

Abstract
Systolic and diastolic myocardial dysfunction has been demonstrated to be associated with an activation of the circulating and local renin–angiotensin–aldosterone system (RAAS), and with a subsequent inappropriately increased production of reactive oxygen species (ROS). While, at low concentrations, ROS modulate important physiological functions through changes in cellular signalling and gene expression, overproduction of ROS may adversely alter cardiac mechanics, leading to further worsening of systolic and diastolic function. In addition, vascular endothelial dysfunction due to uncoupling of the nitric oxide synthase, activation of vascular and phagocytic membrane oxidases or mitochondrial oxidative stress may lead to increased vascular stiffness, further compromising cardiac performance in afterload-dependent hearts. In the present review, we address the potential role of ROS in the pathophysiology of myocardial and vascular dysfunction in heart failure (HF) and their therapeutic targeting. We discuss possible mechanisms underlying the failure of antioxidant vitamins in improving patients’ prognosis, the impact of angiotensin-converting enzyme inhibitors or AT1 receptor blockers on oxidative stress, and the mechanism of the benefit of combination of hydralazine/isosorbide dinitrate. Further, we provide evidence supporting the existence of differences in the pathophysiology of HF with preserved vs. reduced ejection fraction and whether targeting mitochondrial ROS might be a particularly interesting therapeutic option for patients with preserved ejection fraction.
Introduction
Heart failure (HF) is the leading cause of morbidity and mortality in industrialized countries with direct and indirect costs in 2012 across the globe of $108 billion per annum,1 making this condition among the most resource-intensive. Since the incidence of HF increases with age, demographic trends in the world indicate that HF hospitalizations are likely to increase worldwide.
Traditionally, HF has been viewed as a consequence of a reduced ejection fraction (i.e. HF with reduced ejection fraction; HF-rEF) resulting from myocardial infarction (MI), valve disease, arterial hypertension, myocarditis, or other structural or myocardial diseases. More recently, following an improved understanding of cardiac dynamics, it has become clear that ∼50% of patients present with symptoms of HF that are due to diastolic dysfunction despite preserved systolic function (HF with preserved ejection fraction; HF-pEF). Although the prognosis of HF-pEF is apparently less severe than HF-rEF,2 the preserved ejection fraction variety of HF is associated with significant mortality and morbidity early after the first diagnosis (Figure 1).
Figure 1.
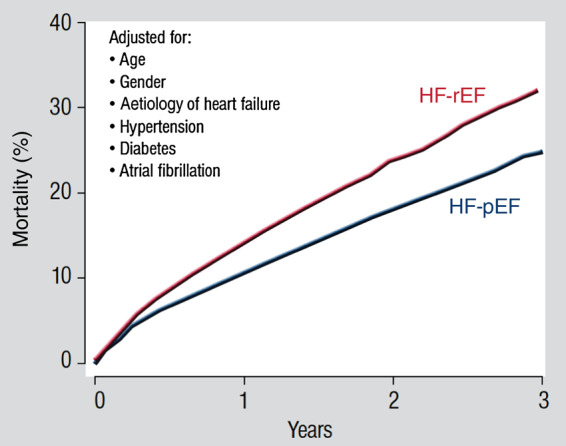
Mortality for patients with heart failure with preserved ejection fraction and heart failure with reduced ejection fraction adjusted for age, gender, aetiology of heart failure, hypertension, diabetes, and atrial fibrillation (with permission from ref.2). Copyright © 2012 Oxford University Press.
The classic clinical syndrome of HF is characterized by fluid retention leading to pulmonary congestion and peripheral oedema (backward failure) and by low cardiac output (forward failure), that combined may cause a severe limitation of exercise capacity. The major pathophysiological cause for HF-rEF is coronary artery disease and myocardial infarction (MI). Following MI, the non-infarcted areas of the heart adapt to the increased wall stress, a process known as cardiac remodelling which involves changes in the structure and function of cardiac myocytes as well as the extracellular matrix in the non-infarcted myocardium. The main mechanisms involved in these adaptive changes involve the activation of the sympathetic nervous system and the renin–angiotensin–aldosterone system (RAAS), which both drive maladaptive ventricular remodelling that produce the HF-rEF phenotype of the failing myocardium.
Notably, activation of the sympathetic nervous system and the RAAS both are associated with increased production of reactive oxygen species (ROS) in cardiovascular tissues. Increased ROS production, in turn, can result in excess oxidative stress, manifest as protein oxidation, lipid peroxidation, and DNA damage, that may contribute to cellular dysfunction. Indeed, a growing body of evidence supports the concept that myocardial systolic and diastolic dysfunction, as well as vascular/endothelial dysfunction, may at least, in part, be a consequence of this increased oxidative stress.
With the present review we wish to introduce the reader first to the complex free radical chemistry, discuss the enzymatic ROS sources that are active in the presence of systolic and diastolic HF and vascular dysfunction, their pathophysiological consequences with respect to the contraction and relaxation of the myocardium and to vascular stiffness and their therapeutic targeting. We will also discuss whether increased oxidative stress in HF-pEF has a different pathophysiology compared with HF-rEF and whether targeting mitochondrial ROS might be a particularly interesting option for patients with HF-pEF.
Free radical chemistry
Reactive oxygen species are oxygen-based chemical species that are able to react rapidly with molecules such as cellular lipids, proteins, and nucleic acids. They comprise a group of highly reactive molecules including the free radicals superoxide (O2·−), hydroxyl radical (·OH), but also non-radical species such as hydrogen peroxide (H2O2) and hypochlorite (OCl−). Superoxide is readily produced by reaction of excess electrons with molecular oxygen, and thus can be considered as a ‘primary’ ROS that with additional acquisition of electrons (reduction), gives rise to the formation of other ROS such as H2O2 and ·OH.
One means whereby superoxide can facilitate subsequent ROS generation is by reacting with Fe3+ to generate Fe2+. This reduced form of iron can then react with H2O2 in the so-called Fenton reaction to produce ·OH (Figure 2). The combined reactions of O2·− and H2O2 to generate ·OH in the presence of iron is known as the Haber–Weiss reaction (see Figure 2). Superoxide can also produce other ROS via its interaction with nitric oxide (·NO). When the two species are formed simultaneously, these two radicals will react with each other at a diffusion-limited rate to form peroxynitrite (ONOO−), a highly reactive intermediate that has important consequences for vascular tone and, as a consequence, ventricular afterload. For example, peroxynitrite produces tyrosine nitration of the prostacyclin synthase that leads to an inhibition of the vasodilator PGI2.3 Similarly, ONOO− can react with and inhibit the activity of the .NO target enzyme, soluble guanylyl cyclase.4 Finally, peroxynitrite oxidizes tetrahydrobiopterin, an important cofactor for the endothelial nitric oxide synthase (eNOS), thereby impairing endothelial ·NO production and even facilitating ROS production from eNOS, a phenomenon known as eNOS uncoupling.5 Collectively, these consequences of ROS production can increase vascular tone resulting in reduced cardiac output and increased myocardial oxygen demand—deleterious effects that can exacerbate the clinical syndrome of HF.
Figure 2.
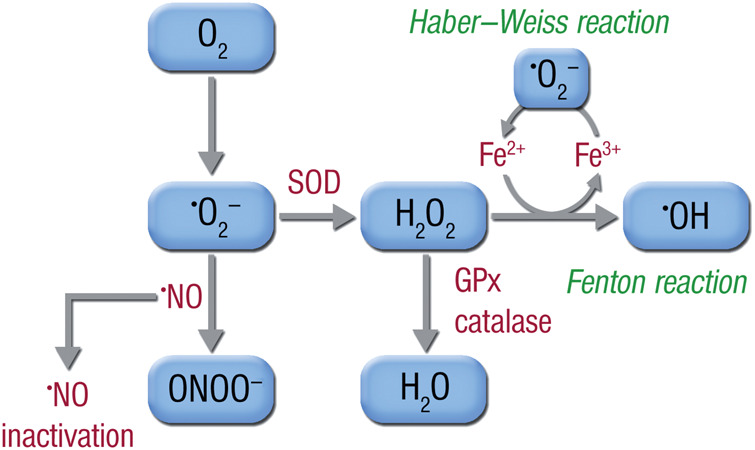
Superoxide (O2·−) is produced from molecular oxygen (O2) and may react with nitric oxide (·NO) to form peroxynitrite (ONOO−), thereby reducing the bioavailability of nitric oxide. Alternatively, superoxide may be dismutated by superoxide dismutases (SODs) to form hydrogen peroxide (H2O2), which is either degraded to water by catalases (CAT) and glutathione peroxidases (GPx), or reacts with free ferrous iron (Fe2+), generated by superoxide from ferric iron (Fe3+), to form hydroxyl radical (·OH) in the Fenton reaction, one of the most reactive species in biological systems. The entire reaction cycle is called the Haber–Weiss reaction or cycle.
