

Abstract
Rotavirus causes severe diarrheal disease in children by broadly dysregulating intestinal homeostasis. However, the underlying mechanism(s) of rotavirus-induced dysregulation remains unclear. We found that rotavirus-infected cells produce paracrine signals that manifested as intercellular calcium waves (ICWs), observed in cell lines and human intestinal enteroids. Rotavirus ICWs were caused by the release of extracellular adenosine diphosphate (ADP) that activated P2Y1 purinergic receptors on neighboring cells. ICWs were blocked by P2Y1 antagonists or CRISPR/Cas9 knockout of the P2Y1 receptor. Blocking the ADP signal reduced rotavirus replication, inhibited rotavirus-induced serotonin release and fluid secretion, and reduced diarrhea severity in neonatal mice. Thus, rotavirus exploited paracrine purinergic signaling to generate ICWs that amplified the dysregulation of host cells and altered gastrointestinal physiology to cause diarrhea.
One Sentence Summary:
Rotavirus triggers extracellular release of ADP from infected cells that dysregulates nearby uninfected cells.
Rotavirus (RV) causes severe diarrhea and vomiting in children worldwide, resulting in ~258 million diarrhea episodes and ~128,000 deaths annually (1). The mechanisms of RV diarrhea are multifactorial and not completely understood. RV infects the enterocytes and enteroendocrine cells at the tip and middle of villi in the small intestine (2–4). Not all cells susceptible to RV are infected, and diarrhea occurs before the onset of histopathologic changes (2, 5–7). During infection, RV dysregulates host cell calcium (Ca2+) signaling pathways to increase cytosolic Ca2+, which is required for RV replication. The RV nonstructural protein 4 (NSP4) drives these changes in Ca2+ homeostasis as both an endoplasmic reticulum-localized viroporin and a secreted enterotoxin (8–10). These perturbations to host Ca2+ signaling activate autophagy, disrupt the cytoskeleton and tight junctions, and trigger fluid secretion pathways (9, 11–14).
A long-held concept in how RV infection causes life-threatening diarrhea is that RV-infected cells release potent signaling molecules that dysregulate neighboring, uninfected cells (6, 15, 16). This notion is based on previous observations of increased signaling molecules during RV infection, including enterotoxin NSP4, prostaglandins (PGE2), and nitric oxide (NO) (9, 17–19). In this theory, enterotoxin NSP4 binds to neighboring, uninfected enterocytes to activate Ca2+-activated chloride channels and cause secretory diarrhea (20–22), and PGE2 and NO further activate fluid secretion processes (23, 24). Simultaneously, dysregulation of neighboring enteroendocrine cells triggers Ca2+-dependent release of serotonin, which stimulates the enteric nervous system both to activate vomiting centers in the central nervous system and to activate secretory reflex pathways in the gastrointestinal tract (4, 7). Thus, this concept of RV-induced diarrhea addresses how limited infection at the middle-to-upper villi may cause widespread dysregulation of host physiology and life-threatening disease. However, this cell-to-cell functional signaling has not been directly observed during a RV infection, and the signaling pathways remain incompletely understood.
Herein we found that RV-infected cells signal to uninfected cells via an extracellular purinergic signaling pathway. This pathway was a dominant driver of observed RV disease processes, including replication, upregulation of PGE2- and NO-producing enzymes, serotonin secretion, fluid secretion, and diarrhea in a neonatal mouse model. Thus, viruses may exploit host paracrine signaling pathways to amplify pathophysiology.
Low multiplicity infection reveals intercellular calcium waves
RV significantly increases cytosolic Ca2+ during infection and disrupts host Ca2+-dependent processes to cause disease (25–27). We used African Green monkey kidney MA104 cells stably expressing the genetically encoded calcium indicator (GECI) GCaMP5G or GCaMP6s to observe changes in cytosolic Ca2+ during RV infection using live-cell time-lapse epifluorescence imaging. We mock- or RV (strain SA114F)-infected MA104-GCaMP cells at a low multiplicity of infection (MOI 0.01) to distinguish Ca2+ changes in infected cells compared to neighboring, uninfected cells. RV-infected cells were identified using immunofluorescence (IF) staining for RV antigen after imaging, and these IF images were superimposed on the time-lapse movies (Fig. 1A). While mock-infected cells had minimal changes in GCaMP fluorescence, RV-infected cells exhibited large fluctuations in fluorescence, and thus cytosolic Ca2+, as discrete signaling events (28). These signaling events increased cytosolic Ca2+ in the RV antigen-positive cell, and we observed successive increases in cytosolic Ca2+ in neighboring, uninfected cells over a time interval of 30 s (Fig. 1A, Movie S1). We quantified the Ca2+-eliciting signal from infected to uninfected cells using a previously described criterion in which changes in normalized relative fluorescence greater than 5% constituted a “Ca2+ spike” (28). To systematically and reproducibly characterize the signals in RV-infected and neighboring, uninfected cells, we visualized the cell monolayer as a grid, similar to previously described analysis techniques (29–31) (Fig. 1B, Fig. S1A). The number and average magnitude of the RV-induced Ca2+ transients decreased in the neighboring 5 (NB+5) and 10 (NB+10) cells (Fig. 1B, blue and purple traces). Quantification of the number and magnitude of Ca2+ spikes of the infected, the NB+5, and the NB+10 cells showed a significant decrease with distance from the infected cell (Fig. 1C,D). Thus, RV-infected cells signaled to surrounding uninfected cells and elicited a Ca2+ signaling response.
Figure 1. Rotavirus-infected cells trigger calcium signaling in neighboring uninfected cells.
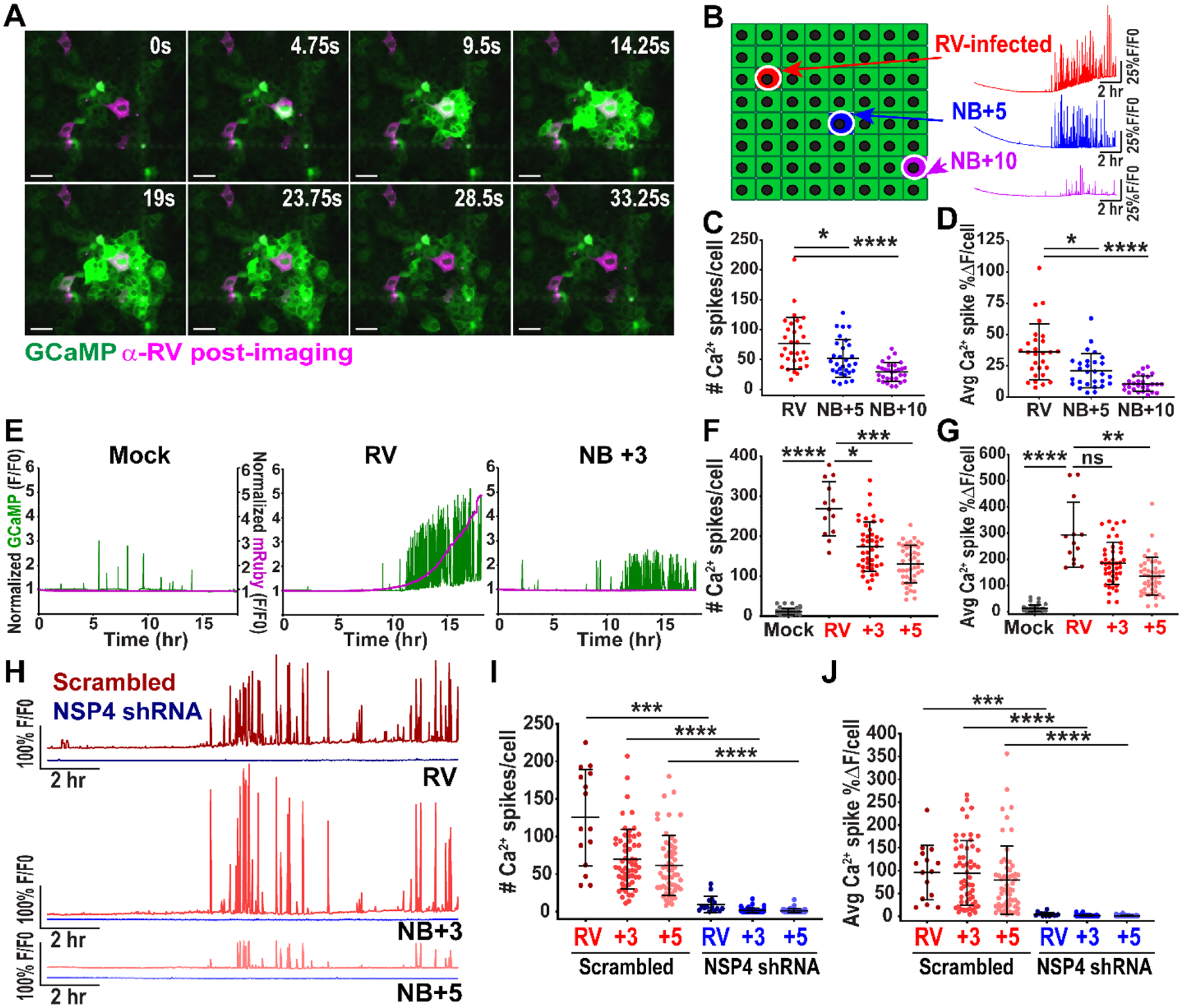
(A) MA104-GCaMP cells infected with rotavirus (RV SA114F) at MOI 0.01 and imaged at ~12 hpi. Cells fixed and immunostained at 24 hpi for RV (pink), images superimposed. Scale bar = 100 μm. (B) Cell monolayer schematic: RV-infected cell (red) and neighboring (NB) cells 5 (blue) and 10 (purple) cells away. Representative single-cell Ca2+ traces with normalized GCamP fluorescence (F/F0). (C) Ca2+ spikes/cell in RV-infected and NB cells, (n=30 cells) and (D) average magnitude of Ca2+ spikes/cell in RV-infected and NB cells (n= 28 cells, data representative of N=3 independent experiments). (E) Representative Ca2+ traces from MA104-GCaMP cells mock- or RV (SA11cl3-mRuby3)-infected and the NB+3 cell imaged 3–21 hpi. Left axis: GCaMP normalized fluorescence (green), Right axis: mRuby3 normalized fluorescence (pink) (F) Ca2+ spikes and (G) average magnitude of Ca2+ spikes/cell in RV-infected, NB +3, and +5 cells (data combined from N=3 independent experiments) (H) Representative Ca2+ traces from MA104-GCaMP-shRNA scrambled or NSP4 cells infected with RV (SA11cl3-mRuby3) and NB+3 and NB+5 cells imaged from ~4–20 hpi. (I) Ca2+ spikes in RV-infected and neighboring (NB) +3 and +5 uninfected cells in scrambled or NSP4 shRNA cells and (K) average magnitude of Ca2+ spikes/cell in RV-infected and NB cells (data combined from N=3 independent experiments). (C-D, F-G, I-K) Kruskal-Wallis with Dunn’s multiple comparisons test used. Data represented as mean ± SD, (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001)
Next we sought to confirm that the Ca2+ signaling in the neighboring cells was not due to RV infection undetected by antibody staining. MA104-GCaMP cells were mock-inoculated or infected with a recombinant SA11 clone 3 strain expressing a mRuby3 fluorescent reporter (SA11cl3-mRuby3) (28), at MOI 0.01 and imaged for ~3–22 hours post-infection (hpi), conditions we used throughout these studies unless noted otherwise. We found no increase in mRuby3 fluorescence in the mock-infected cells, and there was a steep increase in fluorescence in RV-infected cells by hour 6–12 of imaging that correlated with increased Ca2+ signaling (Fig. 1E, Movie S2). Importantly, the NB+3 cell also had robust Ca2+ signaling that paralleled the infected cell, but the lack of increase in mRuby3 fluorescence demonstrated it was not infected (Fig. 1E). There was a similar decrease in the number of Ca2+ spikes and average Ca2+ spike magnitude in the NB+3 and NB+5 cells that correlated with increasing distance from the infected cell (Fig. 1 F,G). Thus, the Ca2+ signaling in the neighboring cell was not due to infection of that cell but due to signals activated by a nearby RV-infected cell.
We next determined whether knockdown of NSP4 would reduce the Ca2+ signals in infected and uninfected cells because RV-induced changes in Ca2+ homeostasis are driven by NSP4. We used MA104-GCaMP cells expressing NSP4-targeted shRNA or a non-targeted scrambled shRNA (28). RV infection of the MA104-GCaMP-shRNA cells with SA114F decreased NSP4 expression by ~95% and significantly decreased Ca2+ signaling (28) (Fig. S1B,C). We infected the MA104-GCaMP cells expressing shRNA2, because they had the greatest percent knockdown, using RV SA11cl3-mRuby3, which has the same NSP4 sequence as RV SA114F. The Ca2+ signaling frequency and Ca2+ spike magnitudes were significantly decreased in the NSP4-targeted shRNA MA104-GCaMP cells, both in RV-infected cells and NB+3 and NB+5 uninfected cells (Fig. 1H–J, Movie S3). Thus, the RV-induced Ca2+ signal required expression of RV NSP4 and dysregulation of host Ca2+ homeostasis in the infected cell.
