

Abstract
Lipid uptake and metabolism are central to the function of organs such as heart, skeletal muscle, and adipose tissue. Although most heart energy derives from fatty acids (FAs), excess lipid accumulation can cause cardiomyopathy. Similarly, high delivery of cholesterol can initiate coronary artery atherosclerosis. Hearts and arteries—unlike liver and adrenals—have non-fenestrated capillaries and lipid accumulation in both health and disease requires lipid movement from the circulation across the endothelial barrier.
This review summarizes recent in vitro and in vivo findings on the importance of endothelial cell (EC) receptors and uptake pathways in regulating FAs and cholesterol uptake in normal physiology and cardiovascular disease.
We highlight clinical and experimental data on the roles of ECs in lipid supply to tissues, heart and arterial wall in particular, and how this affects organ metabolism and function. Models of FA uptake into ECs suggest that receptor-mediated uptake predominates at low FA concentrations, such as during fasting, whereas FA uptake during lipolysis of chylomicrons may involve paracellular movement. Similarly, in the setting of an intact arterial endothelial layer, recent and historic data support a role for receptor-mediated processes in the movement of lipoproteins into the sub-arterial space.
We conclude with thoughts on the need to better understand endothelial lipid transfer for fuller comprehension of the pathophysiology of hyperlipidemia, and lipotoxic diseases such as some forms of cardiomyopathy, and atherosclerosis.
Keywords: triglyceride, fatty acids, CD36, SR-B1, GPIHBP1, lipoproteins, chylomicrons, lipoprotein lipase
Subject codes: Lipids and cholesterol, metabolism, vascular biology, diet and nutrition, atherosclerosis
Introduction
Lipids support energy production and caloric storage, processes essential for cellular structure and growth. In mammals, lipids circulate in the blood as non-esterified (NE) “free” fatty acids (FAs), but mostly (~90%) as esterified FA components of triglycerides (TGs), phospholipids and cholesteryl esters in lipoproteins. In all tissues, except for those with fenestrated capillaries like the liver and adrenals, lipid uptake requires transfer from the circulation across the endothelial cell (EC) barrier, a process coordinated by lipoprotein lipase (LpL), its binding protein glycosylphosphatidylinositol-anchored HDL binding protein 1 (GPIHBP1), cluster of differentiation 36 (CD36) and likely other molecules. This basic function of ECs normally allows optimal heart function and protects against some forms of dilated cardiomyopathy1, 2. This review will focus on EC transport of circulating lipids and how deletions of LpL, GPIHBP1, CD36, and SR-B1 affect EC lipid transport, tissue metabolism and organ function. The biology of metabolically driven cardiac lipotoxicity has been recently reviewed elsewhere3 and in a special issue on cardiovascular disease4. We will also review recent data that implicate ECs in the arterial wall lipid accumulation that initiates atherosclerosis.
The levels of circulating lipids likely regulate the receptors and enzymes involved in their transfer across the EC barrier. During fasting when stored lipids released from adipose tissue and VLDL secreted by the liver are the primary sources of FAs, CD36 facilitates cellular FA uptake acting as a high affinity receptor5, 6. In a well-controlled reaction, LpL releases FAs from circulating dietary TGs. Locally, LpL actions depend on its expression, expression of its inhibitors, and EC expression of GPIHBP1. LpL facilitates FA uptake into high-energy tissues such as the heart and brown fat. During the postprandial period, LpL directs dietary lipids within chylomicrons (CMs) into adipose storage.
Lipolysis within capillaries is well established as the principal route for conversion of CM TGs into NEFAs7 and additional details were provided by the discovery of GPIHBP18 and angiopoietin-like protein (ANGPTL) inhibitors. What is less defined is how NEFAs liberated from different lipoproteins leave the capillaries. Transfer of NEFAs released from TG within VLDL lipids appears partially to require EC CD36 (discussed below). Other receptors, e.g. those recently shown to mediate movement across ECs of the cholesterol-rich lipoproteins LDL and HDL might also transfer VLDL lipids. In contrast, as shown in Figure 1, FAs from CMs could enter the subendothelial space via a yet to be defined receptor, or via non-receptor movement either through or around the cells through channels created by the loosening of EC junctions. Evidence both in vitro 9 and in perfused arteries10 suggest that active lipolysis increases permeability of the EC barrier.
Figure 1. Pathways of FA transfer across ECs.

Left: The concentration of FAs is likely to modulate the pathway mediating their transit from the circulation to subendothelial cells. At low FA levels, as found during fasting, transfer is receptor-mediated involving cell surface CD36. FAs are internalized together with CD36, most likely in vesicles that might deliver the FA to the ER for activation by FATP3 or FATP4, for oxidation or incorporation into cell lipids. How and in what form the FA exits the basolateral EC surface to the subendothelial space is not known and is under investigation. Right: At higher FA concentrations such as locally occurs during lipolysis of triglyceride-rich lipoproteins (TRLs), EC CD36 will be saturated, being mostly internalized and cellular FA uptake would mainly occur via another pathway that we hypothesize involves paracellular flux between ECs. Activation of paracellular flux might occur by promoting loosening of EC junctions through phosphorylation of VE-cadherin by Src, protein kinase C or other kinases influenced by FAs. These processes are likely restricted to capillaries, as ECs of large arteries mostly do not express CD36, GPIHBP1 and do not have associated LpL.
This review will focus on the central processes of vascular lipid delivery to tissue, address current areas of controversy and highlight experimentally tractable findings with observations related to normal tissue function and pathophysiology. We will highlight how new studies of LpL and GPIHBP1 regulation and of the role of EC lipid receptors add to our understanding of the processes regulating sites of tissue lipid uptake. We will conclude by considering the poorly studied biophysical processes that occur within vessels and that likely regulate interaction of lipoproteins with the luminal EC surface.
Endothelial Cell Transport of FAs
FA oxidation supplies most of the energy for chronically working muscles such as heart and diaphragm, in contrast to type 2 or fast twitch muscles that preferentially use glucose. During restricted caloric intake, the sustained energy necessary for the heart requires more FA use as glucose availability becomes limiting11, 12. In the postprandial period, circulating FAs are mostly derived from lipolysis of lipoprotein (CM and VLDL) associated TGs, whereas during fasting NEFAs result both from intracellular lipolysis of adipose TG stores and from lipolysis of VLDL TGs. The first step in movement of NEFAs from the circulation to myocytes or adipocytes requires their uptake by ECs or their paracellular movement between ECs. The relative importance of these pathways is likely affected by concentrations of NEFAs near the EC luminal surface (Figure 1).
Circulating NEFA levels fluctuate throughout the day in parallel with the metabolic state. Most NEFAs are bound to albumin and unbound protein-free FA level in the circulation is determined by the molar ratios of FA to albumin and is very low. Fasting FA : albumin ratios in serum range between 0.3–1.5 and associate with unbound FA levels of 5–15 nM13. Much higher ratios would occur in vicinity of ECs during CM lipolysis. Studies of long chain FA uptake kinetics in intact cells14, 15, reviewed in5, 16, 17, documented saturation of uptake at <10 nM unbound FA and competitive inhibition between different FAs14. The kinetic data suggest that a receptor-mediated process is used for uptake of FAs of more than eight carbons. A similar conclusion was reached in a real time fluorescence microscopy study of FA transfer that included inhibitors and long versus short chain FAs18. Overall, kinetic properties of cellular FA uptake identify a saturable receptor mediated process at low FA: albumin ratios and a non-saturable, presumably non-receptor mediated process at higher FA/albumin ratios.
Receptor mediated FA uptake:
Transporters mediate transmembrane movement of NEFAs while intracellular FA-binding proteins trap the FA and influence cellular lipid accumulation, the combined steps referred to as vectorial transport. Each of these steps can affect tissue concentration of tracers and lipids, but a transporter must be on the plasma membrane, bind to FAs, and lead to rapid cellular FA accumulation.
The best characterized FA transporter is CD36 (Table 1), a member of the CD36 class B scavenger receptor family that includes scavenger receptor-B1 (SR-B1), a lipoprotein binding receptor (see later sections), and the lysosomal integral membrane protein 2 (LIMP2)19. In addition to FAs, CD36 recognizes lipoproteins (VLDL, HDL, and LDL), bacterial lipids, and non-lipid ligands (e.g. thrombospondin-1). The ligands induce CD36 mediated signaling and consequently the protein has pleiotropic cellular effects12, 19–21 that include, as recently reported, the regulation of insulin and AMPK actions22, 23. CD36 fulfills the requirements for a transporter: membrane localization, FA binding, and modulation of cellular FA content. Its expression enhances FA oxidation and FA incorporation into lipids and it translocates to the plasma membrane, acutely in response to insulin, AMPK, leptin, or chronically with excess FA supply24–28, as reviewed29.
Table 1.
| EC surface proteins implicated in uptake of long chain FAs | CD36, Cav-1, α5β1/CD36 complex (adipose tissue specific) Prohibitin–annexin A2-CD36 complex (adipose tissue specific), |
| EC surface proteins implicated in uptake of LDL, SR-B1 also mediates HDL uptake | SR-B1, ALK1, |
| EC FA Uptake regulators | PPARg, PGC1α, VEGF-A, VEGF-B, FATP3, FATP4, Angiopoietin-2 (adipose tissue specific), Meox2/Tcf15, 3 hydroxyisobutyrate |
| Proteins active in lipolysis at the EC surface | LpL, GPIHBP1 |
| Lipolysis regulators | APOC3, APOC2, APOA5, ANGPTL3, ANGPTL4, ANGPTL8, GPIHBP1 |
CD36’s function in FA uptake by rat adipocytes was identified through its binding of a reactive ester of oleic acid, sulfo-N-succinimidyl oleate (SSO) followed by its isolation and cloning30–32. Because SSO is currently commercially available as a specific inhibitor of CD36, it is useful to mention some of its properties. SSO is specific in that it blocks FA uptake without directly affecting other substrates or the metabolic fate of FAs33 and labeled [3H]-SSO or the palmitate ester [3H]-SSP specifically bind CD36 in plasma membranes34,35. However, SSO specificity, due to its long hydrocarbon chain, is dependent on its membrane impermeability. Although we initially used high inhibitor concentrations, as albumin which scavenges SSO was included in the medium, SSO is effective and recommended at low concentrations (10–25uM) 22, 36–38. When using high SSO levels, if membrane integrity is not monitored, the inhibitor will enter cells and interact with proteins other than CD3639.
The crystal structure of the CD36 family member LIMP2 reported in 201319 identified an internal lipid transport tunnel that traverses the length of the proteins in this family and empties in the bilayer’s proximity. This is discussed in more detail in the section where we compare CD36 and SR-B1. SSO binds to lysine 164 in a hydrophobic pocket in the CD36 ectodomain37 that connects with the transport tunnel40. Long chain FAs were associated with crystal CD36 and localized to the intramolecular tunnel41.
CD36 enhances FA uptake and FA oxidation in mice models42–45 and human CD36 polymorphisms associate with defects of lipid handling and metabolic disease (e.g., hyperlipidemia, metabolic syndrome, type 2 diabetes)22, 46–49. Total CD36 deficiency in 9–10% of African Americans and Africans46, 50–52 and in 1%–3% of Asians53, 54 cause’s defective FA uptake by heart, muscle and adipose tissues55–57. Common CD36 variants increase blood FAs58–59 and DNA methylation sites that reduce CD36 expression increase CM remnants60, 61. Genome wide association studies52, 62 relate CD36 expression inversely to HDL and positively to VLDL levels. The latter could reflect a hepatic effect of CD36 deletion, which inhibits liver VLDL secretion in mice63.
The FA transporter protein (FATP) 1 (Table 1) was identified through a functional screening for proteins that facilitate FA accumulation in cells64, 65 and belongs to the SLC27 protein family that activate FAs by CoA modification66. There are at least 6 family members; FATP2–5 are found primarily inside the cell, but FATP1 and FATP6 are detected on the plasma membrane (Cell Atlas and REACTOME databases) and FATP1 translocates to the membrane after insulin67. The FATPs share with the yeast ortholog Fatp1 two motifs required for binding ATP and FAs, and might function, like Fatp1, in vectorial coupling of FA uptake and FA activation, as the CoA-ligase activity traps the FA inside the cell and influences its metabolic partitioning68–70. The effect of gain and loss of function studies in animal models (Table 2) might reflect FATP isoform tissue expression, cellular localization and FA specificity, as reviewed67. FATP1 is most highly expressed in the heart and its overexpression increases heart TG content and produces cardiomyopathy, 71 while overexpression of FATP4 in skeletal muscle increases FA uptake and oxidation72. Deletion of FATP2 in the liver appears to be beneficial by activating PPARα remodeling of lipid metabolism73 (Table 2). FATP3 and FATP4 are expressed in ECs and have been implicated in endothelial FA accumulation, as will be discussed in the following section (endothelial regulation of FA transport). In summary, the FATP family of proteins play an important role in cellular FA uptake, but the nature of this role remains unclear; it might involve interaction with the FA in the plasma membrane, trapping the FA through its activation or interacting with membrane transporters providing vectorial transfer of FA to specific metabolic sites.
