

Abstract
In many types of familial amyotrophic lateral sclerosis (fALS), mutations cause proteins to gain toxic properties that mediate neurodegenerative processes. It is becoming increasingly clear that the proteins involved in ALS, and those responsible for a host of other neurodegenerative diseases, share many characteristics with a growing number of prion diseases. ALS is a heterogenous disease in which the majority of cases are sporadic in their etiology. Studies investigating the inherited forms of the disease are now beginning to provide evidence that some of this heterogeneity may be due to the existence of distinct conformations that ALS-linked proteins can adopt to produce the equivalent of prion strains. In this review, we discuss the in vitro and in vivo evidence that has been generated to better understand the characteristics of these proteins and how their tertiary structure may impact the disease phenotype.
Keywords: Amyotrophic Lateral Sclerosis, Superoxide dismutase-1, TDP-43, prion, strains
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that leads to paralysis and ultimately death of those afflicted with the condition. Although weakness can first appear anywhere in the body, including the respiratory system, most cases manifest in limb or bulbar muscles as a focal weakness before spreading throughout the motor axis to weaken respiratory muscles. This muscle loss is caused by the degeneration of both upper and motor neurons throughout the CNS, and, unfortunately, there are no treatments that slow disease progression or significantly lengthen survival. The prevalence of ALS is about 5 cases per 100,000 with an incidence of 1-2 individuals diagnosed per 100,000 each year, revealing how quickly the disease progresses. The majority of cases are fatal within 2-4 years after diagnosis, but factors such as the site of onset, duration from first symptom to diagnosis, presence of cognitive impairment, and genotype can increase survival to more than 10 years after onset [1, 29]. The motor dysfunction in ALS has also been demonstrated to overlap with frontotemporal dementia (FTD), a disease characterized by the degeneration of the frontal and temporal lobes. Up to 50% of patients with ALS develop frontal lobe dysfunction, whereas approximately 15% of patients with FTD develop motor neuron loss.
Although clinically indistinguishable, ~90% of ALS cases are sporadic and have no known origin (sALS) while ~10% are familial (fALS) and are typically passed down with inheritance. There are now more than 50 genes that have been implicated in ALS pathogenesis, yet many of these have yet to be completely validated. Interestingly, the proteins encoded by these ALS-causing genes have a diverse set of functions, highlighting the complex nature of this disease and suggesting a range of mechanisms that ultimately lead to the degeneration of motor neurons. The first of these mutations to be discovered, and most widely studied, was found in the gene encoding the Cu/Zn superoxide dismutase 1 (SOD1) gene [112], of which there are now more than 180 clinically relevant mutations identified (http://alsod.iop.kcl.ac.uk/). However, mutations in SOD1 account for only 10-20% of all fALS diagnoses. Other genes associated with a significant portion of fALS cases include C9ORF72 and those encoding the 43 kDa transactive response (TAR) DNA-binding protein (TDP-43) and the fused in sarcoma/translocated in liposarcoma (FUS/TLS) protein (http://alsod.iop.kcl.ac.uk/). Together, the identification and characterization of these disease-causing mutations and their proteins have greatly enhanced our understanding of ALS.
Aside from the key neuropathological feature of upper and lower motor neuron loss in cases of ALS, molecular neuropathology has demonstrated the appearance of ubiquitinated skein-like, or dense and round, inclusions in the cytoplasm of motor neurons [83, 85]. In the majority of ALS cases (~97%), including disease caused by repeat expansions in C90RF72, these protein inclusions are immunoreactive for TDP-43. In ALS cases caused by mutations in the genes encoding for SOD1 and FUS, protein inclusions are composed of those gene-products, and, generally, TDP-43 pathology is lacking [94]. Studies of mutant SOD1, TDP43, and FUS in cell and animal models have revealed that they harbor many of the properties of the infectious PrP prion protein (PrPSc) and of other proteins associated with neurodegeneration such as synuclein, tau, and amyloid-β (Aβ). These properties include the protein’s ability to aggregate into non-native structures, template its misfolded conformation, spread from cell-to-cell in culture, spread along neuroanatomically connected regions in vivo, and, in some cases, adopt distinct conformations or “strains” [8, 30, 74, 101, 115, 118, 137]. With the recent advances in imaging, such as cryo-electron microscopy (cryo-EM), along with better in vitro and in vivo tools, we now have the ability directly assess the existence and consequences of conformationally distinct strains of misfolded proteins in neurodegenerative diseases.
In this review, we will discuss the existing evidence linking ALS to prion diseases and emphasize those studies investigating the conformational heterogeneity of SOD1 and other proteins implicated in ALS.
PrP prions and strains
The classical PrP prion diseases are a group of infectious neurodegenerative diseases affecting humans and a wide range of other mammals with the most notable being domesticated cattle and sheep that are consumed by humans. The major neuropathological hallmarks in the central nervous system (CNS) include neuronal loss, spongiform degeneration, gliosis, and the accumulation of amyloid plaques or aggregates consisting of the abnormal, protease-resistant form of the host-encoded prion protein (PrP) [36, 37, 107]. The identification and nature of the causative agent of these diseases was a highly contentious topic that resulted in the recognition of a novel infectious pathogen: a prion – a small proteinaceous infectious particle [18, 106]. Prior to its identification, and based upon the central dogma of molecular biology, the infectious agent was believed to be due to an “unconventional” or “slow” virus [52]. Ultimately, it was the transmission of the scrapie agent to hamsters and its subsequent purification and biochemical characterization that led to the identification of the infectious prion protein (PrPSc) [76, 87, 105].
Though the prion hypothesis was supported by the data, many characteristics of PrPSc existed that perplexed researchers; the most puzzling being the finding that transmission studies with different PrPSc inocula resulted in disease with distinct incubation periods and neuropathological features [77]. At the time, the only explanation for these findings was the existence of a strain-specific nucleic acid as part of the infectious agent. It was a theorized mechanism by J.S. Griffith in which he explained the potential for a pathogenic protein that enciphers its own replication without the need for nucleic acid that led researchers to examine and hypothesize that the structural heterogeneity of PrPSc could account for the disparate symptoms observed in experimental animals [61]. To address this hypothesis, researchers utilized two hamster-adapted strains of the transmissible mink encephalopathy (TME) agent that had been isolated following multiple passages in Syrian golden hamsters [16]. These two isolated strains had vastly different incubation periods and neuropathology and were designated hyper (HY) and drowsy (DY) due to their differing symptoms at the clinical stage of disease [16]. Biochemical studies of HY and DY PrPSc revealed differences in their migration by western blot analysis, their sensitivity to digestion with proteinase K, and their sedimentation in N-lauroylsarcosine [15]. It was also demonstrated that protease-resistant fragments of PrP that were unique to the two strains were maintained upon in vitro conversion of radiolabeled recombinant hamster PrPc (the normally folded cellular version of the protein) to either labeled HY or DY PrPSc [14]. Further studies investigating both human and animal prion diseases using a variety of biochemical and histological techniques also provided persuasive evidence to the origins of PrP prion strains [31, 126]. It is now well-established that strain-specific information is encoded in the tertiary structure of PrPSc and that the protein can adopt a wide variety of conformations [116].
Prion strains are operationally defined by the distinct phenotypic characteristics they display upon infection when certain variables are held constant, such as the dose of the agent, the route of infection, and the genotype of the host [98]. The differences commonly observed when such variables are accounted for include differences among clinical symptoms, incubation periods, the location and severity of neuropathology, and biochemical properties of abnormal PrPSc. Although differences in biochemical properties such as a protein’s sensitivity to protease digestion or denaturation can be explained based on the tertiary structure and the solvent accessibility of its amino acids, it is not well understood how the conformation of a protein can account for variations in clinical symptoms and neuropathology [9].
As mentioned previously, it is now beginning to be understood that proteins involved in other neurodegenerative disorders share many of the same features and characteristics of the PrP protein. This includes the existence of distinct protein conformers or strains. For instance, it has been hypothesized that the various clinical phenotypes among tauopathies, such as Alzheimer’s disease (AD), progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD), were due to different tau strains; however, it wasn’t until recently with the advent of new technologies like cryo-EM, that researchers were able to directly analyze the structure of protein fibrils at the atomic scale. Using this technology, it was directly shown that the tau fibrils isolated from the brains of patients who succumbed to these different diseases had distinct fibril conformations [43, 47, 56, 140]. These studies are just now starting to shed light on the structural heterogeneity that can exist for a given misfolded, pathogenic protein, and it will be quite interesting to explore whether this same phenomenon holds true for other neurodegenerative diseases, such as ALS.
General properties of SOD1
SOD1 is an antioxidant protein whose normal function is to catalyze the dismutation of highly reactive superoxide anions to hydrogen peroxide and dioxygen. It is a ubiquitously expressed and abundant protein mainly localized in the cytosol [33]. Some localization to the mitochondrial intermembrane space and nuclear compartments has also been reported [46, 54]. In its natively folded state, SOD1 exists as an extremely stable 32-kDa homodimer, in which each 153-amino-acid subunit contains one copper and one zinc ion along with an intra-subunit disulfide bond. Post-translational maturation and proper folding of SOD1 to a mature, highly stable dimer first involves the non-chaperone mediated incorporation of zinc (Zn) [51]. The Zn-bound form of SOD1 is then recognized by the copper chaperone for SOD1 (CCS), forming a heterodimer that facilitates copper (Cu) delivery and disulfide bond formation [11, 117]. The disulfide-oxidized holo version of human SOD1 is an extremely stable enzyme that melts at 92°C and resists proteolytic digestion at concentrations up to 1 mg/ml for 30 minutes at 37°C [108, 129]. However, even in the absence of Cu, the incorporation of Zn has shown to significantly alter its secondary structure and stabilize SOD1, so much so that this binding alone is sufficient to allow the formation of its normal quaternary structure [119].
