs are expressed by and active in phagocytic cells, including macrophages, dendritic cells (DCs), and neutrophils. Among these, a population of mononuclear phagocytes (MNPs), marked by the fractalkine receptor CX3CR1, expresses the highest levels of dectin 1, dectin 2, and mincle66 (Figure 2). CX3CR1+ MNPs phagocytose fungal species in the intestines, and then activate T cells.66,67. Intestinal colonization with Candida induces T-cell production of IL17 and IL22 and these Th17 cells control of specific commensal bacteria and fungi in mice and humans47,66,68–71. Activation of Th17 cells by CX3CR1+MNPs in response to mycobiota might help maintain intestinal homeostasis.
In addition to direct interaction with phagocytic cells, fungi alter phagocyte function via their secreted products. C albicans hyphae secrete the toxin canadidalysin, which induces NLRP3 inflammasome-dependent IL1B maturation via K+ efflux in macrophages and DCs65. Furthermore, Candida secrete aspartic proteases such as SAP3 and SAP6, which activate CLR-independent responses by monocytes, macrophages, and DCs via induction of K+ efflux and reactive oxygen species72. Fungal toxins and other soluble factors are absorbed through extension of “balloon-like” protrusions by a population of CX3CR1+ macrophages in the distal colon that protect the epithelium73.
Receptors expressed on cells outside of the immune system, such as ERBB2, EGFR, EPHA2, FIBCD1, and MelLec contribute to the antifungal immune response at mucosal surfaces, with less clear roles in the gut (Figure 2)74–78. In addition to receptor-mediated antifungal responses, C albicans can directly damage of oral and vaginal epithelium via the toxin candidalysin, resulting in production of inflammatory cytokines79,80. Candidalysin has been reported to damage human epithelial colorectal adenocarcinoma cells81, justifying further exploration of the effects of this toxin in the gut.
Balancing the viral and fungal communities
In the absence of an underlying condition or genetic deficiency, intestinal colonization by viruses and fungi might have immune-protective effects. Although it remains to be determined whether a healthy or normal virome exists, recognition of viral nucleic acid clearly has homeostatic effects that promote intestinal barrier function (Figure 3). Mimicking viral infection of the epithelium through administration of a RIG-I ligand offsets the intestinal barrier damage caused by radiation and chemotherapy20, consistent with a previous finding showing that MAVS signaling protects against DSS-mediated colitis82. Based on in vitro experiments, RIG-I recognition of RNA from the bacterial microbiota was originally suggested to protect against DSS82. However, providing mice with a cocktail of antivirals increases susceptibility to DSS83,84. According to one model, enteric viruses are essential for inducing IL15 downstream of RIG-I and MAVS, a cytokine necessary to maintain intra-epithelial lymphocytes with tissue regenerative properties83 (Figure 3).
Figure 3. Recognition of viruses by immune cells fortifies the intestinal barrier.
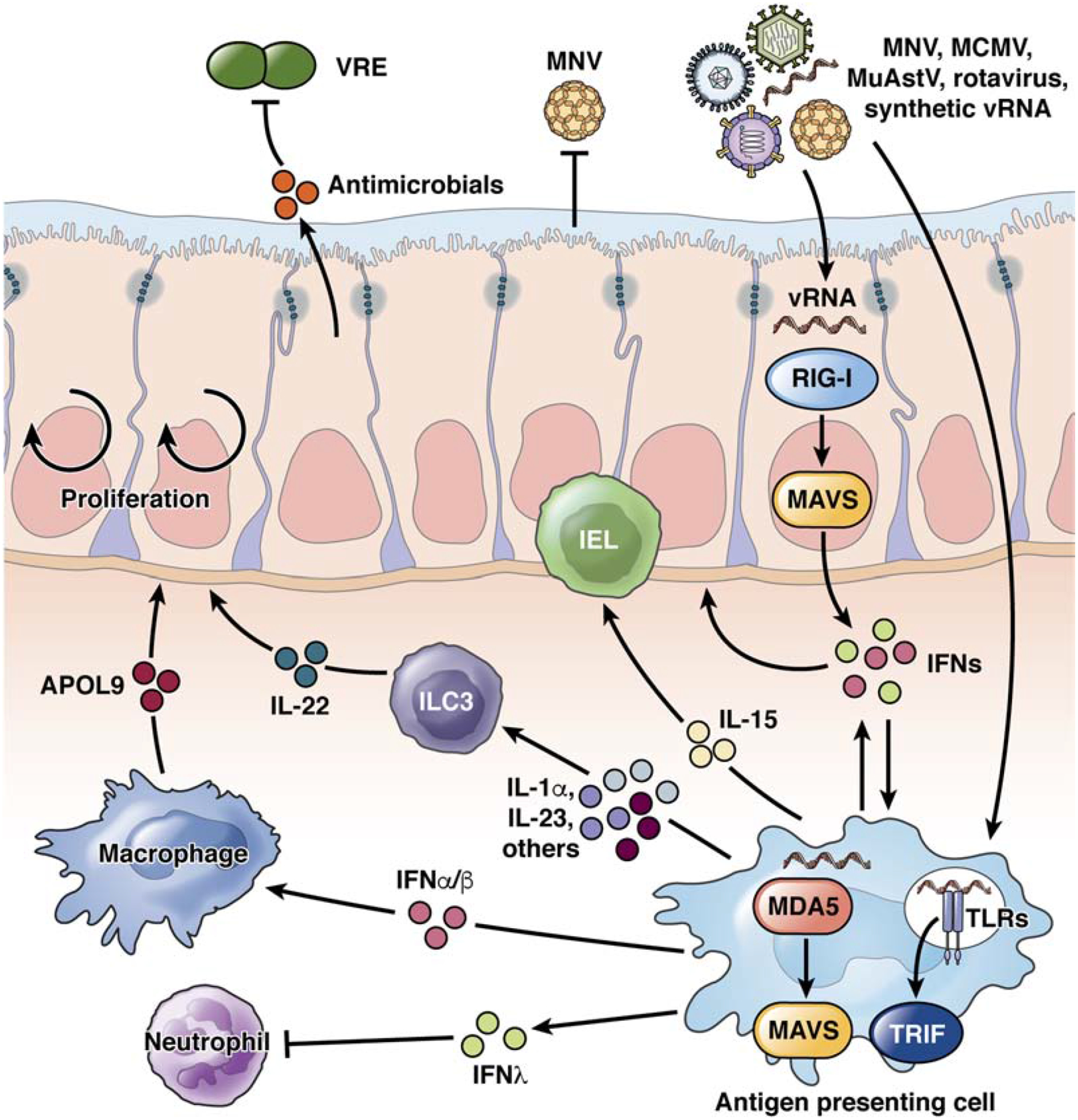
MNV, cytomegalovirus (MCMV), astrovirus (MuAstV) infect myeloid, lymphoid, or epithelial cells, depending on strain. These viruses, heat-killed rotavirus, or synthetic viral RNAs (vRNAs) activate RIG-I or MDA5 in cytosol or TLRs on endosomes of epithelial cells and antigen-presenting cells, which signal through MAVS and TRIF, respectively. These signaling pathways trigger production of type I IFN (IFNA and IFNB), which mediate bi-directional communication between myeloid cells with the epithelium; IFNL, which inhibits neutrophils; IL15, which promotes IEL function; and IL23, which induces ILC3s to produce IL22, increasing resistance to injury and bacteria such as vancomycin resistant Enterococci (VRE). IFNA and IFNB also activate macrophages to produce APOL9, which promotes epithelial cell proliferation and protection from injury.