Multiple antioxidant defence systems are in place to limit the damage of inappropriate ROS production. These include both non-enzymatic and enzymatic systems. The former consist mainly of antioxidants such as glutathione, vitamins C and E, bilirubin, uric acid, and beta carotene. The enzymatic systems include superoxide dismutase (SOD), catalase, peroxiredoxin (Prx), glutathione peroxidase (GPx), and thioredoxin (Trx). These antioxidant defence systems can become overwhelmed under certain circumstances and disease states characterized by persistently high levels of ROS. Indeed, excessive ROS formation can deplete the low-molecular-weight antioxidants and peroxynitrite, in particular, is known to inactivate important antioxidant enzymes such as the mitochondrial MnSOD (by nitration and dityrosine formation)6,7 and thiol-based antioxidant enzymes (by sulfoxidation). In fact, one can find increased 3-nitrotyrosine formation, a marker of excessive peroxynitrite generation in vivo, in the setting of inflammatory,8 cardiovascular,9 and neurodegenerative diseases.10
In general, there is a delicate balance between ROS formation and the inactivation of ROS via the antioxidant systems. Several ROS, particularly H2O2, are known to serve as local signalling molecules,11,12 thus completely quenching ROS could have important adverse consequences via interruption of normal signalling cascades. It is thought that ROS are best suited for compartmentalized signalling and antioxidant defence systems may prevent their ‘leaking’ to unwanted areas resulting in cellular damage.13 When a certain threshold of ROS formation and impaired ROS degradation is reached, redox signalling may be overcome by oxidative stress and the cell can accumulate oxidatively modified structures (e.g. proteins, DNA, lipids)14 leading to disease states.
Enzymatic reactive oxygen species sources in the myocardium and the vasculature
A growing body of evidence suggests that in the setting of HF, ROS production within the myocardium and the vasculature is substantially increased.15–19 This has been shown for several animal models of HF but also for patients with systolic and diastolic dysfunction and clinical signs of congestive HF. Various sources of ROS, all interacting with each other, were identified in HF (Figure 3).
Figure 3.
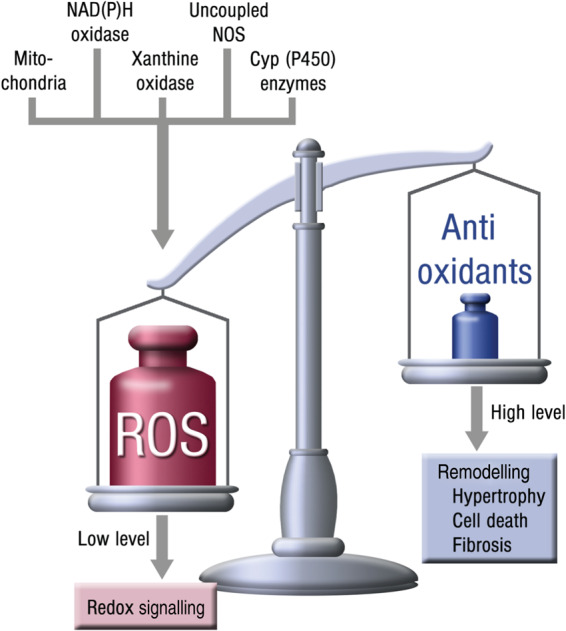
Enzymatic superoxide sources in heart failure.
Mitochondria
In mitochondria, the Krebs cycle generates NADH and FADH2 which donate electrons to the electron transport chain (ETC) (Figure 4). Through sequential redox reactions along the respiratory chain, a proton gradient is formed, which drives ATP production.20
Figure 4.

Interplay between excitation–contraction (EC) coupling, mitochondrial redox state, and oxidative stress in normal and failing hearts. The Krebs cycle produces NADH, which donates electrons (e−) to the electron transport chain (ETC) to promote proton (H+) translocation across the inner mitochondrial membrane (IMM). The proton gradient (ΔpH) is harnessed by the F1Fo ATP synthase to generate ATP. Superoxide anion radicals (·O2−) are produced at the ETC and dismutated to hydrogen peroxide (H2O2) by Mn2+-dependent superoxide dismutase (SOD). Hydrogen peroxide is eliminated by enzymes requiring NADPH (i.e. glutathione peroxidase, GPX; and peroxiredoxin, PRX). Reduced form of NADP is regenerated by enzymes facilitating products of the Krebs cycle (i.e. isocitrate dehydrogenase, IDH; malic enzyme, MDH; and transhydrogenase, THG). Ca2+ is taken up into mitochondria via a Ca2+ uniporter (MCU) and stimulates key enzymes of the Krebs cycle. Ca2+ is exported from mitochondria via a Na+/Ca2+ exchanger (NCLX). During EC coupling, Ca2+ is released from the SR via ryanodine receptors (RyR), and due to the close vicinity of the SR to mitochondria, a mitochondrial Ca2+ microdomain with very high Ca2+ concentrations facilitates Ca2+ uptake into mitochondria. Mitochondrial Ca2+ uptake during increased physiological workload (i.e. during β-adrenergic stimulation) regenerates NADH and NADPH to match ATP supply to demand (through NADH) and regenerate the antioxidative capacity (through NADPH). In heart failure, decreased SR Ca2+ release and increased cytosolic Na+ (in part related to an increased late Na+ current, late INa) impairs mitochondrial Ca2+ uptake, which oxidizes NADH and NADPH and thus, provokes energetic deficit and oxidative stress (all changes in heart failure are marked with red arrows). This oxidizes Ca2+/Calmodulin-dependent protein kinase II (CaMKII) which in turn phosphorylates RyRs, thereby increasing SR Ca2+ leak, and also increases late INa, setting in motion a positive feedback loop of impaired EC coupling with contractile dysfunction and arrhythmias, impaired mitochondrial energetics and oxidative stress. Furthermore, CaMKII phosphorylates histone deacetylase 4 (HDAC4), which promotes cardiac hypertrophy through activation of pro-hypertrophic genes in the nucleus. Potential points of intervention in patients with heart failure are late INa (by ranolazine), improving SR Ca2+ ATPase (SERCA) activity through gene therapy, or by ameliorating mitochondrial ROS emission by Bendavia (SS-31) or MitoQ. GSH, glutathione; TRX, thioredoxin; ΔΨm, mitochondrial membrane potential. NCX, Na+/Ca2+ exchanger; NKA, Na+/K+ ATPase; NHE, Na+/H+ exchanger; OMM, outer mitochondrial membrane.
In the heart, the ATP pool of a cardiac myocyte is consumed in less than a minute, and in situations of exertion, cardiac output (and thus, ATP consumption) can be increased several fold.21,22 Thus, matching ATP supply to demand is critical for normal cardiac myocyte function. One mechanism for ATP supply in the setting of increased demand (e.g. β-adrenergic stimulation) is an accelerated regeneration of oxidized NAD+ to reduced NADH by increasing the amplitude and frequency of cytosolic Ca2+ transients and mitochondrial uptake of Ca2+,23 which in turn activates key dehydrogenases of the Krebs cycle. Thus, typical variations in ATP demand and supply involve considerable modulation of oxidative phosphorylation and mitochondrial activity.
Depending on the respiratory state of the cell, between 0.2 and 2% of electrons can escape the respiratory chain to interact with oxygen thereby non-enzymatically generating O2·− during oxidative phosphorylation. In mitochondria, much of this superoxide is dismutated to H2O2 by Mn-SOD24 and the resulting hydrogen peroxide eliminated by glutathione peroxidase and peroxiredoxin. Notably, these antioxidant processes also require reduced NADPH.25,26 In summary, mitochondrial Ca2+ uptake is not only important to match ATP supply to demand (via NADH),27 but also to regenerate the antioxidant capacity in the mitochondrial matrix (via NADPH).28
Unlike normal cardiac function, studies in HF identified increased O2·− generation from complex I of the respiratory chain, which consequently lead to excessive formation of H2O2 and ·OH29,30 as well as consumption of NAD(P)H. Further work suggested that excess ROS could lead to a feedforward cycle. In this scenario, ROS produced damage to mitochondrial DNA that lead to either down-regulation (and/or mismatched stoichiometry) of respiratory complexes that exhibited reduced respiratory coupling and enhanced electron leakage from the respiratory chain to elevate ROS production.31
Besides these changes in ROS production, an increase in mitochondrial ROS emission might result from the pathological remodelling of cytosolic and mitochondrial ion handling that are hallmarks of HF.25 During a cardiac action potential, Ca2+ enters myocytes triggering further release of Ca2+ from the sarcoplasmic reticulum (SR) and allowing contraction of the myofilaments. In HF, the dysfunction of the SR Ca2+ ATPase (SERCA2a) and an increased leak of Ca2+ from the SR via ryanodine receptors lead to a slowed kinetics of Ca2+ decay in the cytoplasm of myocytes and, therefore, to an impaired relaxation of the cardiac muscle during diastole. To compensate for the slowed uptake of Ca2+ into the SR, the expression and activity of the sarcolemmal Na+–Ca2+ exchanger are up-regulated, resulting however in an increased cytosolic Na+ concentration.32–36
These changes in ionic homeostasis affect excitation–contraction coupling and have adverse effects on mitochondrial Ca2+ handling. Further, they affect mitochondrial energetics and ROS production through a reduced mitochondrial Ca2+ uptake and accelerated Ca2+ extrusion via the mitochondrial Na+/Ca2+ exchanger.23,28,37 The decreased Ca2+ concentration hampers Krebs cycle activation and accounts for oxidation of NADH and NADPH, which may contribute to both an energetic deficit and oxidative stress.28,38 Showing the importance of the maladaptive up-regulation of the Na–Ca exchanger in the pathophysiology of HF, pharmacological inhibition of this enzyme has been shown to restore mitochondrial Ca2+ levels, reducing mitochondrial ROS emission and improving remodelling in an animal model of HF.39 Taken together, the changes in cytoplasmic Ca2+ handling may result in perturbations of mitochondrial Ca2+ and Na+, which in turn limit NADH and NADPH levels needed for antioxidant activity thereby producing a pro-oxidative shift in the redox state of the mitochondrial matrix and an increased mitochondrial ROS emission.25
Since several ion handling proteins are redox-regulated, oxidative stress can further impact excitation–contraction coupling either directly or through the activation of so-called stress kinases, which may contribute to contractile dysfunction and arrhythmias.40 A key mediator in this respect is the Ca2+/calmodulin-dependent protein kinase II (CaMKII), a central regulator of proteins involved in excitation–contraction coupling. The expression and activity of the CaMKII are up-regulated in HF and thought to account for many pathological changes in the failing heart.40 CaMKII is activated by oxidation41 resulting in an increase in late INa as well as an increase in the open probability of ryanodine receptors that can initiate a feedforward mechanism of decreased sarcoplasmic Ca2+ load and release, elevated [Na+]i, mitochondrial ROS emission, and CaMKII activation.42 Similar changes may result from ROS-mediated oxidative activation or inhibition of membrane ion transporters.40 Other than Ca2+ handling, ROS have been shown to have important implications for contractile myofilaments as their Ca2+ sensitivity and elasticity are subject to redox modifications by reactions with titin and other proteins.43,44
Besides affecting excitation–contraction coupling, ROS are also known to contribute to maladaptive structural remodelling of the left ventricle through activation of CaMKII, HDAC4 and various mitogen-activated protein kinase pathways.41,45,46 Finally, ROS are critical activators of the mitochondrial permeability transition pore, inducing apoptosis and/or necrosis.47 Murine models of HF suggest that apoptosis may have an important role in determining the transition to dilated cardiomyopathy.48 Thus, considerable evidence indicates that HF-rEF involves disturbances of cytosolic and mitochondrial ion handling that may account for an increase in mitochondrial ROS levels that has important implications for excitation–contraction coupling, leading to further impairments in myocardial contractility, energetics, and oxidative stress.