Table 2.
| Mouse model | Phenotype |
|---|---|
| FATP1 knockout FATP−/− |
- Reduced skeletal muscle lipid accumulation - Improved insulin sensitivity after lipid challenges |
| FATP1 overexpression | - Increased lipid accumulation in the heart - Cardiomyopathy |
| Skeletal muscle FATP4 overexpression (rats) | - Increased FA uptake/oxidation in skeletal muscle |
| Liver-FATP2 knockdown | - Protection from hepatosteatosis - Improved glucose levels and insulin sensitivity following high-fat diet |
| Knockout of PPARγ in EC (γEC/BM-KO) | - Increased circulating FAs - Protected from high fat diet induced adiposity and insulin resistance |
| Adipose tissue VEGF-A overexpression | - Increased vascularization - Increased thermogenesis and energy expenditure - Protection from obesity and insulin resistance |
| VEGF-B Knockout Vegfb−/− | - Less 14C FA uptake and more 18F-FDG uptake by heart and brown adipose tissue (BAT), and less LDs in heart, oxidative muscle and BAT - Higher body fat mass |
| Diabetic mice with VEGF-B deletion (db/db Vegfb−/−) | - Prevented ectopic lipid deposition - Increased muscle glucose uptake - Maintained normoglycemia |
| Endothelial cell CD36 knockout, EC-Cd36−/− | - Postprandial hypertriglyceridemia - No cardiac accumulation of lipid droplets during fasting - Increased insulin sensitivity - Increased expression of insulin signaling and glucose metabolism genes |
| Gpihbp1Knockout Gpihbp −/− | - Severe hyperchylomicronemia - Reduced post-heparin lipoprotein lipase activity |
| Adipocyte-Angpt2 knockout, Angpt2ΔAd) | - Markedly reduced fatty acid uptake and storage in subcutaneous adipose tissue (SAT - Ectopic lipid accumulation in glucose-consuming organs including skeletal muscle and liver - Systemic insulin resistance |
| ApoC3 knockout ApoC3−/− | - Hypotriglyceridemia |
| APOC3 overexpression | - Increased TG levels |
| Homozygous loss-of-function apoC2 mutant | - Hypertriglyceridemia |
| ApoA5 knockout ApoA5−/− | - Hypertriglyceridemia |
| Cav-1 knockout Cav-1–/– | - Lean - Resistant to high fat diet-induced obesity - High blood FA and TG levels - Impaired albumin transcytosis - Suppressed development of atherosclerosis |
| Endothelial cell- SR-B1 knockout SR-B1ΔEC | - Diminished uptake of LDL into the aorta - Reduced uptake of LDL by aortic wall macrophages - Markedly reduced atherosclerosis in ApoE–/– mice, and mice with genetic or PCSK9-induced LDLR deficiency |
| Endothelial cell SR-B1 overexpression | - Atheroprotective in high fat/high-cholesterol fed C57BL/6 mice and chow diet fed ApoE–/– mice |
| skeletal muscle DGAT1 Overexpression | - Increased myocyte TG - Increased insulin sensitivity |
| Cardiomyocyte DGAT1 overexpression (MHC-Dgat1) | - Increased heart TGs - Reduced lipotoxicity |
| Adipocyte/macrophage-DGAT1 overexpression (aP2-Dgat1) | - Prone to diet-induced obesity - Reduced diet-induced insulin resistance and inflammation |
In addition to the receptor-mediated FA uptake, there is evidence for nonsaturable receptor independent uptake. Long chain FAs can cross from the outer to the inner leaflet of the plasma membrane via a process termed ‘flip-flop’, which has been modeled using protein-free lipid vesicles74, 75. These studies initially suggested that FA transfer does not need protein facilitation. However, the flip-flop rates measured across cellular membranes are slower. In a series of studies76–78 with some monitoring FA transfer using the fluorescent FA binding protein ADIFAB injected inside cells, Kleinfeld and colleagues concluded that flip-flop rates are too slow for FA uptake by adipocytes and cardiomyocytes and protein facilitation is required. Slow FA flip-flop rates are consistent with the need for cells to regulate FA uptake. As FA availability is variable (i.e. in fasting, feeding, exercise, stress, etc.), protein transporters allow cells to regulate FA entry and provide the organism with a mechanism for optimizing FA biodistribution. When unbound FA concentrations are in the low nanomolar range, e.g. during fasting, a high affinity cellular receptor allows FA sparing for cells and tissues with high FA requirements such as heart, muscle and brown fat. In contrast, at high FA concentrations such as during feeding, cellular downregulation of FA receptors would mitigate excess intake. In addition, it is likely that receptor independent FA uptake, which would occur at high FA levels, is also physiologically regulated as proposed in Figure 1.
Endothelial Regulation of FA Transport
Uptake of FAs from the circulation into tissues occurs primarily in capillaries where, unlike in most arteries, the ECs express both CD36 and GPIHBP1. However, we should note this generalization overlooks a newly described subpopulation of aortic ECs that can express these genes79. The effects of myocyte lipid uptake on cellular concentrations of ceramides, diacylglycerols and likely other signaling lipids and the insulin-signaling pathway have been reviewed by others80, 81. Channeling of these lipids via esterification and storage as TG leads to lipid droplets (LDs), which are thought to be non-toxic. This is illustrated by the effect of diacylglycerol acyl transferase (DGAT) overexpression. In muscle, it creates a phenotype similar to that of the athlete paradox, greater myocyte TG and greater insulin sensitivity82. Similarly, TG accumulation in the heart and macrophages due to DGAT overexpression reduces lipid-driven cellular toxicity83, 84. Thus, in addition to uptake, the ability to store the lipid affects how excess supply alters cell function.
Evidence that the ECs affect parenchymal cell lipid accumulation comes from a number of genetic studies where specific EC genes were altered. One of the early reports highlighting importance of the endothelium in regulating tissue FA uptake and metabolism involved EC deletion of the master transcriptional regulator and FA sensor PPARγ. The EC-knockout mice had increased circulating FAs but were protected from high fat diet induced adiposity and insulin resistance, compared to control mice85 (Table 2). Expression of PPARγ gene targets, including CD36, was reduced as expected.
The PPARγ coactivator PGC1α stimulates mitochondrial biogenesis and FA β-oxidation and in addition, co-activates VEGF expression and angiogenesis. Jang et al. tested the possibility that PGC1α might coordinate both pathways by regulating FA flux by ECs.86. Conditioned medium from myotubes overexpressing PGC1α increased FA uptake by human umbilical vein ECs (HUVECs). The active medium factor was identified as 3 hydroxyisobutyrate (3-HIB) generated through PGC1α upregulation of the enzymes of valine catabolism. In vivo, 3-HIB stimulated muscle tissue FA uptake and lipid accumulation and reduced insulin sensitivity86. Although the 3-HIB increase in FA flux was blocked by the CD36 inhibitor SSO it was more sensitive to inhibition by knockdown of FATP4 as compared to that of CD36. In a follow up study the same group reported that EC FATP4 resides in the ER juxtaposed to mitochondria and can use mitochondrial ATP to activate FA and influence EC FA uptake and oxidation87. This study demonstrates the importance of FA activation in EC FA uptake. Other regulators of EC FA transport are the Meox2/Tcf15 heterodimer transcription factors identified using microarray profiling, which determines a microvascular EC signature enriched in genes related to FA uptake including CD36 and LpL. The Meox2/Tcf15 in cardiac ECs upregulate uptake of FAs supplied by albumin or derived from LpL hydrolysis of triglycerides88. The above studies illustrated how altering EC gene expression influences tissue FA uptake and insulin sensitivity; they also highlighted the importance of a specific microvascular EC gene signature that includes LpL, GPIHBP1 and CD36 for FA uptake function79, 88.
Vascular endothelial growth factors (VEGF A-D) and their receptors (VEGFR 1–3) control angiogenesis and maintenance of the vasculature and consequently, can influence nutrient exchange in tissues. Overexpression of VEGF-A in adipose tissue increased vascularization, activated thermogenesis and energy expenditure, and conferred protection from obesity and insulin resistance89, 90. This protection is associated with enhanced clearance of circulating FAs and TGs and the upregulation of LpL in heart and adipose tissues90. Deletion of VEGF-B in mice reduced heart FA uptake and increased glucose uptake. Moreover, VEGF-B was proposed to function in endothelial FA uptake through transcriptional upregulation of vascular-specific FATP3 and FATP491. As deletion of VEGF-B in diabetic mice reversed tissue lipid accumulation and glucose intolerance (Table 2), the authors suggested that targeting VEGF-B could improve muscle insulin sensitivity by impairing endothelial FA transport92. However, other studies did not observe an effect of VEGF-B on FA uptake by ECs86 or with VEGF-B overexpression in heart93.
Microvascular ECs robustly express CD36, while it is sparse in macrovascular cells. This suggests that there is a specific role for this molecule in capillaries. EC-specific CD36 deletion reduced FA uptake into heart and skeletal muscle, assessed in vivo using both tracer kinetics and PET scanning. EC CD36 deficiency in ECs also downregulated expression of PPAR target genes94. Brown fat FA uptake was reduced which might have implications for thermogenesis; both LpL and CD36 affect brown adipose uptake of FAs during cold exposure95.
Capillary ECs from tissues which most actively uptake circulating FAs have high levels of CD36 expression96.These ECs also express LpL and its EC-anchoring protein, GPIHBP1. During fasting, adipose tissue lipases (ATGL and HSL) are activated, which increases blood FA levels and leads to LD accumulation in the heart, muscle and liver. Hearts of EC-Cd36–/– mice, unlike hearts of control mice, do not accumulate LDs during fasting94. Cardiomyocyte CD36 deletion also reduces LD formation, which suggests that this protein affects lipid uptake both in ECs and cardiomyocytes (reviewed in29). EC-Cd36–/– mice have slow clearance of postprandial TGs, likely reflecting LpL inhibition by increased local FAs. In CD36-deficient humans, heart FA uptake was reduced at all FA levels and only at lower FA levels in adipose and muscle tissues57.
The EC-Cd36–/– mice, like the germline Cd36–/– mice, are more insulin sensitive and RNA-Seq data of heart tissue documented increased expression of genes of insulin signaling and glucose metabolism (Table 2). These phenotypes were not observed when CD36 was deleted from cardiomyocytes94, where in contrast, to the effects of EC CD36 loss, inducible CD36 deletion in myocytes resulted in muscle insulin resistance. This effect is likely exclusive of the role of CD36 in FA transport and involves its action to maintain Src phosphorylation of the insulin receptor (IR). As a result, CD36 deletion prevents full activation of IR22. Since CD36 regulation of IR is prevented by saturated FAs, this could be related to FA-induced muscle insulin resistance. Overall, these findings indicate that EC CD36 acts as a gatekeeper of tissue FA delivery and its metabolic effects integrate both its transport and signaling functions.
Tissue Specific Effects due to EC FA Transport
Capillaries and ECs are heterogeneous across organs and it is likely that the effects mentioned above, where targeting of EC FA transport enhanced systemic insulin sensitivity, reflect changes in muscle tissues. Skeletal muscle and heart are the major FA consumers and increasing or decreasing FA uptake would enhance or reduce muscle insulin stimulated glucose disposal. This interpretation is consistent with data suggesting that enhancing EC FA uptake targeted to adipose tissue improves systemic metabolism. Increasing adipose tissue vascularity by overexpression of VEGF-A improved insulin sensitivity while enhancing clearance of circulating FAs and TGs90. A recent study more directly supports this concept. Adipocyte-specific knockout of Angiopoietin-2 (Angpt2) identified it as a regulator of EC-FA uptake in subcutaneous adipose tissue97. Angpt2 interaction with ECs stimulates integrin α5β1 signaling and EC CD36 internalization. The Angpt2 stimulated uptake process was inhibited by knockdown of CD36 and FATP3 but not of FATP4 (unlike in muscle). Inhibition of Angpt2-stimulated EC FA uptake in adipose tissue caused ectopic fat accumulation and promoted insulin resistance, supporting the benefit of upregulating EC FA uptake in subcutaneous fat97. Other data suggested that adipose tissue has a specific EC FA uptake process mediated by a prohibitin–annexin A2 complex that increases cell surface CD36 and FA uptake by ECs and adipocytes98.
In summary, EC uptake of FAs is a highly regulated process that influences tissue metabolism and targets FAs to specific tissues with high nutrient requirements, such as the heart and brown fat. Several important regulators of EC FA uptake (Table 1) work either at the level of gene transcription or acutely through EC receptors. Much remains to be learned about the effects of angiogenic factors on the regulation of the endothelial barrier and how this might affect FA transfer. The role of the EC FATPs, notably FATP3 and FATP4, will need to be investigated further in animal models with targeted EC deletion of these proteins, and information on their functional protein interactions is needed. Understanding the molecular mechanism by which CD36 facilitates FA transfer and how its function might be regulated by interaction with membrane integrins or integrin ligands99 and possibly the FATPs are needed. CD36 contributes to uptake of albumin-bound FAs and of FAs released from VLDL by LpL as shown for heart100 and brown adipose tissue, 95 but it does not contribute to heart CM FA uptake in vivo100. The regulation of lipoprotein lipid uptake by ECs and the receptors likely to be involved are discussed in the following section.
Regulation of Lipoprotein Lipolysis at the EC Luminal Surface
The major sources of cellular lipids other than NEFAs are the circulating lipoproteins that contain TGs, cholesterol, cholesteryl esters and phospholipids. LpL is the rate-limiting enzyme for hydrolysis of TG-rich lipoproteins (TRLs), VLDL and CMs and it associates with the luminal surface of capillary ECs where it generates NEFAs, monoacylglycerols and remnant lipoproteins. LpL is synthesized in parenchymal cells of metabolic tissues like heart, skeletal muscle and adipose tissue and secreted into the sub-endothelial space. The endothelial anchoring protein GPIHBP1, present on the basolateral side of ECs, captures LpL from the interstitial space and transports it across ECs to the capillary lumen, which is the site of LpL’s catalytic action101. Other proteins such as the VLDL receptor might provide a secondary transport system102 and account for the reduced TG levels after heparin injection with GPIBHP1 deficiency. GPIHBP1 may also anchor TG-rich lipoproteins to the endothelial surface and protect LpL from endogenous inhibitors120, 121. Deficiency of LpL in humans causes massive chylomicronemia, low levels of HDL-cholesterol and recurrent pancreatitis. Unlike humans, LpL deficiency is neonatally lethal in mice, likely from hypoglycemia103 and mice at equally high plasma TGs do not develop pancreatitis.
Mice lacking GPIHBP1 develop severe chylomicronemia due to decreased ability of LpL to metabolize circulating TGs8. Surprisingly, on a high fat diet, these mice have lower TG levels, perhaps due to better hepatic TG-rich lipoprotein uptake. Patients with GPIHBP1 deficiency have less functional LpL in the capillary lumen, diminished TG hydrolysis and severe chylomicronemia. Moreover, chylomicronemic patients have been identified who have GPIHBP1 autoantibodies104 that block GPIHBP1’s binding and LpL transport, thus interfering with LpL mediated TG hydrolysis.
Olivecrona105 and others106 had noted many years ago that monomeric LpL was unstable. During enzyme purification from either post-heparin plasma or bovine milk, active LpL aggregated into dimers or multimers; this aggregation likely was aided by the presence of heparin used to stabilize LpL activity. These aggregates are more stable at 37°C, leading to the assumption by the research community that in vivo active LpL required dimers. Beigneux et al.107 reinvestigated this question and showed that active LpL is indeed present as a monomer. They added purified GPIHBP1 to LpL and eluted active and monomeric LpL. They also co-expressed both inactive but GPIHBP1-binding LpL together with active LpL that did not bind GPIHBP1. Dimerization would have been expected to produce double tagged, more active enzyme. However, very little of this mixed dimer was found, and what was found was in the inactive fraction. It is plausible that non-GPIHBP1 binding LpL is less stable, and native LpL has its activity stabilized by GPIHBP1. Additionally, Birrane et al.108 and Arora et al.109 independently solved the crystal structure of LpL bound to GPIHBP1. The derived structures were very similar and will likely provide more in depth structural view of the lipolysis process.