TLR3 and TLR7 agonists that mimic viral double-stranded and single-stranded RNA, respectively, induce production of type I IFNs by colonic plasmacytoid DCs (pDCs), which also protects against DSS84. This same study demonstrated a similar protective effect of inactivated rotavirus, and that TLR3 and TLR7 gene variants are associated with IBD susceptibility in humans. Also, a TLR7 agonist recreates the beneficial effect of MNV infection by enhancing resistance against vancomycin-resistant Enterococcus through IL22 producing ILCs85 (Figure 3). IFNL has an anti-inflammatory effect in the gut by inhibiting reactive oxygen species production and degranulation by neutrophils86. In this study, an antiviral cocktail was used to implicate viral members of the microbiota in IFNL production (Figure 3). As with the other studies that rely on antivirals, it will be important to rule out non-specific effects of the drugs by identifying specific viruses that serve these functions. Astroviruses require consideration. In a recent study, immunocompromised mice were found to be surprisingly resistant to MNV and rotavirus infections, which was attributed to presence of a murine astrovirus strain that provides cross-protection against other viruses through production of IFNL in the gut87 (Figure 3). Murine astrovirus can also protect neonates from enteropathogenic E. coli colonization43. The oral polio vaccine (an attenuated strain of poliovirus) reduces the incidence of diarrhea caused by rotavirus and other agents88, indicating that similar interactions between viruses occur in human gut. So, much like the gut bacterial microbiota increases resistance to colonization by bacterial pathogens, the gut virome promotes resistance against exogenous viral infections.
Virus-induced production of IL22 by ILCs might be important for intestinal barrier function in the presence of pathogenic bacteria. Persistent and non-persistent strains of MNV protect mice given antibiotics from induction of diarrhea by Citrobacter rodentium and vancomycin-resistant Enterococcus24,85 (Figure 3). MNV-induced IL22 led to the survival of newly weaned mice, which otherwise died during C rodentium infection. Young children have multiple viral infections, which might help them from infections with pathogenic bacteria 27. MNV also increases resistance to lung infection by Pseudomonas aeruginosa, although it is not clear how an enteric virus affects other organs89.
Anti-fungal agents promote expansion of filamentous fungi such as Wallemia mellicola, Aspergillus amstelodami, and Epicoccum nigrum, which aggravate intestinal inflammation and allergic airway disease61,67,90,91. The systemic effects of intestinal fungi are not known. Depletion of CX3CR1+ MNPs from the gut but not the lung revealed that these cells detect fungal dysbiosis and increase numbers of Th2 cells and eosinophils to promote lung allergic inflammation67. Furthermore, mono-colonization with C albicans or S cerevisiae, which are recognized by CX3CR1+ MNPs66, supports the establishment of intestinal homeostasis and protects against virus-induced lung inflammation and DSS-induced gut barrier damage92. However, administration of a single antibiotic did not protect mice with intestinal C albicans70. Similarly S cerevisiae was not protective (and even detrimental) in SPF mice93, indicating the pre-existing gut microbiota composition influences intestinal inflammation or protective immunity by intestinal fungi.
Gut-adapted C albicans protected mice from systemic challenge with fungal and bacterial pathogens, including C albicans itself94. This innate immune memory response95,96 was of short duration, required IL6, and was observed in lymphocyte-deficient mice94. However, in the same mice, a C albicans isolate that had not been adapted to the gut protected against subsequent infections with C albicans or Staphylococcus aureus, by inducing a Th17 cell-mediated responses70. These findings indicate that C albicans can induce innate and adaptive immune memory responses, possibly to specific strains of fungi. Altogether, these studies have shown that a balanced gut mycobiota contributes to immune homeostasis, and that intestinal colonization with specific fungi can induce immune memory for specific enteric viruses and bacteria.
Laboratory mice are raised in artificial housing conditions that limit the diversity of microbes they encounter. Releasing laboratory mice into a confined outdoor enclosure that aims to replicate their natural environment increases the diversity and load of intestinal fungi, especially Aspergillus species97,98. This expansion of fungi was associated with increases in peripheral granulocytes, activated T cells, and other signs of immune maturation. Similar observations concerning the gut mycobiota were made in wild mice, which are also infected with viruses that are excluded from institutional vivaria99,100. Deficiencies in gut fungi or microbes, along with other variables relating to exposure to infectious agents100, might account for the inability of the immune system of laboratory mice to fully recapitulate that of adult humans. Mouse colonies with natural mycobiomes or viromes can be maintained in a laboratory environment, allowing researchers to overcome some of these shortcomings99. Findings from studies in which intestinal fungi and viruses have been altered, or specific fungi and viruses have been introduced (such as C albicans and MNV), have revealed unexpected benefits to mice, such as improved resistance to barrier disruption by chemicals and pathogens as mentioned earlier.
IBD
IBD, colorectal cancer, and other gastrointestinal and hepatologic disorders have been associated with altered communities of fungi and viruses6–11,16,17,101–105 . Most sequencing and experimental studies have focused on IBD, for which data from multiple cohorts and experimental models are available. IBD, including Crohn’s disease (CD) and ulcerative colitis (UC), are associated with altered richness and diversity in the gut mycobiota102,103,106,107. Candida (mainly C albicans) are the predominant genera reproducibly identified in fecal or mucosal samples from IBD cohorts18,101–105 (Table 1). Candida species can be intestinal symbionts or pathobionts, but the effects of other fungi are under investigation. These studies are complicated by their abundance in food sources, on skin, or in the surrounding environment18,22,94,108. There have been numerous studies of mycobiota in fecal or mucosal samples from patients with CD or mixed populations of patients with CD and UC, but only few of studies from patients with UC (Table 1).
Table 1.
Viral and Fungal Microbes in the Intestine
| Microbe | Disease or potential therapy | Alterations in fungi or viruses | Immune Responses | References |
|---|---|---|---|---|
| Fungi | ||||
| CD |
Candida expansion and decreased S cerevisiae in several cohorts Increased abundance of M restricta in 1 cohort Malassezia and Aspergillus presence linked to CARD9 S12N in 1 cohort Fungal dysbiosis characterized by decreased Ascomycota and increased Basidiomycota diversity in several cohorts |
Increased C albicans-reactive T cells. Increased ASCA Decreased ASCA in persons with T280M in CX3CR1 |
65,100–102,104,107,162 | |
| UC | Candida expansion in several cohorts | None reported | 100,105,163 | |
| FMT for patients with UC | High gut Candida before FMT associated with clinical response in 1 cohort Decreased Candida abundance after FMT associated with reduced severity in 1 cohort |
Increased anti-Candida antibodies in placebo controls are abrogated in FMT recipients | 119 | |
| FMT for patients with CDI | Gut fungal dysbiosis and expansion of C albicans in patients with CDI. Decreased Candida abundance after FMT, increased Candida abundance after FMT non-responders Increased Saccharomyces and Aspergillus in responders in 1 cohort |
None reported | 153 | |
| Viruses | ||||
| CD | Expansion of Caudovirales phages in several cohorts Increased frequency of anellovirus in a combined cohort Exclusive presence of enteroviruses in 1 cohort Norovirus increases odds of disease in the Swedish National Registry and flares |
None reported | 7,11 6,126–128 | |
| UC | Other eukaryotic viruses display altered presence but potentially cohort-specific or associated with diet Expansion of Caudovirales phages in several cohorts. Increased frequency of anellovirus in several cohorts. Other eukaryotic viruses display altered presence but potentially cohort-specific or associated with diet |
None reported | 7,11,121,125,126 | |
| FMT for pediatric patients with UC | Siphoviridae phages belonging to Caudovirales displayed enhanced engraftment. | None reported | 122 | |
| FMT for patients with CDI | FMT responsiveness associated with engraftment of donor-derived Caudovirales phages, consistent with observation that filtered feces contains therapeutic benefit | None reported | 123,124 | |
| Celiac disease | Patients more likely to have high titers of anti-reovirus antibody Frequent rotavirus infection associated with disease development in a longitudinal study |
IRF1 expression in intestinal mucosa of patients with high anti-reovirus titer | 137,139 |
ASCA, Anti-saccharomyces cerevisiae antibodies; FMT, Fecal microbiota transplantation; CDI, Clostridium difficile infection; IBD, inflammatory bowel disease; CD, Crohn’s disease; UC, ulcerative colitis
Despite confounding factors related to methodology and technology, which have hindered direct comparisons among studies, C albicans and C parapsilosis appear to be consistently increased in fecal or mucosal samples from patients with CD, whereas changes in proportions of C tropicalis, Saccharomyces, and Malassezia spp. vary among cohorts16,18,101−10549 (Figure 4A, B). C albicans is also expanded in fecal samples from patients with C difficile infection (CDI), compared to patients without CDI138 (Figure 4C). Based on limited information, S cerevisiae appears to be reduced in feces of patients with IBD—particularly those with active inflammation102,109. Oral administration of S cerevisiae to mice produced effects ranging from detrimental to neutral to protective during DSS-induced colitis92,93,110.