Evidence for mitochondrial ROS formation in HF comes from experimental work in animals with pressure overload-induced left- and right-ventricular failure.49,50 In fact, recent experimental studies implicate mitochondrial ROS production as a causal feature of pressure-overload and also angiotensin II-induced systolic or diastolic HF.51–53 In these models, mitochondria-targeted antioxidants14,19,54,55 have been shown to prevent HF and cardiomyocyte dysfunction. Collectively, these studies suggested that mitochondria appear to integrate and even amplify ROS signalling from other non-mitochondrial sources in the cell.19 In the following paragraphs, we will focus on other ROS sources and the interplay between ROS sources in various cell types.
NADPH oxidase
This enzyme was initially described in inflammatory cells like neutrophils and macrophages (phagocytic NADPH oxidase) but is known to also exist as seven distinct isoforms that each have distinct tissue distributions. In the cardiovascular system, specific isoforms have been demonstrated in endothelial, smooth muscle, and adventitial cells (vascular NADPH oxidase)56–59 as well as cardiac myocytes60 and NADPH oxidase activity is increased in failing human hearts.16,61 Thus, vascular and myocardial tissue contain isoforms of the NADPH oxidase, suggesting that they could be involved in the regulation of cardiac function.
Over the last decade, evidence indicates that specific isoforms of the NADPH oxidase family have important roles in both the physiology and pathophysiology of the heart. Myocardial cells exposed to physiological stretch activate the sarcolemmal and t-tubule-localized Nox2 NADPH isoform to produce ROS in a process that requires intact microtubules (known as X-ROS signalling). Localized ROS, in turn, sensitize nearby ryanodine receptors in the SR to produce increased Ca2+ entry in the cytoplasm.62,63 This process is called mechano-chemotransduction and also appears to involve nitric oxide synthase as well as Ca2+/calmodulin-dependent protein kinase II (CaMKII),63 raising the possibility that peroxynitrite and methionine oxidation are also involved.64
There is also evidence that Nox2 could be a target of therapy in HF. In murine models of ischaemic HF, genetic deletion of the Nox2 accessory protein, p47phox, improved infarct area and survival,65 whereas gene therapy with Nox2–siRNA in nanoparticles improved cardiac function.66 Whether the vascular or the phagocytic NADPH oxidase located in inflammatory cells prevail in determining the favourable impact of Nox2 inhibition in ischaemia-induced HF remains unclear. In vascular models of oxidative stress, such as angiotensin II infusion,67,68 however, depletion of inflammatory cells attenuates some features of oxidative stress such as endothelial dysfunction, vascular ROS formation, and also arterial hypertension.68 Thus, it is tempting to speculate that inhibition of the phagocytic NADPH oxidase may also have ameliorative effects on HF, but this remains to be investigated.18
The Nox4 isoform of NADPH oxidase has also been described in the myocardium. In contrast to Nox2, Nox4 is an unusual enzyme isoform as it does not require any regulatory subunits and it constitutively produces H2O2 rather than superoxide. In the cell, it is located in the endoplasmic reticulum and perhaps, also in the mitochondria.69–71 Stimuli that activate Nox4 include ischaemia, hypoxia, and adrenergic stimuli, all present and enhanced in the setting of HF. The implications of Nox4 for HF syndromes are not yet clear, but evidence exists for some beneficial as well as negative effects. For example, mice lacking cardiac Nox4 were shown to either exhibit reduced or aggravated maladaptive remodelling in pressure overload-induced HF72 are sensitized to ischaemic myocardial injury,73 perhaps through reductive stress and down-regulation of stress–response pathways such as HIF-1α.73,74 Since Nox4 is also known to promote angiogenesis, its absence in the myocardium could lead to capillary rarefaction and sensitivity to ischaemic injury.75
Crosstalk between mitochondria and the NADPH oxidase
A crosstalk between mitochondria and NADPH oxidases via ROS signalling appears to be of utmost importance for the cardiovascular system.76,77 The release of ROS from the mitochondria into the cytosol may trigger further ROS-induced ROS production mediated by the activation of diverse ‘redox-switches’ within the cell (Figure 5). For example, oxidation of thiol residues in xanthine dehydrogenase promote conversion to its ROS-generating oxidase form78 and oxidative events promote the uncoupling of NO synthase via multiple mechanisms including redox-sensitive kinases, BH4 depletion, and disruption of the enzyme zinc-sulfur complex (summarized in ref.76). These mechanisms may be initiated by the ‘escape’ of mtROS that may occur in a burst-like fashion as a consequence of increased membrane permeability (summarized in refs.25,77), under de-energized conditions in the failing heart.
Figure 5.
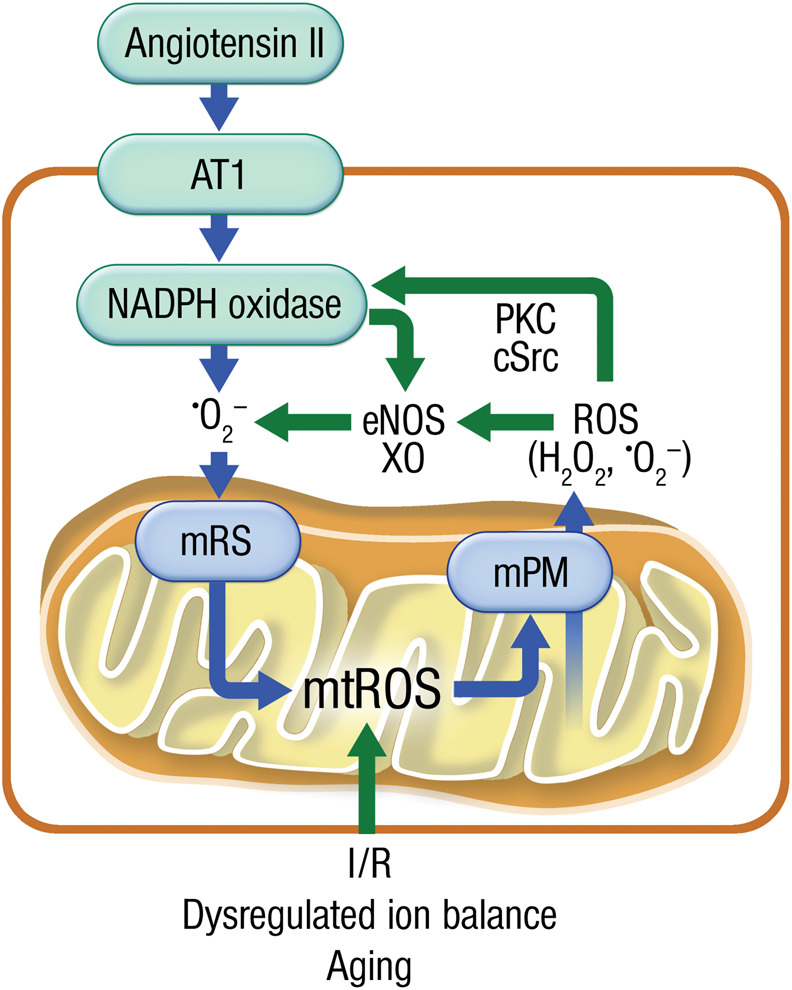
Crosstalk between different cardiovascular ROS sources. Upon agonist-driven activation of NADPH oxidase (e.g. via angiotensin-II-mediated AT1R activation), the enzyme produces superoxide and hydrogen peroxide leading to activation of mitochondrial ROS (mtROS) formation by mitochondrial redox switches (mRS). These mtROS can escape from mitochondria by increased mitochondrial permeability (mPM) involving several pores and channels. In the cytosol these mtROS can activate Nox2 (or Nox1) via redox-sensitive kinases such as protein kinase C (PKC) and tyrosine kinases (cSrc). Likewise Nox- and mitochondria-derived ROS can convert xanthine dehydrogenase to the xanthine oxidase (XO) or lead to eNOS uncoupling further contributing to this vicious circle. Alternatively, this crosstalk starts at the mitochondrial level by excessive mtROS formation during the aging process, a dysregulated ion balance (e.g. in the failing heart) or in response to ischaemia/reperfusion (I/R) damage.