In the last decade, LpL and its posttranslational regulators have attracted attention due to a host of genetic studies showing clear association of plasma TGs with risk of cardiovascular disease (CVD). APOC3, produced by the liver and to a small extent by the intestine, inhibits LpL and is a key regulator of plasma TG metabolism (Table 1). APOC3 associates with ApoB-containing lipoproteins and can be acquired by HDL during lipolysis. APOC3 inhibits LpL activity and also hepatic uptake of remnants110, and its overexpression in mice increased TG levels. In contrast, APOC3-deficient mice have reduced TGs in both wild type and Ldlr−/− and Apoe−/− backgrounds111, 112 (Table 2). The link of APOC3 to CVD risk prompted pharmaceutical development of inhibitors in the form of antisense oligonucleotides (ASOs), interference RNAs and monoclonal antibodies113, 114.
Apolipoprotein C2 (APOC2) is required for LpL activation, as it binds LpL and guides the TG substrate into the active site115, 116. APOC2 mimetic peptides directly activate LpL and inhibit APOC3115. Loss-of-function mutations in APOC2 in humans increase plasma TG levels and can provoke acute pancreatitis117. Mice expressing a homozygous loss-of-function Apoc2 mutant118 have increased plasma TGs, consistent with its role as an LpL cofactor (Table 2).
APOA5 also increases lipolysis but its mode of action is incompletely understood. Mice lacking APOA5 develop hypertriglyceridemia119 (Table 2) and APOA5 deficiency associates with hyperchylomicronemia in humans 120.
Three angiopoietin-like proteins also modulate lipolysis (Table 1). ANGPTL3, primarily synthesized in the liver, inhibits endothelial lipase (EL) and LpL. Its activity requires its association with a second circulating protein, ANGPTL8121. Mutations in ANGPTL3 cause hypolipoproteinemia without steatosis122. ANGPTL4 inhibits LpL primarily in adipose tissue123–125 while the ANGPTL3/8 complex is more effective in muscle126.
EC Uptake and Transcytosis of Lipoprotein Lipids
Differences in gene expression between capillary ECs and most large vessel ECs likely relate to their distinctive functions. As previously mentioned, there is a gene expression signature specific for microvascular ECs that is marked by CD36 and enriched in genes (GPIHBP1, LpL, PPARγ) for TG lipolysis and fatty acid uptake86, 77. In large vessels such as the aorta, the gene expression signature of macrovascular ECs, marked by VCAM1 (vascular cell adhesion molecule 1), is enriched in genes for extracellular matrix organization and integrin cell surface signaling pathways77. However, heterogeneity of ECs is also observed within individual vessels. For example, in the aorta a subset of ECs was found to express the gene signature typical of microvascular ECs, including CD36, LpL and GPIHBP1. These ECs show a distinctive distribution, being more abundant in the greater curvature area of the aortic arch, a non-atherosclerosis prone region, where the Vcam1/CD36 ratio is lower. In contrast, the Vcam1/CD36 ratio increases in areas of lesser curvature, 77 which are more atherosclerosis prone regions and have higher expression of SR-B1170. These findings suggest that although lipid lipolysis is thought to only occur in capillaries, some lipolysis and fatty acid uptake likely occur in the greater curvature areas of the aorta, while more LDL transcytosis occurs in areas of lower curvature. Such a conclusion would not support a role of CD36 in atherosclerosis or the long-held hypothesis that local lipolysis creates either remnants or toxic lipids that promote atherosclerosis127.
Capillary EC uptake of circulating lipids might occur via caveolae-mediated transcytosis. Caveolae are small membrane invaginations (50–80 nm in diameter) present in many cell types and particularly abundant in capillary ECs as well as in adipocytes and smooth muscle cells. Caveolin-1 (Cav-1) is a structural protein required for formation of caveolae in ECs. Mice deficient in Cav-1 (Cav-1–/–) show caveolae loss128, are lean, resistant to high fat diet-induced obesity, but have high blood FA and TG levels129, a phenotype similar to that of the Cd36–/– mouse42, 130 (Table 2). One key function of caveolae (and Cav-1) is regulating transcytosis across the endothelial barrier131. Cav-1–/– mice have impaired transcytosis132 of albumin, which carries FAs and other lipids. Likewise, the function of caveolae in transendothelial LDL transport is supported by the finding that Cav-1 deletion suppressed atherosclerosis development133, 134, while rescue of Cav-1 expression in ECs reversed the phenotype134 (see below). Several pathways link Cav-1 to lipid metabolism135. Cav-1 facilitates assembly of membrane lipid domains (caveolae)136–138 through interaction with cholesterol139 and FAs140. It also translocates to LDs during lipolysis141–144. Sessa and colleagues145 reported that ECs lacking Cav-1 have reduced LD formation that is reversed by rescue of EC Cav-1. They noted that the decrease in LDs associated with a decrease in CD36, consistent with Cav-1 promoting caveolae CD36 localization in the plasma membrane146, 147.
Aside from FAs, the heart, like most tissues, requires a number of other lipoprotein-associated lipids, including cholesterol and retinoids. However, hearts obtain little cholesterol from circulating LDL148 and also do not synthesize it149. Using perfused hearts obtained without the use of heparin, which dissociates LpL from the EC surface, Fielding150 showed that during lipolysis, CMs could supply the hearts with cholesterol. Because uptake of the APOB core protein component of these particles is very low100, it is likely that lipids—including cholesterol, phospholipids and retinoids—liberated during lipolysis move across the EC barrier. Another option is uptake of these lipids via the VLDL receptor, which is expressed by ECs and was reported to mediate greater lipid storage and injury during cardiac ischemia151.
While TG lipolysis accounts for most tissue uptake of lipoprotein-derived FAs, some transfer of lipoprotein TGs across the EC barrier likely occurs in the absence of LpL mediated lipolysis. An example of this is the development of skin eruptive xanthomas with lipid-enriched macrophages associated with genetic LpL deficiency and high plasma TG levels. For more than 50 years, the process underlying this pathology has remained unclear. Electron microscopy studies of skin xanthomas in patients with hyperchylomicronemia and diabetes showed no evidence of CMs within ECs nor defective EC junctions152. Although a similar study in LpL deficient patients has not been reported, it is likely that the pathology is the same. The similarity of the TGs, but not cholesteryl esters, and FAs within CMs and skin xanthomas suggested that CM-derived FAs crossed the EC barrier, but not necessarily as intact CMs. Studies acutely tracking labeled nascent CMs did not find these particles within ECs153, 154. In contrast, Guyton and Klemp155 noted lipid droplets adhering to the luminal membrane and within ECs in rabbits fed a cholesterol and butter diet. Presence of CM-derived lipoproteins within the artery156 also suggests that CM remnants penetrate the artery as or more efficiently than LDL.
The reason for the inconsistent LD findings in ECs remains unclear and suggests the possibility that transfer of lipoproteins into and through ECs is extremely rapid and often completed during the time required to obtain and process tissues for microscopy. A recent study from Guo et al. using confocal microscopy confirmed the presence of EC lipid droplets in the aorta of mice after an oil gavage157. Although they equated these lipid droplets with those found when ECs were incubated with FAs in vitro, aortic ECs are unlikely to have significant local lipolysis, as most aortic ECs do not express GPIHBP1 and fail to approximate CMs and LpL153. However, since a subpopulation of aortic ECs that can express these genes has been recently described79 revisiting the above studies is needed to determine the contribution of this subpopulation to the observed LDs. The uptake pathway for CMs and CM remnants remains unclear (Figure 2). Possibly, CMs non-specifically interact with the EC surface and are internalized by macro-pinocytosis after association with cell surface proteoglycans. Alternatively, specific lipoprotein receptors mediate their uptake possibly by transcytosis, as discussed below for LDL and HDL.
Figure 2. Non-LpL mediated uptake of CMs and VLDL by ECs.

In capillaries, most FAs from TG are liberated by LpL, but with LpL deficiency or in arteries a novel pathway for CM uptake is likely to be present that does not involve CD36. In arteries, this additional pathway leads to uptake of lipids or chylomicrons to form intracellular lipid droplets.
Most tissues obtain cholesterol from LDL and HDL after their transport through or around the endothelial barrier. Movement of both LDL and HDL into tissues has been most intensely studied in the context of atherosclerosis development. HDL must cross the endothelium to mediate athero-protective efflux of cholesterol from lipid-laden macrophages back to the liver158. How LDL and HDL traverse the endothelial barrier remains poorly understood. Early atherosclerotic lesions are overlaid by an intact endothelium, which would preclude the paracellular passage of molecules exceeding 6 nm in diameter159. Electron microscopic studies of rat aorta perfused with LDL clearly show its internalization into cellular vesicles and LDL targeting to the basolateral membrane160. Support for a specific aortic EC LDL transporter is provided by the atheroprotective effect of Cav-1 deletion in APO E knockout mice161, with its rescue in ECs being sufficient to promote lesion progression and LDL infiltration of the aortic wall162,163. These findings strongly suggest that cholesterol-carrying lipoproteins cross the aortic endothelium via a transcytotic route.
Scavenger Receptor-B1.
SR-B1 identified by expression cloning as a CD36-related protein n binds HDL and mediates selective uptake of HDL cholesteryl esters into liver cells164, 165. SR-B1 also functions in peripheral tissues to facilitate efflux of cholesterol to HDL and back to the liver166, 167 (Table 1). To access cholesterol in the intima, HDL and its main protein component, APO A-I, must cross the EC barrier. In vitro, HDL transcytosis across EC monolayers requires SR-B1 and also ABCG1168, while transcytosis of APOA-I alone is mediated by the ATP-binding cassette transporter (ABC)A1. Endothelial-specific overexpression of SR-B1 is athero-protective in high-fat/high-cholesterol fed C57BL/6 mice, as well as in ApoE–/– mice fed a chow diet169. SR-B1 in the lymphatic endothelium helps remove cholesterol by HDL transcytosis170.
SR-B1 also binds modified, as well as native, LDL164 and has recently been implicated in aortic uptake and transcytosis of LDL. Total internal reflection fluorescence (TIRF) microscopy demonstrated that SR-B1 mediates LDL transcytosis in human coronary ECs and HDL competition inhibited transcytosis171. Treatment with physiologic concentrations of estrogen reduces SR-B1 mRNA and protein expression in primary human ECs from men and postmenopausal women and inhibits LDL transcytosis172. The role of endothelial SR-B1 in LDL transcytosis was recently confirmed in vivo. Mice with EC-specific deletion of the receptor exhibit significantly diminished uptake of DiI-labeled LDL into the aorta, as well as reduced uptake of administered LDL by aortic wall macrophages. In culture, LDL transcytosis involves its direct binding to SR-B1 and recruitment by the receptor of the guanine nucleotide exchange factor dedicator of cytokinesis 4 (DOCK4). Endothelial-specific deletion of SR-B1 markedly reduces atherosclerosis in ApoE–/– mice, as well as in mice with genetic or PCSK9-induced LDLR deficiency173. How can SR-B1 deletion and overexpression in the endothelium both be protective? One possibility could entail an effect of overexpression in altering receptor dimerization. Although the functional role of SR-B1 dimerization is not well understood, it could be important for binding specific ligands and, as recently shown, for stabilizing SR-B1 at the plasma membrane174 (see following section).
Other EC receptors have been studied in lipoprotein transcytosis. Systemic endothelial LDL transcytosis is largely independent of the LDL receptor (LDLR), made evident by the lack of effect of its genetic deletion or its PCSK9-mediated degradation on non-hepatic tissue metabolism170, 171. However, LDLR has been reported to participate in LDL transcytosis across the blood brain barrier175. In 2015, a genome-wide iRNA screen identified activin receptor-like kinase 1 (ALK1) as a novel low-affinity, high-capacity LDL receptor, which mediates LDL transcytosis in vitro independent of its kinase activity. Importantly, endothelial-specific deletion of ALK1 reduced DiI-LDL uptake into the aortic endothelium of Ldlr–/– mice176.
CD36 and SR-B1 Structures and Interaction with Lipoproteins
Greater SR-B1 expression in more atherosclerosis prone segments of the aorta170 could underlie the reason some vascular regions associate with more with atherosclerosis, whereas ECs with greater CD36 expression function to augment FA uptake. For this reason, a comparison of their functional structure would be informative. Overall, the two receptors overlap in ligand specificity, although efficiency of binding or transfer might differ; FAs40, 177, modified and native lipoproteins40, 178, phospholipids40, 179, apoptotic cells180, 181, pathogens182–185 and advanced glycation end products186, 187 are ligands for both receptors. Most aortic ECs have low CD36 and Cav1 expression and are not surrounded by cells with LpL expression (myocytes and adipocytes). Thus, TRL lipolysis is unlikely on aortic EC surfaces. In contrast, cardiac and aortic ECs express SR-B1, which could function in lipid uptake by both capillaries and large vessels
The parallel functions of SR-B1 and CD36 in lipid transport reflect structural similarities (Figure 3). Structural modeling identified a cationic enriched apex region in both SR-B1 and CD36 that is absent in LIMP-2188 and mutation of key cationic residues inhibited binding of HDL, oxidized-LDL, acetyl-LDL188 and silica189 to SR-B1 and of oxidized-LDL and NEFAs to CD3637, 188. The electrostatic similarity between the apex regions of SR-B1 and CD36 is in line with the ligand overlap.
Figure 3. Tertiary structures of CD36 and SR-B1.
