

Abstract
Mitochondria contain an autonomous and spatially segregated genome. The organizational unit of their genome is the nucleoid, which consists of mitochondrial DNA (mtDNA) and associated architectural proteins. Here, we show that phase separation is the primary physical mechanism for assembly and size control of the mitochondrial nucleoid (mt‐nucleoid). The major mtDNA‐binding protein TFAM spontaneously phase separates in vitro via weak, multivalent interactions into droplets with slow internal dynamics. TFAM and mtDNA form heterogenous, viscoelastic structures in vitro, which recapitulate the dynamics and behavior of mt‐nucleoids in vivo. Mt‐nucleoids coalesce into larger droplets in response to various forms of cellular stress, as evidenced by the enlarged and transcriptionally active nucleoids in mitochondria from patients with the premature aging disorder Hutchinson‐Gilford Progeria Syndrome (HGPS). Our results point to phase separation as an evolutionarily conserved mechanism of genome organization.
Keywords: biomolecular condensate, genome organization, mitochondrial genome, phase separation, TFAM
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics;
Mitochondrial nucleoids behave as biomolecular condensates, whose formation is primarily driven by interactions between the main packaging protein TFAM and mitochondrial DNA.
Introduction
Mitochondria are the major sites of cellular energy production through oxidative phosphorylation and generation of ATP. Within each mammalian cell, mitochondria contain several hundred copies of a 16.6 kb double‐stranded circular genome (Chen & Butow, 2005; Gustafsson, Falkenberg et al, 2016). Although small in size, the mitochondrial genome is gene dense, encoding essential polypeptides involved in mitochondrial respiration and oxidative phosphorylation (Burger, Gray et al, 2003). Unlike the nuclear genome, mtDNA is not organized by histones, but is packaged by a distinct set of proteins into nucleoprotein complexes to form mt‐nucleoids (Chen & Butow, 2005; Kukat & Larsson, 2013). These structures are typically uniformly ~100 nm in size, each containing 1–2 molecules of mtDNA, and are spatially separated throughout the mitochondrial network (Brown, Tkachuk et al, 2011; Kukat, Wurm et al, 2011). mt‐nucleoids lack any delimiting membranes, yet act as discrete functional units involved in replication and transcription of the mitochondrial genome (Kukat & Larsson, 2013).
The major mt‐nucleoid packaging protein in human cells is the mitochondrial transcription factor A (TFAM) (Chen & Butow, 2005), which binds and compacts mtDNA in vitro into nucleoid‐like structures under dilute conditions (Brewer, Friddle et al, 2003; Kaufman, Durisic et al, 2007; Farge, Mehmedovic et al, 2014; Kukat, Davies et al, 2015). TFAM contains two high mobility group (HMG) domains that each intercalate into the DNA double helix, bending the DNA strand to form a tight U‐turn structure at promoter sequences (Ngo, Kaiser et al, 2011; Rubio‐Cosials, Sydow et al, 2011). Moreover, TFAM can also form loops and can bind cross‐strands of DNA without sequence specificity (Kukat et al, 2015), and these conformations are further stabilized by cooperative TFAM‐TFAM interactions (Farge, Laurens et al, 2012). In human cells, TFAM is present at high enough concentrations to coat the entirety of the circular mtDNA, driving the compaction of mtDNA from ~5 µm in contour length to the ~100 nm mt‐nucleoid (Gustafsson et al, 2016). Beyond the direct binding of TFAM to mtDNA, it remains largely unknown how the higher‐order morphological features of the mitochondrial genome emerge, how they affect function, and how anomalies in the structure of mt‐nucleoids may contribute to disease (Friedman & Nunnari, 2014).
Maintenance of mt‐nucleoid structure is linked to mitochondrial organization and function. Disruption of mitochondrial fusion and fission processes affects mt‐nucleoid size, as seen in knockdown of mitochondrial fission GTPase, Drp1, which leads to clustering of mt‐nucleoids into large assemblages in hyperfused mitochondria (Ban‐Ishihara, Ishihara et al, 2013; Ishihara, Ban‐Ishihara et al, 2015). Similarly, downregulation of the inner mitochondrial membrane protein Mic60/Mitofilin leads to disassembly of the mitochondrial contact site and cristae organizing system (MICOS), ultimately resulting in enlarged, spherical mt‐nucleoids (Li, Ruan et al, 2016). Enlargement and remodeling of mt‐nucleoids are also associated with cellular stress responses. For example, prolonged exposure to DNA intercalating agents leads to altered mt‐nucleoid size distributions (Ashley & Poulton, 2009; Alán, Špaček et al, 2016), and viral infection results in aberrant sizes of mt‐nucleoids (West, Khoury‐Hanold et al, 2015). Given the oxidative environment within mitochondria (Balaban, Nemoto et al, 2005; Sun, Youle et al, 2016) and the absence of protective histone proteins, the mt‐nucleoid‐associated proteins have been hypothesized to contribute to mitochondrial genome integrity (Yakes & Van Houten, 1997; Cadenas & Davies, 2000). Mutations in mtDNA have direct physiological relevance as elevated mutation levels of mtDNA are associated with premature aging phenotypes (Trifunovic, Wredenberg et al, 2004; Kujoth, Hiona et al, 2005). Moreover, mtDNA mutations tend to accumulate over the course of normal aging (Bratic & Larsson, 2013; Sun et al, 2016) and even single point mutations in mtDNA can elicit a myriad of other disease phenotypes (Taylor & Turnbull, 2005).
An emerging organizational principle of non‐membrane bound cellular structures is phase separation (Hyman, Weber et al, 2014). Numerous ribonucleoprotein and nucleoprotein complexes spontaneously self‐assemble into non‐membrane bound cellular bodies, or biomolecular condensates, via phase separation (Banani, Lee et al, 2017). The canonical examples of RNA‐protein bodies include the nucleolus in the nucleus (Brangwynne, Mitchison et al, 2011; Feric, Vaidya et al, 2016) as well as P‐granules (Brangwynne, Eckmann et al, 2009) and stress granules in the cytoplasm (Molliex, Temirov et al, 2015; Guillén‐Boixet, Kopach et al, 2020; Sanders, Kedersha et al, 2020; Yang, Mathieu et al, 2020). In addition, DNA‐protein complexes can phase separate in the nucleus, such as the heterochromatin protein HP1α in the context of heterochromatin (Larson, Elnatan et al, 2017; Strom, Emelyanov et al, 2017), histones to form chromatin domains (Gibson, Doolittle et al, 2019; Sanulli, Trnka et al, 2019), or super‐enhancers which form active transcriptional hubs (Sabari, Dall'Agnese et al, 2018).
Here, we have explored the higher‐order organizational principles of the mitochondrial genome. We demonstrate, based on in vitro and in vivo observations, that mt‐nucleoids self‐assemble via phase separation. We find that the major mt‐nucleoid protein TFAM exerts its architectural role by promoting phase separation via weak, multivalent self‐interactions to generate the multiphasic mt‐nucleoid structure. We also demonstrate that aberrant mt‐nucleoid size is a consequence of phase behavior and is associated with mitochondrial dysfunction in the context of premature aging. Our observations suggest phase separation is an evolutionarily conserved mechanism in genome organization.
Results
Enlarged mitochondrial nucleoids in vivo
During the course of in‐depth analysis of cellular morphological changes in a model system of aging, we noticed the presence of aberrant mitochondria and enlarged mt‐nucleoids in skin fibroblasts from patients with Hutchinson–Gilford Progeria Syndrome (HGPS) (Fig 1). HGPS is a rare, invariably fatal premature aging disorder characterized by multi‐tissue symptoms, including of bone, muscle, skin, and cardiovascular failure. The disease is caused by a point mutation in LMNA resulting in the production of progerin, a dominant negative form of the major architectural protein lamin A (Gordon, Rothman et al, 2014). In line with mitochondrial abnormalities associated with HGPS (Rivera‐Torres, Acín‐Perez et al, 2013; Xiong, Choi et al, 2016), we find that ~70% of advanced HGPS patient cells had a sub‐population of mitochondria that were swollen, spherical in shape and isolated from the surrounding mitochondrial network compared to the typical tubular, elongated mitochondrial networks in control cells (Fig 1A and B; Appendix Fig S1E–K). The extent and number of enlarged mitochondria correlated with disease progression (Fig 1C; Appendix Fig S1K), and several chaperones and proteases of the mitochondrial unfolded protein response (UPRmt) including HSPD1 (mtHSP60), mtHSP10, mtHSP70, ClpP, and LONP1 were enriched in enlarged mitochondria, indicating that the altered mitochondrial morphology is associated with mitochondrial stress (Fig 1J–L; Appendix Fig S1R–W). Exogenous expression of progerin was sufficient to induce in wild‐type cells an increase in the number of aberrant mitochondria that scaled with progerin expression (Appendix Fig S1X–AF).
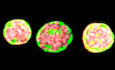
Figure 1. Enlarged mitochondrial nucleoids are prominent in a premature aging disease and can also arise from liquid‐like fusion events under stress.
-
A, BMaximum intensity projections of SIM images of fixed normal (A) and HGPS (B) human skin fibroblasts, where the mitochondria are labeled in magenta with MitoTracker Red, mtDNA in green with anti‐DNA, and the nucleus in blue with DAPI. Scale bar = 5 µm.
-
CBar graph quantifying the number of damaged mitochondria per cell based on high‐throughput imaging of two wild type and four HGPS primary skin fibroblast cell lines. Bars and error bars represent averages ± SEM for n = 3 independent experimental replicates (each experimental replicate had 15 technical replicates each containing 5 fields of view, approximately 2,000–5,000 total cells for each cell line analyzed), where P‐value for the ANOVA test statistic was P < 0.001. For individual pairs, **P < 0.01, ***P < 0.001.
-
D–GThree‐dimensional views of normal mitochondria (D) annotated by white box in (B) and swollen mitochondria (F) annotated by yellow box in (B) and showing TFAM localization in red with anti‐TFAM; the length of the box = 4 µm. (E, G) Normalized intensity distributions of mt‐nucleoids labeled with anti‐DNA and with anti‐TFAM corresponding to images from (D) and (F), respectively.
-
H, ITime‐course experiment of live HGPS cells under phototoxic conditions. Mitochondria were labeled with MitoTracker Deep Red (H, magenta), and mtDNA was labeled with PicoGreen (H,I, green). Scale bar = 2 µm. Arrow heads indicate pairs of mt‐nucleoids that undergo liquid‐like fusion events.
-
J, KSingle z‐slices of SIM images of UPRmt in mitochondria from fixed normal (J) and HGPS (K) human skin fibroblasts, where the UPRmt marker is gray‐scale with anti‐HSPD1 (mtHSP60), and mtDNA is in green with anti‐DNA. Scale bar = 1.5 µm.
-
LImmunofluorescence quantification of UPRmt markers (ClpP, LonPI, mtHSP10, mtHSP60, mtHSP70) in all six primary cell lines reported as normalized intensity values in undamaged and damaged mitochondria from high‐throughput confocal images, where n = 3 independent experimental replicates (each experimental replicate, containing three technical replicates, had a total of 150–600 cells for each cell line and UPRmt marker), and error bars are standard error.
Analysis by high‐resolution Structured Illumination Microscopy (SIM) imaging revealed the presence of enlarged mt‐nucleoids in morphologically aberrant mitochondria of HGPS cells (Fig 1F and G). In these mitochondria, mt‐nucleoids clustered together into structures that were considerably brighter and larger than the typically uniform mt‐nucleoids of ~100 nm found in normal mitochondria (Fig 1D and E). Several mt‐nucleoid markers, including mtDNA and the major mtDNA‐packaging protein, TFAM, were locally enriched in the atypical mitochondria (Fig 1F and G; Appendix Fig S1O and P), while total TFAM and mtDNA levels were not altered in HGPS cells (Appendix Fig S1L–N).
The formation of enlarged mt‐nucleoids is an indication of mitochondrial damage since they could also be induced in vivo when we exposed primary skin fibroblasts to phototoxic stress (Minamikawa, Sriratana et al, 1999) (see Materials and Methods). Within ~10 min, neighboring mt‐nucleoids dynamically fused and relaxed into spherical, droplet‐like structures (aspect ratio ≈ 1, time scale ≈ 20 ± 10 s, Appendix Fig S1Q) > 100 nm in size and up to a few microns in diameter analogous to those seen in damaged, swollen mitochondria of HGPS cells (Fig 1H and I and Movies EV1 and EV2). Similar, but even more pronounced, fusion events were observed in the presence of the intercalator EtBr (Movies EV3 and EV4). The homotypic fusion events between neighboring mt‐nucleoids are consistent with the behavior of coalescing liquid droplets (Hyman et al, 2014; Banani et al, 2017). We conclude that loss of mitochondrial homeostasis results in the inability of mitochondria to maintain mt‐nucleoid size and leads to the coalescence of multiple proximal mt‐nucleoids to form larger, membrane‐less droplets of nucleoprotein complexes in a process that closely resembles the phase separation of many other biomolecular condensates (Hyman et al, 2014; Banani et al, 2017).
TFAM phase separates in vitro into viscoelastic droplets
To explore if phase separation drives mt‐nucleoid assembly, we examined the ability of the major mt‐nucleoid packaging protein TFAM to undergo phase separation in vitro. In line with phase separation behavior, TFAM formed spherical droplets in low‐salt conditions and at protein concentrations of ≥ 5 μM TFAM establishing a second proteinaceous phase, separate from the dilute, aqueous phase (Fig 2A; Appendix Fig S2D). Droplet formation was reversible upon increasing salt concentration (Appendix Fig S2F) and was reduced upon 1,6 hexanediol treatment (Appendix Fig S2G). At 30–60 min post‐mixing, droplets coarsened to sizes of ~1–5 μm and sedimented toward the bottom of the imaging chamber (Fig 2A). The TFAM concentrations required for phase separation in vitro were well within the estimated physiological range inside the mitochondria on the order of ~10 µM (see Materials and Methods) (Kukat et al, 2011).