Conversely, we also know that NADPH oxidase-derived ROS can augment ambient mitochondrial ROS levels via stimulation of ROS production (Figure 5) or simply by depletion of mitochondrial antioxidant systems (e.g. NADH/NADPH). Through this self-amplificating mechanism, focal cytosolic ROS production may be extended across the entire cellular mitochondrial network.79–81 This mechanism may be particularly relevant in HF-pEF, since in a mouse model in which chronic angiotensin II infusion (a canonical Nox2 activator) induced diastolic (but not systolic) dysfunction and fibrosis, and this was completely ablated by mitochondrial overexpression of catalase or a mitochondria-targeted peptide (SS-31; see also below).19,52
Uncoupled endothelial nitric oxide synthase
Heart failure is associated with endothelial dysfunction,82 an abnormality that can be corrected with oral or intra-arterial administration of vitamin C,83 suggesting a role for oxidative stress in this phenomenon. Preclinical studies with experimental animal models of HF (both dilated cardiomyopathy and myocardial infarction) reveal both vascular and myocardial ROS production is up-regulated in these settings.84,85 Further, inhibition of eNOS resulted in a decrease in ROS, demonstrating the existence of an uncoupled enzyme state where ·NO production was replaced by superoxide production.84 Similarly, several other enzymes downstream in the NO cascade (e.g. soluble guanylyl cyclase or cGMP-dependent kinase I) were also inhibited. Thus, ·NO bioactivity was impaired and appeared related to activation of the RAAS, as inhibition with either captopril or the AT1 receptor blocker irbesartan improved the phenotype.85 Collectively, these findings emphasize the role of the activated RAAS in the vascular abnormalities encountered in the setting of HF.
Xanthine oxidase and reactive oxygen species production in heart failure
Recent studies have demonstrated that serum uric acid levels >7 mg/dL are an important prognostic marker for all-cause mortality in HF.86 In this context, serum uric acid levels are known to rise with increased purine catabolism resulting from tissue hypoxia, apoptosis, and/or enhanced or up-regulated xanthine oxidoreductase activity (reviewed in ref.87), raising the possibility that xanthine oxidase could contribute to the pathophysiology of HF. In this regard, increased xanthine oxidase activity and expression have been described in failing myocardium and inhibition of xanthine oxidase appeared to improve coronary and peripheral endothelial dysfunction, and myocardial contractility in HF patients.88,89
Despite these promising effects in smaller randomized trials, the results of a larger multicentre trial have been disappointing. Hare et al. randomized 405 class III or IV HF patients to oxypurinol or placebo and found no significant changes in clinical outcomes such as HF morbidity, mortality, and quality of life, with a non-significant trend in patients with serum uric acid levels >9.5 mg/dL.90 Furthermore, in a very recent trial in patients with HF-rEF, allopurinol failed to improve clinical status, exercise capacity, quality of life, or left ventricular ejection fraction at 24 weeks.91 Therefore, the most direct interpretation is that serum uric acid levels might not have a causal role in HF but rather represent a marker of adverse prognosis indicative of oxidative damage within the myocardium and/or the vasculature.
Nitric oxide deficiency and heart failure with preserved ejection fraction
It is known for many years that ·NO and NOS are having a role in cardiac relaxation. The endothelial and also the neuronal NOS are located in the heart and in particular the neuronal NOS has been demonstrated to be an important modulator of cardiac nitroso balance and function. E.g. it has been demonstrated that nNOS modulates cardiac relaxation via effects on phospholamban phosphorylation.92 Interestingly, in the setting of arterial hypertension, Silberman et al.93 demonstrated that decreased myocardial ·NO levels lead to increased cytosolic calcium and diastolic dysfunction. These adverse effects were corrected by supplementation with BH4, a NOS cofactor, but not NH4 suggesting an uncoupling of NOS as the principle mechanism.93
While oxidative stress is common in patients with both HF-rEF and HF-pEF, different cellular and enzymatic sources of ROS production have been suggested. As recently outlined by Paulus and Tschöpe,94 comorbidities induce a proinflammatory state in patients with HF-pEF (through elevated plasma levels of interleukin-6, soluble ST2, tumour necrosis factor-α, and pentraxin 3) that may induce ROS production in endothelial cells. Through paracrine mechanisms, this may then affect protein kinase G signalling in cardiac myocytes that induce sarcomere stiffness and pro-hypertrophic signalling. In contrast, in HF-rEF, most ROS production may occur in cardiac myocytes and induce maladaptive remodelling through the induction of programmed cell death, autophagy, and fibrosis. Such differential ROS signalling pathways may be one explanation why treatments that improved the outcome of patients with HF-rEF failed in patients with HF-pEF and should be subject to further research that enables us to identify better treatments of either syndrome.
Therapeutic strategies
The important contribution of ROS to the pathophysiology of HF may indicate that therapeutic strategies aimed at reducing oxidative stress within the myocardium and the vasculature may beneficially influence the course of the disease. In the next paragraphs we will discuss why so far simple antioxidant strategies, e.g. with vitamins failed to improve prognosis in patients with HF, discuss new approaches to reduce oxidative stress by the use of mitochondria targeting antioxidants and why ACE inhibitors or the combination therapy such as ISDN/hydralazine may be effective in restoring the impaired redox balance.
Antioxidants/vitamins
Despite a predominant role of ROS in the pathophysiology of HF, large clinical trials have failed to show any benefit of antioxidants such as vitamin E and vitamin C in any cardiovascular disease (reviewed in ref.95). Many explanations have been offered as to why antioxidants have proved ineffective in these studies, a prominent one being that agents such as vitamin E and vitamin C are not targeted to sites of ROS generation that are most important in pathological conditions, such as mitochondria.
In addition, specific ROS often react much more quickly than can be quenched by antioxidants. For example, superoxide combines with .NO to form peroxynitrite in a diffusion-limited reaction (k = 1.9 × 1010 mol L−1 s−1) that is almost 10× faster than its degradation by SOD, suggesting that it is very difficult to prevent peroxynitrite formation. With regard to common antioxidants, superoxide and vitamin C react much slower than .NO, on the order of 105 mol L−1 s−1, indicating that exceptionally high concentrations of this vitamin would be needed in specific tissue or even cell organelle to be effective. Since vitamin E is even less reactive (∼103 mol L−1 s−1) the case is even stronger for its ineffectiveness. Further, the compartimentalization of ROS production makes non-targeted approaches much less effective. Thus, it is not likely that vitamins can scavenge superoxide in order to prevent its interaction with .NO and the subsequent formation of the highly reactive and toxic intermediate peroxynitrite, even when given in high concentrations.
Indeed, although vitamin E effectively scavenges lipid peroxyl radicals, it has been demonstrated to have a rather limited activity against other oxidants such as superoxide, peroxynitrite, and hypochlorous acid that have been implicated to play a major role in the pathophysiology of atherosclerosis.
There is also experimental evidence from animals and patients that suggests that lipid peroxidation occurs even in the presence of vitamin E and higher doses of the vitamin could be harmful (reviewed in ref.95). For example, further attempts to increase the effectiveness with higher doses of vitamin E resulted in worsening of atherosclerosis and vascular function,96 which may be related to the formation of the pro-oxidative vitamin E radical97 (Figure 6A and D). Enrichment of the vitamin E radical in the myocardium may exert negative inotropic effects, which may explain why long-term treatment with vitamin E does not prevent but rather induce HF and acute left heart decompensations as shown for the HOPE98 and HOPE TOO trials99 (Figure 6B and C).
Figure 6.

(A) Effects of Vitamin E high and low concentrations on endothelial function of cholesterol fed animals (with permission from ref.93). Copyright © 1994 American Society for Clinical Investigation. (B) Effects of Vitamin E treatment on the incidence of heart failure (with permission from ref.113). Copyright © 2005 American Medical Association. (C) Effects of Vitamin E treatment on left heart decompensations (with permission from ref.113). Copyright © 2005 American Medical Association. (D) Potential pro-oxidative effects of Vitamin E (with permission from ref.94). Copyright © 1999 Elsevier Science Ltd. All rights reserved.
Another important aspect why antioxidants may fail was presented by Ristow et al.100 The authors demonstrated that antioxidant supplements such as vitamin C and E prevented the molecular regulators of insulin sensitivity and endogenous antioxidant defence induced by physical exercise,100 and exercise was shown to improve symptoms and quality of life in patients with HF-rEF and also HF-pEF.101–103
Angiotensin-converting enzyme inhibitors or AT1-receptor blocker: are they the better ‘antioxidants’?
Angiotensin-converting enzyme inhibitors inhibit kininase II, which leads to increased formation of bradykinin, which in turn enhances stimulation of the endothelial bradykinin receptor B2 prompting the release of ·NO,104 endothelium-derived hyperpolarizing factor and prostacyclin. These actions explain in part the combined vasodilator, antithrombotic, and anti-proliferative effects of ACE inhibitors. By inhibiting angiotensin II formation, they also beneficially influence inflammatory processes in the vessel wall,105,106 prevent smooth muscle proliferation, and prevent the activation of the vascular and phagocytic NADPH oxidase, a dominant vascular source of superoxide.84 By limiting the production of superoxide at its enzymatic source, many issues concerning the free radical scavenging efficacy of antioxidants (i.e. compartimentalization, pro-oxidant effects, etc.) are not a consideration.