ADP and the P2Y1 receptor mediate RV-induced intercellular calcium waves
The pattern of signaling observed between RV-infected cells and uninfected cells was a phenomenon called intercellular calcium waves (ICWs), in which increases in cytosolic Ca2+ occur in an expanding circular pattern from a central initiating cell (31–33). Several molecules have been implicated in ICW propagation and may communicate via gap junctions or through paracrine signaling (33). To test if RV-induced ICWs propagated via gap junctions, we infected MA104-GCaMP cells and treated with DMSO vehicle or the gap junction blockers 18β-glycyrrhetinic acid and TAT-Gap19. There was no significant difference in the Ca2+ spikes in neighboring cells between the mock-, 18β-glycyrrhetinic acid-, or TAT-Gap19-treated cells, supporting that the RV-induced ICWs did not propagate via gap junctions (Fig. 2A). This finding was consistent with a negative scrape loading/dye transfer assay, in which the lack of dye beyond the cells immediately adjacent to the scrape demonstrate a lack of gap junction intercellular communication (34, 35) (Fig. S2A). Next we tested if the extracellular enterotoxin form of NSP4 may be the signaling molecule responsible for propagating the ICWs. We treated RV-infected MA104-GCaMP cells with purified anti-NSP4 rabbit peptide antisera (amino acids 120–147) or anti-NSP4 monoclonal antibody 622 that bind to the enterotoxin domain and neutralize eNSP4-mediated Ca2+ flux (Fig. 2B) (22). Purified M60, a non-neutralizing anti-VP7 monoclonal antibody, was used as a control (36). We did not observe a significant reduction in Ca2+ spikes in the neighboring cells treated with anti-NSP4 antibodies, suggesting that enterotoxin NSP4 was not responsible for the RV-induced ICWs (Fig. 2B, Movie S4). Previous studies found increased PGE2 and NO during RV infection, and thus they potentially mediate RV-induced ICWs (17–19). However, adding PGE2 or the NO donor NOC-7 to MA104-GCaMP cells did not elicit a Ca2+ response, demonstrating that these molecules could not directly transmit the RV-induced ICWs (Fig. S2B–E). Thus, ICW propagation was mediated by an unidentified signaling molecule.
Figure 2. Rotavirus induces intercellular calcium waves by activating purinergic signaling.
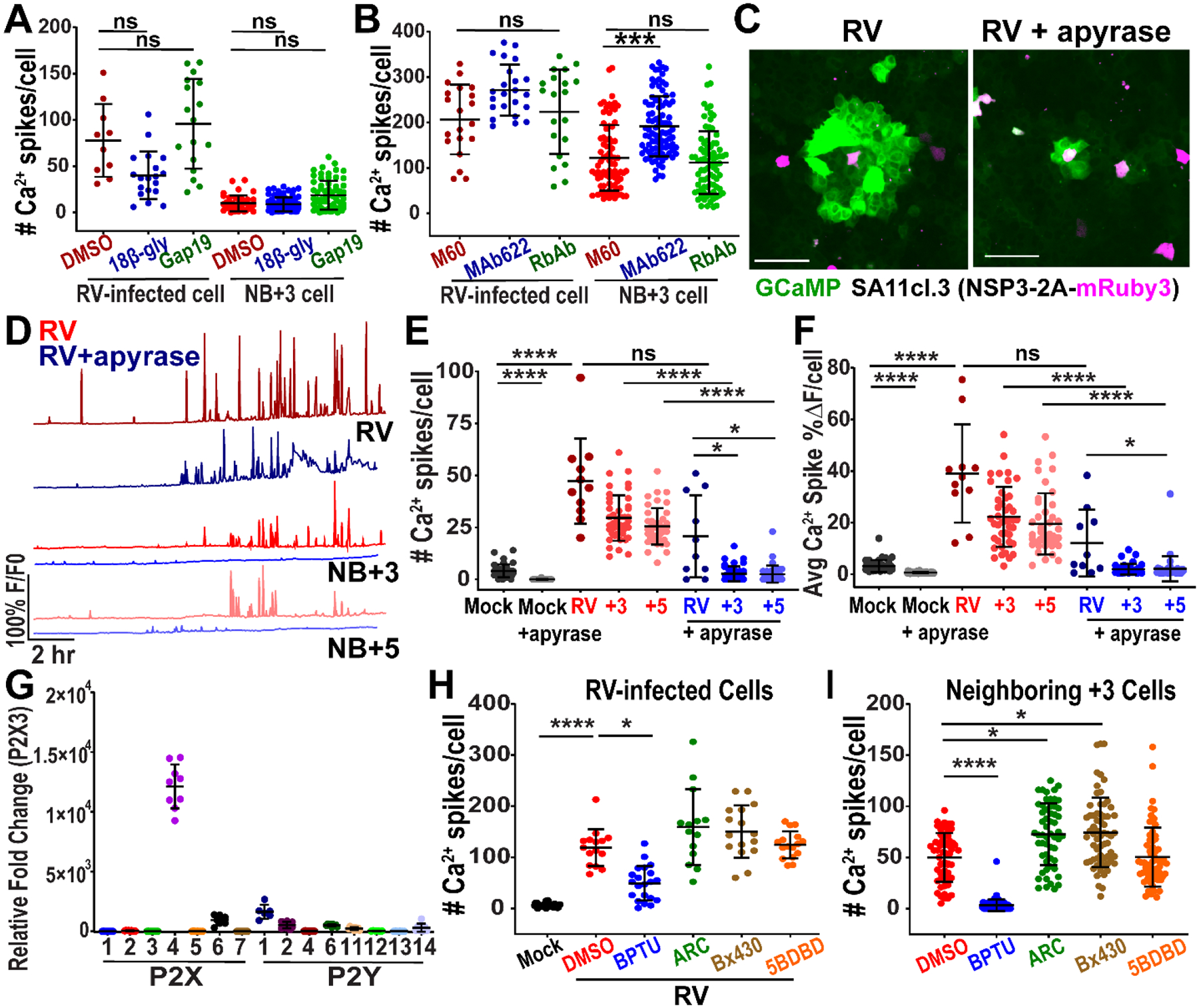
(A-B) Number of Ca2+ spikes in MA104-GCaMP cells in RV-infected (SA11cl3-mRuby3) at MOI 0.01 and neighboring (NB)+3 cell, treated with: (A) DMSO, 30μM 18β-glycyrrhetinic acid (18β-gly), or 50μM TAT-Gap19 (Gap19) and imaged ~8–24 hpi or (B) anti-VP7 M60 MAb, anti-NSP4 MAb 622, or rabbit anti-NSP4 antisera 120–147 (Rb Ab) and imaged ~8–24 hpi. (data combined from N=3 independent experiments) (C) Representative images of intercellular calcium waves in MA104-GCaMP cells infected with RV (SA11cl3-mRuby3) at MOI 0.1 and mock- or 10U/mL apyrase-treated (5 U/mL apyrase VI and 5U/mL apyrase VII) and imaged ~10 hpi with (D) representative Ca2+ traces from ~8–25 hpi. (E) Ca2+ spikes in RV-infected and NB cells. (F) Average magnitude of Ca2+ spikes/cell in RV-infected and NB cells, (data combined from N=3 independent experiments). (G) qPCR of purinergic receptor mRNA normalized to 18S mRNA and fold change relative to P2X3 mRNA transcript levels in MA104 cells, (data combined from N=3 independent experiments) (H-I) Ca2+ spikes in MA104-GCaMP cells RV (SA114F)-infected (H) or neighboring (NB) cells (I) and treated with DMSO, 10μM BPTU, 10μM AR-C 118925XX (ARC), 10μM Bx430, or 10μM 5-BDBD (data combined from N=3 independent experiments). (A-B, E-F, H-I) Kruskal-Wallis with Dunn’s multiple comparisons test used. Scale bar = 100 μm. Data represented as mean ± SD, (*p<0.05, ****p<0.0001).
We then sought to determine whether adenosine triphosphate (ATP) was the signaling molecule responsible for the RV-induced ICWs. ATP is an important extracellular messenger and a common mediator of ICW signaling (33). We mock- or RV (SA11cl3-mRuby3)-infected MA104-GCaMP cells and after infection mock-treated or treated with apyrase, an enzyme that degraded the purines ATP and adenosine diphosphate (ADP). Apyrase-treated wells showed a dramatic decrease in the area of Ca2+ signaling compared to untreated wells (Fig. 2C, Movie S5). With apyrase treatment, the Ca2+ signaling frequency and amplitude were noticeably decreased in the neighboring, uninfected +3 and +5 cells (Fig. 2D), which resulted in a significant decrease in the number of Ca2+ spikes and average Ca2+ spike magnitude in the neighboring +3 and +5 cells, but not in the RV-infected cells (Fig. 2E–F). Thus, extracellular ATP/ADP propagated ICWs in RV infection.
Purinergic signaling, i.e., nucleotides as extracellular signaling molecules, is a key signaling pathway in intestinal physiology and involves multiple receptor subtypes. MA104 cells showed greatest expression of the purinergic receptors P2X4, P2X6, P2Y1, P2Y2, and P2Y14 (Fig. 2G). Relative mRNA expression levels of purinergic receptors did not substantially change with RV infection (Fig. S3A, Table S1). We first tested the ligand-gated ion channel P2X4 and the Gq/11 protein-coupled receptors P2Y1 and P2Y2 because they are strongly activated by ATP or ADP (33). We treated RV(SA11cl3-mRuby3)-infected MA104-GCaMP cells with small molecule inhibitors (Bx430 and 5BDBD for P2X4, BPTU for P2Y1, and AR-C 118925XX for P2Y2). The P2X4 inhibitors Bx430 and 5BDBD or the P2Y2 inhibitor AR-C 118925XX had no effect on RV-induced Ca2+ signaling. The P2Y1 blocker BPTU significantly decreased Ca2+ spikes in both the RV-infected cell and NB+3 cells, and this decrease was dose-dependent (Fig. 2H–I, Fig. S3C, Movie S6). We tested the specificity of BPTU for the P2Y1 receptor and AR-C 118925XX for the P2Y2 receptor, which are strongly activated by ADP and ATP, respectively. The P2Y1 inhibitor BPTU significantly reduced the Ca2+ response in MA104-GCaMP cells from ADP, but not from ATP (Fig. S3D). Similarly, the P2Y2 inhibitor AR-C 118925XX significantly reduced the Ca2+ response from ATP, but not from ADP, supporting their specificity (Fig. S3D).
Finally, we tested if a different RV strain, Rhesus rotavirus (RRV), also elicited ICWs from RV-infected cells by the same mechanism. We mock- or RRV-infected MA104-GCaMP cells (MOI 0.05) and found that RRV-infected cells elicited ICWs that increased cytosolic Ca2+ in uninfected NB+3 and NB+5 cells (Fig. S3E). Treatment with apyrase or BPTU significantly decreased the ICW signals only in the uninfected NB+3 and NB+5 cells (Fig. S3E). Thus, cells infected with RRV also triggered ICWs through extracellular release of ADP and P2Y1 receptor activation, demonstrating this phenomenon was common to multiple RV strains.
RV induces intercellular calcium waves in human intestinal enteroids
MA104 cells are a robust model system to study RV replication and form a simple flat epithelium that facilitates microscopy studies. However, they are neither human nor intestinal in origin. To determine whether RV also causes ICWs in intestinal epithelial cells, we used jejunum-derived human intestinal enteroids (HIEs) stably expressing the GECI GCaMP6s (jHIEGCaMP6s) (28). HIEs are a non-cancerous in vitro model system established from human intestinal epithelial stem cells (37). They recapitulate the different epithelial cell types and support RV infection and replication, which makes them a biologically relevant system to study gastrointestinal pathophysiology (38–40). We infected jHIE-GCaMP6s monolayers with mock inoculum or the human RV strain Ito and imaged at 4 hpi (Fig. 3A, Movie S7). We observed ICWs in both mock- and RV-infected jHIE-GCaMP6s monolayers; however, the areas of Ca2+ signaling were larger and occurred with greater frequency in the RV-infected monolayer than in the mock-infected or “basal” monolayer. To verify infection, we fixed the monolayers at 24 hpi, immunostained for RV antigen, and observed positive staining in the RV-infected monolayers (Fig. 3B). Thus, ICWs occurred under basal conditions in human intestinal epithelial cell models, but infection with RV increased the size of the ICWs.
Figure 3. Rotavirus infection induces intercellular calcium waves in human intestinal enteroids.