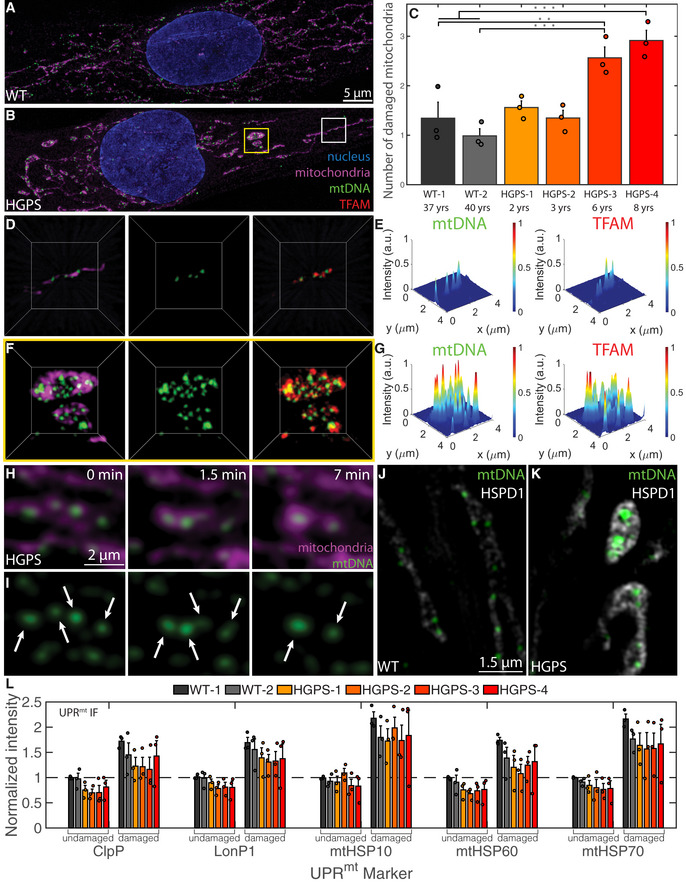
Figure 2. TFAM phase separates into droplets with slow dynamics, driven by many weak, multivalent interactions.
- Phase diagram (left panel) of TFAM under various protein and salt concentrations, where gray dots indicate single/soluble phase, red dots signify two phases/droplets present. DIC image (middle) and maximum intensity projection (right) of TFAM‐DyLight‐594 droplets at 25 µM and 150 mM NaCl in 20 mM Tris–HCl, pH 7.5 30 min after mixing. Scale bar = 5 µm.
- FRAP using 488 and 561 nm light performed on a ~1 µm spot on TFAM droplet 30 min after mixing. Inset shows representative fluorescent image of TFAM‐DyLight‐594 pre‐bleach, immediately post‐bleach, and 25 min post‐bleach. Scale bar = 2 µm. Values represent averages ± SD from n = 15 droplets.
- Aspect ratio of droplet shape as a function of time after contact for a representative droplet. Inset shows fusion images corresponding to the trace at t = 0, 2, 4, and 40 min. Scale bar = 2 µm.
- The partition coefficient of dextran‐FITC into TFAM droplets as a function of dextran average hydrodynamic radius estimated from the molecular weight. Solid line is an exponential fit to the data, where the partition coefficient is giving rise to nm. Inset shows representative images showing localization of dextran‐FITC for Rh ≈ 1, 2 and 25 nm. Scale bar = 10 µm. Values represent averages ± SD from n = 3 experiments (>20 droplets analyzed per condition for each experiment).
- Schematic diagram of mutants with HMG domains in red and intrinsically disordered regions in gray. Yellow and green lines indicate point mutations in L6 and no‐dimer mutants, respectively.
- Phase diagram of mutants at 150 mM NaCl and 20 mM Tris–HCl, pH 7.5 for a range of protein concentrations.
- Fluorescent maximum intensity projections of mutants at 50 µM protein and 150 mM NaCl, 20 mM Tris–HCl, pH 7.5 within 30–60 min after mixing. Scale bar = 2 µm.
We performed a series of biophysical assays on TFAM droplets to assess their material properties within the first 30–90 min after mixing. Photobleaching of a spot revealed slow dynamics with a characteristic time scale of , which corresponded to a diffusivity of . Furthermore, recovery was incomplete, with an immobile fraction of 0.5 ± 0.2, indicative of viscoelastic behavior (Fig 2B, Movie EV5). Similarly, time‐lapse images of TFAM droplets undergoing coalescence events also displayed correspondingly slow dynamics with time scales of τ = 4 ± 0.5 min, giving rise to an inverse capillary velocity of 80 ± 20 s/µm (Fig 2C). Although the droplets had the propensity to relax upon contact, the average aspect ratio upon fusion was 1.36 ± 0.04, which deviated from that of a sphere (AR = 1.0) (Appendix Fig S2H). Introduction of dextran‐FITC of varying sizes as an inert probe to sample the physicochemical environment of the droplets demonstrated that small particles of ≤ 1 nm preferentially accumulated within the droplets, most likely due to electrostatic interactions between negatively charged FITC and positively charged TFAM (Appendix Fig S2C), while increasing probe size resulted in reduced partitioning (Fig 2D). These properties are indicative of a characteristic pore or mesh size of ~1 nm, suggesting the presence of an effective polymer meshwork forming among individual TFAM molecules with markedly slow internal arrangements and signatures of viscoelasticity.
TFAM phase separation is driven by multivalent interactions
TFAM contains two DNA‐binding high mobility group (HMG) domains separated by a disordered linker domain and flanked by an intrinsically disordered C‐tail, together forming a relatively flexible protein chain (Appendix Fig S2A) (Ngo et al, 2011; Rubio‐Cosials et al, 2011). Additionally, TFAM is one of the most highly charged proteins in the mt‐nucleoid (Appendix Fig S2C) and is primarily enriched in positive amino acids distributed throughout the length of the protein (Appendix Fig S2B). To dissect the molecular features of TFAM responsible for phase separation, a set of TFAM mutants was analyzed for phase behavior in vitro (Fig 2E).
In phase separation assays in vitro, the HMGA domain alone failed to form the typical micron‐sized droplets as seen with full‐length TFAM, but assembled into small puncta near the diffraction limit even at high concentrations (Fig 2F and G; Appendix Fig S2I–L). Maintaining half of the protein, either by adding a linker to HMGA (HMGA + linker) or with the structurally analogous HMGB + C‐tail mutant, restored droplet formation, albeit at higher saturation concentrations than full‐length TFAM, suggesting that multivalency seen in the full‐length protein lowers the barrier for phase separation and that the addition of a disordered domain to HMGA promotes phase separation (Fig 2F and G; Appendix Fig S2I–L). Loss of the disordered C‐tail (ΔC‐tail) did not affect the saturation concentration, but influenced the wetting behavior of the droplets on the glass coverslip as indicated by a decreased smoothness along the droplet perimeter (Fig 2F and G; Appendix Fig S2I and J). The change in wetting behavior indicates differences in interfacial tension arising from molecular interactions between the droplet phase and the surroundings. Removal of the HMGA domain (ΔHMGA), which leaves a single HMG domain flanked by two disordered sequences, also resulted in droplet formation, but at slightly higher saturation concentrations. Finally, introduction of six non‐polar residues in the linker region (L6 mutant) (Ngo, Lovely et al, 2014) affected the bulk properties of the droplets as evidenced by the highly non‐spherical morphologies, whereas inclusion of non‐polar residues in the HMGA domain to prevent dimerization (no‐dimer mutant) (Ngo et al, 2014) increased saturation concentrations and produced smaller droplets, underscoring the contribution of multivalent interactions in phase separation of TFAM (Fig 2F and G; Appendix Fig S2I–L). Taken together, these observations suggest a mechanism by which many weak interactions along a flexible backbone of TFAM allow for robust phase separation and that the disordered linker and C‐tail provide flexibility of the biopolymer chain to promote phase separation into prominent droplets.
Formation of TFAM‐mtDNA multiphase condensates in vitro
To examine the interplay of mtDNA and TFAM in phase‐separated mt‐nucleoids, we investigated the in vitro phase behavior of TFAM in the presence of mtDNA (Fig 3; Appendix Fig S3A and B). As expected, mtDNA (0–10 nM or equivalently 0–100 ng/µl) on its own did not phase separate, but only when combined with TFAM at concentrations that support phase separation (≥ 5 µM), mtDNA readily partitioned into droplets. Importantly, the presence of mtDNA significantly affected droplet formation and morphology (Fig 3A; Appendix Fig S3C). Under initial mixing molar ratios of mtDNA/TFAM on the order of 1 × 10−5 – 1 × 10−3, which roughly correspond to the order of magnitude estimates of their physiological ratio (Kukat et al, 2011) (see Materials and Methods), TFAM and mtDNA readily associated into droplets together (Fig 3A; Appendix Fig S3C). At higher mtDNA/TFAM molar ratios (> 0.001), large, phase‐dense droplets were no longer detected, potentially due to saturation behavior (Fig 3A; Appendix Fig S3C). However, at these high mtDNA/protein ratios, microscopic droplet formation could be restored upon addition of the crowder PEG (Appendix Fig S3D). For molar ratios of mtDNA/TFAM ≤ 6 × 10−4, the aspect ratio of the droplets notably increased with increasing ratio of mtDNA/TFAM molar concentrations (Fig 3B). For mtDNA/TFAM molar ratios < 3 × 10−4, the number of droplets (measured ~1 h after mixing) increased with increasing mtDNA/TFAM levels (Fig 3B inset), suggesting that mtDNA can potentiate droplet formation under those conditions, possibly acting as a nucleating agent and paralleling how RNA drives phase separation when added to RNA‐binding proteins (Lin, Protter et al, 2015). ssDNA, dsDNA, and RNA as well as free nucleotides (dNTPs) also partitioned and supported TFAM droplet formation (Appendix Fig S3E–T). Interestingly, ssDNA and dsDNA, considerably longer than the 16.6 kB mtDNA and without any sequence specificity to mtDNA, resulted in even more irregular droplet morphologies (Appendix Fig S3E–T). We conclude that the addition of long, polymerized strands of DNA, irrespective of sequence, leads to favorable interactions between TFAM and DNA, thereby affecting the emergent droplet behavior (Appendix Fig S3E–V). While the aspect ratio of droplet morphologies did indeed vary across conditions tested, the structures that formed were invariably phase‐dense and > 1 µm in diameter, corroborating that the TFAM‐mtDNA in vitro system consistently exhibited phase separation as opposed to other modes of DNA compaction, such as salt‐induced condensation or polymer collapse. These findings demonstrate that the material properties of the droplets depend on DNA/TFAM composition, where increasing DNA leads to more irregular droplet morphologies.
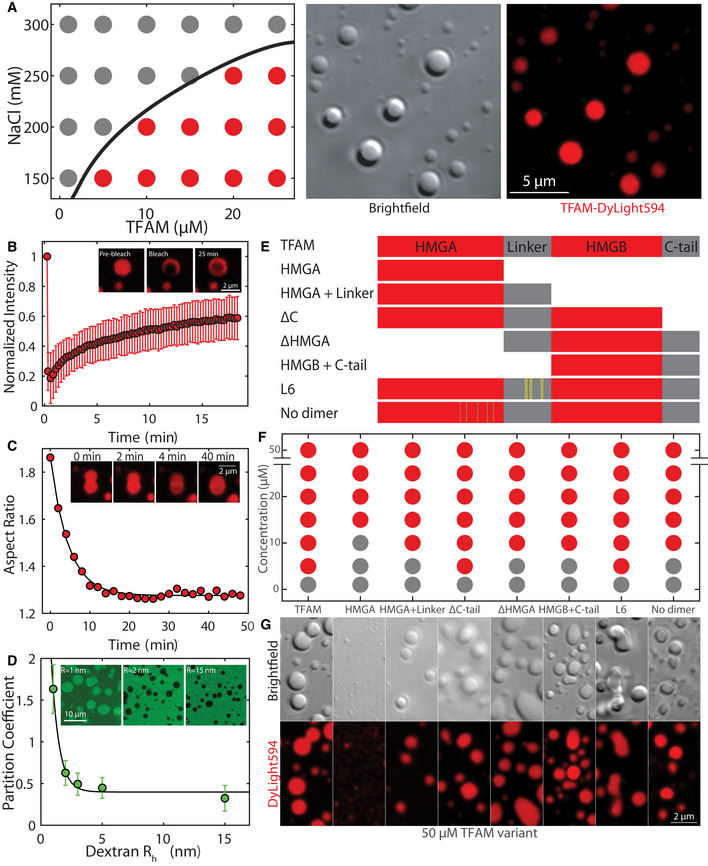
Figure 3. TFAM and mtDNA form heterogenous droplets whose relaxation timescale is set by mtDNA in vitro .
-
APhase diagram of mtDNA versus TFAM denoting single/soluble phase (gray) or two phases/droplets (green). Each point on the phase diagram representing a unique DNA and protein concentration was measured from n = 2–12 independent experiments. Black solid line delineates deduced phase boundary.
-
BAspect ratio as a function of dimensionless concentration (molar concentration mtDNA/molar concentration TFAM). Values represent binned mtDNA/TFAM conditions as measured in (A), and error bars are SEM. Green dashed line is an exponential fit to the data where . Inset: number of TFAM‐mtDNA droplets per field of view as a function of dimensionless concentration. Values represent binned conditions from mtDNA/TFAM conditions measured in (A) and error bars are SEM. Green dashed line is an exponential fit to the data where y = 5,600 – 2,100e (−x/2.1).
-
C–GSIM images of droplets 30 min after mixing with various amounts of mtDNA: 0 nM (C), 0.1 nM (D), 1 nM (E), 4 nM (F), and 10 nM (G). Top row is of TFAM‐DyLight‐594 (red), middle row is of mtDNA‐Alexa 488 (green) and bottom row is the merged image. Scale bar = 2 µm.
-
H, IFRAP experiments on TFAM‐mtDNA droplets at 25 µM TFAM (H, red) and 10 nM mtDNA (I, green). Scale bar = 1 µm.
-
JFRAP recovery curve showing intensity as a function of time for TFAM (red) and mtDNA (green). Black line are single exponential fits, where mobile fraction ≈ 0.60 and characteristic recovery time t ≈ 350 ± 30 s for TFAM (y = 0.73–0.38exp(‐t/350)), and recovery times t >> 15 min for mtDNA (error = 95% CI). Values represent averages ± SD from n = 16 droplets.