Figure 4. Gut fungi in patients with IBD.
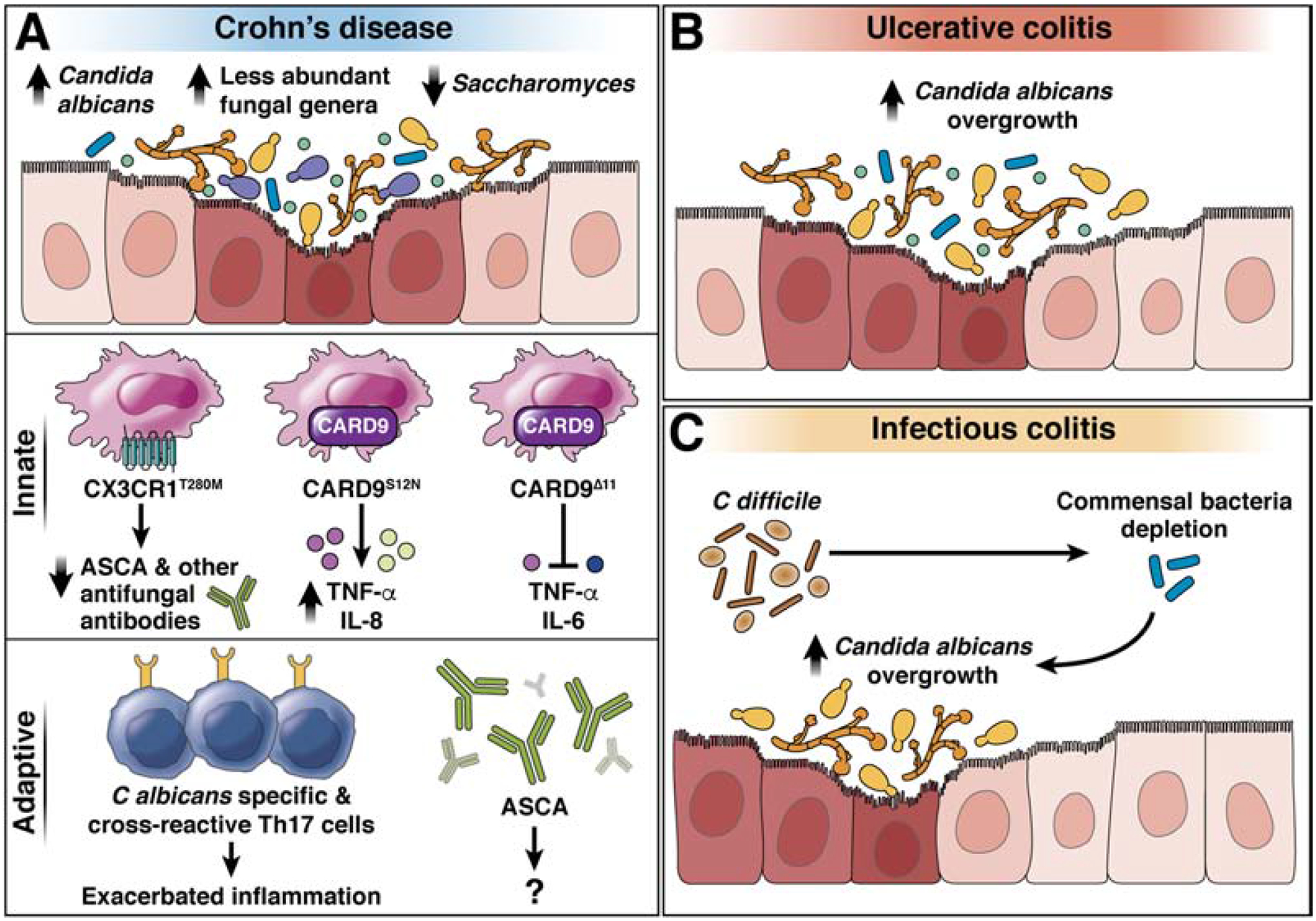
A) Gut fungal dysbiosis, characterized by the overgrowth of opportunistic fungal species, in patients with CD. CD is associated with Candida spp. overgrowth, expansion of less abundant fungal genera such as Malassezia spp., and decrease of Saccharomyces spp. in fecal samples. The T280M mutation in CX3CR1 is associated with decreased ASCA and other antifungal antibodies’ production; the S12N mutation in CARD9 increases production of inflammatory cytokines (TNF, IL8,); the delta 11 deletion in CARD block production of TNF and IL6 to prevent development of CD. Expansion of C albicans-specific and Aspergillus cross-reactive Th17 cells and increase of ASCA antibodies is associated with Crohn’s Disease. Candida albicans overgrowth has been observed in patients with UC (B) and CDI (C).
Malassezia sequences were increased in intestinal washings from patients with CD; the presence of M restricta was associated with S12N mutation in CARD9, which increases risk for CD and UC104. Phagocytes from these patients had increased production of inflammatory cytokines upon fungal stimulation111. Alternatively, the S12NΔ11 truncation mutation in CARD9 disrupts its interaction with the ubiquitin ligase TRIM62; this prevents production of inflammatory cytokines by DCs exposed to ligands from fungi112 (Figure 4A).
Most mechanistic studies have been performed with a limited set of model fungi strains. In contrast, researchers have made progress elucidating strain-specific properties of disease-associated bacterial species113,114. Strain-specific features of IBD-associated fungi might affect development of inflammation. Altogether these studies indicate that patients with IBD have changes in the intestinal mycobiota, and that Candida are consistently associated with an inflamed gut. The roles of specific strains of Candida, other fungal species, and their metabolites contribution to development of bowel inflammation requires further investigation.
Consistent with findings from studies of fecal mycobiota from patients with CD, blood samples from these patients have higher frequencies of C albicans-reactive T cells compared with healthy controls71. So intestinal C albicans might promote inflammation by inducing development of fungal antigen-reactive Th17 cells (Figure 4A). However, patients with CD given the IL17A antibody secukinumab have a higher rate of fungal infections than patients with CD who do not take this drug115. This adverse outcome correlated with pathology features, indicating that IL17 affects fungal communities, possibly in the gut.
Antibodies against S cerevisiae mannan (ASCA) are increased in blood samples from patients with CD or other diseases with a gastrointestinal component, such as autoimmune liver diseases, primary biliary cirrhosis, alcoholic liver disease, and primary sclerosing cholangitis16,116. Although the role of ASCAs in pathogenesis is unclear, their detection identifies individuals who will develop CD within 5 years with high accuracy117. A loss of function mutation (T280M) in CX3CR1 is associated with impaired production of ASCA in patients with CD, and decreased titers of serum IgG against C albicans, C parapsilosis, S cerevisiae, and P kudriavzevii, but not Wallemia or Malassezia spp., which have low presence in the gut66, indicating a link between CX3CR1+ MNPs and ASCA (Figure 4A).
Fecal microbiota transplantation (FMT) was reported to be effective in a subset of patients with UC118. A high abundance of C albicans in feces before FMT was associated with a clinical response (Table 1). Stable titers of IgG against C albicans and decreased Candida abundance in fecal samples after FMT was associated with reduced severity of UC. This observation indicates that FMT acts in part by reducing Candida abundance and containing proinflammatory immune responses by fungi during intestinal inflammation119 (Figure 5A, B).
Figure 5. Gut fungi and viruses in FMT.