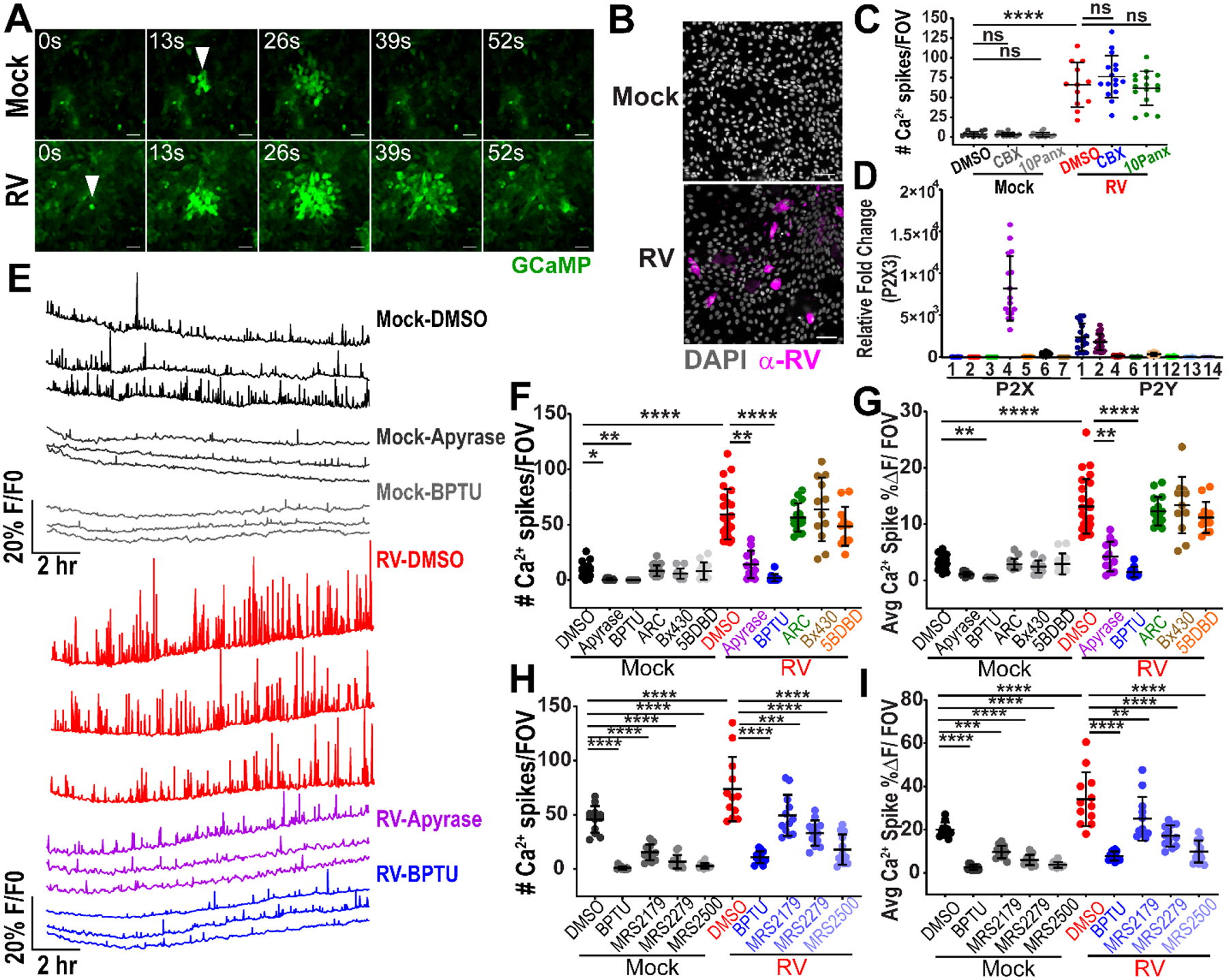
(A) J3 jHIE-GCaMP6s monolayers mock- or RV (Ito)-infected produce intercellular calcium waves originating at a cell (arrowheads), imaged at ~4 hpi. (B) IF images of (J3)HIE-GCaMP6s monolayers mock- or RV-infected, fixed at 24 hpi, and immunostained for RV antigen (pink) and counterstained with DAPI (gray). (C) Ca2+ spikes per field-of-view (FOV), in (J3)HIEs mock- or RV-infected and treated with vehicle, 100 μM carbenoxolone (CBX), or 10 μM 10Panx at 8–22 hpi (data combined from N=3 independent experiments). (D) qPCR of purinergic receptor mRNA normalized to 18S mRNA and fold change relative to P2X3 mRNA transcript levels in (J3)HIE monolayers (data combined from N=3 independent experiments). (E) Representative Ca2+ traces/FOV of (J3)HIE-GCaMP6s monolayers either mock- or RV-infected and treated with DMSO, 100 U/mL apyrase, or 10 μM BPTU between 8.5–22 hpi. Mock- or RV-infected (J3)HIEs, treated with DMSO, apyrase, or purinergic receptor blockers. (F) Ca2+ spikes/FOV in (J3)HIE-GCaMP6s monolayers treated with DMSO, 100U/mL apyrase (50 U/mL apyrase VI and 50U/mL apyrase VII), 10 μM BPTU, 10 μM AR-C 118925XX, 10 μM Bx430, or 10 μM 5-BDBD and (G) average magnitude of Ca2+ spikes/FOV for 8.5–22 hpi, (data combined from N=3 independent experiments) (H) Ca2+ spikes/FOV of patient J2 jHIE-GCaMP6s monolayers mock- or RV-infected and treated with DMSO, 10 μM BPTU, 10 μM MRS2179, 10 μM MRS2279, or 10 μM MRS2500 and (I) average magnitude of Ca2+ spikes/FOV for 8–22 hpi, (data combined from N=3 independent experiments). (C,H,I) One-way ANOVA with Bonferroni’s and (F,G) Kruskal-Wallis with Dunn’s multiple corrections test used. Scale bar = 50 μm. Data represented as mean ± SD, (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001)
Next, we investigated the mechanism of the RV-induced ICWs in the HIEs. To test gap junction transmission, we treated mock- or RV (Ito)-infected jHIE-GCaMP6s monolayers with DMSO vehicle, the glycyrrhetinic acid-derivative carbenoxolone (CBX), or the pannexin blocker 10Panx. We were unable to perform single-cell analysis for these experiments because individual jHIE cells were much smaller and more mobile than MA104 cells. Instead we measured the GCaMP6s fluorescence over the whole field-of-view (FOV) (~455 μm2). While we observed statistically greater Ca2+ spikes in RV-infected than in mock-infected monolayers, CBX or 10Panx did not significantly decrease the number of Ca2+ spikes/FOV in mock- or RV-infected monolayers (Fig. 3C). The scrape loading/dye transfer was also negative in jHIE monolayers, which further demonstrated a lack of gap junction intercellular communication (Fig. S2A). Adding PGE2 or NOC-7 to jHIE-GCaMP6s cells also did not elicit a Ca2+ response (Fig. S2F–I). Thus, RV-induced ICWs in HIEs do not occur via gap junctions or through PGE2/NO, and basal ICWs do not appear to have significant gap junction transmission.
In jHIE monolayers, P2X4, P2Y1, and P2Y2 had the highest expression levels, similar to that of MA104 cells (Fig. 2G, 3D). The relative mRNA expression levels of purinergic receptors did not substantially change with RV (Ito) infection (Fig. S3B, Table S1). To test the involvement of the different purinergic receptors in ICWs, we mock- or RV (Ito)-infected jHIE-GCaMP6s monolayers and treated with DMSO vehicle, apyrase, BPTU, AR-C 118925XX, Bx430, or 5BDBD and found that apyrase and BPTU treatment decreased the Ca2+ signaling and amplitude in RV-infected monolayers (Fig. 3E, Movie S8). Quantification verified a significant decrease in the number of Ca2+ spikes/FOV and average Ca2+ spike magnitude with apyrase or BPTU treatment compared to the DMSO vehicle control in the RV-infected monolayers (Fig. 3F,G). Interestingly, BPTU and apyrase treatment also reduced the number of Ca2+ spikes in mock-inoculated monolayers (Fig. 3F). This suggested there was purinergic receptor-induced Ca2+ signaling in jHIEs under basal conditions that was exploited during RV infection.
Genetic variation may influence whether RV-infection can cause ICWs because jHIEs are human-derived cultures. We created an additional jHIE-GCaMP6s line derived from a different individual (J2). We mock- and RV-infected these jHIE-GCaMP6s monolayers and treated with a panel of P2Y1 receptor blockers (BPTU, MRS2179, MRS2279, and MRS2500). All of the P2Y1 blockers significantly decreased the number of Ca2+ spike/FOV and average Ca2+ spike magnitude when compared to DMSO vehicle control for both the mock- or RV-infected monolayers (Fig. 3H,I). Thus, RV-induced ICWs in intestinal epithelial cells occurred by extracellular ADP signaling and the P2Y1 receptor, the same as in MA104 cells.
CRISPR/Cas9 P2Y1 receptor knockout reduces intercellular calcium waves
The P2Y1 purinergic receptor exhibits greater activation with ADP than with ATP, suggesting that ADP mediated the ICWs in RV infection (41). This was consistent with the reduced cytoplasmic ATP concentrations found in RV-infected cells (42). To test whether ADP was the signaling molecule associated with the ICWs, we infected MA104-GCaMP cells with RV (SA114F) and then treated with apyrase grade VI, which had a high ATPase/ADPase activity ratio, or apyrase grade VII, which had a low ATPase/ADPase activity ratio. Treatment with apyrase grade VII, but not grade VI, significantly decreased the number of Ca2+ spikes and the spike magnitude in RV-infected wells, supporting that ADP was the signaling molecule in RV-induced ICWs (Fig. 4A,B). To further test that the P2Y1 receptor was required for RV-induced ICWs, we genetically knocked out the P2Y1 receptor in MA104-GCaMP cells using lentivirus encoding CRISPR/Cas9 with small-guide RNAs targeted for the P2Y1 receptor gene (MA104-GCaMP-P2Y1ko). Genotyping analysis revealed several mutations in the sequence downstream of the guide RNA in 3 clonal populations of cells (Fig. S4A,B). To test the P2Y1 knockout phenotype, we added ADP to parental and MA104-GCaMP-P2Y1ko cells and found a significantly decreased Ca2+ response in the knockout cells compared to the parental cells, consistent with a lack of P2Y1 receptor function (Fig. 4C).
Figure 4. CRISPR/Cas9 knockout of P2Y1 receptor reduces intercellular calcium waves.
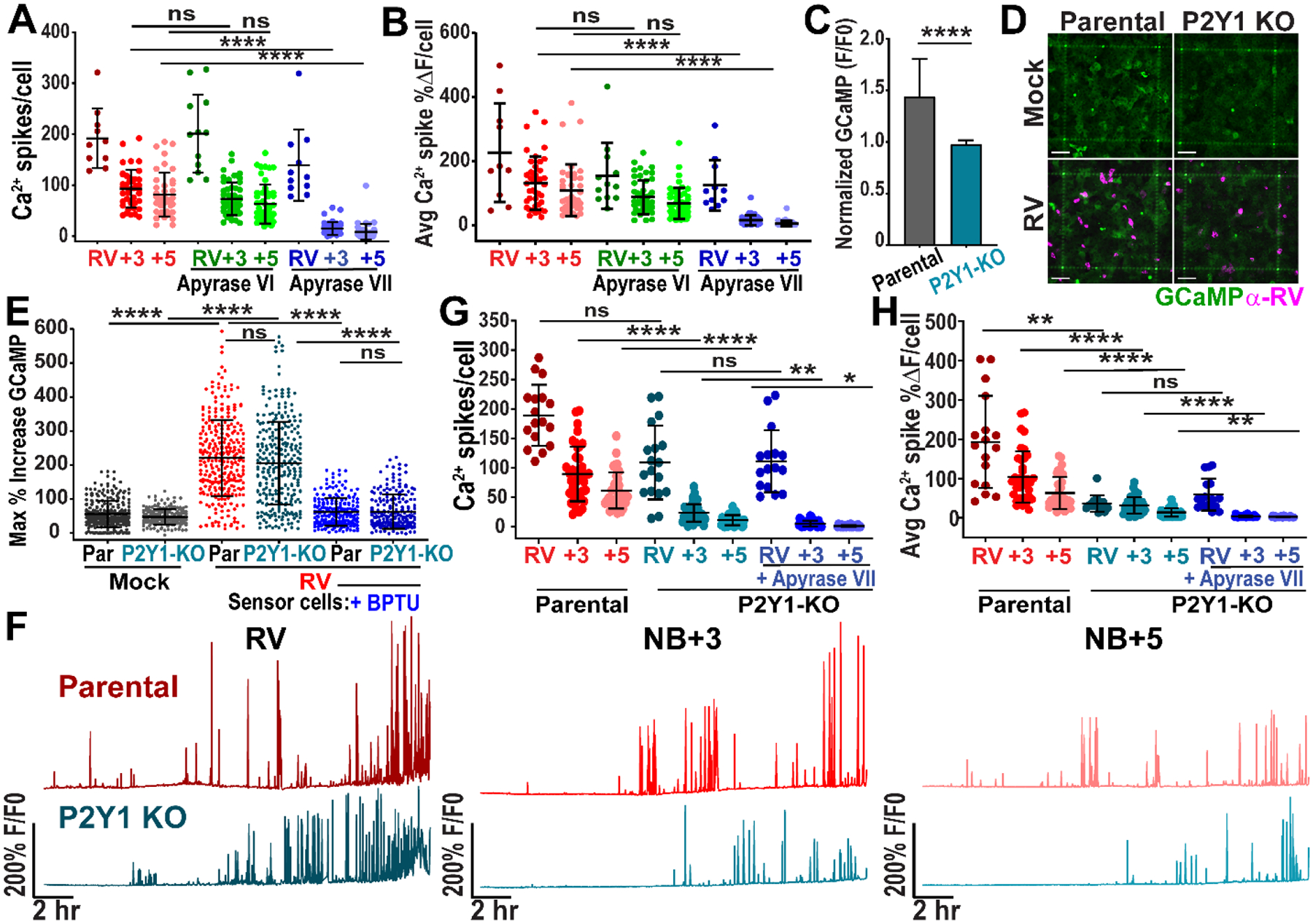
(A-B) MA104-GCaMP cells infected with rotavirus (RV SA114F) at MOI 0.01 and mock- or treated with apyrase grade VI or grade VII, imaged ~3–25 hpi. (A) Ca2+ spikes per RV-infected cell and neighboring (NB) +3 and +5 cells and (B) the average magnitude of Ca2+ spikes/cell in RV-infected and NB cells (data combined from N=3 independent experiments). (C) Normalized GCaMP fluorescence increase in parental or P2Y1 knockout (KO) MA104-GCaMP cells treated with 1 nM ADP (n=27 cells, data combined from N=3 independent experiments, Mann-Whitney statistical test). (D) Parental or P2Y1 knockout MA104-GCaMP monolayers mock- or RV-infected, fixed at 24 hpi, and immunostained for RV antigen (pink). Scale bar = 100 μm. (E) Maximum % increase in GCaMP fluorescence of MA104-GCaMP cells (+/− 10 μM BPTU) after addition of supernatant from mock- or RV (SA114F)-infected parental (Par) or P2Y1 KO MA104-GCaMP cells (MOI 5, harvested ~5–6 hpi). (n≥210 cells per condition, data combined from N=3 independent experiments). (F) Representative Ca2+ traces of parental or P2Y1 knockout cells infected with RV (SA11cl3-mRuby3). (G) Ca2+ spikes/cell in RV-infected or NB+3 or NB+5 cell and (H) average magnitude of Ca2+ spikes/cell (data combined from N=3 independent experiments). (A-B, E, G-H) Kruskal-Wallis with Dunn’s multiple comparisons test used. Data represented as mean ± SD, (*p< 0.05, **p<0.01, ****p<0.0001).