SOD1 was the first dominantly inherited gene described to have a role in ALS pathogenesis [112] and now accounts for ~10-20% of all fALS cases. To date, there are now more than 180 different mutations that have been identified that span the entire length of the gene. These are predominantly single amino-acid residue substitutions; however, deletions, insertions, and C-terminal truncations have also been described [120, 129]. It was initially hypothesized that mutations in SOD1 resulted in the loss of enzymatic activity and toxicity, though many SOD1 variants display normal enzymatic activity. In studies with transgenic mice overexpressing the G93A variant, animals developed disease despite increased levels of enzymatic activity [19, 63, 66]. Additionally, mice lacking endogenous SOD1 do not exhibit ALS-like phenotypes [109]. Ultimately, studies in transgenic models expressing various fALS-linked SOD1 mutants demonstrated that aggregation of the mutant protein was an invariant pathologic feature of mice that developed an ALS-like paralytic disease [23, 72, 134, 135]. Together, these data implicated a gain-of-toxic mechanism for SOD 1-mediated pathogenesis and led researchers to focus on the misfolding of SOD1 as a critical event in disease pathogenesis.
Familial ALS point mutations in SOD1 protein folding may act primarily by slowing the protein’s rate of maturation rather than completely preventing the acquisition of its native conformation. Studies in which the residues involved in Cu binding (H46 and H48) were mutated did not prevent, but slowed the rate of dimerization and intramolecular disulfide bond formation [24]. This was also the case for mutations in the cysteine residues that produce the intramolecular disulfide bond (C57 and C146); even though these mutations prevented the formation of the disulfide bond, they allowed ~50% of the C146R-SOD1 to acquire resistance to proteinase [24]. This data suggests that although post-translational modifications of SOD1 have impacts on the rate of maturation, the inherent properties of the SOD1 sequence determine whether the protein will achieve its native conformation.
Aggregation and conformational templating of SOD1
Although not all of the more than 180 fALS-SOD1 associated mutations have been characterized, those that have were found to result in an increased propensity for SOD1 to adopt misfolded conformations and acquire detergent insolubility [23, 72, 103]. These variants have been primarily investigated by transient overexpression in cell models [103], but in transgenic mouse models overexpressing SOD1 mutants, detergent-insoluble aggregates of SOD1 are also observed [23, 133]. Importantly, similar proteinaceous inclusions immunoreactive for SOD1 are also detected in the CNS of fALS-SOD1 patients, leading to the widely accepted hypothesis that SOD1 aggregation is associated with the etiology of SOD1-linked fALS. Protein aggregation is associated with the pathogenesis in many other neurodegenerative diseases, such as AD, Parkinson’s disease, and Huntington’s disease, in which intracellular inclusions and/or extracellular amyloid fibrils are the main pathological hallmarks. Like SOD1, mutations within the proteins implicated in the pathogenesis of these other diseases result in their misfolding and aggregation. However, unlike SOD1, patients that succumb to the sporadic form of these diseases accumulate proteinaceous inclusions comprised of the wild-type (WT) versions of their associated toxic protein. Although conformational antibodies specific for “misfolded” SOD1 have revealed sparse immunopositive inclusions within the CNS of postmortem sALS patients, the role for WT SOD1 in the pathogenesis of sALS remains controversial [5, 20, 21, 35, 60, 99, 113, 127].
As previously discussed, WT SOD1 is an extremely stable protein due to its post-translational modifications, and preventing these modifications from occurring affects the kinetics of maturation and destabilizes the protein. The apo (metal free) version of SOD1 has been demonstrated to form detergent-insoluble amyloid fibrils under destabilizing conditions such as high temperature, low pH, or in the presence of organic solvents [50, 123]. Moreover, under mild denaturing conditions at 37°C and with constant shaking, it was demonstrated that both WT and mutant SOD1 spontaneously form fibrils [27, 28]. Similar cell-free assays were initially used in the prion field to study the kinetics of PrPc to PrPSc conversion and supported the hypothesis that the PrP prion protein undergoes aggregation via a multistep process referred to as template-dependent polymerization [4, 78]. It was observed that fibrillization initiates through a rate-limiting nucleation step in which PrPSc “seeds” are produced that can then go on to convert the PrPc substrate into additional PrPSc fibrils. More recently, through the optimization of techniques such as protein misfolding cyclic amplification (PMCA) and real-time quaking-induced conversion (RT-QuIC), it has been demonstrated that fibrillization can be greatly enhanced by adding a pre-formed seed, such as PrPSc-containing homogenates, and also by applying sonication or shaking to break up the continually growing fibrils, thereby producing more seeds [4, 114]. The kinetics of SOD1 fibrillization in the cell-free assays closely mirror those involving PrP, indicating a similar mechanism of fibril assembly. Chia et al. demonstrated the ability to induce fibrillization of both recombinant WT and mutant SOD1 under mild denaturing conditions following the addition of homogenates prepared from the spinal cords of transgenic mice overexpressing mutant SOD1 [28]. This study was paramount in demonstrating the ability of SOD1 to act as a seed for fibrillization and was one of the first studies to highlight the similarities of PrP and SOD1.
These cell-free studies then led researchers to investigate the transmission properties of SOD1 in living cells. Using neuroblastoma cell lines, Munch et al. were the first to demonstrate that exogenously added recombinant mutant SOD1 fibrils were able to be internalized via lipid-raft dependent micropinocytosis [91]. Once inside the cells, these SOD1 species were capable of inducing the aggregation of the overexpressed cytosolic mutant SOD1, causing it to adopt a detergent-insoluble conformation [91]. The authors observed that over time, as the cells continued to divide and the internalized exogenous SOD1 aggregates disappeared, the induced aggregates remained abundant for over a month. This finding revealed that SOD1 aggregation in this cell system was a heritable phenotype and/or the induced aggregates were capable of cell-to-cell transmission [91]. Although the induction of SOD1 aggregation in this assay was dependent on the overexpression of the SOD1 substrate, another study revealed the ability for endogenously expressed WT SOD1 to misfold and aggregate upon the transient overexpression of mutant SOD1 [59]. The WT SOD1 protein upon misfolding also acquired detergent insolubility and the ability to infect neighboring cells, either through the association with exosomes or from being released by dying cells [60, 121].
The in vivo demonstration of mutant SOD1 transmission was dependent on identifying transgenic animal lines that were permissive for transmission. Early work in our laboratory examined whether the onset of motor neuron disease (MND) in mice expressing the G93A or G37R fALS variants of SOD1 could be accelerated by injecting tissue homogenates from paralyzed mice of the same genotype into the spinal cords of newborn transgenic mice. In our experience, using small numbers of mice initially, mice expressing these mutants were not permissive; however, we subsequently observed that the onset of MND could be accelerated in mice expressing the G85R fALS variant of SOD1 fused to yellow fluorescent protein (YFP) [7]. When bred to homozygosity, the G85R-SOD1:YFP transgenic mouse lines develop an ALS-like phenotype comprising of paralysis and muscle loss at ~16 months of age [132]. Mice that are hemizygous for the transgene rarely develop disease over their lifespan [7]. It was previously demonstrated for PrP transmission that utilizing a mouse model that expresses the mutant transgene below the threshold level required to spontaneously develop neurodegenerative disease enabled induction of pathology and disease by injecting PrPSc-containing homogenates [67]. Similar to this paradigm, our group prepared spinal cord homogenates from paralyzed mice that overexpressed different SOD1 mutants and injected them into the spinal cords of neonatal (P0) hemizygous G85R-SOD1:YFP mice to determine whether MND could be transmitted. The homogenates were prepared from diseased transgenic mice overexpressing the G93A or G37R SOD1 proteins. Surprisingly, both preparations resulted in the induction of G85R-SOD1:YFP misfolding and aggregation throughout the CNS and the onset of paralysis. Homogenates prepared from mice that did not contain SOD1 aggregates were unable to induce pathology or disease following injection. Similar outcomes were reported by another group that used a line of mice expressing the G85R variant (no tag) at levels that produce disease between 12-16 months. Intraspinal injection of tissue homogenates from paralyzed G85R mice into young adult animals led to a markedly accelerated age to paralysis [17]. Together, these studies demonstrated that mice expressing the G85R variant of SOD1 were permissive for mutant SOD1 prion-like seeding. Subsequent studies in our laboratories demonstrated that mice expressing truncation mutants of SOD1 are also permissive to mutant SOD1 seeding [6].
In other work, we also demonstrated that injection of tissue homogenates unilaterally into the sciatic nerve of G85R-SOD1:YFP mice produces a unilateral paralysis in the ipsilateral hind limb that spreads to the contralateral hind limb; reminiscent of the spreading paralytic features observed in ALS patients [8]. A longitudinal study of these sciatic-nerve-injected mice revealed that the induced G85R-SOD1:YFP aggregates accumulated in the CNS in a predictable manner; first in the ipsilateral spinal cord before spreading to neuroanatomically connected regions of the CNS.
Importantly, recombinant SOD1 fibrils and homogenates prepared from the spinal cords of fALS-SOD1 patients were also capable of transmitting the disease [6]. An equally important negative finding from these transmission studies was observed when homogenates prepared from the spinal cords of transgenic mice used to model other neurodegenerative diseases, such as those for synucleinopathies and tauopathies, were used as inoculum in the G85R-SOD1:YFP injection paradigm [6]. These lines (expressing mutant synuclein or mutant tau) also develop progressive paralytic phenotypes, have extensive spinal cord pathology, and have a massive glial response within the CNS that many believe could have a role in the disease process. However, upon injecting these homogenates, none of the G85R-SOD1:YFP mice developed MND nor accumulated SOD1 inclusions, indicating the specificity of SOD1 prions to induce MND transmission [6].
SOD1 strains
As mentioned previously for PrP prions, the variability observed among prion diseases in regard to clinical symptoms, incubation period, and neuropathology has been attributed to the existence of prion strains, or alternative conformations of the PrPSc protein. Immunohistological studies of mutant SOD1 pathology in transgenic mouse models provided evidence that different fALS mutants of SOD1 produced alternative conformations of misfolded SOD1 [13]. Whether such conformational differences underlie aspects of human disease remains unclear. In both sporadic ALS and SOD1-ALS, muscle loss and paralysis can begin in the arm(s), leg(s), or bulbar region, the degree of upper vs. lower motor neuron involvement can vary, the age of onset is observed anywhere from 25 to 70 years of age, and the course of disease can last from one to more than 20 years [104]. A similar phenotypic variability is also observed in patients harboring mutations within the PRNP prion gene. In PrP prion diseases, particular polymorphisms within PRNP dictate whether the individual develops Creutzfeldt-Jakob disease (CJD), Gerstmann-Straussler-Scheinker disease (GSS), or fatal familial insomnia (FFI) – three diseases that have distinct pathological manifestations [57, 79]. These disparate phenotypes are believed to arise due to different tertiary conformations that the mutant PrP protein adopts, which create distinct strains of prions. Similar data are being observed for tauopathies, in which it is now beginning to be understood, due in large part to the advances in cryo-EM, that distinct tauopathies like AD, PSP, and CBD are caused by distinct conformations that tau fibrils can adopt [41, 42, 47, 140]. Based on these observations for PrP and tau prions, it seems possible that mutations in SOD1 could lead to distinct strains of misfolded protein and account for some of the heterogeneity observed among fALS-SOD1 cases (Fig. 2).