Targeting mitochondrial reactive oxygen species
Recent studies indicate that targeted inhibition of ROS production in certain microdomains holds promise as a therapeutic strategy over the untargeted approach with classical antioxidants (Figure 4). In this regard, targeting mitochondria with their central role in cellular ROS formation has been a topic of great interest, particularly in cardiac myocytes. The Szeto-Schiller peptide (SS-31) is a tetrapeptide that accumulates several thousand-fold in mitochondria and protects from apoptotic cell death.107 While initial studies suggested that SS-31 was an antioxidant,107 more recent studies revealed that it does not directly scavenge ROS, but rather prevents mitochondrial ROS formation via interaction with cardiolipin, an important lipid of the inner mitochondrial membrane critical for proper assembly of the respiratory complexes of the ETC.108 Mitochondrial ROS production oxidizes cardiolipin and makes it dysfunctional, allowing dissociation of respiratory chain complexes which may give rise to further electron leakage promoting O2·− formation. SS-31 directly interacts with cardiolipin and prevents its oxidation, and this presumably interrupts a vicious cycle of cardiolipin oxidation, ROS production and deterioration of ATP production.25,109
The SS-31 peptide, but not the non-targeted N-acetyl cysteine, prevented the development of diastolic dysfunction in response to angiotensin II (a canonical activator of NADPH oxidase). These findings bolster the concept of NADPH oxidase/mitochondrial crosstalk of ROS signalling.51,52,76 and provide a rationale for microdomain-targeted therapies. Furthermore, SS-31 prevented HF development and maladaptive ventricular remodelling in a systolic HF model induced by pressure overload.53 Also, in a large animal model of ischaemic cardiomyopathy, both short- and long-term treatment with SS-31 improved LV function, which was associated with improvement in mitochondrial respiration and ATP production. Under the name Bendavia, the drug is currently being tested clinically in a comprehensive programme involving patients with HF,108 while a phase II study on patients with myocardial infarction showed safety and some promising signs towards improvement of cardiac and renal function (STEMI-EMBRACE trial, presented at ACC, Chicago, March 2015 and110 for the study design).
Another class of compounds that appears attractive for targeting mitochondrial ROS are antioxidants linked to the lipophilic cation TPP+111 that accumulate in the mitochondrial matrix. Linkage of coenzyme Q, a respiratory chain component for ROS detoxification with TPP+ (mitoQ), provided protection in various animal models of cardiovascular and neurodegenerative diseases.25,111 For example, MitoQ improved cardiomyocyte function in a model of acute cardiac volume overload.54 Clinically, MitoQ was safe in patients with neurodegenerative diseases but has not been tested in cardiovascular conditions as yet.111 Interestingly, a recent clinical trial on 420 patients with HF with unconjugated coenzyme Q10 showed a significant improvement of morbidity and mortality,112 but its implications for future HF guidelines are unclear due to the rather small scale of the study.113 The mitochondria-targeted antioxidant mitoTEMPO (as well as mitochondria-specific overexpression of human catalase) have been highly effective in preventing pressure-overload-induced HF in animals.55 Considering the tight interaction between excitation–contraction coupling and mitochondrial energetics, attempts to improve cytosolic and/or mitochondrial ion handling may provide additional benefit by preventing mitochondrial ROS production. Two examples of this could be gene therapy to up-regulate SERCA (CUPID trial) or inhibiting late INa with ranolazine.114 In this context, it is worth noting that both treatment with β-blockers and ACE inhibitors was associated with improvements of excitation–contraction coupling on the cellular level, pinpointing how these drugs may improve energetics and prevent oxidative stress as well (see preceding paragraph).
Cotherapy of hydralazine and nitrates improves the redox balance between nitric oxide and superoxide in patients with chronic heart failure
In patients with coronary artery disease (CAD), treatment with nitroglycerin (GTN) and isosorbide dinitrate (ISDN) cause rapid haemodynamic and anti-anginal tolerance,115 an effect that is also seen in patients with chronic congestive HF where ISDN causes rapid tolerance development with respect to the left ventricular filling pressure.116
An interesting aspect is the interaction between the organic nitrate ISDN and the arteriolar dilator hydralazine in patients with severe HF. Several clinical studies have clearly established that the combination of ISDN with hydralazine is able to improve prognosis117 compared with prazosin in the V-Heft-I and exercise capacity compared with enalapril in V-HeFT II trial.118 Also, the African-American Heart Failure Trial (A-HeFT), a double-blind, randomized, trial demonstrated that the combination of ISDN and hydralazine markedly improved the composite endpoint of the trial, which included death from any cause, first hospitalization for HF and quality-of-life measures.119 One potential mechanism for these observations is that this particular combination of ISDN and hydralazine is rather devoid of tolerance and its associated endothelial dysfunction. Indeed, more recent experimental studies demonstrated that hydralazine is a powerful inhibitor of GTN-induced formation of reactive oxygen species such as superoxide or peroxynitrite (ONOO−) in vitro in the vasculature120 and in vivo,121 thus correcting the disrupted redox balance between superoxide and nitric oxide in the cardiovascular system of patients with chronic congestive HF.122 Dulce et al.123 demonstrated that by reducing sarcoplasmic Ca2+-leak, hydralazine improves calcium cycling and contractility of the myocardium as well as restoring cardiac excitation–contraction coupling. Thus, these drugs in combination exert direct myocardial effects that provide a mechanistic basis for the favourable functional and structural responses in the treatment of congestive HF.
Phosphodiesterase 5 inhibition
According to a recent meta-analysis, the phosphodiesterase 5 (PDE5) inhibitor sildenafil improved haemodynamic parameters in HF-rEF patients when compared with placebo.124 In contrast, administration of sildenafil for 24 weeks did not significantly improve exercise capacity or clinical status compared with placebo.125
Exercise training
Exercise training improves quality of life, symptoms, and possibly the outcome in patients with HF-pEF and HF-rEF. These benefits may be mediated by reducing central and systemic neuroendocrine activation, in particular the RAAS and sympathetic nervous systems. On a molecular level, exercise training may impact not only the heart directly, but also vessels, the kidney, skeletal muscles, and other organs, systemically interconnected through central sympatho-excitatory processes and mediated by reduced β-adrenergic- and angiotensin II-signalling that reduces oxidative stress and increases the bioavailability of NO from eNOS.126
Conclusions
While a large body of evidence supports an association between oxidative stress and vascular dysfunction, the relationship with myocardial contractile dysfunction has only been investigated recently. Reactive oxygen species are important mediators of physiological functions, but in higher concentrations they may modify cardiac metabolism and mechanics, triggering a feedforward mechanism that leads to further worsening of systolic and diastolic function.
Scavenging of ROS with antioxidants has however proved to have no effect in modifying patients’ prognosis. While this evidence does not disprove the role of ROS in cardiovascular pathophysiology, it emphasizes the need of studies addressing ROS-dependent pathophysiology with highly specific interventions targeted at the cellular microcompartiments in which these ROS are produced.
Funding
This work was supported by the Stiftung Mainzer Herz and the Robert Müller Stiftung. Further support was provided by the Center for Translational Vascular Biology (CTVB), the German Center vor Cardiovascular Research (DZHK), and the Center for Thrombosis and Hemostasis (CTH). T.M. is PI oft he DZHK. C.M. is supported by the Deutsche Forschungsgemeinschaft (DFG; Heisenberg Programm and SFB 894), by the Deutsche Herzstiftung (Margret Elisabeth Strauß-Projektförderung), and the Corona Stiftung
Conflict of interest: C.M. received speaker honoraria from Berlin Chemie (Ranolazine) and Stealth Biotherapeutics (Bendavia), and is a scientific advisor to Stealth Biotherapeutics.
