

Abstract
Excipients, considered “inactive ingredients,” are a major component of formulated drugs and play key roles in their pharmacokinetics. Despite their pervasiveness, whether they are active on any targets has not been systematically explored. We computed the likelihood that approved excipients would bind to molecular targets. Testing in vitro revealed 25 excipient activities, ranging from low-nanomolar to high-micromolar concentration. Another 109 activities were identified by testing against clinical safety targets. In cellular models, five excipients had fingerprints predictive of system-level toxicity. Exposures of seven excipients were investigated, and in certain populations, two of these may reach levels of in vitro target potency, including brain and gut exposure of thimerosal and its major metabolite, which had dopamine D3 receptor dissociation constant Kd values of 320 and 210 nM, respectively. Although most excipients deserve their status as inert, many approved excipients may directly modulate physiologically relevant targets.
For most drug products, the major components by mass are not the active pharmaceutical ingredient (API) but rather are excipients; these are classified as “inactive ingredients” in the FDA’s Inactive Ingredients Database (IID; www.fda.gov/Drugs/InformationOnDrugs/ucm113978.htm). Examples of excipients are molecules such as lactose, pectin, and xanthan gum, which stabilize the API in pill form; antioxidants such as propyl gallate that improve shelf life; detergents such as sodium lauryl sulfate that solubilize the API in the gut; and dyes such as FD&C Yellow No. 5 (tartrazine), D&C Red No. 28 (Phloxine B), and FD&C Blue No. 1 (Brilliant Blue FCF) that color medicines so that they can be better dis-tinguished by patients and pharmacists. In many drug formulations, excipients can reach concentrations of hundreds of micromolar to millimolar in the gastrointestinal tract, up to 100 times the concentration achieved by the API.
Despite excipients’ widespread use, their activity on molecular targets has not been systematically investigated; their “inactive” designation derives from gross tolerability studies in animals or from historical precedents. Most approved excipients lack obvious toxicity at allowed concentrations in animal studies. However, the ways in which they interact with molecular targets—and thus how they might perturb pharmacology in a way not visible in whole-animal tests—have remained largely unexplored. A few, such as bithionol [21 CFR 700.11 (1)] and amaranth, formerly known as FD&C Red No. 2 [21 CFR 81.30 (2)], have been removed from use over concerns of photosensitization and tumorigenicity (3), respectively. Some excipients, such as Methylene Blue, are used directly as drugs; others closely resemble known bioactives (Fig. 1A). Thus, although approved excipients may lack gross physiological effects, it is conceivable that some may in fact have specific activity on molecular targets, perturbing their functions and those of the cellular networks in which they are involved. Meanwhile, approved excipients may be swapped in drug formulations (4) as long as they do not affect the pharmacokinetics of the API. This could affect drug activity, as different excipients would be expected to have different target activities, influencing the overall side effects of the medicine.
Fig. 1. Motivation for testing excipient activity, operational workflow, and selected in vitro concentration-response curves.
(A) The similarity between excipients (top) and FDA-approved drugs (bottom). Benzethonium chloride is itself FDA-approved as a topical antiseptic wash. (B) Workflow. More than 600 molecular excipients were screened computationally, and a list of potential protein targets was predicted for each one on the basis of its SEA E-value. A subset of high-ranking excipient-target pairs was tested in vitro. In a second set of experiments, commonly used excipients were experimentally tested against a panel of clinical toxicity targets; these tests were unrelated to the SEA predictions, although sometimes they overlapped. (C) Molecular structures of thimerosal and ethyl mercury. (D) Concentration-response curves of selected excipient-target pairs with activity ranging from low-nanomolar (propyl gallate inhibition of COMT) to mid-micromolar (tartrazine binding to dopamine D1). Red curves represent a reference positive control; blue curves represent excipient binding: (a) propyl gallate and reference compound tolcapone binding to COMT; (b) tartrazine and reference compound (+)-butaclamol binding to DRD1; (c) diethyl phthalate and reference compound RO 20–1724 binding to PDE4D; (d) thimerosal and reference compound (+)-butaclamol binding to DRD3; (e) butylparaben and reference compound L670596 binding to TBXA2R; (f) benzethonium chloride and reference compound potriptyline binding to SLC6A2 (previously known). The D3 binding curve for thimerosal is one representative of replicates in three separate laboratories. Additional dose-response curves are provided in fig. S1.
Predicting and testing excipient activity on in vitro targets
We used a two-part strategy to systematically investigate the activity of approved inactive ingredients against biologically relevant molecular targets. First, we computationally predicted plausible targets for excipients from among 3000 medically relevant proteins, using chemoinformatic (5) inference (6, 7), an approach that has previously been used to predict off-targets and mechanism-of-action targets for drugs and probes (8–10). Second, we empirically screened widely used excipients against a panel of 28 toxicity-related targets regularly used to identify potential clinical adverse events of drug candidates (targets associated with clinical safety) (11), and against several other targets with important roles in drug toxicity or drug activity (such as the organic anion transporter; see below).
The chemoinformatic Similarity Ensemble Approach (SEA; http://sea16.ucsf.bkslab.org) posits that targets with ligands that resemble a bait compound—here, an excipient—may also bind that compound. This approach has been used to predict side effects and mechanism-of-action targets for drugs (12–14). Of the 3296 FDA-approved excipients in the IID, 639 excipients that are well-defined, monomeric molecules—excluding, for instance, compound excipients such as honey and dye mixtures [see http://excipients.ucsf.bkslab.org (15)]— were computationally screened by SEA against 3117 human targets in the ChEMBL database for which ligands are known. From the nearly 2 million possible excipient-target pairs implied, just over 20,000 emerged as plausible, with SEA E-values of 10−5 or better [these E-values, akin to those in BLAST (16–18), reflect the likelihood that a prediction would occur at random; lower E-values are more significant]. Of these, 69 excipient-target pairs were prioritized by visual inspection of the excipient versus the target ligands, eliminating those, for instance, where there was a key physical difference between the excipient “bait” and the ligands of the target (such as a charge group that might be important for binding) that was overlooked in the overall chemoinformatic similarity, as in previous studies (6, 12, 13). These 69 were tested experimentally in vitro in functional assays, typically with full dose-response curves (Fig. 1). Nineteen excipients were found to be active against at least one of 12 targets, including muscarinic acetylcholine receptors, the intestinal organic anion transporter 2B1 (OATP2B1), and catechol O-methyltransferase (COMT), for a total of 25 excipient-target activities, a 36% success rate (characteristic excipients are shown in Table 1; tables S1 to S5 list all 69 predictions, including those that failed to confirm experimentally). Active excipients were counter-screened for colloidal aggregation (19–21), a common source of false positives in vitro; any excipient for which aggregation was observed within an order of magnitude of its target activity was discounted as potentially artifactual (22). On-target activities well above predicted fed-gut concentrations were also excluded [estimated as the maximum allowed dosage into a 250-ml volume (23)].
Table 1. Excipient–off-target predictions with SEA, confirmed by in vitro binding assays.