Fig. 2.
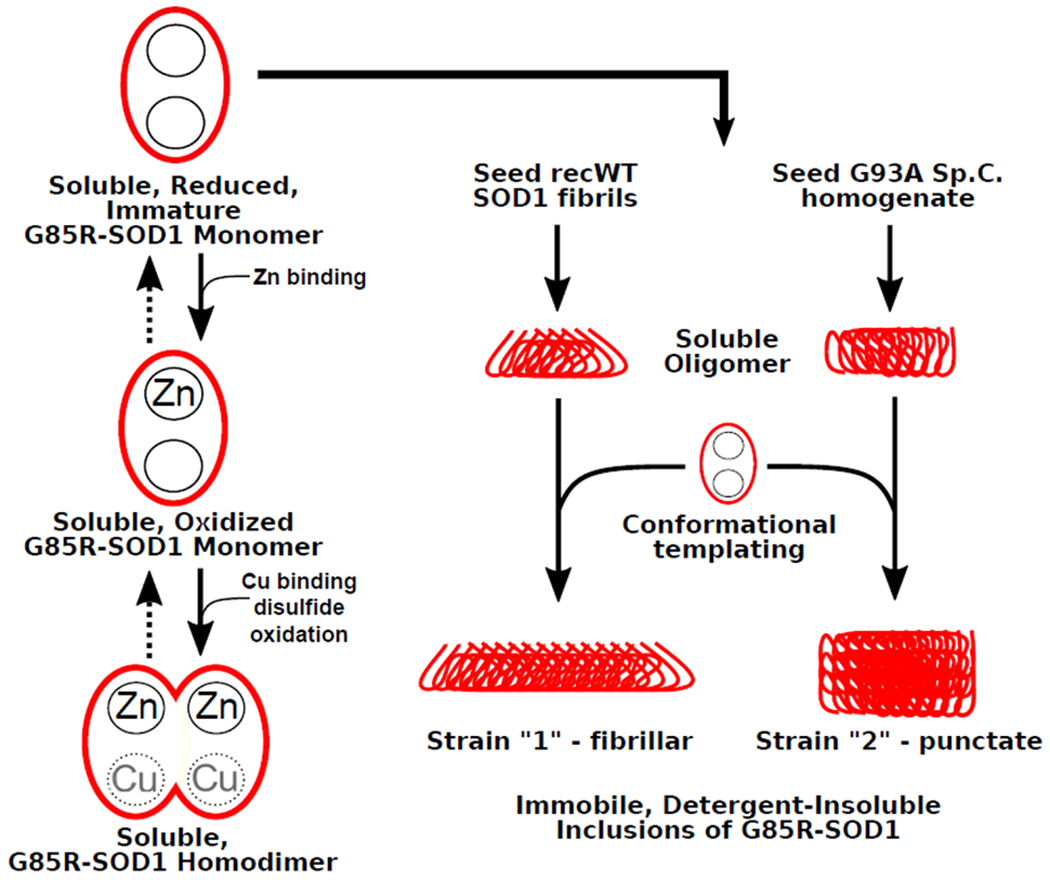
Alternative folding pathways for G85R-SOD1 fALS missense point mutant of SOD1 exposed to misfolded SOD1 seeds. Human G85R-SOD1 exhibits defects in the binding of Zn and Cu (indicated by grayed out Cu) that destabilize native structure [26]. A subset of G85R-SOD1 subunits can acquire Zn and Cu and achieve near native conformation. Immature mutant SOD1 exposed to different SOD1 seed aggregates is susceptible to conformational templating, leading to alternative conformations of misfolded protein. In the example here, seeding with fibrils of recombinant (recWT) WT SOD1 produce a strain that forms fibrillar inclusion pathology while seeding with spinal cord homogenates from paralyzed G93A mice induces punctate pathology [6]. A similar type of diagram could be drawn for G85R SOD1 seeded with tissue homogenates from paralyzed G85R-SOD1 mice (Strain 1) and spinal homogenates from paralyzed D90A-SOD1 mice (Strain 2) [17].
A characteristic of PrP prion strains that has been implicated to impact the course of disease is PrP’s rate of replication and aggregation kinetics [9]. To better understand the aggregation propensity of SOD1 and its relationship to fALS-SOD1 disease features, a number of clinically relevant SOD1 mutants were transiently transfected into human embryonic kidney (HEK293) cells [104]. It was found that the inherent aggregation propensity varied considerably among the more than 30 SOD1 mutants tested and that this property did not correlate with properties such as enzyme activity, protein thermostability, mutation position, or degree of change in protein charge the mutation imparts [104]. Interestingly, though the variability in disease duration among fALS-SOD1 patients could not be explained by the SOD1 mutant’s aggregation propensity, it was observed that those mutations associated with shorter disease durations were more prone to aggregate [104]. Although the differences in the biochemical and biophysical properties of the SOD1 mutants did not correlate within many of the clinical features, these studies demonstrated inherent differences in the characteristics of SOD1 that are enciphered within the amino acid sequence or the tertiary structure it adopts.
The conformational variability among strains of PrP prions has also been demonstrated via immunohistochemistry, using a panel of antibodies that spans the entire length of the protein. This technique is referred to as “epitope mapping” and was used extensively to distinguish experimental and natural sources of scrapie and bovine spongiform encephalopathy (BSE) in sheep and goats [58, 69]. A similar approach has been used to study the different conformational states that misfolded SOD1 can adopt. Using a panel of eight antibodies that span the length of SOD1, Bergh et al. revealed that the detergent-insoluble SOD1 aggregates from various lines of SOD1 transgenic mice differed in their immunoreactivity; WT, G85R, and G93A overexpressing mice accumulated one immunohistochemical profile (Strain A), whereas D90A overexpressing mice accumulated two profiles (Strain A and Strain B) [13]. Interestingly, these structural variants also displayed differences in molecular properties and growth kinetics, and both Strain A and Strain B were different from recombinant SOD1 fibrils generated in vitro under a variety of conditions. The authors also determined whether the Strain A and Strain B structural features would be inherited when passaged in G85R-overexpressing transgenic mice [17]. Spinal cord homogenates from diseased mice expressing G85R (Strain A) or D90A (Strain B) were injected into the spinal cords of G85R overexpressing mice, and following disease onset, epitope mapping was performed on their spinal cords. Supporting the notion of conformation-dependent templated propagation, the induced aggregates had the same immunohistochemical profile as the injected SOD1 prions [17]. Together, this data further demonstrates the structural heterogeneity that misfolded SOD1 can adopt and the consequences of these variations on the course of disease, strongly implicating the existence and similarity of SOD1 strains to PrP strains.
Studies performed in our lab demonstrating the transmissible properties of SOD1, discussed earlier, also provided findings that implicated the existence of SOD1 strains. When recombinant WT SOD1 fibrils or homogenates prepared from paralyzed SOD1-overexpressing mice were injected into the permissive G85R-SOD1:YFP mouse line, distinct pathologies were observed [6, 7]. Mice that succumbed to disease following injection with homogenates prepared from paralyzed G93A-SOD1 mice developed round punctate G85R-SOD1:YFP inclusions, whereas mice injected with recombinant WT SOD1 fibrils developed distinct intracellular skein-like inclusions (Fig. 1). More importantly, when spinal cord homogenates prepared from these pathologically distinct mice were passaged a second time within naïve G85R-SOD1:YFP mice, the pathologic inclusions in the recipient mice retained the characteristics of the original donor mice (Fig. 1). Similarly, we have shown that fibrilized recombinant SOD1 can induce motor neuron disease in G85-SOD1:YFP mice and that mutation of the primary sequence of recombinant SOD1 in these fibrils can influence the morphology of inclusions that develop in paralyzed G85R-SOD1:YFP mice [34]. Therefore, the most plausible explanation for these finding is that the unique SOD1 conformations within these preparations are capable of templating these conformations to naïve G85R-SOD1: YFP protein, manifesting as distinct pathologies. This phenomenon is also observed for PrP prions in which heritable phenotypes are observed upon successive passages and occurs as a result of conformation-dependent templated propagation [16, 126].
Fig. 1.