References
- 1. Cook C Cole G Asaria P Jabbour R Francis DP. The annual global economic burden of heart failure. Int J Cardiol 2014;171:368–376. [DOI] [PubMed] [Google Scholar]
- 2. Meta-analysis Global Group in Chronic Heart Failure. The survival of patients with heart failure with preserved or reduced left ventricular ejection fraction: an individual patient data meta-analysis. Eur Heart J 2012;33:1750–1757. [DOI] [PubMed] [Google Scholar]
- 3. Zou MH Bachschmid M. Hypoxia-reoxygenation triggers coronary vasospasm in isolated bovine coronary arteries via tyrosine nitration of prostacyclin synthase. J Exp Med 1999;190:135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weber M Lauer N Mulsch A Kojda G. The effect of peroxynitrite on the catalytic activity of soluble guanylyl cyclase. Free Radic Biol Med 2001;31:1360–1367. [DOI] [PubMed] [Google Scholar]
- 5. Forstermann U Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 2006;113:1708–1714. [DOI] [PubMed] [Google Scholar]
- 6. MacMillan-Crow LA Crow JP Kerby JD Beckman JS Thompson JA. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc Natl Acad Sci USA 1996;93:11853–11858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. MacMillan-Crow LA Crow JP Thompson JA. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry 1998;37:1613–1622. [DOI] [PubMed] [Google Scholar]
- 8. Kooy NW Lewis SJ Royall JA Ye YZ Kelly DR Beckman JS. Extensive tyrosine nitration in human myocardial inflammation: evidence for the presence of peroxynitrite. Crit Care Med 1997;25:812–819. [DOI] [PubMed] [Google Scholar]
- 9. Turko IV Murad F. Protein nitration in cardiovascular diseases. Pharmacol Rev 2002;54:619–634. [DOI] [PubMed] [Google Scholar]
- 10. Ischiropoulos H Beckman JS. Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J Clin Invest 2003;111:163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rhee SG. Redox signaling: hydrogen peroxide as intracellular messenger. Exp Mol Med 1999;31:53–59. [DOI] [PubMed] [Google Scholar]
- 12. Santos CX Anilkumar N Zhang M Brewer AC Shah AM. Redox signaling in cardiac myocytes. Free Radic Biol Med 2011;50:777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Al Ghouleh I Khoo NK Knaus UG Griendling KK Touyz RM Thannickal VJ Barchowsky A Nauseef WM Kelley EE Bauer PM Darley-Usmar V Shiva S Cifuentes-Pagano E Freeman BA Gladwin MT Pagano PJ. Oxidases and peroxidases in cardiovascular and lung disease: new concepts in reactive oxygen species signaling. Free Radic Biol Med 2011;51:1271–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen AF Chen DD Daiber A Faraci FM Li H Rembold CM Laher I. Free radical biology of the cardiovascular system. Clin Sci (Lond) 2012;123:73–91. [DOI] [PubMed] [Google Scholar]
- 15. Landmesser U Spiekermann S Dikalov S Tatge H Wilke R Kohler C Harrison DG Hornig B Drexler H. Vascular oxidative stress and endothelial dysfunction in patients with chronic heart failure: role of xanthine-oxidase and extracellular superoxide dismutase. Circulation 2002;106:3073–3078. [DOI] [PubMed] [Google Scholar]
- 16. Heymes C Bendall JK Ratajczak P Cave AC Samuel JL Hasenfuss G Shah AM. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol 2003;41:2164–2171. [DOI] [PubMed] [Google Scholar]
- 17. Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest 2005;115:500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ijsselmuiden AJ Musters RJ de Ruiter G van Heerebeek L Alderse-Baas F van Schilfgaarde M Leyte A Tangelder GJ Laarman GJ Paulus WJ. Circulating white blood cells and platelets amplify oxidative stress in heart failure. Nat Clin Pract Cardiovasc Med 2008;5:811–820. [DOI] [PubMed] [Google Scholar]
- 19. Maack C Bohm M. Targeting mitochondrial oxidative stress in heart failure throttling the afterburner. J Am Coll Cardiol 2011;58:83–86. [DOI] [PubMed] [Google Scholar]
- 20. Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961;191:144–148. [DOI] [PubMed] [Google Scholar]
- 21. Mootha VK Arai AE Balaban RS. Maximum oxidative phosphorylation capacity of the mammalian heart. Am J Physiol 1997;272:H769–H775. [DOI] [PubMed] [Google Scholar]
- 22. Balaban RS. Cardiac energy metabolism homeostasis: role of cytosolic calcium. J Mol Cell Cardiol 2002;34:1259–1271. [DOI] [PubMed] [Google Scholar]
- 23. Kohlhaas M Maack C. Calcium release microdomains and mitochondria. Cardiovasc Res 2013;98:259–268. [DOI] [PubMed] [Google Scholar]
- 24. Balaban RS Nemoto S Finkel T. Mitochondria, oxidants, and aging. Cell 2005;120:483–495. [DOI] [PubMed] [Google Scholar]
- 25. Nickel A Kohlhaas M Maack C. Mitochondrial reactive oxygen species production and elimination. J Mol Cell Cardiol 2014;73C:26–33. [DOI] [PubMed] [Google Scholar]
- 26. Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal 2008;10:179–206. [DOI] [PubMed] [Google Scholar]
- 27. Brandes R Bers DM. Intracellular Ca2+ increases the mitochondrial NADH concentration during elevated work in intact cardiac muscle. Circ Res 1997;80:82–87. [DOI] [PubMed] [Google Scholar]
- 28. Kohlhaas M Liu T Knopp A Zeller T Ong MF Böhm M O'Rourke B Maack C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 2010;121:1606–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ide T Tsutsui H Kinugawa S Utsumi H Kang D Hattori N Uchida K Arimura K Egashira K Takeshita A. Mitochondrial electron transport complex i is a potential source of oxygen free radicals in the failing myocardium. Circ Res 1999;85:357–363. [DOI] [PubMed] [Google Scholar]
- 30. Ide T Tsutsui H Kinugawa S Suematsu N Hayashidani S Ichikawa K Utsumi H Machida Y Egashira K Takeshita A. Direct evidence for increased hydroxyl radicals originating from superoxide in the failing myocardium. Circ Res 2000;86:152–157. [DOI] [PubMed] [Google Scholar]
- 31. Ide T Tsutsui H Hayashidani S Kang D Suematsu N Nakamura K Utsumi H Hamasaki N Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res 2001;88:529–535. [DOI] [PubMed] [Google Scholar]
- 32. Hobai IA O'Rourke B. Enhanced Ca(2+)-activated Na(+)-Ca(2+) exchange activity in canine pacing-induced heart failure. Circ Res 2000;87:690–698. [DOI] [PubMed] [Google Scholar]
- 33. Despa S Bers DM. Na(+) transport in the normal and failing heart—remember the balance. J Mol Cell Cardiol 2013;61:2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Armoundas AA Hobai IA Tomaselli GF Winslow RL O'Rourke B. Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ Res 2003;93:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Weber CR Piacentino V III Houser SR Bers DM. Dynamic regulation of sodium/calcium exchange function in human heart failure. Circulation 2003;108:2224–2229. [DOI] [PubMed] [Google Scholar]
- 36. Weisser-Thomas J Piacentino V III Gaughan JP Margulies K Houser SR. Calcium entry via Na/Ca exchange during the action potential directly contributes to contraction of failing human ventricular myocytes. Cardiovasc Res 2003;57:974–985. [DOI] [PubMed] [Google Scholar]
- 37. Maack C Cortassa S Aon MA Ganesan AN Liu T O'Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res 2006;99:172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu T O'Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res 2008;103:279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu T Takimoto E Dimaano VL DeMazumder D Kettlewell S Smith G Sidor A Abraham TP O'Rourke B. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a guinea pig model of heart failure. Circ Res 2014;115:44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wagner S Rokita AG Anderson ME Maier LS. Redox regulation of sodium and calcium handling. Antioxid Redox Signal 2013;18:1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Erickson JR Joiner ML Guan X Kutschke W Yang J Oddis CV Bartlett RK Lowe JS O'Donnell SE Aykin-Burns N Zimmerman MC Zimmerman K Ham AJ Weiss RM Spitz DR Shea MA Colbran RJ Mohler PJ Anderson ME. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008;133:462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bay J Kohlhaas M Maack C. Intracellular Na(+) and cardiac metabolism. J Mol Cell Cardiol 2013;61:20–27. [DOI] [PubMed] [Google Scholar]
- 43. Alegre-Cebollada J Kosuri P Giganti D Eckels E Rivas-Pardo JA Hamdani N Warren CM Solaro RJ Linke WA Fernandez JM. S-glutathionylation of cryptic cysteines enhances titin elasticity by blocking protein folding. Cell 2014;156:1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gao WD Liu Y Marban E. Selective effects of oxygen free radicals on excitation-contraction coupling in ventricular muscle. Implications for the mechanism of stunned myocardium. Circulation 1996;94:2597–2604. [DOI] [PubMed] [Google Scholar]
- 45. Ago T Liu T Zhai P Chen W Li H Molkentin JD Vatner SF Sadoshima J. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell 2008;133:978–993. [DOI] [PubMed] [Google Scholar]
- 46. Burgoyne JR Mongue-Din H Eaton P Shah AM. Redox signaling in cardiac physiology and pathology. Circ Res 2012;111:1091–1106. [DOI] [PubMed] [Google Scholar]
- 47. Halestrap A. Biochemistry: a pore way to die. Nature 2005;434:578–579. [DOI] [PubMed] [Google Scholar]
- 48. Wencker D Chandra M Nguyen K Miao W Garantziotis S Factor SM Shirani J Armstrong RC Kitsis RN. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest 2003;111:1497–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Redout EM Wagner MJ Zuidwijk MJ Boer C Musters RJ van Hardeveld C Paulus WJ Simonides WS. Right-ventricular failure is associated with increased mitochondrial complex II activity and production of reactive oxygen species. Cardiovasc Res 2007;75:770–781. [DOI] [PubMed] [Google Scholar]
