

Abstract
Rationale: Idiopathic pulmonary fibrosis (IPF) is an insidious and fatal interstitial lung disease associated with declining pulmonary function. Accelerated aging, loss of epithelial progenitor cell function and/or numbers, and cellular senescence are implicated in the pathogenies of IPF.
Objectives: We sought to investigate the role of alveolar type 2 (AT2) cellular senescence in initiation and/or progression of pulmonary fibrosis and therapeutic potential of targeting senescence-related pathways and senescent cells.
Methods: Epithelial cells of 9 control donor proximal and distal lung tissues and 11 IPF fibrotic lung tissues were profiled by single-cell RNA sequencing to assesses the contribution of epithelial cells to the senescent cell fraction for IPF. A novel mouse model of conditional AT2 cell senescence was generated to study the role of cellular senescence in pulmonary fibrosis.
Measurements and Main Results: We show that AT2 cells isolated from IPF lung tissue exhibit characteristic transcriptomic features of cellular senescence. We used conditional loss of Sin3a in adult mouse AT2 cells to initiate a program of p53-dependent cellular senescence, AT2 cell depletion, and spontaneous, progressive pulmonary fibrosis. We establish that senescence rather than loss of AT2 cells promotes progressive fibrosis and show that either genetic or pharmacologic interventions targeting p53 activation or senescence block fibrogenesis.
Conclusions: Senescence of AT2 cells is sufficient to drive progressive pulmonary fibrosis. Early attenuation of senescence-related pathways and elimination of senescent cells are promising therapeutic approaches to prevent pulmonary fibrosis.
Keywords: cellular senescence, alveolar type 2 cells, pulmonary fibrosis
At a Glance Commentary
Scientific Knowledge on the Subject
Idiopathic pulmonary fibrosis (IPF) is an insidious and fatal interstitial lung disease associated with declining pulmonary function. Mechanisms involved in the initiation and progression of IPF are poorly defined and most likely multifactorial. Accelerated aging, loss of epithelial progenitor cell function and/or numbers, and cellular senescence are implicated in the pathogenies of IPF.
What This Study Adds to the Field
Our study shows that alveolar type 2 cells isolated from IPF lung tissue exhibit characteristic transcriptomic features of cellular senescence. Furthermore, we establish a novel mouse model of conditional Sin3a loss of function to demonstrate a causal role for p53-dependent cellular senescence of alveolar type 2 cells in progressive pulmonary fibrosis and elucidate potential underlying mechanisms. We provide evidence that early targeting of senescent cells is therapeutic in moderating lung fibrosis in mice and provide a novel model for drug discovery and validation.
Idiopathic pulmonary fibrosis (IPF) is a progressive interstitial lung disease with a median diagnosis age of 66 years and estimated survival of 2–4 years after diagnosis (1). The mechanisms involved in the initiation and progression of IPF are poorly defined and are most likely multifactorial. Recent studies have implicated mechanisms of accelerated aging, including loss of epithelial progenitor cell function and/or numbers and cellular senescence, in IPF pathogenesis (2–8). Indeed, about 20% of IPF cases are familial, among which causative mutations have been identified in genes involved in telomere function, protein folding, and secretion, which impact the function of epithelial cells (9–12). These mutations in the surfactant protein-C gene, whose product is expressed exclusively by alveolar type 2 (AT2) cells, suggest that defects in AT2 cell function play key roles in IPF pathogenesis. Furthermore, disease-associated gene variants defined in genome-wide screens have been linked to defects in host defense and regulation of cellular senescence (13, 14).
Normal maintenance of the alveolar epithelium is accomplished through the proliferation of AT2 cells, a facultative stem cell that produces and secretes pulmonary surfactant in its quiescent state (15). However, stress from genetic or environmental factors can compromise the ability of AT2 cells to transition from quiescent to proliferative states, leading to defective epithelial maintenance. We recently demonstrated that fibrotic regions of IPF explant tissue show both regional depletion of AT2 cells and abnormal activation of multiple pathways related to cellular senescence, including p53 signaling (16). Furthermore, we established that AT2 cells isolated from IPF lung tissue exhibit reduced alveolosphere-forming ability (17). Thus, age-dependent senescence of AT2 cells, accompanied by loss of stem/progenitor cell function, could contribute to progressive lung fibrosis.
Here, we define the senescent phenotype of AT2 cells in lung tissue of patients with IPF and establish a novel mouse model to test the contribution made by AT2 dysfunction in progressive lung fibrosis. To this end, we generated mice with AT2-specific conditional loss of Sin3a, a key component of Sin3–HDAC complex that regulates chromatin structure and gene expression in all eukaryotic cells. Silencing of Sin3a led to defective progenitor cell function and induction of p53-dependent AT2 senescence. We establish that senescence of AT2 cells is sufficient to initiate progressive lung fibrosis that closely resembles pathological remodeling seen in IPF lungs. Fibrosis was diminished either by selective loss of p53 function in AT2 cells, by systematic inhibition of p53, or by ablation of senescent cells by systemic delivery of senolytic drugs. Our data suggest that p53-induced AT2 senescence serves as a proximal driver and therapeutic target in progressive lung fibrosis and provides key evidence to support roles for epithelial dysfunction in both initiation and progression of distal lung fibrosis seen in IPF.
Some of the results of these studies have been previously reported in the form of a preprint (https://doi.org/10.1101/820175).
Methods
Detailed methods are included in the online supplement.
Explant tissue was obtained from patients undergoing transplantation for end-stage IPF (see Table E1 in the online supplement) in compliance with consent procedures accepted by the Internal Review Board of Cedars-Sinai Medical Center.
Mice were handled in accordance with Institutional Animal Care and Use Committee–approved protocols.
Statistical Analysis
One-way ANOVA analysis or Kruskal-Wallis test was used for multiple comparisons. Two-tailed unpaired Student's t test or nonparametric Mann-Whitney test was used for two-group comparisons. Log-rank (Mantel-Cox) test was used as a survival analysis. All analysis was performed in GraphPad Prism 8. Results are presented as the mean ± SEM. P < 0.05 was considered statistically significant.
Results
Senescent Phenotype of AT2 Cells in End-Stage IPF Lung Tissue
The staining of IPF explant tissue for SA-βgal (senescence-associated β-gal) revealed multiple senescent cell types, including epithelial cells (Figure E1A). To better define senescent epithelial cell types of the IPF lung, we performed single-cell RNA sequencing (scRNA-Seq) with fluorescence-activated cell sorting–enriched epithelial cells from either control donor proximal and distal lung tissues or IPF fibrotic lung tissues (hematoxylin and eosin staining in Figures E1B and E1C). The 58,808 single cells passed quality control for further batch correction and unsupervised clustering. The major cell types, including AT1, AT2, club, SCGB3A2+ club-like, ciliated, preciliated, MUC5AC high goblet, MUC5B high goblet, KRT15+ basal (KRT15+ and KRT17+), KRT17+ basal (KRT15− and KRT17+), and ionocyte, were assigned according to their expression of known cell type–specific gene signatures (18, 19) (Figures 1A and E2A–E2C). Unlike recent studies of IPF and control tissue samples, which only sampled distal lung tissue, our dataset includes both proximal airway and distal epithelial control donors. Consistent with other recent reports, we observe increased representation of KRT17+ basal cells and SCGB3A2+ secretory cells as well as reduced representation of AT2 cells in distal tissue of IPF explant lung (18, 20) (Figure E2B). Importantly, novel cell types observed in distal IPF lung tissue share close similarity with counterparts in proximal airways, suggesting that they may arise by proximalization of distal lung tissue. To assess the contribution of epithelial cells to the senescent cell fraction, we compared the core senescence gene list (503 genes) from CSGene database to our scRNA-Seq data and generated a cellular senescence score (21). Cellular senescence score is significantly increased in multiple IPF epithelial cell types (including AT2 cells and KRT17+ basal) compared with donor cell types (Figures 1B and 1C).