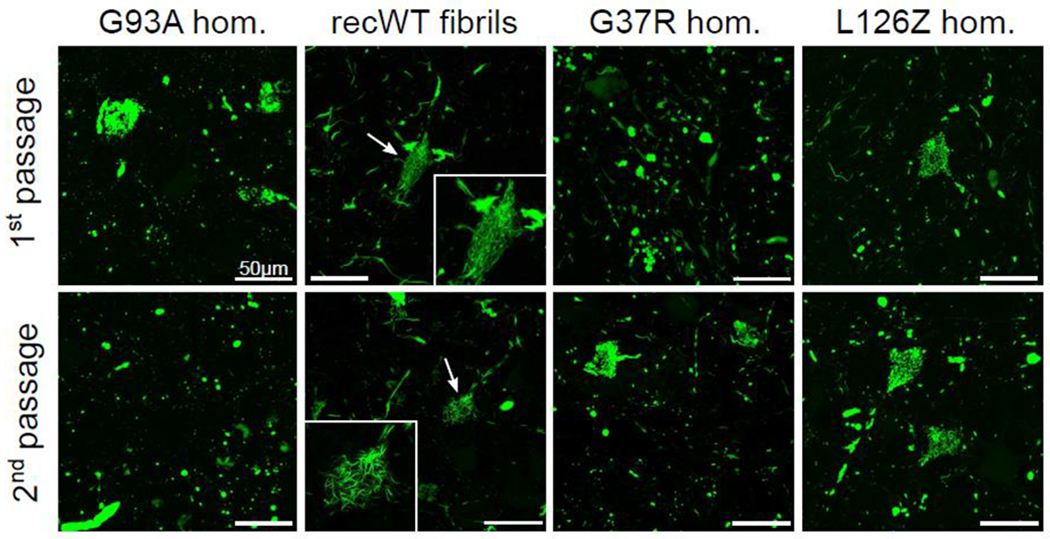
Distinct pathologies induced in G85R-SOD1:YFP inoculated mice. Images of spinal cords from diseased G85R-SOD1:YFP mice injected within their spinal cords at P0 with the first or second passages of the indicated homogenates. Significant differences were observed among those G85R-SOD1:YFP inclusions induced by homogenates from diseased G93A expressing mice versus those induced by recWT fibrils. These differences were retained among secondary passaging and is a strong indication of SOD1 conformational templating. Reprinted by permission from Springer Nature Customer Service Centre GmbH: [Springer] [Acta Neuropathologica] (Distinct conformers of transmissible misfolded SOD1 distinguish human SOD1-FALS from other forms of familial and sporadic ALS, Ayers, J.I., et al., 2016 [6]
These transmission studies also revealed other prion-like characteristics of the misfolded SOD1, including shorter incubation periods upon second passage in recipient G85R-SOD1:YFP mice and increased penetrance [6]. For PrP prion strains, this phenomenon is termed “host adaptation” and is thought to occur due to the greater compatibility between the inocula “seed” and the host substrate when the sequences of both are identical [22]. Notably, in the PO injection paradigm, the second passage of inocula derived from G93A mice produced paralysis at ~3 months of age, whereas inocula derived from G37R mice produced paralysis at ~6 months of age [6]. This difference did not appear to be due to the titer of the SOD1 seeding species, as the seeding dose for both inocula appeared similar when tested in a G85R-SOD1:YFP organotypic spinal cord slice culture assay [6]. Interestingly, fALS-SOD1 patients carrying the G37R-SOD1 mutation have one of the longest disease durations (18.7 ± 11.4 years) among all other fALS-SOD1 mutations, whereas the G93A variant has one of the shortest (2.4 ± 1.4 years). Notably, in the mouse study, we observed a modulation of onset by sequence variant, whereas in human fALS the sequence variation appears to dictate disease duration. Understanding whether different fALS variants of SOD1 produce sequence-specific strain attributes that influence the evolution of disease requires further study with a greater number of SOD1 variants associated with long and short disease durations.
General properties of TDP-43
As mentioned previously, abnormal localization of the 43 kDa transactive response (TAR) DNA-binding protein (TDP-43) is observed in ~97% of all ALS cases, making it the most prevalent pathological hallmark in ALS; however, TDP-43 pathology is not limited to ALS. It is also observed in other neurodegenerative disorders, including frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U) [94], AD [73], chronic traumatic encephalopathy (CTE) [89], and cerebral age-related TDP-43 [92]. This 414 amino-acid protein is encoded by the highly conserved TARDBP gene, and is ubiquitously expressed and localized primarily within the nucleus where it has multiple roles in RNA metabolism, including splicing regulation, trafficking, and degradation [25, 71]. Although mainly localized to the nucleus, the protein contains both a nuclear localization signal (NLS) and a nuclear export signal (NES). It also contains two tandem RNA-recognition motifs (RRM) and a C-terminal low complexity domain (LCD), or prion-like domain (PrLD), that is glycine-rich and appears to be of key importance to the intrinsic aggregation propensity of the protein.
Although little is still known about the aggregation pathway for TDP-43, its N-terminal region has been observed to play a regulatory role in the formation of different types of aggregates while the LCD, under certain experimental conditions, undergoes liquid-liquid phase separation that has been hypothesized to be the precursor to fibril formation [10, 32, 88, 124]. TDP-43 inclusions are hyperphosphorylated, ubiquitinated, and accumulate intracellularly in both upper and lower motor neurons and in some glial cells [94]. In those neurons containing TDP-43 inclusions, the nuclear TDP-43 is depleted, indicating that the loss of its normal function in RNA metabolism may contribute to toxicity [55, 94]. Endogenous nuclear TDP-43 is known to be tightly regulated and appears to be critical for survival. Overexpression in yeast is toxic, and overexpression in the CNS of mice and rats causes neuronal degeneration [70, 125, 139], whereas knockout of TARDBP in mice led to embryonic lethality [80]. However, similar to SOD1, mutations in TARDBP that have been associated with ALS are dominantly inherited, implicating a gain-of-toxic property. TARDBP mutations have been found in ~5% of fALS and ~1% sALS cases, where there is no known family history. There are now more than 50 mutations that have been identified, and they are primarily localized to the C-terminal LCD (http://alsod.iop.kcl.ac.uk/). These findings highlight the uncertainty that still exists as to whether TDP-43 associated toxicity is caused by a gain-of-toxic function, loss-of-toxic function, a combination of the two, or an unknown mechanism.
It is becoming increasingly evident that the C-terminal portion of the protein plays a critical role in the aggregation and toxicity of TDP-43. The PrLD or LCD is found in a number of other proteins, many of which are also RNA/DNA binding proteins and implicated in the pathogenesis of various neurodegenerative diseases [64]. In yeast proteins containing a PrLD, it has been demonstrated that this region can switch from a disordered conformation to a self-templating, cross-β-sheet-rich amyloid-like conformation, and similarly, the PrLD in TDP-43 was found to be intrinsically disordered and crucial for aberrant protein aggregation in vitro and in vivo [3, 71, 84]. Furthermore, toxicity associated with TDP-43 was found to be eliminated when the PrLD was deleted; however, this was observed when the RNA-binding ability of TDP-43 was eliminated as well [3, 70, 131]. Biochemical characterization of the insoluble fractions of ALS patient tissue has also demonstrated that C-terminal fragments of TDP-43 are produced ranging in size from ~25-35 kDa and that these fragments contain a primarily β-sheet-rich conformation and are highly amyloidogenic and cytotoxic [62, 94, 141, 142].
TDP-43 aggregation and conformational templating
Given that TDP43 exhibits biochemical properties that overlap with prions, studies began to elucidate its ability to template misfolding of the normally folded TDP-43 protein. The first demonstration of this utilized recombinant WT and sarkosyl-insoluble aggregates of TDP-43, which were observed to be taken up by cultured cells and also induce the aggregation of endogenous, intracellular TDP-43 [49]. Nonaka et al. then isolated sarkosyl-insoluble TDP-43 from ALS and FTLD-TDP brains and demonstrated that when added to a neuronal cell line overexpressing TDP-43, the cells accumulated phosphorylated, ubiquitinated, and fragmented cytoplasmic inclusions of TDP-43, recapitulating the features of pathological TDP-43 inclusions in the brains of patients [97]. The induced TDP-43 inclusions were also capable of inducing subsequent pathology when re-administered back onto naïve TDP-43 overexpressing neurons. Smethurst et al. also demonstrated the ability for TDP-43 isolates to induce TDP-43 aggregation in vitro through the addition of brain and spinal cord extracts from ALS patients to cells overexpressing TDP-43 [122]. Although modest, they also demonstrated the spread of TDP-43 aggregates to neighboring cells. This spread was also investigated in primary neurons cultured in microfluidic devices and shown to undergo both anterograde and retrograde transport [44]. Together, this data implicates TDP-43 as an additional prion protein capable of inducing misfolding and aggregation via template-dependent polymerization.
Successful studies demonstrating the in vivo transmission of TDP-43 pathology is limited to one recent study by Porta et al. [102]. As discussed previously, the successful demonstration of in vivo transmissibility of prions is very dependent on the animal models available, and, unfortunately, due to the tight regulation of endogenous TDP-43, the majority of TDP-43 transgenic models that overexpress the protein have led to an aggressive toxic phenotype [128]. To circumvent this issue, Porta et al. utilized a doxycycline-regulatable transgenic mouse model expressing a cytoplasmic NLS mutant of human TDP-43 that had been previously described and develops little to no TDP-43 pathology throughout its lifespan [2, 68]. The authors induced transgene expression one week prior to intracerebrally injecting the mice with TDP-43 enriched samples prepared from FTD patients and found the induction of de novo TDP-43 pathology at just one-month post-induction [102]. In addition, the localization of TDP-43 pathology over time indicated the spread of the infectious agent via connected neuroanatomical regions of the brain, similar to propagative spread observed for SOD1 and other prions. Interestingly, injection of these same samples into non-transgenic mice induced aggregation of endogenous mouse TDP-43, albeit at a much lower level.
TDP-43 strains
As mentioned, TDP-43 pathology is observed in the majority of ALS cases along with many other neurodegenerative disorders, including FTD. Cases of FTD that contain TDP-43 inclusions are referred to as FTLD-TDP and account for approximately half of all FTD cases. These cases can also present with different cognitive deficits such as behavioral or language impairments. Because these cases indicate the potential presence of strains, the variation in the distribution and morphology of TDP-43 species has been stratified into at least four subtypes (types A-E) [82, 86]. Type A neuropathology is primarily seen in FTLD-TDP patients that have mutations in the gene encoding progranulin (PGRN) and is defined by abundant dystrophic neurites and crescent or oval shaped neuronal cytoplasmic inclusions (NCIs) primarily in cortical layer 2. Type B is observed most commonly in patients with ALS and FTD and presents with mild levels of NCIs in all cortical layers and displays few short dystrophic neurites. Type C neuropathology is associated with sporadic disease and is observed most often in the semantic dementia variant of FTLD-TDP. TDP-43 pathology in type C is characterized by numerous long dystrophic neurites that are primarily found in cortical layer 2. Type D is observed in patients carrying mutations in the gene for the valosin-containing protein (VCP), which gives rise to inclusion body myopathy with early onset Paget’s disease and FTD (IBMPFTD). The pathology within these patients consists of short dystrophic neurites, lentiform neuronal intranuclear inclusions, and few NCIs within all layers of the cortex. Lastly, type E pathology is associated with a rapidly progressive subtype termed behavioral variant FTD (bvFTD) that has no known genetic cause. TDP-43 pathology in these cases has a wide neuroanatomic distribution that consists of ubiquitin-negative granulofilamentous neuronal inclusions, fine grey matter grains, and oligodendroglial inclusions. In addition to the variation in clinical and pathological phenotypes, hyperphosphorylated, sarkosyl-insoluble TDP-43 fractions isolated from brain homogenates of each of these subtypes reveal distinct western blot banding patterns of the C-terminal fragments, suggesting that different conformations of TDP-43 exist in the brains of these patients [65]. Together, this data demonstrates the variability that exists among ALS and FTLD-TDP patients in respect to their clinical symptoms, TDP-43 pathology, and western blot banding patterns, yet it also reveals the correlation that is observed among five distinct subtypes. This strongly implicates the existence of TDP-43 strains as a cause for some of the variability observed.