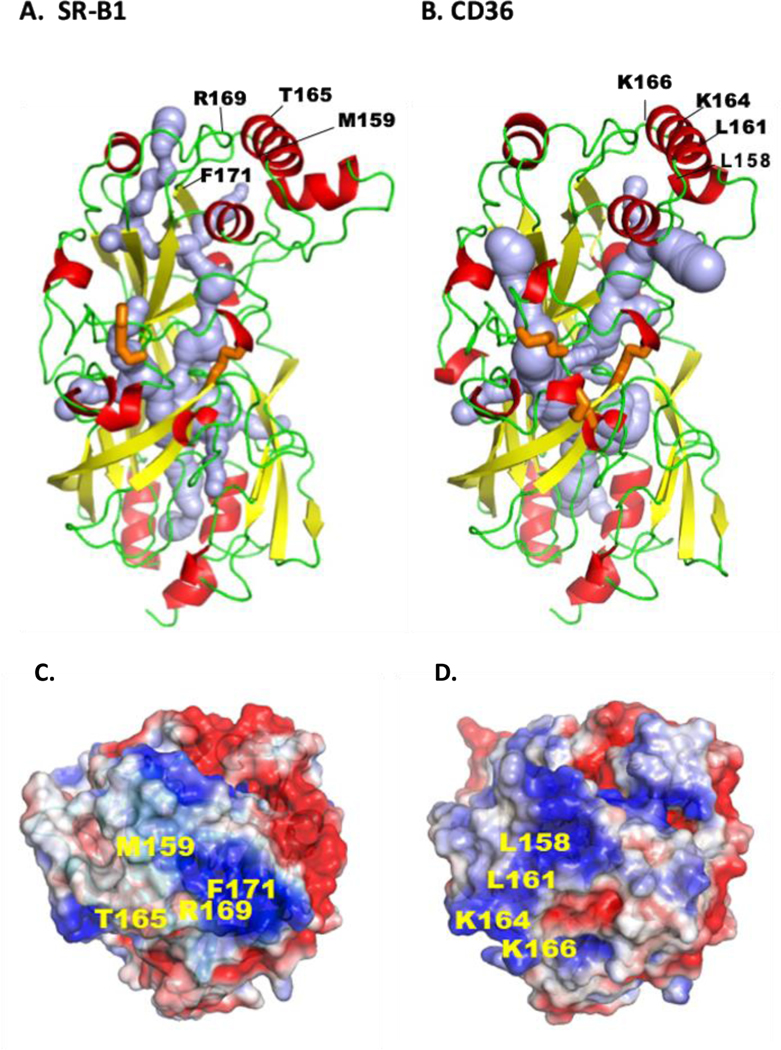
Both receptors are members of the CD36 family that also includes LIMP2, and both play important roles in endothelial lipid transport. The comparison is based on the crystal structures. The ectodomain of human CD36 (A) is derived from its crystallography and homology modeling of human SR-BI (B). For both proteins the α-helices are in red, the β-sheets in yellow and the loops in green. Crystal structure identified a hydrophobic internal lipid transport tunnel (light blue) that traverses the length of these proteins ending at the bilayer proximity. This tunnel can be accessed from the apex region and residues identified to be important for ligand binding are highlighted. C and D show the apex regions’ electrostatic surface potential for SR-B1 (C) and CD36 (D) and residues implicated in binding of LDL and HDL (SR-B1), and FA and oxidized-LDL (CD36) are indicated. The tunnel is surrounded by short α-helices and the disulfide bridges (orange) would stabilize structure and dimensions of the cavity for it to accommodate the lipid ligands. E shows linear sequence of the C-terminal domains for SR-B1 and CD36 highlighting the PDZ binding sequence (red) of SR-B1 and the palmitoylated (green) and ubiquitinated (orange) residues of CD36 important for signaling. The tertiary structures were rendered in PyMol211, cavity prediction used CaverWeb 1.0212 and the electrostatic surface potential map used APBS212 with color gradient of −2 (red) to +2 (blue) kT/e.
Lipid transport by both SR-B1 and CD36 is thought to occur through an internal tunnel. Existence of a hydrophobic “channel” within SR-B1 that facilitates cholesterol ester uptake from HDL particles to the plasma membrane was first proposed by the group of David Williams190. Direct evidence for tunnel existence was provided by crystallography of the ectodomain of the third CD36 family member LIMP-2188. Critical residues for tunnel formation are largely conserved across family members, as are the two cysteine-cysteine bridges that stabilize the structure and the dimensions of the tunnel cavity, which accommodates cholesterol esters and FAs. Crystallography of the ectodomain of CD36 confirmed presence of a hydrophobic internal tunnel as in LIMP-241 and identified long chain FAs within the tunnel (Figure 3). It is postulated that the FA binds to a hydrophobic pocket on the CD36 surface and accesses the internal tunnel, which delivers it to the bilayer40
Scavenger receptor B family members have two transmembrane domains. Both SR-B1191, 192 and CD36193, 194 form dimeric and higher order oligomeric forms. The GxxGxxxAxxG motif in the N-terminal transmembrane domain of both proteins is important for dimerization of both SR-B1195 and CD36196. A leucine zipper motif in the C-terminal transmembrane domain of SR-BI (absent in CD36) is also required197, 198. Oligomerization retains SR-B1 at the plasma membrane; its disruption after mutating the C-terminal leucine zipper motif, but not the N-terminal glycine motif, induces SR-B1 endocytosis and degradation174. The functional role of receptor oligomerization is unclear, but it could regulate interaction with specific ligands, in addition to playing a role in receptor/ligand internalization.
Both SR-B1 and CD36 have short cytoplasmic C-terminal tails that mediate intracellular signaling in response to lipid ligands (Figure 3). Binding of HDL to SR-B1 activates endothelial nitric oxide synthase (eNOS), 199, 200 a process involving Src201, 202 and LKB1203 kinases with downstream activation of AMPK203, Akt201, 203 and MAPK201. The C-tail 47 amino acids of SR-B1 contain a PDZ domain sequence and the adapter protein PDZK1 facilitates SR-B1 interaction with Src200, 202. CD36 also relies on the Src family23, 204–206 and LKB1 kinases23, 207 for downstream activation of AMPK23, 207, Akt22, 208 and MAPK, 204–206 but its smaller C-tail of 13 amino acids has no PDZ binding domain. In macrophages, the adapter protein FcRγ facilitates CD36-Src interaction209.
In summary, structural similarities in the ectodomains of SR-B1 and CD36 may explain the overlap in ligand specificity, and oligomerization may influence ligand interaction and handling. For example, oligomerization mediated retention of SR-B1 at the plasma membrane could facilitate cholesteryl ester uptake from HDL and at the same time prevent LDL transcytosis. If CD36 ligand uptake is mediated by endocytosis, oligomerization could lead to plasma membrane retention. This might allow C-tail association with adapters and the activation of kinases, which like Src, could affect insulin signaling and other cellular functions.
Influence of Lipoprotein Size
In capillaries, TRLs interact with the EC surface for lipolysis to occur. A number of factors, some not fully defined, regulate this process. The radius of the lipoprotein markedly influences its interaction with ECs. Depending on its size, a chylomicron, as compared to a VLDL, is likely to have a 10–100-fold greater chance of interacting with LpL on the EC luminal surface. The size difference, rather than differences in LpL substrate preference, likely determines the more rapid lipolysis of larger lipoproteins. In rapidly flowing blood, it is likely that large CM size particles >200 nm, but not smaller lipoproteins marginate210. Margination would increase their effective concentration along the arterial surface where they can interact with either EC receptors or LpL (Figure 4). Other biochemical processes, exclusive of the physiology of fluid flow, modulate lipolysis, including apolipoproteins and their interaction with LpL, GPIHBP1, proteoglycans or other luminal membrane proteins.
Figure 4. Lipoprotein interaction with endothelial cells differs in arteries and capillaries.

A. In an artery, the rapid blood flow likely causes margination of larger particles, leading to a gradient with more chylomicrons in proximity of the arterial wall. B. This gradient is less likely in capillaries, still chylomicrons would still interact more with lipoprotein lipase (LpL) than the smaller VLDL.
Summary
Great advances over the past decade have provided insight into the regulation of intravascular lipolysis. The discoveries of ANGPTLs and GPIHBP1, and new findings on the importance of APOC3 as well as insight into the structure of LpL have led to the development of clinically relevant TG-reducing therapies. We can expect additional information on molecular regulation of LpL function. However, despite more than 5 decades of research, a number of basic physiologic and pathological processes central to lipid metabolism in ECs are still unclear and under investigation. Recent studies have emphasized the important role of endothelial receptors in regulating tissue lipid uptake and the tissue’s metabolic phenotype. However, the pathways involved in EC interaction with FAs and lipoproteins, important in muscle energy metabolism and insulin sensitivity, adipose lipid storage, vascular function and atherogenesis, remain poorly understood. It is apparent that NEFAs derived from different sources exit the circulation by different routes. The importance of receptors versus possibly paracellular pathways depends on the FA concentration or other circulating factors that could alter EC junctions. SR-B1 and CD36, members of the same gene family, are emerging as central molecules in EC lipid metabolism and vascular biology. We need a better understanding of their interactions with different lipids and lipoproteins and how each receptor regulates transit of its ligands from the lumen. The handling of LDL and HDL by SR-B1 appears to regulate movement of atherogenic lipids into the artery. A second receptor, Alk1, also mediates EC LDL uptake. As global rather than EC-specific SR-B1 deficiency leads to vascular disease, additional information is required to understand the pro-atherogenic processes derived from SR-B1 loss in cells other than ECs. SR-B1 is expressed in both micro- and macro-ECs, whereas CD36 expression along with GPIHBP1 is relatively restricted to capillary ECs. The molecular processes controlling this differentiation are not understood although they promote the distribution of FAs required for normal physiology and are central to tissue energy and growth. Finally, human clinical observations illustrated a lipolysis-independent route that mediates exit of CM lipids from the circulation leading to development of eruptive xanthomas in patients with LpL deficiency. The molecular details of this pathway and its importance for normal physiology are still unclear.
A final note should be included related to the role of factors that affect EC uptake and could not be addressed by this review. Multiple physiologic parameters affect lipoprotein interactions with ECs, including the rate of blood flow, turbulence, viscosity, ionic interactions, and the diameter of the vessel. In vitro studies of EC-lipoproteins interaction often use media that mimics lipoprotein concentrations found in circulating rodent or human blood. These studies do not account for the more than 50-fold difference in rate of blood flow between arteries and capillaries. In rapidly flowing blood, it is likely that large CM size particles >>200 nm, but not smaller lipoproteins marginate210, which increases their effective concentration along the arterial surface where they can either interact with EC receptors or LpL (Figure 4). Smaller lipoproteins and RBCs likely reduce luminal EC surface exposure to HDL and LDL. The effects of these physical influences on lipoprotein/EC interactions have been poorly studied and deserve further investigation.
Acknowledgments
Sources of funding: HL45095 (IJG and NAA), HL73029 (IJG), DK111175 (NAA), AHA Career Development Award 20CDA35320109 (DB).
Disclosures: Dr. Goldberg has received funds for preclinical studies of triglyceride-reducing agents from Ionis and Arrowhead Pharmaceutical. He has served on advisory boards for Esperion and Pfizer.
Non-standard Acronyms and Abbreviations
- Angpt2
Angiopoietin-2
- ANGPTL
Angiopoietin like protein
- APOC
Apolipoprotein C
- Cav-1
Caveolin-1
- CD36
Cluster of differentiation 36
- EC
Endothelial cell
- FA
Fatty acid
- FATP
FA transporter protein
- GPIHBP1
Glycosylphosphatidylinositol-anchored HDL binding protein 1
- HDL
High density lipoprotein
- LD
Lipid droplet
- LDL
Low density lipoprotein
- LIMP2
Lysosomal integral membrane protein 2
- LpL
Lipoprotein lipase
- NEFAs
Non-esterified free fatty acids
- PPAR
Peroxisome proliferator-activated receptor
- SR-B1
Scavenger receptor-B1
- SSO
Sulfo-N-succinimidyl oleate
- TGs
Triglycerides
- VEGF
Vascular endothelial growth factors
- VLDL
Very low density lipoprotein
References:
- 1.Duncan JG, Bharadwaj KG, Fong JL, Mitra R, Sambandam N, Courtois MR, Lavine KJ, Goldberg IJ and Kelly DP. Rescue of cardiomyopathy in peroxisome proliferator-activated receptor-alpha transgenic mice by deletion of lipoprotein lipase identifies sources of cardiac lipids and peroxisome proliferator-activated receptor-alpha activators. Circulation. 2010;121:426–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang J, Sambandam N, Han X, Gross RW, Courtois M, Kovacs A, Febbraio M, Finck BN and Kelly DP. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ Res. 2007;100:1208–17. [DOI] [PubMed] [Google Scholar]
- 3.Sletten AC, Peterson LR and Schaffer JE. Manifestations and mechanisms of myocardial lipotoxicity in obesity. J Intern Med. 2018;284:478–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glatz JFC, Zuurbier CJ and Larsen TS. Targeting metabolic pathways to treat cardiovascular diseases. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165879. [DOI] [PubMed] [Google Scholar]
- 5.Goldberg IJ, Eckel RH and Abumrad NA. Regulation of fatty acid uptake into tissues: lipoprotein lipase- and CD36-mediated pathways. J Lipid Res. 2009;50 Suppl:S86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bastie CC, Hajri T, Drover VA, Grimaldi PA and Abumrad NA. CD36 in myocytes channels fatty acids to a lipase-accessible triglyceride pool that is related to cell lipid and insulin responsiveness. Diabetes. 2004;53:2209–16. [DOI] [PubMed] [Google Scholar]
- 7.Eckel RH. Lipoprotein lipase. A multifunctional enzyme relevant to common metabolic diseases. N Engl J Med. 1989;320:1060–8. [DOI] [PubMed] [Google Scholar]
- 8.Beigneux AP, Davies BS, Gin P, Weinstein MM, Farber E, Qiao X, Peale F, Bunting S, Walzem RL, Wong JS, Blaner WS, Ding ZM, Melford K, Wongsiriroj N, Shu X, de Sauvage F, Ryan RO, Fong LG, Bensadoun A and Young SG. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 2007;5:279–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eiselein L, Wilson DW, Lamé MW and Rutledge JC. Lipolysis products from triglyceride-rich lipoproteins increase endothelial permeability, perturb zonula occludens-1 and F-actin, and induce apoptosis. Am J Physiol Heart Circ Physiol. 2007;292:H2745–53. [DOI] [PubMed] [Google Scholar]
- 10.Rutledge JC, Woo MM, Rezai AA, Curtiss LK and Goldberg IJ. Lipoprotein lipase increases lipoprotein binding to the artery wall and increases endothelial layer permeability by formation of lipolysis products. Circ Res. 1997;80:819–28. [DOI] [PubMed] [Google Scholar]