Examples of marketed drugs containing the excipient, and maximal excipient dosages allowed by the FDA in any single formulation, are also shown. Functions are drawn from the Handbook of Pharmaceutical Excipients (64) except as noted. All drugs listed are administered orally.
| Excipient | Function | Structure | Example marketed drug | Highest excipient amount/dose | Predicted target | SEA score | Ki/IC50 (μM) |
|---|---|---|---|---|---|---|---|
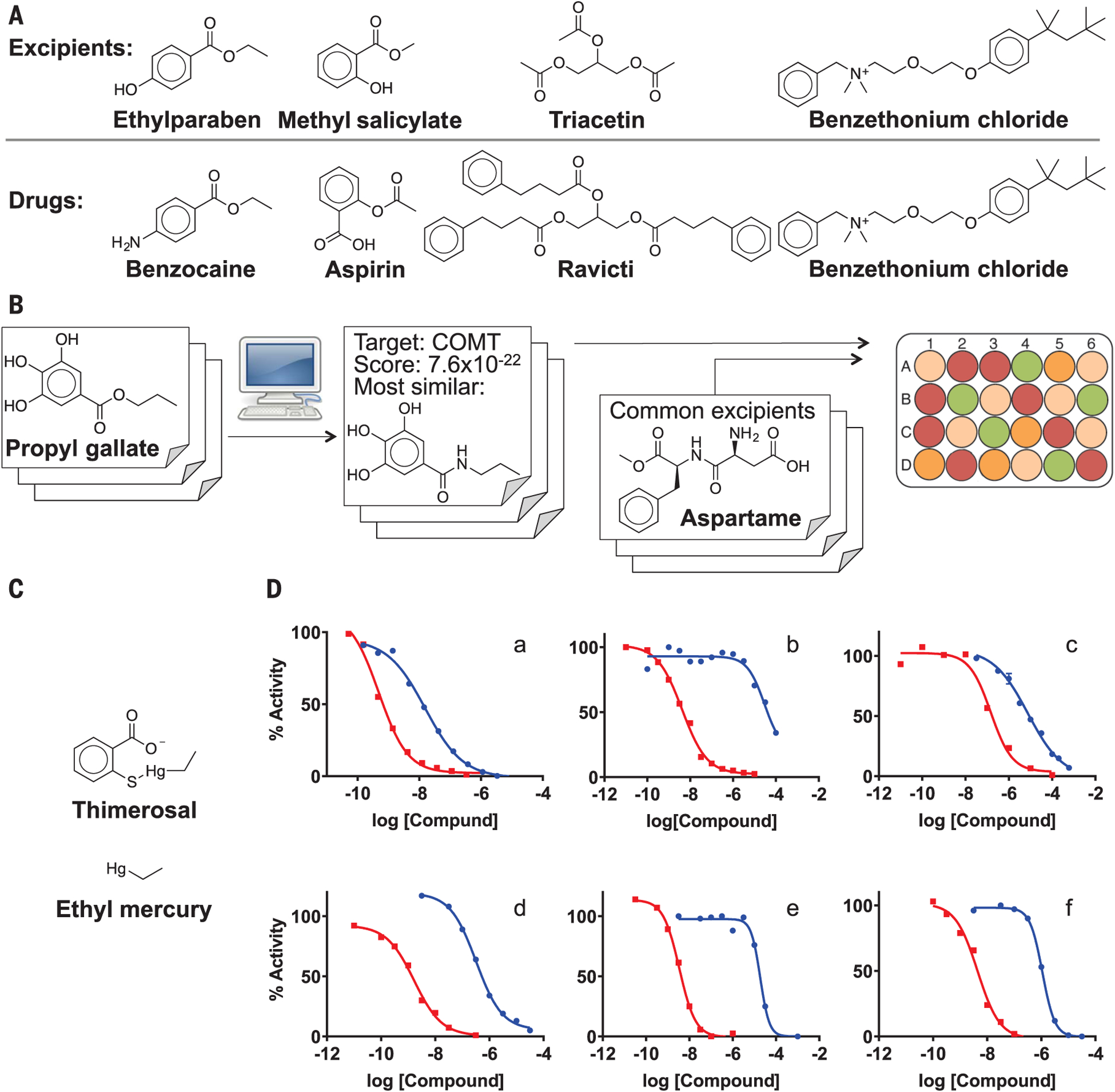
| Butylparaben | Antimicrobial preservative | 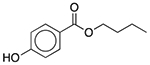 |
Fexofenadine (children’s allergy) | 8 mg/5 ml | CHRM1 | 9.1 × 10−9 | 12 |
| D&C Orange No. 4 | Colorant | 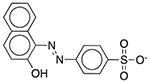 |
AloeGuard Antimicrobial Soap | 0.039 mg | SLCO2B1 | 1.4 × 10−31 | 1.9 |
| D&C Red No. 28 | Colorant | 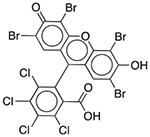 |
Nexium 24HR | 2.5 mg/5 ml | PRMT1 SLC22A6 |
2.6 × 10−19 7.8 × 10−16 |
0.70 0.064 |
| Diethyl phthalate | Film-forming agent; plasticizer; solvent | 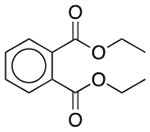 |
Ranitidine | 20.52 mg | PDE4D† | 1.0 × 10−9 | 8.5 |
| Docusate sodium | Surfactant; fecal softener; wetting agent | 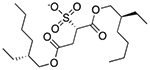 |
Prednisone | 11 mg | SLCO2B1 | 4.7 × 10−34 | 2.3 |
| FD&C Red No. 3 | Colorant | 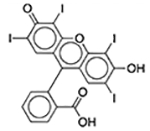 |
Esomeprazole | 2 mg/5 ml | PRMT1 | 3.7 × 10−17 | 0.46 |
| FD&C Red No. 40 | Colorant | 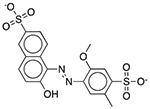 |
Prevacid | 40 mg | SLCO2B1 | 1.4 × 10−27 | 4.7 |
| FD&C Yellow No. 6 | Colorant | 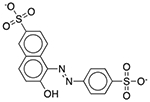 |
Advil | 60.02 mg | SLCO2B1 | 1.4 × 10−31 | 74 |
| Glycyrrhizin | Flavoring agent* | 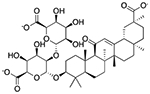 |
Junior ibuprofen | 1.25 mg/5 ml | SLCO1B1 | 3.0 × 10−30 | 13 |
| Propyl gallate | Antioxidant | 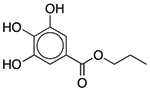 |
Janumet XR | 2 mg | COMT | 7.6 × 10−22 | 0.015 |
| Propylparaben | Antimicrobial preservative | 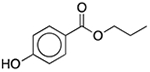 |
Keppra | 200 mg/5 ml | SLCO2B1 | 1.6 × 10−28 | 200 |
| Sodium lauryl sulfate | Surfactant; detergent; emulsifier; skin penetrant; lubricant; wetting agent |  |
Cetirizine | 96 mg | SLCO1B1 SLCO2B1 |
3.7 × 10−34 | 2.9 2.8 |
From (65).
SEA predicted PDE4B and PDE1, neither of which were readily available for testing, but the isozyme PDE4D was.