Next, we sought to determine if knockout of the P2Y1 receptor would also prevent ICWs during RV infection. Fixing and immunostaining for RV antigen at 24 hpi showed positive staining for RV in parental and P2Y1ko cells confirming that the P2Y1 knockout did not prevent infection (Fig. 4D). We next tested whether RV-infected MA104-GCaMP-P2Y1ko cells still released ADP to confirm that the P2Y1 knockout was specific to receptor function and did not affect ADP release, which could be responsible for decreased ICWs. We mock or RV (SA114F)-infected parental or P2Y1ko cells, washed off inoculum, and added ARL 67156, an ecto-ATPase inhibitor, to block degradation of released nucleotides from cellular ectoATPases. Approximately 5–6 hpi, we transferred the mock- or infected-cell supernatant to uninfected MA104-GCaMP cells to measure the Ca2+ response as a “sensor” of released ADP. We found a significantly greater increase in GCaMP fluorescence in RV-infected than in mock-infected supernatants (Fig. 4E). However, there was no significant difference in the extent of that increase between parental- or P2Y1ko-derived supernatants (Fig. 4E). To show specificity of this Ca2+ signal to ADP/P2Y1, we treated the uninfected sensor MA104-GCaMP cells with BPTU, which significantly lessened the increase in GCaMP fluorescence from both parental and P2Y1ko supernatants (Fig. 4E). Thus, the Ca2+ response from both parental- and P2Y1ko-derived supernatants was predominantly due to ADP. Also, there was no significant difference between adding parental- or P2Y1ko-derived supernatants to the BPTU-treated sensor cells, supporting that RV-infected P2Y1ko cells still release ADP (Fig. 4E). Lastly, we tested whether RV-infected P2Y1ko cells had decreased ICWs. We mock- or RV (SA114F)-infected MA104-GCaMP (parental) or MA104-GCaMP-P2Y1ko cells. In the P2Y1ko cells, there was decreased Ca2+ signaling, Ca2+ spikes, and average Ca2+ spike magnitude in the NB+3 and NB+5 cells compared with the parental, though not in the RV-infected cell (Fig. 4F–H, Movie S9). Additional treatment of apyrase VII to the P2Y1ko cells only modestly decreased the Ca2+ spikes in neighboring cells (Fig. 4G–H).
Similarly, we tested the effect of genetic knockout of the P2Y1 receptor on ICWs in RV-infected jHIEs (jHIE-GCaMP6s-P2Y1ko). Genotyping analysis revealed two P2Y1 knockout enteroid populations comprised of an insertional mutation and a deletion in the sequence downstream of the guide RNA (Fig. S4C,D). Phenotypic knockout was confirmed by a reduced Ca2+ response to ADP in jHIE-GCaMP6s-P2Y1ko enteroids compared to parental enteroids (Fig. 5A). We created an additional jHIE-GCaMP6s enteroid line with CRISPR/Cas9 knockout of the P2Y2 receptor (jHIE-GCaMP6s-P2Y2ko). Genotyping analysis of the P2Y2ko enteroids detected two alleles with deletion mutations downstream of the guide RNA sequence (Fig. S5A,B). To test the P2Y2 knockout phenotype, we added non-hydrolyzable ATP-γS to parental and P2Y2ko enteroids and found a significantly decreased Ca2+ response in the P2Y2ko jHIEs (Fig. 5B). As with the P2Y1ko MA104 cells, immunostaining the jHIE monolayers for RV antigen at 24 hpi showed similar levels of positive staining for RV in parental, P2Y1ko, and P2Y2ko enteroids, demonstrating equivalent susceptibility to RV infection (Fig. 5C,D). Mock- or RV (Ito)-infection of the parental, jHIE-GCaMP6s-P2Y1ko, or jHIE-GCaMP6s-P2Y2ko resulted in significantly fewer Ca2+ signaling events, Ca2+ spikes/FOV, and smaller average Ca2+ spike magnitude in the P2Y1 knockout jHIEs, but not in P2Y2 knockout jHIEs (Fig. 5E–G, Movie S10). Interestingly, there also was significantly fewer Ca2+ spikes/FOV and average Ca2+ spike magnitude in the P2Y1 knockout enteroids compared to parental or P2Y2 knockout enteroids in mock-inoculation (Fig. 5F,G). Thus, ADP and the P2Y1 receptor mediated the ICWs in RV infection and reduced basal ICW signaling.
Figure 5. CRISPR/Cas9 knockout of P2Y1 receptor in jHIEs reduces intercellular calcium waves.
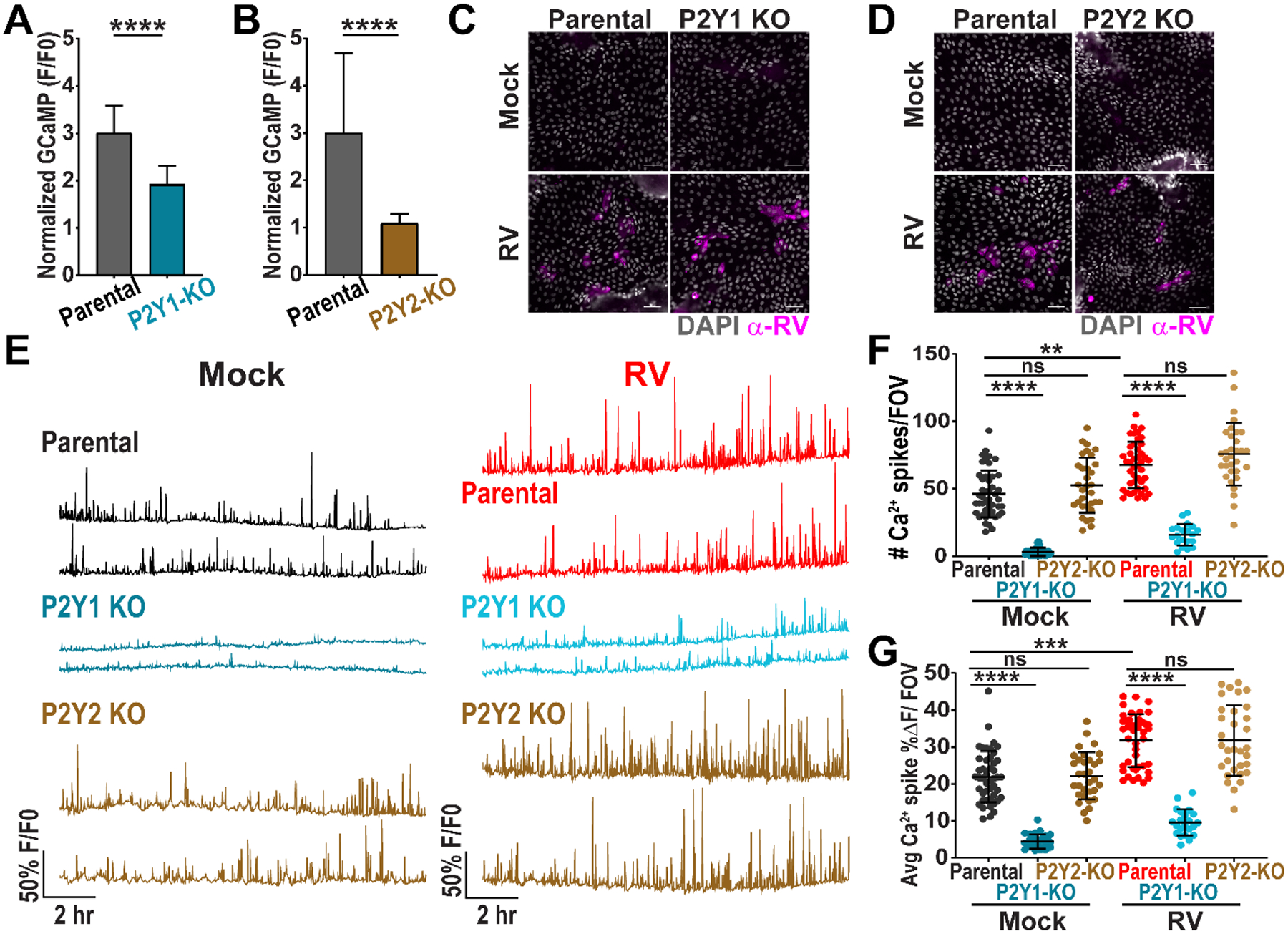
(A-B) Normalized GCaMP fluorescence increase in (A) parental or P2Y1 knockout (KO) (J2)HIE-GCaMP6s monolayers treated with 10 nM ADP (n=75 cells, data combined from N=3 independent experiments) and (B) parental or P2Y2 KO (J2)HIE-GCaMP6s monolayers treated with 10 μM ATP-γS (n=30 cells, data representative of N=3 independent experiments). (C-D) Parental or P2Y1 KO jHIE-GCaMP6s or parental or P2Y2 KO jHIE-GCaMP6s monolayers mock- or RV-infected, fixed at 24 hpi, and immunostained for RV antigen (pink) and counterstained with DAPI (gray). (E) Representative Ca2+ traces per field-of-view (FOV) of mock- or RV-infected parental, P2Y1 KO jHIE-GCaMP6s, or P2Y2 KO jHIE-GCaMP6s monolayers. (F) Ca2+ spikes/FOV and (G) average magnitude of Ca2+ spikes/FOV. (A-B) Mann-Whitney test or (F-G) Kruskal-Wallis with Dunn’s multiple corrections test used. Scale bar = 50 μm. Data represented as mean ± SD, (***p<0.001, ****p<0.0001).
Purinergic signaling increases RV replication and expression of IL-1α, iNOS, and COX2
Extracellular ATP reduces the replication of several viruses through activation of P2X7 receptors, which specifically bind ATP (43, 44). Less is known about the role of ADP in cellular signaling or its effects on viral infection and replication. We first tested the effect of purinergic blockers on RV replication. New infectious yield from RV (SA114F)-infected MA104 cells was significantly decreased when treating cells with apyrase or BPTU after inoculation by ~92% and ~90%, respectively (Fig. 6A). No significant decrease was observed when adding AR-C 118925XX, Bx430, 5BDBD, or the non-selective P2 receptor antagonists suramin and PPADS (Fig. 6A). Furthermore, RV infectious yield was also reduced from RV (SA11cl3-mRuby)-infected MA104-GCaMP-P2Y1ko cells compared to parental MA104-GCaMP cells by ~95% (Fig. 6B). Thus, blocking ADP signaling and the P2Y1 receptor decreased RV replication.
Figure 6. Purinergic signaling contributes to rotavirus disease processes.