To probe how mtDNA localizes within the droplets, we performed SIM imaging on TFAM‐mtDNA droplets containing increasingly higher concentrations of mtDNA. We find that mtDNA is not uniformly distributed, but de‐mixes from TFAM within the droplet (Fig 3C–G), consistent with multiphase behavior seen in other multi‐component phase separating systems, and is reminiscent of the dense fibrillar component and fibrillar center of the nucleolus (Feric et al, 2016). Similar multiphase organization was observed with both ssDNA and dsDNA, but not with dNTPs nor RNA (Appendix Fig S3J–S). To characterize the dynamics of multiphase TFAM‐mtDNA droplets, fluorescence recovery after photobleaching (FRAP) demonstrated that TFAM was able to diffuse within TFAM‐mtDNA droplets with similar recovery behavior as in pure TFAM droplets (Fig 3H and J; Movie EV6). In contrast, on these timescales, mtDNA within the droplets remained strikingly immobile (Fig 3I and J; Movie EV6), suggesting that the mtDNA molecules within the TFAM‐mtDNA droplets determine the time scale for the bulk properties of the droplets, while also explaining the observed non‐spherical shapes at high mtDNA/TFAM ratios. The FRAP data suggest that these droplets represent kinetically arrested states arising from the slow dynamics associated with mtDNA inside the droplet, and as such, the observed dynamics are consistent with the multiphase, heterogenous sub‐structure visualized in these droplets.
TFAM modular domains influence organization of mtDNA in compound droplets in vitro
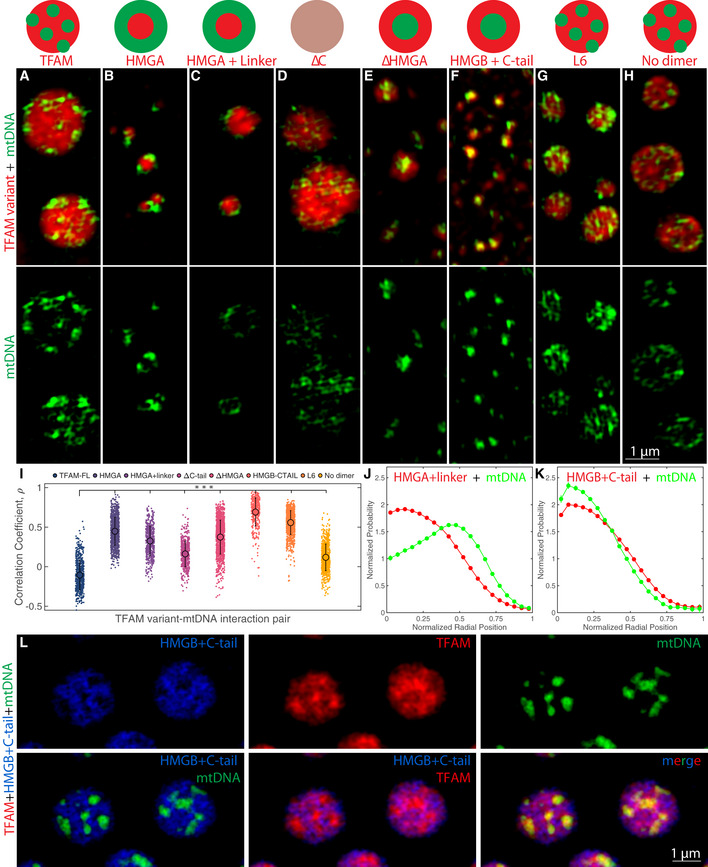
As all of the TFAM mutants were able to phase separate, we next investigated how they interacted with mtDNA in vitro to more directly probe the molecular interactions influencing the phase behavior and observed internal structure. When TFAM mutants were combined in vitro with mtDNA, the resulting droplets significantly varied in their internal organization based on the propensity of each mutant to phase separate on its own (i.e., average droplet size) as well as each mutant’s binding affinity for mtDNA (Fig 4A–I). For example, full‐length TFAM formed larger droplets than most of the mutants and had heterogenous localization of mtDNA (Fig 4A). However, mtDNA was exclusively located along the periphery of the mutants containing only HMGA domains (HMGA and HMGA + linker mutants; Fig 4B, C, I and J). In HMGA‐mutants to which the HMGB domain was appended, such as in the case of ΔC, mtDNA was more diffusely localized within the interior of the droplet (Fig 4D). Upon loss of the HMGA domain, the ΔHMGA and HMGB + C‐tail mutants had inverse localization of mtDNA as mtDNA was primarily located within the interior of the droplets (Fig 4E, F, I and K). The remaining two mutants that retained all functional domains but had altered interactions in the linker (L6, Fig 4G) or prevented dimerization (no dimer, Fig 4H) both showed similar heterogenous localization as full‐length TFAM (Fig 4A and I). These results demonstrate that TFAM has varying degrees of affinity for mtDNA across its modular domains. To directly test this hypothesis, we mixed combinations of full‐length TFAM with equimolar amounts of mutants HMGA + linker or HMGB + C‐tail (each protein at 25 μM) in the presence or absence of mtDNA (Fig 4L; Appendix Fig S4; see legends/Methods for details). For TFAM and HMGA + linker, there was high miscibility, but addition of mtDNA led to exclusively peripheral localization of the droplet relative to the mtDNA, unlike the internal heterogenous organization of full‐length TFAM with mtDNA, suggesting the HMGA + linker mutant dominates the organization in these droplets (Appendix Fig S4). Interestingly, TFAM and HMGB + C‐tail also had high miscibility when mixed together (Appendix Fig S4H), but upon the addition of mtDNA, they de‐mixed within the droplet and TFAM concentrated in microdomains highly enriched in mtDNA and completely devoid of HMGB + C‐tail, which instead formed large domains that enveloped microdomains of TFAM and mtDNA (Fig 4L). This organization reveals that TFAM can outcompete the HMGB + C‐tail mutant for mtDNA, leading to further enrichment of TFAM‐mtDNA domains inside the droplet. These observations are consistent with measurements from the literature which indicate significantly higher binding affinity of the HMGA domain alone compared to the poor binding of the HMGB domain alone (Wong, Rajagopalan et al, 2009). Although TFAM’s HMGA + linker and HMGB + C‐tail mutants are structurally similar, they do not form physicochemically equivalent droplets. Together, these observations paint TFAM as undergoing a multitude of interactions along its backbone that differ in their interaction strengths for DNA and in their propensity to undergo phase separation.
Figure 4. TFAM modular domains influence organization of mtDNA in compound droplets in vitro .
-
A–HSingle z‐slices of SIM images of TFAM mutants with mtDNA, where TFAM full‐length (A), HMGA (B), HMGA + linker (C), ΔC (D), ΔHMGA (E), HMGB + C‐tail (F), L6 (G), and no dimer (H). Final concentrations of TFAM mutants (red) and mtDNA (green) were each 25 µM and ~1 nM, respectively. Schematic diagram represents typical organization of TFAM mutant and mtDNA inside of droplet. Top panel of images is the merge of TFAM mutant and mtDNA channels, and the bottom panel of images is of the mtDNA channel only. Scale bar = 1 µm.
-
ICorrelation coefficient as a function of each TFAM mutant with mtDNA analyzed for the above images, where n = ~200–1,000 droplets. Error bars represent standard deviation (SD), and a one‐way ANOVA test was performed, where ***P < 0.001.
-
J, KRadial distribution functions of the localization of HMGA + linker (J, red) or HMGB + C‐tail (K, red) and mtDNA (green) inside the droplet. Droplet dimensions were normalized where 0 is the center of the droplet and 1 is the periphery of the droplet.
-
LSingle z‐slices of SIM images of compound droplets containing HMGB + C‐tail (blue), TFAM (red), and mtDNA (green). Top row contains individual channels for HMGB + C‐tail, TFAM, and mtDNA, respectively. Bottom panel shows overlays, where the leftmost is of HMGB + C‐tail and mtDNA channels, middle panel is HMGB + C‐tail and TFAM, and rightmost panel is the three‐color overlay. Scale bar = 1 µm.
Phase behavior of TFAM in live cells
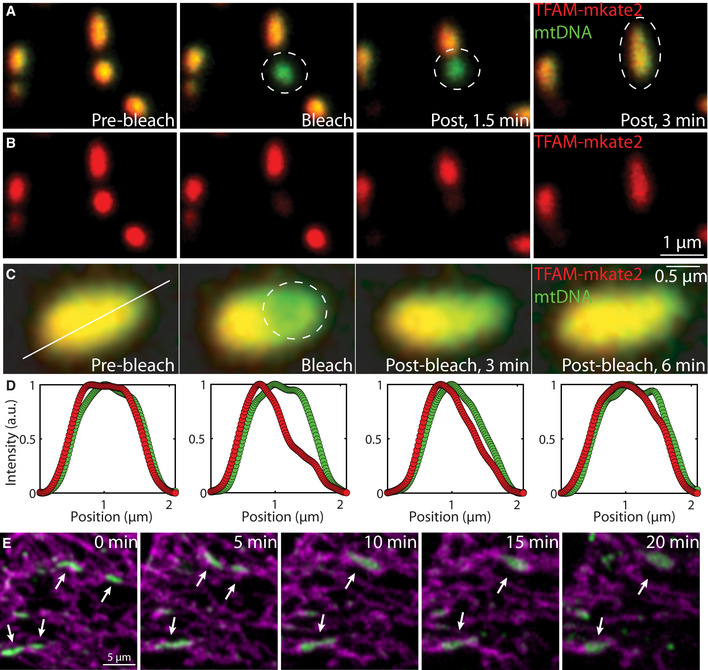
To test if the in vitro phase behavior of TFAM and mtDNA reflects the dynamic properties and structural organization of mt‐nucleoids in vivo, we visualized mt‐nucleoids in HeLa cells using TFAM‐mKate2. In photobleaching experiments, TFAM‐mKate2 exhibited very low recovery (immobile fraction = 0.9 ± 0.3) (Appendix Fig S5A–D; Movie EV7) indicative of limited exchange of TFAM between the mt‐nucleoid and the mitochondrial volume, consistent with the very low concentration of free TFAM (Matsushima, Goto et al, 2010; Lu, Lee et al, 2013). However, in line with in vitro observations, the ability of TFAM to rapidly diffuse within a mt‐nucleoid became evident when a bleached mt‐nucleoid fused with a neighboring unbleached mt‐nucleoid resulting in rapid exchange within the coalescing droplet (Fig 5A and 5B; Movie EV8). Furthermore, overexpression of TFAM‐mKate2 to high levels in HeLa cells led to formation of enlarged mt‐nucleoids (~1 µm) (Appendix Fig S5F and G), and photobleaching a small spot () within these structures (Fig 5C; Appendix Fig S5E; Movie EV9) gave rise to recovery of ~70 ± 20% (percentage of area under the curve recovered at t = 6 min after photobleaching) of signal further indicating relatively high mobility of TFAM within mt‐nucleoids (Fig 5D) and corresponding roughly to diffusivities on the order of (Fig 5D), in agreement with in vitro observations. Moreover, mt‐nucleoids larger than the diffraction limit exhibited classic liquid‐like fusion behavior upon phototoxic and EtBr induced stress, consistent with a phase separation model (Fig 5E; Movie EV10), and existed as discrete structures in sporadically swollen mitochondria in HeLa cells (Appendix Fig S5J). Furthermore, mt‐nucleoids were largely unaffected upon swelling of mitochondria under hypo‐osmotic conditions (Appendix Fig S5H and I), suggesting their components are not diffuse in the mitochondrial matrix, but that the size and shape of mt‐nucleoids are intrinsic properties arising from phase separation. These dynamics and recovery features are comparable to the viscoelastic material properties of TFAM droplets observed in vitro.
Figure 5. Phase behavior of TFAM in live cells.
-
A, BFRAP experiment performed on a small mt‐nucleoid that later undergoes a fusion event in live HeLa cells expressing TFAM‐mKate2. (A) The overlay of TFAM‐mKate2 (red) and of mtDNA labeled with PicoGreen (green) pre‐bleach, bleach, 1.5 and 3 min post‐bleach, and (B) the single‐channel of TFAM‐mKate2 (red). Scale bar = 1 µm.
-
C, DFRAP experiment performed on a R ≈ 0.2 μm spot in an enlarged mt‐nucleoid in live HeLa cells expressing TFAM‐mKate2 (n = 18 mt‐nucleoids), where (C) is the overlay of TFAM‐mKate2 (red) and of mtDNA labeled with PicoGreen (green) pre‐bleach, bleach, 3 and 6 min post‐bleach. The solid white line indicates the pixels that were analyzed for intensity quantification. The dashed white circle denotes the part of the mt‐nucleoid that was bleached. Scale bar = 0.5 µm. (D) The normalized intensity of TFAM‐mKate2 (red) and PicoGreen (green) where the x‐axis corresponds to the solid white line from (C). Recovery, τ, was ~70 ± 20% (percentage of area under the curve recovered at t = 6 min after photobleaching, n = 18 mt‐nucleoids), and error bars represent SD. Diffusion coefficient was estimated as D ~ R2/τ ~ 1 × 10−4 μm2/s.
-
EImages of mt‐nucleoids undergoing liquid‐like fusion events in HeLa cells overexpressing TFAM‐mKate2 after ~30 min exposure to EtBr with phototoxic imaging conditions where mtDNA (PicoGreen, green) and mitochondria (MitoTracker Deep Red, magenta) are shown. Arrow heads point to mt‐nucleoids involved in homotypic fusion events. Scale bar = 5 µm.