Figure 1.
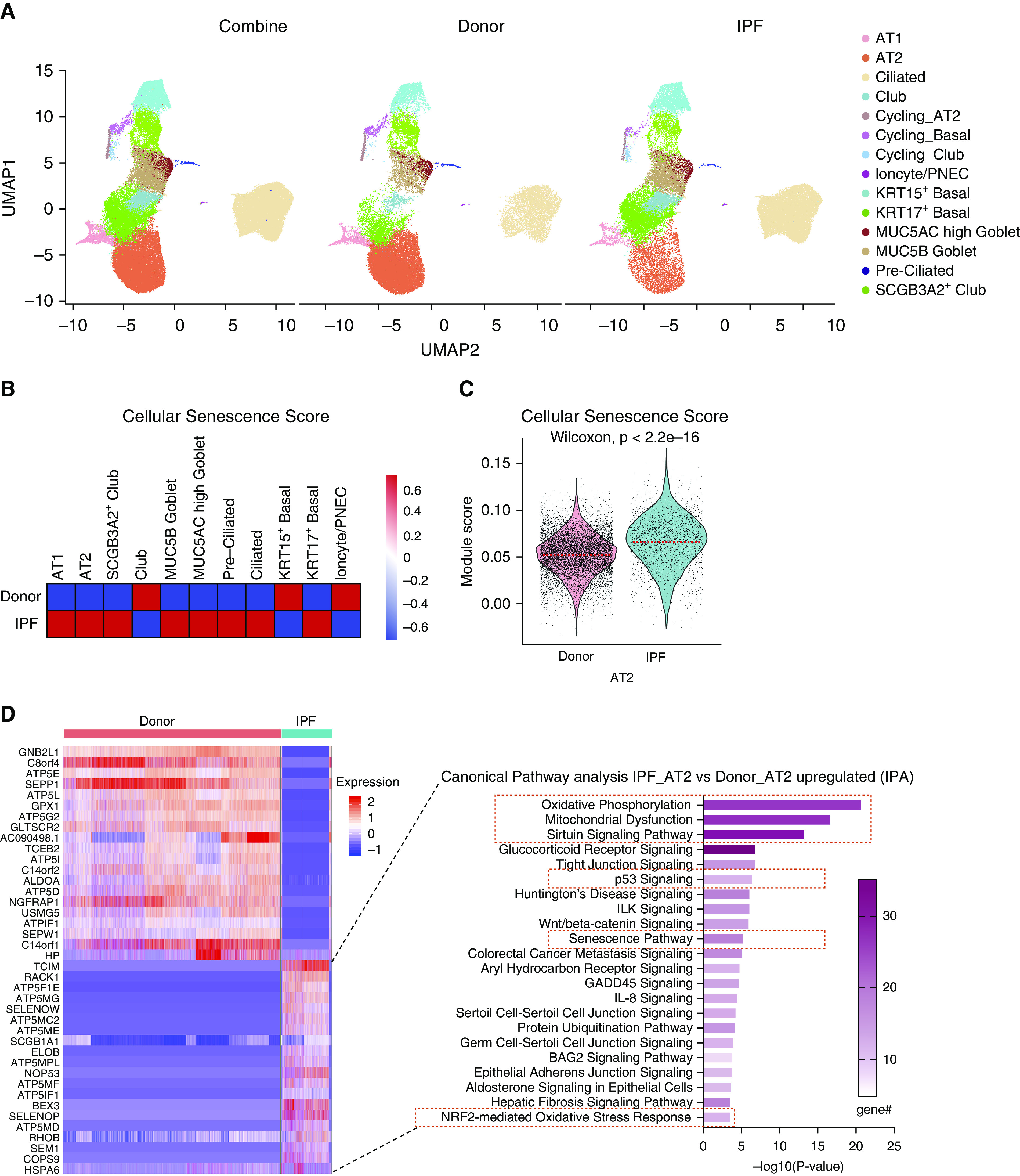
Accumulation of senescent alveolar type 2 (AT2) cells in idiopathic pulmonary fibrosis (IPF) explant tissue. (A) Uniform manifold approximation and projection (UMAP) visualization of cell type clustering (AT1, AT2, club [SCGB3A2+, SCGB1A1+], SCGB3A2+ club-like [SCGB3A2+, SCGB1A1−], ciliated, preciliated [MCIDAS+], MUC5AC high goblet [MUC5AC+, MUC5B+], MUC5B high goblet [MUC5AC−, MUC5B+], KRT15+ basal [KRT5 high, KRT17+, KRT15 high], KRT17+ basal [KRT5 low, KRT17+, KRT15 low], and ionocyte) and cell origin of single-cell RNA sequencing data on isolated epithelial cells from donor lung tissues and fibrotic region of lung tissue from patients with IPF (9 donor patient samples and 11 IPF patient samples). (B) Heat-map visualization of cellular senescence score based on core senescence genes of all epithelial cell types. Color scale represents log-transformed ratio of cellular senescence score. (C) Violin plot visualization of cellular senescence score based on core senescence genes of the AT2 cell subset from human epithelial cell single-cell RNA sequencing. The dashed line indicates the median value of each group. (D) Heat map of the top 20 differential expression genes of the AT2 cell subset comparing IPF with donor and top canonical pathways of genes upregulated in IPF AT2 cells identified by Ingenuity Pathway Analysis; senescence pathway and senescence-related pathways are highlighted. In total, 506 significantly upregulated genes and 1,209 significantly downregulated genes comparing IPF AT2 cells with donor AT2 cells. IPA = Ingenuity Pathway Analysis; PNEC = pulmonary neuroendocrine cell.
To further evaluate the contribution AT2 cells make to the senescent cell fraction, differentially expressed genes within the AT2 cell cluster were calculated using Wilcoxon rank-sum test with a cutoff of P = 0.01 and evaluated by Ingenuity Pathway Analysis (IPA). Senescence and senescence-related pathways featured among the top upregulated pathways in IPF AT2 cells when compared with donor AT2 cells (22–27) (Figure 1D). The expression of core senescence markers, including CDKN1A/p21, CDKN2A/p16, TP53, MDM2, and CCND1 (28, 29), were significantly elevated in IPF AT2 cells when compared with donor AT2 cells (Figures E3A and E3B). These findings were recapitulated among single AT2 cell transcriptomes evaluated in a distinct IPF dataset (6) (GSE122960; Figures E3C–E3F). These observations were validated by bulk RNA-Seq generated from enriched HTII-280+ presumptive AT2 cells isolated from either donor or IPF explants (16) (Figure E3G). Because of the possibility that HTII-280 immunoreactivity may identify cells other than bona fide AT2 cells in IPF lungs, we furthermore validated by immunostaining. We found increased CDKN2A/p16 and CDKN1A/p21 immunoreactivity in SFTPC+ AT2 cells within localized hyperplastic epithelium adjacent to fibrotic regions of IPF lung tissue (Figures E4A–E4D). Immunoreactivity of CDKN2A/p16 and CDKN1A/p21 was also observed among other epithelial and stromal cell types, consistent with the observed distribution of SA-βgal staining among multiple cell types in IPF explant tissue (Figure E1A). These data demonstrate AT2 cells contribute to the total pool of senescent lung cells in IPF, and senescence of AT2 cells is a common pathological feature of IPF observed among patient samples evaluated in our study. Accordingly, we sought to better define the contribution made by senescent AT2 cells toward progressive tissue remodeling observed in fibrotic lung disease.