In a different approach, we experimentally screened 73 commonly used excipients against a panel of 28 targets associated with drug clinical safety (11), and also against other bio-relevant targets including the drug target VMAT2, the well-known ion channel toxicity targets Nav1.5 and Cav1.2, and two transporters with roles in drug pharmacokinetics, BSEP and OATP2B1. Of these, 32 excipients were active against one or more targets, for a total of 109 activities, almost all of which have potency values of 30 μM or less (characteristic excipients are shown in Table 2; table S6 lists all excipients tested against the clinical safety panel). The panel involves both binding and functional assays; for enzymes and transporters functional inhibition was used, whereas for receptors such as 5HT2B and the hERG ion channel, both radioligand binding and functional assays were typically performed. Overall, just over 50% of these activities were measured in functional assays.
Table 2. Excipient-target activities determined in vitro.
The targets are drawn from safety profiling assays generally used for determining off-target effects of drug candidates and marketed drugs. Functions are drawn from the Handbook of Pharmaceutical Excipients (64) except as noted. Examples of marketed drugs containing the excipient, and maximal excipient dosages occurring in any single formulation, are shown. Biochemical and radioligand binding data (IC50) are unformatted; functional data (antagonism/inhibition, IC50; agonism, half maximal effective concentration EC50) are in bold in the Target column. Activity values are accurate to ±20%.
| Excipient | Function | Structure | Example marketed drug | Highest excipient amount/dose | Target | Ki/IC50 Or EC50 (μM) |
|---|---|---|---|---|---|---|
| Aspartame | Sweetener |  |
Purixan | 450 mg† | NR1I2 | 13 |
| Benzethonium chloride | Antimicrobial preservative; antiseptic; disinfectant |  |
Ketalar/ketamine hydrochloride§ | 0.1%‡ | ADORA3 ADRA1A DRD1 DRD3 ESR1 HRH3 PRG OPRM PDE4D PTGS2 ABCB11 SLC18A2 KCNH2 |
5.3 4.7 3.8 1.6 11 27 6.2 6.0 14 24 29 0.18 1.9 |
| Benzoic acid | Antimicrobial preservative; therapeutic agent |  |
Excedrin Migraine | 25 mg/5 ml† |
ABCB11 SLC18A2 |
260 18 |
| Butylparaben | Antimicrobial preservative | 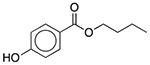 |
Fexofenadine (children’s allergy) | 8 mg/5 ml† |
SLCO2B1 ADRA1A SLC6A2 SLC6A3 TBXA2R SLC18A2 SCN5A |
39 16 20 18 19 6.6 33 |
| Cetylpyridinium chloride | Antimicrobial preservative; antiseptic; surfactant; disinfectant; solubilizer; wetting agent | Dyazide | 1.5 mg† |
ADRA1A DRD1 DRD3 ESR1 HRH3 PRG OPRM1 PDE4D PTGS1 PTGS2 TBXA2R SLC18A2 KCNH2 SCN5A |
6.4 4.8 0.55 18 13 4.5 5.1 13 7.5 2.5 3.4 0.73 1.3 34 |
|
| Chloroxylenol | Antimicrobial preservative; antiseptic; disinfectant | 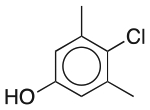 |
Miconazole nitrate | 0.1%‡ |
CHRM1 ADRA1A SLC18A2 |
9.9 14 0.79 |
| Cysteine | Antioxidant* | 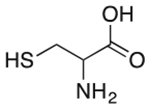 |
Zyban | 16.2 mg† | PTGS2 | 4.7 |
| D&C Red No. 28 | Colorant | 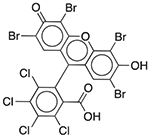 |
Nexium 24HR | 2.5 mg/5 ml† |
SLCO2B1 ADRA1A DRD1 ADORA3 CHRM2 OPRM1 PDE3A PDE4D |
0.60 0.48 0.59 0.48 0.68 0.82 0.13 0.94 |
| Ethylparaben | Antimicrobial preservative | 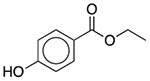 |
Clindamycin (pediatric) | 2 mg/5 ml† | SLC18A2 | 22 |
| FD&C Red No. 3 | Colorant | 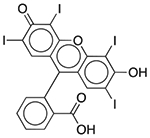 |
Esomeprazole | 2 mg/5 ml† | ACHE PDE3A PDE4D |
0.42 0.092 0.32 |
| FD&C Yellow No. 5 (Tartrazine) | Colorant | 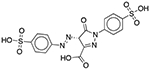 |
Pepcid Complete (tropical flavor) | 652 mg† | DRD1 OPRM1 GABRA1 |
23 16 13 |
| Methylene Blue | Colorant | 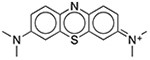 |
Diurex Water Pills† | 1%§ | ACHE CHRM2 HRH3 KCNH2 |
0.090 0.19 0.88 27.5 |
| Oleic acid | Emulsifying agent; skin penetrant |  |
Verapamil | 598.6 mg† |
PTGS1 PTGS2 SLC18A2 |
6.7 3.5 4.7 |
| Palmitic acid | Emulsifying agent; skin penetrant; tablet and capsule lubricant |  |
Captopril | 6 mg† |
PDE3A PDE4D SLC18A2 |
4.1 6.6 4.4 |
| Phenylmercuric acetate | Antimicrobial preservative; antiseptic |  |
- | 0.01%¶ | DRD1 DRD3 SLC6A2 SLC6A3 CHRM1** SLC18A2 KCNH2 |
2.2 0.024 1.7 1.8 8.1 0.36 6.2 |
| Propyl gallate | Antioxidant |  |
Janumet XR | 2 mg† |
ALOX5 PTGS2 TY3H KDM4C FTO |
0.43 7.2 1.9 39 3.0 |
| Propylparaben | Antimicrobial preservative | 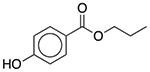 |
Keppra | 200 mg/5 ml† |
SLC6A2 KDR LTA4H |
30 24 260 |
| Sodium lauryl sulfate | Anionic surfactant; detergent; emulsifying agent; skin penetrant; lubricant; wetting agent | 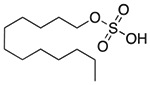 |
Cetirizine | 96 mg† | HTR1A ADORA3 CHRM1 ADRA1A DRD3 PRG PDE4D |
15 15 29 15 18 15 23 |
| Stearic acid | Emulsifying agent; solubilizing agent; tablet and capsule lubricant |  |
Kariva | 1203 mg/5 ml† |
PDE3A PDE4D |
3.1 6.7 |
| Thimerosal | Antimicrobial preservative; antiseptic |  |
Influenza vaccines multidose packaging | 0.03%§ | ADORA3 ADRA2A SLC18A2 DRD1 DRD3 5HT2b** |
1.6 3.8 0.97 2.4 0.32 15 |
| Ethyl mercury | Thimerosal major metabolite |  |
NA | NA | DRD3 DRD5 |
0.21 0.15 |
| Thymol | Antioxidant; antiseptic; cooling agent; disinfectant; flavoring agent; skin penetrant |  |
Listerine† | 0.01%# |
CHRM1 ADRA1A SLC6A2 SLC18A2 |
14 26 28 25 |
From (65).
Oral.
Auricular.
Intravenous.
Topical.
Inhalation.
An agonist effect was observed.