TFAM influences localization and miscibility of mt‐nucleoid components
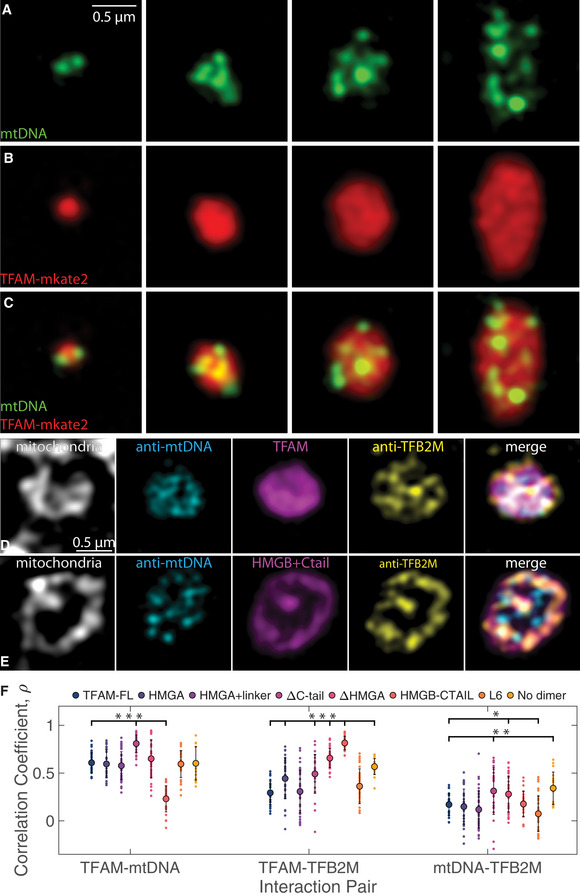
Our in vitro titration experiments suggested correlation between TFAM and mtDNA concentrations and morphology of the phase‐separated structures. To probe the effect of TFAM concentration on mt‐nucleoid organization in vivo, we expressed TFAM‐mKate2 and selected mt‐nucleoids with varying levels of expression for analysis using SIM. Consistent with in vitro titration data, we find an increase of mt‐nucleoid sizes, reaching up to a few microns in length, upon increasing levels of TFAM‐mKate2 (Fig 6A–C; Appendix Fig S6A). In addition, the mtDNA localization within these enlarged mt‐nucleoids had similar multiphase organization as observed in TFAM‐mtDNA droplets in vitro (Fig 6A–C). This behavior was not dependent on the mKate2 tag as overexpression of unlabeled TFAM gave similar results (Appendix Fig S6B and C). This non‐uniform structure is consistent with the layered organization of mt‐nucleoids deduced from biochemical analysis (Bogenhagen, Rousseau et al, 2008).
Figure 6. TFAM mutants influence miscibility and localization of mt‐nucleoid components.
-
A–CSingle z‐slices of SIM images of individual mt‐nucleoids ranging in size after overexpression of TFAM‐mKate2 in a fixed HeLa cell, where mtDNA (A, green, anti‐DNA), and TFAM (B, red, TFAM‐mKate2) and merge (C). Scale bar = 0.5 µm. Each structure represents a single, enlarged mt‐nucleoid containing multiple mtDNA foci found in one mitochondrion.
-
D, ESingle z‐slices of SIM images of individual mt‐nucleoids in single mitochondria from fixed HeLa cells after TFAM‐mKate2 (D) and HMGB + C‐tail‐mKate2 (E) overexpression (magenta), where mitochondria are labeled with MitoTracker Far Red (gray), mtDNA is labeled with anti‐mtDNA (cyan), and TFB2M is labeled with anti‐TFB2M (yellow). Merged images are shown as an overlay of cyan, magenta, and yellow channels only. Each structure represents a single, enlarged mt‐nucleoid containing multiple mtDNA foci found in one mitochondrion. Scale bar = 0.5 µm.
-
FColocalization of channels is shown computed from Pearson’s correlation coefficient for each interaction pair shown for TFAM‐FL and all TFAM mutant constructs overexpressed in HeLa cells (n = 20–40 mt‐nucleoids from 4–5 cells imaged for each mutant construct). One‐way ANOVA analysis shows statistical significance across all mutants for each interaction pair, where P < 2e‐16 for TFAM‐mtDNA interactions, P < 2e‐16 for TFAM‐TFB2M interactions, and P = 1.3e‐8 for mtDNA‐TFB2M interactions. Error bars represent standard deviation (SD). Notation on graph denotes statistical analysis relative to TFAM‐FL based on the Least Significant Difference test for each interaction pair, where *P < 0.05, **P < 0.01, and ***P < 0.001.
We next characterized the effect of TFAM on the multiphase properties of mt‐nucleoids by analyzing the behavior of an additional core mitochondrial transcription factor, TFB2M (Bogenhagen et al, 2008). We find that TFB2M did not mix homogenously with TFAM and DNA, but was preferentially localized toward the mitochondrial inner membrane (Fig 6D) in line with the multi‐component and multiphase nature of mt‐nucleoids. Based on Pearson’s correlation coefficient to assess partitioning within each microphase, TFAM has higher colocalization with mtDNA () than with TFB2M () (Fig 6F). Furthermore, the correlation between TFAM and mtDNA is not 1, suggesting that there are microdomains within the mt‐nucleoid that are enriched in TFAM, consistent with in vitro multiphase behavior.
To relate the in vitro behavior of TFAM mutants to their in vivo properties, we examined them in HeLa cells (Fig 6E; Appendix Fig S6D–O). Notably, the HMGB + C‐tail‐mutant caused a complete inversion in localization of the mutant from the interior, and as a result, the interactions with mtDNA were largely depleted, while the interactions with TFB2M were enriched (,) (Fig 6F). This change in localization reflected differences in protein miscibility of the HMGB + C‐tail mutant between the microphases, demonstrating that the HMGA and linker domains are important for association of TFAM with mtDNA. Other mutants also affected interactions between the mt‐nucleoid components, although to a lesser extent. Specifically, the ΔC‐tail mutant increased colocalization with mtDNA (), which suggests that loss of the C‐tail increased affinity and binding of TFAM and mtDNA, while potentially outcompeting the interactions with other proteins (Fig 6F; Appendix Fig S6F). Several other mutants also had increased affinities for TFB2M, albeit to a lesser extent as the HMGB + C‐tail mutant, including the ΔHMGA mutant (), which approached the HMGB + C‐tail mutant in composition (Fig 6F; Appendix Fig S6G). The partitioning of the mutants in live cells, especially regarding the behavior of the HMGB + C‐tail mutant, is in line with their in vitro behavior (Fig 4; Appendix Fig S4).
Functional features of enlarged mt‐nucleoids
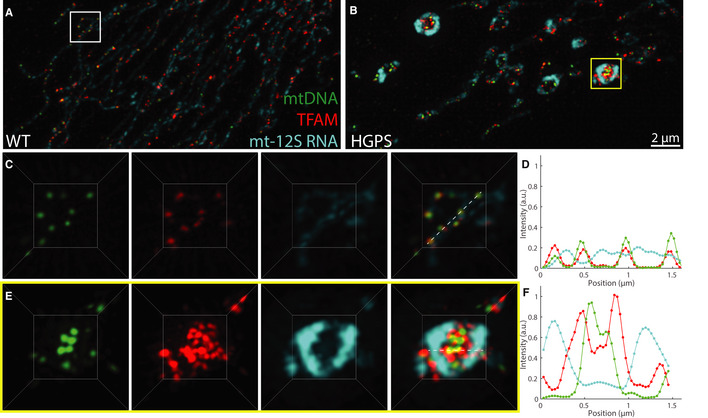
To finally assess whether the phase behavior of mt‐nucleoids is related to mitochondrial function, we measured mitochondrial activities in HGPS cells, which are enriched for enlarged mt‐nucleoids (Fig 7). Single molecule FISH for mt‐12S and mt‐COI RNA demonstrated enrichment of mt‐RNA transcripts in enlarged mt‐nucleoids that were proportional to local TFAM levels (Fig 7A and B; Appendix Fig S7A–F, and M). The expanded volume inherent to the damaged mitochondria made evident that RNA transcripts localized along the perimeter of the mitochondrial membrane, but they did not colocalize with mt‐nucleoids, suggesting that mt‐nucleoids and mt‐RNA exist as distinct structures, and potentially immiscible phases, of (ribo)nucleoproteins (Fig 7C–F). The separation of mtDNA from mt‐RNA transcripts is in line with the weak partitioning and minor effect on morphology of RNA relative to DNA on TFAM droplets in vitro (Appendix Fig S3E–V). Similar increased transcriptional activity of enlarged mt‐nucleoids was evident when nascent transcription was measured by BrU incorporation (Appendix Fig S7G–L and N). Consistently, we did not observe changes in mt‐RNA transcripts on a population level as measured by qPCR, which supports that enlarged mt‐nucleoids remain transcriptionally active, in line with RNA FISH imaging (Appendix Fig S7O).
Figure 7. Functional features of enlarged mt‐nucleoids.
-
A, BMaximum intensity projections of SIM images of fixed normal (A) and HGPS (B) human skin fibroblasts, where mtDNA is in green with anti‐DNA, TFAM is in red with anti‐TFAM, and mt‐12S RNA is in cyan with RNA FISH. Scale bar = 2 µm.
-
CThree‐dimensional views of normal mitochondria annotated by the white box in (A) and showing individual channels and an overlay; box ≈ 3.5 × 3.5 µm.
-
DNormalized intensity distributions of mt‐nucleoids labeled with anti‐DNA (green), anti‐TFAM (red), and mt‐12S RNA FISH (cyan) corresponding to images from (C).
-
EThree‐dimensional views of swollen mitochondria annotated by the yellow box in (B) and showing individual channels and an overlay; box ≈ 3.5 × 3.5 µm.
-
FNormalized intensity distributions of mt‐nucleoids labeled with anti‐DNA (green), anti‐TFAM (red), and mt‐12S RNA FISH (cyan) corresponding to images from (E).
On a population level, the enrichment of enlarged phase‐separated mt‐nucleoids in HGPS patients appeared to be preceded by altered mitochondrial metabolic functions, since defects in mitochondrial respiration were found in cells from both young and old HGPS patients, but morphological defects were more abundant only in the old HGPS patients (Fig 1; Appendix Fig S7P). Specifically, basal mitochondrial respiration, maximal respiration, and reserve capacity were reduced in all HGPS fibroblasts compared to isogenic non‐affected fibroblasts (Appendix Fig S7Q–S), which supports that functional impairment of mitochondrial oxidative phosphorylation and ATP regeneration occurred prior to mt‐nucleoid enlargement. Furthermore, HGPS fibroblasts from older donors had elevated mitochondrial ROS and membrane potential compared to HGPS fibroblasts from young or unaffected proband control fibroblasts (Appendix Fig S7T and U). These observations of mitochondrial dysfunction were consistent with our finding that other sources of stress, such as ROS production from phototoxic stress, were similarly followed by mt‐nucleoid fusion events (Fig 1H and I). These observations demonstrate that loss of mt‐nucleoid size control gives rise to enlarged mt‐nucleoids via phase separation and is associated with mitochondrial dysfunction.
Discussion
Our results show that mitochondrial nucleoids form by phase separation. Our finding that the well‐established architectural mt‐nucleoid protein TFAM has a high propensity for self‐assembly into a separate protein‐rich phase, and combined with mtDNA, generates complex, multiphasic structures in vitro suggest that TFAM is the primary driver of mt‐nucleoid phase separation. In support, we find that the properties of in vitro generated condensates containing TFAM and mtDNA closely mirror in vivo behavior of mt‐nucleoids.
These results provide new insights into the biogenesis of mt‐nucleoids and the function of the major architectural mtDNA‐binding protein TFAM. While the ability of TFAM to bind DNA is well‐established (Wong et al, 2009; Farge et al, 2014; Kukat et al, 2015), binding of TFAM to mtDNA alone does not explain the higher‐order structural features of mt‐nucleoids, such as their observed size and shape, and their dynamic emergent properties, including liquid‐like fusion events and internal rearrangements. The phase behavior of TFAM and mtDNA described here accounts for the observed morphological features of mt‐nucleoids as discrete, heterogenous, non‐membranous entities within mitochondria, their viscoelastic dynamics in vivo, their normally uniform size, and their ability to reach sizes larger than 100 nm upon mitochondrial dysfunction as observed in HGPS cells. This physical phase separation model complements previous self‐assembly studies of TFAM and mtDNA into nucleoid‐like structures under dilute conditions (Wong et al, 2009; Farge et al, 2014; Kukat et al, 2015), by extending them to reveal the importance of protein–protein interactions necessary in driving the self‐assembly of the mitochondrial genome under physiologically more relevant conditions.
Based on our mutational analysis of TFAM, we suggest a model in which weak interactions along the flexible backbone of TFAM promote phase separation into viscoelastic droplets. In the presence of DNA, the HMG domains of full‐length TFAM each bind DNA by intercalating into the helical strand, and together, further stabilize the DNA fiber (Appendix Fig S4B) (Ngo et al, 2011; Rubio‐Cosials et al, 2011). Collectively, TFAM molecules fully coat the DNA fiber, encapsulating it within a meshwork, while interacting with other bound or unbound TFAM molecules to form a phase‐separated condensate. Differential interactions and solubilities of full‐length TFAM with mt‐nucleoid components (mtDNA, TFB2M) likely contribute to the generation of microphases within the mt‐nucleoid droplet, giving rise to their multiphasic nature. Our observation that truncation and modification of TFAM domains lead to pronounced changes in localization within the mt‐nucleoid droplet underscores the role of TFAM‐specific interactions in independently conferring miscibility and determining overall mt‐nucleoid structure. These physical properties may also generate a platform for other mt‐nucleoid‐associated proteins to partition into these dynamic, yet persistent, structures (Bogenhagen et al, 2008) and to exclude other components from the mitochondrial matrix from fully mixing with the mt‐nucleoid.
The observed preferential localization of a TFAM mutant consisting of the HMGB and C‐terminal domains with the mitochondrial membrane suggests that the TFAM N‐terminus interacts with mtDNA in the mt‐nucleoid interior within the mitochondrial matrix, and that the C‐terminus is relatively less soluble and interacts with other mt‐nucleoid‐associated proteins and with the mitochondrial inner membrane, suggestive of TFAM acting as a surfactant. Previous work suggested that mt‐nucleoids interact with membranes (Brown et al, 2011; Kopek, Shtengel et al, 2012), potentially via a tethering mechanism (Chen & Butow, 2005). Our results suggest that the interaction of mt‐nucleoids with membranes is due to the emergent wetting behavior of the condensate on the membrane (Snead & Gladfelter, 2019). Similar observations have been made for contact sites between tethering proteins from various membrane bound organelles, including the mitochondrial mitofusin 1 (Mfn1) tethering protein and Sec61β of the ER membrane (King, Sengupta et al, 2020), and for the condensation of Atg1‐complex droplets, as part of the pre‐autophagosomal structure (PAS), along vacuolar membranes (Fujioka, Alam et al, 2020). It is tempting to speculate that the wetting behavior between the mitochondrial inner membrane and the mt‐nucleoid may not only play a role in regulating the size and diffusion of mt‐nucleoids, but also in functional processes such as replication (Lewis, Uchiyama et al, 2016). In addition, mt‐nucleoids are not the only example of phase separation within the mitochondria, as mitochondria have also been reported to contain various RNA granules, which do not colocalize with mt‐nucleoids. This is further supported by our observation that RNA weakly partitions into TFAM droplets in vitro, suggesting poor miscibility of RNA within the mt‐nucleoid phase.