Although only a handful of in vitro studies have investigated TDP-43’s prion-like characteristics, one of these studies describes results that strongly support the existence of TDP-43 strains. By adding extracts from ALS patients to HEK293 cells overexpressing WT TDP-43, Smethurst et al. demonstrated that a range of morphologically distinct TDP-43 aggregates were observed following immunostaining [122]. These aggregates displayed morphologies reminiscent of those observed in ALS patients – including skein, round, dot-like, and granular inclusions. Although the authors go on to reveal that extracts from these cells could then induce naïve cells through serial passages, they do not describe the resultant TDP-43 morphologies and whether they resemble those from the primary passage, perhaps due to the heterogeneity of the morphologies within a given sample.
Prion properties of other ALS-linked proteins
There is mounting evidence that other proteins implicated to have a role in ALS also have properties akin to prion proteins, though they have not been studied to the extent of SOD1 and TDP-43. These include FUS and the dipeptide repeat (DPR) proteins produced in response to the repeat expansion in C90RF72. FUS was discovered shortly after the identification of TDP-43, and mutations within the gene account for ~4% of fALS patients [81, 130]. Similar to TDP-43, FUS is an RNA binding protein that plays a critical role in transcriptional regulation, RNA metabolism, and, potentially, DNA repair [12, 48, 136]. It also has many of the same domains as TDP-43 including a NES, NLS, and a disordered PrLD; however, unlike TDP-43, the bulk of the identified mutations (predominantly frameshift deletions) have not been observed in the PrLD but rather in an arginine-glycine-glycine (RGG) rich region (http://alsod.iop.kcl.ac.uk/). In patients carrying mutants of FUS, the protein forms cytoplasmic inclusions in neurons and glial cells within the brain and spinal cord whereas the nuclear localization is either partially or completely abrogated [93, 130]. In regard to the characteristics and mechanisms of FUS aggregation and transmissibility, there have only been a couple of studies to date. Nomura et al. demonstrated the increased propensity for purified recombinant FUS containing the G156E mutation, but not WT, to spontaneously form fibrils and revealed the ability for these fibrils to induce the aggregation of the WT FUS protein [96]. In addition, they demonstrated that when co-transfected into rat hippocampal primary neurons, the G156E mutant FUS was capable of recruiting and inducing the aggregation of WT FUS [96]. Another study utilized Drosophila primary neuronal cultures to reveal the ability of mutant forms of FUS to transfer between neurons, revealing another prion-like feature of the protein [45]. There are many additional studies investigating the ability of FUS to form liquid-liquid phase separation and how this contributes to its gain and/or loss of function, including its role in the formation of pathological cytoplasmic inclusions [75, 95, 100]. While these findings have begun to attribute prion properties to FUS, further studies will need to be completed to better understand its characteristics and whether misfolded FUS is capable of acquiring multiple conformations.
An intronic hexanucleotide GGGGCC (G4C2) repeat expansion within the first intron of the C9ORF72 gene is one of the more recently found genes to have a role in ALS and FTD and is now considered to be the most frequent genetic cause of ALS with ~40% of fALS and 5-20% of sALS patients carrying the expansion [38, 110, 111]. In addition to the toxic mechanisms that may arise due to the production of these DPRs [90, 143], there is also the theory that toxicity comes about from the loss of normal C9orf72 function [38] or a gain-of-toxic function caused by the formation of expanded toxic RNA species [39, 53]. Though there is data that has revealed the toxicity associated with five different DPRs produced from translation of the repeat RNA (poly-GR, poly-GA, poly-GP, poly-PA, and poly-PR), there are limited studies investigating their prion-like properties. One such study demonstrated that all of these DPRs, with the exception of poly-PR, was capable of undergoing cell-to-cell transmission via exosome-dependent and exosome-independent pathways in vitro [138].
Conclusion
With an ever-increasing amount of data, it is now becoming obvious that the proteins responsible for many neurodegenerative diseases share numerous properties with PrP, leading some to classify them as prions (Table 1). The ALS-linked proteins discussed here are no exception. Though studies have implicated distinct conformers of these toxic proteins as the basis for disease heterogeneity, the recent advances in technologies like cryo-EM now give researchers the ability to assess the structures of protein fibrils isolated from the brains of patients at the atomic scale. These techniques, as they are now doing for tauopathies, will help to reveal the range of misfolded conformations SOD1 and TDP-43, among others, can adopt and provide insight on the complexity of these diseases. These will be important studies for therapeutic strategies for ALS as they will begin to elucidate how stratified the patient population is and whether targeted therapies to the misfolded protein will be useful among a range of protein conformers.
Table 1.
Prion-like properties of ALS-linked proteins
| Protein/Gene | Seeded Aggregation | Spread | Inducible MND in mice | Strain properties | |||||
|---|---|---|---|---|---|---|---|---|---|
| Cell-free | Cell culture | In vivo | Cell-culture | In vivo | Recombinant protein | Murine tissue lysate | Human tissue lysate | ||
| SOD1 | [27, 28, 50] | [59, 60, 91] | [7] | [59, 60, 91] | [8] | [6] | [7, 17] | [6, 40] | [6, 17] |
| TDP-43 | [49, 71] | [44, 49, 97, 122] | [102] | [44, 97, 122] | [102] | n.d.1 | n.d. | [102] | [65, 102, 122] |
| FUS/TLS | [96] | [96] | n.d. | [45] | n.d. | n.d. | n.d. | n.d. | n.d. |
| C9orf72 | n.d. | n.d. | n.d. | [138] | n.d. | n.d. | n.d. | n.d. | n.d. |
not done
Acknowledgements
This work was supported by a grant from the National Institutes of Neurological Disease and Stroke (1R01NA092788-01).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References
- 1.Al-Chalabi A, Hardiman O (2013) The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol 9: 617–628 Doi 10.1038/nrneurol.2013.203 [DOI] [PubMed] [Google Scholar]
- 2.Alfieri JA, Pino NS, Igaz LM (2014) Reversible behavioral phenotypes in a conditional mouse model of TDP-43 proteinopathies. J Neurosci 34: 15244–15259 Doi 10.1523/JNEUROSCI.1918-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ash PE, Zhang YJ, Roberts CM, Saldi T, Hutter H, Buratti E, Petrucelli L, Link CD (2010) Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum Mol Genet 19: 3206–3218 Doi 10.1093/hmg/ddq230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atarashi R, Moore RA, Sim VL, Hughson AG, Dorward DW, Onwubiko HA, Priola SA, Caughey B (2007) Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat Methods 4: 645–650 Doi 10.1038/nmeth1066 [DOI] [PubMed] [Google Scholar]
- 5.Ayers J, Xu G, Pletnikova O, Troncoso JC, Hart PJ, Borchelt DR (2014) Conformational specificity of the C4F6 SOD1 antibody; low frequency of reactivity in sporadic ALS cases. Acta Neuropathologica Communications 2: 55 Doi 10.1186/2051-5960-2-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ayers JI, Diamond J, Sari A, Fromholt S, Galaleldeen A, Ostrow LW, Glass JD, Hart PJ, Borchelt DR (2016) Distinct conformers of transmissible misfolded SOD1 distinguish human SOD1-FALS from other forms of familial and sporadic ALS. Acta Neuropathol 132: 827–840 Doi 10.1007/s00401-016-1623-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ayers JI, Fromholt S, Koch M, DeBosier A, McMahon B, Xu G, Borchelt DR (2014) Experimental transmissibility of mutant SOD1 motor neuron disease. Acta Neuropathol 128: 791–803 Doi 10.1007/s00401-014-1342-7 [DOI] [PubMed] [Google Scholar]
- 8.Ayers JI, Fromholt SE, O’Neal VM, Diamond JH, Borchelt DR (2016) Prion-like propagation of mutant SOD1 misfolding and motor neuron disease spread along neuroanatomical pathways. Acta Neuropathol 131: 103–114 Doi 10.1007/s00401-015-1514-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ayers JI, Schutt CR, Shikiya RA, Aguzzi A, Kincaid AE, Bartz JC (2011) The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathog 7: e1001317 Doi 10.1371/journal.ppat.1001317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Babinchak WM, Haider R, Dumm BK, Sarkar P, Surewicz K, Choi JK, Surewicz WK (2019) The role of liquid-liquid phase separation in aggregation of the TDP-43 low-complexity domain. J Biol Chem 294: 6306–6317 Doi 10.1074/jbc.RA118.007222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banci L, Bertini I, Cantini F, Kozyreva T, Massagni C, Palumaa P, Rubino JT, Zovo K (2012) Human superoxide dismutase 1 (hSOD1) maturation through interaction with human copper chaperone for SOD1 (hCCS). Proc Natl Acad Sci U S A 109: 13555–13560 Doi 10.1073/pnas.1207493109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belly A, Moreau-Gachelin F, Sadoul R, Goldberg Y (2005) Delocalization of the multifunctional RNA splicing factor TLS/FUS in hippocampal neurones: exclusion from the nucleus and accumulation in dendritic granules and spine heads. Neurosci Lett 379: 152–157 Doi 10.1016/j.neulet.2004.12.071 [DOI] [PubMed] [Google Scholar]
- 13.Bergh J, Zetterstrom P, Andersen PM, Brannstrom T, Graffmo KS, Jonsson PA, Lang L, Danielsson J, Oliveberg M, Marklund SL (2015) Structural and kinetic analysis of protein-aggregate strains in vivo using binary epitope mapping. Proc Natl Acad Sci U S A 112: 4489–4494 Doi 10.1073/pnas.1419228112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bessen RA, Kocisko DA, Raymond GJ, Nandan S, Lansbury PT, Caughey B (1995) Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature 375: 698–700 Doi 10.1038/375698a0 [DOI] [PubMed] [Google Scholar]