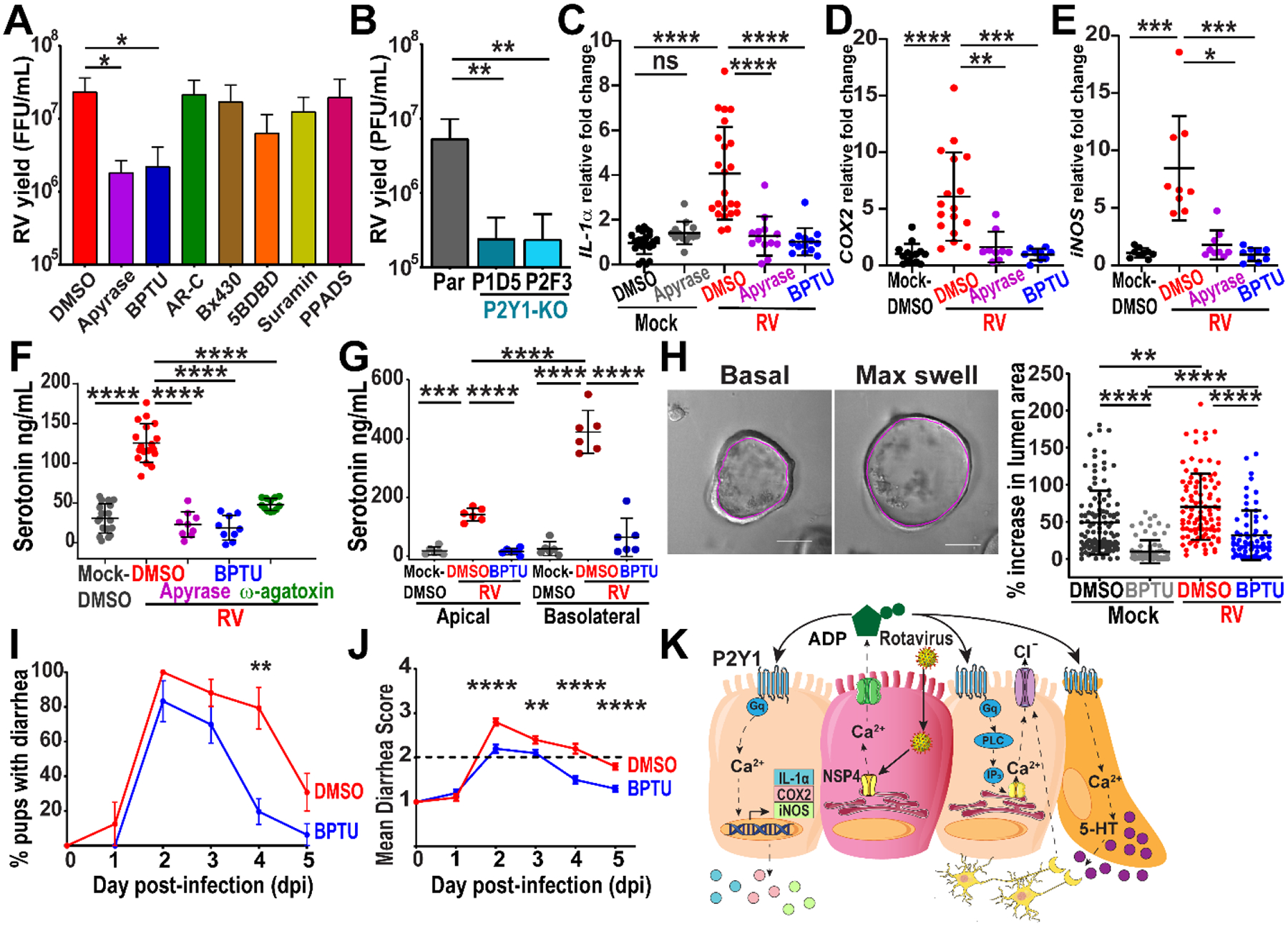
(A) Rotavirus yield from SA114F-infected MA104 cells treated with DMSO vehicle, 10U/mL apyrase, 10μM BPTU, 10μM AR-C 118925XX, 20μM Bx430, 20μM 5-BDBD, 300μM suramin, or 10μM PPADS by fluorescent focus assay (data combined from N=3 independent experiments). (B) Plaque assay yield of RV (SA114F)-infected MA104-GCaMP (Par) or MA104-GCaMP P2Y1 knockout (KO) cells (data combined from N=3 independent experiments). (C-E) qPCR of mRNA transcripts normalized to 18S mRNA transcripts and fold change relative to the Mock-DMSO transcript levels in (J3)HIE monolayers mock- or RV (Ito)-infected and treated with DMSO vehicle, 100U/mL apyrase, or 10 μM BPTU (data combined from N=3 independent experiments). (F-G) Serotonin secretion from RV (Ito)-infected jHIE (F) monolayers and (G) transwells treated with DMSO, 100U/mL apyrase, 10μM BPTU, or 300nM ω-agatoxin (data combined from N=3 independent experiments). (H) Enteroid swelling assay: 3D jHIE-GCaMP6s enteroids mock- or RV (Ito)-infected and treated with DMSO or 10μM BPTU. Cross-sectional area of the internal lumen (pink outline) used for percent increase between basal and max swelling (left panels). Scale bar = 100μm. (n ≥ 68 HIEs per condition, data combined from N=3 independent experiments) (I-K) C57Bl/6J mouse pups with diarrhea infected with Rhesus RV and vehicle- or BPTU-treated and the (J) mean diarrhea score. (data combined from 4 cages of each condition, 26–30 pups total per condition, mean ± SEM) (K) Summary model of RV-induced ICWs mediated by extracellular ADP. (A-B, F-G) One-way ANOVA with Bonferroni multiple comparisons test, (C-J) Kruskal-Wallis with Dunn’s multiple comparisons, or (I-J) Mann-Whitney tests used. (A-H) Data represented as mean ± SD, (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Extracellular ATP is also known to have pro-inflammatory functions and activate components of the innate immune system such as macrophages (45, 46). RV increases expression and release of the inflammatory cytokine interleukin-1α (IL-1α), which is highly expressed in epithelial cells (47, 48). Furthermore, strong cytosolic Ca2+ agonists can induce the secretion of IL-1α independent of inflammasome activation (49). To determine if RV-induced ICWs increase IL-1α, we mock- or RV (Ito)-infected jHIE monolayers and treated with apyrase or BPTU. RV infection significantly increased IL-1α expression levels over mock infection, and treatment with apyrase or BPTU inhibited this increase (Fig. 6C). Thus, RV-induced ICWs and ADP signaling increased IL-1α expression and mediated the inflammatory response from HIEs.
Because increased PGE2 and NO have been found during RV infection, we also investigated if RV-induced ICWs could stimulate their production (17–19). Purinergic signaling stimulates cyclooxygenase-2 (COX2) expression and PGE2 release from Caco-2 and rat-derived IEC-6 cell lines and activates inducible nitric oxide synthase (iNOS) expression and NO production (50, 51). We mock- or RV (Ito)-infected jHIE monolayers and treated with apyrase or BPTU. RV significantly increased both COX2 and iNOS expression levels over mock-infection, and treatment with apyrase or BPTU inhibited this increase (Fig. 6D–E). Both RV-induced ICWs and ADP signaling increased the expression of COX2 and iNOS and thus potentially caused the increased PGE2 and NO observed in RV infections.
Blocking purinergic signaling reduces serotonin and fluid secretion
RV infection causes a multifactorial secretory diarrhea, and two-thirds of the fluid secretion are attributed to enteric nervous system (ENS) activation (7). RV infection stimulates the release of serotonin from enterochromaffin cells to activate the ENS and blocking serotonin decreases diarrhea, making serotonin release a key mechanism for RV-induced diarrhea (4, 52). To determine if RV-induced ADP signaling stimulated serotonin secretion, we mock- or RV (Ito)-infected jHIE monolayers and treated with DMSO vehicle, apyrase, or BPTU. After 24 hr, we found significantly increased serotonin in the supernatant of RV-infected jHIE monolayers compared to mock-infection, which was significantly inhibited by treatment with apyrase or BPTU (Fig. 6F). Furthermore, treatment of RV-infected monolayers with ω-agatoxin, a P-type voltage-gated Ca2+ channel blocker, significantly inhibited the RV-induced secretion of serotonin, supporting that the measured serotonin was from cellular release rather than RV-induced cell death (Fig. 6F). We found similar results when infecting jHIE transwells, a more polarized model of epithelium, with RV. Treatment with BPTU significantly decreased serotonin measured in both the apical and basolateral compartments compared with DMSO vehicle (Fig. 6G). Thus, ADP signaling stimulated serotonin secretion during RV infection.
RV infection also activates secretory pathways in epithelial cells through the stimulation of chloride secretion which drives paracellular movement of water. We tested whether ADP signaling was important for RV-induced epithelial fluid secretion using the enteroid swelling assay, in which an increase in fluid secretion causes an increase in the lumen of three-dimensional (3D) enteroid cultures (38, 53). We mock- or RV-infected 3D jHIE-GCaMP6s enteroids, treated with DMSO vehicle or BPTU, and found that BPTU treatment significantly decreased the percentage of swelling in mock- and RV-infected enteroids (Fig. 6H, Movie S11). Thus, ADP signaling stimulated epithelial fluid secretion in RV infection.
Finally, we determined whether inhibition of the P2Y1 receptor would attenuate RV-induced diarrhea in a neonatal mouse model. BPTU consistently had the most potent inhibition of Ca2+ spikes and average Ca2+ spike magnitude in HIEs (Fig. 3H,I), and binds to an allosteric pocket on the external surface of the P2Y1 receptor (54). We tested a drug treatment model in which 6 to 8-day-old C57Bl/6J pups were orally gavaged with RRV and then gavaged with BPTU or vehicle control on days 1–4 post-infection. RRV is more pathogenic than SA11 in C57Bl/6J pups and causes a moderate diarrhea (55, 56). RRV also causes ICWs similar to SA11 and thus is appropriate for assessing if BPTU treatment can reduce the diarrhea phenotype in vivo (Fig. S3E). Stools were assessed each day and watery, mucus, yellow stools were indicators of diarrhea. Peak diarrhea occurred at 2 dpi in both DMSO- and BPTU-treated pups, but the percentage of pups with diarrhea decreased significantly on 4 dpi (Fig. 6I). BPTU-treated pups had significantly less severe diarrhea as measured by their mean diarrhea score (Fig. 6J). Intestinal cross-sections from the duodenum, jejunum, and ileum of RV-infected pups treated with DMSO or BPTU were relatively similar with no histopathological features at 5 dpi (Fig. S6A). We then tested whether treatment with MRS2500, a water-soluble small molecule P2Y1 receptor blocker with a different mechanism of inhibition from BPTU, would similarly attenuate RV diarrhea (54). In these studies, there were similar percentages of pups with diarrhea on dpi 1–5 (Fig. S6B). However, pups treated with MRS2500 had significantly less severe diarrhea at the peak of diarrhea on dpi 3–4 compared to pups treated with control saline (Fig. S6C). Thus, blocking the P2Y1 receptor decreased diarrhea severity and caused diarrhea to resolve more quickly than the DMSO-treated animals.
The current concept of RV pathogenesis proposes that paracrine signaling from RV-infected cells dysregulates surrounding uninfected cells and contributes to life-threatening diarrhea and/or vomiting. RV elevates cytosolic Ca2+ during infection and increases paracrine signaling molecules such as eNSP4, PGE2, and NO, but functional signaling from RV-infected cells to uninfected cells had not yet been determined (9, 18, 19). Thus, the primary goal of this study was to investigate the role and identify the mechanisms of intercellular signaling during RV infection. Using long-term live-cell fluorescence imaging to detect signaling between infected and uninfected cells, our work described the direct observation of ICWs originating from RV-infected cells and provided experimental proof for the widely-held paracrine signaling idea as summarized in Figure 6K.
Extracellular purinergic signaling is a well-conserved communication system found in single-celled bacteria and protozoa as well as plants and higher order eukaryotes (57). Though this signaling is most commonly ATP, RV activated ADP- and P2Y1 receptor-mediated ICWs, and this was the most dominant Ca2+ signal observed during RV infection. Mechanical stress and cell swelling are two major triggers of ATP secretion from intact, non-neuronal cells (33). Since RV disrupts the actin cytoskeleton and greatly increases intracellular Ca2+ during infection, we speculate that cell shape changes or cellular swelling may be sufficient to cause mechanical stress and ADP release (58, 59). RV exploited ADP signaling in host cells to benefit its replication, but also increased hallmarks of RV disease such as fluid and serotonin secretion. Furthermore, treatment of neonatal mice with two mechanistically different small molecule P2Y1 receptor inhibitors attenuated RV diarrhea. Thus, activation of purinergic signaling was biologically relevant to RV disease. While eNSP4 can induce diarrhea in animal models, here eNSP4 was not involved in ICWs or purinergic signaling and may induce fluid secretion through an alternative pathway (9, 20). Blocking ADP signaling and the P2Y1 receptor reduced the RV-induced expression of COX2 and iNOS that drive production of PGE2 and NO, respectively. This suggested that ICWs and ADP signaling induced production of these additional paracrine signaling molecules. RV-induced IL-1α expression was decreased upon blocking ADP signaling; thus, ADP signaling may also be upstream of other inflammatory responses such as interferon pathways increased in RV infection (47, 60). Activating ICWs and ADP signaling may be a common strategy for enteric viruses to amplify dysregulation of host cells. Thus, targeting the P2Y1 receptor may be an effective host-directed approach to treating viral diarrhea and other inflammatory pathways.
ICWs and extracellular ADP signaling may be a broadly important form of communication in the gastrointestinal epithelium. Basal ICWs in jHIEs were reduced when treated with apyrase and P2Y1 blockers, and P2Y1 receptor knockout enteroids had significantly reduced Ca2+ signaling. Since ATP-mediated ICWs are activated in wound healing responses (61, 62), tonic purinergic signaling may also be important for maintaining epithelial barrier integrity and epithelialto-epithelial cell communication of damage, stretch, and compression forces. Purinergic signaling regulates responses to injury and restitution by mediating the innate immune system (46). Basal, ADP-based ICWs may also have a role in the balance of pro- and anti-inflammatory signaling from the epithelium. Furthermore, extracellular ATP signaling in the gastrointestinal tract stimulates epithelial ion transport (63), suggesting basal ICWs could stimulate fluid secretion and hydrate the mucus layer. Finally, ADP-driven ICWs may mediate communication from the epithelium to the enteric nervous system directly or signal to the lamina propria and endothelium as part of tissue-level homeostasis. ICWs in intestinal epithelium and HIE models have not been widely studied, and the possible physiological roles of their ICWs remain to be characterized. Because the epithelium is the primary interface between the environment and the host, understanding how it coordinates and communicates is critical for developing therapeutics when it becomes dysregulated in disease states.