From the two approaches, 38 “inactive ingredients” emerged with 134 activities against 44 targets (Tables 1 and 2 and tables S1 to S3). These activities ranged from 15 nM to 260 μM; 30 (22%) were submicromolar, with several in the low- to mid-nanomolar range, and another 37% were lower than 10 μM. These activities are more potent than the on-target activities of some small-molecule therapeutic drugs, which have a median affinity of ~20 nM (24).
Excipient cell-based toxicities
Although these activities belie the designation of these ingredients as “inactive,” they do not necessarily imply that the excipients will have activities on organ systems that would mani-fest in health or behavioral changes of an individual. Such systemic disturbances depend on integrated effects on tissues, and on exposure of the excipient to their targets in relevant contexts in the body.
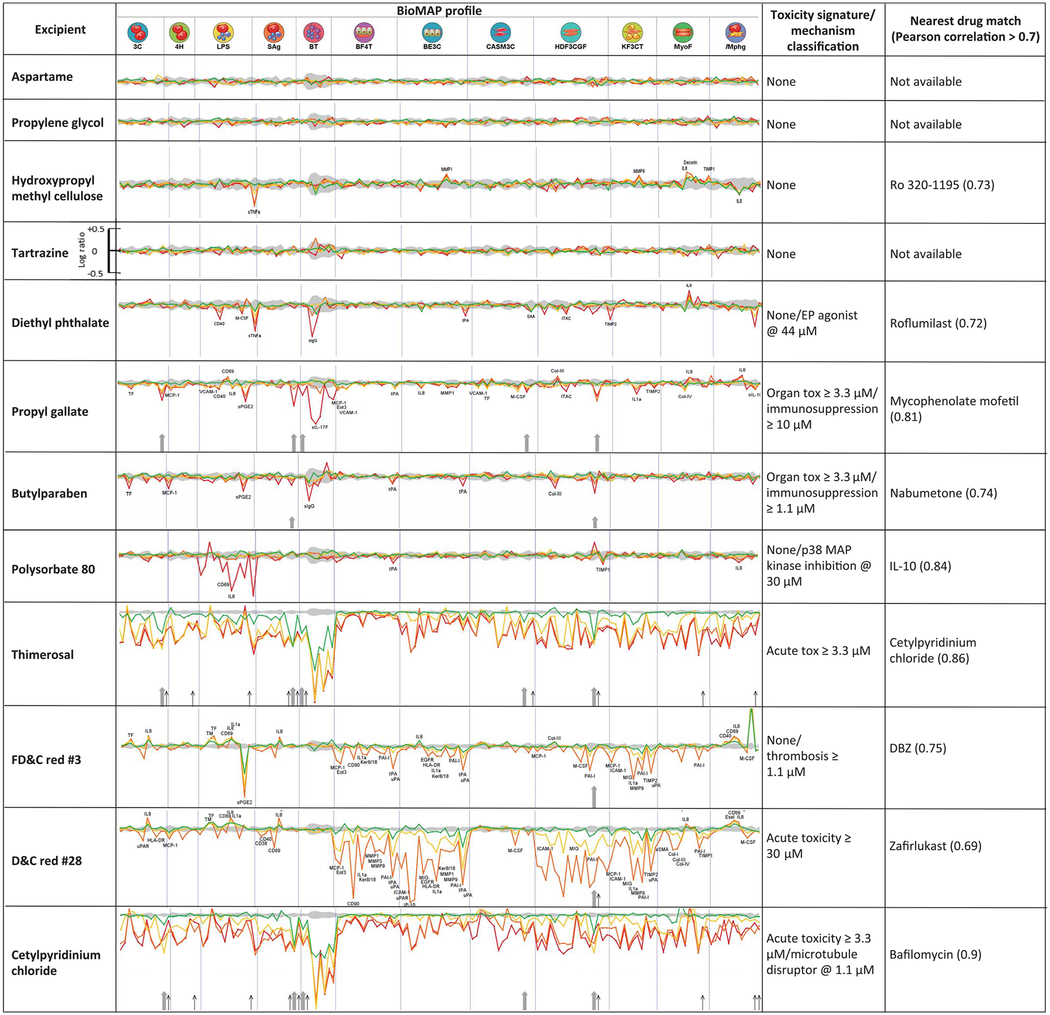
Accordingly, 12 of the target-active excipients were further investigated with the BioMAP Diversity PLUS panel, a suite of cell-based systems widely used to model drug- and chemical-induced effects and toxicology in vascular, lung, skin, and inflammatory tissues, among others (25–27) (see supplementary materials). These 12 were prioritized for testing by several criteria, including their frequency of use in drug formulations and their coverage of different excipient functions (e.g., colorants, antimicrobials, antioxidants, emulsifiers, and surfactants). Although we mostly tested excipients found to be active in the in vitro assays, several inactive ones were also included. Excipients with poor solubility were excluded. Each excipient generated a profile on 148 biomarker readouts in the cells, at four different concentrations (22) (Table 3 and fig. S14).
Table 3. Excipient activity in cell-based model systems.
BioMAP Diversity PLUS profiles for 12 excipients are shown. Each section of the spectrum corresponds to a different cell or tissue system (22). Profiles represent concentration responses across the assays: green, 1.1 μM; yellow, 3.3 μM; orange, 10 μM; red, 30 μM. Upward and downward strokes represent increased and decreased levels, respectively, in log scale (see tartrazine for reference). The gray shaded area represents the baseline response range. The rightmost columns show toxicity signatures and the closest drug matches from the BioMAP database with corresponding correlation between the two profiles. Cytotoxicity is indicated by thin black arrows; antiproliferative effects are marked by gray arrows. Cell types corresponding to assay codes: 3C, venular endothelial cells (VECs); 4H, VECs; LPS, peripheral blood mononuclear cells (PBMCs), endothelial cells (ECs); SAg, PBMCs, ECs; BT, PBMCs, B cells; BF4T, bronchial epithelial cells (BECs), dermal fibroblasts; BE3C, BECs; CASM3C, coronary artery smooth muscle cells; HDF3CGF, fibroblasts; KF3CT, keratinocytes, fibroblasts; MyoF, lung fibroblasts; iMphg, macrophages, VECs. See supplementary materials for detailed explanation. For measurements on all tested excipients, see figs. S2 to S13.