- 50. Kaludercic N Takimoto E Nagayama T Feng N Lai EW Bedja D Chen K Gabrielson KL Blakely RD Shih JC Pacak K Kass DA Di Lisa F Paolocci N. Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ Res 2010;106:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dai DF Chen T Szeto H Nieves-Cintron M Kutyavin V Santana LF Rabinovitch PS. Mitochondrial targeted antioxidant peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol 2011;58:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dai DF Johnson SC Villarin JJ Chin MT Nieves-Cintron M Chen T Marcinek DJ Dorn GW II Kang YJ Prolla TA Santana LF Rabinovitch PS. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res 2011;108:837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dai DF Hsieh EJ Liu Y Chen T Beyer RP Chin MT MacCoss MJ Rabinovitch PS. Mitochondrial proteome remodelling in pressure overload-induced heart failure: the role of mitochondrial oxidative stress. Cardiovasc Res 2012;93:79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gladden JD Zelickson BR Wei CC Ulasova E Zheng J Ahmed MI Chen Y Bamman M Ballinger S Darley-Usmar V Dell'Italia LJ. Novel insights into interactions between mitochondria and xanthine oxidase in acute cardiac volume overload. Free Radic Biol Med 2011;51:1975–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hoshino A Okawa Y Ariyoshi M Kaimoto S Uchihashi M Fukai K Iwai-Kanai E Matoba S. Oxidative post-translational modifications develop LONP1 dysfunction in pressure overload heart failure. Circ Heart Fail 2014;7:500–509. [DOI] [PubMed] [Google Scholar]
- 56. Griendling KK Sorescu D Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res 2000;86:494–501. [DOI] [PubMed] [Google Scholar]
- 57. Lassegue B Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol 2010;30:653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Brandes RP Weissmann N Schroder K. Nox family NADPH oxidases: molecular mechanisms of activation. Free Radic Biol Med 2014;76C:208–226. [DOI] [PubMed] [Google Scholar]
- 59. Lassegue B San Martin A Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res 2012;110:1364–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li JM Gall NP Grieve DJ Chen M Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 2002;40:477–484. [DOI] [PubMed] [Google Scholar]
- 61. Maack C Kartes T Kilter H Schafers HJ Nickenig G Bohm M Laufs U. Oxygen free radical release in human failing myocardium is associated with increased activity of rac1-GTPase and represents a target for statin treatment. Circulation 2003;108:1567–1574. [DOI] [PubMed] [Google Scholar]
- 62. Prosser BL Ward CW Lederer WJ. X-ROS signaling: Rapid mechano-chemo transduction in heart. Science 2011;333:1440–1445. [DOI] [PubMed] [Google Scholar]
- 63. Jian Z Han H Zhang T Puglisi J Izu LT Shaw JA Onofiok E Erickson JR Chen YJ Horvath B Shimkunas R Xiao W Li Y Pan T Chan J Banyasz T Tardiff JC Chiamvimonvat N Bers DM Lam KS Chen-Izu Y. Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling. Sci Signal 2014;7:ra27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Luczak ED Anderson ME. Camkii oxidative activation and the pathogenesis of cardiac disease. J Mol Cell Cardiol 2014;73:112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Doerries C Grote K Hilfiker-Kleiner D Luchtefeld M Schaefer A Holland SM Sorrentino S Manes C Schieffer B Drexler H Landmesser U. Critical role of the NAD(P)H oxidase subunit p47phox for left ventricular remodeling/dysfunction and survival after myocardial infarction. Circ Res 2007;100:894–903. [DOI] [PubMed] [Google Scholar]
- 66. Somasuntharam I Boopathy AV Khan RS Martinez MD Brown ME Murthy N Davis ME. Delivery of Nox2-NADPH oxidase siRNA with polyketal nanoparticles for improving cardiac function following myocardial infarction. Biomaterials 2013;34:7790–7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Guzik TJ Hoch NE Brown KA McCann LA Rahman A Dikalov S Goronzy J Weyand C Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 2007;204:2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wenzel P Knorr M Kossmann S Stratmann J Hausding M Schuhmacher S Karbach SH Schwenk M Yogev N Schulz E Oelze M Grabbe S Jonuleit H Becker C Daiber A Waisman A Munzel T. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation 2011;124:1370–1381. [DOI] [PubMed] [Google Scholar]
- 69. Maejima Y Kuroda J Matsushima S Ago T Sadoshima J. Regulation of myocardial growth and death by NADPH oxidase. J Mol Cell Cardiol 2011;50:408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lee CF Qiao M Schroder K Zhao Q Asmis R. Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circ Res 2010;106:1489–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Block K Gorin Y Abboud HE. Subcellular localization of nox4 and regulation in diabetes. Proc Natl Acad Sci USA 2009;106:14385–14390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kuroda J Ago T Matsushima S Zhai P Schneider MD Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A 2010;107:15565–15570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Matsushima S Kuroda J Ago T Zhai P Ikeda Y Oka S Fong GH Tian R Sadoshima J. Broad suppression of NADPH oxidase activity exacerbates ischemia/reperfusion injury through inadvertent downregulation of hypoxia-inducible factor-1α and upregulation of peroxisome proliferator-activated receptor-α. Circ Res 2013;112:1135–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yu Q Lee CF Wang W Karamanlidis G Kuroda J Matsushima S Sadoshima J Tian R. Elimination of NADPH oxidase activity promotes reductive stress and sensitizes the heart to ischemic injury. J Am Heart Assoc 2014;3:e000555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Craige SM Chen K Pei Y Li C Huang X Chen C Shibata R Sato K Walsh K Keaney JF Jr. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation 2011;124:731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kroller-Schon S Steven S Kossmann S Scholz A Daub S Oelze M Xia N Hausding M Mikhed Y Zinssius E Mader M Stamm P Treiber N Scharffetter-Kochanek K Li H Schulz E Wenzel P Munzel T Daiber A. Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid Redox Signal 2014;20:247–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Daiber A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim Biophys Acta 2010;1797:897–906. [DOI] [PubMed] [Google Scholar]
- 78. Nishino T. The conversion from the dehydrogenase type to the oxidase type of rat liver xanthine dehydrogenase by modification of cysteine residues with fluorodinitrobenzene. J Biol Chem 1997;272:29859–29864. [DOI] [PubMed] [Google Scholar]
- 79. Zorov DB Juhaszova M Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta 2006;1757:509–517. [DOI] [PubMed] [Google Scholar]
- 80. Zorov DB Filburn CR Klotz LO Zweier JL Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 2000;192:1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Aon MA Cortassa S Marban E O'Rourke B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem 2003;278:44735–44744. [DOI] [PubMed] [Google Scholar]
- 82. Drexler H Hayoz D Munzel T Hornig B Just H Brunner HR Zelis R. Endothelial function in chronic congestive heart failure. Am J Cardiol 1992;69:1596–1601. [DOI] [PubMed] [Google Scholar]
- 83. Hornig B Arakawa N Kohler C Drexler H. Vitamin C improves endothelial function of conduit arteries in patients with chronic heart failure. Circulation 1998;97:363–368. [DOI] [PubMed] [Google Scholar]
- 84. Mollnau H Oelze M August M Wendt M Daiber A Schulz E Baldus S Kleschyov AL Materne A Wenzel P Hink U Nickenig G Fleming I Munzel T. Mechanisms of increased vascular superoxide production in an experimental model of idiopathic dilated cardiomyopathy. Arterioscler Thromb Vasc Biol 2005;25:2554–2559. [DOI] [PubMed] [Google Scholar]
- 85. Schafer A Fraccarollo D Tas P Schmidt I Ertl G Bauersachs J. Endothelial dysfunction in congestive heart failure: ACE inhibition vs. angiotensin II antagonism. Eur J Heart Fail 2004;6:151–159. [DOI] [PubMed] [Google Scholar]
- 86. Anker SD Doehner W Rauchhaus M Sharma R Francis D Knosalla C Davos CH Cicoira M Shamim W Kemp M Segal R Osterziel KJ Leyva F Hetzer R Ponikowski P Coats AJ. Uric acid and survival in chronic heart failure: validation and application in metabolic, functional, and hemodynamic staging. Circulation 2003;107:1991–1997. [DOI] [PubMed] [Google Scholar]
- 87. Lee BE Toledo AH Anaya-Prado R Roach RR Toledo-Pereyra LH. Allopurinol, xanthine oxidase, and cardiac ischemia. J Investig Med 2009;57:902–909. [DOI] [PubMed] [Google Scholar]
- 88. Baldus S Mullerleile K Chumley P Steven D Rudolph V Lund GK Staude HJ Stork A Koster R Kahler J Weiss C Munzel T Meinertz T Freeman BA Heitzer T. Inhibition of xanthine oxidase improves myocardial contractility in patients with ischemic cardiomyopathy. Free Radic Biol Med 2006;41:1282–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Baldus S Koster R Chumley P Heitzer T Rudolph V Ostad MA Warnholtz A Staude HJ Thuneke F Koss K Berger J Meinertz T Freeman BA Munzel T. Oxypurinol improves coronary and peripheral endothelial function in patients with coronary artery disease. Free Radic Biol Med 2005;39:1184–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hare JM Mangal B Brown J Fisher C Jr Freudenberger R Colucci WS Mann DL Liu P Givertz MM Schwarz RP Investigators O-C. Impact of oxypurinol in patients with symptomatic heart failure. Results of the OPT-CHF study. J Am Coll Cardiol 2008;51:2301–2309. [DOI] [PubMed] [Google Scholar]