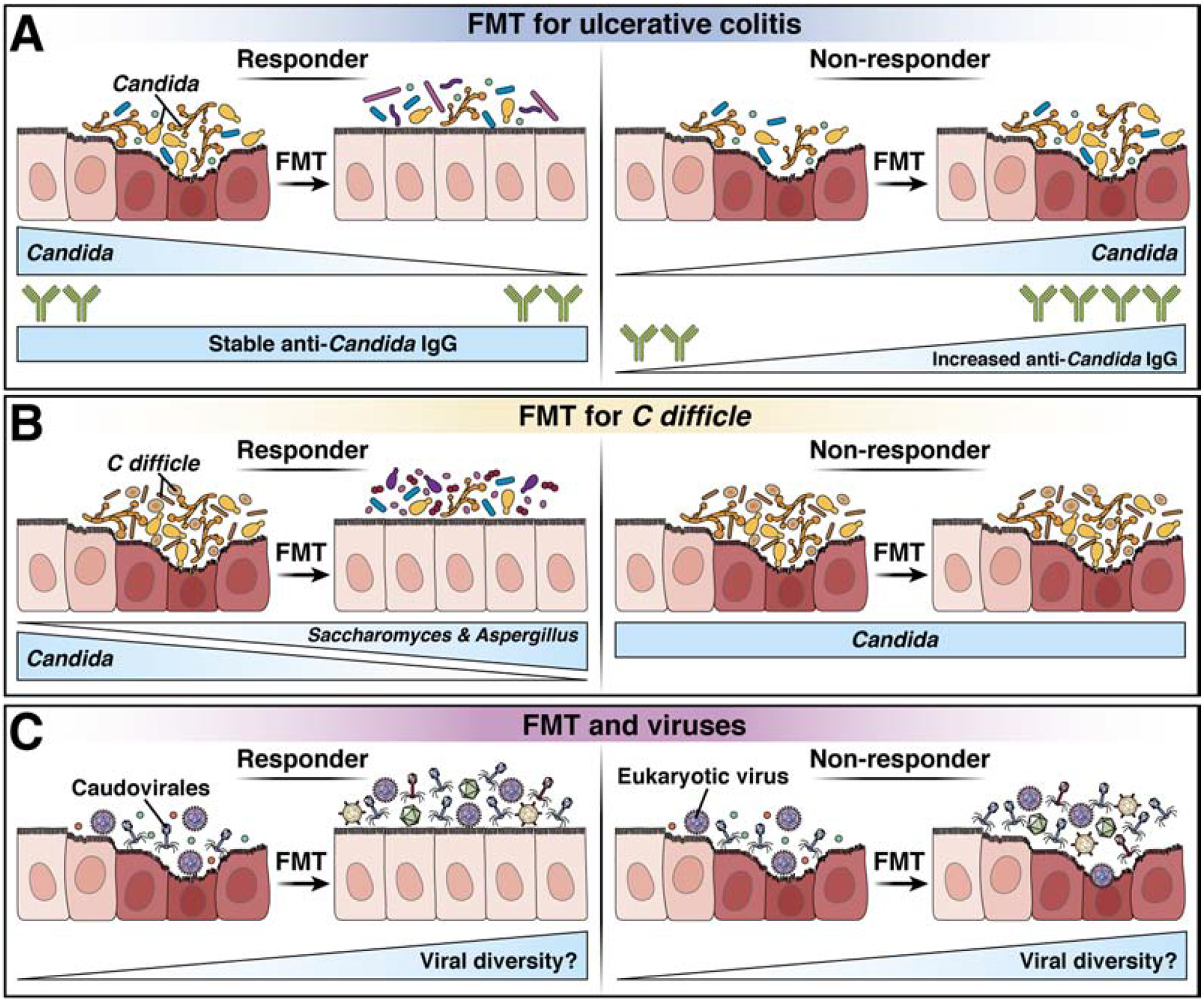
A) In patients with UC, increased C albicans abundance before FMT is associated with a clinical response (top), whereas stable titers of IgG against C albicans in blood samples, and decreased Candida abundance in fecal samples, associate with reduced disease severity. B) In patients with CDI, a high relative abundance of Saccharomyces and Aspergillus in fecal samples after FMT is associated with response to the treatment (top), whereas overrepresentation of C albicans and decreased fungal diversity after FMT associate with non-responsiveness (bottom). C) In patients with CDI, FMT modulates gut phage composition; response to FMT is associated with an increased abundance of Caudovirales taxa. Little is known about the effects of viruses that infect eukaryotic cells. It will be important to determine which viruses are included in donor sample and how FMT affects the viruses already present in the recipient.
Although we have focused on viruses that infect eukaryotic cells, fecal samples from patients with IBD have alterations in phage communities, including increases in the Caudovirales family in samples from CD or UC patients compared to healthy controls7,11,120,121 (Table 1). Siphoviridae of the order Caudovirales were more likely to be transferred to patients with UC during FMT compared with other phages122. Fecal samples from patients with CDI have a high abundance and low diversity of Caudovirales. FMT treatment of CDI alters the phage composition of fecal samples, and response to treatment is associated with increased abundance of donor-derived Caudovirales taxa in recipients123 (Figure 5C). Fecal filtrates from healthy donors were effective in treatment of CDI in 5 patients, suggesting that phage or other components of feces contribute towards the effects of FMT124. It is not clear whether phage infection of bacteria occurs before or after dysbiosis, but nucleic acids from phages induce production of type I IFNs by phagocytes2. Oral inoculation of germ-free mice with phages induced a Th1 cell-mediated immune response, via activation of TLR9 on antigen-presenting cells that internalized viral DNA. This response did not involve type I IFNs, and exacerbated DSS-induced colitis3. Expansion of phages in microbiomes of patients with IBD therefore may contribute to intestinal inflammation.
Viruses that infect eukaryotic cells, such as anelloviruses, are more prevalent in fecal samples from patients with IBD than individuals without IBD7,125. Intestinal mucosal samples from patients with UC have an increased abundance of Hepadnaviridae, whereas samples from patients with CD have an increased abundance of Hepeviridae. Intestinal mucosal samples from patients with UC also have lower levels of Polydnaviridae and Tymoviridae than controls, whereas samples from patients with CD have reduced Virgaviridae abundance126. Some of these viruses are better known for infecting plants and insects, and may have originated from the diet, similar to fungi identified in sequencing experiments. Antecedent norovirus infection is associated with CD in the Swedish National Register, and a separate study demonstrated association between norovirus infection and flares of CD127,128 (Table 1). These finding raise important questions about whether patients with IBD are more susceptible to viral infections or to adverse consequences of infections, such as flares.
Eukaryotic viruses contribute to disease in animal models of IBD. Mice with variants of Atg16l1 associated with increased risk of CD have structural abnormalities in Paneth cells upon persistent infection by MNV129,130. This defect, in which Paneth cells lack their characteristic antimicrobial granules, was observed in patients with CD homozygous for the ATG16L1 variant encoding the T300A substitution129. ATG16L1 is required for autophagy, in which cytosolic material such as damaged mitochondria are sequestered in double-membrane vesicles and targeted to the lysosome for degradation131. MNV-infected mice with this variant of Atg16l1 develop more severe intestinal inflammation and have increased mortality following chemical injury with DSS than wild-type mice130,132.
In the intestinal epithelium, ATG16L1 is required for organelle homeostasis and prevents necroptosis in response to virus-induced tumor necrosis factor (TNF)132. Intestinal organoids generated from individuals homozygous for the ATG16L1 T300A variant are also susceptible to necroptosis in the presence of TNF or cytokines produced by allogeneic Th1 cells133. MNV also exacerbates Th1-associated colitis in Mdr1a−/−, Stat1−/−, and Il10−/− mice134–136. MNV infection therefore induces production of cytokines that normally protect the intestine from bacterial pathogens, but cause damage in tissues that are genetically susceptible. The observation that MNV triggers or exacerbates disease in animal models of IBD lends support to the concept that animal viruses are a relevant component of the gut microbiota.
The induction of IFN and Th1 cell-mediated responses could be a common mechanism by which intestinal viruses contribute to inflammatory diseases. Reovirus infection of mice induces production of IL12B by CD103+CD11B–CD8A+ DCs, which promotes Th1 polarization in the mesenteric lymph nodes (MLNs) and increases reactivity towards dietary antigens, together with a type I IFN response that interferes with differentiation of T-regulatory cells137. A non-persistent strain of MNV induces a similar break in tolerance, and with both viruses, the Th1 cell-mediated response requires IRF1137,138. The observation that the titer of antibodies against reovirus correlates with expression of IRF1 in small intestinal biopsies from patients with celiac disease is consistent with findings from mice137. Frequent rotavirus infections have also been associated with development of celiac disease139.