A Novel Mouse Model of Conditional AT2 Cell Senescence
We next sought to determine whether senescence of AT2 cells drives progressive fibrosis. Our previous work demonstrated endodermal progenitor cells of the developing mouse lung are uniquely dependent on Sin3a; loss of Sin3a in endodermal progenitor cells leads to their senescence and eventual apoptosis, leading to defects in branching morphogenesis (30). To determine whether progenitor function of adult AT2 cells shows a similar dependence on Sin3a, we generated SftpcCreER, Sin3af/f, and RosamTmG mice with conditional Sin3a loss of function in AT2 cells (Sin3a-LOF) (Figure 2A). Near complete loss of Sin3a within AT2 cells 2 weeks after tamoxifen administration was verified by immunoblotting and immunostaining (Figures 2B and E5A).
Figure 2.
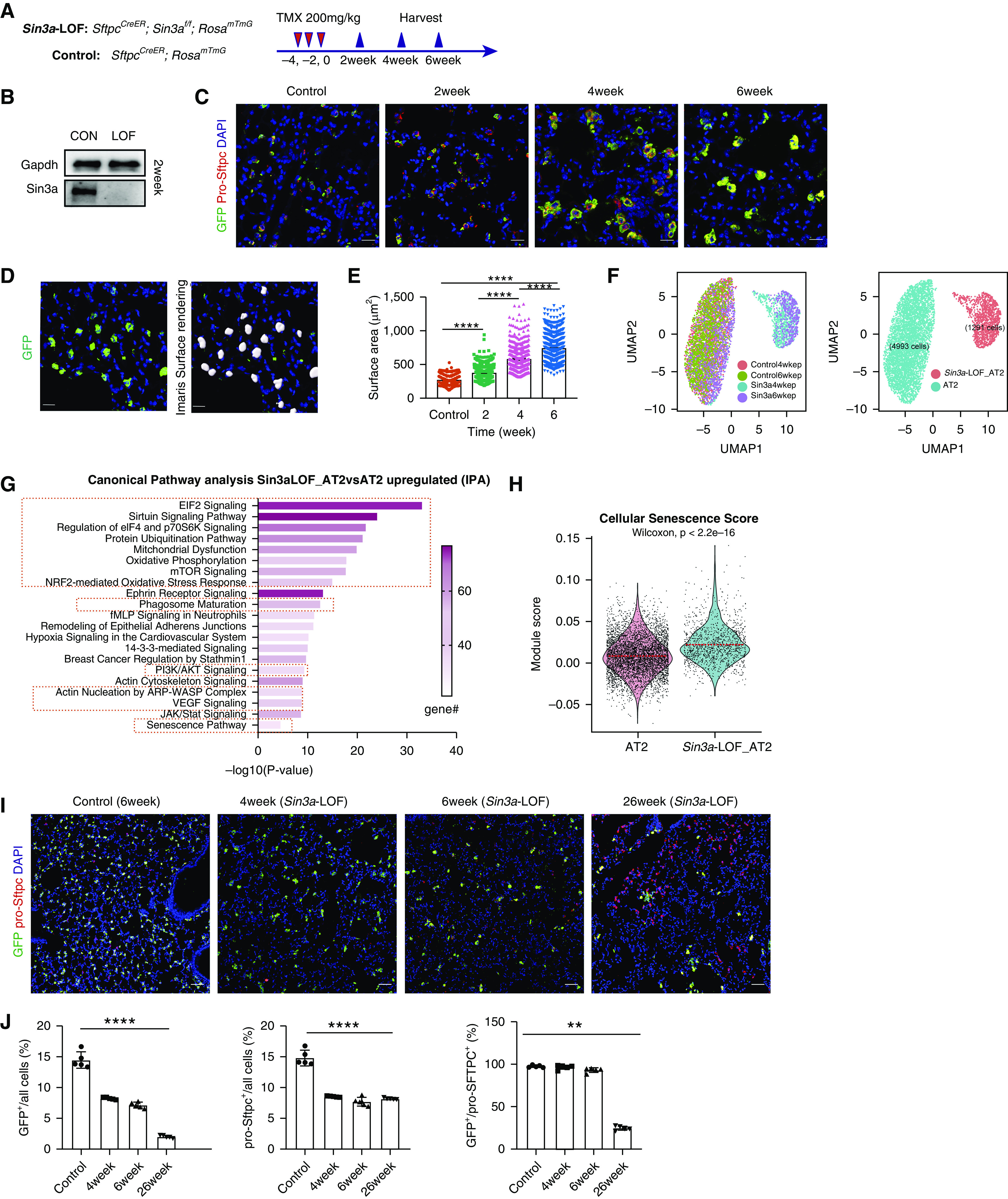
Loss of Sin3a in adult mouse alveolar type 2 (AT2) cells leads to cellular senescence. (A) Schematic outline of experiment design. (B) Western blot detection of Sin3a knockout efficiency in isolated AT2 cells 2 weeks after tamoxifen treatment. (C) Time course of immunofluorescence staining for lineage reporter-GFP (green fluorescent protein) and pro-Sftpc in control and Sin3a-LOF (Sin3a loss of function) lung. Scale bars, 20 μm. (D) Representative immunofluorescence staining of membrane-bound GFP and Imaris surface rendering for surface area quantification. Scale bars, 20 μm. (E) AT2 cell surface area quantification. (F) Uniform manifold approximation and projection (UMAP) visualization of AT2 cell subset, origin, and clustering from mouse epithelial cell single-cell RNA sequencing. (G) Ingenuity Pathway Analysis canonical pathway analysis of genes upregulated in AT2 cells of Sin3a-LOF compared with control; senescence pathway and senescence-related pathways are highlighted. (H) Violin plot visualization of cellular senescence score based on core senescence genes comparing Sin3a-LOF AT2 cells with control AT2 cells. The dashed line indicates the median value of each group. (I) Representative immunofluorescence staining of lineage reporter gene (GFP) and pro-Sftpc, comparing lung tissue of control with Sin3a-LOF at different times after tamoxifen exposure. Scale bars, 20 μm. (J) Quantification of percentage of either lineage reporter GFP+ cells, pro-Sftpc+ cells, or the ratio of GFP+/pro-Sftpc+ cells as a function of total cells within a section of lung lobe of Sin3a-LOF mice over the time course. Control samples are SftpcCreER and RosamTmG mice 6 weeks after tamoxifen treatment. P values were calculated by nonparametric Mann-Whitney test. **P < 0.01 and ****P < 0.0001. CON = control; IPA = Ingenuity Pathway Analysis.