- 15.Bessen RA, Marsh RF (1992) Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. Journal of Virology 66: 2096–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bessen RA, Marsh RF (1992) Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. The Journal of general virology 73 ( Pt 2): 329–334 [DOI] [PubMed] [Google Scholar]
- 17.Bidhendi EE, Bergh J, Zetterstrom P, Andersen PM, Marklund SL, Brannstrom T (2016) Two superoxide dismutase prion strains transmit amyotrophic lateral sclerosis-like disease. J Clin Invest 126: 2249–2253 Doi 10.1172/JCI84360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bolton DC, McKinley MP, Prusiner SB (1982) Identification of a protein that purifies with the scrapie prion. Science (New York, NY) 218: 1309–1311 [DOI] [PubMed] [Google Scholar]
- 19.Borchelt DR, Lee MK, Slunt HS, Guamieri M, Xu ZS, Wong PC, Brown RH, Price DL, Sisodia SS, Cleveland DW (1994) Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proceedings of the National Academy of Sciences of the United States of America 91: 8292–8296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D et al. (2010) Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci 13: 1396–1403 Doi 10.1038/nn.2660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brotherton TE, Li Y, Cooper D, Gearing M, Julien J-P, Rothstein JD, Boylan K, Glass JD (2012) Localization of a toxic form of superoxide dismutase 1 protein to pathologically affected tissues in familial ALS. Proceedings of the National Academy of Sciences of the United States of America: Doi 10.1073/pnas.1115009109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bruce M, Chree A, McConnell I, Foster J, Pearson G, Fraser H (1994) Transmission of bovine spongiform encephalopathy and scrapie to mice: strain variation and the species barrier. Philos Trans R Soc Lond B Biol Sci 343: 405–411 Doi 10.1098/rstb.1994.0036 [DOI] [PubMed] [Google Scholar]
- 23.Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW (1998) Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science (New York, NY) 281: 1851–1854 [DOI] [PubMed] [Google Scholar]
- 24.Bruns CK, Kopito RR (2007) Impaired post-translational folding of familial ALS-linked Cu, Zn superoxide dismutase mutants. EMBO J 26: 855–866 Doi 10.1038/sj.emboj.7601528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buratti E, Baralle FE (2001) Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem 276: 36337–36343 Doi 10.1074/jbc.M104236200 [DOI] [PubMed] [Google Scholar]
- 26.Cao X, Antonyuk SV, Seetharaman SV, Whitson LJ, Taylor AB, Holloway SP, Strange RW, Doucette PA, Valentine JS, Tiwari A et al. (2008) Structures of the G85R variant of SOD1 in familial amyotrophic lateral sclerosis. J Biol Chem 283: 16169–16177 Doi 10.1074/jbc.M801522200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chattopadhyay M, Durazo A, Sohn SH, Strong CD, Gralla EB, Whitelegge JP, Valentine JS (2008) Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc Natl Acad Sci U S A 105: 18663–18668 Doi 10.1073/pnas.0807058105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chia R, Tattum MH, Jones S, Collinge J, Fisher EM, Jackson GS (2010) Superoxide dismutase 1 and tgSOD1 mouse spinal cord seed fibrils, suggesting a propagative cell death mechanism in amyotrophic lateral sclerosis. PLoS One 5: el0627 Doi 10.1371/journal.pone.0010627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiò A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, Traynor BG, On Behalf Of The Eurals C (2009) Prognostic factors in ALS: A critical review. Amyotrophic Lateral Sclerosis 10: 310–323 Doi 10.3109/17482960802566824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M et al. (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 11: 909–913 Doi 10.1038/ncb1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collinge J, Sidle KC, Meads J, Ironside J, Hill AF (1996) Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 383: 685–690 Doi 10.1038/383685a0 [DOI] [PubMed] [Google Scholar]
- 32.Conicella AE, Zerze GH, Mittal J, Fawzi NL (2016) ALS Mutations Disrupt Phase Separation Mediated by alpha-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 24: 1537–1549 Doi 10.1016/j.str.2016.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crapo JD, Oury T, Rabouille C, Slot JW, Chang LY (1992) Copper,zinc superoxide dismutase is primarily a cytosolic protein in human cells. Proceedings of the National Academy of Sciences of the United States of America 89: 10405–10183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crown A, McAlary L, Fagerli E, Brown H, Yerbury JJ, Galaleldeen A, Cashman NR, Borchelt DR, Ayers JI (2020) Tryptophan residue 32 in human Cu-Zn superoxide dismutase modulates prion-like propagation and strain selection. PLoS One 15: e0227655 Doi 10.1371/journal.pone.0227655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Da Cruz S, Bui A, Saberi S, Lee SK, Stauffer J, McAlonis-Downes M, Schulte D, Pizzo DP, Parone PA, Cleveland DW et al. (2017) Misfolded SOD1 is not a primary component of sporadic ALS. Acta Neuropathol 134: 97–111 Doi 10.1007/s00401-017-1688-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dearmond SJ, McKinley MP, Barry RA, Braunfeld MB, McColloch JR, Prusiner SB (1985) Identification of prion amyloid filaments in scrapie-infected brain. Cell 41: 221–235 [DOI] [PubMed] [Google Scholar]
- 37.Dearmond SJ, Yang SL, Lee A, Bowler R, Taraboulos A, Groth D, Prusiner SB (1993) Three scrapie prion isolates exhibit different accumulation patterns of the prion protein scrapie isoform. Proceedings of the National Academy of Sciences of the United States of America 90: 6449–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J et al. (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72: 245–256 Doi 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM et al. (2013) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80: 415–428 Doi 10.1016/j.neuron.2013.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ekhtiari Bidhendi E, Bergh J, Zetterstrom P, Forsberg K, Pakkenberg B, Andersen PM, Marklund SL, Brannstrom T (2018) Mutant superoxide dismutase aggregates from human spinal cord transmit amyotrophic lateral sclerosis. Acta Neuropathol 136: 939–953 Doi 10.1007/s00401-018-1915-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, Crowther RA, Ghetti B, Scheres SHW, Goedert M (2018) Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 561: 137–140 Doi 10.1038/s41586-018-0454-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Falcon B, Zhang W, Schweighauser M, Murzin AG, Vidal R, Garringer HJ, Ghetti B, Scheres SHW, Goedert M (2018) Tau filaments from multiple cases of sporadic and inherited Alzheimer’s disease adopt a common fold. Acta Neuropathol 136: 699–708 Doi 10.1007/s00401-018-1914-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R, Crowther RA, Newell KL, Ghetti B, Goedert M et al. (2019) Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 568: 420–423 Doi 10.1038/s41586-019-1026-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feiler MS, Strobel B, Freischmidt A, Helferich AM, Kappel J, Brewer BM, Li D, Thal DR, Walther P, Ludolph AC et al. (2015) TDP-43 is intercellularly transmitted across axon terminals. J Cell Biol 211: 897–911 Doi 10.1083/jcb.201504057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feuillette S, Delarue M, Riou G, Gaffuri A-L, Wu J, Lenkei Z, Boyer O, Frébourg T, Campion D, Lecourtois M (2017) Neuron-to-Neuron Transfer of FUS in Drosophila Primary Neuronal Culture Is Enhanced by ALS-Associated Mutations. Journal of Molecular Neuroscience 62: 114–122 Doi 10.1007/s12031-017-0908-y [DOI] [PubMed] [Google Scholar]
- 46.Field LS, Furukawa Y, O’Halloran TV, Culotta VC (2003) Factors controlling the uptake of yeast copper/zinc superoxide dismutase into mitochondria. J Biol Chem 278: 28052–28059 Doi 10.1074/jbc.M304296200 [DOI] [PubMed] [Google Scholar]
- 47.Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, Scheres SHW (2017) Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547: 185–190 Doi 10.1038/nature23002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fujii R, Takumi T (2005) TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J Cell Sci 118: 5755–5765 Doi 10.1242/jcs.02692 [DOI] [PubMed] [Google Scholar]
- 49.Furukawa Y, Kaneko K, Watanabe S, Yamanaka K, Nukina N (2011) A seeding reaction recapitulates intracellular formation of Sarkosyl-insoluble transactivation response element (TAR) DNA-binding protein-43 inclusions. J Biol Chem 286: 18664–18672 Doi 10.1074/jbc.M111.231209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Furukawa Y, Kaneko K, Yamanaka K, O’Halloran TV, Nukina N (2008) Complete loss of post-translational modifications triggers fibrillar aggregation of SOD1 in the familial form of amyotrophic lateral sclerosis. J Biol Chem 283: 24167–24176 Doi 10.1074/jbc.M802083200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Furukawa Y, Torres AS, O’Halloran TV (2004) Oxygen-induced maturation of SOD1: a key role for disulfide formation by the copper chaperone CCS. EMBO J 23: 2872–2881 Doi 10.1038/sj.emboj.7600276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gajdusek DC (1977) Unconventional viruses and the origin and disappearance of kuru. Science (New York, NY) 197: 943–960 [DOI] [PubMed] [Google Scholar]
- 53.Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PE, Caulfield T, Daughrity L, Dunmore JH, Castanedes-Casey M, Chew J et al. (2013) Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol 126: 829–844 Doi 10.1007/s00401-013-1192-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gertz B, Wong M, Martin LJ (2012) Nuclear localization of human SOD1 and mutant SOD1-specific disruption of survival motor neuron protein complex in transgenic amyotrophic lateral sclerosis mice. J Neuropathol Exp Neurol 71: 162–177 Doi 10.1097/NEN.0b013e318244b635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Giordana MT, Piccinini M, Grifoni S, De Marco G, Vercellino M, Magistrello M, Pellerino A, Buccinna B, Lupino E, Rinaudo MT (2010) TDP-43 redistribution is an early event in sporadic amyotrophic lateral sclerosis. Brain Pathol 20: 351–360 Doi 10.1111/j.1750-3639.2009.00284.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goedert M, Falcon B, Zhang W, Ghetti B, Scheres SHW (2018) Distinct Conformers of Assembled Tau in Alzheimer’s and Pick’s Diseases. Cold Spring Fiarb Symp Quant Biol 83: 163–171 Doi 10.1101/sqb.2018.83.037580 [DOI] [PubMed] [Google Scholar]