In mouse models of CD and celiac disease, viral infections induce features associated with human disease (such as Paneth cell defects and gluten-reactive lymphocytes). However, viral infections are insufficient to induce overt disease. A requirement for additional environmental triggers could explain how common viruses are associated with diseases in 1% or less of the population. Also, effects of viruses are strain-specific in mouse models of CD and of celiac disease130,137,138. If these observations provide any indication of what happens in humans, it would be a challenge for epidemiology studies to identify viral causes of these diseases. Few studies are equipped to distinguish the presence of a virus at the level of point mutations.
Perhaps the strongest evidence that intestinal viruses promote chronic immune diseases in humans comes from studies of patients with type 1 diabetes. Group B coxsackieviruses and other viruses with fecal to oral transmission have been suspected to contribute to this autoimmune disease, in which T cells attack insulin-producing pancreatic β cells140. In one of the few prospective, longitudinal virome studies, chronic enterovirus B infection and lower incidence of mastadenovirus C infection were associated with development of autoantibodies against insulin141. Episodic enteroviral infection was not associated with disease, indicating that prolonged infections might contribute to development of chronic disorders. Other prospective studies have also associated an altered enteric virome with development of type 1 diabetes, such as lack of early-life infection with small, single-stranded DNA viruses belonging to the Circoviridae family and development of serum antibodies associated with type 1 diabetes44,142. Experiments with the non-obese diabetic (NOD) mouse model of type 1 diabetes found that infection with group B coxsackievirus reduced disease in younger mice, but accelerated disease if introduced into older mice, at a time at which insulitis had already initiated, indicating that infection affects ongoing autoimmune reactions143,144.
Rotaviruses and reoviruses are not considered pancreatropic, but have a similar time-dependent effect on diabetes145–147. In NOD mice, rotavirus accelerates type 1 diabetes by spreading from the gut to the lymph nodes that drain the pancreas, where type I IFNs are produced by pDCs to activate autoreactive lymphocytes147. MNV infection also protects against development of diabetes in NOD mice, which is associated with increases in T-regulatory cells and an altered bacterial microbiota148. Thus, studies of patients with type 1 diabetes and NOD mice have shown that gut viruses can affect extra-intestinal immune responses. Intestinal fungi also have extra-intestinal effects, modulating systemic immune responses as well as immunity in the lung and liver16,61,67,70,71,105,149.
Many patients with COVID-19 have gastrointestinal symptoms. An increasing number of published and unpublished findings are reporting the presence of SARSCoV-2 in stool or intestinal tissue, consistent with the observation that the virus readily replicates in intestinal organoids150–152. The aforementioned experiments in mice predict the presence of the virus, or viral RNA, in the gut would induce an immune response with a broad range of consequences for the host. Additionally, prospective studies are necessary to understand the long-term consequences of SARS-CoV-2 infection for individuals with IBD or other immune-mediated disorders. As our understanding of SARS-CoV-2 pathogenesis improves, we are confident that the relationship between this virus and the gastrointestinal tract will be clarified.
Conclusions
Decades of infectious disease research have shown that fungi and viruses activate innate and adaptive immunity at barrier surfaces. Fungi and viruses are indispensable parts of the human gut microbiota fully integrated within the bacterial community and affect the immune response, health, and development of inflammatory diseases. Despite advances, our understanding on the interactions between these components of the gut microbiota and the mucosal immune system is limited. Deep sequencing-based analysis of gut viruses and fungi indicate the composition of the mycobiome and virome vary considerably among cohorts, but that specific viruses and fungal species are associated with multiple cohorts—these warrant further investigation.
Intestinal viruses and fungi modulate the immune response and can contribute to intestinal inflammation, making them targets of microbiome-based therapies. Broad spectrum antifungal or antiviral drugs have been consistently reported to have detrimental effects on patients. Diets and FMT alter the fungal and viral components of the gut16,123,153, but have other broad effects, making it a challenge to identify any virus-mediated effects on health or disease. Identifying specific fungi or viruses in the intestinal microbiota that contribute to or prevent disease, and any strain-specific targets, would be required to develop therapeutic agents that modify these microbes. Testing pharmacologic agents that alter immune responses to fungi or viruses might also help identify their targets. Nevertheless, considerably more basic research is needed to inform therapeutic approaches in this new area of microbiome science.
Another emerging area of research not covered by this review is interactions among different microbes in the gut. Studies of the oral, vaginal, and lung mucosa provide evidence for viruses-bacteria and fungi-bacteria interactions at these sites. Several studies have begun to explore such interactions in the gut. Many enteric viruses that infect eukaryotic cells require the bacterial microbiota for infection and transmission. Mechanisms include direct effects in which bacteria bind virions to enhance stability and attachment to target animal cells, or indirect effects of bacteria on the mucosal immune system154–158. In contrast to these mechanisms in which bacteria promote viral infection, in mice that are spontaneously resistant to rotavirus infection, segmented filamentous bacteria were found to promote epithelial cell proliferation and expulsion to block virus infectivity159. This shows that the presence of an individual bacterial species can affect the course of an enteric viral infection.
Most studies of interactions between fungi and bacteria in the gut have focused on the detrimental effects of bacteria on fungi, although positive or nutritional relationships have been documented17,22. Lactobacilli in the intestine promote restructuring of the C albicans cell wall and mask β-glucan by producing lactic acid160. Serratia marcescens releases unique anti-fungal effector proteins with activity against Candida species and S cerevisiae161. However, entirely new approaches must be developed to assess trans-kingdom and intra-kingdom interactions among microbes in the intestinal community.
Although intestinal fungi and viruses might be the dark matter of the microbiota, they are a highly immunoreactive component with unrecognized potential. It will be important to learn how these microbes affect each other, and the immune response, to promote health and development of disease.
Acknowledgements
Author contributions: Drs. Iliev and Cadwell jointly drafted the manuscript and approved the final version.
Funding
K.C. has received funding from US National Institute of Health (NIH) grants R01 AI121244, R01 HL123340, R01 DK093668, R01 AI130945, R01 HL125816, and R01 AI140754, and pilot awards from NYU CTSA grant UL1TR001445 from the National Center for Advancing Translational Sciences (NCATS) and NYU Cancer Center grant P30CA016087. K.C. has also been recently supported by Faculty Scholar grant from the Howard Hughes Medical Institute, Kenneth Rainin Foundation, Crohn’s & Colitis Foundation, and is a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Diseases. I.D.I has received funding from US NIH grants R01 DK113136, R01 DK121977, R21 AI146957 and R56 AI137157, the Leona M. and Harry B. Helmsley Charitable Trust, Crohn’s and Colitis Foundation, Pilot Funding from the Center for Advanced Digestive Care (CADC) and the Jill Roberts Institute for Research in IBD, Kenneth Rainin Foundation, and the Irma T. Hirschl Career Scientist Award, and is a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
I.D.I has no conflict of interests to report related to this manuscript. K.C. has consulted for or received an honorarium from Puretech Health, Genentech, Vedanta, and AbbVie, Inc., has received research support from Puretech Health, Pacific Biosciences, and Pfizer, Inc, and holds U.S. patent 10,722,600 and provisional patent 62/935,035.