To better define how Sin3a-LOF impacts the phenotype of AT2 cells, we performed bulk RNA-Seq on AT2 cells isolated from wild-type and Sin3a-LOF mouse lungs 2 weeks after tamoxifen treatment. Differentially expressed genes were determined by DEseq2-test with a cutoff of adjusted P = 0.05 for further IPA canonical pathway analysis. Senescence-related pathways were upregulated in Sin3a-LOF AT2 cells, mirroring that seen in AT2 cells of IPF tissue (Figures E5B–E5E). Changes in the molecular phenotype of Sin3a-LOF AT2 cells were accompanied by dramatic increases in cell size (Figures 2C–2E), a characteristic feature of senescent cells (31). Indeed, 6 weeks after tamoxifen treatment, the average cross-sectional area of individual AT2 cells increased almost threefold in Sin3a-LOF mice (735.5 ± 8.076 μm2) compared with age-matched control donors (263 ± 2.797 μm2) (Figure 2F). This was accompanied by about an 80% reduction in in vitro colony-forming efficiency (Figures E5F–E5H) and a >99% reduction in in vivo proliferative index as assessed by 6-week 5-iodo-2′-deoxyuridine (IdU) incorporation (0.037 ± 0.018% compared with 5.651 ± 0.335%; Figures E6A–E6C) of Sin3a-LOF AT2 cells compared with Sin3a-sufficient control AT2 cells, respectively. These data suggest that Sin3a-LOF leads to permanent cell-cycle arrest of AT2 cells.
To further define the molecular changes accompanying Sin3a loss within AT2 cells, we performed scRNA-Seq on enriched epithelial cells isolated from Sin3a-LOF and control mouse lungs 4 weeks and 6 weeks after tamoxifen treatment. Major cell types were assigned to clusters according to known cell type–specific gene signatures and expression of the GFP lineage reporter (Figures E7A and E7B). AT2 cells defined by expression of Sftpc and GFP were selected for further analysis and formed two distinct clusters (Figure 2F). Because of the low detection of Sin3a transcript, we furthermore investigated the origins of single cells within these clusters (Figures E8A–E8C). The larger AT2 cluster included representation of all cells from Sin3a-sufficient mice and some cells (presumably representing unrecombined cells) from Sin3a-LOF mice. In contrast, the smaller AT2 cluster only included cells derived from Sin3a-LOF mice, which were considered to represent Sin3a-LOF AT2 cells (recombination of both Sin3a alleles). We identified differentially expressed genes using Wilcoxon rank-sum test with a cutoff of P = 0.01 (Figure E7C) and used this gene list to define pathways that were dysregulated by Sin3a-LOF. As seen among AT2 cells of IPF lung, IPA analysis of mouse AT2 cells revealed upregulation of senescence and senescence-related pathways among Sin3a-LOF AT2 cells compared with their Sin3a-sufficient counterparts (32–35) (Figure 2G). Furthermore, cellular senescence score and expression of core senescence markers Cdkn1a/p21, Cdkn2d/p19 (36), Trp53, Mdm2, and Ccnd1 as well as a serial of senescence-associated secretory phenotype (SASP)-related genes were all significantly increased in Sin3a-LOF AT2 cells compared with Sin3a-sufficient counterparts, accompanied by increased SA-βgal signal in Sin3a-LOF lung compared with wild type (Figures 2H and E8D–E8G). These data, together with our earlier demonstration of cell-cycle exit, suggest that loss of Sin3a in adult AT2 cells activates cellular senescence. However, despite the senescent phenotype of Sin3a-LOF AT2 cells, evidence for elevated caspase-3 staining and declining abundance of lineage reporter GFP+ AT2 cells over time suggests these cells are eventually cleared (Figures 2I and 2J and E6D and E6E).
Roles for Sin3a-LOF, AT2 Cell Dysfunction, and Senescence in Progressive Lung Fibrosis
We next sought to determine if AT2 cell dysfunction mediated by Sin3a-LOF impacted lung fibrosis. The 10- to 12-week-old Sin3a-LOF mice were exposed to three doses of tamoxifen. Lung injury and remodeling were assessed over the following 8 weeks (Figure 3A). Sin3a-LOF led to persistent morbidity, as indicated by a decline in body weight, and a significant increase in mortality (Figures 3B and 3C). Histological assessment revealed focal consolidations of matrix deposition initiated at 4 weeks at the lung edges and progressed inward forming extensive interstitial fibrosis at later times (Figure 3D). Importantly, this pattern of fibrosis closely mirrors both the onset and progression of fibrosis seen in human lung (37). However, fibroblastic foci were observed at the 4-week recovery time point (Figure E9) but without evidence of honeycombing seen in the end-stage human IPF lung.
Figure 3.

Loss of Sin3a in alveolar type 2 cells leads to progressive lung fibrosis. (A) Schematic outline of experiment design. (B) Body weight change for tamoxifen-exposed Sin3a-LOF (Sin3a loss of function) and control mice. (C) Survival curve for tamoxifen-exposed Sin3a-LOF and control mice. The P value was calculated by log-rank (Mantel-Cox) survival analysis. (D) Time course of Masson’s trichrome staining of lung tissue from tamoxifen-exposed Sin3a-LOF and control mice. Squares indicate zones of magnified images. Scale bars: top, 1 mm; bottom, 50 μm. (E) Representative immunofluorescence staining of Sin3a-LOF lung tissue 6 weeks after tamoxifen exposure for lineage reporter-GFP (green fluorescent protein), pro-Sftpc, and αSMA. The dashed line represents the boundary between fibrotic and nonfibrotic tissue. E′ and E″ are magnified images of representative regions from either nonfibrotic or fibrotic tissue. Scale bars: E, 200 μm; E′ and E″, 20 μm. (F) Western blot detection of αSMA abundance within lung tissue of Sin3a-LOF mice. (G) Hydroxyproline content of right lung between Sin3a-LOF and control mice 6 weeks after tamoxifen exposure (n = 10 for each group). The P value was calculated by two-tailed Student’s t test. ****P < 0.0001.
We performed whole-lung RNA-Seq to define global changes in gene expression that accompany progressive fibrosis in Sin3a-LOF mice. IPA identified altered pathways involving activation of fibrosis 4 weeks after tamoxifen, which increased in significance at later times (Figure E10A). Paralleling the increases in fibrosis scores was a marked stimulation of multiple collagen and other fibrosis-related genes, such as Fibronectin, Ctgf, and myofibroblast marker αSMA; an elevated signal for αSMA immunoreactivity was seen in fibrotic regions (Figures 3E and 3F and E10B and E10C). Hydroxyproline content, a marker of collagen deposition, was markedly elevated in Sin3a-LOF lungs compared with control (Figure 3G). Collectively, these data suggest AT2 cell dysfunction and associated cellular senescence resulting from loss of Sin3a is sufficient to drive progressive lung fibrosis in a pattern that closely mimics that seen in IPF.
p53 Signaling Pathway Activation in Senescent AT2 Cells
We previously reported that p53 is dramatically upregulated within AT2 cells of IPF explant tissue (12). IPA upstream analysis of total lung RNA-Seq data was used to identify candidate regulators of fibrosis in Sin3a-LOF mice. Trp53 was among the top upstream regulators (Figure E11A). Similarly, we also identified TP53/Trp53 as top upstream regulators by IPA from either bulk RNA-Seq of isolated HTII-280+ cells or AT2 subsets of IPF scRNA-Seq and Sin3a-LOF scRNA-Seq data (Figures E11B and E6D). Thus, these data identify TP53/Trp53 as a critical regulator of fibrosis in both human and mouse epithelial samples.