- 11.Goodpaster BH and Sparks LM. Metabolic Flexibility in Health and Disease. Cell Metab. 2017;25:1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abumrad NA and Goldberg IJ. CD36 actions in the heart: Lipids, calcium, inflammation, repair and more? Biochim Biophys Acta. 2016;1861:1442–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richieri GV and Kleinfeld AM. Unbound free fatty acid levels in human serum. J Lipid Res. 1995;36:229–40. [PubMed] [Google Scholar]
- 14.Abumrad N, Park J and Park CR. Permeation of long-chain fatty acid into adipocytes. Kinetics, specificity, and evidence for involvement of a membrane protein. Journal of Biological Chemistry. 1984;259:8945–8953. [PubMed] [Google Scholar]
- 15.Sorrentino D, Robinson RB, Kiang CL and Berk PD. At physiologic albumin/oleate concentrations oleate uptake by isolated hepatocytes, cardiac myocytes, and adipocytes is a saturable function of the unbound oleate concentration. Uptake kinetics are consistent with the conventional theory. J Clin Invest. 1989;84:1325–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abumrad N, Harmon C and Ibrahimi A. Membrane transport of long-chain fatty acids. Evidence for a facilitated process [In Process Citation]. J Lipid Res. 1998;39:2309–18. [PubMed] [Google Scholar]
- 17.Berk PD. Regulatable fatty acid transport mechanisms are central to the pathophysiology of obesity, fatty liver, and metabolic syndrome. Hepatology. 2008;48:1362–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Storch J, Lechene C and Kleinfeld AM. Direct determination of free fatty acid transport across the adipocyte plasma membrane using quantitative fluorescence microscopy. J Biol Chem. 1991;266:13473–6. [PubMed] [Google Scholar]
- 19.Canton J, Neculai D and Grinstein S. Scavenger receptors in homeostasis and immunity. Nat Rev Immunol. 2013;13:621–34. [DOI] [PubMed] [Google Scholar]
- 20.Silverstein RL and Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal. 2009;2:re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kazerounian S, Duquette M, Reyes MA, Lawler JT, Song K, Perruzzi C, Primo L, Khosravi-Far R, Bussolino F, Rabinovitz I and Lawler J. Priming of the vascular endothelial growth factor signaling pathway by thrombospondin-1, CD36, and spleen tyrosine kinase. Blood. 2011;117:4658–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samovski D, Dhule P, Pietka T, Jacome-Sosa M, Penrose E, Son NH, Flynn CR, Shoghi KI, Hyrc KL, Goldberg IJ, Gamazon ER and Abumrad NA. Regulation of Insulin Receptor Pathway and Glucose Metabolism by CD36 Signaling. Diabetes. 2018;67:1272–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samovski D, Sun J, Pietka T, Gross RW, Eckel RH, Su X, Stahl PD and Abumrad NA. Regulation of AMPK activation by CD36 links fatty acid uptake to beta-oxidation. Diabetes. 2015;64:353–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonen A, Luiken JJ, Arumugam Y, Glatz JF and Tandon NN. Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. J Biol Chem. 2000;275:14501–8. [DOI] [PubMed] [Google Scholar]
- 25.Ring A, Le Lay S, Pohl J, Verkade P and Stremmel W. Caveolin-1 is required for fatty acid translocase (FAT/CD36) localization and function at the plasma membrane of mouse embryonic fibroblasts. Biochim Biophys Acta. 2006;1761:416–23. [DOI] [PubMed] [Google Scholar]
- 26.Samovski D, Su X, Xu Y, Abumrad NA and Stahl PD. Insulin and AMPK regulate FA translocase/CD36 plasma membrane recruitment in cardiomyocytes via Rab GAP AS160 and Rab8a Rab GTPase. J Lipid Res. 2012;53:709–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, Steinbusch LKM, Nabben M, Kapsokalyvas D, van Zandvoort M, Schonleitner P, Antoons G, Simons PJ, Coumans WA, Geomini A, Chanda D, Glatz JFC, Neumann D and Luiken J. Palmitate-Induced Vacuolar-Type H(+)-ATPase Inhibition Feeds Forward Into Insulin Resistance and Contractile Dysfunction. Diabetes. 2017;66:1521–1534. [DOI] [PubMed] [Google Scholar]
- 28.Momken I, Chabowski A, Dirkx E, Nabben M, Jain SS, McFarlan JT, Glatz JF, Luiken JJ and Bonen A. A new leptin-mediated mechanism for stimulating fatty acid oxidation: a pivotal role for sarcolemmal FAT/CD36. Biochem J. 2017;474:149–162. [DOI] [PubMed] [Google Scholar]
- 29.Glatz JFC and Luiken J. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J Lipid Res. 2018;59:1084–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harmon CM, Luce P, Beth AH and Abumrad NA. Labeling of adipocyte membranes by sulfo-N-succinimidyl derivatives of long-chain fatty acids: inhibition of fatty acid transport. The Journal of membrane biology. 1991;121:261–268. [DOI] [PubMed] [Google Scholar]
- 31.Harmon CM and Abumrad NA. Binding of sulfosuccinimidyl fatty acids to adipocyte membrane proteins: Isolation and ammo-terminal sequence of an 88-kD protein implicated in transport of long-chain fatty acids. The Journal of membrane biology. 1993;133:43–49. [DOI] [PubMed] [Google Scholar]
- 32.Abumrad NA, El-Maghrabi MR, Amri E, Lopez E and Grimaldi P. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. Journal of Biological Chemistry. 1993;268:17665–17668. [PubMed] [Google Scholar]
- 33.Coort SL, Willems J, Coumans WA, van der Vusse GJ, Bonen A, Glatz JF and Luiken JJ. Sulfo-N-succinimidyl esters of long chain fatty acids specifically inhibit fatty acid translocase (FAT/CD36)-mediated cellular fatty acid uptake. Mol Cell Biochem. 2002;239:213–9. [PubMed] [Google Scholar]
- 34.Harmon CM and Abumrad NA. Binding of sulfosuccinimidyl fatty acids to adipocyte membrane proteins: isolation and amino-terminal sequence of an 88-kD protein implicated in transport of long-chain fatty acids. J Membr Biol. 1993;133:43–9. [DOI] [PubMed] [Google Scholar]
- 35.Tanaka T and Kawamura K. Isolation of myocardial membrane long-chain fatty acid-binding protein: homology with a rat membrane protein implicated in the binding or transport of long-chain fatty acids. J Mol Cell Cardiol. 1995;27:1613–22. [DOI] [PubMed] [Google Scholar]
- 36.Kuda O, Jenkins CM, Skinner JR, Moon SH, Su X, Gross RW and Abumrad NA. CD36 protein is involved in store-operated calcium flux, phospholipase A2 activation, and production of prostaglandin E2. J Biol Chem. 2011;286:17785–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuda O, Pietka TA, Demianova Z, Kudova E, Cvacka J, Kopecky J and Abumrad NA. Sulfo-N-succinimidyl oleate (SSO) inhibits fatty acid uptake and signaling for intracellular calcium via binding CD36 lysine 164: SSO also inhibits oxidized low density lipoprotein uptake by macrophages. J Biol Chem. 2013;288:15547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sundaresan S, Shahid R, Riehl TE, Chandra R, Nassir F, Stenson WF, Liddle RA and Abumrad NA. CD36-dependent signaling mediates fatty acid-induced gut release of secretin and cholecystokinin. FASEB J. 2013;27:1191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jay AG, Simard JR, Huang N and Hamilton JA. SSO and other putative inhibitors of FA transport across membranes by CD36 disrupt intracellular metabolism, but do not affect FA translocation. J Lipid Res. 2020;61:790–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pepino MY, Kuda O, Samovski D and Abumrad NA. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annual review of nutrition. 2014;34:281–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsieh FL, Turner L, Bolla JR, Robinson CV, Lavstsen T and Higgins MK. The structural basis for CD36 binding by the malaria parasite. Nat Commun. 2016;7:12837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coburn CT, Knapp FF Jr., Febbraio M, Beets AL, Silverstein RL and Abumrad NA. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J Biol Chem. 2000;275:32523–9. [DOI] [PubMed] [Google Scholar]
- 43.Hajri T, Han XX, Bonen A and Abumrad NA. Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36-null mice. J Clin Invest. 2002;109:1381–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonen A, Han XX, Habets DD, Febbraio M, Glatz JF and Luiken JJ. A null mutation in skeletal muscle FAT/CD36 reveals its essential role in insulin- and AICAR-stimulated fatty acid metabolism. American journal of physiology Endocrinology and metabolism. 2007;292:E1740–9. [DOI] [PubMed] [Google Scholar]
- 45.Ibrahimi A, Bonen A, Blinn WD, Hajri T, Li X, Zhong K, Cameron R and Abumrad NA. Muscle-specific overexpression of FAT/CD36 enhances fatty acid oxidation by contracting muscle, reduces plasma triglycerides and fatty acids, and increases plasma glucose and insulin. J Biol Chem. 1999;274:26761–6. [DOI] [PubMed] [Google Scholar]
- 46.Love-Gregory L, Sherva R, Sun L, Wasson J, Schappe T, Doria A, Rao DC, Hunt SC, Klein S, Neuman RJ, Permutt MA and Abumrad NA. Variants in the CD36 gene associate with the metabolic syndrome and high-density lipoprotein cholesterol. Hum Mol Genet. 2008;17:1695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Farook VS, Puppala S, Schneider J, Fowler SP, Chittoor G, Dyer TD, Allayee H, Cole SA, Arya R, Black MH, Curran JE, Almasy L, Buchanan TA, Jenkinson CP, Lehman DM, Watanabe RM, Blangero J and Duggirala R. Metabolic syndrome is linked to chromosome 7q21 and associated with genetic variants in CD36 and GNAT3 in Mexican Americans. Obesity (Silver Spring). 2012;20:2083–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Silverstein RL. Linking Metabolic Dysfunction to Atherosclerosis Via Activation of Macrophage CD36 Gene Transcription by Retinol Binding Protein-4. Circulation. 2017;135:1355–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuasa-Kawase M, Masuda D, Yamashita T, Kawase R, Nakaoka H, Inagaki M, Nakatani K, Tsubakio-Yamamoto K, Ohama T, Matsuyama A, Nishida M, Ishigami M, Kawamoto T, Komuro I and Yamashita S. Patients with CD36 deficiency are associated with enhanced atherosclerotic cardiovascular diseases. J Atheroscler Thromb. 2012;19:263–75. [DOI] [PubMed] [Google Scholar]
- 50.Aitman TJ, Cooper LD, Norsworthy PJ, Wahid FN, Gray JK, Curtis BR, McKeigue PM, Kwiatkowski D, Greenwood BM, Snow RW, Hill AV and Scott J. Malaria susceptibility and CD36 mutation. Nature. 2000;405:1015–6. [DOI] [PubMed] [Google Scholar]
- 51.Love-Gregory L and Abumrad NA. CD36 genetics and the metabolic complications of obesity. Curr Opin Clin Nutr Metab Care. 2011;14:527–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Elbers CC, Guo Y, Tragante V, van Iperen EP, Lanktree MB, Castillo BA, Chen F, Yanek LR, Wojczynski MK, Li YR, Ferwerda B, Ballantyne CM, Buxbaum SG, Chen YD, Chen WM, Cupples LA, Cushman M, Duan Y, Duggan D, Evans MK, Fernandes JK, Fornage M, Garcia M, Garvey WT, Glazer N, Gomez F, Harris TB, Halder I, Howard VJ, Keller MF, Kamboh MI, Kooperberg C, Kritchevsky SB, LaCroix A, Liu K, Liu Y, Musunuru K, Newman AB, Onland-Moret NC, Ordovas J, Peter I, Post W, Redline S, Reis SE, Saxena R, Schreiner PJ, Volcik KA, Wang X, Yusuf S, Zonderland AB, Anand SS, Becker DM, Psaty B, Rader DJ, Reiner AP, Rich SS, Rotter JI, Sale MM, Tsai MY, Borecki IB, Hegele RA, Kathiresan S, Nalls MA, Taylor HA Jr., Hakonarson H, Sivapalaratnam S, Asselbergs FW, Drenos F, Wilson JG and Keating BJ. Gene-centric meta-analysis of lipid traits in African, East Asian and Hispanic populations. PloS one. 2012;7:e50198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miyaoka K, Kuwasako T, Hirano K, Nozaki S, Yamashita S and Matsuzawa Y. CD36 deficiency associated with insulin resistance. Lancet. 2001;357:686–7. [DOI] [PubMed] [Google Scholar]
- 54.Masuda Y, Tamura S, Matsuno K, Nagasawa A, Hayasaka K, Shimizu C and Moriyama T. Diverse CD36 expression among Japanese population: defective CD36 mutations cause platelet and monocyte CD36 reductions in not only deficient but also normal phenotype subjects. Thromb Res. 2015;135:951–7. [DOI] [PubMed] [Google Scholar]
- 55.Yoshizumi T, Nozaki S, Fukuchi K, Yamasaki K, Fukuchi T, Maruyama T, Tomiyama Y, Yamashita S, Nishimura T and Matsuzawa Y. Pharmacokinetics and metabolism of 123I-BMIPP fatty acid analog in healthy and CD36-deficient subjects. J Nucl Med. 2000;41:1134–8. [PubMed] [Google Scholar]
- 56.Tanaka T, Nakata T, Oka T, Ogawa T, Okamoto F, Kusaka Y, Sohmiya K, Shimamoto K and Itakura K. Defect in human myocardial long-chain fatty acid uptake is caused by FAT/CD36 mutations. J Lipid Res. 2001;42:751–9. [PubMed] [Google Scholar]
- 57.Hames KC, Vella A, Kemp BJ and Jensen MD. Free fatty acid uptake in humans with CD36 deficiency. Diabetes. 2014;63:3606–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shibao CA, Celedonio JE, Ramirez CE, Love-Gregory L, Arnold AC, Choi L, Okamoto LE, Gamboa A, Biaggioni I, Abumrad NN and Abumrad NA. A Common CD36 Variant Influences Endothelial Function and Response to Treatment with Phosphodiesterase 5 Inhibition. J Clin Endocrinol Metab. 2016;101:2751–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma X, Bacci S, Mlynarski W, Gottardo L, Soccio T, Menzaghi C, Iori E, Lager RA, Shroff AR, Gervino EV, Nesto RW, Johnstone MT, Abumrad NA, Avogaro A, Trischitta V and Doria A. A common haplotype at the CD36 locus is associated with high free fatty acid levels and increased cardiovascular risk in Caucasians. Hum Mol Genet. 2004;13:2197–205. [DOI] [PubMed] [Google Scholar]