References
- 1.Kim MS, Park EJ, Roh SW & Bae JW Diversity and abundance of single-stranded DNA viruses in human feces. Appl Environ Microbiol 77, 8062–8070 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sweere JM, et al. Bacteriophage trigger antiviral immunity and prevent clearance of bacterial infection. Science 363(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gogokhia L, et al. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host Microbe 25, 285–299 e288 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim ES, et al. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nature medicine (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liang G, et al. The stepwise assembly of the neonatal virome is modulated by breastfeeding. Nature, 10.1038/s41586-41020-42192-41581 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nystrom N, et al. Human enterovirus species B in ileocecal Crohn’s disease. Clin Transl Gastroenterol 4, e38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Norman JM, et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 160, 447–460 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Legoff J, et al. The eukaryotic gut virome in hematopoietic stem cell transplantation: new clues in enteric graft-versus-host disease. Nature medicine 23, 1080–1085 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Nakatsu G, et al. Alterations in Enteric Virome Are Associated With Colorectal Cancer and Survival Outcomes. Gastroenterology 155, 529–541 e525 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Lindfors K, et al. Metagenomics of the faecal virome indicate a cumulative effect of enterovirus and gluten amount on the risk of coeliac disease autoimmunity in genetically at risk children: the TEDDY study. Gut (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clooney AG, et al. Whole-Virome Analysis Sheds Light on Viral Dark Matter in Inflammatory Bowel Disease. Cell Host Microbe 26, 764–778 e765 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Abbas AA, et al. Redondoviridae, a Family of Small, Circular DNA Viruses of the Human Oro-Respiratory Tract Associated with Periodontitis and Critical Illness. Cell Host Microbe 26, 297 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Monaco CL, et al. Altered Virome and Bacterial Microbiome in Human Immunodeficiency Virus-Associated Acquired Immunodeficiency Syndrome. Cell Host Microbe 19, 311–322 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nash AK, et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 5, 153 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Human Microbiome Jumpstart Reference Strains, C., et al. A catalog of reference genomes from the human microbiome. Science 328, 994–999 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li XV, Leonardi I & Iliev ID Gut Mycobiota in Immunity and Inflammatory Disease. Immunity 50, 1365–1379 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richard ML & Sokol H The gut mycobiota: insights into analysis, environmental interactions and role in gastrointestinal diseases. Nat Rev Gastroenterol Hepatol 16, 331–345 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Hallen-Adams HE & Suhr MJ Fungi in the healthy human gastrointestinal tract. Virulence 8, 352–358 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suhr MJ, Banjara N & Hallen-Adams HE Sequence-based methods for detecting and evaluating the human gut mycobiome. Lett Appl Microbiol 62, 209–215 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Fischer JC, et al. RIG-I/MAVS and STING signaling promote gut integrity during irradiation- and immune-mediated tissue injury. Sci Transl Med 9(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffmann C, et al. Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PLoS One 8, e66019 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiers WD, Gao IH & Iliev ID Gut mycobiota under scrutiny: fungal symbionts or environmental transients? Curr Opin Microbiol 50, 79–86 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karst SM, Wobus CE, Lay M, Davidson J & Virgin HW t. STAT1-dependent innate immunity to a Norwalk-like virus. Science 299, 1575–1578 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Kernbauer E, Ding Y & Cadwell K An enteric virus can replace the beneficial function of commensal bacteria. Nature 516, 94–98 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun L, et al. Type I Interferons Link Viral Infection to Enhanced Epithelial Turnover and Repair. Cell Host Microbe (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dennis EA, et al. Cytomegalovirus promotes intestinal macrophage-mediated mucosal inflammation through induction of Smad7. Mucosal Immunol 11, 1694–1704 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neil JA, et al. IFN-I and IL-22 mediate protective effects of intestinal viral infection. Nat Microbiol 4, 1737–1749 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Metzger RN, Krug AB & Eisenacher K Enteric Virome Sensing-Its Role in Intestinal Homeostasis and Immunity. Viruses 10(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Broquet AH, Hirata Y, McAllister CS & Kagnoff MF RIG-I/MDA5/MAVS are required to signal a protective IFN response in rotavirus-infected intestinal epithelium. J Immunol 186, 1618–1626 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Crawford SE, et al. Rotavirus infection. Nat Rev Dis Primers 3, 17083 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee S, et al. A Secreted Viral Nonstructural Protein Determines Intestinal Norovirus Pathogenesis. Cell Host Microbe 25, 845–857 e845 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saxena K, et al. A paradox of transcriptional and functional innate interferon responses of human intestinal enteroids to enteric virus infection. Proc Natl Acad Sci U S A 114, E570–E579 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu S, et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 546, 667–670 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Winkle JA, et al. Persistence of Systemic Murine Norovirus Is Maintained by Inflammatory Recruitment of Susceptible Myeloid Cells. Cell Host Microbe 24, 665–676 e664 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riddle MS, Chen WH, Kirkwood CD & MacLennan CA Update on vaccines for enteric pathogens. Clin Microbiol Infect 24, 1039–1045 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Bok K & Green KY Norovirus gastroenteritis in immunocompromised patients. N Engl J Med 367, 2126–2132 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomov VT, et al. Differentiation and Protective Capacity of Virus-Specific CD8(+) T Cells Suggest Murine Norovirus Persistence in an Immune-Privileged Enteric Niche. Immunity 47, 723–738 e725 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilen CB, et al. Tropism for tuft cells determines immune promotion of norovirus pathogenesis. Science 360, 204–208 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang P, et al. Nlrp6 regulates intestinal antiviral innate immunity. Science 350, 826–830 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ettayebi K, et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 353, 1387–1393 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drummond CG, et al. Enteroviruses infect human enteroids and induce antiviral signaling in a cell lineage-specific manner. Proc Natl Acad Sci U S A 114, 1672–1677 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holly MK & Smith JG Adenovirus Infection of Human Enteroids Reveals Interferon Sensitivity and Preferential Infection of Goblet Cells. Journal of virology 92(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cortez V, et al. Astrovirus infects actively secreting goblet cells and alters the gut mucus barrier. Nature communications 11, 2097 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao G, et al. Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proc Natl Acad Sci U S A 114, E6166–E6175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson SS, et al. Alpha-defensin-dependent enhancement of enteric viral infection. PLoS Pathog 13, e1006446 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gounder AP, et al. Defensins Potentiate a Neutralizing Antibody Response to Enteric Viral Infection. PLoS Pathog 12, e1005474 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Speakman EA, Dambuza IM, Salazar F & Brown GD T Cell Antifungal Immunity and the Role of C-Type Lectin Receptors. Trends Immunol 41, 61–76 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iliev ID, et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 336, 1314–1317 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang TT, et al. Dectin-3 Deficiency Promotes Colitis Development due to Impaired Antifungal Innate Immune Responses in the Gut. Plos Pathogens 12(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Plantinga TS, et al. Early stop polymorphism in human DECTIN-1 is associated with increased candida colonization in hematopoietic stem cell transplant recipients. Clin Infect Dis 49, 724–732 (2009). [DOI] [PubMed] [Google Scholar]
- 51.van der Velden WJ, et al. The incidence of acute graft-versus-host disease increases with Candida colonization depending the dectin-1 gene status. Clin Immunol 136, 302–306 (2010). [DOI] [PubMed] [Google Scholar]