Normally, cells tightly maintain mt‐nucleoids to have a fixed size of ~100 nm, yet various stressors, such as phototoxic stress or age‐associated damage, induce mt‐nucleoid fusion to generate microscopic droplet‐like structures that are considerably larger than > 100 nm. This behavior suggests that phase separation plays a role in mt‐nucleoid size control. The innate phase behavior of mt‐nucleoids favors their fusion to form a large macroscopic phase, while biological activity under normal conditions seems to prevent these events, similar to how hundreds of nucleoli are prevented from fusing in large nuclei of frog eggs via a unique nuclear actin scaffold (Feric & Brangwynne, 2013). The functional benefits of tightly regulating mt‐nucleoid size and the biological mechanisms that mitigate these fusion events in normal cells are yet to be determined.
In line with loss of mt‐nucleoid size control, we find that enlarged mt‐nucleoids in HGPS cells are associated with mitochondrial dysfunction. Our observation that transcriptional levels of mitochondrial genes are unchanged in enlarged mt‐nucleoids is consistent with phase separation: If both small and large droplets arise from the same coordinate on the phase diagram, then the droplets should be compositionally and functionally similar. We find enlarged mt‐nucleoids under conditions of acute or chronic stress such as phototoxicity or as in HGPS cells, which exhibit elevated levels of ROS (Kubben, Zhang et al, 2016). The molecular mechanisms that lead to the formation of enlarged mt‐nucleoids in HGPS cells and their functional relevance to disease remain to be determined. Our finding that the presence of enlarged mt‐nucleoids is limited to cells from older HGPS patients, whereas mitochondrial defects are found in HGPS patients of all ages, suggest that the enlarged mt‐nucleoids emerge as a consequence of aberrant mitochondrial function in HGPS. An intriguing possibility with regard to functional properties of mt‐nucleoids is that phase separation of TFAM may be a protective response toward maintaining mtDNA sequence integrity (Cadenas & Davies, 2000; Bogenhagen, 2012). The formation of a proteinaceous phase of TFAM around mtDNA may act as a physical barrier impeding diffusion of free radicals. Fusion into a single larger, spherical droplet would reduce the total surface area exposed to the surrounding oxidizing environment and increase the distance for ROS to diffuse to reach the mt‐nucleoid core, thereby minimizing the formation of ROS‐induced mutations and deterioration of the mitochondrial genome (Barja & Herrero, 2000). Additionally, phase separation of the mt‐nucleoid components into droplet‐like structures could serve as a platform to recruit and retain necessary mtDNA repair factors in response to damage (Kazak, Reyes et al, 2012).
The phase behavior of mt‐nucleoids may also account for the observation of a high degree of pathogenicity of a remarkably diverse set of mutations in mt‐nucleoid proteins (Suomalainen & Battersby, 2018). For example, a large number point mutations in the linker region of POLG lead to a spectrum of diseases (Chan & Copeland, 2009). These mutations may exert their pathogenic effect not by specifically disrupting the active site of the affected protein, but rather by globally influencing the biophysical interactions that contribute to higher‐order structure and therefore long‐term stability of mt‐nucleoids.
Mitochondria are originally derived from prokaryotes, and nucleoids are an evolutionarily ancient feature used to organize both prokaryotic and mitochondrial genomes (Dillon & Dorman, 2010). In support of a common organizational principle, bacterial genomes are packaged into nucleoid structures that have also been described to behave as fluids (Cunha, Woldringh et al, 2001), and some bacterial nucleoprotein complexes also undergo phase separation (Monterroso, Zorrilla et al, 2019; Remesh, Verma et al, 2020). Unlike TFAM, bacterial architectural nucleoid‐associated proteins, including HU, histone‐like nucleoid structuring protein (H‐NS), factor for inversion stimulation (FIS), and the integration host factor (IHF), lack HMG domains as seen in TFAM, suggesting a multitude of molecular interactions can condense DNA (Kucej & Butow, 2007; Dillon & Dorman, 2010). Interestingly, HMG domains have purely eukaryotic origins as exemplified by how mt‐nucleoids in budding yeast are analogously packaged primarily by the TFAM‐homologue Abf2p, which also contains two HMG domains separated by a shorter linker (Brewer et al, 2003).
Phase separation has been implicated in various levels of organization of the nuclear genome (Iborra, 2007; Larson et al, 2017; Strom et al, 2017; Gibson et al, 2019; Misteli, 2020). Although phase separation cannot account for all aspects of nuclear genome organization (McSwiggen, Mir et al, 2019; Mir, Bickmore et al, 2019; Kantidze & Razin, 2020), several lines of evidence point to a major contribution of phase separation to genome architecture. Purification of nucleosomes in vitro leads to liquid droplet formation via phase separation, and droplet dynamics and organization can be directly modulated by regulatory factors, including linker length and post‐translational modifications (Gibson et al, 2019). In vivo, many chromatin domains are emerging to behave as molecular condensates, including transcriptionally relevant super‐enhancers (Sabari et al, 2018). In fact, RNA polymerase II and many nuclear transcription factors have low‐complexity domains or intrinsically disordered regions, similar to the disordered modular domains of TFAM and which are features known to promote phase separation (Boehning, Dugast‐Darzacq et al, 2018; Boija, Klein et al, 2018; Cho, Spille et al, 2018; Chong, Dugast‐Darzacq et al, 2018). On an even larger scale, phase separation can explain the emergence and maintenance of heterochromatin, as HP1ɑ readily phase separates in vitro and has dynamic, liquid‐like properties in live cells (Larson et al, 2017; Strom et al, 2017; Sanulli et al, 2019). The involvement of phase separation in the organization of diverse genomes from simple mitochondrial and bacterial nucleoids to complex eukaryotic nuclear genomes suggests phase separation is an evolutionarily conserved mechanism in genome organization.
Materials and Methods
Cell culture
Primary human dermal fibroblast cell lines were obtained from the Progeria Research Foundation (PRF). Two of the cell lines were from normal, middle‐aged parents. The remaining four cell lines were from patients that have the classic mutation in LMNA Exon 11, heterozygous c.1824C > T (p.Gly608Gly) and the age at donation sampled early (ages 2 and 3 years) and late (ages 6 and 8 years) stages of the disease. For all experiments, cells were passage matched between P10‐20. Cells were grown according to PRF’s recommended protocols. Briefly, cells were grown in media containing DMEM (Thermo Fisher Scientific, #11960‐044) and supplemented with 15% FBS (Thermo Fisher Scientific, #10437), 1% penicillin‐streptomycin (Thermo Fisher Scientific), and 1% glutaMAX (Thermo Fisher Scientific, #35050‐061) in T25 and T75 ml flasks, and cells were periodically split with trypsin (Thermo Fisher Scientific).
For live‐cell experiments and for pharmacological disruption experiments, HeLa cells (ATCC) were used. Cells were grown in media containing DMEM (Thermo Fisher Scientific, #11960‐044) and supplemented with 10% FBS (Thermo Fisher Scientific, #10437), 1% penicillin‐streptomycin (Thermo Fisher Scientific), and 1% glutamine (Thermo Fisher Scientific).
For viral production, HEK293FT cells were used and grown in media containing DMEM (Thermo Fisher Scientific, #11960‐044) and supplemented with 10% FBS (Thermo Fisher Scientific, #10437), 1% penicillin‐streptomycin (Thermo Fisher Scientific), and 1% glutamine (Thermo Fisher Scientific).
For isolation of mtDNA, immortalized wild‐type human fibroblasts CRL‐1474 cells were used and grown in media containing MEM (Thermo Fisher Scientific) and supplemented with 15% FBS (as purchased), 1% penicillin‐streptomycin (Thermo Fisher Scientific), 1% glutamine, 1% non‐essential amino acids (Thermo Fisher Scientific), and 1% sodium pyruvate (Thermo Fisher Scientific). For overexpression of GFP‐LA and GFP‐Δ50 experiments, immortalized wild‐type human fibroblasts CRL‐1474 cells were used and grown in media containing DMEM and supplemented with 15% FBS, 1% penicillin‐streptomycin, and 1% glutaMAX.
Lentiviral construct and transfection
The sequences for TFAM and mKate2 were sequentially cloned from a previous TFAM lentiviral construct (TFAM, pLenti‐GIII‐CMV‐RFP‐2A‐Puro, ABM) into a pCDH‐CMV backbone (Addgene plasmid #72265) to create the fusion TFAM‐mKate2 construct for lentiviral transfection. TFAM mutants (see below, HMGA (43–122), HMGA + linker (43–152), ΔC (43–222), ΔHMGA (123–246), and HMGB + C‐tail (153–246), L6 and no dimer) were cloned into the pCDH‐CMV‐mKate2 vector containing the original TFAM mitochondrial targeting sequence and cleavage site. GFP‐Lamin A (GFP‐LA) and GFP‐Progerin (GFP‐Δ50) constructs were in a pCDHblast MCSNard vector and were a gift from Nard Kubben(DeBoy, Puttaraju et al, 2017; Kubben, Adriaens et al, 2012).
HEK293 cells were transfected with vectors containing the gene of interest, pMD2g, and pSPAX2 using PEI. Viruses were collected and concentrated with Lenti‐X Concentrator (Takara), flash frozen in liquid nitrogen, and stored in −80C. Frozen aliquots of lentiviruses generated from HEK293 cells were quickly thawed in a 37°C water bath, and lentiviruses were applied to HeLa cells and removed after 24 h.
CRL1474 cells were infected with GFP‐LA and GFP‐Δ50 viruses for 24 h and were sorted based on GFP expression using a standard FACS protocol. Sorted cells were imaged 2–4 weeks after initial infection. To assess the behavior of unlabeled TFAM in vivo, HeLa cells were directly transfected with the original plasmid (TFAM, pLenti‐GIII‐CMV‐RFP‐2A‐Puro, ABM) containing TFAM and mKate2 under separate promoters using FuGene and were fixed after 72 h on 12‐well plates.
Protein constructs and purification
Human_TFAM_NoMTS_pET28 (full‐length TFAM protein), Human_TFAM_noMTS_L6_pET28a+ (L6 mutant), and Human _TFAM_NoMTS_Dimer_pET28a+ (no‐dimer mutant) (Addgene plasmids #34705, #60012, and #60013, respectively) were used. The L6 construct contains the following mutations K136A, H137A, K139A, R140A, K146A, and K147A in the linker region (Ngo et al, 2014). The no‐dimer construct contains the following mutations K95A, Y99F, E106A, E112A, and R116A in the HMGA domain to prevent dimerization (Ngo et al, 2014). All other domain mutants were cloned into a pET28a + expression vector (Novagen) using BamHI and XhoI sites using standard molecular cloning techniques. Specifically, domain mutants consisted of HMGA (43–122), HMGA + linker (43–152), ΔC (43–222), ΔHMGA (123–246), and HMGB + C‐tail (153–246) (Ngo et al, 2011).
All proteins were purified using a slightly modified protocol (Ngo et al, 2011). Briefly, all constructs were transformed in BL21 star (DE3) pRare E. coli. Bacterial cultures (500 ml for full‐length TFAM and 250 ml for each mutant) of Dynamite media plus kanamycin and chloramphenicol were inoculated with each construct and incubated until an OD of 7 was reached. IPTG was added at 0.5 mM to induce protein expression, and the culture was incubated overnight at 16°C at 220 rpm. The culture was centrifuged, and the bacterial pellet was resuspended and lysed in lysis buffer [20 mM Tris–HCl, 500 mM NaCl, pH 8.0]. Cells were mechanically lysed with a microfluidizer and centrifuged for 30 min at 70,000 g at 4°C. Protein was purified using an immobilized metal affinity chromatography (IMAC) column and eluted with elution buffer [20 mM Tris–HCl, 500 mM NaCl, 250 mM Imidazole, pH 8.0] and dialyzed into lysis buffer [20 mM Tris–HCl, 500 mM NaCl, pH 8.0] and stored at 4°C.
To remove any bound nucleic acids, a 5 ml HiTrap Heparin High Performance column (GE Healthcare) was used following a modified protocol (Wong et al, 2009). The protein was first diluted in Heparin buffer (20 mM Tris–HCl, 300 mM NaCl, pH 7.5). The column was pre‐equilibrated with Heparin buffer; the protein was loaded onto the column; the column was washed with 5–10× column volumes of Heparin buffer; and the protein was eluted with a two‐step gradient of 700 mM NaCl followed by 1 M NaCl. Fractions containing purified protein were pooled together. To label the protein, a small fraction of protein (~100 µg) was buffer exchanged into PBS and was labeled with the DyLight‐594 antibody labeling kit (Thermo Fisher). Mutants were labeled with DyLight‐594, DyLight‐488, and DyLight‐650 labeling kits (Thermo Fisher). Typical reaction efficiency was 0.1‐1 moles DyLight per 1 mole of protein as determined by absorption spectra on the DeNovix DS‐11 FX + Spectrophotometer/Fluorometer. Glycerol was added to a final concentration of 10% (vol/vol), and aliquots of labeled and unlabeled protein were separately flash frozen in liquid nitrogen and stored at − 80°C. Specifically, aliquots of 10–20 µl of labeled protein (~1 µg/µl) were frozen while 500 µl of unlabeled protein (~1 µg/µl) were frozen. Prior to buffer exchange, protein was thawed and labeled protein was mixed with unlabeled protein at a ~1:100 mass ratio.