- 57.Goldfarb LG, Petersen RB, Tabaton M, Brown P, LeBlanc AC, Montagna P, Cortelli P, Julien J, Vital C, Pendelbury WW (1992) Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism. Science (New York, NY) 258: 806–808 [DOI] [PubMed] [Google Scholar]
- 58.Gonzalez L, Martin S, Jeffrey M (2003) Distinct profiles of PrP(d) immunoreactivity in the brain of scrapie- and BSE-infected sheep: implications for differential cell targeting and PrP processing. J Gen Virol 84: 1339–1350 Doi 10.1099/vir.0.18800-0 [DOI] [PubMed] [Google Scholar]
- 59.Grad LI, Guest WC, Yanai A, Pokrishevsky E, O’Neill MA, Gibbs E, Semenchenko V, Yousefi M, Wishart DS, Plotkin SS et al. (2011) Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proceedings of the National Academy of Sciences of the United States of America 108: 16398–16403 Doi 10.1073/pnas.1102645108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grad LI, Yerbury JJ, Turner BJ, Guest WC, Pokrishevsky E, O’Neill MA, Yanai A, Silverman JM, Zeineddine R, Corcoran L et al. (2014) Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc Natl Acad Sci U S A 111: 3620–3625 Doi 10.1073/pnas.1312245111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Griffith JS (1967) Self-replication and scrapie. Nature 215: 1043–1044 [DOI] [PubMed] [Google Scholar]
- 62.Guo W, Chen Y, Zhou X, Kar A, Ray P, Chen X, Rao EJ, Yang M, Ye H, Zhu L et al. (2011) An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat Struct Mol Biol 18: 822–830 Doi 10.1038/nsmb.2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX (1994) Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science (New York, NY) 264: 1772–1775 [DOI] [PubMed] [Google Scholar]
- 64.Harrison AF, Shorter J (2017) RNA-binding proteins with prion-like domains in health and disease. Biochem J 474: 1417–1438 Doi 10.1042/BCJ20160499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hasegawa M, Nonaka T, Tsuji H, Tamaoka A, Yamashita M, Kametani F, Yoshida M, Arai T, Akiyama H (2011) Molecular dissection of TDP-43 proteinopathies. J Mol Neurosci 45: 480–485 Doi 10.1007/s12031-011-9571-x [DOI] [PubMed] [Google Scholar]
- 66.Hayward LJ, Rodriguez JA, Kim JW, Tiwari A, Goto JJ, Cabelli DE, Valentine JS, Brown RH Jr. (2002) Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J Biol Chem 277: 15923–15931 Doi 10.1074/jbc.M112087200 [DOI] [PubMed] [Google Scholar]
- 67.Hsiao KK, Groth D, Scott M, Yang SL, Serban H, Rapp D, Foster D, Torchia M, Dearmond SJ, Prusiner SB (1994) Serial transmission in rodents of neurodegeneration from transgenic mice expressing mutant prion protein. Proceedings of the National Academy of Sciences of the United States of America 91: 9126–9130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Igaz LM, Kwong LK, Lee EB, Chen-Plotkin A, Swanson E, Unger T, Malunda J, Xu Y, Winton MJ, Trojanowski JQ et al. (2011) Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J Clin Invest 121: 726–738 Doi 10.1172/JCI44867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jeffrey M, Martin S, Gonzalez L (2003) Cell-associated variants of disease-specific prion protein immunolabelling are found in different sources of sheep transmissible spongiform encephalopathy. J Gen Virol 84: 1033–1045 Doi 10.1099/vir.0.18825-0 [DOI] [PubMed] [Google Scholar]
- 70.Johnson BS, McCaffery JM, Lindquist S, Gitler AD (2008) A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc Natl Acad Sci U S A 105: 6439–6444 Doi 10.1073/pnas.0802082105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD (2009) TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem 284: 20329–20339 Doi 10.1074/jbc.M109.010264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnston JA, Dalton MJ, Gurney ME, Kopito RR (2000) Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 97: 12571–12576 Doi 10.1073/pnas.220417997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM, Petrucelli L, Senjem ML, Knopman DS, Boeve BF et al. (2014) TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol 127: 811–824 Doi 10.1007/s00401-014-1269-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jucker M, Walker LC (2013) Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501: 45–51 Doi 10.1038/nature12481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J et al. (2012) Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149: 753–767 Doi 10.1016/j.cell.2012.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kimberlin RH, Walker C (1977) Characteristics of a short incubation model of scrapie in the golden hamster. The Journal of general virology 34: 295–304 [DOI] [PubMed] [Google Scholar]
- 77.Kimberlin RH, Walker CA (1978) Evidence that the transmission of one source of scrapie agent to hamsters involves separation of agent strains from a mixture. The Journal of general virology 39: 487–496 [DOI] [PubMed] [Google Scholar]
- 78.Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, Caughey B (1994) Cell-free formation of protease-resistant prion protein. NeuroReport 370: 471–474 Doi doi: 10.1038/370471a0 [DOI] [PubMed] [Google Scholar]
- 79.Kovacs GG, Trabattoni G, Hainfellner JA, Ironside JW, Knight RS, Budka H (2002) Mutations of the prion protein gene phenotypic spectrum. J Neurol 249: 1567–1582 Doi 10.1007/s00415-002-0896-9 [DOI] [PubMed] [Google Scholar]
- 80.Kraemer BC, Schuck T, Wheeler JM, Robinson LC, Trojanowski JQ, Lee VM, Schellenberg GD (2010) Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol 119: 409–419 Doi 10.1007/s00401-010-0659-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kwiatkowski TJ Jr., Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T et al. (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323: 1205–1208 Doi 10.1126/science.1166066 [DOI] [PubMed] [Google Scholar]
- 82.Lee EB, Porta S, Michael Baer G, Xu Y, Suh E, Kwong LK, Elman L, Grossman M, Lee VM, Irwin DJ et al. (2017) Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol 134: 65–78 Doi 10.1007/s00401-017-1679-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Leigh PN, Anderton BH, Dodson A, Gallo JM, Swash M, Power DM (1988) Ubiquitin deposits in anterior horn cells in motor neurone disease. 93: 197–203 Doi 10.1016/0304-3940(88)90081-x [DOI] [PubMed] [Google Scholar]
- 84.Liebman SW, Chernoff YO (2012) Prions in yeast. Genetics 191: 1041–1072 Doi 10.1534/genetics.111.137760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lowe J, Lennox G, Jefferson D, Morrell K, McQuire D, Gray T, Landon M, Doherty FJ, Mayer RJ (1988) A filamentous inclusion body within anterior horn neurones in motor neurone disease defined by immunocytochemical localisation of ubiquitin. Neuroscience Letters 94: 203–210 Doi 10.1016/0304-3940(88)90296-0 [DOI] [PubMed] [Google Scholar]
- 86.Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DM, Lee VM (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122: 111–113 Doi 10.1007/s00401-011-0845-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marsh RF, Kimberlin RH (1975) Comparison of scrapie and transmissible mink encephalopathy in hamsters. II. Clinical signs, pathology, and pathogenesis. J Infect Dis 131: 104–110 Doi 10.1093/infdis/131.2.104 [DOI] [PubMed] [Google Scholar]
- 88.McGurk L, Gomes E, Guo L, Shorter J, Bonini NM (2018) Poly(ADP-ribose) Engages the TDP-43 Nuclear-Localization Sequence to Regulate Granulo-Filamentous Aggregation. Biochemistry 57: 6923–6926 Doi 10.1021/acs.biochem.8b00910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW, Perl DP, Hedley-Whyte ET, Price B, Sullivan C et al. (2010) TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 69: 918–929 Doi 10.1097/NEN.0b013e3181ee7d85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C et al. (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339: 1335–1338 Doi 10.1126/science.1232927 [DOI] [PubMed] [Google Scholar]
- 91.Munch C, O’Brien J, Bertolotti A (2011) Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc Natl Acad Sci U S A 108: 3548–3553 Doi 10.1073/pnas.1017275108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nelson PT, Trojanowski JQ, Abner EL, Al-Janabi OM, Jicha GA, Schmitt FA, Smith CD, Fardo DW, Wang WX, Kryscio RJ et al. (2016) “New Old Pathologies”: AD, PART, and Cerebral Age-Related TDP-43 With Sclerosis (CARTS). J Neuropathol Exp Neurol 75: 482–498 Doi 10.1093/jnen/nlw033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Neumann M, Roeber S, Kretzschmar HA, Rademakers R, Baker M, Mackenzie IR (2009) Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol 118: 605–616 Doi 10.1007/s00401-009-0581-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (New York, NY) 314: 130–133 Doi 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- 95.Niaki AG, Sarkar J, Cai X, Rhine K, Vidaurre V, Guy B, Hurst M, Lee JC, Koh HR, Guo L et al. (2020) Loss of Dynamic RNA Interaction and Aberrant Phase Separation Induced by Two Distinct Types of ALS/FTD-Linked FUS Mutations. Mol Cell 77: 82–94 e84 Doi 10.1016/j.molcel.2019.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nomura T, Watanabe S, Kaneko K, Yamanaka K, Nukina N, Furukawa Y (2014) Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J Biol Chem 289: 1192–1202 Doi 10.1074/jbc.M113.516492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nonaka T, Masuda-Suzukake M, Arai T, Hasegawa Y, Akatsu H, Obi T, Yoshida M, Murayama S, Mann DM, Akiyama H et al. (2013) Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep 4: 124–134 Doi 10.1016/j.celrep.2013.06.007 [DOI] [PubMed] [Google Scholar]
- 98.Outram GW (1976) The pathogenesis of scrapie in mice. Frontiers of biology 44: 325. [PubMed] [Google Scholar]
- 99.Pare B, Lehmann M, Beaudin M, Nordstrom U, Saikali S, Julien JP, Gilthorpe JD, Marklund SL, Cashman NR, Andersen PM et al. (2018) Misfolded SOD1 pathology in sporadic Amyotrophic Lateral Sclerosis. Sci Rep 8: 14223 Doi 10.1038/s41598-018-31773-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM et al. (2015) A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 162: 1066–1077 Doi 10.1016/j.cell.2015.07.047 [DOI] [PubMed] [Google Scholar]