|
These effects were compared to more than 4000 drug and chemical reference profiles in the BioMAP database. For example, butylparaben, a widely used excipient coformulated with APIs as varied as acetaminophen, hydrocodone, di-phenhydramine, and fluoxetine, had a cellular efficacy fingerprint suggesting inflammation-related activity. This excipient dose-dependently decreased MCP-1 (monocyte chemoattractant protein 1, or CCL2) and sPGE2 (soluble prosta-glandin E2) in peripheral blood mononuclear cells and venular endothelial cells, whereas its biomarker profile shared six common activities with the anti-inflammatory nabumetone at 30 μM (Pearson correlation r = 0.735) (fig. S2). Meanwhile, propyl gallate, an excipient coformulated in drugs such as Advil Sinus Congestion and Pain, Janumet, ezetimibe, and simvastatin, was antiproliferative in B and T cells, coronary artery smooth muscle cells, endothelial cells, and fibroblasts, at 10 or 30 μM. Propyl gallate had cellular activities that mapped to the immunomodulatory and inflammation-related activities of phenazopyridine (fig. S7) and mycophenolate, anesthetic, and immunosuppressant drugs, respectively. Intriguingly, at lower concentrations, propyl gallate’s profile most resembled that of caffeic acid phenethyl ester, a 15-lipoxygenase inhibitor with anti-inflammatory and immunomodulatory properties, consistent with our observation that propyl gallate is an inhibitor of 5-lipoxygenase with a potency of 430 nM (Table 2). Another interesting example is diethyl phthalate. Notably, diethyl phthalate modulated tumor necrosis factor–α (TNF-α) in a dose-dependent manner (Table 3), as previously observed (28). Furthermore, TNF-α production can be affected by PDE4 inhibition (29)—an in vitro activity of diethyl phthalate determined in this study (concentration of 50% inhibition IC50 = 8.5 μM; Table 1). Finally, the BioMap profile of diethyl phthalate matched that of roflumilast (r = 0.72) (fig. S11), a PDE4 inhibitor used to treat chronic obstructive pulmonary disease (COPD). Several of the excipients that were active in isolated enzyme and receptor assays, including aspartame, propylene glycol, and tartrazine (FD&C Yellow No. 5), had no measurable activity in these cell-based systems.
Systemic exposure of excipients in animal models
Excipients are often exposed at high concentrations to targets in the gut epithelia, including the intestinal uptake transporter OATP2B1, calcium and sodium ion channels, and bile acid transporters; however, for systemic impact they must cross intestinal and metabolic barriers and enter the general circulation. Accordingly, we investigated seven of the more active and widely used excipients for exposure in blood after oral dosing in a rodent model (Table 4 and Fig. 2). Most did not reach blood concentrations high enough to modulate their targets upon oral dosing, which suggests that despite potent target-based and even cell-based activities, they were sequestered in the gut or were rapidly metabolized. Examples include butylparaben, which—although administered at doses up to 8 mg (5 ml) in several medications, implying a concentration of 160 μM, in the fed-state gut—reaches only negligible concentrations in the blood when dosed at 10 mg/kg in the rat, likely reflecting rapid metabolism in the blood. Similarly, FD&C Red No. 3 reaches a peak concentration (Cmax) value of 17 nM only after a 1.0 mg/kg oral dose, making its 92 nM inhibition of phosphodiesterase 3A (PDE3A) less concerning, particularly with a 99% plasma protein binding. The 15 nM COMT inhibitor propyl gallate reaches Cmax values of 5 nM only after a 10 mg/kg oral dose, much higher than its maximum dose of 2 mg in drug formulations, even allowing for allometric scaling. [However, as discussed below, propyl gallate is used in much higher amounts in food, and there is some indication that COMT has a role in gut disorders, where propylparaben will reach much higher concentrations (30).] Moreover, at least therapeutically, most of COMT’s functions are in the central nervous system, and it is likely that brain exposure would be even lower than in the general circulation. Of the orally dosed excipients, only the antiseptic cetylpyridinium chloride, which occurs in mouthwash, reached a blood Cmax that was in the range of its activity against the dopamine D3 receptor (Cmax = 260 nM, IC50 = 550 nM).
Table 4. Pharmacokinetics parameters of excipients.
Shown are pharmacokinetic parameters in rats of excipients after i.v. (1 mg/kg) and oral administration (cetylpyridinium chloride, 1 mg/kg; butylparaben, 10 mg/kg; FD&C Red No. 3, 1 mg/kg; D&C Red No. 6, 1 mg/kg; propyl gallate, 10 mg/kg). Diethyl phthalate and thimerosal pharmacokinetic data were obtained from the literature, as noted. MDCK-LE Papp is the apparent permeability measured in the Madin-Darby canine kidney permeability assay. AUC, area under curve; %F, bioavailability; N.D., not determined; d.n., dose normalized.
| Excipient | Target | IC50 (μM) | MDCK-LE Papp (×10−6 cm/s) | t1/2 from i.v. dose (hours) | i.v. AUC (nM-hour) 0–24 (d.n.) | Oral AUC (nM-hour) 0–last (d.n.) | Oral Cmax (nM) | %F |
|---|---|---|---|---|---|---|---|---|
| Butylparaben | TBXA2R | 19 | 8.3 | N.D.* | N.D.* | N.D.* | <2.6 | N.D.* |
| CetyIpyridinium chloride | DRD3 | 0.55 | <0.5 | 9.7 | 15,972 | 4,052 | 260 | 5.4 |
| FD&C Red No. 3 | PDE3A | 0.092 | <0.5 | 3.4 | 4,826 | 32 | 16.9 | 0.7 |
| D&C Red No. 6 | SLCO1B1 | 3.1 | <1.2 | 6.9 | 30,511 | 776 | 164 | 2.5 |
| Diethyl phthalate | PDE4D2 | 16 | – | 1.0† | 553† | 350† | 253† | N.D.† |
| Propyl gallate | COMT | 0.015 | 11.7 | 4.9 | 6908 | N.D.* | <8.9 | N.D.* |
| Thimerosal | DRD3 | 0.32 | – | 24.2‡ | N.D. | N.D. | 30 to 40 ng/g‡ (70 to 105 nM) | N.D. |
Fig. 2. Time-concentration profiles of excipients administered to rats.