- 60.Masuda D, Hirano K, Oku H, Sandoval JC, Kawase R, Yuasa-Kawase M, Yamashita Y, Takada M, Tsubakio-Yamamoto K, Tochino Y, Koseki M, Matsuura F, Nishida M, Kawamoto T, Ishigami M, Hori M, Shimomura I and Yamashita S. Chylomicron remnants are increased in the postprandial state in CD36 deficiency. J Lipid Res. 2009;50:999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Love-Gregory L, Kraja AT, Allum F, Aslibekyan S, Hedman AK, Duan Y, Borecki IB, Arnett DK, McCarthy MI, Deloukas P, Ordovas JM, Hopkins PN, Grundberg E and Abumrad NA. Higher chylomicron remnants and LDL particle numbers associate with CD36 SNPs and DNA methylation sites that reduce CD36. J Lipid Res. 2016;57:2176–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Frazier-Wood AC, Aslibekyan S, Borecki IB, Hopkins PN, Lai CQ, Ordovas JM, Straka RJ, Tiwari HK and Arnett DK. Genome-wide association study indicates variants associated with insulin signaling and inflammation mediate lipoprotein responses to fenofibrate. Pharmacogenetics and genomics. 2012;22:750–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nassir F, Adewole OL, Brunt EM and Abumrad NA. CD36 deletion reduces VLDL secretion, modulates liver prostaglandins, and exacerbates hepatic steatosis in ob/ob mice. J Lipid Res. 2013;54:2988–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schaffer JE and Lodish HF. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell. 1994;79:427–36. [DOI] [PubMed] [Google Scholar]
- 65.Schaffer JE. Fatty acid transport: the roads taken. American journal of physiology Endocrinology and metabolism. 2002;282:E239–46. [DOI] [PubMed] [Google Scholar]
- 66.Kazantzis M and Stahl A. Fatty acid transport proteins, implications in physiology and disease. Biochim Biophys Acta. 2012;1821:852–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Anderson CM and Stahl A. SLC27 fatty acid transport proteins. Mol Aspects Med. 2013;34:516–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Black PN and DiRusso CC. Vectorial acylation: linking fatty acid transport and activation to metabolic trafficking. Novartis Found Symp. 2007;286:127–38; discussion 138–41, 162–3, 196–203. [DOI] [PubMed] [Google Scholar]
- 69.Digel M, Ehehalt R, Stremmel W and Fullekrug J. Acyl-CoA synthetases: fatty acid uptake and metabolic channeling. Mol Cell Biochem. 2009;326:23–8. [DOI] [PubMed] [Google Scholar]
- 70.Coleman RA. It takes a village: channeling fatty acid metabolism and triacylglycerol formation via protein interactomes. J Lipid Res. 2019;60:490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chiu HC, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, Yamada KA, Brunet S, Xu H, Nerbonne JM, Welch MJ, Fettig NM, Sharp TL, Sambandam N, Olson KM, Ory DS and Schaffer JE. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res. 2005;96:225–33. [DOI] [PubMed] [Google Scholar]
- 72.Nickerson JG, Alkhateeb H, Benton CR, Lally J, Nickerson J, Han XX, Wilson MH, Jain SS, Snook LA, Glatz JF, Chabowski A, Luiken JJ and Bonen A. Greater transport efficiencies of the membrane fatty acid transporters FAT/CD36 and FATP4 compared with FABPpm and FATP1 and differential effects on fatty acid esterification and oxidation in rat skeletal muscle. J Biol Chem. 2009;284:16522–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Perez VM, Gabell J, Behrens M, Wase N, DiRusso CC and Black PN. Deletion of fatty acid transport protein 2 (FATP2) in the mouse liver changes the metabolic landscape by increasing the expression of PPARalpha-regulated genes. J Biol Chem. 2020;295:5737–5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hamilton JA. Transport of fatty acids across membranes by the diffusion mechanism. Prostaglandins Leukot Essent Fatty Acids. 1999;60:291–7. [DOI] [PubMed] [Google Scholar]
- 75.Kamp F, Zakim D, Zhang F, Noy N and Hamilton JA. Fatty acid flip-flop in phospholipid bilayers is extremely fast. Biochemistry. 1995;34:11928–37. [DOI] [PubMed] [Google Scholar]
- 76.Kleinfeld AM. Lipid phase fatty acid flip-flop, is it fast enough for cellular transport? J Membr Biol. 2000;175:79–86. [DOI] [PubMed] [Google Scholar]
- 77.Kampf JP, Parmley D and Kleinfeld AM. Free fatty acid transport across adipocytes is mediated by an unknown membrane protein pump. American journal of physiology Endocrinology and metabolism. 2007;293:E1207–14. [DOI] [PubMed] [Google Scholar]
- 78.Carley AN and Kleinfeld AM. Fatty acid (FFA) transport in cardiomyocytes revealed by imaging unbound FFA is mediated by an FFA pump modulated by the CD36 protein. J Biol Chem. 2011;286:4589–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kalluri AS, Vellarikkal SK, Edelman ER, Nguyen L, Subramanian A, Ellinor PT, Regev A, Kathiresan S and Gupta RM. Single-Cell Analysis of the Normal Mouse Aorta Reveals Functionally Distinct Endothelial Cell Populations. Circulation. 2019;140:147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Samuel VT and Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest. 2016;126:12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chavez JA and Summers SA. A ceramide-centric view of insulin resistance. Cell Metab. 2012;15:585–94. [DOI] [PubMed] [Google Scholar]
- 82.Liu L, Zhang Y, Chen N, Shi X, Tsang B and Yu YH. Upregulation of myocellular DGAT1 augments triglyceride synthesis in skeletal muscle and protects against fat-induced insulin resistance. J Clin Invest. 2007;117:1679–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu L, Shi X, Bharadwaj KG, Ikeda S, Yamashita H, Yagyu H, Schaffer JE, Yu YH and Goldberg IJ. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J Biol Chem. 2009;284:36312–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Koliwad SK, Streeper RS, Monetti M, Cornelissen I, Chan L, Terayama K, Naylor S, Rao M, Hubbard B and Farese RV Jr. DGAT1-dependent triacylglycerol storage by macrophages protects mice from diet-induced insulin resistance and inflammation. J Clin Invest. 2010;120:756–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kanda T, Brown JD, Orasanu G, Vogel S, Gonzalez FJ, Sartoretto J, Michel T and Plutzky J. PPARgamma in the endothelium regulates metabolic responses to high-fat diet in mice. J Clin Invest. 2009;119:110–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jang C, Oh SF, Wada S, Rowe GC, Liu L, Chan MC, Rhee J, Hoshino A, Kim B, Ibrahim A, Baca LG, Kim E, Ghosh CC, Parikh SM, Jiang A, Chu Q, Forman DE, Lecker SH, Krishnaiah S, Rabinowitz JD, Weljie AM, Baur JA, Kasper DL and Arany Z. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat Med. 2016;22:421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ibrahim A, Yucel N, Kim B and Arany Z. Local Mitochondrial ATP Production Regulates Endothelial Fatty Acid Uptake and Transport. Cell Metab. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Coppiello G, Collantes M, Sirerol-Piquer MS, Vandenwijngaert S, Schoors S, Swinnen M, Vandersmissen I, Herijgers P, Topal B, van Loon J, Goffin J, Prosper F, Carmeliet P, Garcia-Verdugo JM, Janssens S, Penuelas I, Aranguren XL and Luttun A. Meox2/Tcf15 heterodimers program the heart capillary endothelium for cardiac fatty acid uptake. Circulation. 2015;131:815–26. [DOI] [PubMed] [Google Scholar]
- 89.Elias I, Franckhauser S, Ferre T, Vila L, Tafuro S, Munoz S, Roca C, Ramos D, Pujol A, Riu E, Ruberte J and Bosch F. Adipose tissue overexpression of vascular endothelial growth factor protects against diet-induced obesity and insulin resistance. Diabetes. 2012;61:1801–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sun K, Wernstedt Asterholm I, Kusminski CM, Bueno AC, Wang ZV, Pollard JW, Brekken RA and Scherer PE. Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proc Natl Acad Sci U S A. 2012;109:5874–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hagberg CE, Falkevall A, Wang X, Larsson E, Huusko J, Nilsson I, van Meeteren LA, Samen E, Lu L, Vanwildemeersch M, Klar J, Genove G, Pietras K, Stone-Elander S, Claesson-Welsh L, Yla-Herttuala S, Lindahl P and Eriksson U. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature. 2010;464:917–21. [DOI] [PubMed] [Google Scholar]
- 92.Hagberg CE, Mehlem A, Falkevall A, Muhl L, Fam BC, Ortsater H, Scotney P, Nyqvist D, Samen E, Lu L, Stone-Elander S, Proietto J, Andrikopoulos S, Sjoholm A, Nash A and Eriksson U. Targeting VEGF-B as a novel treatment for insulin resistance and type 2 diabetes. Nature. 2012;490:426–30. [DOI] [PubMed] [Google Scholar]
- 93.Kivela R, Bry M, Robciuc MR, Rasanen M, Taavitsainen M, Silvola JM, Saraste A, Hulmi JJ, Anisimov A, Mayranpaa MI, Lindeman JH, Eklund L, Hellberg S, Hlushchuk R, Zhuang ZW, Simons M, Djonov V, Knuuti J, Mervaala E and Alitalo K. VEGF-B-induced vascular growth leads to metabolic reprogramming and ischemia resistance in the heart. EMBO Mol Med. 2014;6:307–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bergman M, Manco M, Sesti G, Dankner R, Pareek M, Jagannathan R, Chetrit A, Abdul-Ghani M, Buysschaert M, Olsen MH, Nilsson PM, Medina JL, Roth J, Groop L, Del Prato S, Raz I and Ceriello A. Petition to replace current OGTT criteria for diagnosing prediabetes with the 1-hour post-load plasma glucose>/=155mg/dl (8.6mmol/L). Diabetes Res Clin Pract. 2018;146:18–33. [DOI] [PubMed] [Google Scholar]
- 95.Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, Kaul MG, Tromsdorf UI, Weller H, Waurisch C, Eychmuller A, Gordts PL, Rinninger F, Bruegelmann K, Freund B, Nielsen P, Merkel M and Heeren J. Brown adipose tissue activity controls triglyceride clearance. Nat Med. 2011;17:200–5. [DOI] [PubMed] [Google Scholar]
- 96.Nolan DJ, Ginsberg M, Israely E, Palikuqi B, Poulos MG, James D, Ding BS, Schachterle W, Liu Y, Rosenwaks Z, Butler JM, Xiang J, Rafii A, Shido K, Rabbany SY, Elemento O and Rafii S. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev Cell. 2013;26:204–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bae H, Hong KY, Lee CK, Jang C, Lee SJ, Choe K, Offermanns S, He Y, Lee HJ and Koh GY. Angiopoietin-2-integrin alpha5beta1 signaling enhances vascular fatty acid transport and prevents ectopic lipid-induced insulin resistance. Nat Commun. 2020;11:2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Salameh A, Daquinag AC, Staquicini DI, An Z, Hajjar KA, Pasqualini R, Arap W and Kolonin MG. Prohibitin/annexin 2 interaction regulates fatty acid transport in adipose tissue. JCI Insight. 2016;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Khalifeh-Soltani A, McKleroy W, Sakuma S, Cheung YY, Tharp K, Qiu Y, Turner SM, Chawla A, Stahl A and Atabai K. Mfge8 promotes obesity by mediating the uptake of dietary fats and serum fatty acids. Nat Med. 2014;20:175–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bharadwaj KG, Hiyama Y, Hu Y, Huggins LA, Ramakrishnan R, Abumrad NA, Shulman GI, Blaner WS and Goldberg IJ. Chylomicron- and VLDL-derived lipids enter the heart through different pathways: in vivo evidence for receptor- and non-receptor-mediated fatty acid uptake. J Biol Chem. 2010;285:37976–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Davies BS, Beigneux AP, Barnes RH 2nd, Tu Y, Gin P, Weinstein MM, Nobumori C, Nyren R, Goldberg I, Olivecrona G, Bensadoun A, Young SG and Fong LG. GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. 2010;12:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Obunike JC, Lutz EP, Li Z, Paka L, Katopodis T, Strickland DK, Kozarsky KF, Pillarisetti S and Goldberg IJ. Transcytosis of lipoprotein lipase across cultured endothelial cells requires both heparan sulfate proteoglycans and the very low density lipoprotein receptor. J Biol Chem. 2001;276:8934–41. [DOI] [PubMed] [Google Scholar]
- 103.Merkel M, Weinstock PH, Chajek-Shaul T, Radner H, Yin B, Breslow JL and Goldberg IJ. Lipoprotein lipase expression exclusively in liver. A mouse model for metabolism in the neonatal period and during cachexia. J Clin Invest. 1998;102:893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Beigneux AP, Miyashita K, Ploug M, Blom DJ, Ai M, Linton MF, Khovidhunkit W, Dufour R, Garg A, McMahon MA, Pullinger CR, Sandoval NP, Hu X, Allan CM, Larsson M, Machida T, Murakami M, Reue K, Tontonoz P, Goldberg IJ, Moulin P, Charriere S, Fong LG, Nakajima K and Young SG. Autoantibodies against GPIHBP1 as a Cause of Hypertriglyceridemia. N Engl J Med. 2017;376:1647–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Osborne JC Jr., Bengtsson-Olivecrona G, Lee NS and Olivecrona T. Studies on inactivation of lipoprotein lipase: role of the dimer to monomer dissociation. Biochemistry. 1985;24:5606–11. [DOI] [PubMed] [Google Scholar]
- 106.Ben-Zeev O and Doolittle MH. Determining lipase subunit structure by sucrose gradient centrifugation. Methods Mol Biol. 1999;109:257–66. [DOI] [PubMed] [Google Scholar]
- 107.Beigneux AP, Allan CM, Sandoval NP, Cho GW, Heizer PJ, Jung RS, Stanhope KL, Havel PJ, Birrane G, Meiyappan M, Gill JEt, Murakami M, Miyashita K, Nakajima K, Ploug M, Fong LG and Young SG. Lipoprotein lipase is active as a monomer. Proc Natl Acad Sci U S A. 2019;116:6319–6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Birrane G, Beigneux AP, Dwyer B, Strack-Logue B, Kristensen KK, Francone OL, Fong LG, Mertens HDT, Pan CQ, Ploug M, Young SG and Meiyappan M. Structure of the lipoprotein lipase-GPIHBP1 complex that mediates plasma triglyceride hydrolysis. Proc Natl Acad Sci U S A. 2019;116:1723–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Arora R, Nimonkar AV, Baird D, Wang C, Chiu CH, Horton PA, Hanrahan S, Cubbon R, Weldon S, Tschantz WR, Mueller S, Brunner R, Lehr P, Meier P, Ottl J, Voznesensky A, Pandey P, Smith TM, Stojanovic A, Flyer A, Benson TE, Romanowski MJ and Trauger JW. Structure of lipoprotein lipase in complex with GPIHBP1. Proc Natl Acad Sci U S A. 2019;116:10360–10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gordts PL, Nock R, Son NH, Ramms B, Lew I, Gonzales JC, Thacker BE, Basu D, Lee RG, Mullick AE, Graham MJ, Goldberg IJ, Crooke RM, Witztum JL and Esko JD. ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J Clin Invest. 2016;126:2855–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jong MC, Rensen PC, Dahlmans VE, van der Boom H, van Berkel TJ and Havekes LM. Apolipoprotein C-III deficiency accelerates triglyceride hydrolysis by lipoprotein lipase in wild-type and apoE knockout mice. J Lipid Res. 2001;42:1578–85. [PubMed] [Google Scholar]