- 101.Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van den Haute C, Melki R, Baekelandt V (2015) alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522: 340–344 Doi 10.1038/nature14547 [DOI] [PubMed] [Google Scholar]
- 102.Porta S, Xu Y, Restrepo CR, Kwong LK, Zhang B, Brown HJ, Lee EB, Trojanowski JQ, Lee VM (2018) Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat Commun 9: 4220 Doi 10.1038/s41467-018-06548-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Prudencio M (2009) Implication of Detergent-Insoluble Aggregates of Superoxide Dismutase 1 in Familial Amyotrophic Lateral Sclerosis. 1–252
- 104.Prudencio M, Hart PJ, Borchelt DR, Andersen PM (2009) Variation in aggregation propensities among ALS-associated variants of SOD1: Correlation to human disease. Human molecular genetics 18: 3217–3226 Doi 10.1093/hmg/ddp260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Prusiner SB (1978) An approach to the isolation of biological particles using sedimentation analysis. J Biol Chem 253: 916–921 [PubMed] [Google Scholar]
- 106.Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science (New York, NY) 216: 136–144 [DOI] [PubMed] [Google Scholar]
- 107.Prusiner SB, McKinley MP, Bowman KA, Bolton DC, Bendheim PE, Groth DF, Glenner GG (1983) Scrapie prions aggregate to form amyloid-like birefringent rods. Cell 35: 349–358 [DOI] [PubMed] [Google Scholar]
- 108.Ratovitski T, Corson LB, Strain J, Wong P, Cleveland DW, Culotta VC, Borchelt DR (1999) Variation in the biochemical/biophysical properties of mutant superoxide dismutase 1 enzymes and the rate of disease progression in familial amyotrophic lateral sclerosis kindreds. Human molecular genetics 8: 1451–1460 [DOI] [PubMed] [Google Scholar]
- 109.Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH et al. (1996) Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nature genetics 13: 43–47 Doi 10.1038/ng0596-43 [DOI] [PubMed] [Google Scholar]
- 110.Reisin R (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Yearbook of Neurology and Neurosurgery 2012: 170–172 Doi 10.1016/j.yneu.2012.05.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L et al. (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72: 257–268 Doi 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362: 59–62 Doi 10.1038/362059a0 [DOI] [PubMed] [Google Scholar]
- 113.Saberi S, Stauffer JE, Schulte DJ, Ravits J (2015) Neuropathology of Amyotrophic Lateral Sclerosis and Its Variants. Neurol Clin 33: 855–876 Doi 10.1016/j.ncl.2015.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Saborio GP, Permanne B, Soto C (2001) Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411: 810–813 Doi 10.1038/35081095 [DOI] [PubMed] [Google Scholar]
- 115.Sacino AN, Ayers JI, Brooks MM, Chakrabarty P, Hudson VJ 3rd, Howard JK, Golde TE, Giasson BI, Borchelt DR (2016) Non-prion-type transmission in A53T alpha-synuclein transgenic mice: a normal component of spinal homogenates from naive non-transgenic mice induces robust alpha-synuclein pathology. Acta Neuropathol 131: 151–154 Doi 10.1007/s00401-015-1505-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB (1998) Eight prion strains have PrP(Sc) molecules with different conformations. Nature medicine 4: 1157–1165 Doi 10.1038/2654 [DOI] [PubMed] [Google Scholar]
- 117.Sala FA, Wright GSA, Antonyuk SV, Garratt RC, Hasnain SS (2019) Molecular recognition and maturation of SOD1 by its evolutionarily destabilised cognate chaperone hCCS. PLoS Biol 17: e3000141 Doi 10.1371/journal.pbio.3000141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC, Thorpe JR, Serpell LC et al. (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82: 1271–1288 Doi 10.1016/j.neuron.2014.04.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sea K, Sohn SH, Durazo A, Sheng Y, Shaw BF, Cao X, Taylor AB, Whitson LJ, Holloway SP, Hart PJ et al. (2015) Insights into the role of the unusual disulfide bond in copper-zinc superoxide dismutase. J Biol Chem 290: 2405–2418 Doi 10.1074/jbc.M114.588798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shaw BF, Valentine JS (2007) How do ALS-associated mutations in superoxide dismutase 1 promote aggregation of the protein? Trends Biochem Sci 32: 78–85 Doi 10.1016/j.tibs.2006.12.005 [DOI] [PubMed] [Google Scholar]
- 121.Silverman JM, Fernando SM, Grad LI, Hill AF, Turner BJ, Yerbury JJ, Cashman NR (2016) Disease Mechanisms in ALS: Misfolded SOD1 Transferred Through Exosome-Dependent and Exosome-Independent Pathways. Cell Mol Neurobiol 36: 377–381 Doi 10.1007/s10571-015-0294-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Smethurst P, Newcombe J, Troakes C, Simone R, Chen YR, Patani R, Sidle K (2016) In vitro prion-like behaviour of TDP-43 in ALS. Neurobiol Dis 96: 236–247 Doi 10.1016/j.nbd.2016.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Stathopulos PB, Rumfeldt JA, Scholz GA, Irani RA, Frey HE, Hallewell RA, Lepock JR, Meiering EM (2003) Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis show enhanced formation of aggregates in vitro. Proc Natl Acad Sci U S A 100: 7021–7026 Doi 10.1073/pnas.1237797100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sun Y, Chakrabartty A (2017) Phase to Phase with TDP-43. Biochemistry 56: 809–823 Doi 10.1021/acs.biochem.6b01088 [DOI] [PubMed] [Google Scholar]
- 125.Tatom JB, Wang DB, Dayton RD, Skalli O, Hutton ML, Dickson DW, Klein RL (2009) Mimicking aspects of frontotemporal lobar degeneration and Lou Gehrig’s disease in rats via TDP-43 overexpression. Mol Ther 17: 607–613 Doi 10.1038/mt.2009.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Telling GC, Parchi P, Dearmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB (1996) Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science (New York, NY) 274: 2079–2082 [DOI] [PubMed] [Google Scholar]
- 127.Tokuda E, Takei YI, Ohara S, Fujiwara N, Hozumi I, Furukawa Y (2019) Wild-type Cu/Zn-superoxide dismutase is misfolded in cerebrospinal fluid of sporadic amyotrophic lateral sclerosis. Mol Neurodegener 14: 42 Doi 10.1186/s13024-019-0341-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tsao W, Jeong YH, Lin S, Ling J, Price DL, Chiang PM, Wong PC (2012) Rodent models of TDP-43: recent advances. Brain Res 1462: 26–39 Doi 10.1016/j.brainres.2012.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Valentine JS, Doucette PA, Zittin Potter S (2005) Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem 74: 563–593 Doi 10.1146/annurev.biochem.72.121801.161647 [DOI] [PubMed] [Google Scholar]
- 130.Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P et al. (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science (New York, NY) 323: 1208–1211 Doi 10.1126/science.1165942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Voigt A, Herholz D, Fiesel FC, Kaur K, Muller D, Karsten P, Weber SS, Kahle PJ, Marquardt T, Schulz JB (2010) TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PLoS One 5: e12247 Doi 10.1371/journal.pone.0012247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang J, Farr GW, Zeiss CJ, Rodriguez-Gil DJ, Wilson JH, Furtak K, Rutkowski DT, Kaufman RJ, Ruse CI, Yates JR et al. (2009) Progressive aggregation despite chaperone associations of a mutant SOD1-YFP in transgenic mice that develop ALS. Proceedings of the National Academy of Sciences of the United States of America 106: 1392–1397 Doi 10.1073/pnas.0813045106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang J, Slunt H, Gonzales V, Fromholt D, Coonfield M, Copeland NG, Jenkins NA, Borchelt DR (2003) Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum Mol Genet 12: 2753–2764 Doi 10.1093/hmg/ddg312 [DOI] [PubMed] [Google Scholar]
- 134.Wang J, Xu G, Borchelt DR (2002) High molecular weight complexes of mutant superoxide dismutase 1: age-dependent and tissue-specific accumulation. Neurobiol Dis 9: 139–148 Doi 10.1006/nbdi.2001.0471 [DOI] [PubMed] [Google Scholar]
- 135.Wang J, Xu G, Slunt HH, Gonzales V, Coonfield M, Fromholt D, Copeland NG, Jenkins NA, Borchelt DR (2005) Coincident thresholds of mutant protein for paralytic disease and protein aggregation caused by restrictively expressed superoxide dismutase cDNA. Neurobiol Dis 20: 943–952 Doi 10.1016/j.nbd.2005.06.005 [DOI] [PubMed] [Google Scholar]
- 136.Wang WY, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, Mackenzie IR, Huang EJ, Tsai LH (2013) Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat Neurosci 16: 1383–1391 Doi 10.1038/nn.3514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Watts JC, Giles K, Oehler A, Middleton L, Dexter DT, Gentleman SM, DeArmond SJ, Prusiner SB (2013) Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci U S A 110: 19555–19560 Doi 10.1073/pnas.1318268110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Westergard T, Jensen BK, Wen X, Cai J, Kropf E, Iacovitti L, Pasinelli P, Trotti D (2016) Cell-to-Cell Transmission of Dipeptide Repeat Proteins Linked to C9orf72-ALS/FTD. Cell Rep 17: 645–652 Doi 10.1016/j.celrep.2016.09.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, Smits V, Ceuterick-de Groote C, Van Broeckhoven C, Kumar-Singh S (2010) TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A 107: 3858–3863 Doi 10.1073/pnas.0912417107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhang W, Tarutani A, Newell KL, Murzin AG, Matsubara T, Falcon B, Vidal R, Garringer HJ, Shi Y, Ikeuchi T et al. (2020) Novel tau filament fold in corticobasal degeneration. Nature: Doi 10.1038/s41586-020-2043-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, Lin WL, Tong J, Castanedes-Casey M, Ash P et al. (2009) Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A 106: 7607–7612 Doi 10.1073/pnas.0900688106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhang YJ, Xu YF, Dickey CA, Buratti E, Baralle F, Bailey R, Pickering-Brown S, Dickson D, Petrucelli L (2007) Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci 27: 10530–10534 Doi 10.1523/JNEUROSCI.3421-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH et al. (2013) RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A 110: E4968–4977 Doi 10.1073/pnas.1315438110 [DOI] [PMC free article] [PubMed] [Google Scholar]