Together, these studies identify ADP as the signaling compound that elicited a paracrine signal involved in RV diarrhea and vomiting responses. Reduction of ADP signaling and ICWs decreased many known aspects of RV disease including serotonin release, epithelial fluid secretion, and diarrhea in a neonatal mouse model. Paracrine purinergic signaling and ICWs may be a common strategy among enteric viruses to amplify their dysregulation of host systems. Thus, targeting the P2Y1 receptor may be an effective approach to treating viral diarrhea.
Materials and Methods
Cells and rotaviruses
MA104 cells (African green monkey kidney cells) (ATCC CRL-2378.1) and HEK293T cells (ATCC CRL-3216) were cultured in high glucose DMEM supplemented with 10% fetal bovine serum (FBS) and Antibiotic/Antimycotic (Invitrogen) at 37°C in 5% CO2. The rotavirus human strain Ito, used for all human intestinal enteroid infections, was prepared as previously described (38). Rotavirus strains SA114F and Rhesus RV (RRV) were produced from in-house stocks. Recombinant and SA11 clone 3-mRuby3 was produced previously (28). All viruses were propagated in MA104 cells in serum-free DMEM supplemented with 1 μg/mL Worthington’s Trypsin, and after harvest stocks were subjected to three freeze/thaw cycles and activated with 10 μg/mL Worthington’s Trypsin for 30 min at 37 °C prior to use.
Rotavirus diarrhea mouse model
C57Bl/6J dams with natural litters (purchased from Center for Comparative Medicine, Baylor College of Medicine) were used and housed in standard cages with food and water provided ad libitum. Mouse pups, both male and female at natural ratios, were housed with their dam throughout the experiment. Animal experiments were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee (IACUC), approved protocol AN-6903. All experiments were performed in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Six- or seven-day-old mouse pups were orally gavaged with 1.5 × 107 PFU/mL of Rhesus rotavirus (RRV), a dosage sufficient to cause diarrhea in 100% of pups. The inoculum was mixed with sterile-filtered green food dye (3% by volume) for visualization of the gavage. For BPTU experiments, pups were orally gavaged with BPTU (4 mg/kg) (4 cages, n = 30 pups) or an equivalent volume of DMSO vehicle control (4 cages, n = 26 pups) suspended in pharmaceutical grade corn oil on days 1–4 post-infection. For MRS2500 experiments, pups were orally gavaged with MRS2500 (4 mg/kg) (9 cages, n = 60 pups) suspended in pharmaceutical grade normal saline or an equivalent volume of saline control (8 cages, n = 46 pups) on days 1–4 post-infection. Cages receiving control or drug treatment were assigned at random.
Each day, mouse pups were gently palpated on the abdomen to express and visualize feces. Stools were assessed on a scale between 1–4 for volume, color, and consistency as described previously (9), an average score of ≥2 was considered diarrhea. For scoring consistency, select experiments were scored by an observer blinded to experimental condition. Percentage of pups with diarrhea was calculated as the number of stools with a diarrhea score ≥ 2 divided by the number of stools collected per cage per day. Pups from which a stool sample could not be collected on that day were not counted for percent diarrhea or mean diarrhea severity calculations.
Chemicals
BPTU, AR-C 118925XX, Bx430, 5-BDBD, MRS2179, MRS2279, MRS2500, 10-Panx, suramin, TAT-Gap19, PPADS (Pyridoxalphosphate-6-azophenyl-2’,4’-disulfonic acid), NOC-7, prostaglandin E2, ARL 67156, and ω-agatoxin were purchased from Tocris Bioscience. Apyrase grade VI (catalog no. A6410) and apyrase grade VII (catalog no. A6535) were purchased from Sigma-Aldrich. 18β-glycyrrhetinic acid and carbenoxolone were purchased from Santa Cruz Biotechnology.
Antibodies
To detect RV by IF, we used rabbit anti-RV strain Alabama (12) (1:80,000) and the secondary antibody donkey anti-rabbit IgG H&L AlexaFluor 568 (Invitrogen, A10042) (1:2000). For western blots, we used rabbit anti-NSP4 aa120–147 (64) (1:3000) to detect RV NSP4, mouse anti-GAPDH monoclonal antibody (Novusbio, NB600–502) (1:5000) to detect GAPDH, and secondary antibodies alkaline phosphatase-conjugated goat anti-rabbit IgG or goat anti-mouse IgG (Southern Biotech, 4030–04, 1036–04) (1:2000). For eNSP4-blocking assays, we used purified anti-NSP4 rabbit antisera (120–147) (22), purified anti-NSP4 monoclonal antibody 622 generated against bacterially expressed SA11 NSP4 coiled-coil domain (amino acids 95–146) (65), and purified M60, a non-neutralizing anti-VP7 monoclonal antibody, was used as a control (36). Equal concentrations of antibody, estimated using protein OD280, were added to RV-infected wells after inoculation.
Calcium indicator lentiviruses, cell lines, and enteroids
GCaMP5G (Addgene plasmid #31788), GCaMP6s (Addgene plasmid #40753), and GCEPIA1er (Addgene plasmid #105012) were cloned into pLVX-Puro. RGECO1.2 (Addgene plasmid #45494), R-CEPIA1er (Addgene plasmid #58216), and GCaMP6s were cloned into pLVX-IRES-Hygro. Lentivirus vectors for the GECI constructs were packaged in HEK293T cells as previously described(66) or produced commercially (Cyagen Biosciences, Inc.). MA104-GCaMP5G, MA104-GCaMP5G/RCEPIAer, MA104-RGECO1/GCEPIAer, and MA104-GCaMP6s-shRNA cell lines and the jHIE-GCaMP6s enteroids (enteroid line J3) were generated as previously described (28, 66). Both MA104-GCaMP5G and MA104-GCaMP6s cells were used in these studies; we observed no difference in calcium responses with these two indicators and thus refer to both as MA104-GCaMP. Jejunum (J2) human intestinal enteroid cultures expressing GCaMP6s (in pLVX-IRES-Hygro) were created using lentivirus transduction as described previously and grown in high Wnt3a CMGF+ with 50 μg/mL hygromycin B for selection (67).
CRISPR/Cas9 KO cell lines and enteroids
Lentivirus constructs in pLentiCRISPRv2 vectors were purchased from GenScript Biotech Corp (USA) and were packaged in HEK293T cells as previously described (66). The CRISPR/Cas9-expression vectors encode a puromycin-resistance gene for drug selection. The following small guide RNAs were used: P2Y1-sg1 CTACAGCATGTGCACGACCG and P2Y2-sg1 TTTGTCACCACCAGCGCGCG. MA104-GCaMP6s cells (pLVX-IRES-Hygro) were transduced with the P2Y1 CRISPR/Cas9-expressing construct and at 72 hrs post-transduction the cells were passaged in the presence of 50 μg/mL hygromycin B and 10 μg/mL puromycin to select for co-expression of GCaMP6s and CRISPR/Cas9. The J2-derived jHIE-GCaMP6s(hygro) enteroid cultures described above were transduced with the CRISPR/Cas9-expressing constructs to make two separate enteroid lines and grown with 50 μg/mL hygromycin B and 1 μg/mL puromycin to select for co-expression of GCaMP6s and CRISPR/Cas9.
Genotyping was performed on whole genomic DNA prepared using DirectPCR (Viagen) with Proteinase K (Qiagen) according to manufacturer’s instructions. Genomic DNA was a template to PCR amplify a region surrounding the P2Y1 or P2Y2 sgRNA-target sites using primers listed in Table S2. PCR products were cloned into the pMiniT2.0 vector using the NEB PCR Cloning Kit (New England Biolabs). Five clones for P2Y1ko HIEs and five clones for P2Y2ko HIEs were picked and sequenced to determine the repertoire of alleles present in the HIE cultures. Sanger sequencing was performed by GENEWIZ (USA).
Phenotypic knockout was tested through measurement of relative GFP fluorescence increase after addition of 1 nM ADP for MA104-GCaMP6s-P2Y1ko cells, 10 nM ADP for (J2)HIE-GCaMP6s-P2Y1ko enteroids and 10 μM ATP-γS for (J2)HIE-GCaMP6s-P2Y2ko enteroids compared to parental (J2)HIE-GCaMP6s-P2Y2ko enteroids. Experiments were performed at least 3 times with n ≥ 25 cells per experiment analyzed.
Establishment of HIE cultures
The human intestinal enteroid (HIE) cultures used in this study were generated previously and deposited into the Texas Medical Center Digestive Diseases Center (TMC-DDC) Gastrointestinal Experimental Model Systems (GEMS) Core (37, 38). We used jejunal HIEs cultures J2 and J3, both from adult females, obtained from the GEMS core. Complete media with and without growth factors (CMGF+ and CMGF−, respectively) (37, 38), differentiation media (67), high Wnt3a CMGF+ (hW−CMGF+) (67), and a Fluorobrite-DMEM based differentiation media (FBDiff) were prepared as previously described (28). HIEs were grown in phenol red-free, growth factor-reduced Matrigel (Corning). HIE monolayers were prepared from three-dimensional cultures and seeded into optical-bottom 10-well Cellview chamber slides coated with dilute collagen IV (Sigma) as described previously (68, 69). After 24 hr in CMGF+ and 10 μM Y-27632 Rock inhibitor, differentiation medium was used and changed every day for 4–5 days.
Microscopy and image analysis
For calcium imaging, MA104 cells and HIEs were imaged with a widefield epifluorescence Nikon TiE inverted microscope using a SPECTRAX LED light source (Lumencor) and either a 20x Plan Fluor (NA 0.45) phase contrast or a 20X Plan Apo (NA 0.75) differential interference contrast (DIC) objective. Fluorescence and transmitted light images were recorded using an ORCA-Flash 4.0 sCMOS camera (Hamamatsu), and Nikon Elements Advanced Research v4.5 software was used for multipoint position selection, data acquisition, and image analysis. Images were read-noise subtracted using an average of 10 no-light acquisitions of the camera. Single cells were selected as Regions of Interest (ROI) and fluorescence intensity measured for the experiment. The fluorescence intensity of the whole field-of-view was measured for HIE monolayers. Fluorescence intensity values were exported to Microsoft Excel and normalized to the baseline fluorescence. The number and magnitude of Ca2+ spikes were calculated as described previously (28). Ca2+ signals with a ΔF magnitude of >5% were counted as Ca2+ spikes for MA104-GCaMP cells and J3 HIE-GCaMP6s monolayers, which is based on our previously established threshold for Ca2+ spikes being >3 standard deviations above the mean ΔF. For J2 HIE-GCaMP6s monolayers, the mean Ca2+ transient increase per FOV for mock-infected jHIE monolayers was 5.4% (± 1.0% standard deviation), which was greater than that for MA104 cells or J3 HIEs (28). Thus, using the threshold for Ca2+ spikes being >3 standard deviations above the mean, Ca2+ signals with a ΔF magnitude of >10% were counted as Ca2+ spikes for J2 HIE-GCaMP6s monolayers. Average relative fluorescence increase, or average magnitude of Ca2+ spikes, was calculated as the average percent increase in fluorescence from largest 50 Ca2+ spikes per cell or per field of view.
Long-term calcium imaging
MA104-GECI cells.
Fluorobrite DMEM supplemented with 15mM HEPES, 1X sodium pyruvate, 1X Glutamax, and 1X non-essential amino acids (Invitrogen) was used for fluorescence Ca2+ imaging (FB-Plus). Confluent monolayers of MA104-GCaMP cells in gridded 8-well chamber slides (ibidi) were mock- or RV-infected in FBS-free media for 1 hr at the indicated multiplicity of infection (MOI). The inoculum was removed and replaced with FB-Plus, and for experiments where noted, with DMSO vehicle or drugs at indicated concentrations. For experiments with gap junction blockers, either 30 μM 18β-glycyrrhetinic acid (18β-gly) or 50 μM TAT-Gap19 (Gap19) was added. For experiments with apyrase, either 10 U/mL of apyrase VI, 10 U/mL of apyrase VII, or a mixture of 5 U/mL of apyrase VI and 5 U/mL of apyrase VII was added. For experiments with purinergic receptor blockers, 10 μM BPTU, 10 μM AR-C 118925XX, 10 μM Bx430, or 10 μM 5-BDBD was used or otherwise noted. The slide was mounted into an Okolab microscope stage-top incubation chamber equilibrated to 37°C with a humidified 5% CO2 atmosphere. In each experiment, 3–5 positions per well were selected and imaged every 1 minute for ~3–22 hours post infection. Each imaging experiment was performed at least three times independently, and data shown are representative of 1–3 experiments combined.
GECI HIEs.