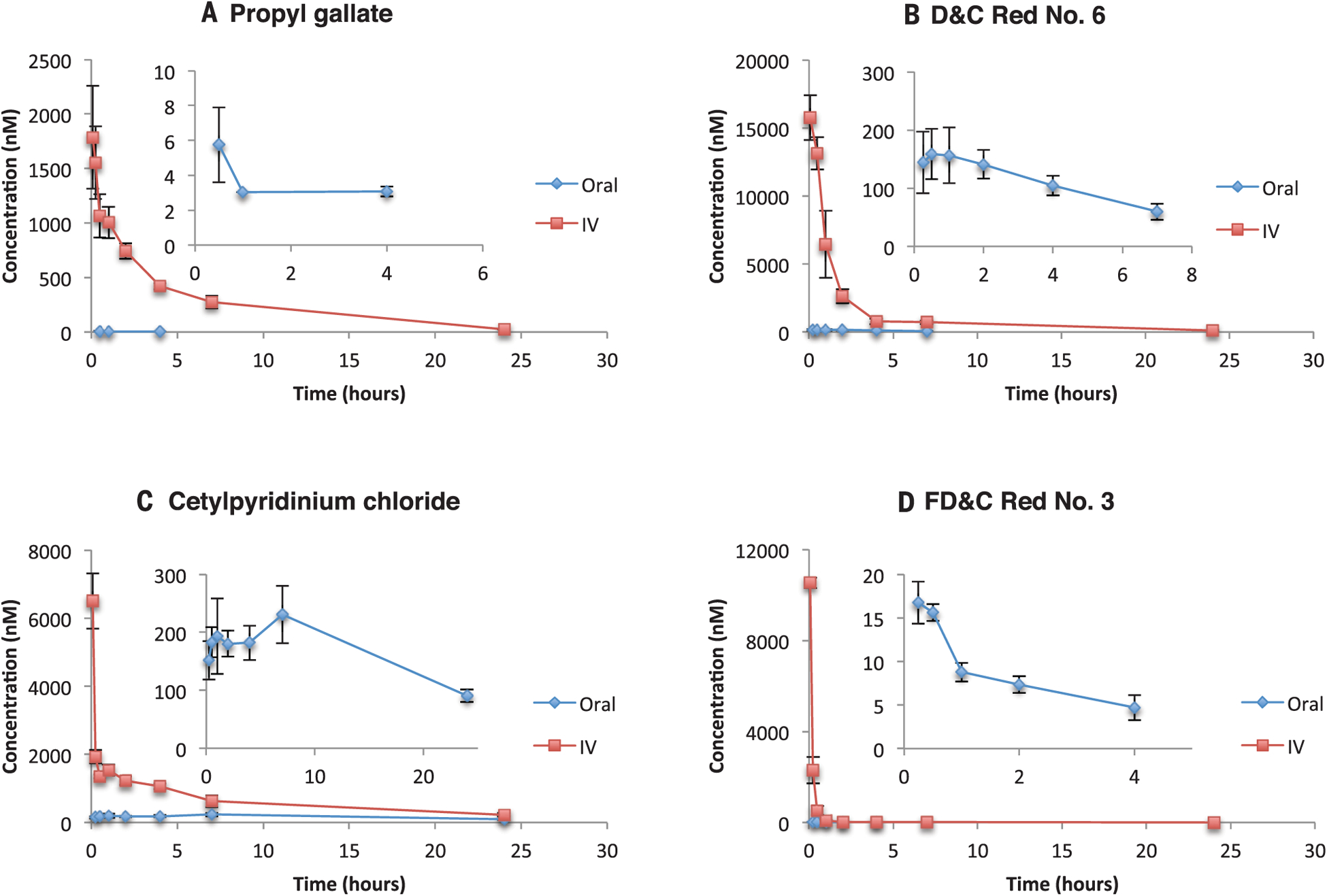
(A to D) Time-concentration curves of blood exposure in rats after i.v. (1 mg/kg) application of excipients and oral administration of propyl gallate (10 mg/kg) (A), D&C Red No. 6 (1 mg/kg) (B), cetylpyridinium chloride (1 mg/kg) (C), and FD&C Red No. 3 (1 mg/kg) (D). Insets are expanded views of the oral administration curves. Error bars denote SD from measurements in three rats.
There are other routes to the systemic circulation, such as excipients in injected drug formulations. One we investigated was thimerosal (www.spectrumchemical.com/MSDS/TH125_AGHS.pdf), an antibacterial present in several parenteral applications, notably vaccines such as those for influenza and diphtheria/tetanus, but also broadly used in ophthalmic solutions (31), antivenom injections (32), and topical applications of dermatological products (33). Thimerosal is well known for its discounted association with autism, after epidemiological studies failed to find causal correlation between pre- and postnatal vaccination and neuropsychological man-ifestations (34–36). Still, because thimerosal is a mercury derivative (Fig. 1C), there has been a move to limit or eliminate it in parenteral formulations, particularly in pediatric vaccines (37, 38). Indeed, thimerosal was removed from childhood vaccines in the United States in 2001 (33). Nonetheless, the molecule is still used in industrial preparation of vaccines, and it continues to occur in multidose adult influenza vaccines in the United States (39, 40) and in multidose vaccines, including for infant vaccination, in the developing world (www.who.int/vaccine_safety/GACVSsymposiumTrack1-Safety-issues-reviewed-during-early21st-centuryRev2.pdf?ua=1). Administered from such multidose formulations, a patient could receive 12.5 to 25 μg of thimerosal per vaccination (38–40).
We find that thimerosal binds to the dopamine D3 receptor with a Kd of 320 nM, whereas its primary metabolite, ethyl mercury, binds to the same receptor with a Kd of ~210 nM and to the dopamine D5 receptor with a Kd of ~150 nM (Table 2 and table S7). Mercury levels after newborn and infant vaccination containing thimerosal reached an average Cmax of 5 ng/ml (25 nM) in the blood, with a calculated half-life t1/2 of 3.7 days and a complete clearance 30 days after vaccination (41). The level of Hg in the stool was higher, reaching an average Cmax of 45 ng/ml (225 nM) (41). Allometric scaling from infant monkeys (42), the only study measuring brain exposure, projected an estimated 13 to 24 ng/g (65 to 120 nM) Cmax of mercury in the brain of human infants, with t1/2 of ~21 days (fig. S15A and table S8). Meanwhile, in a study measuring results after repeated vaccination in human adults (43), the median blood concentration was 0.33 ng/ml (0.17 to 1.3 ng/ml = 0.85 to 6.5 nM) for Hg and 0.14 ng/ml (0.06 to 0.43 ng/ml = 0.7 to 2.2 nM) for ethyl mercury, respectively, with a mean half-life of 5.6 days. Although the blood concentration of Hg in adults was substantially lower than projected for infants, the mercury-containing molecules are expected to concentrate and accumulate in the brain, reaching concentrations of 25 to 30 nM.
Taken together, these observations suggest that thimerosal and ethyl mercury may reach gut and brain concentrations in the human infant that overlap with their affinity (Kd) values for the dopamine D3 receptor (Table 2). For this receptor, the safety margin for the ratio of pharmacokinetic exposure to IC75 is ~1 (i.e., the exposed concentration should be less than concentration required for 75% receptor occupancy) (44). This does not demonstrate in vivo D3 receptor activity of thimerosal and ethyl mercury, even for infants (who are no longer exposed to this excipient in the developed world). Still, the activity on the dopamine D3 receptor, and for ethyl mercury the dopamine D5 receptor—both important targets and antitargets, present both in brain and gut— does make a physiological effect by thimerosal plausible.
Discussion
A key observation from this study is that many “inactive ingredients,” ubiquitous in drug formulations, have direct activities against biologically relevant enzymes, receptors, ion channels, and transporters in vitro. Overall, we observed 134 activities for 38 excipients were observed (Tables 1 and 2 and tables S1 to S3). These activities covered a wide range: 15 nM for propyl gallate on COMT; mid-nanomolar activities for thimerosal at the dopamine D3 receptor, of FD&C Red No. 3 at phosphodiesterase 3A (PDE3A), and for benzethonium chloride at the vesicular monoamine transporter VMAT2; low-micromolar activities for cetylpyridinium chloride and benzethonium chloride at the hERG ion channel; and mid-micromolar activities for butylparaben at the muscarinic receptor CHRM1 and for diethyl phthalate at PDE4D. Many of these enzymes and receptors have crucial roles in neurotransmitter signaling, including COMT, the phosphodiesterases, the VMAT2 transporter, and the dopamine and muscarinic receptors, which have pleiotropic physiological effects and are targeted by multiple therapeutic drugs, whereas the hERG ion channel is notorious for serious adverse drug-related cardiac effects. The median, in vitro activity of all tested excipients that had measurable activity was 5.9 μM. For several excipients, such as propyl gallate, thimerosal, and FD&C Red No. 3, their target-based IC50 values overlap with those of therapeutic drugs (24). At the same time, many of these excipients are administered at much higher doses than the API whose physical behavior and stability they are intended to modulate and protect. Although most will never reach systemic circulation, it is clear, even from this preliminary study, that excipients such as cetylpyridinium chloride and thimerosal do so at concentrations that are high enough to plausibly affect the function of the proteins identified here, all of which are well-accepted therapeutic or toxicity targets.