Given that p53 activation is linked to loss of progenitor cell function and induction of cellular senescence (38, 39), we evaluated p53 activation status in Sin3a-LOF mice and in IPF patient samples. We found p53 signaling was among the top upregulated pathways both in Sin3a-LOF mouse lung and in IPF AT2 cells (Figures 1D and E9A). These findings were confirmed by increased lung p53 protein levels in Sin3a-LOF mice (Figure E11E). Increased p53 protein abundance was also evident in lung tissue of mice 7 and 14 days after bleomycin exposure (Figure E11F). Next, we compared the Kyoto Encyclopedia of Genes and Genomes (KEGG) database p53 pathway activation gene list to AT2 subsets of Sin3a-LOF and human IPF scRNA-Seq data. We observed a significant increase in p53 activation score (Figures 4A and 4B) and increased immunoreactivity and mRNA expression of p53 downstream target Cdkn1a/p21 (CDKN1A/p21) (Figures 4C and 4D, E4C and E4D, and E11G and E11H) in Sin3a-LOF and IPF AT2 cells compared with their matched controls.
Figure 4.

p53 pathway activation in Sin3a-LOF (Sin3a loss of function) alveolar type 2 (AT2) cells. (A) Violin plot visualization of p53 activation score calculated using p53 Kyoto Encyclopedia of Genes and Genomes pathway genes of the mouse single-cell RNA sequencing (scRNA-Seq) AT2 cell subset. The dashed line indicates the median value of each group. (B) Violin plot visualization of p53 activation score of the human scRNA-Seq AT2 cell subset. The dashed line indicates the median expression of each group. (C) Representative immunofluorescence staining of lineage reporter-GFP (green fluorescent protein) and Cdkn1a/p21, comparing control lung tissue with Sin3a-LOF 6 weeks after tamoxifen exposure. Scale bars, 10 μm. (D) Violin plot representation showing relative expression of Cdkn1a/p21 in mouse scRNA-Seq AT2 subset. The dashed line indicates the median expression of each group. (E) Schematic outline of experiment design for p53-LOF studies. (F) Survival curve for Sin3a-LOF and Sin3a/p53-LOF groups 6 weeks after tamoxifen treatment. The P value was calculated by log-rank (Mantel-Cox) survival analysis. (G) Hematoxylin and eosin and Masson’s trichrome staining of Sin3a-LOF and Sin3a/p53-LOF lung tissues 6 weeks after tamoxifen treatment. Squares indicate zones of magnified images. (H) Hydroxyproline content of right caudal lobe in Sin3a-LOF and Sin3a/p53-LOF mice 6 weeks after tamoxifen treatment (n = 10 for each group). P values were calculated by two-tailed Student’s t test. ****P < 0.0001. H&E = hematoxylin and eosin; IPF = idiopathic pulmonary fibrosis; ns = not significant.
To further examine the importance of p53 activation as a downstream mediator of progressive lung fibrosis in Sin3a-LOF mice, we generated SftpcCreER, Sin3af/f, and p53f/f mice (Sin3a/p53-LOF) with conditional loss of both Sin3a and p53 in AT2 cells. The 10- to 12-week-old Sin3a-LOF and Sin3a/p53-LOF mice were exposed to tamoxifen and monitored for 6 weeks (Figure 4E). In contrast to Sin3a-LOF mice, Sin3a/p53-LOF mice were protected from body weight decline, mortality, and loss of AT2 cells (Figure E11I). Inhibition of p53 signaling was also associated with decreased expression of p21 and SA-βgal staining (S12A-D) and reduced fibrosis (Figures 4G and 4H) in Sin3a/p53-LOF lungs compared with those of Sin3a-LOF controls. These results were recapitulated after systemic delivery of Pifithrin-α, a p53 inhibitor, to Sin3a-LOF mice (Figures E13A–E13E). These data demonstrate that silencing of either p53 expression or activity in AT2 cells prevented progressive fibrosis resulting from Sin3a-LOF and suggest p53 activation is necessary to promote AT2 cell dysfunction and lung fibrosis.
Senescence rather than Loss of AT2 Cells Drives Progressive Lung Fibrosis
To determine contributions of AT2 loss versus senescence in progressive fibrosis, we compared fibroproliferative responses between Sin3a-LOF mice and mice in which AT2 cells were ablated by conditional activation of diphtheria toxin A (SftpcCreER, DTAf/f, and RosamTmG [DTA]). The 10- to 12-week-old Sin3a-LOF and DTA mice were exposed to tamoxifen once/week for 8 weeks and evaluated at 4, 6, and 8 weeks after the first exposure (Figure 5A). Loss of AT2 cells in DTA and Sin3a-LOF mice were assessed through measurement of GFP and Sftpc at mRNA and protein levels by either quantitative PCR or immunofluorescence. DTA mice experienced a more than 90% decrease in AT2 cells throughout the time course compared with about a 50% reduction in Sin3a-LOF mice (Figures 5B, E14A–E14C, and E15A–E15C). At the 8-week exposure time point, DTA mice experienced 10% mortality with no significant decline in body weight. In contrast, no Sin3a-LOF mice survived (Figure E14D and E14E). Both Sin3a-LOF and DTA mice showed tamoxifen-dependent parenchymal consolidation and matrix deposition at 4 weeks (Figures 5C and 5E), consistent with a previous report showing that diphtheria toxin–mediated ablation of AT2 cells promotes increased matrix deposition in mice with conditional AT2 cell activation of diphtheria toxin receptor (40). However, despite histopathological evidence of progressive fibrosis seen in Sin3a-LOF mice, DTA mice recovered at the 6- and 8-week exposure time points with no histological evidence of fibrosis (Figures 5C and 5D). Furthermore, DTA mice show only modest increases in hydroxyproline content compared with the dramatic increase observed among Sin3a-LOF mice (Figures 5E and 5F). Together, these data suggest that AT2 cell dysfunction, rather than loss of AT2 cells, leads to progressive pulmonary fibrosis in Sin3a-LOF mice.
Figure 5.