- 52.Thompson A, et al. The protective effect of inflammatory monocytes during systemic C. albicans infection is dependent on collaboration between C-type lectin-like receptors. PLoS Pathog 15, e1007850 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu LL, et al. C-type lectin receptors Dectin-3 and Dectin-2 form a heterodimeric pattern-recognition receptor for host defense against fungal infection. Immunity 39, 324–334 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Wevers BA, et al. Fungal engagement of the C-type lectin mincle suppresses dectin-1-induced antifungal immunity. Cell Host Microbe 15, 494–505 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Xiao Y, et al. Targeting CBLB as a potential therapeutic approach for disseminated candidiasis. Nature medicine 22, 906–914 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wirnsberger G, et al. Inhibition of CBLB protects from lethal Candida albicans sepsis. Nature medicine 22, 915–923 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lamas B, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nature medicine 22, 598–605 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zelante T, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39, 372–385 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Wang T, et al. The Adaptor Protein CARD9 Protects against Colon Cancer by Restricting Mycobiota-Mediated Expansion of Myeloid-Derived Suppressor Cells. Immunity 49, 504–514 e504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Malik A, et al. SYK-CARD9 Signaling Axis Promotes Gut Fungi-Mediated Inflammasome Activation to Restrict Colitis and Colon Cancer. Immunity 49, 515–530 e515 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wheeler ML, et al. Immunological Consequences of Intestinal Fungal Dysbiosis. Cell Host Microbe 19, 865–873 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Borghi M, et al. Pathogenic NLRP3 Inflammasome Activity during Candida Infection Is Negatively Regulated by IL-22 via Activation of NLRC4 and IL-1Ra. Cell Host Microbe 18, 198–209 (2015). [DOI] [PubMed] [Google Scholar]
- 63.Hise AG, et al. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 5, 487–497 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Netea MG, Joosten LA, van der Meer JW, Kullberg BJ & van de Veerdonk FL Immune defence against Candida fungal infections. Nat Rev Immunol 15, 630–642 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Kasper L, et al. The fungal peptide toxin Candidalysin activates the NLRP3 inflammasome and causes cytolysis in mononuclear phagocytes. Nat Commun 9, 4260 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leonardi I, et al. CX3CR1(+) mononuclear phagocytes control immunity to intestinal fungi. Science 359, 232–236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li X, et al. Response to Fungal Dysbiosis by Gut-Resident CX3CR1(+) Mononuclear Phagocytes Aggravates Allergic Airway Disease. Cell Host Microbe 24, 847–856 e844 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Conti HR & Gaffen SL IL-17-Mediated Immunity to the Opportunistic Fungal Pathogen Candida albicans. J Immunol 195, 780–788 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Atarashi K, et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 163, 367–380 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shao TY, et al. Commensal Candida albicans Positively Calibrates Systemic Th17 Immunological Responses. Cell Host Microbe 25, 404–417 e406 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bacher P, et al. Human Anti-fungal Th17 Immunity and Pathology Rely on Cross-Reactivity against Candida albicans. Cell 176, 1340–1355 e1315 (2019). [DOI] [PubMed] [Google Scholar]
- 72.Pietrella D, et al. Secreted aspartic proteases of Candida albicans activate the NLRP3 inflammasome. Eur J Immunol 43, 679–692 (2013). [DOI] [PubMed] [Google Scholar]
- 73.Chikina AS, et al. Macrophages Maintain Epithelium Integrity by Limiting Fungal Product Absorption. Cell 183, 411–428 e416 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu W, et al. EGFR and HER2 receptor kinase signaling mediate epithelial cell invasion by Candida albicans during oropharyngeal infection. Proc Natl Acad Sci U S A 109, 14194–14199 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Swidergall M, Solis NV, Lionakis MS & Filler SG EphA2 is an epithelial cell pattern recognition receptor for fungal beta-glucans. Nat Microbiol 3, 53–61 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ho J, et al. Candidalysin activates innate epithelial immune responses via epidermal growth factor receptor. Nat Commun 10, 2297 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stappers MHT, et al. Recognition of DHN-melanin by a C-type lectin receptor is required for immunity to Aspergillus. Nature (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moeller JB, et al. Modulation of the fungal mycobiome is regulated by the chitin-binding receptor FIBCD1. J Exp Med 216, 2689–2700 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Moyes DL, et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 532, 64–68 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Richardson JP, et al. Candidalysin drives epithelial signaling, neutrophil recruitment, and immunopathology at the vaginal mucosa. Infect Immun (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Allert S, et al. Candida albicans-Induced Epithelial Damage Mediates Translocation through Intestinal Barriers. MBio 9(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li XD, et al. Mitochondrial antiviral signaling protein (MAVS) monitors commensal bacteria and induces an immune response that prevents experimental colitis. Proceedings of the National Academy of Sciences of the United States of America 108, 17390–17395 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu L, et al. Commensal viruses maintain intestinal intraepithelial lymphocytes via noncanonical RIG-I signaling. Nat Immunol (2019). [DOI] [PubMed] [Google Scholar]
- 84.Yang JY, et al. Enteric Viruses Ameliorate Gut Inflammation via Toll-like Receptor 3 and Toll-like Receptor 7-Mediated Interferon-beta Production. Immunity 44, 889–900 (2016). [DOI] [PubMed] [Google Scholar]
- 85.Abt MC, et al. TLR-7 activation enhances IL-22-mediated colonization resistance against vancomycin-resistant enterococcus. Sci Transl Med 8, 327ra325 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Broggi A, Tan Y, Granucci F & Zanoni I IFN-lambda suppresses intestinal inflammation by non-translational regulation of neutrophil function. Nat Immunol 18, 1084–1093 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ingle H, et al. Viral complementation of immunodeficiency confers protection against enteric pathogens via interferon-lambda. Nat Microbiol 4, 1120–1128 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Contreras G Effect of the administration of oral poliovirus vaccine on infantile diarrhoea mortality. Vaccine 7, 211–212 (1989). [DOI] [PubMed] [Google Scholar]
- 89.Thepaut M, et al. Protective role of murine norovirus against Pseudomonas aeruginosa acute pneumonia. Vet Res 46, 91 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim YG, et al. Gut dysbiosis promotes M2 macrophage polarization and allergic airway inflammation via fungi-induced PGE(2). Cell Host Microbe 15, 95–102 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Noverr MC, Noggle RM, Toews GB & Huffnagle GB Role of antibiotics and fungal microbiota in driving pulmonary allergic responses. Infect Immun 72, 4996–5003 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jiang TT, et al. Commensal Fungi Recapitulate the Protective Benefits of Intestinal Bacteria. Cell Host Microbe 22, 809–816 e804 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chiaro TR, et al. A member of the gut mycobiota modulates host purine metabolism exacerbating colitis in mice. Sci Transl Med 9(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tso GHW, et al. Experimental evolution of a fungal pathogen into a gut symbiont. Science 362, 589–595 (2018). [DOI] [PubMed] [Google Scholar]
- 95.Cheng SC, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345, 1250684 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saeed S, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 345, 1251086 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lin JD, et al. Rewilding Nod2 and Atg16l1 Mutant Mice Uncovers Genetic and Environmental Contributions to Microbial Responses and Immune Cell Composition. Cell Host Microbe (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yeung F, et al. Altered Immunity of Laboratory Mice in the Natural Environment Is Associated with Fungal Colonization. Cell Host Microbe (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rosshart SP, et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 365(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Beura LK, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532, 512–516 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lewis JD, et al. Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell Host Microbe 18, 489–500 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sokol H, et al. Fungal microbiota dysbiosis in IBD. Gut 66, 1039–1048 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hoarau G, et al. Bacteriome and Mycobiome Interactions Underscore Microbial Dysbiosis in Familial Crohn’s Disease. MBio 7, e01250–01216 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Limon JJ, et al. Malassezia Is Associated with Crohn’s Disease and Exacerbates Colitis in Mouse Models. Cell Host Microbe (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lang S, et al. Intestinal Fungal Dysbiosis and Systemic Immune Response to Fungi in Patients With Alcoholic Hepatitis. Hepatology (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chehoud C, et al. Fungal Signature in the Gut Microbiota of Pediatric Patients With Inflammatory Bowel Disease. Inflamm Bowel Dis 21, 1948–1956 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ott SJ, et al. Fungi and inflammatory bowel diseases: Alterations of composition and diversity. Scand J Gastroenterol 43, 831–841 (2008). [DOI] [PubMed] [Google Scholar]