Although the systemic exposure of most excipients remains limited at regulated maximum doses in medicines, the wide and chronic use of multiple drugs, sometimes with overlapping excipients [for instance, in elderly populations (45)] could result in higher dosing of excipients than allowed for in any one drug, and thus in increased systemic levels. The widespread occurrence in foods, drinks, and cosmetics of many of the same excipients that occur in drugs may further exacerbate excipient exposure. Indeed, many of the excipients investigated here—including colorants such as tartrazine, FD&C Red No. 40, and FD&C Blue No. 1; preservatives such propyl gallate; and sweeteners such as aspartame—occur in food and drinks in even higher amounts than they do in drugs (for instance, propyl gallate is allowed in foods at an acceptable daily intake of 0.5 mg kg−1 day−1, a 35-mg daily intake for a 70-kg adult, which might bring the COMT effect within the range of that absorbed in the rat study). Altered gut absorption (46) and local effects on the microbiome (47) could exacerbate unexpected effects of target-active excipients.
Although most excipients may not reach relevant systemic exposure, two of the seven for which pharmacokinetics were explored— cetylpyridinium chloride and thimerosal—may do so. No study has demonstrated that thimerosal leads to in vivo toxicities, and we have not done so here. Still, our observation that the affinities of thimerosal and ethyl mercury for the D3 dopamine receptor overlap their pharmacokinetic exposure makes the occurrence of target-based activity at least plausible. Consistent with this view, rats treated with thimerosal postnatally had impaired locomotor function, increased anxiety/neophobia in the open field, and alterations in social behavior (48), all side effects that are well-precedented for dopamine receptor ligands (49, 50). In these rodent studies, immunohistochemistry revealed a decline in striatal D2 receptor density (48), and prenatal treatment with thimerosal impaired the serotonergic and dopaminergic systems in the rat brain (51). We note that two independent studies in nonhuman primates did not find alterations in behavior after thimerosal treatment (52); however, immunohistochemistry experiments were not pursued in either study, and in one study the behavioral tests lacked challenges to dopaminergic components (53).
For excipients with inherent on-target activity, it is plausible that they will lead to different outcomes than the API alone would have, as could different excipients used with the same API. Although a detailed investigation is beyond the scope of this study, such excipient switching is well known. It is allowed as long as switching these “inactive ingredients” can be shown not to compromise the exposure of the API itself, and as long as replacements are in fact as inactive as they are assumed to be (4). If, however, the excipients have their own on-target activity, and if they are exposed to that target, the last assumption breaks down. There may be early indications that this is the case for some drug formulations.
An example is the API levothyroxine (T4), available in Synthroid and in generic formulations, which contains Aluminum Lake dyes and lactose, among other excipients (www.rxabbvie.com/pdf/synthroid.pdf). There has been concern that dye and food allergies, as well as gastrointestinal comorbidities, may arise from these excipients (54, 55). Levothyroxine can also be delivered without these excipients in a soft gel form, containing only water, glycerin, and gelatin (56). The removal of the other excipients appears to aid the absorption of levothyroxine with less dependence on the level of stomach acid and fewer interactions with other medications (57, 58).
A second example is the API ketamine, formulated with the antimicrobial benzethonium chloride as the sole excipient in an intravenous formulation. Benzethonium chloride has well-documented toxicity (59), and we find it to have multiple in vitro activities (Table 2). Ketalar has had substantial reports of ventricular arrhythmias (www.accessdata.fda. gov/drugsatfda_docs/label/2012/016812s039lbl. pdf; https://open.fda.gov/data/faers/). Such cardiac adverse drug reactions often reflect activity on the hERG ion channel (60). Whereas ketamine itself has no detectable hERG activity, we find that benzethonium chloride is a 0.5 μM hERG blocker. Although benzethonium chloride should only reach a concentration of 90 nM in the blood given its dosage with ketamine, this is within the factor of 30 safety window at which one may be concerned about adverse events. We caution that these are just examples of possible adverse effects and are inevitably muddied by several confounds, including those inherent in databases such as the FDA Adverse Event Reporting System (FAERS) (61). Disentangling the possible activities of these and other excipients from the drugs with which they are paired will demand further investigation; no definite conclusions should be drawn from this study.
This work retains several caveats. Perhaps the most important is the lack of systemic exposure for many excipients tested here, even those that have potent in vitro activities. The physical properties of these molecules often preclude gut absorption, or they are rapidly metabolized. Thus, despite potent in vitro activities (Tables 1 and 2), and even activities in cellular assays designed to model organ-level toxicities (Table 3), these excipients effectively remain inert in the body. Also, we emphasize that most excipients examined were inactive even in vitro, at least against the targets prioritized here; a majority of the excipients tested showed no binding, no functional activity, and no toxicity in the assays conducted. Finally, even when we observed on-target binding and cellular toxicity, these were not directly linked to whole-animal toxicity. What this study does is reveal excipient activity on biological targets associated with therapeutic and toxic side effects; it does not demonstrate that even the target active excipients lead to toxic effects. Still, knowing these target-based activities allows investigation of particular excipients for particular effects in a way rarely possible with whole-animal studies alone.
These cautions should not obscure the principal observations from this work: Drug excipients, classified as “inactive ingredients” and often present in large amounts in single-drug formulations, can have substantial activities on medically relevant targets. Excipient exposure may be especially high in populations, such as the elderly, that take multiple medicines together. With affinities that dip to the low- and mid-nanomolar, the in vitro activities of excipients overlap with those of therapeutic drugs (24). Although many excipients do not reach the general circulation, several do, as shown even in this limited, proof-of-concept study. Once they do so, they can have unplanned pharmacology of their own. Most excipients genuinely are inactive and will continue to play crucial roles in drug formulations; by contrast, those excipients that have relevant in vitro activities and that reach substantial systemic concentrations may merit further review, beyond the gross animal physiology previously under-taken in safety studies.
This work suggests a systematic method to identify such active “inactive ingredients,” including the detection of allergenic and immunogenic properties. Replacements are readily available for most excipients, including non-catechol antioxidants for propyl gallate, plant-based colorants for the aromatic azo dyes, and, in the case of thimerosal, simply replacing multidose formulations with single-dose vials (which do not, even now, have thimerosal in them), with relatively minor cost effect (62). However, many of the excipients studied here, such as propyl gallate, diethyl phthalate, butylparaben, and tartrazine (63), also occur as food and cosmetic additives, often in far larger amounts than in drugs; this too merits review.
Supplementary Material
ACKNOWLEDGMENTS
We thank P. Bouchard, Preclinical Safety, NIBR, for support and consultations throughout the project.
Funding: Supported by FDA-CERSI grant FD004979 (K.M.G. and B.K.S.), by NIH grants GM122481 (B.K.S.) and GM71896 (J.J.I.), and by the NIMH Psychoactive Drug Screening Program (B.L.R.).
Footnotes
Competing interests: None.
Data and materials availability: All compounds tested in this work are available from commercial vendors as well as from the authors.
SUPPLEMENTARY MATERIALS
science.sciencemag.org/content/369/6502/403/suppl/DC1
Materials and Methods
Figs. S1 to S15
Tables S1 to S8
References (67–93)
REFERENCES AND NOTES
- 1.Code of Federal Regulations Title 21 (21CFR700.11); www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=700.11.