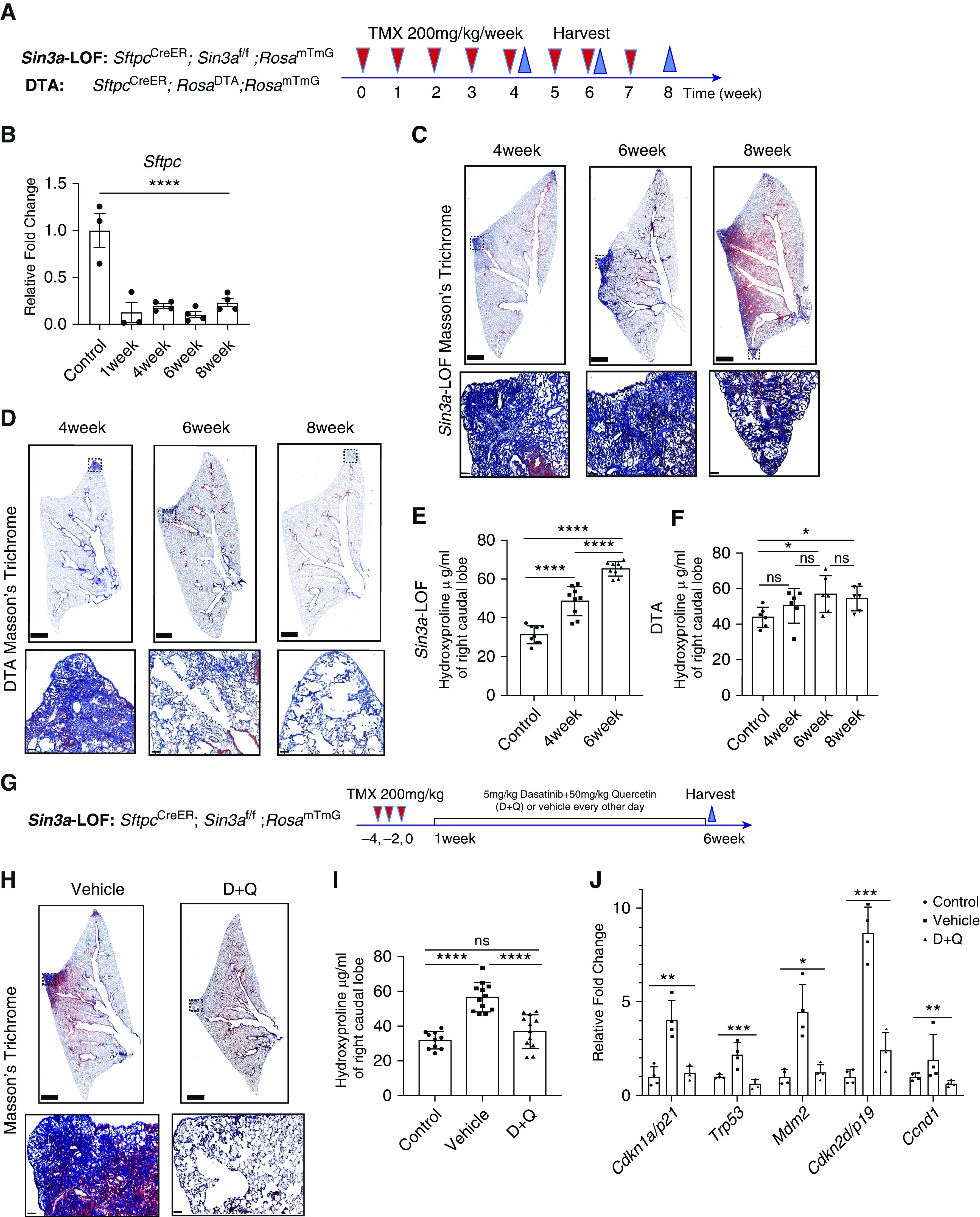
Senescence rather than loss of alveolar type 2 (AT2) cells results in progressive lung fibrosis. (A) Schematic outline of experiment design for AT2 cell ablation. (B) Quantitative PCR of control and diphtheria toxin A (DTA) mouse lung tissues for relative changing expression of Sftpc, indicating AT2 cell ablation efficiency (n > 3). (C) Masson’s trichrome staining of Sin3a-LOF (Sin3a loss of function) mice after repeated tamoxifen treatment. Squares indicate zones of magnified images. Scale bars: top, 1 mm; bottom, 50 μm. (D) Masson’s trichrome staining of DTA mice after repeated tamoxifen treatment. Squares indicate zones of magnified images. Scale bars: top, 1 mm; bottom, 50 μm. (E) Hydroxyproline content in the right caudal lobe of Sin3a-LOF mice after 4 and 6 weeks repeated exposure of tamoxifen and control groups. (F) Hydroxyproline content in the right caudal lobe of DTA mice after 4, 6, and 8 weeks repeated exposure of tamoxifen and control groups. (G) Schematic outline of experiment design for senolytic drug treatment. (H) Masson’s trichrome staining of Sin3a-LOF mice 6 weeks after tamoxifen treatment comparing 5 mg/kg dasatinib and 50 mg/kg quercetin (D+Q) cocktail treatment and vehicle treatment groups. Squares indicate zones of magnified images. Scale bars: top, 1 mm; bottom, 50 μm. (I) Quantitative PCR of isolated AT2 cells from control, vehicle-treated, and D+Q-treated Sin3a-LOF mouse lung tissues for relative changing expression of senescence markers indicating senolytic drug treatment efficiency (n = 4). (J) Hydroxyproline content in right caudal lobe of Sin3a-LOF mice treated with either D+Q or vehicle 6 weeks after tamoxifen treatment (n > 10 for each group). P values were calculated by nonparametric Mann-Whitney test (comparison of two groups) or Kruskal-Wallis test (comparison of more than two groups). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. ns = not significant.
Finally, we tested the hypothesis that senescence of AT2 cells promotes progressive pulmonary fibrosis in Sin3a-LOF mice. We treated 10- to 12-week-old Sin3a-LOF mice with a cocktail of senolytic drugs, dasatinib and quercetin, or vehicle by oral gavage every other day for 5 weeks starting at 1 week after the last dose of tamoxifen treatment (Figure 5G). Senolytic efficiency was approximately 40% as determined by measuring the abundance of lineage-traced AT2 cells in dasatinib and quercetin–treated Sin3a-LOF mice compared with vehicle-treated Sin3a-LOF control mice (Figures E16A–E16D). Senolytic drugs protected Sin3a-LOF mice against body weight decline and mortality (Figures E16E and E16F), blocked histological evidence of fibrosis, normalized hydroxyproline content, and decreased expression of both senescence markers and SA-βgal staining compared with vehicle-treated mice (Figures 5I and 5J and E14G). These data suggest that senescence of AT2 cells is a determinant of progressive fibrosis seen in Sin3a-LOF mice and by inference, a primary driver of tissue remodeling in IPF.
Discussion
Increased cellular senescence has been linked to both disease-associated gene variants and age-dependent loss of epithelial progenitor cell function. However, mechanisms by which cellular senescence drives tissue fibrosis are unclear. Herein, we determine senescence and declining abundance of AT2 cells, facultative stem cells that maintain the epithelial lining in the gas-exchange regions of the lung, are linked processes in end-stage IPF. To define disease mechanisms, we sought to define the contribution of AT2 cell senescence and AT2 cell loss toward initiation and progression of disease. We found that conditional loss of Sin3a within AT2 cells of adult mice induced a program of senescence and eventual apoptotic cell loss. Furthermore, even though either senescence or depletion of AT2 cells led to spontaneous lung fibrosis, fibrosis seen in mice with AT2 depletion was transient and rapidly resolved, whereas conditional senescence promoted progressive interstitial lung fibrosis resembling that seen in human IPF. We establish AT2 senescence, rather than AT2 cell depletion per se, represents a critical determinant of fibrogenesis in epithelial tissues.