For RV infection in monolayers, jHIE-GCaMP6s monolayers were washed once with CMGF- and treated with an inoculum of 50 μL CMGF- plus 30 μL MA104 cell lysate or RV (Ito) and incubated at 37°C and 5% CO2 for 2 hr. Then inoculum was removed, and monolayers were washed once with FB-Diff before adding FB-Diff with DMSO or drugs at indicated concentrations. For experiments with apyrase, a mixture of 50 U/mL of apyrase VI and 50 U/mL of apyrase VII was added. For experiments with purinergic receptor blockers, 10 μM BPTU, 10 μM AR-C 118925XX, 10 μM Bx430, 10 μM 5-BDBD, 10 μM MRS2179, 10 μM MRS2279, or 10 μM MRS2500 was used. Monolayers were transferred to the stage-top incubator for imaging with 4 fields of view chosen per well and imaged every minute for ~7–22 hours post-infection. Each imaging experiment was performed at least three times independently, and data shown are representative of all replicates combined.
Supernatant transfer assay
Confluent monolayers of parental MA104-GCaMP or MA104-GCaMP-P2Y1ko cells in a flat-bottom 24-well cell culture plate (Corning) were mock- or RV-infected in FB-Plus for 1 hr at MOI 5. Inoculum was removed and replaced with 300 μL FB-Plus with 20 μM ARL 67156, an ecto-ATPase inhibitor. Reporter cells consisted of confluent monolayers of parental MA104-GCaMP cells in a flat-bottom 96-well cell culture plate (Greiner Bio One). Reporter cells were incubated for one hour before imaging with or without 10 μM BPTU in 50 μL FB-Plus.
After the onset of intercellular calcium waves (~5–6 hpi, confirmed by epifluorescence microscopy), 50 μL of supernatant was collected from mock- or RV-infected wells and transferred to the reporter cells. Reporter cells were imaged every 2 s for 15–30 s for baseline and then for 1–2 min after addition. Change in GCaMP fluorescence for the 10–15 most responsive cells per FOV were analyzed. Each imaging run was repeated 3 times per experimental condition, and the entire experiment performed at least three times independently. Data shown are representative of 1–3 experiments combined.
Enteroid swelling assay
Jejunum HIEs were split and grown in hW-CMGF+ for 2 days followed by differentiation medium for 1 day. jHIEs were gently washed using ice cold 1XPBS and suspended in inoculum of 50 μL MA104 cell lysate or RV (Ito) diluted with 150 μL CMGF- and incubated for 1 hr. Then HIEs were washed, resuspended in 25% Matrigel diluted in FB-Diff (with DMSO vehicle or BPTU) and pipetted onto 8-well chamber slides (Matek) pre-coated with Matrigel. Imaging positions were chosen so that between 30–50 enteroids were selected per experimental condition. Enteroids were imaged using the stage-top incubator with transmitted light and GFP fluorescence every 2–3 minutes for ~18 hrs. Swelling was determined by measuring the cross-sectional area of the internal lumen at the beginning of the run compared to the time point of maximum swelling and calculating the percent increase. Area measurements were determined using the Nikon Elements AR v4.5 software. Experiments were performed three times independently, and data shown are representative of all replicates combined, n ≥ 68 HIEs per condition.
Scrape loading/dye transfer assay
Lucifer Yellow in Hank’s Buffered Salt Solution (HBSS) (1 mg/mL) or Rhodamine 123 in H2O (1 mg/mL) was diluted 1:100 in FB-Plus and added to MA104 or jHIE cell monolayers. Monolayers were scraped with a 26 g needle and incubated for 5 min at room temperature. Monolayers were then washed 3 times with FB-Plus and imaged. The mechanical disruption causes cells adjacent to the scrape to take up dye, which then is permeable to adjacent cells if there are functional gap junction channels. Images are representative of each experiment performed in triplicate.
Immunofluorescence
MA104 cells and HIEs were fixed using the Cytofix/Cytoperm kit (BD Biosciences) according to manufacturer instructions. Primary antibodies were diluted in 1X Perm/Wash and incubated with cells overnight at 4°C. The next day, the cells were washed three times with 1X Perm/Wash solution and then incubated with corresponding secondary antibodies for 1 hr at room temperature. Nuclei were stained with NucBlue Fixed Cell Stain (Life Technologies) for 5 min at room temperature and washed with 1X PBS for imaging. Experiments were performed three times independently.
Fluorescence focus assay
Fluorescence focus assays (FFA) were performed as described previously with the following modifications (13). After inoculation with RV, MA104 cells were treated with DMSO, 10U/mL apyrase, 10 μM BPTU, 10 μM AR-C 118925XX, 20 μM Bx430, 20 μM 5-BDBD, 300 μM suramin, or 10 μM PPADS. Then, cells and media at 24 hpi were freeze-thawed three times and activated with 10 μg/mL Worthington’s Trypsin for 30 min at 37 °C prior to use. Ten-fold dilutions were used to inoculate MA104 monolayers in 96-well plates for 1 hr and cells were fixed at 20 hpi with ice-cold methanol for ~20 min at 4°C, then washed 3 times with 1X phosphate buffered solution (PBS). Primary rabbit anti-rotavirus antisera (12) was used at 1:100,000 and incubated overnight at 4°C. Monolayers were washed 3x with 1XPBS and then incubated with secondary antibody donkey anti-rabbit AlexaFluor 488 (1:2000) for 1 hr at 37°C and then washed 3x with 1XPBS prior to imaging. FFA experiments were performed three times independently.
Plaque assay
Plaque assays were performed as described previously with the following modifications (70). Briefly, MA104 cells were seeded and grown to confluency in 6 well plates. Wells were infected at 10-fold dilutions in duplicate for 1 hr and media replaced with an overlay of 1.2% Avicel in serum-free DMEM supplemented with DEAE dextran, and 1μg/mL Worthington’s Trypsin, and, for indicated experiments, DMSO vehicle, apyrase, or purinergic receptor blocker drugs (71). The cells were incubated at 37°C/5% CO2 for 48–72 hrs before overlay was removed and cells stained with crystal violet to count plaques. Plaque assays of using MA104-GCaMP and MA104-GCaMPP2Y1ko cells were performed three times independently, and data shown are representative of all replicates combined.
RNA extraction, reverse transcription, and quantitative PCR
Total RNA was extracted from HIE 96-well monolayers or MA104 cells grown to confluency in a 6-well plate using TRIzol reagent (Ambion). In RV-infection experiments, cells were harvested at 24 hpi. Total RNA was treated with Turbo DNase I (Ambion) and cDNA was generated from 250 ng RNA using the SensiFAST cDNA synthesis kit (Bioline). Quantitative PCR was performed using Fast SYBR Green (Life Technologies) with primers either designed in-house or published previously (72) (Table S3) and using the QuantStudio real time thermocycler (Applied Biosciences). Target genes were normalized to the housekeeping gene ribosomal subunit 18s and relative expression was calculated using the ddCT method. Experiments were performed three times independently, and data shown are representative of all replicates combined. For IL-1α, COX2, and iNOS experiments, HIE monolayers were Mock or RV (Ito)-infected and then treated with DMSO, 100U/mL apyrase, or 10 μM BPTU and normalized to 18s mRNA transcripts and relative to the Mock-DMSO condition.
Immunoblot analysis
RV NSP4 protein were detected by immunoblot analysis as previously described, with minor changes as follows (64). Confluent MA104 cell monolayers in 6-well tissue culture plates were mock- or RV (SA114F)-infected at MOI 0.01 and then harvested at 18 hpi. Cells were lysed using a 1X RIPA buffer solution [10 mM Tris-HCL pH 8.0, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl, and 1 tablet complete mini protease inhibitor (Roche)] and passed through a Qiashredder (Qiagen). Lysed cells were boiled for 10 min at 100°C in SDS-PAGE sample buffer and separated on Tris-glycine 4–20% SDS-PAGE gels (BioRad). The detected NSP4 protein band was quantified for gel densitometry measurements using ImageJ software.
Measurement of serotonin release
Serotonin secretion by HIEs following stimulation with RV infection and treatment with DMSO, 100U/mL apyrase, 10 μM BPTU, or 300 nM ω-agatoxin was quantified by ELISA (Eagle Biosciences) according to the manufacturer’s instructions. A standard curve of known serotonin concentrations was plotted against optical density at 450 nm with a limit of detection of 2.6 ng/mL (Infinite F200Pro, Tecan). Experiments in flat monolayers were performed three times independently and experiments in transwells were performed three times independently. Data shown are representative of all replicates combined.
Statistical analysis
Biostatistical analyses were performed using GraphPad Prism (version 8.1) software (GraphPad Inc., La Jolla, CA). Outlier data points were removed using the ROUT method (Q = 1%). Statistical comparisons were made using the Mann-Whitney test, the one-way Analysis of Variance (ANOVA) with Tukey or Bonferroni multiple comparison’s test, or the Kruskal-Wallis test with Dunn’s multiple comparisons test. Parametric or non-parametric tests were used depending on Normality testing of the data sets. Differences between the groups were considered significant at p < 0.05 (*), and the data are presented as mean ± standard deviation unless otherwise noted. All authors had access to the study data, reviewed, and approved the final manuscript.
Supplementary Material
Acknowledgements:
We would like to thank Xi-Lei (Shelly) Zeng and Xiaomin Yu for their help with enteroid cultures and media. Additional thanks to the BCM Center for Comparative Medicine and technicians for their care of the animals.
Funding: This work was supported in part by NIH grants K01DK093657, R03DK110270, R01DK115507 (PI: J. M. Hyser); R01AI080656 and U19AI116497 (PI: M. K. Estes); and R01AI123278 and U01 AI124290 (PI. R. A. Britton). Trainee support for A.C.G. was provided by NIH grants F30DK112563 (PI: A. Chang-Graham) and the BCM Medical Scientist Training Program and support for both A.C.G. and A.C.S. was provided by the Integrative Molecular and Biomedical Sciences Graduate Program (T32GM008231, PI: D. Nelson). Training support for M.A.E. was provided by NIH grant K01DK123195 (PI: M. Engevik). Training support for K.A.E. was provided by NIH grant T32DK07644 (PI: R. Shulman). Training support for H.A.D. was provided by NIH grant F32AI136404 (PI: H. Danhof). Training support for J.T.G. was provided by McNair M.D./Ph.D. Scholars Program at Baylor College of Medicine. This project was also supported in part by PHS grant P30DK056338, which supports the Texas Medical Center Digestive Diseases Center (TMC-DDC) Gastrointestinal Experimental Model Systems (GEMS) Core and the Cellular and Molecular Morphology Core. FACS sorting of cell lines utilized the BCM Cytometry and Cell Sorting Core with funding from the CPRIT Core Facility Support Award (CPRIT-RP180672), the NIH (CA125123 and RR024574), and the expert assistance of Joel M. Sederstrom.
Footnotes
Competing interests: The authors declare no competing financial interests with this work.
Data and materials availability: All data is available in the manuscript or the supplementary materials.