- 108.Witchley JN, et al. Candida albicans Morphogenesis Programs Control the Balance between Gut Commensalism and Invasive Infection. Cell Host Microbe 25, 432–443 e436 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liguori G, et al. Fungal Dysbiosis in Mucosa-associated Microbiota of Crohn’s Disease Patients. J Crohns Colitis 10, 296–305 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sovran B, et al. Enterobacteriaceae are essential for the modulation of colitis severity by fungi. Microbiome 6, 152 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Graham DB & Xavier RJ Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature 578, 527–539 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cao Z, et al. Ubiquitin Ligase TRIM62 Regulates CARD9-Mediated Anti-fungal Immunity and Intestinal Inflammation. Immunity 43, 715–726 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kim SG, et al. Microbiota-derived lantibiotic restores resistance against vancomycin-resistant Enterococcus. Nature 572, 665–669 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Plichta DR, Graham DB, Subramanian S & Xavier RJ Therapeutic Opportunities in Inflammatory Bowel Disease: Mechanistic Dissection of Host-Microbiome Relationships. Cell 178, 1041–1056 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hueber W, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut 61, 1693–1700 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Standaert-Vitse A, et al. Candida albicans is an immunogen for anti-Saccharomyces cerevisiae antibody markers of Crohn’s disease. Gastroenterology 130, 1764–1775 (2006). [DOI] [PubMed] [Google Scholar]
- 117.Torres J, et al. Serum Biomarkers Identify Patients Who Will Develop Inflammatory Bowel Diseases Up to 5 y Before Diagnosis. Gastroenterology (2020). [DOI] [PubMed] [Google Scholar]
- 118.Paramsothy S, et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo-controlled trial. Lancet 389, 1218–1228 (2017). [DOI] [PubMed] [Google Scholar]
- 119.Leonardi I, et al. Fungal Trans-kingdom Dynamics Linked to Responsiveness to Fecal Microbiota Transplantation (FMT) Therapy in Ulcerative Colitis. Cell Host Microbe (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fernandes MA, et al. Enteric Virome and Bacterial Microbiota in Children With Ulcerative Colitis and Crohn Disease. J Pediatr Gastroenterol Nutr 68, 30–36 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zuo T, et al. Gut mucosal virome alterations in ulcerative colitis. Gut 68, 1169–1179 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chehoud C, et al. Transfer of Viral Communities between Human Individuals during Fecal Microbiota Transplantation. mBio 7, e00322 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zuo T, et al. Bacteriophage transfer during faecal microbiota transplantation in Clostridium difficile infection is associated with treatment outcome. Gut 67, 634–643 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ott SJ, et al. Efficacy of Sterile Fecal Filtrate Transfer for Treating Patients With Clostridium difficile Infection. Gastroenterology 152, 799–811 e797 (2017). [DOI] [PubMed] [Google Scholar]
- 125.Tokarz R, et al. Characterization of Stool Virome in Children Newly Diagnosed With Moderate to Severe Ulcerative Colitis. Inflamm Bowel Dis 25, 1656–1662 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ungaro F, et al. Metagenomic analysis of intestinal mucosa revealed a specific eukaryotic gut virome signature in early-diagnosed inflammatory bowel disease. Gut Microbes 10, 149–158 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Axelrad JE, et al. Enteric Infections Are Common in Patients with Flares of Inflammatory Bowel Disease. Am J Gastroenterol 113, 1530–1539 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Axelrad JE, et al. Gastrointestinal Infection Increases Odds of Inflammatory Bowel Disease in a Nationwide Case-Control Study. Clin Gastroenterol Hepatol 17, 1311–1322 e1317 (2019). [DOI] [PubMed] [Google Scholar]
- 129.Cadwell K & Coscoy L The specificities of Kaposi’s sarcoma-associated herpesvirus-encoded E3 ubiquitin ligases are determined by the positions of lysine or cysteine residues within the intracytoplasmic domains of their targets. J Virol 82, 4184–4189 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cadwell K, et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 141, 1135–1145 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Matsuzawa-Ishimoto Y, Hwang S & Cadwell K Autophagy and Inflammation. Annu Rev Immunol 36, 73–101 (2018). [DOI] [PubMed] [Google Scholar]
- 132.Matsuzawa-Ishimoto Y, et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J Exp Med (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Matsuzawa-Ishimoto Y, et al. An Intestinal Organoid-Based Platform That Recreates Susceptibility to T Cell-Mediated Tissue Injury. Blood (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lencioni KC, Seamons A, Treuting PM, Maggio-Price L & Brabb T Murine norovirus: an intercurrent variable in a mouse model of bacteria-induced inflammatory bowel disease. Comp Med 58, 522–533 (2008). [PMC free article] [PubMed] [Google Scholar]
- 135.Basic M, et al. Norovirus triggered microbiota-driven mucosal inflammation in interleukin 10-deficient mice. Inflamm Bowel Dis 20, 431–443 (2014). [DOI] [PubMed] [Google Scholar]
- 136.Bolsega S, et al. Composition of the Intestinal Microbiota Determines the Outcome of Virus-Triggered Colitis in Mice. Front Immunol 10, 1708 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bouziat R, et al. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science 356, 44–50 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bouziat R, et al. Murine Norovirus Infection Induces TH1 Inflammatory Responses to Dietary Antigens. Cell Host Microbe 24, 677–688 e675 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Stene LC, et al. Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: a longitudinal study. Am J Gastroenterol 101, 2333–2340 (2006). [DOI] [PubMed] [Google Scholar]
- 140.Notkins AL, Yoon J, Onodera T & Jenson AB Virus-induced diabetes mellitus: infection of mice with variants of encephalomyocarditis virus, coxsackievirus B4, and reovirus type 3. Advances in experimental medicine and biology 119, 137–146 (1979). [DOI] [PubMed] [Google Scholar]
- 141.Vehik K, et al. Prospective virome analyses in young children at increased genetic risk for type 1 diabetes. Nature medicine 25, 1865–1872 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kim KW, et al. Higher frequency of vertebrate-infecting viruses in the gut of infants born to mothers with type 1 diabetes. Pediatr Diabetes (2019). [DOI] [PubMed] [Google Scholar]
- 143.Serreze DV, Ottendorfer EW, Ellis TM, Gauntt CJ & Atkinson MA Acceleration of type 1 diabetes by a coxsackievirus infection requires a preexisting critical mass of autoreactive T-cells in pancreatic islets. Diabetes 49, 708–711 (2000). [DOI] [PubMed] [Google Scholar]
- 144.Drescher KM, Kono K, Bopegamage S, Carson SD & Tracy S Coxsackievirus B3 infection and type 1 diabetes development in NOD mice: insulitis determines susceptibility of pancreatic islets to virus infection. Virology 329, 381–394 (2004). [DOI] [PubMed] [Google Scholar]
- 145.Graham KL, et al. Rotavirus infection accelerates type 1 diabetes in mice with established insulitis. Journal of virology 82, 6139–6149 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wetzel JD, et al. Reovirus delays diabetes onset but does not prevent insulitis in nonobese diabetic mice. Journal of virology 80, 3078–3082 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pane JA, Webster NL & Coulson BS Rotavirus activates lymphocytes from nonobese diabetic mice by triggering toll-like receptor 7 signaling and interferon production in plasmacytoid dendritic cells. PLoS Pathog 10, e1003998 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pearson JA, et al. Norovirus Changes Susceptibility to Type 1 Diabetes by Altering Intestinal Microbiota and Immune Cell Functions. Front Immunol 10, 2654 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yang AM, et al. Intestinal fungi contribute to development of alcoholic liver disease. J Clin Invest (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Scaldaferri F, et al. The Thrilling Journey of SARS-CoV-2 into the Intestine: From Pathogenesis to Future Clinical Implications. Inflamm Bowel Dis 26, 1306–1314 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhou J, et al. Infection of bat and human intestinal organoids by SARS-CoV-2. Nat Med 26, 1077–1083 (2020). [DOI] [PubMed] [Google Scholar]
- 152.Lamers MM, et al. SARS-CoV-2 productively infects human gut enterocytes. Science 369, 50–54 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zuo T, et al. Gut fungal dysbiosis correlates with reduced efficacy of fecal microbiota transplantation in Clostridium difficile infection. Nat Commun 9, 3663 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kuss SK, et al. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science 334, 249–252 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jones MK, et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 346, 755–759 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kane M, et al. Successful transmission of a retrovirus depends on the commensal microbiota. Science 334, 245–249 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Baldridge MT, et al. Commensal microbes and interferon-lambda determine persistence of enteric murine norovirus infection. Science 347, 266–269 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Grau KR, et al. The intestinal regionalization of acute norovirus infection is regulated by the microbiota via bile acid-mediated priming of type III interferon. Nat Microbiol 5, 84–92 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Shi Z, et al. Segmented Filamentous Bacteria Prevent and Cure Rotavirus Infection. Cell 179, 644–658 e613 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ballou ER, et al. Lactate signalling regulates fungal beta-glucan masking and immune evasion. Nat Microbiol 2, 16238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Trunk K, et al. The type VI secretion system deploys antifungal effectors against microbial competitors. Nat Microbiol 3, 920–931 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Li Q, et al. Dysbiosis of gut fungal microbiota is associated with mucosal inflammation in Crohn’s disease. J Clin Gastroenterol 48, 513–523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mar JS, et al. Disease Severity and Immune Activity Relate to Distinct Interkingdom Gut Microbiome States in Ethnically Distinct Ulcerative Colitis Patients. MBio 7(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]