Cellular senescence can be induced by multiple intrinsic stress-related stimuli through one of two major mechanisms, activation of either p53–p21 and/or p16INK4a–pRB pathways (41). Interestingly, mouse models with conditional loss of Trf1 or Trf2, the telomere-related shelterin complex components, show a less pronounced phenotype than with Sin3a-LOF (4, 42, 43). We show that p53 signaling is one of the top pathways upregulated in IPF AT2 cells compared with donor AT2 cells. Increased expression of p53 downstream target CDKN1A/p21 suggested AT2 cell cycle arrest and senescence may depend on activation of the p53–p21 signaling axis. Certainly, hypertrophic AT2 cells present within IPF explant tissue show strong induction of p21 but also show elevated immunoreactivity for p16. We were able to show Sin3a-LOF in mouse AT2 cells resulted in p53–p21 activation and p53-dependent senescence and fibrosis. Inhibition of p53 mitigates fibrosis resulting from Sin3a-LOF, partially rescues AT2 cell loss, and reduces the level of p21 induction and SA-βgal staining. Though loss of p53 cannot fully block declining AT2 cell abundance because of Sin3a-LOF, inhibition of p53 signaling may rescue fibrosis through abrogation of downstream Tgfβ activation (44–47). Furthermore, decreased expression of p21 in Sin3a-LOF/p53-LOF mice suggests that Sin3a-regulated p53 pathway activation may occur through changes in p53AcK317 and p53AcK379 deacetylation rather than through HDAC-medicated histone modification (48). Although we cannot exclude a role for p16, our data suggest that p53 activation is necessary to induce AT2 senescence leading to progressive pulmonary fibrosis.
SASP has been shown to serve important roles in repair after an acute injury yet is a likely contributor to IPF pathogenesis (49, 50). We show that AT2 cells of the IPF lung and Sin3a-LOF mouse lung present cellular senescence, with increased SASP gene expression, including TGFB1/Tgfb1. The critical role for AT2 senescence with associated gradual apoptotic cell loss in both initiation and progression of fibrosis was suggested by progressive fibrosis of Sin3a-LOF mice but not with AT2 cell ablation and the demonstration that senolytic drugs were protective. Our finding that elimination of senescent AT2 cells from lungs of Sin3a-LOF mice protects from progressive fibrosis supports a critical role for AT2 senescence, rather than simple loss of AT2 cells, in both initiation and progression of IPF. However, the nonspecific nature of a senolytic drug raises possibilities that other mechanisms in addition to depletion of senescent cells may contribute to the observed protective effects.
In summary, we present a novel conditional mouse model of progressive interstitial lung fibrosis in which loss of Sin3a function and resulting senescence of AT2 cells recapitulates key pathological features of IPF. Sin3a-LOF in mouse AT2 cells activates senescence in a p53- and p21-dependent manner, leading to downstream signaling and fibrogenesis. Using this mouse model, we provide evidence that early targeting of senescent cells is of therapeutic benefit in preventing lung fibrosis, a strategy that may have broad potential to mitigate fibrosis in a wide range of epithelial tissues.
Supplementary Material
Acknowledgments
Acknowledgment
The authors thank Dr. William Parks and Stripp Lab members for critical reading of the manuscript and the Cedars-Sinai Biobank for assistance with tissue procurement.
Footnotes
Supported by the California Institute of Regenerative Medicine (LA1-06915), the NIH (4 U01HL110967-05, P01HL108793, R01HL135163, and T32HL134637), Bristol Myers Squibb IDEAL Consortium, and the Parker B. Francis Fellowship Program, UCLA CTSI KL2 (NCATS KL2TR001882).
Author Contributions: C.Y., G.C., and B.R.S. contributed to conceptual design of experiments. C.Y., X.G., T.P., X.L., G.H., and A.M. performed experiments. C.Y. and B.R.S. analyzed data. H.J.S., G.D., S.S.W., J.A.B., P.C., D.J., and P.W.N. provided material and technical assistance. C.Y., P.C., and B.R.S. were involved in manuscript writing.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202004-1274OC on September 29, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.King TE, Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 2.Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017;8:14532. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lehmann M, Korfei M, Mutze K, Klee S, Skronska-Wasek W, Alsafadi HN, et al. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur Respir J. 2017;50:1602367. doi: 10.1183/13993003.02367-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alder JK, Barkauskas CE, Limjunyawong N, Stanley SE, Kembou F, Tuder RM, et al. Telomere dysfunction causes alveolar stem cell failure. Proc Natl Acad Sci USA. 2015;112:5099–5104. doi: 10.1073/pnas.1504780112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minagawa S, Araya J, Numata T, Nojiri S, Hara H, Yumino Y, et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2011;300:L391–L401. doi: 10.1152/ajplung.00097.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med. 2019;199:1517–1536. doi: 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tian Y, Li H, Qiu T, Dai J, Zhang Y, Chen J, et al. Loss of PTEN induces lung fibrosis via alveolar epithelial cell senescence depending on NF-κB activation. Aging Cell. 2019;18:e12858. doi: 10.1111/acel.12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parimon T, Yao C, Stripp BR, Noble PW, Chen P. Alveolar epithelial type II cells as drivers of lung fibrosis in idiopathic pulmonary fibrosis. Int J Mol Sci. 2020;21:2269. doi: 10.3390/ijms21072269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kadota T, Fujita Y, Yoshioka Y, Araya J, Kuwano K, Ochiya T. Emerging role of extracellular vesicles as a senescence-associated secretory phenotype: insights into the pathophysiology of lung diseases. Mol Aspects Med. 2018;60:92–103. doi: 10.1016/j.mam.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 10.Katzen J, Wagner BD, Venosa A, Kopp M, Tomer Y, Russo SJ, et al. An SFTPC BRICHOS mutant links epithelial ER stress and spontaneous lung fibrosis. JCI Insight. 2019;4:e126125. doi: 10.1172/jci.insight.126125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nureki SI, Tomer Y, Venosa A, Katzen J, Russo SJ, Jamil S, et al. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J Clin Invest. 2018;128:4008–4024. doi: 10.1172/JCI99287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borok Z, Horie M, Flodby P, Wang H, Liu Y, Ganesh S, et al. Grp78 loss in epithelial progenitors reveals an age-linked role for endoplasmic reticulum stress in pulmonary fibrosis. Am J Respir Crit Care Med. 2020;201:198–211. doi: 10.1164/rccm.201902-0451OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45:613–620. doi: 10.1038/ng.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moore C, Blumhagen RZ, Yang IV, Walts A, Powers J, Walker T, et al. Resequencing study confirms that host defense and cell senescence gene variants contribute to the risk of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2019;200:199–208. doi: 10.1164/rccm.201810-1891OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123:3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J, et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight. 2016;1:e90558. doi: 10.1172/jci.insight.90558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang J, Zhang Y, Xie T, Liu N, Chen H, Geng Y, et al. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat Med. 2016;22:1285–1293. doi: 10.1038/nm.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv. 2020;6:eaba1972. doi: 10.1126/sciadv.aba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carraro G, Mulay A, Yao C, Mizuno T, Konda B, Petrov M, et al. Single cell reconstruction of human basal cell diversity in normal and IPF lung. Am J Respir Crit Care Med. doi: 10.1164/rccm.201904-0792OC. [online ahead of print] 21 Jul 2020; DOI: 10.1164/rccm.201904-0792OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv. 2020;6:eaba1983. doi: 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao M, Chen L, Qu H. CSGene: a literature-based database for cell senescence genes and its application to identify critical cell aging pathways and associated diseases. Cell Death Dis. 2016;7:e2053. doi: 10.1038/cddis.2015.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hutter E, Renner K, Pfister G, Stöckl P, Jansen-Dürr P, Gnaiger E. Senescence-associated changes in respiration and oxidative phosphorylation in primary human fibroblasts. Biochem J. 2004;380:919–928. doi: 10.1042/BJ20040095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23:303–314. doi: 10.1016/j.cmet.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee SH, Lee JH, Lee HY, Min KJ. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019;52:24–34. doi: 10.5483/BMBRep.2019.52.1.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajesh K, Krishnamoorthy J, Gupta J, Kazimierczak U, Papadakis AI, Deng Z, et al. The eIF2α serine 51 phosphorylation-ATF4 arm promotes HIPPO signaling and cell death under oxidative stress. Oncotarget. 2016;7:51044–51058. doi: 10.18632/oncotarget.10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jung SH, Hwang HJ, Kang D, Park HA, Lee HC, Jeong D, et al. mTOR kinase leads to PTEN-loss-induced cellular senescence by phosphorylating p53. Oncogene. 2019;38:1639–1650. doi: 10.1038/s41388-018-0521-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fulop GA, Kiss T, Tarantini S, Balasubramanian P, Yabluchanskiy A, Farkas E, et al. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. Geroscience. 2018;40:513–521. doi: 10.1007/s11357-018-0047-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barnes PJ, Baker J, Donnelly LE. Cellular senescence as a mechanism and target in chronic lung diseases. Am J Respir Crit Care Med. 2019;200:556–564. doi: 10.1164/rccm.201810-1975TR. [DOI] [PubMed] [Google Scholar]
- 29.Liu B, Yi J, Yang X, Liu L, Lou X, Zhang Z, et al. MDM2-mediated degradation of WRN promotes cellular senescence in a p53-independent manner. Oncogene. 2019;38:2501–2515. doi: 10.1038/s41388-018-0605-5. [DOI] [PubMed] [Google Scholar]
- 30.Yao C, Carraro G, Konda B, Guan X, Mizuno T, Chiba N, et al. Sin3a regulates epithelial progenitor cell fate during lung development. Development. 2017;144:2618–2628. doi: 10.1242/dev.149708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21:1424–1435. doi: 10.1038/nm.4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferbeyre G. Aberrant signaling and senescence associated protein degradation. Exp Gerontol. 2018;107:50–54. doi: 10.1016/j.exger.2017.06.016. [DOI] [PubMed] [Google Scholar]
- 33.Gomez-Cambronero J, Kantonen S. A river runs through it: how autophagy, senescence, and phagocytosis could be linked to phospholipase D by Wnt signaling. J Leukoc Biol. 2014;96:779–784. doi: 10.1189/jlb.2VMR0214-120RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coppé JP, Kauser K, Campisi J, Beauséjour CM. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem. 2006;281:29568–29574. doi: 10.1074/jbc.M603307200. [DOI] [PubMed] [Google Scholar]
- 35.Yun UJ, Park SE, Shin DY. p41-Arc, a regulatory subunit of Arp2/3 complex, can induce premature senescence in the absence of p53 and Rb. Exp Mol Med. 2011;43:389–392. doi: 10.3858/emm.2011.43.7.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sonzogni SV, Ogara MF, Belluscio LM, Castillo DS, Scassa ME, Cánepa ET. p19INK4d is involved in the cellular senescence mechanism contributing to heterochromatin formation. Biochim Biophys Acta. 2014;1840:2171–2183. doi: 10.1016/j.bbagen.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 37.Smith M, Dalurzo M, Panse P, Parish J, Leslie K. Usual interstitial pneumonia-pattern fibrosis in surgical lung biopsies: clinical, radiological and histopathological clues to aetiology. J Clin Pathol. 2013;66:896–903. doi: 10.1136/jclinpath-2013-201442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene. 2013;32:5129–5143. doi: 10.1038/onc.2012.640. [DOI] [PubMed] [Google Scholar]
- 39.McConnell AM, Yao C, Yeckes AR, Wang Y, Selvaggio AS, Tang J, et al. p53 regulates progenitor cell quiescence and differentiation in the airway. Cell Rep. 2016;17:2173–2182. doi: 10.1016/j.celrep.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 40.Sisson TH, Mendez M, Choi K, Subbotina N, Courey A, Cunningham A, et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181:254–263. doi: 10.1164/rccm.200810-1615OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tchkonia T, Kirkland JL. Aging, cell senescence, and chronic disease: emerging therapeutic strategies. JAMA. 2018;320:1319–1320. doi: 10.1001/jama.2018.12440. [DOI] [PubMed] [Google Scholar]
- 42.Povedano JM, Martinez P, Flores JM, Mulero F, Blasco MA. Mice with pulmonary fibrosis driven by telomere dysfunction. Cell Rep. 2015;12:286–299. doi: 10.1016/j.celrep.2015.06.028. [DOI] [PubMed] [Google Scholar]
- 43.Naikawadi RP, Disayabutr S, Mallavia B, Donne ML, Green G, La JL, et al. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight. 2016;1:e86704. doi: 10.1172/jci.insight.86704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Atfi A, Baron R. p53 brings a new twist to the Smad signaling network. Sci Signal. 2008;1:pe33. doi: 10.1126/scisignal.126pe33. [DOI] [PubMed] [Google Scholar]
- 45.Cordenonsi M, Dupont S, Maretto S, Insinga A, Imbriano C, Piccolo S. Links between tumor suppressors: p53 is required for TGF-beta gene responses by cooperating with Smads. Cell. 2003;113:301–314. doi: 10.1016/s0092-8674(03)00308-8. [DOI] [PubMed] [Google Scholar]
- 46.Ji L, Xu J, Liu J, Amjad A, Zhang K, Liu Q, et al. Mutant p53 promotes tumor cell malignancy by both positive and negative regulation of the transforming growth factor β (TGF-β) pathway. J Biol Chem. 2015;290:11729–11740. doi: 10.1074/jbc.M115.639351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kalo E, Buganim Y, Shapira KE, Besserglick H, Goldfinger N, Weisz L, et al. Mutant p53 attenuates the SMAD-dependent transforming growth factor beta1 (TGF-beta1) signaling pathway by repressing the expression of TGF-beta receptor type II. Mol Cell Biol. 2007;27:8228–8242. doi: 10.1128/MCB.00374-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dannenberg JH, David G, Zhong S, van der Torre J, Wong WH, Depinho RA. mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genes Dev. 2005;19:1581–1595. doi: 10.1101/gad.1286905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang WJ, Cai GY, Chen XM. Cellular senescence, senescence-associated secretory phenotype, and chronic kidney disease. Oncotarget. 2017;8:64520–64533. doi: 10.18632/oncotarget.17327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.