Supplementary Materials
Figures S1–S6
Tables S1–S3
Movies S1–S11
References and Notes
- 1.Troeger C, Khalil IA, Rao PC, Cao S, Blacker BF, Ahmed T, et al. , Rotavirus Vaccination and the Global Burden of Rotavirus Diarrhea among Children Younger Than 5 Years. JAMA Pediatr 172, 958–965 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davidson GP, Goller I, Bishop RF, Townley RR, Holmes IH, Ruck BJ, Immunofluorescence in duodenal mucosa of children with acute enteritis due to a new virus. J. Clin. Pathol 28, 263–266 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Starkey WG, Collins J, Wallis TS, Clarke GJ, Spencer AJ, Haddon SJ, et al. , Kinetics, tissue specificity and pathological changes in murine rotavirus infection of mice. J. Gen. Virol 67, 2625–2634 (1986). [DOI] [PubMed] [Google Scholar]
- 4.Hagbom M, Istrate C, Engblom D, Karlsson T, Rodriguez-Diaz J, Buesa J, et al. , Rotavirus stimulates release of serotonin (5-HT) from human enterochromaffin cells and activates brain structures involved in nausea and vomiting. PLoS Pathog 7, 1–10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Little JA, Shadduck Lynn M., Pathogenesis of rotavirus infection. Infect. Immun 38, 755–763 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morris AP, Estes MK, VIII. Pathological consequences of rotavirus infection and its enterotoxin. Am. J. Physiol. Liver Physiol 281, G303–G310 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Lundgren O, Peregrin a T., Persson K, Kordasti S, Uhnoo I, Svensson L, Role of the enteric nervous system in the fluid and electrolyte secretion of rotavirus diarrhea. Science 287, 491–495 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Hyser JM, Collinson-Pautz MR, Utama B, Estes MK, Rotavirus disrupts calcium homeostasis by NSP4 viroporin activity. MBio 1, 1–12 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ball JM, Tian P, Zeng CQ, Morris a P., Estes MK, Age-dependent diarrhea induced by a rotaviral nonstructural glycoprotein. Science 272, 101–104 (1996). [DOI] [PubMed] [Google Scholar]
- 10.Tian P, Estes MK, Hu Y, Ball JM, Zeng CQ, Schilling WP, The rotavirus nonstructural glycoprotein NSP4 mobilizes Ca2+ from the endoplasmic reticulum. J. Virol 69, 5763–5772 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kunzelmann K, Schreiber R, Kmit A, Jantarajit W, Martins JR, Faria D, et al. , Expression and function of epithelial anoctamins. Exp. Physiol 285, 184–192 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Crawford SE, Hyser JM, Utama B, Estes MK, Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase-β signaling is required for rotavirus replication. Proc. Natl. Acad. Sci. U. S. A 109, E3405–13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zambrano JL, Sorondo O, Alcala A, Vizzi E, Diaz Y, Ruiz MC, et al. , Rotavirus Infection of Cells in Culture Induces Activation of RhoA and Changes in the Actin and Tubulin Cytoskeleton. PLoS One 7, e47612 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beau I, Cotte-Laffitte J, Amsellem R, Servin AL, A protein kinase A-dependent mechanism by which rotavirus affects the distribution and mRNA level of the functional tight junction-associated protein, occludin, in human differentiated intestinal Caco-2 cells. J. Virol 81, 8579–8586 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hagbom M, Sharma S, Lundgren O, Svensson L, Towards a human rotavirus disease model. Curr. Opin. Virol 2, 408–418 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Hodges K, Gill R, Infectious diarrhea: Cellular and molecular mechanisms. Gut Microbes 1, 4–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sowmyanarayanan TV, Natarajan SK, Ramachandran A, Sarkar R, Moses PD, Simon A, et al. , Nitric oxide production in acute gastroenteritis in Indian children. Trans. R. Soc. Trop. Med. Hyg 103, 849–51 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Yamashiro Y, Shimizu T, Oguchi S, Sato M, Prostaglandins in the plasma and stool of children with rotavirus gastroenteritis. J. Pediatr. Gastroenterol. Nutr 9, 322–7 (1989). [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez-Diaz J, Banasaz M, Istrate C, Buesa J, Lundgren O, Espinoza F, et al. , Role of Nitric Oxide During Rotavirus Infection. J. Med. Virol 78, 979–985 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Morris AP, Scott JK, Ball JM, Zeng CQ-Y, O’Neal WK, Estes MK, NSP4 elicits age-dependent diarrhea and Ca2+-mediated I- influx into intestinal crypts of CF mice. Am. J. Physiol. Gastrointest. Liver Physiol 277, 431–444 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Ousingsawat J, Mirza M, Tian Y, Roussa E, Schreiber R, Cook DI, et al. , Rotavirus toxin NSP4 induces diarrhea by activation of TMEM16A and inhibition of Na+ absorption. Pflugers Arch. Eur. J. Physiol 461, 579–589 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Seo N, Zeng CQ, Hyser JM, Utama B, Crawford SE, Kim KJ, et al. , Integrins are receptors for the rotavirus enterotoxin. Proc. Natl. Acad. Sci 105, 8811–8818 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujii S, Suzuki K, Kawamoto A, Ishibashi F, Nakata T, Murano T, et al. , PGE 2 is a direct and robust mediator of anion/fluid secretion by human intestinal epithelial cells. Sci. Rep 6, 1–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mourad FH, Turvill JL, Farthing MJG, Role of nitric oxide in intestinal water and electrolyte transport. Gut 44, 143–147 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michelangeli F, Ruiz MC, del Castillo JR, Ludert JE, Liprandi F, Effect of rotavirus infection on intracellular calcium homeostasis in cultured cells. Virology 181, 520–527 (1991). [DOI] [PubMed] [Google Scholar]
- 26.Hyser JM, Estes MK, Annu. Rev. Virol, in press, doi: 10.1146/annurev-virology-100114-054846. [DOI] [PMC free article] [PubMed]
- 27.Crawford SE, Ramani S, Tate JE, Parashar UD, Svensson L, Hagbom M, et al. , Rotavirus infection. Nat. Rev. Dis. Prim 3 (2017), doi: 10.1038/nrdp.2017.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang-Graham AL, Perry JL, Strtak AC, Ramachandran NK, Criglar JM, Philip AA, et al. , Rotavirus Calcium Dysregulation Manifests as Dynamic Calcium Signaling in the Cytoplasm and Endoplasmic Reticulum. Sci. Rep 9 (2019), doi: 10.1101/627877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang W, Segura BJ, Lin TR, Hu Y, Mulholland MW, Intercellular calcium waves in cultured enteric glia from neonatal guinea pig. Glia 42, 252–262 (2003). [DOI] [PubMed] [Google Scholar]
- 30.D’hondt C, Himpens B, Bultynck G, Mechanical stimulation-induced calcium wave propagation in cell monolayers: the example of bovine corneal endothelial cells. J. Vis. Exp, e50443 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanderson MJ, Charles AC, Dirksen ER, Mechanical stimulation and intercellular communication increases intracellular Ca2+ in epithelial cells. Cell Regul 1, 585–96 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ, Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science 247, 470–3 (1990). [DOI] [PubMed] [Google Scholar]
- 33.Leybaert L, Sanderson MJ, Intercellular Ca(2+) waves: mechanisms and function. Physiol. Rev 92, 1359–1392 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Babica P, Sovadinová I, Upham BL, in Gap Junction Protocols, Vinken M, Johnstone SR, Eds. (Springer New York, New York, NY, 2016; 10.1007/978-1-4939-3664-9_9), pp. 133–144. [DOI] [Google Scholar]
- 35.Upham BL, Sovadinová I, Babica P, Gap junctional intercellular communication: A functional biomarker to assess adverse effects of toxicants and toxins, and health benefits of natural products. J. Vis. Exp 2016, 1–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emslie KR, Miller JM, Slade MB, Dormitzer PR, Greenberg HB, Williams KL, Expression of the rotavirus SA11 protein VP7 in the simple eukaryote Dictyostelium discoideum. J. Virol 69, 1747–1754 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato T, Stange DE, Ferrante M, Vries RGJ, Van Es JH, Van Den Brink S, et al. , Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Saxena K, Blutt SE, Ettayebi K, Zeng X, Broughman JR, Crawford SE, et al. , Human Intestinal Enteroids: a New Model To Study Human Rotavirus Infection, Host Restriction, and Pathophysiology. J. Virol 90, 43–56 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Foulke-Abel J, In J, Kovbasnjuk O, Zachos NC, Ettayebi K, Blutt SE, et al. , Human enteroids as an ex-vivo model of host-pathogen interactions in the gastrointestinal tract. Exp. Biol. Med 239, 1124–1134 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zachos NC, Kovbasnjuk O, Foulke-Abel J, In J, Blutt SE, De Jonge HR, et al. , Human enteroids/colonoids and intestinal organoids functionally recapitulate normal intestinal physiology and pathophysiology. J. Biol. Chem 291, 3759–3766 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Léon C, Hechler B, Vial C, Leray C, Cazenave JP, Gachet C, The P2Y1 receptor is an ADP receptor antagonized by ATP and expressed in platelets and megakaryoblastic cells. FEBS Lett 403, 26–30 (1997). [DOI] [PubMed] [Google Scholar]
- 42.Dickman KG, Hempson SJ, Anderson J, Lippe S, Zhao L, Burakoff R, et al. , Rotavirus alters paracellular permeability and energy metabolism in Caco-2 cells. Am. J. Physiol. Gastrointest. Liver Physiol 279, G757–G766 (2000). [DOI] [PubMed] [Google Scholar]
- 43.Zhang C, He H, Wang L, Zhang N, Huang H, Xiong Q, et al. , Virus-Triggered ATP Release Limits Viral Replication through Facilitating IFN-β Production in a P2X7-Dependent Manner. J. Immunol 199, 1372–1381 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Ferrari D, Idzko M, Müller T, Manservigi R, Marconi P, Purinergic Signaling: A New Pharmacological Target Against Viruses? Trends Pharmacol. Sci 39, 926–936 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Zumerle S, Calì B, Munari F, Angioni R, Di Virgilio F, Molon B, et al. , Intercellular Calcium Signaling Induced by ATP Potentiates Macrophage Phagocytosis. Cell Rep 27, 1–10.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cekic C, Linden J, Purinergic regulation of the immune system. Nat. Rev. Immunol 16, 177–192 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Hernández PP, Mahlakõiv T, Yang I, Schwierzeck V, Nguyen N, Guendel F, et al. , Interferon-γ and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nat. Immunol 16, 698–707 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Di Paolo NC, Shayakhmetov DM, Interleukin 1α and the inflammatory process. Nat. Immunol 17, 906–913 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Groß O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, et al. , Inflammasome Activators Induce Interleukin-1α Secretion via Distinct Pathways with Differential Requirement for the Protease Function of Caspase-1. Immunity 36, 388–400 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Ohtani Y, Minami M, Satoh M, Expression of inducible nitric oxide synthase mRNA and production of nitric oxide are induced by adenosine triphosphate in cultured rat microglia. Neurosci. Lett 293, 72–74 (2000). [DOI] [PubMed] [Google Scholar]
- 51.Degagné E, Grbic DM, Dupuis A-A, Lavoie EG, Langlois C, Jain N, et al. , P2Y 2 Receptor Transcription Is Increased by NF-κB and Stimulates Cyclooxygenase-2 Expression and PGE 2 Released by Intestinal Epithelial Cells. J. Immunol 183, 4521–4529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bialowas S, Hagbom M, Nordgren J, Karlsson T, Sharma S, Magnusson K-E, et al. , Rotavirus and Serotonin Cross-Talk in Diarrhoea. PLoS One 11, e0159660 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dekkers JF, Wiegerinck CL, de Jonge HR, Bronsveld I, Janssens HM, Winter-de Groot KM, et al. , A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med 19, 939–945 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Zhang D, Gao ZG, Zhang K, Kiselev E, Crane S, Wang J, et al. , Two disparate ligand-binding sites in the human P2Y1 receptor. Nature 520, 317–321 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mossel EC, Ramig RF, Rotavirus Genome Segment 7 (NSP3) Is a Determinant of Extraintestinal Spread in the Neonatal Mouse. J. Virol 76, 6502–6509 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramig RF, The effects of host age, virus dose, and virus strain on heterologous rotavirus infection of suckling mice. Microb. Pathog 4, 189–202 (1988). [DOI] [PubMed] [Google Scholar]
- 57.Verkhratsky A, Burnstock G, Biology of purinergic signalling: Its ancient evolutionary roots, its omnipresence and its multiple functional significance. BioEssays 36, 697–705 (2014). [DOI] [PubMed] [Google Scholar]
- 58.Brunet JP, Cotte-Laffitte J, Linxe C, Quero a M., Géniteau-Legendre M, Servin A, Rotavirus infection induces an increase in intracellular calcium concentration in human intestinal epithelial cells: role in microvillar actin alteration. J. Virol 74, 2323–2332 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berkova Z, Crawford SE, Blutt SE, Morris AP, Estes MK, Expression of Rotavirus NSP4 Alters the Actin Network Organization through the Actin Remodeling Protein Cofilin. J. Virol 81, 3545–3553 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saxena K, Simon LM, Zeng X-L, Blutt SE, Crawford SE, Sastri NP, et al. , A paradox of transcriptional and functional innate interferon responses of human intestinal enteroids to enteric virus infection. Proc. Natl. Acad. Sci 114, E570–E579 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takada H, Furuya K, Sokabe M, Mechanosensitive ATP release from hemichannels and Ca 2 + influx through TRPC6 accelerate wound closure in keratinocytes. J. Cell Sci 127, 4159–4171 (2014). [DOI] [PubMed] [Google Scholar]
- 62.Sung Y, Sung Z, Ho C, Lin M, Wang J, Yang S, et al. , Intercellular calcium waves mediate preferential cell growth toward the wound edge in polarized hepatic cells. Exp. Cell Res 287, 209–218 (2003). [DOI] [PubMed] [Google Scholar]
- 63.Novak I, Purinergic signalling in epithelial ion transport: regulation of secretion and absorption. Acta Physiol 202, 501–522 (2011). [DOI] [PubMed] [Google Scholar]
- 64.Hyser JM, Zeng CQY, Beharry Z, Palzkill T, Estes MK, Epitope mapping and use of epitope-specific antisera to characterize the VP5* binding site in rotavirus SA11 NSP4. Virology 373, 211–228 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sastri NP, Viskovska M, Hyser JM, Tanner MR, Horton LB, Sankaran B, et al. , Structural Plasticity of the Coiled-Coil Domain of Rotavirus NSP4. J. Virol 88, 13602–13612 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Perry JL, Ramachandran NK, Utama B, Hyser JM, Use of genetically-encoded calcium indicators for live cell calcium imaging and localization in virus-infected cells. Methods (2015), doi: 10.1016/j.ymeth.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chang-Graham AL, Danhof HA, Engevik MA, Tomaro-Duchesneau C, Karandikar UC, Estes MK, et al. , Human intestinal enteroids with inducible neurogenin-3 expression as a novel model of gut hormone secretion. Cell. Mol. Gastroenterol. Hepatol 0 (2019), doi: 10.1016/j.jcmgh.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.VanDussen KL, Marinshaw JM, Shaikh N, Miyoshi H, Moon C, Tarr PI, et al. , Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut 64, 911–920 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ettayebi K, Crawford SE, Murakami K, Broughman JR, Karandikar U, Tenge VR, et al. , Replication of human noroviruses in stem cell – derived human enteroids 5211 (2016), doi: 10.1126/science.aaf5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Estes MK, Graham DY, Gerba CP, Smith EM, Simian rotavirus SA11 replication in cell cultures. J. Virol 31, 810–5 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Matrosovich M, Matrosovich T, Garten W, Klenk H-D, New low-viscosity overlay medium for viral plaque assays. Virol. J 3, 63 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maynard JP, Lee J-S, Sohn BH, Yu X, Lopez-Terrada D, Finegold MJ, et al. , P2X3 purinergic receptor overexpression is associated with poor recurrence-free survival in hepatocellular carcinoma patients. Oncotarget 6 (2015), doi: 10.18632/oncotarget.6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.