Bioinformatics
Disorder predictions on human TFAM were performed using the Predictor of Natural Disordered Regions (PONDR) program (Xue, Dunbrack et al, 2010). Images of the crystal structure of TFAM and DNA were obtained from the RCSB Protein Data Bank (rscb.org) (Berman et al, 2006). Proteins involved in mtDNA replication and transcription were selected from the reported mt‐nucleoid proteome (Bogenhagen et al, 2008), and sequences were obtained from PubMed based on respective accession numbers. Charged residues were identified using a custom‐built Matlab program.
mtDNA purification
Total DNA was extracted directly from CRL‐1474 cells using a DNeasy Blood & Tissue Kit (Qiagen). To obtain mitochondrial DNA, two overlapping fragments of the mtDNA were amplified by long‐range PCR with Takara LA Taq DNA polymerase (Clontech) using two sets of primers (~9 kb fragment: 5′‐AACCAAACCCCAAAGACACC‐3′ and 5′‐GCC AATAATGACGTGAAGTCC‐3′ and ~7 kb fragment: 5′‐TCCCACTCCTAAACACATCC‐3′ and 5′‐TTT ATGGGGTGATGTGAGCC‐3) (Sundaresan, Simpson et al, 2015). To directly label mtDNA, trace amounts of fluorescently labeled nucleotides (ChromaTide Alexa Fluor 488 5‐dUTP, Thermo Fisher Scientific) were added to the PCR reaction [50 µl reaction containing 10 ng of total DNA, 0.2 µM forward primer, 0.2 µM reverse primer, 1 × LA Takara buffer, 400 µM unlabeled dNTPs (each), 2 µM 488‐labeled dUTP (to fluorescently label), 0.5 µl Takara polymerase and PCR‐grade water]. Reactions were performed at 94°C for 1 min, 30 cycles of (i) 94°C at 30 s, (ii) 55°C for 15 s, (iii) 68C for 11 min, followed by 72°C for 10 min and chilled at 4°C. mtDNA fragments were gel extracted after gel electrophoresis and post‐staining with EtBr. mtDNA gel fragments were further purified with Zymoclean Large Fragment DNA Recovery Kit (Zymo Research) and eluted in low‐salt buffer (20 mM Tris–HCl, 0 M NaCl, pH 7.5). Unlabeled mtDNA concentration was measured using a Qubit dsDNA HS assay kit (Molecular Probes) on a Qubit Fluorometer, while labeled mtDNA was measured using a DeNovix (DS‐11 FX+). For mtDNA that was not labeled with fluorescent nucleotides, DAPI was used to visualize DNA and added in the no salt buffer in trace amounts.
Nucleic acids
Single‐stranded DNA from calf, RNA from calf, RNA from yeast, double‐stranded DNA from E. coli, and double‐stranded DNA from calf were obtained (Sigma). Free dNTPs (TaKaRa) were mixed with trace amounts of ChromaTide Alexa Fluor 488‐5‐dUTP (Thermo Fisher Scientific). Nucleic acids and free dNTPs were resuspended in 20 mM Tris, 0 M NaCl, pH 7.5 buffer. To fluorescently label nucleic acids, SYBR Gold (Thermo Fisher Scientific) was added in trace amounts prior to mixing.
Phase separation assays in vitro
Frozen aliquots of protein were thawed at room temperature. Protein solutions were concentrated and buffer exchanged (20 mM Tris–HCl, 500 mM NaCl, pH 7.5) using 0.5 ml Centrifugal filters 3‐ and 10‐kDa (Amicon) using a table top centrifuge at 4°C. Final protein concentration was measured by Bradford Assay using BSA standards (Bio‐Rad) on a DeNovix spectrophotometer. To induce phase separation, concentrated protein solutions were added to low‐salt buffer (20 mM Tris–HCl, 0 M NaCl, pH 7.5) to yield final protein concentrations of 1–50 µM and NaCl concentrations of 150–300 mM. For TFAM‐mtDNA experiments, the low‐salt solution was prepared first, followed by addition of mtDNA (final concentration 1–100 ng/µl) and protein was added last. Solutions were mixed gently and centrifuged prior to imaging. For TFAM‐mtDNA under crowded conditions, 5% PEG (Sigma, MW ~3K) was added to the buffer. For 1,6 hexanediol experiments, 10 and 20% 1,6 hexanediol (Sigma) was added to the buffer.
For high‐throughput assays, ~10–50 µl of protein solution were pipetted to wells of a 384‐well plate (PerkinElmer) and covered with 50–100 µl of mineral oil to prevent evaporation. Pipetting was done either manually or in an automated manner using the ECHO525 liquid handler (Labcyte) into 384 wells. For laser scanning confocal and structured illumination microscopy experiments, 3–6 µl of solution were added to the center of a 4.5 mm diameter × 0.6 mm depth silicone isolator (Grace‐biolabs) on a coverslip and sealed with a glass slide prior to imaging.
Dextran‐FITC (3–5 kDa, 40 kDa, 500 kDa, Sigma) solutions were prepared in 1 mg/ml in water. For partitioning experiments, dextran was first was added to the low‐salt buffer, followed by concentrated protein, to obtain a final concentration of 0.1–0.3 mg/ml. Dextran intensity was measured inside and outside of the droplets to obtain a partition coefficient, which accounted for background intensity. The partition coefficient as a function of dextran size was fit to an exponential to obtain a characteristic length scale.
To capture fusion of droplets, time‐lapse movies were performed at 2‐min intervals for several fields of view on 384‐well plates using high‐throughput confocal microscopy immediately after mixing. Fusion events captured within the first 1–2 h were manually identified, and the aspect ratio as a function of time was quantitatively analyzed using Matlab. The characteristic time scale, τ, and the size of the drop, , were used to estimate the inverse capillary velocity, (Brangwynne et al, 2011).
To determine the mobility within the droplets, a ~1 µm spot was photobleached precisely 30 min after mixing. The fraction recovered was measured as a function of time and normalized to obtain a characteristic time scale, τ (Phair, Gorski et al, 2003).
Live‐cell imaging
For live‐cell imaging, primary skin fibroblasts and HeLa cells were seeded 1–3 days prior to the experiment in 8‐well imaging chambers (Thermo Fisher Scientific). Cells were washed a few times with pre‐warmed complete media lacking phenol red to replace the original cell media prior to imaging. For FRAP experiments, HeLa cells expressing TFAM‐mKate2 were incubated with PicoGreen 0.3% (vol/vol) (Ashley, Harris et al, 2005) and 100 nM MitoTracker Deep Red at least 1 h prior to imaging on the LSM780. For coarsening experiments, HeLa and primary HGPS cells were also pretreated with MitoTracker Deep Red (100 nM), PicoGreen (0.3% vol/vol) for ~1 h, and/or with Ethidium Bromide (1 µg/ml) for ~0.5–1 h prior to imaging with either laser scanning confocal microscopy or Airyscan confocal microscopy (Ashley & Poulton, 2009). To osmotically induce mitochondrial swelling, normal media were replaced with DI water and live cells were immediately imaged.
Immunofluorescence
Cells were seeded in 96‐well or 384‐well plates (PerkinElmer) or in 12‐well dishes 24 h prior to fixation. To label the mitochondrial network, cells were incubated with 100 nM MitoTracker Red/Deep Red (Thermo Fisher Scientific) for 15–30 min at 37°C. Cells were immediately washed with PBS and fixed with 4% PFA for 10 min. Cells were washed and permeabilized with 0.1% Triton X for 10 min. Cells were washed and incubated with primary antibodies in PBST with 5% BSA for 1 h at room temperature or overnight at 4°C. The following antibodies were used: anti‐DNA (EMD Millipore, clone AC‐30‐10), anti‐TOMM20 (Sigma, HPA011562), anti‐TFAM (Sigma, HPA063684), anti‐TFB2M (Sigma, HPA028482), anti‐SSBP1 (Sigma, HPA002866), anti‐TOP1MT (Sigma, HPA001915), anti‐HSPD1 (Sigma, HPA001523), anti‐LONP1 (Proteintech, 154401‐A), anti‐CLPP (Thermo Fisher Scientific, PA5‐52722), anti‐HSP10/EPF (RND Systems, MAB3298), anti‐mtHSP70 (Thermo Fisher Scientific, MA3‐028). Cells were washed 1 × with PBST and 2 × with PBS followed by incubation with secondary antibodies for 1 h at room temperature. The following secondary antibodies were used: anti‐rabbit (Thermo Fisher Scientific, as purchased) and anti‐mouse (Thermo Fisher Scientific, as purchased). Cells were washed 1× with PBST and 2 × with PBS followed by an incubation with DAPI for 30 min. Cells were stored in PBS at 4°C. 96‐well plates were sealed for use in high‐throughput imaging, while coverslips were mounted onto glass slides with VectaShield mounting medium (Vectorlabs) and sealed with nail polish or with ProLong Gold Antifade Mountant (Thermo Fisher Scientific), as purchased, and left to cure for > 24 h for use in super‐resolution imaging. For all high‐throughput microscopy, all immunofluorescent experiments had three technical replicates with > 4 fields of view per well and three experimental replicates. For super‐resolution imaging, at least two experimental replicates were performed on three or more cells per condition.
RNA FISH
Custom Stellaris® FISH Probes were designed against mt‐12S and mt‐COI by utilizing the Stellaris® RNA FISH Probe Designer (Biosearch Technologies, Inc., Petaluma, CA) available online at www.biosearchtech.com/stellaris designer (version 4.2). Primary cells in 96‐well plates or in 12‐well plates were hybridized with the mt‐12S and mt‐COI Stellaris RNA FISH Probe set labeled with Qusar‐670 (Biosearch Technologies, Inc.), following manufacturer’s instructions available online at www.biosearchtech.com/stellarisprotocols for sequential immunofluorescence and RNA FISH. Briefly, cells were fixed in 4% PFA for 10 min and permeabilized with 0.1% Triton X for 10 min. Immunofluorescence was performed first without blocking agents, and cells were fixed again for 10 min with 4% PFA. Manufacturer’s instructions were followed for subsequent FISH labeling and samples were stored in PBS.
BrU incorporation
Cells were incubated with 2.5 mM BrU (Sigma) for 1 h in cell media. Media were removed and replaced with media containing 100 nM MitoTracker Red for 15–30 min to label the mitochondrial network and remove any diffuse BrU (Jourdain, Koppen et al, 2013; Ramos, Motori et al, 2019). Cells were subsequently washed briefly with PBS, fixed 10 min with PFA and permeabilized with 70% ethanol for 1 h at 4°C followed by 10 min of 0.5% Triton X at room temperature. Primary antibodies for anti‐BrdU (Sigma, 11170376001) and anti‐TFAM (Sigma, HPA063684) were applied for 1 h at room temperature followed by secondary antibodies without the use of blocking agents.
Light microscopy techniques
High‐throughput confocal imaging was performed on fixed cells in 96‐ and 384‐well plates (PerkinElmer) and on phase separation assays on 384‐well plates (PerkinElmer) using the fully automated Yokogawa CV7000S spinning disk microscope using a 60× water immersion objective with 405, 488, 561, and 647 nm laser lines as well as with brightfield imaging. Images were acquired as maximum intensity projections of 6–10 z‐slices with 1 µm thickness.
Structured Illumination Microscopy (SIM) was performed on fixed cells and TFAM‐mtDNA droplets using ELYRA PS.1 on an AxioObserver Z1 inverted microscope controlled by ZEN software. A 63×/1.4 NA oil Plan Apochromat objective was used with 405, 488, 561, and 647 nm laser lines.
Laser scanning confocal microscopy was performed using a Carl Zeiss LSM780 inverted microscope controlled by ZEN software and with definite focus. Live cells were imaged using either 63×/1.46 NA oil objective and a 100×/1.46 NA objective for FRAP experiments with 488, 561, and 633 nm laser lines using a CO2/heating control stage insert. TFAM phase‐separated droplets were imaged on a 60 × objective with 594 laser lines and in brightfield/DIC. For in vitro FRAP experiments, a ~1 µm spot corresponding to TFAM‐Dylight‐594 and/or mtDNA‐Alexa 488 was bleached using 488 and 561 nm light within the center of the droplet using maximum laser power and recovery was monitored over a period of 10–30 min. For in vivo FRAP experiments, a ~0.5 µm spot corresponding to TFAM‐mKate2 was bleached using 488 and 561 nm light inside mt‐nucleoids using maximum laser power and recovery was monitored over a period of ~10 min.
Airyscan confocal microscopy was performed using a Carl Zeiss LSM880 inverted confocal microscope controlled by ZEN software and with definite focus. Coarsening experiments in live cells were imaged using a 63×/1.46 NA oil objective with 488, 561, and 633 laser lines and inside of an incubation chamber.
Image analysis
Images and time‐lapse movies were visualized and created using Fiji, Zen, Matlab, and Imaris software. For representation in figures, some images were filtered using standard Gaussian, mean, and/or band pass filters. For Figs 1D–F, 1H–I, 1J–K, 2B–D, 3C and D, 3H and I, 4A–H (green channel only), 5A–E, and 7A–E the same brightness/contrast settings were used across conditions from the same experiment. For Figs 1A and B, 2G, 3E–G, 6A–C, and 6D–E, the same brightness/contrast settings were not used, but rather adjusted for each panel for clarity.
All quantitative image analysis was performed using custom‐built Matlab code. Image analysis of phase‐separated droplets consisted of measuring the average intensity, size, aspect ratio, and number of droplets per field of view. These values were averaged for ≥ 4 fields of view per well.
For analysis of high‐throughput images, nuclear markers, such as DAPI, were used to identify individual cells while cytoplasmic markers, such as MitoTracker Red, were used to segment cells using a watershed algorithm. To quantify the extent of morphological damage to the nucleus, the mean negative curvature of the nucleus was obtained for each cell following a published method (Driscoll, Albanese et al, 2012), and mean intensity within the nucleus was also computed. To detect damaged mitochondria, the mitochondrial network was filtered with a Gaussian filter, segmented and thinned. The mitochondria within each cell were analyzed based on their size, brightness and shape to identify the phenotype of interest: swollen, bright mitochondria. The number of damaged mitochondria per cell was averaged across all cells within a well. All wells were averaged per technical replicate (total of three technical replicates per condition) and then averaged for all experimental biological replicates, where n = 3.