- 91. Givertz MM Anstrom KJ Redfield MM Deswal A Haddad H Butler J Tang WH Dunlap ME LeWinter MM Mann DL Felker GM O'Connor CM Goldsmith SR Ofili EO Saltzberg MT Margulies KB Cappola TP Konstam MA Semigran MJ McNulty SE Lee KL Shah MR Hernandez AF, NHLBI Heart Failure Clinical Research Network. Effects of xanthine oxidase inhibition in hyperuricemic heart failure patients: the xanthine oxidase inhibition for hyperuricemic heart failure patients (EXACT-HF) study. Circulation 2015;131:1763–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhang YH Zhang MH Sears CE Emanuel K Redwood C El-Armouche A Kranias EG Casadei B. Reduced phospholamban phosphorylation is associated with impaired relaxation in left ventricular myocytes from neuronal no synthase-deficient mice. Circ Res 2008;102:242–249. [DOI] [PubMed] [Google Scholar]
- 93. Silberman GA Fan TH Liu H Jiao Z Xiao HD Lovelock JD Boulden BM Widder J Fredd S Bernstein KE Wolska BM Dikalov S Harrison DG Dudley SC Jr. Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation 2010;121:519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Paulus WJ Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 2013;62:263–271. [DOI] [PubMed] [Google Scholar]
- 95. Gori T Munzel T. Oxidative stress and endothelial dysfunction: therapeutic implications. Ann Med 2011;43:259–272. [DOI] [PubMed] [Google Scholar]
- 96. Keaney JF Jr Gaziano JM Xu A Frei B Curran-Celentano J Shwaery GT Loscalzo J Vita JA. Low-dose alpha-tocopherol improves and high-dose alpha-tocopherol worsens endothelial vasodilator function in cholesterol-fed rabbits. J Clin Invest 1994;93:844–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Stocker R. The ambivalence of vitamin E in atherogenesis. Trends Biochem Sci 1999;24:219–223. [DOI] [PubMed] [Google Scholar]
- 98. Yusuf S Sleight P Pogue J Bosch J Davies R Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med 2000;342:145–153. [DOI] [PubMed] [Google Scholar]
- 99. Lonn E Bosch J Yusuf S Sheridan P Pogue J Arnold JM Ross C Arnold A Sleight P Probstfield J Dagenais GR, Hope, Investigators H-TT. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA 2005;293:1338–1347. [DOI] [PubMed] [Google Scholar]
- 100. Ristow M Zarse K Oberbach A Kloting N Birringer M Kiehntopf M Stumvoll M Kahn CR Bluher M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci USA 2009;106:8665–8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dieberg G Ismail H Giallauria F Smart NA. Clinical outcomes and cardiovascular responses to exercise training in preserved ejection fraction heart failure patients: systematic review & meta-analysis. J Appl Physiol 2015; 10.1152/japplphysiol.00904.2014. [DOI] [PubMed] [Google Scholar]
- 102. Edelmann F Gelbrich G Dungen HD Frohling S Wachter R Stahrenberg R Binder L Topper A Lashki DJ Schwarz S Herrmann-Lingen C Loffler M Hasenfuss G Halle M Pieske B. Exercise training improves exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction: results of the Ex-DHF (Exercise training in Diastolic Heart Failure) pilot study. J Am Coll Cardiol 2011;58:1780–1791. [DOI] [PubMed] [Google Scholar]
- 103. Piepoli MF Conraads V Corra U Dickstein K Francis DP Jaarsma T McMurray J Pieske B Piotrowicz E Schmid JP Anker SD Solal AC Filippatos GS Hoes AW Gielen S Giannuzzi P Ponikowski PP. Exercise training in heart failure: from theory to practice. A consensus document of the Heart Failure Association and the European Association for Cardiovascular Prevention and Rehabilitation. Eur J Heart Fail 2011;13:347–357. [DOI] [PubMed] [Google Scholar]
- 104. Porsti I Bara AT Busse R Hecker M. Release of nitric oxide by angiotensin-(1–7) from porcine coronary endothelium: implications for a novel angiotensin receptor. Br J Pharmacol 1994;111:652–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Caspritz G Alpermann HG Schleyerbach R. Influence of the new angiotensin converting enzyme inhibitor ramipril on several models of acute inflammation and the adjuvant arthritis in the rat. Arzneimittelforschung 1986;36:1605–1608. [PubMed] [Google Scholar]
- 106. Soehnlein O Schmeisser A Cicha I Reiss C Ulbrich H Lindbom L Daniel WG Garlichs CD. ACE inhibition lowers angiotensin-II-induced monocyte adhesion to HUVEC by reduction of p65 translocation and AT 1 expression. J Vasc Res 2005;42:399–407. [DOI] [PubMed] [Google Scholar]
- 107. Zhao K Zhao GM Wu D Soong Y Birk AV Schiller PW Szeto HH. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem 2004;279:34682–34690. [DOI] [PubMed] [Google Scholar]
- 108. Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol 2014;171:2029–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Birk AV Liu S Soong Y Mills W Singh P Warren JD Seshan SV Pardee JD Szeto HH. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol 2013;24:1250–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Chakrabarti AK Feeney K Abueg C Brown DA Czyz E Tendera M Janosi A Giugliano RP Kloner RA Weaver WD Bode C Godlewski J Merkely B Gibson CM. Rationale and design of the embrace stemi study: a phase 2a, randomized, double-blind, placebo-controlled trial to evaluate the safety, tolerability and efficacy of intravenous bendavia on reperfusion injury in patients treated with standard therapy including primary percutaneous coronary intervention and stenting for ST-segment elevation myocardial infarction. Am Heart J 2013;165:509–514e507. [DOI] [PubMed] [Google Scholar]
- 111. Smith RA Hartley RC Cocheme HM Murphy MP. Mitochondrial pharmacology. Trends Pharmacol Sci 2012;33:341–352. [DOI] [PubMed] [Google Scholar]
- 112. Mortensen SA. Coenzyme Q10: will this natural substance become a guideline-directed adjunctive therapy in heart failure? JACC. Heart Failure 2015;3:270–271. [DOI] [PubMed] [Google Scholar]
- 113. Ezekowitz JA. Time to energize coenzyme Q10 for patients with heart failure? JACC Heart Fail 2014;2:650–652. [DOI] [PubMed] [Google Scholar]
- 114. Nickel A Löffler J Maack C. Myocardial energetics in heart failure. Basic Res Cardiol 2013;108:358. [DOI] [PubMed] [Google Scholar]
- 115. Elkayam U Kulick D McIntosh N Roth A Hsueh W Rahimtoola SH. Incidence of early tolerance to hemodynamic effects of continuous infusion of nitroglycerin in patients with coronary artery disease and heart failure. Circulation 1987;76:577–584. [DOI] [PubMed] [Google Scholar]
- 116. Elkayam U Roth A Mehra A Ostrzega E Shotan A Kulick D Jamison M Johnston JV Rahimtoola SH. Randomized study to evaluate the relation between oral isosorbide dinitrate dosing interval and the development of early tolerance to its effect on left ventricular filling pressure in patients with chronic heart failure. Circulation 1991;84:2040–2048. [DOI] [PubMed] [Google Scholar]
- 117. Cohn JN Archibald DG Ziesche S Franciosa JA Harston WE Tristani FE Dunkman WB Jacobs W Francis GS Flohr KH Goldman S Cobb FR Shah PM Saunders R Fletcher RD Loeb HS Hughes VC Baker B. Effect of vasodilator therapy on mortality in chronic congestive heart failure. Results of a veterans administration cooperative study. N Engl J Med 1986;314:1547–1552. [DOI] [PubMed] [Google Scholar]
- 118. Cohn JN Johnson G Ziesche S Cobb F Francis G Tristani F Smith R Dunkman WB Loeb H Wong M Bhat G Goldman S Fletcher RD Doherty J Hughes CV Carson P Cintron G Shabetai R Haakenson C. A comparison of enalapril with hydralazine-isosorbide dinitrate in the treatment of chronic congestive heart failure. N Engl J Med 1991;325:303–310. [DOI] [PubMed] [Google Scholar]
- 119. Taylor AL Ziesche S Yancy C Carson P D'Agostino R Jr Ferdinand K Taylor M Adams K Sabolinski M Worcel M Cohn JN, African-American Heart Failure Trial I. Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med 2004;351:2049–2057. [DOI] [PubMed] [Google Scholar]
- 120. Daiber A Oelze M Coldewey M Kaiser K Huth C Schildknecht S Bachschmid M Nazirisadeh Y Ullrich V Mulsch A Munzel T Tsilimingas N. Hydralazine is a powerful inhibitor of peroxynitrite formation as a possible explanation for its beneficial effects on prognosis in patients with congestive heart failure. Biochem Biophys Res Commun 2005;338:1865–1874. [DOI] [PubMed] [Google Scholar]
- 121. Munzel T Kurz S Rajagopalan S Thoenes M Berrington WR Thompson JA Freeman BA Harrison DG. Hydralazine prevents nitroglycerin tolerance by inhibiting activation of a membrane-bound NADH oxidase. A new action for an old drug. J Clin Invest 1996;98:1465–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Hare JM. Nitroso-redox balance in the cardiovascular system. N Engl J Med 2004;351:2112–2114. [DOI] [PubMed] [Google Scholar]
- 123. Dulce RA Yiginer O Gonzalez DR Goss G Feng N Zheng M Hare JM. Hydralazine and organic nitrates restore impaired excitation-contraction coupling by reducing calcium leak associated with nitroso-redox imbalance. J Biol Chem 2013;288:6522–6533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Zhuang XD Long M Li F Hu X Liao XX Du ZM. PDE5 inhibitor sildenafil in the treatment of heart failure: a meta-analysis of randomized controlled trials. Int J Cardiol 2014;172:581–587. [DOI] [PubMed] [Google Scholar]
- 125. Redfield MM Chen HH Borlaug BA Semigran MJ Lee KL Lewis G LeWinter MM Rouleau JL Bull DA Mann DL Deswal A Stevenson LW Givertz MM Ofili EO O'Connor CM Felker GM Goldsmith SR Bart BA McNulty SE Ibarra JC Lin G Oh JK Patel MR Kim RJ Tracy RP Velazquez EJ Anstrom KJ Hernandez AF Mascette AM Braunwald E Trial R. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 2013;309:1268–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Zucker IH Schultz HD Patel KP Wang H. Modulation of angiotensin II signaling following exercise training in heart failure. Am J Physiol Heart Circ Physiol 2015;308:H781–H791. [DOI] [PMC free article] [PubMed] [Google Scholar]