- 112.Masucci-Magoulas L, Goldberg IJ, Bisgaier CL, Serajuddin H, Francone OL, Breslow JL and Tall AR. A mouse model with features of familial combined hyperlipidemia. Science. 1997;275:391–4. [DOI] [PubMed] [Google Scholar]
- 113.Gouni-Berthold I. The role of antisense oligonucleotide therapy against apolipoprotein-CIII in hypertriglyceridemia. Atheroscler Suppl. 2017;30:19–27. [DOI] [PubMed] [Google Scholar]
- 114.Khetarpal SA, Zeng X, Millar JS, Vitali C, Somasundara AVH, Zanoni P, Landro JA, Barucci N, Zavadoski WJ, Sun Z, de Haard H, Toth IV, Peloso GM, Natarajan P, Cuchel M, Lund-Katz S, Phillips MC, Tall AR, Kathiresan S, DaSilva-Jardine P, Yates NA and Rader DJ. A human APOC3 missense variant and monoclonal antibody accelerate apoC-III clearance and lower triglyceride-rich lipoprotein levels. Nat Med. 2017;23:1086–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wolska A, Dunbar RL, Freeman LA, Ueda M, Amar MJ, Sviridov DO and Remaley AT. Apolipoprotein C-II: New findings related to genetics, biochemistry, and role in triglyceride metabolism. Atherosclerosis. 2017;267:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zdunek J, Martinez GV, Schleucher J, Lycksell PO, Yin Y, Nilsson S, Shen Y, Olivecrona G and Wijmenga S. Global structure and dynamics of human apolipoprotein CII in complex with micelles: evidence for increased mobility of the helix involved in the activation of lipoprotein lipase. Biochemistry. 2003;42:1872–89. [DOI] [PubMed] [Google Scholar]
- 117.Ueda M, Dunbar RL, Wolska A, Sikora TU, Escobar MDR, Seliktar N, deGoma E, DerOhannessian S, Morrell L, McIntyre AD, Burke F, Sviridov D, Amar M, Shamburek RD, Freeman L, Hegele RA, Remaley AT and Rader DJ. A Novel APOC2 Missense Mutation Causing Apolipoprotein C-II Deficiency With Severe Triglyceridemia and Pancreatitis. J Clin Endocrinol Metab. 2017;102:1454–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sakurai T, Sakurai A, Vaisman BL, Amar MJ, Liu C, Gordon SM, Drake SK, Pryor M, Sampson ML, Yang L, Freeman LA and Remaley AT. Creation of Apolipoprotein C-II (ApoC-II) Mutant Mice and Correction of Their Hypertriglyceridemia with an ApoC-II Mimetic Peptide. J Pharmacol Exp Ther. 2016;356:341–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kluger M, Heeren J and Merkel M. Apoprotein A-V: an important regulator of triglyceride metabolism. Journal of inherited metabolic disease. 2008;31:281–8. [DOI] [PubMed] [Google Scholar]
- 120.Surendran RP, Visser ME, Heemelaar S, Wang J, Peter J, Defesche JC, Kuivenhoven JA, Hosseini M, Peterfy M, Kastelein JJ, Johansen CT, Hegele RA, Stroes ES and Dallinga-Thie GM. Mutations in LPL, APOC2, APOA5, GPIHBP1 and LMF1 in patients with severe hypertriglyceridaemia. J Intern Med. 2012;272:185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chi X, Britt EC, Shows HW, Hjelmaas AJ, Shetty SK, Cushing EM, Li W, Dou A, Zhang R and Davies BSJ. ANGPTL8 promotes the ability of ANGPTL3 to bind and inhibit lipoprotein lipase. Mol Metab. 2017;6:1137–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stitziel NO, Khera AV, Wang X, Bierhals AJ, Vourakis AC, Sperry AE, Natarajan P, Klarin D, Emdin CA, Zekavat SM, Nomura A, Erdmann J, Schunkert H, Samani NJ, Kraus WE, Shah SH, Yu B, Boerwinkle E, Rader DJ, Gupta N, Frossard PM, Rasheed A, Danesh J, Lander ES, Gabriel S, Saleheen D, Musunuru K, Kathiresan S, Promis and Myocardial Infarction Genetics Consortium I. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. J Am Coll Cardiol. 2017;69:2054–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kersten S, Mandard S, Tan NS, Escher P, Metzger D, Chambon P, Gonzalez FJ, Desvergne B and Wahli W. Characterization of the fasting-induced adipose factor FIAF, a novel peroxisome proliferator-activated receptor target gene. J Biol Chem. 2000;275:28488–93. [DOI] [PubMed] [Google Scholar]
- 124.Yoon JC, Chickering TW, Rosen ED, Dussault B, Qin Y, Soukas A, Friedman JM, Holmes WE and Spiegelman BM. Peroxisome proliferator-activated receptor gamma target gene encoding a novel angiopoietin-related protein associated with adipose differentiation. Mol Cell Biol. 2000;20:5343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim I, Kim HG, Kim H, Kim HH, Park SK, Uhm CS, Lee ZH and Koh GY. Hepatic expression, synthesis and secretion of a novel fibrinogen/angiopoietin-related protein that prevents endothelial-cell apoptosis. Biochemical Journal. 2000;346:603–610. [PMC free article] [PubMed] [Google Scholar]
- 126.Wang Y, McNutt MC, Banfi S, Levin MG, Holland WL, Gusarova V, Gromada J, Cohen JC and Hobbs HH. Hepatic ANGPTL3 regulates adipose tissue energy homeostasis. Proc Natl Acad Sci U S A. 2015;112:11630–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Goldberg IJ, Eckel RH and McPherson R. Triglycerides and heart disease: still a hypothesis? Arterioscler Thromb Vasc Biol. 2011;31:1716–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Razani B and Lisanti MP. Caveolin-deficient mice: insights into caveolar function human disease. J Clin Invest. 2001;108:1553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Razani B, Combs TP, Wang XB, Frank PG, Park DS, Russell RG, Li M, Tang B, Jelicks LA, Scherer PE and Lisanti MP. Caveolin-1-deficient mice are lean, resistant to diet-induced obesity, and show hypertriglyceridemia with adipocyte abnormalities. J Biol Chem. 2002;277:8635–47. [DOI] [PubMed] [Google Scholar]
- 130.Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF and Silverstein RL. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem. 1999;274:19055–62. [DOI] [PubMed] [Google Scholar]
- 131.Frank PG, Pavlides S and Lisanti MP. Caveolae and transcytosis in endothelial cells: role in atherosclerosis. Cell Tissue Res. 2009;335:41–7. [DOI] [PubMed] [Google Scholar]
- 132.Schubert W, Frank PG, Razani B, Park DS, Chow CW and Lisanti MP. Caveolae-deficient endothelial cells show defects in the uptake and transport of albumin in vivo. J Biol Chem. 2001;276:48619–22. [DOI] [PubMed] [Google Scholar]
- 133.Frank PG, Lee H, Park DS, Tandon NN, Scherer PE and Lisanti MP. Genetic ablation of caveolin-1 confers protection against atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:98–105. [DOI] [PubMed] [Google Scholar]
- 134.Fernández-Hernando C, Yu J, Suárez Y, Rahner C, Dávalos A, Lasunción MA and Sessa WC. Genetic evidence supporting a critical role of endothelial caveolin-1 during the progression of atherosclerosis. Cell metabolism. 2009;10:48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Parton RG and Del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nature reviews Molecular cell biology. 2013;14:98–112. [DOI] [PubMed] [Google Scholar]
- 136.Gaus K, Le Lay S, Balasubramanian N and Schwartz MA. Integrin-mediated adhesion regulates membrane order. The Journal of cell biology. 2006;174:725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hoffmann C, Berking A, Agerer F, Buntru A, Neske F, Chhatwal GS, Ohlsen K and Hauck CR. Caveolin limits membrane microdomain mobility and integrin-mediated uptake of fibronectin-binding pathogens. Journal of cell science. 2010;123:4280–4291. [DOI] [PubMed] [Google Scholar]
- 138.Roy S, Luetterforst R, Harding A, Apolloni A, Etheridge M, Stang E, Rolls B, Hancock JF and Parton RG. Dominant-negative caveolin inhibits H-Ras function by disrupting cholesterol-rich plasma membrane domains. Nature cell biology. 1999;1:98–105. [DOI] [PubMed] [Google Scholar]
- 139.Murata M, Peränen J, Schreiner R, Wieland F, Kurzchalia TV and Simons K. VIP21/caveolin is a cholesterol-binding protein. Proceedings of the National Academy of Sciences. 1995;92:10339–10343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Trigatti BL, Anderson RG and Gerber GE. Identification of caveolin-1 as a fatty acid binding protein. Biochemical and biophysical research communications. 1999;255:34–39. [DOI] [PubMed] [Google Scholar]
- 141.Alejandro Fernández-Rojo M, Restall C, Ferguson C, Martel N, Martin S, Bosch M, Kassan A, Leong GM, Martin SD and McGee SL. Caveolin-1 orchestrates the balance between glucose and lipid-dependent energy metabolism: Implications for liver regeneration. Hepatology. 2012;55:1574–1584. [DOI] [PubMed] [Google Scholar]
- 142.Brasaemle DL, Dolios G, Shapiro L and Wang R. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. Journal of Biological Chemistry. 2004;279:46835–46842. [DOI] [PubMed] [Google Scholar]
- 143.Martin S and Parton RG. Caveolin, cholesterol, and lipid bodies. Seminars in cell & developmental biology. 2005;16:163–174. [DOI] [PubMed] [Google Scholar]
- 144.Pol A, Martin S, Fernandez MA, Ferguson C, Carozzi A, Luetterforst R, Enrich C and Parton RG. Dynamic and regulated association of caveolin with lipid bodies: modulation of lipid body motility and function by a dominant negative mutant. Molecular biology of the cell. 2004;15:99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kuo A, Lee MY, Yang K, Gross RW and Sessa WC. Caveolin-1 regulates lipid droplet metabolism in endothelial cells via autocrine prostacyclin–stimulated, cAMP-mediated lipolysis. Journal of Biological Chemistry. 2018;293:973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ring A, Le Lay S, Pohl J, Verkade P and Stremmel W. Caveolin-1 is required for fatty acid translocase (FAT/CD36) localization and function at the plasma membrane of mouse embryonic fibroblasts. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids. 2006;1761:416–423. [DOI] [PubMed] [Google Scholar]
- 147.Mattern HM, Raikar LS and Hardin CD. The effect of caveolin-1 (Cav-1) on fatty acid uptake and CD36 localization and lipotoxicity in vascular smooth muscle (VSM) cells. International journal of physiology, pathophysiology and pharmacology. 2009;1:1. [PMC free article] [PubMed] [Google Scholar]
- 148.Spady DK, Turley SD and Dietschy JM. Rates of low density lipoprotein uptake and cholesterol synthesis are regulated independently in the liver. J Lipid Res. 1985;26:465–72. [PubMed] [Google Scholar]
- 149.Dietschy JM, Spady DK and Stange EF. Quantitative importance of different organs for cholesterol synthesis and low-density-lipoprotein degradation. Biochem Soc Trans. 1983;11:639–41. [DOI] [PubMed] [Google Scholar]
- 150.Fielding CJ. Metabolism of cholesterol-rich chylomicroms. Mechanism of binding and uptake of cholesteryl esters by the vascular bed of the perfused rat heart. J Clin Invest. 1978;62:141–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Perman JC, Bostrom P, Lindbom M, Lidberg U, StAhlman M, Hagg D, Lindskog H, Scharin Tang M, Omerovic E, Mattsson Hulten L, Jeppsson A, Petursson P, Herlitz J, Olivecrona G, Strickland DK, Ekroos K, Olofsson SO and Boren J. The VLDL receptor promotes lipotoxicity and increases mortality in mice following an acute myocardial infarction. J Clin Invest. 2011;121:2625–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Parker F, Bagdade JD, Odland GF and Bierman EL. Evidence for the chylomicron origin of lipids accumulating in diabetic eruptive xanthomas: a correlative lipid biochemical, histochemical, and electron microscopic study. J Clin Invest. 1970;49:2172–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.He C, Weston TA, Jung RS, Heizer P, Larsson M, Hu X, Allan CM, Tontonoz P, Reue K, Beigneux AP, Ploug M, Holme A, Kilburn M, Guagliardo P, Ford DA, Fong LG, Young SG and Jiang H. NanoSIMS Analysis of Intravascular Lipolysis and Lipid Movement across Capillaries and into Cardiomyocytes. Cell Metab. 2018;27:1055–1066 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tott S, Grosicki M, Klimas B, Augustynska D, Chlopicki S and Baranska M. Raman spectroscopic features of primary cardiac microvascular endothelial cells (CMECs) isolated from the murine heart. The Analyst. 2018;143:6079–6086. [DOI] [PubMed] [Google Scholar]
- 155.Guyton JR and Klemp KF. Early extracellular and cellular lipid deposits in aorta of cholesterol-fed rabbits. Am J Pathol. 1992;141:925–36. [PMC free article] [PubMed] [Google Scholar]
- 156.Proctor SD and Mamo JC. Retention of fluorescent-labelled chylomicron remnants within the intima of the arterial wall--evidence that plaque cholesterol may be derived from post-prandial lipoproteins. Eur J Clin Invest. 1998;28:497–503. [DOI] [PubMed] [Google Scholar]
- 157.Kuo A, Lee MY and Sessa WC. Lipid Droplet Biogenesis and Function in the Endothelium. Circ Res. 2017;120:1289–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhang X, Sessa WC and Fernandez-Hernando C. Endothelial Transcytosis of Lipoproteins in Atherosclerosis. Front Cardiovasc Med. 2018;5:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.von Eckardstein A and Rohrer L. Transendothelial lipoprotein transport and regulation of endothelial permeability and integrity by lipoproteins. Curr Opin Lipidol. 2009;20:197–205. [DOI] [PubMed] [Google Scholar]