Within each cell, individual mitochondrial nucleoids labeled with anti‐DNA were detected and analyzed using adapted Matlab Multiple Particle Tracking Code adapted (see https://site.physics.georgetown.edu/matlab/code.html) (Crocker & Grier, 1996). The coordinates of identified mt‐nucleoids were mapped onto the channel containing signal of mt‐nucleoid protein markers, such as TFAM, to correlate integrity intensity of anti‐DNA with that of the associated protein. Similar analysis was performed to quantify RNA transcript levels, where mt‐nucleoids were identified with anti‐TFAM that were later correlated with the intensity in the channel labeled by FISH or anti‐BrU. The mt‐nucleoids were then mapped on to the mitochondrial network to identify if they were in a normal or damaged mitochondrion. Similar analysis was performed on UPRmt markers, except mean intensity of the UPRmt marker for each segmented mitochondrion was used instead of intensity at the mt‐nucleoid. On a per cell basis, the average intensity of mt‐nucleoid markers from damaged mitochondria was compared to the average intensity of mt‐nucleoid markers from undamaged mitochondria. Average values were computed for each technical replicate and then averaged for all experimental replicates. Intensity values were normalized to the average intensity values of the WT‐1 cell line.
Colocalization of mutants was quantified using Pearson’s correlation coefficient for specified pairs of channels using intensity values for all pixels within the brightest z‐plane for each segmented mt‐nucleoid (Dunn, Kamocka et al, 2011). Correlation coefficients were averaged for all mt‐nucleoids (n ≈ 20–40) in 4–5 cells per construct.
Western blot
Cells were collected, washed in PBS, dissolved in 2 × Laemmli Sample Buffer (Bio‐Rad), and denatured for 5–10 min at 95°C and stored at − 20°C. Samples were loaded onto a 10‐well 4–12% Bis‐Tris Protein Gel at 150 V for 1 h and transferred to a membrane via wet transfer at 250 mA for 90 min. Antibodies used were Lamin A/C (Santa Cruz, 376248), β‐actin (Sigma, A2228), TFAM (Sigma, HPA063684), HSPD1 (Sigma, HPA001523), and ATF5 (Abcam, ab60126). Western blots were performed on three‐six independent experimental replicates, and band intensities were quantified using Bio‐Rad Image Lab software.
mtDNA copy number
Total DNA (both genomic and mitochondrial) was purified using the DNeasy Blood and Tissue Kit (Qiagen). DNA concentration was measured using a Denovix. qPCR was performed on 50 ng of DNA in a 20 µl mixture on 96‐well plates using a CFX Real‐Time PCR instrument (Bio‐Rad). Primers for mtDNA tRNALeu(UUR) (5′ CACCCAAGAACAGGGTTTGT‐3′ and 5′ TGGCCATGGGTATGTTGTTAA‐3′) and nuclear 18S rDNA (5′ TAGAGGGACAAGTGGCGTTC‐3′, 5′‐CGCTGAGCCAGTCAGTGT‐3′) were used as previously described (Xiong et al, 2016). Six independent experimental replicates were performed, each with three technical replicates. Bio‐Rad CFX Maestro Software was used to determine the average C t for each experiment. The relative mtDNA copy number was obtained as 2 × 2ΔCt where (Gonzalez‐Hunt et al, 2016).
Estimating concentration of TFAM and DNA in a single mitochondrion
A single mitochondrial nucleoid has a diameter of ~100 nm and is estimated to have 1 molecule of mtDNA and ~1,000 molecules of TFAM (Kukat et al, 2011). Based on these measurements, the volume of a spherical mitochondrial nucleoid is ~5 × 10−22 m3 or ~5 × 10−19 l, which yields a concentration of approximately or ~100 µg/µl. Mitochondrial DNA is roughly 16 kb, which has a molar mass of ~10 Mg/mol, which yields or 30 µg/µl inside a mitochondrial nucleoid. This gives a molar ratio of ~0.001 and a mass ratio of DNA/TFAM ≈ 0.3 within the nucleoid. To determine the diffuse concentration of mtDNA and TFAM if a single mitochondrial nucleoid was fully solubilized in the mitochondrion, we assume a unit mitochondrion to be a cylinder with 1 µm in length and 0.2 µm in radius, which gives a unit volume of ~1 × 10−19 m3 or ~1 × 10−16 l. Assuming all 1,000 molecules of TFAM and 1 molecule of mtDNA are uniformly distributed in the unit mitochondrion, we obtain concentrations of or 0.5 µg/µl and or 0.2 µg/µl, which also gives a molar ratio of ~0.001 and a mass ratio of DNA/TFAM ≈ 0.3 that is comparable to the conditions used in our in vitro experiments.
qPCR analysis
RNA was isolated from primary cells using a RNeasy Plus Micro Kit (Qiagen), and cDNA was obtained from 1 µg of RNA after reverse transcription using an iScript cDNA Synthesis Kit (Bio‐Rad). 10 µl RT–PCR reactions containing a SYBR Green Master Mix (Bio‐Rad) were dispensed using an Echo 525 Liquid Handler (Labcyte) into Hard‐Shell 384‐well PCR plates (Bio‐Rad). qPCR was performed on a CFX384 Touch Real‐Time PCR Detection System (Bio‐Rad), and Ct values were obtained using CFX Maestro Software (Bio‐Rad). Each qPCR reaction had three technical replicates, and data were averaged for three biological replicates (Taylor, Nadeau et al, 2019). 12S and mt‐COI transcripts were analyzed relative to ACTB, ATF5 was normalized relative to TUBB1, and all other UPRmt transcripts were analyzed relative to TBP. Primers are listed in Appendix Table S1.
Mitochondrial oxygen consumption
Cells were plated at 50,000/well in 96‐well Seahorse cell culture plate containing DMEM media with 15% FBS, 2 mM GlutaMAX, and 1 mM Sodium Pyruvate (Gibco). Cells were incubated overnight for 18 h before media were replaced with Seahorse base XF medium containing 2 mM GlutaMAX, 1 mM Sodium Pyruvate, and 25 mM Glucose, at pH 7.4. Cell plates were then placed into a non‐CO2 incubator at 37°C for 1 h for equilibration before running the XF96 Extracellular Flux Analyzer standard mitochondrial stress test. The three injection drugs were administered as follows: Port A) Oligomycin: 2.5 µM; Port B) FCCP 2 µM; Port C) Antimycin A 2 µM. In our studies, the outermost wells in each 96‐well plate were not used as a precaution against hypothetical temperature inequality/edge effects. At the end of seahorse run, the medium was removed, and 50 µl of 1 × RIPA lysis buffer was added to each well, the plate was frozen at − 20°C, then thawed at 4°C on a shaker for 1 h, the lysate was collected into 1.5 ml microcentrifuge tube, spun down, and the supernatant was collected to measure protein concentration by standard BCA method. The determined protein concentrations from each well were used to normalize oxygen consumption rates.
Mitochondrial membrane potential and reactive oxygen species generation
Mitochondrial membrane potential (MMP) and mitochondrial reactive oxygen species (ROS) were measured by a BD FACSCanto II flow cytometer. HGPS cells were plated in 6‐well plate at a confluence of 200 K cells/well. On the day of experiment, cells were washed in PBS and harvested by trypsinization, washed once more with PBS, and resuspended in DMEM without phenol red (Invitrogen). Cells were incubated with dyes tetramethylrhodamine, methyl ester (TMRM) (40 nM for 15 min) to detect MMP, and mitoSOX (3 μM for 30 min) for mitochondrial ROS (all from Life Technologies), fluorescence was determined with flow cytometry from 1 × 104 cells. Data were analyzed using FCS Express 4 software.
Statistical methods
For high‐throughput imaging experiments with fixed cells, three technical replicates were performed, each with ≥ 5 fields of view, and averaged to obtain the measurement for that experiment. These experiments were repeated independently three times (n = 3), and the values for each experiment were averaged. For high‐throughput experiments with in vitro droplets, each condition was assayed per well, with ≥ 9 fields of view. Measurements from each field of view were averaged together per experiment, and each experiment was repeated independently at least three times. For all experiments, measurements were repeated on ≥ 3 days. For FRAP experiments, all FRAP measurements were pooled together and averaged. For mitochondrial functional testing, three experimental replicates (each with technical replicates) were performed for each cell line and averaged. The data for WT and HGPS cell lines were pooled for some measurements as indicated. For all experiments, error was reported as standard deviation or standard error of the mean as indicated. Number of damaged mitochondria, mitochondrial membrane potential, and mitochondrial ROS were compared using a single‐factor analysis of variance (ANOVA) statistical test, and the P‐value for the test statistic was determined using Excel and R and was reported in the figure legend. Pairs of means were determined statistically significant using the two‐sided Fisher’s least significant difference (LSD) method and indicated graphically. P‐values were determined for basal respiration, maximal respiratory capacity, and reserve capacity using a two‐sided Student’s t‐test based on the standard deviation from experimental replicates.
Author contributions
MF performed live/fixed cell microscopy and in vitro experiments and analysis in all main figures and majority of supplementary figures. TGD, JT, DLC, and VAB performed and/or analyzed mitochondrial Seahorse assays, mitochondrial membrane potential and mitochondrial ROS measurements on normal and HGPS cells (Appendix Fig S7P–U). TM and MF planned experiments, discussed results, and wrote the manuscript. All authors revised the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Movie EV9
Movie EV10
Source Data for Appendix
Review Process File
Acknowledgements
We thank the members of the Misteli lab for discussion and help with experimental design; L. Schiltz and A. Schibler for initial help with protein purification; J. Jones and M. Taylor for help with final protein expression and purification at the NIH/NCI/CCR Protein Production Core; T. Karpova and D. Ball for help with Structured Illumination Microscopy and Laser Scanning Confocal Microscopy as part of the NIH/NCI/CCR LRBGE Optical Imaging Core; G. Pegoraro and L. Ozbun for help with high‐throughput imaging and automated liquid handling at the NIH/NCI/CCR High‐Throughput Imaging Facility (HiTIF); and K. M. McKinnon for help with cell sorting as part of the NIH/NCI/CCR FACS Core Facility. Research in the Misteli lab is supported by funding from the Intramural Research Program of the National Institutes of Health (NIH), National Cancer Institute, and Center for Cancer Research (1‐ZIA‐BC010309); MF is supported by a Postdoctoral Research Associate Training (PRAT) fellowship from the National Institute of General Medical Sciences (NIGMS, 1Fi2GM128585‐01); TD, JT, DC, and VB are supported by the NIA Intramural Research Program of the NIH (AG000727).
The EMBO Journal (2021) 40: e107165.
Data availability
No primary datasets have been generated and deposited.