- 2.Code of Federal Regulations Title 21 (21CFR81.30); www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=81.30.
- 3.Boffey PM, “Death of a dye?” New York Times Magazine, 29 February 1976, p. 9. [Google Scholar]
- 4.FDA, Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System: Guidance for Industry (2017); www.fda.gov/media/70963/download.
- 5.Maggiora G, Vogt M, Stumpfe D, Bajorath J, J. Med. Chem 57, 3186–3204 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Keiser MJ et al. , Nat. Biotechnol 25, 197–206 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Hert J, Keiser MJ, Irwin JJ, Oprea TI, Shoichet BK, J. Chem. Inf. Model 48, 755–765 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hopkins AL, Nat. Chem. Biol 4, 682–690 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Paolini GV, Shapland RHB, van Hoorn WP, Mason JS, Hopkins AL, Nat. Biotechnol 24, 805–815 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Besnard J et al. , Nature 492, 215–220 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bowes J et al. , Nat. Rev. Drug Discov 11, 909–922 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Lounkine E et al. , Nature 486, 361–367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keiser MJ et al. , Nature 462, 175–181 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gregori-Puigjané E et al. , Proc. Natl. Acad. Sci. U.S.A 109, 11178–11183 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irwin JJ et al. , Clin. Pharmacol. Ther 101, 320–323 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ, J. Mol. Biol 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 17.Karlin S, Altschul SF, Proc. Natl. Acad. Sci. U.S.A 87, 2264–2268 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pearson WR, J. Mol. Biol 276, 71–84 (1998). [DOI] [PubMed] [Google Scholar]
- 19.McGovern SL, Caselli E, Grigorieff N, Shoichet BK, J. Med. Chem 45, 1712–1722 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Feng BY et al. , J. Med. Chem 50, 2385–2390 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Irwin JJ et al. , J. Med. Chem 58, 7076–7087 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peters JU, J. Med. Chem 56, 8955–8971 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Mudie DM et al. , Mol. Pharm 11, 3039–3047 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Overington JP, Al-Lazikani B, Hopkins AL, Nat. Rev. Drug Discov 5, 993–996 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Kleinstreuer NC et al. , Nat. Biotechnol 32, 583–591 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Berg EL, Polokoff MA, O’Mahony A, Nguyen D, Li X, Int. J. Mol. Sci 16, 1008–1029 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shah F et al. , Cell Chem. Biol 24, 858–869.e5 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Hansen JF et al. , PLOS ONE 10, e0131168 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoshimura T et al. , Gen. Pharmacol 29, 633–638 (1997). [DOI] [PubMed] [Google Scholar]
- 30.Karling P et al. , PLOS ONE 6, e18035 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson-Holt N, Dart JK, Eye 3, 581–587 (1989). [DOI] [PubMed] [Google Scholar]
- 32.Merck & Co. Inc., package insert for Antivenin; www.merck.com/product/usa/pi_circulars/a/antivenin/antivenin_pi.pdf.
- 33.World Health Organization, Mercury in Skin Lightening Products (2019); www.who.int/publications/i/item/WHO-CED-PHE-EPE-19.13.
- 34.Counter SA, Buchanan LH, Toxicol. Appl. Pharmacol 198, 209–230 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Hviid A, Stellfeld M, Wohlfahrt J, Melbye M, JAMA 290, 1763–1766 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Parker SK, Schwartz B, Todd J, Pickering LK, Pediatrics 114, 793–804 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Bigham M, Copes R, Drug Saf 28, 89–101 (2005). [DOI] [PubMed] [Google Scholar]
- 38.FDA, Thimerosal and Vaccines (2018); www.fda.gov/BiologicsBloodVaccines/SafetyAvailability/VaccineSafety/UCM096228.
- 39.National Library of Medicine, Flucelvax Quadrivalent (Prefilled Syringe) label (2018); https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=6688ebee-44bd-4655-99d0-4a54549eb169&audience=consumer%20.
- 40.FDA, Fluzone Quadrivalent Southern Hemisphere—Package Insert (2017); www.fda.gov/media/119856/download.
- 41.Pichichero ME et al. , Pediatrics 121, e208–e214 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Burbacher TM et al. , Environ. Health Perspect 113, 1015–1021 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barregard L et al. , Toxicol. Sci 120, 499–506 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Smith DA, Waterbeemd H. d., Walker DK, in Pharmacokinetics and Metabolism in Drug Design, Mannhold R, Kubinyi H, Timmerman H, Eds. (Wiley, 2001), pp. 121–146. [Google Scholar]
- 45.Charlesworth CJ, Smit E, Lee DS, Alramadhan F, Odden MC, J. Gerontol. A 70, 989–995 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hatton GB, Madla CM, Rabbie SC, Basit AW, Drug Discov. Today 24, 417–427 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Maier L et al. , Nature 555, 623–628 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olczak M, Duszczyk M, Mierzejewski P, Meyza K, Majewska MD, Behav. Brain Res 223, 107–118 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Yorifuji T, Kado Y, Diez MH, Kishikawa T, Sanada S, Arch. Environ. Occup. Health 71, 170–177 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Emilien G, Maloteaux JM, Geurts M, Hoogenberg K, Cragg S, Pharmacol. Ther 84, 133–156 (1999). [DOI] [PubMed] [Google Scholar]
- 51.Ida-Eto M et al. , Brain Dev 35, 261–264 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Curtis B et al. , Environ. Health Perspect 123, 579–589 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gadad BS et al. , Proc. Natl. Acad. Sci. U.S.A 112, 12498–12503 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McMillan M et al. , Drugs R&D 16, 53–68 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi YH et al. , Thyroid 22, 1090–1090 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Institut Biochimique SA, package insert for Tirosint; www.accessdata.fda.gov/drugsatfda_docs/label/2007/022121lbl.pdf. [Google Scholar]
- 57.Ernst FR et al. , Drugs R&D 17, 103–115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seng Yue C, Benvenga S, Scarsi C, Loprete L, Ducharme MP, J. Pharm. Pharm. Sci 18, 844–855 (2015). [DOI] [PubMed] [Google Scholar]
- 59.3 Final Report on the Safety Assessment of Benzethonium Chloride and Methylbenzethonium Chloride. J. Am. Coll. Toxicol 4, 65–106 (1985). [Google Scholar]
- 60.Redfern WS et al. , Cardiovasc. Res 58, 32–45 (2003). [DOI] [PubMed] [Google Scholar]
- 61.Maciejewski M et al. , eLife 6, e25818 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.CDC Vaccine Price List (2019); www.cdc.gov/vaccines/programs/vfc/awardees/vaccine-management/price-list/index.html.
- 63.Elhkim MO et al. , Regul. Toxicol. Pharmacol 47, 308–316 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Rowe RC, Sheskey PJ, Quinn ME, Eds., Handbook of Pharmaceutical Excipients (Pharmaceutical Press, London, 2009). [Google Scholar]
- 65.Leucht S et al. , Lancet 382, 951–962 (2013). [DOI] [PubMed] [Google Scholar]
- 66.Kao ML et al. , Xenobiotica 42, 389–397 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.