- 160.Vasile E, Simionescu M and Simionescu N. Visualization of the binding, endocytosis, and transcytosis of low-density lipoprotein in the arterial endothelium in situ. J Cell Biol. 1983;96:1677–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Frank PG, Lee H, Park DS, Tandon NN, Scherer PE and Lisanti MP. Genetic ablation of caveolin-1 confers protection against atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:98–105. [DOI] [PubMed] [Google Scholar]
- 162.Fernandez-Hernando C, Yu J, Suarez Y, Rahner C, Davalos A, Lasuncion MA and Sessa WC. Genetic evidence supporting a critical role of endothelial caveolin-1 during the progression of atherosclerosis. Cell Metab. 2009;10:48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fernandez-Hernando C, Yu J, Davalos A, Prendergast J and Sessa WC. Endothelial-specific overexpression of caveolin-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2010;177:998–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Acton SL, Scherer PE, Lodish HF and Krieger M. Expression cloning of SR-BI, a CD36-related class B scavenger receptor. J Biol Chem. 1994;269:21003–9. [PubMed] [Google Scholar]
- 165.Kozarsky KF, Donahee MH, Rigotti A, Iqbal SN, Edelman ER and Krieger M. Overexpression of the HDL receptor SR-BI alters plasma HDL and bile cholesterol levels. Nature. 1997;387:414–7. [DOI] [PubMed] [Google Scholar]
- 166.Krieger M. Scavenger receptor class B type I is a multiligand HDL receptor that influences diverse physiologic systems. J Clin Invest. 2001;108:793–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Shen WJ, Azhar S and Kraemer FB. SR-B1: A Unique Multifunctional Receptor for Cholesterol Influx and Efflux. Annu Rev Physiol. 2018;80:95–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rohrer L, Ohnsorg PM, Lehner M, Landolt F, Rinninger F and von Eckardstein A. High-density lipoprotein transport through aortic endothelial cells involves scavenger receptor BI and ATP-binding cassette transporter G1. Circ Res. 2009;104:1142–50. [DOI] [PubMed] [Google Scholar]
- 169.Vaisman BL, Vishnyakova TG, Freeman LA, Amar MJ, Demosky SJ, Liu C, Stonik JA, Sampson ML, Pryor M, Bocharov AV, Eggerman TL, Patterson AP and Remaley AT. Endothelial Expression of Scavenger Receptor Class B, Type I Protects against Development of Atherosclerosis in Mice. Biomed Res Int. 2015;2015:607120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lim HY, Thiam CH, Yeo KP, Bisoendial R, Hii CS, McGrath KC, Tan KW, Heather A, Alexander JS and Angeli V. Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell Metab. 2013;17:671–84. [DOI] [PubMed] [Google Scholar]
- 171.Armstrong SM, Sugiyama MG, Fung KY, Gao Y, Wang C, Levy AS, Azizi P, Roufaiel M, Zhu SN, Neculai D, Yin C, Bolz SS, Seidah NG, Cybulsky MI, Heit B and Lee WL. A novel assay uncovers an unexpected role for SR-BI in LDL transcytosis. Cardiovasc Res. 2015;108:268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ghaffari S, Naderi Nabi F, Sugiyama MG and Lee WL. Estrogen Inhibits LDL (Low-Density Lipoprotein) Transcytosis by Human Coronary Artery Endothelial Cells via GPER (G-Protein-Coupled Estrogen Receptor) and SR-BI (Scavenger Receptor Class B Type 1). Arterioscler Thromb Vasc Biol. 2018;38:2283–2294. [DOI] [PubMed] [Google Scholar]
- 173.Huang L, Chambliss KL, Gao X, Yuhanna IS, Behling-Kelly E, Bergaya S, Ahmed M, Michaely P, Luby-Phelps K, Darehshouri A, Xu L, Fisher EA, Ge WP, Mineo C and Shaul PW. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature. 2019;569:565–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Marques PE, Nyegaard S, Collins RF, Troise F, Freeman SA, Trimble WS and Grinstein S. Multimerization and Retention of the Scavenger Receptor SR-B1 in the Plasma Membrane. Dev Cell. 2019;50:283–295 e5. [DOI] [PubMed] [Google Scholar]
- 175.Dehouck B, Fenart L, Dehouck MP, Pierce A, Torpier G and Cecchelli R. A new function for the LDL receptor: transcytosis of LDL across the blood-brain barrier. J Cell Biol. 1997;138:877–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kraehling JR, Chidlow JH, Rajagopal C, Sugiyama MG, Fowler JW, Lee MY, Zhang X, Ramirez CM, Park EJ, Tao B, Chen K, Kuruvilla L, Larrivee B, Folta-Stogniew E, Ola R, Rotllan N, Zhou W, Nagle MW, Herz J, Williams KJ, Eichmann A, Lee WL, Fernandez-Hernando C and Sessa WC. Genome-wide RNAi screen reveals ALK1 mediates LDL uptake and transcytosis in endothelial cells. Nature communications. 2016;7:13516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wang W, Yan Z, Hu J, Shen WJ, Azhar S and Kraemer FB. Scavenger receptor class B, type 1 facilitates cellular fatty acid uptake. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865:158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Calvo D, Gomez-Coronado D, Lasuncion MA and Vega MA. CLA-1 is an 85-kD plasma membrane glycoprotein that acts as a high-affinity receptor for both native (HDL, LDL, and VLDL) and modified (OxLDL and AcLDL) lipoproteins. Arterioscler Thromb Vasc Biol. 1997;17:2341–9. [DOI] [PubMed] [Google Scholar]
- 179.Kawasaki Y, Nakagawa A, Nagaosa K, Shiratsuchi A and Nakanishi Y. Phosphatidylserine binding of class B scavenger receptor type I, a phagocytosis receptor of testicular sertoli cells. J Biol Chem. 2002;277:27559–66. [DOI] [PubMed] [Google Scholar]
- 180.Ren Y, Silverstein RL, Allen J and Savill J. CD36 gene transfer confers capacity for phagocytosis of cells undergoing apoptosis. J Exp Med. 1995;181:1857–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Murao K, Terpstra V, Green SR, Kondratenko N, Steinberg D and Quehenberger O. Characterization of CLA-1, a human homologue of rodent scavenger receptor BI, as a receptor for high density lipoprotein and apoptotic thymocytes. J Biol Chem. 1997;272:17551–7. [DOI] [PubMed] [Google Scholar]
- 182.Cocquerel L, Voisset C and Dubuisson J. Hepatitis C virus entry: potential receptors and their biological functions. J Gen Virol. 2006;87:1075–84. [DOI] [PubMed] [Google Scholar]
- 183.Stuart LM, Deng J, Silver JM, Takahashi K, Tseng AA, Hennessy EJ, Ezekowitz RA and Moore KJ. Response to Staphylococcus aureus requires CD36-mediated phagocytosis triggered by the COOH-terminal cytoplasmic domain. J Cell Biol. 2005;170:477–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Vishnyakova TG, Bocharov AV, Baranova IN, Chen Z, Remaley AT, Csako G, Eggerman TL and Patterson AP. Binding and internalization of lipopolysaccharide by Cla-1, a human orthologue of rodent scavenger receptor B1. J Biol Chem. 2003;278:22771–80. [DOI] [PubMed] [Google Scholar]
- 185.Barth H, Schnober EK, Neumann-Haefelin C, Thumann C, Zeisel MB, Diepolder HM, Hu Z, Liang TJ, Blum HE, Thimme R, Lambotin M and Baumert TF. Scavenger receptor class B is required for hepatitis C virus uptake and cross-presentation by human dendritic cells. J Virol. 2008;82:3466–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ohgami N, Miyazaki A, Sakai M, Kuniyasu A, Nakayama H and Horiuchi S. Advanced glycation end products (AGE) inhibit scavenger receptor class B type I-mediated reverse cholesterol transport: a new crossroad of AGE to cholesterol metabolism. J Atheroscler Thromb. 2003;10:1–6. [DOI] [PubMed] [Google Scholar]
- 187.Ohgami N, Nagai R, Ikemoto M, Arai H, Miyazaki A, Hakamata H, Horiuchi S and Nakayama H. CD36, serves as a receptor for advanced glycation endproducts (AGE). J Diabetes Complications. 2002;16:56–9. [DOI] [PubMed] [Google Scholar]
- 188.Neculai D, Schwake M, Ravichandran M, Zunke F, Collins RF, Peters J, Neculai M, Plumb J, Loppnau P, Pizarro JC, Seitova A, Trimble WS, Saftig P, Grinstein S and Dhe-Paganon S. Structure of LIMP-2 provides functional insights with implications for SR-BI and CD36. Nature. 2013;504:172–6. [DOI] [PubMed] [Google Scholar]
- 189.Tsugita M, Morimoto N, Tashiro M, Kinoshita K and Nakayama M. SR-B1 Is a Silica Receptor that Mediates Canonical Inflammasome Activation. Cell Rep. 2017;18:1298–1311. [DOI] [PubMed] [Google Scholar]
- 190.Rodrigueza WV, Thuahnai ST, Temel RE, Lund-Katz S, Phillips MC and Williams DL. Mechanism of scavenger receptor class B type I-mediated selective uptake of cholesteryl esters from high density lipoprotein to adrenal cells. J Biol Chem. 1999;274:20344–50. [DOI] [PubMed] [Google Scholar]
- 191.Reaven E, Cortez Y, Leers-Sucheta S, Nomoto A and Azhar S. Dimerization of the scavenger receptor class B type I: formation, function, and localization in diverse cells and tissues. J Lipid Res. 2004;45:513–28. [DOI] [PubMed] [Google Scholar]
- 192.Sahoo D, Darlington YF, Pop D, Williams DL and Connelly MA. Scavenger receptor class B Type I (SR-BI) assembles into detergent-sensitive dimers and tetramers. Biochim Biophys Acta. 2007;1771:807–17. [DOI] [PubMed] [Google Scholar]
- 193.Thorne RF, Meldrum CJ, Harris SJ, Dorahy DJ, Shafren DR, Berndt MC, Burns GF and Gibson PG. CD36 forms covalently associated dimers and multimers in platelets and transfected COS-7 cells. Biochem Biophys Res Commun. 1997;240:812–8. [DOI] [PubMed] [Google Scholar]
- 194.Daviet L, Malvoisin E, Wild TF and McGregor JL. Thrombospondin induces dimerization of membrane-bound, but not soluble CD36. Thromb Haemost. 1997;78:897–901. [PubMed] [Google Scholar]
- 195.Gaidukov L, Nager AR, Xu S, Penman M and Krieger M. Glycine dimerization motif in the N-terminal transmembrane domain of the high density lipoprotein receptor SR-BI required for normal receptor oligomerization and lipid transport. J Biol Chem. 2011;286:18452–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wei P, Sun FD, Zuo LM, Qu J, Chen P, Xu LD and Luo SZ. Critical residues and motifs for homodimerization of the first transmembrane domain of the plasma membrane glycoprotein CD36. J Biol Chem. 2017;292:8683–8693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Chadwick AC, Jensen DR, Hanson PJ, Lange PT, Proudfoot SC, Peterson FC, Volkman BF and Sahoo D. NMR Structure of the C-Terminal Transmembrane Domain of the HDL Receptor, SR-BI, and a Functionally Relevant Leucine Zipper Motif. Structure. 2017;25:446–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sahoo D, Peng Y, Smith JR, Darlington YF and Connelly MA. Scavenger receptor class B, type I (SR-BI) homo-dimerizes via its C-terminal region: fluorescence resonance energy transfer analysis. Biochim Biophys Acta. 2007;1771:818–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Assanasen C, Mineo C, Seetharam D, Yuhanna IS, Marcel YL, Connelly MA, Williams DL, de la Llera-Moya M, Shaul PW and Silver DL. Cholesterol binding, efflux, and a PDZ-interacting domain of scavenger receptor-BI mediate HDL-initiated signaling. J Clin Invest. 2005;115:969–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zhu W, Saddar S, Seetharam D, Chambliss KL, Longoria C, Silver DL, Yuhanna IS, Shaul PW and Mineo C. The scavenger receptor class B type I adaptor protein PDZK1 maintains endothelial monolayer integrity. Circ Res. 2008;102:480–7. [DOI] [PubMed] [Google Scholar]
- 201.Mineo C, Yuhanna IS, Quon MJ and Shaul PW. High density lipoprotein-induced endothelial nitric-oxide synthase activation is mediated by Akt and MAP kinases. J Biol Chem. 2003;278:9142–9. [DOI] [PubMed] [Google Scholar]
- 202.Saddar S, Carriere V, Lee WR, Tanigaki K, Yuhanna IS, Parathath S, Morel E, Warrier M, Sawyer JK, Gerard RD, Temel RE, Brown JM, Connelly M, Mineo C and Shaul PW. Scavenger receptor class B type I is a plasma membrane cholesterol sensor. Circ Res. 2013;112:140–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kimura T, Tomura H, Sato K, Ito M, Matsuoka I, Im DS, Kuwabara A, Mogi C, Itoh H, Kurose H, Murakami M and Okajima F. Mechanism and role of high density lipoprotein-induced activation of AMP-activated protein kinase in endothelial cells. J Biol Chem. 2010;285:4387–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL and Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med. 2000;6:41–8. [DOI] [PubMed] [Google Scholar]
- 205.Moore KJ, El Khoury J, Medeiros LA, Terada K, Geula C, Luster AD and Freeman MW. A CD36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J Biol Chem. 2002;277:47373–9. [DOI] [PubMed] [Google Scholar]
- 206.Chong M, Yin T, Chen R, Xiang H, Yuan L, Ding Y, Pan CC, Tang Z, Alexander PB, Li QJ and Wang XF. CD36 initiates the secretory phenotype during the establishment of cellular senescence. EMBO Rep. 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Habets DD, Coumans WA, El Hasnaoui M, Zarrinpashneh E, Bertrand L, Viollet B, Kiens B, Jensen TE, Richter EA, Bonen A, Glatz JF and Luiken JJ. Crucial role for LKB1 to AMPKalpha2 axis in the regulation of CD36-mediated long-chain fatty acid uptake into cardiomyocytes. Biochim Biophys Acta. 2009;1791:212–9. [DOI] [PubMed] [Google Scholar]
- 208.Pan J, Fan Z, Wang Z, Dai Q, Xiang Z, Yuan F, Yan M, Zhu Z, Liu B and Li C. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3beta/beta-catenin pathway. J Exp Clin Cancer Res. 2019;38:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Heit B, Kim H, Cosio G, Castano D, Collins R, Lowell CA, Kain KC, Trimble WS and Grinstein S. Multimolecular signaling complexes enable Syk-mediated signaling of CD36 internalization. Dev Cell. 2013;24:372–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Carboni E, Tschudi K, Nam J, Lu X and Ma AW. Particle margination and its implications on intravenous anticancer drug delivery. AAPS PharmSciTech. 2014;15:762–71. [DOI] [PMC free article] [PubMed] [Google Scholar]