References
- Alán L, Špaček T, Reguera DP, Jabůrek M, Ježek P (2016) Mitochondrial nucleoid clusters protect newly synthesized mtDNA during Doxorubicin‐and Ethidium Bromide‐induced mitochondrial stress. Toxicol Appl Pharmacol 302: 31–40 [DOI] [PubMed] [Google Scholar]
- Ashley N, Harris D, Poulton J (2005) Detection of mitochondrial DNA depletion in living human cells using PicoGreen staining. Exp Cell Res 303: 432–446 [DOI] [PubMed] [Google Scholar]
- Ashley N, Poulton J (2009) Anticancer DNA intercalators cause p53‐dependent mitochondrial DNA nucleoid re‐modelling. Oncogene 28: 3880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T (2005) Mitochondria, oxidants, and aging. Cell 120: 483–495 [DOI] [PubMed] [Google Scholar]
- Banani SF, Lee HO, Hyman AA, Rosen MK (2017) Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18: 285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban‐Ishihara R, Ishihara T, Sasaki N, Mihara K, Ishihara N (2013) Dynamics of nucleoid structure regulated by mitochondrial fission contributes to cristae reformation and release of cytochrome c. Proc Natl Acad Sci 110: 11863–11868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barja G, Herrero A (2000) Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J 14: 312–318 [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2006) The protein data bank, 1999. In International Tables for Crystallography Volume F: Crystallography of biological macromolecules, pp 675–684. Springer. [Google Scholar]
- Boehning M, Dugast‐Darzacq C, Rankovic M, Hansen AS, Yu T, Marie‐Nelly H, McSwiggen DT, Kokic G, Dailey GM, Cramer P (2018) RNA polymerase II clustering through carboxy‐terminal domain phase separation. Nat Struct Mol Biol 25: 833–840 [DOI] [PubMed] [Google Scholar]
- Bogenhagen DF (2012) Mitochondrial DNA nucleoid structure. Biochim Biophys Acta 1819: 914–920 [DOI] [PubMed] [Google Scholar]
- Bogenhagen DF, Rousseau D, Burke S (2008) The layered structure of human mitochondrial DNA nucleoids. J Biol Chem 283: 3665–3675 [DOI] [PubMed] [Google Scholar]
- Boija A, Klein IA, Sabari BR, Dallagnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM et al (2018) Transcription factors activate genes through the phase‐separation capacity of their activation domains. Cell 175: 1842–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Jülicher F, Hyman AA (2009) Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324: 1729–1732 [DOI] [PubMed] [Google Scholar]
- Brangwynne CP, Mitchison TJ, Hyman AA (2011) Active liquid‐like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc Natl Acad Sci 108: 4334–4339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratic A, Larsson N‐G (2013) The role of mitochondria in aging. J Clin Invest 123: 951–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer LR, Friddle R, Noy A, Baldwin E, Martin SS, Corzett M, Balhorn R, Baskin RJ (2003) Packaging of single DNA molecules by the yeast mitochondrial protein Abf2p. Biophys J 85: 2519–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TA, Tkachuk AN, Shtengel G, Kopek BG, Bogenhagen DF, Hess HF, Clayton DA (2011) Superresolution fluorescence imaging of mitochondrial nucleoids reveals their spatial range, limits, and membrane interaction. Mol Cell Biol 31: 4994–5010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger G, Gray MW, Lang BF (2003) Mitochondrial genomes: anything goes. Trends Genet 19: 709–716 [DOI] [PubMed] [Google Scholar]
- Cadenas E, Davies KJ (2000) Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med 29: 222–230 [DOI] [PubMed] [Google Scholar]
- Chan SS, Copeland WC (2009) DNA polymerase gamma and mitochondrial disease: understanding the consequence of POLG mutations. Biochim Biophy Acta 1787: 312–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XJ, Butow RA (2005) The organization and inheritance of the mitochondrial genome. Nat Rev Genet 6: 815 [DOI] [PubMed] [Google Scholar]
- Cho W‐K, Spille J‐H, Hecht M, Lee C, Li C, Grube V, Cisse II (2018) Mediator and RNA polymerase II clusters associate in transcription‐dependent condensates. Science 361: 412–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong S, Dugast‐Darzacq C, Liu Z, Dong P, Dailey GM, Cattoglio C, Heckert A, Banala S, Lavis L, Darzacq X (2018) Imaging dynamic and selective low‐complexity domain interactions that control gene transcription. Science 361: eaar2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker JC, Grier DG (1996) Methods of digital video microscopy for colloidal studies. J Colloid Interface Sci 179: 298–310 [Google Scholar]
- Cunha S, Woldringh CL, Odijk T (2001) Polymer‐mediated compaction and internal dynamics of isolated Escherichia coli nucleoids. J Struct Biol 136: 53–66 [DOI] [PubMed] [Google Scholar]
- DeBoy E, Puttaraju M, Jailwala P, Kasoji M, Cam M, Misteli T (2017) Identification of novel RNA isoforms of LMNA. Nucleus 8: 573–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon SC, Dorman CJ (2010) Bacterial nucleoid‐associated proteins, nucleoid structure and gene expression. Nat Rev Microbiol 8: 185–195 [DOI] [PubMed] [Google Scholar]
- Driscoll MK, Albanese JL, Xiong Z‐M, Mailman M, Losert W, Cao K (2012) Automated image analysis of nuclear shape: What can we learn from a prematurely aged cell? Aging 4: 119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KW, Kamocka MM, McDonald JH (2011) A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol 300: C723–C742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farge G, Laurens N, Broekmans OD, Van Den Wildenberg SM, Dekker LC, Gaspari M, Gustafsson CM, Peterman EJ, Falkenberg M, Wuite GJ (2012) Protein sliding and DNA denaturation are essential for DNA organization by human mitochondrial transcription factor A. Nat Commun 3: 1–9 [DOI] [PubMed] [Google Scholar]
- Farge G, Mehmedovic M, Baclayon M, van den Wildenberg SM, Roos WH, Gustafsson CM, Wuite GJ, Falkenberg M (2014) In vitro‐reconstituted nucleoids can block mitochondrial DNA replication and transcription. Cell Rep 8: 66–74 [DOI] [PubMed] [Google Scholar]
- Feric M, Brangwynne CP (2013) A nuclear F‐actin scaffold stabilizes ribonucleoprotein droplets against gravity in large cells. Nat Cell Biol 15: 1253–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feric M, Vaidya N, Harmon TS, Mitrea DM, Zhu L, Richardson TM, Kriwacki RW, Pappu RV, Brangwynne CP (2016) Coexisting liquid phases underlie nucleolar subcompartments. Cell 165: 1686–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Nunnari J (2014) Mitochondrial form and function. Nature 505: 335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka Y, Alam JM, Noshiro D, Mouri K, Ando T, Okada Y, May AI, Knorr RL, Suzuki K, Ohsumi Y (2020) Phase separation organizes the site of autophagosome formation. Nature 578: 301–305 [DOI] [PubMed] [Google Scholar]
- Gibson BA, Doolittle LK, Schneider MW, Jensen LE, Gamarra N, Henry L, Gerlich DW, Redding S, Rosen MK (2019) Organization of chromatin by intrinsic and regulated phase separation. Cell 179: 470–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Hunt CP, Rooney JP, Ryde IT, Anbalagan C, Joglekar R, Meyer JN (2016) PCR‐based analysis of mitochondrial DNA copy number, mitochondrial DNA damage, and nuclear DNA damage. Curr Protoc Toxicol 67: 20.11.1–20.11.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon LB, Rothman FG, López‐Otín C, Misteli T (2014) Progeria: a paradigm for translational medicine. Cell 156: 400–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillén‐Boixet J, Kopach A, Holehouse AS, Wittmann S, Jahnel M, Schlüßler R, Kim K, Trussina IR, Wang J, Mateju D (2020) RNA‐induced conformational switching and clustering of G3BP drive stress granule assembly by condensation. Cell 181: 346–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson CM, Falkenberg M, Larsson N‐G (2016) Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem 85: 133–160 [DOI] [PubMed] [Google Scholar]
- Hyman AA, Weber CA, Jülicher F (2014) Liquid‐liquid phase separation in biology. Annu Rev Cell Dev Biol 30: 39–58 [DOI] [PubMed] [Google Scholar]
- Iborra FJ (2007) Can visco‐elastic phase separation, macromolecular crowding and colloidal physics explain nuclear organisation? Theor Biol Med Model 4: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara T, Ban‐Ishihara R, Maeda M, Matsunaga Y, Ichimura A, Kyogoku S, Aoki H, Katada S, Nakada K, Nomura M (2015) Dynamics of mitochondrial DNA nucleoids regulated by mitochondrial fission is essential for maintenance of homogeneously active mitochondria during neonatal heart development. Mol Cell Biol 35: 211–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdain AA, Koppen M, Wydro M, Rodley CD, Lightowlers RN, Chrzanowska‐Lightowlers ZM, Martinou J‐C (2013) GRSF1 regulates RNA processing in mitochondrial RNA granules. Cell Metab 17: 399–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantidze OL, Razin SV (2020) Weak interactions in higher‐order chromatin organization. Nucleic Acids Res 48: 4614–4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman BA, Durisic N, Mativetsky JM, Costantino S, Hancock MA, Grutter P, Shoubridge EA (2007) The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid‐like structures. Mol Biol Cell 18: 3225–3236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazak L, Reyes A, Holt IJ (2012) Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol 13: 659–671 [DOI] [PubMed] [Google Scholar]
- King C, Sengupta P, Seo AY, Lippincott‐Schwartz J (2020) ER membranes exhibit phase behavior at sites of organelle contact. Proc Natl Acad Sci 117: 7225–7235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopek BG, Shtengel G, Xu CS, Clayton DA, Hess HF (2012) Correlative 3D superresolution fluorescence and electron microscopy reveal the relationship of mitochondrial nucleoids to membranes. Proc Natl Acad Sci 109: 6136–6141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubben N, Adriaens M, Meuleman W, Voncken JW, van Steensel B, Misteli T (2012) Mapping of lamin A‐and progerin‐interacting genome regions. Chromosoma 121: 447–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubben N, Zhang W, Wang L, Voss TC, Yang J, Qu J, Liu G‐H, Misteli T (2016) Repression of the antioxidant NRF2 pathway in premature aging. Cell 165: 1361–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucej M, Butow RA (2007) Evolutionary tinkering with mitochondrial nucleoids. Trends Cell Biol 17: 586–592 [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA et al (2005) Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309: 481–484 [DOI] [PubMed] [Google Scholar]
- Kukat C, Davies KM, Wurm CA, Spåhr H, Bonekamp NA, Kühl I, Joos F, Polosa PL, Park CB, Posse V (2015) Cross‐strand binding of TFAM to a single mtDNA molecule forms the mitochondrial nucleoid. Proc Natl Acad Sci 112: 11288–11293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukat C, Larsson N‐G (2013) mtDNA makes a U‐turn for the mitochondrial nucleoid. Trends Cell Biol 23: 457–463 [DOI] [PubMed] [Google Scholar]
- Kukat C, Wurm CA, Spåhr H, Falkenberg M, Larsson N‐G, Jakobs S (2011) Super‐resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci 108: 13534–13539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, Narlikar GJ (2017) Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 547: 236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SC, Uchiyama LF, Nunnari J (2016) ER‐mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 353: aaf5549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ruan Y, Zhang K, Jian F, Hu C, Miao L, Gong L, Sun L, Zhang X, Chen S (2016) Mic60/Mitofilin determines MICOS assembly essential for mitochondrial dynamics and mtDNA nucleoid organization. Cell Death Differ 23: 380–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Protter DS, Rosen MK, Parker R (2015) Formation and maturation of phase‐separated liquid droplets by RNA‐binding proteins. Mol Cell 60: 208–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Lee J, Nie X, Li M, Morozov YI, Venkatesh S, Bogenhagen DF, Temiakov D, Suzuki CK (2013) Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Mol Cell 49: 121–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima Y, Goto Y‐I, Kaguni LS (2010) Mitochondrial Lon protease regulates mitochondrial DNA copy number and transcription by selective degradation of mitochondrial transcription factor A (TFAM). Proc Natl Acad Sci 107: 18410–18415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSwiggen DT, Mir M, Darzacq X, Tjian R (2019) Evaluating phase separation in live cells: diagnosis, caveats, and functional consequences. Genes Dev 33: 1619–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamikawa T, Sriratana A, Williams DA, Bowser DN, Hill JS, Nagley P (1999) Chloromethyl‐X‐rosamine (MitoTracker Red) photosensitises mitochondria and induces apoptosis in intact human cells. J Cell Sci 112: 2419–2430 [DOI] [PubMed] [Google Scholar]
- Mir M, Bickmore W, Furlong EE, Narlikar G (2019) Chromatin topology, condensates and gene regulation: shifting paradigms or just a phase? Development 146: dev182766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T (2020) The Self‐Organizing Genome: Principles of Genome Architecture and Function. Cell 183: 28–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP (2015) Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163: 123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monterroso B, Zorrilla S, Sobrinos‐Sanguino M, Robles‐Ramos MA, López‐Álvarez M, Margolin W, Keating CD, Rivas G (2019) Bacterial FtsZ protein forms phase‐separated condensates with its nucleoid‐associated inhibitor SlmA. EMBO Rep 20: e45946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo HB, Kaiser JT, Chan DC (2011) The mitochondrial transcription and packaging factor TFAM imposes a U‐turn on mitochondrial DNA. Nat Struct Mol Biol 18: 1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo HB, Lovely GA, Phillips R, Chan DC (2014) Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat Commun 5: 3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phair RD, Gorski SA, Misteli T (2003) Measurement of dynamic protein binding to chromatin in vivo, using photobleaching microscopy, Methods in Enzymology pp 393–414. Elsevier; [DOI] [PubMed] [Google Scholar]
- Ramos ES, Motori E, Brüser C, Kühl I, Yeroslaviz A, Ruzzenente B, Kauppila JH, Busch JD, Hultenby K, Habermann BH (2019) Mitochondrial fusion is required for regulation of mitochondrial DNA replication. PLoS Genet 15: e1008085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remesh SG, Verma SC, Chen J‐H, Ekman AA, Larabell CA, Adhya S, Hammel M (2020) Nucleoid remodeling during environmental adaptation is regulated by HU‐dependent DNA bundling. Nat Commun 11: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera‐Torres J, Acín‐Perez R, Cabezas‐Sánchez P, Osorio FG, Gonzalez‐Gómez C, Megias D, Cámara C, López‐Otín C, Enríquez JA, Luque‐García JL (2013) Identification of mitochondrial dysfunction in Hutchinson‐Gilford progeria syndrome through use of stable isotope labeling with amino acids in cell culture. J Proteomics 91: 466–477 [DOI] [PubMed] [Google Scholar]
- Rubio‐Cosials A, Sydow JF, Jiménez‐Menéndez N, Fernández‐Millán P, Montoya J, Jacobs HT, Coll M, Bernadó P, Solà M (2011) Human mitochondrial transcription factor A induces a U‐turn structure in the light strand promoter. Nat Struct Mol Biol 18: 1281 [DOI] [PubMed] [Google Scholar]
- Sabari BR, Dall'Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC (2018) Coactivator condensation at super‐enhancers links phase separation and gene control. Science 361: eaar3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DW, Kedersha N, Lee DS, Strom AR, Drake V, Riback JA, Bracha D, Eeftens JM, Iwanicki A, Wang A (2020) Competing protein‐RNA interaction networks control multiphase intracellular organization. Cell 181: 306–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanulli S, Trnka M, Dharmarajan V, Tibble R, Pascal B, Burlingame A, Griffin P, Gross J, Narlikar G (2019) HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature 575: 390–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead WT, Gladfelter AS (2019) The control centers of biomolecular phase separation: how membrane surfaces, PTMs, and active processes regulate condensation. Mol Cell 76: 295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, Karpen GH (2017) Phase separation drives heterochromatin domain formation. Nature 547: 241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Youle RJ, Finkel T (2016) The mitochondrial basis of aging. Mol Cell 61: 654–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan P, Simpson DA, Sambare C, Duffy S, Lechner J, Dastane A, Dervan EW, Vallabh N, Chelerkar V, Deshpande M (2015) Whole‐mitochondrial genome sequencing in primary open‐angle glaucoma using massively parallel sequencing identifies novel and known pathogenic variants. Genet Med 17: 279 [DOI] [PubMed] [Google Scholar]
- Suomalainen A, Battersby BJ (2018) Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat Rev Mol Cell Biol 19: 77 [DOI] [PubMed] [Google Scholar]
- Taylor RW, Turnbull DM (2005) Mitochondrial DNA mutations in human disease. Nat Rev Genet 6: 389–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SC, Nadeau K, Abbasi M, Lachance C, Nguyen M, Fenrich J (2019) The ultimate qPCR experiment: producing publication quality, reproducible data the first time. Trends Biotechnol 37: 761–774 [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly‐Y M, Gidlöf S, Oldfors A, Wibom RJN (2004) Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429: 417 [DOI] [PubMed] [Google Scholar]
- West AP, Khoury‐Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520: 553–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong TS, Rajagopalan S, Freund SM, Rutherford TJ, Andreeva A, Townsley FM, Petrovich M, Fersht AR (2009) Biophysical characterizations of human mitochondrial transcription factor A and its binding to tumor suppressor p53. Nucleic Acids Res 37: 6765–6783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong ZM, Choi JY, Wang K, Zhang H, Tariq Z, Wu D, Ko E, LaDana C, Sesaki H, Cao K (2016) Methylene blue alleviates nuclear and mitochondrial abnormalities in progeria. Aging Cell 15: 279–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN (2010) PONDR‐FIT: a meta‐predictor of intrinsically disordered amino acids. Biochim Biophys Acta 1804: 996–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakes FM, Van Houten B (1997) Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci 94: 514–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P, Mathieu C, Kolaitis R‐M, Zhang P, Messing J, Yurtsever U, Yang Z, Wu J, Li Y, Pan Q (2020) G3BP1 is a tunable switch that triggers phase separation to assemble stress granules. Cell 181: 325–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Movie EV9
Movie EV10
Source Data for Appendix
Review Process File
Data Availability Statement
No primary datasets have been generated and deposited.